��
����ϸ����������֯����Ҫ������ϸ��-������������������ϸ���ļ���-M1��M2
Ŀ¼
����ϸ��ռ������֯ϸ��������1.5~5%
���Ὣ����ϸ���������ٽ�������״̬
Tumor cells hijack macrophages via lactic acid
M1 and M2 Macrophages: Oracles of Health and Disease.
A Breakthrough: Macrophage-Directed Cancer Immunotherapy
Macrophage arginine metabolism and the inhibition or stimulation of cancer
HIF-1���ľ���ϸ����������Tϸ�����ܲ��ٽ�������չ
IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia
Anatomy of a Discovery: M1 and M2 Macrophages
TAM Tumor-associated macrophages in cancers
TAMeless traitors: macrophages in cancer progression and metastasis
Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing
Modulation of macrophage phenotype by cell shape
Effect of low pH on single skeletal muscle myosin mechanics and kinetics
Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity
Lactic acid upregulates VEGF expression in macrophage~Macrophage induces angiogenesis
Lactate induces expression of TGF-beta2,via THBS-1 (MMP depends on TGF-beta)
GPR81 is a membrane receptor of lactic acid
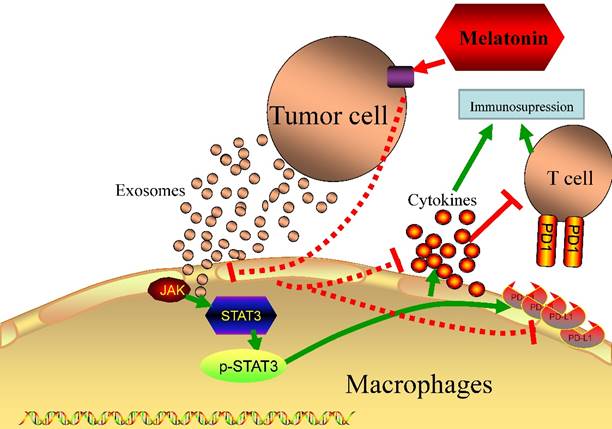
Melatonin has anti-proliferation, antimetastasis, reduced expression of PD-L1 on HCC.
Metabolic coupling and the Reverse Warburg Effect in cancer
15% of heme is degraded in liver by macrophages/KC cells
Images show that co-culture of cancer cells, macrophages and M leads to increased caspase-3 expression of cancer cells. Co-incubations of cancer cells and macrophages only or cancer cells and ferumoxytol only do not lead to significant apoptosis induction.
IJMS | Free Full-Text | Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing
https://www.mdpi.com/1422-0067/18/7/1545
Immune Effectors » Protozoan and Helminth Parasites » Pathogen Profile Dictionary
https://ppdictionary.com/parasites_2.htm
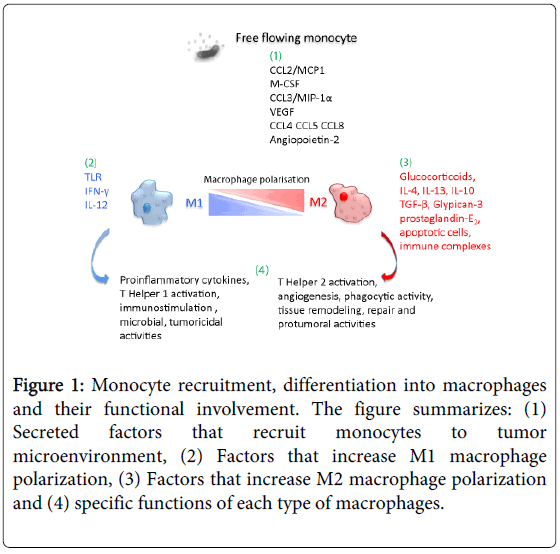
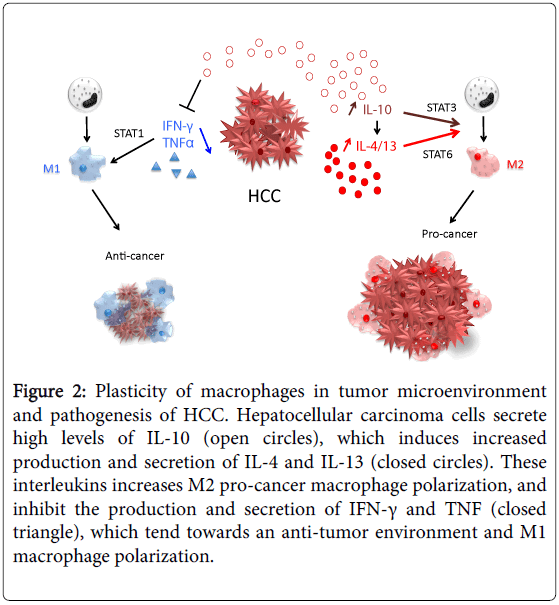
Figure 2: The phenotypic polarization of macrophages in the tumor microenvironment. Macrophages can be schematically classified into two main classes depending on their phenotypic polarization: macrophages mount M1 phenotype in response to M-CSF, INF��, LPS and other microbial products, whereas they differentiate into M2 in the presence of TGF��, VEGF, CCL2, M-CSF, IL-4, IL-10, IL-13, glucocorticoids and immune complexes/TLR ligands. M1 and M2 display different functions. M1 macrophages are able to trigger Th1 immune response and exert cytotoxic activity towards ingested microorganisms and cancer cells. M2 macrophages activate Th2 immune response and promote angiogenesis, tissue remodeling, and tumor progression. Refer to the text for abbreviations.
The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages : Figure 2
https://www.hindawi.com/journals/bmri/2013/187204/fig2/
Bavituximab �C Novel Checkpoint Inhibitor in Phase 3 | Cancer Biology
https://blogs.shu.edu/cancer/2015/02/04/bavituximab-novel-checkpoint-inhibitor-in-phase-3/��
Fig. 1.
In the tumor microenvironment, M1 macrophages secrete antitumor mediators. Cytokines, such as IL-4, IL-10, and IL-13, educate macrophages to promote tumor progression. M2 macrophages and tumor-associated macrophages produce protumor factors. The phenotypes of these macrophages are reversibly interchangeable. MMP, matrix metalloproteinase.
Fig. 2.
Schematic overview of macrophage polarity and insulin resistance. M1 and M2 macrophages are generated from monocytes in response to different stimuli. M1 and M2 macrophages have distinct secretary profiles that oppositely regulate inflammation and impact local/systemic insulin sensitivity.Control of Macrophage Dynamics as a Potential Therapeutic Approach for Clinical Disorders Involving Chronic Inflammation | Journal of Pharmacology and Experimental Therapeutics
http://jpet.aspetjournals.org/content/354/3/240��
Tumor cells hijack macrophages via lactic acid
Tumor-associated macrophages in cancers | SpringerLink
https://link.springer.com/article/10.1007%2Fs12094-015-1373-0��
��
Defining M1 and M2 Macrophages
This classification is based upon macrophage polarization rather than macrophage location.
M1 macrophages are classically activated, typically by IFN-�� or lipopolysaccharide (LPS), and produce proinflammatory cytokines, phagocytize microbes, and initiate an immune response. M1 macrophages produce nitric oxide (NO) or reactive oxygen intermediates (ROI) to protect against bacteria and viruses.
M2 macrophages are alternatively activated by exposure to certain cytokines such as IL-4, IL-10, or IL-13. M2 macrophages will produce either polyamines to induce proliferation or proline to induce collagen production. These macrophages are associated with wound healing and tissue repair.The Difference Between M1 & M2 Macrophages | Astarte Biologics
https://astartebio.com/blog/ask-scientist-whats-difference-m1-m2-macrophages/��
���Ὣ����ϸ���������ٽ�������״̬
Oncoimmunology. 2016 Mar; 5(3): e1014774.
Oscar R Colegio��˹����R����������1,2,3
1Ү³��ѧҽѧԺƤ����ѧ�Ͳ���ѧϵ�����������Ҹ���Ŧ����
2Ү³��ѧҽѧԺҮ³-Ŧ������ֲ���ģ����������Ҹ���Ŧ����
3Ү³��ѧҽѧԺҮ³��֢���ģ����������Ҹ���Ŧ����
ժҪ
�ڴ�������͵������У���������صľ���ϸ���벻��Ԥ���йء�Ȼ�����������ϸ�������������ź���δ�õ��ܺõĶ��塣���ǻع�����������ķ��֣���������Դ���������������صľ���ϸ������Ϊ�ٽ�������״̬�DZ�Ҫ�ͳ�ֵġ�
����
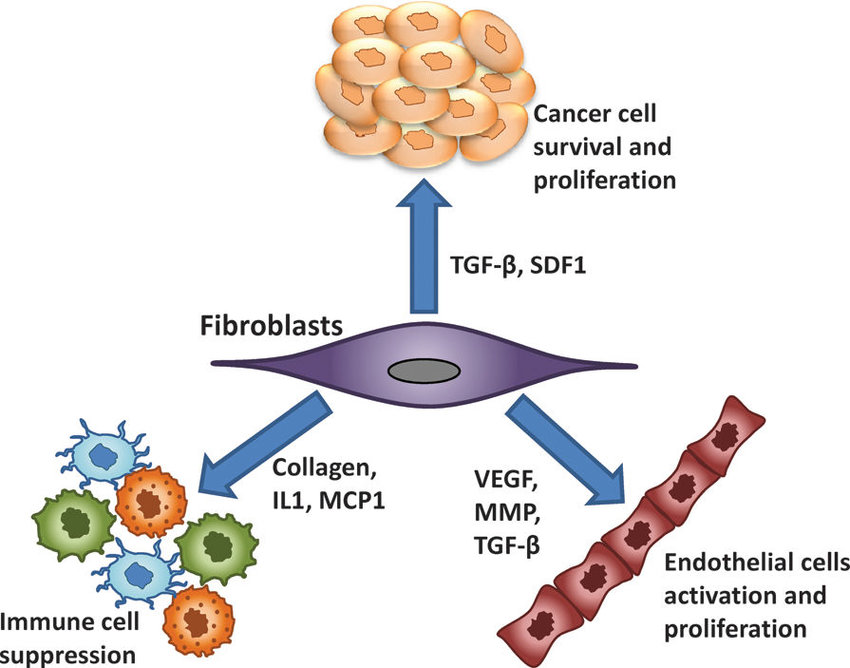
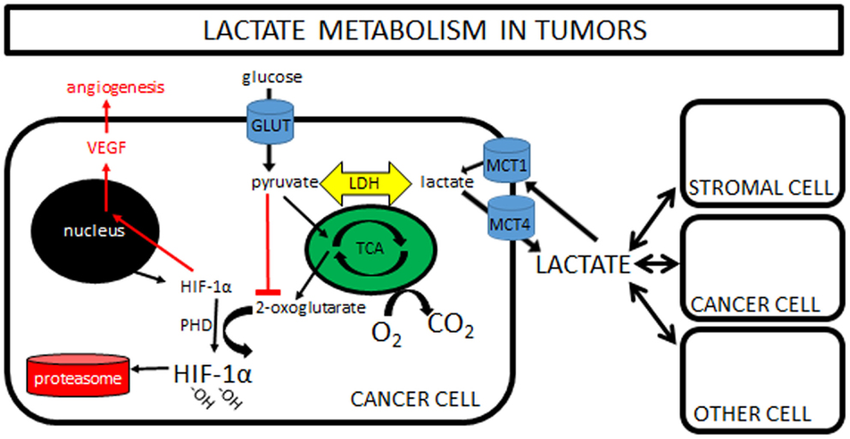
����ϸ����ά����֯��̬ƽ��������������Ҫ�����á�1 Ҫ������Щ���ã�����ϸ�������֪��������֯��ʵ��ϸ���Ĺ���״̬��һ������������̬��ƫ�룬����ϸ���ͱ���ͨ�������������ӻ���������ϸ�����ṩ�ʵ���֧�����������ֳ��쳣��������֯�����ٵ���������������ϸ����ɺ���֯�ṹ��2 ��ˣ���������صľ���ϸ��������̬���ܣ��ɴٽ�����������3-5������صľ���ϸ�����������д������VEGF
Ϊ���о�������ؾ���ϸ��������ϸ��֮����Է��ڹ�ϵ���������ȴ�����˹�ΰ���LLC����B16-F1��ɫ������B16����CT-26.WT��3�ֲ�ͬ��ͬԴ����������ֲģ���б�����������ؾ���ϸ�����᳦����CT16��.6 ����ȷ����ȡ�����������ͣ�������ؾ���ϸ����CD11b + F4 / 80 +��ռ����ϸ���Ĺ̶��ٷֱȣ���Χ�Ӵ�Լ1.5����B16����5.5����LLC��CT26�� ����������ʹ��ӫ�⼤���ϸ����ѡ�������봿ϸ��Ⱥʱ�����Ƿ����������е���������ϸ����ȣ�����ϸ���������б����˴������Ѫ����Ƥ�������ӣ�VEGF���� Judah Folkman���еĵر����о���ʾ����Ѫ���γ������������еĹؼ����á�Ȼ����ͨ����Ϊ��ϸ����VEGF����Ҫ��Դ�����ǵķ��ֱ�������������3�ֲ�ͬ������ģ���У���������صľ���ϸ����VEGF����Ҫ��Դ��
HIF1����������ؾ���ϸ���������յ���VEGF�������������
������֪��ȱ����ͨ��HIF1���յ�VEGF����������Ѫ���γɡ�7 ���ǣ�ijЩ�����������ΰ������������ã���Ѫ���Ժܷḻ����ˣ����Ǽ��裬��������ϸ���Ŀ��������������ڳ����������յ�����ϸ������VEGF��ʹ������LLC��B16��CT26ϸ�������������������Լ������Ե��������ͣ����Ƿ���������������Ե���������ϸ��������Һ�ڳ�����20��O2�����յ�����Դ����ϸ������VEGF��20��O2����ˮƽ��ȱ��ʱ��O2Ϊ0.1�������������ơ����Ƿ����ڳ��������£���������Һ�п��������ӿ��ȶ�������������ϸ���е�HIF1����������ȱ��״̬��һ����Ϊ��ȷ���Ƿ���ҪHIF1�����յ���������Һ��VEGF���յ������Dz����˾���HIF1��ȱ�ݵľ���ϸ����С������ЩС���У���������������Һ��VEGF���յ������ϳ��������ÿ���������ͨ���ȶ�HIF1�������źš���ˣ��ڳ��������£���������Һ����Ϊȱ��������
������ͨ��HIF1���յ�����ϸ���е�VEGF
����ʾ�ڳ����������ȶ�HIF1���ļ������ӡ�8 Ϊ�˼�����������Һ�еĸ������ӣ��������Ȱ���С���зּ�����ȷ��������<3 kDa���ڳ��������£�������4�ֿ������ӿ��ȶ�HIF1�������ᣬ��ͪ�ᣬ���պ�����pH������ȷ�������ӼȲ�������Ҳ��������pH��Ȼ����������ͱ�ͪ�ᡣ���С��ֲ���Otto Warburg���۲쵽����ϸ��ͨ�������ǽͽ��������Ƚ������Ǵ�лΪ���ᡣ9 ���Ʋ⣬���������������ֳ��ϸ�������Ȳ����ͷ��ڣ�Ϊ��ֳϸ�����������ṩ������ϳɵĻ�������ˣ�������֪�������Ƿ��������������Һ�еĻ����źŴ������ӡ�����ȷ������߹����Եİ�ϸ��ϵ�������������в���������Ũ����ߣ��������������Դ�ľ���ϸ����VEGF���յ��й�����ù۲���һ�£�������̼�������Դ�ľ���ϸ���ڳ����������յ�����VEGF��ͼ1������ˣ����������Ϊ����ϸ���ṩ����ϳɵ�ǰ�壬���һ�������Ϊ֧��ϸ���������ϸ�������ź�ת�����ӡ�
ͼ1��
����ϸ����������ؾ���ϸ��֮������Ĵ������Է��ڹ�ϵ��ʾ��ͼ��
�������Խ�����ϸ������ΪM2�������ٽ�״̬��
Ϊ��ȷ���������ϸ������Ŀ��������ӣ����Ǿ۽�VEGF��Ϊ�ؼ�ָ�����Ȼ�����������ı����Ѿ���������صľ���ϸ������������д������������������ѧ�о���δ֪�Ĺ������塣����ȷ��������VEGF�⣬���ỹ�������ܴ̼��Ĺ�����������ϸ��������Arg1��Fizz1��Mgl1��Mgl2�����յ���������ؾ���ϸ����صĸ��㷺�Ļ������⣬��Ұ���ͣ�WT)С����ȣ���û��HIF1��������£�Arg1��Fizz1��Mgl2�ı���ˮƽ�ϵ���
������֪��ϸ������IL-4��IL-13���ھ���ϸ�����յ�M2������10 ��������صľ���ϸ��Ҳ��������M2����ϸ����ص����൫����ȫ��������ˣ���������صľ���ϸ��������Ϊ��Ϊ��ȷ��IL-4��IL-13�ź���������ؾ���ϸ�������յ����Ƿ���ؼ����ã���CT26ϸ��ע�䵽ȱ��IL-4���������С�����ڣ��Ӷ�����������IL -4��IL-13�źŴ�������Ұ����С����ȣ���Щ������������ؾ���ϸ��δ��ʾ����M2��صľ������Arg1�ļ��٣���Fizz1�ı�������м��٣��������ǵ�IL-4��IL-13�źž����DZ���ı��͡��෴��ȱ��HIF1���ľ���ϸ����IL-4�̼�����ʾ����Arg1��Fizz1��Mgl2���յ����١�������������Щ���ֱ���IL-4��IL13�źŴ���������������صľ���ϸ�������ǿ��п��ġ����ǣ������������ỹ����IL-4�յ���M2�����ϸ�����Ͷ���ҪHIF1����
�����Ѿ��ܺõ�ȷ����VEGF����������ѧ�еĹ��ܣ���ARG1�Ĺ����в������ ARG1������ѭ���Ͷష�ϳ��еĹؼ����衣Ϊ��ȷ��ARG1�Ĺ�����Ҫ�ԣ�������Arg1ȱ���ľ���ϸ��������С������3��ʱ������ԼռWTС������������һ�롣Ϊ��ȷ�����ھ���ϸ�������Ƿ���Ҫ���ᣬ����ͨ���ó���ͪ�ἤø��Pkm2����ͬ�����������ǽͽ���أ��Ӷ���������������LLCϸ���� Pkm2�ó������������ٵ����ᣬ���нϵ͵ľ���ϸ��Arg1�����PKM2Ұ����������С��һ�롣��Щ���ڷ��ֱ����������յ�����ϸ��Arg1������������������Ҫ��
��֪��������صľ���ϸ����������չ�к���Ҫ�����ǵķ��ֵ��������ڣ����ᣨͨ�������ǽͽ��ǰ�ϸ���³´�л�ĸ����Ҳ���ž���ϸ���Ĺؼ��ź�ת�����ӵ����ã���������������ȱ������֯��������������£�����ϸ������ͼͨ�������������Ӻ�ø���ָ���֯��̬���Ӷ�����������������
Oncoimmunology. 2016 Mar; 5(3): e1014774.
Lactic acid polarizes macrophages to a tumor-promoting state
Oscar R Colegio1,2,3
1Departments of Dermatology and Pathology, Yale University School of Medicine; New Haven, CT, USA
2Yale-New Haven Transplantation Center, Yale University School of Medicine; New Haven, CT, USA
3Yale Cancer Center, Yale University School of Medicine; New Haven, CT, USA
Abstract
Tumor-associated macrophages have been associated with a poor prognosis in most types of tumors. However, tumor-derived signals that activate macrophages have not been well defined. We review our recent finding that tumor-derived lactic acid is necessary and sufficient to polarize tumor-associated macrophages to a tumor-promoting state.
Introduction
Macrophages play critical roles in the maintenance of tissue homeostasis.1 To perform these roles, macrophages must sense the functional states of the parenchymal cells of the tissues in which they exist. Upon detection of deviation from homeostasis, macrophages must provide appropriate support through the production of growth factors or the phagocytosis of damaged cells. Tumors exhibit many features of abnormally developed tissues and organs, including cellular composition and tissue architecture.2 As such, tumor-associated macrophages perform homeostatic functions that facilitate tumor growth.3-5Lactic acid polarizes macrophages to a tumor-promoting state
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4839384/Judah Folkman, a pioneer in the study of angiogenesis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2268723/��
References
1. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol 2009; 9:259�C70; PMID:19282852; http://dx.doi.org/10.1038/nri2528 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
2. Egeblad M, Nakasone ES, Werb Z.. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 2010; 18:884�C901; PMID:20627072; http://dx.doi.org/10.1016/j.devcel.2010.05.012 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
3. Mantovani A, Allavena P, Sica A, Balkwill F.. Cancer-related inflammation. Nature 2008; 454:436�C44; PMID:18650914; http://dx.doi.org/10.1038/nature07205 [PubMed] [CrossRef] [Google Scholar]
4. Grivennikov SI, Greten FR, Karin M.. Immunity, inflammation, and cancer. Cell 2010; 140:883�C99; PMID:20303878; http://dx.doi.org/10.1016/j.cell.2010.01.025 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
5. Qian BZ, Pollard JW.. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141:39�C51; PMID:20371344; http://dx.doi.org/10.1016/j.cell.2010.03.014 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
6. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014; 513:559�C63; PMID:25043024; http://dx.doi.org/10.1038/nature13490 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
7. Shweiki D, Itin A, Soffer D, Keshet E.. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992; 359:843�C5; PMID:1279431; http://dx.doi.org/10.1038/359843a0 [PubMed] [CrossRef] [Google Scholar]
8. Lu H, Forbes RA, Verma A.. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 2002; 277:23111�C5; PMID:11943784; http://dx.doi.org/10.1074/jbc.M202487200 [PubMed] [CrossRef] [Google Scholar]
9. Warburg O. On the origin of cancer cells. Science 1956; 123:309�C14; PMID:13298683; http://dx.doi.org/10.1126/science.123.3191.309 [PubMed] [CrossRef] [Google Scholar]
10. Gordon S, Martinez FO.. Alternative activation of macrophages: mechanism and functions. Immunity 2010; 32:593�C604; PMID:20510870; http://dx.doi.org/10.1016/j.immuni.2010.05.007 [PubMed] [CrossRef] [Google Scholar]��
Tumor cells hijack macrophages via lactic acid
Vincenzo Bronte
First published: 05 August 2014
Macrophages are among the most abundant cells in the tumor stroma and can contribute to neoplastic growth, invasion and metastatic diffusion by translating instructive signals delivered by transformed cells. These signals comprise soluble factors such as chemokines and cytokines.1 In many cancers, tumor�\associated macrophages (TAMs) are constantly recruited to the tumor environment by the CCL2 chemokine that attracts CCR2+ monocytes circulating in the blood.2 It is generally accepted that the tumor environment polarizes TAMs to express a set of genes common to M2�\type macrophages, a specialized subset intervening in inflammation resolution, tissue remodeling and control of parasitic infections.1 These genes include the neoangiogenesis�\promoter vascular endothelial growth factor (VEGF) and the l�\arginine�\metabolizing enzyme arginase (ARG). A recent paper by Colegio et al.3 in Nature opens a new scenario, showing that TAMs can ��sense�� metabolic changes typical of the malignant state.
The Nobel laureate Otto Heinrich Warburg postulated that glucose cell metabolism was fundamental for tumor progression. The ��Warburg effect�� defines the prevalent energy production in many cancers by a high rate of glycolysis, resulting in lactic acid secretion even in the presence of oxygen (aerobic glycolysis). This marks a straightforward difference with normal cells, where the oxidative breakdown of pyruvate within the mitochondria is the prevalent source of energy.
The presence of hypoxic areas represents another peculiar feature of the anarchic neoplastic growth. The hypoxia�\inducible factor (HIF) is the central mediator of transcriptional responses to hypoxia but it can also be activated by O2�\independent pathways.4 HIF proteins form heterodimeric complexes comprising an O2�\labile ���\subunit (HIF1��, HIF2�� or HIF3��) and a stable �©\subunit (HIF1��). These complexes recognize and bind hypoxia�\responsive elements with a shared RCGTG sequence in target genes. Under normoxic conditions, HIF�\specific prolyl�\hydroxylases modify HIF�� subunits and promote their proteasomal degradation by the von Hippel�CLindau tumor suppressor protein. When cells become hypoxic, posttranslational modification and stabilization of HIF1�� and HIF2�� subunits increase the transcriptional activity.4
Colegio et al.3 demonstrate that tumor�\derived mediators stabilize HIF1�� under normoxic conditions, leading to the transcription of the VEGF and ARG1 genes in macrophages. A heat�\stable factor present in the low�\molecular weight (<3 kDa) fraction of tumor�\conditioned medium was able to activate HIF1��. Unexpectedly, this factor was lactic acid, a byproduct of tumor glycolysis (Figure 1).
Among the enzymes involved in the glycolytic cascade, the M2 isoform of pyruvate kinase (PKM2) is predominant in tumor cell lines and its expression levels correlate directly with lactate production in the tumor environment. Upon release by cancer cells, uptake of lactic acid by macrophages requires its active transport by the monocarboxylate transporter on the cell membrane, a process facilitated by low pH. Once inside TAMs, lactic acid induces an HIF�\1���\dependent, M2�\like transcriptional profile in TAMs (Figure 1).3One gene considered as an emblem of M2 macrophage orientation is ARG1, as TH2�\type cytokines IL�\4 and IL�\13 are potent inducers of its transcription and activity.1,5 However, the role of these cytokines in regulating ARG1 within the tumor is only partially elucidated.2 In gliomas, for example, GM�\CSF released by neoplastic cells can upregulate the IL�\4 receptor in tumor�\associated myeloid cells promoting ARG1 induction by IL�\13.6 Hypoxia, on the other hand, regulates both ARG1 and ARG2 in macrophages, fibroblasts and endothelial cells.7 HIF1�� can control ARG1 and another l�\arginine�\metabolizing enzyme, the inducible isoform of nitric oxide synthase, in TAMs, thus enhancing their immunosuppressive activity on T lymphocytes.8
Colegio et al. demonstrate that lack of IL�\4R did not alter ARG1 expression in TAMs, at least in a lung cancer model, suggesting that the tumor environment can alternatively use lactate to influence M2 polarization. On the other hand, HIF1�� was required for the regulation of some M2 macrophage�\associated genes by IL�\4.3 It is thus conceivable that metabolic signals and cytokines can cooperate to shape TAMs in different tumor types (Figure 1).
Two sets of data in this manuscript point to an in vivo role for lactate in macrophage polarization and ARG1 expression. Tumor cell lines lacking PKM2 grew slower and had a reduced amount of ARG1 mRNA; conversely, co�\injection of cancer cells with macrophages derived from in vitro cultures of bone marrow cells stimulated with lactate grew more rapidly in mice.3
Whereas the role of VEGF in cancer development is well established, positioning arginase intervention requires further studies. Colegio et al.3 show that mice lacking ARG1 in myeloid cells by LysM promoter targeted deletion had a reduced growth of an implanted, subcutaneous tumor. Arginases are metabolic enzymes present in two isoforms that hydrolyze l�\arginine to l�\ornithine and urea.5 ARG1 in myeloid cells, including TAMs, could act as a tumor�\landscaping gene through two main pathways: supporting tumor growth and suppressing antitumor immune responses. Various tumors, both in humans and mice, express ARG isoforms at certain stages of their development, either in tumor�\infiltrating stroma or in the very same neoplastic cells.7 ARG1 activation is able to induce immune suppression by depleting l�\arginine in the microenvironment. Reduction of this semi�\essential amino acid can inhibit T�\cell proliferation through downregulation of CD3�� chain expression in T lymphocytes.2,5 ARG1 could also have a role without the intervention of adaptive immunity, by promoting tumor cell growth and survival. l�\Ornithine, produced downstream of ARG1 activity, is the precursor of polyamines, that is, putresceine, spermidine and spermine, which can act as proliferative signals for mammalian cells. However, only the growth of some transplanted tumors is affected in ARG1 knockout mice (unpublished results), suggesting other potential pathways can bypass the need for ARG1 in the myeloid compartment.
ARG1 might also control tissue remodeling as l�\ornitine can be converted into l�\proline, which is necessary for collagen synthesis.5 However, whereas the absence of ARG1 in the myeloid compartment resulted in prolonged inflammation and a negative effect on matrix deposition during the wound healing process, liver fibrosis was exacerbated in conditional ARG1 knockout mice exposed to Schistosoma mansoni, a pathology dependent on a TH2 lymphocyte response.9 It is conceivable that ARG1�\expressing macrophages might represent a subcategory of M2�\like macrophages, operating as suppressors rather than inducers of Th2�\dependent inflammation and fibrosis.9
Considering that many tumors cannot influence species evolution, as they arise after the peak of the reproductive age, lactate sensing must have a role in other macrophage responses. Is cytosolic lactate sensing a mechanism to program macrophages toward inflammation resolution and regulation of adaptive immunity? This will certainly be of interest for future researches but we can speculate about at least two conditions.
First, lactic acid�\producing bacteria constantly interact with our body and are part of the normal microbiota in the gut and other mucosal surfaces. It is thus conceivable that their fermentation products influence the local macrophage response. Bacterial vaginosis is a common clinical syndrome arising when anaerobic bacteria replace the protective lactic acid�\producing bacteria (mainly species of the Lactobacillus genus). Although the specific role of lactic acid remains to be proven, some Lactobacilli strains can exert an anti�\inflammatory activity, helping to control colitis severity, by regulating M2 orientation and ARG1 activity in macrophages.10
Second, exposure of bone marrow cells to granulocyte�\macrophage colony�\stimulating factor (GM�\CSF) and IL�\6 generates immunosuppressive myeloid cells including macrophages and results in a fast (within 24 h) activation of l�\arginine�\metabolizing enzymes and increased uptake of glucose, which is mainly metabolized by anaerobic glycolysis.11,12 Thus, the main metabolic changes in myeloid cells exposed to cytokines produced by several tumors, such as GM�\CSF and IL�\6, can also lead to the accumulation of endogenous lactate. Lactic acid might thus represent a converging crossroad integrating external and internal milieu to regulate l�\arginine metabolism and polarization in macrophages (Figure 1). However, this altered metabolic state might promote macrophage death. In fact, although macrophages survive in a hypoxic environment, exposure to lactate levels produced by tumors can cause their dismissal and possibly contribute to their continuous replenishment by circulating precursors as well as their spatial distribution within specific areas of tumors.13
Figure 1
Macrophages integrate metabolic and environmental signals to promote tumor growth. Area within dotted rectangle indicates proposed mechanisms of action. ARG, arginase; HIF, hypoxia�\inducible factor; MCT, monocarboxylate transporter; NADH, nicotine adenine dinucleotide, reduced; PKM2, M2 isoform of pyruvate kinase; VEGF, vascular endothelial growth factor.europepmc.org/abstract/MED/25091608
http://europepmc.org/abstract/MED/25091608��
Lactic acid polarizes macrophages to a tumor-promoting state
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4839384/��
��
����ϸ����HIF-1����������Tϸ�����ܲ��ٽ�������չ
Macrophage Expression of HIF-1�� Suppresses T cell Function and Promotes Tumor Progression
1Division of Biological Sciences, University of California, San Diego, La Jolla, California
2Essen University Clinic, Institute for Physiology, Essen, Germany
3Department of Surgery, Medical University of South Carolina, Charleston, South Carolina
4The Scripps Research Institute, Department of Immunology, La Jolla, California
5Department of Pathology, School of Medicine, Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, California
6Laboratory of Gene Regulation, Department of Pharmacology, School of Medicine, University of California, San Diego, La Jolla, California
Corresponding Author: Randall S. Johnson, 9500 Gilman Drive, mail code 0377, La Jolla, CA 92093, ude.dscu@nosnhojsr
ժҪ
Tϸ�����������������������������������������еĹ���ͨ�������ơ�����ʵ�������ֳ��ḻ�ľ���ϸ������͵����������������������Ӱ����������ϸ�������������չ��Ӱ��֪֮���١�����VEGF-A��Ѫ�ܻ�δ�ı䣬���ڽ��������ٰ�С��ģ���о���ϸ����ȱ����Ӧ��ת¼����HIF-1���İ���ȱʧ���������������١�������صľ���ϸ������ͨ�����ֻ����������������Tϸ�������Ƿ���ȱ���������ھ���ϸ��HIF-1������ķ�ʽ��������Ч��ǿ�˾���ϸ���鵼��Tϸ���������á����ǵķ���ͨ���������������յ�Tϸ�����ƣ������������ߵ�����Ӧ��������չ��ϵ������
ȱ�������¾���ϸ������Tϸ����ֳ
Tϸ���Ļ����ֳ����Ӧ������Ӧ���бز����ٵIJ��裬�������ӿ�ԭ������Tϸ���Ŀ�¡Ƶ�ʣ����յ��ֻ�ΪЧӦϸ���ͼ���ϸ����ЧӦϸ�������ϸ���ĺ��������ϸ�������ͷźͿ�ԭ������ϸ�����ԡ����ǵ�����ϸ����Tϸ��������ʵ�����������������Dz����������ϸ����������Tϸ���ڳ����͵�����������ֳ���������ڸòⶨ�У�����ͨ����ʽϸ�������Tϸ�����ѡ�ֵ��ע����ǣ��ڳ����£�ֻ����߱����ľ���ϸ����������Tϸ����ֳ��ͼ3A�CC������ϸ�ζ�����ϸ����Tϸ���ı��������ǣ�������ˮƽ���ͻ���ǿ ����ϸ����Tϸ�����������á���CD3 / 28�̼���Tϸ���ܹ��ڵ�����1������������ֳ��ͼ3B��1��80�����������ǣ�������ϸ��ռ��ϸ������5��ʱ��ϸ�����ڵĽ�չ�ͱ�������ϣ�ͼ3A�CC��1��20�����������ڸñ����µ���������ֳ�ںܴ�̶��ϲ���Ӱ�졣ͼ3C��������һ����Χ�ľ���ϸ����Tϸ�����������ֵ���������������ǿ����֤����������������ǿ����ϸ����Tϸ����ֳ��������������������
ͼ3
ȱ��ʱ����ϸ����Tϸ����ֳ������������ǿ
��A��B��ͨ��ϡ��CFSE������Tϸ����ֳ�Ĵ����Զ�������������Tϸ������CFSE����������ͨ��CD3 / 28������ڣ�A��������B��1����������������C���£���ָ�������Ĺ�����������ϸ��������60h����������ʵ����ͼ�η�ʽ����˵�����ǿ�ľ���ϸ����Tϸ����ֳ���������ã�n = 2������8������ʵ���й۲쵽ȱ��/����ϸ�����������Ƶ����ӣ���Щʵ���漰���־���ϸ��Ⱥ��������פ��Ĥ��3�Σ����ϻ������շ��ģ�2�Σ����������ľ���ϸ����3�Σ�����������߾�ΪS.E.M.ȱ��ʱ����ϸ����T������HIF-1��/ iNOS�����Ե�
Ϊ�˲���HIF-1���ھ���ϸ���鵼��ȱ��Tϸ�������е����ã�������Դ��HIF-1��+ f / + f / LysM-cre +/-������ϸ��HIF KO������ľ���ϸ�������˹�������ͼ4������ͼ4A�У�������ʾ�ڳ����£��ڴ�Χ�ľ���ϸ����Tϸ�����ʣ����ߺͺ��ߣ�֮�䣬������֮�����������û�б仯��Ȼ������ȱ�������£�HIF-1����Ч����ϸ����Tϸ����ֳ�IJ������Ƽ�������Ϊ1:20���ߵı��ʣ���Ұ���;���ϸ���յ���Чϸ������ͣ�͵ı��ʣ�ͼ4A����ɫ����ɫ��CFSEʾ��ͼ4B���� ��ʹ�õ⻯�����ų��������ڹ̶�ʱ����������ռ����Ļ�ϸ����������ͼ4C����ʾ����Ұ���;���ϸ����1:20������Tϸ�������������HIF������Tϸ����30���Ļ���-1���վ���ϸ������Щ��������ڵ��������¾���ϸ���鵼��Tϸ����ֳ������HIF-1�������Եġ�
ͼ4 HIF-1����Ч�ľ���ϸ��������ȱ����������ǿTϸ���������ã���WT����������ȡ����HIF-1���ϵ�����ϸ��iNOS��ˮƽ�����ҿ���ͨ���к;���Ĵ���ϸ�����ӻ��ض���iNOS������������ϣ�A���ڳ�����1�������лTϸ��60Сʱ��CFSEϡ�ͣ�����������������ָ�������ľ���ϸ���� ��B������A��һ����1:20����ϸ����Tϸ�������������Tϸ����CFSEϡ��Һ�Ĵ����Ը���ͼ����Ʒ���Զ���ʵ�飬���Կ���ͨ����A����ͬ����ɫ�������м���������δ�̼���CFSE����Tϸ��������ȱ�������»��յĻ�ɫ��C������Tϸ���У�ռ�����������»��յ�1���İٷֱȣ�20������ϸ����Tϸ��n �� 2�����н������3������ʵ�顣 ��D��������Tϸ�����Ĵ�����CFSEϡ��Һ������WT������Դ�ľ���ϸ��������60h����������ʾ��ϸ�������кͿ��壨����ʾ���壩��iNOS���Ƽ�1400W����3��������ʵ���й۲쵽�����ƵĽ���������Tϸ���ڴ̼���Ѹ�ٲ���Th1��Th2ϸ�����ӣ�����IFN�ú�IL-4������ϸ������Щϸ�����Ӿ��������Ժ���ת¼��Ӧ��������IFN�ô̼�������iNOS���µ�ArgI�������IL-4�̼�������ArgI���µ�iNOS���29����������ϸ��ȱ��ʱ�������ֶ���HIF�����ķ�ʽ������9���������Ѿ��㷺������iNOS��ArgI������Tϸ�����ܣ�30��������iNOS����ͨ��һ�����������Ĺ����������ε��γ���Ѹ����ֹTϸ����ֳ����ArgIͨ���������ľֲ�L-��������������������������-������������ڼ������������в�̫����Ӱ��Tϸ����ֳ��31������Ϊ���Tϸ�������Th1��Th2ϸ�����ӣ��������IFN�ú�IL-12���кͿ�����Բ���Th2��ϸ�������ף�ͨ����IL-4����Ϊ��Ҫ��Tϸ��������ϸ�����ӣ��������IL- 4����Th1�����ļ���
Ϊ�����Ȳ���HIF�б��Th1�յ��Ļ���iNOS�������е����ã�������iNOS���������Ƽ�1400W��������Ļ��ԣ�32������Щʵ��������¼��ͼ4D�С����Կ�����Ұ���;���ϸ����Tϸ���ı���Ϊ1��10����ȱ����������ֹTϸ����ֳ����ɫ�ۼ�����ʹ����ֳ������δ�̼���ϸ���൱����ɫ�ۼ�������IL-4�����кͿ�������ֹTϸ����ֳ��Ȼ������IFN�ú�IL-12�����кͿ���ʹTϸ���Ӿ���ϸ��/������ǿ��ϸ�����ڽ�����ֹ�ͷš����ǵ��������۵����ݣ�������HIF-1����������IFN�õ����������Կ�����IL-4��ȣ����������iNOS�������������á�ʵ���ϣ���1400W����1:10����ϸ����Tϸ�����������ȫ�ָ�ȱ��������ǿ����Tϸ����ֳ��������ͼ4D����ɫ�ۼ���+ 1400W��������iNOS-/-С��ľ���ϸ�������ּ���������ͬ����������Tϸ����ֳ������δ��ʾ����Ӧ��ָ�����������������ϸ���������õ�Tϸ������������ϸ��ͨ�������IFN�á�����ϸ��HIF-1�������������ڵ�ϸ������Tϸ����Ӧ
HIF-1�����ڵ�øArgI��iNOS�����յ�Tϸ��������������18��35-37�������Ƕ���������HIF-1��Ұ���ͻ���ЧС�������PyMT�����ѽ��������ø���Բⶨ������Ԥ�ڵģ�����Ұ����С����ѽ����е�ArgI�������Ÿ��ߣ�ͼ5B����Ұ���������е�iNOS���Խϸߣ�����ϴ�������ͳ��ѧ���壨����δ��ʾ������Щ�����ƺ���ӳ��PyMT /����ϸ����������ͼ5A��������iNOS����Ч���յ�ArgI�������Ƕ����������ĵ�ϸ����Һ�ķ����У����Ƿ��ֽ����CD8 + Tϸ������û�����Ų��죨����δ��ʾ��������Ұ���������д�������������ArgI���ԣ�����ֱ��ȡ��Ұ���ͺ�����HIF-1����Ч����������Tϸ���ķ�Ӧ�Էdz���Ҫ��
Tϸ����Ӧ�Ե�һ��ؼ�ָ���Ǵ̼�����������������-�ã�IFN�ã����������ݱ��������⼤��������IFN����Tϸ��������ϸ������DZ��������أ���Tϸ��ЧӦ�������Ĺؼ�ָ�꣨38-40����Ϊ�˲�����������HIF-1��Ұ���ͺ���Ч����������Tϸ����Ӧ�ԣ��������ÿ�CD3 / 28�����������ϸ����Һ����ǿ�ҵ�pan-Tϸ���̼������IFN�õ�Tϸ�����֡�������Ұ����������Tϸ����ȣ�������HIF-1����Ч�����в���IFN�õ�Tϸ������ �����Ҹ߶��������ӣ�ͼ5C������֤��������ϸ���е�HIF-1������������ϸ������Tϸ���������йء�
Figure 6
Cartoon depicting a model for myeloid, HIF-dependent suppression of T cell function in hypoxic regions of tumors
��������
������Tϸ�����������������������Ʒ�����Ҫ�ϰ����������ƵĻ��ƺܶ࣬����iNOS��ArgI�ľֲ�ø�ٻ��ԣ�12��36��41���������������ʾ������ϸ������Щ��������ø��HIF-1�����ƶ�Tϸ������ֳ�����ͶԴ̼��ķ�Ӧ�Ծ�����Զ��Ӱ�졣��MMTV-PyMT��������ģ�������Է���CD8 + Tϸ����Ӧ��֤�ݣ��ɼ�������������26�����������Ƿ�������HIF-1��ȱʧ��������չ��������й�-���ǣ����ǵ����ݲ��ų���Tϸ����������������������ƣ���Щ�����������յ��������͡�
����Ϣ��HIF-1��������Ч�����У�����Tϸ��������������̬�����������ģ�����ͼ3����������ʵ�ǣ�����iNOS��ArgI�⣬HIF-1��������������������������������ӵı������ͼ4�������������HIF-1������ͨ��������������;������Tϸ����������Ҫ����Ӧ��Ⱦ�����˻�����ļ���ͼ�������ϸ���IJ�������״̬��
��PyMTģ���У���û������HIF-1��������¹۲쵽���Ƶ�����Ѫ���ܶ�ͻ����������HIF���ܵĸ����ԡ����Ѵ�����ϸ����ɾ��VEGF-A���о��У�������Ѫ���й۲쵽�˸�Ϊ���ŵ����ã�42����ȷʵ�����Ƿ�������ϸ����VEGF-A��ȱʧ�ᵼ��������С�ͽ�չ����������ϸ����HIF-1��ȱʧ��ķ����෴��28��������������ģ�;����෴�ı��ͣ���ˣ�HIF-1���鵼��VEGF-A�յ���ȱʧʵ���Ͽ��ܼ����˴˴������ı��͵Ĵ�С�����ǣ����ǵ����ݲ�δ��ʾ������VEGF-A�����ó��й۲쵽������Ѫ���ܶȵ���Ҫ�仯����ȱ�������£�PyMT������Ƥϸ���Ĺ��������¾���ϸ����ArgI����ǿ���յ����ڸòⶨ�У�����δ�ڵ�����ˮƽ�ϼ�iNOS�յ�����ϸ���Ͳ����ɷ��⣬����Tϸ����IFN���������ϸ����iNOS����Ч���յ���֮һ�� IFN���ڼ�������ϸ�����Է�Ӧʱ��Ҫ��Tϸ�����������������ڴ�������MEC������ģ���в�����Tϸ������ˣ��ý��������������ϸ�������Ŀ����������յ�ArgI��ģ��һ�£���iNOS�����������е��������ӣ���������IFN�õ�Tϸ����˲ʱ�յ�������С����о�������iNOS��������������ϸ���б��ﲻ�ѣ�43��44��-��������PyMT MEC��iNOS���յ����ò������ArgI������������Ҫ����������ø����iNOS���������������������á��ھ���ϸ���鵼��Tϸ�����Ƶ���������»��ڸ����ľ���ϸ����Tϸ��������з�����Ҫ���á�
iNOS��ArgIø��������ͬ�İ�����L-������������ı���������������Ҫ����������ø�Ļ��Բ��ܻ���Tϸ���������á�Я�������Ӱ�����ת�˵���2��CAT2��ͻ���С�����ͨ��iNOS��������һ������������ϸ����ͨ��ArgI;���Ĵ�������������Ҫ��45��46��������С���Է������β���֢3���䣨47����һ�鱨��˵��ȫ ����iNOSȱ������Tϸ���������ӣ�48������ֵ��ע����ǣ�Ұ����С����������߱��ֳ�IFN�������ԣ���iNOS��Ч��С����Ȼ�У�49��50����������������ע��������ʵ��L-������Ĵ�лͨ���ᵼ��Tϸ�����ƣ��ٴ�ǿ��������ø��������Tϸ�����ƵĹ۵㣨35��41�����˴��ṩ�����ݱ�������ȱ���������У���ArgI��iNOS������HIF-1���Ŀ��ƿ��յ�Tϸ�����ƣ�ͼ6�н�ģ�����������������������ı�־������ϸ���Ľ���Ҳ���������HIF-1����ȱ���Ŀ����£�����������Tϸ������;�������ӱ������������������ͽ���ϸ��Эͬ���ã��ƻ�����Ӧ�����߷�Ӧ�����⣬���������Ĺ���ϸ������Ӧ������ϵͳ�ľֲ���HIF-1�������Ժ͵�����ǿ�������ã�����HIF���Ƽ����ư�֢������Ҫ���塣������о���������ϸ��HIF-1���������ķ�ʽ�������ã��ٽ������Ѫ�����ɡ����ǵ�����ȷ���������������л�Ծ��HIF-1�������Ե�����ǿ�������� ͨ·����Щ���ݱ��������ֲ�HIF-1�����Ʒ�չΪһ�ֵ������������Ʒ���ϵİ�֢���Ʒ�ʽ��Ŭ�����ȿ��Խ����ϸ��HIF-1���Ĵ��������ã��ֿ��Լ�������ϸ��HIF-1���������������á�
��Macrophage Expression of HIF-1�� Suppresses T cell Function and Promotes Tumor Progression
Abstract
T cells can inhibit tumor growth, but their function in the tumor microenvironment is often suppressed. Many solid tumors exhibit abundant macrophage infiltration and low oxygen tension, yet how hypoxic conditions may affect innate immune cells and their impact on tumor progression is poorly understood. Targeted deletion of the hypoxia responsive transcription factor HIF-1�� in macrophages in a progressive murine model of breast cancer resulted in reduced tumor growth, although VEGF-A and vascularization was unchanged. Tumor associated macrophages can suppress tumor infiltrating T cells by several mechanisms, and we found that hypoxia powerfully augmented macrophage-mediated T cell suppression in vitro in a manner dependent on macrophage expression of HIF-1��. Our findings link the innate immune hypoxic response to tumor progression through induction of T cell suppression in the tumor microenvironment.Macrophages inhibit T cell proliferation under hypoxia
T cell activation and proliferation are essential steps in the adaptive immune response, and increase the clonal frequency of antigen specific T cells, as well as inducing differentiation into effector and memory cells. Subsequent activation of effector or memory cells results in cytokine release and antigen-specific cytotoxicity. Given the infiltration of the experimental mammary tumors with macrophages and T cells, we tested the capacity of T cells co-cultured with macrophages to proliferate under normoxia and hypoxia. In this assay, T cell division can be detected by flow cytometry. Notably, only the highest ratios of macrophages inhibited T cell proliferation under normoxia (Figure 3A�CC). Careful titration of macrophage:T cell ratios, however, revealed reduced oxygen levels augmented T cell inhibition of macrophages. T cells are able to proliferate under hypoxia (1% oxygen) after CD3/28 stimulation (Figure 3B, 1:80 condition). However, when macrophages were 5% of the total cell number, cell cycle progression was markedly blocked (Figure 3A�CC, 1:20 conditions), whereas normoxic proliferation at this ratio was largely unaffected. Figure 3C quantitates this hypoxic potentiation of suppressive capacity over a range of macrophage:T cell ratios, and demonstrates that lowering oxygen tension increases macrophage inhibition of T cell proliferation and viability.Macrophage T suppression in hypoxia is HIF-1��/iNOS dependent
To test the role of HIF-1�� in macrophage-mediated hypoxic T cell suppression, we carried out co-cultures with macrophages derived from HIF-1��+f/+f/LysM-cre+/− (macrophage HIF KO) bone marrow (Figure 4). In Figure 4A, we show that under normoxia, no change in suppressive capacity between the genotypes exists over a wide range of macrophage:T cell ratios (green and black lines). In hypoxia, however, HIF-1�� null macrophages were poor inhibitors of T cell proliferation at 1:20 and higher ratios, ratios where wild type macrophages induced a potent cell cycle arrest (Figure 4A, red and blue lines, CFSE traces Figure 4B). Using propidium iodide exclusion to count viable cells collected during a fixed time and flow rate, we show in Figure 4C that T cells incubated with wild type macrophages at 1:20 have only 30% of the viability of those incubated with an equal number of HIF-1�� null macrophages. These results show that macrophage-mediated suppression of T cell proliferation in hypoxia is HIF-1�� dependent.Figure 4
HIF-1�� null macrophages fail to augment T cell suppression under hypoxia, and WT suppressive effect is dependent on HIF-1�� upregulation of macrophage iNOS and can be blocked by neutralizing classical proinflammatory cytokines or specific iNOS inhibition
(A) CFSE dilution after T cell activation 60h in normoxia or 1% oxygen with the indicated number of macrophages added to the cultures. (B) representative overlays of CFSE dilution in T cells after activation and co-culture at 1:20 macrophage:T cell as in A; samples are from an independent experiment yet can be identified by the same color scheme as in A. Control unstimulated CFSE loaded T cells appear in grey (C) viable T cells recovered in hypoxic conditions as a % of the amount recovered in normoxic conditions at 1:20 macrophage:T cells; n=2, all results representative of 3 independent experiments. (D) representative CFSE dilution of purified T cells after activation and co-culture for 60h with WT bone-marrow derived macrophages with the cytokine neutralizing antibodies (�� to indicate antibody) or the iNOS inhibitor 1400W added as indicated. Similar results were observed in 3 independent experiments.��
Macrophage Expression of HIF-1�� Suppresses T cell Function and Promotes Tumor Progression
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2948598/��
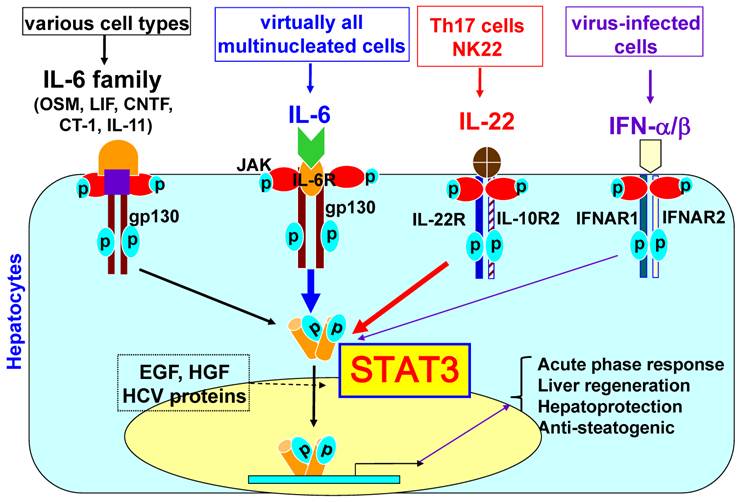
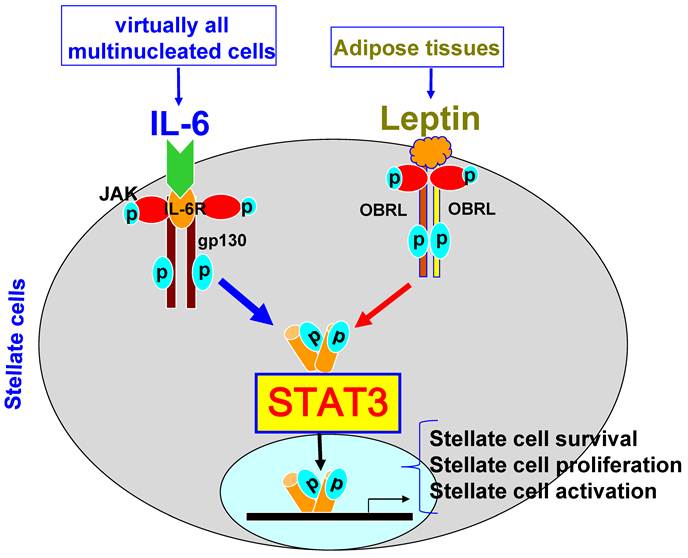
IL-10�Ծ���ϸ��VEGF�����ĵ���ȡ���ھ���ϸ��������ȱ��
IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia
Ѫ����Ƥ��������A��VEGF��������֢�����˺����֯�������е�Ѫ������������Ҫ�����ڲ��������£�VEGF���յ���֯�ƻ���Ѫ�����ɡ�����ϸ������Ӧ��������Ӧһϵ��Э���ź��Դٽ�������֢ʱ�����֧��Ѫ�����ɵ�VEGF�����ݴ̼��IJ�ͬ������ϸ������ı��ʹ��¿ɷ�ΪM1��NOS2 +����M2��������ø-1+����
Abstract
Vascular endothelial growth factor A (VEGF) is critical for vascular remodelling during tissue repair subsequent to inflammation or injury, but under pathological conditions, VEGF induces tissue damaging angiogenesis. Macrophages generate VEGF that supports angiogenesis, when they adapt to their environment and respond with a co-ordinated set of signals to promote or resolve inflammation. Depending on the stimulus, the phenotype of macrophage activation is broadly classified into M1 (NOS2+) and M2 (arginase-1+).In recent studies, IL-10, an anti-inflammatory cytokine that suppresses the M1 phenotype, has been shown to dampen the angiogenic switch and subsequent neovascularisation. However, as we show here, these effects are context dependent. In this study, we have demonstrated that IL-10 inhibits M1 bone marrow-derived macrophages (BMDMs) VEGF, stimulated by LPS/CGS21680 (adenosine A2A receptor agonist), but does not prevent VEGF production from M2 macrophages stimulated with prostaglandin E2 (PGE2). Furthermore, we show that hypoxic-conditioned BMDM generated VEGF was maintained in the presence of IL-10, but was suppressed when concomitantly stimulated with IFN-��. Finally, LPS/PGE2 generated an arginase-1+ M2 macrophage that in addition to generating VEGF produced significant quantities of IL-10. Under these conditions, neither in IL-10 deficient macrophages nor following IL-10 neutralization was VEGF production affected. Our results indicate IL-10 suppressed M1 but not M2 derived VEGF, and that activation signals determined the influence of IL-10 on VEGF production. Consequently, therapies to suppress macrophage activation that as a result generate IL-10, or utilising IL-10 as a potential anti-angiogenic therapy, may result in a paradoxical support of neovascularisation and thus on-going tissue damage or aberrant repair.
