��
�����Ƥϸ��������
Liver sinusoidal endothelial cells and liver regeneration
��
ժҪ
�����Ƥϸ����LSECs��������������Ϊ�����ڸ����˺�ĸ������������������У���ϸ�����ӣ�HGF������Ҫϸ����Դ�Ǹ���״ϸ�������ڸ����˺�HGF���ﱻ��Ϊ����ֳ��LSEC���������ӡ�Ȼ�����³��ֵ����ݱ�����ʹ�����˺�HGF��LSEC����Ҳ���������ӡ��෴������HGF��LSEC��BM SPC���Ĺ�����ϸ�������˺�ļ�������ࡣ�������Ӹ���������BM SPCs�����dz����LSEC�������������ĽǶ���������������
����
HGF��һ��ϸ�����ӣ�����֯�������̼�ϸ��������ϸ���˶�����̬��������ؼ����á� 20����ǰ��Jacquelyn Maher�״μ�������Ƥϸ����LSECs�������ڸ���������֤������״ϸ��������������HGF����Ҫ��Դ�����ڸ������˺�HGF������LSECs���������ӡ� ��1���������ʾbFGF�̼������е�LSEC��ֳ�����̼���ϸ����ֳ������bFGF����������LSEC��ֳ�������Ƹ�ϸ����ֳ���ý����bFGF��VEGF�ڲ��ָ��г��������Ӹ��������Ĺ۲����ϣ���bFGF��������������������Ƥϸ��ָ�����������ĸ���һ�£�2���� VEGF�����ⲻ�̼���ϸ����ֳ������ϸ�������Ƥϸ���������о��������о���֤��VEGFͨ��VEGFR1;���̼���ϸ������ֳ��3������Щ�о�������ͨ��VEGFR1;������ڳ�ϸ�������ĸ���Ƥϸ�����ϵ�HGF������3������ˣ���Щ�������VEGFͨ��VEGFR1�̼��ĸ�����Ƥϸ���ͷ�HGF�ٽ���ϸ����ֳ��3������������һƪ���ı�����VEGFR2�յ��ͻ�������С��ĸ���������������LSECs��Ѫ�ܷ������ӣ�����HGF���ı�����٣�4����Ȼ������������Ƥϸ��һ����LSEC����dz��ٵ�HGF��1,5�����෴��LSECs��BM SPCs���Ĺ�����ϸ�����������˺�Ͳ��ָ��г�����ļ�������ࣨ5��������HGF�� BM SPC��LSEC�Ĵ�С��ͬ�����������ǣ�5��;��ˣ�LSEC���뷽�������Իָ���ֲ�������е�BM SPCs�Լ�LSEC����ˣ������о����ܴ���ؽ���ֲ��BM SPCs�����Թ����ڳ����LSEC��Ŀǰ����LSECs���������������о������BM SPCsļ������������dz���LSECs�ƶ����������ļ�����һ�µģ�������LSEC��������������������費�ܽ��ͼ�����֪�Ĺ۲������������������м������ף���Ϊ�Ѿ�ļ���������BM SPCs�����dz����LSEC�Ǹ�����������Ҫ�������أ�ͼ11����
��
ͼ1
BM SPCs����������
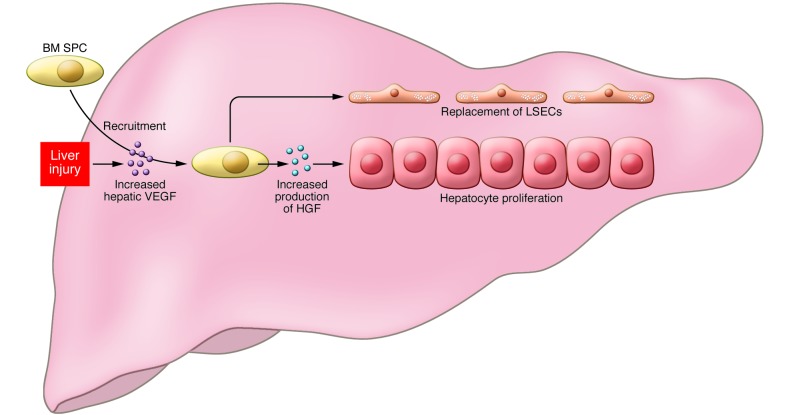
ʾ��ͼ����˸�ϸ����BM SPC��������ϸ�����Ը������Ĺ��ס�������(Liver injury)�յ�����VEGF�������ӣ�����������HGF��BM SPC��ļ��(Recruitment)��ͨ��פ����SPC��LSEC�ٽ�HGF�ı�� HGF�������̼��������и�ϸ������ֳ(Hepatocyte proliferation)�����⣬SPCȡ�����������ڼ䶪ʧ��LSEC��
LSEC��������
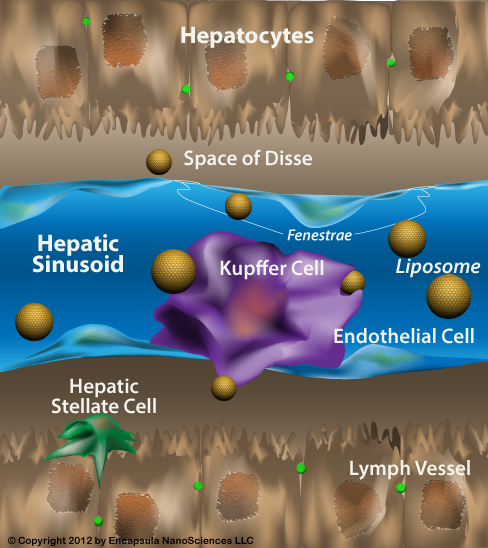
LSEC����̬ѧ�����϶��ص���Ƥϸ���� LSEC��Сϸ�����ڷ���ʱֱ��ԼΪ6.5��m����չ��һ��dz����ĸ���ڲ㡣 LSEC��Ψһ���Ǹ�Ĥ������ȱ������Ĥ���ϵIJ��鶯����Ƥϸ����������ɸ���оۼ���һ�𣬲��Ҵ��Ĵ�С��������LSEC���������ž�������Ҷ��СҶ��������ͬ��6-10���� LSEC�ڹ�����Ҳ�Ƕ�һ���ġ�����鵼���������õĸ���ΪLSEC�ṩ��һ�ָ����ʣ���������ϵͳ�����ѭ���еĽ���Ϳ����Է������ӣ�11,12�����������õ�������Ҫ�����Ǹ�¶�����壬����������Fc������IIb2��12���� LSEC��һЩ�ζ���ҩ��Ͷ������˵����Ŀ�꣨13-20������������ȱѪ�ٹ�ע���ˣ�21-24����
���Ѿ�����������Ѫ�ܴ��е���Ƥϸ������ϸ�����ο�����25-29��������౨���е�һЩ����LSEC�����ڵ���ϸ��������״ϸ����30,31��֮����ڴ��š�����״ϸ���Ļ������ά����������LSEC������ֹ����״ϸ���Ļ����ʹ��ĸ���״ϸ��ʧ�31���� LSEC�ڸ���ά��֮ǰ��չ���ı�ı��ͣ���ΪëϸѪ�ܻ���ëϸ�ܻ���LSECʧȥ��Ԥ������״ϸ�����ʹ��ĸ���״ϸ��ʧ����������ڸ���ά���Ĵ���ģ���У�LSEC��ëϸѪ�ܻ�����תʹ��ĸ���״ϸ��ʧ�������ά������ת���������ڽ��е������ڼ�ëϸѪ�ܻ�����ת��ֹ�˸�Ӳ���Ľ�չ��32�����ڽ����ĸ����У���ϸ������״ϸ��ͨ���ͷ�VEGFά��LSEC�ı��ͣ�30����
��
��������Ƥϸ��һ����LSEC����CD31��Ҳ��ΪPECAM����Ȼ������������Ƥϸ����ͬ������LSEC��CD31���������ϸ���ʣ�������ϸ�� - ϸ�����Ӵ���ϸ�����棨30������������Ƥϸ���γ������Աȵ��ǣ�LSEC������Ϊֹ������Ψһ����CD45��10,33,34������Ƥϸ����CD45��һ�ֵ��͵���Ѫϸ������ CD45���ž���LSEC�ϸ߱������ҶLSEC�ϱ������������СҶ����LSEC�����ڣ�10����
��ϸ��������Ϊ���ԳƷ��ѵ�ϸ��������һ����Ϊ��ϸ������ϸ����һ����Ϊ��ϸ�����ػ�ϸ������ϸ����ͼ2��.2�������ݶ��壬��ϸ���������Ҹ��²������ڵ����ӵ�������ͬ���͵�ϸ������ϸ����Ҳ��Ϊת������ϸ�����ȸ�ϸ��������ϵ�������ػ�ϸ��������ֻ�ܸ������������Ѿ���������������Ƥϸ����ϸ����SPC����BM SPC��פ�������SPC�� BM SPCs��������LSECת��û�й��ף��������˻ָ��г�����ļ����������5,33,35����פ��SPC�������Գ������ݶ���Ĺ��裨5����ļ��������ij�פSPCs��BM SPCs������LSECһ�𱻷��룬����Ⱦ��LSEC�����꣨5�������ݶ���פ��SPCs��Ƶ�ʴ������ŵı����ԣ���Sprague-Dawley���������LSEC��1����Fischer�����7�����ȣ�5�����ڸ����ڣ��Ѿ���������Ϊ��DZ���ϸ���������������ƶ���פ��LSEC��ϸ�������Ҹ��£���ϵ�����Էֻ�����������ֳ��ȷ������������δ�õ�֤ʵ�������һ������Ȼ�ܴ�����Ȼ��������פ��SPC�ı����־��ı�DZ���ϸ���Ĵ��ڱ�����פ����SPC��Դ�ڸ��ƶ���פ��LSEC��ϸ�����ر��ǿ��ǵ�פ����SPC�������Գ������ݶ���Ĺ��衣��Sprague-Dawley�����У�0.1����LSEC���ƶ���פ��LSEC��ϸ����5������������к�ϸ������Ѫ��ϸ����0.01��Ƶ����ȣ��ƶ��ij�פLSEC��ϸ����0.1��Ƶ�ʽϸߣ�36������Ȼ��פLSEC��ϸ���ͳ�פSPC������������������ת�в���LSEC�������ֹ�����δ�õ�֤ʵ��
��
��
ͼ2
��ϸ������ϸ����
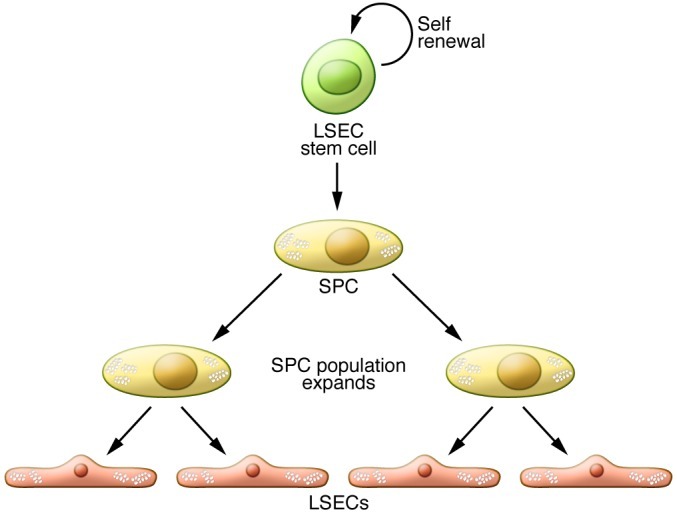
��ϸ�����Ҹ��²�������ϸ������ϸ����ת������ϸ���ȸ�ϸ��������ϵ�������������ϸ�����ػ�ϸ��������ֻ�ܸ�������������������SPCȺ����Բ���LSEC��BM SPC���������˻ָ��г�����ļ�������࣬�Լ�פ�������SPC����������������LSECת���� SPC��Դ�ڸ�ϸ����SPCȺ���ڲ�������LSEC֮ǰ����
��פ��BM SPC��Ϊ��ϸ�������CD133���ԣ�������LSECһ�������DZ�����Ƥϸ�������CD31����Ѫϸ�������CD45��5������פSPC��VEGFR1��VEGFR2�����ԡ���Ȼ��δ���BM SPC�е�VEGFR���������LSEC��VEGFR1��VEGFR2�����Եģ�BM SPCҲ������������פ��SPC���������LSEC�����ֵĿ���ģʽ���������е�SPC�Ƿǿ����ģ�����ļ�������ಢֲ�������γ�������LSEC����ģʽ��5����פ����BM SPC��CD45�Ĵ���ʹ�������ͨ�õ���Ƥ��ϸ����EPC��������������ݶ���CD45���ԡ�
�ڲ��ָ��г������ж������˺�BM SPC����ֳ���ӳ�������������BM SPC��ѭ���Ķ�Ա����2��4����5,35����������Դ��LSEC��������ʱ������ƶ����ӣ��ڲ��ָ��г������3���õ�LSEC 25�������Թ��裬���ж������ж����ʶ����������ĵ�5��͵�14���ֱ�40����70����LSEC�����Թ��裨5,35�����о��Ѿ��ų��˹�����Դϸ��ļ����LSEC�е��ںϣ�33����������ֽ����������ֲ������ǹ���ϸ��������LSEC���ںϡ� BM SPCs�ڸ�����ֲ���γ���ɸ������֯�Ĵ��ף�5,35�����ڲ��ָ��г�����ֲ������BM SPCs�ȳ�פSPCs��ֳ���࣬������LSEC���������ӵ�HGF����Ҫ��Դ��5����
��
��
BM SPCs�Ǹ�����������Ĺؼ�֤�����Թ�������ʵ�顣�ڴ����У����ؽڵ��β���������֫����Լ40�����ܹ��裬���Ҹ���������䵼�����ܰ�ϸ���������Ƽ���40����5��������Щ������Ĵ�����в��ָ��г���ʱ������������40�������ҵ�5���ϸ����ֳ��С�����������������ӳ١��ڲ��ָ��г������1�콫��פSPC�������ע��������Ĵ����������ٽ���ϸ����ֳ��ʹ�������Ļָ���ȫ���������ý������������BM SPC��ɥʧ�����������������������ڱ��о��У����ָ��г������5�����������������������ƺ�ϸ����ֳ��ȱ����ƣ�����LSEC��/��פSPC������HGF��Դ�����ֲ�BM SPC�̼�����������ʧ��ֵ��ע����ǣ��ڲ��ָ��г������3�죨�����ǵ�1�죩��ע��פSPC����輸��û���洦�������SPC��ע�����ڵڶ��ָ�ϸ����ֳ֮ǰ���������ڵ�2���ڴ�������ɡ� LSEC�����ڵ���ֳ�ڲ��ָ��г������1�����ӣ��ڵ�3��ﵽ��ֵ��5,37,38���������BM SPC����������ͨ���ٽ���ϸ����ֳ���̼��������������Ե�3���LSEC��/��SPC����ֳ����������Ѫ������������ġ������LSEC�Ƿ�Ҳ����ֳ�������Ƿ����е���ֳ��������פ����BM SPC���д�ȷ����
�����עפ����SPC�����ڹ����������ȸ���������Ԥ��פ����SPC���������ڸ�������Ȼ���������������������������������������ɱ���������פ����SPC���ܲ���BM SPC����ʧ��ע���ע��פSPCs�̼���ϸ����ֳ����ԭλפ��SPC�Ը�������û���������ף��������Щϸ��ֻҪ���ڸθ�ϸ����̬λ�оͻᱣ����Ծ�ֹ״̬��
��
BM SPCs�Ը����˻ָ��Ĺ���Ҳ���������ж��Ը���ģ���еõ�֤ʵ��33,35������Ұ�ٺϼ��յ������ۺ�����SOS;Ҳ��Ϊ�ξ��������Լ�����ģ�͵��о�������Ұ�ٺϼ���ƻ�LSECs��15,20,33,39����BM SPCs��33���������˵Ļָ��Σ�Լ25����LSEC���Թ��裬����BM SPC������LSEC��Ȼ���������Ұ��Ѻ�СҶ���ij�Ѫ�Ի����ĸ߶ȣ�Ұ�ٺϼ�ʹ������SPCs����������50������ʹѭ��BM SPCs����������95�����������������Ƕ��Լ�����Ұ�ٺϼ������SOS�����������ע��ȫ��ֹ�˸���һ������Ұ�ٺϼ�Ĵ����SOS��������������ص�SOS����ˣ�BM SPCs��LSEC������SOS�Ĺؼ��������ء����仰˵��SOS������LSEC�����˺�BM SPCȱ��������ϡ��������������֪��BM SPCs�Ը�������Ӱ�죬��SOS��BM SPC�������ٲ�������ֹ��¶���������ߵ�����ֳ����������������ĸ�ϸ����ֳ������ܽ���������Ѫϸ����ֲ��������ֲ��֮ǰ�Ӹ������軯�ƿ���SOS�Ļ��ߵĸ߲��������ʡ� BM SPCҲ���ж������Ķ�������������LSEC���ˣ�����BM SPC����ע�������������ˡ�
���ڹ���SPCs��ļ��
����VEGF��Ҳ��ΪVEGFA����Ӧ��������ʽ�ĸ����˺Ͳ��ָ��г��������ӣ�35,37,38,40-43�����÷���Ѻ������õ���VEGF��Ӿ�Ը���Ķ������ˣ�35��������עVEGF���Ը����ж������ˣ�44-46�������Ӹ�ϸ����ֳ��38���Ͳ��ָ��г��������������3,4�����ڶ������˺Ͳ��ָ��г�������VEGF�Ѿ���ʾ������BM SPC�����ļ����ÿ�����裺BM SPC����ֳ��BM SPC��ѭ���Ķ�Ա��BM SPC�ڸ����е�ֲ���Լ�BM�ķֻ��� SPCs���п�LSECs�����������ߣ�35����ͼ��ͼ3��.3������ˣ�����VEGF��BM SPCļ����������ڼ������ҶԸ�������������Ҫ�������յ���VEGFR2ȱ�ݵ�Vegfr2flox / floxС���ڲ��ָ��г����������������4�������ֿ��ܵļ���һ�£�����VEGF��BM SPCsļ��������Ӷ��ٽ�����������������ЩС��ȱ��VEGF- BM SPCs�̼�HGF�ͷţ�3���������ģ���
ͼ3
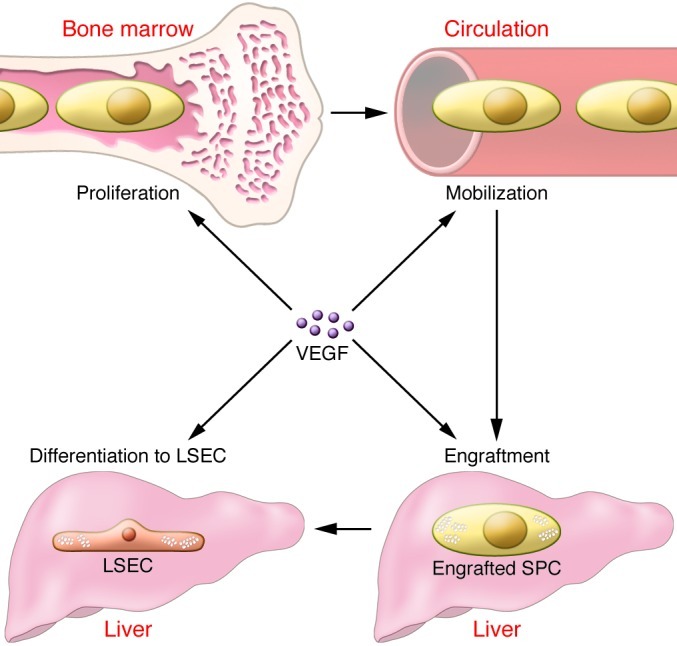
����VEGF�Ǵӹ��赽����ļ��SPC�����ĵ��ڼ���
�ڲ�ͬ��ʽ�ĸ����˺���VEGF���ӡ� VEGF�����Ӷ���BM SPC����ֳ��Proliferation)���ӣ�BM SPC��ѭ���Ķ�Ա(mobilization)��BM SPC�ڸ����е�ֲ��(Engraftment)�Լ��ڸ����˺�ֻ�ΪLSECs(Differentiation to LSEC)�DZ���ġ�
������˿����ԭ
���ڸ������ļ�ƪ���������ṩ�˶Կ��Ƹ��������ź�ͨ·���������ۣ����磬�ο�����47,48�������ڽ��ἰ�뵱ǰ������ص�һЩ��������źŴ���;�������ڸ�������������Щ;����������������ȫ����˿����ԭ������˿����ԭ����������������˿����ԭ�������ϸ���������Ǵ���˿���ѵģ����ҵ�������ע��ʱ�ܹ��յ����������ϸ��DNA�ϳɣ�48�����෴��������˿����ԭ�����ⲻ���д���˿�������ã����Ҳ��������������ϸ��DNA�ϳɺ�������48�������ڸ�����˿����ԭ���ź�ͨ·�ӳٸ�������������������48�������������Wnt2������LSEC���ֵ���˿����ԭ��4����
��Ȼ������������LSEC������ϸ����HGF����HGF�ļ�����Դ���������ڸ�������������״ϸ�������������е�HGF�������������ϸ�������ڸ����˵����Ȼ�̼ģ���б���û�����ӣ�1�������ָ��г�����1Сʱ�ڷ����ĸ���HGF���ӹ����ڴ�ϸ�����ϸ�����ǰ��������ͷ�HGF��48��������ʱ���ԭ��һ�ָ�ϸ����ֳ�Ĵ̼�����������HGF�⣨48�������ָ��г�����3Сʱ��������HGF���ױ������ӣ�49�����Σ�50��������Ƣ��HGF����������ӣ�51�������ָ��ں���HGF��Ը������ĸ��Թ������д�ȷ����
DNA������Ƽ�1��Id1��������ʧ������� - �� - �������ף��������ת¼���ӵĻ���HLD����ij�Ա�γ�������塣 Id1ͨ����ǿHIF-1����52-54�����ȶ��Ժͻ���������VEGF���෴��VEGF / VEGFR2;���ɼ���Id1��4������Id1 - / - С���е��о��Ѿ�֤������Ķ�Ա����EPC��ļ������������Ѫ�����ɺ�������������55�������ָ��г�����ĸ�������Id1 - / - С��������4��������BM SPC�Ķ�Ա��ļ����������EPC�������ƣ�4���� Id1 - / - С���ڲ��ָ��г�����HGF��Wnt2�ı��オ�ͣ�4�����������Id1�Ƿ���VEGF / VEGFR2�̼���Wnt2��HGF�����ת¼���ӣ�����Wnt2��HGF�ĸ�������Ƿ͡����������ڱ���Wnt2��HGF��BM SPCs��ļ�����١���Ȼ�����о�ʹ�ø�ϸ�����˸�����Ƥϸ�����������ܹ�����ģ��������Ƿ���ҪId1�������̼���ϸ����ֳ��4��������Щ����Ľ��������ѵģ���ΪLSEC���ڱ�co�д� - ����Լ4�졣
EGF����ʮ��ָ��Brunner��������������ٷ���ԭ��56��; EGF���ڵ�����ѭ���б���Ϊ�����ڸ�������������EPC����EGF��57������Ӧ��פ����BM SPC�м��EGF������EGF��SPCs�д�������о�����Ҫ���SPC������EGF�ڴٽ��������е����á�
���ϲ����о��˸���VEGF��ļ������HGF�Ϳ��ܻ���Wnt2��BM SPC�е����á�Ȼ�����ø���Ƥϸ�����е������о��Ѿ�֤��VEGF���Դ̼�����ĸ�����Ƥϸ�������ܺ���LSEC�ͳ�פSPCs������HGF���Ӷ��ٽ������ϸ����ֳ��3�����������о���VEGF�յ���HGF�����Ƿ����Գ�פSPC������LSEC�����߶���δ֪�ģ�5�������⣬�в���������˺�LSEC�����HGF�������ӵ���Ҫԭ���Ǹ���VEGF�̼�ļ���ĸ߱���HGF��BM SPCs���߸���VEGF����̼���ֲ��BM��HGF�ı�� SPC�������ߵ���ϡ�
�ٴ����������
�о�����������ĸ����������������Ƴɱ����ر����ƣ�5������Щ���ֵ��ٴ���������ԡ������Щ�۲����������еõ�֤ʵ����ôȫ��������ƻ�BM SPCs��ѡ�������ƽ����������������ٴ�ҽ����һ����Ҫ�������ء�Ȼ������һ�۲�����������ٴ����⡣�����������Ըβ���ѡ��Ĺ���Ⱥ����BM SPCs�Ƿ���Ա����ƣ����ָ��༲���ζ������˶�BM SPCs��ļ��BM SPCs���ź�ͨ·�к�Ӱ�죿���������еĸ��������Ը�ϸ����ֳ��58�����������Ըβ��У���������Ҫ��ϸ����ϸ������ֳ�� BM SPCs��������ϸ����ϸ���ĸ������з�������������ҽѧ�ܴ���������������Ƹ���������BM SPCs������ٽ�BM SPCļ���ĸ����źŴ����ڸ��༲���м��٣���ô��Դ��BM SPCs�Ǹ��ಢ��ֲ���� VEGFʩ���ǰ���IJ��ҿ�����ˮ����ø���;��ļBM SPCs���ź�ͨ·�е����������Ƿ���ʺ����ƣ�
����
BM SPCs�ڲ��ָ��г������ж��Ը����˺�ļ�������ࡣ����Щģ���У�BM SPC������ļ�����ڸ�ϸ����ֳ���������Ļָ��DZ���ġ� BM SPC��LSEC���������ӵ�HGF����Ҫ��Դ��Ȼ�������ǵ����˺���ں���HGF���ӵ�������Դ��HGF����������BM SPCs����Ҫ���ס���ȻBM SPCs��HGF���ܺ���Ҫ����δ�����о���Ҫ��ȷΪʲôBM SPCļ���Ը�������������Ҫ��
VEGF / VEGFR2;�����ڸ������DZ���ġ� Id1�Ǹ�;���б����ת¼���ӣ�����������Ҫ��һ�����塣 VEGF��BM SPCļ����������ڼ������˺����VEGF�����ӵ���BM SPC����ֳ��BM SPC��ѭ���Ķ�Ա�������е�ֲ���Լ�BM SPC��LSEC�ķֻ��� VEGF���̼�LSEC����ϸ�������ӵ�HGF���������LSEC��/��LSEC��ϸ����
�����������г������˺������ļ��Իָ������������ڼ�������ά�ֶ��DZز����ٵġ����õ��˽�BM SPCs�Ը������Ĺ����Լ�BM SPC�����ļ���ĵ���Ӧ��ʹ���Ϊת���о��ķ�������
��
��
Liver sinusoidal endothelial cells and liver regeneration
��
��
Abstract
Liver sinusoidal endothelial cells (LSECs) have long been noted to contribute to liver regeneration after liver injury. In normal liver, the major cellular source of HGF is the hepatic stellate cell, but after liver injury, HGF expression has been thought to increase markedly in proliferating LSECs. However, emerging data suggest that even after injury, LSEC expression of HGF does not increase greatly. In contrast, bone marrow progenitor cells of LSECs (BM SPCs), which are rich in HGF, are recruited to the liver after injury. This Review examines liver regeneration from the perspective that BM SPCs that have been recruited to the liver, rather than mature LSECs, drive liver regeneration.
