靶向T细胞代谢以调节T细胞活化,分化和疾病功能
Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease
概要
越来越清楚的是,代谢重编程在T细胞活化,分化和功能中起关键作用。为此,细胞代谢不仅满足T细胞的能量需求,而且还为其生长和功能提供了关键的底物。此外,代谢物本身正在成为免疫反应的关键调节剂。由于揭示了代谢重编程如何调节免疫功能的细节,已经出现了调节免疫应答的新潜在靶标。
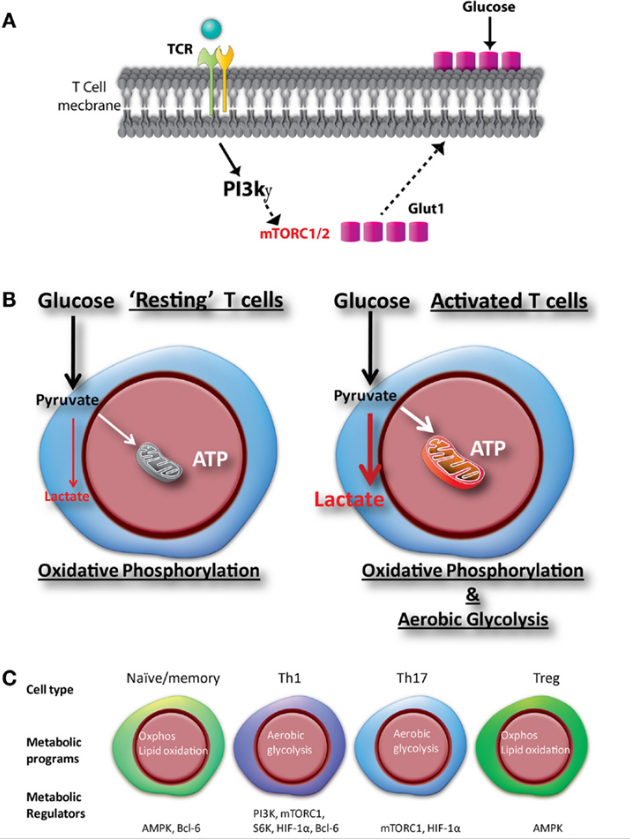
实际上,不同T细胞亚群的不同代谢需求使它们对代谢的药理学抑制剂非常敏感。在这篇综述中,我们将描述新兴策略,其中靶向代谢可以塑造T细胞反应。介绍幼稚CD8 + T细胞向活跃效应细胞的转变以及记忆细胞的发育涉及动态和协调的代谢重编程[1]。这种重编程不是激活的结果,而是与分化和激活过程密切相关。这些代谢变化反映了支持T细胞不同阶段所需的燃料和底物[2,3]。幼稚T细胞和记忆T细胞主要依靠氧化磷酸化和脂肪酸氧化作为燃料。这反映了对能源的低水平但持续的需求;这种细胞是长寿的。或者,效应T细胞具有非常高的能量和合成需求。这些细胞表现得像肿瘤细胞,并开始糖酵解,并采用TCA循环来支持他们对在肝内合成蛋白质,脂质和核酸的需求。同样,对于CD4 + T细胞,已经表明,分化为不同的效应子集伴随着差异代谢编程[4]。最值得注意的是,TH1,TH2和TH17细胞依赖于糖酵解来支持效应子功能,而调节性T细胞(Tregs)更依赖于氧化磷酸化和脂肪酸氧化。
通过了解不同T细胞亚群的代谢需求,我们现在提供了选择性定制免疫应答的有希望的治疗机会。在本综述中,我们将描述靶向代谢的一些具体实例,以调节T细胞活化,分化和疾病功能。靶向T细胞代谢提供了以细胞内在方式真正调节免疫应答的机会。在自身免疫疾病和移植的情况下,抑制效应功能和增强调节性T细胞是至关重要的。或者,靶向代谢也提供了一种有前途的新策略,以增强癌症免疫疗法中的T细胞应答。
要点:
代谢重编程与T细胞活化密切相关;
不同的T细胞亚群具有不同的代谢需求;
基于细胞需求实现靶向代谢的选择性靶向代谢可以抑制和增强T细胞功能;
靶向代谢代表了治疗自身免疫,移植排斥和增强免疫疗法的新方法。
Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease
Proton production and its transporting kinetics in intracellular and extracellular fluid in metabolic tissues. The production of organic acids such as lactic acid and ketone bodies is accelerated by elevating glycolytic anaerobic metabolism and β-oxidation in metabolic cells. Body fluid pH is strictly maintained by buffering systems, efflux across plasma membrane, and acid excretion. Monocarboxylate transporter (MCT) and Na+/H+ exchanger (NHE) contribute to proton extrusion from the cytosol to the extracellular space. In contrast to intracellular fluid and blood containing pH buffers such as Hb (hemoglobin) and albumin, the interstitial fluid pH could be easily reduced by acid stress owing to the limited availability of the buffering factors such as proteins.
