��
��
Sinusoidal Endothelial Cells Prevent Rat Stellate Cell Activation and Promote Reversion to Quiescence
Abstract
Capillarization precedes hepatic fibrosis. We hypothesize that capillarization of sinusoidal endothelial cells (SEC) is permissive for hepatic stellate cell (HSC) activation and therefore permissive for fibrosis. We examined whether freshly isolated SECs prevent activation of HSCs and promote reversion to quiescence, and whether this effect was lost in capillarization. HSCs were cultured alone or co-cultured with differentiated or capillarized SECs. Results: Co-culture with freshly isolated SECs markedly decreased HSC activation after 3 days in culture, but co-culture with capillarized SEC had no effect. Inhibition of nitric oxide (NO) synthesis abolished SEC suppression of HSC activation. Activated HSCs reverted to quiescence when co-cultured with SEC plus vascular endothelial growth factor (VEGF) (that is, with SECs that maintained differentiation), but co-culture with capillarized SECs did not. Reversion of activated HSCs to quiescence in the presence of SECs plus VEGF was abolished by inhibition of NO synthesis. To establish whether there was indeed reversion, activated and quiescent HSCs were counted before and 3 days after adding freshly isolated SECs plus VEGF to activated HSCs, and proliferation was quantified in quiescent HSCs; the stoichiometry demonstrated reversion. Conclusion: Differentiated SECs prevent HSC activation and promote reversion of activated HSCs to quiescence through VEGF-stimulated NO production. Capillarized SECs do not promote HSC quiescence, because of loss of VEGF-stimulated NO production.
Activation of the quiescent hepatic stellate cell (HSC) to an activated, collagen-producing HSC is considered the pivotal event leading to fibrosis. The activated HSC is defined by a variety of phenotypical markers, such as expression of ��-smooth muscle actin (ASMA), type I collagen, tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), and increased F-actin stress fibers. In addition to HSC activation, another change that precedes fibrosis is capillarization, which is defined as the formation of an organized basement membrane in the space of Disse with characteristic changes in the sinusoidal endothelial cell (SEC) phenotype.1-3 Several phenotypical changes have been noted in the capillarized SEC, but the defining morphological change is the loss of the characteristic open fenestrae organized in sieve plates.1
Because capillarization precedes fibrosis,1,2,4,5 this raises the question of whether capillarization itself might predispose to activation of HSCs. A causal link between capillarization and activation of HSC is biologically plausible. Studies over the last 15 years have shown in other vascular beds that endothelial cells and neighboring mural cells (pericytes, vascular smooth muscle cells) maintain each other's phenotype.6-8
In the liver, the SEC phenotype is maintained by paracrine production of vascular endothelial growth factor (VEGF) by hepatocytes and HSCs and autocrine production of VEGF-stimulated nitric oxide (NO).9 The current study examines whether there is reciprocity, that is, whether SECs might maintain and promote the quiescent HSC phenotype, whether this effect is lost when SECs undergo capillarization, and what the mechanism is by which SECs promote the quiescent phenotype of HSC.
Materials and Methods
Reagents
Chemicals were obtained from the Sigma Chemical company (St. Louis, MO) unless stated otherwise.
Cell Isolation
SECs were isolated by collagenase perfusion, iodixanol density gradient centrifugation, and centrifugal elutriation as previously described.10 The average yield of SEC per rat was 88 �� 106 with greater than 98% viability and 97% or greater purity. SEC purity was assessed by fluorescent acetylated low-density lipoprotein followed by a peroxidase stain to reveal any contaminating Kupffer cells.
SEC Phenotype
SEC cultured for 3 days in the presence of VEGF or in the presence of freshly isolated, quiescent HSCs remain fenestrated for 3 days9 and are considered differentiated SECs, whereas SECs cultured alone for 3 days without VEGF lack fenestration,9 the definitive marker of a differentiated or noncapillarized SEC, and are considered capillarized. SECs isolated from thioacetamide-induced cirrhotic liver are also capillarized.
HSCs were isolated by collagenase/pronase digestion and Stractan density gradient centrifugation.11 The yield of HSC is typically between 4 �� 106 and 10 �� 106 with 95% or greater viability and 90% or greater purity; purity is assessed by vitamin A autofluorescence. Hepatocytes were isolated by collagenase perfusion, gravity sedimentation, and Percoll density gradient centrifugation. Viability and purity of hepatocytes is 80% or greater and 95% or greater, respectively; purity is assessed by morphology on light microscopy.
Co-culture Protocol
HSC were plated in the wells of 24-well plates at a density of 130,000/cm2. SECs (400,000 cells/cm2) or hepatocytes (105,000 cells/cm2) were plated in collagen-coated Transwell inserts with 3-��m pore size (Costar, Fisher scientific, Pittsburgh, PA). This allows cells to maintain contact through shared culture medium without mixing of cells. Transwells were added to the culture plates after the SECs or hepatocytes had adhered.
Alpha-Smooth Muscle Actin Staining
HSCs were cultured on glass coverslips and fixed with 10% formal-dehyde (EM Science, Gibbstown, NJ) for 20 minutes at room temperature. Cells were incubated with a mouse monoclonal anti-alpha smooth muscle (ASMA) (1:100), followed by a rabbit anti-mouse immunoglobulin G tetramethylrhodamine isothiocyanate conjugate (1:50, Santa Cruz Scientific, Santa Cruz, CA). Controls were stained with a mouse isotype control antibody, a nonspecific immunoglobulin G2a kappa immunoglobulin (Sigma), as a primary antibody. Slides were examined using a Nikon PCM-2000 confocal microscope with a Nikon Eclipse TE300 microscope with a plan Apo 60��/1.4 aperture oil immersion objective, 543-nm laser, and Simple PCI software from the C-Imaging series from Nikon/Compix Inc (Cranberry Township, PA). Values were obtained by counting the number of ASMA-positive cells in 15 randomly selected fields.
F-Actin Stress Fibers
HSC were grown on glass coverslips, fixed with 2% paraformaldehyde/0.1% glutaraldehyde (Electron Microscopy grade, Polysciences, Warrenton, PA) and incubated with 50 mM ammonium chloride for 5 minutes. Cells were permeabilized with 0.1% Triton X-100, blocked with 5% fetal bovine serum in phosphate-buffered saline, stained with Alexa Fluor-488 phalloidin (Molecular Probes, Eugene, OR), and counterstained with propidium iodide (Molecular Probes). Fluorescence was examined using a Nikon TE 300 Quantum inverted fluorescence microscope, with a plan Apo 60��/1.4 aperture oil immersion objective, a Hamamatsu Orca digital CCD camera (C4742-95-12), and Metamorph Meta imaging version 6.1 software (Universal Imaging Corporation, West Chester, PA). Fluorescence was quantified in 10 fields with exclusion of dead cell fluorescence and normalized to the number of HSCs counted per field.
Immunoblot Analysis
HSC were cultured in a six-well plate for 3 or 6 days. For detection of ASMA and type I collagen, cells were harvested in lysis buffer (20 mM Tris-HCL, pH 7.6, 20 mM NaF, 20 mM ��-glycerophosphate, 0.5 mM Na3VO4, 2.5 mM metabisulfite, 5 mM benzamidine, 1 mM ethylenediaminetetra-acetic acid, 0.5 mM ethylene glycol tetra-acetic acid, 300 mM NaCl, with 10% glycerol, protease inhibitor, and 1% triton X-100). An equal amount of the whole cell protein (100 ��g/lane) was separated by 4% to 12% NuPage Bis-Tris Gel (Invitrogen, Carlsbad, CA) under reducing conditions and transferred to nitrocellulose. Protein was detected by incubating with 1:10,000 monoclonal anti-ASMA and 1:5000 rabbit polyclonal anti-type I collagen (Rockland, Gilbertsville, PA), followed by incubation with a horseradish peroxidase conjugated secondary antibody (Santa Cruz) at 1:10,000. For detection of TIMP-1, cell culture medium was centrifuged at 16,800g for 15 minutes at 4��C. Supernatants were mixed with loading buffer (Invitrogen) and transferred to nitrocellulose. Protein was detected by incubating 1 ��g/mL mouse monoclonal anti-rat TIMP-1 (R & D System Inc, Minneapolis MN) followed by incubation with a horseradish peroxidase- conjugated secondary antibody (Santa Cruz) at 1:1000. The protein was visualized by a commercial chemiluminescent method using the Pierce ECL kit (Amersham Bioscience, Piscataway, NJ).
Nitric Oxide Assay
NO was determined as the sum of nitrite plus nitrates. Escherichia coli nitrate reductase and nicotinamide adenine dinucleotide phosphate, reduced form were used to convert nitrate to nitrite in culture medium, and nitrite was quantified according to the Griess reaction.12 Fifty microliters culture supernatant is mixed with equal volumes of 0.1% N-1-naphthylenediamine hydrochloride and 1% sulfanilamide in 5% H3PO4. After 5 minutes at room temperature, absorbance is measured at 540 nm on a Biorad 3550 microplate reader (Biorad, Hercules, CA).
Thioacetamide Model
Rats (240-260 g males, Sprague Dawley, Harlan, Indianapolis, IN) were treated with thioacetamide 200 mg/kg intraperitoneally three times weekly for 9 weeks.
Tracer Studies
HSCs were plated on glass coverslips in 24-well plates. On the day of isolation (day 0), HSCs were incubated with 5 ��M Vybrant carboxyfluorescin diacetate, succinimidyl ester (CFDA SE) cell tracer (Molecular Probes) for 15 minutes at 4��C. This dye binds irreversibly to intracellular amines, it cannot be transmitted to surrounding cells, and dye fluorescence per cell is reduced by half with each cell division. From day 0 to day 3, HSCs were cultured alone, and from day 3 to day 6 activated HSC were co-cultured with freshly isolated SECs in a Transwell insert with 40 ng/mL VEGF added to the medium.
The number of quiescent and activated HSC were counted in 15 high-power fields on day 3 and day 6. Compact cells with cytoplasmic lipid droplets were considered quiescent HSCs. At the conclusion of the experiment on day 6, cells were fixed with 2% paraformaldehyde. Vybrant CFDA fluorescence was examined using a Nikon TE 300 Quantum inverted fluorescence microscope with a plan Apo 60��/1.4 aperture oil immersion objective, a Hamamatsu Orca digital CCD camera (C4742-95-12), and Metamorph Meta imaging software version 6.1 software (Universal Imaging Corporation, West Chester, PA). Fluorescence was quantified in 10 fields with exclusion of dead cell fluorescence and normalized to the number of HSCs counted in each field.
Statistics
All data, expressed as mean �� standard error of the mean, were from at least three separate experiments. Groups were compared by analysis of variance (ANOVA) with a posteriori contrast by least significant difference; or by Student t test using the Microsoft Excel Analysis ToolPak (Microsoft, Redmond, WA). P < 0.05 was considered significant.
