��
������֢�ǽ��֢֮�յĹؼ���?
Is Chronic Inflammation the Key to Unlocking the Mysteries of Cancer?
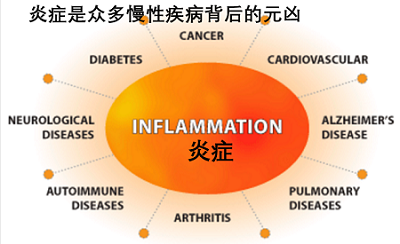
�˽�������֢���ᵼ�����ಡ�������ĺ�Ĭ���������������������ǽ��֢֮�յĹؼ�
����ע:��������������2007��7�µġ���ѧ�����ˡ���־�Ϸ����ģ������������µ��о�������Ѫ���������Ƽ����ܻ�ʹ������ø������Ǹ�С��
5�ڶ���ǰ��һ��ר�ŵ�ø�͵����ʽ������������ǵ�ԭʼ���������ⲿ����Ĺ��������һ������ͻ����һЩ��������ʱ���Ķ���Ⱥ����ǣ����ڹ��ϵ�����ϵͳ�ij�Ա�����Щ�����߽���Ұ����Э���Ĺ�������ϸ�����Ͽ��ף��³���ѧ���أ����߽��������ʺ������������ˡ�һ�������߱���Dz������Ӫ�ͻῪ�������ϸ�������������������ϸ�����������ͻ���������Ϣ��
�����������߷�Ӧ�dz���Ч���������Ľ��������У���������涼�õ��˱���������֪��������ȷ�ģ���Ϊ�о����֣�������������ͬ����������Ϊ�͵ȹ�Ӭ����������������ڳ���5����ǰ�Ĺ�ͬ�����зֻ���
������������ѧ�о���Ա����һ��������ϵͳ�Ĺ�ע��Խ��٣���������Ϊ����һ���������������Դ��κ������Ƥ������ǣ�����С�ķ�϶�����ǶԸ��Ƚ�����Ӧ������ϵͳ�����˼���Ĺ�ע����������ϵͳ���Խ������������������������ʶ���������ߣ�����һ������δѱ��������ϵͳ��ȱ�������ԡ�
�ڹ�ȥ��15����������߱��ܹ�ע����֢�����ı�־�������Ѿ�������Ϊ�����������Լ�����DZ�����أ��������ʪ�Թؽ��Ϳ��������������Ե�������ף���������������֢���Լ����ಡ���з����Ҫɱ�֡������ʮ������������Ҫ��ɱ�ְ�֢����ϵ�Ŀ������ܵ����ϸ����顣����֢�Ͱ�֢֮�����ϵ�Ѿ�ת�Ƶ��о���������ĽΣ�����ʡ����ѧԺ�Ļ��غ�������ҽѧ�о��������ء�A���²���(Robert a . Weinberg)ָ��������������Ҫ�̿��顶��֢����ѧ��(Garland Science, 2006)��ǿ������һ�仯���ص㡣
����ת����ʶ����������״̬��������չ���ڵĹؼ��н��塣��֢��ʼ��һϵ�еĻ���ı䣬��ʹһ��ϸ�����ȸ��ƣ�Ȼ����Ϯ��Χ��֯�����������Ķ���������ʼ�ĵط������գ�һЩ����ϸ�����ܻ���ѣ�����ңԶ�ĵط������µ�����(ת��)����һ���Ѿ��������˺ܳ�ʱ�䡣����֢����ѧ�Һ�����ѧ���Ѿ���ʼ��ʶ���Ӳ�����֯�ݱ䵽��ȫ�İ�����֯��Ҫ����֯���˵�����ϸ���IJ��룬�����ǽٳֵ�������֯��Ϊ�����ͬı�Ͱ��ס�����һЩ�о���Ա�������Ķ��״̬:���������ǵ�ȼ����Ļ����֢������������ȼ����
����д�̿���Ĺ����У�������������һ���쳣ϸ��;��������һ��֧��ϵͳ��һ���������������������ֲ�ͬ������ϸ�����ͺͽ���Ļ�ѧ�źţ��Լ�һ��Ѫ�����硣�����Ĵ�����һ���Ƿ����ٵ�״̬�����Ĵ��ڲ���Ϊ������ѪҺ���ų����ڵĶ��أ�����ֻ������������Ŀ�ġ�
�����¹۵���ζ�ţ���������ÿһ����ϸ�������Dz���Ҫ�ġ��෴�����װ�֢���ƿ��Է�ֹ��ǰϸ�����䣬����ֹ����������ɢ�������Զ�˲�λ����֢����Ҳ���ܹ����������������ҩ���ð��̲�����ø���һ�������Ҳ���Ϊ�����DZ�Ȼ��Ŀ�ꡣ���ݴ�ѧ�ɽ�ɽ��У�İ�֢����ѧ��Lisa M. Coussens���۵�����������ܿ��Ƽ������ȹ������Ȼ�������ǽ���һ�����ʤ������
���������
������֢�Ͱ�֢֮�����ϵ��Ҫ�˽�����������ߵķ�Ӧ���Լ�����֢״̬����̫��ʱ����������������α�ת��Ϊ�ٽ���֢�ġ�����ȵ�һ�Ŷ��Ӻ�������ŵ�ϸ�����ܵ�����һϵ�е����ʺͰ�ϸ���Ļ�ӭ����Щ�����ʺͰ�ϸ�������ڵ�Ӱ�����ж���2��������ѡ�ǡ�
��һ������:��Լ20�����嵰���ʣ���������������Ϊ����������������������ศ��ɣ�
�û�ѧ�����������߷���Ϊԭ�����š��ڲ���ϵͳ���Һ��ס��������ͬʱ���̿���������Ϊ��רҵ������ϸ��-רҵ��ʳϸ�������Ź�����
ȱ���������ǣ���Щ��ľ���ϸ����������ϸ���������ɡ�δ������Ŀ��ˡ��������õ�������Ա������Ȼɱ��ϸ�����ʴ�ϸ������������ϸ�������������Ƕ������߷������������Ѫ�йص�ѪС����ע��Ѫ�ܵ��ڲ�ת�Ƶ�Ƥ�������Ѵ���øֱ����ϸ������ʣ���ϸ���̶����ĵ����ʻ��ʡ�һ�����γɣ�Ƥ����������������֢���̽�����Ȼ������ʱ��֢����ֹͣ���κ�������֢����֯(��������Ƥ��)����Ϊ��ԭ�塢���ػ�������˻ᶼ�ᵼ�¼����������ಡ����֢��
���˵�һ������⣬�����ﻹװ���˶��������������Ӧϵͳѧϰ�����ߵ��ض�����������Ȼ������Ϊɱ��Ŀ�ꡣ���е�������Bϸ����������������ӣ��ܹ��кͲ�ԭ�壬���߱�������Ա���𣬴��⣬Tϸ���������ʹ��Ⱦϸ����ɱ����ڻ�ѧ���ʣ�ֱ��Ӱ����������ϵͳ�Ļ��
��������������֤�ݱ�����������֢��ijЩ���͵������ķ�չ������������Ҫ�����á���֢����֢֮�����ϵһֱ�����ɡ���1863�꣬�����ĵ¹�����ѧ��Rudolf Virchowע��ڶ�����֯�д�����ν���ܰ�ϸ������(��ϸ��)������1978�꣬�����ٴ��о�����������ѧ��Alberto Mantovani�۲쵽����������ϸ�������ھۼ���һЩ������Χ������ҽѧԺ�İ�֢����ѧ��Harold F. Dvorak��1986��˵�������ǡ��������ϵ��˿ڡ���Ȼ������״ȴ�ڱ�������ʮ��ǰ����������ѧ����Ȼ�����Ϊ����ϵͳ������������ԭ�壬���ҿ����������ǰ���쳣ϸ����������ϸ�۲�������Χ���������������벻�������顣
���Եĸ���
��1990���ĩ���ش�ѧ��QUEEN MARY ��֢�о�����Frances Balkwillһֱ���о���Ϊ������������(TNF)��ϸ������(�����������źŷ���), ��������������Ϊ�����ֱ��ע������ʱ���������ӿ���ɱ����ϸ�����������Ե�ˮƽ��TNF�������ǻ�ʱ���������þʹ���ͬ�ˡ�Balkwill��ʵ���ҹر��������TNF����ʹ�������������ֵ�����:�����dzԾ����ǣ�����û�и�Ⱦ������������˵:������İ����ǵ����˸����е�è���������о�TNF���˶����Ż��ˡ�����ԭ��Ϊ����ϸ��������һ�����ư�֢�ķ���ʵ������һ���������������ӡ�
�����ó�С����ֳɿ����ԣ����Բ���ѡ���Թرջ����Ч����������ͻ����֢-��֢����ϵ��Coussens������ͬ��Douglas Hanahan��Zena Werb��1999�걨��˵���ü���İ�֢��������С����û�зʴ�ϸ��(��һ�ֹ�������ϸ��)��չ����û�н�չ����ȫ���������İ�ǰ��֯��2001�꣬�ܸ��w��������(Jeffrey W. Pollard)�����ڰ������ء�����˹̹ҽѧԺ(Albert Einstein College of Medicine)��ͬ���Ƕ������������������Щ����Ļ���������ڻ������ٰ��������������Dz����İ�ǰ��֯��û����ȫ����ԣ���������þ���ϸ���İ�����
�ĺ��ͼƬ��û����ȫ�Ʒ��ɵġ���ʵ�ϣ�����ʾ������ϵͳ��һ��˫�н������Ӻ�ϸ�������磬�����ڴ��Եĸ����ԣ���Ȼ��һ�����:��ʱ����ٽ���֢;����ʱ�������谭������ijЩ���͵���Ȼ����ϸ��������Ȼɱ��ϸ����ʵ���Ͽ��Է�ֹ��������������������������Ϊ����״̬ʱ����������ϸ������������������; ���û����֢�����ǿ��ܻ����Ū�������⣬��֢�������������в���������������ȫ��������ѪԴ��֢����ϵ�в���ȷ��
��Ѱ���������ʱ���о���Ա�����������۽��ھ���ϸ���ϣ�������ϸ�������������еİ�ϸ����ռ������Ҫ��λ�á�����ϸ���ܹ�ɱ������ϸ����������ϵͳ��Tϸ��������������������ȱ�ݵġ����ǣ������º������о���Ա���о��Ѿ���ϸ˵���˾���ϸ������α���ϸ�������½�������������ǵ�����ġ����dz�Ϊϸ�����Ӻ��������ӵĹ������ٽ������ķ�չ��
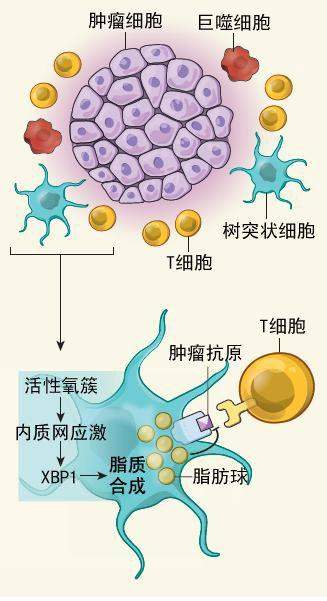
������ϸ����Ϊ����ϸ����ϸ�����������źţ��������ǽ�������ʱ������ϸ����ʼת��Ϊ��ͽ���������ڲ���������ϸ�����������֮�죬���������ǿ�ʼ��ȱ����������ȱ������������ϸ������Ϣ�Ľ��������һ�����̣����µ��ľ���ϸ����Ϊ�����ƶ��ߵĻ����ݡ���֢����ѧ�ҽ���Щ�ۼ���������Χ�������߶���Ϊ������ؾ���ϸ����TAM����
���ڣ�����ѧ���Ѿ��ܹ�����֢�뵥���źŷ��ӵ�ˮƽ��ϵ������Ϊ�방�����ϵ�ṩ�˸�������֤�ݡ����磬������-Kappa B (NF-KB)��һ�ָ��ӵĵ����ʣ���������Ϊ������֢����Ϳ���ϸ�������������ء���������ѧ;���ķ�չ��NF-KB�����������ģ��Ѿ����Ƽ����Ƿ��ֲ����ר��������ҩ�↑�������а���ŵ������������ά���Ͷ���Ħ�ͷ����ա�A�����أ�������Ϊ��������Ԫר�����ϵĶ���
2004��Ү·����ϣ������ѧ��Yinon Ben-Neriah ��Eli Pikarsky�����ǵ�ͬ�±���, ͨ���Ŵ������ó�NF-KB ���ߴ���TNF�źű��رպ��ñ��Ŵ����Ϊ����С���Ե��³ɸΰ������ϰ�ǰ���˲��ᷢչΪ�ΰ����ں���,�кͿ������������������,��ֹ�����ǰ�ĸ�ϸ���ϵ�����;����Ķ�ʧ��ֹ��TNF����һ�����Ӽ������Ӷ�����NF-KB�����ء����NF-KB��ʹ��ǰ��ϸ����ʼ�������������ϸ����������һ����ص��о��У����˶������պ����ڼ��ݴ�ѧʥ���Ǹ��У�ĺ������Ƿ��֣�����NF-KB�����С����Ƴɽ᳦�ף�����ܵ��½᳦����Ҳ�ٽ���ϸ����������������ϸ��(�����ϸ��)�йر�ͨ·��Ҳ����ֹ�����ķ�չ��
����Ϊֹ���������֤�ݱ�������֢����֢֮����ڹ������������֢��ʹ��ǰ��֯ת��Ϊ�����������������ﷴӦҲ���ܲ��뵽����������ת�ƵĹ����С���Ⱦ�����ݸ˾���ϸ����������֢������������θ���ķ��գ��������ײ������ܻᵼ�¸ΰ�������ָ�����ְ�֢����ԭ��Ҳ���ܲ������ɻ����Ӷ��ƻ�DNA��������֢���ܴ�һ��ʼ���漰DNA�ĸı䣬���������о�������֢������DNA�ı�Ļ�
����ת�Ƶ����õİ����Ƚ�ǿ�ң�������о�Ҳ֤ʵ����һ���衣���յ��о�С����4��5�յġ���Ȼ����־�ϱ���˵����֢�������ǰ�֢ϸ���Ļ���仯����̼�����Ƴ�ǰ���ٰ��������ת�ơ��о���������ǰ������������������ϸ��������ϸ�������յ�����ϸ��������ֹת�Ƶĵ����ʵIJ���������ָ������һ������ܽ�����һ�����˷ѽ�ķ��֣����и�����������ǰ���ٴ��̻�죬��ʱ�ƺ���ٽ�ת�ơ����������ȷ�ģ���Ԥ��������֢���ܵ���ת�ơ����ͬʱ��Pollard���о�С���ڶ�С����е�һ���о��з��֣�����ϸ��������������ϸ����Ѫ��Ǩ�ƣ������������͵�ңԶ�ĵط���ͬʱ������·��ͻ�ѧ��Ϣ��
��������ϵͳ��̽����֢���ܵ��°�֢�Ĺ����еõ�����㷺�Ĺ�ע��������������ϵͳһ��������ϵͳ(Tϸ����Bϸ�������Ŀ����������ϸ�����ض�����)�����˲����������֮�Կ�����ʮ����������һֱ��̽����ǿTϸ���Կ���֢�������Ʒ������ܽ����������ʧ����
���⣬һ�����ڸ�����ͼ���Ѿ���ʼ��ʾ�������Ӧ������ϸ��֮��Ĵ��۸��ӵĶԻ������ǿ��ܲ�����Լ����Ĵ������о���֢������о���Ա��������ǵ�����ʱ��������Ҫ������Щ�������ã���ȷ��������Ч��һ���о��������ѳ���������һ���źŷ��ӣ���������������Tϸ��������һ������Ӧ����ϸ�������࣬����ʹ����Tϸ����������[�μ�Lisa Melton�ġ������������ӡ�;����ѧ�����ˡ�,2002��12��)��
���ͬʱ��Coussens������U.C.S.F.��ͬ����2005�귢���ڡ���֢ϸ�����ϵ�һ���о��з��֣������Ϊ����Ƥ������С�����ڵĿ�������Bϸ�����Ƴ����Է�ֹ��֯�ı��Ѫ�����ɣ����Ǽ�����չ��ǰ����������Ϊ��ԭ�幥���ߣ�Bϸ�������Ŀ��壬ͨ��ѪҺѭ������־������ϸ���ѱ�����������ϸ������Ȼ������Ϊ��ǰ��֯�źŵķ�Ӧ�������յ�����ϵͳ�ڰ�֢��չ�к�����һ���������о������������������ο�ʼ�ġ�һ�ֿ������ǣ���ϸ�����ܻ�����������ϸ��(��������ͻ״ϸ��)������Ϣ��Ȼ��Bϸ�����źſ�����TOLL�������йأ���������������������Ϣ�������Ѿ���Ϊ��Ҫ���н�(�μ�Luke A. J. O ' neil�ġ���������Ԥ��ϵͳ��;����ѧ�����ˡ�,2005��1��)��
��֢���ͼ�
��֢������һ�����٣�����������ϸ��������DNAͻ���ϸ������Ҳ������Ϊʲô��ǰ���Ƶ�һЩ����ȡ�������ijɹ���������˵:�����Dzɼ���ϸ����Ȼ�����������н�����ת������������ֲ�붯�����ڡ��������ǻ᳤��С��������������һЩ���顣�����Dz��Ǹ��ӵ���֯������Ȼ������������һ�ַdz����ӵ���֯��
�����ɱ����ϸ��-����ҩ�����ƺͷ����Ʒ���Ŀ�겻ͬ���µķ�������ͨ��������֢��������ҩ�û�о���ϸ������������������ϸ�����룬��ǰ��֯���ܻ��յ����ơ�
�ӱ�����˵����֢���ܻ��Ϊһ�������ڷ�ʪ�Թؽ������Լ�����������һ����֢�Լ���������ס������û��������ԭ����֢�����¿���˹��ѧ����ɭ��֢����(University of Texas m.d. Anderson cancer Center)�Ľ������ɵ¡��Ų���(Raymond DuBois)˵�����ǰ�֢�Ŀ���ҩ���о���Ա�������˼�����������ת�ơ���
һ�ֿ�������֢��ҩ���ɱ������ϸ��(���ɱ���ģ�����������ϸ��)�������������������л��ƵĽ�����������ã�������ҩ����������Եģ�����ÿ��ʹ����ΪԤ����Σ���ߡ����в�ѧ���ٴ��о�������ʹ�÷��������ҩ(�����忹��ҩ)�簢˾ƥ�֣������ӻ�ijЩʵ�������ķ����� ����ѡ���Ե����ǰ�����صIJ������о����ڽ��С�ǰ���������ɷ����忹��ҩ���Ƶĵ��ط��ӡ��ر�������ǰ������E2������ҩ�����������֢������������ͬʱ������Vioxx������NSAIDsһ���θ���������ҩ�����Ѫ�ܸ�����[�μ�Gary Stix�ġ����õ�Ŀ����ʹ������[�μ�]������ѧ�����ˡ�, 2007��1��)�����ǻ��ڿ���ʹ���������ڵ���͡��ҩ�������͵��̴��Ŀ������á�
һЩ���Ʒ����Ѿ����ڡ�ҩ��Avastin����Ѫ������-�ٽ�VEGF�IJ�������������ѧ�ұ����������������������ٽ�Ѫ��������������������Ϊ�����������Լ�����������ҩ��Ҳ���Կ���������Щҩ����ܱ��ϲ�������HIV�����̲�����������ҩ�Fβ�ƣ�Ҳ����Ѫ���������Ƽ���ɱϸ������
TNF�����Ƽ��Ѿ�������������ʪ�ؽ��ס�����������������������Ŀǰ���ڽ���ʵ��������Ѫ�����ٴ����顣�����ʪ�ؽ���Bϸ���ܰ���������Bϸ���ĵ���¡����Rituxan�����ܻ���ֹ�γɹ�����������֢��Ӧ��������ϸ�����Ӻ���ط���(IL-6��IL-8��CCL2��)Ҳ��DZ�ڵİб꣬NF-KBҲ�ǡ�
һЩ���еĻ�������������忹��ҩ�������������������з��ֵģ�����ͨ������NF-KB�����������á����ǣ���Ҫ����ҩʵ���������о����ַ�������ĸ�ѡ�������Ƽ���������������Ե���NF-KB���Ե�ø(��I-KB��ø)��
һ����ѧľ��
һ�����ڿ���һ�ּ��������Ʒ�����һ�ַ���������ľ����Ӣ��л�ƶ��´�ѧ��Claire Lewis��Munitta Muthana�����ǵ�ͬ�������һ��ҩ�ﴫ�ݷ��������þ���ϸ������Ȼ��������ʹ���������ȱ����������������˾���ϸ������һ�������ԵIJ����͵�ȱ��������������Щ����Գ��������绯�ƺͷ��Ƶķ�Ӧ�ܲ��ΪѪҺ��Ӧ���㡣һ������ϸ����������(��ĿǰΪֹ��������������)��ÿһ������ϸ�������ͷ���ǧ������������Ȼ���Ⱦ��ϸ����Ȼ����Щϸ���еĵ����ʼ���ÿ�����������ƻ���Ȼ���������ָʾϸ��-ɱ�����صĺϳɡ�������ϸ������Ǩ�Ƶ�һ���ط����������������£��������������ķ�ʽ���������ķ�չ����Lewis˵��
��֢�Ŀ��ײ��Ե�ȷ���������д���������������ϸ�����γɵ�����ԭ��ķ������ϣ�Ҳ���������ķ��ա�������һ���dz����ӵ����⣬���Ų���˵�������������عر�������ϵͳ����ͻ����������Ⱦ�����⣬�����̲�һ��������������֢�Լ�����ʹ��TNF���ͼ����˺�������Ⱦ�йأ������������ܰ��������⣬����NF-KBͨ·���ܻ���ijЩ������շ���֢������NF-KB��ʱ�ᵼ����֯���ˣ���������֯�쳣�������Ӷ����°�֢��
������ˣ���һ���Ŀ���ҩ�ƺ��Խ����뻯��ҩ������С����Լ��������Լ�����DZ�ڵ���֢�����������˿����仯�ı�־�������¹۲쵽:�����Ƕ��е�����˷��ˡ�������Χ����������������������ǽ�����ͻ��ϸ������ʹ��֢��Ϊһ�������ǹ�ͬ����ļ�����
https://s.click.taobao.com/hYsTINw
��
�����
Is Chronic Inflammation the Key to Unlocking the Mysteries of Cancer?
Understanding chronic inflammation, which contributes to heart disease, Alzheimer's and a variety of other ailments, may be a key to unlocking the mysteries of cancer
By Gary Stix on November 9, 2008
Is Chronic Inflammation the Key to Unlocking the Mysteries of Cancer?
Credit: ESA
Editor's Note: This story, originally printed in the July 2007 issue of Scientific American, is being posted in light of two new studies showing that angiogenesis inhibitors, discussed in this article, may actually make tumors bigger, not smaller.
More than 500 million years ago a set of specialized enzymes and proteins evolved to defend our primitive ancestors against assaults from the outside world. If a microbe breached the shell of some Cambrian-era fauna, the members of this early vintage immune system would stage a savage but coordinated attack on these interlopers��punching holes in cell walls, spitting out chemical toxins or simply swallowing and digesting the enemy whole. Once the invaders were dispatched, the immune battalion would start to heal damaged cells, or if the attacked cells were too badly damaged it would put them to rest.
