肥胖引起的炎性综合征: 脂肪组织细胞因子在与肥胖有关的代谢紊乱中的作用
The Inflammatory Syndrome: The Role of Adipose Tissue Cytokines in Metabolic Disorders Linked to Obesity
Brent E. Wisse——作者的从属关系
代谢、内分泌学、营养学、内科、海景医学中心、华盛顿大学西雅图分校
文摘
肥胖症的代谢作用使这种高度流行的疾病成为糖尿病、高血压和动脉粥样硬化最常见的危险因素之一,而动脉粥样硬化是终末期肾功能衰竭的主要原因。
然而,正如体重指数定义的那样,肥胖本身并不能很好地预测这些疾病的发展,而更有可能出现一系列与肥胖相关的异常,现在被称为代谢综合症。对这一综合征的认识,可以很容易地在临床环境中通过定义腰围、血压、空腹血糖和血脂异常的阈值识别出来,从而可以更早地对这些高危患者进行干预。
系统的胰岛素抵抗被认为是一种可能的因素,将内脏肥胖与不良的代谢后果联系在一起;然而,脂肪组织引起胰岛素敏感性改变的机制仍不清楚。
感染和炎症通常与胰岛素抵抗有关,内脏肥胖与慢性、低度炎症有关,这表明炎症可能是肥胖导致胰岛素抵抗的潜在机制。此外,脂肪组织现在被认为是一种免疫器官,分泌大量的免疫调节因子,似乎是引起胰岛素抵抗的炎症信号的重要来源。
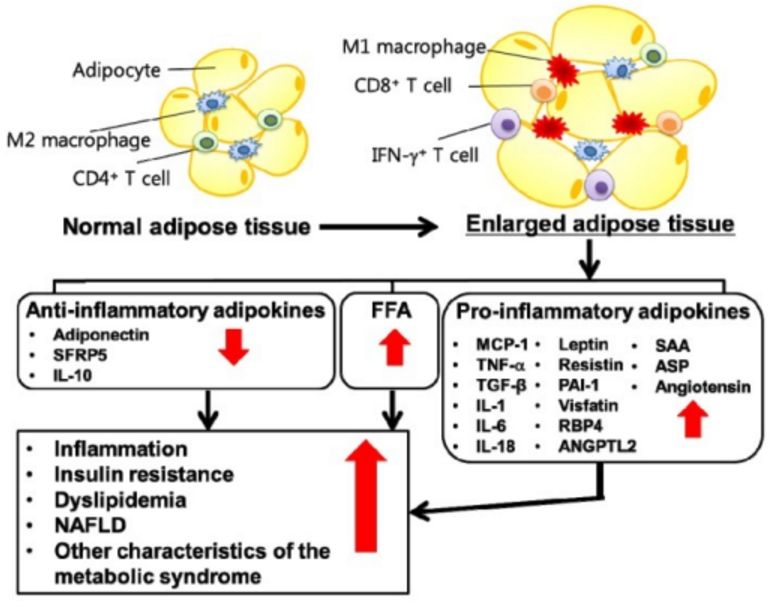
因此,白色脂肪组织(WAT)内的炎症可能是导致代谢综合征的许多病理特征的出现,并导致糖尿病和动脉粥样硬化的重要步骤。
全球肥胖的患病率显著增加,和最近的估计显示,近三分之二的美国成年人超重或肥胖。与此同时,儿童肥胖是在一个更惊人的速度增加,这表明这种流行病不大可能很快减弱。
虽然增加体重、身体质量指数(BMI)所定义的,是一种强大的代谢性疾病风险的预测, 对于任何给定的肥胖程度,遗传和环境因素在2型糖尿病和动脉粥样硬化的表现造成相当大的变化。这些差别的一个可能的解释是,个人用同样的体重指数可能会有截然不同的大量的内脏(也称为“中央”或“腹部”)的脂肪-脂肪组织仓库相关的已知最大的代谢风险。当内脏肥胖是伴随着一系列代谢紊乱,包括胰岛素抵抗、低高密度脂蛋白、甘油三酯升高, 血压升高,预测心血管疾病的风险显著增加。这个不良代谢现在被称为代谢综合征,在美国是一个非常普及的情况,影响近25%的成年人。代谢综合征极大的增加患上2型糖尿病、高血压、高血脂症和心血管疾病的风险。
尽管目前的理论主要关注胰岛素抵抗作为将内脏肥胖与不良代谢变化联系起来的主要因素,但研究表明,代谢综合征的病理生理学不能仅用胰岛素抵抗来解释。除了葡萄糖稳态、血脂异常和血压外,代谢综合征患者还有许多其他病理生理特征。其中慢性全身性炎症是最一致的证据之一,众多炎症标志物与肥胖程度和胰岛素抵抗程度高度相关; 这些炎症标志物中有许多是血管疾病风险的高预测性。
本综述讨论了脂肪内炎症在产生代谢综合征中的潜在作用,重点讨论了脂肪组织分泌的细胞因子,这些细胞因子调节免疫系统,有利于慢性全身性炎症。瘦素的炎症作用, 脂肪特异性细胞因子对能量平衡的控制至关重要,以及经典的促炎细胞因子IL - 6和TNF-α,特别突出显示。
脂肪组织内分泌学
脂肪组织从能量储存的被动器官到体内平衡系统激素调节的主动参与者的概念转化是最近才发生的。
1994年,脂肪组织被证实为瘦素(Leptin)的来源,为脂肪细胞内分泌学研究的新时代打开了大门。在过去的十年中,瘦素的内分泌作用已经扩大到包括调节生殖免疫功能和,和许多其他脂肪组织来源的分子影响葡萄糖稳态,血管生物学、肿瘤发展、脂蛋白代谢和炎症已确定,这些炎症因素表2中列出。
然而,脂肪组织是由多种细胞类型组成的不均匀器官,这一事实引发了关于脂肪细胞与间质/血管和免疫细胞在分泌某些内分泌和旁分泌调节因子方面的真正作用的激烈争论。
事实上,成熟的脂肪细胞似乎缺乏储存泡和其他结构性细胞成分,这些成分通常与内分泌细胞分泌的蛋白质的调节释放有关。此外,由于这些多能细胞的功能特征和转录模式与免疫细胞相似,因此频繁使用前脂肪细胞细胞系在体外研究脂肪细胞生物学也加剧了这一争议。
事实上,未成熟的脂肪细胞在体外和体内都能分化成巨噬细胞。最近使用大规模基因分析描述胖人和瘦人的不同脂肪组织的基因表达,已经开始澄清脂肪细胞作为激素和细胞因子分泌细胞的作用。
令人吃惊的是,在这些研究中, 小鼠中脂肪的增加与脂肪组织中巨噬细胞的大量基因表达高度相关。脂肪细胞体积和体重是脂肪组织内巨噬细胞数量的强大的预测因子,内脏脂肪中巨噬细胞数量比皮下脂肪的关联度更高。
这些骨髓来源的巨噬细胞似乎对未知的信号做出反应,入侵脂肪组织,在肥胖的动物中,这些巨噬细胞往往聚集并形成具有慢性炎症特征的巨型细胞,这表明脂肪组织是活跃的炎症部位。
从脂肪组织的排序细胞的基因表达研究表明巨噬细胞(macrophage) 产生几乎所有TNF-α(肿瘤坏事因子α), 而成熟的脂肪细胞分泌的大部分的瘦素,而IL-6基因在巨噬细胞、脂肪细胞和非巨噬细胞基质血管细胞呈大致均等的表达。重要的是,这些研究表明,巨噬细胞入侵脂肪组织中的脂肪和炎症相关基因表达可能是一个前哨事件,在这些动物的胰岛素抵抗发展之前。
图示:Stuffed fat cells and invading macrophages send out interleukin-6, tumor necrosis factor-alpha, and other signals that cause inflammation.
