°°
Roles of NF-kB in development of Cancer
°°
chemotherapy-induced NF-¶ B activation was dependent on ATM-TRAF6 signaling, and NF-¶ B transcription factor p65 directly regulated the cytokines expression. …œ∫£Ωª¥Û
Degradation of IKK-¶¬ kinase consequent activation of the mTOR pathway;
Activation of the I¶ B kinase (IKK) complex is a prerequisite for NF-¶ B activation.
Akt-mediated regulation of NF¶ B and the essentialness of NF¶ B for the oncogenicity of PI3K and Akt
°°
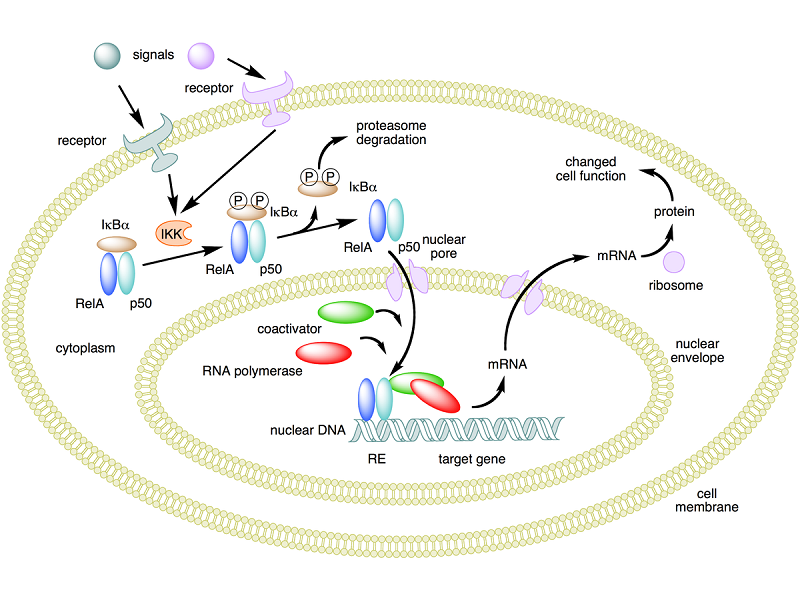
NF-¶ B◊˜”√ª˙¿Ì°£‘⁄∏√Õº÷–£¨“‘Rel∫Õp50µ∞∞◊◊È≥…µƒNF-¶ B“Ï∂˛æ€ÃÂŒ™¿˝°£¥¶”⁄√ªÓ◊¥Ã¨ ±£¨NF-¶ BŒª”⁄”Γ÷÷∆µ∞∞◊I¶ B¶¡∏¥∫œµƒ∞˚÷ »ÐΩ∫÷–°£Õ®π˝’˚∫œƒ§ ÐõƒΩȵº£¨∂ý÷÷œ∏∞˚Õ‚–≈∫≈ø…“‘º§ªÓ√∏I¶ Bº§√∏£®IKK£©°£ IKK∑¥π˝¿¥ª·¡◊À·ªØI¶ B¶¡µ∞∞◊£¨¥”∂¯µº÷¬∑∫ÀÿªØ£¨I¶ B¶¡”ÎNF-¶ Bµƒ∑÷¿Î“‘º∞µ∞∞◊√∏ÃÂ◊Ó÷’ΩµΩ‚I¶ B¶¡µƒ◊˜”√°£»ª∫Û£¨ªÓªØµƒNF-¶ B“◊ŒªµΩœ∏∞˚∫À÷–£¨≤¢”Î≥∆Œ™∑¥”¶‘™º˛£®RE£©µƒÃÿ∂®DNA–Ú¡–Ω·∫œ°£»ª∫Û£¨DNA /NF-¶ B∏¥∫œŒÔƒººØ∆‰À˚µ∞∞◊£¨¿˝»Áπ≤º§ªÓ“Ú◊”∫ÕRNAæ€∫œ√∏£¨’‚–©µ∞∞◊Ω´œ¬”ŒDNA◊™¬º≥…mRNA°£∑¥π˝¿¥£¨mRNA±ª∑≠“Î≥…µ∞∞◊÷ £¨µº÷¬œ∏∞˚π¶ƒÐµƒ∏ƒ±‰°£
NF-¶ B - Wikipedia
https://en.wikipedia.org/wiki/NF-%CE%BAB°°
°°
°°
°°
NF-¶ B, inflammation, immunity and cancer: coming of age | Nature Reviews Immunology
https://www.nature.com/articles/nri.2017.142°°
NF-¶ B addiction and its role in cancer: °Æone size does not fit all°Ø | Oncogene
https://www.nature.com/articles/onc2010566°°
°°
Different roles of Nrf2 and NFKB in the antioxidant ...
https://www.sciencedirect.com/science/article/pii/S0009279718306100
Oct 01, 2018 °§ NFkB is a critical survival factor in lymphopoiesis [10,11]. In several hematologic diseases, constitutive activation of NFkB contributes to abnormal proliferation and survival of transformed cells . NF-kB is sensitive to intracellular redox state and it is modulated by RNS and NOS .
Cited by: 1
Publish Year: 2018
Author: Virginia Rubio, Ana I. Garc®™a-P®¶rez, Angel Herr®¢ez, Jos®¶ C. Diez
Novel Targeted Nano-Parthenolide Molecule against NF-kB in ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6600366
Jun 03, 2019 °§ Nuclear factor kappa B (NF-kB) is a regulator for different biological responses, including cell proliferation and differentiation. In acute myeloid leukemia (AML), constitutive NF-¶ B has been detected in more than 50% of cases, enabling leukemic cells to resist apoptosis and stimulate uncontrolled proliferation.
Cited by: 1
Publish Year: 2019
Author: Noureldien H. E. Darwish, Thangirala Sudha, Kavitha Godugu, Dhruba J. Bharali, Osama Elbaz, Hasan A....
Role of NF-¶ B inhibitor in Acute Myeloid Leukemia
https://www.researchgate.net/publication/322721588_Role_of_NF-kB_inhibitor_in_Acute...
Objective: To investigate the role of NF-¶ B inhibitor in occurence and development of AML. Methods: AML and normal bone marrow samples were collected from 8 AML patients and 8 no
Roles of NF-¶ B in Cancer and Inflammatory Diseases and ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4931664
Mar 29, 2016 °§ Nuclear factor-¶ B (NF-¶ B) is a transcription factor that plays a crucial role in various biological processes, including immune response, inflammation, cell growth and survival, and development. NF-¶ B is critical for human health, and aberrant NF-¶ B activation contributes to development of ...
Cited by: 146
Publish Year: 2016
Author: Mi Hee Park, Jin Tae Hong
Role of NF-kB in hematopoietic stem cells and leukemia ...
grantome.com/grant/NIH/R01-CA144248-05
Leukemia-initiating cells (LIC) in acute myeloid leukemia (AML) represent a small fraction of the total tumor that can re-initiate AML upon transfer to secondary irradiated mice. LIC can be distinguished from normal hematopoietic stem/progenitor cells in bone marrow based on constitutive activation of the Akt and classical NF-?B signaling pathways in LIC.
The role of NF-¶ B in lymphoid malignancies | Request PDF
https://www.researchgate.net/publication/5283568_The_role_of_NF-kB_in_lymphoid...
The transcription factor nuclear factor-¶ B (NF-kB) was discovered 30 years ago and plays an important role in carcinogenesis, cell growth, apoptosis and in immune and inflammatory responses.
NF-kB in development and progression of human cancer ...
https://link.springer.com/article/10.1007/s00428-005-1264-9
Apr 27, 2005 °§ NF-kB plays a well-known function in the regulation of immune responses and inflammation, but growing evidences support a major role in oncogenesis. NF-kB regulates the expression of genes involved in many processes that play a key role in the development and progression of cancer such as proliferation, migration and apoptosis.
Cited by: 804
Publish Year: 2005
Author: Xavier Dolcet, David Llobet, Judit Pallares, Xavier M°°
Molecular Targets of Curcumin and Future Therapeutic Role in Leukemia
°°Scientific Research, Vol.6 No.4, April 2018
Abstract Full-Text HTML XML Download Download as PDF (Size:4217KB) PP. 33-50
DOI: 10.4236/jbm.2018.64003 444 Downloads 1,251 Views Citations
Author(s) Leave a comment Sabika Rafiq1, Muhammad Hassan Raza2, Mehwish Younas1, Fariha Naeem1, Romisha Adeeb1, Javed Iqbal1, Pervez Anwar1, Umara Sajid1, Hafiza Muniba Manzoor3 Affiliation(s)
1Department of Biotechnology, University of Gujrat Sialkot Subcampus, Sialkot, Pakistan.
2Department of Biotechnology & Bioinformatics, International Islamic University, Islamabad, Pakistan.
3Department of Biotechnology, University of Health Sciences Lahore, Lahore, Pakistan.
ABSTRACT
Leukemia is a most prevalent type of cancer around the globe. Due to major side effects of Chemotherapy and radiotherapy, scientists worked to explore the alternative source to treat leukemia. An alternative source for the treatment of leukemia existed in the form of curcumin, a natural phenolic compound extracted from curcuma longa plant. It exhibited anticancer properties reducing the tumor load via apoptosis and cell cycle arrested in various cancer cell lines and controlled tumor proliferation by blocking tumor inducing gene such as FLT3, Akt gene, ROS and NF-¶ B inhibition. At molecular level, curcumin plays a key therapeutic role in protection of normal cells by up regulation of NRF-2 that induces production of cellular antioxidants. It regulates various signaling pathways including NF-KB, JAK/STAT, PI3K/AKT and JNK pathways, thereby affecting cancer initiation, progression, and metastasis. This review described the potential of curcumin for treatment of leukemia; it affects different signaling cascades and their regulation. This study provides a preclinical foundation for future usage of curcumin in the treatment of cancer.
Molecular Targets of Curcumin and Future Therapeutic Role in Leukemia
https://www.scirp.org/Journal/PaperInformation.aspx?PaperID=83874°°
Published: 22 January 2018
NF-¶ B, inflammation, immunity and cancer: coming of age
Koji Taniguchi & Michael KarinDepartment of Microbiology and Immunology, Keio University School of Medicine, 35 Shinanomachi, Shinjyuku-ku, 160®C8582, Tokyo, Japan
Department of Surgery and Science, Graduate School of Medical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, 812®C8582, Fukuoka, Japan
Departments of Pharmacology and Pathology, Laboratory of Gene Regulation and Signal Transduction, School of Medicine, University of California, San Diego, 9500 Gilman Drive, La Jolla, 92093, California, USA
Nature Reviews Immunology volume 18, pages309®C324(2018)Cite this article
Key Points
Inflammation has been recognized as a hallmark of cancer and is known to play an essential role in the development and progression of most cancers, even those without obvious signs of inflammation and infection.
Nuclear factor-¶ B (NF-¶ B), a transcription factor that is essential for inflammatory responses, is one of the most important molecules linking chronic inflammation to cancer, and its activity is tightly regulated by several mechanisms.
Activation of NF-¶ B is primarily initiated by bacterial endotoxins such as lipopolysaccharide and pro-inflammatory cytokines such as tumour necrosis factor and IL-1. NF-¶ B activation occurs in cancer cells and in the tumour microenvironments of most solid cancers and haematopoietic malignancies.
NF-¶ B activation induces various target genes, such as pro-proliferative and anti-apoptotic genes, and NF-¶ B signalling crosstalk affects many signalling pathways, including those involving STAT3, AP1, interferon regulatory factors, NRF2, Notch, WNT®C¶¬-catenin and p53.
