癌症病人都有酸性的体质,是真的吗?为什么癌症有酸性的微环境?
Why does cancer have an acidic microenvironment?
.png)
肿瘤组织呈酸性,这说明了什么?为什么癌细胞更喜欢这样做? 在制造出酸性微环境后,它能得到什么相关的或有利于生存的东西吗?
GPR68, a proton‐sensing GPCR, mediates interaction of cancer‐associated fibroblasts and cancer cells - Wiley - 2018 - The FASEB Journal - Wiley Online Library
https://faseb.onlinelibrary.wiley.com/doi/full/10.1096/fj.201700834R
我在癌症代谢这一领域做了广泛的研究,所以我将尝试把这个问题放在临床背景下,并加入一些非常有趣和积极的数据,来自我自己的专业领域,晚期的乳腺癌治疗;下面的讨论是我最近进行的(待发表的)一次内部审查的高度总结:
微环境酸中毒
微环境酸中毒现象,即微环境酸化,通过这三种主要机制促进肿瘤侵袭:
(1)相邻正常细胞群的破坏
(2)酸诱导细胞外基质降解(ECM)
(3)促进血管生成
所以我们知道增加糖酵解的上调,产酸的增加导致微环境酸中毒促进肿瘤侵袭。因为酸产量的增加会导致局部细胞外液的pH值的显著降低, 正常细胞长时间接触酸性微环境,通过依赖于p53 caspase 3机制导致坏死或凋亡。有证据表明,酸从肿瘤向肿瘤周正常组织的扩散提供了促进肿瘤侵袭的特定机制。因此,正常细胞由于缺乏适应细胞外酸中毒(如p53突变)的机制,无法在微环境酸中毒的条件下存活,而肿瘤群体继续增殖。
人类的临床证据
但更重要的是,最近从Matteo Bellone和同事的证据表明, 酸性本身代表一个免疫逃避机制,这种机制的免疫逃避通过肿瘤微环境的酸化可以针对pH-调控途径的药物,如质子泵抑制剂(如唑(埃索美拉唑)研究中使用)来克服,从而增加T细胞癌症免疫疗法的临床潜力。
这是一个令人兴奋的靶向治疗的新领域: 靶向微环境酸中毒,如上所示,通过使用质子泵抑制剂(PPIs),以减少或逆转促进肿瘤生成、肿瘤侵袭和肿瘤恶化的微环境酸化。
突破导致转移性疾病
我们现在终于有人类临床数据证实这种方法:上海癌症中心的研究人员在中国和在意大利罗马的国立卫生研究院在2012年的SABCS 大会上展示了一个随机试验的结果-这个实验调查了在乳腺癌转移病人中,在使用esomeprazole (Nexium埃索美拉唑)的同时加上高剂量质子泵抑制剂(PPI)疗法能否改善化疗效果。
他们发现,对比没有使用PPI疗法的普通乳腺癌转移患者,PPI疗法显著延长了无进展生存期。
更富戏剧性的是,在哪些恶性程度越高、预后最差的转移性乳腺癌即三阴乳腺癌(TNBC)患者,PPI疗法的使用比没有使用者,无进展生存期几乎延长了三倍。
这些是突破性的发现,如果在III期试验结果进一步获得证实,表示二个:
(1) 一个独一无二的无毒干预能改善转移性肿瘤的生存结果;
(2) 癌症新陈代谢假说的重要确认,特别是在晚期癌症微环境酸中毒作为潜在的治疗目标;
[注:我期望在即将到来的SABCS 2013年会议上有进一步的确认]。
Why does cancer have an acidic microenvironment?
Cancer shows lowered pH, what does this indicate and why cancer cell prefer to do this? Is anything relevant or edge that it can get after making the acidic micro-environment?
_____________________________________________________
I have done extensive research in this area of cancer metabolism so I will try to place the issue in a clinical context, and also add some highly intriguing and positive data from my own field of expertise, advanced breast cancer therapeutics; the discussion below represents a highly distilled summary of a recent internal review I conducted [pending publication]:
MICROENVIRONMENTAL ACIDOSIS
The phenomenon of microenvironmental acidosis, that is, acidification of the microenvironment, is known to facilitate tumor invasion through these three major mechanisms [1]:
(1) DESTRUCTION OF ADJACENT NORMAL CELL POPULATIONS
(2) ACID-INDUCED DEGRADATION OF THE EXTRACELLULAR MATRIX (ECM)
(3) ANGIOGENESIS PROMOTION
So we know that increased acid production from upregulation of glycolysis results in microenvironmental acidosis which facilitates invasion, and which happens because increased acid production leads to significant decreases in local extracellular pH, and prolonged exposure of normal cells to an acidic microenvironment results in either or both necrosis or apoptosis, through p53-dependent and caspase-3-dependent mechanisms [2,3], and there is evidence that diffusion of acid from the tumor into peritumoral normal tissue provides a specific mechanism promoting tumor invasion [4]. Thus, normal cells, by virtue of lacking a mechanism to adapt to extracellular acidosis (like p53 mutation) cannot survive under such conditions of microenvironmental acidosis, whereas tumor populations continue to proliferate.
