��
Vitamin C, a Multi-Tasking Molecule, Finds a Molecular Target in Killing Cancer Cells
Summary
vitamin c decreaase HIF-1 expression in hyroid tumor tissues
Intracellular H2O2 produces a rapid cell death in HL-60 cells
vitamin c is an inhibitor of tyrosinase
vitamin c is an inhibitor of tyrosine kinase
vitamin c enhances antitumor activity via p53 in cancer cells (thru ROS)
vitamin C kills thyroid cancer cells by inhibiting MAPK/ERK and PI3K/AKT pathways via a ROS-dependent mechanism
vitamin C inhibits phosphorilation of IKK kinase of NF-kB complex
vitamin C, free ferrous or ferric iron,kills cancer cells/microbials thru Fenton reaction.
vitamin C mobilizes release of ferric iron��Fe3+) from cellular ferritin.Ferroptosis is a new type of oxidative regulated cell death (RCD) driven by iron-dependent lipid peroxidation.
Anti-angiogenic effect of high doses of ascorbic acid~concentrations of AA higher than 100 mg/dl suppressed capillary-like tube formation on Matrigel for all cells tested and the effect was more pronounced for progenitor cells in comparison with mature cells.
��
The role of quercetin and vitamin C in Nrf2‑dependent oxidative stress production in breast cancer cells
The role of quercetin and vitamin C in Nrf2‑dependent oxidative stress production in breast cancer cells
https://www.spandidos-publications.com/ol/13/3/1965��
��
Can You Overdose On Vitamin C? Can Enough Vitamin C Kill You? - YouTube
https://www.youtube.com/watch?v=w9l0kRbFbIIVitamin C: Oral vs. Intravenous, Immune Effects, Cancer, Exercise Adaptation & More - YouTube
https://www.youtube.com/watch?v=zfJQTyOklnE��
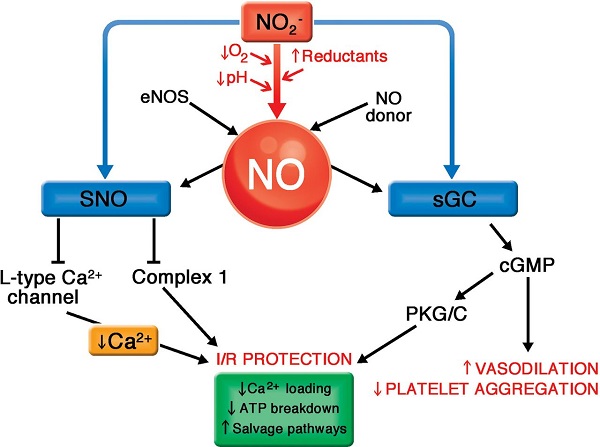
Oxidative stress has been shown to increase the levels of catalytic iron in tissues. A series of electron paramagnetic resonance (EPR) studies demonstrated that ultraviolet light increased the levels of labile iron in skin [186]; heat stress-induced oxidative events increased the level of labile iron in liver [187]; and ischemia reperfusion increased the level of catalytic iron in the heart [188]. In addition, ionizing radiation and some chemotherapeutic drugs have been shown to increase catalytic free iron levels [189�C191]. Since it takes only very low concentrations of catalytic metals to bring about the rapid oxidation of ascorbate [192,193], approaches that increase catalytic iron could potentially enhance the cytotoxicity of pharmacologic ascorbate in vivo [194].
since pharmacological ascorbate is a prodrug for H2O2 generation, there should be many applications where delivery of H2O2 via pharmacological ascorbate may have clinical benefit, such as infectious diseases caused by viruses, bacteria, and other pathogens [10,233].
Ascorbic acid: Chemistry, biology and the treatment of cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3608474/��
The Evolution of Iron Chelators for the Treatment of Iron Overload Disease and Cancer | Pharmacological Reviews
http://pharmrev.aspetjournals.org/content/57/4/547��
Ascorbate and ferritin interactions: Consequences for iron release in vitro and in vivo and implications for inflammation - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0891584918317180��
��
Vitamin C Inhibits NF-��B Activation by TNF Via the Activation of p38 Mitogen-Activated Protein Kinase | The Journal of Immunology
https://www.jimmunol.org/content/165/12/7180��
https://onlinelibrary.wiley.com/doi/abs/10.1002/adma.201904197
React Oxyg Species (Apex). Author manuscript; available in PMC 2018 May 18.
Vitamin C, a Multi-Tasking Molecule, Finds a Molecular Target in Killing Cancer Cells
Robert Li1,2,3,4,5
Author information Copyright and License information Disclaimer
1School of Osteopathic Medicine, Campbell University, Buies Creek, NC 27506, USA
2College of Pharmacy and Health Sciences, Campbell University, Buies Creek, NC 27506, USA
3Virginia Tech-Wake Forest University School of Biomedical Engineering and Sciences, Blacksburg, VA 24061, USA
4Department of Biomedical Sciences and Pathobiology, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA
5Department of Biology, University of North Carolina, Greensboro, NC 27412, USA
Correspondence: ude.llebpmac@ily (Y.R.L.)
��
Abstract
Early work in the 1970s by Linus Pauling, a twice-honored Nobel laureate, led to his proposal of using high-dose vitamin C to treat cancer patients. Over the past several decades, a number of studies in animal models as well as several small-scale clinical studies have provided substantial support of Linus Pauling��s early proposal. Production of reactive oxygen species (ROS) via oxidation of vitamin C appears to be a major underlying event, leading to the selective killing of cancer cells. However, it remains unclear how vitamin C selectively kills cancer cells while sparing normal cells and what the molecular targets of high-dose vitamin C are. In a recent article published in Science (2015 December 11; 350(6266):1391�C6. doi: 10.1126/science.aaa5004), Yun et al. reported that vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting glyceraldehyde 3-phosphate dehydrogenase (GAPDH) through an ROS-dependent mechanism. This work by Yun et al. along with other findings advances our current understanding of the molecular basis of high-dose vitamin C-mediated cancer cell killing, which will likely give an impetus to the continued research efforts aiming to further decipher the novel biochemistry of vitamin C and its unique role in cancer therapy.
Keywords: Cancer cell killing, Cancer therapy, Dehydroascorbate, Dehydroascorbate reductase, Glucose transporter, Glutathione, Glyceraldehyde 3-phosphate dehydrogenase, Glycolysis, Reactive oxygen species, S-Glutathionylation, Vitamin C
1. OVERVIEW
Vitamin C, also known as ascorbic acid or ascorbate, is a water-soluble molecule synthesized endogenously in animals except humans, monkeys, guinea pigs, and several other animal species [1]. Humans lost this capability as a result of a series of inactivating mutations of the gene encoding gulonolactone oxidase (GULO), a key enzyme in vitamin C biosynthesis. Humans normally acquire vitamin C from dietary sources through a substrate-saturable transporting mechanism (see Section 3). Dietary sources of vitamin C are mainly vegetables and fruits, including brussels sprout, broccoli, cauliflower, bell pepper, chili pepper, lettuce, kale, tomato, citrus fruits (e.g., orange, lemon), strawberry, papaya, kiwifruit, pineapple, and mango, among others. Another source is vitamin C supplement.
Oral vitamin C intake produces plasma concentrations that are tightly controlled; once its oral intake exceeds 200 mg daily, it becomes difficult to further increase the plasma concentrations of vitamin C. The maximal plasma concentration attainable by oral intake of vitamin C has been estimated to be approximately 200 mM though the physiological plasma concentrations of vitamin C in healthy humans range from 40 mM to 100 mM. In contrast, intravenous injection of large doses of vitamin C produces millimolar concentrations of plasma vitamin C [2]. So does intraperitoneal injection of large doses of vitamin C in experimental animals. Under physiological conditions, intracellular levels of vitamin C are in the millimolar range. This is due to selective intracellular accumulation via vitamin C transporting system present in the plasma membrane [3] (see Section 3).
The high intracellular concentrations of vitamin C in mammalian tissues suggest its essential roles in maintaining physiological homeostasis and proper functions of organs and systems. In this article, we first examine the novel biochemical properties and functions of vitamin C, and then discuss recent research evidence supporting its potential role in cancer therapy, which lays a basis for understanding the most recent discovery of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a critical molecular target for vitamin C to kill KRAS and BRAF mutant colorectal cancer cells [4]
2. REDOX FORMS OF VITAMIN C
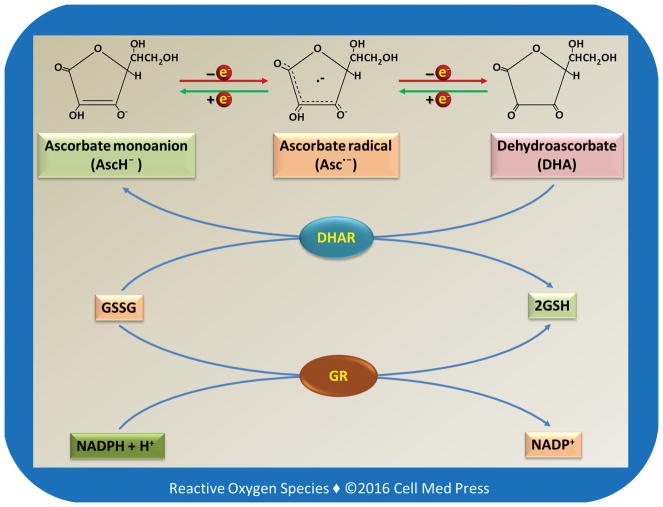
Vitamin C may exist in different redox forms depending on the biological conditions. As illustrated in Figure 1, vitamin C (AscH2) has two ionizable hydroxyl groups. At a physiological pH, vitamin C exists predominantly as a monoanion, i.e., ascorbate monoanion (AscH−). AscH− acts as a reducing agent and is converted to ascorbate radical (Asc•−, also known as semidehydroascorbate) after donation of one electron. After losing another electron, Asc• − is converted to dehydroascorbate (DHA). Asc• − can be reduced back to AscH−. DHA can also be reduced by either one electron to Asc• − or by two electrons to AscH−. The two-electron reduction of DHA to AscH− is catalyzed by dehydroascorbate reductase (DHAR) using the reduced form of glutathione (GSH) as the electron donor. It should be noted that if not specified otherwise, the term vitamin C typically refers to the reduced form of vitamin C, i.e., AscH2 or AscH−.
FIGURE 1
Redox chemistry of vitamin C and its relationship to glutathione
As illustrated, the 2-electron reduction of DHA to vitamin C is catalyzed by dehydroascorbate reductase (DHAR) with GSH as the electron donor. The oxidized form of glutathione (GSSG) is reduced back to GSH by glutathione reductase (GR) using NADPH as an electron donor.