Results
Maintenance of Stellate Cell Phenotype
A much larger proportion of HSCs maintained quiescence when co-cultured with freshly isolated, differentiated SECs compared with either HSCs cultured alone, co-cultured with capillarized SECs, or co-cultured with hepatocytes (Figs. 1, ,2,2, ,6).6). When HSCs were cultured on plastic in homotypic culture, 70.5% �� 5.8% of cells expressed ASMA after 3 days as assessed by confocal microscopy. In contrast, only 29.6% �� 1.8% of HSCs co-cultured together with freshly isolated SEC expressed ASMA after 3 days (HSC cultured alone versus co-culture with freshly isolated SECs, P < 0.01). Previous studies have demonstrated that SECs cultured with quiescent HSCs for 3 days under these conditions maintain fenestrae in sieve plates.9 Two models were used to obtain capillarized SECs. As previously reported,9 SECs cultured alone without exogenous VEGF for 3 days lack fenestrate (Fig. 3). SECs isolated from thioacetamide-treated cirrhotic liver (n = 5) were examined for fenestrae: there was a complete lack of fenestrae and sieve plates in 15 electron microscopy (EM) pictures per liver (Fig. 3). When HSCs were co-cultured with capillarized SECs from in vitro capillarization or from cirrhotic rats (Fig. 3), 79.9% �� 4.0% and 81.2% �� 2.1%, respectively, of HSCs were ASMA positive on confocal microscopy after 3 days (Fig. 1). Similarly, 75.6% �� 5.3% of HSCs co-cultured with hepatocytes for 3 days were ASMA positive on confocal microscopy (Fig. 6).
��
Fig. 1
Alpha-Smooth muscle actin (ASMA) as a marker of HSC activation. This experiment examines whether differentiated or capillarized SECs can prevent stellate cell activation in vitro. ASMA expression by HSCs was examined by confocal microscopy after 3 days. In the left panel, HSCs were cultured alone (open bar) from day 0 (the day of isolation) until day 3, co-cultured from day 0 to day 3 with freshly isolated SECs (black bar), or with SECs allowed to capillarize in vitro over 3 days, which were then added to HSC for 3 days (diagonal hatched bar). In the right panel, from day 0 to day 3, HSCs were cultured alone (open bar) or with capillarized SECs freshly isolated from rats with thioacetamide-induced cirrhosis (vertical hatched bar) (n = 3-8).
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0002.jpg
Fig. 2
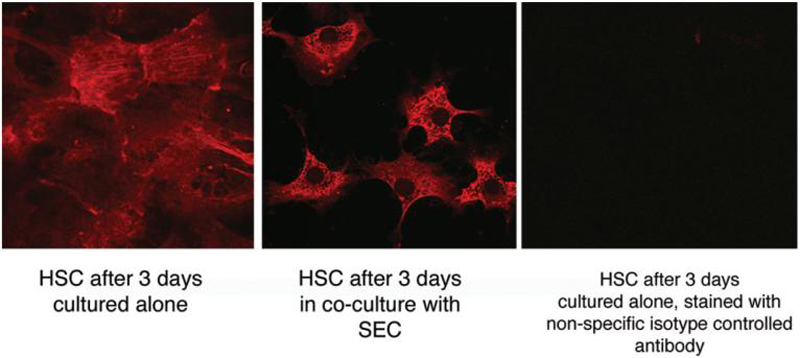
Confocal imaging of ASMA expression of HSCs cultured alone or with SECs for 3 days. The panel on the left shows HSCs cultured alone for 3 days. The panel in the middle shows HSCs cultured with SECs for 3 days. The panel on the right shows a control of HSCs cultured alone for 3 days stained with a nonspecific isotype-controlled primary antibody.
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0003.jpg
Fig. 3
Scanning electron microscopy of SECs. (A) Sinusoidal endothelial cells cultured for 1 day after isolation from normal liver demonstrate fenestrae in sieve plates. (B) Sinusoidal endothelial cells cultured alone for 3 days lack fenestrae. (C) Sinusoidal endothelial cells cultured for 1 day after isolation from thioacetamide-induced cirrhotic liver lack fenestrae.
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0006.jpg
Fig. 6
Regulation of HSC phenotype by nitric oxide. Left panel: The addition of 3 mM L-NAME blocked the paracrine ability of SECs to suppress HSC activation; from left to right: HSC day 3: HSCs cultured alone for 3 days; HSC/L-NAME: HSCs cultured with L-NAME for 3 days; HSC and SEC day 3: HSCs and SECs co-cultured for 3 days; HSC and SEC day 3/L-NAME: HSCs and SECs co-cultured for 3 days with the addition of L-NAME. Right panel: hepatocytes alone do not prevent HSC activation, but when 100 ��M V-PYRRO/NO is added, hepatocytes produce NO and mimic the ability of SEC to prevent HSC activation; from left to right: HSC day 3: HSCs cultured alone for 3 days; HSC and Hep day 3: HSC and hepatocytes co-cultured for 3 days; HSC day 3/V-PYRRO-NO: HSCs cultured alone for 3 days with V-PYRRO-NO; HSC and Hep day 3/V-PYRRO-NO: HSCs and hepatocytes co-cultured for 3 days with V-PYRRO-NO (n = 6).
The effect of co-culture with freshly isolated SECs on HSC activation was also examined using immunoblot for ASMA and TIMP-1 (Fig. 4) or by quantitation of fluorescence of F-actin stress fibers (Fig. 5). HSC expression of ASMA and of TIMP-1 was markedly reduced by co-culture with freshly isolated SECs compared with HSCs cultured alone, but co-culture with SECs obtained from thioacetamide-treated cirrhotic liver did not reduce HSC expression of ASMA or TIMP-1 (Fig. 4). Quantitation of F-actin stress fiber fluorescence demonstrated a 5-log difference when comparing HSCs cultured alone versus co-culture with freshly isolated SECs for 3 days (Fig. 5, P < 0.05). There was no difference in HSC stress fiber formation between HSCs cultured alone compared with HSCs co-cultured with SECs that had been allowed to capillarize in vitro.
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0004.jpg
Open in a separate window
Fig. 4
Immunoblot of HSC expression of ASMA and TIMP-1. This experiment examines whether differentiated or capillarized SECs can prevent stellate cell activation in vitro. Three conditions are examined: HSCs cultured alone, HSCs co-cultured with capillarized SECs, and HSCs co-cultured with differentiated SECs. In each condition, equal numbers of HSCs are plated. (A) Immunoblot for ASMA and TIMP-1. Lane 1: HSCs cultured alone from day 0 (day of HSC isolation) to day 3. Lane 2: HSCs cultured from day 0 to day 3 with SECs isolated from thioacetamide-treated cirrhotic rats, that is, SECs that were capillarized. Lane 3: HSCs cultured from day 0 to day 3 with SECs isolated from control liver, that is, differentiated SECs. Lanes 1 and 2 examine whether differentiated or capillarized SECs, respectively, can prevent HSC expression of activation markers on day 3. (B) Densitometry of ASMA corrected for ��-actin loading (n = 3). P < 0.0001 by ANOVA, P < 0.001 by least significant difference for HSCs co-cultured with normal SECs compared with HSCs alone (n = 3) (C) Densitometry of TIMP-1, corrected for ��-actin loading. P < 0.0001 by ANOVA, P < 0.001 by least significant difference for HSCs co-cultured with normal SECs compared with HSCs alone (n = 3).
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0005.jpg
Fig. 5
Fluorescence of F-actin stress as a marker of HSC activation. The panel on the left shows fluorescence per cell in HSCs cultured alone or with either capillarized SECs or freshly isolated SECs after 3 days in culture. The two panels on the right are photomicrographs of the F-actin stress fibers in HSCs co-cultured for 3 days with either capillarized or freshly isolated SECs (n = 3).
Paracrine Regulation of Stellate Cell Phenotype by Nitric Oxide
To determine whether nitric oxide (NO) contributes to maintenance of HSC phenotype, nitric oxide synthase was inhibited with 3 mM NG-nitro-L-arginine methyl ester (L-NAME). The number of ASMA-positive HSCs in co-culture with SECs in the presence and absence of L-NAME was 64.2% �� 2.1% versus 23.0% �� 2.3% (P < 0.0001). Thus, the addition of L-NAME blocked SEC suppression of HSC activation (Fig. 6), indicating that NO is an essential mediator of the SEC effect. Freshly isolated HSC produce very low amounts of NO, whereas SECs are the major hepatic source of NO13; thus L-NAME is acting on NO produced by SECs.
Hepatocytes did not prevent HSC activation (Fig. 6). However, when V-PYRRO/NO is added to hepatocytes, NO is liberated by P450 cleavage. When HSCs were co-cultured with hepatocytes in the presence of 100 ��M V-PYRRO/NO, that is, when hepatocytes produced NO, HSC activation was prevented (Fig. 6). V-PYRRO/NO in the absence of hepatocytes had no effect on HSC. This further supports the concept that NO plays a major role in suppressing HSC activation.
As described above, differentiated SEC promote quiescence in HSC, but capillarized SECs do not. SECs maintain a differentiated phenotype by day 3 when co-cultured with quiescent HSCs, whereas SECs cultured alone are capillarized by day 3.9 After 3 days in culture, NO in the medium of SECs co-cultured with quiescent HSCs is 40.1 �� 0.3 nmole/million SECs (9.4 �� 0.7 ��M) versus 30.1 �� 2.1 nmole/million cells (7.5 �� 0.5 ��M) from SECs cultured alone that have capillarized by day 3 (n = 3; P < 0.01). Thus, if the ability of differentiated, but not of capillarized, SECs to prevent HSC activation is due to NO, it is due to a difference in NO production of approximately 30%.
SECs Induce Reversal of Stellate Cell Phenotype
HSCs in homotypic culture were allowed to activate on plastic over 3 days. From day 3 to day 6, HSCs were cultured under four different conditions: cultured alone, cultured in the presence of VEGF, in co-culture with SECs freshly isolated on day 3 but without exogenous VEGF, or in co-culture with SECs freshly isolated on day 3 plus exogenous VEGF (Figs. 7--9).9). When HSCs were cultured alone for 3 days followed by 3 days of co-culture with freshly isolated SECs, the SECs were capillarized after 3 days as ascertained by surface expression of CD31 (data not shown); the use of CD31 as a marker of capillarization has been previously validated.9 In contrast, when VEGF is added, SECs do not capillarize.9 By day 6, HSCs co-cultured from day 3 to day 6 with freshly isolated SECs plus VEGF had significantly fewer ASMA-positive cells compared with HSCs cultured alone (Fig. 8; 38.3% �� 5.2% versus 88.7% �� 5.2% ASMA-positive cells; n = 4, P < 0.0001). In contrast, there was no significant decrease in the number of ASMA-positive HSCs on day 6 after culture from day 3 to day 6 with either SECs without VEGF or VEGF alone, when compared with HSCs cultured alone. These findings suggest that SECs that remain differentiated in the presence of VEGF induce reversal of the stellate cell phenotype. Of note, the addition of hepatocytes on day 3 did not reduce the number of ASMA-positive HSCs by day 6 (n = 3, data not shown). Morphology of HSC was also significantly different: HSCs cultured with SECs plus VEGF from day 3 to day 6 regained the appearance of quiescent HSC with compact cytoplasm and fat droplets (Fig. 8), whereas HSC cultured alone, co-cultured with SECs without VEGF, or with VEGF alone maintained F-actin stress fibers (Fig. 8).