Chirag H. Patel1,2 and Jonathan D. Powellcorresponding author1,2
Author information ► Copyright and License information ► Disclaimer
The publisher's final edited version of this article is available at Curr Opin Immunol
See other articles in PMC that cite the published article.
Summary
It is becoming increasingly clear that metabolic reprogramming plays a critical role in T cell activation, differentiation and function. To this end, cellular metabolism not only meets the energetic demands of T cells but also provides critical substrates for their growth and function. Furthermore, metabolites themselves are emerging as key regulators of immune responses. As the details of how metabolic reprogramming regulates immune function are revealed, new potential targets for modulating immune responses have emerged. Indeed, the distinct metabolic demands of different T cell subsets make them exquisitely sensitive to pharmacologic inhibitors of metabolism. In this review, we will describe the emerging strategies whereby targeting metabolism can shape the T cell response.
Introduction
The transition of a naïve CD8+ T cell to an active effector and to the development of memory cells involves dynamic and coordinated metabolic reprogramming [1]. This reprogramming is not a consequence of activation but rather is intimately linked to the differentiation and activation processes. These metabolic changes reflect the fuel and substrates necessary to support different stages of a T cell [2,3]. Both naïve T cells and memory T cells rely primarily on oxidative phosphorylation and fatty acid oxidation for fuel. This reflects the low level yet persistent need for energy; such cells are long-lived. Alternatively, effector T cells have extraordinarily high energetic and synthetic demands. These cells behave like tumor cells and turn up glycolysis and employ the TCA cycle to support their demand for de novo proteins, lipids and nucleic acids synthesis. Likewise, for CD4+ T cells, it has been shown that differentiation in to distinct effector subsets is accompanied by differential metabolic programming [4]. Most notably, TH1, TH2 and TH17 cells rely upon glycolysis to support effector function while regulatory T cells (Tregs) are more dependent on oxidative phosphorylation and fatty acid oxidation.
By appreciating the metabolic requirements of different T cell subsets, we are now provided with a promising therapeutic opportunity to selectively tailor immune responses. In this review, we will describe some specific examples of targeting metabolism to regulate T cell activation, differentiation and function in disease. Targeting T cell metabolism affords the opportunity to truly regulate immune responses in a cell intrinsic manner. In the case of autoimmune diseases and transplantation, it is critical to inhibit effector function and enhance regulatory T cells. Alternatively, targeting metabolism also provides a promising new strategy to enhance T cell responses in immunotherapy for cancer.