HUMAN CLINICAL EVIDENCE
But more importantly, recent evidence from Matteo Bellone and colleagues [5] has shown that acidity per se represents a mechanism of immune escape, and that this mechanism of immune escape via the acidification of the tumor microenvironment can be overcome by drugs targeting pH-regulatory pathways, like PPIs (such as esomeprazole (Nexium) which was used in the study), which can increase the clinical potential of T cell-based cancer immunotherapy.
This is an exciting new area of therapeutic targeting: to target microenvironmental acidosis, which as shown above encourages tumor invasion and malignant transformation, by use of anti-secretory proton pump inhibitors (PPIs), in order to reduce or reverse acidification of the microenvironment and thus pro-tumorigenic and carcinogenic processes.
BREAKTHROUGH RESULTS IN METASTATIC DISEASE
And we now finally have human clinical data in confirmation of this approach: researchers at the Shanghai Cancer Center in China and at the National Institute of Health in Roma, Italy have presented results at the SABCS 2012 Conference of a randomized trial [6] investigating whether the efficacy of chemotherapy could be improved with the addition of high-dose proton pump inhibitor (PPI) therapy, in the form of esomeprazole (Nexium), in metastatic breast cancer patients.
What they found was a significant improvement in profession-free survival (PFS) for the general population of metastatic breast cancer patients who were on PPI therapy compared to those who were not,
and even more dramatically, in those metastatic breast cancer patients who had the most aggressive and prognostically unfavorable breast cancer subtype, namely triple negative breast cancer (TNBC), PFS was improved by almost three-fold over patients not on PPI therapy.
METHODOLOGY OF THIS REVIEW
A search of the PUBMED, Cochrane Library / Cochrane Register of Controlled Trials, MEDLINE, EMBASE, AMED (Allied and Complimentary Medicine Database), CINAHL (Cumulative Index to Nursing and Allied Health Literature), PsycINFO, ISI Web of Science (WoS), BIOSIS, LILACS (Latin American and Caribbean Health Sciences Literature), ASSIA (Applied Social Sciences Index and Abstracts), SCEH (NHS Evidence Specialist Collection for Ethnicity and Health) and SCIRUS databases was conducted without language or date restrictions, and updated again current as of date of publication, with systematic reviews and meta-analyses extracted separately. Search was expanded in parallel to include just-in-time (JIT) medical feed sources as returned from Terkko (provided by the National Library of Health Sciences - Terkko at the University of Helsinki). A further "broad-spectrum" science search using SCIRUS (410+ million entry database) was then deployed for resources not otherwise included. Unpublished studies were located via contextual search, and relevant dissertations were located via NTLTD (Networked Digital Library of Theses and Dissertations) and OpenThesis. Sources in languages foreign to this reviewer were translated by language translation software.
REFERENCES
1. Gatenby RA, Gillies RJ Why do cancers have high aerobic glycolysis? Nat Rev Cancer 2004; 4(11):891-9.
2. Park, H. J., Lyons, J. C., Ohtsubo, T. & Song, C. W. Acidic environment causes apoptosis by increasing caspase activity. Br. J. Cancer 1999; 80, 1892–1897.
3. Williams, A. C., Collard, T. J. & Paraskeva, C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: implications for clonal selection during colorectal carcinogenesis. Oncogene 1999; 18, 3199–3204.
4. Gatenby, R. A. & Gawlinski, E. T. A reaction-diffusion model of cancer invasion. Cancer Res. 1996; 56, 5745–5753.
5. Bellone M, Calcinotto A, Filipazzi P, De Milito A, Fais S, Rivoltini L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. OncoImmunology 2012; 2:e22058.
6. Hu X, Wang B, Sun S, et al. Intermittent High Dose Proton Pump Inhibitor Improves Progression Free Survival as Compared to Standard Chemotherapy in the First Line Treatment of Patients with Metastatic Breast Cancer. Abstracts: Thirty-Fifth Annual CTRC-AACR San Antonio Breast Cancer Symposium-- Dec 4-8, 2012; San Antonio, TX. Cancer Res December 15, 2012; 72(24 Supplement): P6-11-01.
7. De Milito A, Marino ML, Fais S. A rationale for the use of proton pump inhibitors as antineoplastic agents. Curr Pharm Des 2012; 18(10):1395-406.
8. Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy.
Trends Pharmacol Sci 2013; 34(7):393-400.
Why does cancer have an acidic microenvironment? https://www.researchgate.net/post/Why_does_cancer_have_an_acidic_microenvironment
January, 2018
GPR68, a proton‐sensing GPCR, mediates interaction of cancer‐associated fibroblasts and cancer cells
Department of Pharmacology, University of California, San Diego, La Jolla, CA 92093, USA.