IL-10 regulation of macrophage VEGF production is dependent on macrophage polarisation and hypoxia - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0171298510000914��
Figure 3: Immunosuppression in tumor microenvironment. Tumors secrete various factors such as VEGF, IL-6, IL-10, TGF- , Fas-L, and IDO, all of which promote the accumulation of heterogeneous populations of tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), or immature DCs. These immunosuppressive cells inhibit antitumor immunity by various mechanisms, including depletion of arginine and elaboration of reactive oxygen species (ROS) and nitrogen oxide (NO). The tumor microenvironment also promotes the accumulation of regulatory T cells (Tregs) that suppress CD8+ CTL function through secretion of IL-10 or TGF- from Tregs and tumors.
Regulation of Tumor Immunity by Tumor/Dendritic Cell Fusions : Figure 3
https://www.hindawi.com/journals/jir/2010/516768/fig3/��
Tumor-associated macrophages in cancers
��
1.Department of OncologyThe Third Affiliated Hospital of Soochow UniversityChangzhouChina
2.Department of Tumor Biological TreatmentThe Third Affiliated Hospital of Soochow UniversityChangzhouChina
3.Jiangsu Engineering Research Center for Tumor ImmunotherapyChangzhouChina
Tumor-associated macrophages (TAMs) are major component of leukocytic infiltrate of tumors and play important roles in progression and regression of tumors. Tumor microenvironment determines the mutual conversion between M1 and M2 macrophages. In many kinds of tumors, M2 type macrophages are of the majority in TAMs and promote tumor progression and metastasis. The dynamic balance and interaction between TAMs and tumor cells have important effects on the occurrence and development of tumor. TAMs in malignant tumors are useful for clinical diagnosis and may provide a novel target for cancer treatment.Tumor-associated macrophages in cancers | SpringerLink
https://link.springer.com/article/10.1007%2Fs12094-015-1373-0��
Anatomy of a Discovery: M1 and M2 Macrophages
Charles Dudley Mills1,*
Author information Article notes Copyright and License information Disclaimer
1Biomedical Consultants, Marine on St. Croix, MN, USA
Edited by: Uday Kishore, Brunel University London, UK
Reviewed by: John P. Vasilakos, 3M Company, USA; Cordula M. Stover, University of Leicester, UK
*Correspondence: Charles Dudley Mills, Biomedical Consultants, 16930 197th Street, Marine on St. Croix, MN 55047, USA, ude.nmu@200sllim��
Front Immunol. 2015; 6: 212.
Anatomy of a Discovery: M1 and M2 Macrophages
Charles Dudley Mills1,*
1Biomedical Consultants, Marine on St. Croix, MN, USA
Abstract
M1 and M2 macrophage-type responses kill or repair in vivo. The unique ability of macrophages to make these polar opposite type of responses provides primary host protection and maintains tissue homeostasis throughout the animal kingdom. In humans and other higher animals, M1 and M2-type macrophage responses also initiate and direct T cells/adaptive immunity to provide additional protection such as Th1 (cytotoxic) or Th2 (antibody-mediated) type responses. Hence, macrophages were renamed M1 and M2 to indicate the central role of macrophages/innate immunity in immune systems. These findings indicate that the long held notion that adaptive immunity controls innate immunity was backward: a sea change in understanding how immune responses occur. The clinical impact of M1/kill and M2/repair responses is immense playing pivotal roles in curing (or causing) many diseases including infections, cancer, autoimmunity, and atherosclerosis. How M1/M2 came to be is an interesting story that, like life, involved Direction, Determination, Discouragement, and Discovery.
Keywords: macrophages, innate immunity, M1, M2, wound, cancer, Th1/Th2
Introduction
A revolution in immunology is underway. Macrophages and innate immunity are now known to be the primary host defense in all animals (1). It had long been thought that adaptive responses (T and B cells) direct innate immunity (2�C6). Immunology had it backward. Why? I am reminded of the humorous phrase: ��If you hear the sound of hooves, don��t look for zebras.�� That is, look for the obvious (Figure (Figure1).1). Immunology overlooked animal anatomies. Macrophages were the first ��immune�� cells to appear in evolution, are present in virtually all tissues, and far outnumber other leukocytes (7�C9). Despite these anatomical signposts, most immunologists (from the time of Jenner in the 1700s) have been on a quest for the holy grail of immunology: specificity. One cannot blame them. Specific vaccines have resulted in the elimination of world disease scourges such as smallpox and polio.
Figure 1
It is useful sometimes to recognize the obvious rather than look for more complicated explanations in science, or in life.
Meanwhile, macrophages were mainly viewed as ��trash disposal units�� serving at the bequest of the T and B cells and hidden ��under the sink�� (10). One might say an ��Adaptive Dictator�� was in charge (9).
Macrophages were also an enigma. They displayed the stupefying polar-opposite abilities to inhibit proliferation (e.g., kill pathogens) or to promote proliferation (e.g., repair wounds). How could this be? The kill and repair paradox turned out to be based on the elegantly simple and fascinating ability of macrophages to metabolize arginine to either nitric oxide (NO) or ornithine, respectively (11�C17). As important as this discovery was, macrophages held another big secret: one that would fundamentally change our understanding of how immune responses occur.
Macrophages�� unique abilities to kill or repair were found in sterile inflammation, where there were no pathogens (foreign antigens), and also in mice without T (or B) cells (14, 16). These observations helped overturn the long-held belief that adaptive responses were necessary to ��activate�� or ��alternatively activate�� macrophages (3, 18�C20). The importance and independence of innate immunity are highlighted by the oft-overlooked fact that >95% of animals do not have T cells and survive happily in a sea of pathogens, earthworms being an example (7, 8). How? Macrophages! They can kill pathogens within hours. Rapid killing of pathogens is necessary. One bacterium can become the mass of a human in about 4 days, while a T (or B) cell can only become about 16 cells in 4 days. Thus, mathematical considerations alone indicate that clonal proliferation of lymphocytes cannot serve as the primary host defense; this is the job of macrophages throughout the animal kingdom (1). Moreover, in higher animals (e.g., vertebrates) that do have T cells, kill or repair type macrophages (or dendritic cells1) necessarily direct T cells to make Th1 or Th2-type responses, respectively (16, 21�C23).
Together, these and other results about macrophages have caused a fundamental change in our understanding about how immune systems operate. Macrophages/innate immunity initiates and directs virtually all immune responses, including T and B cells/adaptive immunity (1, 9). Hence, I specifically renamed macrophages M1 and M2 to highlight that they, not T cells, are the core of immune systems (16). Of course, once given innate direction by macrophages, the different types of Th1 or Th2-type responses that result can further elevate (or inhibit) M1- or M2-type macrophage responses (1). The macrophage ��revolution�� did not happen overnight, and is continuing. But how macrophages came out from ��under the sink�� to occupy the epicenter of immunology is an interesting story that resembles life itself: one of Direction, Determination, Discouragement, and finally Discovery. It is about the horses, not zebras, of immunology.
Setting a Course of Study: Cancer and Immunology
My path to the study of macrophages took awhile. When I entered graduate school in 1974 at the University of Chicago, immunology was pretty new. There was only one immunology course available and few textbooks; so, learning came mainly from reading journals such as the Journal of Immunology or Journal of Experimental Medicine. I came to believe that the next great immunologic triumph would be more specific vaccines. Having drawn blood in a hospital, as an undergraduate at Syracuse University, patients dying of cancer made a profound impression on me. So, immunology and cancer became my focus: my Direction. At this time, cancers appeared to be ��foreign�� like pathogens (24�C26) and viruses were also implicated (27, 28). But, the antigens on cancer cells were weak; they did not readily elicit specific T (or B) cell responses (29).
Having decided I wished to study immunology, I joined Robert Hunter��s lab at the University of Chicago because he was investigating why some antigens were more immunogenic (elicit an immune response) than others in hopes of augmenting anti-cancer and other immune responses (30). While thinking about a Ph.D. project, I realized that animal bodies, as a whole, are ��negatively charged�� (proteins, sugars, cells, etc.). For example, the electrophoretic separation of most proteins is possible because they migrate from the anode (-) to the cathode (+) at different rates. Cells and other molecules must repel, not stick together, in order to move, to flow. Therefore, I proposed that if a protein antigen was modified to be ��positively charged,�� it would ��stick�� in the body longer and elicit a stronger immune response as illustrated in Figure Figure2.2. It worked (31).
Figure 2
Injection of bovine serum albumin, chemically modified to be ��positively charged,�� into a mouse caused it to be retained longer at the site of injection, and stimulated a stronger T cell mediated immune response.
However, not a lot of people were interested in what made antigens immunogenic in the 1970s (recall the Adaptive Dictatorship), and my manuscript to the Journal of Immunology was rejected: a lesson in Discouragement. Along the way, I learned that humor is a useful way to deal with Discouragement. The south side of Chicago was more ethnically ��diverse�� than where I had lived. The black friends I developed there had the best sense of humor of any group I have encountered. They used humor artfully to diffuse the increased societal Discouragement they typically faced compared to white boys like me.
Investigating how the ��charge�� of an antigen affects its immunogenicity may seem far removed from the title of this paper. However, studying the biochemistry of antigens and how the immune system handles them provided me with important tools that would help later in figuring out how immune systems operate.
With continued excitement that cancer was ��foreign�� and with training in what makes antigens immunogenic (particularly in vivo), I continued in my Determination to boost anti-cancer responses. I joined Bob North��s lab as a postdoc at the Trudeau Institute. Back at Chicago, I had become interested in cytolytic T lymphocytes (CTL) mainly because of Zinkernagel and Doherty��s work, and because Frank Fitch��s lab next door was measuring them (32, 33). Bob, Earl Dye, and I found out that we could use adjuvants (e.g., C. parvum or LPS) to augment tumor-specific CTL responses in vivo that handily caused tumor rejection (34, 35). This was exciting news. The NIH took notice and began clinical cancer trials trying to boost ��killer�� lymphocytes (36).
However, a major crack in the ��cancer vaccine�� armor was becoming apparent to me. It had been reported that mice deficient in T cells did not have an increased incidence of cancer (37). It had also been recently proposed that the immune system could stimulate cancer growth (38). Too, the ongoing NIH clinical cancer immunotherapy trials themselves needed therapy: they did not work (39). The T cell-mediated ��immunosurveillance�� theory of cancer thus seemed wrong (40): another potential Discouragement. However, I was lucky to be at the Trudeau Institute because the studies there mostly focused on understanding diseases processes in vivo: an approach I would continue to use. In addition, macrophage ��activation�� had been discovered there (18, 41) that opened my eyes to another cellular element of the immune system. I also found most interesting the recent observations that macrophages were required for T cells to be activated (42, 43). My postdoctoral studies thus added breadth to my immunologic knowledge that would soon become an advantage: as Pasteur said, ��Chance favors the prepared mind.��
Exploring Macrophages and Solving Their Enigmatic Kill or Repair Abilities
Because of increasing doubts about the ��foreignness�� of cancer, my introduction to macrophages (and moving to Brown University), I adjusted my Direction to focus on the ��trash disposal units�� of the immune system. I was also going to learn that collaborating with people whose expertise is very different than one��s own can be important. I have come to call it ��cross-fertilization.�� I teamed up with surgeons Michael Caldwell and Jorge Albina (and Jeff Shearer) who studied wound metabolism, far different from my expertise in immunology. We found macrophages to be the majority leukocytes in sterile wounds, and that they produced the growth/repair-promoting molecule, ornithine (a precursor of polyamines and collagen), that aids in healing (14). But as I previously mentioned, I had learned from studies at the Trudeau Institute that macrophage ��activation�� was necessary to kill bacterial pathogens (18).
How could one cell perform the polar-opposite activities of growth inhibition (killing pathogens) and growth promotion (healing wounds)? This was vexing indeed. Solving this paradox would eventually lead to the discovery of M1/kill and M2/repair-type macrophages. Not yet, however, as there was still work to be done: Determination.
Pursuing wound healing further, we found that macrophages produced so much ornithine in wounds that they markedly and specifically depleted the substrate, arginine, in vivo. Could low arginine concentrations in inflammation be important? As I mentioned, I focused on studying immune responses in vivo. However, dissecting cellular physiology and functions is sometimes better studied in vitro. Having some skills in biochemistry and contemporary tissue culture techniques, I was able to test the hypothesis that low arginine concentrations negatively impact leukocyte functions. Since macrophages were the predominant leukocytes in sterile wounds, we harvested some resident rat peritoneal macrophages and cultured them in varying concentrations of arginine. Opposite from our hypothesis, the more arginine we added to macrophages, the more their functions declined after a few days. We shelved these experiments, thinking we were dealing with some undecipherable in vitro artifact. Whereas, this seemed another potential Discouragement, I got ��lucky.��
While perusing the current Journal of Immunology in 1987, I came upon an article by John Hibbs and colleagues reporting that macrophages kill tumor cells using arginine: and only arginine (12).
Wow (I will use Wow throughout to highlight those rare and wonderful ��realization�� moments).
I realized that the reason our experiments of adding arginine to macrophages decreased (not increased) their functions was that we were unknowingly adding the ��fuel�� macrophages use to kill, and that the mysterious arginine-derived molecule also killed the macrophages (13). Within months, the arginine-derived killer molecule would be determined to be NO (44). It was a gas (both literally and figuratively humor intended), because now there was an answer to the enigmatic ability of macrophage to kill or repair. Macrophages have the unique ability to metabolize arginine to either make a ��Stop�� signal or a ��Go�� signal, as illustrated by the traffic light in Figure Figure33 [(13), reviewed in Ref. (9, 17)].
Figure 3
Macrophages have both iNOS and arginase enzymes that can convert arginine to NO or ornithine, respectively. Products of each reaction inhibit the opposing reaction, promoting preferential NO or ornithine production.
Go to:
Macrophage Kill and Repair Activities in Wounds and Tumors
The discovery that macrophages could make either a Stop signal (NO) or a Go signal (ornithine) from arginine was amazing to me. But, were these polar-opposite activities physiologically important? We immediately set about determining if and when macrophages made these Stop or Go molecules in vivo. Recall that we already knew that macrophages in healing wounds were making the growth-promoting molecule ornithine. So, we examined if macrophages were also making NO in wounds. They did, but only for a few days after wounding (to kill pathogens if introduced) as shown in Figure Figure44 (14). I was now convinced that these dual arginine-based kill or repair pathways in macrophages were important in vivo.
Figure 4
(A, C) Following wounding, there is a 1-2 day ��burst�� of killer NO (measured as Citrulline and NO2) in vivo, followed by (B, D) macrophages metabolizing arginine to the growth-promoting repair molecule, ornithine (and urea), as healing proceeds. From Ref. (14). Copyright 1990. The American Association.
In parallel with studying macrophages in wounds, I was continuing my cancer studies using an intraperitoneal tumor model. This site allowed me to look at the cellular and molecular events going on inside growing tumors, or in tumors being rejected. I found that macrophages inside growing tumors primarily made ornithine, just like macrophages in healing wounds. Notably, macrophages in growing tumors only made ornithine; there was no initial ��burst�� of NO as observed following wounding. In marked contrast, macrophages inside rejecting tumors (preimmunized mice) made a lot of NO (and there was a strong intratumor CTL response and IFN-�� production) (45). Thus, macrophages inside growing tumors make a molecule (ornithine) that promotes proliferation, and macrophages inside rejecting tumors make a molecule (NO) that inhibits proliferation as shown in Figure Figure55.
Figure 5
(C, D) Macrophages in a growing tumor (naïve) make growth-promoting ornithine (and urea). (A, B) Macrophages in a rejecting tumor (immune) make killer NO (and citrulline). From Ref. (45). Copyright 1992. The American Association of Immunologists, Inc.
Wow These seminal results in 1990 and 1992 convinced me that macrophage arginine-based repair or kill responses were not only important in vivo, but involved with the growth or rejection of cancer: my original Direction.
Involvement of Macrophage Kill or Repair Activities in Many Diseases
The findings that macrophages make proliferation-promoting ornithine during inflammation where cells are growing (healing wounds or cancer), or make proliferation-inhibiting NO where cells are being killed caused me to re-double my Determination to studying these macrophage responses in diseases. My family and I moved to the University of Minnesota where a great new lab complex had been constructed for Mike Caldwell, Jeff Shearer, and me. The breadth of immunologic knowledge I had acquired about macrophages at the Trudeau Institute and collaborations with people whose expertise was different than mine would continue to be fruitful.
Along the way, there were some funding and other difficulties: Discouragement. For example, as I did not publish a lot of papers, funding agencies were perennially ��reminding�� me of this (instead of focusing on citation impact). But my Direction and Determination remained with macrophages.
Damn the torpedoes, full speed ahead! James Farragut, Civil War, 1864.
Having realized from our earlier studies that when macrophages make the gas NO it non-specifically kills everything nearby, I began to wonder if macrophage kill/NO or repair/ornithine responses were involved in other disease processes. For example, it had been observed during several chronic infections that macrophages inhibit specific T (or B) cell responses through ��suppressor�� activity (46, 47). Knowing this, and having the tools to enhance or inhibit NO production, we were able to show that macrophage ��suppressor�� activity (measured in vitro) could largely be attributed to their production of NO (48). It was also revealed that the presence of red blood cells blocked the NO-mediated suppressor activity (NO binds avidly to hemoglobin) (17). But, there are myriad differences between in vitro leukocyte reactions and how the immune system operates in vivo, as I have recently discussed (49). I knew that in rejecting tumors (mentioned earlier, Figure Figure5)5) that there were both specific CTL and macrophages making a lot of NO (45). This model system allowed me to test if macrophage NO also inhibited T cells in vivo. We implanted Alzet Pumps containing N-g-monomethyl-l-arginine (iNOS/NO inhibitor) inside rejecting tumors. Doing so elevated the tumor-specific CTL response (50). Thus, NO was thus not simply beneficial against tumors (or pathogens), but also immunoregulatory. If overproduced, NO could inhibit beneficial immune responses in vivo [reviewed in Ref. (17)]. In a related connection, we knew from our earlier studies in wounds that tissue disruption causes a short ��burst�� of NO production as shown in Figure Figure44 (14). It is now clear that this is an evolutionarily old response that most animals have which serves to ��sterilize�� the area (in case pathogens are introduced) �C something I have termed the ��Damage Danger�� response (9). It happened upon a surgery resident at the University of Minnesota who was working with the noted transplant surgeon David Sutherland. They were trying to figure out how to improve ��islet�� transplantation (groups of insulin-producing �� cells from the pancreas) for diabetes treatment. As in a wound, we found that injecting islets also caused a short local burst of NO. We were able to show that inhibiting this rapid NO response increased the efficiency of cellular transplantation (51). In another study, we found that ��-cell destruction in autoimmune diabetes was also associated with overproduction of macrophage NO and was regulated by insulin (52).
The aforementioned results greatly expanded the ��universe�� of macrophage NO in vivo from that of a host protective molecule to an immunoregulatory molecule and a non-specific tissue-damaging element if overproduced. Subsequent studies have verified the powerful two-edged sword nature of macrophage NO (and ornithine) in many infectious and autoimmune diseases (9, 17, 53�C57), as we had originally observed in wounds and tumors (14, 45). Of particular note, overproduction of macrophage NO appears to be causative in atherosclerosis (58�C60). Thus, the balance between the macrophage killer (NO) and repair (ornithine) responses now seemed important in both of the two major health problems of modern man: cancer and atherosclerosis.
Wow Stay tuned; it gets even better.
The Road to M1 and M2 Macrophages
T cells determine immunity: Or do they?
While I was busy studying macrophages, most immunologists continued to view ��immunity�� in humans (higher animals) from a T cell/adaptive immunity perspective. For example, it had been shown that different strains of mice vary tremendously in their susceptibility to infectious agents. In particular, C57Bl/6 mice were much more resistant to Leishmania than were Balb/c mice (61). The difference in resistance correlated with the ability of C57Bl/6 mouse T cells to produce a lot of IFN-�� during infection that activates macrophages to kill the parasite [by now NO was known to be important in killing intracellular pathogens (62)]. In contrast, Balb/c T cells made more IL-4 that did not stimulate NO production, but instead stimulated antibody production, which was ineffective against the parasite. The IFN-�� dominant T cell response came to be known as Th1, while the IL-4 dominant response was called Th2 (2). That hosts mounted very different T cell responses to Leishmania was an exciting development because it seemed to explain differences in disease susceptibilities.
But my immunology experiences had taught me that correlation is not causation. Recall which leukocytes are the most abundant in animals �C macrophages �C not T cells. The saying that, ��If you hear the sound of hooves, don��t look for zebras�� was about to take on an important new meaning.
Macrophage responses vary between individuals independent of T cells
Knowing there were major differences in the T cell responses of different mouse strains to Leishmania, I wondered if the macrophage killer and repair responses I was studying also varied. We harvested resident tissue macrophages from C57Bl/6 and Balb/c mice (and a few other strains), and compared their abilities to make the killer molecule NO or the repair molecule ornithine. Note: unlike most, I used resident macrophages, not ��elicited�� macrophages. Though the cell yield was much lower (more mice needed), it allowed me to look at ��resting�� macrophages. Resident C57Bl/6 macrophages were much easier to stimulate to make NO (with IFN-�� or LPS) than were Balb/c macrophages. Furthermore, LPS stimulated NO production by C57Bl/6 macrophages, but instead caused increased ornithine production by Balb/c macrophages (16). Thus, using the same stimulus C57Bl/6 mouse macrophages could produce a growth-inhibiting molecule while Balb/c made a growth-promoting molecule. This was very interesting. Also, because the stimuli used were not specific to Leishmania the results suggested that differences in macrophage responses between mouse strains were general phenomena. Having an amino acid analyzer available (because of our interest in metabolism), importantly made direct measurement of the arginine-derived kill (NO) and repair (ornithine) molecules possible: a point I will discuss later. We made our own serum-free culture media for these experiments because it was known that serum contains high levels of TGF-�� (mainly from lysed platelets) that strongly inhibits macrophage NO production (17). Serum-free media also allowed us to show that macrophages make TGF-��, and when they are stimulated to make NO, TGF-�� production goes down, as shown in Figure Figure6.6. Subsequent studies have confirmed that TGF-�� is a key cytokine that regulates the balance between macrophage NO and ornithine production (1, 16, 17, 63�C66).
Figure 6
Dominant NO production by C57B/6 macrophages compared to Balb/c macrophages. Also, NO production is inversely proportional to macrophage TGF-�� production. From Ref. (16). Copyright 2000. The American Association of Immunologists, Inc.
The differences observed in the responsiveness of C57Bl/6 and Balb/c macrophages to LPS or IFN-�� suggested that resistance to Leishmania might involve macrophages. To rule out the influence of T (or B) cells, I compared the ability of C57Bl/6 or Balb/c Nude or SCID macrophages to make NO or ornithine. The results were breathtaking. C57Bl/6 Nude or SCID macrophages made a lot of NO while Balb/c Nude or SCID macrophages did not, just like their normal counterparts (16).
Major Wow
The propensity of macrophages to make killer or repair responses was independent of T (or B) cells. Could this also mean that differences in macrophages between individuals (not T cells) determine susceptibility to Leishmania or other diseases?
The discovery and the importance of M1 and M2 macrophages
As part of investigating macrophage kill or repair responses in different mouse strains, I also wondered whether the propensity of C57Bl/6 or Balb/c T cells to make IFN-�� (Th1) or IL-4 (Th2), respectively, was only true in Leishmania infection. It was not. When I stimulated C57Bl/6 or Balb/c spleen cells with Con A (polyclonal stimuli), they made more IFN-�� or IL-4, respectively. Thus, C57Bl/6 and Balb/c mice had a general propensity to make Th1- or Th2-type cytokines. But why? To answer this question, I designed an experiment that perhaps only an immunologist/immunogeneticist could enjoy. We harvested C57Bl/6 �� Balb/c F1 spleen cells and depleted the macrophages and red blood cells. Then, we mixed the F1 lymphocytes with macrophages from SCID C57Bl/6 or SCID Balb/c mice and added Con A. C57Bl/6 SCID macrophages caused the T cells to make a Th1-type response (IFN-��) and Balb/c SCID macrophages caused the same type of T cells to make a Th2-type response (TGF-��) (16). Note: these experiments were possible because F1 T cells do not recognize either parent as foreign. Differences in macrophage responses alone could explain the ability of different mice to mount Th1- or Th2-type responses and in turn their susceptibility to diseases. Macrophages direct T cells as illustrated in Figure Figure77.
Figure 7
Macrophages from C57Bl/6 mice make M1-dominant (NO) responses while Balb/c are M2-dominant (ornithine). M1- or M2-dominant responses stimulate Th1- or Th2-type responses that can further amplify cellular/CTL and M1, or antibody-type and M2 responses. From Ref. (1) with permission from S. Karger AG, Basel.
Discovery Wow
Because of their polar-opposite kill and repair activities, the independence of these responses from T cells, and that these types of responses stimulated Th1- or Th2-type responses, I renamed macrophages M1 and M2 to highlight the importance of innate immunity over adaptive immunity (16). M1/inhibit and M2/heal responses and their impacts on inflammation and immunity are illustrated in Figure Figure8.8. The long-held belief that ��zebras�� (T cells) were necessary to ��activate�� or ��alternatively activate�� macrophages was incorrect and even backward (3�C6, 19, 20). The adaptive dictator had been overthrown. The horses/macrophages were the center of the immune ��solar�� system. Anatomy was proven correct after all.
Figure 8
Macrophages initiate and direct other immune responses. For example, M1-type responses (e.g., through IL-12 and antigen presentation) direct T cells to become cytolytic T cells and produce IFN-�� that further elevates M1 activity. In contrast, M2-type macrophages cause T cells to produce molecules like IL-4 and TGF-�� that cause B cells to produce antibody and elevate M2 responses. From Ref. (1) with permission from S. Karger AG, Basel.
M1 and M2 Macrophage Responses Defined
Causative functions and molecules that affect health
As described, M1 and M2 macrophage responses were originally defined in vivo by the preferential production of the causative functional molecules NO or ornithine which inhibit or promote proliferation. Since then M1 or M2 macrophages responses have been shown to occur in concert with certain other molecules that can aid in characterization. As shown in Figure Figure9,9, M1 responses are linked with IL-12 and IL-8/CCL production, and cell surface expression of CD 80 or 86 that attract or killer cells like neutrophils and/or stimulate Th1 responses such as CTL and further M1-type activation. M2 responses are associated with TGF-��, and growth factor production (e.g., VEGF or EGF), cell surface expression of CD163 or 206, and the propensity to stimulate Th2 responses such as antibody production and further amplification of M2-type responses, as illustrated in Figure Figure7.7. Macrophages also make TNF-��, IL-6, IL-1, IL-10, NADPH oxidases, and metalloproteinases. However, these molecules are produced by many macrophage populations and are not as clearly diagnostic of M1 or M2-type responses as NO or ornithine or the other molecules listed in Figure Figure99.
Figure 9
Cytokines and other molecules associated with M1/inhibit or M2/heal-type responses. Certain products (middle) have been associated with both M1 and M2-type responses and can be thought of as general ��inflammatory�� cytokines or factors. From Ref. (1) with permission from S. Karger AG, Basel.
Other markers of macrophages
As mentioned above, in addition to molecules that are closely linked to M1 and M2-type macrophage responses, macrophages produce a variety of other what can be called ��inflammatory�� molecules. However, as mentioned earlier with T cells and Leishmania resistance, correlation is not causation. In this regard, some refer to M1 or M2-type responses as ��pro-inflammatory�� or ��anti-inflammatory.�� But, this practice is misleading. For example, M2-type responses dominate in wounds as shown in Figure Figure4.4. As anyone knows a wound is hardly ��anti-inflammatory.�� Wherever macrophages accumulate, there is inflammation. So, molecules like IL-1 or IL-6 are more diagnostic of the presence of macrophages rather than of M1 or M2-type responses. In turn, the use of these inflammation-type markers by some laboratories has lead to classifying macrophage populations as M1 or M2-type that are not. In a related vein, techniques like transcriptomics and FACS (67�C69) are creating ever-enlarging lists of other ��markers�� being used in analysis of macrophage populations, and individual laboratories often use their own particular markers. Not surprisingly then, these variations in the ��metrics�� used has created confusion in trying to classify macrophage populations. For example, various different names have been proposed for macrophages such as: M2 a, b, c; type II; or regulatory macrophages (69�C71). But, such ��subsets�� do not have distinct functions associated with them in vivo like M1/kill or M2/repair. To try and address this confusion, a new ��nomenclature�� was recently suggested to classify macrophages (72). However, the nomenclature suggested is also not based on functions, but mainly on what cytokine or factor was added to macrophages in vitro. In this connection, the various combinations of different cytokines, agonists, or markers that can be employed in stimulating or analyzing macrophages are very high. But the number of macrophage functions is small.
Specifically, macrophages have four core functions called SHIP [sample, heal (M2), inhibit (M1), and present (antigen)] as shown in Figure Figure10.10. Therefore, to best understand macrophage populations it is important to stay focused on analyzing them by functions, such as SHIP, that are known to affect health as recently discussed (1, 49).
Figure 10
Macrophages have four basic SHIP functions [sample, heal, inhibit and present (antigen)] that allow them to recognize pathogens or injury, and respond directly (or indirectly by presenting antigens) to engender responses that provide optimal host protection. From Ref. (1) with permission from S. Karger AG, Basel.
Heterogeneity and plasticity are not macrophage functions
Infections, cancer, or other inflammatory conditions are ever evolving as disease protection or progression occurs. This fact and that macrophages have the unique ability to drastically change their physiology to protect hosts necessarily means that macrophage populations are heterogeneous. Plasticity, a term I coined in 2001 (17), was later popularized by my now deceased friend, Bob Stout, and his wife Jill Suttles (73). Plasticity is a useful word to describe the unique ability of macrophages to change their functions. Beyond this, some have posited that macrophages are like a ��color wheel�� (74). But, it is important to note that heterogeneity, plasticity, or color wheels are not functions that affect health. For example, as we saw, M2-type macrophages inside tumors promote tumor growth while M1-type inhibits tumor growth as illustrated in Figure Figure55 (45). These findings have since been verified in many human cancers (75�C78). Therefore, if one has cancer, one would wish to decrease intratumor M2-type and increase M1-type macrophages. Heterogeneity, plasticity, or color wheels will not stop cancer. So, though such terms are useful in describing the fungibility of macrophage populations, again, advancing health will only come from knowing what macrophage functions are by measuring them, so they can be modulated as needed (49).
Parallel Elements of the ��4Ds�� in Scientific Investigations and Life
As I said at the outset, events in biomedical research can resemble life itself: there is Direction, Determination, Discouragement, and Discovery.
I feel most fortunate in having an upbringing that allowed me to become a scientist. I have met many people along the way with towering intelligence, but who did not have such an advantage and who work at difficult manual labor jobs. I was also fortunate in being influenced early in my Direction by seeing the devastation that cancer can bring, and in picking immunology to study cancer. Regarding Determination, many people work hard and I am not unique. But in science, one should not ��fall in love�� with one��s ideas. As described here, Wow-type moments often come through serendipity: when one must trust the results and abandon existing hypotheses. Recognizing that cancer was not overtly ��foreign��, and focusing on macrophages/innate immunity was one of those moments for me. One cannot know everything about an immense field such as immunology. However, I think cultivating a breadth of knowledge helped prepare me for times when, ��Chance favors the prepared mind��, such as elucidating the arginine-based dual M1/inhibit or M2/heal functions of macrophages (16, 17). As I mentioned earlier, my belief in the importance of macrophages was bolstered by the findings that they were necessary stimulators of T cells (21, 22). Also, toll receptors were identified on macrophages in the 1990s (79�C81) that provided additional support for my concept that macrophage responses are independent of T cells, and also initiate immune responses. As a sidebar here, I worked with Ralph Steinman some when I was a postdoc at the Trudeau Institute in the early 1980s and enjoyed his company. He is credited with discovering dendritic cells (21). It may well be from me having a lack of folds in my cerebrum, but I have always found it simpler to consider dendritic cells as a subset of macrophages (17). So, if you are dendritic cell ��fan,�� you could substitute those words for macrophages in some places in this treatise. But, it does not change the larger point that M1/M2-type macrophages have the unique ability to display polar opposite kill or repair responses and that innate immunity directs adaptive immunity. I will leave the macrophage versus dendritic cell discussion to others (22, 23).
As I said earlier, there was Discouragement along the way. As anyone in biomedical research knows, funding one��s work can be difficult and frustrating. In my own case, I knew deep down I had found something beautiful about the immune system in the Discovery of M1/inhibit and M2/heal-type macrophages in 2000. But dogma can be slow to change, and I did not get an important NIH grant renewed. Having had my own lab for many years, I did not wish to work for someone else, and I left the University of Minnesota to do other things for a while. One very satisfying thing I did was coach my sons and daughter��s basketball teams. I feel sports teach important life lessons, such as fair play, and being gracious in victory or defeat.
In the mid 2000s, my M1/M2 Discovery started to be appreciated. Of course, medical research is very competitive; so, it was not surprising that some tried to rewrite history about macrophage subsets (69, 70). In particular, it is ��curious�� that some reviews about M1 or M2-type tumor-associated macrophages (82, 83) somehow overlook the seminal studies that elucidated the M1 growth-inhibiting and M2 growth-promoting macrophages in tumors and wounds [e.g., Figures Figures44 and and5;5; (14, 45)]. Like I mentioned about sports and fair play, it is appropriate to try and acknowledge other��s studies. In this connection, Zouhair Atassi recognized that I was the origin of the M1/M2 macrophage concept, and asked me to write a review for Critical Reviews in Immunology in 2012 that I entitled, ��M1and M2 Macrophages: Oracles of Health and Disease�� (9).
The M1/M2 concept has fundamentally changed our understanding of what ��immunity�� is by showing the biochemical bases for the unique abilities of macrophages to kill or repair, and that macrophages/innate immunity initiate and direct immune responses throughout the animal kingdom, including adaptive immunity in humans (1, 7, 8, 16). M1/M2 has not only stood the test of time, but thousands of publications indicate that interest in macrophages/innate immunity and clinical applications are ever increasing. I apologize for not mentioning many good results about M1 and M2 macrophages here, but readers should be able to track down studies of interest from the reference list. I am particularly gratified that there is great potential for the successful immunotherapy of cancer by modulating M2 into M1-type macrophages (76, 84): my original Direction. Indeed, Science magazine referred to 2013 as the year of immunotherapy. Some examples of the myriad diseases where the powerful two-edged sword nature of M1 or M2-dominant macrophage responses can be beneficial or detrimental are illustrated in Figure Figure11.11. Macrophages are indeed the oracles of health or disease.
Figure 11
M1/inhibit or M2/heal-dominant macrophage responses (or mixtures) can each be beneficial or detrimental depending on the disease circumstance. For example, M1/Th1-dominant responses are required to fight many infections (left). But, M1/Th1 responses are also causative of destructive inflammatory conditions in the brain and in atherosclerosis (right).
I hope you got a ��charge�� (humor from my Ph.D. earlier) out of ��Anatomy of a Discovery�� and experience your own Wows.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I am fortunate to have associated with colleagues with Direction and Determination across the broad biomedical landscapes to partner with, and which expanded the breadth of my immunologic knowledge. Klaus Ley is one recent example. I have also had superior students and employees to pose good questions, carry out experiments, and share humor to diffuse Discouragement. Finally, having old and new friends and a great family is the biggest Discovery of all.
Footnotes
1Macrophages and dendritic cells are both derived from ��myeloid�� precursor cells. Whether dendritic cells are a unique cell type or a specialized type of macrophage is less important than the bigger point that both are innate-type cells that can present antigens to T cells. So, for convenience, ��macrophage�� will be used throughout here. Readers are directed elsewhere for discussions about macrophages and dendritic cells (21�C23).Anatomy of a Discovery: M1 and M2 Macrophages
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4419847/M1 and M2 Macrophages: Oracles of Health and Disease. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/23428224Charles D Mills | PhD | Independent Researcher | Immunology
https://www.researchgate.net/profile/Charles_Mills2A Breakthrough: Macrophage-Directed Cancer Immunotherapy | Cancer Research
https://cancerres.aacrjournals.org/content/76/3/513��
��
Crit Rev Immunol. 2012;32(6):463-88.
M1 and M2 Macrophages: Oracles of Health and Disease.
Mills CD1.