This inflammatory immune response worked so well that many aspects of it have been preserved during the protracted aeons of evolution. We know this to be true because studies have found that we share many of the same immune genes as the lowly fruit fly��and vertebrates and invertebrates diverged from a common ancestor in excess of half a billion years ago.
ADVERTISEMENT
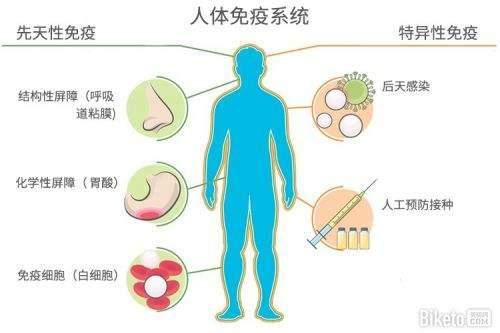
For years, immunology researchers have paid relatively little attention to this thuggish innate immune system, basically thinking of it as a crew of biochemical bouncers that pummel anything able to penetrate the tiniest opening in a living being��s skin or shell. They lavished their attention, instead, on the more advanced adaptive immune system, which can marshal antibodies and other weaponry that identify and then target an intruder with a specificity lacking in the untamed innate system.
In the past 15 years, innate immunity has come into its own. Inflammation, its hallmark characteristic, has gained recognition as an underlying contributor to virtually every chronic disease��a list that, besides obvious culprits such as rheumatoid arthritis and Crohn��s disease, includes diabetes and depression, along with major killers such as heart disease and stroke. The possibility of a link with a third major killer��cancer��has received intensive scrutiny in this decade. ��The connection between inflammation and cancer has moved to center stage in the research arena,�� notes Robert A. Weinberg of the Massachusetts Institute of Technology��s Whitehead Institute for Biomedical Research, who has highlighted the changing emphasis in a revision of his leading textbook, The Biology of Cancer (Garland Science, 2006).
This transformation recognizes that the immune inflammatory state serves as a key mediator of the middle stages of tumor development. Cancer begins with a series of genetic changes that prompt a group of cells to overreplicate and then invade surrounding tissue, the point at which true malignancy begins. Eventually some tumor cells may break off and establish new growths (metastases) at distant sites. That much has been understood for a long time. But cancer biologists and immunologists have begun to realize that the progression from diseased tissue to full-blown invasive cancer often requires cells that normally participate in healing cuts and scrapes to be diverted to the environs of the premalignant tissue, where they are hijacked to become co-conspirators that aid and abet carcinogenesis. As some researchers have described the malignant state: genetic damage is the match that lights the fire, and inflammation is the fuel that feeds it.