��
Introduction
HGF is a cytokine that plays a crucial role in tissue regeneration, stimulating cell growth, cell motility, and morphogenesis. More than 20 years ago, Jacquelyn Maher first hypothesized that liver sinusoidal endothelial cells (LSECs) contribute to liver regeneration, demonstrating that hepatic stellate cells serve as the major source of HGF in normal liver but that after liver injury, HGF expression increased markedly in LSECs (1). It was shown subsequently that bFGF stimulates LSEC proliferation in the liver but not hepatocyte proliferation, and that inhibition of bFGF inhibits LSEC proliferation but not hepatocyte proliferation. This result, coupled with the observation that bFGF or VEGF increases liver weight after partial hepatectomy, whereas bFGF inhibition impairs liver regeneration, is consistent with the concept that endothelial cells direct liver regeneration (2). VEGF does not stimulate hepatocyte proliferation in vitro, but in vivo studies or co-culture studies of hepatocytes with liver endothelial cells have demonstrated that VEGF stimulates proliferation of hepatocytes through the VEGFR1 pathway (3). These studies also demonstrated upregulation of HGF gene expression through the VEGFR1 pathway in sinusoidal lining cells or isolated liver endothelial cells (3). Thus these results suggested that VEGF promotes hepatocyte proliferation through VEGFR1-stimulated release of HGF from liver endothelial cells (3). Finally, a recent paper has suggested that impaired liver regeneration in mice with inducible genetic ablation of VEGFR2 is due to diminished expression of angiocrine factors, including HGF, from LSECs (4). However, like other endothelial cells, LSECs express very little HGF (1, 5). In contrast, bone marrow progenitor cells of LSECs (BM SPCs), which are recruited to the liver after injury and after partial hepatectomy (5), are rich in HGF. BM SPCs are the same size as LSECs and share surface markers (5); therefore, LSEC isolation methods can also recover BM SPCs engrafted in the liver along with LSECs. Thus, in vitro studies could mistakenly attribute the properties of engrafted BM SPCs to mature LSECs. Many current findings on LSECs and liver regeneration are consistent with the hypothesis that BM SPCs recruited to the liver, rather than mature LSECs, drive liver regeneration, whereas the alternate hypothesis, that mature LSECs drive liver regeneration, cannot account for several known observations. This Review examines the literature from the perspective that BM SPCs that have been recruited to the liver, rather than mature LSECs, are the major drivers of liver regeneration (Figure (Figure11).
��
��
Figure 1
BM SPCs and liver regeneration.
Schematic depicting the contributions of liver cells and BM SPCs to liver regeneration. Liver injury induces increased hepatic VEGF expression, which drives recruitment of HGF-rich BM SPCs and promotes expression of HGF by resident SPCs and LSECs. HGF, in turn stimulates the proliferation of hepatocytes in liver regeneration. In addition, SPCs replace LSECs that were lost during injury.
��
LSECs and their progenitors
LSECs are unique endothelial cells, both morphologically and functionally. LSECs are small cells, with a diameter of around 6.5 ��m when isolated, that are stretched out into a very thin layer lining the hepatic sinusoids. LSECs are the only mammalian endothelial cells that combine non-diaphragmed fenestrae with the lack of a basement membrane. The fenestrae are clustered together in sieve plates, and the size and number of fenestrae differ in LSECs from the periportal, midlobular and centrilobular regions of the liver (6�C10). LSECs are also functionally unique. The high activity of receptor-mediated endocytosis provides LSECs with a high-rate, high-capacity system to clear colloids and soluble waste macromolecules from the circulation (11, 12). The three main receptors for endocytosis are the mannose receptor, the scavenger receptor, and the Fc�� receptor IIb2 (12). LSECs are the initial target of injury for some hepatotoxic drugs and toxins (13�C20) and are susceptible to ischemia-reperfusion injury (21�C24).
As has been described for endothelial cells and pericytes in other vascular beds (refs. 25�C29 are a few of many such reports), crosstalk exists between LSECs and the neighboring pericytes, the hepatic stellate cells (30, 31). Activation of hepatic stellate cells leads to fibrosis. Healthy LSECs prevent the activation of hepatic stellate cells and inactivate activated hepatic stellate cells (31). LSECs develop an altered phenotype preceding hepatic fibrosis that is called capillarization. Capillarized LSECs lose the ability to prevent hepatic stellate cell activation and inactivate activated hepatic stellate cells. In a rat model of hepatic fibrosis, reversal of capillarization of LSECs inactivates activated hepatic stellate cells and accelerates reversal of fibrosis, whereas reversal of capillarization during an ongoing insult prevents progression of cirrhosis (32). In healthy liver, hepatocytes and hepatic stellate cells maintain the phenotype of LSECs through release of VEGF (30).
Like other endothelial cells, LSECs express CD31 (also known as PECAM). However, unlike most endothelial cells, the CD31 expression of normal LSECs is restricted to the cytoplasm rather than to the cell surface at cell-cell junctions (30). Also in stark contrast to other endothelial cells, LSECs are the only endothelial cells reported to date that express CD45 (10, 33, 34), a classic hematopoietic cell marker. CD45 is highly expressed on periportal LSECs, less strongly expressed on midlobular LSECs, and absent on centrilobular LSECs (10).
