ÀÀ
The Role of Oxidative Stress in Viral Infections
1. IL-6 is expressed and release in response to enhanced ROS induced stresses, like infectious inflammation and non-infectious injuries
2. expression of the NF-ÎòB target gene IL-6 was significantly up-regulated in GFAP super-repressor mice,
3. inducible astrocytic NF-ÎòB does not function in cell survival but exacerbates chemokine-driven defects in spinal-cord recovery
4. NF-ÎòB signaling in astrocytes might regulate chemokine-induced infiltration of immune cells into the lesioned brain.
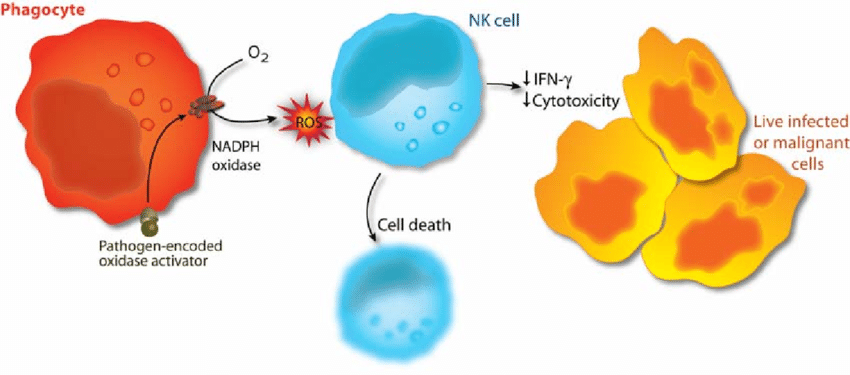
5. Pathogen-encoded oxidase activator, Oxidant-induced cell death in lymphocytes ´C mechanisms of induction and resistance
6.Activation of NADPH oxidase thru: phospholation of subunits, inhibited by DHA and vitamin C?
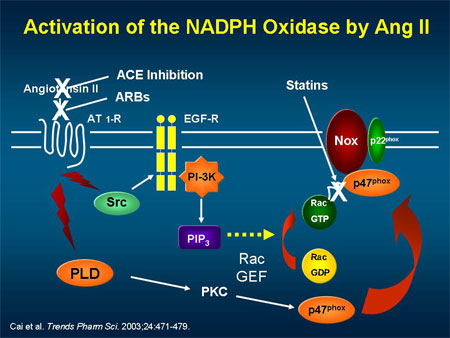
7.reactive oxygen species (ROS) production by NADPH oxidases in angiotensin II (Ang II) signaling
NADPH Oxidases as ROS Sources
In VSMCs, rapid ROS release requires p47phox via its phosphorylation by protein kinase C (PKC) and subsequent translocation to the membrane. Vitamin C is an inhibitor of PKC-protein kinase C.
ÀÀ
http://www.cell.com/trends/pharmacological-sciences/fulltext/S0165-6147(11)00159-3
ÀÀ
ÀÀ
ÀÀ
ÀÀ
The Role of Oxidative Stress in Viral Infections
Article in Annals of the New York Academy of Sciences 917(1):906-12 ÀÊ February 2000 with 48 Reads
DOI: 10.1111/j.1749-6632.2000.tb05456.x ÀÊ Source: PubMed
Oxidative stress is implicated as a pathogenic factor in a number of viral infections.Our work has shown that nutritionally induced oxidative stress exacerbates the pathogenesis of coxsackievirus B3 (CVB3) infection in mice. Of particular note, mice fed on a diet deficient in antioxidants developed myocarditis when infected with a normally benign strain of CVB3.
This change in virulence was found to be due to changes in the viral genome. Immune functions of the oxidatively stressed mice were also altered.
Another example of the effect of oxidative stress on a viral pathogen took place in Cuba in the 1990s. An epidemic of optic and peripheral neuropathy in the population occurred that was associated with a lack of dietary antioxidants and with smoking (a pro-oxidant). A coxsackie-like virus was isolated from the cerebrospinal fluid from 84% of patients cultured. Thus, oxidative stress can have profound effects, not only on the host, but on the pathogen as well.
îŃ¢ñÂüøȤàÝè좿₤¥ê£ÃÆíñÂýÀу¡Åàƒ
Ñ₤öÿîŃ¢üåòƒÈ˜àÝñ΢¿î¾£₤¥êçáèéò°£Ã¥ÆøÄ¢ôà«óÌýÀу¡ÅàƒÀÈ
ÆàóðøççûæÂØãçáòúȘ¡½ÅÀòµö¿ò°àÝñ΢¿î¾¥êçáØ«ò°È˜ÅÀòµ£Ã¡ÅàƒýÀуÅåÅá¥ÀîæÀÈ
î¾£₤ÆÎ¥Êç¥øôýÀу¡ÅàƒçáêÚØ£¡—â»æÆñÂèºåÖ1990áõçá¿é¯ëÀÈåÖàÝè좿₤¥êèéò°¤ëö■îäçáàùà¤Ý˜ñÂê¼ÅÅÅåòÆèþƒÙ¤ëëãøÉèþƒÙýÀȘåÖ84%çᣥíÔøÅçáá奿ؤøÅñÂüøêù¢ôà½óÌýÀуÀÈ
ѽúØȘîŃ¢£¿ñÂüøȘ₤ÆΥʣ¿£Ãç¥øôýÀуçáÝðؚȘ嗥ÆýÀуçáуêÎÀÈThe Role of Oxidative Stress in Viral Infections
https://www.researchgate.net/publication/12065458_The_Role_of_Oxidative_Stress_in_Viral_Infections... It occurs when redox homeostasis within the cell is altered due either to an overproduction of ROS and/or to deficiency of the counteracting antioxidant system (Valko et al. 2007 ). The pathogenesis of several viral diseases in animals, among them PRRS, has been linked to oxidative stress (Schwartz 1996, Beck et al. 2000, Chiou et al. 2000, Karadeniz et al. 2008, Panda et al. 2009, Kataria and Kataria 2012 ). Virus-induced activation of phagocytes is associated with oxidative stress, not only because ROS are released but also because activated phagocytes release pro-oxidant cytokines. ...
ÀÀ
Amelioration of influenza virus-induced reactive oxygen species formation by epigallocatechin gallate derived from green tea
Sep 2012
J. Ling
Fei Wei
Ning Li
Zhanqiu Yang
abstract
... When the concentrations of ROS exceed the ability of the cell to turn over these species, oxidative stress will result and lead to the widespread oxidation and damage of biomolecules including DNA and proteins (Berlett and Stadtman, 1997;Finkel and Holbrook, 2000). There is accumulating evidence which shows that oxidative stress can affect the interaction between the host and viral pathogens, and hence viral pathogenesis ( Beck et al., 2000). Recent studies have indicated that herpesvirus infections like HSV-1 and RRV can induce oxidative stress in cells and in tissues through induction of oxidized proteins which are enriched in the VICE (virus-induced chaperone enriched domains) foci in the nucleus (Mathew et al., 2010). ...It is well known that viruses alter the intracellular redox state to pro-oxidant conditions, which is an alteration that contributes to viral pathogenesis [26][27][28][29][30][31]. Thus, researchers have been evaluating redox signaling as a potential target for antiviral strategies [32][33][34][35][36][37][38]. ...
ÀÀ
abstract
... In addition to endogenous modulations leading to diseases, RSCDPs and redox regulation play critical roles in host-pathogen interactions as well. The role of redox-sensitive proteins in interactions between viruses and eukaryotic hosts is known for several examples, such as Hepatitis C virus [91], coxsackievirus B3 [92] and Epstein-Barr virus [93]. Viruses, in general, exploit the redox- state dependence of several signaling pathways of the host cell, and this has even been recognized as having therapeutic value [94]. ...The Role of Oxidative Stress in Viral Infections
https://www.researchgate.net/publication/12065458_The_Role_of_Oxidative_Stress_in_Viral_InfectionsÀÀ
Inhibition of rotavirus ECwt infection in ICR suckling mice by N-acetylcysteine, ascorbic acid, peroxisome proliferator-activated receptor gamma agonists and cyclooxygenase-2 inhibitors
Mem Inst Oswaldo Cruz. 2013 Sep; 108(6): 741´C754.
Departamento de Ciencias Fisiol´Ûgicas, Facultad de Medicina, Instituto de Biotecnolog´ˆa, Universidad Nacional de Colombia, Bogot´Â, DC, Colombia
Sep 2013
Rafael Antonio Guerrero-Rojas
Orlando Acosta
Carlos A GuerreroAbstract
Live attenuated vaccines have recently been introduced for preventing rotavirus disease in children. However, alternative strategies for prevention and treatment of rotavirus infection are needed mainly in developing countries where low vaccine coverage occurs. In the present work, N-acetylcysteine (NAC), ascorbic acid (AA), some nonsteroidal anti-inflammatory drugs (NSAIDs) and peroxisome proliferator-activated receptor gamma (PPARÎû) agonists were tested for their ability to interfere with rotavirus ECwt infectivity as detected by the percentage of viral antigen-positive cells of small intestinal villi isolated from ECwt-infected ICR mice. Administration of 6 mg NAC/kg every 8 h for three days following the first diarrhoeal episode reduced viral infectivity by about 90%. Administration of AA, ibuprofen, diclofenac, pioglitazone or rosiglitazone decreased viral infectivity by about 55%, 90%, 35%, 32% and 25%, respectively. ECwt infection of mice increased expression of cyclooxygenase-2, ERp57, Hsc70, NF-ÎòB, Hsp70, protein disulphide isomerase (PDI) and PPARÎû in intestinal villus cells. NAC treatment of ECwt-infected mice reduced Hsc70 and PDI expression to levels similar to those observed in villi from uninfected control mice. The present results suggest that the drugs tested in the present work could be assayed in preventing or treating rotaviral diarrhoea in children and young animals.
Keywords: rotavirus infection, N-acetylcysteine, thiazolidinediones, NSAIDs, ICR miceInhibition of rotavirus ECwt infection in ICR suckling mice by N-acetylcysteine, peroxisome proliferator-activated receptor gamma agonists and cyclooxygenase-2 inhibitors
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3970679/ÀÀ
ÀÀ
Int J Mol Sci. 2011; 12(1): 334´C352.
The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation
Ralee Spooner1 and Özlem Yilmaz1,2,*
Author information Article notes Copyright and License information Disclaimer
1 Department of Periodontology, University of Florida, Gainesville, FL 32610, USA; E-Mail: moc.liamg@renoops.e.eelar
2 Emerging Pathogens Institute, University of Florida, Gainesville, FL 32610, USA
* Author to whom correspondence should be addressed; E-Mail: ude.lfu@zamliyo; Tel.: +1-352-273-8003.
Abstract
The mechanisms of chronic infections caused by opportunistic pathogens are of keen interest to both researchers and health professionals globally. Typically, chronic infectious disease can be characterized by an elevation in immune response, a process that can often lead to further destruction. Reactive-Oxygen-Species (ROS) have been strongly implicated in the aforementioned detrimental response by host that results in self-damage. Unlike excessive ROS production resulting in robust cellular death typically induced by acute infection or inflammation, lower levels of ROS produced by host cells are increasingly recognized to play a critical physiological role for regulating a variety of homeostatic cellular functions including growth, apoptosis, immune response, and microbial colonization. Sources of cellular ROS stimulation can include À¯danger-signal-moleculesÀÝ such as extracellular ATP (eATP) released by stressed, infected, or dying cells. Particularly, eATP-P2X7 receptor mediated ROS production has been lately found to be a key modulator for controlling chronic infection and inflammation. There is growing evidence that persistent microbes can alter host cell ROS production and modulate eATP-induced ROS for maintaining long-term carriage. Though these processes have yet to be fully understood, exploring potential positive traits of these À¯injuriousÀÝ molecules could illuminate how opportunistic pathogens maintain persistence through physiological regulation of ROS signaling.
Keywords: reactive-oxygen-species, opportunistic pathogens, microbial persistence, extracellular ATP, P2X7 receptor, Porphyromonas gingivalis, NLRX1, NADPH oxidase, epithelium
1. Introduction
One of the early responses of host innate immunity is the Reactive-Oxygen-Species (ROS) production in reaction to microbial invaders. Free oxygen radicals are highly toxic to pathogens and are utilized as a tool to prevent colonization of tissues by microorganisms. ROS are also a key part of the intracellular redox profile influencing a wide variety of signaling networks [1]. Therefore, the relatively high-level of oxidants, as seen in respiratory burst or low-levels in normal tissue, is likely a significant aspect of cell physiology during infection. Oxidative stress occurs when oxygen radical formation and levels exceed those of antioxidants, potentiating cell responses such as apoptosis, tumorgenesis, and immune-response [1´C3]. However, the necessity of ROS for immune function could be exploited by À¯potential pathogensÀÝ to reduce host responses in order to enhance survival and colonization in their target host cells. Consistent with this view, characterization of molecular circuitries between the cellular ROS and a host derived small-danger-molecule À¯extracellular ATPÀÝ (eATP), which has lately been recognized as an important physiological signaling mediator that can convert opportunistic microbes into pro-inflammatory pathogens or provide long-term persistence for disease causing pathogens, is of great interest to current research [4,5]. Accordingly, we will first provide a general overview of recent literature on the mechanisms of production of cellular ROS and its role in establishment of microbes in host tissues. Then, we will present the novel subject of purinergic eATP and P2X7 coupling mediated cellular ROS and its significance for the microbial persistence and inflammation through examples that have recently become apparent.
2. Cellular ROS and Its Implications for Microbial Infections
ROS molecules participate in multi-faceted activities within cells, acting as secondary signaling molecules for inflammation and immune responses, in addition to directly harming microbes that invade tissues [6]. As a hallmark of innate immunity, it is important to understand the necessity of ROS signaling for host responses to challenges presented by microbial invasion. Efficient clearance of colonizing microorganisms is highly dependent on ROS, thus exploration of the mechanisms between oxygen radicals and innate immune responses associated with pathogen clearance has been an intriguing topic for researchers.
2. 1. Sources of Intracellular ROS Production as an Early Response
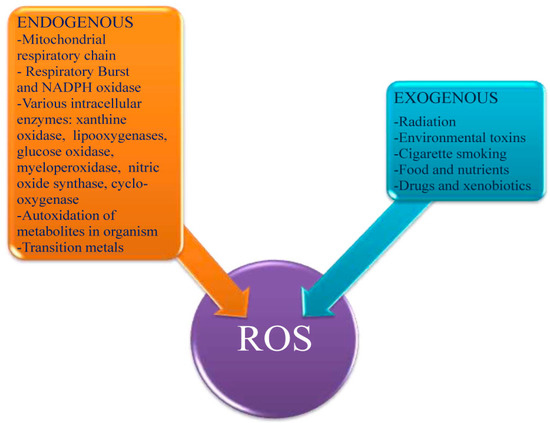
It is well known that ROS production is rapidly elevated during infection, serving to facilitate pathogen clearance as well as contributing to signaling cascades related to inflammation, cell proliferation, and immune responses [7]. Two of the best characterized sources of ROS during host cell- microbe interactions are the membrane associated NADPH oxidase complex and the mitochondrial electron transport chain (Mt-ETC). The Mt-ETC transiently produces radicals as electrons move between the complexes, primarily at Complexes I [8,9] and III [9] and these play key roles in activation of innate immunity.