Biomedical Consultants, 16930 197th St. N, Marine, MN 55047, USA. Mills002@umn.edu
Abstract
The purpose of immunology is simple. Cure or prevent disease. M1/M2 is useful because it is simple. M1/M2 describes the two major and opposing activities of macrophages. M1 activity inhibits cell proliferation and causes tissue damage while M2 activity promotes cell proliferation and tissue repair. Remarkably, the molecules primarily responsible for these "Fight" (NO) or "Fix" (Ornithine) activities both arise from arginine, and via enzymatic pathways (iNOS and arginase) that down regulate each other. The names M1 and M2 were chosen because M1 and M2 macrophages promote Th1 and Th2 responses, respectively. Products of Th1 and Th2 responses (e.g., IFN-��, IL-4) also down regulate M2 and M1activity, respectively. Thus, M1/M2 demonstrated the importance of Innate Immunity and how it is linked to Adaptive Immunity in a beautifully counterbalanced system. "Civilization" and increased longevity present new disease challenges such as cancer and atherosclerosis that do not display classical "foreign" antigens. And, these diseases are often associated with (or caused by) M1- or M2- type responses that were formerly useful for fighting infections, but now are inappropriate in our increasingly "germ-free" societies. In turn, there is considerable potential for modulating M1 or M2 Innate responses in modern diseases to achieve better health. Finally, since M1 and Th1 (or M2 and Th2) often work in concert to produce characteristic immune responses and disease pathologies, it is recommended that Immune Type 1 or 2 (IT1, IT2) would be a simpler and unifying terminology going forward.
M1 and M2 Macrophages: Oracles of Health and Disease. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/23428224��
A Breakthrough: Macrophage-Directed Cancer Immunotherapy
Charles D. Mills, Laurel L. Lenz and Robert A. Harris
DOI: 10.1158/0008-5472.CAN-15-1737 Published February 2016
Abstract
Successful immunotherapy of cancer is becoming a reality aided by the realization that macrophages play an important role in the growth or regression of tumors. Specifically, M2/repair-type macrophages predominate in human cancers and produce growth-promoting molecules that actively stimulate tumor growth in much the same way they help wounds heal. However, modulating M2/repair-type macrophages to M1/kill-type can slow or stop cancer growth. The effects involve direct activity of M1 kill-type as well as the ability of M1-type macrophages to stimulate Th1-type cytotoxic T cells and other effector cells. Macrophage responses can also predict cancer susceptibility; individuals with a high M1/kill to M2/repair ratio are less prone. That macrophages/innate immunity can be modulated to play a central role in directly or indirectly combating cancer is a breakthrough that seems likely to finally make successful immunotherapy of cancer a reality. Cancer Res; 76(3); 513�C6. ©2016 AACR.
Background to Cancer Immunology
Cancer is the most dreaded disease of modern man. Despite billions of dollars spent on finding ��cures,�� cancer kills people at about the same rate it did 50 years ago. The immune system has long been thought to have the potential to slow cancer. Virchow is widely credited with first describing ��white�� cells in tumors, which he called lymphoreticular cells (1), sometimes comprising >50% of the tumor mass. Metchnikoff named such cells ��Big Eaters�� (macrophages) because he observed them engulfing dead cells or pathogens. However, the role of macrophages in tumors was mostly overlooked while investigators tried to identify tumor-specific anticancer responses and to create specific ��cancer vaccines.�� Paul Erhlich was an early proponent of this idea around the turn of the 20th century (1, 2). He and others noticed that cancers could not be transplanted between individuals, suggesting ��foreignness�� such as that with pathogens. However, it was soon realized that normal cells or organs could also not be transplanted: there were ��allogeneic�� differences between individuals that stimulate strong rejection responses by the immune system.
Nonetheless, the concept that cancer was ��foreign�� continued to be enticing to immunologists because of the spectacular successes of specific vaccines against disease scourges, such as smallpox and polio. Dr. William B. Coley and a few others obtained some successes against human cancer in the early 20th century by injecting mixtures of bacteria called ��Coley's Toxin�� (3). However, why this occurred was not clear, and the treatments were dangerous and mostly abandoned. Knowledge of the role of the immune system in cancer was hindered by the fact that tumors died with their hosts. The development of inbred mice and tissue culture techniques were major advances because they allowed tumors to be serially transplanted between individuals or maintained in the laboratory for study. Tumors were identified in mice that have antigens recognized by tumor-specific T cells, and that could be specifically rejected with ��cancer vaccines�� (4). In addition, bolstering hope for specific cancer vaccines was the observations of viruses in cancers, which, if common, could provide a target against which specific T cells or antibodies could be directed (5). Optimism remained high that cancer vaccines would be the next great immunologic triumph.
However, subsequent difficulties in identifying tumor-specific antigens recognized by the immune system in most human cancers stimulated investigators to take a fresh look at how anticancer immune defenses might be boosted. The answer was hiding in plain site. It was the leukocyte long known to predominate in cancer: the macrophage (1, 6).
Observations Trump Optimism in Cancer Immunology
Amidst optimism about specific cancer vaccines were four key observations suggesting that the relationship between the immune system and cancer was not at all as envisaged.
First, there is little or no evolutionary pressure for humans to develop anticancer defenses. Animals succeed/advance mainly by breeding, which enables the retention of desirable, heritable qualities.
Because most cancer occurs after breeding age has been attained, its absence is not an evolutionary survival advantage (6). Second, in the 1970s, it was observed that mice deficient in T cells did not exhibit overall increases in the incidence of cancer (7). Third, it was demonstrated that immune responses could stimulate tumor growth (8). Fourth, though many researchers studied ��immunogenic�� tumors in mice, most spontaneous murine tumors did not possess tumor-specific antigens. Similarly, because few human tumors expressed recognizable tumor-specific antigens, attempts by the NCI and others to stimulate specific ��killer�� lymphocytes in vitro or in vivo against patients' cancers have only been successful with certain tumors such as melanoma (9).
In a related vein, genomic technologies have provided new optimism that unique mutations will be identified in cancer that may allow personalized cancer treatments with drugs or through boosting the immune system. However, so far few targetable differences in cancer have been observed, and the expense involved in this approach is likely to be prohibitive for the general population.
The foregoing observations suggest that tumor-specific antigens on human tumors are rare, and/or there is something unknown about the immune system that prevents immunologic responses from occurring that could inhibit cancer growth. Current evidence suggests that both these conclusions are true. Here, we will focus on new observations that tumor growth�Cpromoting macrophages predominate in cancer but can be modulated into tumor growth�Cinhibiting macrophages, resulting in successful cancer immunotherapy.
Macrophages Predominate in Cancers and Wounds and Promote Growth
Investigations into the activity of macrophages in sterile wounds and developing tumors revealed important similarities. In both circumstances, the macrophages present produce a large quantity of the growth-promoting molecule ornithine (a precursor of polyamines required for cell proliferation; refs. 10, 11; reviewed in ref. 12). Although macrophages had been shown in the 1960s to be ��activated�� by T cells in vivo and to be necessary for defense against many bacteria (13), macrophages in either sterile wounds or growing tumors did not exhibit killing activity (10, 11). At this time, it was unclear how macrophages killed pathogens. In the late 1980s, John Hibbs and colleagues discovered that macrophages kill pathogens and cancer cells through the production of nitric oxide (NO; ref. 14). Fascinatingly, macrophages produce both growth-inhibiting NO and growth-promoting ornithine via the enzymatic conversion of arginine through inducible nitric oxide synthase or arginase, respectively (6, 15). In contrast with a growing tumor, macrophages inside a tumor being rejected produced prodigious quantities of NO (11). These seminal observations provided the biochemical explanation for the unique ability of macrophages to kill or repair, depending on the circumstance (reviewed in 12). Macrophage populations that inhibit growth or kill are now called M1-type, and those that promote growth and repair are called M2-type (16). Most relevant to cancer, the results demonstrated that macrophages inside growing tumors actively promote growth: findings roundly verified in human tumors (12, 17, 18). Tumor-associated macrophages have since been demonstrated to produce other growth-promoting molecules in addition to ornithine, including VEGF, EGF, and TGF�� as illustrated in Fig. 1A (12).
Figure 1.
A, tumor growth is accompanied by the preferential accumulation of M2/repair-type macrophages. Such macrophages promote growth and metastasis through their production of growth-promoting molecules and intercellular matrices. B, macrophage-innate conversion from M2 to M1-type (MIC1) can directly cause tumor rejection. C, if tumor-specific antigens are present, macrophage-adaptive conversion from M2 to M1-type (MAC1) can directly (non-specifically) and indirectly (specifically) cause tumor rejection.
Direct Macrophage/Innate Effects on Cancer
As discussed above, most cancers are primarily populated by M2/repair-type macrophages, but if instead M1/kill activity is enhanced, locally tumor inhibition is observed (11, 18, 19). Because many cancers do not display distinct tumor antigens, both the growth promotion and the growth inhibition occur via ��innate�� mechanisms (11, 20). Macrophage tumor growth promotion can occur because the tumor environment provides signals that inhibit M1/kill-type activation, such as PGE2 or TGF�� (17, 18). Although such signals are believed to be important in suppressing macrophages, M1/kill-type activity may also not be stimulated because of the absence of ��toll��-like or specific tumor antigens as in a sterile wound (10). The tumor environment or the lack of activating stimuli also seems to play a role in limiting the activation of other innate responses, such as natural killer cells that can kill tumor cells directly, or augment M1/kill-type activation through IFN�� production (20).
Whereas this intratumor circumstance may seem foreboding, recent evidence indicates that M2/repair-type macrophages can be modulated to M1/kill-type, and such activation is sufficient on its own to cause tumor rejection (11, 19, 20). The importance of direct macrophage activity in cancer outcomes is supported by observations that animals with M1/kill-dominant responses (e.g., C57Bl/6) exhibit a decreased tumor incidence when compared with M2/repair-dominant mice (e.g., Balb/c; refs. 12, 16, 21). Lower animals such as invertebrates (without T or B cells) also exhibit low cancer incidences, which is consistent with an important role of macrophages/innate immunity in inhibiting tumor appearance or growth (22�C24). Figure 1B illustrates how macrophage innate conversion therapy to M1/kill responses (or MIC1) can result in tumor regression. As will be evident in the following section, new knowledge indicates that proper modulation of intratumor macrophages is also necessary to direct T or B cells toward tumoricidal responses that can occur whether or not tumor-specific antigens are present.
Macrophages/Innate Immunity Indirectly Influences Cancer and Other Immune Responses
In addition to their direct tumor-promoting activity discussed above, other key discoveries about macrophages have indicated that their influence on immune responses to cancer and other diseases is much greater than was previously thought. In particular, it became known in the 1970s that macrophages are necessary to present antigens to T cells (25). Toll and other receptors were then identified on macrophages, indicating that they can directly and specifically recognize pathogens (26)��something T cells are unable to do. Subsequently, it was demonstrated that the respective polar-opposite macrophage M1/NO/kill and the M2/ornithine/repair responses both occurred in mice devoid of T cells (16), which importantly established the independence of innate from adaptive immunity. As mentioned, some animals (e.g., C57Bl/6 mice) exhibit M1/kill-dominant macrophage responses associated with lower cancer incidence, whereas others (e.g., Balb/c) are M2/repair dominant. Perhaps most pertinent to anticancer responses, it was also discovered that M1-dominant macrophages stimulated naïve T cells to make a Th1/cytotoxic response, whereas those exhibiting M2-dominant responses stimulated a Th2-type response associated with antibody production (16). This was the reason macrophages were specifically termed M1 and M2. Some prefer the term ��dendritic cells�� for myeloid-derived cells that direct T-cell responses. However, the most salient point is that either leukocyte can direct T- and B-cell responses, so here we will use the term ��macrophage�� for both and leave that debate to others (27).
Regardless of the terminology, an important and promising new observation for cancer immunotherapy (as well as immunology in general) is that macrophages not only direct T- or B-cell responses, but can also do so in the presence or absence of specific antigens. In particular, M2-type macrophages, through innate signals such as TGF�� and IL10, induce T cells into Treg and other T-cell type responses without anticancer activity (16, 18). In contrast, M1-type macrophages activate Th1-type responses that can further amplify M1/killer-type activity through the production of IFN�� (12). Such Th1-type activity can inhibit cancer because macrophage-derived NO is nonspecific in its killing activity once generated (14, 28). In addition, if specific tumor antigens are present, macrophage-directed adaptive immunity can result in the stimulation of tumor-specific cytotoxic T cells. This macrophage adaptive conversion therapy, or MAC1 (Fig. 1C), has the additional advantage that cytotoxic T cells recognize and kill tumor cells directly, preventing collateral damage by macrophage killing, and also protective T- or B-cell memory can be engendered (12).
Thus, the realization that macrophages/innate immunity plays pivotal roles in directing cancer outcomes, either directly or by nonspecifically influencing T- and B-cell functions, in addition to the potential to activate specific anticancer defenses (if suitable antigens are present), is opening up new approaches to cancer immunotherapy.
The Bright Future of Macrophage-Directed Therapy for Eliminating Cancer
Evidence reviewed herein indicates that modulating macrophage responses is a breakthrough that will facilitate successful immunotherapy. There are still hurdles to overcome. For example, earlier attempts at stimulating macrophages/innate immunity (typified by ��Coley's Toxin��) were accompanied by dangerous side effects (3) that have also been observed in more recent attempts at immunotherapy (9). However, an increased understanding of the mediators involved in such side effects and an armamentarium of new drugs should allow the positive effects of elevating M1/kill and other anticancer innate responses to be manifest while minimizing undesirable effects. Although increasing M1/kill responses through macrophage-innate or adaptive conversion therapy (MIC1 or MAC1) is beneficial against cancer, it is also recognized now that overzealous M1/kill�CTh1 cytotoxic responses contribute to (or cause) atherosclerosis and other chronic inflammatory conditions (6, 12, 29). Therefore, in cancer and in other conditions, it will be important to be mindful of the powerful two-edged nature of macrophage responses for optimal results.
The biggest triumphs of immunology to date have been against infectious diseases. An exciting new chapter is beginning. Macrophage-based immunotherapy will help ameliorate cancer and other diseases via more natural, effective and less-toxic and disabling means than chemotherapy, drugs, or surgery.A Breakthrough: Macrophage-Directed Cancer Immunotherapy | Cancer Research
https://cancerres.aacrjournals.org/content/76/3/513��
��
Macrophage arginine metabolism and the inhibition or stimulation of cancer
Article
Nov 1992
Charles D Mills
Jeffry D Shearer
R Evans
M D Caldwell
The potential of the immune system to inhibit or stimulate tumor growth is a vivid example of the "two-edged sword" nature of immune responses. Our results provide evidence that this dual capacity can be attributed, in part, to the dual pathways of arginine metabolism exhibited by intratumor macrophages. Specifically, i.p. tumor rejection in P815-p...
Macrophage Arginine Metabolism to Ornithine/Urea or Nitric Oxide/Citrulline: A Life or Death Issue
Article
Full-text available
Feb 2001
Charles D Mills
Macrophages can metabolize arginine to nitric oxide in quantities that inhibit pathogens or nearby host cells. They can instead metabolize arginine to ornithine (a precursor of polyamines and collagen) in quantities that stimulate pathogens or nearby host cells. Macrophages are essentially the only circulating cells that can make these life or deat...
View
Question - What could be the effective LPS dose M1 polarization?
Charles D Mills
Mar 2016
Answer
A 'two-signal' model of macrophage 'activation' developed in the late 20th century. It believed IFN-g primed and LPS triggered. Virtually all of the data that supported that idea were obtained by culturing macrophages in FBS-containing media. FBS contains enough TGF-b to strongly inhibit M1/kill/NO polarization. Getting rid of TGF-b revealed that either IFN-g or LPS is sufficient for M1-type activation. Additionally, several investigators used Balb/c mice which are more difficult to M1-type activate. C57Bl/6 in contrast readily responds to either IFN-g or LPS alone for M1-type activation. Good luck. Papers attached. Charlie
View
Question - Are there any specific cell surface markers for M1 and M2 macrophages?
Charles D Mills
Mar 2016
Answer
Hi Shilpa:
Understand that macrophages are not chameleons: able to change their 'colors' as necessary. If macrophages commit to kill, they make molecules, like NO, that kill everything nearby, including the macrophages themselves. Innate kill molecules are non-specific unlike adaptive responses, like CTL that can bind to and kill specifically without killing themselves or surrounding targets. So, the whole idea of bi directional 'plasticity' in macrophages welled from people who don't fully understand macrophage biology/biochemistry. Mosser et al is on source of thinking that macrophages are like a 'color wheel', akin to chameleons. M2/repair to M1/kill: yes. The other way, no. I hope helpful. Charlie Mills
A Breakthrough: Macrophage-Directed Cancer Immunotherapy
Article
Jan 2016
Charles D Mills
Laurel L Lenz
Robert Harris
Successful immunotherapy of cancer is becoming a reality aided by the realization that macrophages play an important role in the growth or regression of tumors. Specifically, M2/repair-type macrophages predominate in human cancers and produce growth-promoting molecules that actively stimulate tumor growth in much the same way they help wounds heal....
View
Question - Are there any specific cell surface markers for M1 and M2 macrophages?
Charles D Mills
Sep 2015
Answer
Hi Chad:
Thanks for your interesting post. Phagocytosis was the basis for Metchnikov naming them "Big Eaters" (macrophages), of course, in the late 1800's. Classical activation (as originally elucidated by Mackaness and colleagues in vivo at the Trudeau Institute) has long been associated with increased phagocytic activity, and increased killing ability through the production of reactive oxygen and nitrogen species. So, temporally, it would seem that "killer"/M1 macrophage activity is linked to phagocytic activity often. Of course, phagocytosis is not required to classically activate macs; they can get revved up by Toll and other receptors (e.g., binding LPS). So, in this sense I'm not sure I could support M1-type as non phagocytic versus M2-type as a stand alone way to distinguish macs. Too, eating by macrophages is not really a "functional" activity because it is what happens after they eat that really matters. Do they digest/eliminate what they eat and/or do they kill it with O or N species.? Of note: M1/kill activity results in the production of Nitric Oxide which is a gas that freely diffuses in all directions killing everything nearby - it is non-specific. This includes killing the macs that make it (reviewed in the Anatomy of a Discovery paper I posted earlier). So, those that have posited bidirectional "plasticity" (e.g., Mosser), I feel do not fully understand the biology of macs. Likely, M1 activation normally results in an end stage cell that "takes one for the team" (dies in battle). Thus, M2s (resident macs) can become M1s, but not normally vice versa. Actually, I coined the term "plasticity" in 2001 to indicate that macs can change, and that all populations of macs are evolving mixtures as inflammation proceeds in one way or the other. Getting back to phagocytosis, certainly M2s eat. Whether on a cell per cell basis they eat more than M1 activated, or that this is a valid way to distinguish M1 and M2 activity, I'm not so sure. After all, what we agree on is that assessing macrophages by their health-impacting effects (be it wound healing or pathogen elimination) is the most valuable metric. Finally, Alex Carrel in the early 1990s proposed that macrophage could "transform" into fibroblasts. I believe this is true in some regard, though what "flavor" of fibroblast they become is unknown. This seems to occur in inflammation that is "sterile", like wound healing, or in vitro if there is no M1-type activation. So, if this is true, M2s are the ones that transform. Fibroblasts lose most ability to phagocytose, like macs. So in this circumstance, M2s evidently have less, not more phagocytic activity. Check out Figure 4 of my attached original M`/M2 paper. Mac morphology changes dramatically depending on the culture conditions. If interested, there are some references to macrophage conversion to fibroblast-like cells in my 2012 review attached. Anyway, those are some thoughts about the complex "lives" that macs lead. I hope helpful. Charlie
View
��Charles D Mills | PhD | Independent Researcher | Immunology
https://www.researchgate.net/profile/Charles_Mills2��
TAMeless traitors: macrophages in cancer progression and metastasis
Shweta Aras & M Raza Zaidi
British Journal of Cancer volume 117, pages1583�C1591 (2017)and Department of Medical Genetics and Molecular Biochemistry, Fels Institute for Cancer Research and Molecular Biology, Lewis Katz School of Medicine at Temple University, Philadelphia, 19140, PA, USA
Abstract
Macrophages are conventionally classified into M1 and M2 subtypes according to their differentiation status and functional role in the immune system. However, accumulating evidence suggests that this binary classification system is insufficient to account for the remarkable plasticity of macrophages that gives rise to an immense diversity of subtypes. This diverse spectrum of macrophage subtypes play critical roles in various homeostatic and immune functions, but remain far from being fully characterised. In addition to their roles in normal physiological conditions, macrophages also play crucial roles in disease conditions such as cancer. In this review, we discuss the roles tumour-associated macrophages (TAMs) play in regulating different steps of tumour progression and metastasis, and the opportunities to target them in the quest for cancer prevention and treatment.
Concluding remarks
The conventional binary model of macrophage polarisation is becoming antiquated and a newer model of polarisation spectrum is coming into conception, which focuses on involvement of an array of differentiated macrophages in various immunoregulatory disorders as well as cancers. Moreover, it is now well-established that apart from tumour cell-derived factors, stromal microenvironmental factors play a substantial role in tumourigenesis and TAMs constitute as key players in this phenomenon by regulating various steps of tumour initiation, progression, and metastasis. Since the abrogation of immunosuppressive macrophages in the tumour microenvironment enhances anti-tumour response, targeting TAMs is rapidly emerging as a promising therapeutic strategy for cancer patients.��
Main
Macrophages constitute a prominent set of immune cells that are phagocytic in nature and are present in almost all tissues. In general, they are differentiated cells of mononuclear origin and display specific phenotypic characteristics. In mice, macrophages show surface expression of markers such as CD11b, F4/80 and colony-stimulating factor-1 receptor (CSF-1R) and do not express Gr1; whereas, in humans, macrophages show expression of CD68, CD163, CD16, CD312 and CD115 (Qian and Pollard, 2010).
Macrophages are an incredibly diverse set of cells, constantly altering their functional state in response to environmental stimuli. They undergo the ��polarisation�� process wherein they express different surface markers and functional programs in response to microenvironmental stimuli such as the cytokines and other signalling mediators. According to the binary polarisation concept, there exist two polarisation states of macrophages: Classically activated macrophages (M1) produce pro-inflammatory cytokines and reactive oxygen/nitrogen species, which are crucial for host defence and tumour cell killing and, therefore, are considered as ��good�� macrophages. The alternatively activated macrophages (M2) produce anti-inflammatory cytokines and are involved in the resolution of inflammation. These are considered ��bad�� macrophages because they not only suppress the destructive immunity against parasites and tumour cells, but also promote angiogenesis and matrix remodelling, which make the tumour microenvironment conducive to tumour progression and metastasis (Huang and Feng, 2013).
Macrophages not only perform vital functions in normal physiological conditions such as development, wound healing, infection and maintenance of tissue homeostasis, but are also involved in a variety of disease conditions such as autoimmune disorders, atherosclerosis and tumourigenesis (Wynn et al, 2013). Tumourigenesis is a highly complex, multi-step process and many findings provide strong evidence for the role of specific subsets of macrophages in tumourigenesis. These macrophages are commonly referred to as ��tumour-associated macrophages (TAMs)��. Tumour-associated macrophages were thought to closely resemble the M2 phenotype; however, findings discussed in this review suggest that this binary polarisation model is going obsolete, and there exists a whole spectrum of TAM phenotypes that are yet to be discovered and fully characterised. Tumour-associated macrophages also contribute to many steps of tumourigenesis, such as transformation, tumour cell proliferation, angiogenesis, invasion and metastasis. In this review, we summarise various physiological and functional aspects of TAMs, as well as their roles in regulating various steps of tumour initiation, progression and metastasis. This review will help shed light on the potential of TAMs as prognostic biomarkers for various cancers, as well as ways to target them for therapeutic interventions.
The two faces of macrophages
The role of macrophages in tumourigenesis has been controversial. Macrophages have conventionally been considered to be anti-tumourigenic in nature, play tumour-suppressive roles, and illustrate a significant link between high density and better prognosis, particularly in the case of colorectal cancer. For instance, Ong et al showed that macrophages in a colorectal cancer model are pro-inflammatory, and inhibited the growth of tumour cells by secreting chemokines to attract T cells, thereby priming an anti-tumour type-1 inflammatory response (Ong et al, 2012). It was also reported that when stimulated with TLR ligands, anti-CD40 or interferon-�� (IFN-��), TAMs show anti-tumoural functions, provided CD47 expression on cancer cells does not inhibit tumour cell phagocytosis (Jaiswal et al, 2009). Moreover, pro-inflammatory macrophages were also shown to have tumour suppressive effects via production of reactive oxygen species and reactive nitrogen intermediates (Vicetti Miguel et al, 2010). However, many subsequent studies challenged this notion and indicated that macrophages also display pro-tumourigenic properties. In a mouse model of breast cancer, Lin et al showed that a homozygous null mutation of a gene encoding the macrophage growth factor, colony stimulating factor-1 (CSF-1), not only reduced macrophage infiltration but also completely abolished tumour progression and metastasis. On the contrary, overexpression of this CSF-1 protein increased the rate of tumour progression and metastasis (Lin et al, 2001). Moreover, inhibition of the CSF-1 receptor signalling pathway by virtue of a small molecule inhibitor abrogated infiltration of TAMs and enhanced recruitment of CD8+ T cells, thereby reducing cervical and mammary tumour growth (Strachan et al, 2013). According to Shree et al, cathepsin-expressing macrophages protected against chemotherapy-induced tumour cell death in breast cancer, and cathepsin inhibition significantly reversed this phenomenon (Shree et al, 2011). Recently, Gordon et al, showed that both mouse and human TAMs express programmed cell death protein 1 (PD-1), thereby negatively regulating their phagocytic activity against tumour cells. Blockade of this PD-1/PD-L1 axis restores phagocytic activity by these TAMs, reduces tumour growth and lengthens survival of mice, strongly suggesting a pro-tumourigenic potential of these TAMs (Gordon et al, 2017).
Macrophages are multifaceted and highly plastic in their characteristics. The classically activated M1 macrophages are stimulated by microbial substrates such as lipopolysaccharide, toll-like receptor ligands and cytokines such as IFN-��, and are involved in Th1 type of responses. Once activated, M1 macrophages are characterised by secretion of pro-inflammatory cytokines such as interleukins IL6, IL12, IL23 and tumour necrosis factor-��, and have a strong microbicidal and tumouricidal functions. Phenotypically, they express high levels of major histocompatibility complex class II (MHC-II), CD68, and CD80 and CD86 costimulatory molecules.
The alternatively activated M2 macrophages are stimulated by IL4 and IL13, secrete IL10, transforming growth factor-�� (TGF-��), and chemokines, and are involved in tissue remodelling and tumour progression (Fan et al, 2016). Phenotypically, M2 macrophages express low levels of MHC-II and feature expression of CD163 (Barros et al, 2013), CD200R membrane glycoprotein (Jaguin et al, 2013) as well as high levels of MGL1 and MGL2, which are members of the macrophage galactose type C-lectin family (Raes et al, 2005). A genetic profile for M2 macrophages showed upregulation of the genes arginase 1 (Arg1), MMR (Mrc1), resistin-like molecule �� (FIZZ1), and chitinase-like protein Ym1 (Raes et al, 2002). Tumour-associated macrophages in the tumour microenvironment exhibit M2-like polarisation state of macrophages with pro-tumourigenic functions because they express a series of markers, such as CD163, the Fc fragment of IgG, C-type lectin domains, and heat shock proteins, some of which are commonly expressed in M2-macrophages. Moreover, acquisition of an M2-like phenotype is also caused by secretion of tumour-derived cytokines such as IL4, IL10, and IL13 (Sica et al, 2002; Sakai et al, 2008) (Figure 1).
Macrophage differentiation and their role in tumourigenesis. Classically activated (M1) macrophages are activated by IFN-��, LPS or TLR ligands, secrete pro-inflammatory cytokines and play tumouricidal roles. Alternatively activated (M2) macrophages are activated by IL-4 and IL-13, secrete anti-inflammatory cytokines IL-10 and TGF-�� and play tumourigenic roles. Tumour-associated macrophages (TAMs) display M2-like phenotype and exhibit pro-tumourigenic features.
The classification conundrum
A large body of research clearly suggests that the historical binary classification of macrophages is grossly oversimplified, and represents two extremes of their activation states. In view of some recent findings about macrophage activation, the classical M1/M2 polarisation model seems to be obsolete as it fails to fully account for the complexity of the macrophage activation process. Xue et al recently showed that by virtue of highly specific and standardised stimulation of human macrophages, the current M1/M2 paradigm can be expanded into a ��spectrum model�� (Xue et al, 2014). This model suggests that due to the presence of a network of transcriptional regulators, there exists a spectrum of differentiated macrophages, many of which are yet to be fully discovered.
A recently published report identified a few other categories of macrophages with molecular phenotypes that do not fit the conventional M1 or M2 types, but have been involved as main players in various human pathologies. For example, the antigen CD169 (Siglec-1) is highly expressed on and is reported as a marker of one macrophage subpopulation found in bone marrow, lymph node, liver, and spleen. Although the information about the signalling pathway involved in the activation of CD169+ macrophages is imprecise, CD169+ macrophages are mainly involved in erythropoiesis and immune regulation (Chow et al, 2013). Another non-M1/M2 subtype of macrophages expresses T-cell receptor (TCR). T-cell receptor is required for antigen recognition, and several reports have suggested the presence of murine and human TCR+ macrophages, especially TCR����+ and TCR�æ�+ macrophages, in inflammatory and infectious diseases (Chavez-Galan et al, 2015). Georgoudaki et al recently identified a novel subtype of TAMs with an M2-like immunosuppressive gene profile expressing a novel receptor ��macrophage receptor with collagenous structure�� or ��MARCO�� in mouse tumour models of mammary carcinoma, colon cancer and B16 melanoma (Georgoudaki et al, 2016).
Another example of non-M1/M2-type macrophages is IFN-��-secreting macrophages. Interferon-�� forms an important constituent of the innate immune defence system and secretion of IFN-�� by human NK cells and T cells in response to interleukins has been long established. However, IFN-�� secretion by macrophages is still controversial owing to the doubts about contamination of macrophages by NK or T cells. However, a growing body of evidence suggests that macrophages are capable of secreting IFN-�� both in vitro and in vivo. For instance, Darwich et al showed that at single cell level, human macrophages secrete IFN-�� after induction with interleukins IL-12 and IL-18 (Darwich et al, 2009). Robinson et al showed a similar phenomenon of induction of IFN-�� secretion post Mycobacterium tuberculosis infection of human macrophages (Robinson et al, 2010). We have also identified macrophages secreting IFN-��, which promoted melanoma growth in an allograft mouse model (Zaidi et al, 2011) (Figure 2).
The ��binary�� vs ��spectrum�� model of macrophage polarisation. Recent evidence strongly suggests that the conventional model of binary polarisation of macrophages into M1 and M2 subtypes is oversimplified and the molecular profile of several newly discovered subtypes of macrophages do not fit either phenotype. The spectrum model of macrophage polarisation suggests that there exist various subtypes of differentiated macrophages by virtue of an intricate network of transcriptional regulators, which participate in many homeostatic as well as pathological functions.
Given the importance of macrophages in homeostatic and pathological conditions, a thorough investigation of the multiple factors in normal and diseased microenvironments is absolutely warranted to dissect the mechanisms of macrophage activation, plasticity, and polarisation.
TAMs in tumour microenvironment
Tumour-associated macrophages originate from the circulating peripheral blood monocytes, which are derived from the bone marrow. These monocytes are recruited to the tumour tissues and then differentiate locally in response to a variety of cytokines, chemokines, and growth factors produced by the stromal and tumour cells in the tumour microenvironment. For instance, the chemokine CCL2 and macrophage colony-stimulating factor were shown to recruit inflammatory monocytes to the tumour site, and then differentiate into TAMs in response to IL-4, IL-10, IL-13 and other cytokines in the tumour microenvironment and promote tumour metastasis (Qian et al, 2011). Another report suggested that hypoxia-inducible chemotactic factors such as the CXCR4 ligand CXCL12 and Angiopoietin-2 (Ang-2) promote recruitment of Tie2-expressing monocytes in hypoxic areas of tumours and differentiate them into Tie2-expressing macrophages (Murdoch et al, 2007). Some other microenvironmental factors such as CSF-1, CCL2, IL6, vascular endothelial growth factor (VEGF-A) and platelet-derived growth factor (PDGF) have also been involved in infiltration of monocytes to the tumour sites (Balkwill, 2004; Joyce and Pollard, 2009).
It is now well-established that the majority of malignant tumours contain macrophages as a major component of their tumour microenvironment. Upon stimulation, these TAMs secrete a wide variety of cytokines, growth factors, inflammatory substrates and proteolytic enzymes that play major roles in cancer progression (Figure 3). Moreover, clinicopathological studies have shown that there also exists a strong correlation between increased macrophage density and poor prognosis in lung, hepatocellular carcinoma, renal cell carcinoma (Komohara et al, 2011) and breast cancer (Campbell et al, 2011; Mahmoud et al, 2012; Medrek et al, 2012). Moreover, according to a recent report by Wang et al, macrophages were also shown to play a critical role in melanoma resistance to BRAF inhibitors (Wang et al, 2015). Therefore, TAM infiltration can potentially be used as a prognostic marker of clinical outcomes for many cancers and can be potentially targeted for cancer prevention or treatment.
��
Given the importance of macrophages in homeostatic and pathological conditions, a thorough investigation of the multiple factors in normal and diseased microenvironments is absolutely warranted to dissect the mechanisms of macrophage activation, plasticity, and polarisation.
TAMs in tumour microenvironment
Tumour-associated macrophages originate from the circulating peripheral blood monocytes, which are derived from the bone marrow. These monocytes are recruited to the tumour tissues and then differentiate locally in response to a variety of cytokines, chemokines, and growth factors produced by the stromal and tumour cells in the tumour microenvironment. For instance, the chemokine CCL2 and macrophage colony-stimulating factor were shown to recruit inflammatory monocytes to the tumour site, and then differentiate into TAMs in response to IL-4, IL-10, IL-13 and other cytokines in the tumour microenvironment and promote tumour metastasis (Qian et al, 2011). Another report suggested that hypoxia-inducible chemotactic factors such as the CXCR4 ligand CXCL12 and Angiopoietin-2 (Ang-2) promote recruitment of Tie2-expressing monocytes in hypoxic areas of tumours and differentiate them into Tie2-expressing macrophages (Murdoch et al, 2007). Some other microenvironmental factors such as CSF-1, CCL2, IL6, vascular endothelial growth factor (VEGF-A) and platelet-derived growth factor (PDGF) have also been involved in infiltration of monocytes to the tumour sites (Balkwill, 2004; Joyce and Pollard, 2009).
It is now well-established that the majority of malignant tumours contain macrophages as a major component of their tumour microenvironment. Upon stimulation, these TAMs secrete a wide variety of cytokines, growth factors, inflammatory substrates and proteolytic enzymes that play major roles in cancer progression (Figure 3). Moreover, clinicopathological studies have shown that there also exists a strong correlation between increased macrophage density and poor prognosis in lung, hepatocellular carcinoma, renal cell carcinoma (Komohara et al, 2011) and breast cancer (Campbell et al, 2011; Mahmoud et al, 2012; Medrek et al, 2012). Moreover, according to a recent report by Wang et al, macrophages were also shown to play a critical role in melanoma resistance to BRAF inhibitors (Wang et al, 2015). Therefore, TAM infiltration can potentially be used as a prognostic marker of clinical outcomes for many cancers and can be potentially targeted for cancer prevention or treatment.
��TAMs in angiogenesis
In many cancers, benign-to-malignant transition is associated with a significant increase in vascularisation, a process known as angiogenesis, which provides cancer cells nutrients and oxygen to allow them to multiply, invade and metastasise. This process of forming new vasculature is highly complex and TAMs are one of the major contributors in this process (Lin and Pollard, 2007). According to a recent report, quantitative analysis and assessment of the spatial associations between TAMs and tumour neovasculature demonstrated the great significance and close association of TAMs and tumour angiogenesis during cervical cancer development and progression (Jiang et al, 2016). Neovascularisation is induced when TAMs secrete pro-angiogenic factors, such as VEGF, adrenomedullin (ADM), PDGF, TGF-�� and matrix metalloproteinases (MMPs). For instance, VEGF-A was reported to contribute to neoangiogenesis and macrophage recruitment at the tumour site in a mouse model of skin carcinogenesis (Linde et al, 2012). Moreover, TAMs were shown to sense hypoxia in avascular areas within tumours and release VEGF-A, a very potent pro-angiogenic factor (Laoui et al, 2014). Another report on Merkel cell carcinoma, a highly malignant neuroendocrine tumour of the skin, shows that TAMs express high levels of VEGF-C, which promotes lymphovascularisation (Werchau et al, 2012; Matsumoto-Okazaki et al, 2015). These reports strongly demonstrate the role of VEGF-producing TAMs in angiogenesis and tumour progression. Chen and colleagues found that infiltrating TAMs produced ADM when co-cultured with melanoma cells. There was also a significant improvement in endothelial cell proliferation and tube formation with ADM and this effect was abrogated upon administration of neutralising ADM antibody in vitro, suggesting the pro-angiogenic action of TAM-derived ADM (Chen et al, 2011b).
Apart from VEGFs and ADM, MMPs were also shown to be expressed by TAMs and involved in angiogenesis. According to a recent report, MMP9 secretion by TAMs recruited into the tumour site, in response to osteopontin signalling in melanoma, induced angiogenesis and tumour growth. These reports suggest that MMP9 aids tumour progression by remodelling the extracellular matrix and by promoting neoangiogenesis (Kale et al, 2015). Macrophage differentiation and chemotaxis is regulated by growth factors such as CSF1. Studies involving TAM depletion in breast tumours using Csf1-null mutation displayed substantial reduction in angiogenic potential and tumour burden, suggesting that these macrophages are required for angiogenesis. Additionally, this TAM depletion was reversed upon rescuing CSF1 in breast epithelium. Furthermore, overexpression of CSF1 in wild-type mice led to premature accumulation of macrophages in lesions and a dramatic increase in angiogenesis. It has also been demonstrated that most TAM depletion strategies using liposome-encapsulated clodronate inhibits angiogenesis in tumour models (Gazzaniga et al, 2007; Halin et al, 2009).
Hypoxia is a major contributor in angiogenesis. Dual staining of hypoxia and macrophage markers reveals massive infiltration of TAMs in hypoxic/necrotic regions of the tumour. This massive recruitment of TAMs is usually facilitated by chemokines like CCL2, CCL5, VEGF, CSF-1, semaphorin 3A (SEMA3A), endothelin, eotaxin and oncostatin M. Once macrophages arrive in these tumour compartments, their migration is halted via hypoxia-dependent mechanisms. Pro-tumoural functions of macrophages are then facilitated by a hypoxia-dependent transcription factor HIF1a, which induces expression of a large set of genes associated with angiogenesis such as VEGF (Henze and Mazzone, 2016). This pro-tumoural function of hypoxic TAMs was validated by Casazza et al, wherein they showed that macrophage-specific genetic deletion of Nrp-1, a binding partner of hypoxia induced TAM attractant Semaphorin 3A (Sema3A), prevented macrophage entry into the hypoxic region and ablated pro-angiogenic and immunosuppressive functions of TAMs, thereby inhibiting tumour growth and metastasis (Casazza et al, 2013).Role of TAMs in tumourigenesis. Different roles of TAMs in promoting tumour invasion and metastasis, along with the specific markers, are depicted. Tumour microenvironmental cues educate the macrophages to adopt a specific phenotype and perform distinct roles contributing towards tumourigenesis.
��
AMs in migration and invasion The potential of tumour cells to invade and metastasise depends on the tumour microenvironment. Since TAMs constitute a major component of the tumour microenvironment, they play a crucial role in facilitating these processes. TAMs primarily promote tumour cell invasion and metastasis via secretion of matrix metalloproteinases, serine proteases, and cathepsins, which alter the composition of the ECM by modifying cell�Ccell junctions and promoting basal membrane disruption. For instance, high levels of cathepsin protease activity are induced in the majority of macrophages in the microenvironment of pancreatic islet cancers, mammary tumours, and lung metastases during malignant progression. Furthermore, TAM-secreted cathepsins B and S were critical for promoting pancreatic tumour growth, angiogenesis, and invasion in vivo, and also markedly enhanced the invasiveness of cancer cells in culture (Gocheva et al, 2010). According to a recent report, STAT3 and STAT6 were shown to synergistically promote cathepsin secretion by macrophages, thereby enhancing tumour invasion and metastasis. Genetic deletion of Stat3 and Stat6 impaired tumour development and invasion in vivo. Together, these findings demonstrate that STAT3 and STAT6 cooperate in macrophages and enhance tumour progression in a cathepsin-dependent manner (Yan et al, 2016). Recently, Baghel and colleagues showed that a macrophage-derived protein MIP-1�� potentiated cancer cell invasion and metastasis via upregulation of MYO3A gene within breast cancer cells. Moreover, there was also a significant correlation between higher expression of this protein and poor survival of breast cancer patients, thereby validating the findings (Baghel et al, 2016). A novel real-time multiphoton imaging system developed by Wyckoff et al to investigate the metastatic nature of tumour cells demonstrated that invasion of breast cancer cells occurred in association with TAMs in mammary tumours, which is in agreement with the notion that TAMs support tumour invasion and metastasis (Wyckoff et al, 2007). In another clinicopathological study on breast carcinoma by Yang et al, the infiltration densities of TAMs were significantly higher in breast cancer patient specimens as compared to adjacent normal tissue (Yang et al, 2015). Moreover, in pancreatic tumours, targeting TAMs by inhibiting either the myeloid cell receptors colony-stimulating factor-1 receptor (CSF1R) or chemokine (C-C motif) receptor 2 (CCR2) decreased the number of tumour-initiating cells (TIC) and inhibited metastasis (Mitchem et al, 2013). A more recent study demonstrated that Warburg metabolism in tumour-conditioned macrophages promoted vascularisation, augmented extravasation of tumour cells from blood vessels, and metastasis in human pancreatic ductal adenocarcinoma. Furthermore, inhibition of glycolysis in TAMs with a competitive inhibitor disrupted this metastatic phenotype, reversing the observed increases in TAM-supported angiogenesis, extravasation, and epithelial�Cmesenchymal transition (EMT) (Penny et al, 2016). TAMs have also been reported to play crucial role in ovarian cancer growth, invasion and metastasis. Using an established mouse model for epithelial ovarian cancer, Yin et al showed that TAMs promote spheroid formation and tumour growth by secreting EGF. Activation of EGFR on tumour cells by EGF in turn upregulated VEGF/VEGFR signalling in surrounding tumour cells to support tumour cell proliferation and migration. Pharmacological blockade or antibody neutralisation of EGFR in TAMs abrogated spheroid formation and ovarian cancer progression in mouse models. These findings suggest that EGF secreted from TAMs plays a critical role in promoting early metastasis of ovarian cancer (Yin et al, 2016). Moreover, TAMs were also shown to promote invasion via toll-like receptor signalling in patients with ovarian cancer (Ke et al, 2016). TAMs in epithelial�Cmesenchymal transition Epithelial�Cmesenchymal transition plays a critical role in tumour progression and metastasis wherein polarised epithelial cells change their phenotype to motile mesenchymal cells. Recent studies have shown that TAMs are one of the orchestrators of this process, which involves loss of cell�Ccell contact and acquisition of a migratory phenotype. Epithelial�Cmesenchymal transition is characterised by suppression of epithelial markers such as E-cadherin, and upregulation of mesenchymal markers, including Vimentin, Slug, Snail, Fibronectin, zinc-finger E-box binding homeobox 1 (ZEB1), ZEB2, and ��-smooth muscle actin, as a result of which the cells acquire the ability to migrate and invade, leading to tumour progression and metastasis. Regulation of EMT is mediated by many growth factors and cytokines such as TGF-��, forkhead box protein M1 (FoxM1), hepatocyte growth factor, EGF, NF��B, Notch, and Wnt (Zhang et al, 2015). A growing body of evidence has shown the important contribution of TAMs in EMT. For instance, Liu et al showed that M2-polarised TAMs promoted EMT in pancreatic cancer cells, partially through the TLR4/IL-10 signalling pathway (Liu et al, 2013). According to another report, TAMs promote cancer stem cell (CSC)-like properties via TGF-��1-induced EMT in hepatocellular carcinoma (Fan et al, 2014). TAMs in intravasation and extravasation Intravasation is the process by which tumour cells enter a local blood vessel, and it is one of the important steps in the cascade of events leading to metastasis. Macrophages have been shown to enhance the ability of cancer cells to intravasate. Multiphoton intravital imaging techniques have shown that macrophages are located at the periphery of the tumour and their density decreases towards the centre wherein they are localised to the blood vessels and assist tumour cells to intravasate into the bloodstream (Sidani et al, 2006; Condeelis and Weissleder, 2010). Mechanistically, tumour cells secrete CSF1, which stimulates macrophages to produce EGF that in turn activates migration of the tumour cells (Wyckoff et al, 2004). Epidermal growth factor and CSF1 induce formation of invadopodia in cancer cells and podosomes in TAMs, structures that degrade extracellular matrix and facilitate intravasation (Condeelis and Pollard, 2006). In case of breast cancer patients, mammary TAMs secrete CCL18 which in turn triggers integrin clustering on cancer cells. This results in adherence of these cells to extracellular matrix and promotes intravasation (Chen et al, 2011a). While in circulation, platelets form aggregates with tumour cells and protect them from cytotoxic immune cell recognition. Platelets escort tumour cells in the circulation to the site of extravasation, where they help tumour cells exit the circulation into secondary organs. According to a recent study, platelets promote extravasation of tumour cells and metastatic seeding through ATP-dependent activation of the endothelial P2Y2 receptor, which opens the vessel barrier (Schumacher et al, 2013). Qian et al used an intact ex vivo lung imaging system and showed that tumour cells interacting with macrophages showed a higher percentage of extravasation, whereas depletion of macrophages using L-clodronate significantly reduced the number of tumour cells undergoing extravasation (Qian et al, 2009). TAMs in the seed and soil paradigm The seed and soil hypothesis, also known as the organ tropism hypothesis, proposed a concept that before metastatic colonisation, the primary tumour secretes factors that prepares a pre-metastatic niche at a distant site to become receptive for subsequent metastasis. It is characterised by accumulation of bone marrow (BM)-derived cell types, increased numbers of fibroblasts, secreted oncoproteins, and cytokines. Kaplan et al showed that BM-derived VEGFR+ cells arrive at the distant pre-metastatic site well before the primary tumour cells arrive. Furthermore, depleting VEGFR1+ cells by antibody-mediated inhibition of VEGFR1 signalling or receptor mutation interfered with the formation of these pre-metastatic clusters and inhibited metastasis (Kaplan et al, 2005). Erler et al showed that the expression of copper-dependent amine oxidase called lysyl oxidase (LOX), which is secreted by hypoxic breast tumour cells and is a major target of hypoxia-inducible factor (HIF) signalling, stabilises the ECM network by cross-linking collagen IV in the basement membranes at the pre-metastatic sites. This crosslinking facilitates myeloid cell recruitment and subsequent tumour cell colonisation. Moreover, LOX ablation prevents the formation of such sites and inhibits metastatic growth (Erler et al, 2009). A more recent report by Wang et al showed that colorectal carcinoma cells secrete VEGF-A, which stimulates TAMs to produce CXCL1 in the primary tumour. Elevation of CXCL1 in premetastatic liver tissue recruited CXCR2-positive myeloid-derived suppressor cells (MDSC) to form a premetastatic niche that ultimately promoted liver metastases (Wang et al, 2017). Thus, TAMs play a critical role in preparing the premetastatic niche, recruitment and retention of circulating tumour cells at the metastatic site, and fostering their growth. Interaction with cancer stem cells Cancer stem cells are specific subpopulations of cells within tumours that exhibit stem cell-like properties and have the potential to initiate tumours by undergoing self-renewal and differentiation. Owing to the evidence of pivotal roles of stromal cells such as TAMs, the interaction between TAMs and CSCs has become an exciting area of research. Recent studies have tried to analyse functional roles of TAMs in regulating tumour promoting activities of CSCs through complex molecular networks comprised of cytokines, chemokines and growth factors (Jinushi et al, 2012). Several studies have reported interaction of macrophages with CSCs in many tumour models. Yi et al showed a positive correlation between infiltration of macrophages and glioma initiating cancer stem cells or GICSCs. TAMs were localised in higher densities in areas with a higher number of GICSCs with close contacts between the two cell types. Moreover, in glioma tissue, the secretion of CSC-derived chemoattractants such as CCL2, CCL5, VEGF-A was much higher, facilitating recruitment of macrophages (Yi et al, 2011). Another report suggested that CSCs in glioma tissue induced macrophage infiltration and polarisation into M2 phenotype because these macrophages secreted a large number of cytokines, such as TGF-��1 and IL-10, and facilitated immunosuppression (Wu et al, 2010). Jinushi et al showed that TAMs interact with CSCs and promote their tumourigenic potential via production of milk fat globule-epidermal growth factor�CVIII (MFG-E8) and IL-6 through coordinated activation of the STAT3 and sonic hedgehog pathways. Interestingly, CSC population is the major population promoting the production of MFG-E8 and IL-6 from macrophages, suggesting that they impart macrophages with the ability to produce tumourigenic factors such as MFG-E8 and IL-6 (Jinushi et al, 2011). TAMs in immunosuppression The role TAMs play in immunosuppression to promote tumour progression has been widely investigated. TAMs are involved in immunosuppression either by directly inhibiting the CD8+ T-cell response through direct cell�Ccell interaction with T cells or by secreting immunosuppressive cytokines and proteases such as IL-10, TGF-��, Arginase-1, and prostaglandins, which inhibit T-cell activation and proliferation. For instance, macrophages express PD-L1/PD-L2 and CD80/CD86, which are the ligands of the inhibitory receptors programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA4), respectively. Activation of these receptors by their ligands results in inhibition of TCR signalling and T-cell cytotoxic function (Kuang et al, 2009; Ojalvo et al, 2009). Recently, it was shown that hypoxia also plays an important role in immunosuppression. TAMs found in hypoxic regions of the tumour upregulate PD-L1 expression via HIF-1�� signalling and consequently induce T-cell suppression (Doedens et al, 2010; Noman et al, 2014). Another report suggested that, as compared to MDSCs, macrophages produced higher levels of anti-inflammatory factors and were more immunosuppressive, facilitating tumour immunoevasion in multiple murine models of breast cancers (Fang et al, 2017). CAFs were also shown to induce accumulation of TAMs and secretion of IL-10, TGF-��, and Arginase-1 in oral squamous cell carcinoma and promote an immunosuppressive microenvironment by suppressing T-cell proliferation (Takahashi et al, 2017). TAM-derived cytokines also play an important role in immunosuppression. For instance, TFG-�� was shown to inhibit the anti-tumour activity of CD8+ T cells by downregulating the expression of cytolytic genes (Thomas and Massague, 2005). Moreover, IL-10 which is expressed by TAMs, CD8+ T cells, and tumour cells is an important cytokine in the tumour microenvironment and plays anti-inflammatory, immunosuppressive role that favours tumour escape from immune surveillance. TAM-derived IL-10 suppresses the expression of IL-12, which is considered as a potential anti-tumour cytokine (Matsuda et al, 1994). TAMs as therapeutic targets Several reports have provided strong evidence that TAMs are crucial components of the tumour microenvironment and TAM infiltration is strongly associated with poor prognosis and survival rates in cancer patients. Based on these findings, targeting TAMs is emerging as an attractive strategy for therapeutic intervention (Chanmee et al, 2014; Yang and Zhang, 2017). It is now well-established that chemoattractants in tumour microenvironment facilitate massive infiltration of macrophages in tumours. Hence, depletion or inhibition of TAM recruitment by modulating levels of these chemoattractants may serve as an effective strategy. Indeed, pharmacological inhibition, neutralising monoclonal antibodies, or genetic mutation of chemoattractants such as CCL2, VEGFR2, CSF-1R depleted TAM infiltration and reduced tumour growth. For instance, in case of CSF1R, the humanised monoclonal antibody RG7155 potently inhibited CSF1R dimerisation and also induced a striking reduction in the CSF1R+CD163+ macrophage population within tumour tissues (Ries et al, 2014). Moreover, PLX3397, a potent tyrosine kinase inhibitor of CSF1R decreased macrophage infiltration thereby enhancing the efficacy of immunotherapy (Mok et al, 2014). Differentiation of pro-tumourigenic M2 to the anti-tumour M1 phenotype is rapidly emerging as a new therapeutic approach. Activation of TLR3/Toll-IL-1 receptor domain-containing adaptor molecule 1 by Poly (I:C) rapidly enhanced secretion of pro-inflammatory cytokines and accelerated M1 macrophage polarisation (Shime et al, 2012). Emerging evidence has indicated that abnormalities in tumour vasculature alter the tumour microenvironment and influences tumour progression and responses to cancer therapy. The re-education of TAMs within the tumour could restore normal vasculature and block the pro-tumourigenic effects of TAMs. Indeed, polarisation from an M2 to M1 phenotype suppressed mammary tumour growth and angiogenesis in vivo (Zhang et al, 2013). According to another report, histidine-rich glycoprotein inhibited tumour growth and metastasis by inducing macrophage polarisation and vessel normalisation via downregulation of the placental growth factor (Rolny et al, 2011).