The microenvironment of pancreatic ductal adenocarcinoma (PDAC 胰腺导管腺癌) is characterized by a dense fibrotic stroma (desmoplasia) generated by pancreatic cancer‐associated fibroblasts (CAFs) derived from pancreatic stellate cells (PSCs) and pancreatic fibroblasts (PFs). Using an unbiased GPCRomic array approach, we identified 82 G‐protein‐coupled receptors (GPCRs) commonly expressed by CAFs derived from 5 primary PDAC tumors. Compared with PSCs and PFs, CAFs have increased expression of GPR68 (a proton‐sensing GPCR), with the results confirmed by immunoblotting, The Cancer Genome Atlas data, and immunohistochemistry of PDAC tumors. Coculture of PSCs with PDAC cells, or incubation with TNF‐α, induced GPR68 expression. GPR68 activation (by decreasing the extracellular pH) enhanced IL‐6 expression via a cAMP/PKA/cAMP response element binding protein signaling pathway. Knockdown of GPR68 by short interfering RNA diminished low pH‐induced production of IL‐6 and enhancement of PDAC cell proliferation by CAF conditioned media. CAFs from other gastrointestinal cancers also express GPR68. PDAC cells thus induce expression by CAFs of GPR68, which senses the acidic microenvironment, thereby increasing production of fibrotic markers and IL‐6 and promoting PDAC cell proliferation. CAF‐expressed GPR68 is a mediator of low‐pH‐promoted regulation of the tumor microenvironments, in particular to PDAC cell‐CAF interaction and may be a novel therapeutic target for pancreatic and perhaps other types of cancers.
TNF‐α increases GPR68 expression in PSCs
PDAC cells and PSCs have bidirectional crosstalk (29, 30). PDAC cells can activate PSCs and increase their proliferation and migration; TGF‐β1 is a driver of PSC activation (31, 32). We used a Transwell coculture system to study interaction between PDACs and PSCs. Coculture of PSCs with AsPC‐1, BxPC‐3, or MIA PaCa‐2 cells increased the expression of GPR68, α‐SMA (ACTA2), and IL‐6 (Fig. 3A). Incubation of PSCs with TGF‐β1 increased expression of ACTA2 and COL1A1 but did not change GPR68 expression (Fig. 3B). By contrast, PSCs incubated with TNF‐α had 18‐fold greater expression of GPR68 and 31‐fold greater expression of IL‐6 but not ACTA2 or COL1A1 (Fig. 3B). These results suggest that regulation of GPR68 expression in PSCs depends on TNF‐α but may be independent of TGF‐β‐promoted transformation of PSCs to myofibroblasts. Indeed, adding TNF‐α‐neutralizing antibody to the coculture system diminished the PDAC‐induced increase in GPR68 expression in PSCs (Fig. 3C). In addition, MG132, IKK2 inhibitor VI, JSH‐23, and CAY10512, which all inhibit NF‐κB, reduced the TNF‐α‐promoted increase in GPR68 expression in PSCs (Fig. 3D). Together, the results imply that PDAC cells release TNF‐α, which acts via NF‐κB in PSCs to increase GPR68 expression, a pathway for GPR68 induction akin to that of monomac 6 cells (33). To test whether hypoxia up‐regulated GPR68 expression (34), we cultured PSCs in 1% O2 for 48 h and found a 1.7‐fold increase in GPR68 expression compared with that of normoxia‐cultured PSCs, but hypoxia did not significantly change myofibroblast marker expression (Supplemental Fig. S2C).