3. TRANSPORT OF VITAMIN C FROM DIETARY SOURCES INTO CELLS AND SUBCELLULAR ORGANELLES
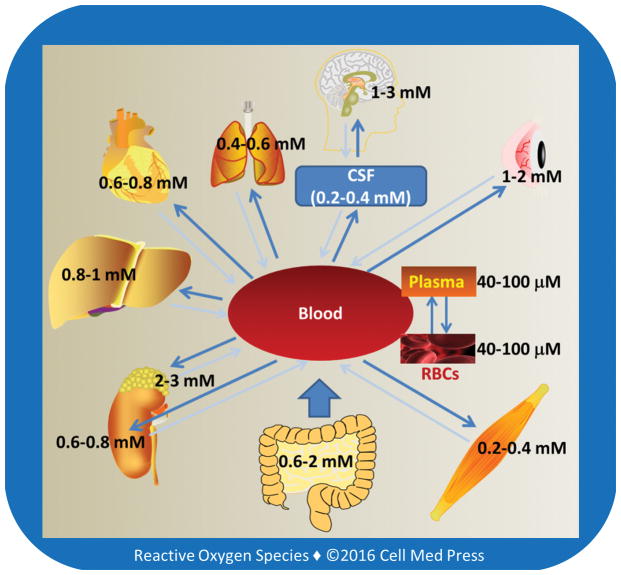
The highest tissue concentrations of vitamin C are found in the brain and in neuroendocrine tissues especially adrenal gland, which may range from 1 mM to 3 mM (Figure 2). These concentrations are 15�C50 times higher than those in the plasma [5, 6], pointing to the existence of active transporting mechanisms. Early seminal work by Emmanuel J. Diliberto and coworkers showed that adrenomedullary cells accumulated vitamin C through a saturable and energy-dependent process and that the newly taken-up vitamin C was also secreted from the cells through specific transporter mechanisms [7�C10]. It is now well-established that vitamin C enters and accumulates in neurons and other types of cells via two different transporting systems, as described below.
FIGURE 2
Tissue levels of vitamin C
Human red blood cells (RBCs) express a high number of glucose transporter (GLUT1), but have no sodium-dependent vitamin C transporters (see Section 3.1), and the intracellular concentration of vitamin C in these cells is similar to that in the plasma. The concentration of vitamin C in the cerebrospinal fluid is ~5�C10 times higher than that in the plasma. Larger arrows indicate the main direction of vitamin C transportation. As shown, vitamin C accumulates in organs and tissues and the high tissue concentrations are due to high intracellular levels of vitamin C, usually in the millimolar range.
3.1. Sodium-Dependent Vitamin C Transporters
The reduced form of vitamin C is transported into cells via sodium-dependent vitamin C transporters (SVCT1 and SVCT2) (Figure 3). SVCT1 (the product of the SLC23A1 gene in humans) is mainly expressed in intestinal and renal epithelial cells, where it helps to mediate absorption and re-absorption of the vitamin, respectively. SVCT2 (the product of the SLC23A2 gene in humans) is found in cells of most other tissues, including the brain. Both SVCT1 and SVCT2 mediate high affinity, sodium- and energy-dependent transport of vitamin C into cells and are essential to establish steep concentration gradients of vitamin C across the plasma membrane [5]. SVCTs, particularly SVCT2, may also transport vitamin C from the cytosol into the mitochondrial matrix [11].
FIGURE 3
Transport of vitamin C and DHA into cells and mitochondria
As illustrated, in extracellular milieu vitamin C is oxidized to DHA, likely due to the presence of transition metal ions, especially copper ions. DHA is reduced to vitamin C by DHAR in the cytosol, and DHA in the mitochondrial matrix is reduced to vitamin C by the electrons derived from the mitochondrial electron transport chain (METC).
3.2. Transport of Oxidized Vitamin C by Glucose Transporters
There are two classes of glucose transporting machineries involved in glucose homeostasis in the body: (1) the facilitated transporters or uniporters (commonly known as glucose transporters and abbreviated as GLUTs) and (2) the active transporters or symporters, namely, sodium-glucose transporters (SGLTs, also known as Na+/glucose cotransporters or symporters). The energy for active glucose transport is provided by the sodium gradient across the cell membrane, which is maintained by the Na+/K+ ATPase (also known as Na+/K+ pump) [12]. There are 12 members of the human SGLT gene family, including cotransporters for sugars, anions, vitamins, and short-chain fatty acids. The two most well-known members of SGLT family are SGLT1 and SGLT2, with SGLT1 being expressed in intestinal epithelial and renal proximal tubular cells and SGLT2 expressed predominantly in renal proximal tubular cells. The Na+-electrochemical gradient provided by the Na+/K+ ATPase is utilized to transport glucose into cells against its concentration gradient across the luminal membrane of cells lining the small intestine and the proximal tubules of the kidneys. SGLTs are unable to transport either reduced or oxidized forms of vitamin C. In contrast, several GLUTs (see below) are able to transport oxidized vitamin C.
3.2.1. Transport of Oxidized Vitamin C from Extracellular Milieu into Cells
Fourteen GLUT proteins are expressed in the human, and they include transporters for substrates other than glucose, such as fructose, myoinositol, and urate. The primary physiological substrates for at least half of the 14 GLUT proteins are either uncertain or completely unknown. The four well-established GLUT isoforms, namely, GLUTs 1�C4, have been demonstrated to have distinct regulatory and/or kinetic properties that reflect their specific roles in cellular and whole body glucose homeostasis [13, 14]. Several GLUTs, such as GLUTs 1, 3, and 4, have also been shown to transport the oxidized form of vitamin C (i.e., DHA) from extracellular milieu into cells (Figure 3).
Interestingly, among all cells, human erythrocytes express the highest level of GLUT1. However, glucose transport actually decreases during human erythropoiesis despite a more than 3-log increase in GLUT1 transcripts. In contrast, GLUT1-mediated transport of DHA is dramatically enhanced during human erythropoiesis. Stomatin, an integral erythrocyte membrane protein, is responsible for regulating the switch from glucose to DHA transport [15].
Once inside the cells, DHA is reduced to vitamin C via the action of DHAR using GSH as the electron donor (Figure 1). In this reaction, GSH is oxidized to glutathione disulfide (GSSG), which is in turn reduced to GSH by glutathione reductase (GR) using NADPH as the reducing equivalent. NADPH is produced primarily from the pentose phosphate pathway. Although DHA at physiological levels can be reduced to vitamin C by the DHAR/GSH system without causing significant depletion of GSH, DHAR/GSH-catalyzed reduction of large amounts of DHA may cause GSH depletion and thereby accumulation of reactive oxygen species (ROS). In this context, GSH is a major cellular defense in the detoxification of ROS.
3.2.2. Transport of Oxidized Vitamin C from the Cytosol into the Mitochondrial Matrix
Certain GLUT isoforms are also expressed in the mitochondrial inner membrane. GLUT1 is the most extensively studied GLUT isoform in terms of mediating the transport of DHA into the mitochondrial matrix [16, 17]. Once transported into the matrix, DHA is reduced to vitamin C primarily by the mitochondrial electron transport chain (METC) [18] (Figure 3). A recent study suggested that GLUT10 might also be expressed in mitochondria participating in transporting DHA from the cytosol into the mitochondrial matrix [19].
4. VITAMIN C AS A MULTI-TASKING MOLECULE
Since its isolation from adrenal glands by Albert Szent-Györgyi (1893�C1986) in 1928 [20], vitamin C has gradually gained a reputation of being a multi-tasking molecule possessing a wide range of distinct biological activities. Of note, Albert Szent-Györgyi won the Nobel Prize in Physiology and Medicine in 1937 for his discoveries of vitamin C and the catalysis of fumaric acid. The multi-tasking activities of vitamin C can be summarized into the following four categories: (1) as a cofactor for various enzymes; (2) as an antioxidant at physiological doses; (3) as a potential pro-oxidant at pharmacological doses; and (4) other emerging novel activities.
4.1. Vitamin C as a Cofactor for Various Enzymes
Vitamin C serves as a cofactor for at least 8 enzymes in mammalian species, including humans. The most notable ones are proline hydroxylase and lysine hydroxylase, which are involved in collagen synthesis. The other enzymes for which vitamin C acts as a cofactor are involved in carnitine synthesis, catecholamine synthesis, peptide amidation, and tyrosine metabolism [18]. Due to its critical role in collagen synthesis, deficiency of vitamin C compromises the integrity of blood vessels, leading to scorbutic gums and pinpoint hemorrhage, characteristic manifestations of vitamin C deficiency.
4.2. Vitamin C as an Antioxidant
The redox chemical properties of vitamin C are responsible for its antioxidant as well as potential pro-oxidant activities. As depicted in Figure 1, the two-electron reduction of DHA to AscH− is catalyzed by DHAR using GSH as the electron donor. This two-electron reduction reaction brings together two highly prevalent intracellular antioxidant molecules: vitamin C and GSH, both of which are present in millimolar concentrations inside the cells. Indeed, vitamin C and GSH cooperate closely to maintain the redox homeostasis of mammalian cells [21]. In addition, the METC, especially the complex III, may also reduce DHA to AscH− [22]. The in vivo significance of this mitochondria-dependent reduction is, however, unclear. As aforementioned, SVCT2 may participate in transporting vitamin C from the cytosol into the mitochondrial matrix. Together, the above findings may help explain the high concentrations of vitamin C in mitochondria and its role in maintaining the redox status of this major ROS-generating organelle. Due to its high instability, DHA, if not rapidly reduced, undergoes hydrolytic ring opening to form 2,3-diketogulonic acid.
In many in vitro systems, vitamin C has been found to scavenge various reactive oxygen and nitrogen species (ROS/RNS), and protect cells from oxidative damage. Vitamin C is able to regenerate a-tocopherol and coenzyme Q from a-tocopherol radical and coenzyme Q radical, respectively, and thereby playing a role in maintaining the antioxidant activities of a-tocopherol and coenzyme Q, two important lipophilic antioxidants in cells. It has recently been demonstrated that vitamin C is also able to reduce 1-Cys peroxiredoxin [23]. Peroxiredoxin is critical for the detoxification of hydrogen peroxide and peroxynitrite. As noted above, vitamin C and GSH cooperate to act as an efficient dual-antioxidant system in mammalian cells [21].
In addition to the above activities involved in detoxifying ROS/RNS, vitamin C is found to inhibit the expression of NADPH oxidase subunit p47phox induced by inflammatory insults, thereby decreasing the formation of ROS from this important cellular source [24]. The inducible expression of inducible nitric oxide synthase (iNOS) in septic mice is also inhibited by vitamin C [25]. It is unlikely that vitamin C directly inhibits the enzyme activity of either NADPH oxidase or iNOS. The reduced expression of the above enzymes or enzyme subunits by vitamin C most likely results from its modulation of cellular redox signaling that is involved in the inducible expression of the above two ROS/RNS-generating enzyme systems.
Interestingly, vitamin C also induces heme oxygenase-1 (HO-1), an enzyme with potent antioxidative and anti-inflammatory activities [26]. While the mechanism involved in the induction of HO-1 remains to be determined, it is likely that vitamin C provokes a pro-oxidant state in cells that subsequently activates HO-1 gene expression. Indeed, HO-1 is known as an oxidative stress-responsive gene, and as described next, vitamin C at pharmacological doses behaves as a pro-oxidant.
4.3. Vitamin C as a Potential Pro-oxidant
Under certain conditions, such as in the presence of redox active metal ions (especially copper ions), vitamin C may behave as a potent pro-oxidant, giving rise to ROS, damaging DNA, and causing protein glycation. Indeed, increased oxidative DNA damage occurs in individuals consuming a large amount of vitamin C [27]. Vitamin C induces decomposition of lipid hydroperoxide to genotoxins even in the absence of redox active metal ions [28]. In a humanized mouse model, vitamin C is found to mediate chemical aging of lens crystallins via the Maillard reaction [29]. In experimental animals, administration of pharmacological doses of vitamin C causes formation of vitamin C radical and ROS in the extracellular fluid and decreases growth of aggressive tumor xenografts [30]. Pharmacological doses of vitamin C have been employed as a potential therapeutic modality for cancer patients, especially those with advanced cancers [31�C33] (see Section 5 below). The pro-oxidant activity of vitamin C also makes it an effective agent for killing drug-resistant Mycobacterium tuberculosis [34]. Hence, the pro-oxidant activities of vitamin C may exert either detrimental or beneficial effects dependent on the physiological and pathophysiological conditions.