填充脂肪细胞和入侵的巨噬细胞发出白细胞介素-6、肿瘤坏死因子-阿尔法,以及其他导致炎症的信号。
因此,小鼠体重增加与巨噬细胞浸润脂肪和脂肪组织促炎信号的表达有关。值得注意的是,这些炎症变化在内脏脂肪(沉积在内脏的脂肪危害最大)中最为明显,似乎先于代谢综合征的其他特征,包括葡萄糖稳态受损。
通过引入免疫细胞作为脂肪组织释放的炎性介质来源,以及脂肪细胞功能和激素分泌的旁分泌调节因子,已经显著改变了现有的脂肪组织内分泌学范式,从而有可能控制由过度肥胖引起的代谢变化。旨在阐明吸引巨噬细胞向脂肪转移的信号并剖析脂肪组织、巨噬细胞和脂肪细胞之间相互交流的旁分泌机制的研究,有可能确定可能减少脂肪诱导炎症的治疗靶点,并可能降低与肥胖相关的代谢风险。
脂肪和免疫系统
虽然巨噬细胞由于肥胖而浸润脂肪是令人惊讶的,但是脂肪组织和免疫系统之间的相互作用被很好地描述了,各种各样的理论被提出来试图解决为什么这种联系应该存在的问题。
白色脂肪组织和骨髓具有胚胎起源,中间胚层和前脂肪细胞是一种强大的吞噬细胞,在基因表达的形态和模式上都类似于巨噬细胞。此外,成熟的脂肪细胞具有分泌细胞因子和激活补体级联的能力,就像单核免疫细胞一样。将脂肪和免疫系统联系起来的一种推理是基于脂肪细胞激素大剂量在控制能量稳态和免疫中的作用。
在禁食/饥饿期间,当血浆瘦素水平下降时,下丘脑的神经通路会导致食欲增加,能量消耗减少,试图恢复身体脂肪储备。此外,血浆瘦素(Leptin)的减少会减少甲状腺激素的分泌并抑制生殖轴,这两种作用都能在营养贫乏的时期节省能量。然而,饥饿也会抑制免疫系统,导致胸腺细胞的凋亡增加,单核细胞的增殖减少,这种影响可以通过在没有重新进食的情况下单独使用瘦素完全逆转。
由于维持免疫功能被估计占每日能量消耗的15%,在这里,瘦素的作用也可以被认为有助于减少整体能量消耗;因此,脂肪组织对免疫的调节可以被视为脂肪在能量稳态中内分泌作用的延伸。
关于脂肪/免疫系统协调的第二个论据是基于目的论的。在果蝇中,脂肪体是控制先天免疫系统的器官,分泌到淋巴系统分子中,以摄取的病原体为目标。同样,在脊椎动物中,脂肪可能是唤醒先天免疫系统并触发急性期反应的关键警报系统之一,这是抵御细菌感染的第一道防线。当然脂肪组织瘦素的产量大幅增加在急性感染和脂肪可能导致循环白介素升高,TNF-αIL-1β,以及许多其他因素主要免疫系统或直接参与防御病原生物体。脂肪组织是否在控制人体免疫功能中起着重要作用的问题尚未得到最终的答案;然而,脂肪组织可以详细说明急性炎症信号,提示脂肪的炎症能力的失调可能在触发或延续代谢综合征的慢性炎症中起作用。
虽然急性和慢性炎症在许多方面不同,但在急性感染期间所见的代谢扰动与已知的代谢综合征特征有许多共同的特点。最重要的是,胰岛素抵抗和高血糖在这两种情况都很常见,高甘油三酯血症、脂肪分解障碍和非酯化脂肪酸的增加也是如此。瘦素, IL - 6, TNF-α, 急性期反应物C反应蛋白(CRP)以及其他循环炎症标记物在两种情况下升高。
此外,与人类相比,啮齿类动物的肥胖和急性感染都与下丘脑-垂体-肾上腺轴的激活和循环糖皮质激素水平的升高有关。在急性感染期间,这些代谢变化被认为是适应性的,为大脑节省燃料,激活细菌细胞表面的补体级联,剥夺细菌的铁和其他重要的微量营养素,等等。但是和其他适应性生理过程一样,当这些急性变化变成慢性的时候,可能会导致病理结果。
将代谢综合征相关的慢性炎症与急性炎症反应等同起来显然太过轻率,但它强调了一个有趣的可能性:免疫/炎症因子可能是与肥胖和急性疾病相关的代谢紊乱的根源。对特定细胞因子的生物反应可能在急性和慢性炎症状态之间存在差异,而控制这种差异反应的机制通常仍不清楚。
似乎很明显,虽然类似的细胞因子调节,肥胖和急性炎症综合征的非代谢特性有很大的不同和多个机制可能占了这个变化,包括血浆细胞因子浓度的差异,可溶性细胞因子受体和受体拮抗剂,平衡,和抗炎细胞因子网络在组织层面,和细菌细胞壁的产物,只列出许多潜在变量中的几个。这两种情况之间一个有趣的代谢差异值得注意,那就是食欲的控制。
慢性炎症和急性感染通常与厌食症和恶病质有关,据信这是细胞因子介导的,而肥胖/代谢综合征患者的食欲并不明显减少,尽管与厌食症相关的细胞因子的循环浓度升高。因此,尽管可能存在代谢综合征是由炎症反应引起的失败,或许更具体,脂肪组织内炎症反应在慢性刺激作用下变得不适应的,具体的细节cytokine-mediated效果和各种炎症介质的机械作用仍有待定义清楚。以下部分总结三个关键的潜在角色脂肪来源细胞因子—瘦素,IL-6,t,TNF-α-in导致代谢综合症的表现。
瘦素
脂肪细胞来源的荷尔蒙瘦素对能量平衡非常重要,把一个重要的信息-脂肪储存的损耗或积累传送到大脑。虽然确定为一个经典肽激素,四个α螺旋域折叠结构使瘦素最类似于细胞因子如- 2。此外,瘦素受体(leptin receptor, lepr)与1型细胞因子受体具有显著的同源性;因此,瘦素在很多方面更适合被认为是一种细胞因子。虽然瘦素在控制能量稳态的作用正日益被详尽确定,目前尚不清楚是否瘦素在腹部肥胖引起的炎症综合征的作用。显然,血清瘦素浓度与体内脂肪含量成正比; 因此,患有代谢综合征的肥胖个体通常具有较高的循环瘦素浓度。
然而,肥胖者似乎对瘦素的下丘脑效应有抵抗力; 因此,为了减少食欲和增加能量消耗而设计的分解代谢途径没有被激活,多余的体重得以维持。
尽管许多瘦素的作用是直接在下丘脑神经元的结果, 功能性瘦素受体(长篇或leprb)也发现在许多组织在中枢神经系统(CNS)之外,包括免疫细胞。重要的是,被认为有助于下丘脑瘦素抵抗的机制,如血脑屏障运输缺陷,在周围组织中没有被证实,也没有研究证明肥胖个体的外周瘦素抵抗。因此,免疫细胞可能在肥胖个体中增加了瘦素的影响, 根据动物实验, 一个信号可以激活先天免疫系统和同源免疫系统转向促炎Th1 T细胞群的优势,同时减少调节性Th2表型。
虽然在啮齿动物和人类的急性感染中,瘦素信号增强的免疫调节特性被认为是有益的,但长期来看,这种促炎性转移可能是有害的。事实上,非常肥胖、瘦素缺乏的小鼠,尽管有所有的代谢风险因素,但都可以防止动脉粥样硬化,这表明这种激素可能直接导致血管疾病的风险。
此外,在一项针对人类的前瞻性研究中,在控制了人体测量和代谢风险因素后,循环瘦素浓度被证明是预测心血管事件的独立风险因素。因此,长期高水平的瘦素,就像在肥胖者身上看到的那样,有可能导致动脉粥样硬化的进展。
然而,这些数据与以下事实有些不一致:女性的瘦素水平通常高于男性,而心血管疾病风险则较低。因此,尽管血清瘦素浓度增加,通过对免疫系统的直接影响,很明显可能触发促炎状态,但迄今为止,只有啮齿类动物研究的间接数据表明,这种细胞因子导致了与代谢综合征相关的慢性低级别炎症。
IL - 6
大多数细胞因子主要作为旁分泌或自分泌因子发挥作用。然而,IL-6并不常见,因为它是一种真正的内分泌细胞因子,这意味着该细胞因子的大多数细胞靶点离释放位点较远,IL-6的作用与血清浓度有关(41)。在脂肪组织内,脂肪细胞和巨噬细胞都分泌IL-6,而检测血清IL-6浓度动静脉增加的研究清楚地显示,脂肪组织库净分泌IL-6,表明脂肪约占人体循环IL-6浓度的30%。与瘦素一样,脂肪组织的IL-6的产生随着脂肪含量的增加而增加,而循环的IL-6浓度与体脂和胰岛素抵抗的百分比高度相关。然而,与瘦素一样,体外研究表明,对于给定重量的脂肪组织,皮下脂肪产生的IL-6要多于内脏脂肪,因此削弱了这种细胞因子与内脏脂肪依赖性代谢综合征之间的联系。