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0007.jpg
Open in a separate window
Fig. 7
Reversal of HSC phenotype. In this experiment, HSCs are allowed to activate over 3 days (days 0-3) by culturing alone on plastic. From day 3 to day 6, HSCs are cultured either: alone, with SEC (which capillarize over the course of the next 3 days), with VEGF, or with VEGF plus SEC (that remain differentiated because of the presence of VEGF. (A) Immunoblot for ASMA and type I collagen. Lane 1 examines HSCs cultured alone for 6 days. Lanes 2-4 examine whether HSCs that are activated by day 3 can revert to quiescence by day 6. Lane 2, HSCs cultured alone from day 0 (the day of HSC isolation) to day 3 followed by co-culture from day 3 to day 6 with SECs isolated on day 3 (note: SECs will capillarize when cultured for 3 days with activated HSCs). Lane 3, HSCs cultured alone from day 0 to day 3, followed by the addition of VEGF from day 3 to day 6. Lane 4, HSCs cultured alone from day 0 to day 3, then from day 3 to day 6 with the addition of VEGF plus co-culture with SECs isolated on day 3 (note: SECs remain differentiated when cultured for 3 days with VEGF). (B) Densitometry of ASMA corrected for ��-actin loading. P < 0.0001 by ANOVA, P < 0.001 by least significant difference for HSC co-cultured with SECs plus VEGF compared with HSCs alone (n = 3). (C) Densitometry of type I collagen, corrected for ��-actin loading; P < 0.005 for ANOVA, P < 0.005 by least significant difference for HSCs co-cultured with SECs plus VEGF compared with HSCs alone (n = 3).
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0008.jpg
Fig. 8
SECs with VEGF induces reversal of stellate cell phenotype. The top panel shows the percentage of HSCs that express ASMA determined on confocal microscopy, and the lower panels demonstrate representative photomicrographs of HSCs. From left to right in the upper and lower panels: HSC day 6: HSCs cultured alone for 6 days; HSC day 0-6, SEC day 3-6: HSCs cultured alone from day 0 to day 3, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 (note: SECs cultured with activated HSCs for 3 days will capillarize); HSC day 0-6, VEGF day 3-6: HSCs cultured alone from day 0 to day 6, with the addition of VEGF to the medium from day 3 to day 6; HSC day 0-6, SEC + VEGF day 3-6: HSCs cultured alone for 3 days, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 plus VEGF (note: SECs cultured for 3 days with VEGF remain differentiated) (n = 4-6).
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0009.jpg
Open in a separate window
Fig. 9
Reversal of activated HSC to quiescence as demonstrated by loss of F-actin stress fiber fluorescence. Quantitation of F-actin fluorescence per cell demonstrates a marked drop in fluorescence when HSCs are in co-culture with SECs plus VEGF compared with the other conditions. From left to right: HSC day 6: HSCs cultured alone for 6 days; HSC day 0-6, SEC day 3-6: HSCs cultured alone from day 0 to day 3, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 (note: SECs cultured with activated HSCs for 3 days will capillarize); HSC day 0-6, VEGF day 3-6: HSCs cultured alone from day 0 to day 6, with the addition of VEGF to the medium from day 3 to day 6; HSC day 0-6, SEC & VEGF day 3-6: HSCs cultured alone for 3 days, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 plus VEGF (note: SECs cultured for 3 days with VEGF remain differentiated; n = 3).
The effect of SECs plus VEGF on HSC reversal was also examined by immunoblot for ASMA and type I collagen expression (Fig. 7) and by detection of F-actin stress fibers in HSCs (Fig. 9). Co-culture with capillarized SECs or the addition of VEGF to HSCs did not reduce the expression of ASMA or type I collagen (Fig. 7A, lanes 2 and 3, and Fig. 7B, C) compared with HSCs cultured alone for 6 days (Fig. 7A lane 1, and Fig. 7B, C). In contrast, there was a significant reduction in ASMA and type I collagen in HSCs co-cultured with SEC plus VEGF from day 3 to day 6 (Fig. 7A, lane 4, and Fig. 7B, C) There was a 7-log drop in F-actin stress fiber fluorescence in HSCs cultured from day 3 to day 6 with SECs plus VEGF compared with HSCs cultured alone (Fig. 9). There was no effect of co-culture with capillarized SECs or the addition of VEGF on HSC F-actin stress fiber expression (Fig. 9).
Role of Nitric Oxide in Reversal of the Activated HSC Phenotype
To determine whether NO plays a role in reversal of activated HSC to a quiescent phenotype, L-NAME was added to the co-culture system (Fig. 10). HSCs were cultured alone from day 0 to day 3 and then cultured with freshly isolated SECs and VEGF with or without L-NAME. L-NAME abolished the reduction in ASMA-positive HSCs seen in the co-culture of SECs with VEGF. The stability of L-NAME over 3 days was confirmed by dissolving L-NAME in culture medium, leaving it in an incubator for 3 days, and then comparing the ability to suppress NO production with that of freshly dissolved L-NAME; L-NAME activity was stable for 3 days (data not shown).
An external file that holds a picture, illustration, etc.
Object name is nihms-88899-f0010.jpg
Open in a separate window
Fig. 10
Effect of L-NAME on ASMA-positive HSCs in the presence of SECs and VEGF. From left to right: HSC day 6: HSCs cultured alone for 6 days; HSC day 0-6, SEC day 3-6: HSCs cultured alone from day 0 to day 3, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 (note: SECs cultured with activated HSCs for 3 days will capillarize); HSC day 0-6, SEC and VEGF day 3-6: HSCs cultured alone for 3 days, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 plus VEGF (note: SECs cultured for 3 days with VEGF remain differentiated); HSC day 0-6, SEC, VEGF, and L-NAME day 3-6: HSCs cultured alone for 3 days, followed by co-culture from day 3 to day 6 with SECs isolated on day 3 plus VEGF. The addition of L-NAME to the co-culture with SECs and VEGF blocks nitric oxide synthase and prevents the decrease in ASMA-positive HSCs (n = 6).
As described, freshly isolated SECs added to HSCs on day 3 were capillarized by day 6 unless VEGF was added. NO concentration was 32.1 �� 0.6 nmol/million cell (7.1 �� 0.9 ��M) on day 6 in the medium after co-culture of HSCs with SECs without exogenous VEGF from day 3 to day 6 and was 48.8 �� 3.2 nmol/million cells (10.0 �� 2.1 ��M) on day 6 in the medium after co-culture of HSC with SEC plus VEGF from days 3 to 6 (n = 3; P < 0.01).
Reversal of Stellate Cell Phenotype
Studies were done to determine whether the decrease in activated HSCs on day 6 after co-culture from day 3 to day 6 with SECs plus VEGF was indeed attributable reversion to a quiescent phenotype (Table 1). There were 7 times more quiescent HSCs per high-power field on day 6 than on day 3 and less than half the number of activated HSC. There are two possible explanations for the increase in quiescent HSCs and the decrease in activated HSCs. One possibility is that activated HSCs reverted to a quiescent phenotype. The second possibility is that half the activated HSCs underwent apoptosis (half the number of activated HSCs were present on day 6) and that the quiescent HSCs proliferated. The average number of quiescent HSCs per high-power field increased from 2.5 on day 3 to 17.5 on day 6. According to the formula, doublings = log(final number/starting number)/log 2 = log (17.5/2.5)/log 2 = log7/log 2 = 2.8. Thus, the quiescent HSCs would have to undergo an average of 2.8 doublings to account for the number of quiescent HSC found on day 6.
Table 1
Quantitation of Quiescent and Activated HSCs
Average Cell No/HPF Quiescent HSC Activated HSC Total
HSC day 3 2.5 �� 0.1 10.2 �� 0.2 12.8 �� 0.3
HSC day 6/SEC day 3/VEGF 17.5 �� 1.4 4.4 �� 0.3 21.7 �� 1.6
HSCs were cultured alone on plastic from day 0 to day 3. From day 3 to day 6, HSCs were co-cultured with SECs isolated on day 3 plus VEGF. HSCs were counted on day 3 and day 6 in 15 randomly selected high-power fields (HPF). Quiescent HSCs were considered compact cells with cytoplasmic lipid droplets (n = 3).
To test these two possibilities, cell division of quiescent HSCs was tracked with a fluorescent dye. The dye fluorescence on day 3 in quiescent HSCs was the same as on day 0, indicating that none of the quiescent HSCs had divided by day 3. On day 6, half of the quiescent HSCs had undergone two doublings, and half of the quiescent HSCs present had never divided, for an average of 1 doubling, that is, less than the average 2.8 doublings needed if the explanation was apoptosis and proliferation of the quiescent HSCs present on day 3. Thus, reversion of activated HSCs best accounts for the findings.
Discussion
The data presented here demonstrate that SECs prevent HSC activation and also promote reversion of activated HSCs to a quiescent phenotype, whereas capillarized SECs lose this effect. Coupled with the in vivo observation in humans and in animal models that capillarization precedes fibrosis,1,2,4,5 the findings in this study suggest that capillarization of sinusoidal endothelial cells may be permissive for HSC activation and fibrosis. The current study examined whether the decrease in the number of activated HSCs and the increase in quiescent HSCs in the presence of SECs with VEGF was indeed attributable to reversion of activated HSCs to the quiescent phenotype. Enumeration of quiescent and activated HSCs and quantitation of proliferation of quiescent HSCs showed that only reversion of activated HSCs could account for the stoichiometry.
NO production by SECs was necessary to maintain the quiescent HSC phenotype. A study by Langer et al.14 examined whether NO generated by SECs might induce apoptosis of activated HSCs.14 HSC apoptosis was not significantly increased over background at 5 ��MNOdelivered by an exogenous NO donor, but was induced in a dose-dependent fashion with NO concentrations ranging from 50 to 500 ��M.14 In the current study, NO derived from SECs co-cultured for 3 days with quiescent HSCs or cultured with VEGF was 9.5 and 10 ��M, respectively. These NO concentrations are within the same order of magnitude as those found in vivo in the rat hepatic vein13 and 5-fold to 50-fold lower than the concentrations that induced HSC apoptosis.14
In normal liver, paracrine production of VEGF by hepatocytes and HSCs stimulates NO by SECs, and VEGF-stimulated NO production by SECs is essential for maintenance of differentiated SECs.9 In capillarization before cirrhosis, paracrine production of VEGF is markedly decreased, VEGF stimulation of NO is lost, and only basal NO production persists.15 Capillarized SEC did not maintain a quiescent HSC phenotype and did not promote reversion of activated HSC to a quiescent phenotype in co-culture. NO concentration in the medium of capillarized SECs was 7 to 7.5 ��M, that is, 25% to 30% lower than NO production by differentiated SECs either cultured with VEGF or co-cultured with a cellular source of VEGF, and this reflects the decrease in NO production when VEGF stimulation is lost.9 Thus, the current study suggests that VEGF-stimulated NO production is also necessary for preservation of the quiescent HSC pheno-type. Another change in capillarized SECs that promotes HSC activation is expression of fibronectin containing an extra type III domain (EIIIA fibronectin).16
Interdependence in maintenance of phenotype between a differentiated SEC and a quiescent HSC is consistent with studies from Dr. Eli Keshet's laboratory. In these studies, a transgenic system is used for conditional switching of a soluble VEGF receptor in the liver. Secretion of the soluble VEGF receptor blocks VEGF, which leads to loss of SEC fenestration and a marked increase in ASMA-positive stellate cells; these changes are fully reversible with lifting of the VEGF blockade (E. Keshet, personal communication).