��Concluding remarks
The conventional binary model of macrophage polarisation is becoming antiquated and a newer model of polarisation spectrum is coming into conception, which focuses on involvement of an array of differentiated macrophages in various immunoregulatory disorders as well as cancers. Moreover, it is now well-established that apart from tumour cell-derived factors, stromal microenvironmental factors play a substantial role in tumourigenesis and TAMs constitute as key players in this phenomenon by regulating various steps of tumour initiation, progression, and metastasis. Since the abrogation of immunosuppressive macrophages in the tumour microenvironment enhances anti-tumour response, targeting TAMs is rapidly emerging as a promising therapeutic strategy for cancer patients.TAMeless traitors: macrophages in cancer progression and metastasis | British Journal of Cancer
https://www.nature.com/articles/bjc2017356��
Int. J. Mol. Sci. 2017, 18(7), 1545; https://doi.org/10.3390/ijms18071545
Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing
by Mark Hesketh, Katherine B. Sahin, Zoe E. West and Rachael Z. Murray *OrcID
The Institute for Health and Biomedical Innovation, School of Biomedical Sciences, Faculty of Health, Queensland University of Technology, Brisbane QLD 4059, Australia
Abstract: Macrophages and inflammation play a beneficial role during wound repair with macrophages regulating a wide range of processes, such as removal of dead cells, debris and pathogens, through to extracellular matrix deposition re-vascularisation and wound re-epithelialisation. To perform this range of functions, these cells develop distinct phenotypes over the course of wound healing. They can present with a pro-inflammatory M1 phenotype, more often found in the early stages of repair, through to anti-inflammatory M2 phenotypes that are pro-repair in the latter stages of wound healing. There is a continuum of phenotypes between these ranges with some cells sharing phenotypes of both M1 and M2 macrophages. One of the less pleasant consequences of quick closure, namely the replacement with scar tissue, is also regulated by macrophages, through their promotion of fibroblast proliferation, myofibroblast differentiation and collagen deposition. Alterations in macrophage number and phenotype disrupt this process and can dictate the level of scar formation. It is also clear that dysregulated inflammation and altered macrophage phenotypes are responsible for hindering closure of chronic wounds. The review will discuss our current knowledge of macrophage phenotype on the repair process and how alterations in the phenotypes might alter wound closure and the final repair quality.
Keywords: macrophage; monocyte; wound healing; fibrosis; chronic wound; diabetes; chronic venous disease
Figure 1. M1 and M2 polarisation of macrophages. Monocytes can be classically or alternatively activated to form M1 and M2 macrophages respectively. M1 macrophages can also differentiate into M2 macrophages through local cues and after efferocytosis. The M1 phenotype is pro-inflammatory, phagocytic and bactericidal, while the M2 macrophages act to switch off inflammation and regulate re-vascularisation and wound closure.
Figure 2. The M1 to M2 switch is dysregulated in chronic wounds and, unlike in acute wounds, macrophages are unable to phagocytose neutrophils. (A) In the early inflammatory stage of an acute wound, macrophages phagocytose spent neutrophils. In the later stages, having performed this role, macrophages switch phenotype and are predominantly M2 macrophages; (B) In a chronic wound, macrophages are predominantly M1 that are unable phagocytosis spent neutrophils. This leads to the recruitment of more macrophages and an increase in inflammation.
��
IJMS | Free Full-Text | Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing | HTML
https://www.mdpi.com/1422-0067/18/7/1545/htm��
Arginine is an Ornithine agonist, while lysine inhibits Ornithine metabolism. Ornithine supplements, like arginine may be useful in a variety of diseases. Ornithine releases growth hormone and may be of value for growth-impaired children and athletes in training.
UREA CYCLE AMINO ACIDS; Arginine; Ornithine - DCNutrition.com
www.dcnutrition.com/amino-acids/urea-cycle-amino-acids-arginine-ornithine/��
Orchestration of Metabolism by Macrophages
Figure 1. Orchestration of Metabolism by Macrophages
Italicized words indicate macrophage-mediated functions related to metabolic activities. Red, associated diseases. Inset: Selected metabolic features of polarized macrophages. See the main text for details.Iron Metabolism
Macrophages play an important role in iron homeostasis by recycling iron through phagocytosis of senescent red blood cells and rendering it available for processes like erythropoiesis. Mouse and human macrophage polarization is associated with differential regulation of iron metabolism (Cairo et al., 2011). IL-4-activated macrophages expressed high levels of CD163 and CD94 (heme uptake), low Ferritin (iron storage), and high Ferroportin (iron export). The high levels of intracellular heme pool together with induction of heme oxygenase result in production of CO, which has immunosuppressive activity. In contrast, IFN��-activated macrophages show a CD163lowFerritinhighFerroportinlow phenotype. Thus, M2-polarized macrophages are set in an iron-export mode that supports immunoregulation, promotion of matrix remodeling, and cell proliferation. Conversely, M1-polarized macrophages are set in an iron-retention mode that supports bacteriostatic and tumoristatic activity. Proteins like Hepcidin and human hemochromatosis protein (HFE) contribute to the iron homeostasis in macrophages by degrading ferroportin, which results in inhibition of iron release (Cairo et al., 2011). Thus, iron handling has emerged as a key property of macrophage polarized activation with broad implications in immunopathology.Diseases Related to Iron Metabolism
In chronic venous leg ulcers, evidence in mice and patients suggested that iron overloading skews macrophages toward an unrestrained proinflammatory M1-like phenotype that sustains tissue damage and impairs wound healing (Sindrilaru et al., 2011). Macrophage-derived ROS was suggested to mediate DNA damage, fibroblast cellular senescence, and defective tissue repair in this disease. It is tempting to speculate that similar mechanisms may underlie M2-like skewing in burnt patients.
Hereditary hemochromatosis is a genetically determined iron overload disease, associated with a defect in the hemochromatosis gene, Hfe. In fact, in macrophages from Hfe knockout mice, lower intracellular iron correlated with a reduced inflammatory cytokine response to LPS and an impaired innate immune response, as observed in patients with defects in iron homeostasis (Cairo et al., 2011).��
Orchestration of Metabolism by Macrophages: Cell Metabolism
https://www.cell.com/cell-metabolism/fulltext/S1550-4131(12)00015-0Orchestration of Metabolism by Macrophages - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1550413112000150��
Modulation of macrophage phenotype by cell shape
Frances Y. McWhorter,a,b,1 Tingting Wang,a,b,1 Phoebe Nguyen,a,b Thanh Chung,a,b and Wendy F. Liua,b,c,2
Author information Copyright and License information Disclaimer
Departments of aBiomedical Engineering and
cChemical Engineering and Materials Science, and
bThe Edwards Lifesciences Center for Advanced Cardiovascular Technology, University of California, Irvine, CA, 92697
SIGNIFICANCE
Macrophages are central regulators of the immune response during infection and wound healing, and their behavior is largely thought to be regulated by soluble factors in the microenvironment. Here, we find that adhesive cues are also critical for regulating the proinflammatory vs. prohealing state of these cells. We use engineered cell culture substrates to control cell shape and find that elongation of cells promotes a prohealing phenotype. Moreover, we identify key molecular pathways that are responsible for transducing physical cues to the biochemical cues that govern this response. Together, these studies suggest an important role for cell shape in regulating macrophage function.
ABSTRACT
Phenotypic polarization of macrophages is regulated by a milieu of cues in the local tissue microenvironment. Although much is known about how soluble factors influence macrophage polarization, relatively little is known about how physical cues present in the extracellular environment might modulate proinflammatory (M1) vs. prohealing (M2) activation. Specifically, the role of cell shape has not been explored, even though it has been observed that macrophages adopt different geometries in vivo. We and others observed that macrophages polarized toward different phenotypes in vitro exhibit dramatic changes in cell shape: M2 cells exhibit an elongated shape compared with M1 cells. Using a micropatterning approach to control macrophage cell shape directly, we demonstrate here that elongation itself, without exogenous cytokines, leads to the expression of M2 phenotype markers and reduces the secretion of inflammatory cytokines. Moreover, elongation enhances the effects of M2-inducing cytokines IL-4 and IL-13 and protects cells from M1-inducing stimuli LPS and IFN-��. In addition shape- but not cytokine-induced polarization is abrogated when actin and actin/myosin contractility are inhibited by pharmacological agents, suggesting a role for the cytoskeleton in the control of macrophage polarization by cell geometry. Our studies demonstrate that alterations in cell shape associated with changes in ECM architecture may provide integral cues to modulate macrophage phenotype polarization.Modulation of macrophage phenotype by cell shape
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3808615/��
Effect of low pH on single skeletal muscle myosin mechanics and kinetics
Abstract
Acidosis (low pH) is the oldest putative agent of muscular fatigue, but the molecular mechanism underlying its depressive effect on muscular performance remains unresolved. Therefore, the effect of low pH on the molecular mechanics and kinetics of chicken skeletal muscle myosin was studied using in vitro motility (IVM) and single molecule laser trap assays. Decreasing pH from 7.4 to 6.4 at saturating ATP slowed actin filament velocity (Vactin) in the IVM by 36%. Single molecule experiments, at 1 ��M ATP, decreased the average unitary step size of myosin (d) from 10 �� 2 nm (pH 7.4) to 2 �� 1 nm (pH 6.4). Individual binding events at low pH were consistent with the presence of a population of both productive (average d = 10 nm) and nonproductive (average d = 0 nm) actomyosin interactions. Raising the ATP concentration from 1 ��M to 1 mM at pH 6.4 restored d (9 �� 3 nm), suggesting that the lifetime of the nonproductive interactions is solely dependent on the [ATP]. Vactin, however, was not restored by raising the [ATP] (1�C10 mM) in the IVM assay, suggesting that low pH also prolongs actin strong binding (ton). Measurement of ton as a function of the [ATP] in the single molecule assay suggested that acidosis prolongs ton by slowing the rate of ADP release. Thus, in a detachment limited model of motility (i.e., Vactin ∼ d/ton), a slowed rate of ADP release and the presence of nonproductive actomyosin interactions could account for the acidosis-induced decrease in Vactin, suggesting a molecular explanation for this component of muscular fatigue.
Keywords: acidosis, fatigue, velocity, laser trap
Effect of low pH on single skeletal muscle myosin mechanics and kinetics
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2493560/��
Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity
imageXenia Geeraerts1,2, imageEvangelia Bolli1,2, imageSarah-Maria Fendt3,4* and imageJo A. Van Ginderachter1,2*
1Laboratory of Myeloid Cell Immunology, VIB Inflammation Research Center, VIB, Ghent, Belgium
2Laboratory of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Brussels, Belgium
3Laboratory of Cellular Metabolism and Metabolic Regulation, VIB Center for Cancer Biology, VIB, Leuven, Belgium
4Laboratory of Cellular Metabolism and Metabolic Regulation, Department of Oncology, KU Leuven and Leuven Cancer Institute (LKI), Leuven, Belgium
��Macrophages are not only essential components of innate immunity that contribute to host defense against infections, but also tumor growth and the maintenance of tissue homeostasis. An important feature of macrophages is their plasticity and ability to adopt diverse activation states in response to their microenvironment and in line with their functional requirements. Recent immunometabolism studies have shown that alterations in the metabolic profile of macrophages shape their activation state and function. For instance, to fulfill their respective functions lipopolysaccharides-induced pro-inflammatory macrophages and interleukin-4 activated anti-inflammatory macrophages adopt a different metabolism. Thus, metabolic reprogramming of macrophages could become a therapeutic approach to treat diseases that have a high macrophage involvement, such as cancer. In the first part of this review, we will focus on the metabolic pathways altered in differentially activated macrophages and link their metabolic aspects to their pro- and anti-inflammatory phenotype. In the second part, we will discuss how macrophage metabolism is a promising target for therapeutic intervention in inflammatory diseases and cancer.
Introduction
Immunometabolism is a fast evolving field, which determines the metabolic machinery of immune cells and investigates how changing their metabolic phenotype affects immune cell function. It is known that the microenvironment shapes the metabolism of cells, which in turn contributes to their functionality. Environmental signals involved in metabolic regulation are cytokines, growth factors, oxygen levels, and nutrient availability. There is a growing evidence that immune cells in a specific microenvironment, such as inflamed tissue or tumors, reprogram their metabolic phenotype to fulfill cellular needs, such as survival, growth, and proliferation, or to carry out specific effector functions, such as phagocytosis and cytokine production. By changing the metabolism of immune cells, in particular, macrophages, it will be possible to modulate their function, which would be useful in diseases with a high macrophage commitment. Hence, understanding immune cell metabolism and its regulation will be essential to use metabolism as a therapeutic target to affect disease outcome (1�C3).
In this review, we provide an overview of the current knowledge concerning the metabolism of differentially activated macrophages. We will discuss recent findings on macrophage metabolism in the context of cancer and inflammatory diseases such as obesity and atherosclerosis. Furthermore, we will comment on promising metabolic targets for therapeutic purposes and approaches to reprogram macrophage metabolism in particular diseases.
Figure 1. M1 macrophage metabolism. M1 macrophage metabolism is characterized by enhanced aerobic glycolysis, converting glucose into lactate. M1 macrophages have an increased flux through the pentose phosphate pathway (PPP), generating NADPH, used for the generation of the anti-oxidant glutathione (GSH) and the inflammatory mediators nitric oxide (NO) and reactive oxygen species (ROS). In M1 macrophages, the tricarboxylic acid (TCA) cycle is broken in two places, leading to the accumulation of succinate and citrate. While the accumulation of succinate leads to HIF-1�� stabilization and the transcription of pro-inflammatory and glycolytic genes, citrate is used for the generation of fatty acids, NO, ROS, and the synthesis of itaconate. Another aspect of M1 macrophage metabolism is the conversion of l-arginine to NO and l-citrulline. All important metabolic reactions present in M1/M2 macrophages are shown in black, reactions shown in gray are absent or less pronounced. The metabolic pathways strongly upregulated by M1/M2 macrophage polarization are highlighted by a colored shadow, the width of the shadow illustrates the weight of a particular pathway in the macrophage activation state. All metabolic enzymes are indicated in green. Dotted lines represent impaired metabolic reactions. Abbreviations: ��-KG: ��-ketoglutarate; AASS: aspartate�Carginosuccinate shunt pathway; ACLY: ATP-citrate lyase; CAD: cis-aconitate decarboxylase; CIC: mitochondrial citrate carrier; ETC: electron transport chain; FAS: fatty acid synthase; GLUT: glucose transporter; HK: hexokinase; IDH: isocitrate dehydrogenase; iNOS: inducible nitric oxide synthase; LDH: lactate dehydrogenase; MCT: monocarboxylate transporter; ME: malic enzyme; OAA: oxaloacetate; PEP: phosphoenolpyruvate; PDH: pyruvate dehydrogenase; PFK: phosphofructokinase; SDH: succinate dehydrogenase; SLC: solute carrier.
��
Figure 2. M2 macrophage metabolism. M2 macrophages mainly produce ATP through an oxidative TCA cycle coupled to oxidative phosphorylation (OXPHOS). To fuel the TCA cycle, M2 macrophages rely on fatty acid oxidation (or ��-oxidation) and glutamine metabolism. Furthermore, M2 macrophages show a lowered glycolysis and pentose phosphate pathway (PPP). Moreover, M2 macrophages convert l-arginine into urea and l-ornithine, which serves as precursor for l-proline production. All important metabolic reactions present in M1/M2 macrophages are shown in black, reactions shown in gray are absent or less pronounced. The metabolic pathways strongly upregulated by M1/M2 macrophage polarization are highlighted in orange. All metabolic enzymes are indicated in green. Dotted lines represent impaired metabolic reactions. Abbreviations: ��-KG: ��-ketoglutarate; ARG: arginase; CD: cluster of differentiation; CPT: carnitine palmitoyl transferase; ETC: electron transport chain; LAL: lysosomal acid lipase; PEP: phosphoenolpyruvate; PFK: phosphofructokinase; SLC: solute carrier.
Figure 3. Metabolic reprogramming of tumor-associated macrophages (TAM) toward an antitumoral phenotype might affect tumor growth. Tumors are highly infiltrated by tumor-infiltrating immune cells, with TAM being amongst the most abundant ones. Within the same tumor, the co-existence of two distinct TAM subpopulations has been shown: M2-like protumoral TAM and M1-like antitumoral TAM. The TAM phenotype depends on the stage of tumor development, leading to a majority of M2-like TAM in late stage tumors which stimulate tumor growth, angiogenesis, invasion, metastasis, suppression of antitumor immunity, and mediation of therapy resistance. Strategies that metabolically reprogram protumoral M2 TAM into an antitumoral M1 phenotype, without depleting the full TAM population, could reduce tumor growth and metastasis and allow re-establishment of conventional cancer therapies.
A study concerning TAM metabolism by Colegio and colleagues indicated that tumor-derived lactate is necessary to polarize TAM toward a protumoral M2 phenotype. Stimulation of bone marrow-derived macrophages with lactate was sufficient to induce the expression of the M2-related genes Vegf, Arg1, Relma, Mgl1, and Mgl2. Interestingly, stabilization of HIF-1�� by tumor-derived lactate was the actual driving force for this M2 polarization. Moreover, by inducing Vegf and ARG-1, lactate fulfills a key role in shaping the protumoral phenotype of TAM. While Vegf induces angiogenesis, ARG-1 contributes to tumor growth by the generation of polyamines which act as a proliferative signal for mammalian cancer cells (81). This study proves that nutrients in the tumor microenvironment, such as lactate, can affect the phenotype of infiltrating immune cells, such as TAM.
esearch by Penny and colleagues demonstrated that in vitro generated macrophages, cultured with tumor-conditioned media from a pancreatic ductal adanocarcinoma (PDAC) cell line, have a pronounced metabolic preference for aerobic glycolysis. In comparison to control macrophages, PDAC TAM showed enhanced angiogenesis and cancer cell extravasation, hence inducing metastasis. Treating PDAC TAM with 2-deoxyglucose, an inhibitor of the first glycolytic enzyme HK2, blocked the pro-metastatic TAM phenotype (82). This study links pancreatic TAM metabolism to aerobic glycolysis, which is in contrast to our current understanding of M2 macrophage metabolism, as extensively described before, claiming their lowered glycolysis and preference for oxidative mitochondrial metabolism.
��Frontiers | Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2017.00289/full��
Immunity. Author manuscript; available in PMC 2017 Mar 15.
Macrophages and iron metabolism
1Instituto Gulbenkian de Ci��ncia, Rua da Quinta Grande, 6, 2780-156 Oeiras, Portugal
2University of Maryland 2413 ANSC, Bldg 142 College Park, Maryland 20742, USA
Abstract
Iron is a transition metal that due to its inherent ability to exchange electrons with a variety of molecules is essential to support life. In mammals, iron exists mostly in the form of heme, enclosed within an organic protoporphyrin ring and functioning primarily as a prosthetic group in proteins. Paradoxically, free iron also has the potential to become cytotoxic when electron exchange with oxygen is unrestricted and catalyzes the production of reactive oxygen species. These biological properties demand that iron metabolism is tightly regulated such that iron is available for core biological functions whilst preventing its cytotoxic effects. Macrophages play a central role in establishing this delicate balance. Here, we review the impact of macrophages on heme-iron metabolism and, reciprocally, how heme-iron modulates macrophage function.
Macrophages and iron metabolism
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4794998/��
Immunol Rev. 2015 Mar; 264(1): 182�C203.
Macrophage defense mechanisms against intracellular bacteria
1Department of Internal Medicine VI, Infectious Disease, Immunology, Rheumatology, Pneumology, Medical University of Innsbruck, Innsbruck, Austria
2Cellular Microbiology, Priority Area Infections, Research Center Borstel, Borstel, Germany
3Department of Immunology, London School of Hygiene and Tropical Medicine, London, UK
4German Centre of Infection Research, TTU-TB, Borstel, Germany
Macrophages and neutrophils play a decisive role in host responses to intracellular bacteria including the agent of tuberculosis (TB), Mycobacterium tuberculosis as they represent the forefront of innate immune defense against bacterial invaders. At the same time, these phagocytes are also primary targets of intracellular bacteria to be abused as host cells. Their efficacy to contain and eliminate intracellular M. tuberculosis decides whether a patient initially becomes infected or not. However, when the infection becomes chronic or even latent (as in the case of TB) despite development of specific immune activation, phagocytes have also important effector functions. Macrophages have evolved a myriad of defense strategies to combat infection with intracellular bacteria such as M. tuberculosis. These include induction of toxic anti-microbial effectors such as nitric oxide and reactive oxygen intermediates, the stimulation of microbe intoxication mechanisms via acidification or metal accumulation in the phagolysosome, the restriction of the microbe's access to essential nutrients such as iron, fatty acids, or amino acids, the production of anti-microbial peptides and cytokines, along with induction of autophagy and efferocytosis to eliminate the pathogen. On the other hand, M. tuberculosis, as a prime example of a well-adapted facultative intracellular bacterium, has learned during evolution to counter-balance the host's immune defense strategies to secure survival or multiplication within this otherwise hostile environment.
Macrophage defense mechanisms against intracellular bacteria
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4368383/��
��
��
19 internationally renown research teams affiliated with Inserm, CNRS and Aix-Marseille University
Our transversal and multidisciplinary approach of cancer research translates in our scientific projects exploring the fundamental bases of tumourigenesis from the molecular and functional point of view in integrated experimental models - oncogenes and tumour suppressors, signalling and cell polarity, regulation of the epigenome, genome instability, DNA replication repair, cellular stress, tumour-host interactions and anti-tumoural immunity �C, and their applications in potential new diagnostic and therapeutic strategies for the main pathologies of focus of the hospital.
The originality of the CRCM also lies in its capacity to address major clinical issues (resistance to treatment, prognostic prediction, anti-metastasis strategies��) thanks to our interactive translational and clinical programmes which bring together medical doctors, pharmacists and scientists.Research Teams: Centre de Recherche en Canc��rologie de Marseille
http://crcm.marseille.inserm.fr/en/researchteams/��
Macrophage response to lactic acid Tumor cells hijack macrophages via lactic acid
Article in Immunology and Cell Biology 92(8) �� August 2014
Immunology and Cell Biology focuses on the general functioning of the immune system in its broadest sense, with a particular emphasis on its cell biology. Areas that are covered include but are not limited to: Cellular immunology, Innate and adaptive immunity, Immune responses to pathogens,Tumour immunology,Immunopathology, Immunotherapy, Immunogenetics, Immunological studies in humans and model organisms (including mouse, rat, Drosophila etc)
... In cancer, the tumor cells are mostly unrecognized as antigens because a dominant anti-inflammatory response driven by the tumor cells suppresses any antitumoral immune response and promotes tumor progression and dissemination (immunosuppression). In fact, tumors are called wounds that do not heal, because the tumor hijacks the wound healing machinery and uses it to promote itself[1,2]. In contrast, in autoimmune diseases, self-tolerance is broken and the inflammatory response is activated in excess against the host tissue cells, which express autoantigens that are misrecognized and attacked by the immune system, gradually leading to permanent tissue damage. ...
... Thus, neighboring stroma cells, particularly macrophages, are exposed to increased levels of lactate, which is actively transported into them. Lactate contributes to macrophage polarization by stabilizing HIF-1a and inducing expression of typical M2phenotype markers like VEGF and arginase-I (ARG-I)[59], so that tumor-associated macrophages (TAMs) sense the metabolic changes in tumor cells and respond to them in a proangiogenic manner[1]. In autoimmune diseases, the increased infiltration of leukocytes into the inflamed site increases the demand for oxygen beyond the available supply. ...Macrophage response to lactic acid Tumor cells hijack macrophages via lactic acid
https://www.researchgate.net/publication/264502410_Macrophage_response_to_lactic_acid_Tumor_cells_hijack_macrophages_via_lactic_acid��
Microbial carcinogenesis: Lactic acid bacteria in gastric cancer
... In addition, other studies substantiated that high levels of lactate suppresses TCR-stimulated cytokine release (IFN-�� µ��»¾, TNF-�� µ��»¼, and IL-2) and prompts partial damage of lytic granules exocytosis in CTLs by selectively downregulating the MAPKs p38 and JNK/c-Jun signaling pathways [81]. Moreover, tumor-derived lactate enhances arginase-1 (ARG1) expression in tumor-associated macrophages (TAMs), hindering T-cell activity and prolif- eration [144], inhibiting antitumor immune responses and promoting tumor growth [145, 146]. Lately, Colegio et al. [145] demonstrated that, under normoxic conditions, lactate stabilizes HIF-1�� µ��»¼, resulting in ARG1 and VEGF gene expression in macrophages.��
Vitamin K-3 and K-5 are inhibitors of tumor pyruvate kinase M2
Pyruvate kinase M2 (PKM2) is a rate-limiting enzyme of aerobic glycolysis in cancer cells and plays important roles in cancer metabolism and growth. Here we show that vitamin K(3) and K(5) (VK(3) and VK(5)) are relatively specific PKM2 inhibitors. VK(3) and VK(5) showed a significantly stronger potency to inhibit PKM2 than to inhibit PKM1 and PKL, 2 other isoforms of PK dominantly expressed in most adult tissues and liver. This study combined with previous reports supports that VK(3) and VK(5) have potential as adjuvant for cancer chemotherapy.... Previously, it has been revealed that naphthaquinones including shikonin, vitamin K 3 and vitamin K 5 are effi- cient inhibitors of glycolysis (9,10). Inhibition of the rate of glycolysis is toxic to cancer cells (11). ...
... Inhibition of the rate of glycolysis is toxic to cancer cells (11). Pyruvate kinase M2 (PKM2) was demonstrated to be a target responsible for the inhibited glycolytic rate in previous studies (9)(10)(11)(12). Replacing PKM2 with PKM1 partially reduced the death of cancer cells induced by naphthaquinones with increased pyruvate kinase activity (10). ...
... The reaction solution contained 50 mM HEPES (pH 7.5), 0.2 mM NADH and 2 mM pyruvate. Relative LDH activity was calculated at 25˚C by comparing the change in absorbance at 340 nm between vehicle controls [200 ng/µl cell extract treated with 1% (v/v) DMSO] and naphthaquinone incubated groups once the cell extract was added to the reac- tion solution, as previously described (9,10). ...
... Events that negatively impact tumorigenesis can also reduce PKM2 function. Vitamins K3 and K5 inhibit tumorigenesis along with potently inhibiting PKM2 activity [38]. Butyrate displays anticolon cancer effects along with the inhibition of PKM2 expression in neoplastic but not nontumor colon tissues [39]. ...
Vitamin K-3 and K-5 are inhibitors of tumor pyruvate kinase M2 | Request PDF
https://www.researchgate.net/publication/51864291_Vitamin_K-3_and_K-5_are_inhibitors_of_tumor_pyruvate_kinase_M2��
Vitamin K5 is an efficient photosensitizer for ultraviolet A light inactivation of bacteria
Photodynamic treatment combining light and a photosensitizer molecule can be an effective method to inactivate pathogenic bacteria. This study identified vitamin K5 as an efficient photosensitizer for ultraviolet light A (UVA)-induced bacterial inactivation. Six bacterial species, Bacillus cereus (vegetative form), Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Staphylococcus epidermidis, Klebsiella pneumoniae, and two species of antibiotic-resistant bacteria, Pseudomonas aeruginosa* and Staphylococcus aureus*, were suspended in aqueous solutions with or without vitamin K5 and exposed to UVA irradiation. UVA irradiation (5.8 J cm-2) with vitamin K5 (1600 ��mol l-1) reduced the colony forming units (CFU) of these bacteria by three to seven logs. Antibiotic resistant bacteria were also susceptible to the bactericidal effects of UVA and vitamin K5 combination treatment. Inactivation of bacteria in human plasma required higher doses of UVA light and vitamin K5. UVA irradiation (30 J cm-2) with vitamin K5 (2000 �� mol l-1) reduced Escherichia coli and Staphylococcus aureus spiked into human plasma by seven logs CFU/ml. Reactive oxygen species, such as superoxide anion radicals and hydroxyl radicals, were found to be generated in vitamin K5 aqueous solution after UVA irradiation, suggesting these oxygen species may mediate the inactivation of the bacteria.Vitamin K-3 and K-5 are inhibitors of tumor pyruvate kinase M2 | Request PDF
https://www.researchgate.net/publication/51864291_Vitamin_K-3_and_K-5_are_inhibitors_of_tumor_pyruvate_kinase_M2��
Tannic acid directly targets pyruvate kinase isoenzyme M2 to attenuate colon cancer cell proliferation
September 2018 �� Food & Function
Abstract Tannic acid (TA), a naturally occurring polyphenolic acid that is primarily found in grapes and green tea, exhibits potent antioxidant and anticarcinogenic characteristics. However, the underlying molecular mechanisms and targets of TA, which are responsible for cancer prevention, remain elusive. In the present study, we used TA-functionalized magnetite nanoparticles to identify pyruvate kinase isoenzyme M2 (PKM2) as the direct target of TA. We report that TA selectively inhibits the pyruvate kinase activity of PKM2, rather than protein kinase activity and PKM2 expression, to suppress colorectal cancer (CRC) cell proliferation. Furthermore, we had discovered that lysine residue 433 (K433) is a selective druggable site. Through direct binding to lysine residue 433, TA triggers the dissociation of PKM2 tetramers and further blocks the metabolic activity of PKM2. Notably, TA has no effect on PKM1 activity as TA does not bind to it. Taken together, these findings show that TA is worthy of consideration as a promising PKM2 inhibitor for the prevention of CRC.
��Tannic acid directly targets pyruvate kinase isoenzyme M2 to attenuate colon cancer cell proliferation | Request PDF
https://www.researchgate.net/publication/331818258_Tannic_acid_directly_targets_pyruvate_kinase_isoenzyme_M2_to_attenuate_colon_cancer_cell_proliferation��
The role of pyruvate kinase M2 in anticancer therapeutic treatments (Review)
(PDF) The role of pyruvate kinase M2 in anticancer therapeutic treatments (Review)
https://www.researchgate.net/publication/336220292_The_role_of_pyruvate_kinase_M2_in_anticancer_therapeutic_treatments_Review��
Pyruvate kinase M2 fuels multiple aspects of cancer cells: from cellular metabolism, transcriptional regulation to extracellular signaling
Ming-Chuan Hsu & Wen-Chun Hung
Molecular Cancer volume 17, Article number: 35 (2018) Cite this article
Abstract
Originally identified as a metabolic enzyme that catalyzes the transfer of a phosphate group from phosphoenolpyruvate (PEP) to ADP in the glycolytic pathway, pyruvate kinase M2-type (PKM2) has been shown to exhibit novel biological activities in the nucleus and outside the cells. Although cell-based studies reveal new non-canonical functions of PKM2 in gene transcription, epigenetic modulation and cell cycle progression, the importance of these non-canonical functions in PKM2-mediated tumorigenesis is still under debate because studies in genetically modified mice do not consistently echo the findings observed in cultured cancer cells. In addition to regulation of gene expression, the existence of PKM2 in exosomes opens a new venue to study the potential role of this glycolytic enzyme in cell-cell communication and extracellular signal initiation. In this review, we briefly summarize current understanding of PKM2 in metabolic switch and gene regulation. We will then emphasize recent progress of PKM2 in extracellular signaling and tumor microenvironment reprogramming. Finally, the discrepancy of some PKM2��s functions in vitro and in vivo, and the application of PKM2 in cancer detection and treatment will be discussed.
Pyruvate kinase M2 fuels multiple aspects of cancer cells: from cellular metabolism, transcriptional regulation to extracellular signaling | Molecular Cancer | Full Text
https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-018-0791-3
��GcMAF UPDATE: With A Tried & True Cancer Cure That Activates Cancer-Killing Macrophages Having Been Removed From The Market, Are There Any Natural Alternatives?
Posted June 4, 2018: by Bill Sardi
We live in an era of cancer immunotherapy. Oncologists once vehemently denied cancer could ever be overcome by the human immune system. A report in the British Medical Journal dated May 24, 1969 stated: ��immunotherapy in patients with advanced disease is doomed to disappointment.��i
Over 50 years ago, even in the face of Dr. Chester Southam injecting millions of cancer cells into elderly patients (without their consent) to prove the human immune system is capable of resisting cancer when intentionally induced, Dr. Southam was unfairly demonized on ethical grounds and cancer immunotherapy was roundly denounced.ii Yet today immunotherapy is the most promising weapon in the fight against cancer.iiiWhite blood cells called macrophages that literally seek out and engulf cancer cells are considered to be the major player in controlling tumor-generated inflammation.iv Uncontrolled inflammation leads to the development of new blood vessels (angiogenesis) that feed tumor growth.v In turn, that leads to the release of malignant tumor cells into the blood circulation that facilitates the spread of cancer (metastasis), which is considered the mortal form of the disease.vi
While there are pro-inflammatory macrophages, in a healthy state the liver naturally produces a macrophage activation factor (Gc protein Macrophage Activating Factor or GcMAF) to stimulate macrophages that digest cancer cells but limit inflammation. But an undesirable enzyme, nagalase, prevents internal GcMAF synthesis and degrades GcMAF that naturally protects from macrophage-orchestrated destruction, i.e. disengages a key factor in cancer immunity.viiGcMAF cancer cure announced in 2008
Having broken the story about GcMAF (Gc protein macrophage activating factor) in 2008 with biochemist Timothy Hubbell, I have been a careful observer of events following the revelation that a blood protein (GcMAF) can inhibit an enzyme (nagalase) that disarms macrophages, a white blood cell that literally digests cancer cells. Our original report about GcMAF has faded into obscurity as concerned parties, recognizing unusual number of deaths among physicians who dared to bring GcMAF into use clinical use, began to bury our report for our own protection. You have to go to the Wayback Machine to find it.viii
Any agent that inhibits nagalase would unleash macrophages to do their job of removing pathogens and malignancies from the blood circulation. Healthy adults are reported to produce GcMAF and have low levels of nagalase (between 0.32 and 0.95 nanomole/min/milligram). It cannot be said that exogenously instilled GcMAF is unproven when works continuously to activate macrophages and keep cancer from spreading.
Successful GcMAF private enterprise put under by regulators
Immuno Biotech (UK; Isle of Guernsey)/ First Immune (Sweden)ix in Europe gleaned GcMAF from blood plasma and offered it for sale worldwide under the leadership of David Noakes, whose company has been derailed by a collaboration of health authorities in Europe including Interpol and at the present time Mr. Noakes is up for a court appearance on charges that could put him in jail.x Members of the Immuo Biotech scientific staff are also charged with criminal conduct.xi
Regulators put Immuno Biotech out of business for being an unlicensed facility and not meeting Good Manufacturing Practices. In other words, their paperwork wasn��t conformant. The actual GcMAF therapy was never evaluated. There were no serious side effects reported from First Immune��s GcMAF injectable product to this writer��s knowledge.Treating cancer with disproven remedies and dismissing unproven ones
We now face the inexplicable dilemma that ineffective and even toxic cancer treatments are approved for use while a natural blood plasma protein that protects from cancer in healthy individuals via macrophage activation is deemed to be unproven and therefore the public should be warned away from it (but how can that be when it is working daily in billions of healthy people?).
I obtained injectable GcMAF to evaluate firsthand and provided it to an end-stage pancreatic cancer patient whose tumor markers rapidly normalized and scans revealed only remnants of remaining cancer.
Confronted with cures, the news media buries the story
Many testimonials of cures were reported and Mr. Noakes actually invited BBC News to an event to speak to cured patients who had unsuccessfully undergone conventional cancer treatments and were basically left for dead. BBC chose to air a negative story about Immuno Biotech rather than broadcast interviews from patients who had successfully overcome cancer with GcMAF.xii
In October of 2016 I wrote this comment on by blog:
BBC News Slams Unlicensed Cancer Cure. But is it all as it seems?
GcMAF is produced internally in the human body. It is vitamin D binding protein with a sugar molecule knocked off. It activates the human immune system to kill off cancer cells and it gained attention in 2010. [National Health Federation 2009]
Today gcMAF (gc protein macrophage activating factor) is being slammed as an unlicensed and overpriced cancer cure by BBC News. [BBC News Oct 16, 2016]
Said to be ��scientifically extremely dubious�� by Cancer Research UK, the criticism is undeserved if for no other reason than it is the way the human body self-controls cancer. It is backed by independent research. [Best Practices Research Clinical Endocrinology Metabolism Oct 2015]True, there are inconsistencies in the initial gcMAF research published by Nobuto Yamamoto. [Cancer Immunology Immunotherapy Dec 2014]
But a private European-based company, Immuno Biotech, has documented shrinkage of tumor volume with GcMAF. [Anticancer Research July 2014] GcMAF has also been shown to reduce a key enzyme (nagalase) associated with aggressive growth of tumors. [Oncoimmunology Aug 1, 2013]
If gcMAF is some sort of fraud, why did the Department of Defense sponsor a study on its ability to inhibit the growth of prostate cancer cells, which by the way was accomplished independent activation of macrophages? [PLoS One Oct 18, 2010]
Immuno Biotech, the European-based company that is producingGcMAF commercially is hiding nothing. First Immune has sponsored 31 research papers on the subject. [First Immune] Goleic, the olive oil-based GcMAF product is meticulously produced and undergoes 9 tests, including two for sterility and three for activity and potency. [Immunocentre] Aside from a mild fever, there have been no reported side effects. The cancer cell killing activity of GcMAF can be viewed online. [YouTube]
GcMAF unavailable today
Today the only readily available source of GcMAF is an oral colostrum-based (first milk) dietary supplement sold online that is encapsulated and bottled in the USA from GcMAF powder obtained from Japan.xiii Its effectiveness is estimated to be marginable given less than flattering remarks by users.
Researchers don��t give up
Researchers continue to delve into nagalase inhibition as a ��key to curing cancer.�� They surmise and predict that: ��the use of gene silencing methods, reduced expression of nagalase and consequently reduced cancer cell invasion capability can be achieved.�� To that end, a natural enzyme inhibitor could be utilized.
Enzyme inhibitors
It is well established that polyphenols derived from turmeric spice (curcumin), grapes (resveratrol), pomegranate, apple peel (quercetin) and tea leaves (catechin) are natural enzyme inhibitors, though they remain untested for nagalase inhibition.
Plant polysaccharides (sugar-like molecules)
Botanical sources of polysaccharides are well documented to control inflammation and activate macrophages.xiv Oligosaccharides derived from mushroomsxv and algae (spirulina, blue green algae) have received considerable attention in this regard.xvi��
Herbal extracts of oligosaccharides (sugar-like molecules in a chain) such as found in Panax ginsengxvii and Boswellia from Frankincensexviii, serve as other examples.
Fucoidan and nagalase
Fucoidan is a seaweed extract rich in polysaccharides that is widely offered as a cancer remedy and immune booster. At high concentrations fucoidan has been demonstrated to inhibit nagalase in a lab dish with tumor cells that have high nagalase activity.xix
Use of a decoy molecule: chondroitin sulfate
Researchers also report that a decoy molecule that interferes with nagalase could be used as a competitive/alternative inhibitor of GcMAF, thus reducing the degradation of GcMAF. This is apparently how Gc protein works in part by binding Tochondroitin sulfate.xx Chondroitin sulfate is internally produced within the matrix of cartilage in the human body and is a constituent of connective tissue.
There are macrophages that increase inflammation. However, internally made chondroitin sulfate sustains anti-inflammatory macrophages.xxi Supplemental chondroitin would presumably do the same.
The provision of chondroitin sulfate to laboratory mice is reported to reduce inflammation and subsequently reduce atherosclerotic arterial plaque and has application in cancer-induced inflammation.xxii
The combination of supplemental chondroitin sulfate and glucosamine is reported to reduce a marker of inflammation (C-reactive protein) by 23% in healthy humans and therefore reduce the risk for cancer.xxiiiChondroitin sulfate activates cancer-destructive macrophages in a lab dish.xxiv The application of chondroitin sulfate in cancer control appears to have been ignored.
