��
The role of macrophages in influenza A virus infection
ROS sets the stage for macrophage differentiatio
When Alveoli Macrophage is Strong, Infection and Inflammation is resolved without rendering for systemic inflammation
1. AM��s are critical in the protection against influenza virus-driven morbidity and mortality.
2. nonproductive infection of M��s may have a role in controlling the virulence of influenza virus infections.
3. Induction of an antiviral state, and in particular interferons, in response to a virus infection can also suppress M�� and neutrophil functions. IFN-�� is produced early in response to viral infection, mainly by M��s and NK cells, and later, when the adaptive responses begin, by CD8 T cells.
4. bronchial IM��s (BIM��s)��lack of IM��s in the alveolar�Ccapillary lung interstitium during normal healthy steady-state conditions�� Gene expression analysis of these BIM��s clearly revealed the presence of monocyte-specific markers
5. . Macrophage function ranges from highly proinflammatory to wound healing
6.ROS sets the stage for macrophage differentiation,ROS production is important in M2 but not M1 macrophage differentiation. Interestingly, pre-treating monocytes with the antioxidant butylated hydroxyanisole (BHA) prior to differentiation inhibits M2 but not M1 polarization M2 but not M1 polarization. TAM differentiation may be a critical target, as BHA administration reduced TAM numbers as well as levels of TAM markers. BHA has no effects on proliferation of three tumor cell lines in vitro. harvard school of public health
�����ǻ������ѣ�BHA)�������嶡��-4-�ǻ������ѡ������������ѣ����BHA��Ϊ���ֳɷ֣�3-BHA��2-BHA���Ļ�������ʽΪC11H16O2����Է�������Ϊ180.25�������ǻ������ѵĿ����������������ų���ԭ�������֬�Զ�������ʵ�ֵġ�7. Macrophage derived Nitric oxide acts as cysteine protease inhibitor, inhibiting viral processing, thus has antiviral effect.
��
��
All macrophages take various forms (with various names) throughout the body and are designated as histiocytes, Kupffer cells, Hofbauer cells, alveolar macrophages and microglia, among others. Despite heterogeneity, tissue-resident macrophages are derived from three sources: yolk sac, foetal liver and hematopoietic stem cells in the bone marrow [7].
Major biological activities of macrophage include phagocytosis, antigen presentation and the release of cytokine (pro-inflammatory/anti-inflammatory mediators), antibacterial substances and enzymes that remodel the extracellular matrix [8]. Macrophages attract and activate other cells of the adaptive immune system, in particular T cells, to sites of chronic inflammation. Further, macrophages are able to sense the time at which an injury is terminated and thus start the resolution process of inflammation and the control of the healing phase [9].Today, an important role of monocytes/macrophages has been shown for the persistence or spread of more than 35 viruses belonging to 13 different families.
Monocytes and Macrophages as Viral Targets and Reservoirs
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6163364/��
ROS sets the stage for macrophage differentiation | Cell Research
https://www.nature.com/articles/cr201388��
Macrophage differentiation is often accompanied by morphological changes, for instance, regarding the differentiation into classically activated M1 or alternatively activated M2 macrophages. M1 macrophages can display a round appearance, while M2 macrophages are more elongated (27�C29).
Alveolar Epithelial Cells Are Critical in Protection of the Respiratory Tract by Secretion of Factors Able To Modulate the Activity of Pulmonary Macrophages and Directly Control Bacterial Growth | Infection and Immunity
https://iai.asm.org/content/81/1/381��
��
��
The role of macrophages in influenza A virus infection
��Marlynne Q. Nicol
Bernadette M. DutiaUniversity of Edinburgh,
The importance of macrophages in the control of infections has long been documented, but macrophages have also been shown to contribute to severe influenza A virus infections. Macrophage function ranges from highly proinflammatory to wound healing and regulatory and a picture of diverse subsets with considerable plasticity in function and phenotype is emerging. Within the lung three subsets of macrophage populations have been identified: resident alveolar macrophages, interstitial macrophages and exudate-derived macrophages. Here we review model systems and techniques for defining macrophage function in vivo and discuss macrophage infection in vitro. The use of detailed phenotypic approaches and techniques to dissect the role of individual macrophage subsets in vivo promises rapid advances in this area of research.Influenza viruses cause acute respiratory infections characterized by fever, nasal secretions, cough, high temperature, aching joints and general malaise. Seasonal influenza A virus (IAV) infections account for 500,000 deaths worldwide annually [1] and are associated with considerable morbidity [2�C4]. The possible emergence of a virulent pandemic influenza virus is a major threat to human populations and the current circulation of highly pathogenic H5N1 strains, as well as the recently emerged H7N9 strain is a cause of concern. Understanding the mechanisms of IAV pathogenesis is critical in devising strategies to deal with infection caused by these viruses.
Studies using animal models show that infection with different IAV subtypes results in differences in cellular infiltrate, temporal changes in virus load, cytokine and chemokine levels, and pathological outcome [5�C7]. The ability of different strains to infect and replicate in the lower respiratory tract, that is, within the lung itself, rather than being confined to the upper respiratory tract, also effects clinical outcome and is associated with severe infections [8]. Evidence is emerging that events early in infection are key in determining the course of infection and the outcome [9]. Epithelial cells in the respiratory tract are the primary target cells for IAV infection [10], but other cell types, including macrophages (M��s), can be infected [11]. M��s are innate sentinel cells responsible for eliciting early immune responses and their infection is likely to alter early cytokine and chemokine responses, causing dysregulation in what is normally a tightly controlled system. A survey of current literature indicates that not all strains of IAV can infect and/or replicate in lung M��s but, although it is clear that there are viral subtype differences, experimental procedures for defining M�� populations also contribute to the conclusions that have been reached. Thus, there is much still to be learnt about the interactions between lung M��s, IAV and the roles that M��s play in the pathogenicity of IAV infections.
M�� populations in the lung
The airway is a nonsterile environment constantly exposed to inhaled pathogens or foreign particles, dust and allergens that have not been cleared by the mucocillary machinery of the nasopharynx. The M�� population within the lung is a heterogeneous dynamic population that consists of at least three cell types �C the alveolar M��s (AM��s) found within the airways (bronchi, bronchioles and alveoli), the interstitial M��s (IM��s), and exudate-derived M��s (EM��s), which are recruited to the lung in response to inflammatory challenges. While it has become clear over the past 10 years that these three populations are phenotypically and functionally distinct and can be differentiated by morphological, phenotypic and functional characteristics, there is a considerable body of literature that does not recognize this, and it is only recently that the origins and functions of these different populations have begun to be teased apart and their individual roles during lung inflammation elucidated.
Alveolar M��s are the primary sentinel cell within the lung and reside in the lumen of the alveolus �C that is, the airspace �C and it is estimated that there is at least one AM�� per alveolus at homeostasis [12]. AM��s are thought to be long-lived cells [13] with an estimated turnover of around 40% in a year [14] that can persist, even during inflammation. AM��s can be exposed to 1010 particles per day [15] and the challenge is to mount an appropriate response that limits deleterious inflammation without damaging the delicate alveolar capillary membrane or interfering with normal gas exchange. Increasing evidence suggests that such tissue-specific M��s are not, as previously thought, terminally differentiated M��s from a blood monocyte origin, but are in fact derived from a separate pool. Fate mapping studies show the prenatal origin of AM��s, confirming that they are independent of blood monocytes [16] and that, at least in uninfected states, they replicate in situ to maintain the population [17,18]. This was reviewed recently by Davies et al. [19].
M��s have been classified into phenotypically distinct groupings termed classically activated M1 M��s or alternatively activated M2 M��s, proinflammatory and anti-inflammatory, respectively. M1 M��s are defined by the production of high levels of cytokines TNF-��, IL-1��, IL-12 and iNOS in response to IFN-�� and express surface markers such as MHC class II, CD80, CD86 and CCR2, whereas M2 M��s produce IL-10, IL-1Ra, arginase 1, FIZZ and YM1/2, and upregulate M�� mannose receptors (M�� mannose receptor, MR and CD206), Dectin-1 and CD200R in response to IL-4 and IL-13 (reviewed in [20,21]). Expression of different STAT transcription factors and SOCS proteins has also been associated with the M1 (STAT1, SOCS2) and M2 (STAT3/STAT6, SOCS3) phenotypes [22]. Interestingly, influenza can induce temporal changes in SOCS proteins in vitro (SOCS1, SOCS3) [23�C25] and in vivo, where they are significantly decreased up to 8 days postinfection in a murine model [nicol et al., unpublished data]. Based on elevated expression of receptors that recognize PAMPs, DAMPs, C-type lectins and scavenger receptors (TLRs, NOD-like receptors, RIG-like receptors, CD206 and MARCO) [19,26�C28], AM��s at steady state are believed to be alternatively activated, a phenotype thought to be important in maintaining homoeostasis and dampening unnecessary inflammatory responses in the lung. However, there is also evidence for transcription of proinflammatory cytokines in these cells [20,29].
Alternatively, activated M2 M��s are predominantly anti-inflammatory, involved in tissue repair [21,30] or resolution of inflammation, secrete regulatory cytokines such as IL-10 [31], and have poor antigen-presenting capacity [32]. Indeed, AM��s at steady state are known to be poor at antigen presentation [33]. Polarizing human AM��s ex vivo to an M2 phenotype resulted in increased levels of the mannose receptor CD206 [34] and examination of bronchoalveolar lavage (BAL) fluid showed between 20 and 50% had a CD206 M2-like phenotype [35,36]. Similarly, mouse alveolar M��s have high expression levels of this receptor [37,38]. Characterization of healthy human AM��s, however, is complicated by the limited availability of cells from healthy donors and the consequent use of cells from those with underlying disease that may not be representative of a normal phenotype. Furthermore, differences in ex vivo handling of cells for example, culturing for various periods of time, can markedly change the cell phenotype.