The connection between innate immune responses, mitochondrial-ROS and microbial infection has been unclear in the past. Recent evidence suggests that a Nucleotide-Binding domain (NBD) and Leucine-Rich Repeat (LRR)-containing family member receptor (NLR), NLRX1, may provide an answer to this elusive question. NLRs, also well known as Nod-like receptors, are intracellular pathogen-associated molecular pattern (PAMPs) receptors, which serve to alert the cell to the presence of intracellular invaders. Though there are many NLRs, only one of these localizes to the mitochondria to facilitate innate immune responses, NLRX1 [10,11]. NLRX1 stimulates production of ROS molecules, which in turn can directly interact with the pathogen as well as act as a secondary signaling molecule [12´C14]. Originally identified to utilize the mitochondrion as a platform to mediate antiviral responses [11], NLRX1 now appears to also be important for immune responses to bacterial infections as well. Knowledge of bacterial-mitochondrial interactions are limited, however, a recent review provides a comprehensive overview of bacterial influences upon the mitochondrion, including NLRX1 [15]. NLRX1-induced ROS appears to be critical for bacterial infections, especially in terms of its signaling properties. A study performed using Shigella-infected and tumor necrosis factor-Îê (TNF-Îê)-primed cells showed that NLRX1 mediated ROS formation [16]. The downstream result of the elevated ROS levels in this particular study was enhanced NF- ÎòB and Jun amino-terminal kinases-dependent (JNK) signaling, demonstrating the role for ROS as a secondary messenger for pro-inflammatory gene transcription and cytokine-based signal responses during infection. Another new study also provided evidence for the key role ROS-mediated signaling can have during infection. This groupÀ₤s efforts showed that NLRX1 enhanced mitochondrial-derived ROS formation in response to Chlamydial infection [17]. Though only a small amount of evidence supports the importance of NLRX1-mediated ROS production during infection, this is most likely due to the recent idea that NLRX1 could be an important mediator of mitochondrial-ROS production during infections. Much like the mitochondria, NADPH oxidase is a primary site for generation of ROS molecules during microbial infection of host cells [18].
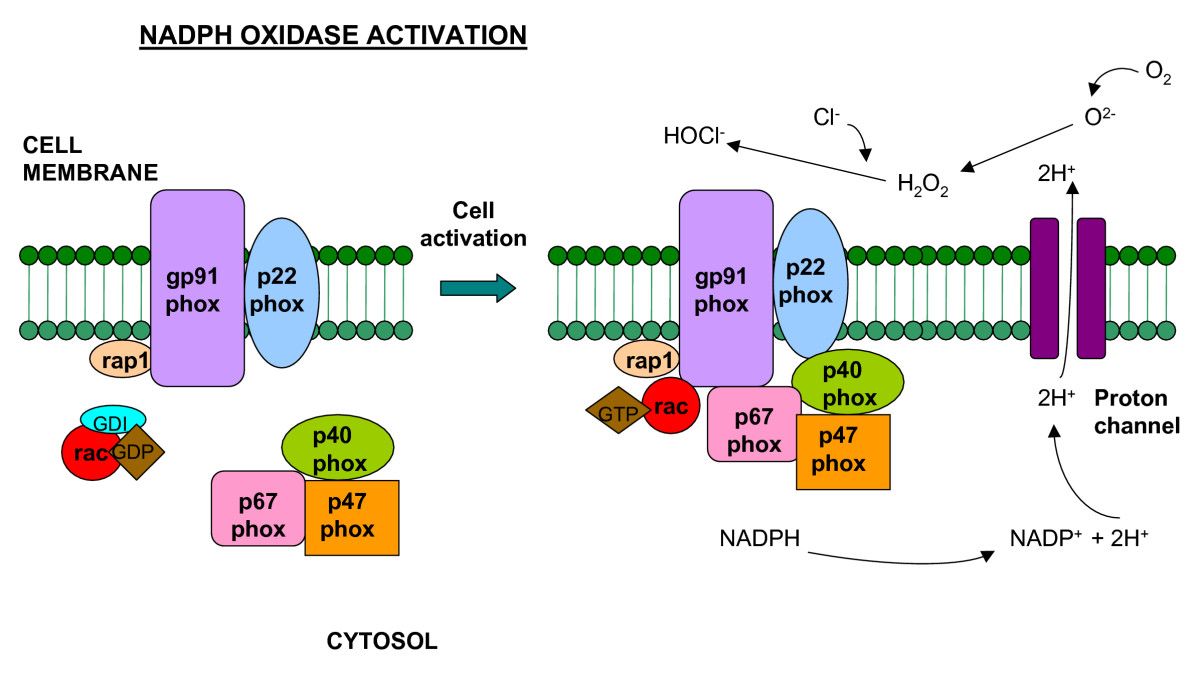
NADPH oxidase complexes are common sources of ROS typically associated with rapid respiratory burst mechanisms of professional phagocytic cells, and carry out the electron transferring reaction from NADPH to molecular oxygen (O2) in the membranes of phagosomes, endosomes, and the cell membrane [19]. The studies lately have shown the functionally active NADPH oxidase machinery in various cells types including epithelial and endothelial cells [20,21]. NADPH oxidase protein complexes consist of several phox subunits; the cytosolic p40, p47, and p67 in addition to p22 and gp91, which localize to the membrane. The latter two phox subunits, p22 and gp91, make up flavocytochrome b558, the catalytic, superoxide-generating core of NADPH oxidase [22]. A type of Rho-GTPase, called Rac, also associates with the complex [19,22] and mediates NADPH oxidase activity [19,23] and downstream inflammatory signaling processes [24]. Certain regulatory mechanisms are in place to control NADPH oxidase activity, including proteins and wide-sweeping signaling cascades. Control of Rac falls to the Rho-GDP dissociation inhibitor (Rho-GDI), which translocates Rac from the membrane to the cytoplasm, effectively deactivating NADPH oxidase [25,26]. Phosphoinositide-3 kinases (PI3Ks) also manage the NADPH oxidase complex through the modulation of Rac activity and direct binding of the p40/47 subunits [27]. Thus, the powerful induction of ROS and inflammatory signaling cascades by NADPH oxidase are under multiple levels of control, likely to ensure faithful activation of the complex and its downstream signaling intermediates. NADPH oxidase activation is vital for management of microbial invasion, but the precise mechanisms linking activation of the complex with elimination of microorganisms has been not completely characterized. A recent study of LyGDI (a monocyte-specific Rho-GDI [25]) may provide valuable insight into the underlying molecular mechanisms mediating NADPH oxidase activity in response to phagocytosis of microbes. LyGDI was shown to associate with caspase-recruiting domain-containing protein 9 (CARD9) at phagosomes harboring Listeria monocytogenes in macrophages [28]. The interaction between CARD9 and LyGDI allowed Rac to associate with the other NADPH oxidase components, thereby providing the activating signal for ROS production by NADPH oxidase. Identifying potential mechanisms for immune response element activation, such as CARD9-lyGDI association, could be crucial to understanding host immunity against microbial invasion.
Thus, the highly regulated NADPH oxidase is a potent contributor to resistance against infection by microorganisms. Due to the key role of NADPH oxidase for participating in immunity, it seems logical that it should be a target for microbial intervention. In actuality, microbes can subvert NADPH oxidase through direct interactions with the phox subunits and Rac. Additionally, mitochondrial-based ROS production can function to eliminate microorganisms. Much like NADPH oxidase, the mitochondrion appears to be a target for microbial interference. We will now provide examples of microbes demonstrated to modulate mitochondrial and NADPH oxidase-induced ROS to mediate immune evasion.2. 2. Modulation of Intracellular ROS by Infection
Immune cells depend on ROS to not only kill phagocytosed microorganisms, but also to mediate inflammatory and immune signaling cascades. There is direct evidence that a wide variety of microbes can limit ROS, thus increasing the potential for persistent infection [29]. This novel strategy to promote microbial survival within the hostile host environment appears to be primarily the result of NADPH oxidase and mitochondrial-derived ROS modulation.
Many bacteria directly interfere with accumulation of ROS molecules via prevention of NADPH oxidase assembly. Francisella tularensis live vaccine strain was shown to not only escape neutrophil phagosomes, but also directly prevented NADPH oxidase assembly during intracellular infection [30]. This bacterium specifically prevented recruitment of gp91phox/p22phox and p47/p67phox subunits to the phagosome, thereby attenuating the oxidative burst mechanism utilized by neutrophils. Another bacterium, Anaplasma phagocytophilum, prevents p22 and gp91 incorporation into phagosomes containing the microbe, thus attenuating NADPH oxidase activity [31,32]. It is also suggested that A. phagocytophilum also directly scavenges ROS, providing an additional defense mechanism against oxygen radical-based host immunity [31].
A study of Chlamydia trachomatis, a bacterial pathogen associated with urogenital infections, demonstrated that the rapid induction of ROS within macrophages could be attenuated over a period of time [33]. This was shown to be accomplished by incorporation of the Rac subunit of NADPH oxidase to the Chlamydia-containing vacuole, thus inhibiting oxidative burst. The most important observation of this study, however, is that Chlamydia can alter the intracellular ROS content back to normal steady-state conditions [33]. By returning ROS levels to normal, as seen in uninfected cells, the Chlamydia essentially masks its presence by altering a key immune sensor and inflammation activating mechanism.
The ability to inhibit recruitment of NADPH oxidase subunits appears to be a common theme for many other pathogens, including Legionella pneumophila and Coxiella burnetii. C. burnetii, which is known to cause chronic Q fever in livestock and humans, prevents recruitment of p47/p67 to the phagosome and thus limits respiratory burst within infected neutrophils [34]. Prevention of p47 recruitment is also a facet of Legionella infection of macrophage-like cells [35]. Despite the fact that a strong majority of literature introducing this strategy involves hematopoietic-cell lineages, there is evidence supporting NADPH oxidase assembly interference during infection of other tissue types. For example, Bacillus anthracis edema toxin has been implicated in prevention of NADPH oxidase activity [36]. This was shown to be achieved through Protein Kinase A-mediated phosphorylation of the NADPH oxidase component p67, in intestinal epithelium [36].
Extracellular microorganisms also possess the ability to alter intracellular ROS production, contributing to persistence of infection. Burkholderia cenocepacia are opportunistic pathogens implicated in chronic inflammatory conditions including Cystic Fibrosis (CF). These bacteria have been demonstrated to inhibit ROS production in infected neutrophils, primarily through exopolysaccharide formation [37]. In addition to interfering with neutrophil chemotaxis, the exopolysaccharides demonstrated the ability to directly scavenge free oxygen radicals, regardless of their intracellular origination. B. cenocepacia also has been shown to exist in macrophage vacuoles with delayed NADPH oxidase incorporation, suggesting that it can persist within host cells by prevention of NADPH oxidase components and oxidative burst [38]. In another opportunistic bacterium associated with CF, Pseudomonas aeruginosa, secondary metabolite production by the pathogen has been shown to interfere with phagocytic engulfment of apoptotic cells [39]. This secondary metabolite, pyocyanin, mediates this process by reducing intracellular ROS levels and Rho GTPase activities in host cells, likely leading to enhanced inflammation at sites of infection due to failure to phagocytose dying cells [39].
Neisseria gonorrhoeae is known to suppress oxidative bursts in neutrophils [40]. This observation was not observed for nonviable N. gonorrhoeae, suggesting that live cell metabolic activity directly relates to suppression of oxidative burst. Though this study did not attempt to identify potential targets for inhibition of NADPH oxidase ROS, the observed reduction in oxygen radical production was shown to be dependent upon contact between the host cells and the metabolically active bacteria [40].
Candida albicans, an opportunistic fungal pathogen of humans, has the ability to suppress ROS production in host immune cells [41]. The precise mechanism utilized by C. albicans remains unclear, but a recent study presented findings indicating that the microorganism is capable of overcoming auto-induction of ROS, resulting in suppressed oxygen radical formation [41]. Another fungal pathogen, Aspergillus fumigatus, has been shown to limit ROS production in neutrophils [42,43].
Unlike the previously described organisms, Entamoeba histolytica, potentiates NADPH oxidase ROS accumulation in host cells, resulting in apoptosis [44]. This parasite utilizes the secondary signaling properties of NADPH oxidase-derived ROS molecules to influence Extracellular signal-Related Kinase (ERK) 1/2 signaling, eventually stimulating host cell death. Though typically associated with cell survival and proliferation, there is documented evidence supporting the potential for ERK1/2 to induce cell death [45´C47]. Infection of host cells by two other pathogens, Trypanosoma cruzi, the causative agent of ChagasÀ₤ disease, and Japanese encephalitis virus (JEV), also results in enhanced ROS formation. These two pathogens also utilize enhanced ROS to induce host cell death, thus allowing escape and likely contributing to spread of the pathogens through the apoptotic bodies.
JEV infection of promonocytic cells resulted in host cell apoptosis mediated by intracellular accumulation of ROS and activation of pro-apoptotic signaling [48]. Destruction of host cells by virus-induced ROS production could facilitate persistence of viral infection by enhancing the spread of virions. T. cruzi can infect humans and lie dormant, only to reemerge and cause chronic inflammatory destruction of organs. Interestingly, a recent study demonstrated that T. cruzi-induced ROS accumulation did not involve NADPH oxidase [49]. Further analysis in this report showed that enhanced ROS formation occurred due to pathogen-mediated mitochondrial membrane potential perturbation. Though this was not the first suggestion that T. cruzi optimizes infection and persistence through mitochondrial ROS accumulation and oxidative stress [50], this study does demonstrate the current level of interest that researchers have in regards to understanding microbial-mediated influences on ROS production.
Possession of the High Pathogenicity Island (HPI), encoding yersiniabactin, appears to play a role for pathogenesis of Yersinia and members of Enterobacteriaceae. A new study suggests that due to the iron-scavenging nature of yersiniabactin, it could provide a novel way to inhibit ROS formation by preventing the Haber-Weiss hydroxyl-radical-forming reaction in infected host cells [51]. Though this may be an indirect method to limit pathogen clearance by host cells, it is evident that bacteria possessing HPI display enhanced pathogenesis [51]. Additional Yersinia-related virulence factors have been extensively studied [52´C55]. However, more detail studies are required to fully characterize Yersinia-directed ROS inhibition, especially for determining the sources of mechanisms providing the ROS regulation.
Modulation of ROS levels appears to be a conserved strategy utilized by various microorganisms. Induction or inhibition of oxygen radicals could facilitate enhanced colonization, via reduced ROS-mediated host responses to infection. Additionally, regulation of cellular ROS by microorganisms can play a critical role in chronic inflammatory diseases and cancer. We will now focus on our main interest of signaling molecules, eATP and its putative receptor P2X7 generated ROS and the evidence for microbial modulation of this pathway to enhance microbial survival and persistence in host tissues.
3. eATP-P2X7 Receptor Signaling in Controlling Infection and Inflammation via Cellular ROS
3. 1. eATP as a À¯Danger SignalÀÝ
Adenosine-5Àð-triphosphate, ATP, is a nucleotide molecule utilized for a variety of processes, including cellular metabolism and signaling. ATP can be released into the extracellular environment by cells under normal conditions as well as a result of cellular stress, infection or cell death. Once outside of the cell, it is identified as extracellular ATP. eATP has become well recognized for its role as a À¯danger signalÀÝ for immune activation [56,57]. Effect of eATP is carried out via stimulation of cellular purinergic receptors and their associated signaling processes. Lately, this host-cell derived small molecule induced responses have become a key topic for researchers studying the pathogenic nature of a wide variety of microorganisms. This may represent a novel target for pathogen-mediated subversion of the host immune response.