Stem cells are defined as cells that divide asymmetrically, producing one daughter cell that is a stem cell and one daughter cell that is either a progenitor cell or a specialized cell (Figure (Figure2).2). By definition, stem cells can self-renew and give rise indefinitely to more cells of the same type. Progenitor cells, also known as transit-amplifying cells, are more lineage committed than stem cells, give rise to specialized cells, and can only replicate a limited number of times. Two populations of liver sinusoidal endothelial cell progenitor cells (SPCs) have been identified: BM SPCs and resident or intrahepatic SPCs. BM SPCs do not contribute to normal LSEC turnover, but are recruited to the liver after injury or partial hepatectomy (5, 33, 35). Resident SPCs are not derived from the bone marrow in the adult rodent (5). Resident SPCs and BM SPCs recruited to the liver are isolated along with mature LSECs, ��contaminating�� LSEC isolates (5). Marked variability exists in the frequency of resident SPCs in rodents, varying from 1% of all LSECs in Sprague-Dawley rats to 7% in Fischer rats (5). Within the liver, a putative resident LSEC stem cell has been identified based on its characteristics as a label-retaining cell. The definitive functional characteristics of self-renewal, lineage-specific differentiation, and serial repopulation have not been demonstrated, so this designation remains tenuous. However, the presence of a label-retaining cell with the surface markers of the resident SPC suggests that resident SPCs derive from this putative resident LSEC stem cell, especially given that resident SPCs do not come from the bone marrow in the adult rodent. In the Sprague-Dawley rat, 0.1% of all LSECs are putative resident LSEC stem cells (5). The 0.1% frequency of the putative resident LSEC stem cell is high when compared with the 0.01% frequency of hematopoietic stem cells among the nucleated cells in the bone marrow (36). Although the logical function for the resident LSEC stem cell a3nd resident SPC would be to give rise to LSECs in normal turnover, such functionality has yet to be demonstrated.
��
��
Figure 2
Stem and progenitor cells.
Stem cells self-renew and give rise to progenitor cells. Progenitor cells or transit-amplifying cells are more lineage committed than stem cells, give rise to additional progenitor cells or to specialized cells, and can only replicate a limited number of times. Two populations of liver SPCs can give rise to LSECs: BM SPCs, which are recruited to the liver after injury or partial hepatectomy, and resident or intrahepatic SPCs, which contribute to normal LSEC turnover. SPCs derive from stem cells, and the SPC population expands before giving rise to the mature LSECs.
Both resident and BM SPCs are positive for the progenitor cell marker CD133 and, like the LSEC, they express the endothelial cell marker CD31 and the hematopoietic cell marker CD45 (5). The resident SPC is positive for VEGFR1 and VEGFR2. While VEGFR expression in BM SPCs has not yet been examined, given that LSECs are positive for VEGFR1 and VEGFR2, BM SPCs may be as well. The resident SPC has a fenestration pattern that is indistinguishable from mature LSECs, whereas SPCs in the bone marrow are non-fenestrated but develop the normal LSEC fenestration pattern after recruitment to and engraftment in the liver (5). The presence of CD45 on resident and BM SPCs distinguishes them from the more generic endothelial progenitor cell (EPC) that is by definition CD45 negative.
After partial hepatectomy or toxic injury, proliferation of BM SPCs increases more than two-fold and mobilization of BM SPCs to the circulation increases two- to four-fold (5, 35). The number of LSECs of bone marrow origin increases over time: 25% of LSECs are bone marrow derived on day 3 after partial hepatectomy, whereas 40% and 70% of LSECs are bone marrow derived on days 5 and 14, respectively, after a toxic dose of dimethylnitrosamine (5, 35). Studies have ruled out fusion in LSECs after recruitment of the bone marrow�Cderived cells (33), so this result is true engraftment rather than fusion of a bone marrow cell with existing LSECs. BM SPCs develop fenestrae organized in sieve plates after engraftment in the liver (5, 35). After partial hepatectomy, BM SPCs that engraft in the liver proliferate much more than resident SPCs and are the major source of increased HGF in the LSEC fraction (5).
The key evidence that BM SPCs are necessary for liver regeneration comes from experiments with bone marrow irradiation. In rats, the two hind limbs from ankle to hip contain about 40% of the total bone marrow, and irradiation of this area results in suppression of peripheral leukocyte count by almost 40% (5). When these irradiated rats undergo partial hepatectomy, liver regeneration is impaired by 40%, and minimal hepatocyte proliferation on day 5 suggests that liver regeneration is not merely delayed. Infusion of either resident SPCs or bone marrow into the irradiated rats on day 1 after partial hepatectomy markedly promotes hepatocyte proliferation and completely normalizes restoration of liver weight. This result demonstrates that liver regeneration is impaired following bone marrow irradiation due to the loss of BM SPCs. The suppression of liver regeneration proportionate to bone marrow suppression and the lack of hepatocyte proliferation on day 5 after partial hepatectomy in this study indicate that LSECs and/or resident SPCs and other sources of HGF cannot compensate for the loss of BM SPC stimulation of liver regeneration. Of note, infusion of resident SPCs or bone marrow on day 3 (rather than day 1) after partial hepatectomy has little benefit, suggesting that SPC infusion must occur prior to the second round of hepatocyte proliferation, which is completed by day 2 in the rat. Proliferation within the LSEC fraction is increased by day 1 after partial hepatectomy and peaks by day 3 in the rat (5, 37, 38). This suggests that the early influx of BM SPCs stimulates liver regeneration by promoting hepatocyte proliferation, whereas proliferation of LSECs and/or SPCs from day 3 on is required for angiogenesis in the regenerating liver. Whether the mature LSEC also proliferates or whether all of the proliferation is attributable to resident and BM SPCs remains to be determined.
If infusion of resident SPCs can rescue liver regeneration after bone marrow irradiation, resident SPCs might be expected to contribute to liver regeneration. However, as noted above, suppression of liver regeneration is proportionate to the amount of bone marrow irradiated, indicating that resident SPCs do not compensate for the loss of BM SPCs. The finding that infusion of resident SPCs stimulates hepatocyte proliferation, but that resident SPCs in situ do not contribute significantly to liver regeneration, suggests that these cells remain relatively quiescent as long as they are in the liver stem cell niche.
The contribution of BM SPCs to recovery from liver injury has also been demonstrated in two models of toxic hepatitis (33, 35). Studies in the monocrotaline-induced model of sinusoidal obstruction syndrome (SOS; also referred to as hepatic venoocclusive disease) have demonstrated that monocrotaline damages both LSECs (15, 20, 33, 39) and BM SPCs (33). During the recovery phase of injury, around 25% of LSECs are of bone marrow origin, demonstrating marked repair of LSECs by BM SPCs. However, at the height of sinusoidal denudation and centrilobular hemorrhagic necrosis, monocrotaline decreases the number of SPCs in the bone marrow by 50% and reduces the number of circulating BM SPCs by 95%. Bone marrow irradiation elicits severe SOS from a subtoxic dose of monocrotaline, whereas infusion of bone marrow completely prevents SOS in rats given a dose of monocrotaline that would otherwise cause severe SOS. Thus, repair of LSEC injury by BM SPCs is a key determinant of SOS. In other words, SOS is due to a combination of injury to LSECs and lack of repair by BM SPCs. Taken together with what we now know about the effect of BM SPCs on liver regeneration, diminished repair by BM SPCs in SOS will not only prevent repopulation of the denuded sinusoid, but also impair hepatocyte proliferation needed for liver regeneration. This likely explains the high case lethality in patients who develop SOS from high-dose myeloablative chemotherapy prior to hematopoietic cell transplantation (bone marrow transplantation). BM SPCs also repair LSEC injury after a toxic dose of dimethylnitrosamine, and infusion of BM SPC attenuates this injury (35).