虽然从不同的脂肪组织的IL-6分泌的不同调节还有待确定,体液(胰岛素,糖皮质激素),神经(交感神经系统活动)和旁分泌(IL-1βTNF-α)信号都调节脂肪产生IL – 6, 确定IL - 6生产从内脏脂肪增加的因素仍然是一个重要的研究目标。虽然IL-6是一种高度多效的细胞因子,激素对许多组织都有影响,但对肝脏、骨髓和内皮细胞的影响被认为是导致肥胖代谢的最重要因素。
循环的IL-6是控制肝脏急性期反应的最重要的因素,是对组织损伤或感染的快速、协调的生理反应,目的是招募宿主防御机制,清除受损细胞,包含病原体,开始组织修复(45)。
在许多正的和负的急性相反应物中,可能最被认可的是CRP,它是pentraxin家族的一员,依附于受损细胞的质膜,通过激活补体级联导致细胞死亡。大量的流行病学数据连接CRP冠状动脉事件,动脉粥样硬化疾病,发展为2型糖尿病。显然,CRP是代谢风险的最强标志之一,而且可能直接参与导致动脉粥样硬化病变和心脏事件的动脉细胞壁机制。由于肝脏产生的CRP受循环IL-6控制,而且在工业化国家,血清IL-6浓度的唯一最重要决定因素是全身脂肪,因此,这种脂肪组织细胞因子很可能对与代谢综合征相关的慢性全身炎症疾病有重要作用。虽然增加的CRP是IL-6作用的最公认的标志,但许多其他IL-6依赖因素可能导致心血管风险。另一种急性期反应物——纤维蛋白原的增加是由IL-6介导的,血小板数量和血小板活性的增加都是由IL-6介导的,所有这些都会增加血栓形成的风险(50)。内皮细胞和血管平滑肌细胞是IL-6作用的靶点,导致粘附分子表达增加,局部肾素-血管紧张素通路激活,均有利于血管壁炎症和损伤。
在中枢神经系统中,IL-6是一种强大的分解代谢剂,它能减少食物的摄入,增加能量的消耗。
神经元和胶质细胞释放的IL- 6的表达和释放似乎对这种细胞因子对能量平衡的作用是必须的。但目前尚不清楚血清循环中IL-6如何控制中央性IL-6的产生,虽然存在IL-6透过血脑屏障进入大脑的机制。重要的是,具有基因缺失的IL-6的小鼠会出现成年期肥胖,这表明这种细胞因子参与了能量平衡的慢性生理调节,而IL-6信号的减少与体重增加有关。
因此,如果脂肪组织分泌的IL-6通过对中枢神经系统的内分泌作用而促进能量平衡,那么就可能引发肥胖诱导的IL-6抵抗状态,就像肥胖对瘦素和胰岛素信号传导的影响所描述的那样。
这一假设也表明,与肥胖相关的脂肪组织IL-6分泌增多可能是一种调节机制,试图纠正超重,实现负能量平衡,类似肥胖中瘦素增加的假设。因此,IL-6对肝脏和内皮细胞产生的系统性炎症可能是在肥胖和中央性IL-6抵抗的情况下适当升高IL-6水平的一个意想不到的后果。
因此,内分泌细胞因子IL-6可能是脂肪组织促炎信号的中介; 然而,旨在阻断IL-6作用的策略仍需作为代谢综合征的治疗手段进行评估。重要的是,限制IL-6对肝脏和内皮细胞的影响可能会严重地损害急性感染的正常宿主反应,而如果外周血IL-6的分泌对控制能量平衡的下丘脑区域提供负反馈,则防止脂肪组织分泌IL-6可能会加剧肥胖。
TNF-α
TNF-α在肥胖引发的系统性炎症反应已经被广泛的研究。巨噬细胞产生了脂肪组织中几乎所有的TNF-α. 在肥胖者中TNF-α mRMA含量和TNF-α的产生增加。反过来,循环中的TNF-α浓度也随肥胖和胰岛素抵抗的增加而增加。
然而,测量动静脉差异的一项研究显示皮下脂肪组织仓库没有净分泌TNF-α,虽然在体外研究表明内脏脂肪组织比皮下脂肪产生更多TNF-α。从内脏脂肪净分泌TNF-α进入血液循环还没有记录。因此,目前尚不清楚肥胖者是否TNF-α直接从脂肪组织分泌的升高血清TNF-α浓度。
体内研究使循环TNF-α的作用解释复杂化,
两个啮齿动物研究使用嵌合TNF-α受体(58)或超表达的可溶性TNF-α受体片段导致肥胖老鼠显著改善胰岛素抵抗,而其他研究在肥胖大鼠或在肥胖2型糖尿病患者,使用抗TNF-α抗体没有影响胰岛素的作用。
尽管如此,显然TNF-α作为旁分泌因子在肥胖引发的炎症反应可能有一个重要的作用,独立于循环细胞因子的浓度。脂肪组织内,TNF-α通过胰岛素受体(IR)和胰岛素受体底物1(IRS-1)
的丝氨酸磷酸化(失活),导致脂肪细胞胰岛素抵抗。这两个导致减少phosphoinositol-3-kinase激活-控制胰岛素的代谢作用的基本第二信使信号(图1)(62)。由TNF-α激活受体与胰岛素信号细胞内途径仍有待建立,但被认为涉及NF-κB和/或物信号。
IR和IRS-1激活的损伤也被证明发生在骨骼肌,它的提出,来自脂肪中巨噬细胞的TNF-α通过旁分泌机制可能会导致肌肉胰岛素抵抗。令人吃惊的是,这种胰岛素抵抗机制并不发生在肝细胞, 尽管肝脏可能会是内脏脂肪释放的TNF-α的主要靶标。尽管如此,在啮齿动物体内的一项研究显示TNF-α 被清除时肝的胰岛素敏感性改善,表明该细胞因子也可影响肝脏中关键的胰岛素依赖型通路。
Figure 1. Schematic overview of the potential interaction between the second messenger pathways activated by insulin and by TNF-α signaling
图示被胰岛素和TNF-α 信号激活的第二信使通路的可能相互影响
TNF-α可能导致胰岛素抵抗的第二个机制是通过脂肪溶解的诱导和刺激肝脂肪生成,导致血液游离脂肪酸(FFA)水平的升高。然而,大多数的TNF-α对脂质代谢的影响是在急性炎症或相对大剂量静脉注射TNF-α条件下研究的,鉴于肥胖者中远为温和的TNF-α浓度,使人怀疑这种机制的对代谢综合征的病理生理学的相关性。
最近,第三个潜在机制TNF-α影响胰岛素抵抗已被确认。由脂肪细胞分泌的脂联素被TNF-α信号大幅的减少,脂联素似乎是胰岛素敏感性的关键调节者,可能解释脂肪中TNF-α的旁分泌作用可能引起系统性胰岛素抵抗。
然而,预测TNF-α在代谢综合征的意义最近明显降温,因为TNF-α 或TNF-α受体遗传性缺失的小鼠对保护小鼠肥胖时体重增加、高血糖和胰岛素抵抗只有有限的作用。因此,尽管TNF-α是一种巨噬细胞来源的炎症因素,通过旁分泌和可能的内分泌机制导致脂肪和肌肉组织的胰岛素抵抗,其他炎性分子可以减弱TNF-α信号; 因此,TNF-α信号拮抗剂在治疗代谢综合征的可行性仍不清楚。
控制炎症信号传导的细胞内通路
尽管炎症可以明显影响胰岛素受体(IR)和胰岛素受体底物(IRS)信号,直到最近,细胞内细胞因子如TNF-α如何防止激活这些分子的机制仍然未知。
使用胰岛素抵抗体外模型的实验表明,丝氨酸/苏氨酸激酶可以使IR和IRS分子磷酸化,从而阻止酪氨酸磷酸化激活。虽然已知细胞因子激活细胞内的丝氨酸/苏氨酸激酶,相当大的注意力已经集中在抑制剂κB激酶(IKK)复合物作为胰岛素抵抗调控者(图1)。
这种酶复合物最出名的是触发NF-κB途径至关重要的作用,一个至关重要的炎症细胞因子信号的第二信使系统。当激活时,IL-1β和TNF-α受体招募激活IKK复合物的激酶。IKK2, 是IKK复合物的关键分解亚单位,磷酸化抑制剂κB, 导致细胞质伴侣分子降解,并允许转录因子NF-κB进入细胞核和激活炎症靶点基因。
体外研究表明,高剂量水杨酸盐可以抑制IKK复合物的活性。此外,在19世纪晚期,大剂量的阿司匹林已经被描述为糖尿病的一种治疗方法,并且显著降低了糖尿病患者的高血糖和糖苷三磷酸。
这二条精明的并列的信息导致许多最近的研究得出NF-κB途径导致胰岛素抵抗的作用的结论。使用大剂量阿司匹林,与其他非甾体类抗炎化合物不同, 在肥胖的啮齿动物体内和在体外TNF-α预处理的脂肪细胞(78),使胰岛素受体激活和下游胰岛素信号分子激活,逆转胰岛素抵抗。