All known hallmarks of cancer involve NF-¶ B activation. In addition to enhancing cancer cell proliferation and survival, NF-¶ B and inflammation promote genetic and epigenetic alterations, cellular metabolic changes, the acquisition of cancer stem cell properties, epithelial-to-mesenchymal transition, invasion, angiogenesis, metastasis, therapy resistance and the suppression of antitumour immunity.
The prevalence of NF-¶ B activation in cancer-related inflammation makes it an attractive therapeutic target with the potential for minimal side effects.
Abstract
Fourteen years have passed since nuclear factor-¶ B (NF-¶ B) was first shown to serve as a molecular lynchpin that links persistent infections and chronic inflammation to increased cancer risk. The young field of inflammation and cancer has now come of age, and inflammation has been recognized by the broad cancer research community as a hallmark and cause of cancer. Here, we discuss how the initial discovery of a role for NF-¶ B in linking inflammation and cancer led to an improved understanding of tumour-elicited inflammation and its effects on anticancer immunity.
°°NF-¶ B, inflammation, immunity and cancer: coming of age | Nature Reviews Immunology
https://www.nature.com/articles/nri.2017.142°°
The Notch/Hes1 Pathway Sustains NF-¶ B Activation through CYLD Repression in T Cell Leukemia
Cancer Cell, Sept 14, 2010Program of Cellular and Molecular Medicine, Children's Hospital, and Immune Disease Institute, Harvard Medical School, Boston, MA 02115, USA
Summary
It was previously shown that the NF-¶ B pathway is downstream of oncogenic Notch1 in T cell acute lymphoblastic leukemia (T-ALL). Here, we visualize Notch-induced NF-¶ B activation using both human T-ALL cell lines and animal models. We demonstrate that Hes1, a canonical Notch target and transcriptional repressor, is responsible for sustaining IKK activation in T-ALL. Hes1 exerts its effects by repressing the deubiquitinase CYLD, a negative IKK complex regulator. CYLD expression was found to be significantly suppressed in primary T-ALL. Finally, we demonstrate that IKK inhibition is a promising option for the targeted therapy of T-ALL as specific suppression of IKK expression and function affected both the survival of human T-ALL cells and the maintenance of the disease in vivo.
Highlights
NF-¶ B activity is active and essential in established Notch-dependent T-ALL leukemias
Notch through Hes1 sustains NF-¶ B activity by repressing the deubiquitinase CYLD
Inhibition of IKK/NF¶ B activity reduces leukemic load and increases survival in mice
Significance
Although aberrant activation of the Notch pathway is a recurring event in T-ALL, the downstream signaling effects of this activation are poorly defined. Our work demonstrates that the Notch-Hes1-CYLD-IKK axis plays a crucial role in this type of leukemia, and that blocking NF-¶ B (or the IKK kinase) is sufficient to eliminate leukemic cells carrying activating Notch mutations. These findings suggest that NF-¶ B targeting could be a key component of successful future T-ALL clinical trials.
Introduction
NF-¶ B is an important regulator of cell survival, proliferation and differentiation and is frequently involved in malignant transformation (Karin, 2006). NF-¶ B is mainly activated in response to proinflammatory stimuli such as TNF¶¡ or IL1¶¬, components of pathogenic bacteria and viruses, or after TCR signaling in T-lymphocytes. Regardless of the activation of specific receptors that associate to different effectors, canonical NF-¶ B signaling invariably leads to the activation of I¶ B kinase (IKK) complex composed by two catalytic and one regulatory subunits called IKK¶¡, IKK¶¬, and NEMO (IKK¶√), respectively. The main target of activated IKK complex is the NF-¶ B inhibitor, I¶ B, that it is rapidly phosphorylated in specific serine residues thus targeting the protein for ubiquitination, leading to proteasomal degradation. Degradation of I¶ B is sufficient to induce the nuclear translocation of the transcription factor that results in specific gene activation (Vallabhapurapu and Karin, 2009). I¶ B¶¡ itself is one of the main transcriptional targets for NF-¶ B activity thus providing an efficient mechanism for signal termination (Hoffmann et al., 2002). However, other mechanisms are required to maintain the nonactive state of NF-¶ B in the absence of stimulation but also to attenuate the signal after chronic stimulation of the pathway. They involve the deubiquitination enzymes A20 and CYLD, which remove the activator K63 ubiquitin chains from different elements of the NF-¶ B signalosome (Brummelkamp et al., 2003, Reiley et al., 2007, Trompouki et al., 2003, Wertz et al., 2004).
Despite the huge amount of gathered data on NF-¶ B activation, not much is known about the crosstalk of NF-¶ B with other signaling pathways, including pathways that control tissue differentiation like the Notch cascade. Notch receptors are crucial regulators of cell differentiation in multiple systems (reviewed by Egan et al., 1998). Activation of Notch by Jagged or Delta ligands results in the cleavage of the intracellular fragment of the receptor by a ¶√-secretase activity (Mumm et al., 2000). Once released from the extracellular part of the molecule, the intracellular cleaved Notch translocates to the nucleus to bind the ubiquitous RBPj¶ factor and to activate transcription of a number of target genes, including the Hes family of transcription factors (Iso et al., 2003, Jarriault et al., 1995). The classical function of Hes family proteins is the transcriptional repression of prodifferentiation genes by direct interaction with specific sequences of their promoters or by the association with other bHLH factors and combinatorial DNA binding (Kageyama and Ohtsuka, 1999, Kuroda et al., 1999). The Notch pathway was recently shown to be oncogenic in a wide variety of cancers, most notably in T cell acute lymphoblastic leukemia (T-ALL), a devastating pediatric tumor arising by the transformation of hematopoietic progenitor cells. In this disease, the majority of patients harbor either activating mutations of Notch1 or inactivating mutations affecting negative regulators of the Notch pathway (Aifantis et al., 2008).
Recent evidence has suggested an interaction between Notch and NF-¶ B, more specifically in the context of cell transformation. Crosstalk between the Notch and NF-¶ B pathways have been proposed in human T-ALL and in mouse models of T cell leukemia (Screpanti et al., 2003, Shin et al., 2006, Vilimas et al., 2007). More specifically, we have previously shown that Notch activation can induce the expression of a large fraction of classical NF-¶ B gene targets in T cell progenitors. We have also demonstrated that this Notch-induced NF-¶ B activation requires both the transcriptional activity of Notch and the function of the IKK complex (Vilimas et al., 2007). However, these studies failed to provide a definitive demonstration that this interaction can be translated into a new therapeutic tool. Moreover, neither the mechanism explaining the ability of Notch to impinge on IKK (and thus NF-¶ B) activation nor the importance of IKK activation for the maintenance of T-ALL in vivo was addressed. These key issues could introduce NF-¶ B inhibition as an effective therapy for the treatment of T-ALL and are all addressed here.The Notch/Hes1 Pathway Sustains NF-¶ B Activation through CYLD Repression in T Cell Leukemia: Cancer Cell
https://www.cell.com/cancer-cell/fulltext/S1535-6108(10)00302-8#%20°°
Published: 20 December 2010
NF-¶ B addiction and its role in cancer: °Æone size does not fit all°Ø
M M Chaturvedi, B Sung, V R Yadav, R Kannappan & B B AggarwalDepartment of Experimental Therapeutics, Cytokine Research Laboratory, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Oncogene volume 30, pages1615®C1630(2011)Cite this article
Abstract
Activation of nuclear factor (NF)-¶ B, one of the most investigated transcription factors, has been found to control multiple cellular processes in cancer including inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance and radioresistance. NF-¶ B is constitutively active in most tumor cells, and its suppression inhibits the growth of tumor cells, leading to the concept of °ÆNF-¶ B addiction°Ø in cancer cells. Why NF-¶ B is constitutively and persistently active in cancer cells is not fully understood, but multiple mechanisms have been delineated including agents that activate NF-¶ B (such as viruses, viral proteins, bacteria and cytokines), signaling intermediates (such as mutant receptors, overexpression of kinases, mutant oncoproteins, degradation of I¶ B¶¡, histone deacetylase, overexpression of transglutaminase and iNOS) and cross talk between NF-¶ B and other transcription factors (such as STAT3, HIF-1¶¡, AP1, SP, p53, PPAR¶√, ¶¬-catenin, AR, GR and ER). As NF-¶ B is °Æpre-active°Ø in cancer cells through unrelated mechanisms, classic inhibitors of NF-¶ B (for example, bortezomib) are unlikely to mediate their anticancer effects through suppression of NF-¶ B. This review discusses multiple mechanisms of NF-¶ B activation and their regulation by multitargeted agents in contrast to monotargeted agents, thus °Æone size does not fit all°Ø cancers.
Introduction
In the previous century, many research initiatives were focused on understanding the genetic etiology of human tumorigenesis. However, cancer remains a major health problem and is responsible for one in eight deaths worldwide (Garcia et al., 2007). Also, an estimated 55% increase in cancer incidence is expected by the year 2020 (Warren et al., 2008).
Cancer is an extremely complex disease (Roukos, 2010). Large-scale analysis of genes has shown that the types of mutations that occur in various cancers are highly heterogeneous (Hudson et al., 2010). Only a minority of cancers are caused by germline mutations, whereas the vast majority (∼90%) are linked to somatic mutations and environmental factors (Aggarwal et al., 2009). The mutations found in the cancer cell genome accumulate over the lifetime of the cancer patient. Mutation rate increases in the presence of substantial exogenous mutagenic exposures, such as tobacco smoke carcinogens, naturally occurring toxic chemicals present in some foods or various forms of radiation including ultraviolet light. These exposures are associated with increased rates of lung, liver and skin cancer, respectively (Grivennikov et al., 2010). Moreover, these agents induce inflammatory responses, suggesting a strong association between inflammation, nuclear factor (NF)-¶ B and cancer (Mantovani et al., 2008; Aggarwal and Gehlot, 2009; Grivennikov et al., 2010).
Currently, 18 485 genes and 464 139 tumors have been sequenced, resulting in a cataloge of 20 090 unique mutations, which have been curated in the Catalogue of Somatic Mutations in Cancer (http://www.sanger.ac.uk/cosmic, v47 release, 24 May, 2010). Studies of the genetic complexities of breast, colorectal, pancreatic and brain cancers have revealed that cancer genomes are highly complex (Bignell et al., 2010; Ding et al., 2010; Pleasance et al., 2010a), with a range of 48®C101 somatic alterations in each tumor, depending on the cancer type (Jones et al., 2008; Parsons et al., 2008; Bell, 2010). Within a given cancer type, there is considerable intertumor heterogeneity, resulting in large number of altered genes. However, this complexity is reduced significantly by considering the biological pathways, rather than the altered gene themselves (Wood et al., 2007; Verhaak et al., 2010). For example, 12 core biological processes or pathways appear to be deregulated in most pancreatic tumors, although precisely how this deregulation is achieved varies from tumor to tumor (Jones et al., 2008). This suggests alterations in the complex network of signaling pathways (Ledford, 2010), and has very clear practical implications for the development of targeted therapeutics, as it is less likely that a drug targeting just one mutated gene or one particular pathway alone could be effective for treating any type of cancer.
The area of research involving NF-¶ B has grown tremendously in the past decade. This is evident from the fact that although NF-¶ B was discovered only 25 years ago (Sen and Baltimore, 1986) and is one of ∼2000 estimated transcription factors in humans (Lander et al., 2001; GuhaThakurta, 2006), ∼10% of research articles listed in PubMed on the subject of transcription factors are associated with NF-¶ B. Furthermore, of the more than 39 000 articles published about NF-¶ B, about 19 000 are associated with tumors and cancers underscoring the importance of this transcription factor in cancer studies.