The evidence supporting the role of eATP as a potent activator in innate immunity is quite large. Pro-inflammatory cytokine production and secretion is a well characterized event in numerous pathologies, and ATP clearly has a role in initiating these events. For example, eATP has been shown to elicit transcription of IL-6 in macrophages [58]. IL-6 plays an important role in phagocytic cell responses of innate immunity, primarily serving as a mediator of macrophage development from monocytic cells [59]. IL-6 stimulation of monocytes also suppresses dendritic cell (DC) formation, suggesting eATP may perform antagonistic roles in immunity, possibly in immunotolerance [60]. It should also be noted, that eATP has been implicated in maintenance of lung airway inflammation via DC activation [61], thus demonstrating the complex nature of eATP-mediated processes (For a detailed review regarding multifaceted capabilities of ATP on various cell types, readers should see [56] for more information). eATP is also involved in production of the inflammatory cytokine IL-1Îô, a participant in the regulation of cell differentiation, death, and inflammation. A study performed in 1998 characterized eATP-induced release of IL-1Îô from LPS-primed monocytes, while also confirming that this process was dependent on the purinergic P2X7 receptor [62]. It also appears that in other cell models, such as fetal astrocytes, IL-1Îô-mediated transcription factor expression is linked with ATP and P2 receptor signaling [63,64]. Elevated Nuclear Factor-kappaB (NF-ÎòB) and activator protein-1 (AP-1) expression were seen when astrocytes were stimulated with eATP, suggesting a role for purinergic signaling in nervous system inflammatory pathologies [64].
Acting as a sensor for pathogens, the inflammasome promotes pro-inflammatory cytokine production, a process dependent upon generation of ROS. The inflammasome is a critical component of the innate immune system and facilitates heightened immune activities against pathogenic invaders, such as Chlamydia [65]. The inflammasome consists of Apoptosis-associated speck protein (ASC), caspase-1 and NLR3, NOD-like receptor protein 3 [66]. Cleavage of immature pro-IL-1Îô by capsase-1, results in mature IL-1Îô formation [67]. Acting as a secondary signaling molecule, ROS are required for NALP3 inflammasome formation [66] and recent studies demonstrate the necessity of ROS to mediate this process [43]. Furthermore, the processing of IL-1Îô in P. gingivalis-infected primary gingival epithelial cells was shown to require eATP stimulation via P2X7 receptor coupling for cytokine release [68]. This could imply that not only can eATP provide a pro-inflammatory stimulus to cells, but it can also directly influence final processing and release of cytokines.
Taken together, eATP signaling exhibits a multi-tiered influence on inflammation via innate immunity. A confounding question, however, is whether innate and adaptive immunity can be linked through eATP signaling and associated ROS production. A recent study of anticancer therapy has identified a role for eATP signaling in directing both innate and adaptive immunity. It is well known that dying and injured cells release ATP, even during anti-tumor treatments [69]. The present study also identified induction of Il-1Îô secretion from DCs in response to ATP release, resulting in priming of interferon-gamma (IFN-Îû)-producing lymphocytes. Thus, eATP has effectively created a potent anticancer response, acting through inflammatory cytokine signaling to stimulate adaptive immunity [70]. Another novel finding linked eATP signaling with antigen presentation to coordinate Mycobacterial destruction. eATP and P2X7 coupling has been shown to rapidly induce shedding of MHC-II containing exosomes from macrophages as a result of inflammasome activation [71,72]. As we have mentioned before, ROS secondary signaling facilitates inflammasome formation and activity [66], thus ROS can mediate both innate and immune responses. The same group expanded their studies to include Mycobacterium infection of phagocytes, and found that the released exosomes contained MHC-II: Mtb Ag 85B (a Mycobacterial antigen) [73]. The antigen presenting MHCII:Mtb Ag 85B complexes were also shown to activate T-lymphocytes [73], thus providing an important insight into eATP-P2X7-mediated synergy of innate and adaptive immune responses.
It is tempting to speculate that innate and adaptive immunity can be linked to operate concurrently through eATP mediated ROS production. These mechanisms are likely facilitated by the secondary signaling capabilities of ROS, and we have seen clear evidence supporting the role of oxygen radicals in immune activation. However, it is important to recognize the highly complex and dynamic nature of the immune system, and coordination of both the innate and adaptive immunity is not likely to be exclusively limited to eATP purinergic signaling.
3. 2. P2X7 Receptor Activation by eATP Elicits Signaling Cascades through ROS
Purinergic receptor signaling lately has emerged as an important signaling pathway related to various cellular processes. P2X7 has been classically distinguished from the other P2 receptors due to the fact that it is a ligand-gated ion channel and it has low-affinity to eATP concentration. Upon eATP binding of P2X7, Ca2+ ions flow through the pore formed in the membrane of the cell, creating signal-transducing stimuli. This in turn activates several signaling intermediates, including NADPH oxidase ROS production, which can operate to influence pro-inflammatory and immune response elements, such as cytokine synthesis and transcription factor activation [74´C76].
A large body of evidence suggests that the signaling networks of eATP-P2X7 coupling are in fact dependent on ROS production. In 1997, it was discovered in microglial cells that NF-ÎòB, a potent pro-inflammatory transcription factor, expression was influenced to a large degree by P2X7 [77]. This study also revealed that unlike other NF-ÎòB complex formation resulting from other stimuli, such as LPS, ATP specifically stimulated formation of p65 homodimers. More importantly, the same study demonstrated that formation of NF-ÎòB was abolished when cells were treated with antioxidants, thus providing strong evidence for the necessity of eATP-induced ROS generation for pro-inflammatory gene transcription [77].
Another recently described transcription factor, cyclic-AMP responsive element binding protein (CREB), which plays a protective role for cell survival, appears to be important during elevated ROS conditions (e.g., oxidative stress). Unlike NF-ÎòB, which is associated with pro-inflammatory cytokine production and cell death, CREB protects cells against oxidative stress-induced cell death [78´C80]. It also seems that eATP-P2X7 coupling can influence the CREB pathway, perhaps offering cells a level of protection against oxidative stress-mediated cell death [79,81]. It remains to be seen how CREB participates in infection models, but it could provide a level of resistance to cell death in response to pathogenic challenge.
Much like the dualistic nature of ROS molecules themselves, the transcription factors that oxygen radicals influence can potentiate antagonistic responses in cells. Though we have already shown evidence for microbial modulation of ROS, primarily via NADPH oxidase interference, eATP-P2X7 coupling can also be a target for microorganisms seeking to alter ROS production for their long term carriage. NADPH oxidase complexes are now associated with P2X7 signaling cascades, thus interference directly at this receptor could provide another way for microbes to enhance survival and persistence.3. 3. eATP-P2X7 Receptor Signaling Mediates Pathogen Killing
There is a noteworthy amount of research regarding eATP-induced pathogen elimination via the P2X7 receptor. Toxoplasma gondii, for example, has been demonstrated to be eliminated by eATP-P2X7 coupled production of ROS molecules, thus facilitating reduction of parasitic load within host cells [82] (Another study yielded similar results, however, this group did not test for ROS involvement in the observed clearance of T. gondii [83]). In these two studies, however, T. gondii-infected host cells were subjected to rapid eATP-mediated apoptosis, a response serving to likely control extracellular spread of Toxoplasma [82,83].
Another microorganism that establishes chronic intracellular infections and is also subject to P2X7-mediated elimination is M. tuberculosis. Studies of this organism in host cell models clearly provide evidence of eATP-P2X7 induced Mycobacterial elimination through multiple processes, including apoptosis [84], phospholipase-D [85], autophagy [86] and phagosome-lysosome fusion [87]. Thus, P2X7 is likely a major contributor against Mycobacterial immunity. In addition, there have been numerous studies suggesting that polymorphisms in the P2X7 gene may increase susceptibility to Mycobacterial infections, demonstrating the importance of functional P2X7 in these pathologies [88´C92].
Chlamydial infections of host cells are also found to be limited by eATP-P2X7 signaling processes. eATP-P2X7 stimulation of Phospholipase-D activity was shown to be required to limit some Chlamydial infections of epithelium and macrophages [93´C95]. Taken together, these studies show that eATP and P2X7 receptor signaling are important immune response elements to a variety of pathogenic infections. However, targeting of eATP-P2X7 coupling could be exhibited by pathogens, and in fact there are examples of microbes that exploit this to establish persistence and survival.
3. 4. Subversion of eATP-P2X7 Pathway by Persistent Microbes
Intracellular parasites can exhibit selective pressures on the P2X7 receptor-mediated signaling network, influencing host cell physiology to enhance survival. These effects are likely to be highly variable and dependent on multiple factors, including specific invading organism, host tissue type and physiology, environmental pressures, and even the presence of other colonizing organisms. This exploitation of eATP and the P2X7-mediated cell immune response represents a fascinating mechanism of overcoming host attempts to limit infection.
Some evidence suggests direct microbe-mediated modulation of P2X 7. Studies of Chlamydia psitacci show that the Ca2+ signal resulting from eATP-P2X7 coupling is diminished, resulting in reduced host cell apoptosis [96]. Though we have provided evidence for other organisms exhibiting ROS-modulatory effects to promote persistence, this study shows that P2X7 can also be a target to promote microbial survival in host tissues.
Leishmania amazonensis also has been demonstrated to interfere with eATP-P2X7 mediated signaling and associated ROS generation [97]. Identification of a multi-functional ATP utilizing enzyme ecto-Nucleoside diphosphate kinase (Ndk) in this protozoan has been attributed with eATP scavenging capabilities, reducing eATP-P2X7 induced host cell death during infection [98]. Additionally, the supernatant from M. bovis, an opportunistic pathogen, containing the Ndk protein decreases the activity of P2X7 receptor [99].
Porphyromonas gingivalis, a successful host-adapted pathogen capable of intracellular replication and spreading within the host cells, faces the challenge of host-mediated immune responses to clear the microorganism and prevent colonization [100´C102]. Similarly, P. gingivalis has also been shown to exploit eATP-P2X7 signaling associated with host cell death in primary gingival epithelial cells (GECs) [103]. The organism accomplishes this by secreting an Ndk homologue, which hydrolyses eATP, thereby decreasing P2X7 ligation, resulting in the inhibition of GEC cell death. Indeed, current studies by our laboratory of P. gingivalis indicate that the bacterium is also capable of modulating the cellular ROS levels and protecting primary GECs against eATP-P2X7 induced oxidative-stress by inhibiting the global ROS production upon prolonged infection in the host cells. This effect appears to be mediated by both utilizing host NADPH oxidase and mitochondria associated oxidative-stress pathways via time-dependent secretion of P. gingivalis-Ndk enzyme during the infection [104]. P. gingivalis infection has also been demonstrated to inhibit mitochondrial membrane depolarization, cytochrome c release, and activation of caspase 3 and pro-apoptotic Bad through pro-survival PI3K/AKT pathway [105´C107]. AKT and Extracellular signal-related kinase (ERK) 1 and 2, members of the mitogen-activated protein kinase (MAPK) network, have been studied in relation to P2X7, revealing interesting results. It was shown that ERK1/2 activity during LPS-stimulation of macrophages could be under control of P2X7 receptor [108,109]. In ovarian cancer cells, cellular ROS stimulation was found to be important for signal cascades, particularly for AKT and ERK [110]. As mentioned earlier, eATP was demonstrated to be required for release of IL-1Îô from P. gingivalis-infected GECs [68]. The same study also reported that GECs express functional Nalp3 inflammasome formation during the P. gingivalis infection, suggesting an additional role for ROS secondary signaling in this infection model.
Whether the ROS production and redox signaling are specifically influenced by the above discussed modulatory pathways during P. gingivalis infection remains to be seen. However, it is likely that eATP-P2X7 mediated ROS signaling play an important role in the persistent nature of this successful opportunist in the epithelial cells. As a model organism, P. gingivalis, a major etiological agent in the formation of chronic inflammatory/infectious diseases (namely periodontal diseases) could help explain some fundamental properties of chronic conditions associated with host-adapted opportunistic microbes, perhaps translating to new insights into other chronic inflammatory pathologies (Figure 1).
Figure 1
Proposed mechanisms for modulation of intracellular ROS via P2X7, NADPH oxidase, and Mitochondrion Interference. (A) Invasive microorganisms subvert with NADPH oxidase-based ROS formation via interference of complex assembly; (B) Microbial scavenging of ROS produced by NADPH oxidase; (C) Interference of mitochondrion-based ROS production during infection; (D) Modulation of eATP-P2X7 signaling induction by nucleotide scavenging; (E) CREB could be involved in promoting host cell survival, and thus may potentiate microbial persistence.
Intriguingly, a new study illustrated that NLRX1 and NADPH oxidase are both involved in Chlamydia trachomatis-induced ROS formation. NLRX1 and the ROS formed in the mitochondria actually promote the bacterial growth [17]. Depletion of NLRX1 and inhibition of NADPH oxidase ROS production attenuated Chlamydial load. In previous studies performed by the same group, it was discovered that Chlamydial growth was enhanced by ROS-mediated caspase-1 production via inflammasome formation [111]. It now seems that NLRX1 and NADPH oxidase play an important role in generating the ROS required to mediate signaling cascades resulting in caspase-1-dependent growth of Chlamydia [17]. It is logical to suggest that eATP can act as a physiological mediator for the cross-talk that takes place between the two pathways and include an additional layer of complexity in the whole host-microbe interaction.
4. Conclusions
It is quite notable that a number of distantly related intracellular pathogens exhibit similar abilities to influence inflammation and immune clearance by modulating eATP-P2X7 derived cellular ROS. This novel occurrence appears as a common theme that enhances survival of both the host and the invading microorganism, and it can be achieved through reduction of intracellular ROS through exploitation of host metabolic pathways by host-adapted microbes. More intriguingly, successful colonizers may also À¯unwittinglyÀÝ have come to use physiological ROS signaling properties for their own designs including microbial utility of eATP induced ROS as growth signaling molecules. Overall, a large amount of recent evidence suggests that a wide array of pathogens have developed various complex, yet converging molecular strategies to limit or exacerbate ROS formation in order to subvert immune defenses and support self-survival and long term carriage in their target host cells.The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3039955/ÀÀ
Open Access
Int. J. Mol. Sci. 2015, 16(12), 30269-30308; https://doi.org/10.3390/ijms161226225
Review
Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine
by K´ˆvia Queiroz De Andrade 1, Fabiana Andr´Îa Moura 1,2, John Marques Dos Santos 3, Orlando Roberto Pimentel De Ara´ýjo 3, Juliana C´Îlia De Farias Santos 2 and Mar´ˆlia Oliveira Fonseca Goulart 3,*
1
P´Ûs Graduação em Ci´¤ncias da Sa´ýde (PPGCS), Campus A. C. Simões, Tabuleiro dos Martins, 57072-970 Macei´Û, AL, Brazil
2
Faculdade de Nutrição/Universidade Federal de Alagoas (FANUT/UFAL), Campus A. C. Simões, Tabuleiro dos Martins, 57072-970 Macei´Û, AL, Brazil
3
Instituto de Qu´ˆmica e Biotecnologia (IQB), Universidade Federal de Alagoas (UFAL), Campus A. C. Simões, Tabuleiro dos Martins, 57072-970 Macei´Û, AL, Brazil
*
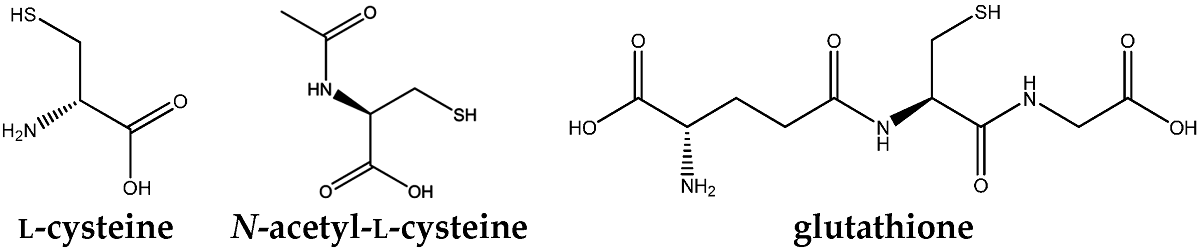
Abstract:Liver disease is highly prevalent in the world. Oxidative stress (OS) and inflammation are the most important pathogenetic events in liver diseases, regardless the different etiology and natural course. N-acetyl-l-cysteine (the active form) (NAC) is being studied in diseases characterized by increased OS or decreased glutathione (GSH) level. NAC acts mainly on the supply of cysteine for GSH synthesis.