缺乏IKK2基因的一个副本(IKK2 + /−) 的突变小鼠,在低脂和高脂饮食改善胰岛素抵抗,这杂合突变培育成瘦素缺失 ob / ob的老鼠能够降低高血糖和改善葡萄糖耐量,证明这些肥胖动物建立IKK复杂是一个至关重要的丝氨酸/苏氨酸激酶,当被激活时, 通过减少刺激IR和IRS-1活化,导致胰岛素抵抗。
然而,这一结论的说服力最近受到基因小鼠模型的质疑,该模型的肌肉特别缺乏IKK2活性,
即使在高脂肪饮食中也不会产生胰岛素抵抗(79)。因此,IKK复合物可能是多个
细胞因子激活的丝氨酸/苏氨酸激酶之一,可引起胰岛素抵抗。
同时,最近的证据指出JNK作为另一个TNF-α激活的激酶可能参与正常的IR和IRS-1功能的减少。
由于多种促炎性细胞因子可能对胰岛素信号传导有类似的作用,共同的的细胞因子诱导的第二信使通路可能是治疗代谢综合征和2型糖尿病的极好治疗靶点。
然而,这些细胞因子诱导的通路在骨骼肌、脂肪组织、肝脏和中枢神经系统中的单独作用仍有待确定,信号通路中的冗余问题需要克服。此外,系统性抑制炎症信号的化合物的潜在免疫抑制作用,在设计药物以阻止肥胖慢性炎症的有害影响的努力中,肯定仍然是一个重大的关注。
其他脂肪组织来源的炎性分子
许多其他炎症介质都是由脂肪组织(表2)产生并潜在地分泌到血液循环中(表2)。其中,脂联素、抵抗素、促酰基刺激蛋白、肾素-血管紧张素系统的组成部分可能是导致慢性炎症、胰岛素抵抗和心血管疾病风险的最重要因素。
结论
慢性炎症是代谢综合征的一个共同特征,炎症信号可能起源于内脏脂肪组织,因为这个脂肪库在长期的正能量平衡的作用下膨胀。脂肪细胞和巨噬细胞在脂肪分泌大量激素和细胞因子,可能导致代谢综合征常见的特征性病理生理改变,脂肪组织中的局部炎症可能是导致系统性胰岛素抵抗和系统性炎症的前哨事件-代谢综合征的二个基本特征。
引发脂肪组织膨胀的事件尚不清楚,但很可能具有重要的遗传成分,在相当肥胖的个体之间可能会造成代谢风险的变异。
单个细胞因子在引起代谢综合征的病理生理特征方面的作用仍有争议;然而;急性炎症所引发的代谢变化,在许多方面模拟代谢综合征,表明循环细胞因子,无论它们是来自脂肪组织还是外周血免疫细胞,可能对肌肉、肝脏和内皮细胞有类似的代谢作用,同时可能对免疫功能有不同的影响。
此外,脂肪组织衍生的细胞因子,如IL-6和瘦素,也可能直接参与血管内皮细胞的激活和炎症,这两种变化都可能导致动脉粥样硬化的进展。
对脂肪细胞和巨噬细胞之间相互作用的认识揭示了脂肪细胞源性因子调节局部免疫反应和巨噬细胞源性细胞因子改变脂肪细胞分化和代谢反应的新范式。
随着未来的研究继续提供有关炎症和新陈代谢之间的联系的新信息,确定治疗代谢综合征的新治疗方案的潜力仍然很大,鼓励全球努力减少与这一高度流行疾病有关的发病率。
美国肾脏病学会©2004
The Inflammatory Syndrome: The Role of Adipose Tissue Cytokines in Metabolic Disorders Linked to Obesity
Brent E. Wisse
- Author Affiliations
Division of Metabolism, Endocrinology and Nutrition, Department of Medicine, Harborview Medical Center, University of Washington, Seattle, Washington
Correspondence to Dr. Brent E. Wisse, Harborview Medical Center, 325 Ninth Avenue, Box 359757, Seattle, WA, 98104-2499. Phone: 206-341-4620; Fax: 206-731-8522; E-mail: bewisse@u.washington.edu
Next Section
Abstract
The metabolic effects of obesity have made this highly prevalent disease one of the most common risk factors for diabetes, hypertension, and atherosclerosis, the leading causes of end-stage renal failure. However, obesity per se, as defined by body mass index, is less predictive of the development of these diseases than is the presence of a constellation of obesity-related abnormalities now known as the metabolic syndrome. Recognition of this syndrome, which can readily be identified in clinical settings using defined threshold values for waist circumference, BP, fasting glucose, and dyslipidemia, allows for earlier intervention in these high-risk patients. Systemic insulin resistance has been implicated as one possible factor that links visceral obesity to adverse metabolic consequences; however, the mechanism whereby adipose tissue causes alterations in insulin sensitivity remains unclear. Infection and inflammation are commonly associated with insulin resistance, and visceral obesity is associated with a chronic, low-grade inflammatory state, suggesting that inflammation may be a potential mechanism whereby obesity leads to insulin resistance. Moreover, adipose tissue is now recognized as an immune organ that secretes numerous immunomodulatory factors and seems to be a significant source of inflammatory signals known to cause insulin resistance. Therefore, inflammation within white adipose tissue may be a crucial step contributing to the emergence of many of the pathologic features that characterize the metabolic syndrome and result in diabetes and atherosclerosis.