A variant of capillarization called pseudocapillarization is seen with aging.17,18 Future studies will need to examine whether aging-related capillarization causes similar changes to disease-related capillarization. If so, this may explain why age is a risk factor for fibrosis in hepatitis C19 and in nonalcoholic fatty liver disease.20-22
In summary, the data presented here demonstrate that differentiated SECs prevent HSC activation and promote reversion of activated HSC to quiescence. The paracrine effect of differentiated SECs on HSC phenotype requires NO production by the SEC. Capillarized SECs lose the ability to maintain quiescence of HSC and to promote reversion and this may be attributable to the decline in NO production that occurs with loss of responsiveness to VEGF. One implication of these findings is that approaches to reverse capillarization might promote resolution of fibrosis.
��
Hepatology. Author manuscript; available in PMC 2009 Sep 1.
Author information Copyright and License information Disclaimer
University of Southern California (USC) Keck School of Medicine Division of Gastrointestinal and Liver Diseases and the USC Research Center for Liver Diseases, Los Angeles, CA
corresponding authorCorresponding author.
Address reprint requests to: Laurie D. DeLeve, M.D., Ph.D., USC Keck School of Medicine, 2011 Zonal Avenue, HMR 603, Los Angeles, CA 90033. E-mail: ude.csu@eveled; fax: 323-442-3238.
Sinusoidal Endothelial Cells Prevent Rat Stellate Cell Activation and Promote Reversion to Quiescence https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2695448/
��
Journal of Hepatology
Volume 66, Issue 1, January 2017, Pages 212-227
Journal of Hepatology
Review
Liver sinusoidal endothelial cells: Physiology and role in liver diseases
Author links open overlay panelJohannePoisson12†SaraLemoinne34†ChantalBoulanger12FrançoisDurand567RichardMoreau567DominiqueValla567Pierre-EmmanuelRautou12567
Show more
https://doi.org/10.1016/j.jhep.2016.07.009Get rights and content
Summary
Liver sinusoidal endothelial cells (LSECs) are highly specialized endothelial cells representing the interface between blood cells on the one side and hepatocytes and hepatic stellate cells on the other side. LSECs represent a permeable barrier. Indeed, the association of ��fenestrae��, absence of diaphragm and lack of basement membrane make them the most permeable endothelial cells of the mammalian body. They also have the highest endocytosis capacity of human cells. In physiological conditions, LSECs regulate hepatic vascular tone contributing to the maintenance of a low portal pressure despite the major changes in hepatic blood flow occurring during digestion. LSECs maintain hepatic stellate cell quiescence, thus inhibiting intrahepatic vasoconstriction and fibrosis development. In pathological conditions, LSECs play a key role in the initiation and progression of chronic liver diseases. Indeed, they become capillarized and lose their protective properties, and they promote angiogenesis and vasoconstriction. LSECs are implicated in liver regeneration following acute liver injury or partial hepatectomy since they renew from LSECs and/or LSEC progenitors, they sense changes in shear stress resulting from surgery, and they interact with platelets and inflammatory cells. LSECs also play a role in hepatocellular carcinoma development and progression, in ageing, and in liver lesions related to inflammation and infection. This review also presents a detailed analysis of the technical aspects relevant for LSEC analysis including the markers these cells express, the available cell lines and the transgenic mouse models. Finally, this review provides an overview of the strategies available for a specific targeting of LSECs.
Previous article in issueNext article in issue
Keywords
Liver sinusoidal endothelial cellsCapillarizationEndothelial dysfunctionCirrhosisLiver regenerationAngiogenesisDrug delivery systemEndothelium
Liver sinusoidal endothelial cells: Physiology and role in liver diseases - ScienceDirect https://www.sciencedirect.com/science/article/pii/S0168827816303336
��
Liver sinusoidal endothelial cells: Physiology and role in liver diseases
Author links open overlay panelJohannePoisson12†SaraLemoinne34†ChantalBoulanger12FrançoisDurand567RichardMoreau567DominiqueValla567Pierre-EmmanuelRautou12567
1
INSERM, UMR-970, Paris Cardiovascular Research Center �C PARCC, Paris, France
2
Universit�� Paris Descartes, Sorbonne Paris Cit��, Paris, France
3
INSERM, UMRS 938, Centre de Recherche Saint-Antoine, Universit�� Pierre et Marie Curie Paris 6, Paris, France
4
Service d��h��patologie, Hôpital Saint-Antoine, APHP, Paris, France
5
Service d��h��patologie, DHU Unity Hôpital Beaujon, APHP, Clichy, France
6
INSERM, UMR-1149, Centre de Recherche sur l��inflammation, Paris-Clichy, France
7
Universit�� Denis Diderot-Paris 7, Sorbonne Paris Cit��, 75018 Paris, France
Received 24 May 2016, Revised 5 July 2016, Accepted 7 July 2016, Available online 14 July 2016.
crossmark-logo
Show less
https://doi.org/10.1016/j.jhep.2016.07.009Get rights and content
Summary
Liver sinusoidal endothelial cells (LSECs) are highly specialized endothelial cells representing the interface between blood cells on the one side and hepatocytes and hepatic stellate cells on the other side. LSECs represent a permeable barrier. Indeed, the association of ��fenestrae��, absence of diaphragm and lack of basement membrane make them the most permeable endothelial cells of the mammalian body. They also have the highest endocytosis capacity of human cells. In physiological conditions, LSECs regulate hepatic vascular tone contributing to the maintenance of a low portal pressure despite the major changes in hepatic blood flow occurring during digestion. LSECs maintain hepatic stellate cell quiescence, thus inhibiting intrahepatic vasoconstriction and fibrosis development. In pathological conditions, LSECs play a key role in the initiation and progression of chronic liver diseases. Indeed, they become capillarized and lose their protective properties, and they promote angiogenesis and vasoconstriction. LSECs are implicated in liver regeneration following acute liver injury or partial hepatectomy since they renew from LSECs and/or LSEC progenitors, they sense changes in shear stress resulting from surgery, and they interact with platelets and inflammatory cells. LSECs also play a role in hepatocellular carcinoma development and progression, in ageing, and in liver lesions related to inflammation and infection. This review also presents a detailed analysis of the technical aspects relevant for LSEC analysis including the markers these cells express, the available cell lines and the transgenic mouse models. Finally, this review provides an overview of the strategies available for a specific targeting of LSECs.
Previous article in issueNext article in issue
Keywords
Liver sinusoidal endothelial cellsCapillarizationEndothelial dysfunctionCirrhosisLiver regenerationAngiogenesisDrug delivery systemEndothelium
Journal of Hepatology
Volume 66, Issue 1, January 2017, Pages 212-227
Introduction
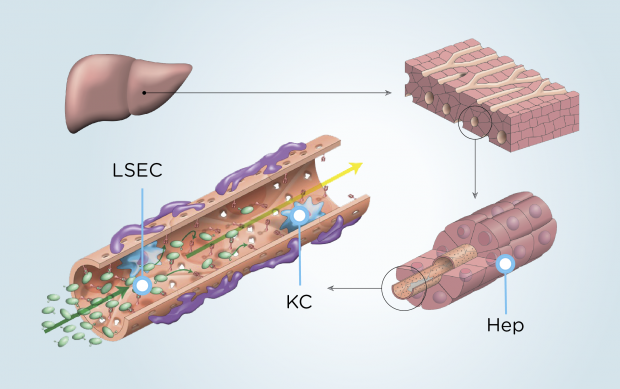
The vascular endothelium, representing the interface between blood and other tissues, is not only a physical barrier, but contributes to different physiological and pathological processes, including hemostasis/thrombosis, metabolites transportation, inflammation, angiogenesis and vascular tone [1]. Liver sinusoidal endothelial cells (LSECs) form the wall of the liver sinusoids and represent approximately 15 to 20% of liver cells but only 3% of the total liver volume [2]. LSECs are highly specialized endothelial cells. They have a discontinuous architecture meaning that fusion of the luminal and abluminal plasma membrane occurs at other sites than cell junctions, in areas called ��fenestrae��. This review focuses on the role of LSECs in physiological conditions and their involvement in liver diseases.
LSECs in the normal liver
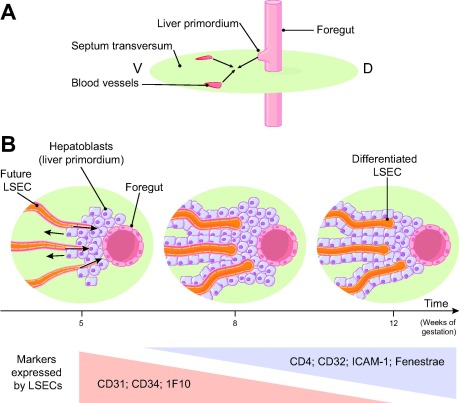
Formation of sinusoids during embryogenesis
As illustrated in Fig. 1, an early structural differentiation of hepatic sinusoids occurs between gestational weeks 5 and 12 in human embryos [3]. During that period, LSECs gradually loose cell markers of continuous endothelial cells including platelet endothelial adhesion molecule-1 (PECAM-1, also called cluster of differentiation (CD)31), CD34 and 1F10 antigen, and acquire markers of adult sinusoidal cells including CD4, CD32 and the intracellular adhesion molecule-1 (ICAM-1). This differentiation of LSECs is regulated by hepatoblasts, both via the vascular endothelial growth factor (VEGF) they release and via direct intercellular interactions [4], [5].
��
Fig. 1. Formation of sinusoids during human embryogenesis. (A) Frontal section of an embryo showing the formation of an outgrowth of the foregut (endoderm), called the liver primordium, which extends into the septum transversum (mesoderm), in which blood vessels are developing. V, ventral; D, dorsal. (B) Transversal section of the embryo showing the liver primordium (i.e., hepatoblasts arranged in thick cords separated by vascular spaces) growing into the septum transversum. The hepatic sinusoids are progressively established. First, the endothelial lining is continuous with a basement membrane (pink region) and no fenestrations. Around gestation week 12, fenestrations appear initially with diaphragms. These diaphragms disappear during development [15], [170], [171].