Altering tumor pH to effect a cure: sodium bicarbonate (baking soda)
Yet another intriguing new avenue of investigation is alteration of pH (acid/alkaline balance). If the pH of the area surrounding tumors could be locally increased towards alkalinity the nagalase enzyme would cease to function and macrophages could do their job of digesting malignant cells.xxv
This topic has recently gained widespread public attention in the news media that alteration of pH by use of oral sodium bicarbonate (yes, baking soda) may serve to abolish the acidic environment surrounding tumor cells that provokes dormant cancer cells out of hiding and makes them vulnerable to treatment. Baking soda also increases activity of T-cells (thymus cells), another type of white blood cell.xxvi��
Tumor cells voraciously derive energy from sugar rather than oxygen as normal healthy cells do. As sugar devourers these malignant cells expel lactic acid, which creates an acidic barrier around tumors. When chemotherapy was combined with an infusion of sodium bicarbonate, tumor cell eradication increased from 63% to 100%! Researchers surmise that alteration of the tumor environment towards alkalinity might increase natural tumor cell die-off (apoptosis) by tumor pH alone.xxvii It is well established that tumor cells hijack macrophages by expelling lactic acid.xxviii
The problem is the sodium bicarbonate was injected into the tumor areas in the animal experiments, which rules out home use. Will oncologists begin to utilize this simple pre-cure? And will it release macrophages to effect a cure on its own? If so it would be known as the ten-cent cancer cure.Given that oncologists have treatments but no cures for cancer, patients are left to try natural remedies in an unguided fashion. Fortunately, the remedies mentioned above are relatively safe (unlikely to result in hospitalization or the demise of the patient) and can be utilized as nutritional support rather than advertised as a cure. Whether complete cures will be reported with their use is unknown. But given the lack of alternatives, a desperate cancer patient may use these remedies with little if any risk or interference with conventional cancer treatment and the potential of a high upside.
2018 Bill Sardi, Knowledge of Health, Inc. Used with permission by Townsend Letter for Doctors & Patients.GcMAF UPDATE: With A Tried & True Cancer Cure That Activates Cancer-Killing Macrophages Having Been Removed From The Market, Are There Any Natural Alternatives?
https://knowledgeofhealth.com/gcmaf-update-natural-alternatives/��
References
i Fairley GH, Immunity to malignant disease in man. British Medical Journal, volume 2 (5655), pages 467-73, May 24, 1969.
ii 1962: Dr. Chester Southam injected live cancer cells into 22 elderly patients. Alliance for Human Research Protection.
iii Healy M, Immunotherapy is the newest weapon in the first against cancer. Los Angeles Times, Oct 13, 2017.
iv Solinas G, et al, Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal Leukocyte Biology, Vol. 86, pages1065-73, Nov 2009.
v Ono M., Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Science, Aug. 2008.
vi Bielenberg, DR, et al, The contribution of angiogenesis to the process of metastasis. Cancer Journal, vol. 21, pages 267-73, July 2015.
vii McCarty MF, Overview of macrophage activating factor and the nagalase assay��potentional for control of micrometastasis or early primary cancer. Semantic Scholar 2013.
viii Sardi, B, Hubbell T, Cancer Cured For Good, National Health Federation. October, 2008.
ix First Immune: https://gcmaf.se
x Regulator warns against GcMAF made in unlicensed facility in Cambridgeshire. February 3, 2015, at Gov.UK
xi Immuno Biotech staff appear in court linked with GcMAF profits. Bailiwick Express, January 16, 2018.
xii Unlicensed blood drug GcMAF still for sale. BBC News, Sept. 27, 2015.
xiii GcMAF Colostrum, Fractal Health. Amazon.com
xiv Schepetkin IA, Quinn MT, Botanical polysaccharides: macrophage immunomodulation and therapeutic potential. International Immunopharmacology, vol. 6, pages 317-33, March 2006
xv Friedman M, Mushroom Polysaccharides: Chemistry and Antiobesity, Antidiabetes, Anticancer, and Antibiotic Properties in Cells, Rodents, and Humans. Foods, vol. 5, page 80, 2016
xvi
xvii Jiao L, et al, Anti-tumour and immunomodulatory activities of oligosaccharides isolated from Panax Ginseng C.A. Meyer. International Journal Biological Macromolecules, vol. 65, Pages 229-33, April 2014.
xviii Boswellin Water Soluble (Polysal) 70% polysaccharides), Sabinsa.com
xix Bakunina I, et al, The effect of Fucoidan from brown alga focus evanescence on the activity of a-N-Acetylgalactosaminidase of human colon carcinoma cells. Marine Drugs, May 2018.
xx Ruggerio, M, et al, Is chondroitin sulfate responsible for the biological effects attributed to the GC protein-derived Macrophage Activating Factor (GcMAF)?. Medical Hypotheses, Vol. 94, pages 1256-31, Sept. 2016.
Xxi Corradetti, B, et al, Immune tuning scaffold for the local induction of a pro-regenerative environment. Scientific Reports Dec. 5, 2017.
xxii Melgar-Lesmes, P, et al, Treatment with chondroitin sulfate to modulate inflammation and atherogenesis in obesity. Atherosclerosis, volume 245, pages 82-87, Feb. 2016.
xxiii Navarro SL, Randomized trial of glucosamine and chondroitin supplementation on inflammation and oxidative stress biomarkers and plasma proteomics profiles in healthy humans. PLoS One, Feb 26, 2015.
xxiv Stephenson EL, Chondroitin sulfate proteoglycans as novel drivers of leucocyte infiltration in multiple sclerosis. Brain, vol. 4, pages 1094-1100, April 1, 2018.
xxv Saburi, E, et al, Is a-N-acetylgalactosaminidase the key to curing cancer? A mini-review and hypothesis. Journal of the Balkan Union of Oncology, Nov-Dec 22 (6): 1372-77.
xxvi Blanchard S, Drinking baking soda could help cure cancer: kitchen ingredient makes hard-to-reach tumour cells easier to target with drugs, study finds. Daily Mail, May 31, 2018; How might baking soda boost cancer therapy. Ludwig Cancer Research, May 31, 2018.
xxvii Zhang H, Will cancer cells be defeated by sodium bicarbonate? Science China Life Sciences, vol. 60, pages 326-28, 2017.
xxviii Bronte, V, Tumor cells hijack macrophages via lactic acid. Immunology & Cell Biology, vol. 92, pages 647-49, 2014.
Posted in Cancer, Dietary Supplements ; No Comments »
Comments are closed.
Popular Articles
The Modern-Day Zinc Deficiency Epidemic
Controlling Atrial Fibrillation Without Drugs
Is This The Cause Of Your Heart Palpitations?
What A Day To Start A Health Radio Show
Common Symptoms Of Nutrient Deficiencies��
Nagalase enzyme
Nagalase is an extracellular matrix-degrading enzyme that is (increased) secreted by cancerous cells in the process of tumour invasion. It also is an intrisic component of envelope protein of various virions, such as HIV and the influenza virus.
Nagalase Blood Test - GcMAF.se
gcmaf.se/nagalase-blood-test/
Nagalase Blood Test: Too Good To Be True? - SelfHacked
https://selfhacked.com/blog/nagalase
Sep 16, 2019 �� High Nagalase Levels 1) Viral Infections. Nagalase levels are increased during viral infections; 2) Cancer. Since cancer cells release this enzyme, people with prostate, cervical, breast,... 3) Autism. Nagalase activity is increased in children with autism [ 2 ]. 4) Lupus. Compared to healthy ...
Author: Helen Quach, BS (Biochemistry)
Nagalase Testing - Living Love Mindfulness Medicine
https://livinglovecommunity.com/2017/02/06/nagalase-testing-nagalase-care
Nagalase is an enzyme found in the body and it has a role to play in breaking down the sugar we take in our food into other forms that can be utilized in the body in the struggle for survival. However, nagalase is a short form for the incredibly long scientific name it represents: N-acetyl-Galactosaminidase.Oncoimmunology. 2013 Aug 1; 2(8): e25769.
��GcMAF (or Gc protein-derived macrophage activating factor) is a protein produced by modification of vitamin D-binding protein.[1]
Biochemically, GcMAF results from sequential deglycosylation of the vitamin D-binding protein (the Gc protein), which is naturally promoted by lymphocytes (B and T cells).[3] The resulting protein may be a macrophage activating factor (MAF).[3] MAFs are lymphokines that control the expression of antigens on the surface of macrophages, and one of their functions is to make macrophages become cytotoxic to tumors.[4]GcMAF - Wikipedia
https://en.wikipedia.org/wiki/GcMAF��
GC protein-derived macrophage-activating factor decreases ��-N-acetylgalactosaminidase levels in advanced cancer patients
1Macro Innovations Ltd.; Cambridge, UK
2Department of Experimental and Clinical Medicine; University of Firenze; Firenze, Italy
3Immuno Biotech Ltd.; Guernsey, UK
4Reno Integrative Medical Center; Reno, NV USA
*Correspondence to: Stefania Pacini, Email: ti.ifinu@inicap.ainafets
Abstract
��-N-acetylgalactosaminidase (nagalase) accumulates in the serum of cancer patients and its activity correlates with tumor burden, aggressiveness and clinical disease progression.The administration of GC protein-derived macrophage-activating factor (GcMAF) to cancer patients with elevated levels of nagalase has been associated with a decrease of serum nagalase activity and with significant clinical benefits.
Here, we report the results of the administration of GcMAF to a heterogeneous cohort of patients with histologically diverse, advanced neoplasms, generally considered as ��incurable�� diseases. In most cases, GcMAF therapy was initiated at late stages of tumor progression. As this is an open-label, non-controlled, retrospective analysis, caution must be employed when establishing cause-effect relationships between the administration GcMAF and disease outcome. However, the response to GcMAF was generally robust and some trends emerged. All patients (n = 20) presented with elevated serum nagalase activity, well above normal values. All patients but one showed a significant decrease of serum nagalase activity upon weekly GcMAF injections. Decreased nagalase activity was associated with improved clinical conditions and no adverse side effects were reported. The observations reported here confirm and extend previous results and pave the way to further studies aimed at assessing the precise role and indications for GcMAF-based anticancer immunotherapy.
Keywords: cancer, complementary medicine, immunotherapy, macrophages, Vitamin D
GC protein-derived macrophage-activating factor decreases ��-N-acetylgalactosaminidase levels in advanced cancer patients
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3812199/��
NAGALASE TESTING �C WHAT IS NAGALASE AND WHY YOU NEED TO KNOW ABOUT IT?
by Dr. Miles Nichols
Nagalase is an enzyme found in the body and it has a role to play in breaking down the sugar we take in our food into other forms that can be utilized in the body in the struggle for survival. However, nagalase is a short form for the incredibly long scientific name it represents: N-acetyl-Galactosaminidase. Nagalase has specialized in splitting off a specific sugar molecule from other large molecules. This molecule is known as N-acetyl galactosamine. Nagalase splits this molecule from Vitamin D-binding protein (DBP) that can be found in serum and is also known as Gc-protein.
The important thing about nagalase is that the scientific community has discovered nagalase levels are increased in patients with tumors (cancer) 1, 2. Furthermore, nagalase depresses the activity of the immune system, and this activity helps cancer cells to grow and tumors to metastasize (spread) to other body organs and sites.
How does nagalase increase tumor cell formation and spread?
Nagalase causes immunosuppression by preventing the formation of a molecule that stimulates the immune system. This molecule is known as Gc-MAF and is derived from Gc-protein 3, 4. Gc protein has three sugar molecules, N-acetyl galactosamine, galactose and sialic acid. Galactose and sialic acid are removed from Gc-protein and the resultant molecule has been observed to have an effect on macrophages, which are immune cells that go around the body destroying invading microorganisms and consuming abnormal cells (for example cancer cells). It has been postulated that Gc-MAF is important in defending against tumor cell growth and spread5, 6. By preventing the formation of Gc-MAF, nagalase leaves the body without a vital defense structure in place, making it more vulnerable to cancer.
Is there a link between Nagalase and chronic viruses?
Increased nagalase activity has been linked with chronic viruses such as influenza viruses, and it has been established that nagalase activity resides on the outer envelope protein (Hemagglutinin) of the influenza virion. Nagalase levels are also increased in people living with HIV, as it is part of the gp120 protein of HIV.
What is the Nagalase test?
A test for nagalase levels is a quantitative test that seeks to establish the prevailing levels of nagalase in your body. When your levels are found to be within the normal range, then you are healthy and the chances of developing cancer are relatively low. However, if your levels are above the normal range for the lab conducting the test, then this means that there has been increased tumor cell activity in your body. This is a harbinger for cancer. Nagalase levels can also increase in other diseases, but this increase has mostly been described for cancer.
Testing will involve taking a blood sample from you and isolating the enzyme from the sample. Enzyme activity of the enzyme will then be determined quantitatively. It is also important to note that when your nagalase levels have declined from a previous high level, this could be interpreted as a success in the current therapeutic intervention being undertaken. This is especially so if the doctors managing you have been monitoring Gc-MAF therapy. Gc-MAF therapy involves introducing synthetic (artificially produced) Gc-MAF into the body in order to stimulate the immune system. This works because the artificial Gc-MAF cannot be inactivated by the nagalase enzyme in your blood.
The Nagalase test is a great blood test for finding very early signs of cancer and/or chronic viral infections, and this will improve chances of early diagnosis and management.
Nagalase Testing �C What is Nagalase and Why You Need to Know About It? - Living Love Mindfulness Medicine
https://livinglovecommunity.com/2017/02/06/nagalase-testing-nagalase-care/��
��
How Vitamin D Stops Cancer Stem Cells - DrJockers.com
https://drjockers.com/vitamin-d-stops-cancer-stem-cells/��
Vitamin D Vital to GcMAF
Researchers propose that supplementing 20,000 IU of D3 daily may be a potent and safe therapy to limit systemic inflammation and inhibit the development of cancer (8). GcMAF is a powerful immune supporting protein that demands a steady supply of D3. GcMAF activity is essential to a healthy immune system and can also assist in the destruction of tumors entirely. (7)
GcMAF Protects You Against Cancer
Epidemiological studies support that people who are deficient in vitamin D have a higher risk for developing an immune-related condition like chronic infections, metabolic complications such as type 1 diabetes, and autoimmune diseases (3).
Receiving and maintaining a healthy dose of vitamin D daily supports the synthesis of GcMAF. This is a tremendous therapeutic strategy to prevent cancer cell growth from occurring in the prostate. Urokinase plasminogen activator (uPAR) is a receptor found in the prostate which is found to support tumor metastasis in its presence (10). Evidence shows that GcMAF inhibits the presence of this pro-cancer receptor and therefore inhibits cancerous activity.
GcMAF has also been shown in many studies to reduce concentrations of nagalase. Cancer cells produce the enzyme nagalase which promotes viral infections and suppresses activated macrophage activity from destroying infected cells. Patients with high levels of nagalase include individuals with systemic lupus erythematosus (SLE), melanoma, as well as prostate, breast, colorectal, and pancreatic cancer. GcMAF stimulates the natural defense pathways of the immune system, reduces nagalase production and therefore supports healthy macrophage activity. (10)
GcMAF Therapy Destroys Cancer
Neither physicians nor scientists alike completely understood the critical role and benefits that optimal vitamin D3 levels provide to human physiology and pathology until the past decade. There has been zero evidence suggesting any toxicity concerns for the use of D3 to modulate GcMAF activity and suppress cancer since research from 2007. What we know today is that vitamin D elevates GcMAF concentrations and provides the following anti-cancer mechanisms: (11)
Boost macrophage activity
Increase in lymphocytes to normal levels
Increases platelet and red blood cell count
Stimulate cancer cell apoptosis (��cell death��)
25% Reduction in tumor size in less than 1 week
How Vitamin D Stops Cancer Stem Cells - DrJockers.com
https://drjockers.com/vitamin-d-stops-cancer-stem-cells/
��Macrophage chemoattractants secreted by cancer cells: Sculptors of the tumor microenvironment and another crucial piece of the cancer secretome as a therapeutic target
��
Highlights
•
Chemokines are critical modulators in the tumor milieu.
•
Cancer cells release chemokines that can promote tumor progression and metastasis.
•
Chemokines derived from cancer cells are potential targets for cancer immunotherapy.
Abstract
Beyond their essential role in leukocyte homing in the context of inflammation, chemokines orchestrate the host response to cancer progression. Chemokines are key accelerators in the amplification of inflammatory signals and metastasis in the distal zone of tumors, indicating possible immune editing of tumor cells in the microenvironment. This review summarizes the main macrophage-attracting chemokines secreted from cancer cells and how these mediators can be targeted to improve cancer immunotherapy in multiple cancer types.
Macrophage chemoattractants secreted by cancer cells: Sculptors of the tumor microenvironment and another crucial piece of the cancer secretome as a therapeutic target - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S1359610119300528��
Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer
Background
Tumours rapidly ferment glucose to lactic acid even in the presence of oxygen, and coupling high glycolysis with poor perfusion leads to extracellular acidification. We hypothesise that acidity, independent from lactate, can augment the pro-tumour phenotype of macrophages.
Methods
We analysed publicly available data of human prostate cancer for linear correlation between macrophage markers and glycolysis genes. We used zwitterionic buffers to adjust the pH in series of in vitro experiments. We then utilised subcutaneous and transgenic tumour models developed in C57BL/6 mice as well as computer simulations to correlate tumour progression with macrophage infiltration and to delineate role of acidity.
Results
Activating macrophages at pH 6.8 in vitro enhanced an IL-4-driven phenotype as measured by gene expression, cytokine profiling, and functional assays. These results were recapitulated in vivo wherein neutralising intratumoural acidity reduced the pro-tumour phenotype of macrophages, while also decreasing tumour incidence and invasion in the TRAMP model of prostate cancer. These results were recapitulated using an in silico mathematical model that simulate macrophage responses to environmental signals. By turning off acid-induced cellular responses, our in silico mathematical modelling shows that acid-resistant macrophages can limit tumour progression.
Conclusions
This study suggests that tumour acidity contributes to prostate carcinogenesis by altering the state of macrophage activation.Buffering tumour-secreted acids alters TAM phenotype in vivo and reduces tumour progression
To determine whether tumour acidity was a contributing factor to the phenotype of TAMs in vivo, we treated TRAMP-C2 subcutaneously injected mice with 200 mM ad lib NaHCO3 as an accepted experimental approach to neutralise tumour acidity. As shown in Supplemental Fig. S4A�CC, systemic sodium bicarbonate raised the intratumoural pH but with no effect on the growth of the established tumours. This provided us the opportunity to evaluate whether tumour acidity had a direct impact macrophage phenotype under constant tumour volume. In addition, analysis of myeloid cell infiltration by flow cytometry revealed no significant differences (Supplemental Fig. S4D). This provided another opportunity to test the polarising effect of acidity independent from changes in the number of immune cells. Accordingly, we then analysed the impact of buffering tumour acidity on macrophage activation using NanoString profiling and RT-PCR quantification of the selected genes in sorted TAMs. As shown in Fig. 4a, buffering tumour acidity increased the NanoString-derived ��inflammation score��, denoting a shift towards a pro-inflammatory phenotype. There were also decreases in the expression of major TAMs markers, including Arg1 and Fcgr2b (Fig. 4b, Supplemental Table S3). In a separate set of experiments, we also observed a significant reduction in Cd206 and Arg1 by single reaction RT-PCR (Fig. 4c). In agreement with this, quantitative image analysis of formalin-fixed sections showed a significant drop in the density of CD206 positivity in bicarbonate-treated tumours compared to untreated controls (Supplemental Fig. S4E). We second examined the TRAMP transgenic prostate model, which allowed us to test the effect of buffering tumour acidity over extended timescale (32 weeks). In this model, macrophage infiltration but not SMA+ fibroblasts corresponded with tumour progression, with the highest infiltration coincident with loss of fibromuscular tunica, disease progression from prostatic intraepithelial neoplasia lesions to high-grade adenocarcinomas and invasion (Fig. 4d�Cf). Algorithm-generated segmentation used to quantify those cell types is shown in Supplemental Fig. S4F. In addition, representative images are shown in Supplemental Fig. S4G. To investigate the role of pH, we treated TRAMP mice with 200 mM ad lib NaHCO3 for 28 weeks, starting at 4 weeks of age. Prostate tissue isolated from buffered TRAMP mice showed lower infiltration of F4/80+ macrophages into the stromal compartment compared to controls (Fig. 4g and Supplemental Fig. S4H). Furthermore, increasing tumour pH normalised prostate interglandular structure, decreased the relative percentage of the stromal compartment and reduced tumour incidence as compared with control (Fig. 4h, i). Together these results indicate that the acidic microenvironment contributes to the pro-tumour polarisation state of TAMs as well as tumour progression.
Lactic acid produced by tumour cells was reported earlier to polarise macrophages into an M2-like phenotype, with Arg1 expression by macrophages essential for lung cancer and melanoma growth.15 In addition, Carmona-Fontaine et al. have demonstrated that lactate cooperates with hypoxia to induce the expression of ARG1 in macrophages. Through the employment of an agent-based model, they also showed that hypoxia-responsive macrophages induce faster tumour growth.42
Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer | British Journal of Cancer
https://www.nature.com/articles/s41416-019-0542-2��
Lactic acid delays the inflammatory response of human ...
https://www.sciencedirect.com/science/article/pii/S0006291X1500011X
Lactic acid (LA) accumulates under inflammatory conditions, e.g. in wounds or tumors, and influences local immune cell functions. We previously noted inhibitory effects of LA on glycolysis and TNF secretion of human LPS-stimulated monocytes.��
Lactic acid delays the inflammatory response of human monocytes
Highlights
•
Lactic acid broadly delays LPS-induced gene expression in human monocytes.
•
Expression of important monocyte effector molecules is affected by lactic acid.
•
Interference of lactic acid with TLR signaling causes the delayed gene expression.
•
The profound effect of lactic acid might contribute to immune suppression in tumors.
Abstract
Lactic acid (LA) accumulates under inflammatory conditions, e.g. in wounds or tumors, and influences local immune cell functions. We previously noted inhibitory effects of LA on glycolysis and TNF secretion of human LPS-stimulated monocytes. Here, we globally analyze the influence of LA on gene expression during monocyte activation. To separate LA-specific from lactate- or pH-effects, monocytes were treated for one or four hours with LPS in the presence of physiological concentrations of LA, sodium lactate (NaL) or acidic pH. Analyses of global gene expression profiles revealed striking effects of LA during the early stimulation phase. Up-regulation of most LPS-induced genes was significantly delayed in the presence of LA, while this inhibitory effect was attenuated in acidified samples and not detected after incubation with NaL. LA targets included genes encoding for important monocyte effector proteins like cytokines (e.g. TNF and IL-23) or chemokines (e.g. CCL2 and CCL7). LA effects were validated for several targets by quantitative RT-PCR and/or ELISA. Further analysis of LPS-signaling pathways revealed that LA delayed the phosphorylation of protein kinase B (AKT) as well as the degradation of I��B��. Consistently, the LPS-induced nuclear accumulation of NF��B was also diminished in response to LA. These results indicate that the broad effect of LA on gene expression and function of human monocytes is at least partially caused by its interference with immediate signal transduction events after activation. This mechanism might contribute to monocyte suppression in the tumor environment.
Lactic acid delays the inflammatory response of human monocytes - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0006291X1500011X��
Blocking CD47 Innate Checkpoint Control for Cancer Treatment | Cancer Biology
https://blogs.shu.edu/cancer/2016/09/07/blocking-cd47-innate-checkpoint-control-for-cancer-treatment/��
��
CD47 promotes cell growth and motility in epithelial ovarian cancer
Author links open overlay panelChiu-LinWangabeMing-JieLinaChia-
Highlights
•
CD47 was positively and significantly correlated with tumor histology and grade.
•
CD47 promotes cell growth, migration and invasion.
•
CD47 may be a useful surface marker and offer a novel therapeutic option in epithelial ovarian cancer.
Abstract
Endometriosis is considered a high risk factor for the development of ovarian carcinoma, including clear cell and endometrioid malignancies. The mechanism by which endometriosis-associated ovarian cancer (EAOC) avoids anti-tumor immune surveillance by macrophages remains unclear, but CD47 is a very important immune checkpoint for macrophage phagocytosis. Therefore, we collected 36 clinical ovarian samples and detected the protein profile of CD47 by immunohistochemistry and analyzed the correlation with clinical pathological features using statistical software. We found that CD47 expression was relatively higher in patients with EAOC compared with the normal group. High CD47 expression was positively and significantly correlated with histology (P = 0.007) and tumor grade (P = 0.002). We also found that CD47 overexpression promotes cancer cell growth and motility in the TOV-112D and TOV-21G cell lines. Silencing CD47 and anti-CD47 mAb inhibit cancer cell growth and motility in cancer cell lines. Together, these results demonstrate that CD47 in EAOC may be a useful surface marker and offer a novel therapeutic option by targeting CD47 in ovarian cancer.CD47 promotes cell growth and motility in epithelial ovarian cancer - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0753332219309989��
��
Is CD47 an innate immune checkpoint for tumor evasion?
Abstract
Cluster of differentiation 47 (CD47) (also known as integrin-associated protein) is a ubiquitously expressed glycoprotein of the immunoglobulin superfamily that plays a critical role in self-recognition. Various solid and hematologic cancers exploit CD47 expression in order to evade immunological eradication, and its overexpression is clinically correlated with poor prognoses. One essential mechanism behind CD47-mediated immune evasion is that it can interact with signal regulatory protein-alpha (SIRP��) expressed on myeloid cells, causing phosphorylation of the SIRP�� cytoplasmic immunoreceptor tyrosine-based inhibition motifs and recruitment of Src homology 2 domain-containing tyrosine phosphatases to ultimately result in delivering an anti-phagocytic����don��t eat me����signal. Given its essential role as a negative checkpoint for innate immunity and subsequent adaptive immunity, CD47-SIRP�� axis has been explored as a new target for cancer immunotherapy and its disruption has demonstrated great therapeutic promise. Indeed, CD47 blocking antibodies have been found to decrease primary tumor size and/or metastasis in various pre-clinical models. In this review, we highlight the various functions of CD47, discuss anti-tumor responses generated by both the innate and adaptive immune systems as a consequence of administering anti-CD47 blocking antibody, and finally elaborate on the clinical potential of CD47 blockade. We argue that CD47 is a checkpoint molecule for both innate and adaptive immunity for tumor evasion and is thus a promising target for cancer immunotherapy.
Is CD47 an innate immune checkpoint for tumor evasion? | Journal of Hematology & Oncology | Full Text
https://jhoonline.biomedcentral.com/articles/10.1186/s13045-016-0381-z��
��
Induction of triglyceride accumulation and mitochondrial ...
https://www.nature.com/articles/srep33732
Sep 20, 2016 �� Lactic acid is a product of glycolysis in cells and the production is enhanced under hypoxia conditions. Lactic acid exists in the sodium salt (i.e. sodium lactate) form in the body in most ...
Cited by: 10
Publish Year: 2016
Author: Jingquan Sun, Jingquan Sun, Xin Ye, Minhao Xie, Jianping Ye
Proc Natl Acad Sci U S A. 2015 Nov 10; 112(45): E6215�CE6223.
Medical Sciences
HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells
aDepartment of Breast Surgery, The First Affiliated Hospital of Xi��an Jiaotong University Health Science Center, Xi��an, Shaanxi 710061, China;
bInstitute for Cell Engineering, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205;
cMcKusick-Nathans Institute of Genetic Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205;
dDepartment of Oncology, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205;
eDepartment of Pediatrics, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205;
fDepartment of Medicine, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205;
gDepartment of Radiation Oncology, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205;
hDepartment of Biological Chemistry, The Johns Hopkins University School of Medicine, Baltimore, MD, 21205SIGNIFICANCE
Uncontrolled cell proliferation and abnormal blood vessel formation result in regions of breast cancers that are hypoxic (deprived of oxygen). Hypoxia-inducible factors (HIFs) stimulate the expression of genes that enable cancer cells to invade and metastasize, leading to patient mortality. In this paper, we report that HIFs stimulate the production of CD47, a protein on the cell surface that enables cancer cells to avoid destruction by macrophages. CD47 is also important for maintaining cancer stem cells, which are a small population of cells that are required for the formation of primary tumors and metastases. Reduction of HIF activity or CD47 levels in breast cancer cells led to increased killing by macrophages and depletion of cancer stem cells.
Keywords: antitumor immunity, immune evasion, tumor-initiating cells, tumor microenvironment, ��don��t eat me�� signal
ABSTRACT
Increased expression of CD47 has been reported to enable cancer cells to evade phagocytosis by macrophages and to promote the cancer stem cell phenotype, but the molecular mechanisms regulating CD47 expression have not been determined. Here we report that hypoxia-inducible factor 1 (HIF-1) directly activates transcription of the CD47 gene in hypoxic breast cancer cells. Knockdown of HIF activity or CD47 expression increased the phagocytosis of breast cancer cells by bone marrow-derived macrophages. CD47 expression was increased in mammosphere cultures, which are enriched for cancer stem cells, and CD47 deficiency led to cancer stem cell depletion. Analysis of datasets derived from thousands of patients with breast cancer revealed that CD47 expression was correlated with HIF target gene expression and with patient mortality. Thus, CD47 expression contributes to the lethal breast cancer phenotype that is mediated by HIF-1.INTRODUCTION
The pathogenesis of breast cancer reflects not only the consequence of somatic mutations that dysregulate oncogenes and tumor suppressor genes, but also the effect of the changing tumor microenvironment, particularly the development of intratumoral hypoxia. The median pO2 within primary breast cancers is 10 mmHg (1.4% O2) compared with 65 mmHg (9.3% O2) in normal breast tissue (1). Exposure of breast cancer cells to reduced O2 availability induces the activity of hypoxia-inducible factors (HIFs), which are heterodimeric transcriptional activators, consisting of an O2-regulated HIF-1��, HIF-2��, or HIF-3�� subunit and a constitutively expressed HIF-1�� subunit, that control the expression of hundreds of target genes (2). Increased expression of the HIF-1�� subunit, detected by immunohistochemistry in primary breast cancer biopsies, is associated with a significantly increased risk of metastasis and mortality (2).
Preclinical studies in mouse models have demonstrated that loss of HIF-1�� or HIF-2�� expression impairs the metastasis of breast cancer cells to axillary lymph nodes (3), lungs (4, 5), and bone (6, 7). Specific HIF target genes have been identified that are induced by hypoxia in breast cancer cells and promote critical steps in the metastatic process, including stromal cell recruitment (8, 9), cancer cell migration (10), invasion and intravasation (11�C15), margination and extravasation (5), and premetastatic niche formation (12, 16). Increased expression of HIF target genes in primary breast cancers is associated with increased patient mortality (17, 18).
To give rise to a primary tumor, a tumor relapse, or a metastatic tumor, a breast cancer cell must possess two important characteristics: first, the cell must avoid destruction by the immune system and, second, the cell must possess stem-cell�Clike properties. Hypoxia induces the breast cancer stem cell (CSC) phenotype (19, 20) through functional and physical interactions of HIF-1 with the coactivator TAZ (20, 21) and by HIF-dependent expression of pluripotency factors (22). Hypoxia also induces immune evasion by several HIF-dependent mechanisms (23, 24). A major mechanism by which cancer cells evade the innate immune system is by expression of CD47, which is a cell-surface protein that interacts with signal regulatory protein �� (SIRP��) on the surface of macrophages to block phagocytosis (25, 26). Expression of calreticulin (CRT) on the surface of cancer cells is the primary trigger for phagocytosis by binding to low-density lipoprotein-related protein (LRP) on the surface of macrophages (27). The prophagocytic signal triggered by CRT�CLRP ligation is counterbalanced by the antiphagocytic signal triggered by CD47�CSIRP�� ligation (28). Analysis of circulating tumor cells isolated from the blood of patients with breast cancer revealed that CD47 expression identified a subpopulation of cells with the capability to generate tumor xenografts in mice (29). Here, we report that CD47 expression is induced in a HIF-dependent manner when human breast cancer cells are exposed to hypoxia. Modest inhibition of CD47 expression was sufficient to increase the phagocytosis of breast cancer cells and decrease the number of breast CSCs. Human breast cancer database analysis revealed that high CD47 expression is correlated with increased HIF target gene expression and decreased patient survival.RESULTS
Hypoxia Induces Increased CD47 Expression in a HIF-Dependent Manner.
Breast cancers are classified based on their expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2); they are also classified according to expression of a 50-mRNA signature (PAM50) into luminal A, luminal B, HER2-enriched, basal-like, and normal-like subgroups (30). We analyzed the effect of hypoxia (1% O2 for 24 h) on CD47 mRNA levels in six different human breast cell lines: MCF10A is an immortalized but nontumorigenic mammary epithelial line; MCF7 is ER+PR+ and tumorigenic but nonmetastatic; HCC1954 is HER2+ and tumorigenic but nonmetastatic; and MDA-MB-231, MDA-MB-435, and SUM159 are ER−PR−HER2− (i.e., triple negative), tumorigenic and metastatic. Reverse transcription (RT) and quantitative real-time PCR (qPCR) assays revealed that expression of CD47 mRNA was significantly induced by hypoxia in MCF7, HCC1954, MDA-MB-435, and SUM159 cells but not in MCF10A or MDA-MB-231 cells (Fig. 1A).
CD47 Is a Direct HIF-1 Target Gene.
To determine whether HIF-1 directly regulates CD47 gene transcription, the human genome sequence was searched for matches to the consensus HIF binding site 5��-(A/G)CGTG-3�� (32) that were located within DNase I-hypersensitive domains at the CD47 locus. Chromatin immunoprecipitation (ChIP) assays revealed that a DNA sequence, which encompassed 5��-GCGTG-3�� at −239 bp (site 1) and 5��-CACGC-3�� (5��-GCGTG-3�� on the antisense strand) at −200 bp (site 2), relative to the CD47 transcription start site (Fig. 2A, Top), was enriched by immunoprecipitation of chromatin from hypoxic MCF7, HCC1954, and SUM159 cells with HIF-1�� or HIF-1�� antibodies (Fig. 2B), indicating that hypoxia induces direct binding of HIF-1 to the CD47 promoter.
HIF Deficiency Increases the Phagocytosis of Breast Cancer Cells.
We observed decreased expression of CD47 on the surface of HIF-deficient human breast cancer cells (Fig. 1 F and G), leading us to hypothesize that HIF inhibition may promote phagocytosis of breast cancer cells by macrophages. To test this hypothesis, we performed in vitro phagocytosis assays on HCC1954 and MCF7 subclones. The breast cancer cells were labeled with the fluorescent dye CFSE, exposed to 20% or 1% O2 for 24 h, cocultured with bone marrow-derived macrophages for 2 h, incubated with allophycocyanin (APC)-labeled F4/80 antibody, and analyzed by flow cytometry to detect APC+CFSE+ cells, which represent macrophages that have phagocytosed breast cancer cells (Upper Right quadrant [Q2] of scatter plots in Fig. 3A). Phagocytosis was significantly increased in MCF7-DKD and HCC1954-DKD cells compared with the respective NTC subclone, under both hypoxic and nonhypoxic conditions (Fig. 3 B and C). Hypoxia modestly increased phagocytosis of both NTC and DKD subclones, which suggests the existence of a prophagocytic signal that is induced by hypoxia in a HIF-independent manner.��
CD47 Deficiency Increases the Phagocytosis of Breast Cancer Cells.
To determine whether CD47 mediates protection of breast cancer cells against phagocytosis, we transfected SUM159 cells with an expression vector encoding one of five different shRNAs targeting CD47. Remarkably, we were not able to establish stable subclones that expressed three of the shRNAs and expression of the other two shRNAs (shCD47-2 and shCD47-4) caused relatively modest (50�C75%) inhibition of CD47 mRNA (Fig. 4A) and cell-surface protein (Fig. 4B) expression. However, phagocytosis was significantly increased in the CD47-knockdown subclones (Fig. 4C) and CD47 levels were inversely correlated with the extent of phagocytosis. Taken together, the data presented in Fig. 4 demonstrate that CD47 expression protects breast cancer cells against phagocytosis by bone marrow-derived macrophages.
CD47 Promotes the Breast CSC Phenotype.
CD47 is preferentially expressed on bladder, liver, and pancreatic CSCs compared with the bulk cancer cells (non-CSCs) (33�C36). To investigate whether CD47 plays a role in breast CSCs, we first analyzed CD47 mRNA levels in SUM159 cells, which were cultured as standard adherent monolayers or as nonadherent spheroids (mammospheres), which are highly enriched for CSCs (37). CD47 mRNA levels were twofold higher in mammosphere cultures (Fig. 5A). Exposure of breast cancer cells to hypoxia for 3 d induces increased mammosphere formation in a HIF-1���Cdependent manner (20). Knockdown of CD47 expression significantly decreased mammosphere formation, with the greatest reduction observed in the shCD47-4 subclone (Fig. 5B), which had the greatest inhibition of CD47 expression (Fig. 4 A and B). Analysis of aldehyde dehydrogenase (ALDH) activity in breast cancer cells also identifies a subpopulation that is enriched for CSCs (38) and the percentage of ALDH+ cells increases in response to hypoxia (19, 20). Hypoxia markedly increased the percentage of ALDH+ cells in the NTC subclone, whereas knockdown of CD47 expression significantly decreased the percentage of ALDH+ CSCs at both 20% and 1% O2 (Fig. 5C). Taken together, the data presented in Fig. 5 demonstrate that CD47 expression plays an important role in promoting the breast CSC phenotype. We and others have previously reported that knockdown of HIF-1�� blocks hypoxia-induced enrichment of breast CSCs, as determined by ALDH expression or mammosphere formation assay (19, 20). Thus, CD47 loss-of-function phenocopies HIF-1�� loss of function with respect to CSC maintenance.
CD47 Expression Is Associated with HIF Target Gene Expression and Patient Mortality.
To test whether the results obtained from the analysis of breast cancer cell lines are relevant to patients with breast cancer, we analyzed gene expression data from 1,040 primary human breast cancer samples that are publicly available in The Cancer Genome Atlas (TCGA) database (39). To determine whether HIFs regulate CD47 gene expression in human breast cancers, we compared CD47 mRNA levels with the levels of P4HA1, P4HA2, ANGPTL4, SLC2A1, CXCR3, VEGFA, PLOD1, PLOD2, L1CAM, and MET mRNA, which are all products of genes that are HIF regulated in breast cancer cells (18, 20). CD47 mRNA levels, like those of other HIF target genes, were most highly expressed in breast cancers of the basal-like molecular subtype (Fig. 6A). Statistical analysis revealed that CD47 mRNA levels were significantly correlated with the levels of 8 of 10 mRNAs encoded by HIF target genes (Fig. 6B), which is comparable to correlations between members of this group (10, 15, 20). Expression of CD44, which is a marker of breast CSCs (40) and a HIF target gene (41), was also significantly correlated with CD47 expression in the 1,040 breast cancers (Fig. 6B).HIF target gene expression in human breast cancers is significantly associated with patient mortality (18). To determine if CD47 mRNA expression was also a prognostic factor in human breast cancer, we used two published datasets that contain both gene expression and patient survival data (42, 43). Patients were stratified into two groups based on whether CD47 mRNA levels in the primary tumor were greater or less than the median value. Increased CD47 mRNA expression levels were associated with a significantly decreased probability of overall survival in the two independent datasets, which together comprised 1,954 patients with breast cancer (Fig. 6C).
DISCUSSION
Growing evidence indicates that in various human cancers, CD47 expression is required to avoid innate immune surveillance and elimination by phagocytosis. Blocking the interaction of CD47 with its receptor, SIRP��, on macrophages enables phagocytosis and inhibits tumor growth (35, 36, 44�C46). However, the molecular mechanisms regulating the expression of CD47 by cancer cells have not been delineated. In this study, we demonstrate that hypoxia, which is a critical microenvironmental stimulus in advanced breast cancers, induced HIF-dependent expression of CD47, leading to decreased phagocytosis of cancer cells by macrophages and induction of the breast CSC phenotype, which promote cancer progression and patient mortality (Fig. 6D).
Despite the recent interest in targeting CD47 for cancer therapy (46), remarkably little is known about the transcriptional regulation of the CD47 gene. The results presented here represent to our knowledge the first identification of a transcriptional regulator of CD47 expression in breast cancer cells and further studies are required to determine whether HIF-1 cooperates with other transcription factors that are induced by the tumor microenvironment, such as CREB, NF-��B, SMAD2, or STAT3. Increased NF-��B activity was observed in hepatocellular carcinoma cells that developed sorafenib resistance (36), which is of interest because the antiangiogenic effects of sorafenib induce intratumoral hypoxia that causes HIF-1�Cdependent induction of breast CSCs (19). Immunosuppressive functional interactions of HIF-1 and CREB have been proposed in T cells (47), but may also occur in cancer cells.
Intratumoral hypoxia is a common finding in breast cancer and HIFs activate the transcription of a large battery of genes encoding proteins that promote multiple steps in tumor progression, including tumor growth and vascularization, stromal cell recruitment, extracellular matrix remodeling, premetastatic niche formation, cell motility, margination and extravasation of circulating tumor cells, and CSC specification/maintenance (2). Here, we find that HIFs also play an important role in immune evasion by activating the expression of CD47, an antiphagocytic signal. In three different breast cancer cell lines, which represent luminal/ER+ (MCF7), HER2-enriched (HCC1954), and basal-like/triple-negative (SUM159) subtypes of breast cancer, the expression of CD47 mRNA and protein was induced by hypoxia in a HIF-dependent manner. A recent study reported that CD47 protein was detected in 5% of hormone receptor positive, HER2− breast cancers (48). Our bioinformatic analysis suggests that CD47 expression is likely to be higher among triple-negative breast cancers, most of which fall into the basal-like molecular subtype that is characterized by increased expression of the HIF transcriptome (18, 39).
Hypoxia did not induce increased CD47 expression in MCF10A or MDA-MB-231 cells. This heterogeneous transcriptional response of breast cell lines to hypoxia is commonly observed. For example, analysis of mRNAs encoding lysyl oxidase (LOX) and LOX-like proteins (LOXL1�C4) revealed that hypoxia induced increased expression of LOX and LOXL4 in MDA-MB-231, whereas LOXL2 expression was induced in MDA-MB-435 cells (12). We also observed heterogeneity with respect to requirements for HIF-�� subunits, as CD47 induction required only HIF-1�� in MCF7 cells, but both HIF-1�� and HIF-2�� in HCC1954 cells. Similarly, we previously reported that hypoxia-induced LOXL2 expression required only HIF-1�� in MDA-MB-435 cells, whereas expression of LOX and LOXL4 in MDA-MB-231 cells required HIF-1�� and HIF-2�� (12). Heterogeneity at the level of gene expression is recognized as a major obstacle to successful cancer therapy. However, in the cases described above, treatment with a HIF inhibitor, such as acriflavine, successfully blocked all hypoxia-induced gene expression (Fig. 1D).
Cells express varying levels of prophagocytic and antiphagocytic signaling proteins, and it is the integration of both signals that determines whether the target cell will be phagocytosed. The relatively modest changes in CD47 cell surface levels that resulted from expression of shRNA targeting HIF-1�� and HIF-2�� or CD47 led to significant changes in phagocytosis, indicating a fine balance between pro- and antiphagocytic signaling, such that a hypoxic tumor microenvironment may significantly tip the balance toward immune evasion. Several prophagocytic signals have been identified, including cell-surface expression of phosphatidylserine and CRT (49, 50). CRT is required for the phagocytosis of cancer cells when CD47�CSIRP�� interaction is blocked (28). CRT expression was induced by exposure of cardiomyocytes to hypoxia and reoxygenation (51), suggesting that the increased phagocytosis of NTC and DKD subclones after hypoxic exposure may be due to HIF-independent CRT expression, but further studies are required to test this hypothesis. A recent report demonstrated that CD47 blockade also promotes T-cell�Cmediated elimination of immunogenic tumors (52) and HIF-1 is known to inhibit effector T cells through the production of adenosine (23, 47), suggesting that HIF inhibitors may improve the response to CD47 blockade both by inhibiting CD47 expression and by disinhibiting T-cell�Cmediated antitumor immunity.
Two findings in our study suggested that hypoxia-induced expression of CD47 plays an important role that is independent of its antiphagocytic function. First, although CD47 mRNA and protein expression were induced by hypoxia in all three breast cancer cell lines analyzed, increased CD47 cell-surface expression and decreased phagocytosis were only induced by hypoxia in SUM159 cells. These data suggest that hypoxia-induced CD47 may fulfill another function that does not require cell-surface expression of the protein. Second, attempts at generating CD47-deficient cells were only successful in establishing subclones with modest knockdown of CD47, suggesting that expression of the protein was required for clonal expansion. Using two established assays for breast CSCs, we demonstrated that CD47 expression was increased in breast CSCs (relative to bulk cancer cells) and was required for breast CSC maintenance, as decreased CD47 expression led to significantly reduced numbers of CSCs in a dose-dependent manner. Further studies are required to determine whether CD47 must be expressed at the cell surface to contribute to the maintenance of breast CSCs.
To generate a primary, recurrent, or metastatic tumor, a breast cancer cell must avoid immune destruction and give rise to both CSCs and differentiated breast cancer cells. A recent study found that among circulating tumor cells in the blood of women with metastatic breast cancer, the cells that were capable of initiating tumors when injected into mice expressed the cell surface proteins CD47, CD44, and MET (29). Remarkably, as in the case of CD47, the expression of CD44 and MET is induced by hypoxia in a HIF-dependent manner (41, 53). Furthermore, our analysis of gene expression data from over 1,000 human breast cancers revealed that CD47 expression was significantly correlated with the expression of CD44, MET, and other HIF target genes (Fig. 6B). We also found that CD47 mRNA expression in primary human breast cancers was significantly associated with patient mortality in two large and independent datasets (Fig. 6C). Similar results were recently reported regarding the immunohistochemical detection of CD47 protein expression in breast cancer biopsies (48).