As NF-¶ B is the key transcription factor involved in the inflammatory pathway, NF-¶ B is constitutively active in most cancers (Table 1), and many of the signaling pathways implicated in cancer are likely to be networked to the activation of NF-¶ B (Figure 1) (Karin, 2009; Grivennikov et al., 2010; Grivennikov and Karin, 2010). Mammalian NF-¶ B is a family of transcription factors that includes five members: RelA/p65, c-Rel, RelB, NF-¶ B1 (p50) and NF-¶ B2 (p52) (Ghosh and Karin, 2002; Vallabhapurapu and Karin, 2009). The primary regulation of the NF-¶ B pathway is through the association of NF-¶ B complexes with their inhibitor, I¶ B proteins. There are multiple human I¶ B proteins, including I¶ B¶¡, I¶ B¶¬, I¶ Bɛ and I¶ B¶∆. In addition, the precursors p50 and p52 and the full-length proteins p105 and p100 also function as I¶ B proteins. The principle inactive form of the NF-¶ B complex is a p50®Cp65 (RelA)®CI¶ B¶¡ trimer, primarily located in the cytoplasm. In the classic or canonical pathway, in response to various external stimuli, I¶ B¶¡ is phosphorylated at Ser 32 and Ser 36 by the I¶ B¶¡ kinase (IKK). This promotes K-48 ubiquitination of I¶ B¶¡ by the SCF®C¶¬TrCP complex and its degradation by the proteasome. The released NF-¶ B dimer (p50®Cp65, which is also phosphorylated by IKK) is translocated in the nucleus, where it binds to its cognate response elements in promoters to activate the transcription of responsive genes (Vallabhapurapu and Karin, 2009). At the NF-¶ B responsive promoters, the p65 subunit of NF-¶ B is further modified by acetylation and methylation, and it interacts with additional coactivators (Werner et al., 2005). A second, non-canonical or alternative pathway involves activation of the p100®CRelB complex to p52®CRelB in response to specific extracellular signals (Senftleben et al., 2001). Unlike its response to I¶ B¶¡, p100, after phosphorylation at Ser 866 and Ser 870, undergoes limited processing to generate p52, also regulated by SUMOylation (Vatsyayan et al., 2008)......
NF-¶ B mutation and cancer
The p65 transactivator subunit of NF-¶ B, RelA, was recognized earlier as the potential oncogene (Gilmore, 2003). However, the mutations that confer Rel-A, c-Rel, or other NF-¶ B proteins with oncogenic potentialities are not abundant and are mainly limited to lymphoid malignancies. REL gene amplifications have been detected frequently in many types of human B-cell, and to a lesser extent T cell, lymphoma (reviewed in (Gilmore et al., 2004; Courtois and Gilmore, 2006)). Chronic lymphocytic leukemia, the most common adult leukemia, is currently incurable with conventional chemotherapeutic agents. Rel-A is a prognostic marker and a therapeutic target in this disease (Pepper et al., 2009; Lopez-Guerra and Colomer, 2010).
Similarly, the NFKB2 gene undergoes structural alterations in certain T-cell lymphomas, chronic lymphocytic leukemias, myelomas and B-cell lymphomas (Courtois and Gilmore, 2006). In Hodgkin's lymphoma, mutations or deletions in the I¶ B¶¡ gene have been described (Cabannes et al., 1999). The Bcl-3 gene, another family member of I¶ B¶¡, is overexpressed and translocated in B-cell leukemia (Martin-Subero et al., 2007; Chapiro et al., 2008). Similarly, amplification of the c-Rel gene is reported in several types of B-cell lymphoma (Pileri et al., 2003). Mutations in other NF-¶ B genes, such as NEMO, result more frequently in immunological disorders (Courtois and Gilmore, 2006).
In lymphoid malignancies, the dysregulation of NF-¶ B activation results in aberrant expression of target gene proteins such as cyclin D1, cyclin D2, c-myc, c-myb, BCL2 and BCL-XL that regulate cell proliferation or survival, as well as cytokines such as interleukin (IL)-2, IL-6 and CD40-L that regulate growth and proliferation of lymphocytes. Thus, constitutively active NF-¶ B has been implicated in various lymphoid malignancies (Aggarwal and Gehlot, 2009). Acute myeloid leukemia involves the activation of RTK Flt3, N-Ras and K-Ras, which activate NF-¶ B through the Akt pathway (Stirewalt and Radich, 2003; Tenen, 2003). As can be seen in Table 1, although NF-¶ B genes are not directly mutated in most cases, mutations in key genes such as RAS, phosphatidylinositol 3-kinase (PI3K)/Akt1, TP53 and EGFR affect the cellular processes that are known to involve the activation of NF-¶ B in some capacity (Grivennikov et al., 2010).°°
Multiple pathways of activation of NF-¶ B and targeting NF-¶ B for cancer prevention and therapy
As alluded to in previous sections, although the NF-¶ B pathway constitutes the central pathway regulating inflammation and cancer, multiple signaling pathways are operating at any given time in any given cancer (for an elaborate review, refer to Staudt (2010)). First, the NF-¶ B pathway is activated by diverse stimuli that are networked and regulated by many other pathways (for example, EGFR/HER2-PI3K-Akt-IKK¶¡, TP53, PTEN, Akt-mTOR, G-protein-coupled receptor-RAS-RAF-Akt and Wnt®C¶¬-catenin), including canonical and non-canonical pathways (Figure 1). Another altogether different mechanism was shown for the activation of I¶ B¶¡ involving tyrosine phosphorylation (Imbert et al., 1996; Singh et al., 1996), brought about by syk kinase induced by H2O2 (Takada et al., 2003). In another study, EGFR tyrosine kinase activity induced Tyr 42 phosphorylation of I¶ B¶¡ (Sethi et al., 2007). H2O2-induced Tyr phosphorylation does not require degradation of I¶ B¶¡ for activation of NF-¶ B (Imbert et al., 1996; Tang et al., 2006); however, pervanadate-induced Tyr-phosphorylated I¶ B¶¡ does undergo degradation (Mukhopadhyay et al., 2000). Of interest, hypoxia-induced activation of NF-¶ B in fetal lung fibroblasts also involved phosphorylation of I¶ B¶¡ at Tyr 42 residues (Wright et al., 2009). Therefore, it is likely that a hypoxic tumor environment may activate NF-¶ B through such a mechanism.
Second, the activated NF-¶ B (p50®Cp65) cooperates and interacts with many other transcription factors to form on and off promoter complexes, such as STAT3 (Grivennikov and Karin, 2010), SP1(Perkins et al., 1993) HIF-1¶¡ (Scortegagna et al., 2008) and so on (Figure 1). Because of the networking of the pathways inhibiting one step or one the pathway, activates the alternative route of activation. This can be exemplified by the fact that as I¶ B¶¡ is degraded by proteasomal pathway, the proteasomal inhibitors are developed to inhibit the NF-¶ B pathway. Velcade (bortezomib) is such a drug already approved by Food and Drug Administration for the treatment of multiple myeloma. However, recent reports indicate that velcade now induces NF-¶ B, instead of inhibiting as was thought by inducing calpane (Li et al., 2010) and caspase-dependent (Hideshima et al., 2009b) mechanisms. In a similar way, IKK¶¬ inhibitor MLN120B blocks the canonical pathway and growth of MM cell lines but does not inhibit the non-canonical NF-¶ B pathway (Hideshima et al., 2009a). Therefore, inhibitors that block more than one step in the pathway or inhibit multiple pathways would be more effective for the treatment of cancer. Figure 4 shows cancer drugs approved by Food and Drug Administration, although not conceived as NF-¶ B inhibitors, these drugs can suppress NF-¶ B activation. For instance, EGFR kinase inhibitors have been shown to suppress NF-¶ B activation induced by epidermal growth factor (Sethi et al, 2007).
°°
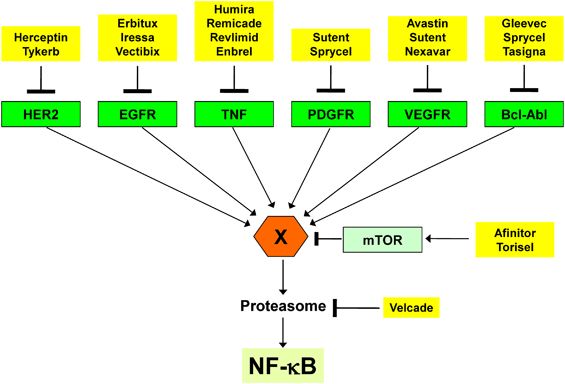
Suppression of NF-kB activation by the Food and Drug Administration-approved drugs for cancer therapy. Molecular targets for many Food and Drug Administration-approved drugs for treatment of cancer are shown. Although these drugs act through their defined molecular targets, they inhibit NF-¶ B via pathway(s) that are not well defined. The X indicates an intermediate that could be either IKK or its upstream activators, which may be different for different pathways. EGFR, epidermal growth factor receptor; mTOR, mammalian target of rapamycin; PDGF, platelet-derived growth factor receptor; TNF, tumor necrosis factor; VEGFR, vascular endothelial cell growth factor receptor.
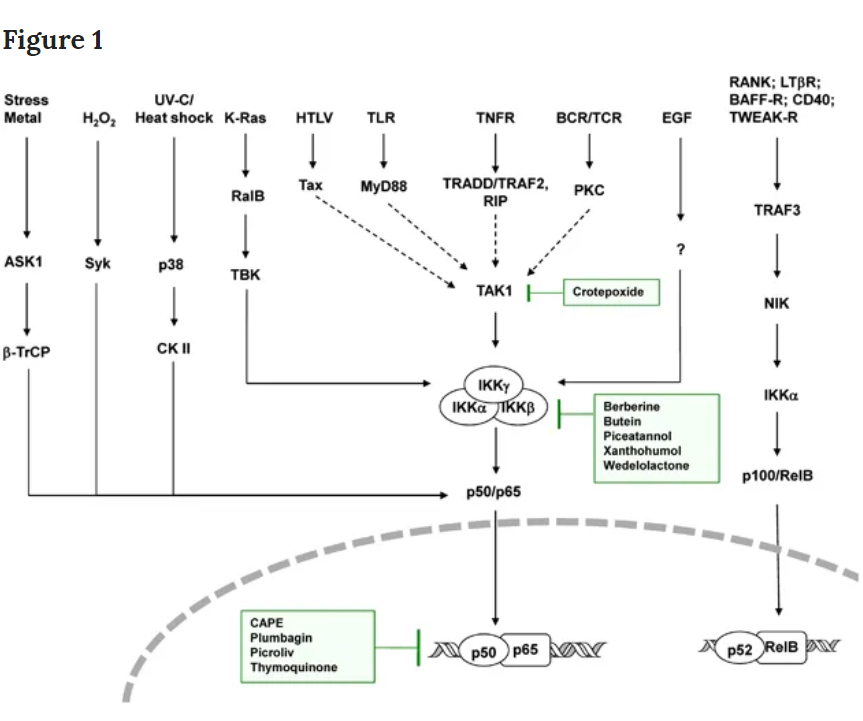
Signaling network of NF-¶ B activation in cancer. Various pathways of NF-¶ B activation in cancers are shown. The sites of action of some phytochemicals are also indicated in the boxes. The network converges at three major sites (IKK kinase such as TAK1, IKK itself and the p50®Cp65 heterodimer). ASK1, apoptosis signal-regulating kinase 1; BAFF-R, TNF family member B cell-activating factor-receptor; CK II, casein kinase II; HTLV, human T-lymphotropic virus; LT¶¬R, lymphotoxin-¶¬ receptor; MyD88, myeloid differentiation primary response gene 88; RalB, RAS-like protein B; RANK, receptor activator for nuclear factor ¶ B; RIP, receptor interacting protein; Syk, spleen tyrosine kinase; TAK1, transforming growth factor (TGF)-¶¬ activating kinase 1; TBK, TRAF family member-associated NF-¶ B activator (TANK)-binding kinase; TLR, toll-like receptor; TNFR, tumor necrosis factor receptor; TRADD, TNFR1-associated death domain; TRAF, TNF-receptor-associated factor; ¶¬-TrCP, ¶¬-transducin repeat-containing protein; TWEAK-R, TNF-related weak inducer of apoptosis-receptor; UV, ultra violet.