一月,2018
GPR68是一种质子感应GPCR,可调节癌症相关的成纤维细胞与癌细胞的相互作用
美国加利福尼亚大学圣地亚哥分校药理学系,拉荷亚,加利福尼亚州92093。
胰腺导管腺癌(PDAC)的微环境特征是由胰腺星状细胞(PSCs)和胰腺成纤维细胞(PFs)衍生的胰腺癌相关成纤维细胞(CAF)产生致密的纤维化基质(异型增生)。使用无偏GPCRomic阵列方法,我们鉴定了源自5种原发性PDAC肿瘤的CAF通常表达的82个G蛋白偶联受体(GPCR)。与PSC和PF相比,CAF增加了GPR68(质子感应GPCR)的表达,免疫印迹,癌症基因组图谱数据和PDAC肿瘤的免疫组织化学证实了这一结果。将PSC与PDAC细胞共培养或与TNF-α孵育可诱导GPR68表达。 GPR68激活(通过降低细胞外pH)通过cAMP / PKA / cAMP反应元件结合蛋白信号传导途径增强了IL-6的表达。通过短干扰RNA敲低GPR68可以降低低pH诱导的CA-6条件培养基诱导的IL-6产生并增强PDAC细胞增殖。其他胃肠道癌的CAF也表达GPR68。因此,PDAC细胞通过CAF诱导GPR68的表达,从而感觉到酸性微环境,从而增加纤维化标记物和IL-6的产生并促进PDAC细胞增殖。表达CAF的GPR68是低pH值促进的肿瘤微环境调节的介体,尤其是PDAC细胞CAF相互作用,并且可能是胰腺癌和其他类型癌症的新型治疗靶标。
TNF-α增加PSC中GPR68的表达
PDAC单元和PSC具有双向串扰(29,30)。 PDAC细胞可以激活PSC,并增加其增殖和迁移。 TGF-β1是PSC激活的驱动器(31、32)。我们使用了Transwell共培养系统来研究PDAC和PSC之间的相互作用。 PSC与AsPC-1,BxPC-3或MIA PaCa-2细胞共培养可增加GPR68,α-SMA(ACTA2)和IL-6的表达(图3A)。用TGF-β1孵育PSC可以增加ACTA2和COL1A1的表达,但不会改变GPR68的表达(图3B)。相比之下,与TNF-α孵育的PSC的GPR68的表达高18倍,IL-6的表达高31倍,而ACTA2或COL1A1则没有(图3B)。这些结果表明,对PSC中GPR68表达的调节取决于TNF-α,但可能与TGF-β促进的PSC向成肌纤维细胞的转化无关。确实,在共培养系统中添加TNF-α中和抗体可以减少PDAC诱导的PSC中GPR68表达的增加(图3C)。此外,MG132,IKK2抑制剂VI,JSH-23和CAY10512均抑制NF-κB,降低了TNF-α促进了PSC中GPR68表达的增加(图3D)。总之,这些结果暗示PDAC细胞释放TNF-α,它通过PSC中的NF-κB来增加GPR68的表达,GPR68的诱导途径类似于monomac 6细胞(33)。为了测试缺氧是否上调了GPR68的表达(34),我们在1%O2中培养了PSC 48小时,发现GPR68的表达量比正常缺氧的PSC增加了1.7倍,但缺氧并没有明显改变成肌纤维细胞标志物表达式(补充图S2C)。
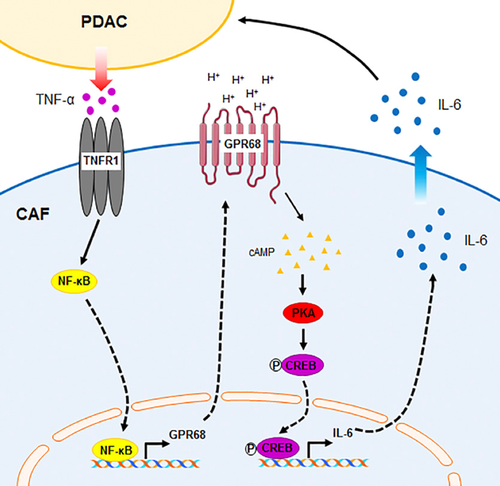
图7 PDAC细胞-CAF相互作用和GPR68的示意图。 PDAC细胞释放TNF-α,从而增加PSC / CAF的GPR68表达。细胞外质子通过cAMP / PKA / CREB途径激活GPR68并增加IL-6表达。从CAF中释放/分泌的IL-6可以刺激PDAC增殖。
Schematic summary of PDAC cell—CAF interaction and GPR68. PDAC cells release TNF‐α, which increases GPR68 expression by PSCs/CAFs. Extracellular protons activate GPR68 and increase IL‐6 expression via the cAMP/PKA/CREB pathway. IL‐6 released/secreted from CAFs can stimulate PDAC proliferation.
PDAC细胞-CAF相互作用和GPR68的示意图。 PDAC细胞释放TNF-α,从而增加PSC / CAF的GPR68表达。细胞外质子通过cAMP / PKA / CREB途径激活GPR68并增加IL-6表达。从CAF中释放/分泌的IL-6可以刺激PDAC增殖。
GPR68, a proton‐sensing GPCR, mediates interaction of cancer‐associated fibroblasts and cancer cells - Wiley - 2018 - The FASEB Journal - Wiley Online Library
https://faseb.onlinelibrary.wiley.com/doi/full/10.1096/fj.201700834R
GPR68: An Emerging Drug Target in Cancer
Department of Pharmacology, University of California, San Diego, La Jolla, CA 92093, USA.
GPR68 (or ovarian cancer G protein-coupled receptor 1, OGR1) is a proton-sensing G-protein-coupled receptor (GPCR) that responds to extracellular acidity and regulates a variety of cellular functions. Acidosis is considered a defining hallmark of the tumor microenvironment (TME). GPR68 expression is highly upregulated in numerous types of cancer. Emerging evidence has revealed that GPR68 may play crucial roles in tumor biology, including tumorigenesis, tumor growth, and metastasis. This review summarizes current knowledge regarding GPR68-its expression, regulation, signaling pathways, physiological roles, and functions it regulates in human cancers (including prostate, colon and pancreatic cancer, melanoma, medulloblastoma, and myelodysplastic syndrome). The findings provide evidence for GPR68 as a potentially novel therapeutic target but in addition, we note challenges in developing drugs that target GPR68.