We have recently demonstrated that exposure of breast cancer cells to hypoxia or chemotherapy induces the breast CSC phenotype through multiple HIF-dependent molecular pathways (18, 20, 54). Induction of CD47 expression provides another mechanism by which hypoxia induces the CSC phenotype. In addition to interacting with SIRP��, CD47 engages in several other functional interactions (55) and further studies are required to determine the molecular mechanism by which the expression of CD47, localized to the cell surface or perhaps an intracellular compartment, promotes the specification and/or maintenance of CSCs. Genetic or pharmacologic inhibition of HIF activity reduced the percentage of CSCs (18�C20, 56), which may be due in part to the inhibition of CD47 expression. Increased expression of HIF-1�� mRNA or protein, or HIF target gene mRNAs, in primary breast cancer biopsies is associated with increased patient mortality (2, 18). Our data suggest that addition of HIF inhibitors to current treatment regimens may improve outcome in such high-risk patients in part by attacking CSCs and disinhibiting innate immunity.
PNAS Plus: HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4653179/��
Hypoxia induces the breast cancer stem cell phenotype by ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4833258
Apr 05, 2016 �� In this study, we report that exposure of breast cancer cells to hypoxia (i.e., reduced O 2 availability), which is a critical feature of the tumor microenvironment, induces N 6-methyladenosine (m 6 A) demethylation and stabilization of NANOG mRNA, thereby promoting the breast cancer stem cell (BCSC) phenotype.
Cited by: 242
Publish Year: 2016
Author:��
Chemotherapy-Induced Ca2+ Release Stimulates Breast Cancer ...
https://www.cell.com/cell-reports/fulltext/S2211-1247(17)30165-1
Breast cancer stem cells (BCSCs) play a critical role in tumor recurrence and metastasis. Exposure of breast cancer cells to chemotherapy leads to an enrichment of BCSCs. Here, we find that chemotherapy induces the expression of glutathione S-transferase omega 1 (GSTO1), which is dependent on hypoxia-inducible factor 1 (HIF-1) and HIF-2 ...
Highlights
•
Exposure of breast cancer cells to carboplatin induces HIF-dependent GSTO1 expression
•
GSTO1 interacts with ryanodine receptor 1 to increase intracellular Ca2+ levels
•
Ca2+ triggers PYK2 �� SRC �� STAT3 signaling, leading to breast cancer stem cell enrichment
•
GSTO1 knockdown blocks cancer stem cell enrichment, tumor initiation, and metastasis��
Although most patients with BC initially respond to cytotoxic chemotherapy, objective clinical responses, which are typically evaluated by tumor shrinkage, often do not translate into a substantial improvement in overall survival (Huff et al., 2006). This paradox may due in part to the counter-therapeutic effect of chemotherapy on BCSCs. Recent studies have indicated that HIFs are required for BCSC enrichment induced by paclitaxel or gemcitabine (Samanta et al., 2014).
��
Introduction
Breast cancer (BC) is the most common cancer in women, with approximately 250,000 new cases and 40,000 deaths in the United States in 2016 (Siegel et al., 2016). Chemotherapy is used in selected early-stage and many advanced breast cancers (Anampa et al., 2015). Many patients initially benefit from chemotherapy but experience recurrence, metastasis, and ultimately death (Abdullah and Chow, 2013). This is particularly true for triple-negative BCs (TNBCs), which lack expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). BC stem cells (BCSCs), which are defined by their infinite proliferative potential and tumor initiating properties, contribute critically to tumor recurrence and metastasis (Pece et al., 2010, Oskarsson et al., 2014). BCSCs have increased chemoresistance through a variety of mechanisms, including increased drug inactivation, increased expression of drug transporter proteins, and enhanced DNA repair activity (Dean et al., 2005). Chemotherapy may eliminate the bulk of cancer cells within a tumor but leave behind BCSCs that are the source of recurrent and metastatic disease. Thus, novel treatment options that target BCSCs are needed.
Hypoxia-inducible factors (HIFs) are associated with resistance to chemotherapy in BC (Flamant et al., 2010, Samanta et al., 2014). HIFs are transcription factors consisting of a highly regulated HIF-1�� or HIF-2�� subunit and a constitutively expressed HIF-1�� subunit (Semenza, 2003). HIFs play important roles in promoting specification and/or maintenance of the BCSC phenotype in response to hypoxia (Xiang et al., 2014, Zhang et al., 2015, Samanta et al., 2016, Zhang et al., 2016a, Zhang et al., 2016b). Chemotherapy also induces enrichment of BCSCs in a HIF-dependent manner (Samanta et al., 2014). We recently reported that chemotherapy induces HIF-dependent activation of the glutathione biosynthesis pathway, leading to increased intracellular glutathione levels. Glutathione inhibits MEK-ERK signaling through copper chelation, leading to nuclear localization of FoxO3 and transcriptional activation of the NANOG gene, which encodes a pluripotency factor that specifies the BCSC phenotype (Lu et al., 2015).
Glutathione also contributes to drug resistance through detoxification of a wide range of exogenous and endogenous compounds (Ramsay and Dilda, 2014), which is catalyzed by glutathione S-transferases (GSTs). GSTs are a superfamily of phase II enzymes that catalyze the conjugation of glutathione with electrophilic compounds (Townsend and Tew, 2003). Seven classes of cytosolic GSTs (A, M, O, P, S, T, and Z) have been identified in mammalian cells. Several GSTs have non-enzymatic roles relevant to drug resistance through protein-protein interactions and regulation of signaling pathways (Adler et al., 1999, Cho et al., 2001, Laliberte et al., 2003). In this study, we demonstrate a role for GSTO1 in specification of the BCSC phenotype in response to chemotherapy that is independent of its enzymatic activity.
Cited by: 57
Publish Year: 2017
Affiliations
Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA��
Hypoxia-Induced Resistance to Chemotherapy in Cancer ...
https://link.springer.com/chapter/10.1007/978-3-030-12734-3_9
Jun 15, 2019 �� Chemotherapy resistance can either be an intrinsic property of malignant cells developed prior to therapy, or acquired following exposure to anti-cancer drugs. Given the impact of drug resistance to the overall poor survival of cancer patients, there is an urgent need to better understand the molecular pathways regulating this malignant phenotype.
Author: Lori M. Minassian, Tiziana Cotechini, Erin Huitema, Charles H. Graham
Publish Year: 2019A major barrier to the successful management of cancer is the development of resistance to therapy. Chemotherapy resistance can either be an intrinsic property of malignant cells developed prior to therapy, or acquired following exposure to anti-cancer drugs. Given the impact of drug resistance to the overall poor survival of cancer patients, there is an urgent need to better understand the molecular pathways regulating this malignant phenotype. In this chapter we describe some of the molecular pathways that contribute to drug resistance in cancer, the role of a microenvironment deficient in oxygen (hypoxia) in malignant progression, and how hypoxia can be a significant factor in the development of drug resistance. We conclude by proposing potential therapeutic approaches that take advantage of a hypoxic microenvironment to chemosensitize therapy-resistant tumours.
��
Intermittent hypoxia induces a metastatic phenotype in ...
https://www.nature.com/articles/s41388-018-0259-3
Louie E, Nik S, Chen J-s, Schmidt M, Song B, Pacson C, et al. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and ...��
Hypoxia�\inducible factors: coupling glucose metabolism and ...
https://www.embopress.org/doi/full/10.15252/embj.201695204
Dec 22, 2016 �� HIF�\1 has been implicated in chemotherapy resistance (Unruh et al, 2003), but recent studies have demonstrated that exposure of breast cancer cells to cytotoxic chemotherapy induces HIF�\1 activity and increases the percentage of BCSCs in a HIF�\dependent manner (Cao et al, 2013; Samanta et al, 2014; Lu et al, 2015).��
Cancer Stem Cells Under Hypoxia as a ... - SpringerLink
https://link.springer.com/article/10.1007/s40139-013-0035-6
Jan 11, 2014 �� Exposure of cancer cells and tissues to hypoxia, or sub-atmospheric concentrations of oxygen (<21 % O 2), stimulates a variety of stress responses that bias the cells toward a self-preserving, anti-apoptotic phenotype, properties that are found in CSCs. Despite major advances in our understanding of hypoxia, CSCs, and their interrelated nature ...��
Flavonoid Apigenin Is an Inhibitor of the NAD+ase CD38 ...
https://diabetes.diabetesjournals.org/content/62/4/1084
Apr 01, 2013 �� We confirmed that quercetin is a CD38 inhibitor in vitro and in cells. Importantly, we demonstrate that apigenin(�۲��أ� is a novel inhibitor of CD38 in vitro and in vivo. Treatment of cells with apigenin or quercetin inhibits CD38 and promotes an increase in intracellular NAD + levels.
Cited by: 157
Publish Year: 2013��
CST - CD38 Antibody
https://www.cellsignal.com/products/primary-antibodies/cd38-antibody/14637
Since CD38 is the primary mammalian NAD + glycohydrolase(NADase) responsible for NAD + metabolism, CD38 may be a valuable therapeutic target for treatment of metabolic diseases regulated by NAD +-dependent pathways (7,8). CD38 has also been considered a possible therapeutic target for antibody-mediated therapy for myeloma and chronic lymphocytic ...
Brand: Cell Signaling Technology
Category: Primary Antibodieshttps://www.spandidos-publications.com/10.3892/ijo.2018.4651
��
Regulation of NAD+ glycohydrolase activity by NAD ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1217703
Sep 15, 1996 �� Abstract. NAD+ glycohydrolase (NADase; EC 3.2.2.5) is an enzyme that catalyses hydrolysis of NAD+ to produce ADP-ribose and nicotinamide. Its physiological role and the regulation of its enzymic activity have not been fully elucidated. In the present study, the mechanism of self-inactivation of NADase by its substrate, NAD+,...��
CD38 affects the biological behavior and energy metabolism of nasopharyngeal carcinoma cells
Department of Oncology, Hunan Provincial Tumor Hospital, Xiangya School of Medicine, Central South University, Changsha, Hunan 410006, P.R. China, Department of Gynecology and Obstetrics, The Third Xiangya Hospital, Central South University, Changsha, Hunan 410078, P.R. ChinaABSTRACT
Nasopharyngeal carcinoma (NPC) is the most common malignant tumor type in Southern China and South‑East Asia. Cluster of differentiation (CD)38 is highly expressed in the human immune system and participates in the activation of T, natural killer and plasma cells mediated by CD2 and CD3 through synergistic action. CD38 is a type II transmembrane glycoprotein, which was observed to mediate diverse activities, including signal transduction, cell adhesion and cyclic ADP‑ribose synthesis. However, the significance of CD38 in NPC biological behavior and cellular energy metabolism has not been examined. In order to elucidate the effect of CD38 on the biological behavior of NPC cells, stable CD38‑overexpressed NPC cell lines were established. It was demonstrated that CD38 promoted NPC cell proliferation with Cell Counting Kit‑8 and colony formation assays. It was also indicated that CD38 inhibited cell senescence, and promoted cell metastasis. Furthermore, it was determined that CD38 promoted the conversion of cells to the S phase and decreased the content of reactive oxygen species and Ca2+. Additionally, cell metabolism assays demonstrated that CD38 increased the concentration of ATP, lactic acid, cyclic adenosine monophosphate and human ADP/acrp30 concentration in NPC cells. To investigate the possible mechanism, bioinformatics analysis and mass spectrometry technology was used to determine the most notably changing molecule and signaling pathways, and it was determined and verified that CD38 regulated the metabolic‑associated signaling pathways associated with tumor protein 53, hypoxia inducible factor‑1�� and sirtuin 1. The present results indicated that CD38 may serve a carcinogenic role in NPC by regulating metabolic‑associated signaling pathways.
Introduction
Nasopharyngeal carcinoma (NPC) refers to malignant tumors that occur on the top and sidewall of the nasopharyngeal cavity (1,2). NPC is one of the most prevalent malignant tumor types in China since 2002-2014 (3,4). Risk factors for NPC include Epstein-Barr virus infection (5), genetic factors, intake of salted food and cigarette smoking (6-9). Female patients accounted for ~30% of cases from 1983-2008 in Hong Kong, indicating that NPC is more common in males (10,11). Currently, radiation therapy is the preferred method used in the treatment of NPC, due to the majority of the NPC cases being poorly differentiated carcinomas (12). The treatment methods of NPC also include surgical treatment and chemotherapy, to improve patient survival rate (13-16).
Nutritional competition among cells can affect the growth, survival and functions. Different cells have different nutrient intake conditions and energy metabolism phenotypes. Therefore, the state of cells is associated with energy metabolism (17). Cluster of differentiation (CD)38 is located on chromosome 4 and was originally observed as a lymphocyte surface antigen. The molecular weight is 45 kDa (18). A number of studies demonstrated that CD38 was a transmembrane protein with receptors, extracellular enzymes and other functions (19-21). CD38 is primarily considered as a Type II membrane protein with a small N-terminal tail protruding into the cell and a catalytically active C-terminus located extracellularly (22). CD38 is not only expressed in T lymphocytes, but also expressed in other immune cells, including plasma cells, natural killer cells, monocytes and dendritic cells (23). CD38 is an important target in the treatment of multiple myeloma (24). In particular, the CD38 targeted therapies exhibit single-agent activity in early-phase clinical trials (25-27).
Although numerous studies have been conducted on CD38, there is relatively limited research regarding the energy metabolism of CD38 in NPC (28). The purpose of the present study was to determine the expression level of CD38 in NPC, investigate the role of CD38 in the biological behavior and cell metabolism of NPC cells, and to investigate the possible mechanisms.
CD38 affects the biological behavior and energy metabolism of nasopharyngeal carcinoma cells
https://www.spandidos-publications.com/10.3892/ijo.2018.4651��
Frontiers | CD38 in Adenosinergic Pathways and Metabolic ...
https://www.frontiersin.org/articles/10.3389/fimmu.2019.00760
Apr 24, 2019 �� Tumor microenvironments are rich in extracellular nucleotides that can be metabolized by ectoenzymes to produce adenosine, a nucleoside involved in controlling immune responses. Multiple myeloma, a plasma cell malignancy developed within a bone marrow niche, exploits adenosinergic pathways to customize the immune homeostasis of the tumor. CD38, a multifunctional protein that ��
Cited by: 1
Publish Year: 2019
Author: Alberto L. Horenstein, Cristiano Bracci, Fabio��
Tumor cell-derived lactate induces TAZ-dependent ...
https://www.ncbi.nlm.nih.gov/pubmed/28604752
Oct 19, 2017 �� PD-L1 expression is upregulated in many solid tumors including lung cancer and functions predominantly in lactate-enriched tumor microenvironments. Here, we provided evidence for PD-L1 induction in response to lactate stimulation in lung cancer cells. Lactate-induced PD-L1 induction was mediated by its receptor GPR81.
Cited by: 53
Publish Year: 2017
��Tumor cell-derived lactate induces TAZ-dependent ...
https://www.nature.com/articles/onc2017188
Jun 12, 2017 �� PD-L1 expression is upregulated in many solid tumors including lung cancer and functions predominantly in lactate-enriched tumor microenvironments. ... (PD-1/PD-L1) has initiated a ��
Cited by: 53
Publish Year: 2017
Author: J Feng, H Yang, Y Zhang, H Wei, Z Zhu, B Zhu, M YaOncogene. 2017 Oct 19;36(42):5829-5839. doi: 10.1038/onc.2017.188. Epub 2017 Jun 12.
Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells.
Feng J1, Yang H2, Zhang Y3, Wei H4, Zhu Z5, Zhu B6, Yang M7, Cao W7, Wang L1, Wu Z3,8.
Author information
1
Medical Oncology, The First Affiliated Hospital of Wannan Medical College, Wuhu, China.
2
The Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Tianjin Lung Cancer Center and Institute, Tianjin Medical University General Hospital, Tianjin, China.
3
Anhui Province Key laboratory of Active Biological Macro-molecules Research, Wannan Medical College, Wuhu, China.
4
Department of Central Laboratory, Wannan Medical College, Wuhu, China.
5
School of Pharmacy, Wannan Medical College, Wuhu, China.
6
School of Forensic Medicine, Wannan Medical College, Wuhu, China.
7
School of Anesthesiology, Wannan Medical College, Wuhu, China.
8
School of Preclinical Medicine, Wannan Medical College, Wuhu, China.
Abstract
The clinical success of immunotherapy that inhibits the negative immune regulatory pathway programmed cell death protein 1/PD-1 ligand (PD-1/PD-L1) has initiated a new era in the treatment of metastatic cancer. PD-L1 expression is upregulated in many solid tumors including lung cancer and functions predominantly in lactate-enriched tumor microenvironments. Here, we provided evidence for PD-L1 induction in response to lactate stimulation in lung cancer cells. Lactate-induced PD-L1 induction was mediated by its receptor GPR81. The silencing of GPR81 signaling in lung cancer cells resulted in a decrease in PD-L1 protein levels and functional inactivation of PD-L1 promoter activity. In addition, GPR81-mediated upregulation of PD-L1 in glucose-stimulated lung cancer cells that recapitulates the enhanced glycolysis in vivo was dependent on lactate dehydrogenase A (LDHA). We also demonstrated that activation of GPR81 decreases intracellular cAMP levels and inhibits protein kinase A (PKA) activity, leading to activation of the transcriptional coactivator TAZ. Interaction of TAZ with the transcription factor TEAD was essential for TAZ activation of PD-L1 and induction of its expression. Furthermore, we found that lactate-induced activation of PD-L1 in tumor cells led to reduced production of interferon-�� and induction of apoptosis of cocultured Jurkat T-cell leukemia cells. Our findings reveal an unexpected role of lactate in contributing to tumor cell protection from cytotoxic T-cell targeting and establishes a direct connection between tumor cell metabolic reprograming and tumor evasion from the immune response.��
������ 2017��10��19��; 36��42����5829-5839�� doi��10.1038 / onc.2017.188�� EPUB 2017��6��12�ա�
����ϸ����Դ��������ͨ��GPR81�յ��˷ΰ�ϸ����TAZ�����Ե�PD-L1�ϵ���
��J1����H2����Y3��κH4����Z5����B6����M7����W7����L1����Z3,8��
������Ϣ
1��
�ߺ�������ҽѧԺ������һҽԺҽѧ�����ơ�
2
���ҽ�ƴ�ѧ��ҽԺ���ΰ����ĺ��о���������зΰ�ת������������������ص�ʵ���ң����
3
����ҽѧԺ������ʡ�������������о��ص�ʵ���ң��ߺ���
4
����ҽѧԺ����ʵ���ң��ߺ���
5
����ҽѧԺҩѧԺ���ߺ���
6
����ҽѧԺ��ҽѧԺ���ߺ���
7
����ҽѧԺ����ѧԺ���ߺ���
8
����ҽѧԺ����ҽѧԺ���ߺ���
����
�����������ߵ���;��������ϸ����������1 / PD-1���壨PD-1 / PD-L1���������Ʒ����ٴ��ɹ�������ת�ư����Ƶ��¼�Ԫ�� PD-L1�����ڰ����ΰ����ڵ�����ʵ�������ϵ���������Ҫ�ڸ�����������������з������á�����������ṩ�˶Էΰ�ϸ��������̼���PD-L1�յ���֤�ݡ������յ���PD-L1�յ�����������GPR81�鵼�ġ��ΰ�ϸ����GPR81�ź�ת���ij�Ĭ����PD-L1����ˮƽ���ͺ�PD-L1�����ӻ��Թ���ʧ����⣬GPR81�鵼�������Ǵ̼��ķΰ�ϸ����PD-L1���ϵ�������������ǿ���ǽͽ⣬����������������øA��LDHA�������ǻ�֤����GPR81�ļ���ή��ϸ����cAMP��ˮƽ�������Ƶ���øA��PKA���Ļ��ԣ��Ӷ�����ת¼����������TAZ�ļ�� TAZ��ת¼����TEAD������ö���TAZ����PD-L1���յ������������Ҫ�����⣬���Ƿ��֣������յ�������ϸ����PD-L1�ļ���¸�����-�õIJ��������Լ��յ���������Jurkat Tϸ����Ѫ��ϸ�����������ǵķ��ֽ�ʾ�������ڴٽ�����ϸ������ϸ������Tϸ������������з����˳������ϵ����ã���������ϸ����л�ر��������Ӧ���ӱ�����֮�佨����ֱ����ϵ��Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28604752��
Eat 'em up: Next-generation therapeutic helps immune cells detect, destroy cancer
Supramolecule provides a double whammy to knock out cancer's 'eat-me-not' signaling, keep macrophages on the attack
Date:
July 2, 2018
Source:
Brigham and Women's Hospital
Summary:
1 Investigators have developed a therapeutic that delivers a double whammy to knock out both mechanisms.
FULL STORY
Macrophages -- immune cells that engulf and digest particles and pathogens -- provide a first line of defense against bacteria and viruses and can also help destroy cancer cells. Macrophages play a paradoxical role, with M1 macrophages rousing the immune system to action and M2 macrophages quelling inflammation. Researchers have found that cancer cells evade destruction by macrophages in two ways -- by converting cells to become docile, M2 macrophages, and by sending out an "eat me not" signal that tricks M1 macrophages into letting them be. Investigators from Brigham and Women's Hospital have developed a therapeutic that delivers a double whammy to knock out both mechanisms. In preclinical models, the new approach has yielded promising results. The team's findings are published today in Nature Biomedical Engineering.
"Clinicians are increasingly realizing that one drug or a one-size-fits-all approach is not enough when combatting cancer, and that a combination immunotherapy, such as blocking two distinct targets in the same immune cell, is the future of immuno-oncology. Our approach capitalizes on this concept," said co-corresponding author Ashish Kulkarni, PhD, a former instructor in the Division of Engineering in Medicine at BWH and assistant professor in the Department of Chemical Engineering at University of Massachusetts, Amherst.
Kulkarni and colleagues have previously reported on the design and engineering of supramolecules -- therapeutics that are built from component molecules that click together like building blocks. To reinvigorate macrophages, the team designed a supramolecule that could block the "don't eat me" signal that cancer cells can produce and simultaneously inhibit signaling that converts macrophages to M2 subtype.
The researchers tested the supramolecular therapeutic in animal models of aggressive forms of breast cancer and skin cancer, comparing their drug directly with a drug currently available in the clinic. Mice that were untreated formed large tumors by Day 10. Mice treated with currently available therapies showed decreased tumor growth. But mice treated with the new supramolecular therapy had complete inhibition of tumor growth. The team also reported an increase in survival and a significant reduction in metastatic nodes.
"We can actually see macrophages eating cancer cells," said co-corresponding author Shiladitya Sengupta, PhD, BWH associate bioengineer and assistant professor of medicine at Harvard Medical School, citing confocal microscopy images published in the paper that show macrophages (red) engulfing cancer cells (green).
The researchers plan to continue testing the new therapy in preclinical models to evaluate safety, efficacy and dosage. The supramolecular therapy they have designed has been licensed and they hope to move the therapeutic into clinical trials in the years ahead should preclinical testing continue to show promise.
Funding for this work was provided by a Department of Defense Breakthrough Award (BC132168), an American Lung Association Innovation Award (LCD-259932-N), an NCI UO1 (CA214411), a National Cancer Institute of the National Institutes of Health (P50CA168504) and Hearst Foundation/Brigham and Women's Hospital Young Investigator Award.
Eat 'em up: Next-generation therapeutic helps immune cells detect, destroy cancer: Supramolecule provides a double whammy to knock out cancer's 'eat-me-not' signaling, keep macrophages on the attack -- ScienceDaily
https://www.sciencedaily.com/releases/2018/07/180702111158.htm��
New study shows that iron oxide nanoparticles inhibit tumour growth
(Nanowerk Spotlight) The intravenous iron-replacement product ferumoxytol and other iron oxide nanoparticles are being used for treating iron deficiency, as contrast agents for magnetic resonance imaging, and as drug carriers.
In a new study, for the first time, researchers have shown an intrinsic therapeutic effect of ferumoxytol on the growth of early mammary cancers and lung cancer metastases in liver and lungs. They showed that ferumoxytol can activate the immune system to attack cancer cells.
"This is the first description of an intrinsic therapeutic effect of iron oxide nanoparticles against cancer," Saeid Zanganeh, PhD, a postdoctoral scholar in the Department of Radiology, Molecular Imaging Program at Stanford University, and first author of a paper on this work (Nature Nanotechnology, "Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues"), tells Nanowerk.
"Our data have broad implications for diagnostic and therapeutic nanoparticle applications," says Morteza Mahmoudi, an assistant professor at Tehran University of Medical Sciences, who heads the Laboratory of Nano-Bio Interactions, and co-author of the paper. "Since ferumoxytol is FDA-approved for intravenous treatment of iron deficiency, it could be applied 'off label' to protect the liver in patients from metastatic seeds and potentiate TAMs-modulating cancer immunotherapies."��
Combining ferumoxytol and macrophages leads to cancer cell apoptosis through the Fenton reaction
Combining ferumoxytol and macrophages leads to cancer cell apoptosis through the Fenton reaction. RAW264.7 macrophages were co-cultured with MMTV-PyMT cancer cells in a transwell system for 24 h, with or without ferumoxytol (2.73 mg ml�C1). Macrophages in the upper chamber and cancer cells in the lower chamber were separated by a 0.4-µm-sized microporous membrane, which allowed for exchange of molecules, but not cells. Cancer cells in the lower chamber were stained for caspase expression using green fluorescent protein (GFP)-conjugated antibodies against active caspase 3 (green). F-actin and the cell nuclei were counterstained with phalloidin rodamine (red) and DAPI (blue), respectively. Images show that co-culture of cancer cells, macrophages and ferumoxytol leads to increased caspase-3 expression of cancer cells. Co-incubations of cancer cells and macrophages only or cancer cells and ferumoxytol only do not lead to significant apoptosis induction. Scale bars, 20 µm. (© Nature Publishing Group) (click on image to enlarge)
Previous studies have described the use of iron oxide nanoparticles for targeted delivery of therapeutics (see for instance our previous Nanowerk Spotlight: "Smart cancer nanotheranostics"). In these studies, the nanoparticles served as carriers of therapeutic drugs to the tumor microenvironment.
Conversely, in this new study, the scientists observed an intrinsic therapeutic effect of iron oxide nanoparticles themselves.
"Our proposed approach to suppress liver metastases by repetitive systemic ferumoxytol administrations should be achievable without significant impact on iron load or liver toxicity," notes Zanganeh.
He adds that, due to the large surface area and chemically defined surface structure, iron oxide nanoparticles such as ferumoxytol can be conjugated or functionalized to numerous ancillary molecules to further amplify the desired immune-modulating properties.
The scientists explain that ferumoxytol could be locally delivered to unresectable tumors via interventional procedures, to micrometastases in confined spaces �C e.g. peritoneal seeds. Or to tumor resection margins at the end of a tumor surgery with positive resection margins, in order to induce a pro-inflammatory reaction against cancer cells and suppress tumor growth in the interval between a tumor surgery and initiation of postsurgical chemotherapy or irradiation.
On the other hand, patients with primary tumors that typically metastasize to the liver could receive protective ferumoxytol medications to prevent early metastatic seeds.
In the next stage of their study, the researchers will be more focused on the evaluations of nanoparticle-drug combinations. Potential clinical applications of ferumoxytol-mediated pro-inflammatory immune responses could entail potentiating the efficacy of other M1-activating cancer immunotherapies, such as anti-CD47 mAbs or antibodies blocking IL-4 or IL-13 signaling.New study shows that iron oxide nanoparticles inhibit tumour growth
https://www.nanowerk.com/spotlight/spotid=44691.php��
Control of Macrophage Dynamics as a Potential Therapeutic Approach for Clinical Disorders Involving Chronic Inflammation
Journal of Pharmacology and Experimental Therapeutics September 2015,University of Tokyo Hospital, Japan
Abstract
Macrophages are a well recognized player of both innate and adaptive immunity and have emerged as a key regulator of systemicmetabolism, hematopoiesis, vasculogenesis, apoptosis, malignancy, and reproduction. Such pleiotropic roles of macrophages are mirrored by their protean features. Upon environmental. challenges, macrophages redistribute and differentiate in situ and contribute to the multiple disease states by exerting protective and pathogenic effects. The environmental challenges include cytokines, chemokines, lipid mediators, and extrinsic insults, such as food and pathogenic bacteria. In addition, homeostasis and the activation state of macrophages are influenced by various metabolites from a commensal microbe that colonizes epithelial and mucosal surfaces, such as the lungs, intestines, and skin. In this review, we describe macrophage differentiation, polarization, and various functions in chronic disease states, including chronic inflammatory bowel disease, tumorigenesis, metabolism and obesity, and central nervous system demyelinating disorders. Controlling the macrophage dynamics to affect the pathologic states is considered to be an important therapeutic approach for many clinical disorders involving chronic inflammation.��
Fig. 1.
In the tumor microenvironment, M1 macrophages secrete antitumor mediators. Cytokines, such as IL-4, IL-10, and IL-13, educate macrophages to promote tumor progression. M2 macrophages and tumor-associated macrophages produce protumor factors. The phenotypes of these macrophages are reversibly interchangeable. MMP, matrix metalloproteinase.��
Fig. 2.
Schematic overview of macrophage polarity and insulin resistance. M1 and M2 macrophages are generated from monocytes in response to different stimuli. M1 and M2 macrophages have distinct secretary profiles that oppositely regulate inflammation and impact local/systemic insulin sensitivity.Involvement of Macrophages in Tumor Development
The tumor microenvironment contains various inflammatory cells and mediators (Mantovani et al., 2008; Hanahan and Weinberg, 2011; Coussens et al., 2013). These inflammatory cells play an essential role in tumor pathogenesis. Macrophages are in the tumor microenvironment and can show both protumor and antitumor phenotypes in the different settings. An array of evidence supports the concept of functional plasticity and heterogeneity of macrophages (Mosser and Edwards, 2008; Murray et al., 2014). Most mouse tissue-resident macrophages originate from the yolk sac progenitors, with some exceptions, such as the intestine (Wynn et al., 2013). These tissue-resident macrophages self-maintain independent of adult bone marrow (Wynn et al., 2013) and recruit additional macrophages from circulating bone marrow monocytes in response to pathogens or following injury. Tumor cells also recruit bone marrow monocytes by secreting chemokines, such as colony stimulating factor (CSF) 1 and MCP-1 (Noy and Pollard, 2014). Thus, both yolk sac�Cderived macrophages and bone marrow�Cderived macrophages reside in the tumor microenvironment. Both tissue resident macrophages and circulating bone marrow monocytes express high levels of the CSF1 receptor (CSF1R) and circulating bone marrow monocytes express high levels of CCR2. Thus, macrophages in the tumor microenvironment are highly heterogeneous. Intriguingly, these macrophages appear to behave differently. In a mouse glioma model, resident yolk sac�Cderived microglia and recruited bone marrow�Cderived macrophages are present in the tumor microenvironment and these cells show distinct responses to antimacrophage therapies (Pyonteck et al., 2013).
Inflammatory conditions link tumor initiation and progression. Chronic infection, such as Helicobacter pylori, in the stomach generates a pool of monocytes that could cause persistent inflammation, resulting in the initiation of tumorigenesis (Cordon-Cardo and Prives, 1999; Peek and Blaser, 2002; Karin et al., 2006; Ferreira et al., 2008; Krueger et al., 2013; Hatakeyama, 2014). Chronic IBD, such as Crohn��s disease and ulcerative colitis, increases the risk of colorectal cancer (Balkwill, 2004; Balkwill et al., 2005; Grivennikov et al., 2009; Grivennikov and Karin, 2010; Terzic et al., 2010; Foersch and Neurath, 2014). Macrophages are involved in the inflammatory responses that promote tumorigenesis by producing a proinflammatory cytokine, such as IL-6, TNF-��, and IFN-��, and by secreting growth factors that induce epithelial cell growth and promote cancer-associated mutations (Mantovani et al., 2008; Grivennikov et al., 2012; Noy and Pollard, 2014). In general, M1-type macrophages activated by IFN-�� or microbial products show tumoricidal potential (Fig. 1). IFN-�� primed macrophages enhance the capacity of the production of proinflammatory cytokines, such as IL-12 and IFN-��, and ROS. These mediators have the potential to kill tumor cells. These macrophages also promote Th1 responses by producing various mediators, such as IL-12, C-X-C motif ligand (CXCL) 9, and CXCL10 (Gordon, 2003; Martinez et al., 2006; Biswas and Mantovani, 2010). Given that Th1 responses are involved in antitumor responses (Tsung et al., 1997; Biswas and Mantovani, 2010), promotion of Th1 responses by M1 macrophages may provide a positive feedback loop in antitumor mechanisms.
Fig. 1.
Download figureOpen in new tabDownload powerpoint
Fig. 1.
In the tumor microenvironment, M1 macrophages secrete antitumor mediators. Cytokines, such as IL-4, IL-10, and IL-13, educate macrophages to promote tumor progression. M2 macrophages and tumor-associated macrophages produce protumor factors. The phenotypes of these macrophages are reversibly interchangeable. MMP, matrix metalloproteinase.
On the other hand, macrophages observed in the established metastatic tumors lose tumoricidal activity but promote tumor progression (Biswas and Mantovani, 2010; Qian and Pollard, 2010). M2 macrophages produce protumor factors, such as IL-8, vascular endothelial growth factor (VEGF), and matrix metalloproteinases. In addition, tumor-associated macrophages, regulatory macrophages and myeloid cells, and myeloid-derived suppressor cells suppress antitumor immunity by secreting IL-10, TGF-��, programmed death-ligand 1, and arginase 1. These populations also promote tumor growth (Fig. 1) (de Visser et al., 2005; Nardin and Abastado, 2008; Sierra et al., 2008; Andreu et al., 2010; Yang et al., 2011). Besides tumor growth, these protumor macrophages facilitate tumor survival, tumor cell invasion, metastasis, and angiogenesis and skew effective T-cell responses by secreting multiple factors, such as VEGF, Wnt, matrix metalloproteinases, cathepsins, and IL-10 (de Visser et al., 2005; Sinha et al., 2005; Nardin and Abastado, 2008; Sierra et al., 2008; DeNardo et al., 2009; Andreu et al., 2010; Gocheva et al., 2010; Qian and Pollard, 2010; Wong et al., 2010; Coussens et al., 2013; Yang et al., 2014). Tyrosine kinase with Ig-like loops and epidermal growth factor homology domain-2 (Tie2)�Cexpressing monocytes/macrophages increase the invasive properties of glioma cells by secreting high levels of geratinase enzymatic proteins (Gabrusiewicz et al., 2014). During the acquisition of malignancy, the tumor microenvironment appears to become a Th2-type immune suppressive environment. Malignant epithelial cells polarize macrophages to an M2-like phenotype (Hagemann et al., 2005, 2006). The cytokines IL-4, IL-10, and IL-13 produced by immune cells, such as Th2 cells and B cells, and/or tumor cells induce M2 polarization of macrophages. Furthermore, growth factors, such as CSF1 and granulocyte macrophage�CCSF, which are produced by tumor cells or helper T cells, promote polarization of macrophages in protumoral phenotypes (Lin et al., 2002; Su et al., 2014). Thus, macrophages are educated in the tumor microenvironment to have a protumor phenotype.
These protumor macrophages are re-educated to be antitumor macrophages (Stout et al., 2009). IFN-��, CpG-DNA, and IL-10 antibody switch macrophages from an M2-like protumor phenotype to an M1 phenotype (Guiducci et al., 2005; Duluc et al., 2009). By inhibition of nuclear factor�C��B and signal transducer and activator of transcription 3, protumor macrophages become cytotoxic to tumor cells and promote regression of tumors in vivo (Kortylewski et al., 2005; Hagemann et al., 2008). Along similar lines, Notch signaling induces antitumor activity in macrophages by promoting M1 polarization (Wang et al., 2010). Thus, the phenotypes of macrophages in the tumor environment are highly plastic (Fig. 1).
Macrophages have a key role in both promoting and preventing tumor progression. Because tumor-associated macrophages promote resistance to various cancer therapies, macrophages constitute an attractive cellular target for cancer therapy in combination with chemotherapy or other strategies. Since CSF1 is the major lineage regulator of macrophages, CSF1 signaling through its receptor CSF1R is a potential target for therapy. Tumor cells secrete a large amount of CSF1 (Lin et al., 2001), leading to recruitment of macrophages in the tumor regions. High expression levels of CSF1 have been shown to correlate with poor prognosis in various cancers (Groblewska et al., 2007; Mroczko et al., 2007; Zhu et al., 2008). CSF1-CSF1R inhibition is effective in reducing the number of macrophages in a mouse gastrointestinal stromal tumors model (Cohen et al., 2013) and enhances the antitumor effects of VEGF-targeted therapies (Priceman et al., 2010). Inhibition of CSF1-CSF1R also induces macrophages to be reprogrammed. These reprogrammed macrophages show an enhanced antigen presentation, thereby promoting antitumor T-cell responses in pancreatic cancer (Zhu et al., 2014). Androgen blocking therapy in combination with CSF1R inhibitors improves macrophage infiltration and enhances tumor disruption, thereby sustaining a more durable therapeutic response in a prostate cancer model (Escamilla et al., 2015). In addition, genetic deletion of CSF1 or inhibitions of CSF1-CSF1R inhibits tumor progression in various tumor settings. Inhibitory molecules of CSF1R are now in clinical trials for cancer therapy (Ries et al., 2014). CSF1 also increases expression of Tie2 in tumor-associated macrophages. Tie2-expressing macrophages are thought to be involved in angiogenesis. Tumor cells also produce chemokine CCL2. CCR2 is highly expressed in circulating monocyte and inflammatory macrophages (Qian et al., 2011). CCL2 neutralization inhibits metastasis by reducing recruitment of monocytes. The CCL2-CCR2 interface is likely to be an attractive therapeutic target as cessation of CCL2 inhibition accelerates breast cancer metastasis by releasing monocytes from the bone marrow, enhancing cancer cell mobilization and promoting angiogenesis (Bonapace et al., 2014).
Cancer cells secrete a large amount of small vesicles, known as exosomes, compared with normal cells (Th��ry et al., 2002). Exosomes contain many proteins, microRNA, mRNA, and lipids derived from the parental cells. Exosomes also express the parental cell�Cderived surface adhesion molecules and specific surface markers. For example, ovarian cancer cell�Cderived exosomes specifically express epithelial cell adhesion molecules (Peng et al., 2011; Liang et al., 2013). The exosomes derived from cancer cells have been reported to show distinct features from those derived from normal cells. The tumor cell�Csecreted exosomes affect the behavior of macrophages within the tumor microenvironment (Ludwig and Giebel, 2012). Both exosomes secreted by normal cells and cancerous cell lines targeted macrophages, whereas only cancer-derived exosomes induced nuclear factor�C��B activation in macrophages, resulting in the secretion of proinflammatory cytokines, such as IL-6, TNF��, and CCL2 through activation of TLR2 on macrophages (Chow et al., 2014). MicroRNAs within the cancer cell�Cderived exosomes trigger the TLR7/TLR8-mediated prometastatic inflammatory response (Valeri et al., 2014). In addition, cancer cell�Cderived exosomes activate distant macrophages. Because exosomes can be transferred to other cells located in both the peripheral region and distant area to modulate the immune system and facilitate tumor progression, exosomes can be a new potential therapeutic target for macrophage-targeted cancer therapy.
In vaccine strategies, tumor antigen-conjugated antibodies or dead tumor cells are treated to activate antitumor immune cells, such as natural killer T cells or cytotoxic T cells (Banchereau and Palucka, 2005). Tumor cell�Cassociated antigens activate antigen-specific CD8 T cells in lymph nodes. These activated CD8 T cells elicit an antitumor immune response. In the activation of tumor-directed adaptive immune responses, macrophages function as antigen-presenting cells. CD169+ macrophages reside at the subcapsular sinus in the lymph node and take up dead tumor cells or tumor cell corpses, thereby crosspresenting tumor antigens to CD8 T cells (Asano et al., 2011). In a human study, high levels of CD169+ macrophages are associated with a favorable clinical prognosis in colorectal carcinoma (Ohnishi et al., 2013). Thus, CD169+ sinus macrophages contribute to the generation and activation of tumor-specific T-cell immunity and seem to be an attractive antitumor approach for cancer therapy.
Control of Macrophage Dynamics as a Potential Therapeutic Approach for Clinical Disorders Involving Chronic Inflammation | Journal of Pharmacology and Experimental Therapeutics
http://jpet.aspetjournals.org/content/354/3/240��
Int J Biol Sci 2017; 13(6):723-734. doi:10.7150/ijbs.19642
Exosomes from Melatonin Treated Hepatocellularcarcinoma Cells Alter the Immunosupression Status through STAT3 Pathway in Macrophages
1. Department of Oncology, the First Affiliated Hospital of Anhui Medical University, Hefei 230022, Anhui, China;
2. Department of Pharmacy, the First Affiliated Hospital of Anhui Medical University, Hefei 230022, Anhui, China;
3. Institute of Clinical Pharmacology, Anhui Medical University, Hefei 230032, Anhui, China;
4. Institute for Liver Diseases of Anhui Medical University, Hefei 230032, Anhui, China.
Abstract
Immunosuppression is a significant factor in the progression of tumor invasion and metastasis. Melatonin, a well-known hormone, has certain cytotoxic and immune regulatory effects to inhibit tumor function. Exosomes are small membrane vesicles released by many kinds of cells, which contain different macromolecules, such as mRNAs and microRNAs (miRNAs), and proteins that can mediate communications between cells. Tumor-derived exosomes may cause immunosuppression, however, it is unknown whether melatonin can attenuate an immunosuppressive status by altering the function of tumor-derived exosomes. In the present study, we evaluated the effects of hepatocellularcarcinoma-derived exosomes (Exo-con) and exosomes derived from hepatocellularcarcinoma cells treated with 0.1 mM melatonin (Exo-MT), on the expression of inflammatory factors and programmed death ligand 1(PD-L1) by co-culturing Exo-con and Exo-MT, respectively, with macrophages differentiated from THP-1 cells or RAW264.7 cells. Our in vitro results indicate that Exo-MT can downregulate the expression of PD-L1 on macrophages while Exo-con can upregulate the expression of PD-L1 through flow cytometry and immunofluorescence analysis. In addition, Exo-con upregulates the secretion of cytokines, such as IL-6, IL-10, IL-1��, and TNF-�� in macrophages. Accordingly, Exo-MT could attenuate the high expression of these inflammatory cytokines. Furthermore, in vivo experiments confirmed the results found in vitro. PD-L1 expression and cytokine secretion were lower in the Exo-MT group compared with those in the Exo-con group. Working to identify a specific mechanism, our research shows that Exo-MT decreases STAT3 activation compared to the Exo-con group. In summary, we found exosomes from melatonin treated hepatocellularcarcinoma cells alters the immunosupression status through STAT3 pathway in macrophages. Our study may provide a new avenue to investigate the mechanisms of melatonin in regulating an immunosuppressive status.
��Keywords: Hepatoma cell, melatonin, exosomes, PD-L1, cytokines, macrophages.
Figure 8
Schematic diagram of different exosomes regulate the function of macrophages. Tumor delivered exosomes can increase the PD-L1 expression and cytokines secretion in macrophages to cause immunosupressive status through the STAT3 signal pathway, while exosomes derived from melatonin treated tumor cells could attenuate the immunosuppressive status.��
Exosomes from Melatonin Treated Hepatocellularcarcinoma Cells Alter the Immunosupression Status through STAT3 Pathway in Macrophages
https://www.ijbs.com/v13p0723.htm��
Signal Transducer and Activator of Transcription 3 in Liver Diseases: A Novel Therapeutic Target
Hua Wang1, Fouad Lafdil2,3, Xiaoni Kong1, Bin Gao1 Corresponding address
1. Laboratory of Liver Diseases, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892, USA;
2. Laboratory of Liver Pathophysiology, INSERM, U955, Cr��teil, F-94000 France;
3. Universit�� Paris-Est, Facult�� de M��decine, UMR-S955, Cr��teil, F-94000 France.
Go File import instruction
Abstract
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is activated by many cytokines and growth factors and plays a key role in cell survival, proliferation, and differentiation. STAT3 activation is detected virtually in all rodent models of liver injury and in human liver diseases. In this review, we highlight recent advances of STAT3 signaling in liver injury, steatosis, inflammation, regeneration, fibrosis, and hepatocarcinogenesis. The cytokines and small molecules that activate STAT3 in hepatocytes may have therapeutic benefits to treat acute liver injury, fatty liver disease, and alcoholic hepatitis, while blockage of STAT3 may have a therapeutic potential to prevent and treat liver cancer.
Keywords: STAT3, STAT3 signaling, liver injury, liver diseases
Figure 1
STAT3 signaling in hepatocytes. Hepatocytes express high levels of gp130, which is a common signal chain for IL-6 and IL-6 family cytokines, high levels of IL-6 receptors and various corresponding receptors for IL-6 family cytokines. IL-6 family cytokines include leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), oncostatin M (OSM), cardiotrophin-1 (CT-1), and IL-11. The ligation of these cytokines (IL-6 and IL-6 family cytokines) with their corresponding receptors leads to the dimerization of gp130, followed by dimerization of gp130-associated Janus kinases (JAKs) and phosphorylation of JAKs and gp130. This receptor-kinase complex then recruits and phosphorylates cytoplasmic protein STAT3. Phosphorylated STAT3 forms a dimer, translocates into the nuclei and subsequently induces transcription of many genes. Hepatocytes also express high levels of IL-22R1 and IL-10R2 for IL-22 signaling. IL-6, IL-6 family cytokines, and IL-22 predominantly activate STAT3, but also induce a weak activation of other STATs and MAP kinases. Human hepatocytes express high levels of IFNAR1 and functional IFNAR2c (while mouse and rat hepatocytes predominantly express inhibitory IFNAR2a and poorly respond to IFN-�� stimulation). IFN-��/�� predominately induce STAT1 activation in primary human hepatocytes but also induce strong STAT3 activation. Activated STAT3 induces transcription of many genes that play important roles in inducing acute phase responses, promoting hepatocyte survival and liver regeneration, and ameliorating fatty liver.��
Figure 2
STAT3 signaling in Kupffer cells. Kupffer cells express high levels of IL-10R1 and IL-10R2. The ligation of IL-10 with IL-10R1 and IL-10R2 leads to prolonged activation of STAT3, thereby inhibiting inflammatory responses. In contrast, the ligation of IL-6 with IL-6R and gp130, which are expressed at high levels on Kupffer cells, leads to transient activation of STAT3, followed by the induction of inflammatory responses. STAT3 activation induces expression of SOCS3, which in turns inhibits IL-6 activation of STAT3, but does not inhibit IL-10 signaling.