In this rewriting of the textbooks, a tumor is not just a clump of aberrant cells; it also includes a support system, a tumor microenvironment, which encompasses a multitude of varying immune cell types and crisscrossing chemical signals, along with a network of blood vessels. The tumor assumes the status of an outlaw organ that exists not to pump blood or rid the body of toxins but to serve only its own ends.
This new view implies that rooting out every last cancer cell in the body might not be necessary. Anti-inflammatory cancer therapy instead would prevent premalignant cells from turning fully cancerous or would impede an existing tumor from spreading to distant sites in the body. Cancer sufferers might then be able to survive, in the same way that new drugs have let HIV patients live longer. ��I don��t think a cure is necessarily the goal. It doesn��t need to be,�� comments Lisa M. Coussens, a cancer biologist at the University of California, San Francisco. ��If you can manage the disease and live your natural life span, that��s a huge win.��
ADVERTISEMENT
Multiple Lines of Defense
Comprehension of the link between inflammation and cancer requires knowing how the body reacts to invaders��and how normal healing is then subverted into promoting cancer when the inflammatory state lasts too long. After you step on a nail, the bacteria that invade the sole of your foot receive a welcome from an array of proteins and white blood cells that resemble rejects from central casting for the movie Creepshow 2. Just one example: Some 20 complement proteins, so called because they complement other bodily defense mechanisms, chemically spritz pathogens until the invaders explode into a big protoplasmic mess. While the complement system slimes the area, an assemblage known in immunology textbooks as professional phagocytes��literally ��expert eating cells����goes to work.
Lacking table manners, these Pac-Man-like macrophages and neutrophils proceed to engulf and consume the uninvited guests. Other members of the attack brigade include natural killer cells, mast cells and eosinophils. Healing represents more than launching an offensive against invaders. Blood platelets involved with clotting migrate to the break in the skin from an inner layer infused with blood vessels. Enzymes direct the repair of the extracellular matrix, the protein-based mortar in which the cells are immobilized. A scab forms, the skin grows back and the whole process of inflammation ends. Sometimes, though, inflammation does not stop. Any tissue (not just skin) that is chronically inflamed because of the persistent presence of pathogens, toxins or genetic damage helps to spur illness, from heart disease to cancer.
Beyond this first layer of defense, vertebrates are equipped with additional weaponry. The adaptive system learns an invader��s specific molecular signature and then uses it as a target for killing. Among the protagonists are B cells, which produce antibody molecules able to neutralize pathogens or mark them for destruction, and T cells, which prompt infected cells to kill themselves or secrete chemicals that direct the activities of other immune players.
In recent years a body of evidence has accumulated to show that chronic inflammation can play an important role in the progression of some types of tumors from a premalignant state to full-blown disease. A link between cancer and inflammation has long been suspected. In 1863 the prominent German pathologist Rudolf Virchow noted the presence of so-called lymphoreticular infiltrate (white blood cells) in malignant tissue. As early as 1978 Alberto Mantovani of Humanitas Clinical Institute and the University of Milan had observed that innate immune cells tend to congregate around some tumors. Cancer biologist Harold F. Dvorak of Harvard Medical School remarked in 1986 that tumors are ��wounds that do not heal.�� The status quo, though, lay elsewhere. Even a decade ago many biologists still hewed to the idea that the immune system serves not only to eliminate pathogens but to ferret out cells that are the abnormal precursors of cancer. But a closer look at the microenvironment surrounding tumors found the unexpected.
Hunting Pigeons
In the late 1990s Frances Balkwill of the Institute of Cancer at Queen Mary, University of London, had been doing research on a cytokine (a hormonelike immune signaling molecule) known as tumor necrosis factor (TNF), which was named for its ability to kill cancer cells when administered directly into a tumor at high levels. But when TNF lingers as a chronic, low-level presence in the tumor, it acts very differently. Balkwill��s lab turned off the TNF gene in mice so that the rodents could not produce the protein: to their surprise, the mice did not contract tumors. ��That really put us as the cat among pigeons,�� she recalls. ��All the people who were working on TNF as an anticancer agent were horrified. This cytokine they thought was a treatment for cancer was actually working as an endogenous tumor promoter.��
ADVERTISEMENT
The ready availability of knockout mice, in which the effects of selectively switching off genes could be tested, helped to highlight the cancer-inflammation link. Coussens and her U.C.S.F. colleagues Douglas Hanahan and Zena Werb reported in 1999 that mice engineered with activated cancer genes but without mast cells (another type of innate immune cell) developed premalignant tissue that did not progress to full malignancy. In 2001 Jeffrey W. Pollard and his co-workers at the Albert Einstein College of Medicine described mice that were genetically engineered to be susceptible to breast cancer tumors but that produced precancerous tissue that did not turn fully malignant unless it enlisted the assistance of macrophages.