Worldwide, the prevalence of obesity is increasing dramatically, and recent estimates show that nearly two thirds of the U.S. adult population is now either overweight or obese (1). Meanwhile, pediatric obesity is increasing at an even more alarming rate, suggesting that this epidemic is unlikely to abate soon (2). Although increasing body weight, as defined by body mass index (BMI), is a powerful predictor of metabolic disease risk, (3) genetic and environmental factors cause considerable variability in the manifestation of type 2 diabetes and atherosclerosis for any given degree of obesity (4). One potential explanation for these differences is that individuals with the same BMI may have vastly different amounts of visceral (also referred to as “central” or “abdominal”) fat, the adipose tissue depot known to be associated with the greatest metabolic risk (5,6⇓). When visceral obesity is accompanied by a constellation of metabolic derangements, including insulin resistance, low HDL, elevated triglycerides, and raised BP (7), the predicted cardiovascular disease risk is increased significantly (8). This adverse metabolic profile is now referred to as the metabolic syndrome and is a highly prevalent condition in the United States, affecting nearly 25% of all adults (9). Clinically, the identification of these high-risk, obese patients has benefited from the recent publication of definitions by the National Cholesterol Education Program expert panel and the World Health Organization (10,11⇓). Although the criteria differ slightly (Table 1), the respective definitions both serve the clinical function of identifying obese patients who are at greater risk of developing comorbid metabolic conditions, including type 2 diabetes, hypertension, hyperlipidemia, and cardiovascular disease (12), allowing for earlier and more directed interventions. Although current theories focus on insulin resistance as the prime factor linking visceral obesity with adverse metabolic changes (7), studies suggest that the pathophysiology of the metabolic syndrome cannot be explained by insulin resistance alone (13). Beyond glucose homeostasis, dyslipidemia, and BP, many other pathophysiologic features have been characterized in individuals with the metabolic syndrome. Among these, evidence of chronic systemic inflammation is one of the most consistent (14), and numerous inflammatory markers are highly correlated with the degree of obesity and insulin resistance (15); many of these inflammatory markers are, in turn, highly predictive of vascular disease risk (16).
View this table:
In this window In a new window
Table 1. Metabolic syndromea
This review discusses the potential role of inflammation within fat in generating the metabolic syndrome, focusing on cytokines secreted by adipose tissue that modulate the immune system in favor of chronic systemic inflammation. The inflammatory roles of leptin, a fat-specific cytokine crucial to the control of energy balance, as well as the classical proinflammatory cytokines IL-6 and TNF-α, are highlighted specifically.
Adipose Tissue Endocrinology
The conceptual transformation of adipose tissue from a passive organ of energy storage to an active participant in hormonal regulation of homeostatic systems occurred relatively recently. In 1994, adipose tissue was identified as the source of the hormone leptin, opening the door for a new era of research focused on adipocyte endocrinology (17). In the past decade, the endocrine role of leptin has expanded to include regulation of reproduction (18) and immune function (19), and numerous other adipose tissue-derived molecules that have an impact on glucose homeostasis, vascular biology, tumor development, lipoprotein metabolism, and inflammation (20) have been identified, and these inflammatory factors are listed in Table 2. However, adipose tissue is an inhomogeneous organ that consists of a variety of cell types, a fact that has prompted significant debate as to the true role of the adipocyte versus stromal/vascular and immune cells in secreting some of these endocrine and paracrine regulators. In fact, mature adipocytes seem to lack the storage vesicles and other structural cellular components usually associated with regulated release of secreted proteins by endocrine cells. In addition, the frequent use of pre-adipocyte cell lines to study adipocyte biology in vitro has added to this controversy because the functional characteristics (21) and transcriptional patterns of these multipotent cells are similar to immune cells.
In fact, immature fat cells can transdifferentiate into macrophages both in vitro and in vivo. (22). Recent studies using large-scale genetic analyses to characterize gene expression patterns in adipose tissue from a variety of obese and lean mice have begun to clarify the role of the adipocyte as a hormone and cytokine secreting cell (23,24⇓). Surprising, in these studies, increasing adiposity in mice correlates very highly with the adipose tissue expression of a large cluster of genes characteristically expressed by macrophages, and both adipocyte size and total body weight are strong predictors of the number of mature macrophages found within adipose tissue, the correlation being even stronger for visceral than for subcutaneous fat. These bone marrow-derived macrophages seem to invade fat in response to as-yet-unknown signals and in obese animals tend to aggregate and form giant cells characteristic of chronic inflammatory disorders, suggesting that adipose tissue is a site of active inflammation. Gene expression studies on sorted cells from adipose tissue revealed that macrophages produce almost all TNF-α, whereas mature adipocytes secrete the majority of leptin and roughly equal IL-6 gene expression was found within macrophages, adipocytes, and nonmacrophage stromal-vascular cells. Importantly, these studies suggest that macrophage invasion of fat and inflammation-related gene expression in adipose tissue may be a sentinel event, preceding the development of insulin resistance in these animals. Therefore, weight gain in mice is associated with infiltration of fat by macrophages and elaboration of proinflammatory signals from adipose tissue. Notably, these inflammatory changes are most marked within visceral fat, the fat depot associated with greatest metabolic risk, and seem to precede other features of the metabolic syndrome, including impaired glucose homeostasis.