GPR68:癌症中的新兴药物靶标
美国加利福尼亚大学圣地亚哥分校药理学系,拉荷亚,加利福尼亚州92093。
GPR68(或卵巢癌G蛋白偶联受体1,OGR1)是质子敏感的G蛋白偶联受体(GPCR),可响应细胞外酸度并调节多种细胞功能。酸中毒被认为是肿瘤微环境(TME)的标志性特征。 GPR68表达在多种类型的癌症中高度上调。最近的证据表明,GPR68可能在肿瘤生物学中起关键作用,包括肿瘤发生,肿瘤生长和转移。这篇综述总结了有关GPR68的当前知识,包括其在人类癌症(包括前列腺癌,结肠癌和胰腺癌,黑色素瘤,成髓细胞瘤和骨髓增生异常综合征)中的表达、调控、信号通路、生理作用及其调控的功能。这些发现为GPR68作为潜在的新型治疗靶点提供了证据,但此外,我们注意到开发靶向GPR68的药物面临的挑战。
Keywords: GPR68; RNA-seq; acidosis; proton-sensing GPCRs; tumor microenvironment.GPR68: An Emerging Drug Target in Cancer - PubMed
https://pubmed.ncbi.nlm.nih.gov/30696114/#:~:text=GPR68%20%28or%20ovarian%20cancer%20G%20protein-coupled%20receptor%201%2C,is%20highly%20upregulated%20in%20numerous%20types%20of%20cancer.
Vitamin C is a kinase inhibitor: dehydroascorbic acid inhibits IkappaBalpha kinase beta.
Program in Molecular Pharmacology and Chemistry, Box 451, Memorial Sloan-Kettering Cancer Center, New York, NY 10021, USA.
Reactive oxygen species (ROS) are key intermediates in cellular signal transduction pathways whose function may be counterbalanced by antioxidants. Acting as an antioxidant, ascorbic acid (AA) donates two electrons and becomes oxidized to dehydroascorbic acid (DHA). We discovered that DHA directly inhibits IkappaBalpha kinase beta (IKKbeta) and IKKalpha enzymatic activity in vitro, whereas AA did not have this effect. When cells were loaded with AA and induced to generate DHA by oxidative stress in cells expressing a constitutive active IKKbeta, NF-kappaB activation was inhibited. Our results identify a dual molecular action of vitamin C in signal transduction and provide a direct linkage between the redox state of vitamin C and NF-kappaB signaling events. AA quenches ROS intermediates involved in the activation of NF-kappaB and is oxidized to DHA, which directly inhibits IKKbeta and IKKalpha enzymatic activity. These findings define a function for vitamin C in signal transduction other than as an antioxidant and mechanistically illuminate how vitamin C down-modulates NF-kappaB signaling.
Front Physiology. 2013 Dec 5;
Acidic tumor microenvironment and pH-sensing G protein-coupled receptors
The tumor microenvironment is acidic due to glycolytic cancer cell metabolism, hypoxia, and deficient blood perfusion. It is proposed that acidosis in the tumor microenvironment is an important stress factor and selection force for cancer cell somatic evolution. Acidic pH has pleiotropic effects on the proliferation, migration, invasion, metastasis, and therapeutic response of cancer cells and the function of immune cells, vascular cells, and other stromal cells. However, the molecular mechanisms by which cancer cells and stromal cells sense and respond to acidic pH in the tumor microenvironment are poorly understood. In this article the role of a family of pH-sensing G protein-coupled receptors (GPCRs) in tumor biology is reviewed. Recent studies show that the pH-sensing GPCRs, including GPR4, GPR65 (TDAG8), GPR68 (OGR1), and GPR132 (G2A), regulate cancer cell metastasis and proliferation, immune cell function, inflammation, and blood vessel formation. Activation of the proton-sensing GPCRs by acidosis transduces multiple downstream G protein signaling pathways. Since GPCRs are major drug targets, small molecule modulators of the pH-sensing GPCRs are being actively developed and evaluated. Research on the pH-sensing GPCRs will continue to provide important insights into the molecular interaction between tumor and its acidic microenvironment and may identify new targets for cancer therapy and chemoprevention.
Keywords: GPR132 (G2A); GPR4; GPR65 (TDAG8); GPR68 (OGR1); acidosis; cancer; proton-sensing G protein-coupled receptors; tumor microenvironment.