��
Regulation of recruitment of bone marrow SPCs
Hepatic VEGF (also known as VEGFA) increases in response to many forms of liver injury and after partial hepatectomy (35, 37, 38, 40�C43). Knockdown of hepatic VEGF with antisense oligonucleotides can exacerbate toxic injury to the liver (35), whereas infusion of VEGF can ameliorate toxic injury (44�C46) and increases hepatocyte proliferation (38) and liver weight after partial hepatectomy (3, 4). After both toxic injury and partial hepatectomy, hepatic VEGF has been shown to regulate each step of BM SPC recruitment to the liver: proliferation of BM SPCs, mobilization of BM SPCs to the circulation, engraftment of BM SPCs in the liver, and differentiation of BM SPCs to fenestrated LSECs lining the sinusoids (35) (Figure (Figure3).3). Thus, hepatic VEGF is a central regulator of BM SPC recruitment and is critical to liver regeneration. The finding that inducible VEGFR2-deficient Vegfr2flox/flox mice have impaired liver regeneration after partial hepatectomy (4) is consistent with two possible hypotheses: that hepatic VEGF recruits BM SPCs to the liver and thereby promotes liver regeneration, and that these mice lack VEGF-stimulated HGF release by BM SPCs (3) (see below).
��
��
��
Figure 3
Hepatic VEGF is a central regulator of recruitment of SPCs from the bone marrow to the liver.
Hepatic VEGF increases after disparate forms of liver injury. The increase in VEGF is necessary for increased proliferation of BM SPCs, mobilization of BM SPCs to the circulation, engraftment of BM SPCs in the liver, and differentiation to LSECs after liver injury.
��
Hepatic mitogens
Several excellent reviews on liver regeneration provide an in-depth discussion of the signaling pathways that control liver regeneration (for example, refs. 47, 48). This section will only mention a few concepts that are relevant to the current Review. Multiple signaling pathways contribute to liver regeneration, and these pathways can be driven by complete or incomplete mitogens (Hepatic mitogens). Complete mitogens are mitogenic in hepatocyte culture in vitro and able to induce liver enlargement and hepatocyte DNA synthesis when injected in vivo (48). In contrast, auxiliary mitogens are not mitogenic in vitro and do not cause hepatocyte DNA synthesis and liver enlargement in vivo (48). Ablation of the signaling pathways for auxiliary mitogens delays liver regeneration rather than abolishing it (48). Wnt2 has recently been reported as being a mitogen derived from the LSEC fraction (4).
��
Although this Review focuses on HGF from LSECs and their progenitors, several sources of HGF may contribute to liver regeneration. Hepatic stellate cells have much higher HGF expression in normal liver than do other liver cells, but expression does not increase in the carbon tetrachloride model of liver injury (1). The increase in hepatic HGF that occurs within one hour of partial hepatectomy is attributed to release of HGF from pericellular and extracellular glycosaminoglycans (48). Because of the timing, stimulation of the first round of hepatocyte proliferation is attributed to this pool of HGF (48). Three hours after partial hepatectomy, HGF protein expression increases in the liver (49) and HGF gene expression is increased in lung (50), kidney, and spleen (51). The respective contributions of the various intrahepatic and extrahepatic HGF pools to liver regeneration remain to be defined.
Inhibitor of DNA binding 1 (Id1) is a dominant-negative helix-loop-helix protein that can form heterodimers with members of the basic HLD family of transcription factors. Id1 activates VEGF by enhancing the stability and activity of HIF-1�� (52�C54), and conversely the VEGF/VEGFR2 pathway may activate Id1 (4). Studies in Id1�C/�C mice have demonstrated impaired mobilization and recruitment of bone marrow EPCs, leading to impaired tumor angiogenesis and tumor growth (55). Liver regeneration after partial hepatectomy is impaired in Id1�C/�C mice (4), suggesting an impaired mobilization and recruitment of BM SPCs that is analogous to the EPC findings (4). Id1�C/�C mice have decreased expression of HGF and Wnt2 after partial hepatectomy (4), but it is unclear whether Id1 is a transcription factor for VEGF/VEGFR2-stimulated expression of Wnt2 and HGF or whether decreased hepatic expression of Wnt2 and HGF are solely due to decreased recruitment of BM SPCs expressing Wnt2 and HGF. While this study used an in vitro two-week co-culture model of hepatocytes with human liver endothelial cells to examine whether Id1 expression is required to stimulate hepatocyte proliferation (4), interpretation of these results is difficult, as LSECs only survive in standard co-culture for about 4 days.
EGF is a complete mitogen that is produced by duodenal Brunner��s glands (56); secretion of EGF into the portal circulation is thought to contribute to liver regeneration. Given that EPCs express EGF (57), EGF expression should also be examined in resident and BM SPCs. If EGF is abundantly expressed in SPCs, studies will need to examine the contribution of SPC-derived EGF in the promotion of liver regeneration.
The section above examines the role of hepatic VEGF in recruiting BM SPCs that express HGF and perhaps also Wnt2. However, in vitro studies with liver endothelial cells have demonstrated that VEGF stimulates production of HGF from isolated liver endothelial cells (presumably containing both LSECs and resident SPCs), which promotes hepatocyte proliferation in vitro (3). Whether the VEGF-induced increase in HGF in the in vitro studies comes from resident SPCs, mature LSECs, or both is not known (5). In addition, it is unclear whether the major reason for increased expression of HGF in the LSEC fraction after liver injury is due to hepatic VEGF-stimulated recruitment of BM SPCs that highly express HGF, high hepatic VEGF expression that stimulates expression of HGF in engrafted BM SPCs, or a combination of both.
��
Clinical implication and questions
Studies demonstrate that liver regeneration in the rat is suppressed proportionately to the suppression of bone marrow (5). The clinical implication of these findings is clear. If these observations are confirmed in humans, then global bone marrow suppression or selective suppression of BM SPCs will impair liver regeneration, an important consideration for clinicians. However this observation raises numerous clinical questions. Suppression of select bone marrow populations in chronic liver disease can occur, but are BM SPCs one of the populations that can be suppressed? What effect do various liver diseases and hepatotoxic insults have on BM SPCs and on the signaling pathways that recruit BM SPCs? Liver regeneration in normal liver comes from hepatocyte proliferation (58), but in chronic liver disease, liver regeneration requires proliferation of hepatocyte progenitors. Do BM SPCs play a role in liver regeneration that is dependent on hepatocyte progenitors? Can regenerative medicine create BM SPCs that could be used therapeutically to promote liver regeneration? If hepatic signaling that promotes BM SPC recruitment is diminished in liver disease, would exogenous BM SPCs home to the liver and engraft? VEGF administration is costly and can be complicated by water retention; will other steps in the signaling pathway that recruits BM SPCs be more amenable to therapy?