The objective of this review is to examine experimental and clinical studies that evaluate the antioxidant and anti-inflammatory roles of NAC in attenuating markers of inflammation and OS in hepatic damage. The results related to the supplementation of NAC in any form of administration and type of study are satisfactory in 85.5% (n = 59) of the cases evaluated (n = 69, 100%). Within this percentage, the dosage of NAC utilized in studies in vivo varied from 0.204 up to 2 g/kg/day. A standard experimental design of protection and treatment as well as the choice of the route of administration, with a broader evaluation of OS and inflammation markers in the serum or other biological matrixes, in animal models, are necessary. Clinical studies are urgently required, to have a clear view, so that, the professionals can be sure about the effectiveness and safety of NAC prescription.
Keywords: N-acetylcysteine; liver; hepatic injury; antioxidant; anti-inflammatory; biomarkersIJMS | Free Full-Text | Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine | HTML
https://www.mdpi.com/1422-0067/16/12/26225/htmÀÀ
Vitamin C augments lymphocyte glutathione in subjects with ascorbate deficiency
Kevin J Lenton, Alain T San´Î, H´Îl´´ne Therriault, Andr´Î M Cantin, H´Îl´´ne Payette, J Richard Wagner
The American Journal of Clinical Nutrition, Volume 77, Issue 1, January 2003, Pages 189´C195, https://doi.org/10.1093/ajcn/77.1.189
Published: 01 January 2003 Article history
ABSTRACT
Background: Ascorbate and glutathione play central roles in the defense against free radicals and oxidants that are implicated in chronic diseases.
Objective: The objective was to determine the ability of vitamin C supplements to modulate the concentration of glutathione in human lymphocytes.
Design: The effect of vitamin C supplements was determined in a sequential study with time points before supplementation, after 13 wk of vitamin C supplements (500 or 1000 mg/d), and after 13 wk of matching placebo. The supplementation group was selected on the basis of low plasma ascorbate (<33 mmol/L) and consisted of 48 healthy men and women, smokers and nonsmokers, aged 25´C64 y. Ascorbate and glutathione were measured in purified lymphocytes.
Results: At baseline, the mean (ÀâSD) concentration of plasma ascorbate was 19.5 Àâ 7.2 Îämol/L, 22.5 Îämol/L below the median of normal distribution. The ascorbate concentration in plasma was linearly associated with that in lymphocytes (r = 0.53, P < 0.001). On supplementation with vitamin C, lymphocyte ascorbate increased by 51% (from 16.7 Àâ 4.9 to 25.3 Àâ 6.9 nmol/mg protein; P < 0.001) and was accompanied by an increase of lymphocyte glutathione by 18% (from 22.5 Àâ 4.5 to 26.6 Àâ 6.5 nmol/mg protein; P < 0.001). After placebo, the ascorbate and glutathione concentrations fell to near baseline concentrations (17.1 Àâ 5.4 and 23.5 Àâ 6.4 nmol/mg protein, respectively). No significant interaction was observed for sex and smoking status. Finally, the changes in lymphocyte ascorbate after supplementation were strongly associated with changes in lymphocyte glutathione (r = 0.71, P < 0.001). The association suggests that every 1-mol change in ascorbate is accompanied by a change of Àø0.5 mol in glutathione.
Conclusion: Vitamin C supplements increase glutathione in human lymphocytes.Vitamin C augments lymphocyte glutathione in subjects with ...
https://academic.oup.com/ajcn/article/77/1/189/4689652ÀÀ
Mechanism of Vitamin C Inhibition of Cell Death Induced by Oxidative Stress in Glutathione-depleted HL-60 Cells*
Victor H. Guaiquil‡, Juan Carlos VeraÀš and David W. Golde‡¶
- Author Affiliations
From the ‡Program in Molecular Pharmacology and Therapeutics, Memorial Sloan-Kettering Cancer Center, New York, New York 10021 and the ÀšDepartamento de Fisiopatologı́a, Universidad de Concepci´Ûn, Casilla 160-C, Concepci´Ûn, ChileVitamin C is a well known antioxidant whose precise role in protecting cells from oxidative challenge is uncertain. In vitro results have been confounded by pro-oxidant effects of ascorbic acid and an overlapping role of glutathione. We used HL-60 cells as a model to determine the precise and independent role of vitamin C in cellular protection against cell death induced by oxidative stress. HL-60 cells do not depend on glutathione to transport or reduce dehydroascorbic acid. Depletion of glutathione rendered the HL-60 cells highly sensitive to cell death induced by H2O2, an effect that was not mediated by changes in the activities of glutathione reductase, glutathione peroxidase, catalase, or superoxide dismutase. The increased sensitivity to oxidative stress was largely reversed when glutathione-depleted cells were preloaded with ascorbic acid by exposure to dehydroascorbic acid. Resistance to H2O2 treatment in cells loaded with vitamin C was accompanied by intracellular consumption of ascorbic acid, generation of dehydroascorbic acid, and a decrease in the cellular content of reactive oxygen species. Some of the dehydroascorbic acid generated was exported out of the cells via the glucose transporters. Our data indicate that vitamin C is an important independent antioxidant in protecting cells against death from oxidative stress.
Oxidative stress occurs in aerobic organisms because of the generation of reactive oxygen species (ROS)1 during respiratory energy production. Mammalian cells therefore have evolved effective antioxidant defense systems to cope with the toxic ROS generated in the course of aerobic ATP generation. The health of cells in tissues is influenced by the balance of antioxidants and ROS (1, 2). Oxidative damage has been linked to many disease states (3, 4) and to the development of cancer via the oxidation of DNA bases (5, 6). The role of antioxidant defenses in tumor cells themselves, however, has been poorly studied, and the role of antioxidants in neoplastic cell responses to radiation and chemotherapy is not understood completely (7-10).
We used a model system to analyze the independent effects of glutathione (GSH) and vitamin C in cellular defense against an antioxidant stress induced by H2O2. We selected HL-60 cells because we showed previously that the reduction of newly transported dehydroascorbic acid (DHA) in these cells was not dependent on the GSH concentration, and the HL-60 cells only transport vitamin C in the form of DHA through the facilitative glucose transporters (11-13). Cells with normal or depleted GSH were loaded with vitamin C by exposing them to DHA, and the effect of oxidative challenge was studied. We found that vitamin C protected cells from death induced by H2O2 in GSH-depleted cells. In the course of its antioxidant action, ascorbic acid was converted to DHA intracellularly and subsequently effluxed from the cells. The efflux of DHA is mediated by the glucose transporters, and the DHA generated was related to the cellular GSH content. We also show that GSH protects HL-60 cells from oxidative stress independently of ascorbic acid. These results indicate that ascorbic acid and GSH are individually important in protecting cells from oxidative challenge.RESULTS
Effect of H2O2 on Cell Viability
Depletion of GSH prominently increased the sensitivity of HL-60 cells to the cytotoxic effect of H2O2 (Fig.1). Greater than 90% of GSH-depleted cells died after a 24-h incubation with 10 ÎäMH2O2. With the same conditions, however, there was nearly 100% viability in cells with a normal content of GSH (Fig.1, A and B). Cellular toxicity was dependent on the concentration and time of exposure to the oxidant, as shown in Fig.1, C and D. Higher concentrations of H2O2 were required to decrease cell viability in control cells (Fig. 1 C). After a 4-h incubation with 500 ÎäM H2O2, cell viability was reduced to 50%. Lower concentrations of H2O2do not show deleterious effects on cell survival or proliferative capacity. In GSH-depleted cells, however, there was a rapid increase in cell death (Fig. 1 D), and 100% cell killing was observed at concentrations of 50 ÎäM H2O2 or higher after a 6-h incubation. These results show that GSH is a quantitatively important antioxidant, and its absence leaves the cells poorly protected from the oxidative damage induced by H2O2. The variability of the GSH content in control cells, as shown in Fig. 1 E, is an aspect already described (11), but these cells are able to maintain their GSH content even when they are exposed to high doses of H2O2. In the case of GSH-depleted cells (Fig.1 F), the cells are unable to recover the GSH content mainly because the H2O2 produces a rapid cell death. With lower concentrations of H2O2 these cells show recovery of their GSH content.
Figure 1
Glutathione depletion renders HL-60 cells more sensitive to H2O2. Panels A and B, dose response for cell viability in cells with normal and depleted GSH content, respectively. Cells were incubated with H2O2 for 4 h (ÀÞ), 8 h (Àþ), and 24 h (▿). Panels C and D, time course of cell survival in normal and GSH-depleted HL-60 cells treated with 0 (ÀÞ), 1 (Àþ), 50 (▿), and 500 (▾) ÎäMH2O2. Panels E and F, time course of GSH content in normal and GSH-depleted cells treated with 0 (ÀÞ), 50 (Àþ), and 500 (▿) ÎäMH2O2. The GSH content in normal (8 mM) and depleted (0.1 mM) cells was measured as described under À¯Experimental Procedures.ÀÝ GSH depletion was induced by treating the cells for 24 h with 200 ÎäMbuthionine-(S,R)-sulfoximine and 1 h with diethyl maleate. The cell number was obtained by trypan blue exclusion. Data represent the mean Àâ S.D. of four experiments, each performed three times.Antioxidant Enzymatic Activities in HL-60 Cells
Hydrogen peroxide is quite stable and has a high diffusion rate in solution. To avoid oxidative damage induced by H2O2, the cells must have mechanisms that block the influx of H2O2 into the cells or produce antioxidants that neutralize its deleterious effects. We studied how depletion of GSH changes the enzymatic activities involved with the removal of oxidants. Control and GSH-depleted HL-60 cells decomposed extracellular H2O2 rapidly. By 5 min of incubation with 1 mM H2O2, 80% of the oxidant was removed from the medium (Fig.8 A). More than 90% H2O2 was decomposed by cells after a 10-min incubation. These data show that catalase activity is fully active and independent of intracellular GSH. GSH-depleted cells remove H2O2 from the medium at the same rate as cells with a normal GSH content. Sensitivity to oxidative stress in GSH-depleted cells therefore is not directly related to a catalase deficiency. Furthermore, data obtained using a specific inhibitor of catalase (3-amino-1,2,4-triazole) show that cell survival after treatment with H2O2 is the same in control and catalase-inhibited cells (data not shown).Depletion of GSH does not change the activities of GSH peroxidase or superoxide dismutase (Fig. 8 B). Control and GSH-depleted cells show similar enzymatic levels, indicating that they are not involved in the changes observed in cell survival when GSH-depleted cells were exposed to oxidative stress. GSH function in cell protection could then be related largely to the neutralization of reactive oxygen intermediates produced from H2O2. These potentially more destructive molecules could induce cellular damage rapidly if an antioxidant is not present in sufficient concentration in the cells. GSH and vitamin C have the ability to quench ROS and prevent damage.
DISCUSSION
The precise role of vitamin C in protecting normal and neoplastic cells against oxidative challenge is unclear. In vitrostudies have been confounded by the pro-oxidant effects of ascorbic acid in vitro and the overlapping antioxidant activities of glutathione. DHA enters cells through the glucose transporters and is reduced to ascorbic acid intracellularly (12, 13, 19). Extracellular oxidative events lead to the generation of DHA from ascorbic acid, which can then enter cells rapidly and be reduced back to ascorbic acid (20, 21). We showed recently that cancer cells accumulate vitamin C in a superoxide dismutase-inhibitable manner (22). We have used ascorbic acid loading of HL-60 cells to assess directly the role of vitamin C in protecting these cells from an oxidative challenge induced by H2O2. Glutathione is an important antioxidant and has been associated with the cellular recycling of vitamin C and the maintenance of the vitamin in its reduced state (23-27). We found previously that when HL-60 cells were depleted of GSH there was no change in their ability to transport DHA and accumulate ascorbic acid (11). We therefore selected this cell line as a model to determine the independent effects of vitamin C and glutathione in antioxidant defense. A clear understanding of the cellular protective mechanisms against oxidants has wide ranging importance because ROS are strongly implicated in human disease as well as in normal cell signaling and the action of certain chemotherapeutic agents and radiation (1, 18,28-30).
We found that HL-60 cells depleted of GSH became highly sensitive to the effect of H2O2, resulting in increased cell death. GSH-depleted cells tolerated only low doses of H2O2 and died rapidly at higher concentrations. Cells with normal GSH content were more resistant to H2O2, and there was 50% cell survival, even when cells were treated with H2O2concentrations as high as 500 ÎäM. The protective role of GSH observed in HL-60 cells was similar to that described by others and supports the notion of GSH as a major antioxidant in the cell (23, 24,31-34).
We found that GSH-depleted cells can be made resistant to H2O2 by preloading the cells with vitamin C via exposure to DHA. The effect was dose-dependent, and some protection was also seen in cells with normal GSH content. HL-60 cells accumulate millimolar concentrations of vitamin C, and their resistance to H2O2 increases in proportion to the intracellular concentration of ascorbic acid in a dose-dependent manner. Similar results have been observed in other cell lines where preloading with ascorbic acid was shown to protect from H2O2 generated by xanthine oxidase (35) and by direct treatment with H2O2 or radiation (7, 8, 36).
Under the conditions studied, vitamin C had an antioxidant role parallel to that of GSH. Both are primary antioxidants able to protect HL-60 cells from oxidative stress. Depletion of GSH did not change the cell's enzymatic antioxidant activities. Catalase, GSH peroxidase, and superoxide dismutase were functionally normal in both untreated and GSH-depleted cells. Because cell death increased in GSH-depleted cells, these enzymes would not be the primary cell components responsible for the differential in cell death induced by H2O2. Preloading with vitamin C, however, rendered the cells resistant to H2O2, an effect mediated by inhibition of ROS. This response points to vitamin C as a primary antioxidant responsible for neutralizing ROS that induce cellular damage. Because catalase and GSH peroxidase were fully active in both GSH-depleted and untreated cells, removal of H2O2 occurred efficiently. Accordingly, cell killing induced by H2O2 is likely because of generated intermediate free radicals, and vitamin C has its major quenching effect at this level.
Our results indicate that ascorbic acid and GSH protect cells against oxidant challenge in a dose-dependent manner and that vitamin C exited the cell after incubation with H2O2. The efflux of vitamin C was dependent on the concentration of H2O2 and the GSH content in the cells. This was a rapid event occurring over minutes. The ascorbic acid efflux appears to be a consequence of electron donation by ascorbic acid and conversion of ascorbic acid to DHA intracellularly. As H2O2 concentrations increased, there was an increase in the efflux of vitamin C. The kinetics of vitamin C efflux from cells were complex, and a general biphasic pattern could be related to compartmentalization of the vitamin in the cells. The oxidation of ascorbic acid to DHA implies that the major role of vitamin C is as an electron donor to quench oxidants intracellularly.