Almost 15 years ago, we showed that nutraceuticals such as curcumin effectively inhibits TNF-¶¡-induced activation of NF-¶ B through inhibition of I¶ B¶¡ phosphorylation (Singh and Aggarwal, 1995). Subsequently, curcumin was shown to inhibit not only IKK kinase activity but also p65 phosphorylation at serine 536, and p65 acetylation (Aggarwal et al., 2006); and p300®CHAT (Balasubramanyam et al., 2004). Like curcumin, anacardic acid was found to suppress NF-¶ B activation by inhibition of HAT (Sung et al., 2008). Interestingly, several nutraceuticals such as berberine, butein, piceatannol, xanthohumol and wedelolactone, have been shown to directly bind to IKK¶¬ through cysteine 179 (Kobori et al., 2004; Pandey et al., 2007, 2008; Harikumar et al., 2009; Son et al., 2010). Other nutraceuticals such as picroliv, thymoquinone, xanthohumol and plumbagin were found to inhibit NF-¶ B activity through directly binding to Cys 38 in p65 (Anand et al., 2008; Sethi et al., 2008; Harikumar et al., 2009). Sesquiterpene lactones were also found to suppress NF-¶ B through direct interaction with Cys 38 in p65 (Garcia-Pineres et al., 2001). Recently, we showed that crotepoxide inhibits the NF-¶ B pathway by inhibiting TAK1 directly, thus chemosensitizing tumor cells through inhibition of the proinflammatory pathway (Prasad et al., 2010). Thus, it is clear that various nutraceuticals can suppress NF-¶ B-mediated inflammatory pathways and this may be linked to their chemopreventive potential.
Conclusions
Extensive sequence-based analysis of the genome of various cancers has revealed that the driver mutations in a wide variety of genes fall into pathways of multiple signal transduction networks. Therefore, it is less likely that targeting just one mutated gene or one particular pathway could be effective alone for treating any type of cancer. Of interest, the inflammatory linkage and constitutive activation of NF-¶ B has emerged as one of the attractive targets for intervention and treatment of cancer. However, NF-¶ B itself is activated by diverse stimuli and by highly networked pathways, suggesting the need for a multitargeted approach. Nutraceuticals derived from fruits, vegetables and spices that target multiple steps in the NF-¶ B pathways are emerging as promising agents for the prevention and treatment of cancers.NF-¶ B addiction and its role in cancer: °Æone size does not fit all°Ø | Oncogene
https://www.nature.com/articles/onc2010566°°
°°
—◊÷¢«˝∂صƒ–° Û∞◊—™≤°ƒ£–Õ
Role of NF-¶ B inhibitor in Acute Myeloid Leukemia
÷–π˙“Ω—ß“Ω—ß‘∫ –≠∫Õ“Ω—ß‘∫—™“∫≤°—–æøÀ˘ —™“∫≤°“Ω‘∫
÷–π˙ µ—È—™“∫—ß‘”÷æ 2016
Objective: To investigate the role of NF-¶ B inhibitor in occurence and development of AML.Methods: AML and normal bone marrow samples were collected from 8 AML patients and 8 normal persons. The expression of NF-¶ B signaling pathway genes was detected by NF-¶ B PCR array. Then, AML mouse model was constructed to test the role of NF-¶ B inhibitor in AML.
Results: The NF-¶ B signal pathway was activated in AML patients. The up-regulated genes, EDARADD, TNFSF14, could activate the NF-¶ B signal pathway, IL6 could regulate the inflammatory signal. The down-regulated genes, TNFRSF 10B, TNFRSF1A, could lead to cell apoptosis. the AML mouse model was constructed successfully. Then administration of NF-¶ B inhibitor reduced the inhibition of leukemia niche to the normal hematopoietic stem cells (HSCs), promoted the HSC to enter into cell cycle.
Conclusion: The NF-¶ B signal pathway is activated in AML cells. AML mouse model is constructed successfully. NF-¶ B inhibitor has a potential to treat AML and promotes the HSC to enrter into cell cycle.
(PDF) Role of NF-¶ B inhibitor in Acute Myeloid Leukemia
https://www.researchgate.net/publication/322721588_Role_of_NF-kB_inhibitor_in_Acute_Myeloid_Leukemia°°
Role of NF-kB in hematopoietic stem cells and leukemia-initiating cell formation
Klug, Christopher A.
University of Alabama Birmingham, Birmingham, AL, United States
Constitutive activation of the classical NF-?B pathway, which principally involves RelA/p65, c-Rel, and p50, has been observed in human acute myeloid leukemia (AML) where constitutive NF-?B DNA-binding activity has been detected in myeloid blasts and in CD34? hematopoietic progenitor cells in 47-100% of all cases. In AML, constitutive NF-?B activity could be a direct downstream consequence of activating mutations affecting the PI3 kinase pathway, which is also constitutively active in the majority of AML cases and induces NF-?B through Akt signaling. In mice, constitutive activation of Akt through inducible deletion of the Akt regulatory phosphatase, PTEN, results in rapid hematopoietic stem cell (HSC) loss from bone marrow and development of a transplantable, lethal myeloproliferative disease (MPD) and AML. These studies and others have shown that constitutive activation of Akt and NF-?B distinguishes leukemia-initiating cells (LIC) from normal HSC in bone marrow. To address whether NF-?B activation in HSC is sufficient to induce LIC formation and promote MPD/AML, we expressed a constitutively active allele of I?B kinase beta (Ikk?SS/EE) in normal HSC using a retroviral vector. Analysis of mice reconstituted with Ikk?SS/EE -expressing cells showed that constitutive NF-?B activity promoted relatively rapid HSC loss from bone marrow that, unlike activated Akt, was not accompanied by the development of MPD/AML. NF-?B-mediated HSC loss was likely due to differentiation and loss of HSC self-renewal potential since there was not evidence of increased HSC apoptosis or mobilization to the spleen. Furthermore, blocking NF-?B activation in HSC by conditional deletion of the Ikk regulatory subunit, NEMO, resulted in a substantial decrease in one of the earliest multipotential progenitor subsets (c-Kit?Sca-1? cells) and an increase in absolute numbers of more primitive c-Kit?Sca-1? (KLSF) cells. NEMO loss also completely blocked proliferation of KLSF cells in vitro, which further supports an essential role for NF-?B in promoting the earliest HSC differentiation event. Together, these results demonstrate that constitutive activation of the classical NF-?B pathway, as an initiating event in AML, is not sufficient to maintain HSC or to promote development of LIC associated with AML. The primary goal of this proposal will be to test the central hypothesis that NF-?B functions in normal HSC to couple proliferation and differentiation associated with the first HSC cell division and to maintain viability of LIC subsequent to other oncogenic changes that preserve HSC self-renewal in the presence of activated NF-?B. To test this hypothesis, we will pursue the following specific aims: (1), Determine the function of NF-?B in the regulation of normal HSC maintenance, proliferation, and differentiation (2), Define the contribution of NF-?B to Akt-stimulated leukemogenesis, LT-HSC loss from bone marrow, and to leukemia-initiating cell formation and (3), Characterize whether recurrent cytogenetic abnormalities found in AML, including the translocations AML1-ETO, inv(16), or NUP98-HOXA9, can preserve HSC self-renewal in the presence of activated Akt or NF-?B.
Public Health Relevance
Leukemia-initiating cells (LIC) in acute myeloid leukemia (AML) represent a small fraction of the total tumor that can re-initiate AML upon transfer to secondary irradiated mice. LIC can be distinguished from normal hematopoietic stem/progenitor cells in bone marrow based on constitutive activation of the Akt and classical NF-?B signaling pathways in LIC. This proposal will examine the role of NF-?B in normal hematopoietic stem/progenitor cell function and test whether activation of NF-?B is sufficient to initiate and/or maintain LIC activity in AML.Role of NF-kB in hematopoietic stem cells and leukemia-initiating cell formation - Christopher Klug
http://grantome.com/grant/NIH/R01-CA144248-05°°
Blood Cells Mol Dis. Author manuscript; available in PMC 2009 Jan 1.
Published in final edited form as:
Blood Cells Mol Dis. 2008; 41(1): 73®C76.