Transcriptional analysis of freshly isolated human AM��s showed them to have a pro- and not an anti-inflammatory profile, with the proinflammatory signature being diminished over time in culture [39]. In fact, they showed levels of proinflammatory cytokines higher than those observed in M-CSF-differentiated monocyte-derived M��s (MDMs), which are often used as a model for tissue M��s [40]. An interesting dichotomy is that GM-CSF is known to differentiate MDMs into an M1-like proinflammatory phenotype, yet is also known to be essential for the AM�� phenotype [41,42], which is supposedly anti-inflammatory. Furthermore, GM-CSF has also been shown to increase numbers of AM��s in mice [43,44]. Similarly, the phenotype of EM��s may be heterogeneous. Infiltrating monocytes after an H3N2 challenge of C57BL/6 mice showed increased levels of both M1 (iNOS) and M2 (arginase 1) expression in CD11cneg/low CD11bhigh cells [45]. In FACS-sorted murine AM��s [45], this mix of M1- and M2-like phenotypes was observed after a lipopolysaccharide (LPS) challenge of C57BL/6 mice, where LPS is classically used to elicit an M1 response [20,46]. Thus, a picture is emerging of a complex heterogeneous cell population that can change rapidly in response to specific challenges and has the ability to grade danger signals and mount appropriate responses. Moreover, as more sophisticated techniques to isolate and identify specific biologically distinct cell populations and increasing numbers of markers are identified, it is clear that AM��s do not fit neatly into either of the M1 or M2 groupings.
Defining lung M�� populations
Understanding M��s in specific micro-anatomical niches is challenging, but several recent studies have tried to address this using more extensive characterization of phenotypes [37,47]. One key problem with studying lung M��s is the need to differentiate between AM��s, IM��s and EM��s. AM��s, which constitute more than 95% of the cells in the airspaces [45] are inherently autofluorescent [45,48]. In a comparison with peritoneal M��s and dendritic cells (DCs) [49,50], AM��s had a fluorescence emission that peaked in the FITC and PE fluorophore region (�� 450�C600 nm) [45], leading to high background noise with the addition of exogenous fluorophores. In contrast, IM��s, which are smaller with reduced granularity, have lower autofluorescence [37,51]. Analysis of human AM��s is additionally hindered by a wider spectrum of autofluorescence [52], with variation in intensity between subjects [53] and is further complicated by factors such as cigarette smoke hydrocarbons. However, this inherent autofluorescence can also be used as a tool in gating strategies. With the advent of multiparametric flow cytometric analysis, more sophisticated fluorophores and the use of conjugates that emit at higher wavelengths (>�� 660 nm) where the background autofluorescence is greatly diminished, better separation of lung M��s in steady state and disease models can be achieved [45].
Applying more stringent flow cytometry gating strategies coupled with multiple phenotypic markers, it is possible to begin to identify resident AM��s and tease apart subtle differences between this population and interstitial and infiltrating M��s. The ��-integrin CD11c is the most commonly used marker for AM��s [17,45,54�C55], with resident AM��s defined as CD11chighCD11b-. CD11c is also expressed on DCs [27,56] but DCs are also CD11b+. EM��s, in comparison, are CD11clow and CD11bhigh but, over time, they can upregulate CD11c and downregulate CD11b [13]. Some studies, however, have shown weak expression of CD11b on AM��s with increased expression of CD11b on resident CD11chighCD11b+ AM��s in mouse models exposed to LPS [13] or on CD11chighLy6C-CD11b+ AM��s following infection with influenza virus [45]. Ly6C is expressed on circulating monocytes [50,56] but its expression is downregulated upon maturation in tissues [54]. These results illustrate the plasticity of the system and highlight the difficulties in distinguishing populations. CD11chighCD11b+ intermediate AM�� phenotypes have been associated with chronic lung inflammation [45] and other chronic lung conditions such as chronic obstructive pulmonary disorder (COPD), which are characterized by increased numbers of M2 AM��s [57], as defined by increased expression of the M�� mannose receptor CD206 as well as CD163 and CD204 scavenger receptors associated with an M2 phenotype [20,58�C59]. Therefore it is still difficult to determine whether the M��s in these conditions are indeed infiltrating or in fact result from changes in expression in subsets of residential AM��s.
Other markers to define lung M�� populations include the sialic acid (SA) binding immunoglobulin superfamily lectin SiglecF. Siglecs are a family of receptors expressed on immune cells whose functions appear to mediate inhibitory signals through interaction with sialylated carbohydrate ligands [60]. SiglecF expression has been shown on mouse AM��s [37,61�C62] with differing expression levels used to differentiate between resident CD11chighSiglecFhighGR-1low M��s and exudate CD11chighSiglecFlowGR-1int M��s [62]. GR-1 is expressed on differentiated granulocytes and is widely used as a marker for neutrophils [63,64]. A combination of using fluorophores outwith the autofluorescence range along with more specific gating strategies and combinations of phenotypic surface markers will enable more accurate identification of cells. Expression of these and other markers on the three lung M�� populations is summarized in Table 1.
Another approach that has been used to identify lung M�� populations is the utilization of the phagocytic ability of the AM��s. Cell-labeling dyes such as PKH26 administered intranasally [13,54,70] are taken up by resident lung phagocytes and can be used to identify resident versus recruited M��s during subsequent infections by flow cytometry. PKH26 (PKH26-phagocytic cell linker) is instilled directly into lungs and forms fluorescent aggregates that accumulate in phagocytic cell compartments, not only in M��s. These aggregates are stable for 2�C3 weeks and can differentiate distinct lung cell populations through the analysis of mean fluorescence intensity: PKHhigh is thought to represent resident AM��s, with PKHlow recruiting mononuclear phagocytes [13] or interstitial M��s. This method also allows for monitoring dynamic changes in M��s in the lung, but the use of fluorescent dyes to label autofluorescent AM��s may be problematic and care must be taken with this approach. Furthermore, the addition of an exogenous agent to the lung has the potential to activate and change the M��s.
Transgenic mice are also being used as tools in order to dissect the role of M��s during infection. The MacGreen mice express EGFP under control of the CSF-1 receptor promoter, meaning that M��s can be readily identified within tissues [71,72]. However, as CSF-1 receptor expression is widespread in M�� subsets as well as being expressed in other polymorphs [73], this does not enhance the definition of individual subsets. Additional transgenic models of interest include the Csf2-/- mice, which lack AM��s as defined by autofluorescent CD45+CD11c+Siglec-F+ cells [74], and the CD169 diphtheria toxin receptor-transgenic mice, which allow specific depletion of F4/80++CD11b- AM��s [75]. The use of these models is discussed later.
These studies illustrate a range of techniques for the characterization of M�� populations in the lung and highlight the difficulties in defining specific phenotypes. However, much progress has been made and there is now a greater understanding of the plasticity of the M�� populations and an appreciation that M��s in the lung can change dynamically in response to the specific environment. It is clear that AM��s and EM��s can express both pro- and anti-inflammatory genes and receptors and may not be easily characterized by surface phenotypic markers alone. Incorporating additional functional analysis will probably establish a more accurate assessment of the M�� in question. In addition, many studies routinely use the murine model in order to study the lung; however, care must be taken when directly extrapolating these data to humans, as there are differences between the species that exist in the levels of some key M1 or M2 markers (iNOS and arginase-1) when M��s are polarized with IFN-�� or IL-4 [76].��
Influenza virus infection in the lung
Productive infection of alveolar epithelial cells (AECs) in the lung results in the release of new virus into the lumen of the lung and leads to cell death and subsequent breaches in the epithelial barrier. It is clear that epithelial and AM�� cells interact and that this interaction is complex and mediated through both soluble factors and cell�Ccell interactions [77]. Infection of these AECs or their destruction will change this cross-talk and alter the dynamics of the system. However, counter to this, infection of AM��s will also lead to dysregulation of this relationship.
Infection of the respiratory epithelium is determined in part by the viral hemagglutinin (HA) binding to SA on the cell surface. SA is predominantly ��-2,6 linked in the upper respiratory tract in humans [78,79] and both ��-2,3 and ��-2,6 linked in the lower respiratory tract [80�C82], while mice have predominantly ��-2,3 and little ��-2,6 [80,83�C84]. The ability of the various HAs to bind preferentially to ��-2,3- or ��-2,6-linked SA is thought to be important in determining host specificity of different virus strains [85]. However, human viruses thought to have tropism for ��-2,6-linked SA grew to similar titers in mice to the titers reached by virus adapted to bind to ��-2,3-linked SA [86]. The presence of SA alone is clearly not the only factor determining entry into and replication in cells and surface molecules, such as the calcium-dependent C-type lectins [87], including the M�� mannose receptor (CD206) [88] and the M�� galactose-type lectin [89,90], have been implicated in virus entry. SA, as well as other potential entry receptors, is present on the surface of AM��s and, therefore, they should be viable targets for infection. Human AM��s have been shown to express ��-2,3 [91], while the murine airway M��s obtained by bronchoalveolar lavage express ��-2,6 [92,93]. Although factors in addition to the presence of receptors alone will likely limit infection, AM��s are one of the first cells to encounter viruses, yet the 'outcome' of the infection of these cells is still not clear (Figure 1A).
M�� infection in vivo & in vitro
The permissiveness of M��s to infection with influenza virus has been studied in vitro [11,25,91,93�C100], in vivo and ex vivo (lung explant tissue) [101] using a variety of influenza virus subtypes and sources of M��s: human, ferret [102], porcine [103] and murine. Immunohistochemical techniques have demonstrated the presence of viral proteins in lung M��s in vivo (Figure 1B) [6,86,104�C105], and infection of mice with a GFP reporter virus showed that viral-encoded proteins could be found in both AM��s and monocytes [106]. These studies do not differentiate between infected cells and the presence of infected material in phagocytic cells. However, in vitro culture of M��s extracted from whole mouse lungs resulted in the production of infectious virus [100], indicating the presence of replicating virus in lung M��s.