��
Figure 3
STAT3 signaling in hepatic stellate cells. Hepatic stellate cells express high levels of the long form of the leptin receptor (OBRL). The ligation of leptin with OBRL induces activation of STAT3, leading to stellate cell activation, proliferation, and survival. IL-6 can also stimulate stellate cell survival, proliferation, and activation via binding of IL-6R and gp130 on stellate cells.Signal Transducer and Activator of Transcription 3 in Liver Diseases: A Novel Therapeutic Target
https://www.ijbs.com/v07p0536.htm��
Tumor-Associated Macrophages, Inflammation and Pathogenesis of Hepatocellular Carcinoma
Journal of Molecular and Genetic Medicine 2014
Matthieu Lewis1,2 and Aksam J Merched1,2,3*
1INSERM Liver Research Group, U1053, Bordeaux, France
2Liver Research Group, Unit��s Mixtes de Recherche 1053, Bordeaux University, Bordeaux, France
3Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, USA
Figure 1: Monocyte recruitment, differentiation into macrophages and their functional involvement. The figure summarizes: (1) Secreted factors that recruit monocytes to tumor microenvironment, (2) Factors that increase M1 macrophage polarization, (3) Factors that increase M2 macrophage polarization and (4) specific functions of each type of macrophages.
Studies show that in and around the HCC microenvironment, TAMs are mainly polarized towards the M2 macrophage phenotype that helps promote tumor growth and survival [24]. Understanding which factors polarize the tumor-associated macrophages in one or the other direction is critical in order to better understand the molecular interactions that class these immune cells into the M1 cytotoxic or M2 pro-tumor sub-types.
��
Figure 2: Plasticity of macrophages in tumor microenvironment and pathogenesis of HCC. Hepatocellular carcinoma cells secrete high levels of IL-10 (open circles), which induces increased production and secretion of IL-4 and IL-13 (closed circles). These interleukins increases M2 pro-cancer macrophage polarization, and inhibit the production and secretion of IFN-�� and TNF (closed triangle), which tend towards an anti-tumor environment and M1 macrophage polarization.
These interleukins, as seen above, favor M2 macrophage polarization. The balance between activation of STAT3/STAT6 versus STAT1, which are controlled by IFN-�� and interleukin concentrations, finely regulate macrophage expression and polarization [38]. The over-expression of the three interleukins (IL-4/13 and IL-10) activates M2 polarization leading to Th2 immune response and inhibits the cytotoxic Th1 immune response. Particularly, IL-10 has an important role in M2 polarization; not only does it activate more M2 promoting cytokines (IL-4 and IL-13), but also it inhibits certain Th1 and cytotoxic promoting factors such as IFN�� and IFN-�� [39] and TNF-�� (Figure 2)[40]. Such events occur as a consequence of cancer progression, immuno-bypassing and result in poor prognostic values.
The phenotypes of M1 and M2 macrophages can be characterized by their gene expression and transcriptional activities. M1 macrophages activate efficient immune effector cells with cytotoxic activities through T Helper 1 immune response. They are capable of eradicating tumor and microorganism cells, presenting high amounts of antigen and produce high amounts of T-cell stimulatory cytokines [30]. M2 macrophages, on the other hand, have a contrasting phenotype. Their phenotype can be characterized by the poor capability to present antigens, therefore inhibiting T cell recognition along with immunity against the pathological HCC cells. Also, the alternatively activated profile can be identified by the low expression of differentiation-associated macrophage antigens, such as carboxypeptidase M and CD51, but also tumor necrosis factor (TNF) and IL-12 [24]. Macrophages carrying low expressions of these factors favor tumor progression and proliferation as TNF promotes cancer regression and IL-12 promotes a cytotoxic, pro-inflammatory T Helper 1 immune response. In addition, M2 cells overexpress certain pro-survival factors such as Arginase I [41]. High levels of the enzyme Arginase I act as an adaptor mechanism to avoid the buildup of toxic nitric oxide (NO) in the tumor environment. It does so by bypassing NO production and producing polyamine and citrulline instead [41]. In addition, M2 macrophages express high levels of IL-10 and TGF- �� in order to maintain the M2 polarization state [23]. Finally, M2 macrophages express chemokines CCL17, CCL22 and CCL24 to promote T Helper 2 immune response, favoring angiogenesis, tissue remodeling and tissue repair [32].Tumor-Associated Macrophages, Inflammation and Pathogenesis of Hepatocellular Carcinoma | OMICS International
https://www.omicsonline.org/open-access/tumorassociated-macrophages-inflammation-and-pathogenesis-of-hepatocellular-carcinoma-1747-0862-1000132.php?aid=33381��
Classical versus alternative macrophage activation: the Ying and the Yang in host defense against pulmonary fungal infections
��
Predominant macrophage activation during pulmonary fungal infections includes M1 and M2a phenotypes. M1 macrophages arise in the presence of interferon (IFN)-�� produced by T helper type 1 (Th1)-type T cells, natural killer cells and activated antigen-presenting cells (not shown). These macrophages are generally microbicidal via the production of reactive oxygen and nitrogen species and promote the generation of a Th1-type immune response. M2a macrophages arise in the presence of cytokines interleukin (IL)-4 and/or IL-13 and are generally not efficient killers of invading pathogens. Instead, M2a macrophages produce growth factors and components of the extracellular matrix and promote the generation/maintenance of Th2-type immune responses. Macrophages have a unique plasticity as polarized macrophages are capable of changing their functional phenotype as the cytokine microenvironment changes. Arg1, arginase 1; CXCL, C-X-C motif chemokine ligand; FIZZ-1, found in inflammatory zone 1; MR, mannose receptor; NOS2, nitric oxide synthase 2; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TNF, tumor necrosis factor.
Classical versus alternative macrophage activation: the Ying and the Yang in host defense against pulmonary fungal infections | Mucosal Immunology
https://www.nature.com/articles/mi201465��
[Frontiers in Bioscience 14, 4848-4861, June 1, 2009]
Transforming growth factor-beta signaling and tumor angiogenesis
Evangelia Pardali, Peter ten Dijke
Department of Molecular Cell Biology, Leiden University Medical Center, Leiden, The Netherlands
Note: lactic acid induces overexpression of TGF-beta in tumor microenvironment
Figure 2. Working model on the role of TGF-beta in tumor angiogenesis.Normal epithelial cells are sensitive to TGF-beta -mediated growth inhibition, however oncogenic initiation results in an increase in TGF-beta expression within the tumor micro-environment and subsequent loss of TGF-beta inhibitory growth control. The tumor-promoting effects of TGF-beta are mediated by regulation of tumor growth, angiogenesis and immune system. TGF-beta can induce angiogenesis directly by acting on the vasculature and indirectly by inducing expression of pro-angiogenic factors (VEGF, PDGF) and extracellular matrix components (MMPs). Finally, TGF-beta can induce epithelial to mesenchymal transition (EMT) of the tumour cells and production of MMPs, which contribute to extracellular matrix degradation, resulting in migration, invasion and tumor metastasis.[Frontiers in Bioscience 14, 4848-4861, June 1, 2009]
http://www.bioscience.org/2009/v14/af/3573/figures.htmLactate-Modulated Induction of THBS-1 Activates Transforming Growth Factor (TGF)-beta2 and Migration of Glioma Cells In Vitro
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3815307/��
Cancer-associated fibroblasts in hepatocellular carcinoma.
Citation: Kubo N, Araki K, Kuwano H, Shirabe K.
World J Gastroenterol 2016; 22(30): 6841-6850��
Figure 1 Microenvironment to be formed with cancer-associated fibroblasts and tumor-associated macrophages. Cancer-associated fibroblasts (CAFs) mainly located at the tumor marginal zone and secreted the various cytokines such as HGF and crosstalk with hepatocellular carcinoma cells and stimulate the tumor progression, invasion and metastasis through the epithelial mesenchymal transition. TIMP-1 suppressed the tumor cell apoptosis via SDF-1/PI3K/AKT signaling. Angiogenesis is occured by the angiogenic factors including VEGF, PDGF, ang-1 and ang-2 secreted by CAFs. TAMs and CAFs make the microenvironment to the immunosuppressive condition to create favorable microenvironment for tumor progression. TAM: Tumor associated macrophage.
Cancer-associated fibroblasts in hepatocellular carcinoma
https://www.wjgnet.com/1007-9327/full/v22/i30/WJG-22-6841-g001.htm��
Effect of cancer associated fibroblasts (CAF) on tumor and other stromal cells. CAFs can promote tumorigenesis directly through multiple mechanisms, including increased angiogenesis, proliferation, invasion, and inhibition of tumor cell death. These effects are mediated through the expression and secretion of numerous growth factors, cytokines, proteases, and extracellular matrix proteins.
Published: 20 March 2019
Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer
Rongsheng Zhang, Fan Qi, Fei Zhao, Geng Li, Shengli Shao, Xiaochao Zhang, Lifei Yuan & Yongdong Feng
Cancer Research Institute, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 430030, China
Abstract
Cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs) are important components of the tumor microenvironment, which have been reported to localize in colorectal carcinomas where they promote tumor progression. One of the crucial effects they exerted is immune-suppression, which was reported recently, however, the overall mechanism has not been fully addressed. In this study, it was shown that TAMs were enriched in colorectal cancer, and their infiltration was associated with VCAM-1 expression. Human colorectal cancer-derived CAFs can promote the adhesion of monocytes by up-regulating VCAM-1 expression in colorectal cancer cells. Furthermore, CAFs can attract monocytes by secreting IL-8 rather than SDF-1 and subsequently promote M2 polarization of macrophages, which synergize with CAFs in suppressing the functioning of natural killer (NK) cells. It was also found that CAFs promoted M2 macrophages recruitment in tumor tissue in vivo, and after VCAM-1 knocking-down in tumor cells or depletion of macrophages, the pro-tumor effect of CAFs was partly abolished, but no change was observed in NK cells infiltration. Collectively, the findings in this work show that TAMs and CAFs function synergistically in the tumor microenvironment and have the capacity to regulate NK cells in colorectal cancer and this presents a novel mechanism.Normal stroma consists of various connective tissues that act like a supportive framework for tissues and organs. Among all the stromal components, fibroblasts are essential to synthesize and deposit the extracellular matrix (ECM) by producing a variety of collagens and fibronectin (2). In addition, they are indispensable for the formation of the basement membrane which separates the epithelium from the stroma by secreting laminin and type IV collagen (3). They are also an ample source of various soluble paracrine and autocrine growth factors that regulate the growth of the surrounding cells as well as themselves (4). Interestingly, fibroblasts not only maintain the integrity of the ECM and basement membrane, in some cases, they also contribute to the ECM remolding by secreting proteases such as matrix metalloproteinase(MMPs) which effectively degrade ECM (5). Another important role of fibroblasts is wound healing.
Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer | Cell Death & Disease
https://www.nature.com/articles/s41419-019-1435-2��
Cancer-associated fibroblasts in tumor microenvironment �C Accomplices in tumor malignancy
Cellular Immunology
Volume 343, September 2019, 103729
Highlights
•
CAF is the major stromal cell type in many tumor microenvironments.
•
CAFs enact field cancerization by promoting extratumoral oxidative stress.
•
CAFs interact with tumor-infiltrating immune cells to promote cancer progression.
•
CAFs can influence the directionality and migration of cancer cells.
•
Targeting different subpopulations of CAFs led to unexpected cancer outcome.
Abstract
There is much cellular heterogeneity in the tumor microenvironment. The tumor epithelia and stromal cells co-evolve, and this reciprocal relationship dictates almost every step of cancer development and progression. Despite this, many anticancer therapies are designed around druggable features of tumor epithelia, ignoring the supportive role of stromal cells. Cancer-associated fibroblasts (CAFs) are the dominant cell type within the reactive stroma of many tumor types. Numerous previous studies have highlighted a pro-tumorigenic role for CAFs via secretion of various growth factors, cytokines, chemokines, and the degradation of extracellular matrix. Recent works showed that CAFs secrete H2O2 to effect stromal-mediated field cancerization, transform primary epithelial cells, and aggravate cancer cell aggressiveness, in addition to inflammatory and mitogenic factors. Molecular characterization of CAFs also underscores the importance of Notch and specific nuclear receptor signaling in the activation of CAFs. This review consolidates recent findings of CAFs and highlights areas for future investigations.
Fig. 1. CAFs and its activating factors. Upon activation by appropriate signals or mediators, such as TGF-�� and NRs, NFs at the primary site are activated to CAFs (p-CAFs) and can contribute to chemoresistance, metastasis, and invasion. Circulating CAFs (c-CAFs) are detected in the vasculature. However, their origin remains unclear and it is still unknown if c-CAFs extravasate at the secondary tumor site. CAFs associated with secondary tumors are also known as m-CAFs. Similarly, their origin remains to be determined, i.e. they arise from fibroblasts at the secondary tumor site or have migrated from the primary tumor site.Cancer-associated fibroblasts in tumor microenvironment �C Accomplices in tumor malignancy - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0008874917302228��
Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-�� and TGF-��
Christina H. Stuelten, Stacey DaCosta Byfield, Praveen R. Arany, Tatiana S. Karpova, William G. Stetler-Stevenson, Anita B. Roberts
Journal of Cell Science 2005 118: 2143-2153; doi: 10.1242/jcs.02334
��We used 2D-cocultures employing fibroblasts of different genetic backgrounds and MCF10A-derived human breast epithelial cells of increasingly malignant potential to investigate tumor-stroma interactions in breast cancer and to identify possible signaling pathways involved. Tumor cells induced expression of matrix-metalloproteinase 9 (MMP-9) in fibroblasts in a pattern dependent on the degree of their malignancy. In-situ zymography localized the main gelatinolytic activity around stromal cells in cocultures and xenografted tumors. Use of Smad3 knockout fibroblasts, small molecule inhibitors, and neutralizing antibodies showed that MMP-9 expression was induced by tumor cell-derived TNF-�� and TGF-��, dependent on Smad-, Ras-, and PI3-kinase-signaling, and likewise modulated by subsequent HGF- and EGF-signaling. Together, our results indicate that MMP-9 levels in tumor fibroblasts are regulated by a complex tumor-stroma cross-talk, involving multiple ligands and cellular signaling pathways.
Fig. 5.
Model of MMP-9 induction in fibroblasts by breast-cancer cells. Tumor-cell-derived TGF-�� and TNF-�� directly induce expression of MMP-9, which is augmented indirectly by the effects of these cytokines on HGF- and EGF-signaling.Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-�� and TGF-�� | Journal of Cell Science
https://jcs.biologists.org/content/118/10/2143.figures-only��
Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth
��
We have previously demonstrated that enhanced aerobic glycolysis and/or autophagy in the tumor stroma supports epithelial cancer cell growth and aggressive behavior, via the secretion of high-energy metabolites. These nutrients include lactate and ketones, as well as chemical building blocks, such as aminoacids (glutamine) and nucleotides. Lactate and ketones serve as fuel for cancer cell oxidative metabolism, and building blocks sustain the anabolic needs of rapidly proliferating cancer cells. We have termed these novel concepts the "Reverse Warburg Effect", and the "Autophagic Tumor Stroma Model of Cancer Metabolism".We have also identified a loss of stromal caveolin-1 (Cav-1) as a marker of stromal glycolysis and autophagy. The aim of the current study was to provide genetic evidence that enhanced glycolysis in stromal cells favors tumorigenesis. To this end, normal human fibroblasts were genetically-engineered to express the two isoforms of pyruvate kinase M (PKM1 and PKM2), a key enzyme in the glycolytic pathway. In a xenograft model, fibroblasts expressing PKM1 or PKM2 greatly promoted the growth of co-injected MDA-MB-231 breast cancer cells, without an increase in tumor angiogenesis. Interestingly, PKM1 and PKM2 promoted tumorigenesis by different mechanism(s). Expression of PKM1 enhanced the glycolytic power of stromal cells, with increased output of lactate.stromal cells, with increased output of lactate. Analysis of tumor xenografts demonstrated that PKM1 fibroblasts greatly induced tumor inflammation, as judged by CD45 staining. In contrast, PKM2 did not lead to lactate accumulation, but triggered a "pseudo-starvation" response in stromal cells, with induction of an NF-��B-dependent autophagic program, and increased output of the ketone body 3-hydroxy-buryrate. Strikingly, in situ evaluation of Complex IV activity in the tumor xenografts demonstrated that stromal PKM2 expression drives mitochondrial respiration specifically in tumor cells. Finally, immuno-histochemistry analysis of human breast cancer samples lacking stromal Cav-1 revealed PKM1 and PKM2 expression in the tumor stroma.
��
Pyruvate kinase expression (PKM1 and PKM2) in cancer associated fibroblasts drives tumor growth. Cancer cells secrete hydrogen peroxide (H2O2) which initiates oxidative stress in adjacent stromal fibroblasts. This, in turn, drives the activation of two main transcription factors, namely HIF1 and NFkB. NFkB is the master regulator of the innate immune response, resulting in the secretion of inflammatory cytokines. HIF1-a stabilization induces autophagy, mitophagy, and aerobic glycolysis. As a consequence, PKM1 and PKM2 expression results in the production of excess L-lactate and ketone bodies (3-hydroxy-butyrate) that are extruded via a mono-carboxylate transporter, known as MCT4. These high-energy nutrients are then taken up and recycled by cancer cells using another mono-carboxylate transporter, namely MCT1. Thus, L-lactate and ketones produced by stromal fibroblasts are transferred to cancer cells and used as fuel for mitochondrial oxidative metabolism (OXPHOS), producing large amounts of ATP. This model explains the anti-cancer activity of Metformin, a diabetic drug, that mechanistically functions as a mitochondrial "poison," as it is a specific inhibitor of mitochondrial complex I. Complex I is a key part of the electron transport system for generating ATP. Note also that a loss of stromal Caveolin-1 (Cav-1) occurs, as it is degraded by stromal autophagy. Loss of stromal Cav-1 further perpetuates oxidative stress, and is a powerful biomarker for predicting early tumor recurrence, metastasis, and drug-resistance, as well as poor clinical outcome in breast cancer patients.
��
F15: Figure 15. Two-compartment tumor metabolism is fueled by the autophagy-senescence transition (AST). In this model, cancer cells secrete hydrogen peroxide (H2O2), which induces oxidative stress in neighboring normal fibroblasts. Oxidative stress in stromal fibroblasts is then sufficient to confer the cancer-associated fibroblast phenotype, resulting in autophagy, mitophagy, and a shift toward aerobic glycolysis. Autophagy also drives the onset of senescence, via the autophagy-senescence transition. Autophagic-senescent fibroblasts then produce high-energy nutrients (L-lactate, ketone bodies, glutamine, and free fatty acids), which ��fuel�� mitochondrial metabolism (OXPHOS and ��-OX; oxidative phosphorylation and ��-oxidation) in adjacent cancer cells, resulting in the onset of anabolic tumor growth. This simple model could explain why chronological aging is one of the most significant risk factors for the development of cancer. AST, autophagy-senescence transition.��
Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production.
Senescent fibroblasts are known to promote tumor growth. However, the exact mechanism remains largely unknown. An important clue comes from recent studies linking autophagy with the onset of senescence. Thus, autophagy and senescence may be part of the same physiological process, known as the autophagy-senescence transition (AST). To test this hypothesis, human fibroblasts immortalized with telomerase (hTERT-BJ1) were stably transfected with autophagy genes (BNIP3, CTSB or ATG16L1). Their overexpression was sufficient to induce a constitutive autophagic phenotype, with features of mitophagy, mitochondrial dysfunction and a shift toward aerobic glycolysis, resulting in L-lactate and ketone body production. Autophagic fibroblasts also showed features of senescence, with increased p21(WAF1/CIP1), a CDK inhibitor, cellular hypertrophy and increased ��-galactosidase activity. Thus, we genetically validated the existence of the autophagy-senescence transition. Importantly, autophagic-senescent fibroblasts promoted tumor growth and metastasis, when co-injected with human breast cancer cells, independently of angiogenesis. Autophagic-senescent fibroblasts stimulated mitochondrial metabolism in adjacent cancer cells, when the two cell types were co-cultured, as visualized by MitoTracker staining. In particular, autophagic ATG16L1 fibroblasts, which produced large amounts of ketone bodies (3-hydroxy-butyrate), had the strongest effects and promoted metastasis by up to 11-fold. Conversely, expression of ATG16L1 in epithelial cancer cells inhibited tumor growth, indicating that the effects of autophagy are compartment-specific. Thus, autophagic-senescent fibroblasts metabolically promote tumor growth and metastasis, by paracrine production of high-energy mitochondrial fuels. Our current studies provide genetic support for the importance of "two-compartment tumor metabolism" in driving tumor growth and metastasis via a simple energy transfer mechanism. Finally, ��-galactosidase, a known lysosomal enzyme and biomarker of senescence, was localized to the tumor stroma in human breast cancer tissues, providing in vivo support for our hypothesis. Bioinformatic analysis of genome-wide transcriptional profiles from tumor stroma, isolated from human breast cancers, also validated the onset of an autophagy-senescence transition. Taken together, these studies establish a new functional link between host aging, autophagy, the tumor microenvironment and cancer metabolism.
��
F15: Figure 15. Two-compartment tumor metabolism is fueled by the autophagy-senescence transition (AST). In this model, cancer cells secrete hydrogen peroxide (H2O2), which induces oxidative stress in neighboring normal fibroblasts. Oxidative stress in stromal fibroblasts is then sufficient to confer the cancer-associated fibroblast phenotype, resulting in autophagy, mitophagy, and a shift toward aerobic glycolysis. Autophagy also drives the onset of senescence, via the autophagy-senescence transition. Autophagic-senescent fibroblasts then produce high-energy nutrients (L-lactate, ketone bodies, glutamine, and free fatty acids), which ��fuel�� mitochondrial metabolism (OXPHOS and ��-OX; oxidative phosphorylation and ��-oxidation) in adjacent cancer cells, resulting in the onset of anabolic tumor growth. This simple model could explain why chronological aging is one of the most significant risk factors for the development of cancer. AST, autophagy-senescence transition.Figure 15. Two-compartment tumor metabolism is fueled b | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3383590_cc-11-2285-g15&req=4��
Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1‑phase arrest
Authors: Woo Hyun Park
Affiliations: Department of Physiology, Medical School, Research Institute for Endocrine Sciences, Chonbuk National University, Jeonju, Jeollabuk 54907, Republic of Korea
Abstract
Hydrogen peroxide (H2O2) is frequently applied to cultured cells to induce oxidative stress. The present study investigated the molecular and cellular effects of exogenous H2O2 on Calu‑6 and A549 lung cancer cells. Based on MTT assays, H2O2 inhibited the growth of Calu‑6 and A549 cells with IC50 values of ~50 and 100 µM at 24 h, respectively. Cells treated with H2O2 demonstrated a considerable G1‑phase arrest of the cell cycle. H2O2 dose‑dependently augmented the numbers of dead (trypan blue‑positive) and Annexin V‑FITC‑stained cells in these cells, which was accompanied by the reduction of Bcl‑2 and pro‑caspase‑3 levels, as well as the upregulation of caspase‑3 and ‑8 activities. In addition, H2O2 triggered the failure of mitochondrial membrane potential (MMP; ����m). However, relatively higher doses of H2O2 did not raise the percentages of sub‑G1 cells in these cell lines. All the tested caspase inhibitors (Z‑VAD for pan‑caspases, Z‑DEVD for caspase‑3, Z‑IETD for caspase‑8 and Z‑LEHD for caspase‑9) decreased the percentages of sub‑G1 and Annexin V‑FITC‑stained cells in the H2O2‑treated Calu‑6 and A549 cells. However, caspase inhibitors did not significantly prevent the loss of MMP (����m) in H2O2‑treated lung cancer cells. In conclusion, H2O2 inhibited the growth of Calu‑6 and A549 lung cancer cells through cell death and G1‑phase arrest. H2O2‑induced cell death resulted from necrosis, as well as caspase‑dependent apoptosis.
Figure 7. - Schematic diagram representing the proposed cellular effects of H2O2 in lung cancer cells. H2O2, hydrogen peroxide; MMP (����m), mitochondrial membrane potential.
Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1‑phase arrest
https://www.spandidos-publications.com/or/40/3/1787��
Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development
a
Department of Medical Oncology Thomas Jefferson University, Philadelphia, PA
b
Drexel University, Philadelphia, PA
c
Department of Cell Biology, Anatomy and Pathology, Thomas Jefferson University, Philadelphia, PA
d
Department of Otolaryngology, Thomas Jefferson University, Philadelphia, PA
abstract
Glucose is a key metabolite used by cancer cells to generate ATP, maintain redox state and create biomass. Glucose can be catabolized to lactate in the cytoplasm, which is termed glycolysis, or alternatively can be catabolized to carbon dioxide and water in the mitochondria via oxidative phosphorylation. Metabolic heterogeneity exists in a subset of human tumors, with some cells maintaining a glycolytic phenotype while others predominantly utilize oxidative phosphorylation. Cells within tumors interact metabolically with transfer of catabolites from supporting stromal cells to adjacent cancer cells. The Reverse Warburg Effect describes when glycolysis in the cancer-associated stroma metabolically supports adjacent cancer cells. This catabolite transfer, which induces stromal-cancer metabolic coupling, allows cancer cells to generate ATP, increase proliferation, and reduce cell death. Catabolites implicated in metabolic coupling include the monocarboxylates lactate, pyruvate, and ketone bodies. Monocarboxylate transporters (MCT) are critically necessary for release and uptake of these catabolites. MCT4 is involved in the release of monocarboxylates from cells, is regulated by catabolic transcription factors such as hypoxia inducible factor 1 alpha (HIF1A) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-��B), and is highly expressed in cancer-associated fibroblasts. Conversely, MCT1 is predominantly involved in the uptake of these catabolites and is highly expressed in a subgroup of cancer cells. MYC and TIGAR, which are genes involved in cellular proliferation and anabolism, are inducers of MCT1. Profiling human tumors on the basis of an altered redox balance and intra-tumoral metabolic interactions may have important biomarker and therapeutic implications. Alterations in the redox state and mitochondrial function of cells can induce metabolic coupling. Hence, there is interest in redox and metabolic modulators as anticancer agents. Also, markers of metabolic coupling have been associated with poor outcomes in numerous human malignancies and may be useful prognostic and predictive biomarkers.Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0093775417301331��
Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment.
Cancer cells show a broad spectrum of bioenergetic states, with some cells using aerobic glycolysis while others rely on oxidative phosphorylation as their main source of energy. In addition, there is mounting evidence that metabolic coupling occurs in aggressive tumors, between epithelial cancer cells and the stromal compartment, and between well-oxygenated and hypoxic compartments. We recently showed that oxidative stress in the tumor stroma, due to aerobic glycolysis and mitochondrial dysfunction, is important for cancer cell mutagenesis and tumor progression. More specifically , increased autophagy/mitophagy in the tumor stroma drives a form of parasitic epithelial-stromal metabolic coupling. These findings explain why it is effective to treat tumors with either inducers or inhibitors of autophagy, as both would disrupt this energetic coupling. We also discuss evidence that glutamine addiction in cancer cells produces ammonia via oxidative mitochondrial metabolism. Ammonia production in cancer cells, in turn, could then help maintain autophagy in the tumor stromal compartment. In this vicious cycle, the initial glutamine provided to cancer cells would be produced by autophagy in the tumor stroma. Thus, we believe that parasitic epithelial-stromal metabolic coupling has important implications for cancer diagnosis and therapy, for example, in designing novel metabolic imaging techniques and establishing new targeted therapies. In direct support of this notion, we identified a loss of stromal caveolin-1 as a marker of oxidative stress, hypoxia, and autophagy in the tumor microenvironment, explaining its powerful predictive value. Loss of stromal caveolin-1 in breast cancers is associated with early tumor recurrence, metastasis, and drug resistance, leading to poor clinical outcome.The reverse Warburg effect.
F2: The reverse Warburg effect. (a) Via oxidative stress, cancer cells activate two major transcription factors in adjacent stromal fibroblasts (hypoxia-inducible factor (HIF)1�� and NF��B). This leads to the onset of both autophagy and mitophagy, as well as aerobic glycolysis, which then produces recycled nutrients (such as lactate, ketones, and glutamine). These high-energy chemical building blocks can then be transferred and used as fuel in the tricarboxylic acid cycle (TCA) in adjacent cancer cells. The outcome is high ATP production in cancer cells, and protection against cell death. ROS, reactive oxygen species. (b) Homotypic cultures (upper panels) of MCF7 cells (right) and hTERT-fibroblasts (left) were immunostained with a mitochondrial membrane antibody (red). Note that mitochondrial mass is lower in mono-cultures of MCF7 cells compared to fibroblasts. However, co-culture of MCF7 cells with fibroblasts (lower panel) induces a dramatic increase in mitochondrial mass in the 'central MCF7 cell colony', outlined by the dotted white oval. In contrast, mitochondrial mass is decreased in co-cultured fibroblasts. Panel (b) was modified and reproduced with permission from [41,78].The reverse Warburg effect. (a) Via oxidative stress, c | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3236330_bcr2892-2&req=4��
LACTATE METABOLISM IN TUMORS
Lactate metabolism in tumors: a simplified cartoon showing lactate being shuttled to and from cancer cells and its potential role as a signaling molecule in driving angiogenesis. Increasing pyruvate inhibits formation of 2-oxoglutarate, with the net effect of less degradation of HIF-1�� in the proteasome and increased VEGF and angiogenesis. Note that pyruvate can be increased by hypoxia ��backing up�� the TCA cycle, or by importation of lactate via MCT1s. Some lactate shuttling likely is present between cancer cells and (1) other cancer cells within the tumor, (2) tumor stromal cells, and/or (3) non-tumor cells both local and distant from the tumor. Note that the traditional ��Warburg Effect�� describes tumors relying heavily on glucose uptake (via GLUTs) with subsequent lactate exportation (via MCT4s) in normoxia. A ��Reverse Warburg Effect�� describes lactate production from stromal cells, which is then taken up and used by local cancer cells. Some unique combination of these pathways is likely present within each tumor, highlighting the need for further in vivo experimentation. (Note that MCTs can transport lactate either direction; MCT1s are typically expressed in cells importing lactate, while MCT4s are expressed in cells exporting lactate.)
��
3. The heme-HO system in macrophages
cited from The macrophage heme-heme oxygenase-1 system and its role in inflammation - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0006295218300674
3.1. Role of HO-1 in macrophages for heme detoxification and iron recycling
Initial evidence for a functional role of HO-1 in iron recycling has come from a HO-1 knockout mouse model, which exhibited signs of anemia and iron overload [33]. Subsequently, similar findings have also been reported for a patient with genetic HO-1 deficiency [34]. Remarkably, HO has been shown to exhibit constitutively high levels of activity and gene expression in liver [35], [36], [37] and spleen [29], [38] tissue macrophages under physiological conditions. More detailed studies in genetically HO-1 deficient mice indicated that lack of HO-1 renders macrophages sensitive to cell death following erythrocyte exposure in vitro, which may explain the loss of tissue macrophages in liver and spleen of these animals [39]. Consistent with this notion, bone marrow transplantation led to a repopulation of these tissues with wild-type macrophages, particularly in the liver and rescued iron reutilization defects [40]. Furthermore, heme-induced HO-1 activity in monocytes has also been shown to regulate the iron exporter ferroportin, which plays a pivotal role for iron recycling [41]. Hence, HO-1 activity in tissue-specific macrophages appears to play a fundamental role in iron recycling and erythropoiesis.
In pathophysiological conditions, high levels of free heme released from cell-free Hb, myoglobin or other intracellular hemoproteins, can have detrimental effects. The central iron atom of the heme molecule can cause excess ROS production via Fenton chemistry. Moreover, heme has potential cytotoxic effects via lipid peroxidation, protein cross-linking and DNA damage [14], [42]. In particular, the endothelial layer of blood vessels is highly sensitive to pro-inflammatory activation [18], [43] and toxicity [44] by heme. Mammalians possess specific protective mechanisms to counteract the toxicity of Hb and heme via the serum proteins haptoglobin (Hp) and hemopexin (Hx) [7], [45], [46], [47]. Hp binds cell-free Hb, which is released from hemolytic erythrocytes and after exhaustion of Hp binding capacity, excess free heme is scavenged by Hx [48]. Interestingly, plasma levels of Hp and Hx have been shown to inversely correlate with mortality in sepsis patients [49], [50].
HO-1 in macrophages appears to play an important role for the clearance of Hb-Hp and heme-Hx complexes [51]. Hb-Hp complexes are specifically taken up by monocytes [52] and macrophages via a CD163-mediated receptor endocytosis-dependent mechanism [53], [54], [55] (depicted in Fig. 2). By contrast, heme-Hx complexes are taken up via LRP1/CD91, the expression of which is not restricted to macrophages [56]. In turn, sequestered heme is enzymatically degraded via HO-1 in macrophages into the protective molecules CO and biliverdin/bilirubin [51]. Moreover, Hb-Hp complex-dependent up-regulation of HO-1 activity is linked to an increased expression of ferritin in macrophages [53] to protect against iron-induced toxicity. A previous report, which has implicated that increased expression of Hp and Hx in liver and kidney [39] was not sufficient to protect HO-1 knockout mice against heme-induced toxicity, might also be in line with this notion. In summary, HO-1 in macrophages may act as a heme-degrading funnel, which is utilized by the serum proteins Hp and Hx to remove free Hb and heme from the circulation, whilst mediating beneficial effects via heme degradation products.
Fig. 1. HO-1 modulated anti-inflammatory macrophages as therapeutic approach. Macrophages obtained from the indicated sources can be treated ex vivo with the various pharmacological inducers of HO-1 or can be genetically engineered to overexpress HO-1 leading to polarization into anti-inflammatory macrophages. These cells can restore homeostasis by virtue of their regenerative capacity and IL-10 secretion.
The macrophage heme-heme oxygenase-1 system and its role in inflammation - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0006295218300674��
Degradation of Heme
��
Degradation begins inside macrophages of the spleen, which remove old and damaged (senescent) erythrocytes from the circulation.
RBCs are engulfed by cells of the reticuloendothelial system. The globin is recycled into amino acids, which in turn are catabolized into intermediates of the citric acid cycle and fatty acid oxidation.
Heme is oxidized; the heme ring is opened by heme oxygenase. The oxidation occurs on a specific carbon, producing the linear tetrapyrrole biliverdin, ferric iron (Fe 3+), and CO.
In the next reaction, second bridging methylene is reduced by biliverdin reductase, producing bilirubin. The green pigment is thus converted to the red-orange bilirubin.
Bilirubin is then transported in the serum by albumin to the liver, where it is conjugated with glucuronate by bilirubin glucuronyl transferase and excreted in the bile.
In the intestine, bilirubin is deconjugated and converted to urobilinogen and stercobilin.