The PI-3kinase pathway in hematopoietic stem cells and leukemia-initiating cells: a mechanistic difference between normal and cancer stem cells
Ömer H. Yilmaz and Sean J. Morrison
Howard Hughes Medical Institute, Department of Internal Medicine, and Center for Stem Cell Biology, Life Sciences Institute, University of Michigan, Ann Arbor, Michigan, 48109-2216
Abstract
The identification of cancer stem cells in leukemia, breast, brain, colon, and other cancers suggests that many tumors are maintained by stem cells in much the same way as normal tissues are maintained. Because cancer stem cells share remarkable phenotypic and functional similarities with normal stem cells, it may be difficult to identify therapeutic approaches to kill cancer stem cells without killing the normal stem cells in the same tissue. Yet in certain tissues, like the hematopoietic system and gut epithelium, this will be critical as regenerative capacity in these tissues is acutely required for life. Components of the PI-3kinase pathway, including Akt, mTor and FoxO are critical regulators of both normal stem cell function and tumorigenesis. Intriguingly, inactivation of some pathway components, like Pten, has opposite effects on normal hematopoietic stem cells (HSCs) and leukemia-initiating cells. This raises the possibility that drugs targeting this pathway could be more effective at eliminating cancer stem cells while being less toxic against normal stem cells.Figure 1
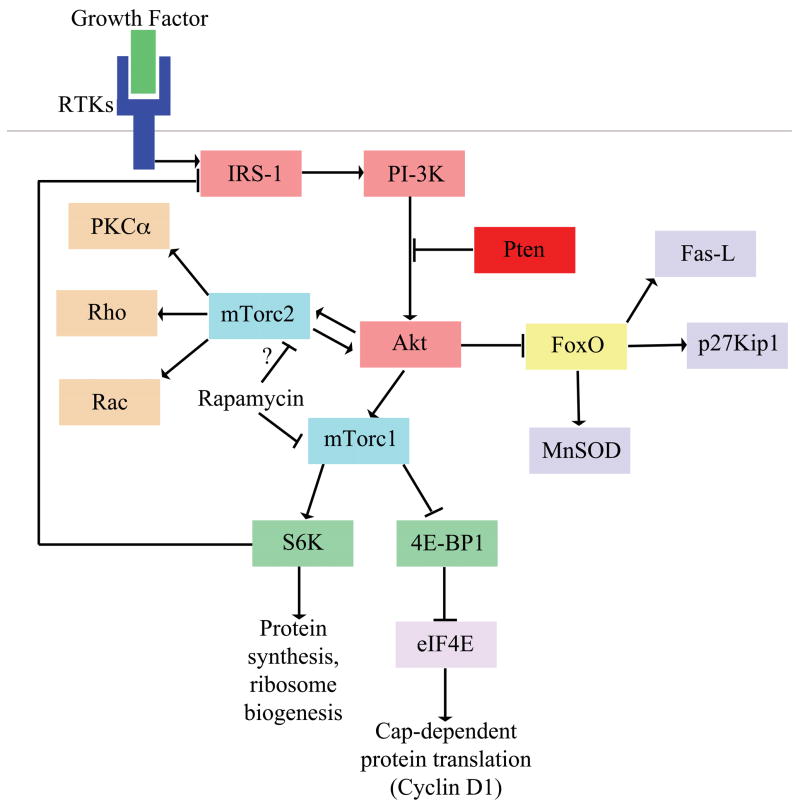
The PI-3kinase pathway and possible mechanisms of HSC depletion
Activated receptor tyrosine kinases (RTKs) signal through scaffolding adaptors (e.g. IRS-1) that activate PI-3kinase. Pten suppresses the activation of Akt. Deletion of Pten leads to the hyperactivation Akt, loss of FoxO function, and activation of the mTorc1 and mTorc2 complexes. Important target genes of FoxO include regulators of cell death (e.g. Bim-1 and Fas-L), cell cycle progression (e.g. p21Cip1 and p27Kip1), and ROS detoxification (e.g. catalase and MnSOD). There are multiple pathways downstream of activated mTorc1, including S6 kinase (which regulates protein synthesis and ribosome biogenesis) and 4E-BP1 (which regulates cap-dependent translation). Rapamycin treatment reduces the activation of S6K, reduces the inhibition of 4E-BP and rescues the function of Pten-deficient HSCs. Although Akt activation leads to mTorc1 activation, mTorc1 activation can reduce Akt activation by a negative feedback mechanism involving S6K. Rapamycin treatment can disrupt this negative feedback mechanism and therefore increase Akt signaling, which affects the many effectors that lie downstream of Akt. Downstream targets of mTorc2 include kinases such as PKC¶¡ Rho, Rac, and Akt. Prolonged rapamycin treatment has been demonstrated to also inhibit assembly of the mTorc2 complex by sequestering mTor, leading to decreased Akt activation. This finding raises the possibility that rapamycin treatment may also restore FoxO function in Pten-deficient HSCs by inhibiting mTorc2. Abbreviations (RTKs, receptor tyrosine kinases; IRS-1, insulin receptor substrate-1; PI-3K, phosphatidylinositol-3-OH kinase; Fas-L, Fas ligand; MnSOD, manganese superoxide dismutase; mTorc1, mTor complex 1; mTorc2, mTor complex 2; 4E-BP1, eukaryotic translation initiation factor 4E binding protein 1; eIF4E, eukaryotic translation initiation factor 4E; S6K, S6 kinase; PKC¶¡, protein kinase C alpha)
Introduction
Stem cells possess two defining characteristics: the ability to self-renew and the capacity to differentiate. In recent years, a number of major cancers including acute myeloid leukemia [1; 2], breast cancer [3], brain cancers [4; 5; 6], and colon cancer [7; 8] have been shown to follow a cancer stem cell model in which cancer cells are hierarchically organized. In each of these cancers, a small population of cancer stem cells appears uniquely capable of forming new tumors and transferring disease. These cancer stem cells both self-renew to form more cancer stem cells as well as differentiate to form phenotypically diverse cancer cells with limited proliferative potential.
Leukemic stem cells are defined as cells that can initiate leukemia after transplantation into healthy recipient mice. Unfractionated human acute myelogenous leukemia (AML) cells are inefficient at transferring disease to immunocompromised mice. However, by using a flow-cytometer to isolate phenotypically distinct subsets of leukemia cells, leukemia-initiating cells can be enriched among CD34+CD38-; AML cells that represent 0.2®C4% of leukemia cells [1]. Other leukemia cells are depleted for the ability to transfer disease. This proved that leukemias follow a cancer stem cell model where some leukemia cells have a greater capacity to transfer disease than others [1; 2; 9; 10]. A recent study has found that leukemia-initiating cells represent a surprisingly high proportion of leukemia cells in various mouse models of leukemia [11]. This raises the possibility that the frequency of leukemia-initiating cells among human AMLs has been underestimated, presumably as a result of the xenogeneic immune response they encounter after transplantation into immunocompromised mice. Nonetheless, AMLs that arise after Pten deletion in mice also follow a cancer stem cell model, with leukemia-initiating cells being 400-fold enriched among cells that express hematopoietic stem cell (HSC) markers [12]. These data indicate that at least some mouse leukemias follow a cancer stem cell model in which leukemia-initiating cells are highly enriched among cells that express markers similar to normal HSCs, just as observed among human AMLs [1; 2]. Many, but perhaps not all, mouse and human AMLs thus follow a cancer stem cell model in which cells capable of extensive proliferation and leukemia initiation represent a minority of leukemia cells.Phenotypic and functional similarities between normal and cancer stem cells
A remarkable finding in cancer biology is that normal stem cells and cancer stem cells often share phenotypic and functional similarities. This has been best characterized in the hematopoietic system where HSCs and leukemia-initiating cells, or leukemic stem cells (LSCs) express many of the same cell surface markers. In addition to these phenotypic similarities, there are also remarkable similarities in the pathways that regulate self-renewal. The best example is the polycomb family transcriptional repressor, Bmi-1. Bmi-1 promotes the self-renewal of both normal HSCs and LSCs, in part by repressing the Ink4a and Arf tumor suppressors [13; 14; 15; 16; 17]. Bmi-1 is not required for the formation of HSCs or LSCs, but is required for their maintenance [13; 18]. Thus, in the absence of Bmi-1, HSCs and LSCs can form, but they fail to transfer hematopoiesis or leukemia upon transplantation. Beyond Bmi-1, there are also a number of other regulators of self-renewal in HSCs that have been implicated in leukemogenesis [19; 20; 21; 22]. The phenotypic and functional similarities among HSCs and LSCs suggest that it may be difficult to target LSCs without also killing HSCs.
Pten deletion has opposite effects on HSCs and LSCs
Pten (Phosphatase and tensin homologue) is a phosphatase that negatively regulates signaling through the phosphatidylinositol-3-OH kinase (PI-3kinase) pathway, attenuating proliferation and survival signals. Pten is the second most frequently mutated gene in human cancers (after p53), and is inactivated by a variety of mechanisms in some leukemias [23; 24; 25]. Furthermore, the PI-3kinase pathway is usually over-activated in a variety of malignancies including leukemia.
To test its effect on HSC function, we conditionally deleted Pten from adult HSCs [12]. Loss of Pten in HSCs led to myeloproliferative disease within days and transplantable AMLs and acute lymphoblastic leukemias (ALLs) within weeks. Pten-deficiency had no discernable effect on HSC differentiation or survival but caused HSCs to go into cycle. This caused a transient increase in HSC numbers; however, by three weeks after Pten deletion HSCs became depleted. Consistent with this, Pten-deficient whole bone marrow cells or purified HSCs were able to engraft in irradiated mice and give rise to all types of blood cell lineages, but only for the first several weeks after injection. The levels of reconstitution declined over time, and recipient mice were rarely long-term multilineage reconstituted by Pten-deficient cells. Longitudinal studies of mice that were chimeric for Pten-deficient and wild-type HSCs showed that the loss of HSCs over time reflected a cell-autonomous requirement for Pten in the maintenance of HSCs [12]. Similar results were independently obtained by Li and colleagues [26]. HSCs thus require Pten to maintain quiescence and to self-renew over time.
In contrast to this requirement for Pten in the maintenance of HSCs, LSCs arose and expanded in number after Pten deletion. The LSCs were transplantable and could be enriched among cells that expressed HSC markers [12]. Most mice died with AML and ALL within 6 weeks of Pten deletion.
The observation that Pten deletion had opposite effects on normal HSCs and LSCs raised the possibility that by targeting this pathway it would be possible to eliminate LSCs without affecting normal HSCs. Pten deletion leads to increased activation of Akt and mTor, the mammalian Target of rapamycin. These signaling kinases have multiple roles within the cell that include promoting proliferation, survival, protein translation, ribosome biogenesis, and glycolysis [27]. To test whether the effects of Pten deletion were mediated by mTor activation, we administered rapamycin, a potent and specific inhibitor of mTor, to Pten-deleted mice. Rapamycin not only eliminated LSCs and maintained the health of mice, but it also rescued the depletion of Pten-deficient HSCs. Pten-deficient HSCs could even give long-term multilineage reconstitution of irradiated mice as long as the mice were maintained on rapamycin. This demonstrated that both the expansion of LSCs and the depletion of normal HSCs were mediated by increased mTor activation. Thus, many of the effects of Pten-deletion were mediated by increased mTor activation. By targeting mTor, LSCs could be eliminated while normal HSC function was rescued.
Although Pten is frequently deleted in many kinds of cancer and the PI-3kinase pathway is usually over-activated in leukemia, Pten is rarely deleted in leukemia [28; 29; 30]. Moreover, inherited germline mutations in Pten [31] are associated with hamartomas and a high risk for breast, thyroid, and endometrial cancers but not an increased risk of leukemia [32]. These observations raise the question of why Pten is rarely deleted in leukemia and why inherited mutations in Pten predispose to many other cancers but not leukemia. Our results offer a potential explanation: HSCs are efficiently depleted after Pten deletion. This means that spontaneous mutations that lead to a loss of Pten from HSCs will lead to the depletion of these HSCs before they have an opportunity to progress to leukemia. As a result, Pten deletion would not represent an efficient path to leukemic transformation. What remains unknown is whether downstream hematopoietic progenitors are also eliminated after Pten deletion, or whether this response is stem cell specific. Nonetheless, it may not be as necessary to deplete downstream progenitors that lose Pten, as the half-lives of these cells are generally much shorter than HSCs, limiting their opportunity to accumulate the additional mutations required for leukemogenesis.
One prediction of this hypothesis is that tissues that give rise to cancers that commonly exhibit Pten deletion should also have stem cells that can tolerate Pten deletion without being eliminated. There is some evidence that this is the case. Pten deletion in both neural stem cells [33] and prostate stem cells [34] leads to sustained increases in self-renewal and stem cell numbers, in contrast to the effect we observed in the hematopoietic system. Furthermore, Pten deletion is frequently observed in brain tumors [35] and prostate cancer [36]. These observations suggest that Pten deletion is an efficient path to transformation only in tissues in which the stem/progenitor cells can tolerate Pten deficiency. Possible mechanisms by which Pten deletion might lead to the loss of certain stem cells are addressed in the next section.
Our observation that rapamycin selectively eliminated LSCs while not harming HSCs suggests that rapamycin and its analogues may be used to treat cancers that exhibit increased PI-3kinase pathway activation. A subset of patients with refractory/relapsed AML responded favorably to a rapamycin analogue [37; 38]. Nonetheless, trials that have tested rapamycin analogues as single agents in a variety of cancers have generally yielded disappointing results [39; 40; 41]. This raises the question of whether our studies of Pten deficient mice can provide any insight into how rapamycin analogues can be used more effectively against cancer. When rapamycin was administered immediately after Pten deletion, it was extremely effective at preventing the generation or maintenance of LSCs: as long as these mice were maintained on rapamycin, they remained healthy with no histological evidence of hematopoietic neoplasms [12]. However, when rapamycin was started weeks after Pten deletion when mice already had leukemia, rapamycin was effective at reducing LSCs and prolonging the life of mice; however, all of these mice eventually died with AML and ALL [12]. It is not clear whether this reduced response to rapamycin in advanced leukemias reflects the accumulation of additional mutations that reduce rapamycin sensitivity, or whether rapamycin simply has a better opportunity to decimate the LSC pool when the pool size is much smaller. Nonetheless, one possibility raised by these results is that rapamycin analogues will yield more encouraging results when used in patients with minimal residual disease.