The existing paradigm of adipose tissue endocrinology has been changed significantly by introducing immune cells as a source of inflammatory mediators released from adipose tissue, as well as a paracrine regulator of adipocyte function and hormone secretion, thereby potentially controlling the metabolic changes that result from excess adiposity. Research designed to elucidate the signals that attract macrophages to fat and dissect the paracrine mechanisms whereby adipose tissue macrophages and adipocytes communicate is likely to identify therapeutic targets that may decrease fat-induced inflammation and perhaps diminish the metabolic risk associated with obesity.
Fat and the Immune System
Although that macrophages infiltrate fat as a result of obesity is surprising, interactions between adipose tissue and the immune system are well described, and various theories have been put forth to try to address the question of why such a link should exist (19). White adipose tissue and bone marrow share an embryologic origin, the mesoderm, and pre-adipocytes are potent phagocytes that resemble macrophages in both morphology and patterns of gene expression (21). In addition, mature adipocytes share the ability to secrete cytokines and activate the complement cascade much like mononuclear immune cells. One line of reasoning connecting fat and the immune system is based on the role of the adipocyte hormone leptin in the control of energy homeostasis and immunity. During fasting/starvation, when plasma leptin levels decline, neural pathways in the hypothalamus cause appetite to increase and energy expenditure to decrease, an attempt to restore body fat stores (25). In addition, the fall in plasma leptin diminishes thyroid hormone production (26) and inhibits the reproductive axis, both effects that save energy during nutritionally lean times (27). However, starvation also inhibits the immune system, resulting in increased apoptosis in the thymus and decreased mononuclear cell proliferation, effects that can be entirely reversed by administering leptin alone in the absence of refeeding (28). Because maintaining immune function has been estimated to account for as much as 15% of daily energy expenditure (29), here, too, leptin’s role may be taken as contributing to reducing overall energy expenditure; thus, the regulation of immunity by adipose tissue can be seen as an extension of the endocrine role of fat in energy homeostasis.
The second argument for fat/immune system coordination is rooted in teleology. In Drosophila, the fat body is the organ that governs the innate immune system, secreting into the lymphatic system molecules that target ingested pathogens (30). Similarly, in vertebrates, fat may be one of the crucial alarm systems that rouses the innate immune system and triggers the acute-phase response, the first line of defense against bacterial infection. Certainly the adipose tissue production of leptin increases dramatically during acute infection (31), and fat may contribute to elevated circulating IL-6, TNF-α, and IL-1β, as well as numerous other factors that prime the immune system or participate directly in the defense against pathogenic organisms. The question of whether adipose tissue plays a significant role in controlling immune function in humans has yet to be answered conclusively; however, that adipose tissue can elaborate acute inflammatory signals suggests that dysregulation of the inflammatory capability of fat could play a role in triggering or perpetuating the chronic inflammation characteristic of the metabolic syndrome. Although acute and chronic inflammation are dissimilar in many ways, the metabolic perturbations seen during acute infection share many common features with the known characteristics of the metabolic syndrome. Most important, insulin resistance and hyperglycemia are common to both conditions, as are hypertriglyceridemia, impaired lipolysis, and increases in nonesterified fatty acids. Leptin, IL-6, TNF-α, the acute-phase reactant C-reactive protein (CRP) as well as other circulating inflammatory markers are elevated in both conditions. In addition, in rodents, more clearly than in humans, both obesity and acute infections are associated with an activation of the hypothalamic-pituitary-adrenal axis and increased circulating glucocorticoid levels. During acute infections, these metabolic changes are thought to be adaptive, sparing fuel for the brain, activating the complement cascade on bacterial cell surfaces, depriving bacteria of iron and other vital micronutrients, etc., but as with other adaptive physiologic processes, pathologic consequences may result when these acute changes become chronic. Equating metabolic syndrome-associated chronic inflammation with acute inflammatory responses to infection is clearly too facile, yet it highlights the intriguing possibility that immune/inflammatory factors may be at the root of the metabolic perturbations associated with both obesity and acute illness. The biologic response to a given cytokine may differ between acute and chronic inflammatory states, and the mechanism governing such differential responses often remains obscure. Clearly, although similar cytokine mediators seem to be involved, the nonmetabolic features of obesity and acute inflammatory syndromes are vastly different and multiple mechanisms may account for this variability, including differences in plasma cytokine concentration, presence of soluble cytokine receptors and receptor antagonists, balance of pro- versus anti-inflammatory cytokine networks at the tissue level, and presence of bacterial cell wall products, to name only a few of the many potential variables. One intriguing metabolic dissimilarity between these two conditions is worthy of mention, namely the control of appetite. Both chronic inflammatory conditions and acute infections are usually associated with anorexia and cachexia, which are believed to be cytokine mediated (32), whereas appetite in individuals with obesity/metabolic syndrome is not obviously diminished, despite elevations in the circulating concentrations of cytokines associated with anorexia. Therefore, although the possibility exists that the metabolic syndrome is caused by an inflammatory response gone awry and, perhaps more specific, an inflammatory response within adipose tissue that becomes maladaptive when chronically stimulated, the details of specific cytokine-mediated effects and the mechanistic role of various inflammatory mediators remains to be defined clearly. The following sections summarize the potential roles of three key adipose tissue-derived cytokines—leptin, IL-6, and TNF-α—in contributing to the manifestations of the metabolic syndrome.
Leptin
The adipocyte-derived hormone leptin is a critical mediator of energy balance that relays information regarding the depletion or accumulation of fat stores to the brain (25,33⇓). Although identified as a classic peptide hormone, the four α helix domains in the folded structure make leptin most similar to cytokines such as IL-2. Moreover, the leptin receptor (lepr) bears significant homology to type 1 cytokine receptors; therefore, the hormone leptin is in many ways more appropriately identified as a cytokine. Although the role of leptin in controlling energy homeostasis is increasingly well defined (25,33⇓), it remains unclear whether leptin plays a role in the inflammatory syndrome caused by abdominal obesity. Clearly, serum leptin concentrations rise in proportion to body adiposity (34); therefore, obese individuals with the metabolic syndrome generally have higher circulating leptin concentrations. However, obese individuals seem to be resistant to the hypothalamic effects of leptin (33); therefore, the catabolic pathways designed to reduce appetite and increase energy expenditure are not activated and excess body weight is maintained. Although many of leptin’s effects result from a direct action of leptin on hypothalamic neurons, the functional leptin receptor (long-form or leprb) is also found on many tissues outside the central nervous system (CNS), including immune cells (35,36⇓). Importantly, the mechanisms that are thought to contribute to hypothalamic leptin resistance (33), e.g., defective blood-brain barrier transport, are either moot or unproved in peripheral tissues, and no studies have documented peripheral leptin resistance in obese individuals. Therefore, immune cells may well be subject to increased leptin effects in obese individuals, a signal that, on the basis of animal studies, may serve to activate the innate immune system and shift the cognate immune system toward a predominance of a proinflammatory Th1 T cell population while reducing the regulatory Th2 phenotype (19). Although the immunomodulatory properties of increased leptin signaling are thought to be beneficial during acute infections in both rodents (37) and humans (38), chronically, this proinflammatory shift may be deleterious. In fact, very obese, leptin-deficient mice are protected from atherosclerosis despite all of the metabolic risk factors, suggesting that this hormone may contribute directly to the risk of vascular disease (39). Moreover, in a prospective study in humans, circulating leptin concentration was shown to be an independent risk factor in predicting cardiovascular events after anthropometric and metabolic risk factors had been controlled for (40). Therefore, chronically elevated concentrations of leptin, as seen in obese individuals, may potentially predispose to progression of atherosclerosis. These data, however, are somewhat incongruent with the fact that women generally have higher leptin levels than men (34) while having lower cardiovascular disease risk. Therefore, although increased serum leptin concentrations, through direct effects on the immune system, clearly may trigger a proinflammatory state, as yet only circumstantial data from rodent studies suggest that this cytokine contributes to the chronic low-grade inflammation associated with the metabolic syndrome.