The extent to which influenza viruses replicate in M��s in vivo and the role that this plays in pathogenesis are still areas of debate. A number of in vitro studies have looked at the replication of seasonal, pandemic and H5N1 viruses in M�� populations, including AM��s [91,93�C94,96�C100] and MDMs [97,100,107], and this subject was reviewed recently by Short et al., who concluded that a systematic study in which the ability of a number of strains of virus to replicate in M��s from different sources (e.g., human or murine AM��s vs MDMs) was needed in order to understand the conditions that lead to productive infection [108]. Current evidence indicates that highly pathogenic viruses, such as some strains of H5N1 and the 1918 pandemic H1N1 virus, can replicate in AM��s and MDMs, but in the majority of cases, M�� infections are nonproductive and do not result in the release of infectious virus particles.
The barrier to productive virus infection and replication in M��s is not thought to be at entry per se, as similar levels of viral NP protein, as determined by immunohistochemistry in human AM��s, were detected when comparing the highly pathogenic H5N1 virus A/HongKong/483/97 (HK/483) and the seasonal H1N1 virus A/HongKong/54/98, yet only the highly pathogenic infection produced any significant virus titers in culture supernatants [91]. When the ability of viruses representing 16 different HA subtypes to replicate in a M�� cell line (RAW264.7) was assessed, only a subset of H5 subtypes were able to replicate and produce infectious virus. Comparison of the H5N1 HK/483 strain, which replicates efficiently in murine AM��s as well as in RAW264.7s, with the 2009 pandemic H1N1 A/California/04/2009 (CA/09) strain showed that while infection of RAW264.7 cells with CA/09 H1N1 and HK/483 H5N1 resulted in >97% of cells staining positive for NP viral protein within 30 min of infection, this number diminished more rapidly in the CA/09 infection, reducing to 43% within 90 min compared with 89% in the HK/483-infected cells [99]. Thus, the initial stages of entry and infection are not blocked in the H1N1 infection, but the viral antigen is rapidly lost in the CA/09-infected cells. In addition, no vRNA or cRNA was detected by PCR, and NS1 protein, which must be synthesized de novo in infected cells, could not be detected in the CA/09-infected cells. As this restriction in the H1N1 virus appears to be early in infection after entry, the authors hypothesized that the viral HA, which is involved in the fusion of the viral membrane with late endosomes and the release of viral capsid into the cytoplasm, might play a role. Using a reverse genetics approach, a CA/09 H1N1 virus with an HK/483 HA gene was shown to replicate in RAW264.7 cells, albeit with slightly reduced kinetics when compared with the HK/483 parental virus [99]. The authors speculate that the ability of the H5 viruses to replicate in M��s may be either due to use of an alternative receptor that allows entry into a productive pathway or to these HAs having different fusion characteristics and more readily allowing the release of viral ribonucleoproteins into the cytoplasm of the cell.
Other studies have addressed the question of virus replication in M��s by comparing different M�� populations. Direct comparison of infection of human AM��s with MDMs from the same donor showed that, for a highly pathogenic H5N1 strain, AM��s were less readily infected than MDMs and more infectious virus was produced from MDMs than from AM��s [97]. Similar results were found for seasonal virus and the 2009 H1N1 pandemic strain. Thus, it would appear that MDMs, which may represent the EM�� population, are more permissive than AM��s.
The role of M�� infection in pathogenesis
The relationship between the ability to infect and replicate in M��s and the pathogenicity of different virus strains is intriguing. The productive infection of AM��s by highly pathogenic viruses may explain, in part, the differences in pathogenesis observed between high- and low-pathogenicity viruses, but this is unlikely to be the whole explanation. Infection of M��s with seasonal IAV strains results in cell activation and the production of cytokines and chemokines, including TNF-��, type I interferons, IL-1, IL-6 and CC chemokines. Following the infection of humans with the highly pathogenic H5N1 strains that appeared in Hong Kong in 1997, a number of studies showed that infection of human MDMs with H5N1/97 resulted in higher expressions of proinflammatory cytokines, including TNF-��, IFN-��, RANTES (CCL5), MIP-1�� (CCL3), MIP-1�� (CCL4), MCP (CCL2) and IP-10 (CXCL10), than were produced by infection with seasonal strains [109,110]. Elevated levels of proinflammatory cytokines are associated with highly pathogenic virus infection in vivo [6] and, as M��s are major producers of these molecules, this leads to the hypothesis that infection of M��s results in overproduction of inflammatory cytokines.
Investigations of the patterns of expression of inflammatory mediators in infected AM��s, however, do not completely reflect those seen in MDMs. Van Riel et al. compared AM��s and MDMs from the same donor and found that infection of AM��s by H5N1 did not result in higher levels of TNF-�� or more infectious viruses than those produced in response to infection with a seasonal strain or the 2009 pandemic H1N1 [97]. AM��s were more readily infected by H5N1 than by a seasonal H3N2 or the 2009 pandemic virus, but produced less infectious virus than MDMs. Yu et al. also showed that MDMs produced more TNF-��, IP-10 (CXCL10) and RANTES (CCL5) than AM��s (although not from the same donor), with H5N1 producing higher levels of cytokines than H1N1 in both cell types [91]. The use of different strains of H5N1 may account for the differences in replication, but both clearly identify a difference in response between AM��s and MDMs.
A recent microarray study of H1N1 A/PR/8/34 (PR8) infection in AM��s showed that type I and type III interferons were among the top 25 genes to be upregulated with this infection [25]. Interestingly, the study also showed that mRNAs for M�� scavenger receptors (Dectin 1, MSR1, CD36, MRC1 and MARCO) were downregulated by infection, and infected AM��s showed decreased the phagocytic ability for zymosan, an important functional change induced by virus infection. Production of infectious virus is not required for transcriptional changes in AM��s, but there is a requirement for live virus, as UV inactivation abolishes the induction of most mediators. An exception is IP-10 (CXCL10), which is robustly induced upon infection of human AM��s with inactivated PR8 or seasonal H3N2 virus [25]. This suggests that virus transcription is necessary for the induction of most cytokines and chemokines.
Other mechanisms by which infection of M��s can contribute to disease are beginning to be addressed by both in vivo and in vitro studies. Release of TRAIL by EM��s was shown to contribute significantly to AEC apoptosis in mice infected with IAV [111]. The recent demonstration that IFN-�� (which is rapidly produced after PR8 infection of murine AM��s) resulted in the upregulation of TRAIL expression by AM��s provides a direct link between virus infection of AM��s and pathogenesis [62]. In this model, administration of antibodies to type I interferons prevented AEC apoptosis, confirming the role of type I interferons in vivo. There is some evidence that these findings extend to a clinical situation, as TRAIL expression was increased in AM��s from patients hospitalized with pdH1N1 virus [62]. However, TRAIL mRNA levels were not increased in the post-mortem lung tissue from fatal 2009 pdH1N1 cases [112], whereas FasL mRNA, which is also associated with apoptosis, was elevated.
Are alveolar M��s depleted after influenza virus infection?
The number of M��s in the lung increases during IAV infection (Figure 1Biii), but is believed to return to preinfection homeostatic numbers following the resolution of the infection. Given that AM��s are phenotypically and functionally different from EM��s, the fate of the individual cell populations during and after infection is likely to be important in the recovering lung. In a model system where recruitment of M��s to the lung is mediated by LPS, the numbers of AM��s remain constant throughout, with the increase being due to recruited EM��s. The decrease in M�� numbers on resolution is brought about by FasL-mediated apoptosis of recruited EM��s [13]. These authors also showed that FasL mediated the decrease in M�� numbers following IAV infection, but did not distinguish between EM��s and resident AM��s, so it is not clear whether both populations are affected. An obvious difference between LPS challenge and IAV infection is that IAV can infect and cause apoptosis of M��s [113], so the effect of this on the different populations must be considered. As discussed earlier, comparison of MDMs with AM��s showed that AM��s were less susceptible to infection than MDMs and produced fewer infectious viruses; hence, it might seem unlikely that the AM�� population is more affected by IAV infection than the EM�� population. However, Ghoneim et al. recently provided evidence that AM��s, defined as CD11chighF4/80highCD11b-, were depleted after infection of mice with PR8 [54]. Depletion could be demonstrated at as early as 3 days postinfection, and by 7 days, there was a 90% decrease in the numbers of resident AM��s. Moreover, they found a significantly higher number of dead AM��s in the infected group, with death due to a secondary necrotic process, while no difference was seen in the IM�� populations. Such an impact on the AM�� population is likely to have a significant impact on secondary bacterial infections [54], which is a common problem in influenza virus infections [114]. Others, however, have not described changes in the AM�� numbers post-influenza virus infection [45,70]. These differences are likely to relate to the different methods used to define M�� populations, and conclusions are awaited from further studies.
The role of M��s in infection: friend or foe?
The critical role of AM��s in response to a respiratory infection with influenza has been shown in depletion studies using dichloromethylene-bisphophonate (clodronate)-loaded liposomes. The liposomes are ingested by phagocytes, resulting in the release of clodronate, which causes cell death [115]. Clodronate liposome depletion of M��s in mice [6,94] and pigs [103] resulted in higher viral loads and increased disease severity, implying a role for M��s in controlling disease severity. However, these studies could not distinguish between the contributions of different lung M�� populations and, moreover, the effects of clodronate on other cell populations or toxicity cannot be ruled out. The route of clodronate administration is also important, with intranasal or intratracheal routes eliminating AM��s, while the intravenous route also eliminated interstitial M��s [47]. However, it is likely that other pulmonary cell subsets, such as infiltrating monocytes or DCs, will also be affected [75]. It is therefore critically important to use a range of appropriate phenotypic markers in order to monitor depletion. Recently, other studies using transgenic mice have, however, confirmed that AM��s have a critical role in preventing severe infections. Infection of GM-CSF-deficient (Csf2-/-) mice with a sublethal dose of PR8 influenza virus resulted in increased disease severity [74]. GM-CSF is critical for the maturation of AM��s, characterized as CD45+CD11c+SiglecF+ with high autofluorescence, and AM��s are completely absent in the lungs of these mice. Reconstitution of AM��s through neonatal transfer of wild-type AM�� progenitors in these mice restored protection from lethal disease. Although the phenotype of DCs in the lungs of these mice shows lower expression of CD103 than wild-type DCs, DC numbers are similar to the wild-type and the mice have intact T- and B-cell responses. In addition, transgenic mice with lung-restricted overexpression of GM-CSF have more AM��s than wild-type mice and are resistant to influenza virus-induced mortality. This resistance is abrogated by the depletion of lung M��s with clodronate, but not by the depletion of T or B cells or neutrophils [44]. These authors and others also demonstrated that intranasal administration of GM-CSF protects against lethal influenza infection [43].