Rapamycin analogues may also provide more promising results in combination with other agents. Rapamycin as a single agent showed only a modest effect when used to treat lymphomas in mice over-expressing Akt [42]. But, when these mice were treated with doxorubicin (a DNA intercalating agent) together with rapamycin, most of these mice achieved remissions lasting more than 60 days [42]. Additional data suggest that NF-kB, a transcription factor that regulates cell survival, can mediate rapamycin resistance and that concomitant inhibition of NF-kB and mTor increases the death of cancer cells [43]. These data suggest that rapamycin may sensitize tumors with activated mTor to other chemotherapeutics.The PI-3kinase pathway, FoxO, and cellular senescence
What mechanisms are responsible for HSC depletion after Pten deletion? One obvious candidate is a senescence response. A prior study demonstrated that Pten deletion led to a p53-mediated senescence response in prostate epithelium progenitor cells [44]. The possibility that HSCs are depleted by a potent senescence response after Pten deletion is appealing because this could also potentially explain why LSCs do not exhibit this response: secondary mutations that occur during the progression of Pten-deficient cells to leukemia could inactivate this senescence response in leukemia cells. This raises the question of whether HSCs induce a senescence response after Pten deletion, and whether this is mediated by p53, Ink4a/Arf, or other gene products.
Our data suggest that whatever pathways lead to the depletion of HSCs after Pten deletion, that this response occurs downstream of mTor because it can be rescued by rapamycin treatment. However, the PI-3kinase pathway is a highly branched pathway, with many outputs and feedback loops, making it difficult to predict exactly which signal induces HSC depletion. mTor exists in two distinct complexes that function downstream and upstream of Akt: mTorc1 and mTorc2 [45; 46]. mTorc1 is downstream of Akt, controls the downstream effectors S6 kinase (S6K, which regulates ribosome biogenesis) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1, which regulates protein translation), and is rapamycin sensitive [46]. mTorc2 activates Akt and is rapamycin insensitive [46]. Although Akt activation leads to mTorc1 activation, mTorc1 activation can reduce Akt activation by negative feedback mechanisms. Rapamycin treatment increases Akt signaling by disrupting the negative feedback inhibition of activated S6K on the PI-3kinase-Akt pathway ([47]; Figure 1). Rapamycin treatment can therefore activate Akt and the many effectors that lie downstream of Akt on pathways that parallel mTorc1. However, recent data suggest that in a small subset of cancers, including some AMLs, prolonged rapamycin treatment not only inhibits the mTorc1 complex but also prevents the assembly of the mTorc2 complex, thus blocking the activation of Akt [48; 49]. It is possible that prolonged rapamycin treatment inhibits Akt instead of further activating it in Pten-deficient HSCs. This raises the general question of whether HSC depletion is initiated by effectors that lie downstream of mTorc1, such as S6K or 4E-BP1, or parallel pathways that lie downstream of Akt.
One set of effectors that lie downstream of Akt are the FoxO transcription factors that are inactivated upon phosphorylation by Akt. FoxO transcription factors regulate cell proliferation, cell survival, and promote the expression of enzymes that detoxify reactive oxygen species (ROS), like manganese superoxide dismutase and catalase [50]. As a result, ROS levels go up after Akt activation and persistent activation of Akt can lead to cell death due to ROS damage. Conditional inactivation of FoxO1, FoxO3, and FoxO4 leads to increased ROS levels in HSCs and to the subsequent loss of HSCs [51]. Like Pten-deficient bone marrow cells, FoxO-deficient bone marrow cells were unable to stably engraft irradiated recipient mice. It is likely that FoxO3a is the most important regulator of HSCs as FoxO3a deletion by itself leads to HSC depletion [52]. Treatment of FoxO-deficient mice with the anti-oxidant N-acetylcystine (NAC) restored HSC quiescence and at least partially rescued the depletion of primitive hematopoietic progenitors, though it was not tested whether the long-term multilineage reconstitution capacity of bone marrow cells was rescued [51]. It is unlikely that FoxO proteins mediate all of the effects of Pten deletion because Pten deletion is much more leukemogenic than deletion of FoxO1, FoxO3, and FoxO4 [53]. Nonetheless, FoxO genes could be one important mediator of the effects of Pten deletion on HSCs. Additional studies will be required to address the relationship between the mTor and FoxO pathways.Conclusion
The cancer stem cell model predicts that cancer therapies must destroy cancer stem cells in order to be effective [54; 55]. Since cancer stem cells and normal stem cells often share remarkable phenotypic and functional similarities, these therapies may be toxic to normal HSCs. The different responses of LSCs and normal HSCs to Pten deletion offers the possibility of eliminating LSCs without harming normal HSCs by targeting this pathway. This provides proof-of-principle that functional differences between cancer stem cells and normal stem cells can be identified and therapeutically exploited. Ultimately, by developing a sophisticated understanding of stem cell self-renewal it may be possible to identify new classes of therapies that are more effective against cancer and less toxic to normal stem cells.The PI-3kinase pathway in hematopoietic stem cells and leukemia-initiating cells: a mechanistic difference between normal and cancer stem cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2517145/°°
Understanding Pancreatic Cancer Blog
using information to our advantage
Understanding Pancreatic Cancer Blog
Cancer Stem Cell Model
1 Reply
Driving treatment decisions on 10 Nov 2015
Scientists use cancer models understand cancer. These models describe the rules cancer follows ®C how it starts, grows, metastasizes, and ultimately how it can be killed. Reviews of models generate new treatment ideas that models indicate should be successful.
Models of how cancer works drive the direction of cancer funding, research, prevention and treatment decisions. Faulty models lead to research producing ineffective treatments. Some researchers say that is happening now.
This post introduces the cancer stem cell model. The cancer stem cell model is not widely accepted but is gaining traction and a share of research money. It was developed as an alternative to the clonal evolution model to explain treatment failures.
Cancer Stem Cell Model
The cancer stem cell (CSC) model is an alternate model to explain the origin and diversity of cancer ®C and why past treatments have failed2. This model says that some (perhaps all) cancers are driven by a small number of treatment-resistant cancer cells with stem cell-like properties3,5. Stem cells have a slower life cycle and thus are largely unaffected by traditional chemotherapies that disrupt rapidly-dividing cells.6
Cancer Stem Cells Accumulate Mutations
Cancer Stem Cells Accumulate Mutations
[The Cancer Stem Cell model was hard to research. There are many papers and web seminars that present their research as, °∞this is the way it is ®C no disputes°±. Sorting out what is commonly accepted or not takes a lot of review and I°Øm certain I don°Øt have it all correct. This is the best I°Øve come up with.]
A cancer stem cell is very long-living, can accumulate genetic mutations over its lifetime, and then produce a nearly unlimited supply of cancer cells containing these mutations. Just as in the clonal evolution model, these cancer cells could continue to generate new mutations and divide uncontrollably.
[Whether CSC°Øs are actual adult stem cells that have become cancerous or are normal cells that have acquired °∞stem-like°± properties is under active investigation.]
Tumor Growth under the Cancer Stem Cell Model
Tumor Growth under the Cancer Stem Cell Model
The key to the cancer stem cell model though, is the small colony of cancer stem cells (red cell above) that regenerate cancer cells but are killed differently than normal cancer cells.5 Killing off the stem cells will result in the eventual dissipation of the tumor as it can no longer regenerate.
Cancer Stem Cell Model on Chemotherapy
Cancer stem cell model response to chemotherapy treatments
Cancer stem cell model response to chemotherapy treatments
Accumulate Mutations
In the CSC model, a long-lived stem cell accumulates the cancer-causing mutations. It is believed that the property of a long lifetime allows it to accumulate all these mutations. A normal cell, with its much shorter lifespan, would be unlikely to accumulate enough mutations. The key difference is that at the core of the tumor is a CSC, sometimes called a tumor-initiating cell.
[I°Øm couching the discussion with phrases above like °∞it is believed°±, but if you read the stem cell theory papers, these expressions of doubt are not presented. Some stem cell theorists say that a normal cell gains mutations and becomes stem cell-like to drive the cancer.]
Tumor Growth
The CSC is the major producer of all the new cancer cells3. The normal cancer cells (NCS) have limited cell division capability (just like normal cells). The CSC can continue to mutate which also results in tumor heterogeneity.
Treatment
As treatment begins, susceptible cells are again destroyed. However, CSC°Øs, which are slowly dividing, are not susceptible to chemotherapy3,5. To make matters worse, the CSC°Øs, acting like stem cells, see the tumor°Øs tissue damage and do what all stem cells do ®C regenerate new tissue (more CSC°Øs!). The result could again be a smaller tumor, but with an even larger concentration of CSC°Øs. In this model, what looks like good news on a CT scan (a smaller tumor) is really bad for the future.
[From a patient perspective, the CSC and clonal evolution model behave the same on scans, but the resulting tumor is very different. If the CSC model is correct, then in the long run, giving treatments that don°Øt kill the CSC°Øs is a bad thing to do.]
Treatment Holiday
After treatment, driven by many more CSC°Øs, the tumor growth accelerates.
[Question for Researchers: How does this square up with patients who have long-lasting remissions to chemotherapy?]
Cancer Stem Cell Model on Chemotherapy and Stem Cell Therapy
Under the CSC theory, the correct treatment protocol is to target the CSC°Øs themselves. Forget about the other cancer cells. Once the CSC°Øs are gone, the normal cancer cells cannot keep going by themselves and eventually perish. You can use a normal chemotherapy agent in addition to the CSC treatment in order to hasten the demise.
Cancer stem cell model response to chemotherapy and stem cell treatments
Cancer stem cell model response to chemotherapy and stem cell treatments
Treatment
As treatment begins, susceptible cells are again destroyed. In theory, stem cell therapy eliminates the CSC°Øs. Careful targeting of the CSC°Øs must be done to make sure that normal stem cells are not affected ®C which would probably be devastating to the patient.
Treatment Holiday
After treatment, with no CSC°Øs to replenish them, the normal cancer cells eventually die off.5
Controversy
The cancer stem cell model is not embraced by many cancer experts. The primary evidence is based on a set of experiments that break a tumor down and separate the tumor cells into different types. When 20,000 (or so) tumor cells of one type are transplanted into mice, a tumor does not take hold. When just 200 tumor cells of another type are transplanted, the tumor grows. This second set of tumor cells are considered CSC°Øs because they initiated human cancer growth in the mice. Experiments like these identify tumor cells with stem-like properties.
Identifying Cancer Stem Cells
Identifying Cancer Stem Cells
Skeptical researchers say that a demonstration of human tumor cell growth in immunodeficient mice is insufficient. Growing human cancer cells in a mouse is too dissimilar an environment to provide proof that these are cancer stem cells.5
It should also be noted that testicular cancer that is curable with chemotherapy alone. If cancer stem cells were involved, this would not be possible. So apparently CSC°Øs do not drive all cancers.
Another View of the Cancer Stem Cell Model
The figure below presents another way to look at tumor growth in the cancer stem call model. Each colored area represents a cell colony with a specific set of mutations. Time progresses to the right. The height of each colored area represents the quantity of cells in the colony. New mutations are represented by stars and may originate from any established colony. The figure shows that these new mutations only originate from the tumor°Øs cancer stem cells (dark red) and then compete for space and resources with other colonies.