The embryological origin of LSECs is still a matter of debate. Initial observational studies described capillaries progressively surrounded by growing cords of hepatoblasts in the septum transversum, suggesting that LSECs derive from the septum transversum mesenchyme, a part of the mesoderm [3], [6], [7]. However, recent cell lineage experiments performed in mice showed that the septum transversum gives rise to mesothelial cells, hepatic stellate cells, portal fibroblasts, and perivascular mesenchymal cells, but not to LSECs [8]. A part of LSECs rather derives from a common progenitor to endothelial and blood cells, called the ��hemangioblast��, as attested by overlapping expression of hematopoietic and endothelial cell markers by LSECs and by fate tracing experiments [9], [10], [11], [12], [13], [14]. These progenitor cells form veins crossing the septum transversum, i.e., vitelin veins [15], umbilical veins or cardinal veins and then LSECs [16], [17]. Another part of LSECs derives from the endocardium of the sinus venosus, a compartment of the primitive cardiac tube [18]. These two embryological origins might explain the heterogeneity of the markers expressed by LSECs in adults.
LSECs renewal
Although specific data are lacking, we can speculate that in a physiological state LSECs are quiescent, i.e., with a low proliferation rate and a long life span, similar to endothelial cells from large vessels [19]. LSECs renewal differs in physiological and in pathological conditions. Three cell types contribute to LSEC renewal, namely mature LSECs, intrahepatic or resident sinusoidal endothelial cell progenitors, and bone marrow derived sinusoidal endothelial cell progenitors [20]. Mature LSECs can self-proliferate in normal conditions, when stimulated with growth factors such as VEGF and FGF (fibroblast growth factor) [20], [21]. Resident sinusoidal endothelial cell progenitors represent 1 to 7% of the LSECs of a normal rodent liver and probably contribute to LSECs regeneration [20]. Bone marrow derived sinusoidal endothelial cell progenitors do not participate in LSEC turnover in a normal liver [22]. By contrast, after liver injury, these cells are the main drivers of liver regeneration [20], [22]. Indeed, a subtoxic dose of monocrotalin, a toxic agent for LSECs, elicits liver injury only when bone marrow is suppressed. In addition, infusion of bone marrow cells after a toxic dose of monocrotalin almost fully corrects liver lesions [23].
Hepatic blood flow regulation
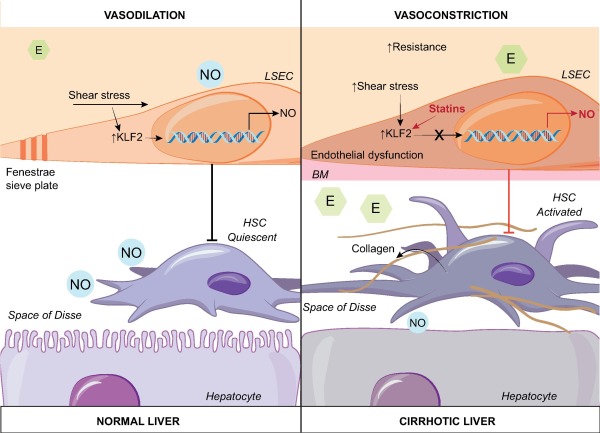
Liver sinusoids have a dual blood supply, receiving blood flow from the portal vein (70%) and the hepatic artery (30%) [24]. Blood pressure equalizes in the sinusoid and blood is then drained into the hepatic veins and the inferior vena cava. Despite major circadian changes in hepatic blood flow due to digestion, hepatic venous pressure gradient remains at 4 mmHg or less in a normal individual, attesting a fine regulation of hepatic vascular tone [25]. Intrahepatic shear stress is recognized as a main driver of hepatic blood flow regulation [26]. Shear stress is a frictional force applied by blood flow on endothelial surface [26]. It is proportional to flow intensity and to blood viscosity and inversely proportional to the cubic radius of the vessel [26]. Intrahepatic shear stress has never been directly measured in human or animal. Its evaluation is indeed difficult since the radius of sinusoids is very small and varies within the liver. Moreover, viscosity is hard to estimate in this specific area and also varies with hemodilution. In normal conditions, in the liver like in other vascular beds, the endothelium is able to generate vasodilator agents in response to increased shear stress in order to attenuate the increase in blood pressure. The loss of this property is called endothelial dysfunction. An endothelial specific transcription factor induced by prolonged shear stress, called Kruppel-like factor 2 (KLF2) mediates this effect of shear stress [27]. KLF2 induces the endothelial upregulation of vasodilating agents including nitric oxide (NO) [28] (Fig. 2). Shah and colleagues previously demonstrated that LSECs are the main source of NO in the normal liver through endothelial nitric oxide synthase (eNOS) activation by shear stress [29]. KLF2 also induces the downregulation of vasoconstrictive molecules including endothelin-1 [28]. Other molecules released by LSECs regulating blood flow include the vasodilating agent carbon monoxide (CO) and the metabolites of the cyclooxygenase (COX) pathway (thromboxane A2, Prostacyclin) [30]. All these molecules act in a paracrine manner on hepatic stellate cells localized in the space of Disse [31]. Healthy LSECs maintain hepatic stellate cell quiescence, thus inhibiting their vasoconstrictive effect [34]. The concept that hepatic stellate cell activation induces sinusoid constriction is based on their expression of molecules found in smooth muscle cells including ��SMA, on their position wrapped around the exterior of LSECs and on the ex vivo observation of their ability to contract [32], [33]. Although still controversial, LSEC could also regulate blood flow by swelling, thus creating an inlet and an outlet sphincter [32]. Kupffer cells possess contractile proteins as well, but their role in the regulation of hepatic blood flow remains controversial [32]. In contrast to most vascular beds where blood flow is mostly regulated by smooth muscle cells, in the liver, smooth muscle cells play a limited role since, although present in hepatic arterioles, they are only found in limited numbers in portal venules [32].
Key point
In a normal liver, differentiated LSECs are gatekeepers of fibrogenesis by maintaining hepatic stellate cells in their inactivated state. LSECs regulate sinusoidal blood flow through their action on hepatic stellate cells and thus maintain a low portal pressure.
Fig. 2. Role of liver sinusoidal cells (LSECs) in chronic liver diseases. In normal conditions, LSECs maintain hepatic stellate cell quiescence through a NO-dependent pathway as long as they are differentiated [101]. Exposure of LSECs to a physiological shear stress activates the transcriptor factor KLF2 leading to the release of vasodilating agents including nitric oxide (NO) and to the downregulation of vasoconstrictive molecules including endothelin-1. In a cirrhotic liver, LSECs become capillarized, meaning that they lose their fenestrae and a basement membrane appears. Capillarized LSECs permit hepatic stellate cell activation and thus production of collagen and of fibrosis. This change is associated with an endothelial dysfunction meaning that increased shear stress no longer leads to vasodilation but rather to vasoconstriction and thus to increased intrahepatic resistance. Simvastatin restores the vasoprotective effect of KLF2 and improves HSC phenotype through a NO-dependent pathway (the effect of simvastatin appears in red) [102]. BM, basement membrane; E, endothelin; HSC, hepatic stellate cell; KLF2, Kruppel-like factor 2; LSEC, liver sinusoidal cell; NO, nitric oxide.
LSECs, a selective barrier
LSECs are positioned at an interface. On their sinusoidal side, they are exposed to the highly oxygenated arterial blood mixed with the portal blood derived from the gut and the pancreas containing nutrients, bile acids, and hormones including insulin and glucagon. On the abluminal side, they interact with hepatic stellate cells and hepatocytes that are crucial for protein, lipid and glucose metabolism. LSECs thus represent a permeable barrier allowing exchanges but also active uptake and degradation of molecules [35].
Fluid exchange through fenestrae
Like endothelial cells located in other exchange territories, such as the glomeruli, the spleen and the bone marrow, LSECs are highly permeable [36]. The association of fenestrae, absence of diaphragm and lack of basement membrane make them the most permeable endothelial cells of the mammalian body [24]. These fenestrae are organized in clusters termed sieve plates [37]. LSEC fenestrae have a diameter ranging from 50 to 150 nm [2], [37], [38]. Their size and number varies depending on their localization in the liver, with larger but fewer fenestrae per sieve plate in the periportal region and smaller but more numerous fenestrae per sieve plate in the centrilobular region [37], [39]. This distribution could be related to the progressive decrease in oxygen tension along the lobule accompanied with an increasing need for oxygen exchange [36]. Alternatively, this distribution could be a marker of LSEC maturation as they spread along the lobule [39]. Fenestrae are not static structures. Their number and size varies in physiological conditions like fasting that decreases the number but increases the size of the fenestrae [40] and in pathological conditions [36], [39], [41], [42], [43]. Using super-resolution optical microscopy, Mönkemöller and colleagues recently showed that sieve plates are surrounded and separated by microtubuli and that each fenestrae within a sieve plate is surround by actin filaments [38]. Cytoskeleton is thus of great importance for the LSECS fenestrations. Fifteen years ago, LSEC fenestrations were thought to be sort of caveolae [44]. Caveolae are uncoated plasma membrane invaginations found in lipid-ordered domains of cell membranes called lipid rafts. Caveolin is a major structural protein of caveolae. Although caveolin-1 has been observed in LSECs fenestrations [44], caveolin-1 knockout mice have normal fenestrations [45]. In addition, Svistounov and colleagues [46], [47] described the ��sieve-raft crosstalk��, where fenestrations are formed in reduced lipid-raft regions of endothelial cells. Thus, fenestrations are not dependent on caveolin-1 and are different structures from caveolae.
In a normal liver, LSECs retain blood cells in the vessels, while molecules, such as metabolites, plasma proteins, pharmaceutical drugs, lipoproteins and small chylomicron remnants, viruses (<200 nm) and exosomes can access the space of Disse to be taken up by hepatocytes and hepatic stellate cells [2], [38], [48]. There is no significant osmotic and hydrostatic pressure gradient across the normal liver sinusoids [41], [49]. Small molecules and gasses freely diffuse through the fenestrae, so that the space of Disse contains a para-vascular part of the plasma volume. In addition, as blood cells squeeze into the sinusoids, they massage the endothelial cells and further mix plasma and space of Disse fluids [49]. Larger molecules, may also cross LSEC by a process called permselectivity or ��sieving��, namely the restricted transport of large molecules due to their deformation capacity through membrane pores [41]. The fluid present in the space of Disse is drained into hepatic lymphatics, then into hepatic hilum lymphatics, cisterna chili, thoracic duct and eventually the central venous circulation, successively [50]. The fluid formed in excess gains free access to the Glisson��s capsule on the liver surface [49]. Contrary to the mesentery, the liver is thus leaky to large molecules including proteins. This explains why ascites related to post-sinusoidal obstruction, such as cardiac failure or Budd-Chiari syndrome, is protein rich while ascites resulting from cirrhosis is not [50].