Acidic tumor microenvironment and pH-sensing G protein-coupled receptors - PubMed
https://pubmed.ncbi.nlm.nih.gov/24367336/
Extracellular acidosis differentiates pancreatitis and pancreatic cancer in mouse models using acidoCEST MRI
Differentiating pancreatitis from pancreatic cancer would improve diagnostic specificity, and prognosticating pancreatitis that progresses to pancreatic cancer would also improve diagnoses of pancreas pathology. The high glycolytic metabolism of pancreatic cancer can cause tumor acidosis, and different levels of pancreatitis may also have different levels of acidosis, so that extracellular acidosis may be a diagnostic biomarker for these pathologies. AcidoCEST MRI can noninvasively measure extracellular pH (pHe) in the pancreas and pancreatic tissue. We used acidoCEST MRI to measure pHe in a KC model treated with caerulein(黄蛙素), which causes pancreatitis followed by development of pancreatic cancer. We also evaluated the KC model treated with PBS, and wild-type mice treated with caerulein or PBS as controls. The caerulein-treated KC cohort had lower pHe of 6.85–6.92 before and during the first 48 h after initiating treatment, relative to a pHe of 6.92 to 7.05 pHe units for the other cohorts. The pHe of the caerulein-treated KC cohort decreased to 6.79 units at 5 weeks when pancreatic tumors were detected with anatomical MRI, and sustained a pHe of 6.75 units at the 8-week time point. Histopathology was used to evaluate and validate the presence of tumors and inflammation in each cohort. These results showed that acidoCEST MRI can differentiate pancreatic cancer from pancreatitis in this mouse model, but does not appear to differentiate pancreatitis that progresses to pancreatic cancer vs. pancreatitis that does not progress to cancer.
Extracellular acidosis differentiates pancreatitis and pancreatic cancer in mouse models using acidoCEST MRI
https://repository.arizona.edu/handle/10150/636360
Ceruletide 黄蛙素 蛙皮素
From Wikipedia, the free encyclopedia
Ceruletide (INN), also known as cerulein or caerulein, is a ten amino acid oligopeptide that stimulates smooth muscle and increases digestive secretions. Ceruletide is similar in action and composition to cholecystokinin. It stimulates gastric, biliary, and pancreatic secretion; and certain smooth muscle. It is used in paralytic ileus and as diagnostic aid in pancreatic malfunction. It is used to induce pancreatitis in experimental animal models.
The tree frog Litoria caerulae, formerly named Hyla caerulae.
Ceruletide was discovered and its structure elucidated in 1967 by Australian and Italian scientists from dried skins of the Australian green tree frog (Litoria caerulea, formerly Hyla caerulea). Its amino acid sequence is Pglu-Gln-Asp-Tyr[SO3H]-Thr-Gly-Trp-Met-Asp-Phe-NH2.[1][2]
Induction of pancreatitis
Ceruletide upregulates pancreatic acinar cell intercellular adhesion molecule-1 (ICAM-1) proteins through intracellular upregulation of NF-κB. Surface ICAM-1 in turn promotes neutrophil adhesion onto acinar cells enhancing pancreatic inflammation.[3] In addition to promoting the inflammatory cell reaction to acinar cells, ceruletide induces pancreatitis through dysregulation of digestive enzyme production and cytoplasmic vacuolization, leading to acinar cell death and pancreatic edema. Ceruletide also activates NADPH oxidase, a source of reactive oxygen species contributing to inflammation, as well as the Janus kinase/signal transducer, another inflammation inducer.[4]
Ceruletide(INN),也称为cerulein或caerulein,是一种十氨基酸的寡肽,可刺激平滑肌并增加消化分泌。 Ceruletide在作用和组成上与胆囊收缩素相似。它刺激胃,胆和胰腺的分泌;和某些平滑肌。它用于麻痹性肠梗阻,并作为胰腺功能障碍的诊断辅助手段。它用于在实验动物模型中诱发胰腺炎。
树蛙Litoria caerulae,原名Hyla caerulae。
澳大利亚和意大利的科学家于1967年从澳大利亚绿树蛙(Litoria caerulea,原名Hyla caerulea)的干燥皮肤中发现了Ceruletide,并阐明了其结构。其氨基酸序列为Pglu-Gln-Asp-Tyr [SO3H] -Thr-Gly-Trp-Met-Asp-Phe-NH2。[1] [2]
诱发胰腺炎
Ceruletide通过细胞内NF-κB的上调来上调胰腺腺泡细胞间粘附分子1(ICAM-1)蛋白。反过来,表面ICAM-1会促进中性粒细胞粘附到腺泡细胞上,从而增强胰腺炎症。[3]除了促进对腺泡细胞的炎性细胞反应外,蓝藻肽还通过消化酶生成失调和细胞质空泡化而诱发胰腺炎,从而导致腺泡细胞死亡和胰腺水肿。 Ceruletide还激活NADPH氧化酶(一种引起炎症的活性氧源)以及另一种炎症诱导剂Janus激酶/信号转导子。[4]
https://en.wikipedia.org/wiki/Ceruletide
Chronic Metabolic Acidosis Destroys Pancreas
Peter Melamed and Felix Melamed
Biotherapy Clinic of San Francisco
One primary reason for the current epidemic of digestive disorders might be chronic metabolic acidosis, which is extremely common in the modern population. Chronic metabolic acidosis primarily affects two alkaline digestive glands, the liver, and the pancreas, which produce alkaline bile and pancreatic juice with a large amount of bicarbonate. Even small acidic alterations in the bile and pancreatic juice pH can lead to serious biochemical/biomechanical changes. The pancreatic digestive enzymes require an alkaline milieu for proper function, and lowering the pH disables their activity. It can be the primary cause of indigestion. Acidification of the pancreatic juice decreases its antimicrobial activity, which can lead to intestinal dysbiosis. Lowering the pH of the pancreatic juice can cause premature activation of the proteases inside the pancreas with the potential development of pancreatitis.The acidification of bile causes precipitation of the bile acids, which irritate the entire biliary system and create bile stone formation. Aggressive mixture of the acidic bile and the pancreatic juice can cause erratic contractions of the duodenum’s walls and subsequent bile reflux into the stomach and the esophagus. Normal exocrine pancreatic function is the core of proper digestion. Currently, there is no effective and safe treatment for enhancing the exocrine pancreatic function. Restoring normal acid-base homeostasis can be a useful toolfor pathophysiological therapeutic approaches for various gastrointestinal disorders. There is strong research and practical evidence that restoring the HCO3 - capacity in the blood can improve digestion.