Chemotherapy treatment is effective on the normal cancer cells, but has the opposite effect on cancer stem cells which multiply in response to the tissue damage. After treatment ends, a larger number of cancer stem cells are present to begin tumor regrowth.
Eventually, one of the colonies acquires the ability to metastasize and migrates to another organ.
Cancer Stem Cell Model with Effective Chemotherapy
Cancer Stem Cell Model with Effective Chemotherapy
Summary
The cancer stem cell model is an alternate explanation for tumor growth and response to treatments. It was devised to try to explain failures of treatments based on the correctness of the clonal evolution model. The correctness of the cancer stem cell model is hotly debated.
Publication counts of Cancer Stem Cell papers started in 2004 and have been on a steep rise, indicating that it is an active topic. For comparison, publication counts for the clonal evolution model are shown in orange below.Cancer Stem Cell Model - Understanding Pancreatic Cancer Blog https://www.pancanology.com/cancer-stem-cell-model/
°°
Evolution of the Cancer Stem Cell Model
Cell, Stem Cell
Antonija Kreso
John E. DickPrincess Margaret Cancer Centre, University Health Network, Toronto, Ontario M5G 1L7, Canada and Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 1A8, Canada
Open ArchiveDOI:https://doi.org/10.1016/j.stem.2014.02.006
Genetic analyses have shaped much of our understanding of cancer. However, it is becoming increasingly clear that cancer cells display features of normal tissue organization, where cancer stem cells (CSCs) can drive tumor growth. Although often considered as mutually exclusive models to describe tumor heterogeneity, we propose that the genetic and CSC models of cancer can be harmonized by considering the role of genetic diversity and nongenetic influences in contributing to tumor heterogeneity. We offer an approach to integrating CSCs and cancer genetic data that will guide the field in interpreting past observations and designing future studies.
°°
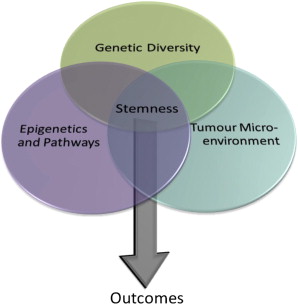
Figure 1Stemness as a Guiding Principle that Governs Therapeutic Response
Three fields in biology°™cancer genetics, epigenetics, and microenvironment°™are coming together to provide increasing clarity to the processes that determine stemness and in turn influence clinical outcome. These three factors can influence stemness simultaneously, but they can also act independently over time. Through evolutionary time, different forces can impact a cell°Øs stemness properties and thereby shape tumor progression and therapeutic response.
°°
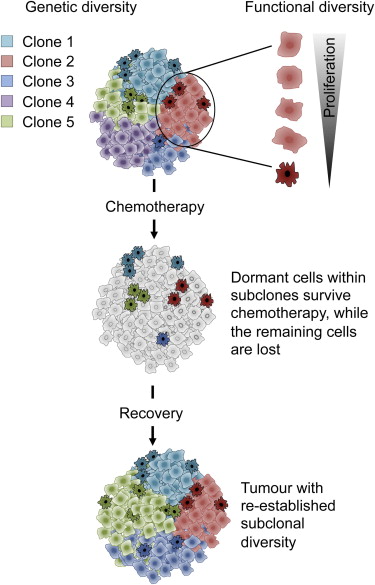
Figure 3Functional Diversity between Cells within Subclones Impacts Response to Therapy
Each clone (depicted by the different colors) contains a mixture of cells that vary with respect to their stemness and/or proliferative ability, including relatively dormant cells. Together these factors represent the functional diversity present within single genetic subclones. Chemotherapy can reduce tumor burden by eliminating the highly proliferative cells within subclones, while sparing the relatively dormant cells; following therapy, these cells can seed a new cancer. Thereby, subclonal diversity can be altered with chemotherapy and can allow for the selection of cells with additional genetic mutations that confer a survival advantage. Not depicted in the diagram is the concept that chemotherapy-resistant cells can exist before treatment and can be selected following chemotherapy. Thus, chemotherapy can introduce new mutations to confer treatment resistance, but it can also select preexisting cells that accumulated mutations, which confer chemotherapy resistance during the long evolution of the tumor before it was diagnosed.Evolution of the Cancer Stem Cell Model: Cell Stem Cell https://www.cell.com/cell-stem-cell/fulltext/S1934-5909(14)00057-5
°°
Nuclear lamina defects cause ATM-dependent NF-¶ B ...
https://www.ncbi.nlm.nih.gov/pubmed/23019125
Oct 15, 2012 °§ Nuclear lamina defects cause ATM-dependent NF-¶ B activation and link accelerated aging to a systemic inflammatory response. Osorio FG(1), B®¢rcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, L®Æpez-Ot®™n C.
Cited by: 149
Publish Year: 2012
Author: Fernando G. Osorio, Clea B®¢rcena, Clara Soria-Valles, Andrew J. Ramsay, F®¶lix de Carlos, Juan Cobo, ...
Nuclear initiated NF-¶ B signaling: NEMO and ATM take ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3193401
Dec 28, 2010 °§ While ATM-dependent phosphorylation of NEMO is an essential step in DSB-induced NF-¶ B activation 54, the requirement for ATM kinase activity in any additional step(s) in the pathway remains unclear. A NEMO-S85A mutant that cannot be phosphorylated by ATM is efficiently SUMO1 modified, but fails to be monoubiquitinated and exported to the cytoplasm following cell exposure to VP16. 1.3E2 cells expressing this mutant NEMO °≠
Cited by: 215
Publish Year: 2011
Author: Shigeki Miyamoto°°
Blocking ATM-dependent NF-¶ B pathway overcomes ... - Leukemia
https://www.nature.com/articles/s41375-019-0458-0
Apr 02, 2019 °§ Inhibition of ATM-dependent NF-¶ B pathway can sensitize ALL to chemotherapeutics, providing a new strategy to eradicate residual chemo-resistant ALL cells. Bone marrow °≠
Author: Ya-Li Chen, Chao Tang, Meng-Yi Zhang, Wen-Li Huang, Yan Xu, Hui-Yin Sun, Fan Yang, Li-Li Song, He Wa...
Author: Ya-Li Chen
Publish Year: 2019
Role of nuclear factor-¶ B-mediated inflammatory pathways ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3141285
Jun 01, 2011 °§ As the disease of cancer progresses, patients develop multiple symptoms that can impair their function and quality of life. The symptoms common in cancer patients include cachexia, anorexia, fatigue, depression, neuropathic pain, anxiety, cognitive impairment, sleep disorders and delirium (acute confusion state).
Cited by: 140
Publish Year: 2011
Author: Subash C Gupta, Ji Hye Kim, Ramaswamy Kannapp°°
Article
Published: 02 April 2019
Acute lymphoblastic leukemia
Blocking ATM-dependent NF-¶ B pathway overcomes niche protection and improves chemotherapy response in acute lymphoblastic leukemia
Ya-Li Chen, Chao Tang, [°≠]Bin-Bing S. Zhou
Leukemia volume 33, pages2365®C2378(2019)Cite this articleKey Laboratory of Pediatric Hematology and Oncology Ministry of Health and Pediatric Translational Medicine Institute, Shanghai Children°Øs Medical Center, Shanghai Collaborative Innovation Center for Translational Medicine and Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine (SJTU-SM), 200025, Shanghai, China
Abstract
Bone marrow (BM) niche responds to chemotherapy-induced cytokines secreted from acute lymphoblastic leukemia (ALL) cells and protects the residual cells from chemotherapeutics in vivo. However, the underlying molecular mechanisms for the induction of cytokines by chemotherapy remain unknown. Here, we found that chemotherapeutic drugs (e.g., Ara-C, DNR, 6-MP) induced the expression of niche-protecting cytokines (GDF15, CCL3 and CCL4) in both ALL cell lines and primary cells in vitro. The ATM and NF-¶ B pathways were activated after chemotherapy treatment, and the pharmacological or genetic inhibition of these pathways significantly reversed the cytokine upregulation. Besides, chemotherapy-induced NF-¶ B activation was dependent on ATM-TRAF6 signaling, and NF-¶ B transcription factor p65 directly regulated the cytokines expression. Furthermore, we found that both pharmacological and genetic perturbation of ATM and p65 significantly decreased the residual ALL cells after Ara-C treatment in ALL xenograft mouse models. Together, these results demonstrated that ATM-dependent NF-¶ B activation mediated the cytokines induction by chemotherapy and ALL resistance to chemotherapeutics. Inhibition of ATM-dependent NF-¶ B pathway can sensitize ALL to chemotherapeutics, providing a new strategy to eradicate residual chemo-resistant ALL cells.Blocking ATM-dependent NF-¶ B pathway overcomes niche protection and improves chemotherapy response in acute lymphoblastic leukemia | Leukemia https://www.nature.com/articles/s41375-019-0458-0
ATM serine/threonine kinase
ATM serine/threonine kinase
ATM serine/threonine kinase, symbol ATM, is a serine/threonine protein kinase that is recruited and activated by DNA double-strand breaks. It phosphorylates several key proteins that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis. Several of these targets, including p53, CHK2, BRCA1, NBS1 and H2AX are tumor suppressors.
ATM serine/threonine kinase - Wikipedia
https://en.wikipedia.org/wiki/ATM_serine/threonine_kinase°°
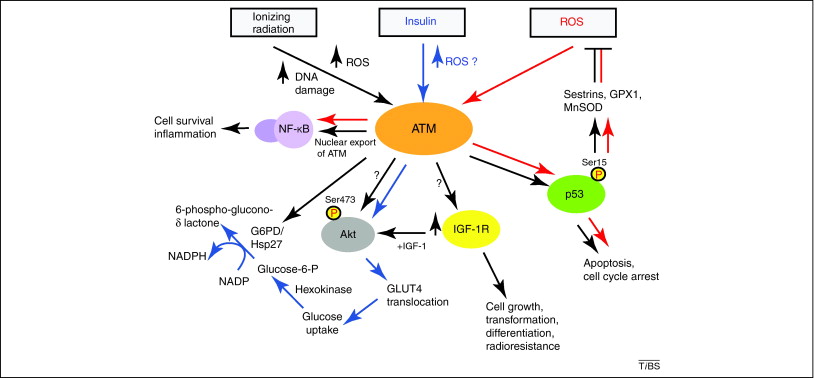
The ATM protein kinase and cellular redox signaling: beyond the DNA damage response
Scott Ditch
Tanya T. Paull
Published:November 11, 2011DOI:https://doi.org/10.1016/j.tibs.2011.10.002
The ataxia®Ctelangiectasia mutated (ATM) protein kinase is best known for its role in the DNA damage response, but recent findings suggest that it also functions as a redox sensor that controls the levels of reactive oxygen species in human cells. Here, we review evidence supporting the conclusion that ATM can be directly activated by oxidation, as well as various observations from ATM-deficient patients and mouse models that point to the importance of ATM in oxidative stress responses. We also discuss the roles of this kinase in regulating mitochondrial function and metabolic control through its action on tumor suppressor p53, AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR) and hypoxia-inducible factor 1 (HIF1), and how the regulation of these enzymes may be affected in ATM-deficient patients and in cancer cells.
The ATM protein kinase and cellular redox signaling: beyond the DNA damage response: Trends in Biochemical Sciences
https://www.cell.com/trends/biochemical-sciences/fulltext/S0968-0004(11)00161-7°°
Inflammation, the microenvironment and chronic lymphocytic ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3046264
In cancer a unique microenvironmental organization is instrumental in the development and thriving of malignant cells: chronic inflammation exposes cells to growth factors, newly formed vessels provide nutrients and immune tolerance avoids immune-mediated elimination.