Key point
LSECs act as a selective barrier, since exchanges occur through fenestrae as well as by transcytosis and LSEC scavenging functions.
Endocytic capacity
LSECs have one of the highest endocytic capacity in the human body [51]. This property combined with a strong lysosomal activity give LSECs the ability to clear waste from the blood, as part of the ��dual-cell principle�� of waste clearance. This principle states that the mononuclear system represents the professional phagocyte, eliminating large particles, and that the scavenger endothelial cells, including LSECs, represents the professional pinocyte, clearing soluble macromolecules and small particles through endocytic receptors [52]. This property can be used to specifically target LSECs. LSEC endocytosis also contributes to the transfer of molecules from the sinusoids to the space of Disse, a process called transcytosis [35]. Endocytosis by LSECs implies different high affinity endocytosis receptors, including scavenger receptors (SR-A, SR-B and SR-H), mannose receptor and Fc gamma-receptor IIb2 [51], [52]. The SRs mediate endocytosis of polyanionic molecules, such as oxidized and acetylate low-density lipoproteins (oxLDL and acLDL), advanced glycation end products and waste products (hyaluronan, chondroitin sulfate or N-terminal propeptides of procollagen (I, III)). The main SRs of LSECs are SR-H/stabilin-1 and SR-H/stabilin-2. Stabilin1/2 double-knockout mice show only a mild liver fibrosis without liver dysfunction but a severe renal glomerular fibrosis [53], suggesting that stabilin-1 and 2 are major liver endocytic receptors implicated in the clearance of molecules toxic mainly for the kidney. The mannose receptors are not specific of LSECs and bind a wide range of glycoproteins and microbial glycans, such as collagen alpha chains (I, II, III, IV, V, XI), tissue plasminogen activator regulating fibrinolytic activity, and lysosomal enzymes that are recruited for further use in LSEC [54]. Thus, they have a role both in immunity and in glycoprotein homeostasis [52]. The Fc gamma-receptor IIb2 is the only Fc gamma-receptor expressed by LSECs and mediates the clearance of small circulating immune complexes; LSEC play a role in vascular immunity through this receptor [51], [52].
Technical aspects for the study of LSECs
Markers of LSEC
Key point
There is no unique specific marker of LSECs, apart from their fenestrae devoid of diaphragm in the absence of basement membrane. A combination of markers is thus mandatory for their identification.
Identification and isolation of LSECs is a major challenge for the understanding of liver physiology and diseases. However, technical barriers as well as a lack of consensual specific LSEC markers explain that LSECs populations differ between research groups, which limits the interpretation and the comparison of the results.
Features used to identify LSECs include: (a) their high and rapid endocytic capacity, using labeled formaldehyde-treated serum albumin, collagen alpha chains or acLDL. As other cells, including Kupffer cells, also have endocytic capacities, labeled molecules have to be incubated in small amount and for a short period of time to be specific for LSECs [55]. (b) Fenestrae without diaphragm and organized in sieve plates, using electron microscopy. Although this feature is the only one specific of LSECs, it has some limitations. First, the distribution of the fenestration varies along the lobule [37]. Second, LSEC isolation methods, including liver perfusion and cell preparation for electron microscopy, dilate fenestrae and might even create holes in cell surface [55]. Third, fenestrae rapidly disappear when LSEC are cultured as a monolayer of cells, out of their environment [56]. This loss of fenestration associated with basement membrane synthesis and modification of the expression of surface markers is called capillarization. Capillarization not only happens in cultured LSEC but also in vivo in most liver diseases [56]. (c) surface markers [24] (Table 1). Some markers are common to other endothelial cells and some to hematopoietic cells. No single marker is specific for LSECs and a combination is required. For instance, Ding and colleagues considered that LSEC are VEGFR3+ CD34− VEGFR2+ VE-Cadherin+ FactorVIII+ CD45−[57], while Lalor and colleagues selected CD31+, LYVE-1+, L-SIGN+, Stabilin-1+, CD34−, PROX-1− cells [56]. CD31, CD45 and CD33 deserve specific comments. CD31 (PECAM-1) is an intercellular adhesion molecule classically expressed at the surface of endothelial cells, but also of several leukocytes [58]. The expression of CD31 by LSECs is controversial. Several studies reported CD31 positivity of LSECs in liver slices analyzed by immunohistochemistry or in cultured cells permeabilized before staining [55]. Conversely, for the isolation of LSECs using flow cytometry, LSECs are considered as CD31 negative, CD31 positive cells being arterial and venous endothelial cells as well as capillarized LSECs. An electron microscopic analysis reconciled these results by showing that CD31 is located intracellularly shortly after establishing LSEC cultures, but, when fenestrae disappear few days later, CD31 becomes expressed at the cell surface like in other endothelial cells [59]. CD45 is a hematopoietic cell marker, expressed by leucocytes. LSECs are usually described as CD45−, and liver CD45+ cells are often considered as Kupffer cells. However, the reality may be more complex, as LSEC CD45 positivity appears to depend on the localization and the differentiation of LSECs [24], [39]: bright CD45 positivity is found in periportal area where LSECs have less fenestration, while CD45 negativity appears to predominate in centrilobular areas where LSECs are more differentiated with more fenestrae.
Table 1. Liver sinusoidal endothelial cell markers.
∗Also expressed by hepatic stellate cells and myofibroblasts; ∗∗correlates with fenestration and corresponds to SE-1 in rats [63]; $controversial [55].
AcLDL, acetylated low-density lipoprotein; Ag, antigen; CD, cluster of differentiation; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin; EC, endothelial cells; ICAM, intracellular adhesion molecule; L-SIGN, liver specific intercellular adhesion molecule-3 grabbing non-integrin; LDL, low-density lipoprotein; LYVE, lymphatic vessel endothelial hyaluronan acid receptor; MHC, major histocompatibility complex; R, receptor; SR, scavenger receptors; VAP, vascular adhesion protein-1; vWF, von Willebrand factor.
Knowledge of LSEC markers helps understanding some drug adverse effects. For instance, Mylotarg® (gemtuzumab ozogamicin), a drug used for acute myeloid leukemia, consists of a humanized antibody anti-CD33, linked to a potent antitumor antibiotic (calicheamicin). CD33 is expressed on the surface of acute myeloid leukemia cells, but also of LSECs likely explaining the high prevalence of hepatic sinusoidal obstruction syndrome following this treatment [60].
LSECs culture
Key point
When cultured, primary LSECs rapidly lose their specific phenotype. However, human and murine immortalized LSECs lines have been successfully developed.
As mentioned above, obtaining a pure culture of primary LSECs is challenging because of the lack of specific markers of these cells. LSEC isolation protocols are detailed elsewhere [61], [62]. The culture of LSECs has at least four particularities. First, cultured LSEC tend to lose their typical phenotype. In order to prevent this dedifferentiation, several methods have been developed. Co-culture with hepatocytes and fibroblasts rather than with hepatocytes alone allows LSECs to maintain their phenotype for up to 2 weeks [63]. Extracellular matrix coating mimicking the space of Disse and its modifications in pathology can also be used, e.g., low-density basement membrane-like matrix imitating normal conditions, and interstitial type matrix (fibril-forming collagen) imitating cirrhosis [63]. The addition of VEGF to the medium or the use of hepatocyte-conditioned medium can also prevent LSECs dedifferentiation [56], [64], [65]. Second, when cultured alone, LSECs undergo apoptosis within 2 days [63]; methods preventing dedifferentiation also prevent cell death. Third, serum supplementation is toxic for LSECs [55]. Fourth, in the normal liver, LSECs are exposed to an oxygen pressure decreasing along the liver lobule from 90 to 30 mmHg [66]; oxygen level is thus lower than in atmospheric conditions where oxygen pressure is 160 mmHg; actually, LSEC are particularly sensitive to hyperoxia and to the resulting oxidative stress [67]; survival of primary LSECs is improved under 5% oxygen instead of the commonly used 20% [51], [66].
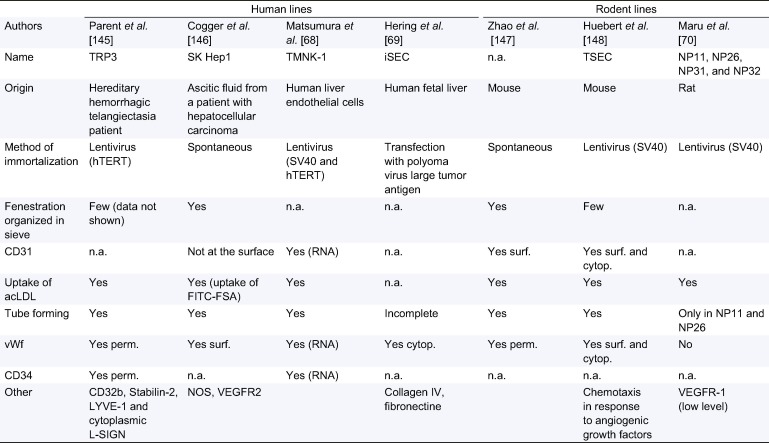
To overcome the difficulties of culturing primary LSECs, several teams have developed human and murine immortalized LSECs lines. However, the first immortalized lines, obtained by viral transfection such as M1LEC, had no fenestrae [68], [69], [70], [71], [72]. Subsequently, several humans and murine immortalized LSEC lines have been developed. As summarized in Table 2, these cell lines display many characteristics of LSEC. Each cell line has particular advantages making it more appropriate for specific studies. For instance, TSECs are adequate for angiogenesis analyses and Sk Hep1 for fenestration. However, the fact that these cell lines are immortalized implies that they may react differently from primary cells in response to stress. Therefore, a confirmation of the findings using primary cells is useful.
Table 2. Liver sinusoidal endothelial cell lines features.
acLDL, acetylate low-density lipoproteins; Cytop., cytoplasmic; L-SIGN, liver specific intercellular adhesion molecule-3 grabbing non-integrin; LYVE, lymphatic vessel endothelial hyaluronan acid receptor; FITC, fluorescein isothiocyanate; FSA, formaldehyde-treated serum albumin; n.a., not available; Perm., Permeabilized; Surf., Surface; Spont., Spontaneous; VEGFR, vascular endothelial growth factor receptor; vWF, Von Willebrand factor.