��
Conclusions
BM SPCs are recruited to the liver after partial hepatectomy and toxic liver injury. Recruitment of BM SPCs to the liver is essential for hepatocyte proliferation and restoration of liver weight in these models. BM SPCs are the major source of increased HGF in the LSEC fraction. However, HGF may not account for the essential contribution of BM SPCs, given that there are other sources of increased intrahepatic and extrahepatic HGF after injury. Although HGF from BM SPCs is probably important, future studies will need to define why BM SPC recruitment is essential for liver regeneration.
The VEGF/VEGFR2 pathway is essential for liver regeneration. Id1 is an essential transcription factor in this pathway, but its role needs further definition. VEGF is a central regulator of BM SPC recruitment: the increase in hepatic VEGF after injury leads to proliferation of BM SPCs, mobilization of BM SPCs to the circulation, engraftment in the liver, and differentiation of BM SPCs to LSECs. VEGF also stimulates increased HGF expression in the cells of the LSEC fraction, i.e., mature LSECs and/or the LSEC progenitors.
Liver regeneration is essential to both acute restoration of liver mass after resection or injury and to maintenance of liver mass during chronic injury. A better understanding of the contribution of BM SPCs to liver regeneration and the regulation of BM SPC recruitment to the liver should make this a fertile field for translational research.
��
J Clin Invest. 2013 May 1; 123(5): 1861�C1866.
Laurie D. DeLeve
Author information Copyright and License information Disclaimer
This article has been cited by other articles in PMC.
��
Published online 2013 May 1. doi: [10.1172/JCI66025]
PMCID: PMC3635729
PMID: 23635783
Liver sinusoidal endothelial cells and liver regeneration https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3635729/
��
��
Gastroenterology. 2012 Apr;142(4):918-927.e6. doi: 10.1053/j.gastro.2011.12.017. Epub 2011 Dec 16.
Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats.
Xie G1, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA, Deleve LD.
Author information
1
Division of Gastrointestinal and Liver Disease, University of Southern California Research Center for Liver Disease, Los Angeles, California 90033, USA.
Abstract
BACKGROUND & AIMS:
Capillarization, characterized by loss of differentiation of liver sinusoidal endothelial cells (LSECs), precedes the onset of hepatic fibrosis. We investigated whether restoration of LSEC differentiation would normalize crosstalk with activated hepatic stellate cells (HSC) and thereby promote quiescence of HSC and regression of fibrosis.
METHODS:
Rat LSECs were cultured with inhibitors and/or agonists and examined by scanning electron microscopy for fenestrae in sieve plates. Cirrhosis was induced in rats using thioacetamide, followed by administration of BAY 60-2770, an activator of soluble guanylate cyclase (sGC). Fibrosis was assessed by Sirius red staining; expression of ��-smooth muscle actin was measured by immunoblot analysis.
RESULTS:
Maintenance of LSEC differentiation requires vascular endothelial growth factor-A stimulation of nitric oxide-dependent signaling (via sGC and cyclic guanosine monophosphate) and nitric oxide-independent signaling. In rats with thioacetamide-induced cirrhosis, BAY 60-2770 accelerated the complete reversal of capillarization (restored differentiation of LSECs) without directly affecting activation of HSCs or fibrosis. Restoration of differentiation to LSECs led to quiescence of HSCs and regression of fibrosis in the absence of further exposure to BAY 60-2770. Activation of sGC with BAY 60-2770 prevented progression of cirrhosis, despite continued administration of thioacetamide.
CONCLUSIONS:
The state of LSEC differentiation plays a pivotal role in HSC activation and the fibrotic process.
Copyright © 2012 AGA Institute. Published by Elsevier Inc. All rights reserved.
Division of Gastrointestinal and Liver Disease, University of Southern California Research Center for Liver Disease, Los Angeles, California 90033, USA.
��
Comment in
Liver sinusoidal endothelial cells in disease--and for therapy? [J Hepatol. 2013]
Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. - PubMed - NCBI https://www.ncbi.nlm.nih.gov/pubmed/22178212/
��
��
Circ Res. 2018 Aug 3;123(4):477-494. doi: 10.1161/CIRCRESAHA.118.313237.
Emerging Roles of Vascular Endothelium in Metabolic Homeostasis.
Pi X1, Xie L1, Patterson C2.
Author information
1
From the Section of Athero & Lipo, Department of Medicine, Cardiovascular Research Institute, Baylor College of Medicine, Houston, TX (X.P., L.X.).
2
University of Arkansas for Medical Sciences, Little Rock (C.P.).
Abstract
Our understanding of the role of the vascular endothelium has evolved over the past 2 decades, with the recognition that it is a dynamically regulated organ and that it plays a nodal role in a variety of physiological and pathological processes. Endothelial cells (ECs) are not only a barrier between the circulation and peripheral tissues, but also actively regulate vascular tone, blood flow, and platelet function. Dysregulation of ECs contributes to pathological conditions such as vascular inflammation, atherosclerosis, hypertension, cardiomyopathy, retinopathy, neuropathy, and cancer. The close anatomic relationship between vascular endothelium and highly vascularized metabolic organs/tissues suggests that the crosstalk between ECs and these organs is vital for both vascular and metabolic homeostasis. Numerous reports support that hyperlipidemia, hyperglycemia, and other metabolic stresses result in endothelial dysfunction and vascular complications. However, how ECs may regulate metabolic homeostasis remains poorly understood. Emerging data suggest that the vascular endothelium plays an unexpected role in the regulation of metabolic homeostasis and that endothelial dysregulation directly contributes to the development of metabolic disorders. Here, we review recent studies about the pivotal role of ECs in glucose and lipid homeostasis. In particular, we introduce the concept that the endothelium adjusts its barrier function to control the transendothelial transport of fatty acids, lipoproteins, LPLs (lipoprotein lipases), glucose, and insulin. In addition, we summarize reports that ECs communicate with metabolic cells through EC-secreted factors and we discuss how endothelial dysregulation contributes directly to the development of obesity, insulin resistance, dyslipidemia, diabetes mellitus, cognitive defects, and fatty liver disease.
Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. - PubMed - NCBI https://www.ncbi.nlm.nih.gov/pubmed/30355249
��
.png)