4.4. Other Emerging Novel Activities of Vitamin C
Vitamin C reduces ferric ion to ferrous ion and thus facilitates iron absorption in the duodenum. Recent studies suggest a role for vitamin C in nucleic acid and histone demethylation, as well as proteoglycan deglycanation. Due to its role in hydroxylation reactions, vitamin C participates in downregulating hypoxia-inducible transcription factor-1a (HIF-1a). In this regard, proline hydroxylation targets HIF-1a for ubiquitin-mediated degradation [35]. Vitamin C may also play a role in stem cell biology. It enhances the reprogramming efficiency of mouse and human fibroblasts transduced with three (Oct4/Klf4/Sox2) or four (Oct4/Klf4/Sox2/cMyc) factors. It also alleviates cell senescence by p53 repression and may accelerate reprogramming by synergizing with epigenetic regulators [36, 37]. A more recent study has identified a novel function of vitamin C in promoting Tet-mediated generation of 5-hydroxyme-thylcytosine (5-hmC), suggesting that the availability of vitamin C may have a profound effect on many cellular functions dictated by DNA demethylation and that vitamin C may act as a critical mediator of the interface between the genome and environment [38, 39]. Moreover, vitamin C at pharmacological doses was shown to inhibit angiogenesis, leading to suppression of tumorigenesis in animal models [40, 41]. These atypical effects of vitamin C along with its unique pro-oxidative activities described above may contribute to the ever-increasing biological functions of this old vitamin in health and disease, and provide more opportunities for employing this multi-tasking molecule for disease intervention, including cancer therapy.
5. VITAMIN C AND CANCER THERAPY
As aforementioned, vitamin C at millimolar concentrations has recently been found to result in marked production of ROS causing selective killing of various types of cancer cells in diverse in vitro and in vivo models [30, 42, 43]. These observations are in line with early clinical studies showing a therapeutic activity of high-dose vitamin C in cancer patients. This section describes the findings related to the utilization of intravenous high-dose vitamin C in the therapeutic intervention of malignancies so as to provide a rationale for continued basic research to understand the molecular basis of vitamin C-based cancer therapy.
5.1. Historical Overview and Current Status
5.1.1. Early Clinical Studies by Ewan Cameron and Linus Pauling versus the Trials at the Mayo Clinic
Early studies conducted by Ewan Cameron and Linus Pauling in the 1970s suggested that large doses (10 g/day via intravenous infusion for about 10 days and orally thereafter) of vitamin C were effective in increasing survival and improving quality of life of terminal cancer patients [31, 32]. Similar findings were also reported by clinical trials in Japan [44]. However, subsequent trials on large doses of vitamin C given orally (10 g/day) at the Mayo Clinic did not reach the same conclusion [45, 46], and the routes of administration (intravenous infusion versus oral intake) may explain the discrepant results. Indeed, as described earlier (Section 1), oral vitamin C intake produces plasma concentrations that are tightly controlled, and the maximal plasma concentration attainable by oral intake of vitamin C has been estimated to be ~200 mM. In contrast, intravenous administration of pharmacological doses of vitamin C results in millimolar plasma concentrations [2], and as stated above (Section 4.3), millimolar concentrations of vitamin C have been shown to selectively kill cancer cells via an ROS-dependent mechanism in experimental models.
5.1.2. Recent Clinical Studies
Recently, Sebastian J. Padayatty and coworkers reported three well-documented cases of advanced cancers in which patients had unexpectedly long survival times after receiving high doses of vitamin C via intravenous administration [47]. Although the findings from this study supported a possible therapeutic efficacy of high intravenous doses of vitamin C in cancer, other possibilities could not be excluded to account for the unexpectedly prolonged survival of the patients. A phase I clinical trial reported in 2008 that high-dose intravenous vitamin C (0.4�C1.5 g/kg body weight, three times weekly) was well-tolerated, but failed to demonstrate an anticancer activity when administered to patients with previously treated advanced malignancies [48]. A clinical study reported in 2014, involving 11 patients with acute myeloid leukemia (AML) excluding acute promyelocytic leukemia (APL), i.e., non-APL AML, reported a limited benefit provided by adding pharmacological doses of vitamin C (1 g/day over 30 min after arsenic trioxide, 5 days a week for 5 weeks) to the standard arsenic trioxide regimen [49].
A phase I�CII clinical trial in 2015, involving 14 patients with advanced cancer receiving standard chemotherapy, reported that high-dose intravenous vitamin C was safe and generally well tolerated. The pre- and post-chemotherapy pharmacokinetic profiles suggested that tissue uptake of vitamin C increased after chemotherapy, with no increase in urinary oxalic acid excretion. Three patients with different types of cancer experienced unexpected transient stable disease, increased energy, and functional improvement after adding the high-dose vitamin C to standard chemotherapy [50]. Although this phase I�CII trial along with the other small-scale clinical studies described earlier neither proved nor disproved the value of high-dose vitamin C in cancer therapy, it did provide practical information and indicate a feasible way to evaluate this plausible but unproven therapy, and if carried out in sufficient numbers, simple studies like this one could identify specific clusters of cancer type, chemotherapy regimen, and high-dose vitamin C in which exceptional responses occur frequently enough to justify appropriately focused large-scale clinical trials [50].
On the other hand, continued efforts aiming to characterize the pharmacokinetic profiles of high-dose vitamin C in cancer patients may lead to the development of optimal effective dosage regimens for cancer treatment. In this regard, a phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous vitamin C in patients with advanced cancer concluded that vitamin C administered intravenously at 1 g/min for 4 consecutive days per week for a total of 4 weeks produced up to 49 mM vitamin C in patients�� blood and was well tolerated. As such, the authors of the study recommended a dose of 70�C80 g/m2 for future clinical studies on vitamin C cancer therapy [51].
More recently, a pharmacokinetic study of vitamin C in prostate cancer patients suggested that the elimination of vitamin C from the body followed first-order elimination kinetics throughout the dosing range with supra-physiological concentrations. The target dose of 60 g of intravenous vitamin C produced a peak plasma vitamin C concentration of 20.3 mM. Elimination half-life was 1.87 hr, volume of distribution was 0.19 L/kg, and clearance rate was 6.02 L/hr. Thus the relatively fast first-order elimination with a half-life of about 2 hr made it impossible to maintain vitamin C concentrations in the potentially cancer cell-killing range after infusion stopped in prostate cancer patients with normal kidney function [52]. The study thus proposed a regimen with a bolus loading dose followed by a maintenance infusion based on the calculated clearance to ensure the plasma concentrations of vitamin C was maintained at high (millimolar) concentrations for a prolonged period of time, which might be required for the vitamin to effectively kill cancer cells in the patients [52]. Hence, continued pharmacokinetic studies in cancer patients would enhance our understanding of how high-dose vitamin C behaves in cancer patients. Likewise, mechanistic studies in experimental models will shed more light on the molecular basis of high-dose vitamin C-based cancer therapy as well as guide the clinical research design.
5.1.3. Millimolar Vitamin C Acts as a Pro-Oxidant in Experimental Cancer Models
In the interstitial fluid surrounding tumor cells, millimolar concentrations of vitamin C exert local pro-oxidant effects by mediating hydrogen peroxide formation in experimental models [30, 42, 43]. More recently, data from experimental models showed that millimolar vitamin C, acting as a pro-oxidant, induced DNA damage and depleted cellular adenosine triphosphate (ATP), activated the ataxia telangiectasia mutated (ATM)/adenosine monophosphate-activated protein kinase (AMPK) pathway, and resulted in mammalian target of rapamycin (mTOR) inhibition and death in ovarian cancer cells. The combination of parenteral vitamin C with the conventional chemotherapeutic agents carboplatin and paclitaxel synergistically inhibited ovarian cancer in mouse models and reduced chemotherapy-associated toxicity in patients with ovarian cancer [53]. Hence, experimental studies have provided strong evidence for an important role of oxidative stress in vitamin C-mediated anticancer activity in animal models [54, 55]. In line with this notion, use of ROS-inducing agents other than vitamin C for cancer treatment has also received much attention in recent years [56]. However, a fundamental question remains: what makes vitamin C selectively toxic to cancer cells? The elegant work by Yun et al. [4] suggests that DHA is the pharmaceutically active agent of high-dose vitamin C therapy that induces oxidative stress, and that the selective toxicity of vitamin C to tumor cells stems from high GLUT1 expression combined with KRAS or BRAF oncogene-induced glycolytic addiction. This addiction leads to an energetic crisis and cell death upon inhibition of GAPDH by DHA-induced oxidative stress.
5.2. Discovery of the ��DHA�CGLUT1�CROS�CGAPDH�� Machinery in Vitamin C-Induced Cancer Cell Killing
Over 50% of human colorectal cancers carry either KRAS or BRAF mutations and are often refractory to epidermal growth factor receptor-targeting drugs. In their recent article published in Science [4], Yun et al. reported that cultured human colorectal cancer cells harboring KRAS or BRAF mutations were selectively killed when exposed to high concentrations (1�C2 mM) of vitamin C. They showed that this selectivity was due to increased uptake of DHA via GLUT1 which is overexpressed in the cancer cells. Increased DHA uptake and the subsequent intracellular reduction by DHAR led to GSH depletion and thereby accumulation of ROS (Figure 1). The resulting oxidative stress led to inhibition of GAPDH in the highly glycolytic KRAS or BRAF mutant cells, causing an energetic crisis and cell death not seen in KRAS and BRAF wild-type cells [4]. Importantly, this novel mechanism of action operated also in in vivo animal models [4].
5.2.1. Uptake of DHA via GLUT1
Given that GLUT1 levels in KRAS and BRAF mutant cells are elevated, Yun et al. hypothesized that the increase in DHA uptake could disrupt redox homeostasis and compromise cellular viability. To test this hypothesis, Yun et al. used a panel of isogenic colorectal cancer cell lines harboring wild-type or mutant alleles of KRAS (HCT116 and DLD1) or BRAF (VACO432 and RKO). They first observed that in culture media, vitamin C was oxidized to DHA with a half-life of ~70 min, and this oxidation could be prevented by adding GSH to the culture media [4]. Oxidation of vitamin C in phosphate-buffered saline (PBS) or culture media has been well-documented in the literature [57]. It has long been demonstrated that trace amounts of transition metal ions, especially copper ions, present in PBS or culture media catalyzes the oxidation of vitamin C to produce DHA and ROS [58]. Prevention of vitamin C oxidation in culture media by adding GSH could be due to the chelation of transition metals ions, especially copper ions by GSH. In this regard, GSH binds copper ions preventing copper ion-mediated redox reactions [59].
By using [14C]-radiolabeled vitamin C, Yun et al. observed that adding GSH to the media to prevent oxidation of vitamin C to DHA abrogated [14C]-vitamin C uptake. Furthermore, [14C]-vitamin C uptake was significantly decreased in both HCT116 and VACO432 cells treated with a GLUT1-specific inhibitor, STF31, and in GLUT1-knockout cells. Glucose also competed with DHA for uptake in the colorectal cancer cells. These results led Yun et al. to conclude that the colorectal cancer cells preferentially import DHA, rather than vitamin C, and that this uptake is mediated by GLUT1 [4]. Indeed, as discussed earlier in Section 3.2, GLUT1 is the best characterized GLUT isoform for mediating the transport of DHA from extracellular milieu into the cytosol as well as that from the cytosol into the mitochondrial matrix.