Mouse models
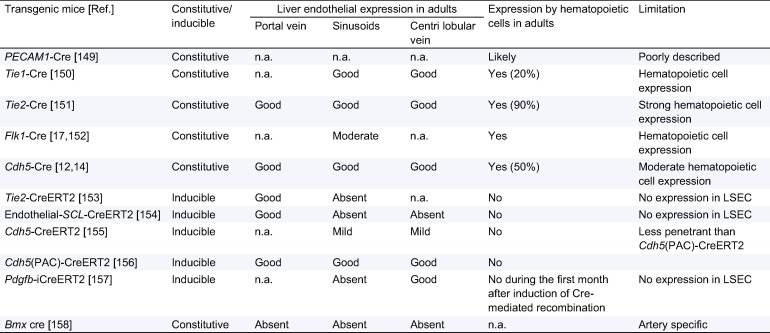
Transgenic mice using the Cre/Lox system can be very useful to study the properties of LSECs in vivo. Briefly, Cre-recombinase, which can be regulated by a tissue-specific promoter, excises essential loxP-flanked (����floxed����) genes via intrachromosomal recombination to generate so called conditional knockouts, i.e., knockouts specifically affecting tissues where the promoter is expressed. Several models with an endothelial cell expression of the Cre-recombinase have been developed and are summarized in Table 3. Mice with a constitutive expression of the Cre-recombinase appeared first. However, the expression of the Cre-recombinase is not restricted to endothelial cells, especially in adult mice, as recombination also occurs in hematopoietic cells. Indeed, early embryonic endothelial and hematopoietic cells arise from a common embryonic precursor called the hemangioblast [14]. This limitation can be overcome by performing a transplantation of wild-type bone marrow together with a clodronate mediated Kupffer cell depletion [73]. Indeed, in the absence of clodronate treatment, 2 months after bone marrow transplantation, 85% of the Kupffer cells are still derived from the recipient [74]. Myeloablation conditionings required for bone marrow transplantation might however alter LSEC function. Another way to overcome the concomitant expression of the Cre-recombinase in endothelial and hematopoietic cells is to use transgenic lines where Cre expression is induced in adult endothelial cells after tamoxifen administration. In that case, there is no expression of the transgene in hematopoietic cells.
Table 3. Characteristics of transgenic mice available to study the properties of liver endothelial cells in vivo.
Recombination was classified as good (>66%), moderate (33�C66%), mild (5�C33%); absent (<5%) based on data provided in the articles describing each model for all mouse lines but Tie2-Cre, Pdgfb-iCreERT2 and Cdh5 (PAC)-CreERT2. Indeed, these last 3 lines were independently and thoroughly analyzed and compared back to back using mT/mG reporter mice by the group of C James, Pessac, France (Kilani et al., unpublished). Cdh5-CreERT2 mice were also analyzed using mT/mG reporter by our group (unpublished data). Regarding LSEC expression, caution is needed since in all cases LacZ staining was performed without immunohistochemistry. Cells considered as LSEC were thus sinusoidal cells. They may be LSEC but also may be Kupffer cells.
LSEC, liver sinusoidal endothelial cell; n.a., information not available.
LSECs in liver diseases
Chronic liver diseases
LSECs play a key role in chronic liver disease initiation and progression, through four processes: sinusoid capillarization, angiogenesis, angiocrine signals and vasoconstriction.
Key point
The loss of the specific phenotype of LSECs, including the disappearance of the fenestrae, the development of a basement membrane, and the appearance of specific markers is called capillarization and is an early even in chronic liver injury. When capillarized, LSECs lose their capacity to inactivate hepatic stellate cells, thus promoting fibrogenesis and intrahepatic vasoconstriction.
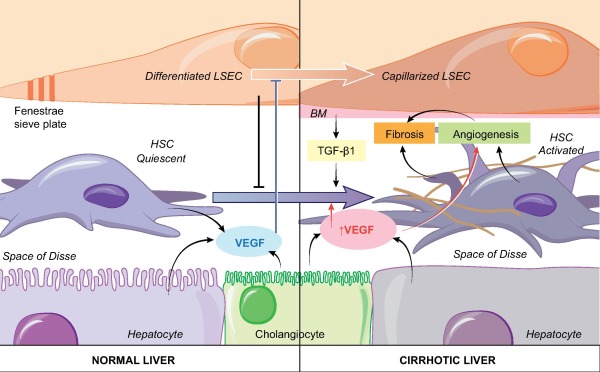
Capillarization of LSECs, also called dedifferentiation, occurs following liver injury in animal models as well as in patients [75], [76], [77], [78], [79], [80]. Capillarization is an early event since it precedes the activation of hepatic stellate cells and macrophages and the onset of liver fibrosis, suggesting that it could be a preliminary step necessary for fibrogenesis [76], [81], [82]. The mechanisms of capillarization and the cross talk between LSECs and hepatic stellate cells has been reviewed elsewhere [83]. Briefly, LSECs are able to maintain hepatic stellate cells quiescent as long as they are differentiated so that differentiated LSECs are gatekeepers of fibrosis [34], [84]. VEGF contributes to the maintenance of LSEC differentiation (Fig. 3). The role of LSECs in fibrosis regression is less clear. Indeed, in experimental models, restoration of LSEC differentiation in vivo promotes regression of mild fibrosis [34], [85]. However, immunohistochemical analysis of paired liver biopsies from 38 hepatitis C virus patients with cirrhosis, before and after antiviral treatment, revealed that sinusoid capillarization persists despite the regression of cirrhosis. LSEC differentiation is thus not crucial for fibrosis regression in this setting [86].
Fig. 3. A dual role of VEGF in chronic liver disease progression. In physiological conditions, VEGF released by hepatocytes, cholangiocytes and HSC, maintains LSEC differentiation (blue arrow) and consequently HSC quiescence. VEGF is thus anti-fibrogenic. During fibrogenesis, liver expression of VEGF increases. These high VEGF levels have a pro-fibrogenic action (red arrows) by inducing liver angiogenesis and by activating HSC. The activation of HSC results from a direct action of VEGF on HSC and from the release of TGF-��1 by capillarized LSECs. BM, basement membrane; HSC, hepatic stellate cell; LSEC, liver sinusoidal cell; VEGF, vascular growth factor; TGF-��1, transforming growth factor ��1.
Angiogenesis is defined by the development of new vessels from preexistent vessels [87]. Hepatic angiogenesis occurs during liver fibrogenesis and these two processes are closely linked [88], [89]. Liver fibrosis enhances angiogenesis and, in turn, liver angiogenesis aggravates liver fibrosis, as attested by the anti-fibrotic effect of most anti-angiogenic agents in animal models of liver fibrosis [90], [91]. However, analysis of the relationships between angiogenesis and fibrogenesis is not straightforward since most tools used to inhibit angiogenesis also act on fibrogenesis. For instance, VEGF, the master regulator of angiogenesis, is also implicated in fibrogenesis (Fig. 3) [87], [92], [93], [94], [95]. Besides LSECs, endothelial progenitor cell (EPC), i.e., endothelial cells derived from bone marrow, also contribute to liver angiogenesis, as reviewed elsewhere [96], [97].
LSECs also regulate fibrosis by releasing angiocrine signals. This latter term refers to the paracrine factors produced by endothelial cells that maintain organ homeostasis, balance the self-renewal and differentiation of stem cells and orchestrate organ regeneration and tumor growth. A recent landmark study demonstrated that LSECs release divergent angiocrine signals balancing liver regeneration and fibrosis. After acute liver injury, activation of CXCR7-Id1 pathway in LSECs stimulates production of hepatic-active angiocrine factors leading to liver regeneration. By contrast, chronic injury causes persistent FGFR1 activation in LSECs that perturbs CXCR7-Id1 pathway and favors a CXCR4-driven pro-fibrotic angiocrine response, thereby provoking liver fibrosis. Therefore, in response to injury, differentially primed LSECs deploy divergent angiocrine signals to balance liver regeneration and fibrosis [98].
Endothelial dysfunction occurs early in chronic liver disease, even before fibrosis and inflammation take place, and persists in advanced cirrhosis [84], [99], [100] (Fig. 2). The mechanisms of endothelial dysfunction have been reviewed elsewhere and are summarized in Fig. 2[83], [84]. Importantly, pharmacologic strategies improving LSECs in chronic liver diseases, including statins, decrease liver fibrosis, endothelial dysfunction and portal pressure [101], [103], [104].
Role of LSECs in hepatocellular carcinoma
Hepatocellular carcinoma (HCC) most often emerges in the context of chronic liver disease. The development of HCC is thought to be a multistep process from precancerous lesions (low then high grade dysplastic nodule) to early and advanced HCC [105]. Dysplastic nodules receive blood supply preferentially via the portal vein similarly to regenerative nodules of cirrhosis. A switch to prominent arterial blood supply occurs at the stage of early HCC [106]. Then, angiogenesis results in a highly vascularized tumor and promotes tumorigenesis and the development of metastasis. HCC is associated with changes in endothelial cells within and around the tumor.
Endothelial cells present within HCC sequentially lose during tumor progression LSECs markers, including stabilin-1, stabilin-2, LYVE-1 and CD32b, as observed both in murine HCC models and in human HCC [107]. Moreover, as compared to LSECs from a healthy human liver, endothelial cells derived from human HCC have a higher expression of integrins, lower expression of ICAM-1, and exhibit higher angiogenic, procoagulant and fibrinolytic capacities [108].
LSECs in the peritumoral tissue also undergo changes as HCC progresses including the loss of the LSEC markers stabilin-2 and CD32b [107]. In a mouse tumor xenograft model, peritumoral liver tissue displays a higher microvascular density and expression of the proangiogenic genes, interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) than the model tumoral tissue [109]. In the same line, peritumoral endothelial cells isolated from patients with HCC proliferate more when cultured with IL-6 and soluble IL-6R than tumoral endothelial cells. IL-6 is secreted by peritumoral endothelial cells in response to hypoxia while IL-6R is secreted by macrophages, present in large number in the peritumoral liver tissue during tumoral progression. These data suggesting a major role of peritumoral endothelial cells in HCC progression echo the previous observation that gene expression in the nontumoral liver from patients with HCC has a higher prognostic value of than gene expression in HCC [110].
LSEC and liver regeneration following acute liver injury or partial hepatectomy
Key point
LSECs are implicated in liver regeneration following acute liver injury or partial hepatectomy since they renew from LSECs and/or LSEC progenitors, they sense the shear stress changes resulting from surgery and interact with platelets and inflammatory cells.
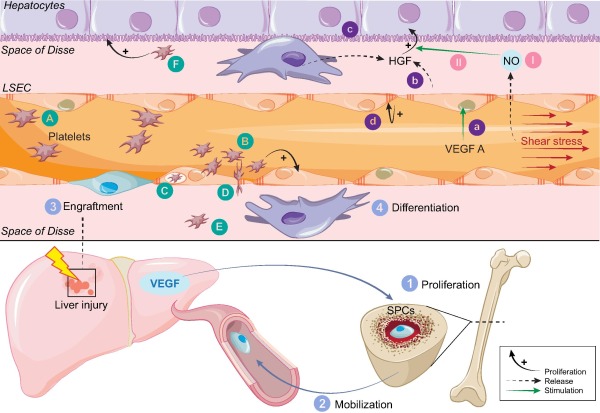
Liver regeneration following acute liver injury or partial hepatectomy is a complex process where LSECs play a key role. LSECs sense the major changes in shear stress resulting from resection. They proliferate, and orchestrate the harmonious regeneration of the different cell types by interacting with sinusoidal progenitor cells, platelets and inflammatory cells (Fig. 4).
