¡¡
¡¡
¡¡
Journal of the American College of Cardiology
Volume 59, Issue 9, February 2012
Update on Myocarditis
Dr. Ingrid Kindermann, Klinik f¨¹r Innere Medizin III, Kardiologie, Angiologie und Internistische Intensivmedizin, Universitätsklinikum des Saarlandes, Kirrberger Strasse 1, Homburg/Saar 66421, GermanyAbstract
Myocarditis is an inflammatory disease of the heart frequently resulting from viral infections and/or post-viral immune-mediated responses. It is one of the important causes of dilated cardiomyopathy worldwide. The diagnosis is presumed on clinical presentation and noninvasive diagnostic methods such as cardiovascular magnetic resonance imaging. Endomyocardial biopsy remains the gold standard for in vivo diagnosis of myocarditis. The therapeutic and prognostic benefits of endomyocardial biopsy results have recently been demonstrated in several clinical trials. Although remarkable advances in diagnosis, understanding of pathophysiological mechanisms, and treatment of acute myocarditis were gained during the last years, no standard treatment strategies could be defined as yet, apart from standard heart failure therapy and physical rest. In severe cases, mechanical support or heart transplantation may become necessary. There is some evidence that immunosuppressive and immunomodulating therapy are effective for chronic, virus-negative inflammatory cardiomyopathy. Further investigations by controlled, randomized studies are needed to definitively determine their role in the treatment of myocarditis.
Key Words
heart failureinflammatory cardiomyopathymyocarditis
Myocarditis is an inflammatory disease of the myocardium caused by different infectious and noninfectious triggers (Table 1). In 1995, myocarditis was defined by the World Health Organization (WHO)/International Society and Federation of Cardiology (ISFC) as an inflammatory disease of the heart muscle, diagnosed by established histological, immunological, and immunohistochemical criteria (1). Myocarditis often results from common viral infections and post-viral immune-mediated responses. With the development of new molecular techniques such as polymerase chain reaction (PCR) and in situ hybridization, the spectrum of most frequently detected viruses in endomyocardial biopsies (EMB) shifted from classic enteroviruses and adenovirus to mainly parvovirus B19 (PVB19) and human herpesvirus 6 (2,3). In European studies, mainly PVB19 was detected in patients with biopsy-proven myocarditis (4¨C6). Whether and why there are geographic differences concerning the distribution of different virus species in myocarditis are currently debated (7). Local and temporal epidemiological differences of virus infections have to be considered, as well as different diagnostic procedures (8). The discussion, whether PVB19 is an innocent bystander or a pathological agent and whether quantification of virus load is a helpful approach, is ongoing (9).
¡¡
Table 1
Etiology of Myocarditis
In patients with human immunodeficiency virus infection, myocarditis was observed in >50% of performed autopsies (10). Furthermore, myocarditis can be triggered by nonviral infections, for example, with Borrelia burgdorferi (Lyme disease), Corynebacterium diphtheriae, or Trypanosoma cruzi (Chagas disease) (11). Numerous medications like antipsychotics (e.g., clozapine [12]), antibiotics (penicillin, ampicillin, sulfonamides, tetracyclines), and antiphlogistics (e.g., mesalamine [13]) can induce hypersensitivity eosinophilic myocarditis, which commonly is reversible after withdrawal of the causative agent. Eosinophilic-lymphocytic myocarditis may also occur after smallpox vaccination (14). Systemic autoimmune diseases such as Churg-Strauss syndrome (15) or hypereosinophilic syndrome (Loeffler's disease) (16) can be associated with eosinophilic myocarditis. In case of cardiac sarcoidosis (17) and giant cell myocarditis (18), which are rare causes of inflammatory myocardial disease, early diagnosis and treatment initiation will significantly improve prognosis.
Myocarditis is regarded as a precursor of dilated cardiomyopathy (DCM), which is currently the most frequent reason for heart transplantation (19). Post-mortem data identify myocarditis in 8.6% to 12% of cases of sudden death in young adults (20). Long-term follow-up studies in patients with acute myocarditis have documented the development of DCM in 21% of patients over a mean follow-up period of 3 years (21).
Pathophysiology of Myocarditis
The pathophysiology of myocarditis in humans is not completely understood. Murine models of enteroviral myocarditis suggest that the course of viral myocarditis is characterized by 3 phases (Fig. 1) (22). First, the entry of the virus into the myocytes is mediated through a specific receptor. Coxsackieviruses of group B and some adenoviruses use a common transmembrane receptor (coxsackievirus and adenovirus receptor [CAR]) for internalization of the viral genome into the myocyte (23). Coxsackieviruses utilize the deflecting decay accelerating factor (DAF) and adenoviruses special integrins (¦Áv¦Â3 and ¦Áv¦Â5) as coreceptors. In the absence of CAR expression on cardiac myocytes, viral infection and inflammation does not occur (24). In explanted hearts of patients with DCM, higher CAR expression was demonstrated than in the myocardium of patients with other heart diseases or healthy hearts (25). Whether increased human CAR expression is a predisposing factor for facilitating viral myocarditis has to be shown in future studies.
Figure 1
Time Course of Viral Myocarditis
Time course of viral myocarditis in 3 phases (derived from murine models). The acute phase of myocarditis takes only a few days, whereas the subacute and chronic phase covers a few weeks to several months.
Modified from Kawai (22).
After viral entry acute injury of the myocytes, induced by virus replication leads to myocyte necrosis, exposure of intracellular antigens (e.g., cardiac myosin), and activation of the host's immune system, which is characterized by the invasion of natural killer cells and macrophages followed by T lymphocytes (Fig. 2). The acute phase of myocarditis takes only a few days. After the acute phase of virus-induced injury, the second phase is characterized by (auto)immune reactions. This subacute phase, which covers few weeks to several months, is defined by activated virus-specific T lymphocytes, which may target the host's organs by molecular mimicry. Cytokine activation (tumor necrosis factor-alpha, interleukin [IL]-1 and -6) and antibodies to viral and cardiac proteins may aggravate cardiac damage and cause impairment of the contractile function. In most patients with myocarditis, immune response declines with virus elimination, and left ventricular (LV) function recovers without sequelae. However, in some murine models and probably in patients, (auto)immune processes persist independently of detection of virus genome in the myocardium and lead to the chronic phase, which is characterized by myocardial remodeling and development of DCM (26).
Figure 2
Pathophysiology of Viral Myocarditis
Pathophysiology of viral myocarditis: after viral entry, virus replication leads to acute injury of the myocytes (acute myocarditis) and to activation of the host's immune system (subacute myocarditis). IFN = interferon; IL = interleukin; TNF = tumor necrosis factor.
Clinical Presentation and Diagnosis of Myocarditis
The clinical manifestation of myocarditis varies with a broad spectrum of symptoms ranging from asymptomatic courses to presentations with signs of myocardial infarction to devastating illness with cardiogenic shock. Chest pain, cardiac arrhythmias, and acute or chronic heart failure (HF) can occur during the course of the disease (4). Hence, the diagnosis of myocarditis based on the clinical presentation alone is usually not possible.
Biomarkers and virus serology
Biomarkers (such as troponins or creatine kinase) lack specificity, but may help to confirm the diagnosis of myocarditis (27,28). In patients with acute myocarditis, serum concentrations of troponin I and T are elevated more frequently than creatine kinase myocardial band fraction (29), and higher levels of troponin T have been shown to be of prognostic value. Nonspecific serum markers of inflammation including leukocytes and C-reactive protein can be elevated in case of acute myocarditis (28,29), but normal values do not exclude an acute myocardial inflammatory process (30).
The utility of virus serology in patients with suspected myocarditis remains unproven. Mahfoud et al. (30) investigated the diagnostic value of virus serology in comparison to analyses of EMB including viral genome detection in patients with clinically suspected myocarditis. Only in 5 of 124 patients (4%) there was serological evidence of an infection with the same virus that was detected by nested PCR in EMB. This result indicates that virus serology should not be commonly used for the diagnosis of myocardial infection in patients with suspected myocarditis. The findings can be explained by the fact that patients are referred for diagnostics and medical treatment with a significant delay from the onset of the initial infection, potentially ranging from some weeks to a few months, when the acute phase of viral myocarditis has already resolved. Moreover, the diagnostic value of serology is also limited in that most viruses involved in the pathogenesis of myocarditis are highly prevalent in the population, for example >70% of the population in Germany have been tested seropositive for PVB19 immunoglobulin G antibodies (31). The interpretation of antibody assays is also complicated by other confounders such as reactivation or reinfection (e.g., in case of herpesvirus infections) or by cross reactions, which have been described for infections with Epstein-Barr virus or enterovirus.
Electrocardiogram
The electrocardiogram (ECG) is widely used as a screening tool despite low sensitivity (32). The ECG findings in patients with myocarditis vary from nonspecific T-wave and ST-segment changes to ST-segment elevation mimicking an acute myocardial infarction (27,33). Also, atrial or ventricular conduction delays as well as supraventricular and ventricular arrhythmias can occur in patients with inflammatory heart disease. The presence of Q waves or a new left bundle branch block are associated with higher rates of cardiac death or heart transplantation (34). Recently, the prognostic role of ECG parameters was investigated in patients with suspected myocarditis (35). The ECG recorded at the time of EMB were related to cardiac outcome during long-term follow-up. A QTc prolongation >440 ms, an abnormal QRS axis, and ventricular ectopic beats were associated with poor clinical outcome. A prolonged QRS duration of ¡Ý120 ms was found to be an independent predictor for cardiac death or heart transplantation. Hence, the ECG represents an easily available tool for risk stratification in patients with suspected myocarditis.
Echocardiography
There are no specific echocardiographic features of myocarditis. However, echocardiography allows the evaluation of cardiac chamber sizes and wall thickness as well as systolic and diastolic function in patients with myocarditis. It is one of the most important tools to rule out other causes of HF such as valvular heart disease or other cardiomyopathies (hypertrophic or restrictive cardiomyopathy). Especially before an EMB procedure, echocardiography is needed to exclude pericardial effusion and intracavitary thrombi, which have been noted in up to 25% of patients (36). The assessment of different echocardiographic parameters is also of prognostic relevance. Patients with fulminant myocarditis often have normal cardiac chamber sizes with an increased septal thickness secondary to acute myocardial edema, whereas patients with acute myocarditis have marked left ventricular dilation and normal wall thickness (37).
Cardiovascular magnetic resonance
Cardiovascular magnetic resonance (CMR) imaging has evolved as a noninvasive and valuable clinical tool for the diagnosis of myocarditis. In particular, the initial changes in myocardial tissue during the first phase of myocardial inflammation represent attractive targets for a successful CMR-based imaging approach. The T2-weighted edema imaging is routinely used as a tool for evaluating the presence of ¡°acute myocardial inflammation¡± (Figs. 3A and 3B) (38,39). Moreover, ECG-triggered T1-weighted images are obtained both before and within the first minutes after gadolinium-diethylenetriaminepentacetate (Gd-DTPA) infusion. Hence, this sequence has been entitled ¡°myocardial early gadolinium enhancement¡± (40). Several studies have confirmed the diagnostic value of this sequence, although it is prone to artefacts that decrease specificity (38). Finally, a T1-weighted segmented inversion-recovery gradient-echo sequence (41) was shown to be superior to others used for contrast-enhancement as it improved the difference in signal intensity between myocardial regions with (diseased) and those without (healthy) Gd-DTPA accumulation, thereby leading to a much better contrast. This method is known as ¡°late gadolinium enhancement (LGE) imaging.¡± In case of myocarditis, LGE imaging revealed 2 common patterns of myocardial damage: either an intramural, rimlike pattern in the septal wall or a subepicardial (patchy) distribution in the free LV lateral wall (Figs. 3C and 3D) (42). However, LGE imaging does not allow to differentiate between acute and chronic inflammation, but represents damaged myocardium. Hence, interpretation of the stage of the illness depends largely on the clinical context. Moreover, the value of LGE imaging for successful diagnosis of myocarditis seems to be related to the histological degree and extent of inflammation (43).