IL-6
Most cytokines function predominantly as paracrine or autocrine factors. However, IL-6 is unusual in that it is a true endocrine cytokine, meaning that most cellular targets of this cytokine are distant from the site of release and the effects of IL-6 are correlated with the serum concentration (41). Within adipose tissue, both adipocytes and macrophages secrete IL-6 (23), and studies measuring arteriovenous increases of serum IL-6 concentration have clearly shown net secretion of IL-6 from adipose tissue depots, suggesting that fat accounts for roughly 30% of circulating IL-6 concentrations in humans (42). Like leptin, production of IL-6 by adipose tissue increases with increasing adiposity, and circulating IL-6 concentrations are highly correlated with percentage of body fat (43) and with insulin resistance (44). However, like leptin, in vitro studies have shown that for a given weight of adipose tissue, subcutaneous fat produces more IL-6 than visceral fat, thereby weakening the link between this cytokine and the visceral fat-dependent metabolic syndrome. Although the differential regulation of IL-6 secretion from different fat depots and from distinct adipose tissue cell types remains to be determined, humoral (insulin, glucocorticoids), neural (sympathetic nervous system activity), and paracrine (IL-1β, TNF-α) signals all have been shown to regulate IL-6 production from fat, and determining the factors that increase IL-6 production from visceral fat remains an important research goal. Although IL-6 is a highly pleiotropic cytokine, with hormonal effects on many tissues, the effects on the liver, bone marrow, and endothelium are thought to be most significant in contributing to the metabolic effects of obesity.
Circulating IL-6 is the single most important factor controlling the hepatic acute-phase response, the rapid, coordinated physiologic reaction to tissue damage or infection designed to recruit host defense mechanisms, eliminate damaged cells, contain pathogens, and begin tissue repair (45). Of the many positive and negative acute-phase reactants, perhaps the most recognized is CRP, a member of the pentraxin family that attaches to the plasma membrane of damaged cells causing cell death through activation of the complement cascade (46). A large volume of epidemiologic data connect CRP to coronary events, atherosclerotic disease, and progression to type 2 diabetes (47,48⇓). Clearly, CRP is one of the strongest markers of metabolic risk and, in addition, may participate directly in the arterial cell wall mechanisms leading to atherosclerotic lesions and cardiac events (49). Because CRP production by the liver is governed by circulating IL-6 and because, in industrialized countries, the single most important determinant of serum IL-6 concentration is whole-body adiposity, it is likely, therefore, that this adipose tissue cytokine contributes significantly to the chronic systemic inflammatory disorder associated with the metabolic syndrome. Although increased CRP is the most recognized marker of IL-6 action, numerous other IL-6-dependent factors may contribute to cardiovascular risk. Increases of fibrinogen, another acute-phase reactant, are mediated by IL-6, as are increases in both platelet number and platelet activity, all of which would contribute to the risk of clot formation (50). Moreover, endothelial cells and vascular smooth muscle cells are targets of IL-6 action, resulting in increased expression of adhesion molecules and activation of local renin-angiotensin pathways, both modifications that favor vascular wall inflammation and damage (51).
In the CNS, IL-6 is a powerful catabolic agent that leads to decreased food intake and increased energy expenditure, based on numerous studies involving the administration of IL-6 into the cerebral ventricles (52). The expression and release of IL-6 by neurons and glial cells seems to be essential for the effects of this cytokine on energy balance, but it remains unclear to what extent central IL-6 production is controlled by circulating IL-6 in the serum, although transport mechanisms seem to exist to deliver IL-6 across the blood-brain barrier (52). Importantly, mice with a genetic deletion of IL-6 develop adult-onset obesity, suggesting that this cytokine is involved in the chronic physiologic regulation of energy balance and that decreased IL-6 signaling is associated with weight gain (53). Therefore, if IL-6 secretion by adipose tissue contributes to energy homeostasis through an endocrine action on the CNS, then one could invoke a state of obesity-induced IL-6 resistance, much as described for the effects of obesity on leptin and insulin signaling. This supposition also suggests the possibility that increased adipose-tissue IL-6 secretion associated with obesity may be a regulatory mechanism attempting to correct excess body weight and achieve negative energy balance, as hypothesized for obesity-related increases in leptin. The systemic inflammation resulting from IL-6 effects on liver and endothelium therefore could be an unintended consequence of appropriately elevated IL-6 levels in the face of obesity and central IL-6 resistance.
The endocrine cytokine IL-6, therefore, is a likely mediator of proinflammatory signaling from adipose tissue; however, strategies designed to block IL-6 action remain to be evaluated as treatments of the metabolic syndrome. Importantly, limiting IL-6 effects on liver and endothelium may well impair the normal host response to acute infection, whereas preventing IL-6 secretion from adipose tissue could potentially worsen obesity if peripheral IL-6 secretion provides negative feedback to hypothalamic areas that govern energy balance.
TNF-α
The role of TNF-α in the systemic inflammatory response triggered by obesity has been studied extensively (54). Within adipose tissue, macrophages account for nearly all TNF-α production (23), and both TNF-α mRNA content and TNF-α production increase in adipose tissue of obese individuals (55). Circulating TNF-α concentration, in turn, also rises with increasing obesity and correlates with insulin resistance (56). However, a study measuring arteriovenous differences showed no net secretion of TNF-α from subcutaneous adipose tissue depots (42), and although in vitro studies have shown that visceral adipose tissue produces more TNF-α than subcutaneous fat (57), net secretion of TNF-α from visceral fat into the circulation has not yet been documented. Therefore, it remains unclear whether TNF-α secretion from adipose tissue directly accounts for the elevated serum TNF-α concentration seen in obesity. In vivo studies have complicated the interpretation of the role of circulating TNF-α, as two rodent studies using chimeric TNF-α receptor (58) or overexpression of a soluble TNF-α receptor fragment (59) both resulted in significant improvement of insulin resistance in obese rats, whereas other studies using anti-TNF-α antibodies had no effect on insulin action in obese rats (60) or in obese individuals with type 2 diabetes (61). Nonetheless, TNF-α clearly may have an important role as a paracrine factor in the inflammatory response triggered by obesity independent of circulating concentration of the cytokine. Within adipose tissue, TNF-α causes adipocyte insulin resistance through serine phosphorylation (inactivation) of both the insulin receptor (IR) and insulin receptor substrate 1 (IRS-1), both of which result in diminished activation of phosphoinositol-3-kinase, the essential second messenger signal that governs most of insulin’s metabolic effects (Figure 1) (62). The intracellular pathways activated by the TNF-α receptor that interact with insulin signaling remain to be established but are thought to involve NF-κB and/or JNK signaling (63,64⇓). Impairment of activation of IR and IRS-1 has also been shown to occur in skeletal muscle (65), where it is proposed that TNF-α derived from macrophages within the fat depots that lie between myocytes may result in muscle insulin resistance via a paracrine mechanism (23). Surprising, this mechanism of insulin resistance does not occur in hepatocytes, even though the liver would presumably be the prime target for TNF-α released by visceral fat (66). Nonetheless, one in vivo study in rodents did show an improvement in hepatic insulin sensitivity when TNF-α was neutralized, suggesting that this cytokine may also have an impact on critical insulin-dependent pathways in the liver (59).