Fig. 4. Liver sinusoidal cell (LSECs) and liver regeneration following acute liver injury or partial hepatectomy. Following liver injury, liver expression of VEGF increases, leading to the proliferation of bone marrow sinusoidal progenitor cells (SPC) (1), to their mobilization to the circulation (2), their engraftment in the sinusoids (3) and their differentiation in mature LSECs (4). VEGF A stimulates liver regeneration trough LSECs (a) leading to HGF production (b), hepatocyte proliferation (c) and LSECs proliferation (d) [20]. Increased shear stress associated with liver resection induces LSECs derived nitric oxide (NO) (I), which increase the effect of HGF on hepatocytes proliferation (II). Platelets are rapidly recruited in the liver after liver surgery (A). They adhere to LSECs and stimulate secretion of key molecules involved in hepatocytes (F) and LSECs (B) proliferation and survival. Platelets can also be endocytosed by LSECs (C), or trapped in the space of Disse (E), a migration facilitated by the increased size of fenestration associated with liver surgery (D). Abbreviations: HSC, hepatic stellate cell; NO, nitric oxide; LSEC, liver sinusoidal cell; HGF, hepatocyte growth factor; VEGF, vascular growth factor; VEGFR, vascular growth factor receptor; SPC, sinusoid progenitor cells.
After an acute liver injury or a partial hepatectomy, LSECs play a central role in liver regeneration through a dynamic regulation of the balance between hepatocytes proliferation and vascular proliferation. There is an asynchronism between hepatocyte and LSEC proliferation. In the early phase (at day 2), non-proliferative LSECs activate hepatocytes proliferation by two complementary mechanisms: (a) the downregulation of the hepatocyte growth inhibitor TGF-��, through the downregulation of the Tie2 receptor antagonist, angiopoiteine-2 [111]; and (b) the secretion of hepatotropic cytokines, Wnt and hepatocyte growth factor (HGF), through the upregulation of the transcription factor Id1 via the VEGFR2/VEGFA pathways [57]. Following liver resection, the portal flow per gram of tissue immediately increases, enhancing the shear stress on LSECs [112], [113]. In response to this increased shear stress, LSECs release NO that sensitizes hepatocytes to HGF [112], [114]. Shear stress is thus a key inducer of liver regeneration. However, when resection is excessive, exaggerated shear stress can damage LSECs and lead to hemorrhagic necrosis [112]. Limiting shear stress could be a potential strategy to prevent post-hepatectomy liver failure as suggested by the beneficial effect of portosystemic shunts, splenectomy or splenic artery embolization in murine models and in patients with large liver resections [112], [115], [116], [117], [118], [119], [120], [121]. A less invasive surgical intervention is being tested in a prospective trial (NCT02390713), using a pneumatic ring to modulate the diameter of the portal vein and thus the post-hepatectomy shear stress. New promising molecules decreasing shear stress to prevent post-hepatectomy liver failure and small-for-size-syndrome have been proposed including the vasodilator olprinone, a phosphodiesterase III inhibitor [122], [123] currently tested in a prospective trial (NCT00966745).
In the second phase following hepatectomy (at day 4), LSECs begin to proliferate, via the upregulation of angiopoietin-2 and VEGFR2/VEGFA pathways [111]. VEGFR2 is a classical mediator of the mitogenic and the angiogenic effect of VEGFA. The role of VEFGA/VEGFR1 pathway is more controversial than that of the VEGFR2 pathway. Le Couteur et al. described that VEGFR1 activation in LSECs after liver injury, can paracrinally induce hepatocyte proliferation, without LSEC proliferation and protects parenchymal cells from the injury [124].
Liver regeneration not only implicates liver cells but also circulating cells including sinusoidal progenitor cells, platelets and inflammatory cells. The role of sinusoidal progenitor cells in liver regeneration has been recently reviewed elsewhere and is summarized in Fig. 4[20]. Briefly, liver injury induces increased hepatic VEGF expression, which drives recruitment of hepatocyte growth factor-rich bone marrow sinusoidal progenitor cells and promotes expression of HGF by resident sinusoidal progenitor cells and LSECs. HGF in turn stimulates the proliferation of hepatocytes in liver regeneration. In addition, sinusoidal progenitor cells replace LSECs that were lost during injury. The role of the interaction between LSECs and platelets in liver regeneration is summarized in Fig. 4[125]. Following liver injury, platelets are recruited to and trapped within the liver, where they adhere to LSEC. Subsequent platelet activation results in the release of platelet granules, which stimulate hepatocyte proliferation. Platelets activate LSECs, leading to the secretion of growth factors, such as IL-6 [125]. Finally, LSECs and hepatocytes can also internalize platelets, but the effects of this alternate process on liver regeneration remain to be explored. The improvement in survival following subtotal liver resection in rats and mice obtained by the induction of thrombocytosis by thrombopoietin injection, splenectomy or platelet-rich plasma transfusion illustrates importance of platelets in liver regeneration [126], [127], [128]. The endothelial-monocyte interaction is also implicated in liver regeneration. Indeed, circulating monocytes are recruited in the injured liver and stimulate parenchymal but also endothelial regeneration. LSECs regulate the infiltration of monocyte in the liver through a destabilization of VE-Cadherin junction and through adhesive molecule expression [129].
Lastly, liver regeneration also depends on the existence of lesion related to ischemia reperfusion. Mechanisms of ischemia reperfusion injury have been reviewed previously and will not be detailed here [130].
Inflammation and infection
LSECs regulate liver inflammation in two manners. First, LSECs are a barrier separating the blood from the rest of liver, and thus restrict or enable the entry of circulating leucocytes into the liver tissue. The detailed mechanisms of the interactions between leucocytes and LSECs have been previously reviewed [131]. Briefly, LSECs express ICAM-1 and vascular adhesion protein-1 (VAP-1), allowing adhesion of leucocytes to the endothelium. During inflammation, expression of ICAM-1 increases and expression of vascular cell adhesion molecule-1 (VCAM-1) and CD31 are induced, leading to the transendothelial migration of leucocytes. Stabilin-1 has also been reported to promote transendothelial migration of leucocytes, preferentially regulatory T cells [132]. Second, LSECs can modulate lymphocytes behavior. In physiological conditions, antigen presentation by LSECs leads to tolerance induction in CD8+ cells [133]. LSECs can also induce differentiation of T cells into immunosuppressive regulatory T cells (Treg) that are functional in vitro and in vivo[134]. As an application, the selective delivery of autoantigen peptides to LSECs in vivo using a polymeric nanoparticle carrier can efficiently prevent and treat an animal model of autoimmunity, by increasing the number of Treg [135]. In inflammatory conditions, LSECs also tend to have an anti-inflammatory action since they increase the expression of the anti-inflammatory cytokine IL-10 in Th1 cells via the Notch pathway [136].
But LSECs can also be targeted by pathogens. Due to their scavenging ability, LSECs can capture circulating viruses via the expression of lectin at their surface and in turn induce infection of hepatocytes, as observed for hepatitis B and hepatitis C viruses [137], [138]. Lectin expressed by LSECs is not only involved in regulation of the entry of viruses but also in the regulation of their clearance by modulating functions of T cells as it has been shown for adenovirus [139]. LSECs can also be infected with CMV (cytomegalovirus) which upregulates ICAM-1 and CXCL10 expression, thus favoring CD4 T cell transendothelial migration. Migration of effector memory T cells through CMV-infected LSECs is associated with a change in memory T cells phenotype towards an activated phenotype facilitating hepatic inflammation, while regulatory T cells transmigrating retain a suppressive phenotype, favoring virus persistence [140]. This change in endothelial cells towards a proinflammatory phenotype induced by CMV might explain why acute CMV infection can trigger portal vein thrombosis [141]. The effect of CMV on LSECs and lymphocytes may also be of particular interest in the setting of liver transplantation where CMV infection may favor acute rejection, a disease characterized by endotheliitis.
LSECs can also be infected with bacteria as electron microscopy studies revealed Bartonella bacilli in LSECs associated with angiomatosis and peliosis hepatitis [142]. The fact that LSECs can be targets for pathogens with an impact on the local environment might explain why nodular regenerative hyperplasia develops in patients with primary hypogammaglobulinemia, a condition frequently associated with intra-sinusoidal lymphocytic infiltration. Immunodeficiency might favor infection of LSECs with pathogens, leading to a change in their phenotype towards a proinflammatory and prothrombotic phenotype and eventually to sinusoid obstruction [143].
LSEC and ageing
Pseudocapillarization refers to changes in the liver sinusoidal endothelium related with ageing. Electron microscopy analyses showed that ageing is associated with a 50% increase in the thickness of LSECs, a 50% reduction in the number of LSEC fenestrae and the formation of a basement membrane with perisinusoidal fibrosis and central vein fibrosis [42], [82]. These changes decrease porosity and endocytic capacity of LSECs. Consequently clearance of chylomicron remnants is impaired leading to post prandial triglyceridemia, which could participate to atherosclerosis development in older individuals [42], [51]. Moreover, these LSEC changes can induce hepatocytes hypoxemia decreasing oxidative drug metabolism and possibly promoting adverse drug reactions [42], [51], [144].
Conclusion
In conclusion, LSECs have a unique highly permeable phenotype allowing the passage of certain but not all molecules and cells. They also have a very special localization at the interface between blood cells on the one side and hepatocytes and hepatic stellate cells on the other side. LSECs are in constant interaction with other liver cells [83]. LSECs are implicated in most liver diseases including chronic liver disease initiation and progression, hepatocellular carcinoma development and progression, liver regeneration following acute liver injury or partial hepatectomy, liver ageing and liver lesions related to inflammation and infection. This role in most liver diseases makes them an attractive therapeutic target. Data summarized in Table 4 suggest a promising place to specific LSECs targeting. Such cell-specific approaches may limit the adverse effects associated with systemic drug delivery.
Table 4. Drug delivery system to LSEC in vivo.
∗ % of expression in liver cells; $Express as % of body distribution; £Express as % of injected dose.
Ab, antibody; Aco-HAS, cis-aconitylated human serum albumin; ALT, alanine aminotransferase; CCLs, lipid-coated cationic lipoplexes; Hep, hepatocytes; HA, hyaluronic acid; I/R, ischemia reperfusion; ICAM, intracellular adhesion molecule; KC, Kupffer cells; LSECs, liver sinusoidal endothelial cells; MEND, multifunctional type nano device; n.a., not available; NQ, not quantified; ODN, antisense oligodeoxynucleotides; PEG, polyethylene glycol; PLL-g-HA, hyaluronate-grafted poly(L-Lysine) copolymer; SA, stearylamine; SAPLs, stabilized antisense lipid particles; siRNA, small interfering RNA; STR-KLRG, sterylated killer cell lectin-like receptor subfamily G; Treg, regulatory T cells; YSK05, pH-sensitive cationic lipid [169].
Liver sinusoidal endothelial cells: Physiology and role in liver diseases - ScienceDirect https://www.sciencedirect.com/science/article/pii/S0168827816303336
��