Figure 3
MRI Findings in Patients With Myocarditis
Cardiac magnetic resonance imaging (MRI) images of a young patient presenting with acute chest pain syndrome due to acute myocarditis. (A) Long-axis and (B) short-axis T2-weighted edema images demonstrating focal myocardial edema in the subepicardium of the left midventricular lateral wall (red arrows). Corresponding (C) long-axis and (D) short-axis T1-weighted late gadolinium enhancement images demonstrate presence of typical late gadolinium enhancement in the subepicardium of the left midventricular lateral wall and the basal septum (red arrows).
Each individual CMR method has individual advantages but also disadvantages in the diagnosis of myocarditis. Consequently, the combination of these methods is currently regarded as the most appropriate noninvasive approach with the highest sensitivity and specificity (38,40). Because there is a high diagnostic conformity between CMR-based and biopsy-based results, it seems to be reasonable to initially perform CMR in patients with clinically suspected myocarditis and/or nonischemic cardiomyopathy (43). However, if the diagnosis of myocarditis is merely based on the CMR study, then detailed information about the degree of inflammation, the presence of special forms of myocarditis (e.g., giant cell or eosinophilic myocarditis, which require specific therapies), or the presence and type of virus is not available. In addition, less severe forms of myocarditis may not be detected by CMR because of its limited spatial resolution as compared to EMB.
Endomyocardial biopsy
The gold standard in diagnosis of myocarditis is still the EMB. According to the Dallas criteria, acute myocarditis is defined by lymphocytic infiltrates in association with myocyte necrosis (Figs. 4A and 4B). Borderline myocarditis is characterized by inflammatory infiltrates without evidence of myocyte necrosis (44). The Dallas criteria are limited by the high interobserver variability in interpreting biopsy specimens (in particular with regard to borderline myocarditis) and because noncellular inflammatory processes cannot be detected (45). Thus, immunohistochemistry (Figs. 4B and 4D) is gaining further acceptance in the diagnosis of myocarditis. Monoclonal antibodies allow the characterization and localization of the mononuclear cell infiltrates: for example, CD3 for T cells, PGM1 (CD68) for activated macrophages, and human leukocyte antigen (HLA)-DR-¦Á to assess HLA class II expression in professional antigen-presenting immune cells (26). With the use of these immunohistological methods the number of EMB revealing myocarditis markedly increased (46). According to the World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies, EMB is considered to be inflamed by immunohistochemical detection of focal or diffuse mononuclear infiltrates (T lymphocytes and macrophages) with >14 cells/mm2, in addition to enhanced expression of HLA class II molecules (1). Molecular biological detection of cardiotropic viruses can be performed by nested PCR/real time-PCR from EMB (47). In situ hybridization techniques allow the identification of cell types replicating viral genomes as shown for PVB19 and enterovirus in Figures 4E and 4F. Because of the lack of available facilities and clinical experience, EMB appears to be infrequently used to diagnose myocarditis. However, when performed by experienced interventionalists, left and right ventricular EMB are safe procedures, with a major complication rate of <1% (48). Recent studies demonstrated not only the diagnostic but also the prognostic value of EMB in patients with suspected myocarditis (4).
Figure 4
Histopathological, Immunohistological, and Molecular Biological Findings in Hearts of Patients With Myocarditis
Histology and immunohistology of (A, B) acute myocarditis and (C, D) chronic myocarditis. In acute myocarditis, numerous necrotic myocytes (A, arrows) are associated with mononuclear cell infiltrates including CD3+ T cells (B), whereas in chronic myocarditis, inflammatory cells such as CD68+ macrophages (D) are mainly present in areas with fibrosis (C, blue staining). (E, F) Radioactive in situ hybridization reveals PVB19 nucleic acid in endothelial cells of an arteriole in a patient with chronic myocarditis (E), whereas enterovirus ribonucleic acid is detected in several myocytes (F).
Treatment of Myocarditis
Although treatment of myocarditis should be focused on the causal pathophysiology, the effect of a specific causative therapy has only been confirmed in a few studies on inflammatory heart diseases such as sarcoidosis and giant cell myocarditis. Because of the high incidence of LV dysfunction, evidence-based HF therapy is mandatory in these patients. As no clinical trials of HF therapy in patients with myocarditis have been performed, only data from animal models can be consulted.
Specific treatment
Specific types of myocarditis based on autoimmunity are treated with immunosuppression, for example, in patients with giant cell myocarditis or cardiac sarcoidosis. In case of giant cell myocarditis, combined treatment with immunosuppressants (cyclosporine and corticosteroids with or without azathioprine or muronomab-CDs) may improve the poor prognosis, and yield a median survival time of 12 months compared with 3 months for untreated affected patients (18,49). Nevertheless, a minority of patients require mechanical circulatory support or heart transplantation within 1 year.
Withdrawal of immunosuppression can results in recurrent and sometimes fatal giant cell myocarditis. In case of cardiac sarcoidosis, early immunosuppressive therapy with high-dose corticosteroids has been associated with improved cardiac function (17). The prognosis of patients with treatment is variable, with a 5 year survival ranging from 60% to 90% (50). Specific treatment options for viral myocarditis are not established yet.
Heart failure therapy
As no pathogen-specific therapy of viral myocarditis has been shown to improve survival free of HF, for now treatment is symptomatic and based on the clinical presentation. Fortunately, most cases of myocarditis are mild (21,51,52). Pharmacological treatment of HF should be initiated according to the current guidelines (53). Standard HF regime including beta-blockers, diuretics, angiotensin-converting enzyme (ACE) inhibitors or angiotensin-II receptor blockers (ARBs) should be initiated according to the New York Heart Association (NYHA) functional class.
ACE Inhibitors and ARBs
By early initiation of renin-angiotensin blockade, chronic maladaptive cardiac remodeling can be attenuated, and the progression to dilated cardiomyopathy can be reduced. In mice models, the ACE inhibitor captopril as well as the ARBs losartan and olmesartan significantly reduced inflammation, necrosis, and fibrosis in experimental autoimmune or virus-induced myocarditis (54¨C57).
In rats with DCM caused by experimental autoimmune myocarditis, olmesartan treatment significantly improved left ventricular function and ameliorated the progression of cardiac remodeling (58). Treatment with different ACE inhibitors and ARBs in animal models may also down-regulate the potential autoimmune component of the disease without increasing the levels of the infectious agents that may have initiated myocarditis (59).
Diuretics
Diuretics are used to prevent or to treat fluid overload. Torsemide reduced the progression of myocarditis to DCM in a rat model of inflammatory cardiomyopathy by decreasing fibrosis, myocyte sizes, and myocardial protein levels of transforming growth factor-beta-1, collagen III, and aldosterone synthase, beyond its renal effects (60).
Beta-Blockers
Beta-blocker treatment should be avoided in the acute phase of decompensated HF and in the very early treatment of fulminant myocarditis (53). Beta-blockade improves ventricular function, reduces hospital admission for worsening HF, and increases survival. Experimental data suggest that the type of beta-blocker has an impact in inflammatory cardiomyopathy. Carvedilol was shown to be cardioprotective in rats with autoimmune myocarditis by suppression of inflammatory cytokines and its antioxidant properties, whereas metoprolol and propranolol were not (61). Metoprolol administration exerted deleterious effects in acute murine coxsackievirus B3 myocarditis showing significantly increased inflammation and necrosis as well as mortality compared to the placebo group (62). However, the underlying mechanism was not identified. In encephalomyocarditis virus inoculated mice, administration of epinephrine exacerbated myocarditis and increased mortality whereas treatment with propranolol decreased myocardial necrosis and infiltration of inflammatory cells as well as gene suppression of tumor necrosis factor-alpha, IL-6, and IL-10. Consequently, a reduced severity of myocarditis and a decreased mortality resulted. In patients with suspected myocarditis, there is evidence that lack of beta-blocker treatment is associated with poor outcome (4).
Aldosterone Antagonists
Administration of aldosterone antagonists is recommended for systolic HF patients with persistent NYHA functional class II to IV symptoms. Aldosterone antagonists reduced hospital admission for worsening HF and increased survival in addition to established HF therapy (53). Anti-inflammatory effects of eplerenone on murine viral myocarditis were shown by inhibition of mast cell-derived proteinases and resulted in an improvement of myocardial remodeling by suppressing fibrosis (63).
Cardiac Glycosides
Cardiac glycosides reduced morbidity in patients with symptomatic systolic HF in NYHA functional class II to IV. High doses of digoxin increased myocardial production of pro-inflammatory cytokines and worsened myocardial injury in virus-infected mice (64). Digoxin may limit the maximal tolerated dose of beta-blocker due to bradycardia or heart block. Therefore, digoxin should be avoided in patients suffering from acute HF induced by viral myocarditis.
Calcium-Channel Blockers
Calcium-channel blockers are not generally recommended in the management of acute HF (53). However, in a murine model of congestive HF induced by viral myocarditis, amlodipine appeared to have a protective effect against myocardial injury in mice by inhibition of over-production of nitric oxide (65). The effects of pranidipine versus amlodipine were analyzed in rats with HF induced by autoimmune myocarditis. Pranidipine and amlodipine ameliorated the progression of left ventricular dysfunction and cardiac remodeling (66).
Nonsteroidal anti-inflammatory drugs and colchicine
Nonsteroidal anti-inflammatory drugs (NSAIDs) and colchicine are applied for anti-inflammatory treatment of pericarditis (67) as a ¡°nonspecific¡± anti-inflammatory therapy, whereas there is no indication for application in patients with myocarditis. In murine models of acute viral myocarditis, indomethacin and NSAIDs increased inflammation and mortality (68,69). Therefore, NSAIDs in the lowest required dose are reserved for patients with perimyocarditis in whom LV function is clearly normal and have prominent chest pain from pericarditis.
Physical activity
In acute myocarditis, avoidance of aerobic physical activity is indicated in addition to pharmacological therapy (70,71). In a murine model of coxsackievirus B3 myocarditis, sustained exercise increased mortality and induced a suppression of T lymphocytes (72). Myocarditis is a relevant cause of sudden death in young athletes (73,74). In 2005, the 36th Bethesda Conference Task Forces recommended that athletes with probable or definite evidence of myocarditis should be withdrawn from all competitive sports for at least 6 months and may return to training and competition if LV function and cardiac dimensions have returned to normal and if there are no clinically relevant arrhythmias (74). The duration of abstinence from competitive sports after recovery from acute myocarditis is still a matter of debate. In patients with stable HF after previous history of myocarditis, physical exercise is recommended (70).
Pacemaker and implantable cardiac defibrillator
Temporary pacemaker insertion is indicated for patients with acute myocarditis who present with symptomatic atrioventricular (AV) block II or III. Lyme carditis patients can have varying degrees of AV conduction abnormalities (75). Persistent AV block III is rare, but necessitates permanent pacing. In Chagas disease, conduction defects with a progression to complete heart block, and life-threatening ventricular arrhythmias are common (11). Because of dyssynchrony, chronic right ventricular pacing should be avoided in patients with restricted LV function, and implantation of a biventricular pacemaker should be considered (76). Insertion of an implantable cardiac defibrillator (ICD) in patients with myocarditis is indicated after cardiac arrest due to ventricular fibrillation or after symptomatic ventricular tachycardia. Cardiac resynchronization therapy with defibrillator function is indicated for patients with impaired LV function (LV ejection fraction ¡Ü35%) and left bundle branch block in NYHA functional class II to IV (76). However, premature implantation of an ICD or a cardiac resynchronization therapy/ICD system should be avoided in patients with inflammatory cardiomyopathy as LV function may improve significantly with guideline-based HF therapy.
Because of the worse prognosis, pacemaker or ICD implantation may be considered early in patients with sarcoidosis or giant cell myocarditis, if second- or third-degree AV block or ventricular arrhythmias have been documented (17,18).
Mechanical circulatory support, heart transplantation
For patients with cardiogenic shock due to acute fulminant myocarditis who deteriorate despite optimal medical treatment, mechanical circulatory support or extracorporeal membrane oxygenation may be required to bridge the patient to recovery or heart transplantation (27). Despite the severe initial presentation, these patients have a good prognosis, with >60% to 80% survivors and a high rate of recovery of native ventricular function (77,78). Aggressive therapy with mechanical circulatory support systems is warranted and should be considered early for patients with fulminant acute myocarditis when maximal pharmacological therapy failed.