mTOR integrates signals from the immune microenvironment
Upon TCR engagement, the outcome of antigen recognition is determined by the integration of signals from the immune microenvironment [5,6]. Through genetic deletion of mTOR and components of the mTOR signaling pathway, our group and others have identified a critical role for mTOR in regulating T cells activation, differentiation and function [7]. CD4+ T cells lacking mTOR fail to become effector cells but instead activation promotes the generation of Tregs [8]. Likewise, T cells selectively lacking the mTORC1 activator Rheb fail to become Th1 or Th17 cells but still can become TH2 cells [9]. On the other hand, T cells lacking the mTORC2 scaffold protein Rictor fail to become Th2 cells yet can be readily differentiated into TH1 and TH17 cells [9,10]. Interestingly, inhibiting mTORC1 activity through the genetic deletion of the scaffolding protein Raptor appears to have a much more profound effect on T cell function disabling TH1, TH2 and even Tregs [11,12]. What has emerged most recently, is the ability of mTOR to regulate T cell differentiation and function is tightly linked to the role of mTOR in promoting metabolic reprogramming [13]. Indeed, mTOR activation promotes glycolysis, fatty acid synthesis and mitochondrial biogenesis. As such, targets upstream and downstream of the mTOR signaling pathway are potential therapeutic targets [7]. For example, the drug rapamycin was initially developed as an “immunosuppressive” agent due to its ability to slow down T cell proliferation [14]. Subsequently, it has been shown that rapamycin can promote Treg generation and T cell anergy [15,16]. However, in a different context, rapamycin has been shown to promote robust responses to vaccination by enhancing CD8+ T cell memory generation [17]. Likewise, deletion of the mTORC1 inhibitory protein TSC2 leads to enhanced mTORC1 activity and consequently increased effector function [18]. Consequently, the pharmacologic or genetic targeting of TSC2 might prove to be a robust means of enhancing anti-tumor immunity.
Targeting T cells through Amino Acids
Cellular growth and function are highly dependent on having an adequate source of amino acids. In addition to being an essential building block for protein synthesis, amino acids also provide an important backbone for de novo nucleotide synthesis [19]. Furthermore, amino acids also act as an essential metabolic fuel source feeding into multiple pathways. Upon T cell activation, there is an immediate uptake of amino acids such as glutamine and leucine that is critical for proper metabolic reprogramming [20,21]. Indeed, influx of branched chain amino acids such as leucine are critical for mTORC1 activation.
Expression of critical amino acid transporters involved in glutamine (SLC1A5) and leucine (SLC7A5/SLC3A2 heterodimer) is relatively low in naïve or resting T cells [20,21]. However, within hours of T cell activation, the expression of these transporters is significantly increased. Just as CD69 and CD44 expression are upregulated upon T cell activation, so too are these crucial transporters. This increase in expression is mediated both by the “immunologic” transcription factor NFAT as well as the “metabolic” regulator Myc [22]. This mechanism of regulation epitomizes the coordinated nature of metabolic reprogramming with T cell activation.
Not surprisingly, recent studies have shown that the modulation of glutamine or leucine levels either through downregulation of transporters or deprivation of amino acids themselves affects T cell activation and function [20,21]. Glutamine is converted to glutamate by glutaminase, and then enters the TCA cycle as α-KG. At this point, the nitrogens and carbons from glutamine are used for de novo nucleotide synthesis as well as the generation of aspartate (which can be further metabolized to pyrimidines). Down modulation of glutamine and leucine metabolism has been shown to suppress the differentiation of Th1 and Th17 effector T cells while maintaining Treg differentiation [20,21]. This decrease in amino acid metabolism resulted in reduced mTORC1 activity and less Myc expression leading to a defect in the upregulation of the metabolic machinery required for differentiation.
The critical role of glutamine metabolism in promoting a potent effector T cell response can be demonstrated using small molecule inhibitors. DON (6-Diazo-5-oxo-L-norleucine) is a glutamine antagonist that potently inhibits glutamine-dependent metabolism [23]. Studies from our lab have demonstrated that when combined with other metabolic inhibitors, DON can potently inhibit T cell responses [24]. DON treatment in combination with the inhibitor of glycolysis 2-DG and the diabetes drug metformin (which inhibits mitochondrial complex I and promotes fatty acid oxidation) markedly inhibits CD4+ and CD8+ T cell effector responses. However, due to the distinct metabolic demands, this therapy reciprocally enhances the generation of antigen specific regulatory T cells. In models of allograft skin and heart transplantation, the metabolic therapy markedly promoted allograft acceptance. In addition, inhibiting glutamine metabolism with DON has been shown to effectively inhibit severe inflammation and to promote survival in mouse models of cerebral malaria and viral encephalitis [25,26].