The altered picture does not completely overturn the old one. In fact, it reveals that the immune system functions as a double-edged sword. The network of molecules and cells, second in complexity only to the brain, remains a paradox: sometimes it promotes cancer; other times it hinders disease. Some types of innate immune cells, such as natural killer cells, can actually protect against tumor growth. Others may nurture a malignancy only when the microenvironment is ��polarized�� into an inflammatory state; when not, they may blot it out. Inflammation, moreover, produces tumors in many organs, but not all��and its link to blood-borne cancers is not well characterized.
When looking for culprits, researchers have often focused their microscopes on macrophages, which occupy a meaningful spot among the white blood cells in the tumor microenvironment. The macrophages are capable of killing tumor cells or sending out an alarm to T cells of the adaptive immune system that something is amiss. But work by Pollard and other researchers has detailed how macrophages are ��reeducated�� by cancer cells to do their bidding. They become factories for cytokines and growth factors that nurture tumor development.
Turning the macrophages into traitors begins when tumor cells send out help signals that attract cells that become macrophages once they reach the tumors. Inside the tumors, proliferating cells grow so quickly that they begin to die for lack of oxygen. A combination of hypoxia and messages from the tumor cells initiates a process whereby the newly arrived macrophages assume their bad-body identity as tumor promoters. Cancer biologists give the name tumor-associated macrophages to these mutineers that congregate in and around the tumor.
Biologists have now been able to follow the inflammation link down to the level of individual signaling molecules, providing harder evidence for a connection to carcinogenesis. For example, nuclear factor-kappa B (NF-KB) is a complex of proteins that acts as a master switch for turning inflammation genes on and for controlling cell death. As biological pathways go, NF-KB��s is world-famous, having been discovered and patented for use in drug development by scientific stars that include Nobelists David Baltimore and Phillip A. Sharp and having subsequently become the object of multimillion-dollar patent litigation.
ADVERTISEMENT
In 2004 Yinon Ben-Neriah and Eli Pikarsky of the Hebrew University of Jerusalem and their colleagues reported that mice engineered to develop hepatitis (which can cause liver cancer) contracted precancerous lesions that did not progress to full malignancy when NF-KB was curtailed through a genetic alteration or when the proinflammatory TNF signaling molecule was shut off. In the latter group, a neutralizing antibody blocked TNF and prevented it from binding to a receptor on the premalignant liver cells; loss of the receptor prevented the TNF from triggering a molecular cascade that turns on the NF-KB master switch. Blocking NF-KB prompted the precancerous liver cells to initiate apoptosis, or programmed cell death. In a related finding that year, Michael Karin and his collaborators at the University of California, San Diego, found that inhibiting NF-KB in mice engineered to develop colitis, which can lead to colon cancer, also promoted apoptosis. And shutting down the pathway in inflammatory cells, such as macrophages, deterred tumor development as well.
So far the clearest evidence of a link between cancer and inflammation is the data demonstrating that inflammation encourages the conversion of precancerous tissue to full malignancy for many cancers. But the biological response may also be involved in initiating the disease and in advancing metastasis. Infections with Helicobacter pylori bacteria induce inflammation that greatly increases the risk of gastric cancer, and the hepatitis C virus can bring on liver cancer, to name just two cancers. Pathogens may also generate free radicals, which can damage DNA. But although inflammation may be involved from the outset, few studies have shown yet that an inflammatory condition actually alters DNA to provide the initiating spark.
The case for a role in metastasis is stronger��and recent studies lend credence to this hypothesis. Karin��s group reported in the April 5 Nature that inflammation, not genetic changes in cancer cells, spurs metastasis in mice engineered to acquire prostate cancer. The research suggests that a cytokine produced by inflammatory cells near a prostate tumor induces tumor cells to decrease production of a protein that blocks metastasis. This result, Karin notes, may explain the puzzling observation that cutting into tumors, such as for a prostate biopsy, sometimes seems to encourage metastasis. If he is correct, the inflammation generated by the intervention could be at fault. Around the same time, Pollard��s group reported in Cancer Research on a study in mice that observed that macrophages accompany breast tumor cells in their migration toward blood vessels that will transport them to remote sites, all the while sending chemical messages to their partners.
The innate immune system has received the most attention in explorations of how inflammation might cause cancer. As with innate immunity, the adaptive immune system��the T cells and antibodies produced by B cells that target specific molecules on invading cells��contributes to pathology or may also fight against it. For decades, immunotherapies designed to enhance T cell responses against cancer have been explored, though often with disappointing results.
Furthermore, an emerging picture has begun to reveal an intricate cross talk between innate and adaptive immune cells that may participate in the promoting of malignant disease. Researchers working on cancer vaccines may need to take account of these interactions in designing their treatments if they are ever to prove effective. One study showed that ovarian tumors produce a signaling molecule that serves to attract regulatory T cells, a subclass of adaptive immune cells responsible for quieting other T cells [see ��Subduing Suppressors,�� by Lisa Melton; Scientific American, December 2002].