Given the increased expression of GLUT1 in the mutant cells, Yun et al. next determined whether KRAS or BRAF mutations influenced vitamin C uptake. They observed that the mutant lines took up significantly more [14C]-vitamin C than did their wild-type counterparts. Moreover, they found KRAS and BRAF mutant cells imported DHA faster than they did [14C]-vitamin C, consistent with the observation that vitamin C must first be oxidized to DHA to enter cells through GLUT1. These results led to the conclusion that GLUT1 is the primary means of vitamin C uptake in the colorectal cancer cells and that elevated GLUT1 expression in KRAS or BRAF mutant cells drives increased DHA uptake [4].
5.2.2. Selective Toxicity of Vitamin C to Cells with Mutant KRAS or BRAF Alleles
In view of the augmented uptake of DHA by the mutant lines, Yun et al. then determined if the increased uptake of DHA in KRAS and BRAF mutant cells could affect their survival and growth. They observed that 24 to 48 hr of vitamin C treatment inhibited KRAS and BRAF mutant cell growth and colony formation, but with reduced effects on their wild-type counterparts. They further showed that vitamin C could selectively kill the KRAS and BRAF mutant cells even in the presence of physiological concentrations (5�C10 mM) of glucose. In line with the effect of GSH on oxidation of vitamin C to DHA, they found that vitamin C-mediated cancer cell killing was prevented in the presence of exogenously added GSH [4].
5.2.3. Dependence of Vitamin C Toxicity on both High GLUT1 Expression and Glycolytic Addiction
The next question asked by Yun et al. was: could accumulation of high-level of DHA alone be enough for causing cancer cell death? To answer this question, Yun et al. determined if vitamin C was also cytotoxic to colorectal cancer cells with mutations of oncogenes other than KRAS or BRAF, and in wide-type cells overexpressing GLUT1. Unlike KRAS or BRAF, they found that the PIK3CA (another frequently mutated oncogene) genotype did not exhibit augmented sensitivity to vitamin C-mediated cytotoxicity. Importantly, Yun et al. found that although overexpression of GLUT1 in wild-type cells increased vitamin C uptake, it did not sensitize wild-type cells to vitamin C-mediated cytotoxicity. Together, these findings suggested that high GLUT1 expression alone, without oncogene-induced metabolic reprogramming (i.e., glycolytic addiction), is not sufficient to make cells susceptible to vitamin C-induced cytotoxicity [4].
5.2.4. Vitamin C-Induced Cancer Cell Killing: From in vitro Cultured Cells to in vivo Animal Models
To investigate the in vivo relevance of the above findings, Yun et al. next determined if high-dose vitamin C treatment could affect the growth of KRAS and BRAF mutant colorectal cancer cells in mice. To this end, Yun et al. treated the mice bearing established xenografts derived from parental HCT116 and VACO432 cell lines twice a day via intraperitoneal injection of a high-dose vitamin C (4 g/kg body weight) or PBS as the vehicle control for 3�C4 weeks. They found that vitamin C treatment significantly reduced tumor growth relative to vehicle control treatment. KRAS and BRAF wild-type isogenic HCT116 and VACO432 cell lines were unable to form xenograft tumors in mice [4]. This finding by Yun et al. is in line with previous studies showing that high-dose vitamin C inhibits tumor growth in diverse animal models (see section 5.1.3).
To directly test the impact of Kras mutation on the sensitivity of tumors to high-dose vitamin C treatment, Yun et al. generated a transgenic model of intestinal cancer, driven by either Apc mutation, or combined Apc and Kras (G12D) mutations. They elegantly demonstrated that whereas Apcflox/flox mice showed no difference in polyp burden after vitamin C treatment, Apcflox/flox/KrasG12D mice had significantly fewer and smaller small intestine polyps, confirming that vitamin C selectively affected Kras mutant tumors. Consistent with the in vitro experiments in the colorectal cancer cell lines, they found that tumors from Apcflox/flox/KrasG12D mice showed higher GLUT1 expression and greater vitamin C uptake than did tumors from Apcflox/flox mice [4]. Collectively, these findings by Yun et al. indicate that the same high-dose vitamin C-dependent cancer cell killing machinery also operates in animal models. Clinical research on high-dose vitamin C therapy in colorectal cancer patients with KRAS mutations is warranted. In this context, a recent study reported a decrease in serum level of Ras protein in cancer patients following high-dose vitamin C treatment [60].
5.2.5. GAPDH as a Molecular Target in Vitamin C-Induced Cancer Cell Killing
Yun et al. then carried out metabolomics studies and found that vitamin C treatment led to inhibition of GAPDH in the KRAS and BRAF mutant cells [4]. They observed that vitamin C caused GSH depletion and ROS accumulation in the mutant lines, which might be responsible for GAPDH inhibition. Considering the dependence of cancer cells with KRAS or BRAF mutations on glycolysis for energy production and survival, Yun et al. next determined if inhibition of GAPDH, an important enzyme in glycolysis, by vitamin C caused energy crisis in the mutant cells [4]. They found that vitamin C treatment caused a rapid decrease in the glycolytic rate in KRAS and BRAF mutant cells, but not in the wild-type cells. Consequently, they found that vitamin C induced a significant drop in ATP levels, with a concomitant increase in AMP levels and activation of AMPK in the KRAS and BRAF mutant cells. The study then demonstrated that the above effects and cell death in the mutant lines were blocked by N-acetylcysteine [4], an antioxidant compound that increases cellular GSH [61], suggesting an oxidative stress mechanism underlying vitamin C-dependent cytotoxicity to the mutant colorectal cancer cells. Notably, Yun et al. showed that supplementing drinking water with N-acetylcysteine over the course of vitamin C treatment abolished the ability of vitamin C to reduce xenograft growth of KRAS mutant cells [4], suggesting that the aforementioned oxidative machinery also operates in vivo.
5.2.6. Vitamin C-Induced S-Glutathionylation of GAPDH
GAPDH is one of the most prominent cellular targets of oxidative modifications when ROS/RNS are formed during metabolism and under stress conditions, and the Cys152 in the active site of the enzyme is subject to redox modifications, leading to enzyme inactivation [62, 63]. Via immunoprecipitating endogenous GAPDH and blotting with an antibody that recognizes S-glutathionylation under nonreducing conditions, Yun et al. observed that vitamin C treatment caused 2�C4-fold increases in the GAPDH S-glutathionylation levels in both KRAS and BRAF mutant lines [4]. Protein S-glutathionylation, the reversible formation of mixed disulfides between glutathione and low-pKa cysteinyl residues, not only is a cellular response to mild oxidative/nitrosative stress, but also occurs under basal (physiological) conditions, and likely participates in redox signaling [64]. The increased GAPDH S-glutathionylation levels in vitamin C-treated KRAS and BRAF mutant cells demonstrated by Yun et al. suggested oxidative modifications of GAPDH. Importantly, direct measurement of the GAPDH activity in lysates of vitamin C treated KRAS and BRAF mutant cells by Yun et al. revealed a significant inhibition (by 50%) of GAPDH and co-treatment with N-acetylcysteine blocked the inhibition [4]. It remained unclear if vitamin C-induced inhibition of GAPDH occurred exclusively due to S-glutathionylation. In this regard, multiple studies suggested that S-glutathionylation of GAPDH led to enzyme inhibition [62, 63]. On the other hand, some other studies using purified human GAPDH and [35S]-labelled GSSG showed that S-glutathionylation of GAPDH did not result in inactivation of the enzyme. Rather, the direct oxidation of GAPDH with hydrogen peroxide was responsible for inhibition of the catalytic activity of the enzyme [65].
5.2.7. Vitamin C-Induced NAD+ Depletion
The metabolomics studies by Yun et al. revealed a 19-fold increase in the accumulation of GAPDH substrate glyceraldehyde-3-phosphate (GAP) in vitamin C-treated mutant cells [4]. This led Yun et al. to examine the changes in the levels of NAD+, the substrate for GAPDH to catalyze the oxidation of GAP to form 1,2-bisphosphoglycerate (BPG) (Figure 4). Indeed, they found that vitamin C treatment resulted in NAD+ depletion. They also showed that vitamin C treatment caused activation of poly(ADP-ribose) polymerase (PARP) likely through oxidative DNA damage. This led Yun et al. to conclude that NAD+ depletion in vitamin C-treated cells may be due to the activation of PARP and the subsequent consumption of NAD+ [4]. Yun et al. then showed that vitamin C-induced cell death was partially rescued by pretreatment with a PARP inhibitor, olaparib, or a cell-permeable NAD+ precursor, nicotinamide mononucleotide. These results led Yun et al. to conclude that in KRAS and BRAF mutant cells vitamin C-induced intracellular ROS accumulation compromises the function of GAPDH by both oxidative modifications of the enzyme and the depletion of its cofactor NAD+, eventually resulting in an energetic crisis and cell death [4].
FIGURE 4
Schematic illustration of the molecular and cellular pathways involved in high-dose vitamin C-mediated selective cytotoxicity to cancer cells that both overexpress GLUT1 and have become addicted to glycolysis for energy production
GAP and BPG denote glyceraldehyde 3-phosphate and 1,3-bisphosphoglycerate, respectively. This scheme is based on Ref. [4].
5.2.8. The Big Picture and Implications of the Study by Yun et al
The elegant study by Yun et al. [4] depicted an important big picture for high-dose vitamin C to selectively kill cancer cells while sparing normal cells (Figure 4). Two critical factors/conditions seem to determine this selectivity of vitamin C-mediated cancer cell killing: (1) the selective accumulation of DHA in cancer cells via a GLUT1-dependent transport mechanism, leading to intracellular GSH depletion and ROS accumulation, and (2) ROS-mediate inhibition of the glycolytic enzyme GAPDH in the background of oncogene-induced metabolic reprogramming (i.e., dependence on glycolysis for survival), resulting in ATP depletion and cell death. The KRAS and BRAF mutant colorectal cancer cells possess the above 2 critical factors and thereby show high susceptibility to high-dose vitamin C-mediated cytotoxicity. It can be further speculated that other types of cancer cells that meet the above 2 conditions would likely be also selectively killed by high-dose vitamin C. Hence, studies are warranted to determine if this unified machinery of ��DHA�CGLUT1�CROS�CGAPDH�� found by Yun et al. also operates in other types of cancer cells harboring mutations in oncogenes other than KRAS and BRAF. Since cancer cells typically exhibit altered status of ROS homeostasis and susceptibility to oxidative stress, future studies are also needed to understand how the intrinsic ROS generating and detoxifying systems in cancer cells impact the high-dose vitamin C-dependent tumoricidal activity. Moreover, continued efforts aiming to decipher the molecular basis of other ROS-inducing agents in cancer therapy will likely expand the paradigm of ROS biology and medicine [56, 66].
Acknowledgments
This work was supported by a grant (CA192936) from the U.S. National Cancer Institute/NIH.