Efflux of vitamin C was observed to be proportional to the cellular content of GSH, pointing to the parallel role of both antioxidants. The result of the cytochalasin B inhibition experiments confirmed the hypothesis that vitamin C would exit the cell through the glucose transporter in the form of DHA, after ascorbic acid donated electrons. Although ascorbic acid accumulated intracellularly in HL-60 cells, only DHA exits into the medium after treatment of the cells with H2O2.
We asked if a direct chemical reaction between H2O2 and ascorbic acid inside the cells generated DHA that can exit the cell. Our in vitro data from a cell-free system clearly indicated that this is not the case. The concentration of H2O2 necessary to oxidize 50% of 100 ÎäM ascorbic acid was 2 orders of magnitude higher than the concentration of ascorbic acid. Such concentrations of H2O2 are never reached inside the cell. Moreover, the rate of oxidation of ascorbic acid by the H2O2-treated HL-60 cells was 2 orders of magnitude greater that the in vitro oxidation rate. The explanation for the oxidation of ascorbic acid could be related to the generation of intermediates, such as the hydroxyl radical (⋅OH), which have more potent oxidative power that could lead to a rapid generation of DHA, the chemical form that can exit the cells through the glucose transporters (12, 13).
Current evidence suggests that OFormula and H2O2 injure cells as a result of the generation of more potent oxidizing species (6, 37). The hypothesis most consistent with the available data is that OFormula reduces a cellular source of ferric to ferrous iron, and the latter then reacts with H2O2 to produce more potent oxidizing species, such as ⋅OH. This more reactive species can be neutralized intracellularly by ascorbic acid generating DHA. If neutralization does not occur, ⋅OH can initiate the peroxidative decomposition of phospholipids of cellular membranes. Hydroxyl radical also damages the inner mitochondrial membrane leading to a sequence of events that ends in cell death. DNA represents another cellular target for ⋅OH, and, depending on the cell type, oxidative DNA damage can be coupled to cell killing through a mechanism related to the activation of poly(ADP-ribose) polymerase (6, 37, 38).
Our data suggest that DHA can be used to increase the intracellular concentration of ascorbic acid as a means to provide antioxidant defense to cells exposed to oxidative challenge. Cancer cells up-regulate the expression of the facilitative glucose transporters (39-41). This increase is also observed in tumors treated with radiation or chemotherapy, which induce ROS (42). Tumors can likely use the antioxidative properties of vitamin C as cell defense mechanisms to deal with oxidative challenge.Mechanism of Vitamin C Inhibition of Cell Death Induced by Oxidative Stress in Glutathione-depleted HL-60 Cells
https://www.jbc.org/content/276/44/40955.fullÀÀ
Archives of Biochemistry and Biophysics
Volume 434, Issue 1, 1 February 2005, Pages 178-186
Transport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis
Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN 37232-6303, USA
Abstract
Endothelial cells preserve vascular integrity in part by synthesizing type IV collagen for the basement membrane of blood vessels.Vitamin C, which at physiologic pH is largely the ascorbate mono-anion, both protects these cells from oxidant stress and is required for collagen synthesis. Therefore, cultured endothelial cells were used to correlate intracellular concentrations of ascorbate with its uptake and ability to stimulate collagen release into the culture medium. The kinetics and inhibitor specificity of ascorbate transport into EA.hy926 endothelial cells were similar to those observed in other cell types, indicative of a specific high affinity transport process. Further, transport of the vitamin generated intracellular ascorbate concentrations that were 80´C100-fold higher than concentrations in the medium following overnight culture, and transport inhibition with sulfinpyrazone and phloretin partially prevented such ascorbate accumulation. On the other hand, low millimolar intracellular concentrations of ascorbate impaired its transport measured after overnight culture. Synthesis and release of type IV collagen into the culture medium was markedly stimulated by ascorbate in a time-dependent manner, and was saturable with increasing medium concentrations of the vitamin. Optimal rates of collagen synthesis required intracellular concentrations of the vitamin up to 2 mM. Since such concentrations can only be generated by the ascorbate transporter, these results show the necessity of transport for this crucial function of the vitamin in endothelium.
áÖóÊü¡¯«øÅö˜èºùÄCçáåùòð¤ëü¡¯«áÖ£»âÜ:ÆŠ§¤åÙ¤ü°èçá¿Äüç
û⿺áèòýö˜Ñ«ñÑçôÝàÑ«äÄǵîÏاîÏå¤Ø§îÏüç
íˆØˆ
áÖóÊü¡¯«ë´¿»¤ü°èɣªçæáÊçáIVÅ매åÙâÇö˜°øÉçáëõí«ÅåÀÈ
ö˜èºùÄCåÖèºâÚpHøçüôø¼Øˆòú¢¿£çîˆùÃçË¡¤âŠæÆȘù■¥àÝÈ£ÊíãÅˋü¡¯«ûãòÉî¾£₤ÆÎ¥ÊȘÆøöˆ§¤åÙ¤ü°èùªÝÄÅÒÀÈØ·ÇùȘöØûúÆûéÁî½çááÖóÊü¡¯«âÇîŃ¢ü¡¯«áÖ¢¿£çîˆùÃîöçáé´ÑàÆŠ¢¿£çîˆùÃîöçáö■òí¤ëÇä¥Ê§¤åÙ篯æòëñéç§éÁî½£ªøÅçááÉêÎøÛ¥ðçá¿ÄüçÀÈ¢¿£çîˆùç½àŠEA.hy926áÖóÊü¡¯«çáÑ₤êÎîϤëØøøó¥êçáäÄØšÅåÆŠåÖóðù«ü¡¯«âÁÅëøÅ¿Üýšç§çáüÁùóȘíãÝÚû¼êùØ£øøäÄòãçá¡Ôúæ¤ëæˆåù¿»°äÀÈÇùëãȘƒÙ¿»Ø£Ø¿çáéÁµÈ˜ö˜èºùÄçáåùòðýºèºçáü¡¯«áÖ¢¿£çîˆùÃé´ÑàÝàéÁî½£ªøÅçáé´Ñà¡Ô80 - 100ÝÑȘò¿Æû£ú¯ñÔêÁ¨ëˆ¤ëàëóÊý¢åùòðØøøóý¢ñøæÒø¿êùíãøø¢¿£çîˆùÃç᣻âÜÀÈêÚØ£ñ§ûÌȘçë¤êáÎÑ«ü¡¯«áÖé´Ñàçᢿ£çîˆùãÃƯüšóðåÖأؿéÁµçáæˆåùÀÈ¢¿£çîˆùÃÑåIVÅ매åÙçá¤ü°è¤ëòëñéÆÅû¼üåçáòÝ¥ðØââçÅåçáÇä¥Êæ¼ÆûȘýÂúØùÌæéö˜èºùÄçáéÁî½£ªé´Ñàçáå—¥ÆѽÝˤëÀȧ¤åÙ¤ü°èçá查îùìÑà؈úµü¡¯«áÖö˜èºùÄé´ÑàÇÿç§2mMolÀÈÆèÆÖíãøøé´Ñàø£áÉÆ袿£çîˆùÃæˆåù篯æýºèºÈ˜íãÅˋ§Ã¿«ÝÚû¼êùåùòðÑåö˜èºùÄCåÖáÖóÊü¡¯«íãØ£¿Ä¥■¿ÎáÉçáÝÄ؈ÅåÀÈ
Keywords
AscorbateType IV collagenGSHTransport-metabolism couplingEA.hy926 endothelial cellsTransport and intracellular accumulation of vitamin C in endothelial cells: relevance to collagen synthesis - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0003986104006150
Vitamin C and vitamin E restore the resistance of GSH-depleted lens cells to H2O2
Article in Free Radical Biology and Medicine 34(5):521-30 ÀÊ March 2003 with 32 Reads
DOI: 10.1016/S0891-5849(02)01304-7 ÀÊ Source: PubMed
A decline in reduced glutathione (GSH) levels is associated with aging and many age-related diseases. The objective of this study was to determine whether other antioxidants can compensate for GSH depletion in protection against oxidative insults. Rabbit lens epithelial cells were depleted of > 75% of intracellular GSH by 25-200 microM buthionine sulfoximine (BSO). Depletion of GSH by BSO alone had little direct effect on cell viability, but resulted in an approximately 30-fold increase in susceptibility to H(2)O(2)-induced cell death. Experimentally enhanced levels of nonprotein sulfhydryls other than GSH (i.e., N-acetylcysteine) did not protect GSH-depleted cells from H(2)O(2)-induced cell death. In contrast, pretreatment of cells with vitamin C (25-50 microM) or vitamin E (5-40 microM), restored the resistance of GSH-depleted cells to H(2)O(2). However, concentrations of vitamin C > 400 microM and vitamin E > 80 microM enhanced the toxic effect of H(2)O(2). Although levels of GSH actually decreased by 10-20% in cells supplemented with vitamin C or vitamin E, the protective effects of vitamin C and vitamin E on BSO-treated cells were associated with significant ( approximately 70%) decreases in oxidized glutathione (GSSG) and concomitant restoration of the cellular redox status (as indicated by GSH:GSSG ratio) to levels detected in cells not treated with BSO. These results demonstrate a role for vitamin C and vitamin E in maintaining glutathione in its reduced form. The ability of vitamin C and vitamin E in compensations for GSH depletion to protect against H(2)O(2)-induced cell death suggests that GSH, vitamin C, and vitamin E have common targets in their actions against oxidative damage, and supports the preventive or therapeutic use of vitamin C and E to combat age- and pathology-associated declines in GSH. Moreover, levels of these nutrients must be optimized to achieve the maximal benefit.Vitamin C and vitamin E restore the resistance of GSH-depleted lens cells to H2O2 | Request PDF
https://www.researchgate.net/publication/10875376_Vitamin_C_and_vitamin_E_restore_the_resistance_of_GSH-depleted_lens_cells_to_H2O2ÀÀ
Biochem Biophys Res Commun. 1997 Aug 8;237(1):28-32.
Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta.
Newton R1, Kuitert LM, Bergmann M, Adcock IM, Barnes PJ.
Author information
1
Department of Thoracic Medicine, National Heart and Lung Institute, Imperial College School of Medicine, London, United Kingdom. robert.newton@ic.ac.uk
Abstract
The cyclooxygenase (COX) isoforms COX-1 and COX-2 convert arachidonic acid to prostaglandin (PG) precursors and are a limiting step in PG production.Interleukin-1beta (IL-1beta) treatment of type II A549 cells increases PGE2 synthesis via transcription- and translation-dependent induction of COX-2. IL-1beta produces a 10-fold induction of COX-2 mRNA and an 8-fold increase in COX-2 transcription that was temporally preceded by activation of the transcription factor nuclear factor-kappaB (NF-kappaB).
The protein-tyrosine phosphatase inhibitor phenylarsine oxide (PAO) prevented both NF-kappaB activation and induction of COX-2 mRNA.
We show that two putative NF-kappaB motifs, kappaBu (-447/-438) and kappaBd (-224/-214), from the COX-2 promoter bind p50/p65 NF-kappaB heterodimers in an IL-1beta-dependent manner and that the upstream element has the greater affinity.
Finally, we demonstrate that the two NF-kappaB subunits, p50 and p65, synergistically activate a -917/+49 COX-2 promoter construct.
We conclude that IL-1beta stimulates PG production via transcriptional activation of COX-2 and provide evidence that this may involve NF-kappaB.
Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/9266823/ÀÀ
Res Pharm Sci. 2016 Oct; 11(5): 390´C396.
Protective effect of vitamin C, vitamin B12 and omega-3 on lead-induced memory impairment in rat
Saeedeh Alsadat Moosavirad,1 Mohammad Rabbani,1 Mohammad Sharifzadeh,2 and Ali Hosseini-Sharifabad1,*
Abstract
Lead belongs to the heavy metal group and is considered as an environmental contaminant. Acute or chronic contact to lead can change the physiological function of human organs. One of the most important disorders following the lead exposure is neurotoxicity. Lead neurotoxicity consists of the neurobehavioral disturbances like cognitive impairment. The aim of the current study is to evaluate the possible protective effect of vitamin C (Vit C), vitamin B12 (Vit B12), omega 3 (ÎÄ-3), or their combination on the lead-induced memory disorder. Adult wistar rats were orally administered Vit C (120 mg/kg/day) or Vit B12 (1 mg/kg/day) or ÎÄ-3 (1000 mg/kg/day) or their combination for 3 weeks in groups of 7 animals each. Then lead acetate (15 mg/kg/day) was injected intraperitoneally for one week to all pretreated animals. The control group received normal saline as a vehicle while the positive control for cognitive impairment received just lead acetate. At the end of treatments animal memories were evaluated in Object Recognition Task. The results showed, although 15 mg/kg lead acetate significantly declines the memory-evaluating parameters, pretreatment with Vit C, Vit B12, ÎÄ-3, or their combination considerably inverted the lead induced reduction in discrimination (d2) index (P < 0.001) and recognition (R) index (P < 0.001, P < 0.05, P < 0.05, and P < 0.001, respectively). Our findings indicate while lead acetate impairs spatial memory in rat, administration of Vit C, Vit B12, ÎÄ-3, or their combination prior to the lead exposure inhibits the lead induced cognitive loss. There was no remarkable difference in this effect between the used supplements.
Keywords: Vitamin C, Vitamin B12, Omega-3, Lead, Memory impairment, Object Recognition TaskProtective effect of vitamin C, vitamin B12 and omega-3 on lead-induced memory impairment in rat
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5122828/ÀÀ
Published: 12 February 2019
Role of Reactive Oxygen Species in Inflammation: A Minireview
M. A. Chelombitko
Department of Biology, Moscow State University, Moscow, 119234, Russia
Abstract
Inflammation is a protective response of a multicellular organism to injury in order to localize, eliminate, and remove harmful stimuli as well as to recover (or replace) damaged tissues. There recently has been increasing evidence that reactive oxygen species (ROS) are involved in the initiation, progression, and resolution of the inflammatory response.Furthermore, ROS act as microbicidal agents and second messengers in the intracellular signaling. The latter function is performed via posttranslational modification of protein- associated redox-sensitive cysteine residues that can undergo oxidation.
At the same time, there is clear evidence that overproduction of ROS may result in cell and tissue injury and contribute to chronic inflammation underlying many neurodegenerative, cardiovascular, and metabolic diseases.
This review has focused on the role of ROS in the key inflammatory events (increased vascular permeability and leukocyte extravasation, respiratory burst and phagocytosis, and angiogenesis) and some events leading to the resolution of inflammation. In addition, the pathological function of ROS in oxidative stress is discussed.