Investigational treatment options
Because mechanism-based therapy of myocarditis is not proven, different approaches have been investigated in clinical studies in recent years. More than 20 treatment trials have been reported, using immunosuppressive, immunomodulating, or anti-inflammatory agents as well as immunoadsorption therapy (Tables 2 and 3).⇓ Immunosuppressive therapy has been evaluated in the trials listed in the following text, and in many smaller studies, but has not become a standard in therapy of inflammatory cardiomyopathy. One of the largest randomized, controlled treatment trials, the Myocarditis Treatment Trial (79), failed to show a benefit from immunosuppressive therapy additional to HF therapy. There was neither a difference in mortality nor an improvement of LV ejection fraction after 1 year of treatment with prednisone with either azathioprine or cyclosporine versus placebo. These results might be due to a lack of consensus in interpretation of EMB findings. However, no immunohistology for the detection of inflammatory cells and no molecular biological analyses of EMB were used for the detection of infectious agents. Thereby, patients with cardiac viral infection might have been treated with immunosuppressive agents, which could have increased virus replication and damaged the myocardium.
Table 2
Treatment Trials of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy, Randomized Controlled Studies
Table 3
Treatment Trials of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy, Nonrandomized Controlled/Uncontrolled and Randomized Uncontrolled Studies
The majority of treatment studies used the Dallas criteria for histological classification of EMB. As mentioned in the preceding text, there is an ongoing debate indicating that the Dallas criteria are not suitable for the diagnosis of this inflammatory disease because of the variation in histological interpretation and the inability of detection of noncellulary mediated inflammation (45). Intermediate data from the ESETCID (European Study of Epidemiology and Treatment of Inflammatory Heart Disease) study (80) showed that inflammation was eradicated in 59% of the patients treated with immunosuppressive agents; however, it also vanished spontaneously in 40% of the placebo group. The high rate of spontaneous improvements in patients with acute inflammatory cardiomyopathy (81) is not considered in many treatment trials. To detect modest (but real) differences in treatment, further placebo controlled treatment studies are needed to reduce this major limitation in therapy assessment. The validity of the listed trials is limited by there frequently being no adequate immunohistological and molecular biological analysis of EMB, and in several trials, no control groups were implemented.
An algorithm outlining a proposed diagnostic and therapeutic approach in patients with suspected myocarditis is pictured in Figure 5.
Figure 5
Proposed Diagnostic and Therapeutic Algorithm for Suspected Myocarditis
Proposed diagnostic and therapeutic algorithm for patients with suspected acute myocarditis considering biomarkers, cardiac magnetic resonance imaging (cMRI), and endomyocardial biopsy (EMB). Bi-VAD = biventricular assist device; Circ. = circulatory; ECMO = extracorporeal membrane oxygenation; LV = left ventricular; LVAD = left ventricular assist device.
Immunoglobulin Treatment
The rationale to use immunoglobulin in viral myocarditis results from their antiviral and immunomodulating effects. In recent onset of myocarditis or DCM, there was no difference in LV function in patients receiving intravenous immunoglobulin and patients given placebo (82). However, children with acute myocarditis showed an improvement of LV function and survival in the first year after treatment (83).
Immunoadsorption
The target of immunoadsorption is the elimination of anticardiac antibodies against various cardiac cell proteins, which have been identified in patients with DCM and myocarditis (84). There is evidence that removal of circulating antibodies by immunoadsorption in DCM improved cardiac function (84) and clinical and humoral markers of HF severity (exercise capacity, N-terminal pro¨CB-type natriuretic peptide (NT-pro¨CBNP) [85,86]) as well as hemodynamic parameters (cardiac and stroke volume index, systemic vascular resistance) (87). Furthermore, immunoadsorption decreased myocardial inflammation (85). In patients with inflammatory cardiomyopathy, LV systolic function improved after protein A immunoadsorption (88). Currently a multicenter, randomized, double-blind, prospective study on the effects of immunoadsorption on cardiac function in 200 patients with DCM is ongoing (NCT00558584). First results are expected in 2011 and 2012.
Immunosuppressive Treatment
Treatments with immunosuppressive agents (cyclosporine, prednisolone, azathioprine) in acute myocarditis have shown controversial results (Tables 2 and 3) (79,80). In chronic DCM, azathioprine and prednisone resulted in an improvement of LV function and NYHA class (89,90). The TIMIC (Immunosuppressive Therapy in Patients With Virus Negative Inflammatory Cardiomyopathy) study (91) was the first randomized, placebo-controlled trial in which all EMB were studied for inflammation by histological and immunohistological criteria. Molecular biological analyses were performed in all biopsy specimens to exclude viral infection. A significant improvement of LV ejection fraction and a decrease in LV dimensions resulted from immunosuppressive therapy with prednisone and azathioprine.
Antiviral Treatment
The rationale to use antiviral drugs results from the knowledge that most common cases of myocarditis are induced by viral infections. In murine coxsackievirus B3-induced myocarditis, interferon (IFN)-beta and IFN-alpha2 therapy protected myocytes against injury and decreased inflammatory cell infiltrates. However, only IFN-beta resulted in an elimination of cardiac viral load (92). Treatment with IFN-beta in patients with myocardial enteroviral or adenoviral persistence and LV dysfunction showed an elimination of viral genomes in all patients and an improvement of LV function in 15 of 22 patients (6). In the subsequent placebo-controlled, randomized, double-blind, Europe-wide multicenter BICC (Betaferon in patients with chronic viral cardiomyopathy) study, 143 patients with inflammatory DCM and confirmed myocardial viral infection were treated with Betaferon (IFN-beta-1b) versus placebo (93). Treatment with Betaferon reduced significantly viral load (enteroviruses) in the myocardium; however, complete viral elimination (PVB19) was not achieved in all patients. A variety of parameters were evaluated, but only the NYHA functional class and patient global assessment improved.
Prognosis and Outcome
The prognosis of patients with myocarditis depends on clinical presentation, different clinical parameters, and EMB findings. Patients with acute myocarditis and preserved LV ejection fraction have a good prognosis with a high rate of spontaneous improvement without sequelae (36). Patients with fulminant viral myocarditis and hemodynamic compromise at presentation have an excellent long-term prognosis and are more likely to experience complete recovery than patients with acute myocarditis (81), if aggressive pharmacological and/or mechanical circulatory support is initiated early during the fulminant phase. In patients with cardiac sarcoidosis or giant cell myocarditis, prognosis depends probably on an early initiated treatment (immunosuppressive therapy or heart transplantation).
Among clinical markers NYHA functional class, right ventricular dysfunction, elevated pulmonary artery pressure, and syncope are able to predict survival free from cardiac death or heart transplantation (36). Other clinical risk factors in patients with suspected myocarditis are low systolic, diastolic, and mean arterial blood pressures as well as high heart rate, as demonstrated by Mahfoud et al. (in review). A prolonged QRS duration ¡Ý120 ms has also been shown to predict for cardiac death or heart transplantation in patients with suspected myocarditis (35).
The prognostic value of EMB findings has been long controversial because of the lack of specific treatment options (48). Since 2007, a consensus statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology recommends EMB in patients with suspected specific myocardial disorders with unique prognosis and specific treatment recommendations (94).
Further studies to investigate the utility of novel tools for the analysis of EMB were recommended. In a study by Kindermann et al. (4), the prognostic role of EMB, with detailed analysis of myocardial specimens including immunohistochemical staining for characterization of inflammation and molecular pathological analysis for detection of viral genome, was examined in 181 patients with suspected myocarditis. Immunohistological evidence of inflammatory infiltrates in the myocardium (with or without evidence of viral genome detection) was demonstrated to predict cardiovascular death and the need for heart transplantation (Fig. 6). Neither the histopathological Dallas criteria nor the detection of viral genome was a predictor of poor outcome. A risk stratification approach based on biopsy results, clinical findings, and drug treatment demonstrated that patients in NYHA functional class III or IV with positive immunohistology and without beta-blocker therapy have the poorest prognosis, with a 5-year transplantation-free survival rate of only 39% (Fig. 6).
Figure 6
Freedom From Cardiac Death and HTx by Endomyocardial Biopsy Findings, IH Results
Immunohistology (IH) evidence of inflammatory infiltrates in the myocardium (IH positive) predicts cardiovascular death and the need for heart transplantation (HTx).
Conclusions
Myocarditis is an under-diagnosed cardiac disease resulting from a broad range of infectious, immune, and toxic causes. Affected patients may recover, develop DCM, or die. Although remarkable advances in diagnosis, understanding of pathophysiological mechanisms, and treatment of myocarditis have been achieved during the last years, standard treatment strategies remain limited to evidence-based HF therapy in the most cases. Immunomodulating and immunosuppressive therapy have been effective, particularly in a single-center trial (TIMIC study), only in chronic, virus-negative inflammatory cardiomyopathy. Immunosuppression therapy is beneficial for acute giant cell myocarditis and sarcoidosis, and for patients with acute myocarditis associated with autoimmune diseases, for example, lupus myocarditis. There is some evidence that antiviral therapies and antimicrobial agents may have a beneficial therapeutic effect, but controlled, adequately powered, randomized studies are needed to determine their role in treatment of myocarditis.