ABBREVIATIONS
AMP adenosine monophosphate
AMPK adenosine monophosphate-activated protein kinase
ATP adenosine triphosphate
BPG 1,2-bisphosphoglycerate
DHA dehydroascorbate
DHAR dehydroascorbate reductase
GAP glyceraldehyde 3-phosphate
GAPDH glyceraldehyde 3-phosphate dehydrogenase
GLUT glucose transporter
GR glutathione reductase
GSH reduced form of glutathione
GSSG glutathione disulfide
HO-1 heme oxygenase-1
iNOS inducible nitric oxide synthase
METC mitochondrial electron transport chain
PARP poly(ADP-ribose) polymerase
RNS reactive nitrogen species
ROS reactive oxygen species
SGLT sodium-glucose transporter
SVCT sodium-dependent vitamin C transporterVitamin C, a Multi-Tasking Molecule, Finds a Molecular Target in Killing Cancer Cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5959041/��
J Cancer. 2019; 10(3): 757�C764.
Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis
Mingtong Ye,3,# Nengzhi Pang,1,# Ting Wan,4 Yuanling Huang,1 Tianyi Wei,3 Xuye Jiang,1 Yujia Zhou,1 Yufeng Huang,2 Hainan Yang,2 Zhenfeng Zhang,2,✉ and Lili Yang1,✉
1Department of Nutrition, Guangdong Provincial Key Laboratory of Food, Nutrition and Health, School of Public Health, Sun Yat-Sen University, Guangzhou, Guangdong, PR China.
2Department of Radiology, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, PR China.
3The First Women and Children's Hospital of Huizhou, Huizhou, Guangdong, PR China.
4Huizhou First People's Hospital, Huizhou, Guangdong, PR China.
Abstract
Switching aerobic respiration to anaerobic glycolysis of cancer cells plays an important role in development and progression of acquired resistance.Since vitamin C enabled the inhibition of glycolysis of cancer cells, and erlotinib-resistant sub-line of HCC827 (ER6 cells) switched its metabolic features to higher glycolysis for survival, we hypothesize that vitamin C is able to inhibit glycolysis of ER6 cells.
In this study, we found that both reduced vitamin C and oxidized vitamin C (DHA) could selectively suppress the viability of ER6 cells. DHA was efficient in inhibiting glycolysis and leading to energy crisis, which could be one mechanism for overcoming drug resistance to erlotinib of ER6 cells. Our data suggest that applying DHA could be a novel treatment strategy for NSCLC with acquired resistance to targeted therapy.
Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6360421/
J Biol Chem. 2014 Feb 7;289(6):3339-51. doi: 10.1074/jbc.M113.538157. Epub 2013 Dec 26.
The hypoxia-inducible factor renders cancer cells more sensitive to vitamin C-induced toxicity.
Tian W1, Wang Y, Xu Y, Guo X, Wang B, Sun L, Liu L, Cui F, Zhuang Q, Bao X, Schley G, Chung TL, Laslett AL, Willam C, Qin B, Maxwell PH, Esteban MA.
Author information
1
From the Key Laboratory of Regenerative Biology of the Chinese Academy of Sciences and Guangdong Provincial Key Laboratory of Stem Cells and Regenerative Medicine, South China Institute for Stem Cell Biology and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Guangzhou 510530, China.
Abstract
Megadose vitamin C (Vc) is one of the most enduring alternative treatments for diverse human diseases and is deeply engrafted in popular culture. Preliminary studies in the 1970s described potent effects of Vc on prolonging the survival of patients with terminal cancer, but these claims were later criticized. An improved knowledge of the pharmacokinetics of Vc and recent reports using cancer cell lines have renewed the interest in this subject. Despite these findings, using Vc as an adjuvant for anticancer therapy remains questionable, among other things because there is no proper mechanistic understanding. Here, we show that a Warburg effect triggered by activation of the hypoxia-inducible factor (HIF) pathway greatly enhances Vc-induced toxicity in multiple cancer cell lines, including von Hippel-Lindau (VHL)-defective renal cancer cells. HIF increases the intracellular uptake of oxidized Vc through its transcriptional target glucose transporter 1 (GLUT1), synergizing with the uptake of its reduced form through sodium-dependent Vc transporters. The resulting high levels of intracellular Vc induce oxidative stress and massive DNA damage, which then causes metabolic exhaustion by depleting cellular ATP reserves. HIF-positive cells are particularly sensitive to Vc-induced ATP reduction because they mostly rely on the rather inefficient glycolytic pathway for energy production. Thus, our experiments link Vc-induced toxicity and cancer metabolism, providing a new explanation for the preferential effect of Vc on cancer cells.2014��2��7��;289(6):3339-51��doi: 10.1074 / jbc.M113.538157��2013��12��26�ա�
ȱ���յ�����ʹ��ϸ����ά����c�յ��Ķ��Ը����С�
��һ����һ����һ����X����B����l����L������ׯQ����X��ʩ��G����TL, Laslett AL, Willam C����B, Maxwell PH, Esteban MA��
������Ϣ
1�й���ѧԺ��������ѧ�ص�ʵ���ҡ��㶫ʡ��ϸ��������ҽѧ�ص�ʵ���ң���������ҽҩ�����о�Ժ���ϸ�ϸ������ѧ������ҽѧ�о���������510530��
ժҪ
�����ά����C (Vc)�����ƶ������༲����־õ�����Ʒ�֮һ����ֲ�������Ļ��С�20����70����ij����о�������Vc���ӳ����ڰ�֢�������淽���ǿ�����ã�����Щ˵�������ܵ�����������Vc��ҩ������ѧ�Ľ�һ���˽�����ʹ�ð�ϸ��ϵ�ı������¼��������Ƕ���һ�������Ȥ����������Щ���֣�ʹ��Vc��Ϊ�������Ƶĸ�������Ȼ��������ģ���Ϊû���ʵ��Ļ����ϵ����⡣���������֤���˵����յ�����(HIF);��������������WarburgЧӦ�����ǿ��vc�Զ��ְ�֢ϸ��ϵ�Ķ��ԣ�����von Hippel-Lindau (VHL)ȱ�ݵ�����ϸ����HIFͨ����ת¼�е�������ת�˵���1 (GLUT1)��������Vc�İ�����ȡ��ͨ����������Vcת�˵���Эͬ�仹ԭ��ʽ����ȡ���ɴ˲�����ϸ���ڸ�ˮƽ��Vc��������Ӧ���ʹ�����DNA���ˣ�����ͨ������ϸ����ATP�������´�л˥�ߡ�hif����ϸ����vc�յ���ATP�����ر����У���Ϊ������Ҫ�������൱��Ч���ǽͽ�;����������������ˣ����ǵ�ʵ�齫Vc�յ��Ķ����방֢��л��ϵ������ΪVc��ϸ��������ЧӦ�ṩ���µĽ��͡�
KEYWORDS:
Cancer; DNA Damage; Hypoxia-inducible Factor (HIF); Oxidative Stress; Renal Cancer; Vitamin C; Warburg EffectThe hypoxia-inducible factor renders cancer cells more sensitive to vitamin C-induced toxicity. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/24371136��
Oxid Med Cell Longev. 2019 Dec 24;2019:7286737. doi: 10.1155/2019/7286737. eCollection 2019.
Pro- and Antioxidant Effects of Vitamin C in Cancer in correspondence to Its Dietary and Pharmacological Concentrations.
Pawlowska E1, Szczepanska J2, Blasiak J3.
Author information
1
Department of Orthodontics, Medical University of Lodz, 92-216 Lodz, Poland.
2
Department of Pediatric Dentistry, Medical University of Lodz, 92-216 Lodz, Poland.
3
Department of Molecular Genetics, Faculty of Biology and Environmental Protection, University of Lodz, 90-236 Lodz, Poland.
Abstract
Vitamin C is an antioxidant that may scavenge reactive oxygen species preventing DNA damage and other effects important in cancer transformation. Dietary vitamin C from natural sources is taken with other compounds affecting its bioavailability and biological effects.High pharmacological doses of vitamin C may induce prooxidant effects, detrimental for cancer cells. An oxidized form of vitamin C, dehydroascorbate, is transported through glucose transporters, and cancer cells switch from oxidative phosphorylation to glycolysis in energy production so an excess of vitamin C may limit glucose transport and ATP production resulting in energetic crisis and cell death. Vitamin C may change the metabolomic and epigenetic profiles of cancer cells, and activation of ten-eleven translocation (TET) proteins and downregulation of pluripotency factors by the vitamin may eradicate cancer stem cells.
Metastasis, the main reason of cancer-related deaths, requires breakage of anatomical barriers containing collagen, whose synthesis is promoted by vitamin C.
Vitamin C induces degradation of hypoxia-inducible factor, HIF-1, essential for the survival of tumor cells in hypoxic conditions. Dietary vitamin C may stimulate the immune system through activation of NK and T cells and monocytes. Pharmacological doses of vitamin C may inhibit cancer transformation in several pathways, but further studies are needed to address both mechanistic and clinical aspects of this effect.
Copyright © 2019 Elzbieta Pawlowska et al.
PMID: 31934267 PMCID: PMC6942884 DOI: 10.1155/2019/7286737Pro- and Antioxidant Effects of Vitamin C in Cancer in correspondence to Its Dietary and Pharmacological Concentrations. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31934267��
FASEB J. 2000 Jul;14(10):1352-61.
Stimulation of the pentose phosphate pathway and glutathione levels by dehydroascorbate, the oxidized form of vitamin C.
Puskas F1, Gergely P Jr, Banki K, Perl A.
Author information
1
Departments of Medicine, Pathology, and. Microbiology and Immunology, State University of New York Health Science Center, College of Medicine, Syracuse, New York 13210, USA.
Abstract
Ascorbic acid, or vitamin C, generally functions as an antioxidant by directly reacting with reactive oxygen intermediates and has a vital role in defenses against oxidative stress. However, ascorbic acid also has pro-oxidant properties and may cause apoptosis of lymphoid and myeloid cells.The present study shows that dehydroascorbate, the oxidized form of vitamin C, stimulates the antioxidant defenses of cells, preferentially importing dehydroascorbate over ascorbate. While 200-800 microM vitamin C caused apoptosis of Jurkat and H9 human T lymphocytes, pretreatment with 200-1000 microM dehydroascorbate stimulated activity of pentose phosphate pathway enzymes glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, and transaldolase, elevated intracellular glutathione levels, and inhibited H(2)O(2)-induced changes in mitochondrial transmembrane potential and cell death. A 3. 3-fold maximal glutathione elevation was observed after 48 h stimulation with 800 microM dehydroascorbate.
In itself, dehydroascorbate did not affect cytosolic or mitochondrial reactive oxygen intermediate levels as monitored by flow cytometry using oxidation-sensitive fluorescent probes. The data reveal a novel mechanism for increasing glutathione levels through stimulation of the pentose phosphate pathway and identify dehydroascorbate as an antioxidant for cells susceptible to the pro-oxidant and proapoptotic properties of vitamin C.
PMID: 10877828 DOI: 10.1096/fj.14.10.1352Stimulation of the pentose phosphate pathway and glutathione levels by dehydroascorbate, the oxidized form of vitamin C. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/10877828��
Nutrients. 2019 Dec 6;11(12). pii: E2997. doi: 10.3390/nu11122997.
Vitamin C Inhibits Triple-Negative Breast Cancer Metastasis by Affecting the Expression of YAP1 and Synaptopodin 2.
Gan L1,2, Camarena V1, Mustafi S1, Wang G1,3.
Author information
1
John P. Hussman Institute for Human Genomics, Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
2
State Key Laboratory of Animal Nutrition, College of Animal Science and Technology, China Agricultural University, Beijing 100193, China.