Figure
View larger version:
In this page In a new window
Download as PowerPoint Slide
Figure 1. Schematic overview of the potential interaction between the second messenger pathways activated by insulin and by TNF-α signaling.
A second mechanism whereby TNF-α may contribute to insulin resistance is through elevations in circulating FFA levels caused by the induction of lipolysis and stimulation of hepatic lipogenesis (67). However, most of the effects of TNF-α on lipid metabolism have been studied under conditions of acute inflammation or with relatively high-dose intravenous TNF-α administration (68), casting doubt on the relevance of this mechanism to the pathophysiology of the metabolic syndrome given the more modest elevations in TNF-α seen in obesity.
More recently, a third potential mechanism whereby TNF-α influences insulin resistance has been identified. Adiponectin secretion by adipocytes is potently reduced by TNF-α signaling (69), and this hormone seems to be a crucial mediator of insulin sensitivity, potentially explaining how paracrine effects of TNF-α within fat could cause systemic insulin resistance.
However, predictions regarding the significance of TNF-α in the metabolic syndrome have been tempered significantly by the fact that mice with genetic deletions of either TNF-α or the TNF-α receptors demonstrate only modest protection from weight gain, hyperglycemia, and insulin resistance when obese (70,71⇓). Therefore, although TNF-α is a macrophage-derived inflammatory factor that contributes to insulin resistance in adipose tissue and muscle via paracrine and potentially endocrine mechanisms, other inflammatory molecules may be able to compensate for the absence of TNF-α signaling; thus, the viability of antagonists of TNF-α signaling in the treatment of the metabolic syndrome remains unclear.
Intracellular Pathways that Control Inflammatory Signaling
Although inflammation can clearly impair IR and IRS signaling, until recently, the intracellular mechanism whereby cytokines such as TNF-α prevent activation of these molecules has remained unknown. Experiments using in vitro models of insulin resistance have demonstrated that serine/threonine kinases can phosphorylate both IR and IRS molecules such that activation by tyrosine phosphorylation is prevented (72). Although cytokines are known to activate a number of intracellular serine/threonine kinases, considerable attention has now focused on the inhibitor κB kinase (IKK) complex as a mediator of insulin resistance (Figure 1). This enzyme complex is best known for a vital role in triggering the NF-κB pathway, a crucial second messenger system for inflammatory cytokine signaling (73). When activated, IL-1β and TNF-α receptors recruit kinases that activate the IKK complex. IKK2, the key catalytic subunit of the IKK complex, phosphorylates inhibitor κB, causing this cytoplasmic chaperone molecule to be degraded and allows the transcription factor NF-κB to translocate into the nucleus and activate inflammatory target genes (74). In vitro studies have shown that the activity of the IKK complex can be inhibited by high-dose salicylate administration (75). Moreover, high-dose aspirin was already described as a treatment of diabetes in the late 1800s (76) and dramatically decreases hyperglycemia and glycosuria in patients with diabetes (77). The astute juxtaposition of these two pieces of information led to a number of recent studies demonstrating conclusively a role for the NF-κB pathway in causing insulin resistance. High-dose aspirin administration, unlike other nonsteroidal anti-inflammatory compounds, reversed insulin resistance and normalized the activation of the insulin receptor and downstream insulin signaling molecules in obese rodents in vivo and in TNF-α-treated adipocytes in vitro (78). That mutant mice lacking one copy of the IKK2 gene (Ikk2+/−) had improved insulin sensitivity on both low-fat and high-fat diets and that this heterozygous mutation bred into leptin-deficient ob/ob mice was able to reduce hyperglycemia and improve glucose tolerance in these obese animals established that the IKK complex is a critical serine/threonine kinase that, when activated, results in insulin resistance by diminishing insulin-stimulated IR and IRS-1 activation. However, the strength of this conclusion was questioned recently by a genetic mouse model specifically lacking IKK2 activity only in muscle that did not develop insulin resistance even on a high-fat diet (79). Therefore, the IKK complex may be one of multiple cytokine-activated serine/threonine kinases that can cause insulin resistance, and recent evidence points to JNK as another TNF-α-activated kinase that may be involved in diminishing normal IR and IRS-1 function (63,80⇓). As multiple proinflammatory cytokines may have similar effects on insulin signaling, the shared, cytokine-induced second messenger pathways may be excellent therapeutic targets for the treatment of metabolic syndrome and type 2 diabetes. However, the individual role of these cytokine-induced pathways in skeletal muscle, adipose tissue, liver, and the CNS remains to be determined, and the issue of redundancy within signaling pathways will need to be overcome. Moreover, the potential immunosuppressive effects of compounds that globally dampen inflammatory signaling must remain a significant concern in the effort to design drugs to block the deleterious effects of chronic inflammation in obesity.
Other Adipose Tissue-Derived Inflammatory Molecules
Numerous other inflammatory mediators are produced and potentially secreted into the circulation by adipose tissue (Table 2). Among these, adiponectin (81), resistin (20), acylation-stimulating protein (82), and components of the renin-angiotensin system (83) may be the most important in contributing to chronic inflammation, insulin resistance, and cardiovascular disease risk.
Previous Section
Next Section
Conclusion
Chronic inflammation is a common feature of the metabolic syndrome, and inflammatory signals may originate within visceral adipose tissue as this fat depot expands in response to chronic positive energy balance. Both adipocytes and macrophages within fat secrete numerous hormones and cytokines that may contribute to the characteristic pathophysiologic changes seen in the metabolic syndrome, and local inflammation within adipose tissue may be the sentinel event that causes systemic insulin resistance and systemic inflammation, two of the cardinal features of the metabolic syndrome. The inciting event that causes adipose tissue to become inflamed as it expands remains unknown but will likely have a significant genetic component potentially accounting for some of the variance in metabolic risk between equivalently obese individuals. The contribution of individual cytokines in causing the pathophysiologic features of the metabolic syndrome remains controversial; however; that the metabolic alterations triggered by acute inflammation mimic the metabolic syndrome in many ways suggests that circulating cytokines, whether they are derived from adipose tissue or peripheral blood immune cells, may have similar metabolic effects on muscle, liver, and endothelium while potentially having differential effects on immune function. In addition, adipose tissue-derived cytokines, such as IL-6 and leptin, may also participate directly in endothelial cell activation and inflammation in the vascular bed, both changes that could contribute to the progression of atherosclerosis. Recognition of the interaction between adipocytes and macrophages within fat exposes a new paradigm whereby adipocyte-derived factors modulate local immune responses and macrophage-derived cytokines alter adipocyte differentiation and metabolic responses. As future studies continue to provide new information regarding links between inflammation and metabolism, the potential to identify new therapeutic options for the treatment of the metabolic syndrome remains high, encouraging the global effort to reduce the morbidity associated with this highly prevalent disease.
© 2004 American Society of Nephrology
The Inflammatory Syndrome: The Role of Adipose Tissue Cytokines in Metabolic Disorders Linked to Obesity http://jasn.asnjournals.org/content/15/11/2792.full
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)
.png)