ADVERTISEMENT
Meanwhile Coussens and her colleagues at U.C.S.F. found in a 2005 study published in Cancer Cell that the removal of antibody-making B cells from mice engineered to be prone to skin cancer prevented the tissue changes and angiogenesis that are prerequisites for disease progression. In their normal role as pathogen fighters, B cell�Cproduced antibodies circulate through the bloodstream and mark viruses and bacteria for destruction by innate immune cells. In response to a signal from precancerous tissue, however, the antibodies induce the innate system to collaborate in cancer development. An open research question is how this process starts. One possibility suggests that a cancer cell may send a message to innate immune cells, perhaps dendritic cells, that then activate B cells. Signaling may involve toll-like receptors, which have emerged as prominent intermediaries in innate immune messaging [see ��Immunity��s Early Warning System,�� by Luke A. J. O��Neill; Scientific American, January 2005].
Cancer Blockers
The recognition that cancer is more like an organ than just a clump of cells with DNA mutations in cell nuclei may also explain why some of the previous approaches to chemotherapy have met with limited success. ��People have taken cells and then transformed them in culture and stuck them into animals,�� Pollard says. ��They grow as little balls. They do certain things there. But they are not complex tissues, whereas a naturally occurring tumor is a very complex tissue.��
Instead of just killing cancer cells��the goal of current drug therapies and radiation��new approaches may supplement existing drugs by slowing inflammation. Without the involvement of macrophages and other innate cells, the premalignant tissue would remain in check.
Cancer could, in essence, become a chronic disease akin to rheumatoid arthritis, another inflammatory condition. ��Keep in mind almost no one dies of primary cancer,�� says Raymond DuBois, provost of the University of Texas M. D. Anderson Cancer Center and a researcher of anti-inflammatory agents for cancer. ��A patient almost always dies from a metastasis.��
A pharmaceutical against chronic inflammation represents a more alluring proposition than massacring malignant cells (and, unavoidably, healthy ones), a consequence of existing chemotherapies. Taken alone, such an agent might be benign enough to use every day as a preventive for high-risk patients. Epidemiological and clinical studies have shown some promise for the use of nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin to stave off the onset of some solid tumors. Investigations continue on more selective blocking of the production of prostaglandins, the regulatory molecules that are curtailed by NSAIDs. In particular, drugs that inhibit production of prostaglandin E2 may curb inflammation and tumor growth, while avoiding the cardiovascular side effects of drugs such as Vioxx and the gastrointestinal problems of the earlier class of NSAIDs [see ��Better Ways to Target Pain,�� by Gary Stix; Scientific American, January 2007]. The anti-inflammatory effects of the ubiquitous statins used to lower cholesterol are also being contemplated.
Some treatment options already exist. The drug Avastin inhibits production of the angiogenesis- promoting VEGF, although oncologists must contend with other molecules in the tumor microenvironment that promote blood vessel growth. Drugs developed for more familiar inflammatory diseases may also fight cancer��and these medicines might be combined into HIV-like drug cocktails, that also include angiogenesis inhibitors and cell-killing agents.
Inhibitors of TNF have received approval for treatment of rheumatoid arthritis, Crohn��s disease and other disorders and are now in clinical trials for both solid tumors and blood cancers. The drug Rituxan, a monoclonal antibody that represses B cells in rheumatoid arthritis and B cell lymphoma, might prevent the inflammatory response that fuels formation of solid tumors. Other cytokines and related molecules (IL-6, IL-8 and CCL2, among others) are also potential targets, as is NF-KB.
Some existing compounds, including NSAIDs and even one found in the spice turmeric, exert at least some of their effects by inhibiting NF-KB. But major pharmaceutical laboratories are investigating highly selective inhibitors of this molecular linchpin, many of them targeted at the enzymes (such as I-KB kinase) that regulate NF-KB activity.
A Chemical Trojan
One group is contemplating a radically ambitious treatment, a molecular Trojan horse of sorts. Claire Lewis and Munitta Muthana of the University of Sheffield in England and their colleagues have designed a drug delivery scheme that takes advantage of the natural attraction of macrophages to the oxygen-starved areas in tumors. They have engineered macrophages to deliver a therapeutic virus to hypoxic tumor regions, which respond poorly to conventional treatments such as chemotherapy and radiation because of an insufficient blood supply. Once the macrophages arrive in a tumor (grown in culture so far), each one releases thousands of copies of the virus, which then infect the cancer cells, after which a protein in those cells activates the therapeutic gene in each virus. This action then directs synthesis of a cell-killing toxin. ��The macrophage is migrating into a site and doing what we want it to do rather than driving tumor development in a normal way,�� Lewis says.
The exact outlines of an anti-inflammatory strategy against cancer have yet to be elucidated. Tweaking immune cells that form a defensive barrier against pathogens bears its own risks. ��It��s a very complicated issue,�� DuBois notes. ��If you magically shut down the immune system, you will have problems with opportunistic infections, just like with AIDS.�� Use of TNF blockers in other inflammatory disorders has been linked to tuberculosis and other infections, even potentially lymphoma. Moreover, inhibiting the NF-KB pathway can paradoxically promote cancer in some instances. Constraining NF-KB can at times lead to tissue damage and a process of abnormal regeneration of that tissue that can foster cancer.
Still, it seems likely that a new generation of anti-inflammatory agents will join the chemotherapeutic arsenal. Chronic diseases��and their underlying inflammatory conditions��are hallmarks of an aging population. ��We��re all a little bit overinflamed,�� Pollard observes. Treating the smoldering embers that surround the tumor rather than just mutant cells could make cancer a disease we can live with.
https://www.scientificamerican.com/article/chronic-inflammation-cancer/
��
.png)


.png)