3
Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Abstract
Vitamin C supplementation has been shown to decrease triple-negative breast cancer (TNBC) metastasis. However, the molecular mechanism whereby vitamin C inhibits metastasis remains elusive. It has been postulated that vitamin C reduces the levels of HIF-1��, the master regulator of metastasis, by promoting its hydroxylation and degradation.Here, we show that vitamin C at 100 µM, a concentration achievable in the plasma in vivo by oral administration, blocks TNBC cell migration and invasion in vitro.
The protein level of HIF-1�� remains largely unchanged in cultured TNBC cells and xenografts, partially due to its upregulated transcription by vitamin C, suggesting that HIF-1�� unlikely mediates the action of vitamin C on metastasis. Vitamin C treatment upregulates the expression of synaptopodin 2 and downregulates the expression of the transcription coactivator YAP1, both genes in the Hippo pathway. The changes in SYNPO2 and YAP1 expression were subsequently validated at mRNA and protein levels in cultured TNBC cells and xenografts. Further experiments showed that vitamin C treatment inhibits F-actin assembly and lamellipodia formation, which correlates with the changes in SYNPO2 and YAP1 expression.
Overall, these results suggest that vitamin C inhibits TNBC metastasis by affecting the expression of SYNPO2 and YAP1. Vitamin C may thus have a potential role in the prevention and treatment of TNBC metastasis.
KEYWORDS:
F-actin; HIF-1��; YAP1; hippo pathway; lamellipodia; metastasis; synaptopodin 2; triple-negative breast cancer; vitamin CVitamin C Inhibits Triple-Negative Breast Cancer Metastasis by Affecting the Expression of YAP1 and Synaptopodin 2. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31817810��
Pharmacokinetic studies have found that the peak concentration of vitamin C achievable in blood plasma following oral ingestion is about 220 ��mol/L, whereas at least 15 mmol/L is achievable following IVC administration [ 7 ].
www.ncbi.nlm.nih.gov/pmc/articles/PMC5981254/��
In Silico Approach to Inhibition of Tyrosinase by Ascorbic Acid using molecular docking simulations
Abstract
Current evidence suggests that endogenous dopamine may act as a neurotoxin following its oxidation to an o-quinone and reaction with cellular thiols, which are neutoxic, which may occur spontaneously or via reaction with tyrosinase or some other enzymes.Tyrosinase (E.C. 1.14.18.1) with two cupper ions coordinated by three histidines is a bifunctional enzyme that catalyses both the hydroxylation of tyrosine to L-DOPA and the consequent oxidation of the resulting catechol-containing species to an o-quinone. Therefore, tyrosinase may play a role in neuromelanin formation in the brain and could be central to dopamine neurotoxicity by contributing to the neurodegeneration associated with Parkinson's disease.
In the present study, inhibitory effect of ascorbic acid against tyrosinase has been investigated and it has shown a remarkable inhibitory effect in in vitro assays. Then, the in silico-based experiments established through molecular docking calculations and scoring, docking search algorithm, and data plotting indicated that ascorbic acid is strong inhibitor of tyrosinase by interacting with four amino acid units (histidine 263, serine 282, phenylalanine 264, and valin 283) in the active site of the enzyme. The compound also had two long distant hydrogen bindings with Cu1 and Cu2 with distances of 3.57 and 3.41 Å, respectively, through its O5 atom.
https://www.researchgate.net/publication/263014823_In_Silico_Approach_to_Inhibition_of...
��
Int J Mol Sci. 2009 Jun; 10(6): 2440�C2475.
Published online 2009 May 26. doi: 10.3390/ijms10062440
PMCID: PMC2705500
PMID: 19582213
An Updated Review of Tyrosinase Inhibitors
Te-Sheng Chang
Author information Article notes Copyright and License information Disclaimer
Abstract
Tyrosinase is a multifunctional, glycosylated, and copper-containing oxidase, which catalyzes the first two steps in mammalian melanogenesis and is responsible for enzymatic browning reactions in damaged fruits during post-harvest handling and processing. Neither hyperpigmentation in human skin nor enzymatic browning in fruits are desirable. These phenomena have encouraged researchers to seek new potent tyrosinase inhibitors for use in foods and cosmetics. This article surveys tyrosinase inhibitors newly discovered from natural and synthetic sources. The inhibitory strength is compared with that of a standard inhibitor, kojic acid, and their inhibitory mechanisms are discussed.
Biosynthetic pathway of melanin [1�C4]. TYR, tyrosinase; TRP; tyrosinase related protein; dopa, 3,4-dihydroxyphenylalanine; DHICA, 5,6-dihydroxyindole-2-carboxylic acid; DHI, 5,6-dihydroxyindole; ICAQ, indole-2-carboxylic acid-5,6-quinone; IQ, indole-5,6-quinone; HBTA, 5-hydroxy-1,4-benzothiazinylalanine.
��
An Updated Review of Tyrosinase Inhibitors
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2705500/��
Enhanced anticancer activity of vitamin C via p53 in cancer cells
2012,
Ascorbate is an important natural antioxidant that can selectively kill cancer cells at pharmacological concentrations. Despite its benefit, it is quite difficult to predict the antitumor effects of ascorbate, because the relative cytotoxicity of ascorbate differs between cancer cell lines. Therefore, it is essential to examine the basis for this fundamental disagreement. Because p53 is activated by DNA-damaging stress and then regulates various cellular conditions, we hypothesized that p53 can sensitize cancer cells to ascorbate. Using isogenic cancer cells, we observed that the presence of p53 can affect ascorbate cytotoxicity, and also reactivation of p53 can make cancer cells sensitive to ascorbate. p53-dependent enhancement of ascorbate cytotoxicity is caused by increased reactive oxygen species generation via a differentially regulated p53 transcriptional network. We also found that transcriptionally activated p53 was derived from MDM2 ubiquitination by ascorbate and subsequently its signaling network renders cancer cells more susceptible to oxidative stress. Similar to the p53 effect on in vitro ascorbate cytotoxicity, inhibition of tumor growth is also stronger in p53-expressing tumors than in p53-deficient ones in vivo. This is the first observation that ascorbate cytotoxicity is positively related to p53 expression, activating its transcriptional network to worsen intracellular oxidative stress and consequently enhancing its cytotoxicity. Based on our study, reactivation of p53 may help to achieve more consistent cytotoxic effects of ascorbate in cancer therapies.
Enhanced antitumor activity of vitamin C via p53 in Cancer cells | Request PDF
https://www.researchgate.net/publication/230670580_Enhanced_antitumor_activity_of_vitamin_C_via_p53_in_Cancer_cells��
Theranostics
. 2019 Jun 9;9(15):4461-4473. doi: 10.7150/thno.35219. eCollection 2019.
Vitamin C Kills Thyroid Cancer Cells Through ROS-dependent Inhibition of MAPK/ERK and PI3K/AKT Pathways via Distinct Mechanisms
Xi Su 1, Zhen Shen 2, Qi Yang 1, Fang Sui 1, Jun Pu 1, Jingjing Ma 1, Sharui Ma 1, Demao Yao 3, Meiju Ji 4, Peng Hou 1
Affiliations
1Key Laboratory for Tumor Precision Medicine of Shaanxi Province and Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China.
2Department of Otolaryngology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China.
3Department of Surgery, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China.
4Center for Translational Medicine, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China.
Abstract
Background:Vitamin C has been demonstrated to kill BRAF mutant colorectal cancer cells selectively. BRAF mutation is the most common genetic alteration in thyroid tumor development and progression; however, the antitumor efficacy of vitamin C in thyroid cancer remains to be explored.
Methods: The effect of vitamin C on thyroid cancer cell proliferation and apoptosis was assessed by the MTT assay and flow cytometry. Xenograft and transgenic mouse models were used to determine its in vivo antitumor activity of vitamin C. Molecular and biochemical methods were used to elucidate the underlying mechanisms of anticancer activity of vitamin C in thyroid cancer.
Results: Pharmaceutical concentration of vitamin C significantly inhibited thyroid cancer cell proliferation and induced cell apoptosis regardless of BRAF mutation status. We demonstrated that the elevated level of Vitamin C in the plasma following a high dose of intraperitoneal injection dramatically inhibited the growth of xenograft tumors. Similar results were obtained in the transgenic mouse model. Mechanistically, vitamin C eradicated BRAF wild-type thyroid cancer cells through ROS-mediated decrease in the activity of EGF/EGFR-MAPK/ERK signaling and an increase in AKT ubiquitination and degradation. On the other hand, vitamin C exerted its antitumor activity in BRAF mutant thyroid cancer cells by inhibiting the activity of ATP-dependent MAPK/ERK signaling and inducing proteasome degradation of AKT via the ROS-dependent pathway.
Conclusions: Our data demonstrate that vitamin C kills thyroid cancer cells by inhibiting MAPK/ERK and PI3K/AKT pathways via a ROS-dependent mechanism and suggest that pharmaceutical concentration of vitamin C has potential clinical use in thyroid cancer therapy.
Keywords: BRAF mutation; PI3K/AKT pathway; ROS; Vitamin C; thyroid cancer.Vitamin C Kills Thyroid Cancer Cells Through ROS-dependent Inhibition of MAPK/ERK and PI3K/AKT Pathways via Distinct Mechanisms - PubMed
https://pubmed.ncbi.nlm.nih.gov/31285773/��
J Cancer. 2019; 10(3): 757�C764.
Published online 2019 Jan 1. doi: 10.7150/jca.28087
PMCID: PMC6360421
PMID: 30719175
Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis
Mingtong Ye,3,# Nengzhi Pang,1,# Ting Wan,4 Yuanling Huang,1 Tianyi Wei,3 Xuye Jiang,1 Yujia Zhou,1 Yufeng Huang,2 Hainan Yang,2 Zhenfeng Zhang,2,✉ and Lili Yang1,✉
Author information Article notes Copyright and License information Disclaimer
1Department of Nutrition, Guangdong Provincial Key Laboratory of Food, Nutrition and Health, School of Public Health, Sun Yat-Sen University, Guangzhou, Guangdong, PR China.
2Department of Radiology, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, PR China.
3The First Women and Children's Hospital of Huizhou, Huizhou, Guangdong, PR China.
4Huizhou First People's Hospital, Huizhou, Guangdong, PR China.
Switching aerobic respiration to anaerobic glycolysis of cancer cells plays an important role in development and progression of acquired resistance. Since vitamin C enabled the inhibition of glycolysis of cancer cells, and erlotinib-resistant sub-line of HCC827 (ER6 cells) switched its metabolic features to higher glycolysis for survival, we hypothesize that vitamin C is able to inhibit glycolysis of ER6 cells. In this study, we found that both reduced vitamin C and oxidized vitamin C (DHA) could selectively suppress the viability of ER6 cells. DHA was efficient in inhibiting glycolysis and leading to energy crisis, which could be one mechanism for overcoming drug resistance to erlotinib of ER6 cells. Our data suggest that applying DHA could be a novel treatment strategy for NSCLC with acquired resistance to targeted therapy.