¡¡
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (KFO 196 to Dr. Böhm and SFB-TR 19 to Drs. Kandolf and Klingel) and by the Ministry of Science and Economy of the State of the Saarland (Drs. Böhm and Kindermann). The authors have reported they have no relationships relevant to the contents of this paper to disclose.Abbreviations and Acronyms
AVatrioventricularCARcoxsackievirus and adenovirus receptorCMRcardiovascular magnetic resonance imagingDCMdilated cardiomyopathyEMBendomyocardial biopsyHFheart failureHLAhuman leukocyte antigenICDimplantable cardiac defibrillatorIFNinterferonILinterleukinLGElate gadolinium enhancementLVleft ventricularNSAIDnonsteroidal anti-inflammatory drugPCRpolymerase chain reactionPVB19parvovirus B19
Update on Myocarditis | JACC: Journal of the American College of Cardiology
http://www.onlinejacc.org/content/59/9/779¡¡
Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10
K Folkers, S Vadhanavikit, and S A Mortensen
PNAS February 1, 1985 82 (3) 901-904; https://doi.org/10.1073/pnas.82.3.901
Abstract
The tissue levels of coenzyme Q10 (CoQ10) in endomyocardial biopsy samples and blood from 43 patients with cardiomyopathy were determined by steps of extraction, purification, and HPLC. The biopsy samples were obtained from the patients after a routine heart catheterization. Six patients were of class I, 18 of class II, 11 of class III, and 8 of class IV (classified according to guidelines of the New York Heart Association). True control biopsies of healthy hearts are not available for ethical reasons, but the data of the four classes by severity of disease may be justifiably compared. Patients of class IV had lower (P less than 0.01) levels of CoQ10 than those of class I. Patients of classes III and IV had a lower (P less than 0.0001) level than those of classes I and II. Biopsy samples were obtained from five patients after treatment with CoQ10 for 2-8 months. The increases of CoQ10 levels ranged from 20% to 85%; the mean value was higher (P less than 0.02) than before treatment. Blood deficiencies also increase with severity of disease, but not as markedly as for the biopsies. These data reveal a myocardial deficiency of CoQ10, which is higher with increasing severity of disease and is reduced by therapy. This biochemistry correlates with the effective treatment of cardiomyopathy with CoQ10.Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10 | PNAS
https://www.pnas.org/content/82/3/901.long?utm_source=TrendMD&utm_medium=cpc&utm_campaign=Proc_Natl_Acad_Sci_U_S_A_TrendMD_1¡¡
Response of patients in classes III and IV of cardiomyopathy to therapy in a blind and crossover trial with coenzyme Q10
P H Langsjoen, S Vadhanavikit, and K Folkers
PNAS June 1, 1985 82 (12) 4240-4244; https://doi.org/10.1073/pnas.82.12.4240
Abstract
Coenzyme Q10 (CoQ10), a biochemically established redox component of respiration including the coupled mechanisms of electron transfer and oxidative phosphorylation, is naturally present in the human myocardium. A double-blind and double-crossover trial has been conducted by administering CoQ10 and a matching placebo orally to two groups of patients having class III or IV cardiomyopathy (classification according to criteria of the New York Heart Association). Group A received CoQ10 and then placebo; group B received placebo and then CoQ10. Blood levels of CoQ10 and cardiac function were determined at 0 and 4 weeks (control stabilization period) and at 16 and 28 weeks (after the 12-week CoQ/placebo-treatment periods). For group A, significant increases in CoQ10 blood levels and cardiac function occurred during CoQ10 treatment and then decreased during crossover to placebo. For group B, there was no change in CoQ10 blood levels and cardiac function during placebo treatment, but increases in both parameters occurred in crossover to CoQ10. These patients, steadily worsening and expected to die within 2 years under conventional therapy, generally showed an extraordinary clinical improvement, indicating that CoQ10 therapy might extend the lives of such patients. This improvement could be due to correction of a myocardial deficiency of CoQ10 and to enhanced synthesis of CoQ10-requiring enzymes.Response of patients in classes III and IV of cardiomyopathy to therapy in a blind and crossover trial with coenzyme Q10 | PNAS
https://www.pnas.org/content/82/12/4240.long?utm_source=TrendMD&utm_medium=cpc&utm_campaign=Proc_Natl_Acad_Sci_U_S_A_TrendMD_0¡¡
Silencing the CSF-1 Axis Using Nanoparticle Encapsulated siRNA Mitigates Viral and Autoimmune Myocarditis
Ingmar Sören Meyer1,2†, Carl Christoph Goetzke3,4†, Meike Kespohl3,4, Martina Sauter5, Arnd Heuser6, Volker Eckstein7, Hans-Peter Vornlocher8, Daniel G. Anderson9,10,11, Jan Haas1,2, Benjamin Meder1,2, Hugo Albert Katus1,2, Karin Klingel5, Antje Beling3,4*† and Florian Leuschner1,2*†
1Internal Medicine III, University Hospital Heidelberg, Heidelberg, Germany
2DZHK (German Centre for Cardiovascular Research), Partner Site Heidelberg-Mannheim, Heidelberg, Germany
3Institute of Biochemistry, Charit¨¦ - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany
4DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Berlin, Germany
5Cardiopathology, Institute for Pathology and Neuropathology, University Hospital Tuebingen, Tuebingen, Germany
6Max-Delbrueck-Center for Molecular Medicine Berlin, Berlin, Germany
7Internal Medicine V, University Hospital Heidelberg, Heidelberg, Germany
8Axolabs GmbH, Kulmbach, Germany
9David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, United States
10Cardiovascular Division, Department of Medicine, Brigham and Women's Hospital, Boston, MA, United States
11Department of Chemical Engineering, Massachusetts Institute of Technology (MIT), Cambridge, MA, United States
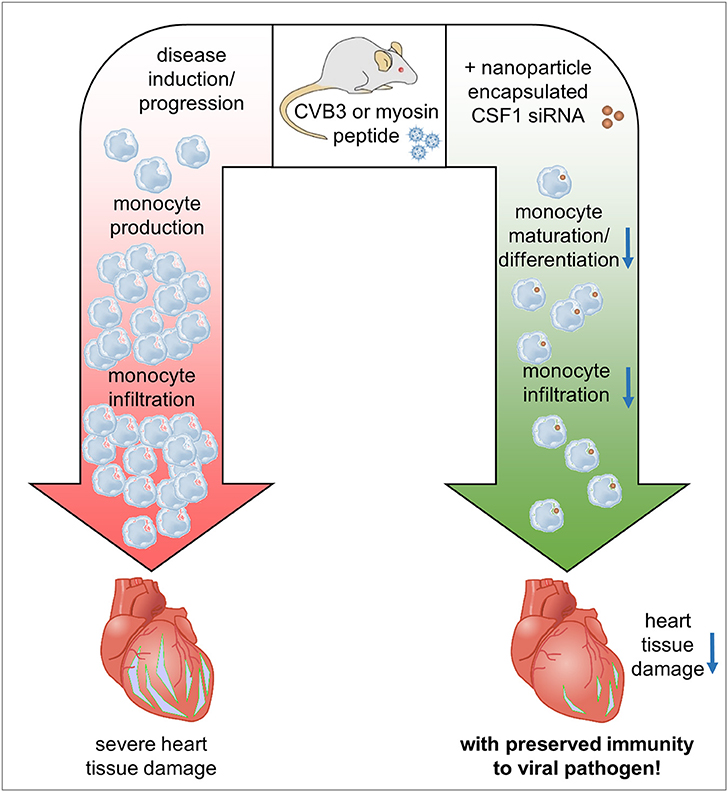
Myocarditis is an inflammatory disease of the heart muscle most commonly caused by viral infection and often maintained by autoimmunity. Virus-induced tissue damage triggers chemokine production and, subsequently, immune cell infiltration with pro-inflammatory and pro-fibrotic cytokine production follows. In patients, the overall inflammatory burden determines the disease outcome. Following the aim to define specific molecules that drive both immunopathology and/or autoimmunity in inflammatory heart disease, here we report on increased expression of colony stimulating factor 1 (CSF-1) in patients with myocarditis. CSF-1 controls monocytes originating from hematopoietic stem cells and subsequent progenitor stages. Both, monocytes and macrophages are centrally involved in mediating tissue damage and fibrotic scarring in the heart. CSF-1 influences monocytes via engagement of CSF-1 receptor, and it is also produced by cells of the mononuclear phagocyte system themselves. Based on this, we sought to modulate the virus-triggered inflammatory response in an experimental model of Coxsackievirus B3-induced myocarditis by silencing the CSF-1 axis in myeloid cells using nanoparticle-encapsulated siRNA. siCSF-1 inverted virus-mediated immunopathology as reflected by lower troponin T levels, a reduction of accumulating myeloid cells in heart tissue and improved cardiac function. Importantly, pathogen control was maintained and the virus was efficiently cleared from heart tissue. Since viral heart disease triggers heart-directed autoimmunity, in a second approach we investigated the influence of CSF-1 upon manifestation of heart tissue inflammation during experimental autoimmune myocarditis (EAM). EAM was induced in Balb/c mice by immunization with a myocarditogenic myosin-heavy chain-derived peptide dissolved in complete Freund's adjuvant. siCSF-1 treatment initiated upon established disease inhibited monocyte infiltration into heart tissue and this suppressed cardiac injury as reflected by diminished cardiac fibrosis and improved cardiac function at later states. Mechanistically, we found that suppression of CSF-1 production arrested both differentiation and maturation of monocytes and their precursors in the bone marrow. In conclusion, during viral and autoimmune myocarditis silencing of the myeloid CSF-1 axis by nanoparticle-encapsulated siRNA is beneficial for preventing inflammatory tissue damage in the heart and preserving cardiac function without compromising innate immunity's critical defense mechanisms.
Myocarditis and its sequela, dilated cardiomyopathy, are leading causes of heart failure and sudden death in young adults (1). While various agents may provoke cardiac inflammation, viral infections are the most common trigger of myocardial inflammation in the Western world. Although various viruses are putative invaders of heart tissue, most of our knowledge on disease pathology comes from infection with enteroviruses, in particular CoxsackievirusB3 (CVB3). CVB3 had been reported among the most prevalent pathogens causing viral myocarditis in North America and Europe in the past (2, 3). Mouse models using different strains with divergent susceptibility for cardiotropic CVB3 elegantly reflect human disease with highly diverse disease outcome (4, 5). The hereditary susceptibility involves a certain immune-anchored genetic phenotype leading either to altered virus control and/or to induction of deleterious immunopathology (6¨C8). Severe virus-induced inflammation can result in a subsequent loss of self-tolerance against cardiac proteins, which contributes to additive auto-destructive activity of infiltrating cells and exaggerates heart tissue damage (9, 10). Cardiac myosin is such a crucial autoantigen in both human and murine virus-induced myocarditis (9). Administration of cardiac myosin or its pathogenic epitope in combination with an adjuvant induces experimental autoimmune myocarditis (EAM) in mice, a model that mimics certain aspects of myocarditis and heart failure in humans (11).
Treatment options for patients with myocarditis are sparse and both conventional immunosuppressive as well as anti-viral approaches have not yielded the desired results in clinical trials (12). Recent data suggest that it is not the presence and/or replicative activity of invading viruses in the myocardium that determines outcome, but the virus-triggered abundance of infiltrating leukocytes is an independent risk factor (13, 14). At the acute state of myocarditis in mice, the majority of accumulating leukocytes in inflamed heart tissue are CD11b+ monocytes and macrophages (15, 16). Consistently, the presence of CD68+ macrophages is a diagnostic hallmark for human myocarditis (3). Infiltration of immune cells is cytokine/chemokine-dependent. Consistent with previous findings (17), we have demonstrated that not CVB3-mediated cytotoxicity itself, but the overwhelming cytokine response initiated by viral PAMPs is responsible for disease severity. Lower pro-inflammatory cytokine/chemokine production during the early phase of infection paralleled in reduced inflammatory heart tissue damage and protected mice from cardiac failure (14). As monocytes and macrophages are key players that secrete pro-inflammatory and pro-fibrotic cytokines thereby exacerbating acute and chronic inflammatory injury during myocarditis (4, 18), effector molecules that modulate their differentiation, activity, and cytokine secretion might be putative drug targets for myocarditis. We have previously described the precise targeting of inflammatory monocytes and their precursors by optimized lipid nanoparticles which were encapsulated with siRNA directed against CCR2 or CD115 (19, 20). Injection of mice with these nanoparticles resulted in rapid blood clearance, accumulation in spleen and bone marrow, and localization to monocytes (19).
Here, we demonstrate RNA sequencing data obtained from endomyocardial biopsies of patients with myocarditis indicating a significantly increased production of Colony Stimulating Factor 1 (CSF-1). The development of monocytes depends on CSF-1 (21) and its receptor CSF-1R/CD115. CSF-1 can be expressed and produced by various cells including monocytes themselves (22). Local production of CSF-1 stimulates tissue-resident macrophage proliferation and reduces apoptosis, thereby influencing cellular survival (23). CSF-1R is expressed on monocytes, macrophages, dendritic cells and their precursors, including ¡°granulocyte-macrophage progenitors¡± (GMP), ¡°monocyte-macrophage DC progenitors¡± (MDP) and ¡°common monocyte progenitors¡± (cMoP) (24, 25). CSF-1 receptor signaling is a well-described mechanism that leads to monocyte production from progenitors and stimulates mature monocytes screwing them into a pro-inflammatory state (26). Based on this, we hypothesized that disruption of the CSF-1 axis in myeloid cells attenuates heart muscle inflammation and the resulting organ damage during myocarditis. Using mouse models of CVB3-induced myocarditis and experimental autoimmune myocarditis, we have found that silencing of CSF-1 upon treatment of mice with CSF-1 siRNA encapsulated nanoparticles substantially mitigated inflammatory heart muscle damage leading to less fibrosis formation and improved heart muscle function without the risk of exacerbating direct viral pathology.