Similarly, depletion of AM��s by the administration of diphtheria toxin intraperitoneally to CD169�Cdiphtheria toxin receptor-transgenic mice, which results in the transient removal of AM��s, led to increased virus loads, lung pathology and inflammation, and ultimately, these mice had to be euthanized [75]. As well as being expressed in AM��s, CD169 (Siglec1) [68] is found in other tissue M��s. However, administration of diphtheria toxin intratracheally, removing only lung AM��s prior to intranasal infection with influenza, resulted in indistinguishable morbidity, suggesting that the observed pathology is primarily the result of the depletion of the lung AM��s. Overall, the data support a conclusion that AM��s are critical in the protection against influenza virus-driven morbidity and mortality.
Other roles for M��s in influenza infections
It has been suggested that nonproductive infection of M��s may have a role in controlling the virulence of influenza virus infections. That is, M��s may be able to act as a 'sink' that absorbs virus and prevents productive infection of epithelial cells. Support for this comes from studies by Tate et al., who showed that a virus that readily infected M��s in vitro was less pathogenic in vivo than a virus with a limited ability to infect M��s [94]. Depletion of M��s in vivo increased the virulence of these M��-tropic viruses, providing evidence that the ability to infect M��s effectively attenuated the virus. In this case, the restriction on M�� infection was related to different levels of glycosylation of the HA molecules, where limited glycosylation led to decreased binding to the M��s. More recently, Schneider et al. have suggested a similar mechanism [74]. They used microarray analysis in order to show that sorted AM��s from PR8-infected mice had elevated expression levels of interferon-induced genes, including IFITM3, which prevents virus infection by blocking the release of virus components from the endosomes [116�C118]. These two very different mechanisms highlight the complexity of the M���Cvirus interactions.
Long-term consequences of M�� infection with influenza
Influenza infection causes significant morbidity and mortality and infection often predisposes individuals to subsequent secondary bacterial infections that can ultimately be the predominant cause of death [119]. AM��s in the lung are important for surveillance and clearance of pathogens, such as bacteria, and as discussed, there is evidence that infection with influenza results in changes in airway M�� populations [45], depletion of resident AM��s [54] and changes in M�� function (discussed below). This will result in an environment that aids the establishment of secondary pneumococcal infections [54,120]. Influenza-mediated destruction of the AECs, which is key in the maintenance of a steady state in the lung [121,122], will likely also play a role in bacterial invasiveness and susceptibility to secondary bacterial infections [123]. However, bacterial infections still occurred in mice when the virus caused minimal damage to the AECs [124]. The mechanisms underlying the interactions between coinfecting pathogens are complex [114] and discussed at length in a recent review by McCullers [125].
Influenza infection within the lung has been shown to alter many pathways that are essential for pathogen recognition and clearance (Figure 1) [25], increasing levels of CD200R, the receptor for the negative regulatory ligand CD200 on murine AM��s [65], as well as decreasing levels of MARCO scavenger receptor [70], impairing the ability of AM��s to respond to and phagocytose bacteria, resulting in subsequent bacterial outgrowth. Interestingly, in mice lacking CD200 or MARCO, there was a 'better outcome' of influenza virus infection with faster clearance of virus [65,126], implying that removal of the negative regulation of AM��s was beneficial. However, this came at a price, with increased numbers of M��s and inflammatory cytokines, which ultimately caused pathology. Alterations in the expression levels of receptors that are important for the recognition and killing of bacteria have been shown to persist for significant periods [127], possibly altering the lung threshold [128] to any subsequent challenge, leading to decreased responsiveness of murine AM��s to TLR ligands, as well as decreased cytokine production and dysregulation in neutrophilia [129] even months after clearance of virus [127,130]. In addition, noninfectious allergic inflammation has also been shown to result in subsequent sensitivity to TLR ligands due to altered AM��s [131].
Induction of an antiviral state, and in particular interferons, in response to a virus infection can also suppress M�� and neutrophil functions. IFN-�� is produced early in response to viral infection, mainly by M��s and NK cells, and later, when the adaptive responses begin, by CD8 T cells. Although IFN-�� is not required for influenza virus clearance in vivo [132], it is produced during infection and can alter the phagocytic capacity of M��s. For example, treatment of AM��s with IFN-�� has been shown to inhibit the phagocytosis of Streptococcus pneumonia [70]. More recently, pharmacologic inhibition of IFN-�� in influenza-infected mice resulted in decreased morbidity and lower S. pneumonia bacterial loads [133]. Cytokines such as IL-10 are important during the resolution of inflammation, although increased levels in the the lungs after influenza infection have been associated with enhanced susceptibility to pneumococcal pneumonia [134,135]. However, studies in IL-10-/- mice infected with influenza showed minimal differences from the wild-type mice in terms of clearing S. pneumonia [70,134], although they clear virus more quickly, possibly due to early adaptive immunity [136,137]. Increased levels of alternatively activated AM��s, defined by the expression of prototypic alternative genes Arg-1, Ym1 and FIZZ [29], resulted in increased susceptibility to secondary challenge with S. pneumonia [138]. Therefore, it is clear that influenza virus infection and the host response to this pathogen results in alterations in lung effector functions that can have sustained impairments, which are important when looking at subsequent bacterial (or viral) exposure in the lung.Changes in the lung M�� populations after virus infection are also mimicked in acute and chronic lung conditions [76,139] when the lung can become particularly susceptible to bacterial infections [140,141], although studies are more limited on the outcome of subsequent viral challenges. However, depression of the innate response may also be beneficial to subsequent challenge [142]. Indeed, some studies have shown that the infection of mice with MHV-68, a murine gammaherpesvirus, protected mice from Listeria monocytogenes and Yersinia pestis [143] and increased survival to a subsequent influenza challenge [144]. Importantly, the authors demonstrated a role for AM��s, where adoptive transfer of AM��s from MHV-68-infected mice, which showed upregulation of MHC class II, resulted in decreased levels of influenza virus [144]. It is intriguing that a persistent latent infection can modulate the outcome of this secondary challenge. Our previous observation that human AM��s from chronically HIV-infected patients have decreased innate immune responses to TLR ligands [145,146] is consistent with sustained long-term changes in lung innate cells during ongoing infections. It is clear that infectious (and noninfectious) insults to the lung can result in long-term alterations in lung M��s, either through changes to resident AM��s or changes in the phenotypes of EM��s, which can dictate downstream responses to heterologous challenges within the lung.
Conclusion
While it is clear that M��s have a critical role in the pathogenesis of influenza virus infections, many important questions remain to be answered. These include: what is the block on virus replication in M��s and how is this overcome by some strains of the virus? What virus gene products and host responses are involved in this process? What happens to M��s during infection and resolution? For each of these questions, infection of the different M�� populations will need to be investigated. Tools for identifying specific M�� populations are becoming available and these, together with systematic approaches to understanding virus gene function, will likely provide answers to these questions and identify novel targets for therapeutic interventions.��
The role of macrophages in influenza A virus infection - Edinburgh Research Explorer
https://www.research.ed.ac.uk/portal/en/publications/the-role-of-macrophages-in-influenza-a-virus-infection(5dedd668-5cb2-47ff-bf92-4600e08805f1).html.....
Alveolar M��s are the primary sentinel cell within the lung and reside in the lumen of the alveolus �C that is, the airspace �C and it is estimated that there is at least one AM�� per alveolus at homeostasis [12]. AM��s are thought to be long-lived cells [13] with an estimated turnover of around 40% in a year [14] that can persist, even during inflammation. AM��s can be exposed to 1010 particles per day [15] and the challenge is to mount an appropriate response that limits deleterious inflammation without damaging the delicate alveolar capillary membrane or interfering with normal gas exchange. Increasing evidence suggests that such tissue-specific M��s are not, as previously thought, terminally differentiated M��s from a blood monocyte origin, but are in fact derived from a separate pool. Fate mapping studies show the prenatal origin of AM��s, confirming that they are independent of blood monocytes [16] and that, at least in uninfected states, they replicate in situ to maintain the population [17,18]. This was reviewed recently by Davies et al. [19].
Depleting macrophages by the inhalation of CL caused a profound inhibition of the early release of inflammatory cytokines into the airways after RSV infection and lessened the activation and recruitment of NK cells. Despite the virtual abolition of early inflammatory mediator release and a rise in viral load at day 4, there was no change in the weight loss, lung function deterioration, or T-cell recruitment that characterizes the later stages of RSV infection. In view of the known viral sensing, proinflammatory, and immunomodulatory effects of AM, depletion seemed to have remarkably little effect on these responses.
A number of studies have observed a very early release of cytokines and chemokines after RSV infection similar to that seen here (6). Our data suggest that this release is AM dependent. This is supported by other studies that show that the activation of NF-��B signaling pathways, which are key in initiating many proinflammatory responses, in the lungs of mice infected with RSV was entirely dependent on the presence of AM (7).The role of macrophages in influenza A virus infection | Future Virology
https://www.futuremedicine.com/doi/10.2217/fvl.14.65��
��
Figure 1.