Targeting T Cells through Glycolysis
As mentioned above, critical to the generation and function of both CD4+ and CD8+ effector cells is the upregulation of glycolysis in the presence of abundant oxygen; the so called Warburg physiology first described for tumor cells [27]. Nonetheless, it is important to remember that oxidative phosphorylation (OXPHOS) is still increased and plays an important role in effector T cells. During glycolysis, cells generate a net gain of 2 ATP molecules for fuel. However, the end product of glycolysis, pyruvate provides an important substrate for the TCA cycle. Additionally, OXPHOS generates reactive oxygen species (ROS), which actually can promote antigen-induced cellular signaling through modulation of NFAT [28]. Again, this observation demonstrates the interconnection between metabolism (the generation of ROS) and T cell activation (activation of NFAT).
Upon initial antigen-induced activation, PI3K activation through co-stimulation leads to the upregulation of surface GLUT1 to facilitate enhanced glucose influx [29]. This upregulation of GLUT1 is critical for T cell function, as genetic deletion of GLUT1 markedly inhibits effector T cells [30]. Concomitant with increased expression of glucose transporters is the upregulation of key glycolytic enzymes [22]. Again, this metabolic reprogramming occurs simultaneously with T cell activation and is facilitated by mTOR as it promotes the expression of the metabolic transcription factors Myc and HIF-1α.
Given the importance of increased glycolysis in the activation of effector T cells, several studies have investigated the ability of 2-DG to suppress autoimmune disease. In one such study, 2-DG was found to markedly diminish disease in a model of EAE [31]. As predicted, from a metabolic perspective, this strategy not only inhibited pathogenic Th17 cells but also enhanced the generation of Treg cells as Th17 cells depend on glycolysis while Treg cells preferably rely on OXPHOS and fatty acid oxidation (FAO) for fuel [4]. Likewise, in a model of systemic lupus erythematosus (SLE), dysregulated metabolism of autoreactive CD4+ T cells was critical for disease pathogenesis [32]. Autoreactive CD4+ T cells displayed higher basal glycolysis compared to normal CD4+ T cells thereby presenting glycolysis as a potential therapeutic target. Inhibition of glycolysis with 2-DG, in combination with metformin was able to reduce disease burden and progression by repressing persistent CD4+ T cell activation and reducing CD4+ T cells in the spleen.
Another potential approach to controlling autoimmune disease is to regulate pyruvate metabolism [33]. The conversion of pyruvate to lactic acid is critical for continued robust metabolism, as this leads to replenishing the cell's supply of NAD+. Alternatively, pyruvate can enter the TCA cycle and is converted to acetyl-CoA. The conversion of pyruvate to acetyl-CoA is facilitated by pyruvate dehydrogenase (PDH), which is inhibited by the PDH kinases (PDHK). Therefore, inhibiting PDHK presents an opportunity to promote Treg generation with enhanced OXPHOS and to inhibit effector T cells by suppressing glycolysis. Indeed, inhibition of PDHK1 with dichloroacetate (DCA) in mice markedly diminished disease pathology in EAE [33]. This decrease in disease was characterized by diminution of TH17 cells and increased Tregs. In addition to PDHK, inhibition of the glycolytic regulatory enzyme PFKFB3 using 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3-PO) has been employed as a means of suppressing pathogenic immune responses in vivo [34]. One such study examined the effect of inhibiting glycolysis on GVHD. It was found that treating mice with 3-PO after bone marrow transplantation significantly decreased GVHD and improved overall survival by suppressing cytokine secreting effector cells and proliferation of T cells.
Interestingly, it has become clear that enhanced glycolytic reprogramming is not only necessary to provide fuel and substrates for effector function but also to directly influence effector mechanisms. LDHA is the enzyme responsible for the conversion of pyruvate to lactic acid [35]. Deletion of LDHA leads to markedly diminished TH1 differentiation and function. Mechanistically, this is due to shunting of acetyl-CoA into the TCA cycle thus diminishing the amount of acetate available for epigenetic regulation. That is, deletion of LDHA leads to diminished Th1 generation via epigenetic regulation. In models of autoimmunity, LDHA deletion in T cells markedly diminished the disease phenotype. In addition to this epigenetic mechanism, glycolysis has also been shown to regulate effector cytokine production via post-translational mechanisms. Upon T cell activation, when glycolysis is active, GAPDH is engaged with its substrates promoting the generation of 1,3-bisphosphoglycerate from glyceraldehyde-3-phosphate. However, when glycolysis is decreased or inhibited free GAPDH molecules inhibit translation of Ifng by binding to its 3′ UTR [36]. Thus, targeting glycolysis can inhibit T cell effector function by both transcriptional and translational mechanisms.