Materials and Methods
Study Approval
All subjects gave written informed consent in accordance with the Declaration of Helsinki. The study protocol was approved by the ethic committee of the Medical Faculty¡ªUniversity of Heidelberg¡ªproject 390/2011 ¡°Central biobank of Department Internal Medicine III for research on molecular and genetic markers in patients with cardiovascular disease.¡±
RNA-Seq Analysis, Read Processing and Mapping
Patient enrollment and biomaterial processing for RNA-seq analysis of heart biopsies was performed as previously described (27). In detail, biopsy specimens were obtained from the apical part of the free LV wall during cardiac catheterization using a standardized protocol. Biopsies of 1- to 2-mm diameter were immediately washed in ice-cold saline (0.9% NaCl), transferred and stored in liquid nitrogen until RNA extraction. After diagnostic workup of the biopsies (histopathology), the remaining material was used to isolate RNA with an Allprep Kit (Qiagen). RNA purity and concentration were determined using the Bioanalyzer 2100 (Agilent Technologies) with a Eukaryote Total RNA Pico assay for RNA from biopsies. Sequencing libraries were generated using the TruSeq Stranded Total RNA Sample Preparation Kit with Ribo-Zero Human/Mouse/Rat from Illumina, adhering to the standard protocol of the kit. Sequencing was performed using 2 ¡Á 75 bp paired end sequencing on an Illumina HiSeq2000 instrument. For transcriptome analysis, raw read files were mapped with STAR v2.4.1c5 using GRCh37/hg19 and the Gencode 19 gene model (http://www.gencodegenes.org/). Read counts were generated with help of subread's feature counts program 6 (subread version 1.4.6.p1), using uniquely mapped reads only (28). Normalization was performed with help of rlog-normalization (29). RNA seq data were deposited to the public repository Gene Expression Omnibus (GEO) - NCBI, accession number GSE120567. RNA seq data for DCM patients are partially demonstrated in (30).
Differential Gene Expression- and Gene Set Enrichment Analysis
Differential gene expression analysis of RNA-seq data was carried out within the RStudio framework using the edgeR package (31). Gene set enrichment analysis was performed with KEGG gene sets.
Histology and Immunohistochemistry
Human endomyocardial biopsy tissue and murine tissue was stained as described elsewhere (32). For AVM, paraffin embedded organ tissue sections were stained with hematoxylin/eosin (HE) or Masson's trichrome according to standard protocols. Immunohistochemical stains for CSF-1 (rabbit polyclonal, abcam), T lymphocytes (CD3 and CD4) and mononuclear phagocytes (Mac-3) was performed as previously described (32). For EAM, hearts were excised 30 days after primary immunization. Hearts were rinsed in PBS, fixed in 10% formaline for 24 h and embedded in paraffin. Serial 5 ¦Ìm sections were stained with Masson's trichrome staining to quantify fibrotic tissue formation. Severity of EAM was evaluated according to a 6-tier scoring system as previously described (19, 20). All slides were counterstained with hematoxylin. Sections were mounted with Pertex mounting media (Medite). Slides were viewed with a Zeiss Axioskop 40 microscope.
Candidate Identification of CSF-1 siRNA
Lysates of several murine cell lines were tested on CSF1 expression. NIH-3T3 cells showed high CSF1 expression, are readily to be transfected, and were therefore used in candidate identification experiments. siRNA loaded lipid-based nanoparticles were generated by Axolabs GmbH (Kulmbach, Germany) as previously described (19). siRNA targeting CSF-1 receptor (CSF-1R, CD115) is described elsewhere (33). To generate siRNAs that target the CSF-1 transcript, NIH 3T3 cells were transfected with siRNAs targeting CSF-1 or non-targeting control siRNA complexed with Lipofectamine2000 Transfection Reagent at 5 and 50 nM final concentration in quadruplicates. Values for CSF-1 were normalized to GAPDH and related to the mean value of three different control siRNAs (100% expression). Optimal siRNA concentration yielding most efficient knockdown of CSF-1 production was obtained with RNA transfections starting at 100 nM in 6-fold dilution steps down to 10 fM. CSF-1 siRNAs that showed the best knockdown in both the dual concentration screen and the concentration response curve screen were used for nanoparticle encapsulation and in vivo experiments.
Induction of Acute Viral Myocarditis (AVM) and Experimental Autoimmune Myocarditis (EAM)
For induction of AVM, 5¨C7 weeks old male A.BY/SnJ mice were infected intraperitoneally (i.p.) with 5 ¡Á 105 PFU CVB3 (cardiotropic Nancy strain) provided by Klingel (15) and Rahnefeld et al. (32). Original breeding stocks for A.BY/SnJ mice were purchased from the Jackson Laboratory. For EAM, male BALB/c were purchased from Janvier (Saint-Berthevin, France). Myocarditis was induced by subcutaneous injection of an emulsion containing 150 ¦Ìg myosin peptide SLKLMATLFSTYASAD (PSL GmbH, Heidelberg, Germany) supplemented with complete Freund's adjuvant (CFA) (Sigma-Aldrich, Taufkirchen, Germany) and 5 mg/ml Mycobacterium tuberculosis H37Ra (Sigma-Aldrich, Taufkirchen, Germany). Directly after the initial immunization, mice were injected with 500 ng pertussis toxin (Sigma-Aldrich, Taufkirchen, Germany) i.p. Seven days after the primary immunization, mice received a second subcutaneous injection of 150 ¦Ìg myosin peptide supplemented with CFA and complemented with Mycobacterium tuberculosis. All mice were housed under standard laboratory conditions with a 12-h light-dark cycle and access to water and food ad libitum. For AVM, the protocol was approved by the Committee on the Ethics of Animal Experiments of Berlin State authorities [G0034/16]. EAM experimental protocols were approved by the institutional review board of the University of Heidelberg, Germany, and the responsible government authority of Baden-W¨¹rttemberg, Germany (project number 35-9185.81/G-209/12). All mouse studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the German animal welfare act, which is based on the directive of the European parliament and of the council on the protection of animals used for scientific purposes. All efforts were made to minimize suffering.
In vivo Silencing of CSF-1 During AVM and EAM
The optimal CSF-1¨Ctargeted siRNA (siCSF-1) was scaled up for in vivo studies. For viral myocarditis, mice were intravenously treated with 0.5 mg/kg nanoparticle encapsulated siLUC or siCSF-1 immediately prior to CVB3 infection and 2, 4, and 6 days after infection. For EAM, nanoparticle treatment started 14 days after the primary immunization with myosin peptide. Animals received four i.v. injections of 0.5 mg/kg siCSF-1 or siLuciferase (LUC)-nanoparticles (control siCD115) per week.
Evaluation of Knockdown Efficacy of siCSF-1
Male BALB/c mice received single injections of 1.5 mg/kg lipid-based nanoparticle containing either siLUC or siCSF-1 on three consecutive days. Animals were sacrificed 24 h after the third injection. Bone marrow cells were isolated and prepared for flow cytometry-based sorting of monocytes, which were identified as Lin−(CD90;B220;CD49b;NK1.1;Ly6G,Ter119);F4/80−; CD11c−; CD11b+. Cell sorting was performed on a FACS ARIAII (BD Bioscience, Heidelberg, Germany). RNA from sorted cells was isolated using Trizol (Life Technologies, Darmstadt, Germany). Knockdown efficacy was evaluated using quantitative real-time PCR. Gene expression was normalized to HPRT. The following primers were used: CSF-1: TCCCATATGTCTCCTTCCATAAA (fwd), GGTGGAACTGCCAGTATAGAAAG (rev); CD115: CGAGGGAGATCTCAGCTACA (fwd), GACTGGAGAAGCCACTGTCC (rev). HPRT: GTCAACGGGGGACATAAAAG (fwd), TGCATTGTTTTACCAGTGTCAA (rev). For the AVM model, spleen tissue was isolated 8 days after virus inoculation and tissue homogenization was performed using a lysis buffer containing 20 mM HEPES, 1 % (v/v) Triton X-100, 4 mM EDTA, 1 mM EGTA, 5 mM TCEP, 50 mM NaF, 5 mM NaPP, 2 mM Na-o-vanadate and Complete® protease inhibitor cocktail (Roche). Western blot analysis was performed following standard procedures. After blocking with 5% milk/PBS-Tween at 4 ¡ãC overnight, membranes were probed with the primary antibody ¦Á-CD115 (ab32633, Cell Signalling) and ¦Á-actin (Merck Millipore). The bound primary antibodies were detected using IRDye800CW labeled goat anti-mouse secondary antibodies in conjunction with an Odyssey CLx infrared imaging system (Li-Cor Biosciences, Bad Homburg, Germany).
Echocardiography
Cardiac function and morphology of mice with AVM were assessed with a VisualSonics Vevo 770 High-Frequency Imaging System with the use of a high-resolution (RMV-707B; 15¨C45 MHz) transducer during anesthesia with 1.5¨C2% isoflurane. Temperature and ECG were continuously monitored. For the EAM model, echocardiography was performed in conscious animals on a VisualSonics Vevo 2100 30 days after the first immunization. Standard imaging planes, M-mode, and functional calculations were obtained. For AVM, the parasternal long-axis four-chamber view of the left ventricle (LV) was used to guide calculations of percentage fractional shortening, ventricular dimensions and volumes. M-mode echocardiographic images were recorded at the level of the papillary muscles from the parasternal short-axis view. An experienced reader blinded to treatment performed all measurements. Ejection fraction (EF) and fractional shortening (FS) were calculated based on M-mode measurements.
Flow Cytometry
Flow cytometric analysis was performed 8 days after infection in AVM and 21 days after the first immunization in EAM. Single cell suspension of bone marrow, spleen and heart tissue were prepared as previously described (34). Hearts were flushed with PBS and homogenized in RPMI 1,640 medium (Biochrom) containing 10% (v/v) fetal calf serum (FCS) (Biochrom), 1% (v/v) penicillin/streptomycin (Pan Biotech), 30 mM HEPES, 0.1 % (w/v) collagenase type 2 (Worthington) and 0.015% (w/v) DNase I (Sigma-Aldrich) at 37¡ãC at 800 rpm for 30 min. Afterwards, 10 mM EDTA was added. Cells were washed with PBS and passed through a 70 ¦Ìm cell strainer as described in reference (35). For the identification of myeloid cells, cell suspensions were stained with a cocktail of PE-conjugated anti-mouse antibodies targeting hematopoietic lineage markers (B220 for B cells (RA3-6B2, BD Bioscience), CD90.2 for T cells (53-2.1, BD Bioscience), CD49b for NK cells (DX5, eBioscience), NK-T/NK Cell Antigen for NK cells (U5A2-13, BD Bioscience) and Ter-119 for erythroid cells (TER-119, BD Bioscience)) and fluorescent-dye conjugated antibodies against the following cell surface markers: CD45.2 (104, Brilliant Violet 711™, BioLegend), CD11b (M1/70, PE-CF594, BD Bioscience), Ly6G (1A8, PerCP/Cy5.5, BioLegend), Ly6C (HK1.4, Pacific Blue™, BioLegend), CD11c (N418, Brilliant Violet 510™, BioLegend), I-A[b] (AF6-120.1, FITC, BD Bioscience) and F4/80 (BM8, APC, BioLegend). Cells were stained in PBS containing 2% FCS, 2 mM EDTA for 20 min at 4¡ãC. For the identification of lymphoid cells, cell suspensions were stained with fluorescent-dye conjugated anti-mouse antibodies against CD45.2 (104, Brilliant Violet 711™, BioLegend), CD3e (145-2C11, PerCP/Cy5.5, BioLegend), CD4 (RM4-5, V500, BD Bioscience), CD8a (53-6.7, Pacific Blue™, BD Bioscience), B220 (RA3-6B2, FITC, BioLegend) and CD19 (6D5, APC, BioLegend). The antibody staining was followed by a cell viability stain (Fixable Viability Dye eFluor® 780, eBioscience) according to the manufacturer's protocol.
Monocytes were identified as Linlow (CD90;B220;CD49b;NK1.1;Ly6G,Ter119), F4/80low, CD11clow, CD11bhigh; or Fixable Viability Dyelow, CD45.2high, CD11bhigh, (B220, CD90.2, CD49, NK-T/NK Cell Antigen, Ter-119)low, Ly6Glow, SSClow, F4/80low and CD11clow and further differentiated according to Ly6C-expression. Inflammatory monocytes express high levels of Ly6C and patrolling monocytes express low levels of Ly6C. Macrophages were identified as Fixable Viability Dyelow, CD45.2high, CD11bhigh, Linlow, Ly6Glow, SSClow, F4/80high and CD11clow/high. Dendritic cells were identified as Fixable Viability Dyelow, CD45.2high, CD11bhigh, Linlow, CD11chigh and MHC IIhigh (compared to isotype control). Neutrophils were identified as Fixable Viability Dyelow, CD45.2high, CD11bhigh, Linlow, Ly6Ghigh and SSChigh. B cells were gated as Fixable Viability Dyelow, CD45.2high, CD3low, B220high and CD19high. T cells were gated as Fixable Viability Dyelow, CD45.2high, B220low, CD3high and either CD4high or CD8high. For identifying proliferating GMPs mice received two s.c. injections of 1 mg/kg Bromdesoxyuridin (BrdU) 12 and 24 h before the animals were sacrificed. BrdU was stained using BrdU flow kit (BD Biosciences). Proliferating GMPs were identified as (CD90;B220;CD49b;NK1.1;Ly6G;CD11b;CD11c;IL-7R;Sca-1)low and (CD117;CD34;CD16/32;BrdU)high.