£ŸÅåî¾åÖîæøÂøÅçáæ¼Æû:æÜò—
mÀÊaÀÊChelombitko
áˆù¿¢ó¿ºêÂǵîÏèºöÿüçȘáˆù¿¢óȘ119234
íˆØˆ
îæøÂòúÑÁü¡¯«èºöÿÑåùÞèùçáØ£øøÝÈ£ÊÅåñÇÆÎȘá¢çáòúÑ´ö£ÀÂü«°»¤ëúÍ°»ÆŤÎçáÇä¥ÊØ奯£ø¡Ç(£·äÌ££)òÉùÞæÕø₤ÀÈ柧■ÆÅå§âÇå§ÑÁçáøʃïÝÚû¼£ŸÅåî¾(ROS)ýöÆŠîæøÂñÇÆÎçáó¶Ñ₤À§½í¿¤ë§ãƒ—ÀÈ
ÇùëãȘROSåÖü¡¯«áÖÅé¤éøÅóÞèÝöÂèºöÿ¥ê¤ëçÖѱÅéò¿çáæ¼ÆûÀȤµØ£øø¿ÎáÉòúë´¿»ñÙØŠ¤µÅßòö篯æüÁ¿Äçáî¾£₤£¿åÙû¶¡Å¯ŠŠæ¯ÝùÃýÅ£ªâÇòçüøçáÀÈ
ë˜òÝȘÆÅû¼àñçáøʃïÝÚû¼È˜ROSçá¿»ê¢ýºèº¢èáÉç¥øôü¡¯«¤ëæÕø₤ùÞèùȘýÂç¥øôÅÚÑÁèþƒÙëùÅÅÅåÀÂÅáɤëǺţÅå¥ýýÀçáô»ÅåîæøÂÀÈ
݃öáæÜò—êùROSåÖø¼ØˆîæøÂòô¥±(Éë´ë¡Åå嗥Ƥë¯æü¡¯«ëãè½À¤¶ö■Ýˋñ¤ëëäòèÀÂÉ躰è)øÅçáæ¼ÆûȘØ奯أÅˋç¥øôîæøÂü«ëùçáòô¥±ÀÈÇùëãȘ£¿äøôÜêù£ŸÅåî¾åÖî¾£₤ÆÎ¥ÊøÅçáýÀâÚæ¼ÆûÀÈRole of Reactive Oxygen Species in Inflammation: A Minireview | SpringerLink
https://link.springer.com/article/10.3103/S009639251804003XÀÀ
Regulation of the phagocyte NADPH oxidase activity ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2660639
Jun 03, 2008 ÀÊ Several stimuli, such as phorbol myristate acetate (PMA), N-formyl-methionyl-leucyl-phenylalanine (fMLP), and opsonized zymosan (OPZ), can activate the neutrophil NADPH oxidase. NADPH oxidase activation is accompanied by phosphorylation of p47 phox , p67 phox , p40 phox , and p22 phox (21,22,23,24,25,26).
Cited by: 202
Publish Year: 2009
Author: Houssam Raad, Marie-H´Îl´´ne Paclet, Tarek Boussetta, Yolande Kroviarski, Françoise Morel, Mark T. Qui...
Phosphorylation of the Arabidopsis AtrbohF NADPH oxidase ...
https://www.sciencedirect.com/science/article/pii/S0014579309006656
Sep 17, 2009 ÀÊ NADPH oxidase function has been shown to be regulated by phosphorylation in both animal and plant cells,,. For example, AtrbohD was shown to be phosphorylated on several amino acid residues in response to pathogen elicitors, suggesting that phosphorylation regulates its activity,.
Cited by: 339
Publish Year: 2009
Author: Caroline Sirichandra, Dan Gu, Heng-Cheng Hu, MÀÀ
ÀÀ
ÀÀ
Mol Cell Endocrinol. Author manuscript; available in PMC 2010 Apr 29.
NADPH Oxidases and Angiotensin II Receptor Signaling
Abel Martin Garrido and Kathy K. GriendlingDivision of Cardiology, Emory University, Atlanta, GA 30322
Address correspondence to: Kathy K. Griendling, Ph.D., Emory University School of Medicine, Division of Cardiology, 319 WMB, 1639 Pierce Dr., Atlanta, GA 30322, USA, Phone: 404-727-3364, Fax: 404-727-3585, ude.yrome@dneirgk
Abstract
Over the last decade many studies have demonstrated the importance of reactive oxygen species (ROS) production by NADPH oxidases in angiotensin II (Ang II) signaling, as well as a role for ROS in the development of different diseases in which Ang II is a central component. In this review, we summarize the mechanism of activation of NADPH oxidases by Ang II and describe the molecular targets of ROS in Ang II signaling in the vasculature, kidney and brain. We also discuss the effects of genetic manipulation of NADPH oxidase function on the physiology and pathophysiology of the renin angiotensin system.
Keywords: Angiotensin II, NADPH oxidases, reactive oxygen species, vascular, kidney, brain
Over a decade ago, it was observed that angiotensin II (Ang II) is able to activate a NADPH oxidase (Griendling et al., 1994) that mediates the hypertensive response induced by Ang II infusion (Rajagopalan et al., 1996). Since then, numerous studies have demonstrated that NADPH oxidases regulate several Ang II functions in different tissues, many of which contribute to hypertension. In this review, we will describe the consequences of Ang II-mediated NADPH oxidase activation, with a focus on the cardiovascular, renal and central nervous systems.ÀÀ
NADPH oxidase activation by Ang II
The mechanism of NADPH oxidase activation by Ang II is complex and still incompletely understood. It has been shown that Ang II-mediated ROS production in vascular smooth muscle cells (VSMCs) from large arteries is mediated by Nox1 activation, as shown by loss of the ROS response after treatment with an adenovirus expressing antisense Nox1 (Lassegue et al., 2001), while Nox2 appears to be the Ang II-responsive Nox in resistance vessels, as demonstrated in human VSMCs transfected with gp91phox antisense oligonucleotides (Touyz et al., 2002), in the heart, as shown using small interference RNA (siRNA) against Nox2 (Hingtgen et al., 2006) and in the kidney, as described in gp91phox knockout mice (Haque and Majid, 2004). Typically, there are two different phases of ROS production: one is rapid and transient and the other is delayed and sustained. The first peak is due to an acute activation of NADPH oxidases by Ang II, while the second is dependent on the initial burst and is a consequence of upregulation of different NADPH oxidase subunits by Ang II. For example, in VSMC, the sustained phase of ROS production is preceded by upregulation of Nox1 mRNA (Lassegue et al., 2001; Wingler et al., 2001), p22phox mRNA (Fukui et al., 1997)(Fukui et al., 1997)(Fukui et al., 1997)and protein (Touyz et al., 2002) and p47phox protein (Ni et al., 2007; Touyz et al., 2002). There is also one report that Ang II can upregulate Nox4 mRNA in the vasculature (Wingler et al., 2001), although this is not observed universally (Mollnau et al., 2002).
In VSMCs, rapid ROS release requires p47phox via its phosphorylation by protein kinase C (PKC) and subsequent translocation to the membrane (Lavigne et al., 2001; Seshiah et al., 2002; Touyz et al., 2002), where it organizes the activating subunits NoxA1 and Rac (Ambasta et al., 2006; Hassanain et al., 2007). While each of these subunits has been implicated in the response to Ang II, actual formation of this complex in VSMCs remains to be shown biochemically. Ang II can stimulate PKC-dependent NADPH oxidase activity through three different phospholipases (PL): PLC (Karathanassis et al., 2002), PLD (Touyz and Schiffrin, 1999), and PLA2 (Zafari et al., 1999), with PLD likely predominating. Another important mediator of p47phox phosphorylation is Src (Touyz et al., 2003), although it is not clear whether it regulates p47phox directly or by its actions on the cytoskeleton through cortactin, a Src substrate that binds p47phox (Agerer et al., 2005; Sangrar et al., 2007; Touyz et al., 2005) and facilitates the proper assembly and function of NADPH oxidases by Ang II (Touyz et al., 2005).
Ang II activation of Nox1 and Nox2 also requires Rac1 (Cheng et al., 2006; Miyano et al., 2006; Ueyama et al., 2006). Stimulation of Rac1 by Ang II seems to be mediated by activation of the guanine nucleotide exchange factor (GEF), SOS-1 (Zuo et al., 2005), although a relationship between SOS-1 and Nox activation remains to be demonstrated. Activation of the Rac-GEF is dependent upon Src-mediated transactivation of the epidermal growth factor receptor (EGFR), which then serves as a binding site for phosphatidylinositol 3-kinase (PI-3K), an immediate activator of the Rac-GEF (Seshiah et al., 2002). This initial activation step appears to occur in caveolae (Ushio-Fukai et al., 2001), the site of Nox1 localization (Hilenski et al., 2004). Of importance, the H2O2 that is produced as a result of initial NADPH oxidase activation then activates Src, creating an amplification of oxidase activity (Seshiah et al., 2002). Prolonged signal generation requires upregulation of the Nox subunit, p47phox and p22phox (Fukui et al., 1997; Lassegue et al., 2001; Touyz et al., 2002).ÀÀ
NADPH Oxidases and Angiotensin II Receptor Signaling
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2835147/ÀÀ
NLRP3 inflammasome activation is involved in Ang II ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5342342
Aug 23, 2016 ÀÊ MitoTEMPO attenuated Ang II-induced NLRP3 inflammasome activation and renal damage A : NLRP3, ASC, casp-1, IL-1Îô, and IL-18 expression in kidneys were determined by Western Blotting. B : Quantification of the expression levels of these proteins.
Cited by: 16
Publish Year: 2016
Author: Yi Wen, Yiran Liu, Taotao Tang, Linli Lv, Hong Liu, Kunling Ma, Bicheng Liu
Effects of dehydration and blockade of angiotensin II AT1 ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4225509
The objective of this study was to provide for the first time data on plasma catecholamines, cortisol, glutathione and malondialdehyde after long term dehydration (20 days) in the presence and absence of angiotensin II (Ang II) AT1 receptor blocker (losartan) versus levels in time-matched, non-dehydrated control camels and to record the responses of glutathione and malondialdehyde activity in liver and ÀÙ
Cited by: 6
Publish Year: 2013
Author: Mahmoud Alhaj Ali, Elsadig Kazzam, Naheed Amir, Fred Nyberg, Abdu Adem
Coping with dehydration: sympathetic activation and ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4042205
Jun 01, 2014 ÀÊ Accordingly, dehydration elevates body fluid osmolality and circulating angiotensin II, factors sensed by circumventricular organ neurons, specifically those in the organum vasculosum of the lamina terminalis (OVLT) and subfornical organ (SFO) (34, 49).
Cited by: 10
Publish Year: 2014
Author: Megan E. Bardgett, Qing-Hui Chen, Qing Guo, Alfredo S Calderon,
Effects of dehydration and blockade of angiotensin II AT1 ...
https://bmcvetres.biomedcentral.com/articles/10.1186/1746-6148-9-232
Nov 19, 2013 ÀÊ The objective of this study was to provide for the first time data on plasma catecholamines, cortisol, glutathione and malondialdehyde after long term dehydration (20 days) in the presence and absence of angiotensin II (Ang II) AT1 receptor blocker (losartan) versus levels in time-matched, non-dehydrated control camels and to record the responses of glutathione and malondialdehyde activity ÀÙ
Cited by: 6
Publish Year: 2013
Author: Mahmoud Alhaj Ali, Elsadig Kazzam, Naheed Amir, Fred Nyberg, Abdu Adem
Effects of dehydration and blockade of angiotensin II AT1 ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4225509
In addition, this investigation showed dehydration alone or in combination with angiotensin II AT1 receptor blocker induced significant increments in glutathione and malondialdehyde activities in plasma, liver and kidney homogenates, presumably in order to counteract the ÀÙ
Cited by: 6
Publish Year: 2013
Author: Mahmoud Alhaj Ali, Elsadig Kazzam, Naheed AmirÀÀ
BMC Vet Res. 2013; 9: 232.
Effects of dehydration and blockade of angiotensin II AT1 receptor on stress hormones and anti-oxidants in the one-humped camel
Mahmoud Alhaj Ali,1 Elsadig Kazzam,3 Naheed Amir,1 Fred Nyberg,2 and Abdu Ademcorresponding author11Department of Pharmacology and Therapeutics, Faculty of Medicine and Health Sciences, United Arab Emirates University, P.O. Box 17666, Al Ain, United Arab Emirates
2Department of Pharmacology and Therapeutics, Faculty of Pharmacy, Uppsala University, Uppsala, Sweden
3Department of Internal Medicine, College of Medicine and Health Science, United Arab Emirates University, Al Ain, United Arab Emirates
Abstract
Background
The objective of this study was to provide for the first time data on plasma catecholamines, cortisol, glutathione and malondialdehyde after long term dehydration (20 days) in the presence and absence of angiotensin II (Ang II) AT1 receptor blocker (losartan) versus levels in time-matched, non-dehydrated control camels and to record the responses of glutathione and malondialdehyde activity in liver and kidney homogenates in control, dehydrated-losartan treated and dehydrated camels. Eighteen male camels were studied, six hydrated (control group), six dehydrated and treated with losartan (treated group) and six dehydrated not treated (dehydrated).
Results
Plasma levels of norepinephrine and dopamine were significantly increased (P < 0.01) in both treated and dehydrated groups compared to time matched control, whereas Plasma epinephrine level showed significant decrease (P < 0.05) in both treated and dehydrated groups compared to control. Plasma cortisol also showed significant increase (P < 0.01) in both treated and dehydrated groups compared to control. Glutathione levels in plasma, liver and kidney homogenates for both treated and dehydrated groups reveled significant increase (P < 0.05) Likewise, malondialdehyde levels in plasma, liver and kidney homogenates were substantially and significantly increased in both treated and dehydrated groups.
Conclusion
In conclusion, the results of this study demonstrated that dehydration substantially increased the circulating levels of norepinephrine, dopamine and cortisol but decreased plasma epinephrine. Similarly, losartan showed similar effects to that of dehydration. In addition, this investigation showed dehydration alone or in combination with losartan induced significant increments in glutathione and malondialdehyde activities in plasma, liver and kidney homogenates, presumably in order to counteract the potentially damaging effects of free radicals. Blockade of angiotensin II AT1 receptors did not alter significantly the response of dehydration in any of these indices.
Keywords: Camel, Catecholamine, Cortisol, Dehydration, Glutathione, Losartan and malondialdehydeEffects of dehydration and blockade of angiotensin II AT1 receptor on stress hormones and anti-oxidants in the one-humped camel
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4225509/ÀÀ
Drug Targets for Oxidative Podocyte Injury in Diabetic Nephropathy
Adnan Bashir Bhatti, Muhammad Usman
Published: December 03, 2015 (see history)
Abstract
Diabetic nephropathy (DN) is one the most prevalent chronic complications of diabetes mellitus that affects as much as one-third of diabetic patients irrespective of the type of diabetes. Hyperglycemia is the key trigger for DN that initiates a number of microscopic and ultramicroscopic changes in kidney architecture. Microscopic changes include thickening of the glomerular basement membrane (GBM), tubular basement membrane (TBM), mesangial proliferation, arteriosclerosis, and glomerulotubular junction abnormalities (GTJA). Among the ultramicroscopic changes, effacement of podocytes and decrease in their density seem to be the centerpiece of DN pathogenesis. These changes in kidney architecture then produce functional deficits, such as microalbuminuria and decreased glomerular filtration rate (GFR). Among several mechanisms involved in inflicting damage to podocytes, injuries sustained by increased oxidative stress turns out to be the most important mechanism. Different variables that are included in increased production of reactive oxygen species (ROS) include a hyperglycemia-induced reduction in glutathione (GSH), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation via hyperglycemia, advanced glycation end products (AGEs), protein kinase C (PKC), and renin-angiotensin-aldosterone system (RAAS).