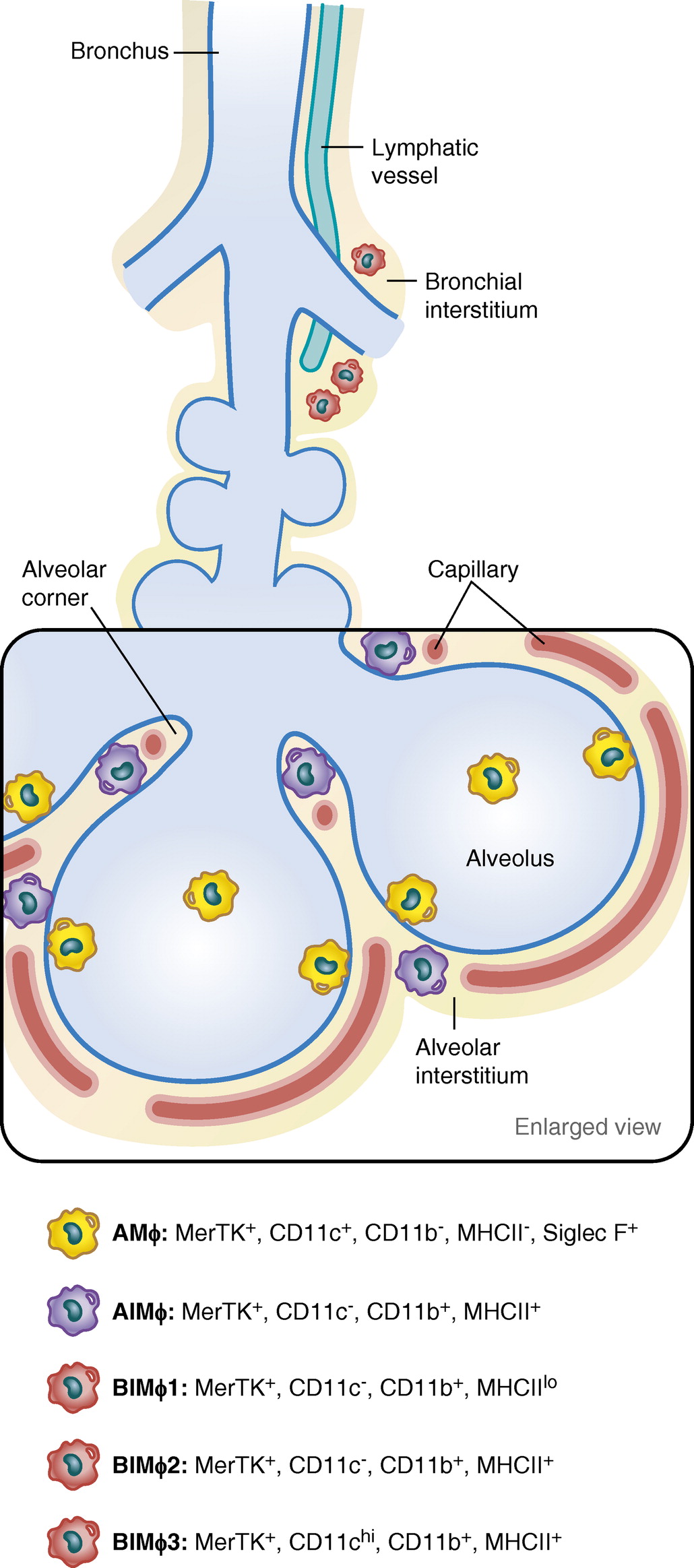
Graphic representation of resident macrophages present in the lung. In the alveolus, sessile (attached) and nonsessile (unattached) alveolar macrophages (AM��s) are shown. Alveolar interstitial macrophages (AIM��s) and bronchial IMs (BIM��s) represent M��s residing in alveolar and bronchial interstitium, respectively. Phenotypic cell surface markers used to identify and characterize the specific M��s are shown. Illustration by Jacqueline Schaffer.Lung Interstitial Macrophages Redefined: It Is Not That Simple Anymore | American Journal of Respiratory Cell and Molecular Biology
https://www.atsjournals.org/doi/full/10.1165/rcmb.2017-0158ED��
��
J Virol. 2008 May; 82(9): 4441�C4448.
Published online 2008 Feb 20. doi: 10.1128/JVI.02541-07
PMCID: PMC2293049
PMID: 18287232
Alveolar Macrophages Are a Major Determinant of Early Responses to Viral Lung Infection but Do Not Influence Subsequent Disease Development▿
Philippa K. Pribul,1,† James Harker,1,† Belinda Wang,1 Hongwei Wang,2 John S. Tregoning,1 J��rgen Schwarze,2 and Peter J. M. Openshaw1,*
Author information Article notes Copyright and License information Disclaimer
Department of Respiratory Medicine, the Centre for Respiratory Research and the MRC & Asthma UK Centre in Allergic Mechanisms of Asthma, National Heart and Lung Institute, Imperial College London, St. Mary's Campus, London W2 1PG, United Kingdom,1 Centre for Inflammation Research, University of Edinburgh, the Queen's Medical Research Institute, 47 Little France Crescent, Edinburgh EH16 4TJ, United Kingdom2
*Corresponding author. Mailing address: Department of Respiratory Medicine, Paddington Campus of Imperial College, Norfolk Place, London W2 1PG, United Kingdom. Phone: 44 20 7594 3854. Fax: 44 20 7262 8913. E-mail: ku.ca.lairepmi@wahsnepo.p
Macrophages are abundant in the lower respiratory tract. They play a central role in the innate response to infection but may also modulate excessive inflammation. Both macrophages and ciliated epithelial cells respond to infection by releasing soluble mediators, leading to the recruitment of innate and adaptive effector cells. To study the role of lung macrophages in acute respiratory viral infection, we depleted them by the inhalation of clodronate liposomes in an established mouse model of respiratory syncytial virus (RSV) disease. Infection caused an immediate local release of inflammatory cytokines and chemokines, peaking on day 1, which was virtually abolished by clodronate liposome treatment. Macrophage depletion inhibited the activation (days 1 to 2) and recruitment (day 4) of natural killer (NK) cells and enhanced peak viral load in the lung (day 4). However, macrophage depletion did not affect the recruitment of activated CD4 or CD8 T cells, weight loss, or virus-induced changes in lung function. Therefore, lung macrophages play a central role in the early responses to viral infection but have remarkably little effect on the adaptive response occurring at the time of peak disease severity.Macrophages are key effector cells of the innate immune response to pathogen invasion but are also thought to have an immune-suppressive effect in the lung, limiting excess inflammation (12). Normal resting alveolar macrophages (AM) produce low levels of inflammatory cytokines and are less actively phagocytic than their counterparts in other tissues, possibly due to lower levels of the phagocytic receptor CD11b (12). AM activation causes increased phagocytosis and production of numerous proinflammatory cytokines, including tumor necrosis factor alpha (TNF-��), interleukin-6 (IL-6), and IL-8 (2). They are critical in determining the outcome of a number of respiratory infections, playing a role in controlling the replication and spread of both viruses, e.g., influenza virus (36), and bacteria, e.g., Mycobacterium tuberculosis (21, 22).
Respiratory syncytial virus (RSV) is a nonsegmented, negative-strand RNA virus of the family Paramyxoviridae. It is the leading cause of infant hospital admissions, causing 70% of bronchiolitis hospitalizations in the developed world (10). The relative contribution of the adaptive and innate responses to the pathogenesis of RSV disease is unclear. The importance of T cells in RSV disease has been extensively studied in the mouse model, where skewed Th1/Th2 responses are associated with different forms of lung disease (30). In humans, severe infantile bronchiolitis is associated with markers of Th2 immunity (1, 23), and RSV-specific T cells producing gamma interferon (IFN-��), IL-4, and IL-5 have been detected in children with RSV, with IL-4 and IL-5 being detected only in those children with severe bronchiolitis (31, 20). In the 1960s, the vaccination of children with formalin-inactivated RSV caused exacerbated illness when the children subsequently became infected with RSV (18, 24). A recent study of fatal cases of RSV bronchiolitis in infants has shown that death is associated with a lack of evidence of adaptive immune responses to infection, such as an absence of recruitment of cytotoxic T cells and lymphocyte-derived cytokines, robust viral replication, and increased apoptosis within bronchiolar epithelial cells (39). In addition, single-nucleotide polymorphism screening of infants hospitalized for severe RSV bronchiolitis shows that polymorphisms in genes associated with innate immunity, such as IFNA2 and NOS2, as well as the known polymorphisms in the adaptive pathways have strong associations with RSV disease in childhood (17).
Liposome-encapsulated dichloro-methylene-diphosphonate (clodronate) is taken up by phagocytic cells in vitro and in vivo. The liposome bilayers are disrupted in the lysosome by phospholipases, allowing the escape of clodronate into the cell; once the clodronate has accumulated sufficiently, the macrophage is irreversibly damaged and dies by apoptosis (38). In mice, the administration of clodronate liposomes (CL) can therefore be used to selectively deplete macrophages (37). The depletion of macrophages from the lungs is associated with an increased pulmonary immune response characterized by dendritic cell (DC) trafficking (16, 35). Macrophage depletion of mice followed by RSV infection has been shown to result in increased viral titers (3); however, the role that macrophages play in initiating and modulating the immune responses and disease after RSV infection has not been fully elucidated.
To delineate the role of macrophages in the immune response to RSV infection, we depleted macrophages by the intranasal administration of CL prior to infection and characterized both the innate and adaptive immune responses and their effects on viral replication and disease. While macrophage depletion strongly inhibited the immediate release of inflammatory mediators and the activation and recruitment of natural killer (NK) cells after viral infection, it had little effect on the adaptive response or overt disease.��
DISCUSSION
Depleting macrophages by the inhalation of CL caused a profound inhibition of the early release of inflammatory cytokines into the airways after RSV infection and lessened the activation and recruitment of NK cells. Despite the virtual abolition of early inflammatory mediator release and a rise in viral load at day 4, there was no change in the weight loss, lung function deterioration, or T-cell recruitment that characterizes the later stages of RSV infection. In view of the known viral sensing, proinflammatory, and immunomodulatory effects of AM, depletion seemed to have remarkably little effect on these responses.
A number of studies have observed a very early release of cytokines and chemokines after RSV infection similar to that seen here (6). Our data suggest that this release is AM dependent. This is supported by other studies that show that the activation of NF-��B signaling pathways, which are key in initiating many proinflammatory responses, in the lungs of mice infected with RSV was entirely dependent on the presence of AM (7).