For the assessment of quantitative data, 123 count eBeads (eBioscience) were used according to manufacturer's protocol. Data were acquired on a FACS Verse (BD Biosciences, Heidelberg, Germany) or on a LSR II (BD Bioscience) and analyzed with FlowJo v10.0 software (FLOWJO, Ashland, United States). Reported cell numbers were normalized to the weight of total hearts, yielding the number of respective cell fraction per mg tissue.
Determination of Viral Load in Heart Tissue
Plaque assays were performed in triplicates on sub-confluent green monkey kidney cell monolayers as described recently (32). In situ hybridization of CVB3 RNA was performed using probes generated with the DIGoxigenin (DIG) RNA labeling kit (Roche) and the pCVB3-R1 plasmid. Plasmid cDNA was linearized with SmaI (36); all other steps were conducted as previously described (37). DIG-labeled CVB3 RNA was detected using a horseradish-peroxidase-conjugated DIG antibody (Roche 1:100). HistoGreen (Linaris) was used as a substrate. All slides were counterstained with hematoxylin.
High-Sensitive (hs)-Troponint (TnT)
Blood was sampled by facial vein puncture and collected in a heparinized capillary. Thereby obtained plasma was diluted 1:15 in PBS. hs-TnT was determined by the electrochemiluminescence method (ECLIA; Elecsys 2010 analyzer) according to the method described in reference (38).
Statistics
Statistical analysis of the data was performed in GraphPad Prism v6.00/v.700 for Windows (GraphPad Software, La Jolla, California, United States). Logarithmic data (virus titer, semi-quantitative RNA quantification) measured on a linear scale was transformed logarithmically prior to data plotting and data analysis. Data summary is indicated on plots as mean ¡À SD unless stated otherwise. Unpaired t-tests were used for two group comparisons. If samples had unequal variances (determined by an F-test), an unpaired t-test with Welch's correction was used. For multiple group comparison unequal variance versions of ANOVA (1-way or 2-way ANOVA) were performed followed by a Sidak-Holm's multiple comparison test. The significance threshold for all tests was set at the 0.05 level.
Results
Increased Abundance of CSF-1 in Heart Muscle Tissue During Myocarditis/Inflammatory Cardiomyopathy
RNA-seq analyses revealed that myocarditis results in a diverse transcriptional response in the patient's heart tissue. We observed 1963 differentially expressed genes in biopsies taken from patients with acute myocarditis vs. patients with a non-inflammatory dilated cardiomyopathy (DCM) (Figure 1A). Gene set enrichment analysis (GSEA) revealed that a decent amount of differentially expressed genes participates in inflammatory processes (especially cytokines and cytokine receptors) and differentiation of hematopoietic cell lineages (Table 1). Myocarditis leads to a massive infiltration with immune cells into heart tissue. Monocytes represent the most prominent leukocyte population both during virus-mediated and experimental autoimmune myocarditis (16, 32). Monocyte production and maturation is strongly dependent on CSF-1 and CSF-1R, and, both effector molecules were identified in two gene sets mentioned above. Our data indicate a pronounced up-regulation particularly of CSF-1 and CSF-1R in endomyocardial specimen from patients with myocarditis/inflammatory cardiomyopathy (Figure 1B). Immunohistochemical stain of heart tissue from patients with myocarditis revealed CSF-1 expressing cells only within inflammatory foci, with a strong focus on mononuclear immune cells (Figure 2A). Altogether, these data argued toward a significant contribution of monocytes/macrophages to the cardiac CSF-1 expression, which we found in patients with inflammatory heart disease.
FIGURE 1
www.frontiersin.org
Figure 1. Transcriptome analysis of endomyocardial specimen from patients with dilated cardiomyopathy and myocarditis. RNA-Sequencing data from endomyocardial biopsies obtained from patients with clinically diagnosed myocarditis or dilated cardiomyopathy as a control were analyzed for relative expression of different gene sets. (A) RNA-seq analyses revealed differential expression of 1963 genes. Heatmap depicts top 500 differentially expressed genes hierarchically clustered by using Euclidean distance measures. (B) CSF-1 and CSF-1R expression taken from RNA-seq data. Unpaired t-tests were used; p-values are indicated on the graph and significant differences (p < 0.05) are marked with blue color.
TABLE 1
www.frontiersin.org
Table 1. Top 10 C2 curated gene sets (KEGG Database) significantly enriched in human biopsies.
FIGURE 2
www.frontiersin.org
Figure 2. CSF-1 production in cardiac tissue during viral myocarditis. Paraffin-embedded tissue sections from endomyocardial biopsies that had been obtained from patients with acute myocarditis were stained by immunohistochemistry. (A) Representative micrographs stained with an anti-CSF-1 antibody [left column] or with a secondary antibody only [right column] are depicted. top: scale bar = 120 ¦Ìm; bottom: scale bar = 60 ¦Ìm. (B) Heart tissue sections were obtained from CVB3-infected A.BY/SnJ mice on day 8 p.i. Representative micrographs of anti-CSF-1 stained heart tissue are shown [scale bar = 36 ¦Ìm]. (C) In addition, cardiac sections from mice were double-stained with an antibody directed against Mac-3 (red) [left: scale bar = 36 ¦Ìm; center: scale bar = 12 ¦Ìm] and against CSF-1 (green). As control, Mac-3 stained tissue sections were counterstained omitting the anti-CSF-1 directed antibody [right: scale bar = 12 ¦Ìm].
Local production of CSF-1 stimulates tissue-resident macrophage proliferation and reduces apoptosis (23). In addition to this function and a direct role of CSF-1 during monocyte development, it might also influence pro-fibrotic processes under inflammatory conditions. Since inflammation and fibrosis are hallmarks of inflammatory heart disease, we aimed to investigate the pathophysiological influence of CSF-1 with respect to manifestation of inflammatory heart tissue injury. First, we determined CSF-1 production in a mouse model of CVB3-induced myocarditis. We performed immunohistochemical stains to evaluate CSF-1 abundance during viral myocarditis. Consistent with our findings in patients, CSF-1 production was increased within inflammatory foci at the acute state of myocarditis in mice (Figure 2B). Since monocytes/macrophages represent the major infiltrating cell population in acute myocarditis, it is very likely that these cells are also involved in CSF-1 production. By double labeling immunohistochemistry we found CSF-1 protein expression in a part of Mac-3 positive mononuclear phagocytes within the cardiac inflammatory lesions (Figure 2C).
Nanoparticle-Encapsulated siRNA Effectively Downregulates CSF-1 Production in Monocytes
CSF-1 can be expressed and produced by various cells including monocytes themselves (22). siRNA encapsulated in lipid-based nanoparticles has been shown to effectively downregulate target genes in monocytes and their lineage progenitors (19, 20). Furthermore, in vivo knockdown of CSF-1R and monocyte depletion with nanoparticle-encapsulated siRNA has recently been demonstrated for ischemic heart disease (33). Thus, in order to investigate the pathophysiological function of CSF-1 production by monocytes/macrophages on inflammatory tissue damage during myocarditis, we decided to use a nanoparticle-encapsulated siRNA approach to target CSF-1 production in myeloid cells. To identify siRNAs leading to highly efficacious suppression of CSF-1 production, 24 different siRNAs targeting CSF-1 were investigated regarding their influence on CSF-1 mRNA levels in vitro (Figure 3A). Six different siRNAs, which yielded optimal in vitro suppression of CSF-1 mRNA production, were further investigated for their knockdown efficacy. Next, we screened the respective CSF-1 directed siRNA regarding to the concentration-dependent knockdown efficacy (Figure 3B) and selected the most efficacious siRNA for in vivo nanoparticle studies. Naive BALB/c mice were intravenously inoculated with 1 mg/kg of this nanoparticle-encapsulated siRNA (termed siCSF-1) on three consecutive days. Expression of CSF-1 was found to be effectively downregulated in monocytes that were sorted from spleen of siCSF-1 treated mice and further evaluated by quantitative PCR analysis (Figure 3C). Since pathogens are frequently involved in the pathogenesis of myocarditis (5), as a next step we set up an experimental approach to decipher the CSF-1 axis using nanoparticle-encapsulated siRNA in a mouse model of virus-mediated myocarditis. A.BY/SnJ mice with high hereditary susceptibility for the development of acute viral myocarditis (AVM) were treated with siCSF-1 or respective controls directly prior to infection with cardiotropic CVB3 (Nancy). siCSF-1 treatment was repeated every other day until mice were sacrificed 8 days after infection at the respective peak of infiltration in heart muscle (Figure 3D) (6¨C8). Following this protocol, we monitored the abundance of CSF-1 receptor levels in spleens of infected mice, which allowed us to conclude on the efficiency of siCSF-1 treatment during infection. Consistent with virus-mediated mobilization of monocytes/macrophages from bone-marrow sources, viral infection resulted in increased CSF-1R levels in the spleen. In siCSF-1-treated mice, we found reduced CSF-1R levels being indicative of suppressed myeloid cell mobilization upon siCSF-1 injection during AVM (Figure 3E).
FIGURE 3
www.frontiersin.org
Figure 3. Suppression of CSF-1 production by siRNA-encapsulated nanoparticles. (A) NIH-3T3 cells were transfected with 24 siRNA candidates directed against CSF-1. siRNA #7 was most efficient to reduce CSF-1 mRNA expression both at 5 and 50 nM and was selected for further studies. (B) CSF-1-directed siRNA # 7 was titrated and the respective CSF-1 knockdown efficacy was determined. (C) Knockdown efficacy of nanoparticle-encapsulated CSF-1 candidate siRNA pool 7 after injection into naive Balb/c mice (n = 3). (D) A.BY/SnJ mice were intravenously treated with nanoparticle encapsulated siRNA targeting either luciferase (n = 7 siLUC, gray color) or CSF-1 siRNA #7 (n = 8 siCSF-1, green color) directly prior to CVB3 inoculation. siRNA treatment was repeated after 2, 4, and 6 days. (E) The overall efficacy of siCSF-1 treatment during AVM as indicated by the presence of CSF-1-R)-positive cells was monitored by Western blot analysis of spleen tissue homogenates (n = 6 mice per group) 8 day after virus inoculation. CSF-1R (high molecular weight band) and the cytoplasmic domain of CSF-1R (around 55 kDa) are depicted. Fluorescence was quantified by the Image Studio Lite Ver 5.2 software. Signal intensity was normalized to actin and is depicted as relative expression levels compared to siLUC-treated mice in the bar graph. Unpaired t-tests were used; p-values are indicated on the graph and significant differences (p < 0.05) are marked with blue color.
siRNA-Mediated Knockdown of CSF-1 Attenuates Virus-Mediated Pathology
Since we found reduced mobilization of monocytes/macrophages in siCSF-1-treated mice during CVB3 infection, this mouse model allowed us to delineate the pathophysiological role of CSF-1 production particularly by monocytes/macrophages during viral myocarditis. First, we questioned whether siCSF-1 treatment manipulated the viral load during AVM. The viral burden as reflected by the amount of infectious viral particles (Figure 4A) was not substantially influenced by siCSF-1 in heart tissue at the acute state of infection. Thus, targeting the CSF-1 axis represents a safe approach regarding to control of virus dissemination and replication in A.BY/SnJ mice. During virus-mediated myocarditis, there is a strong spatial-temporal relation between virus-induced cellular injury and the emergence of inflammatory foci in heart tissue (39). Likewise, viral genome abundance as detected by CVB3 in situ hybridization was spatially connected with high-grade inflammation and most impressive in siLUC-treated mice (Figure 4B). Since CVB3 does not only target the heart, but also replicates in the pancreas, we also determined the magnitude of virus-induced pancreas destruction and found similar tissue injury in both siLUC- and siCSF-1-treated groups (Figure S1).