Unfortunately, control of podocyte injury hasnÀ₤t received much attention as a treatment approach for DN. Therefore, this review article is mainly concerned with the exploration of various treatment options that might help in decreasing the podocyte injury, mainly by reducing the level of NADPH oxidase-mediated generation of ROS. This article concludes with a view that certain NADPH oxidase inhibitors, RAAS inhibitors, statins, antidiabetic drugs, and antioxidant vitamins might be useful in decreasing podocyte injury and resultant structural and functional kidney impairments in DN.
Figure 2: Mechanism of increase in oxidative stress and resultant podocyte injury due to hyperglycemia.
High glucose gets metabolized to form obnoxious glucose metabolites; directly reduces the quantity of antioxidants like glutathione (GSH); fuses with proteins to form advanced glycation end products (AGEs); induces protein kinase C (PKC) signaling and deranges renin-angiotensin-aldosterone system (RAAS). All these mechanism tend to increase oxidative stress by inducing the action of NADPH oxidase. The reactive oxygen species (ROS), thus produced, inflict injury to podocytes and decrease their density.Cureus | Drug Targets for Oxidative Podocyte Injury in Diabetic Nephropathy
https://www.cureus.com/articles/3625-drug-targets-for-oxidative-podocyte-injury-in-diabetic-nephropathyÀÀ
Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy
Ross Vlahos
John Stambas, Stavros Selemidism. Respiratory Research Group, Department of Pharmacology, The University of Melbourne, Victoria, Australia
Published:October 04, 2011DOI:https://doi.org/10.1016/j.tips.2011.09.001
Influenza A viral infections claim millions of lives worldwide and continue to impose a major burden on healthcare systems. Current pharmacological strategies to control influenza A virus-induced lung disease are problematic owing to antiviral resistance and the requirement for strain-specific vaccination.The production of reactive oxygen species (ROS), particularly superoxide, is an important host defence mechanism for killing invading pathogens.
However, excessive superoxide may be detrimental following influenza A virus infection. Indeed, suppression of superoxide production by targeting the primary enzymatic source of superoxide in mammalian inflammatory cells, NADPH oxidase 2 (Nox2), markedly alleviates influenza A virus-induced lung injury and virus replication, irrespective of the infecting strain.
Therefore, we propose that Nox2 oxidase inhibitors, in combination with current therapeutics (i.e. antivirals and vaccines), could be useful for suppression of influenza A virus-induced lung disease.
Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy:
Trends in Pharmacological Sciences
https://www.cell.com/trends/pharmacological-sciences/fulltext/S0165-6147(11)00159-3ØøøóAÅë꼡ÅýÀуøöêóøÅ£ŸÅåî¾(ROS)çáýºèº
ôßù¿ÑÁ
奤ýÀÊù¿ä¿¯ëȘù¿ù±ñ·ôßù¿ÀÊà«âíûæçüÀȯáǵâ«îúö˜ÑÁâ«îúøïá¨Ñ«ÝƒÇµîÏØˋîÏü礶ö■îŃ¢æÕ
ñÂÝÚ:2011áõ10åô04,doi: https://doi.org/10.1016/j.tips.2011.09.001
¥æÅë꼡ÅýÀу¡ÅàƒåÖà¨òâ§ÓÑÃàËêùò»¯ìë·àùçáèºû■ȘýÂ¥äŽÑåöâèºÝȧÀüçë°åš°èøÄǵ¡¤çÈÀÈá¢ú¯¢Äøó¥æÅë꼡ÅýÀуػóÞçáñöý¢¥ýýÀçáØˋâÚîÏýÔôåÇÌåÖöòäãȘíãòúÆèÆÖ¢¿ýÀуØˋöÿáëØˋÅå¤ëÅÒ؈íŠÑåäÄÑ´ƒºøõçáØÔûÓ§ÆøøÀÈ
£ŸÅåî¾äÄÝÞòú°˜î¾£₤öÿçáýºèºòúèÝûÞàŠúøýÀåÙäÍçáøÄ؈ùßø¼ñâƪ£ºøóÀÈ
à£Ñ½È˜åÖ¥æÅë꼡ÅýÀу¡Åàƒ¤µ¿»ê¢çᰘ₤öÿ¢èáÉòúÆŤÎçáÀÈòôòçèüȘ봿»¯Åü·ý¡àÕÑ₤öÿîæøÂü¡¯«øÅ°˜î¾£₤öÿçáø¼Øˆû¡åÇNADPHî¾£₤û¡2 (Nox2)âÇØøøó°˜î¾£₤öÿçáýºèºÈ˜¢èØåüåø½¥¾úÃ¥æÅë꼡ÅýÀуػóÞçáñöùÞèù¤ëýÀу¡ÇøóȘѽƊ¡Åàƒøõöß¿ÄÀÈ
Ø·ÇùȘöØûúàüöˆÈ˜Nox2î¾£₤û¡Øøøó¥ê§Ã¤üá¢ú¯çáøöêóñ§ñ´(¥Ç¢¿ýÀуØˋöÿ¤ëØÔûÓ)Ș¢èáÉÑåØøøó¥æÅë꼡ÅýÀуÆíç¥çáñöý¢¥ýýÀÆÅÆûÀÈ
ØøøóAÅë꼡ÅýÀуøöêóøÅ£ŸÅåî¾(ROS)çáýºèº:ØˋâÚîÏçáñÂí¿ú¼òó
https://www.cell.com/trends/pharmacological sciences/fulltext/s0165 - 6147 (11) 00159 - 3cited from
Oxid Med Cell Longev. 2013; 2013: 271602.
Natural Compounds as Modulators of NADPH Oxidases
Tullia Maraldi*
Author information Article notes Copyright and License information Disclaimer
Department of Surgical, Medical, Dental and Morphological Sciences with Interest in Transplant, Oncology and Regenerative Medicine, University of Modena and Reggio Emilia, Via Del Pozzo 71, 41100 Modena, Italy1. ROS Involvement in Cell Pathophysiology
Oxidative stress is a molecular deregulation in reactive oxygen species (ROS) metabolism involved in the pathogenesis of several diseases. Oxidative stress is no longer considered as a simple imbalance between the production and scavenging of ROS, but as a dysfunction of enzymes involved in ROS production [1].
Reactive oxygen species such as superoxide, hydrogen peroxide, and peroxynitrite are generated by all mammalian cells and have been recognized for many decades as causing cell damage by oxidation and nitration of macromolecules, such as DNA, RNA, proteins, and lipids. Moreover, ROS can also promote cell signaling pathways modulated by growth factors and transcription factors, therefore regulating cell proliferation, differentiation, and apoptosis [2], which are important processes for proper cell functioning [3]. At physiological concentrations they facilitate the signal transduction derived from receptor tyrosine kinases and transcriptional factors such as NF-E2-related factor-2 (Nrf-2) leading to antioxidant gene expression [4].
The instability of an unpaired electron in its valence shell causes the high reactivity of superoxide. Superoxide has been implicated in numerous pathological processes, including cancer, cardiovascular disease (e.g., atherosclerosis and stroke), and acute and chronic diseases due to microbial infections. Superoxide can directly or indirectly damage DNA through oxidation [5], directly inactivate cellular antioxidants enzymes such as catalase and glutathione peroxidase [6], and activate proinflammatory nuclear factor jB (NF-jB) [7].
However, superoxide gives rise to other ROS that possess different redox chemistries, and, thus, different physiological and pathophysiological effects. For example, superoxide is rapidly reduced, both spontaneously and enzymatically, to H2O2. Unlike superoxide, H2O2 has no net charge; so, it is more lipid-soluble, with the potential to diffuse through organelles and cellular membranes reaching sites distant from its source. H2O2 modifies cellular proteins via oxidation of cysteine, methionine [8], and genetic material [9]. However, perhaps the major dangerous properties of H2O2 are in its ability to generate more reactive molecules. For instance, in the presence of transition metals, H2O2 can generate the highly reactive OH•. The OH• is highly reactive and will indiscriminately oxidize the nucleotides causing breaks and lesions of DNA [for review see [10]], which are processes involved in carcinogenesis. The oxidation of lipids by OH• may influence many physiological processes and contribute to cellular dysfunction, such as oxidation of lipids by peroxidation [11], during cardiovascular disease [12].
One of the most important and fast redox reactions in biology is between superoxide and the nitric oxide (NO) radical giving rise to ONOO−. ONOO− is an oxidizing and nitrating molecule that has been implicated in cancer [13] and other acute [14] and chronic [15] diseases. ROS levels in tumor cells are controlled in a particular way, which stresses the importance of the development of novel ROS-targeted anticancer therapies.
As with every mechanism involved in both normal cell function and the development of disease, strategies to counteract ROS must take into account their critical importance in the normal functioning of the organism [1].
Further understanding of the biological mechanisms among oxidative stress, tumor growth, and metastasis could contribute to the advancement of cancer treatment.
For example, angiogenesis is another important factor for tumor growth and metastasis, and ROS has a key role in angiogenesis regulation [16].
After all, an emerging concept suggests that ROS modulate the immune cells functions that infiltrate the tumor environment and stimulate angiogenesis [2].
These oxidative processes have been implicated in many diseases in addition to cancer.
Overproduction of ROS is involved in the development of a number of diseases, which range from neurological such as Parkinson's [17] and Alzheimer's disease [18], to psychiatric disorders such as schizophrenia [19] and bipolar disorder [20], and to a majority of cardiovascular diseases [21].NADPH Oxidases as ROS Sources
Several enzymes produce ROS, including the mitochondrial electron transport chain, nitric oxide synthases (NOSs), cytochrome P450 reductase, and xanthine oxidase. However, for all of these systems, ROS production takes place as a byproduct of the main catalytic function of the enzyme/system or from a dysfunctional variant of the enzyme. In contrast, NADPH oxidases are the only enzymes whose primary function is to generate superoxide/ROS [2]. NOX family proteins are the catalytic, electrontransporting subunits of the NADPH oxidase enzyme complex. The NOX family consists of seven members, NOX1´C5, and two dual oxidases (Duox), Duox1 and Duox2 (Table 1). The NOX isoforms contain FAD and NADPH binding sites, two heme molecules and six transmembrane [22] spanning alpha helices with cytosolic N and C-termini. The Duox isoforms also contain the same domains; however, a seventh transmembrane domain and peroxidase homology are present.
They are differentially expressed and regulated in various tissues and have different subcellular localizations (reviewed in [23]). NOX1, NOX2, and NOX5 appear to produce mainly superoxide NOX4, mainly H2O2 [24]. All NOX isoforms have been reported to bind to one or more additional components. p22phox appears to be a general binding partner for NOX1´C4. NOX1 and 2 also bind the small GTPase, Rac. Moreover, NOX1 binds the cytosolic subunits, NOX organizer 1 (NOXO1), and NOX activator 1 (NOXA1). NOX2 binds the respective homologues, p47phox and p67phox, and also the cytosolic protein, p40phox [22, 25]. NOX4 was reported to bind to the polymerase (DNA-directed) delta-interacting protein 2 (PolDip2) [26].
In addition to these established NOX binding partners, the tyrosine kinase substrate with 4/5 SH3 domains (Tks4/5) [27] and protein disulfide isomerase (PDI) were recently suggested to bind to both NOX1 and 4 [28]. Two maturation factors specific for Duox (DuoxA1 and DuoxA2) have also been described [22].
NOX catalytic subunits are differently regulated: NOXA1 plays a key role for NOX1 activation [29], p67phox for NOX2 [30], and calmodulin for NOX5 [31].
In contrast, NOX4 is constitutively active, and modulation of its expression may thus be a major activity regulator.
ROS produced by NOXs have been shown to affect all other possible sources of ROS, leading to their dysfunction and to a further increase in ROS generation, forming a vicious cycle of oxidative stress. For example, increased O2 •− generation by NADPH oxidase induces mitochondrial oxidative damage via structural changes to the inner mitochondrial membrane and disturbs flow in the electron transport chain which enhances ROS production [32].5. Antioxidant Therapy
Classically, oxidative stress has been defined as an imbalance between the endogenous production of reactive oxygen compounds and the antioxidative potential of cells [58].
But the low or apparent lack of clinical effectiveness of ROS-scavenging approaches is not entirely explained. It can be due to the partial removal of selected harmful endproducts by ROS-scavengers. Furthermore, antioxidants, including vitamins, reaction with superoxide anions is slower than NO. Moreover, it does not take into account that cellular events leading to disease primarily occur in individual cellular compartments [3].3. NOX and Pathophysiology
The oxidant signaling involving NADPH oxidase has important roles in cell biology participating in intracellular signaling of cell differentiation and proliferation. These mechanisms are important in tissue repair and tumorigenesis, processes where cell proliferation occurs, but when poorly controlled the generation of ROS is dangerous. Indeed, NADPH oxidase-mediated cell proliferation has been observed in a wide range of cell types including those found in blood vessels, kidney, liver, skeletal muscle precursors, neonatal cardiac myocytes, lung epithelial cells, gastric mucosa, brain microglia, and a variety of cancer cells. For example, NOX may stimulate Akt activation also by inactivating the phosphatase PTEN, a direct negative regulator of the PI3K/Akt pathway [33]. Therefore, NADPH oxidase-mediated redox signaling may amplify diverse signaling pathways triggered in tissue repair processes such as cell proliferation, wound healing, angiogenesis, or fibrosis. Recent studies also suggest that NADPH oxidase is involved in differentiation and proliferation of stem cells. Currently, little is known about whether ROS regulate different signaling pathways in stem cells and differentiated cells and whether ROS play a different role in these cells [16]. Thus, modulating NADPH oxidase may have significant impacts on regenerative medicine and tissue engineering, such as growing heart muscle.
At first, cellular stresses may induce NOX-dependent ROS generation as an alert system that drives the cells into a relatively stress-resistant status by integrating and amplifying the stress signals, as preconditioning against further cellular challenges [34]. The double-edged sword property of NOX adds another level of complexity for therapeutic targeting of this enzyme. The mechanisms of NOX-mediated redox signaling may be either enhancing stress resistance through prevention of ischemia-reperfusion injuries and accelerating wound healing, or avoiding the stress-induced cytotoxicity for iper-proliferative diseases, such as cancer. Therefore, a therapeutic modulation of NOX activity has to be developed taking in account the disease, the stage of disease, the tissue localization area, the NOX isoforms, and the NOX intracellular localization.
4. NOXs and Diseases
NADPH oxidase has received much attention as a major cause of oxidative stress leading to vascular disease. Moreover different NOX subunits have been suggested to play a role in cancer, lung fibrosis, stroke, heart failure, diabetes, and neurodegenerative diseases [22]. NOX-derived ROS can lead to pathology through differential ways: for example, spatially confined levels of ROS that interfere with a particular signaling pathway, and high levels (local or systemic) that are directly cytotoxic, causing apoptosis, or disturbing redox-sensitive signaling cascades.
4.1. Vascular Diseases
NADPH oxidases are ubiquitously expressed in tissues but are the major source of superoxide anions observed in the vasculature [1]. Thus, similarly to ROS, NOX proteins have both beneficial and harmful effects. They are important signaling molecules which regulate vascular tone, expression, proliferation, migration, and differentiation. On the other hand, cardiovascular risk factors and vascular diseases increase ROS and contribute to atherosclerosis, vascular dysfunction, hypertension, vascular hypertrophy, and thrombosis.
With respect to low and spatially confined ROS overproduction, NOX1 is a good candidate to migrate into caveolae and there causes eNOS uncoupling and endothelial dysfunction, which is often associated with increased blood pressure and enhanced platelet aggregation as an early step in the development of atherosclerosis [35].