Cited by: 15
Publish Year: 2011
Author: Federico Caligaris-Cappio
The Role of Inflammation in Leukaemia | Springer for ...
https://rd.springer.com/chapter/10.1007/978-3-0348-0837-8_13
May 13, 2014 °§ Kristinsson SY, Bjorkholm M, Hultcrantz M, Derolf ÅR, Landgren O, Goldin LR (2011) Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes.
Cited by: 7
Publish Year: 2014
Author: Janusz Krawczyk, Michael O°ØDwyer, Ronan T Swords, Ciara Freeman, Francis J. Giles
Targeting inflammatory pathways in chronic lymphocytic ...
https://www.sciencedirect.com/science/article/abs/pii/S1040842813001625
Herein we review the role chronic inflammation plays in the initiation and progression of CLL. The robust production of inflammatory cytokines and chemokines accompanied by activation of intra-cellular pro-inflammatory pathways, and the presence of somatic mutations that activate pro-inflammatory signaling pathways, suggest that chronic inflammation plays a pathophysiological role in this disease.
Cited by: 13
Publish Year: 2013
Author: Uri Rozovski, Michael J. Keating, Zeev Estrov°°
LPS”’µºµƒPKM2±Ì¥Ô“˝∆¿“∞±À·105µƒÕ¨ ±¡◊À·ªØ£¨∆‰≥Ã∂»”Αˆº”µƒ±Ì¥ÔÀÆ∆Ωœýµ±ªÚ∏¸∏þ°£’‚÷÷¡◊À·ªØΩˆ‘⁄1–° ±∫ÛæÕ∫Ð√˜œ‘£¨‘⁄48–° ±∫Û”’µº◊Ó«ø£®Õº1A£¨÷–º‰£¨œý∂‘¥¯«ø∂»‘ˆº”¡À10.3±∂£©°£±˚Õ™À·º§√∏M2µ˜Ω⁄Hif-1¶¡...
°°
°°
LPS-induced expression of PKM2 causes concurrent phosphorylation of Tyrosine 105 to an extent comparable or greater to the increased expression levels. This phosphorylation is evident after just 1 hr, with the strongest induction after 48 hr (Figure 1 A, middle, 10.3-fold increase in relative band intensity).
Pyruvate Kinase M2 Regulates Hif-1¶¡ ... - ScienceDirect
www.sciencedirect.com/science/article/pii/S1550413114005567
°°
°°
Lipopolysaccharide induces inflammation and facilitates lung metastasis in a breast cancer model via the prostaglandin E2-EP2 pathway
°°
Affiliations: Cancer Therapy and Research Center, Shandong Provincial Hospital, Shandong University, Jinan, Shandong 250021, P.R. China
Inflammation is a potent promoter of tumor metastasis.°°
The aim of the present study was to explore the function of systemic inflammation in the formation of lung metastasis of breast cancer cells in a mouse model. BALB/c mice were injected intraperitoneally with lipopolysaccharide (LPS) in order to establish an inflammatory animal model and 4T1 murine breast cancer cells were injected through the tail vein to induce lung metastasis. The levels of proinflammatory cytokines were evaluated by ELISA. Metastases on the surface of the lungs were counted and histologically analyzed by hematoxylin and eosin staining.
°°
Angiogenesis in the lungs was examined by CD31 immunofluorescence. Mouse pulmonary endothelial cells (MPVECs) were isolated and used to assay endothelial tube formation and determine the protein expression levels of vascular endothelial growth factor (VEGF) in vitro. Serum levels of VEGF and prostaglandin E2 (PGE2), the number and size of metastatic lesions, and the expression levels of cyclooxygenase‑2 were significantly greater in the lungs of LPS‑treated mice, as compared with those in control mice threated with phosphate‑buffered saline. Blood vessel density was also markedly increased in the LPS‑treated mice.
°°
These increases were reversed by treatment with celecoxib. In vitro, the protein expression levels of VEGF produced by the PGE2‑treated cells were significantly increased in a concentration‑dependent manner. In addition, the production of VEGF was increased in response to treatment with the PGE2 receptor (EP2) agonist ONO‑AE1‑259‑01; however, this increase was abrogated by treatment with AH6809, an EP2 receptor antagonist. Treatment with PGE2 or VEGF alone promoted the tube formation of MPVECs and this effect was reversed by treatment with celecoxib. These results demonstrated that PGE2 may regulate the release of VEGF by MPVECs through the EP2 receptor pathway and thereby promoted pulmonary angiogenesis and breast cancer metastasis in a mouse model.
°°
÷¨∂ýëծπ˝«∞¡–œŸÀÿE2-EP2Õææ∂‘⁄»ÈœŸ∞©ƒ£–Õ÷–”’µº—◊÷¢≤¢¥ŸΩ¯∑Œ◊™“∆
—◊÷¢ «÷◊¡ˆ◊™“∆µƒ”––ߥŸΩ¯º¡°£
±æ—–æøµƒƒøµƒ «‘⁄–° Ûƒ£–Õ÷–ÃΩÀ˜»´…Ì—◊÷¢‘⁄»ÈœŸ∞©œ∏∞˚∑Œ◊™“∆–Œ≥…÷–µƒπ¶ƒÐ°£ BALB / c–° Û∏πƒ§ƒ⁄◊¢…‰÷¨∂ý룮LPS£©“‘Ω®¡¢—◊–‘∂،ԃ£–Õ£¨≤¢Õ®π˝Œ≤æ≤¬ˆ◊¢…‰4T1 ۻȜŸ∞©œ∏∞˚“‘”’µº∑Œ◊™“∆°£Õ®π˝ELISA∆¿π¿¥Ÿ—◊œ∏∞˚“Ú◊”µƒÀÆ∆Ω°£º∆ ˝∑Œ±Ì√Ê…œµƒ◊™“∆≤¢Õ®π˝À’ƒææ´∫Õ Ô∫Ï»æ…´Ω¯––◊È÷Ø—ß∑÷Œˆ°£
Õ®π˝CD31√‚“þ”´π‚ºÏ≤È¡À∑Œ÷–µƒ—™πÐ…˙≥…°£∑÷¿Î–° Û∑Œƒ⁄∆§œ∏∞˚£®MPVEC£©£¨≤¢”√”⁄ÃÂÕ‚≤‚∂®ƒ⁄∆§πеƒ–Œ≥…∫Õ»∑∂®—™πЃ⁄∆§…˙≥§“Ú◊”£®VEGF£©µƒµ∞∞◊±Ì¥ÔÀÆ∆Ω°£”ÎLPS¥¶¿Ìµƒ–° Ûœý±»£¨LPS¥¶¿Ìµƒ–° Ûµƒ∑Œ÷–—™«ÂVEGF∫Õ«∞¡–œŸÀÿE2£®PGE2£©µƒÀÆ∆Ω£¨◊™“∆–‘≤°±‰µƒ ˝¡ø∫Õ¥Û–°“‘º∞ª∑—ı∫œ√∏2µƒ±Ì¥ÔÀÆ∆Ωœ‘◊≈∏¸∏þ°£ª∫≥—ŒÀÆ°£æ≠LPS¥¶¿Ìµƒ–° Ûµƒ—™πÐ√Ð∂»“≤œ‘◊≈‘ˆº”°£°°
”√»˚¿¥ŒÙ≤º÷Œ¡∆ø…“‘ƒÊ◊™’‚–©‘ˆº”°£‘⁄ÃÂÕ‚£¨æ≠PGE2¥¶¿Ìµƒœ∏∞˚≤˙…˙µƒVEGFµ∞∞◊±Ì¥ÔÀÆ∆Ω“‘≈®∂»“¿¿µ–‘∑Ω Ωœ‘◊≈‘ˆº”°£¡ÌÕ‚£¨∂‘PGE2 Ðã®EP2£©º§∂غ¡ONO‑AE1‑259‑01¥¶¿Ì∫Û£¨VEGFµƒ≤˙…˙‘ˆº”°£µ´ «£¨Õ®π˝ π”√EP2 ÐÃÂÞ◊øπº¡AH6809ø…“‘œ˚≥˝’‚÷÷‘ˆº”°£µ•∂¿”√PGE2ªÚVEGF÷Œ¡∆ø…¥ŸΩ¯MPVECµƒπЖŒ≥…£¨∂¯”√»˚¿¥ŒÙ≤º÷Œ¡∆ø…ƒÊ◊™’‚÷÷◊˜”√°£’‚–©Ω·π˚±Ì√˜£¨PGE 2ø…ƒÐÕ®π˝MP2Õ®π˝EP2 ÐÃÂÕææ∂µ˜Ω⁄MPVECµƒVEGF Õ∑≈£¨¥”∂¯¥ŸΩ¯¡À–° Ûƒ£–Õ÷–µƒ∑Œ—™πÐ…˙≥…∫ջȜŸ∞©◊™“∆°£°°
Lipopolysaccharide induces inflammation and facilitates lung metastasis in a breast cancer model via the prostaglandin E2-EP2 pathway
https://www.spandidos-publications.com/10.3892/mmr.2015.3258°°
°°
Int J Cancer. Author manuscript; available in PMC 2010 Dec 15.
Published in final edited form as:
Int J Cancer. 2009 Dec 15; 125(12): 2863®C2870.
Akt-mediated regulation of NF¶ B and the essentialness of NF¶ B for the oncogenicity of PI3K and Akt
Dong Bai, Lynn Ueno, and Peter K. Vogt*
The Scripps Research Institute, Department of Molecular and Experimental Medicine, 10550 N. Torrey Pines Road, La Jolla, CA 92037, USA
Abstract
The serine/threonine kinase Akt (cellular homolog of murine thymoma virus akt8 oncogene), also known as PKB (protein kinase B), is activated by lipid products of phosphatidylinositol 3-kinase (PI3K). Akt phosphorylates numerous protein targets that control cell survival, proliferation and motility. Previous studies suggest that Akt regulates transcriptional activity of the nuclear factor-¶ B (NF¶ B) by inducing phosphorylation and subsequent degradation of inhibitor of ¶ B (I¶ B). We show here that NF¶ B-driven transcription increases in chicken embryonic fibroblasts (CEF) transformed by myristylated Akt (myrAkt). Accordingly, both a dominant negative mutant of Akt and Akt inhibitors repress NF¶ B-dependent transcription. The degradation of the I¶ B protein is strongly enhanced in Akt-transformed cells, and the loss of NF¶ B activity by introduction of a super-repressor of NF¶ B, I¶ BSR, interferes with PI3K- and Akt-induced oncogenic transformation of CEF. The phosphorylation of the p65 subunit of NF¶ B at serine 534 is also upregulated in Akt-transformed cells. Our data suggest that the stimulation of NF¶ B by Akt is dependent on the phosphorylation of p65 at S534, mediated by IKK (I¶ B kinase) ¶¡ and ¶¬. Akt phosphorylates IKK¶¡ on T23, and this phosphorylation event is a prerequisite for the phosphorylation of p65 at S534 by IKK¶¡ and ¶¬. Our results demonstrate two separate functions of the IKK complex in NF¶ B activation in cells with constitutive Akt activity: the phosphorylation and consequent degradation of I¶ B and the phosphorylation of p65. The data further support the conclusion that NF¶ B activity is essential for PI3K- and Akt-induced oncogenic transformation.
Keywords: NF-¶ B, PI-3 kinase, Akt, IKK, I¶ B, oncogenic transformation°°
Akt-mediated regulation of NF¶ B and the essentialness of NF¶ B for the oncogenicity of PI3K and Akt
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2767458/°°
°°