FIGURE 4
www.frontiersin.org
Figure 4. Depletion of CSF-1 attenuates virus-mediated pathology. Mice with AVM were subjected to CSF-1 siRNA treatment as indicated in Figure 3D. (A) Infectious virus particles were determined in heart tissue homogenates by plaque assay. Data summary is mean ¡À SEM. A student¡®s t-test was conducted and the p-value is shown. (B) To localize viral RNA in infected heart tissue, in situ hybridization for the detection of the CVB3 genome was performed and slides were counterstained with hematoxylin/eosin. Representative micrographs are depicted (scale bar = 60 ¦Ìm). During viral infection, mice were monitored for body weight (C) and body temperature (D) at the indicated points in time. Dots represent mean ¡À SEM. Repeated measurements versions of two-way ANOVA were performed followed by a Sidak- Holm multiple comparison procedure. P values are indicated (blue color indicates p < 0.05; only significant results are depicted on the graph). (E) To assess injury of cardiomyocytes prior to peak of inflammation, blood was sampled 5 days after infection by facial vein puncture and high sensitive (hs) troponin T plasma levels were determined. Obtained results were normalized to the results obtained with blood samples from non-infected siLUC-treated mice and are depicted as fold changes. Data summary is mean + SD. Repeated measurements two-way ANOVA was performed followed by a Sidak-Holm's multiple comparison test and the p value is depicted. (F) After sacrificing mice 8 days after infection, heart tissue sections were stained with hematoxylin/eosin. (G) To quantify cell infiltration, single cell suspensions of heart tissue obtained from naive mice (uninfected mice that did not receive siRNA treatment; white bars, n = 4) as well as AVM and siRNA-treated mice (siLUC: gray squares, n = 7; siCSF-1: green squares, n = 8) were stained with CD45 antibodies to quantify total leukocyte count in the heart. (H) Cardiac function was assessed by echocardiography prior to CVB3 infection in A.BY/SnJ mice (baseline) by an experienced and blinded investigator. Mice were allocated to respective groups: siLUC and siCSF-1. In all CVB3-infected mice, echocardiography was repeated 8 days after CVB3 infection (siLUC n = 16; siCSF-1 n = 17 mice). Data were analyzed regarding putative alteration during AVM in the respective treatment groups (day 8 after infection vs. baseline measurements of the same cohort). Relative changes of stroke volume, heart rate and cardiac output compared to baseline measurements were calculated for each group and these fold changes are depicted for siLUC and siCSF-1-treated groups. One-sample t-tests were performed to compare baseline measurements and values obtained 8 days after infection. All p values are depicted, p < 0.05 are in blue color.
Next, CVB3-infected mice that received siCSF-1 or siLUC as a control were followed for global signs of acute infection. siCSF-1 treatment profoundly attenuated overall virus-induced pathology as represented by significantly less pronounced body weight reduction (Figure 4C) and only a minor loss of body temperature during infection in comparison to controls that received siLUC (Figure 4D). Overall diminution of virus-mediated pathology under siCSF-1 influence was further corroborated by a significant reduction of cardiac troponin T serum levels as a heart-specific sign of tissue damage (Figure 4E). Cardiac troponin T at an early state of myocarditis might reflect both direct virus-induced cytotoxicity and tissue destruction by innate mediators of the immune response. In line with this, analysis of heart tissue obtained from siCSF-1 treated animals sacrificed 8 days p.i. revealed distinct differences. Histological staining of heart tissue (Figure 4F) demonstrated a profound myocarditis in siLUC-treated A.BY/SnJ mice and in contrast to that only moderate signs of myocarditis after siCSF-1 treatment. Since viral injury of cardiomyocytes provokes an inflammatory response that significantly contributes to tissue damage and functional impairment of the heart (32), next we quantified infiltration with CD45+ immune cells into hearts from siCSF-1 and siLUC-treated mice by flow cytometry. We found a significant reduction of infiltrating leucocytes in siCSF-1 treated mice (Figure 4G), thus indicating reduced inflammatory organ damage under suppression of the CSF-1 axis. Following up on observed systemic and heart-tissue specific responses to siCSF-1 treatment, siCSF-1 effects on cardiac performance were assessed by echocardiography during the inflammatory peak of viral myocarditis. In siLUC-treated, infected A.BY/SnJ mice, both the stroke volume and cardiac output were significantly reduced in comparison to baseline measurements (Figure 4H) and Table S1. Consistent with its heart-directed effects, siCSF-1 treatment mitigated these detrimental changes and CVB3 infection in this group resulted only in minor, non-significant reduction of cardiac performance (Table S1).
siRNA-Mediated Knockdown of CSF-1 Diminishes Immune Cell Infiltration During Acute Viral Myocarditis
As a next step, we aimed to determine whether siCSF-1 specifically influenced infiltration with monocytes/macrophages or whether other immune cells were affected as well. Immunohistochemistry using antibodies directed against marker proteins for myeloid (Mac3) and T cells (CD3 and CD4) indicated reduced infiltration of these respective immune cell populations in siCSF-1-treated mice during AVM (Figures 5A,B). These findings were corroborated by the results obtained from a quantitative flow cytometry-based analysis of the different immune cell populations in infected mouse hearts. We detected 756 ¡À 63 CD11b+/lineage− cells in siLUC- vs. 273.3 ¡À 30.8 CD11b+/lineage− cells/mg heart tissue in siCSF-1 treated mice (p < 0.0001) and 198 ¡À 34 T cells in siLUC- vs. 95 ¡À 12 T cells/mg heart tissue in siCSF-1 treated mice (p = 0.02; Figures 5C,E). The vast majority of infiltrating myeloid cells belonged to the pool of inflammatory monocytes (Figure 5D). Consistent with the influence of CSF-1 on monocyte recruitment and differentiation, inflammatory monocytes were highly significantly reduced in infected mouse hearts upon siCSF-1 treatment. siCSF-1 also led to a significant reduction of patrolling monocytes, macrophages and dendritic cells (Figure 5D), which might all originate from inflammatory monocytes. In correspondence to previous reports (15), the pool of invading T cells during AVM was majorly comprised of CD4+ T cells. Comparable to siCSF-1-induced effects on myeloid cell infiltration, we also found a significant reduction of the CD4+ T cell count in heart tissue upon siCSF-1-treatment (Figure 5E).
FIGURE 5
www.frontiersin.org
Figure 5. Influence of CSF-1 on immune cell infiltration into heart tissue during AVM. To further differentiate immune cell infiltration, immunohistochemical stain for (A) mononuclear phagocytes using antibodies directed against Mac-3 and (B) T-cells using antibodies directed against CD3 and CD4 were performed (scale bars = 60 ¦Ìm). Further differentiation by flow cytometry (Figure S2) was performed. (C) Total infiltrating myeloid cells (identified as CD45+, CD11b+, lymphoid lineage− life single cells) were quantified. (D) Myeloid cells were further differentiated according to the gating strategy depicted in Figure S2A. (E) Equally, lymphoid cells were further analyzed. Representative flow cytometry dot blots of siLUC- and siCSF-1-treated groups are depicted. Unpaired t-tests were performed between siLUC- and siCSF-1-treated groups and p-values are shown. Significant differences (p < 0.05) are marked with blue color.
siRNA-Mediated Knockdown of CSF-1 Mitigates Inflammatory Heart Tissue Damage in a Mouse Model of Experimental Autoimmune Myocarditis
siCSF-1 treatment impressively improved cardiac function upon mitigating the inflammatory damage response during acute viral myocarditis. A high inflammatory disease burden at the acute state might directly translate into the manifestation of a chronic functional impairment. Long-term sequela of acute myocarditis involve cardiac remodeling processes with substantial fibrosis formation and a reduction of systolic cardiac function (4). Experimental autoimmune myocarditis (EAM) represents an excellent model that enables researchers to follow inflammatory disease progression from acute to chronic states of myocardial dysfunction (40). Therefore, we investigated the pathophysiological role of CSF-1 also in EAM, and started siCSF-1 treatment upon manifestation of myosin-heavy chain-directed autoimmunity 14 days after the first immunization (Figure 6). This application strategy might also be considered as a therapeutic regime starting upon manifestation of acute disease. Similar to the AVM model, intravenous siCSF-1 nanoparticle application was repeated every 48 h for a 1-week course. First, we performed a quantitative flow cytometry-based analysis of inflammatory monocytes in the injured hearts directly after this siCSF-1 treatment period. As expected from our results obtained in AVM, we indeed found a reduced number of inflammatory Ly6Chigh monocytes in siCSF-1-treated animals compared to the siLUC-treated controls during EAM (Figure 6B). To investigate whether CSF-1 promotes monocyte development from bone-marrow sources during EAM, we next evaluated the influence of CSF-1 knockdown on granulocyte-monocyte progenitor (GMP) cell numbers. siRNA-mediated knockdown of CSF-1 led to markedly increased numbers of GMPs in the bone marrow during EAM (Figure 6C). However, proliferation rates of GMPs did not differ significantly between siLUC- and siCSF-1-treated groups (Figure 6D). To further validate that CSF-1 knockdown leads to an arrest of progenitor cells in the bone marrow, we measured myeloid cell numbers in the blood and found reduced numbers 21 days after EAM induction (Figure 6E).
FIGURE 6
www.frontiersin.org
Figure 6. CSF-1 silencing during EAM diminishes infiltration of inflammatory monocytes into injured mouse hearts. (A) EAM was induced by inoculation of myosin peptide in conjunction with Freud's adjuvant and mice were boosted after 7 days. Nanoparticle-encapsulated CSF-1 siRNA #7 (Figure 3) was investigated regarding to its potential to manipulate EAM in comparison to siLUC (control). Therefore, mice were treated with 0.5 mg/kg siRNA intravenously every other day starting 14 days after EAM induction, and mice were sacrificed 21 days after the first immunization. (B) Representative dot plots (left) and enumeration (right) of inflammatory Ly6Chi monocytes in heart tissue of siLUC- and siCSF-1-treated mice (n = 8). (C) Representative dot plots (left) and quantification of granulocyte-monocyte progenitors (GMPs) (right) found in bone marrow of siLUC- and siCSF-1-treated mice during EAM (n = 12). (D) Representative FACS plots (left) and quantification (right) of BrdU incorporation in GMPs of the bone marrow in siLUC- and siCSF-1 treated mice (n = 8). (E) Representative FACS density plots (left) and quantification of monocytes/macrophages (right) in the blood of siLUC- and siCSF-1- treated mice (n = 7). Unpaired t-tests were used. p-values are indicated on the graph and significant differences (p < 0.05) are marked with blue color.
Based on our finding of reduced infiltration with inflammatory monocytes upon siCSF-1 treatment, next we followed mice for a total of 30 days after EAM induction and determined the formation of fibrotic scars as an integral hallmark of cardiac remodeling at this chronic disease state (Figure 7A). Knockdown of CSF-1 production resulted in a significant reduction of fibrosis formation in the heart muscle as indicated by quantitative assessment of Masson's trichrome stains (Figure 7B). Consistent with this reduction of long-term, inflammation-mediated tissue damage in the siCSF-1 group, functional investigation of cardiac performance by echocardiography revealed an improved ejection fraction in siCSF-1-treated animals compared to siLUC-treated control animals (Figure 7C). Chronic disease in siLUC-treated mice was mirrored by a substantial reduction of cardiac contractility. As a proof of principle, we also tested whether knockdown of CSF-1R expression could exert similar effects during disease course. Therefore, upon establishment of autoimmune myocarditis mice were treated with a nanoparticle-encapsulated siRNA that specifically targets CSF-1R (33). Formation of cardiac fibrosis assessed 30 days after EAM induction was found to be diminished in comparison to siLUC-treated controls, and was reduced to similar levels as achieved by siCSF-1-treatment (Figure 7B). Likewise, echocardiographic imaging of siCSF-1R-treated animals demonstrated significant improvement of cardiac performance as indicated by higher left ventricular ejection fraction in comparison to siLUC nanoparticle-treated animals (Figure 7C). Altogether, our data demonstrate that silencing the CSF-1 axis hampers monocyte development and substantially mitigates inflammatory heart tissue damage both in a mouse model of viral and autoimmune-mediated myocarditis. Moreover, initiation of CSF-1 knockdown upon manifestation of inflammatory tissue damage attenuates the manifestation of debilitating long-term sequela of acute inflammatory injury, and this parallels in preserved systolic contractility of the heart muscle (Figure 8).