慢性代谢性酸中毒摧毁胰腺
彼得·梅拉梅德(Peter Melamed)和费利克斯·梅拉梅德(Felix Melamed)
旧金山生物治疗诊所
当前的消化系统疾病流行的主要原因之一可能是慢性代谢性酸中毒,这在现代人群中极为普遍。慢性代谢性酸中毒主要影响两个碱性消化腺,肝脏和胰腺,它们会产生碱性胆汁和胰液以及大量碳酸氢盐。即使胆汁和胰液pH值发生微小的酸性变化,也可能导致严重的生化/生物力学变化。胰腺消化酶需要碱性环境才能发挥其正常功能,而降低pH则会破坏其活性。这可能是消化不良的主要原因。胰液的酸化会降低其抗微生物活性,从而导致肠道营养不良。降低胰液的pH值可能会引起胰腺内蛋白酶的过早活化,并可能引起胰腺炎。胆汁酸化会引起胆汁酸沉淀,从而刺激整个胆道系统并形成胆结石。酸性胆汁和胰液的激烈混合会导致十二指肠壁收缩不稳,随后胆汁反流到胃和食道中。正常的外分泌胰腺功能是正常消化的核心。目前,尚无有效且安全的治疗来增强胰腺外分泌功能。恢复正常的酸碱稳态可以是各种胃肠道疾病的病理生理治疗方法的有用工具。有大量的研究和实践证据表明,恢复血液中的HCO3-容量可以改善消化。
Chronic Metabolic Acidosis Destroys Pancreas | Insight Medical Publishing
https://pancreas.imedpub.com/chronic-metabolic-acidosis-destroys-pancreas.php?aid=164
Stimulation of Pancreatic Growth by Secretin, Caerulein, and Pentagastrin
Endocrinology, Volume 106, Issue 1, 1 January 1980,University of Texas, Medical school at Houston
The primary hormonal regulators of pancreatic exocrine secretion are secretin and cholecystokinin. In addition, cholecystokinin is known to stimulate the growth of the exocrine pancreas. The current study examines whether secretin also alters pancreatic growth by itself and whether it influences the trophic response to caerulein, a structural and functional homolog of cholecystokinin. Rats were injected every 8 h for 7 days with either saline, secretin (50 U/kg), caerulein (320 ng/kg), or a combination of the above doses of secretin and caerulein. After sacrificing the animals, the pancreas was isolated, and the in vitro incorporation of [3H]thymidine into DNA was measured. The pancreas was weighed, and the pancreatic content of DNA and RNA were also determined. Secretin and caerulein administered alone both caused significant increases in DNA synthesis and the pancreatic concentrations of both RNA and DNA. The effects of the two hormones on total pancreatic RNA and DNA showed potentiation when combined. The combination of secretin and caerulein increased pancreatic weight significantly more than the sum of the effects of the agents given alone. In all three cases, the RNA and DNA ratios were significantly increased, indicating that hypertrophy as well as hyperplasia occurred. In a similar experiment, pentagastrin (250 μg/kg) also stimulated pancreatic growth. Secretin, when combined with pentagastrin, had no further effect or slightly reduced the trophic effects of pentagastrin. The effect of secretin on the pancreas contrasted dramatically with its effects on the oxyntic gland mucosa of the stomach. Secretin by itself had no trophic action on the stomach and completely inhibited the increase in DNA synthesis and DNA and RNA content stimulated by pentagastrin. In conclusion, secretin, like cholecystokinin and gastrin, produces both pancreatic hyperplasia and hypertrophy and may function in the regulation of the growth of this organ as well as its secretion. The most important aspect of the trophic action of secretin may be its ability to potentiate the effects of cholecystokinin. (Endocrinology106: 323, 1980)
Secretin,Caerulein和Pentagastrin刺激胰腺生长
内分泌学,第106卷,第1期,1980年1月1日,