Some urobilinogen is reabsorbed and excreted as urobilin in the urine. Most urobilinogen is oxidized in the feces to stercobilin which gives feces its color.Heme Degradation | Biochemistry | Microbe Notes
https://microbenotes.com/heme-degradation/��
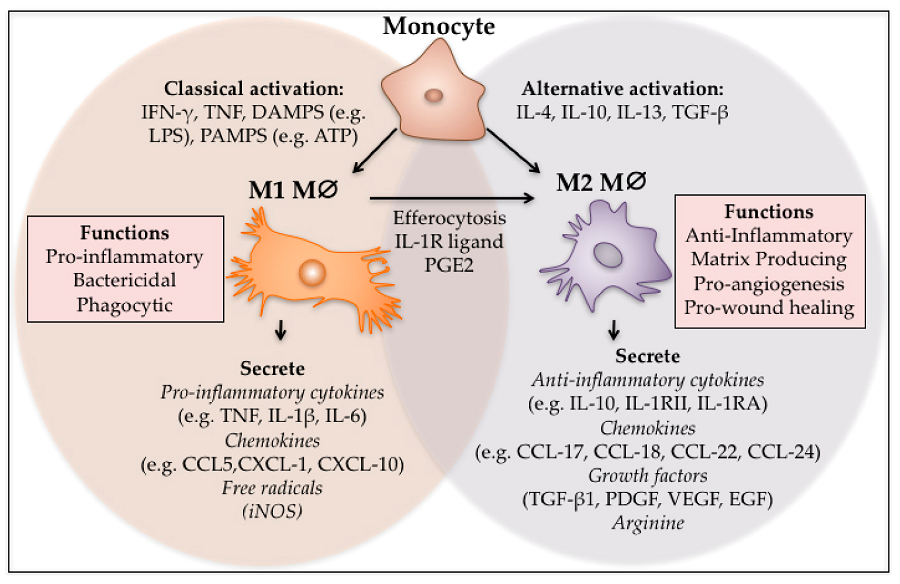
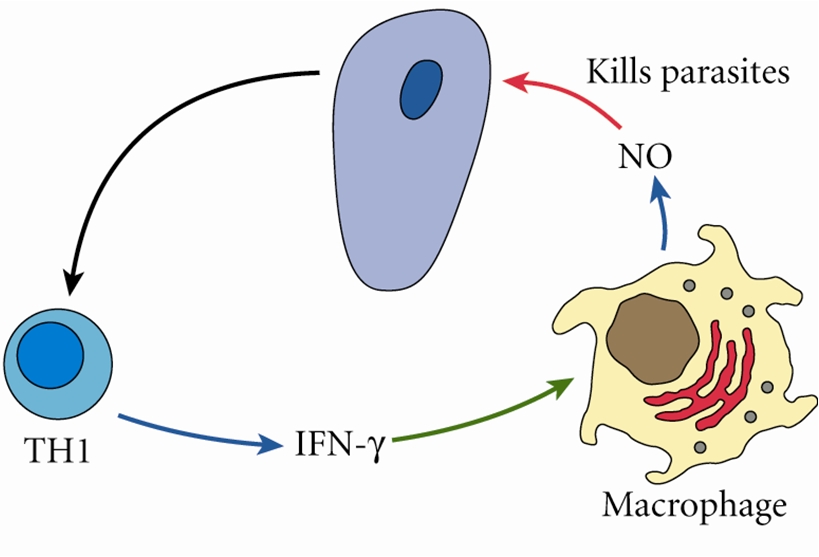
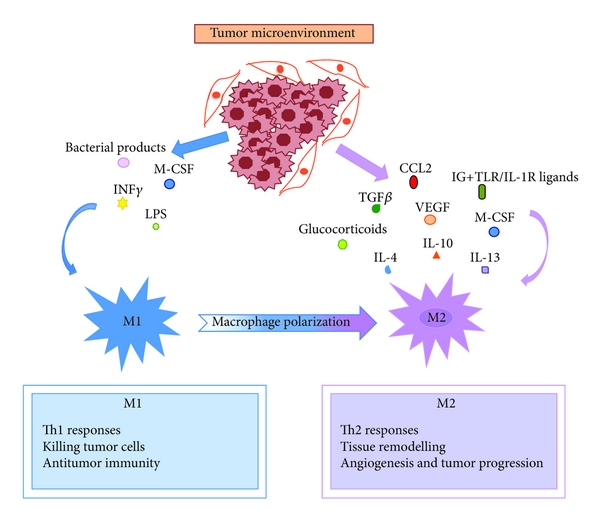
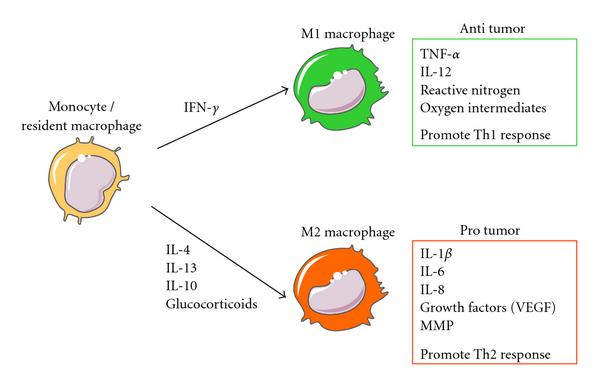

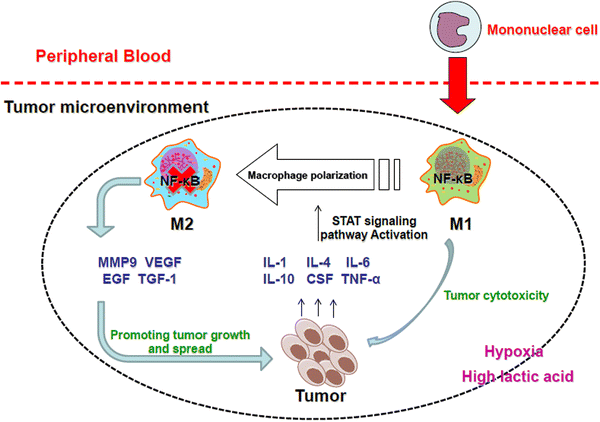
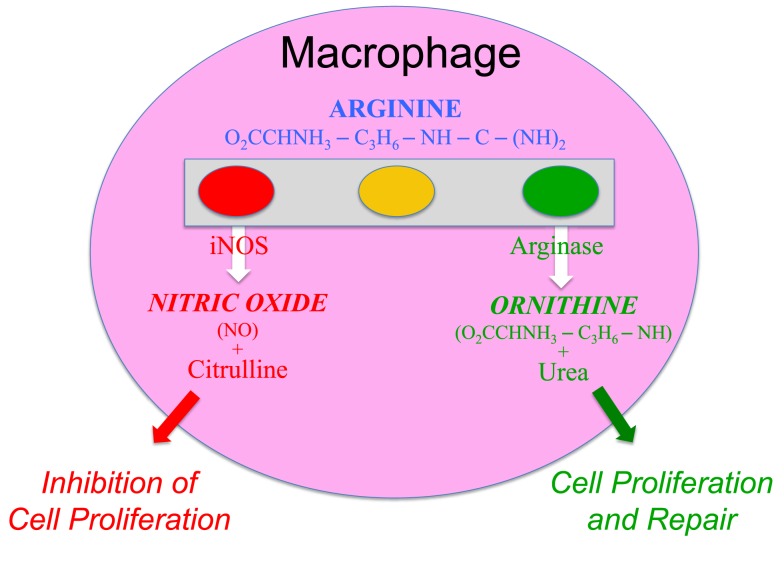

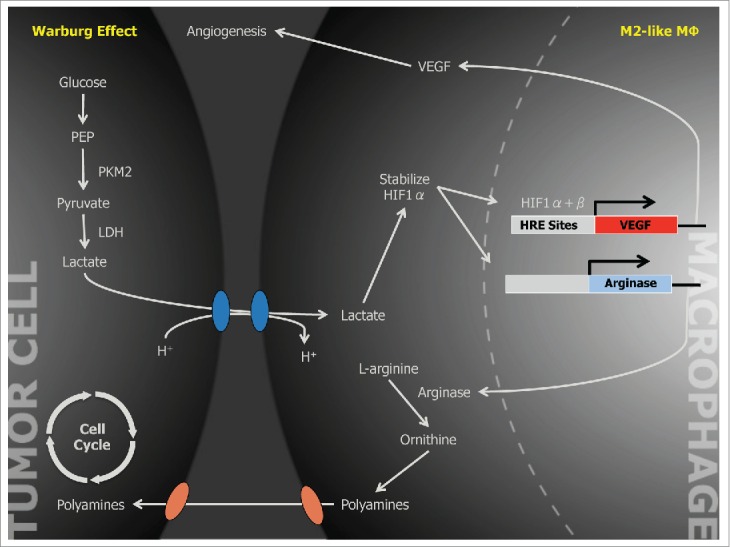
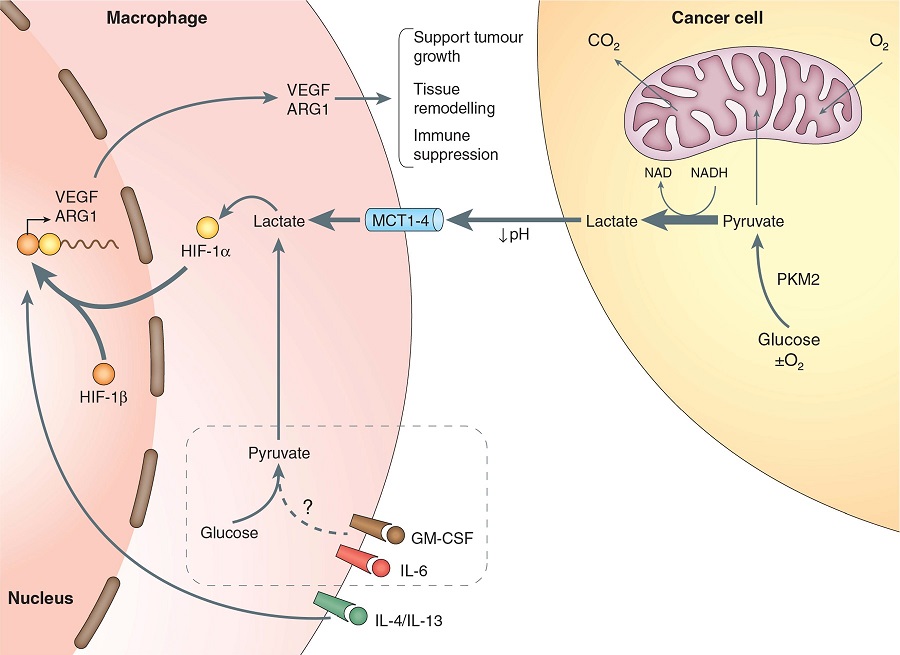

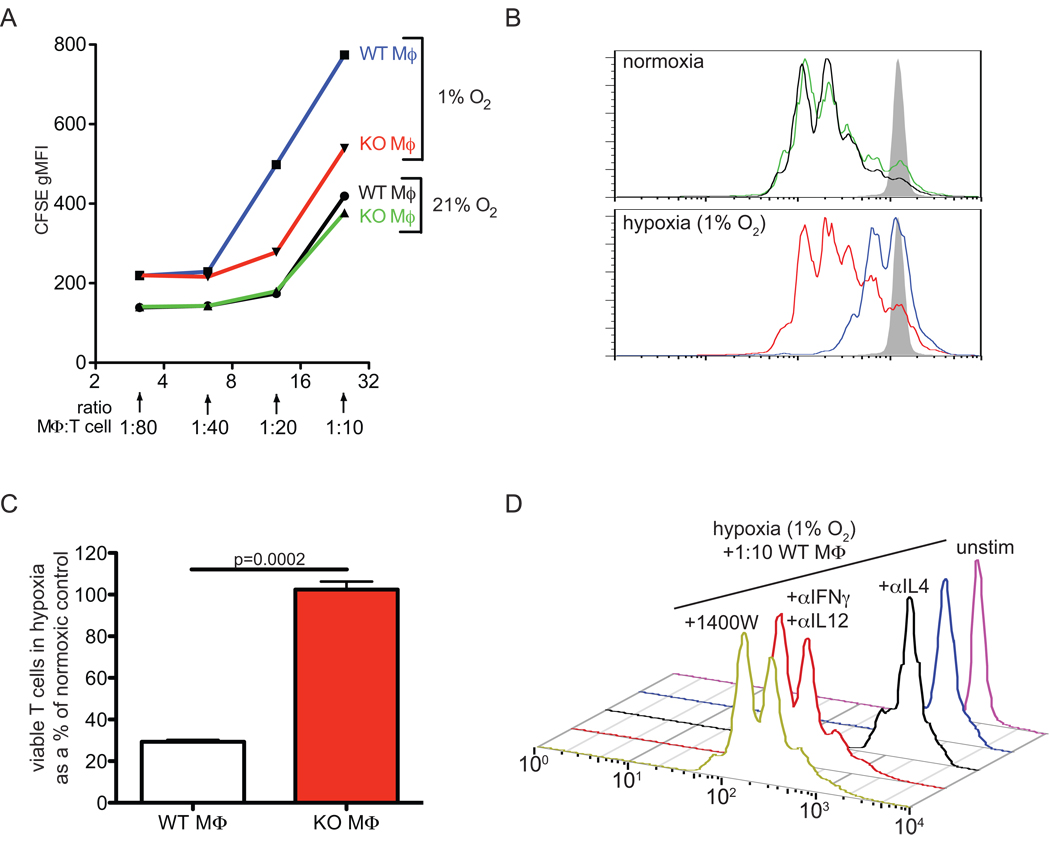
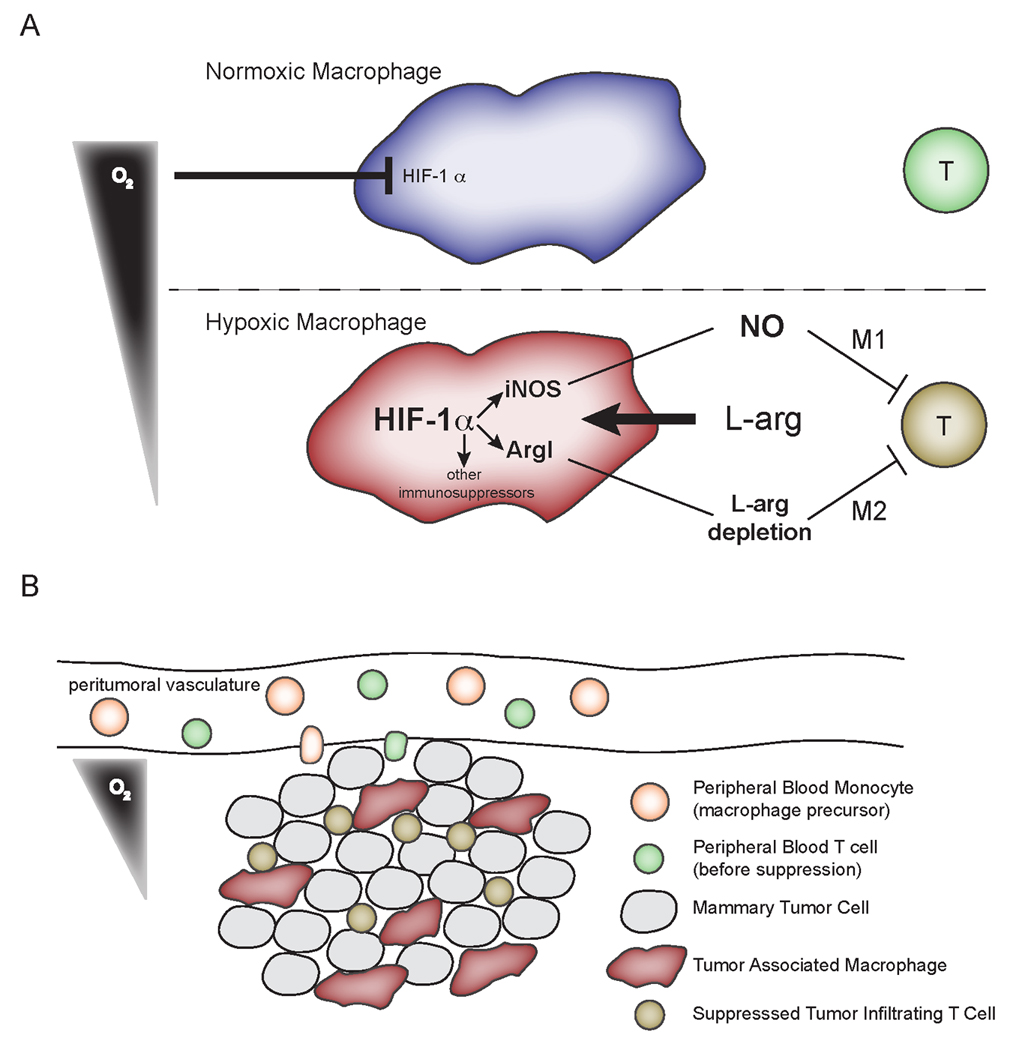
.jpg)

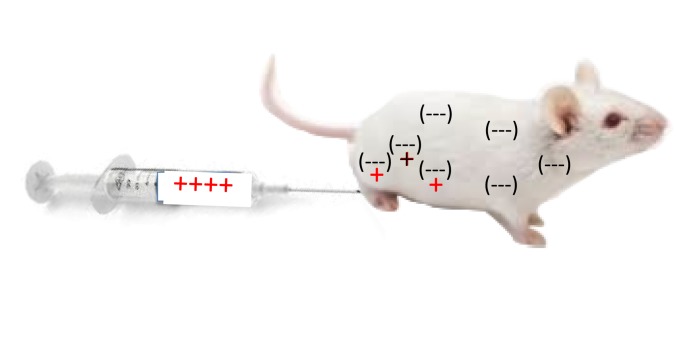

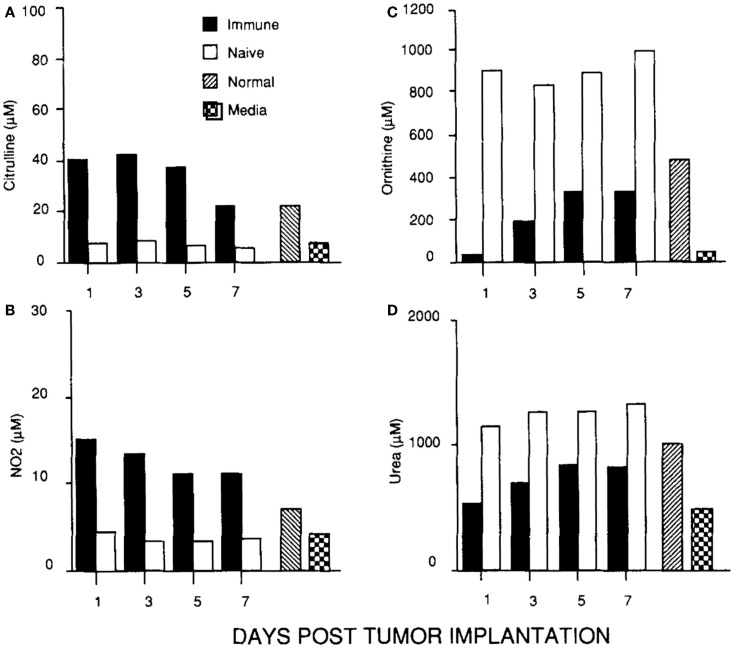
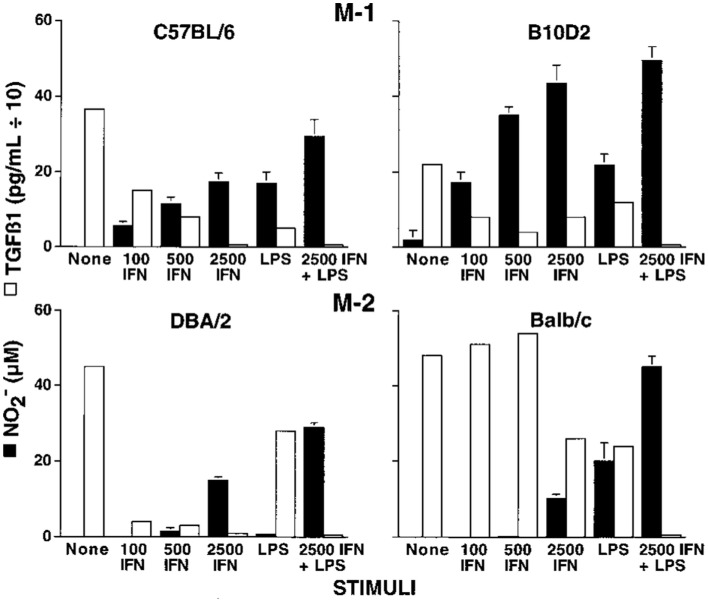
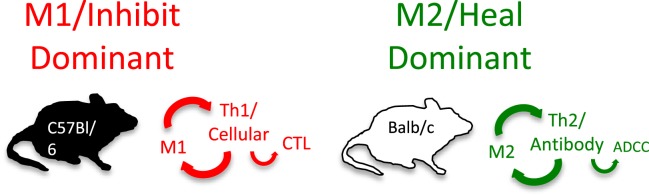
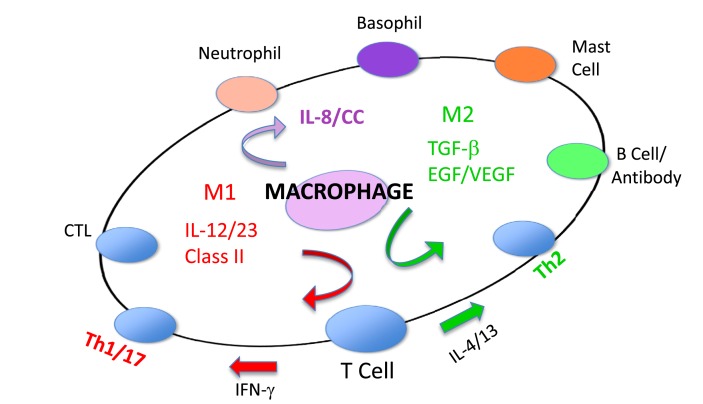

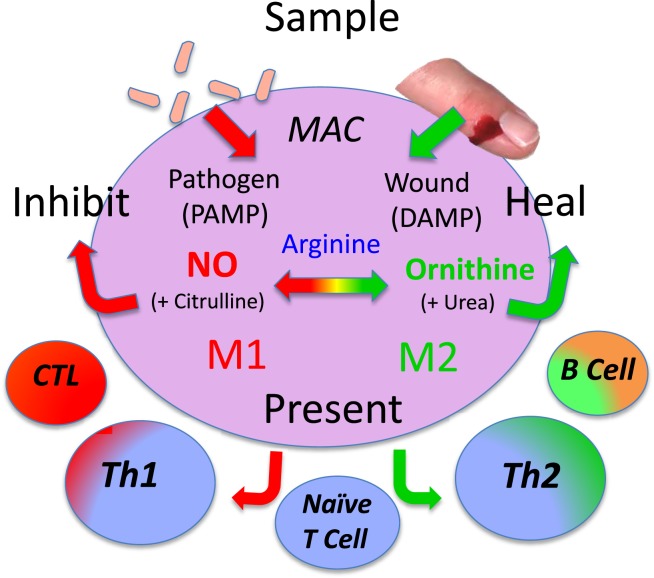
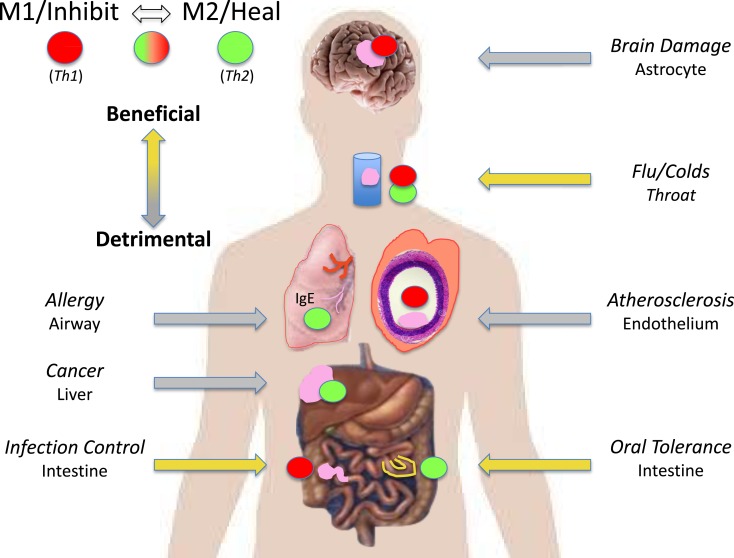

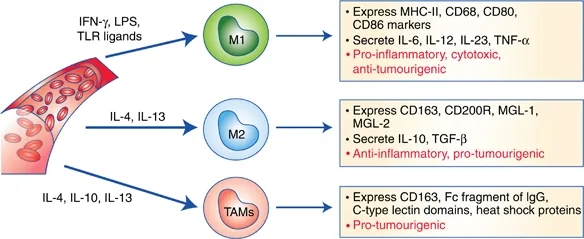
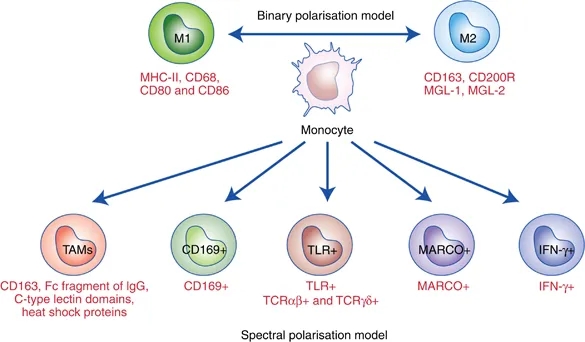
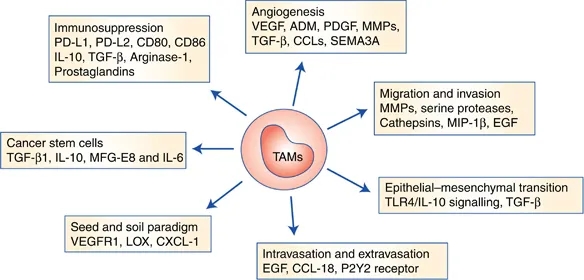
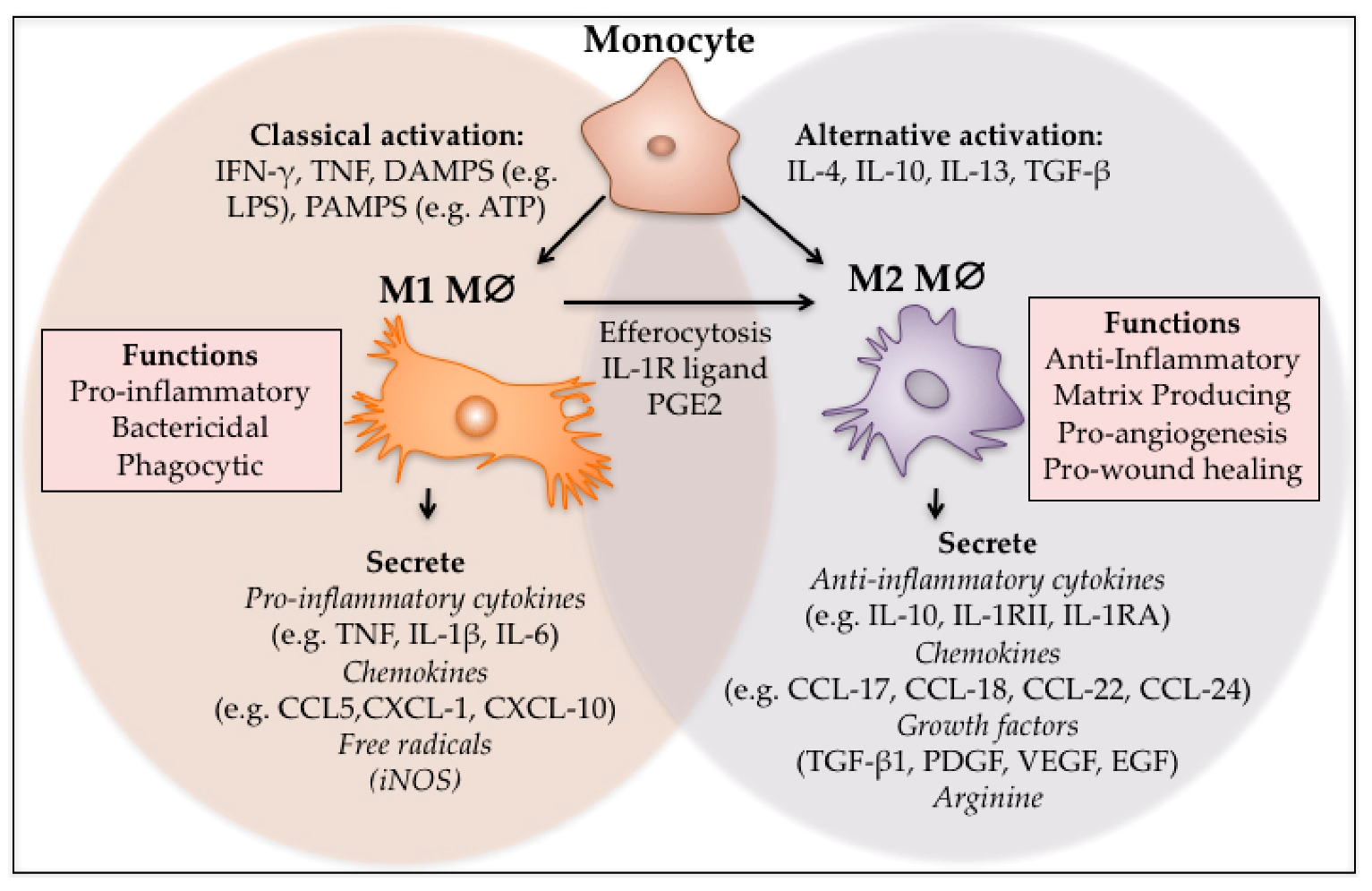
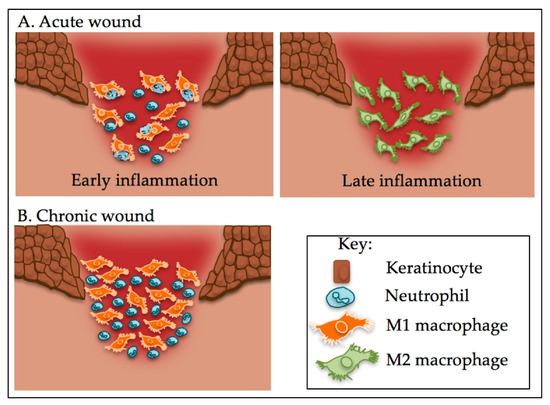

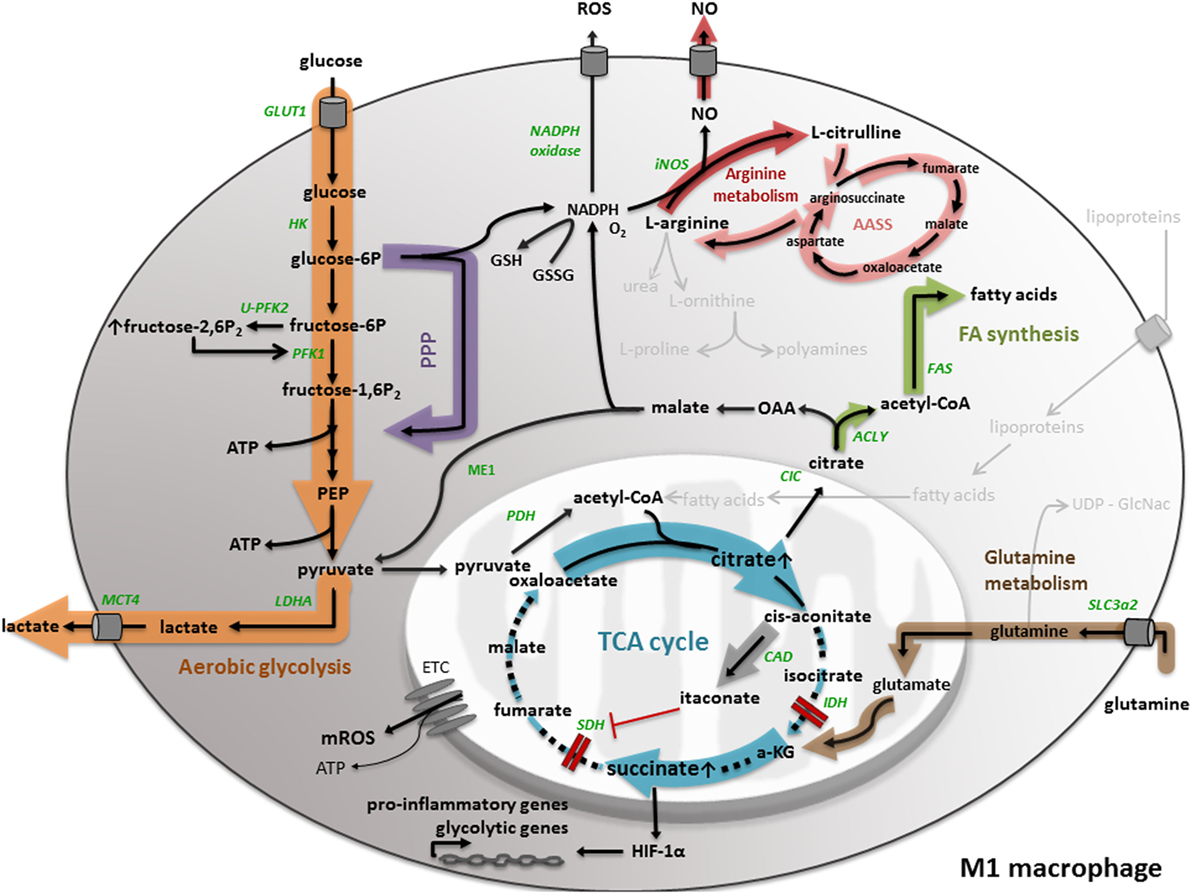
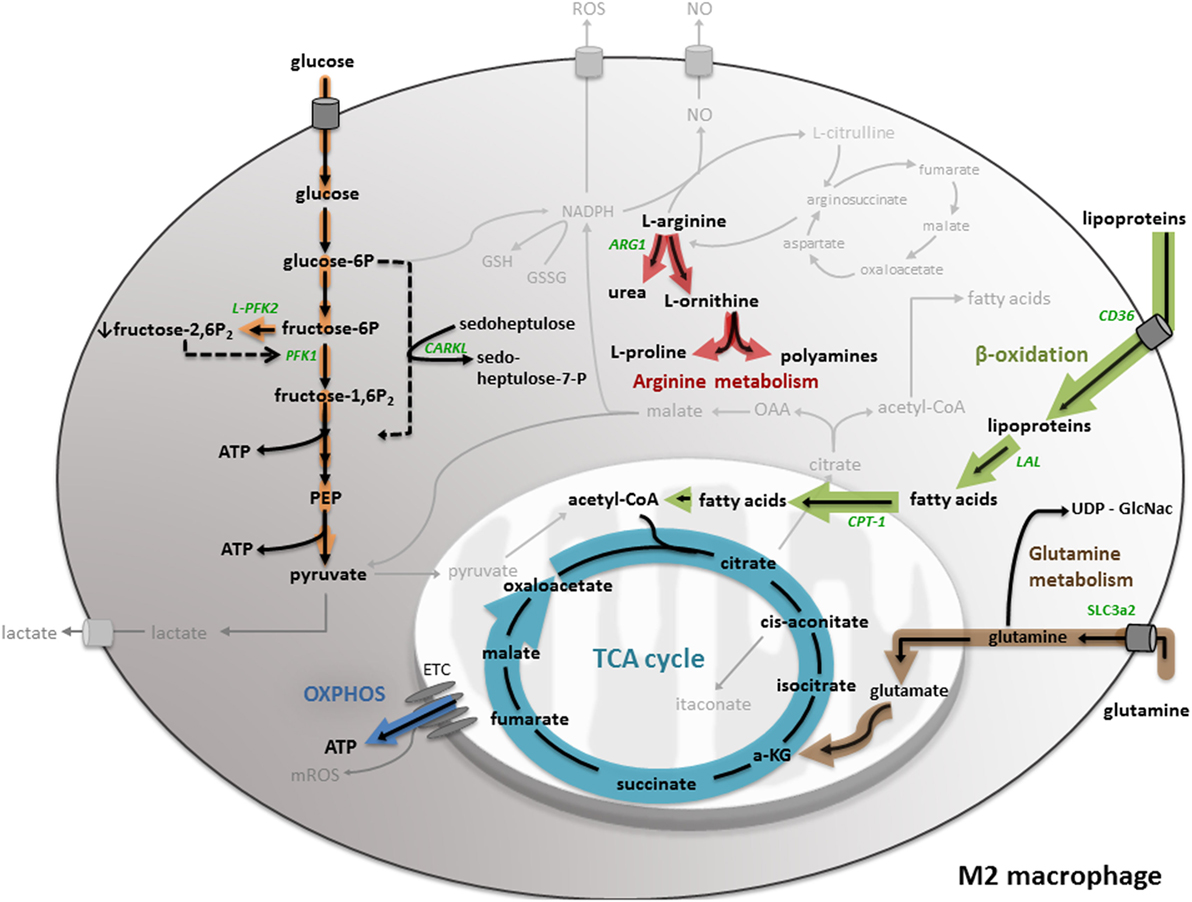
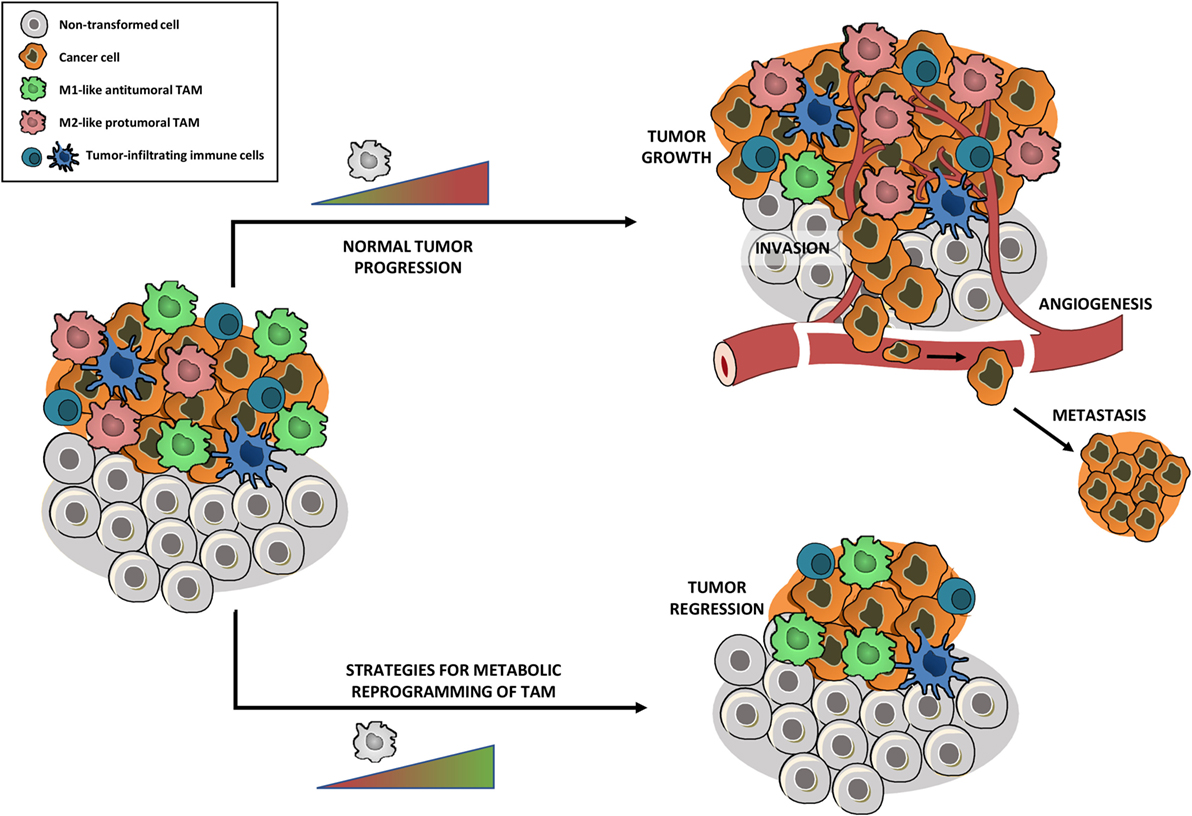
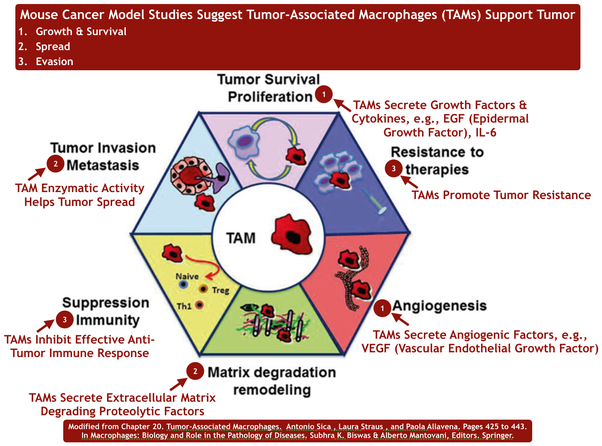
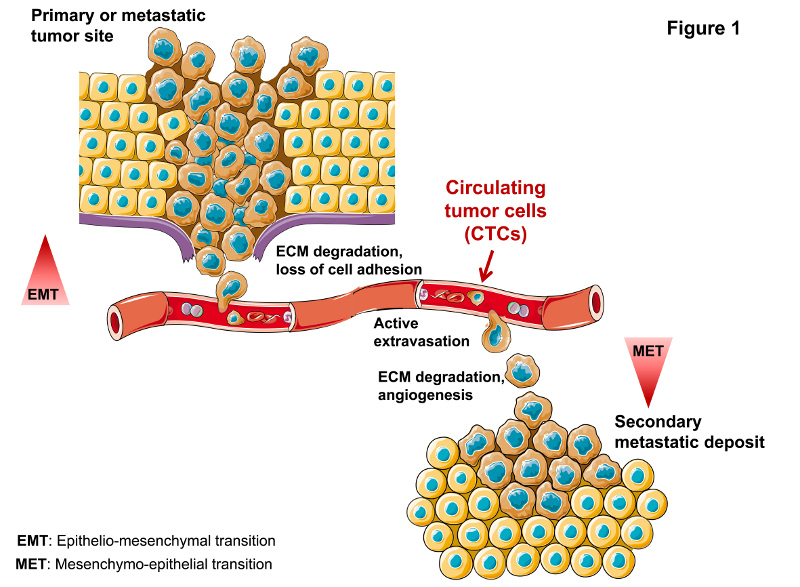
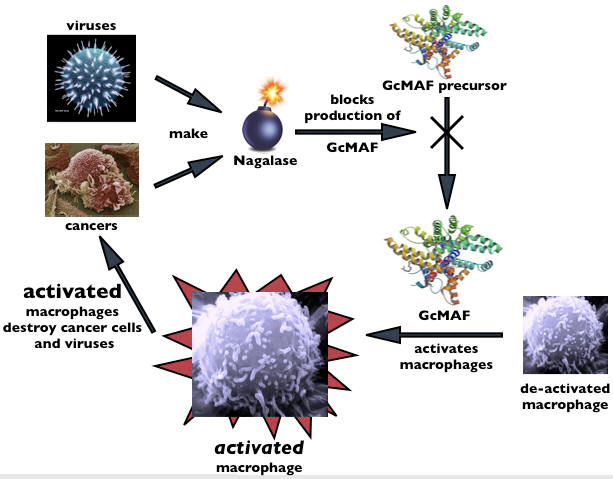
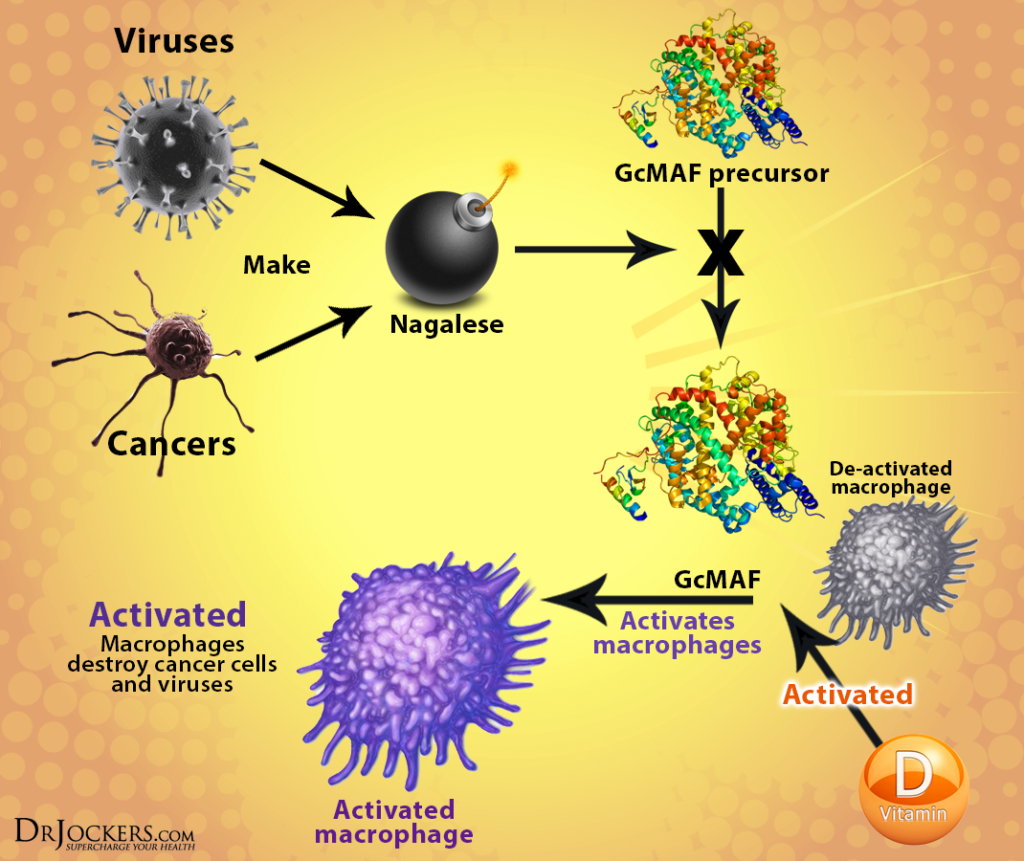
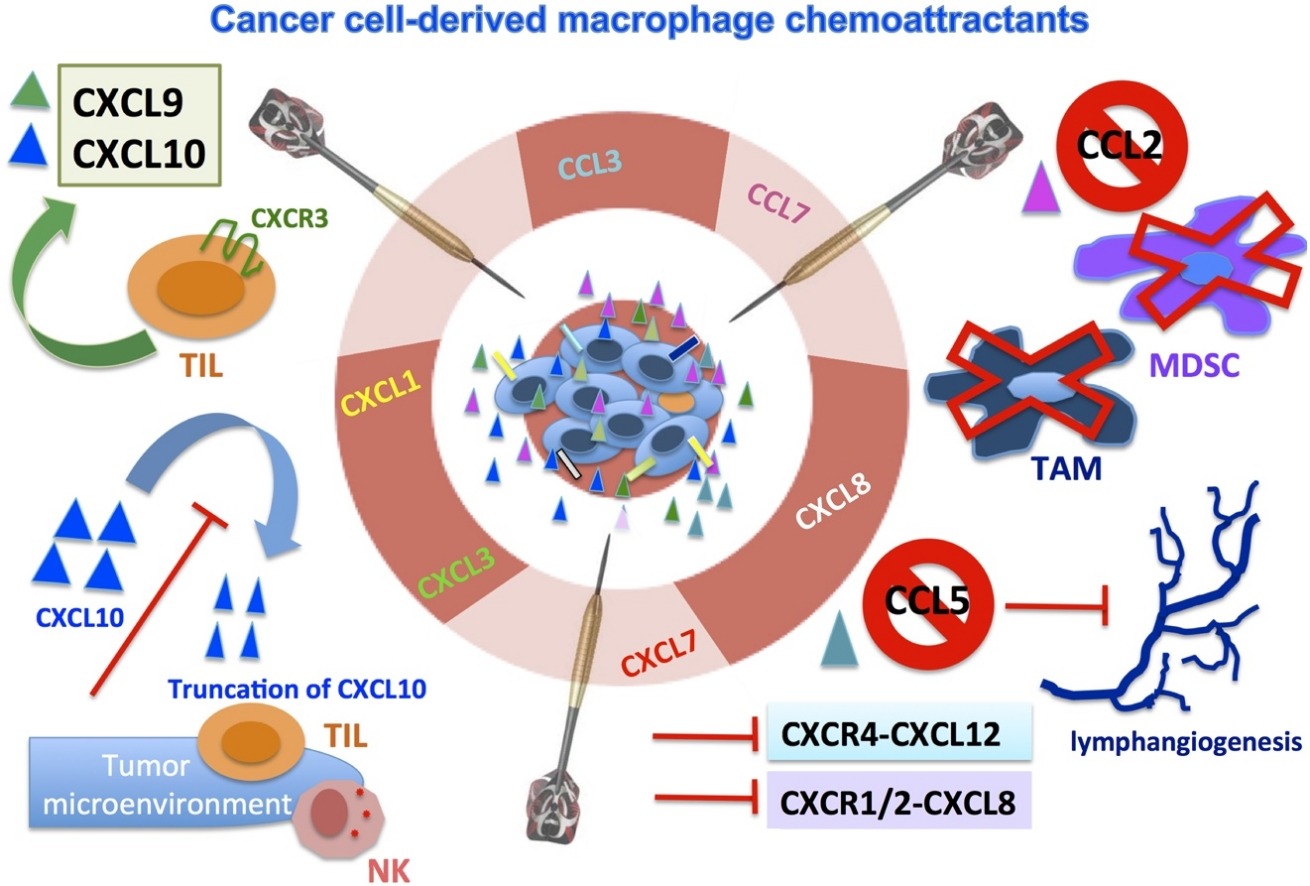
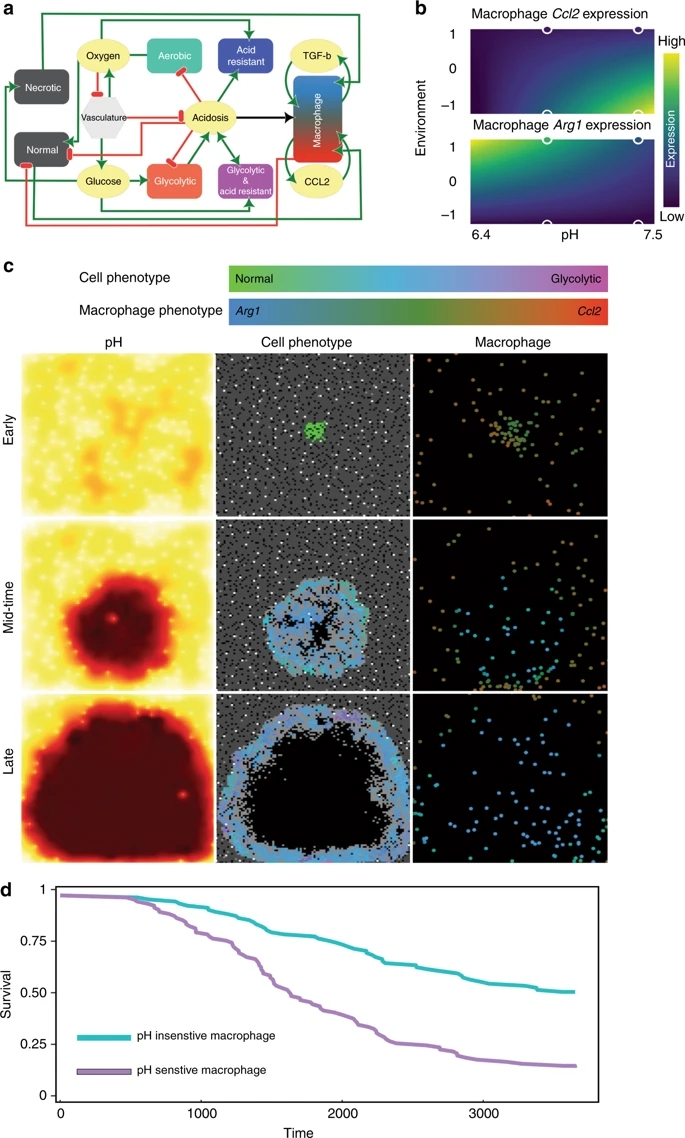
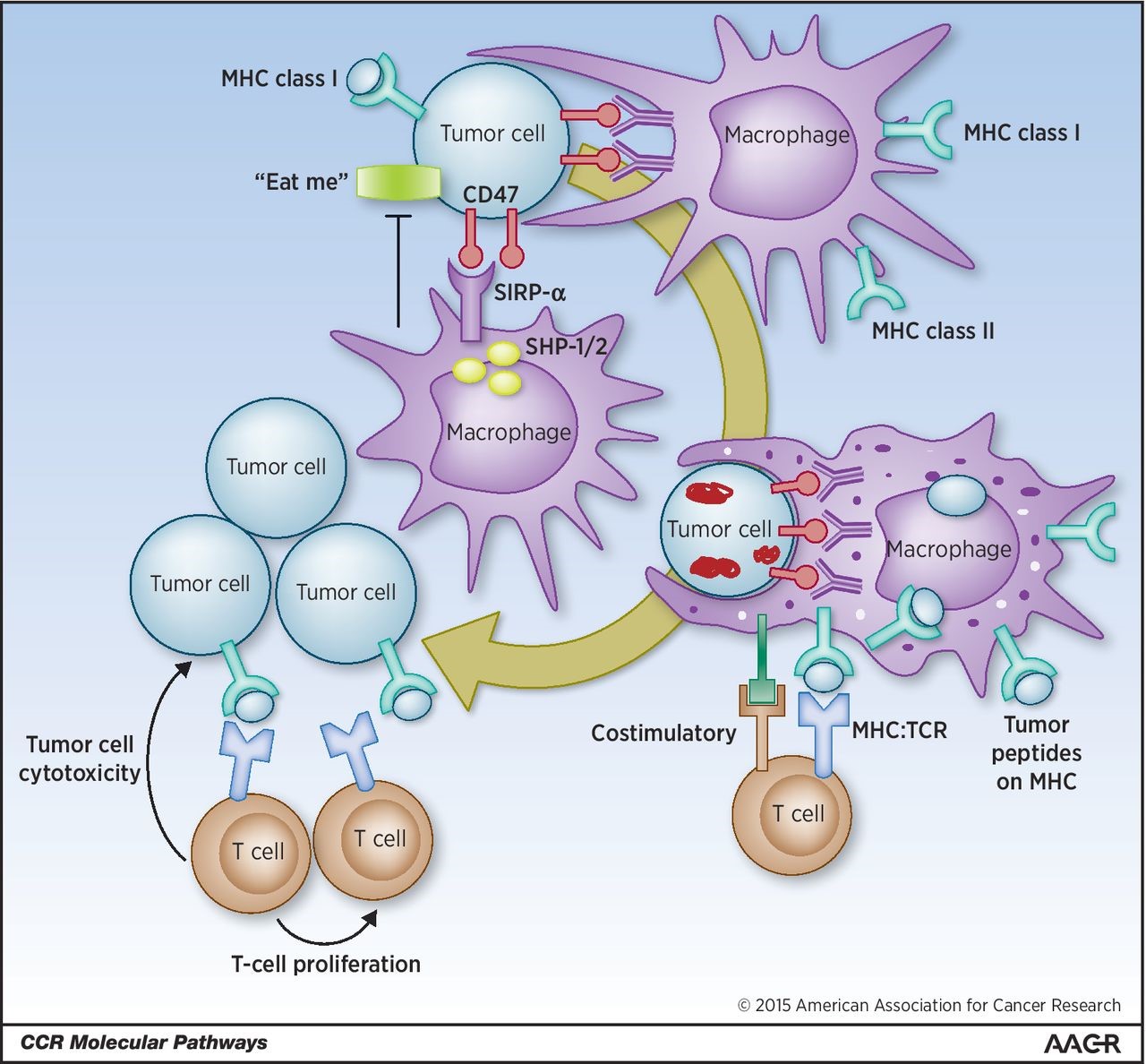
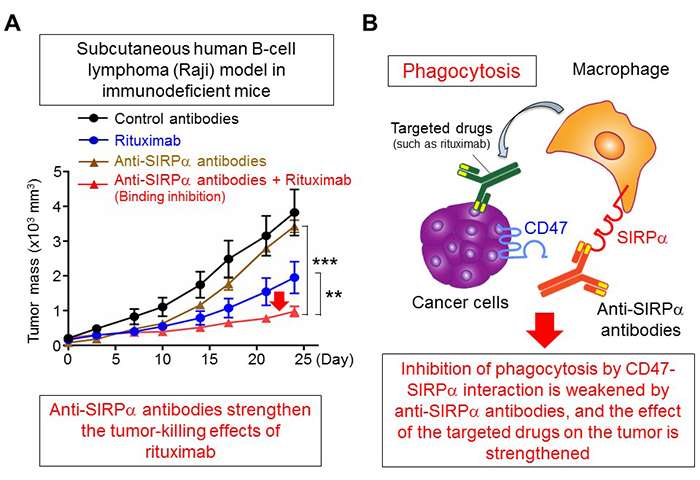
.jpg)