While targeting glycolysis is emerging as a means of inhibiting immune responses in the setting of autoimmune disease and transplantation rejection, this strategy has also been used to enhance anti-tumor immunity by promoting long-lived memory cells [37]. Adoptive Cellular Therapy (ACT) using expanded tumor-specific T cells has shown promise as an effective form of immunotherapy for cancer. It is now clear that the success of such an approach is dependent on the transfer of long-lived memory cells. One approach to enrich for memory cells prior to transfer is to expand tumor specific CD8+ T cells ex vivo in the presence of 2-DG [37]. Recall that memory T cells rely on OXPHOS while effector T cells require increased glycolysis. Indeed, 2-DG conditioned ACT cells switch to an OXPHOS-based metabolic program over glycolysis and display a transcriptional program associated with long-lived memory cells. In a model of ACT for melanoma, 2-DG conditioned T cells demonstrated superior persistence in vivo. More importantly, this approach led to more robust anti-tumor control and improved overall survival due to an increase of cytokine secreting antigen specific T cells in the tumor compared to unconditioned T cells [37].
Altering the Mitochondria
While glycolysis and other metabolic processes occur in the cell's cytoplasm, many processes are compartmentalized within the mitochondria. A key role of the mitochondria is the support it provides to the TCA cycle through the generation of electron carriers for entry into the electron transport chain (ETC) as well as for the generation of important intermediates that can be funneled to other processes [38]. The movement of electrons through the ETC generates substantial amounts of ATP as part of the OXPHOS program, but also makes ROS.
While highly glycolytic effector T cells generate lactate from pyruvate, they will also divert pyruvate to the mitochondria for entry into the TCA cycle as acetyl-CoA. However, cells with lower glycolytic rates such as memory T cells, utilize fatty acid oxidation (FAO) to support the TCA cycle and its OXPHOS program [3]. Inhibiting mTOR signaling with rapamycin or activating the AMPK signaling pathway with metformin are two ways known to promote FAO [39]. Alternatively, inhibiting glycolysis with 2-DG causes a reduction in ATP generation, leading to activation of AMPK and thereby FAO. As previously described, the combination of 2-DG and metformin as part of the triple therapy to treat allograft transplantation suppressed the effector T cell response while effectively inducing Tregs presumably by promoting FAO [24]. Indeed, it has been shown that the presence of exogenous fatty acids can effectively inhibit T effector cell function and generation while preserving the generation of regulatory T cells [4]. Likewise, etomoxir, an inhibitor of CPT1α, a critical enzyme required for FAO, suppressed regulatory T cell generation without affecting effector T cell function.
Although most studies have focused on the critical role of glycolysis in promoting effector T cell generation and function, it has become clear that mitochondrial-directed metabolism also plays an important role. By studying a model of GVHD, Ferrara and colleagues revealed a role for FAO [40]. In this study, inhibition of FAO and mitochondrial metabolism using etomoxir effectively suppressed the allogenic T cell response, leading to a better clinical response. Targeting mitochondrial metabolism also proved effective in decreasing pathology in a model of SLE [32]. Metformin, in addition to promoting FAO, is an inhibitor of Complex I of the ETC. In this study, metformin along with 2-DG led to a decrease in the activation of T cells leading to disease. Recall our group found that adding metformin to DON and 2-DG was the most effective means of inhibiting allograft rejection [24]. Indeed, as monotherapy, metformin was an ineffective inhibitor of effector T cell function. We interpret these observations to suggest that in the setting of compromised glycolysis (by 2-DG) or glutamine metabolism (by DON), the effectiveness of blocking ETC with metformin is markedly increased.