胰腺外分泌的主要激素调节剂是促胰液素(secretin)和胆囊收缩素(cholecystokinin)。另外,已知胆囊收缩素可刺激外分泌胰腺的生长。目前的研究研究了促胰液素是否本身也改变了胰腺的生长,以及它是否影响对胆囊收缩素(一种胆囊收缩素的结构和功能同源物)的营养反应。每8小时给大鼠注射7天的盐水,促胰液素(50 U / kg),黄蛙素caerulein (320 ng / kg)或上述剂量的促胰液素和黄蛙素的组合。处死动物后,分离胰腺,并测量[3 H]胸苷体外掺入DNA。称重胰腺,并测定胰腺的DNA和RNA含量。单独施用的促胰液素和caerulein均引起DNA合成以及RNA和DNA的胰腺浓度显着增加。两种激素对总胰腺RNA和DNA的作用在组合时显示出增强作用。促胰液素和黄蛙素的组合增加的胰腺重量明显大于单独给予的药物作用的总和。在所有这三种情况下,RNA和DNA的比例均显着增加,表明发生了肥大和增生。在类似的实验中,五肽胃泌素(250μg/ kg)也刺激胰腺生长。促胰液素与五肽胃泌素联合使用时,没有进一步的作用,或者稍微降低了五肽胃泌素的营养作用。促胰液素对胰腺的作用与其对胃氧化性腺粘膜的作用形成鲜明对比。促胰液素本身对胃没有营养作用,并且完全抑制了由五肽胃泌素刺激的DNA合成以及DNA和RNA含量的增加。总之,促胰液素与胆囊收缩素和胃泌素一样,会引起胰腺增生和肥大,并可能在调节该器官的生长及其分泌中起作用。促胰液素营养作用最重要的方面可能是其增强胆囊收缩素作用的能力。 (内分泌学106:323,1980)
Caerulein-induced acute pancreatitis in mice that …翻译此页
https://bmcgastroenterol.biomedcentral.com/articles/10...
2006. 5. 15. · The cystic fibrosis (CF) mouse pancreas has constitutively elevated expression of the Reg/PAP cell stress genes (60-fold greater Reg3α, and 10-fold greater PAP/Reg3β and Reg3γ). These genes are suggested to be involved in protection or recovery from pancreatic injury. To test this idea the supramaximal caerulein model was used to induce acute pancreatitis in wild type and CF mice.
Cited by: 18
Publish Year: 2006
Author: Oxana Norkina, Rolf Graf, Philippe Appenzeller, Robert
Tuft Cells Inhibit Pancreatic Tumorigenesis in Mice by …翻译此页
https://pubmed.ncbi.nlm.nih.gov/32717220
In mice with KRAS-induced pancreatic tumorigenesis, loss of tuft cells accelerates tumorigenesis and increases the severity of caerulein-induced pancreatic injury, via decreased production of PGD 2 . These data are consistent with the hypothesis that tuft cells are a metaplasia-induced tumo …
Study shows how mutant KRAS initiates pancreatic cancer …翻译此页
https://www.bioworld.com/articles/500006-study-shows-how...
2020. 11. 10. · Single-cell genomic analysis has shown that mutations in the KRAS oncogene co-opt a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells, initiating pancreatic ductal adenocarcinoma (PDAC), Chinese researchers reported in the November 3, 2020, edition of Nature Cancer. This study finding has important implications for the use of 'cancer-specific ...
Loss of Sirt2 increases and prolongs a caerulein-induced …翻译此页
https://www.nature.com/articles/s41598-018-34792-y
2018. 11. 7. · However, severe and recurrent episodes can develop chronic pancreatitis, leading to permanent inflammation of the pancreas and an increased risk of pancreatic cancer. Caerulein …
Cited by: 3
Publish Year: 2018
Author: Songhua Quan, Daniel R. Principe, Angela E. Dean, Seong Hoon Park, Paul J. Grippo, David R Gius, Nob...
Stimulation of Pancreatic Growth by Secretin, Caerulein, …翻译此页
https://academic.oup.com/endo/article/106/1/323/2592959
1980. 1. 1. · The primary hormonal regulators of pancreatic exocrine secretion are secretin and cholecystokinin. In addition, cholecystokinin is known to stimulate the growth of the exocrine pancreas. The current study examines whether secretin also alters pancreatic growth by itself and whether it influences the trophic response to caerulein, a structural and functional homolog of cholecystokinin.
Cited by: 242
Publish Year: 1980
Author: Artur B. Dembinski, Leonard R. Johnson

.png)
.png)
.png)