In contrast with our findings that the marked reduction of proinflammatory mediators in the airways did not affect the overall disease, the targeted removal of individual mediators does lead to reduced disease. CCL3−/− mice have reduced RSV inflammation (6), the use of depleting antibody to remove TNF reduces weight loss associated with RSV infection (15), and neutralizing the function of CCL5 by the administration of Met-RANTES (a competitive inhibitor) reduces both the CD4 and CD8 responses to RSV (4). In addition, non-IFN-responsive, STAT-1 knockout mice show increased illness and Th2-skewed disease (5). These studies describe the effects of systemic and presumably complete removal of either the factor or the signaling, whereas we specifically depleted AM and found altered cytokine levels in the BAL cells but not the lungs. This reflects a greater (80%) reduction in the number of AM in the BAL cells than the 50% reduction in the number of macrophages in the lung tissue. Therefore, it is possible that the inflammatory mediators in the surrounding lung tissue and alternative sites, such as the lymph nodes and spleen, contribute to the disease seen in the current study.
DC are thought to ��license�� NK cells, potentiating their activation and cytotoxicity (25, 26). Here we show in naïve lungs, where DC are scarce, that AM are required to both recruit and activate NK cells in response to RSV infection. Recently, human macrophages have been shown to be able to activate NK cells by a mechanism that involves contact-mediated signaling through the immune synapse (28). The loss of this signaling may explain the loss of NK cell activation that we observed. It has been shown that IFN-�� production by resident cells in the liver promotes MIP-1�� production and subsequent NK cell migration (32, 33). Therefore, it seems likely that the loss of chemokine production, such as with MIP-1��, may also be critical in determining NK cell recruitment to the lungs.
We have previously shown that NK cells are a major source of early IFN-�� during viral infection (13). In addition, they play an essential role in RSV immunity along with specific T cells, as the depletion of both NK cells and CD8 T cells led to the dissemination of virus from the lungs to the lymph nodes (14). This suggests that the loss of early (days 1 to 4) IFN-�� seen after depletion is most likely due to reduced NK cell recruitment; furthermore, the loss of NK cells may also explain the increased viral titer on day 4. AM could also potentially be directly antiviral; they are the first cells to encounter pathogens in the airways, acquiring the vast majority of inhaled particles by efficient phagocytosis (16). Although viral titers in the lung on days 1 and 2 were unaffected by macrophage depletion, suggesting that initial RSV replication takes place mainly in epithelial cells, at the peak of replication on day 4, depletion led to a higher viral load. This points to a role for AM in controlling antiviral activity against RSV infection. Virus was cleared by day 8 in both normal and macrophage-depleted mice; later clearance of virus is associated with effective CD8 and antibody responses, both of which were unaffected by CL treatment.
In addition to their direct cytotoxic role, NK cells also have been shown to strongly influence the subsequent CD8 T-cell response via their cytokine secretion (13). Although IFN-�� and NK cells were reduced up to day 4 in our study, subsequent T-cell numbers were not altered with the CL treatment. In addition to the normal T-cell response to RSV, weight loss and lung function were unaffected by AM depletion. T-cell infiltration correlated well with these indicators of disease after RSV infection (Fig. (Fig.1),1), and exacerbated CD8 T-cell responses have been strongly associated with both measures of disease, in both RSV (9) and influenza A virus (27) infection.
The dual role of macrophages in both regulation and inflammation may explain why, despite decreasing inflammatory mediator release, AM depletion has no effect on the adaptive immune response to infection and therefore disease. AM suppress the migration (16) and antigen presentation capacity (11) of DC. AM removal has been shown to increase the trafficking of antigen toward the lymph nodes (16); this may be the case in RSV infection. Such increased antigen transport may be due to the presence of enhanced DC numbers in the lungs during the early stages of infection in macrophage-depleted mice. Our results support those of Wijburg et al., showing that DC are the main APC for the induction of virus-specific T-cell responses, since these responses were still effectively induced in the absence of AM (40). The increased viral burden, and antigen load, on day 4 postinfection may also promote enhanced T-cell recruitment, compensating for the loss of inflammatory signals. NK cells may also play a role in the suppression of T-cell responses (29, 34), and therefore, the decrease in NK cell recruitment following AM depletion may allow an increase in the T-cell response.
In conclusion, the depletion of lung macrophages dampens the innate response to RSV infection and increases the peak viral load but does not change weight loss or lung function, as parameters of disease. Therefore, our findings support a role for T-cell-mediated factors in RSV disease. However, in infants with diminished adaptive responses to viral infection, macrophages and the innate responses that they control could be critical in controlling viral load in the lung.Alveolar Macrophages Are a Major Determinant of Early Responses to Viral Lung Infection but Do Not Influence Subsequent Disease Development
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2293049/��
Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS
Murielle Gr��goire, Fabrice Uhel, Mathieu Lesouhaitier, Arnaud Gacouin, Marion Guirriec, Frederic Mourcin, Erwan Dumontet, Arnaud Chalin, Michel Samson, Laure-Line Berthelot, Adrien Tissot, Mallorie Kerjouan, St��phane Jouneau, Yves Le Tulzo, Karin Tarte, Jaroslaw W. Zmijewski, Jean-Marc Tadi��
European Respiratory Journal 2018 52: 1702590; DOI: 10.1183/13993003.02590-2017
ArticleFigures & DataInfo & Metrics PDF
Abstract
Exaggerated release of neutrophil extracellular traps (NETs) along with decreased NET clearance and inability to remove apoptotic cells (efferocytosis) may contribute to sustained inflammation in acute respiratory distress syndrome (ARDS). Recent studies in experimental models of ARDS have revealed the crosstalk between AMP-activated protein kinase (AMPK) and high-mobility group box 1 (HMGB1), which may contribute to effectiveness of efferocytosis, thereby reducing inflammation and ARDS severity.
We investigated neutrophil and NET clearance by macrophages from control and ARDS patients and examined how bronchoalveolar lavage (BAL) fluid from control and ARDS patients could affect NET formation and efferocytosis. Metformin (an AMPK activator) and neutralising antibody against HMGB1 were applied to improve efferocytosis and NET clearance.
Neutrophils from ARDS patients showed significantly reduced apoptosis. Conversely, NET formation was significantly enhanced in ARDS patients. Exposure of neutrophils to ARDS BAL fluid promoted NET production, while control BAL fluid had no effect. Macrophage engulfment of NETs and apoptotic neutrophils was diminished in ARDS patients. Notably, activation of AMPK in macrophages or neutralisation of HMGB1 in BAL fluid improved efferocytosis and NET clearance.
In conclusion, restoration of AMPK activity with metformin or specific neutralisation of HMGB1 in BAL fluid represent promising therapeutic strategies to decrease sustained lung inflammation during ARDS.
Tweetable abstract @ERSpublications
click to tweet
Restoration of AMPK activation and specific inhibition of HMGB1 could reduce lung inflammation during human ARDS
Introduction
Acute respiratory distress syndrome (ARDS) is an acute inflammatory lung injury characterised by a hypoxaemic respiratory failure following a disruption of the endothelial�Cepithelial barrier, alveolar damage and pulmonary oedema [1, 2]. In spite of significant advances in critical care, antibiotics and lung ventilation strategies, effective therapeutic interventions to diminish the severity of lung injury and mortality among ARDS patients are not available [3�C5]. Neutrophils are the first line of innate immune response, producing antibacterial peptides, reactive oxygen species, cytokines and other inflammatory mediators [6]. Neutrophils are also able to release neutrophil extracellular traps (NETs), a unique mechanism of DNA deployment into the extracellular milieu [7, 8]. Although these functions are important to target microbial agents, exaggerated and prolonged activation of neutrophils could contribute to the development of acute lung injury (ALI) [9�C11]. In particular, the lifespan of neutrophils is prolonged during ARDS and several studies have shown a deleterious impact associated with delayed apoptosis of neutrophils [9, 12�C14]. Similar to substantial production of inflammatory mediators, neutrophil-driven excessive NET formation can worsen inflammation, in particular in sterile inflammatory conditions [15�C17]. Therefore, time-dependent neutralisation of apoptotic cells, especially apoptotic neutrophils, and clearance of NETs have appeared to be important steps in the resolution phase and recovery from lung injury, since effective removal of dying cells (known as efferocytosis) plays a crucial role in the maintenance of tissue homeostasis [18]. Macrophage phagocytic function is typically associated with engulfment of dying cells; however, less is known about the mechanisms involved in NET clearance [19�C21]. Besides the recently described benefit of DNase I in experimental sepsis, the role of macrophages in the clearance of NETs, including in conditions associated with development and resolution of ARDS, has not been determined [22].
AMP-activated protein kinase (AMPK) is a serine-threonine protein kinase that functions as a crucial metabolic sensor and regulates cellular energy production and expenditure [23]. Recent studies indicate that AMPK activation also has a potent anti-inflammatory effect. In addition, AMPK activation can stimulate macrophage efferocytosis, along with neutrophil and macrophage capacity to ingest bacteria [24, 25]. However, inflammatory conditions are accompanied by a reduced activity of AMPK in macrophages, in neutrophils and in lung tissue. Restoration of AMPK activity could be an interesting approach to increase efferocytosis and would be likely to decrease inflammatory lung injury in humans, as already reported in mouse models of ALI [25, 26]. Moreover, high-mobility group box 1 (HMGB1), an alarmin that may promote inflammation, has been involved in the development of severe ARDS and has been shown to inhibit efferocytosis [27�C29].
We thus designed the present study to investigate the ability of neutrophils and macrophages to regulate lung inflammation in patients with ARDS. Our first objectives were to evaluate the survival of neutrophils and their ability to produce NETs. Secondly, we studied macrophage capacity to engulf apoptotic cells and NETs. Finally, two potential therapeutic targets, AMPK and HMGB1, were investigated for their ability to restore efferocytosis and NET clearance, and thus to reduce persistent inflammation and decrease lung injury in patients with ARDS.