Keywords: Drug resistance, oxidized vitamin C (DHA), energy homeostasis, glycolysis.Oxidized Vitamin C (DHA) Overcomes Resistance to EGFR-targeted Therapy of Lung Cancer through Disturbing Energy Homeostasis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6360421/��
The Association Between Ascorbate and the Hypoxia-Inducible Factors in Human Renal Cell Carcinoma Requires a Functional Von Hippel-Lindau Protein
Hypoxia-inducible transcription factors (HIFs) drive angiogenesis and cancer cell growth, contributing to an aggressive tumor phenotype. HIF-�� protein levels and activity are controlled at the post-translational level by HIF hydroxylases. Hydroxylated HIF-�� is recognized by the von Hippel Lindau (VHL) tumor suppressor and targeted for degradation. The HIF hydroxylases are members of the iron and 2-oxoglutarate-dependent dioxygenases, which require ascorbate as cofactor for activity. Clear cell renal cell carcinomas (ccRCC) harbor mutations in the VHL gene, whereas papillary RCC (pRCC) have a functional VHL. These natural occurring VHL variants in RCC enable the testing, in clinical samples, of the hypothesis that ascorbate modulates HIF-�� levels through its role as a cofactor for the HIF hydroxylases. We measured ascorbate, HIF-1��, and HIF-2�� protein and HIF downstream targets BNIP3, CA9, cyclin D1, GLUT1, and VEGF (combined to generate the HIF pathway score) in VHL-defective ccRCC (n = 73) and VHL-proficient pRCC human tumor tissue (n = 41). HIF and ascorbate levels were increased in ccRCC and pRCC tumors compared to matched renal cortex. HIF-1 and total HIF pathway activation scores were decreased with higher ascorbate in pRCC tumors (Spearman r = −0.38, p < 0.05 and r = −0.35, p < 0.05). This was not evident for ccRCC tumors. In mechanistic studies in vitro, ascorbate influenced HIF-1 activity in VHL-proficient, but not VHL-defective ccRCC cells. Our results indicate that ccRCC, which lacks a functional VHL, does not respond to ascorbate-mediated modulation of the HIF response. This contrasts with the demonstrated association between ascorbate content and the HIF pathway observed in pRCC and other tumors with a functional VHL. The results support a role for ascorbate as a modulator of HIF activity and tumor aggression in cancer types with a functional hypoxic response.
Conclusion
Our data in pRCC tumors adds further evidence for an association between ascorbate and the HIF-pathway in cancer. The comparison of VHL-defective ccRCC and VHL-proficient pRCC tumors supports the role of ascorbate as a vital cofactor of dioxygenase enzymes that target HIF degradation via VHL. Our data provides no evidence to support ascorbate intervention for ccRCC, which represent the majority of RCC patients.Frontiers | The Association Between Ascorbate and the Hypoxia-Inducible Factors in Human Renal Cell Carcinoma Requires a Functional Von Hippel-Lindau Protein | Oncology
https://www.frontiersin.org/articles/10.3389/fonc.2018.00574/full��
J Transl Med. 2008; 6: 50.
Anti-angiogenic effect of high doses of ascorbic acid
Nina A Mikirova,corresponding author#1 Thomas E Ichim,#2 and Neil H Riordan#2
1Bio-Communications Research Institute, Wichita, Kansas, USA
2Medistem Laboratories Inc, Chandler, Arizona, USA
Abstract
Pharmaceutical doses of ascorbic acid (AA, vitamin C, or its salts) have been reported to exert anticancer activity in vitro and in vivo. One proposed mechanism involves direct cytotoxicity mediated by accumulation of ascorbic acid radicals and hydrogen peroxide in the extracellular environment of tumor cells. However, therapeutic effects have been reported at concentrations insufficient to induce direct tumor cell death. We hypothesized that AA may exert anti-angiogenic effects. To test this, we expanded endothelial progenitor cells (EPCs) from peripheral blood and assessed, whether or not high dose AA would inhibit EPC ability to migrate, change energy metabolism, and tube formation ability. We also evaluated the effects of high dose AA on angiogenic activities of HUVECs (human umbilical vein endothelial cells) and HUAECs (human umbilical arterial endothelial cells). According to our data, concentrations of AA higher than 100 mg/dl suppressed capillary-like tube formation on Matrigel for all cells tested and the effect was more pronounced for progenitor cells in comparison with mature cells. Co-culture of differentiated endothelial cells with progenitor cells showed that there was incorporation of EPCs in vessels formed by HUVECs and HUAECs. Cell migration was assessed using an in vitro wound healing model. The results of these experiments showed an inverse correlation between AA concentrations relative to both cell migration and gap filling capacity. Suppression of NO (nitric oxide) generation appeared to be one of the mechanisms by which AA mediated angiostatic effects. This study supports further investigation into non-cytotoxic antitumor activities of AA.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2562367/
��
Ferroptosis, a new form of cell death: opportunities and challenges in cancerhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC6441206Mar 29, 2019 �� Background. Ferroptosis was first proposed by Dixon as a novel cell death in 2012 [].Unlike autophagy and apoptosis, ferroptosis is an iron-dependent and reactive oxygen species (ROS)-reliant cell death with characteristics mainly of cytological changes, including decreased or vanished mitochondria cristae, a ruptured outer mitochondrial membrane, and a condensed mitochondrial ��
- Cited by: 61
- Publish Year: 2019
- Author: Yanhua Mou, Jun Wang, Jinchun Wu, Dan He,
�й�����ʡ��ɳ������·87�ţ����ϴ�ѧ����ҽԺҽѧ��ѧ�о������й�������������������ѧ�ص�ʵ���ң�410008��3���ϴ�ѧ����ҽѧԺ��������ҽԺ��������ҽԺ����ɳ410008��
Ferroptosis is a novel type of cell death with distinct properties and recognizing functions involved in physical conditions or various diseases including cancers. The fast-growing studies of ferroptosis in cancer have boosted a perspective for its usage in cancer therapeutics. Here, we review the current findings of ferroptosis regulation and especially focus on the function of ncRNAs in mediating the process of cell ferroptotic death and on how ferroptosis was in relation to other regulated cell deaths. Aberrant ferroptosis in diverse cancer types and tissues were summarized, and we elaborated recent data about the novel actors of some "conventional" drugs or natural compounds as ferroptosis inducers in cancer. Finally, we deliberate future orientation for ferroptosis in cancer cells and current unsettled issues, which may forward the speed of clinical use of ferroptosis induction in cancer treatment.
��������һ�����͵�ϸ�����������ж��ص����ʺ�ʶ���ܣ��漰������״���������֢���ڵĸ��ּ����������������о���tosis�ڰ�֢�е�Ӧ�����������ڰ�֢�����е�ǰ������������ǻع������������õ��ڵ����·��֣��ر��עncrna�ڽ鵼ϸ�����������������е����ã��Լ���������������������ϸ�������Ĺ�ϵ�����������˲�ͬ��֢���ͺ���֯�л���������ã���������һЩ����ͳ��ҩ�����Ȼ��������Ϊ��֢���������յ���������Ϊ����������ݡ�������Ƕ�����ϸ�������´���δ����չ�����Ŀǰ��δ�����������������ۣ�����ܻ�ӿ����´��յ������������е��ٴ�Ӧ�á���
2020 Jun 20;9(6):1505.doi: 10.3390/cells9061505.Ferroptosis and Cancer: Mitochondria Meet the "Iron Maiden" Cell Death��
Anna Martina Battaglia 1, Roberta Chirillo 1, Ilenia Aversa 1, Alessandro Sacco 1, Francesco Costanzo 1 2, Flavia Biamonte 1 31Department of Experimental and Clinical Medicine, "Magna Graecia" University of Catanzaro, 88100 Catanzaro, Italy.AbstractFerroptosis is a new type of oxidative regulated cell death (RCD) driven by iron-dependent lipid peroxidation. As major sites of iron utilization and master regulators of oxidative metabolism, mitochondria are the main source of reactive oxygen species (ROS) and, thus, play a role in this type of RCD. Ferroptosis is, indeed, associated with severe damage in mitochondrial morphology, bioenergetics, and metabolism. Furthermore, dysregulation of mitochondrial metabolism is considered a biochemical feature of neurodegenerative diseases linked to ferroptosis. Whether mitochondrial dysfunction can, per se, initiate ferroptosis and whether mitochondrial function in ferroptosis is context-dependent are still under debate. Cancer cells accumulate high levels of iron and ROS to promote their metabolic activity and growth. Of note, cancer cell metabolic rewiring is often associated with acquired sensitivity to ferroptosis. This strongly suggests that ferroptosis may act as an adaptive response to metabolic imbalance and, thus, may constitute a new promising way to eradicate malignant cells. Here, we review the current literature on the role of mitochondria in ferroptosis, and we discuss opportunities to potentially use mitochondria-mediated ferroptosis as a new strategy for cancer therapy.��ϸ�������방֢:�����������������ӡ�ϸ������
Anna Martina Battaglia 1, Roberta Chirillo 1, Ilenia Aversa 1, Alessandro Sacco 1, Francesco Costanzo 1 2, Flavia Biamonte 1 3
1��ϣ����̹����ѧʵ�����ٴ�ҽѧϵ���������̹����88100��
ժҪ
��������һ�����͵���������ϸ������(oxidative regulated cell death, RCD)������������֬�ʹ�����������������������Ҫ����λ���������л����Ҫ�������ӣ��ǻ�����(ROS)����Ҫ��Դ���ڸ���RCD�з������á�������ȷʵ����������̬�������ܺ��³´�л���������йء����⣬�������лʧ������Ϊ�������´���ص��������Լ��������������������幦���ϰ������Ƿ����������������֢���Լ�����������֢�е������幦���Ƿ��뻷���йأ�Ŀǰ�Դ������顣��ϸ�����۸�ˮƽ������ROS���Դٽ����л���������ֵ��ע����ǣ���ϸ���Ĵ�л����ͨ�������Զ������õ��������йء���ǿ�ұ����������ÿ����Ǵ�лʧ���һ����Ӧ�Է�Ӧ����ˣ����ܹ���һ���µ���ϣ���ķ������Ը�������ϸ�����ڴˣ����ǻع���Ŀǰ�й�����������ϸ������֢�е����õ����ף�������������������鵼����ϸ������֢��Ϊ��֢�����²��ԵĿ����ԡ���
Keywords: ROS; cancer; cell death; ferroptosis; iron; mitochondria.Ferroptosis and Cancer: Mitochondria Meet the "Iron Maiden" Cell Death - PubMed��
J Biomed Science. 2017 Oct 30;
Expression of hypoxia inducible factor 1�� and 2�� and its association with vitamin C level in thyroid lesions
Background: Cells adapt to hypoxia by transcriptional induction of genes that participate in regulation of angiogenesis, glucose metabolism and cell proliferation. The primary factors mediating cell response to low oxygen tension are hypoxia inducible factors (HIFs), oxygen-dependent transcription activators. The stability and activity of the �� subunits of HIFs are controlled by hydroxylation reactions that require ascorbate as a cofactor. Therefore, deficiency of intracellular vitamin C could contribute to HIFs overactivation. In this study, we investigated whether vitamin C content of human thyroid lesions is associated with HIF-1�� and HIF-2�� protein levels.
Methods: Expression of HIF-1�� and HIF-2�� as well as vitamin C content was analyzed in thyroid lesions and cultured thyroid carcinoma cell lines (FTC-133 and 8305c) treated with hypoxia-mimetic agent (cobalt chloride) and ascorbic acid. The expression of HIFs and hypoxia-induced glucose transporters were determined by Western blots while quantitative real-time PCR (qRT-PCR) was performed to detect HIFs mRNA levels. Ascorbate and dehydroascorbate levels were measured by HPLC method.