FIGURE 7
www.frontiersin.org
Figure 7. siRNA-mediated knockdown of CSF-1 during acute EAM attenuates the development of chronic disease states. (A) EAM induction and siRNA treatment was conducted as shown in A. Mice were sacrificed after 30 days. (B) Representative Masson's trichrome stains of heart tissue sections obtained 30 days after EAM induction are depicted (left: scale bar = 150 nm). Fibrosis was scored microscopically (n = 8 for siLUC and siCSF-1 as well as n = 7 for siCSF-1R). (C) Heart function was evaluated 30 days after the initial immunization by echocardiography. Representative M-mode echocardiographic images are shown during late state EAM. Calculated left ventricular ejection fraction (EF) (n = 8 for siLUC and siCSF-1 as well as n = 7 for siCSF-1) is shown. One-way-ANOVA was performed. Since ANOVA was significant, a Sidak-Holm-multiple comparison was performed. p-values of multiple comparison are indicated. Blue color indicates p < 0.05.
FIGURE 8
www.frontiersin.org
Figure 8. Graphical synopsis: The CSF-1 axis is induced in patients with acute myocarditis. Based on the preponderant role of CSF-1 for monocyte differentiation/maturation and the disease-modifying function of monocytes during the course of inflammatory heart disease, we investigated how silencing of CSF-1 in monocytes/macrophages using nanoparticle encapsulated siRNA influences heart tissue damage during the onset of acute viral myocarditis and upon manifestation of acute inflammation in an autoimmune myocarditis model. Silencing of the myeloid CSF-1 axis was beneficial for preventing inflammatory tissue damage in the heart and preserving cardiac function at acute and chronic disease states without compromising innate immunity's critical defense mechanisms.
Discussion
One of the major causes of heart failure particularly in young patients is myocarditis. Direct viral cytotoxicity stimulates infiltration and immune response activation leading to pathogen clearance and resolution of organ damage. Nevertheless, in immune-genetically predisposed individuals there is also an adverse scenario where pathogen-induced immune response activation subsequently induces overwhelming inflammatory cytokine response and detrimental immunopathology or autoimmune processes, both leading to cardiac remodeling and fibrotic scarring. It appears to be a slim line between the induction of inflammation to fight the virus and exaggerated immune responses that begin to be deleterious. We here downregulated an important branch of the innate immunity¡ªthe development of monocytes/macrophages¡ªby siCSF-1 treatment for approximately a week during the acute phase of a viral infection, yet found no impairment on pathogen control. In fact, silencing CSF-1 production impressively mitigated acute inflammatory heart tissue damage and attenuated the development of a debilitating long-term sequela of acute inflammatory injury to the heart. This nanoparticle-mediated immune-modulation improved the course of disease both in acute viral myocarditis and autoimmune inflammatory heart disease and importantly, did not adversely influence viral burden and clearance of infectious particles. As it might be hypothesized that attenuating inflammatory activity could allow for enhanced virus-induced cell death, this is not what we observed with siCSF-1 treatment. Our finding is in line with previous data: out of 46 studies that intervened the immune response, more than 90% found no adverse effect on viral load (4). In line with this, we could recently demonstrate that inhibition of cellular proteolysis in immune cells by the immunoproteasome inhibitor ONX 0914 is highly efficient to reverse high grade AVM and this protective effect was attributed to maintained immune cell homeostasis, but not to direct antiviral aspects (14).
A.BY/SnJ mice that were used in this study for AVM are characterized by a high hereditary susceptibility to CVB3-induced cardiomyopathy (6, 15, 41). Upon viral infection of cardiomyocytes, heart tissue injury during the first days is mainly attributed to direct virus-induced cytotoxicity, while the activation of local type I interferon responses (T1IFN) is substantially hampered in this strain (7, 8). This phase of ongoing viral replication is accompanied and succeeded by the recruitment of cells of the innate immune response such as NK cells and monocytes (42). Later on, infiltration with immune cells of the adaptive immune response such as T and B cells follows (15). Analysis of the composition of leukocytes in the heart revealed that 8 days after viral infection a general reduction in invading immune cells was observed which suggests that hampering early responders, such as monocytes e.g., by reduced production of lymphocyte-attracting chemokines might dampen the infiltration and activation of subsequent populations as well. Although we found an overall reduction of CD4+ T cells in the heart in siCSF-1 treated mice, we cannot conclude on the influence of silencing the CSF-1 axis on CD4+ T cell differentiation e.g., into regulatory T cells or Th1 and Th17 cells. From our data we cannot conclude on possible additional effects e.g., of regulatory CD4+ T cells that are known to mitigate the inflammatory tissue damage in the heart (43, 44). CSF-1 facilitates myeloid cell differentiation, monocyte survival, and macrophage proliferation. It was recently shown that CSF-1R also plays a role in splenic monocytopoiesis (33). Monocytes are also important producers of CSF-1 themselves. Hume and MacDonald suggested that modulation of the CSF-1 axis may be beneficial under pathological conditions (45). We here show that siRNA-mediated knockdown of CSF-1 in monocytes and its imminent precursors leads to a significant reduction of inflammatory Ly6Chi monocyte numbers in the inflamed heart. We speculate that this observation results from arresting the CSF-1-dependent monocyte/myeloid cell development at precursor stages. This hypothesis is supported by detection of increased GMP cell numbers in the bone marrow in siCSF-1-treated animals during EAM. Although increased cell numbers may also arise from increased proliferation rates, this situation seems to be unlikely, since the percentages of proliferating GMPs in the bone marrow of siCSF-1- and siLUC-treated animals did not differ. Consistently, the numbers of myeloid cells were reduced in the blood stream of siCSF-1-treated animals during EAM. In agreement with this and the fact that other than in siLUC-treated mice enhanced monocyte counts were not observed in the blood in siCSF-1-treated mice during AVM (data not shown), we found a reduction of CSF-1R abundance in the spleen during AVM in the siCSF-1 group. The spleen itself is a source for monocytes with local production and from which they are recruited to the site of inflammation (34). CSF-1R is expressed throughout the mononuclear phagocyte system, which is primarily composed of monocytes and macrophages (22, 46). A reduction of CSF-1R expression might also be indicative for reduced numbers of monocytes and macrophages. These findings further underscore our assumption of a halted monocyte/macrophage production due to CSF-1 knockdown during inflammation. Mice that carry a deleterious mutation in CSF-1 develop characteristic skeletal malformation, caused by defective osteoclasts. In the context of an inflammatory disease, such as atherosclerosis however, deletion of CSF-1 results in dramatically reduced atherosclerotic plaque size (47, 48). After myocardial infarction an upregulation of CSF-1 is observed in ischemic areas for more than 5 days after the injury (49). In this context, CSF-1 also appears to exhibit indirect effects by regulating chemokine production (49). In the absence of CSF-1, GM-CSF controls the differentiation of selected macrophage subsets and may lead to an enhancement of macrophage lineage numbers (50). Under certain conditions, GM-CSF induces an inflammatory program marked by increased IL-1, IL-6 and TNF-¦Á secretion, whereas the presence of CSF-1 suppresses the production of pro-inflammatory signals (51). Others have reported that administration of CSF-1 in EAM between days 21 and 29 after disease induction ameliorated cardiac fibrosis and left ventricular dysfunction by preventing the accumulation of fibroblasts (52). Notably, no effects were observed in the above mentioned study when CSF-1 was administered at later stages. These conflicting results stress the importance of timing in CSF-1 modulation. The reduced numbers of inflammatory monocytes due to silencing myeloid CSF-1 from days 14 to 21 observed in this study may outweigh the beneficial effects of CSF-1 in the inflamed heart at these early stages. In addition, macrophages have been shown to release pro-fibrotic cytokines such as TGF-¦Â, which causes the differentiation of fibroblasts to myofibroblasts and a massive deposition of extracellular matrix proteins such as collagens (53). Since we see a reduction of myeloid cells during myocarditis in response to siCSF-1 treatment and this includes macrophages, we propose that thereby achieved reduction of pro-fibrotic signals most likely contributes to lower scar formation as observed at advanced states of EAM.
It has been reported that other cell types beyond cells of the myeloid lineage are capable of CSF-1 expression under inflammatory conditions, including fibroblasts and endothelial cells (54, 55). These cells may also influence monocyte production/proliferation during myocarditis. Nevertheless, at the acute state of virus-mediated myocarditis, we observed that Mac-3 positive cells like monocytes/macrophages, which infiltrate the heart, may represent the main producers of CSF-1. Many studies using different pathogenic models of bacterial, viral and fungal infections have highlighted the importance and requirement of TNF- and NO-producing monocytes as facilitators of the resolution of an infection (56). Also, during inflammation bone-marrow derived monocytes can mature into macrophages (56) and macrophage-depletion results in increased virus titers in infected mouse hearts (17). Therefore, it was somewhat surprising at the first glance that depletion of monocytes upon siCSF-1 treatment actually improved virus-mediated pathology. We found no experimental evidence that virus dissemination, control and elimination had been adversely affected by such immune-modulating intervention. Since virus titers were not significantly influenced by siCSF-1-treatment, the impressively reduced inflammatory injury and preserved cardiac function in this group is not influenced by direct virus-mediated effects. These data suggest that virus-induced inflammation can be segregated from pathways that promote and limit virus infection and CSF-1-induced monocyte maturation. Monocytes can be directly activated by CVB3 infection resulting in the production of pro-inflammatory cytokines (57). Thereby, these cells can contribute to a strong inflammatory response and eventual tissue damage in the myocardium. Further experimental evidence for an adverse function of myeloid immune cells comes from macrophage depletion studies, where myocardial injury and formation of cardiac fibrosis were substantially diminished despite and in clear contrast to increased viral burden during AVM (17). Interestingly and in line with our findings in siCSF-1 treated A.BY/SnJ mice, macrophage depletion did not adversely affect clearance of infectious virus particles (17). We conclude that attenuated innate immune cell mobilization and hampered CSF-1-driven differentiation of innate myeloid cells in siCSF-1-treated mice directly suppresses cytotoxicity induced by cytokine production and/or infiltration with lymphocytes in viral myocarditis. Although there is a clear causal relationship between T1IFN-mediated suppression of viral load in infected cardiac cells and attenuation of inflammation and chronic tissue damage (8, 32), several publications including work of our group support the concept that¡ªindependent of direct virus-induced cell injury¡ªparticularly monocytes and macrophages are important players in inflammation and chronic organ damage in response to Coxsackievirus infection (4, 14, 58, 59).
¡¡¡¡
¡¡
¡¡
¡¡
¡¡
¡¡
Taken together, modulation of the CSF-1 axis in the myeloid cell lineage with siRNAs at early stages has beneficial acute and long-term effects in both viral and autoimmune myocarditis. Our data support the notion that particularly infiltrating myeloid cells contribute to acute and chronic functional impairment in inflammatory heart disease. Since pathogen control was not influenced upon suppression of the CSF-1 axis in myeloid cells, this study yields important insights for translating the pathophysiological role of CSF-1 from animal models to putative novel therapeutic targets for patients with inflammatory heart disease.¡¡
Frontiers | Silencing the CSF-1 Axis Using Nanoparticle Encapsulated siRNA Mitigates Viral and Autoimmune Myocarditis | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2018.02303/full¡¡

.jpg)