Materials and methods
Patients
This study was conducted in the medical intensive care unit (ICU) of Rennes University Hospital (Rennes, France). The study protocol was approved by the local ethics committee (number 14.38). Because of the observational nature of the study, a non-opposition form was provided to families and patients. Patients with the Berlin criteria for ARDS were consecutively enrolled and compared with patients who underwent bronchoscopy with normal bronchoalveolar lavage (BAL) in the department of pulmonary medicine (control patients) [30].
Bronchoalveolar lavage
BAL was performed within 2 days of initiation of mechanical ventilation in ARDS patients, or in an outpatient setting for control participants. BAL fluid was obtained by centrifugation and cell population differentials were determined on cytospin slides after May�CGr��nwald�CGiemsa staining.
Cytokine quantification
Interleukin (IL)-6, IL-8, CCL2, CXCL10, plasminogen activator inhibitor (PAI)-1 (ELISA Duoset; R&D Systems, Abingdon, UK) and HMGB1 (ELISA; IBL International GmbH, Hamburg, Germany), were quantified in the BAL fluid by ELISA.
Cell isolation and culture
Human primary bronchial epithelial cells (BECs) were obtained from lung donor trachea or bronchi of the Cohort Of Lung Transplantation (COLT; trial registered at Clinicaltrials.gov, identifier NCT00980967). Tissues were dissociated overnight at 4��C with collagenase in HEPES-buffered RPMI medium (both from Sigma-Aldrich, St Louis, MO, USA). BECs were cultured in cnT17 medium (CELLnTEC Advanced Cell systems AG, Bern, Switzerland) containing penicillin and streptomycin, on plates coated with human type IV collagen (Sigma-Aldrich).
Blood samples were obtained from ARDS patients within the first hours following BAL, or from healthy donors. Neutrophils were purified as previously described [31]. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque density gradient (Eurobio, Courtaboeuf, France). PBMCs were incubated in RPMI 1640 containing 7% fetal calf serum (FCS) and 1% penicillin-streptomycin at 37��C. After 1 h, non-adherent cells were removed by washing with complete medium. Human monocyte-derived macrophages (HMDMs) were then derived from adherent monocytes by culture with 20 ng��mL−1 macrophage colony-stimulating factor (M-CSF; R&D Systems) for 5 days. Purity of HMDMs was >80% and evaluated by flow cytometry.
Apoptosis and necrosis assay
BECs were cultured for 24 h in 50% cnT17 medium and 50% BAL fluid or normal saline solution (Fresenius Kabi, S��vres, France). BEC apoptosis and necrosis were assessed by flow cytometry using annexin V (Cell Signaling Technology, Danvers, MA, USA) and DAPI (4ʹ,6-diamidino-2-phenylindole; Life Technologies, Grand Island, NY, USA).
Circulating neutrophils purified from ARDS patients or healthy donors were cultured for 24 h in 50% RPMI/7% FCS and 50% BAL fluid or saline. Neutrophil apoptosis and necrosis were assessed by flow cytometry using a phycoerythrin-conjugated active caspase-3 apoptosis kit (Becton Dickinson, San Jose, CA, USA) and fluorescein isothiocyanate (FITC) anti-CD66b monoclonal antibody (mAb) (Beckman Coulter, Miami, FL, USA) for apoptosis. Annexin V and DAPI were used to measure necrosis.
NET release quantification
Neutrophils were incubated for 30 min in 50% RPMI/7% FCS and 50% BAL fluid or saline. When indicated, BAL fluids were neutralised beforehand with an anti-HMGB1 mAb (IBL International GmbH) or isotype control for 2 h. Neutrophils were then labelled with 5 µmol��L−1 Sytox blue (Invitrogen, Carlsbad, CA, USA) in RPMI/0.5% FCS with or without DNase I (200 IU��mL−1; Roche, Basel, Switzerland), seeded in Costar 96-well black plates (Corning Costar Corporation, Cambridge, MA, USA) and stimulated or not with 10 µmol��L−1 phorbol myristate acetate (PMA; Sigma-Aldrich) for 3 h at 37��C. The release of NETs (termed NETosis) was quantified by measuring fluorescence with a microplate fluorescence reader (Varioskan, ThermoFisher Scientific, Waltham, MA, USA).
NET isolation and phagocytosis by macrophages
Neutrophils from ARDS patients or healthy donors were incubated in RPMI with 25 nmol��L−1 PMA for 2 h at 37��C. After centrifugation, NETs were quantified in the supernatant by measuring fluorescence using Sytox blue (5 µmol��L−1). HMDMs were allowed to attach in Costar 96-well black plates for 3 h in 50% RPMI/7% FCS and 50% BAL fluid or saline, then Sytox blue-labelled purified NETs were added. After incubation for 2 h at 37��C, HMDMs were washed and NET phagocytosis was assessed by fluorescence quantification. The NET engulfment ratio was determined as the ratio of fluorescence of HMDMs having phagocytised NETs to the fluorescence of HMDMs alone. When indicated, HMDMs were incubated with an anti-HMGB1 neutralising antibody or isotype control for 3 h, or with metformin for 2.5 h (500 µmol��L−1; Sigma-Aldrich).
Immunofluorescence stainings
For NET imaging, purified neutrophils were immobilised on slides coated with poly-d-lysine (Sigma-Aldrich), and incubated with 50% RPMI/7% FCS and 50% BAL fluid from control or ARDS patients for 3 h. Cells were fixed with 4% paraformaldehyde (PFA; Antigenfix Diapath, Martingo, Italy). Coverslips were mounted with Mowiol including Sytox blue (5 µmol��L−1).
For phagocytosis imaging, HMDMs were derived from monocytes on chamber coverslips with M-CSF (20 ng��mL−1) for 5 days. HMDMs were then incubated for 3 h with RPMI containing neutrophil-isolated NETs or not. Cells were fixed with 4% PFA and labelled with anti-neutrophil elastase mAb (Dako, Carpinteria, CA, USA) followed by Alexa Fluor 488 anti-mouse secondary antibody (Jackson ImmunoResearch, Ely, UK), and Texas Red-X Phalloidin (Life Technologies) for actin. Coverslips were mounted with Mowiol including TO-PRO-3 (1 µmol��L−1; Life Technologies).
For efferocytosis assays, HMDMs derived on chamber coverslips were incubated for 3 h with BAL fluid from control or ARDS patients, with or without 500 µmol��L−1 metformin for 2.5 h. When indicated, BAL fluid was pre-treated with an anti-HMGB1 neutralising antibody or isotype control. Efferocytosis was evaluated by adding 106 carboxyfluorescein succinimidyl ester (CFSE)-labelled apoptotic neutrophils. After incubation at 37��C for 1 h, cells were washed and fixed with 4% PFA. The efferocytosis index was determined on 300 cells as the percentage of HMDMs containing at least one ingested apoptotic neutrophil.
For all imaging, slides were examined with a SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). Digital images were processed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Western blot
(Phospho)-AMPK Western blotting was performed using mouse anti-AMPK�� or rabbit anti-phospho-AMPK�� antibodies (Cell Signaling Technology), followed by horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Actin was blotted as loading control, using mouse anti-��-actin (Sigma-Aldrich) and HRP-conjugated anti-mouse secondary IgG. Blots were quantified using ImageJ software.
Statistical analysis
Quantitative variables are expressed as mean��sd or median (interquartile range) when indicated, and qualitative variables as number (percentage). Continuous variables were compared using the nonparametric Mann�CWhitney U-test or Wilcoxon test for matched pairs, as appropriate. Analyses were performed with GraphPad Prism 6.2 (GraphPad Software, La Jolla, CA, USA).
Results
NET formation in ARDS patients may contribute to lung injury
Among ARDS BAL leukocytes, neutrophils were the predominant cell population, whereas the majority of BAL leukocytes in controls are macrophages (online supplementary figure S1a and b). The characteristics of the ARDS patients and control subjects are provided in table 1.
View inlineView popup
TABLE 1
Patient characteristics
Several soluble factors implicated in the development of lung injury, including the pro-inflammatory cytokine IL-6, and CXCL10, CCL2 and IL-8 chemokines were significantly increased in ARDS patients (online supplementary figure S1c) [13, 32]. We also found significantly increased levels of PAI-1, implicated in downregulating efferocytosis in animal models of ALI (figure 1a) [33]. Because HMGB1 has been shown to promote NET release in experimental ALI, we also examined this possibility in ARDS patients [34]. We found that HMGB1 was significantly increased in BAL fluid of ARDS patients versus controls (figure 1b). Subsequent analysis revealed substantial amounts of cell-free DNA in the BAL fluid of ARDS patients, suggesting that HMGB1 accumulation is accompanied by an enhanced NETosis (figure 1c). Furthermore, BAL fluid from ARDS patients was found to induce lung epithelial cell injury, which could be related to NETs (figure 1d�Cg).
FIGURE 1
Download figureOpen in new tabDownload powerpoint
FIGURE 1
Characteristics of the bronchoalveolar lavage (BAL) fluid from acute respiratory distress syndrome (ARDS) patients may contribute to lung epithelial cell injury. a) Quantification of plasminogen activator inhibitor (PAI)-1 (n=19) and b) high-mobility group box 1 (HMGB1) (n=6) by ELISA in BAL fluid from control or ARDS patients. c) BAL fluid from ARDS patients contained high levels of neutrophil extracellular traps (NETs). Quantification of NETs by fluorescence measurement after Sytox blue staining in BAL fluid from control (n=8) and ARDS patients (n=8). MFI: mean fluorescence intensity. d, e) BAL fluid from ARDS patients induced lung epithelial cell apoptosis and necrosis. d) Human primary bronchial epithelial cells (BECs) were treated with 50% normal saline solution (NaCl), or with BAL fluid from control or ARDS patients for 24 h. e) Apoptosis and necrosis were measured using flow cytometry. f, g) NETs induced lung epithelial cell apoptosis and necrosis. f) BECs were treated with 50% of RPMI/0.5% fetal calf serum (FCS) or NETs for 24 h. g) Apoptosis and necrosis were measured using flow cytometry. In all graphs, horizontal bars represent medians. The Mann�CWhitney U-test was used to compare protein and NET quantification and the Wilcoxon test was used for BEC apoptosis. *: p<0.05; **: p<0.01; ***: p<0.001; ns: nonsignificant.