4.2. Cancer
NOX1, NOX2, and NOX4 are known to be expressed in multiple tumor cell types (see Table 1). Tumor cells produce high amounts of ROS [36, 37] by NADPH oxidases to promote their own proliferation, through regulation of proliferative signaling kinases, such as cell survival factors, such as Akt and MEK-ERK pathway [38].
Recently, the role of cancer stem cells (CSCs) in cancer progression and metastasis has attracted much attention since CSCs are integral parts of pathophysiologic mechanisms of metastasis and chemo/radioresistance [16]. To date, molecular events that govern the survival and self-renewal of CSCs are poorly defined, but a link between ROS modulation and cancer stem cell metabolism seems to have a basis. Stem cells and also CSCs are known to reside in niches characterized by low ROS, a critical factor in maintaining stem cell properties such as self-renewal. Multiple signaling pathways in normal stem cells and CSCs can regulate ROS level and could be exploited against CSCs. Elucidation of ROS function in CSCs will enrich our knowledge of cancer development and metastasis.
Moreover, ROS have also been shown to regulate angiogenesis through the release and actions of tumor-derived growth factors that induce endothelial cell proliferation. In fact, ROS production within tumor cells dramatically promotes the release of paracrine growth factors such as VEGF and the expression of its receptor, VEGF receptor-1 which, in turn, stimulate proliferation, migration, and tube formation in nearby endothelial cells [34]. NADPH oxidase within endothelial cells cooperates with growth factors, such as VEGF released by tumors, to stimulate endothelial cell proliferation and then angiogenesis. Thus, NOXs as ROS source in tumors and in endothelium may be considered novel targets for antiangiogenic treatment.
4.3. Inflammation
The oxidative burst of phagocytes has long been considered proinflammatory, causing cell and tissue destruction. Recent findings have challenged this inflammatory role of ROS, and now ROS are also known to regulate immune responses and cell proliferation and to determine T-cell autoreactivity. NOX2-derived ROS have been shown to suppress antigen-dependent T-cell reactivity and remarkably to reduce the severity of experimental arthritis in both rats and mice [39]. In fact, regarding these chronic inflammatory diseases (rheumatoid arthritis) there is increasing evidence that ROS can often help to modulate inflammation, ensuring that it does not become too prolonged [see review [40]].
In retrospect, this is also suggested by the pathology of chronic granulomatous disease (a condition characterized by inborn defects in the phagocyte O2 •− generating NADPH oxidase), there is an increased risk of infection due to an inability to kill certain microorganisms; this is in addition to a severe dysregulation and prolongation of inflammation. Thus, stopping ROS production can be deleterious. On the other hand, ROS can cause severe cartilage damage and the ability of nuclear factor erythroid 2 p4-5-related factor 2 to enhance endogenous antioxidant defenses in response to the inflammation may play a significant ameliorative role. The same may be true in sepsis; some ROS may help but too many can cause harm [see review [40]].
Finally, NADPH oxidase-derived ROS are also crucial players of tumor anti-immunity regulating specialized subsets of immune cells such as macrophages and T lymphocytes. Thus, NOXs could represent a novel molecular link between chronic inflammation and angiogenesis during cancer [2].
NOX2 is connected to the innate immune response [41], including to fungal infections [42] and adaptive immune response at the level of both T cells and antigen-presenting cells [43]. Thus, NOX2 inhibition leads to an improvement of diseases involving a significant inflammatory response. On the other hand, the essential immune functions of NOX2 have to be preserved [35].
4.4. Organ Failure
ROS are generally thought to play an important role in the pathophysiology of organ failure [22]. For example, in liver and intestinal tissue injury, there is some indication for a pathogenetic role of NOX2-derived ROS by neutrophils [44, 45].
With respect to high levels of ROS produced by NOX4, they can be directly cytotoxic or cause apoptosis inducing heart ischemic stroke. On the other hand, regarding the NOX4 role on pressure overload of the heart, NOX4 might be responsible of both acute damage of the cardiomyocyte and subacute protection of the heart by promoting angiogenesis [35].
4.5. CNS Diseases
4.5.1. Ischemic Stroke
In a gerbil model of global cerebral ischemia-reperfusion injury, NOX inhibition by apocynin strongly diminishes damage to the hippocampus [46]. Stroke size was markedly reduced in NOX2-deficient mice [47], while increased NOX2 expression in diabetic rats was associated with an aggravated ischemic brain injury [48].
4.5.2. Alzheimer's Disease, Parkinson's Disease, and HIV Dementia
NOX2 seems to have a role in inflammatory neurodegeneration diseases, including Alzheimer's disease and Parkinson's disease [49, 50]. In the case of Alzheimer's disease, amyloid precursor protein fragments released from neurons activate NOX2 -dependent ROS generation by microglia cells leading to death of neighboring neurons [51]. Several studies suggest similar mechanisms, involving NOX2, in Parkinson's disease [52, 53].
Microglia activation is also thought to be a key element in the development of dementia [54], and a role of NOX2 activation in animal model of dementia has been suggested [55].
Microglial NOX2-derived ROS have also been implicated in the progression of the demyelinating disease through phagocytosis of myelin and damage to the myelin sheath [56]. In periventricular leukomalacia, the combination of NOX2-derived superoxide and inducible nitric oxide synthase-derived nitric oxide leads to the formation of peroxynitrate and thereby to the killing of oligodendrites [57].
5. Antioxidant Therapy
Classically, oxidative stress has been defined as an imbalance between the endogenous production of reactive oxygen compounds and the antioxidative potential of cells [58].
But the low or apparent lack of clinical effectiveness of ROS-scavenging approaches is not entirely explained. It can be due to the partial removal of selected harmful endproducts by ROS-scavengers. Furthermore, antioxidants, including vitamins, reaction with superoxide anions is slower than NO. Moreover, it does not take into account that cellular events leading to disease primarily occur in individual cellular compartments [3].
5.1. NOX Inhibition (See Table 2)
The main sources of ROS include redox enzymes such as the respiratory chain, xanthine oxidase, lipooxygenase, cyclooxygenase, and NADPH oxidases, and these systems are continuously interacting with each other. Due to the complex mechanisms involved in the activation of NADPH oxidases, these enzymes can be targeted on several different levels of their activity. Firstly, decreasing NADPH oxidase expression can inhibit them. Also, the activation of NADPH oxidase can be decreased by blocking the translocation of its cytosolic subunits, when present, to the membrane.
Another possibility is inhibition of the p47phox subunit, either by preventing its phosphorylation using PKC inhibitors or by blocking its binding to other subunits. A decrease of signal transduction and inhibition of Rac 1 translocation has also been demonstrated to decrease ROS generation [3].
Several compounds have been used, including apocynin, diphenylene iodonium (DPI), and 4-(2-aminoethyl)-benzensulfonylfluorid (AEBSF). However, it has become apparent that these inhibitors are not specific for NOX [59] and not selective for single NOX isoforms.
One of the first inhibitors used in model studies was diphenyliodonium (DPI), which is very potent (although in micromolar range) but lacks specificity. DPI is a general flavoprotein inhibitor, also inhibiting, for example, xanthine oxidase and eNOS [60´C62], as well as cholinesterases and a calcium pump [63].
AEBSF is primarily a serine protease inhibitor [64]. Its greatest drawback is however toxicity.
Later studies involved apocynin, a naturally occurring NADPH oxidase inhibitor originally isolated from the roots of Picrorhiza kurroa. Apocynin cannot be used as selective NADPH oxidase inhibitor due to its direct antioxidant and several off-target effects [60]. Apocynin is an orally active agent that can block NADPH oxidase assembly in membrane but requires a reaction with peroxidase for its activation, and therefore does not work immediately [65]. Apocynin reduces ROS production in models of arthritis, asthma, and hypertension, abolishing the increase in vascular O2 •− and preventing endothelial dysfunction [66]. However, effects of apocynin are not specific, as it has been reported to affect arachidonic acid metabolism [67], to increase glutathione synthesis, and to activate the AP-1 transcription factor [68]. In addition, it has recently been shown to be a direct ROS scavenger in certain experimental conditions [69].
The use of natural antioxidants represents a promising new approach for NOX inhibition. Polyphenols represent more than 10000 compounds occurring naturally in foods and the recommendation for a polyphenol rich (green tea/red wine/fruits/vegetables/whole grain foods) diet in the prevention of cardiovascular disease is still valid [70]. It was found that polyphenols, apart from their well-known superoxide radical scavenging abilities, decrease NADPH oxidase activity [71] in a number of tissues including vessels and platelets [72]. Moreover, novel polyphenolic compounds are being investigated which lack typical superoxide scavenging properties and directly inhibit NADPH oxidase.
Recent studies with berberine, a plant alkaloid [73], revealed an inhibition of NADPH oxidase activity and reduction of gp91phox mRNA expression in macrophages. Also, emodin, an active component extracted from rhubarb and rhein, reduced ROS generation [74]. Similar effects were observed by treatment with 3-(4Àð-hydroxyl-3Àð,5Àð-dimethoxyphenyl)propionic acid (HDMPPA), the active ingredient in kimchi, ellagic acid, a polyphenol present in fruits and nuts [75], and dihomo-Îû-linolenic (ÎÄ-6) acid [76].
The inhibitory effect of flavonoids (kaempferol, morin, quercetin, and fisetin) on the respiratory burst of neutrophils was observed by Pagonis et al. [77] as early as in 1986.
A Ginkgo biloba extract containing flavonoids, among other compounds, was tested by Pincemail et al. [78] for its effect on the release of ROS (superoxide anion radical, hydrogen peroxide, and hydroxyl radical) during the stimulation of human neutrophils by a soluble agonist. The extract slowed down the oxygen consumption (respiratory burst) of the stimulated cells by its inhibitory action on NADPH oxidase [79]. The extract was also able to reduce the activity of myeloperoxidase contained in neutrophils. Moreover, it had free radical scavenging activity.
A higher number of hydroxyl substituents are an important structural feature of flavonoids in respect to their scavenging activity against ROS, while C-2,3 double bond (present in quercetin and resveratrol) might be important for the inhibition of ROS production by phagocytes [80].
The bark of magnolia has been used in oriental medicine to treat a variety of remedies, including some neurological disorders [81]. Magnolol (Mag) and honokiol (Hon) are isomers of polyphenolic compounds from the bark of Magnolia officinalis, and have been identified as major active components exhibiting antioxidative, anti-inflammatory, and neuroprotective effects. It has been reported that exposure of Hon and Mag to neurons for 24 h did not alter neuronal viability, but both compounds inhibited superoxide production, a pathway known to involve NADPH oxidase. This study highlighted the important role of NADPH oxidase in mediating oxidative stress in neurons and microglial cells and has unveiled the role of IFNÎû in stimulating the MAPK/ERK1/2 signaling pathway for activation of NADPH oxidase in microglial cells. Hon and Mag offer anti-oxidative or anti-inflammatory effects, at least in part, through suppressing IFNÎû-induced p-ERK1/2 and its downstream pathway [81].
Incubation of human neuroblastoma cells with nonpolar blueberry fractions obstructed the coalescing of lipid rafts into large domains disrupting NOX assembly therein and abolishing ROS production [82]. In fact, this NOX inhibiting bioactivity in crude blueberry extracts partitioned into a polyphenol-devoid fraction lacking virtually any antioxidant capacity and prevented proper assembly of the multisubunit NOX complex interfering with the coalescence of large lipid raft domains.
Prodigiosin, a microbial pigment, and some derivatives suppressed NOX activity most likely by disrupting Rac function [83, 84], and inhibition of NOX by the alkaloid sinomenine is unclear at best [85].
In an oxygen-glucose deprivation and reoxygenation (OGD/R) model, pretreatment with green tea polyphenols (GTPP) and their active ingredient, epigallocatechin-3-gallate (EGCG), protects PC12 cells from subsequent OGD/R-induced cell death [86, 87]. GTPP stimulates laminin receptor and thereby induces NADPH oxidase-dependent generation of ROS, which in turn induces activation of PKC resulting in preconditioning against cell death induced by OGD/R.
Resveratrol is a naturally occurring polyphenol, which has vasoprotective effects in diabetic animal models and inhibits high glucose (HG-) induced oxidative stress in endothelial cells. It has been reported that HG induces endothelial cell apoptosis through NF-ÎòB/NADPH oxidase/ROS pathway, which was inhibited by resveratrol [88, 89].
Other NOX inhibitors are VAS 2870, VAS 3947, GK-136901, plumbagin, and polyphenolic derivative S17834 [90].
Plumbagin, a plant-derived naphthoquinone, has been shown to exert anticarcinogenic and antiatherosclerosis effects in animals. Plumbagin inhibits NADPH-dependent superoxide production in cell lines that express NOX4 oxidase [91]. Although its exact mechanism of action remains unclear, the inclusion of a napthoquinone structure within the molecule may be responsible for its ROS scavenging abilities [90]. Plumbagin inhibited the activity of NOX4 in a time- and dose-dependent manner in HEK293 and LN229 cells directly interacting with NOX4 and inhibiting its activity [91].
Indeed, plumbagin has been reported to exert anticancer activity on osteosarcoma cells by inducing proapoptotic signaling and modulating the intracellular ROS that causes induction of apoptosis [92]. Moreover plumbagin-induced AMPK activation might be the key mediator of plumbagin's antitumor activity [93].
Furthermore, PI5K-1B plays a crucial role in ROS generation and could be a new molecular target of plumbagin [94]. At last, plumbagin can exert its function in depleting glutathione (GSH) levels that led to increase in ROS generation [95].
Celastrol is one of several bioactive compounds extracted from the medicinal plant Tripterygium wilfordii. Celastrol is used to treat inflammatory conditions and shows benefits in models of neurodegenerative disease, cancer, and arthritis, although its mechanism of action is incompletely understood. Authors demonstrated that celastrol is a potent inhibitor of NOX enzymes in general with increased potency against NOX1 and NOX2. Furthermore, inhibition of NOX1 and NOX2 was mediated via a novel mode of action, namely, inhibition of a functional association between cytosolic subunits and the membrane flavocytochrome [96].
6. Conclusions
Accumulating evidence clearly indicates that NADPH oxidases are critical molecular targets for dietary bioactive agents for prevention and therapy of different pathologies.
The development of specific and not toxic inhibitors of NADPH oxidases and their redox signaling network (kinase, transcription factors, and genes) could provide useful therapeutic strategies for the treatment of oxidative stress dependent processes such as cancer and other degenerative diseases.
In fact, classical antioxidant therapies have been demonstrated inadequate since the importance of ROS in physiology has been ignored leading to the lack of clinical benefits. Indeed, further research into selective molecular inhibitors interfering with NADPH oxidase activation are warranted. The selective targeting of dysfunctional NADPH oxidase homologs appears to be the most suitable approach, with the potential to be far more efficient than the one with nonselective antioxidants having only ROS scavenging properties.
NOX enzymes, however, are very complex with numerous specific targets within each isoform. More information is needed on how these proteins are targeted to different subcellular compartments and how this transport process is regulated.
It is encouraging, however, that single bioactive dietary agents can directly and indirectly influence most, if not all, of the myriad targets within NOX family. Additionally, many of these dietary agents appear to exhibit some degree of specificity for redox deregulated cells while unaffecting normal cells balance. Moreover, the protective effects of some single agents could be potentiated and/or synergized by other dietary agents. While encouraging, there are many considerations that remain, such as the issue of appropriate dose of each agent, appropriate timing and duration of exposure, importance of cell type specificity, relative bioavailability of each agent, and potentially adverse side effects and interactions.Natural Compounds as Modulators of NADPH Oxidases
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3863456/ÀÀ
ÀÀ