Long-lived memory cells rely upon OXPHOS and FAO for fuel. Since this metabolic program is strongly dependent on the cell's mitochondria, it is not surprising that the abundance and the organization of the mitochondria are instrumental in generating proper memory cells [41,42]. To this end, the importance of mitochondrial morphology, modulated through mitochondrial fusion and fission, in influencing T cell differentiation by dynamically altering the metabolic program has recently been described [43]. By culturing cells with a “fusion promoter” and “fission inhibitor”, CD8+ T cells adopt a memory phenotype, characterized by more FAO and OXPHOS. To improve the efficacy of ACT, these observations were exploited by generating antigen specific CD8+ T cells in effector promoting conditions with IL-2 and with a “fusion promoter” and a “fission inhibitor”. These metabolically enhanced CD8+ T cells in ACT improved the anti-tumor response due to greater effector function.
Not surprisingly, it has been observed that mitochondrial dysfunction leads to poor immune responses to tumors or to chronic viral infections [44,45]. As such, improving mitochondrial fitness presents an opportunity to enhance immunity.
PGC1α is a transcription factor that promotes mitochondrial biogenesis and function. In as much as PGC1α is regulated by Foxo1, enhanced Foxo1 activation (through the inhibition of mTORC2) can promote the generation of memory CD8+ T cells [18,46]. Pharmacologically or genetically enhancing PGC1α represents a potential strategy for enhancing the robustness of T cell responses. Likewise, it has been shown that enforced overexpression of PGC1α in a model of ACT, leads to more robust anti-tumor responses with better overall survival [44]. The increase in mitochondrial biogenesis with enforced PGC1α resulted in improved metabolic fitness and effector cytokine function compared to control CD8+ T cells.
Conclusion
While metabolic programming and pathways can appear daunting, particularly to the immunologist, a simple paradigm is emerging. Figure 1, which is by no means comprehensive, illustrates some of the pathways and targets discussed in this review. First, metabolism is intimately linked to T cell activation, differentiation and function. Second, targeting metabolism can selectively enhance or inhibit specific T cell subsets. To this end, based on experimental observations we have put forth the concept of “Cellular selectivity based upon demand”. That is, metabolic inhibitors which ostensibly might affect all cells of the body, will demonstrate relative selectivity for the cells which have the most demand. For example, an inhibitor of glycolysis (all cells can employ glycolysis) will robustly and preferentially affect cells (i.e. effector T cells) that have the greatest glycolytic need. As a result, compounds that target metabolism are proving to have strong therapeutic indexes. As such, this regulation can be exploited to both enhance and inhibit T cell responses to treat autoimmunity, transplant rejection and enhance immunotherapy. Current immunosuppressive regimens typically contain calcineurin inhibitors and steroids. Unlike calcineurin inhibitors which block tolerance induction including the generation of Tregs, metabolic therapy provides a platform to promote tolerance. Likewise, unlike steroids and calcineurin inhibitors which cause adverse effects on systemic metabolism (high glucose or high triglycerides) metabolic therapy abrogates such effects. Also, unlike conventional immunotherapy which leads to enhanced reactivation of herpes viruses such as CMV, metabolic therapy can inhibit viral replication [47]. To this end, while the risk of developing cancer is increased by many current immunosuppressive regimens, metabolic therapy can actually be employed to treat cancer. Thus, the robustness of targeting metabolism in terms of regulating immune responses as well as a potentially vastly improved safety profile provides us with the opportunity to change treatment paradigms for autoimmunity, inflammation and transplant rejection alike.
An external file that holds a picture, illustration, etc.
Object name is nihms873626f1.jpg
Figure 1
Targeting metabolism to regulate T cell function. In as much as T cell activation, differentiation and function is intimately linked to metabolism, targeting metabolism is emerging as a novel means of regulating T cell responses. This figure (as with the text) is meant to be instructive rather than comprehensive. Red highlights metabolic inhibitors that have been successfully employed to modulate T cell function. In blue are potential therapeutic targets based on the role of metabolic programs as discussed.
Highlights
Metabolic reprogramming is intimately linked to T cell activation
Different T cell subsets have different metabolic demands
Selectivity of targeting metabolism is achieved based on cellular demand
Targeting metabolism can both inhibit and enhance T cell function
Targeting metabolism represents a novel means to treat autoimmunity, transplant rejection and enhance immunotherapy
Go to:
Acknowledgments
Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5554728/
.png)
.png)
.png)