Results: We found an inverse correlation between vitamin C level and HIF-1�� but not HIF-2�� expression in thyroid lesions. These results agree with our in vitro study showing that vitamin C induced a dose - dependent decrease of HIF-1�� but not HIF-2�� protein level in thyroid cancer cells FTC-133 and 8305C. The decreased HIF-1�� expression was correlated with reduced expression of hypoxia-related glucose transporter 1 (GLUT1) in thyroid cancer cells.
Conclusion: The results demonstrate that HIF-1�� activation is associated with vitamin C content in thyroid lesions. Our study suggests that high tumor tissue ascorbate level could limit the expression of HIF-1�� and its targets in thyroid lesions.
Keywords: HIF-1��/ HIF-2��/ vitamin C/GLUT1/ thyroid cancer.Expression of hypoxia inducible factor 1�� and 2�� and its association with vitamin C level in thyroid lesions - PubMed
https://pubmed.ncbi.nlm.nih.gov/29084538/��
High-dose vitamin C therapy for KRAS/BRAF mutant cancers
, by Lewis Cantley and Jihye Yun
More than 80 years ago, the biochemist Otto Warburg observed that cancer cells consume more glucose and produce more lactate even in the presence of ample oxygen as compared with normal cells. This phenomenon, called aerobic glycolysis or the Warburg effect, has been exploited for visualizing tumors in the clinical setting by imaging their uptake of the radiolabeled glucose analog, [18F] fluoro-2-deoxyglucose (FDG), via Positron Emission Tomography (PET). Although the exact mechanism by which glucose reprograming contributes to tumorigenesis remains unclear, numerous genetic and pharmacological studies showed that this metabolic switch can be essential for cancer survival and proliferation. Thus, targeting glycolysis may offer cancer patients a more selective strategy to treat cancer.
More than half of colorectal cancers (CRCs) harbor activating mutations in KRAS or BRAF, yet those cancers are the most refractory to current targeted therapies. Our group and others showed that oncogenic mutations in KRAS or BRAF contribute to the Warburg effect and the addiction to glucose in part by upregulating a glucose transporter, GLUT1, that allows cancer cells to take up glucose efficiently. These data suggest a strategy for targeting KRAS or BRAF mutant cancer by exploiting the selective expression of GLUT1 and the metabolic liability that comes with increased reliance on glycolysis. Indeed, by targeting these unique features in these cancer cells, we recently showed that high dose vitamin C could selectively kill KRAS or BRAF mutant CRC cells.
Interestingly, GLUT1 not only transports glucose but also transports dehydroascorbic acid (DHA), the oxidized form of vitamin C. Subsequently, we observed that CRC cells harboring KRAS or BRAF mutations uniquely increased uptake of DHA via GLUT1 when treated with vitamin C. The increased DHA uptake in mutant cells produced oxidative stress, increasing the level of reactive oxygen species (ROS) in cells because intracellular DHA was rapidly reduced back to vitamin C at the expense of glutathione (GSH), a master antioxidant in cells. In turn, we found that elevated ROS activated poly (ADP-ribose) polymerase (PARP), a DNA repair enzyme, consuming large amounts of cellular NAD+ as its cofactor. The depletion of NAD+ resulted in the inactivation of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) because GAPDH requires NAD+ as a cofactor. Inhibiting GAPDH in highly glycolytic KRAS or BRAF mutant cells ultimately led to an energy crisis and cell death not seen in their KRAS and BRAF wild-type counterparts. Finally, we showed that high-dose vitamin C therapy reduced both the number and size of tumors in KRAS or BRAF mutant mice as compared to mice without these mutations, confirming that vitamin C selectively targets KRAS or BRAF mutant tumors in mouse models of colon tumors. In short, ascorbate functions as a ��Trojan horse�� through its conversion to DHA, and insidiously enters cancer cells via GLUT1 to promote the generation of intracellular ROS, which ultimately kills the cancer cell.
Although our study showed that DHA is the pharmaceutically active agent, reduced ascorbate (not directly DHA) is used for preclinical and clinical anti-cancer studies because ascorbate has a significantly longer plasma half-life than DHA (>60 min vs a few min). Moreover, the reduced ascorbate can be efficiently oxidized to DHA in tumors�� extracellular fluids where high level of ROS usually exists. Given that KRAS and BRAF mutations are not only restricted to colorectal cancer, vitamin C may be benefit other types of tumors. For example, 90% of pancreatic cancers and approximately 30% of lung cancers have KRAS mutations. These KRAS mutant tumors also have high GLUT1 expression and are linked to altered glucose metabolism similar to CRC. Therefore, high dose vitamin C may benefit other types of tumors harboring KRAS/BRAF mutations. Based on these results, Weill Cornell Medicine is currently conducting a Phase II clinical trial to examine the effects of intravenous high dose vitamin C in the treatment of KRAS-mutant cancers, and Sun Yat-sen University Cancer Center in China are conducting placebo-controlled, randomized Phase III clinical trials in colorectal cancer patients in combination of chemotherapy.
While KRAS and BRAF mutations are certainly two of the most frequently mutated oncogenes in human cancer, they are not the only mutations known to affect glucose metabolism and sensitivity towards ascorbate therapy. For example, we and others have found that renal cancer cells (RCC) with loss of VHL (Von Hippel-Lindau), a tumor suppressor that destabilizes HIF1A via ubiquination, are significantly sensitive to ascorbate treatment compared to VHL-proficient cells. RCC-VHL null cells have increased HIF1A transcriptional activity, which not only increases GLUT1 expression, but also deregulates many other glycolytic enzymes to induce metabolic reprograming. Additionally, cancers with increased levels of DNA damage, such as those that have been treated with radiation or those with mutations in BRCA genes, are more reliant on DNA repair mediated by PARP. Pharmacologic vitamin C might selectively impair such cancers by depriving them of the NAD+ necessary for PARP activity.�����ά����C����KRAS / BRAFͻ�䰩֢High-dose vitamin C therapy for KRAS/BRAF mutant cancers
, by Lewis Cantley and Jihye Yun
80����ǰ�����ﻯѧ�Ұ��С��ֲ���Otto Warburg���۲쵽��������ϸ����ȣ���ϸ����ʹ�ڳ��������������Ҳ���ĸ���������ǣ���������������ᡣ��������Ϊ�����ǽͽ��WarburgЧӦ���ѱ�����ͨ�������ӷ���ϲ�ɨ�裨PET���Է����ǵ�������������[18F]��-2-���������ǣ�FDG������ȡ���г��Ӷ����ٴ��Ͽ��ӻ������������в�����������ر�̴ٳ�����������ȷ�л��ƣ��������Ŵ�ѧ��ҩ��ѧ�о����������ִ�лת�������������������ɢ����������Ҫ����ˣ������ǽͽ����Ϊ��֢�����ṩ���ư�֢�ĸ���ѡ���ԵIJ��ԡ�
����һ��Ľ�ֱ������CRC������KRAS��BRAF�еĻͻ�䣬����Щ��֢�Ե�ǰ�İ����Ʒ������Ρ����ǵ��о�С��������о���Ա������KRAS��BRAF�е��°���ͻ�䲿��ͨ���ϵ�������ת�˵���GLUT1���ٽ�WarburgЧӦ�Ͷ������ǵij��Ӷ�ʹ��ϸ����Ч���������ǡ���Щ���ݱ�����ͨ������GLUT1��ѡ���Ա���Ͷ��ǽͽ��������������³´�л����������KRAS��BRAFͻ�䰩�IJ��ԡ�ȷʵ��ͨ�������Щ��ϸ���е���Щ�����������������������������ά����C����ѡ����ɱ��KRAS��BRAFͻ��CRCϸ����
��Ȥ���ǣ�GLUT1����ת�������ǣ�����ת��ά����C��������ʽ���⿹��Ѫ�ᣨDHA����������ǹ۲쵽����KRAS��BRAFͻ���CRCϸ����ά����C������ͨ��GLUT1���ص�������DHA�����ա�ͻ��ϸ����DHA�����ղ�������Ӧ����������ϸ���л�������ROS����ˮƽ����Ϊϸ����DHAѸ�ٻ�ԭ��ά����C���������ģ�GSH����ϸ���е���Ҫ���������������������Ƿ������ߵ�ROS����ľۣ�ADP-���ǣ��ۺ�ø��PARP����һ��DNA��ø�����Ĵ�����ϸ��NAD +��Ϊ�丨���ӡ� NAD +�ĺĽߵ��¸���ȩ3-��������ø��GAPDH��ʧ���ΪGAPDH��ҪNAD +��Ϊ�����ӡ��ڸ߶��ǽͽ��KRAS��BRAFͻ��ϸ��������GAPDH���յ�������Σ����ϸ��������������KRAS��BRAFҰ���Ͷ�Ӧ�����ǿ������ġ�������DZ�������û����Щͻ���С����ȣ�����ά����C���ƿɼ���KRAS��BRAFͻ��С���������������ʹ�С���Ӷ�֤ʵά����C�ڽ᳦����С��ģ����ѡ������KRAS��BRAFͻ�������������֮������Ѫ��ͨ��ת��ΪDHA��������ľ���������ã���ͨ��GLUT1����ؽ��방ϸ���Դٽ�ϸ����ROS�����ɣ�����ɱ����ϸ����
�������ǵ��о�����DHA��ҩ����Լ�������ԭ�ԵĿ���Ѫ���Σ�������ֱ��DHA�������ٴ�ǰ���ٴ������о�����Ϊ����Ѫ���ε�Ѫ����˥�ڱ�DHA���öࣨ> 60���ӱȼ����ӣ� �����⣬��ͨ�����ڸ�ˮƽROS������ϸ����Һ�У���ԭ�Ŀ���Ѫ�������Ч������ΪDHA�����ǵ�KRAS��BRAFͻ�䲻�����ڽ�ֱ������ά����C�����������������͵����������磬90�������ٰ��ʹ�Լ30���ķΰ�����KRASͻ�䡣��ЩKRASͻ������Ҳ���и�GLUT1���������CRC���ƣ��������Ǵ�л�ı��йء���ˣ�������ά����C������������������KRAS / BRAFͻ���������������Щ������������ζ�ҽѧ����Ŀǰ���ڽ���II���ٴ����飬�Լ�龲��ע������ά����C������KRASͻ���Ͱ�֢�е����ã��й�����ɽ��ѧ��֢�������ڽ��а�ο�����գ��ڴ����������ϻ��Ƶ����III���ٴ����顣
����KRAS��BRAFͻ�����������֢������������°�����ͻ�䣬�����Dz�������֪��Ӱ�������Ǵ�л�ͶԿ���Ѫ���Ʒ������Ե�Ψһͻ�䡣���磬���Ǻ������˷��֣���ʧVHL������ϸ����RCC����Von Hippel-Lindau����һ�������������ӣ���ͨ�����ػ�����ʹHIF1Aʧȥ�ȶ��ԣ��뿹VHL��ϸ����ȣ��Կ���Ѫ�����Ʒdz����С� ȱ��RCC-VHL��ϸ���������ӵ�HIF1Aת¼���ԣ��ⲻ��������GLUT1�ı�����һ���������������ǽͽ�ø�ĵ��أ����յ���л�ر�̡����⣬DNA����ˮƽ���ߵİ�֢�������ѽ��ܷ������ƻ�BRCA����ͻ��İ�֢����������PARP�鵼��DNA����ҩ����ά����C���ܻ����PARP�����������NAD +���Ӷ�ѡ���Ե����֢��
https://www.cancer.gov/research/key-initiatives/ras/ras-central/blog/2020/yun-cantley-vitamin-c��
.jpg)