Neutrophils of ARDS patients enhanced capacity to produce NETs
The cell-free DNA found in BAL fluid could be a result of DNA release from necrotic cell death. However, we found that neutrophils in BAL of ARDS patients had a relatively low apoptotic index (data not shown) and also that circulating neutrophils of ARDS patients presented an increased capacity to produce NETs ex vivo, compared to healthy donors (figure 2a and b). In these experiments, NETosis was measured after stimulation of neutrophils with PMA. NET formation was also used to examine whether BAL fluid from control or ARDS patients influences NET deployment. When compared to BAL fluid from control patients, BAL fluid from ARDS patients effectively increased spontaneous NET release from either control or ARDS neutrophils (figure 2c�Cf).
FIGURE 2
Download figureOpen in new tabDownload powerpoint
FIGURE 2
NETosis (release of neutrophil extracellular traps (NETs)) was enhanced in peripheral blood-derived neutrophils from acute respiratory distress syndrome (ARDS) patients, and increased by bronchoalveolar lavage (BAL) fluid mediators. a, b) Neutrophils from healthy donors or ARDS patients were treated with 10 µmol��L−1 phorbol myristate acetate (PMA) for 3 h before NET quantification by Sytox blue fluorescence (5 µmol��L−1). b) Free DNA (NETs) production was compared in neutrophils from healthy donors (n=5) and ARDS patients (n=5). rMFI: ratio of mean fluorescence intensity. c�Ce) Neutrophils from healthy donors or ARDS patients were treated with BAL fluid from control or ARDS patients for 3 h before NET quantification by Sytox blue fluorescence. rrMFI: ratio of ratio of mean fluorescence intensity. d) Quantification of NET production by healthy donor neutrophils after incubation with control or ARDS BAL fluid (n=6). e) NET production by ARDS neutrophils was quantified after incubation with control or ARDS BAL fluid (n=5). f) Fluorescence microscopy images showing NET formation from a representative ARDS patient after 3-h incubation with control or ARDS BAL fluid. Neutrophil DNA was stained with Sytox blue. In all graphs, horizontal bars represent medians. The Mann�CWhitney U-test was used for comparisons. *: p<0.05.
Neutrophils of ARDS patients show increased viability
In inflammatory conditions, like in ARDS, neutrophils are known to acquire a prolonged viability. To determine neutrophil viability, apoptotic indices were measured after 24 h of neutrophil culture. The amounts of apoptotic neutrophils were significantly lower in circulating neutrophils from ARDS patients compared to healthy donors. This result confirmed that viability of neutrophils is increased in ARDS patients (figure 3a). In subsequent experiments, we examined the effect of BAL fluids on neutrophil viability. As shown in figure 3c, BAL fluid from ARDS patients, unlike that from control patients, increased the viability of healthy donor circulating neutrophils. The apoptotic percentage was even further decreased after exposure of ARDS circulating neutrophils to ARDS BAL fluid (figure 3d). However, to ensure that specific constituent(s) of BAL fluid from ARDS patients increased neutrophil viability, we also explored necrosis rate and found a trend towards diminished neutrophil necrosis when exposed to BAL fluid from ARDS patients (figure 3e).
FIGURE 3
Download figureOpen in new tabDownload powerpoint
FIGURE 3
The proportion of apoptotic peripheral blood-derived neutrophils was decreased in acute respiratory distress syndrome (ARDS) patients, spontaneously and after incubation with bronchoalveolar lavage (BAL) fluid. a) Proportion of active caspase-3+ apoptotic cells among CD66b+ neutrophils from healthy donors (n=9) or ARDS patients (n=10), assessed by flow cytometry after 24-h ex vivo culture. b) Neutrophils from healthy donors or ARDS patients were treated with 50% normal saline solution (NaCl), or with BAL fluid from control or ARDS patients for 24 h before neutrophil apoptosis or necrosis measurement. Proportion of active caspase-3+ apoptotic cells among CD66b+ neutrophils from c) healthy donors, assessed by flow cytometry after 24-h ex vivo culture with NaCl (n=8), or control (n=10) or ARDS (n=10) BAL fluid, and from d) ARDS patients, assessed by flow cytometry after 24-h ex vivo culture with NaCl (n=10), or control (n=10) or ARDS (n=13) BAL fluid. e) Proportion of annexin V+/DAPI+ necrotic neutrophils from ARDS patients, assessed by flow cytometry after 24-h ex vivo culture with control (n=7) or ARDS (n=7) BAL fluid. In all graphs, horizontal bars represent medians. The Mann�CWhitney U-test was used for comparisons. *: p<0.05; **: p<0.01; ns: nonsignificant.
HMDMs from ARDS patients have diminished ability to phagocytose NETs and apoptotic neutrophils
The ability of macrophages to neutralise apoptotic neutrophils plays a central role in termination and resolution of inflammatory conditions. Recent studies also indicate that macrophages are involved in clearance of NETs [20, 35]. As shown in figure 4a�Cd, there was a significant reduction in both NET uptake and apoptotic neutrophil efferocytosis by HMDMs from ARDS patients versus healthy donors. Moreover, a similar decrease in phagocytic ability was observed upon exposure of HMDMs from healthy donors to BAL fluid obtained from ARDS patients (figure 4e). Even further reduction in phagocytic index was found in ARDS HMDMs treated with ARDS BAL fluid (figure 4f). In contrast, BAL fluid from control patients had no impact on efferocytosis (figure 4d versus 4e and 4f). This finding also suggests that reduced efferocytosis was mediated by soluble components in the lung fluid of ARDS patients.
FIGURE 4
Download figureOpen in new tabDownload powerpoint
FIGURE 4
The ability of human monocyte-derived macrophages (HMDMs) to engulf neutrophil extracellular traps (NETs) and apoptotic neutrophils was reduced during acute respiratory distress syndrome (ARDS). a) Engulfment of Sytox blue-labelled NETs by HMDMs from healthy donors (n=6) and ARDS patients (n=6). The NET engulfment ratio was defined as the ratio of fluorescence emitted by HMDMs that had phagocytosed NETs to the fluorescence emitted by HMDMs alone. b) Immunofluorescence images showing HMDMs in the process of engulfing NETs. HMDMs were incubated with NETs purified from neutrophils and these were internalised by HMDMs. The HMDM actin was stained with phalloidin (red), DNA with Sytox blue (blue), and NET neutrophil elastase with antibodies (green). c�Cf) The efferocytosis capacities of HMDMs were decreased by ARDS, both spontaneously and after incubation with bronchoalveolar lavage (BAL) fluid. c) HMDMs were incubated with RPMI/fetal calf serum (FCS) or BAL fluid for 3 h before adding carboxyfluorescein succinimidyl ester (CFSE)-labelled apoptotic neutrophils for 1 h. The efferocytosis index was defined as the number of HMDMs that phagocytosed apoptotic neutrophils relative to the number of HMDMs that did not. Efferocytosis index of d) HMDMs from healthy donors (n=5) versus ARDS patients (n=10), e) healthy donor macrophages cultured with control (n=8) or ARDS (n=8) BAL fluid, and f) macrophages from ARDS patients cultured with control (n=8) or ARDS (n=8) BAL fluid. In all graphs, horizontal bars represent medians. The Mann�CWhitney U-test was used for comparisons. *: p<0.05.
The effects of AMPK and HMGB1 on efferocytosis and NET clearance
To determine factor(s) that are affecting NETosis, efferocytosis and NET engulfment, we first examined the impact of HMGB1 (figure 5a). We did not observe any significant effect of HMGB1-neutralising antibody on NET formation (figure 5b). In contrast, we found that an anti-HMGB1 antibody increased the clearance of apoptotic neutrophils by ARDS HMDMs (figure 5c). However, it had no effect on NET uptake (figure 5d). The ability of HMGB1 to affect efferocytosis is consistent with previous studies in a murine model of inflammatory organ injury, in particular linking HMGB1 release into the extracellular milieu with diminished clearance of apoptotic cells [27�C29].
FIGURE 5
Download figureOpen in new tabDownload powerpoint
FIGURE 5
Inhibition of high-mobility group box 1 (HMGB1) and activation of AMP-activated protein kinase (AMPK) increased neutrophil extracellular trap (NET) engulfment and efferocytosis by human monocyte-derived macrophages (HMDMs). a) Bronchoalveolar lavage (BAL) fluid from acute respiratory distress syndrome (ARDS) patients was treated for 2 h with an anti-HMGB1 (��-HMGB1) or isotype control antibody (��-IgY) before incubation with neutrophils from healthy donors or HMDMs from ARDS patients for 3 h. b) NET production by healthy donor neutrophils (n=8). rrMFI: ratio of ratio of mean fluorescence intensity. c) HMDM efferocytosis index was determined after 1-h contact with apoptotic neutrophils (n=6). The efferocytosis index was defined as the number of HMDMs that phagocytosed apoptotic neutrophils relative to the number of HMDMs that did not. d) NET engulfment by HMDMs from ARDS patients (n=6) was determined after 2-h contact with neutrophil-derived NETs. The NET engulfment ratio was defined as the ratio of fluorescence emitted by HMDMs that had phagocytosed NETs to the fluorescence emitted by HMDMs alone. e) HMDMs from ARDS patients were incubated with BAL fluid from control or ARDS patients for 3 h before being treated with or without metformin for 2.5 h. Representative Western blots and quantitative analysis of phospho (p)-AMPK, total AMPK and actin from ARDS patient HMDMs incubated with f) control or ARDS BAL fluid, and with g) ARDS BAL fluid and metformin or medium alone. h) HMDM efferocytosis index determined after 1-h contact with apoptotic neutrophils (n=7). i) Engulfment by HMDMs of Sytox blue-stained NETs, determined after 2 h (n=8). In all graphs, horizontal bars represent medians. The Wilcoxon test was used to compare effects of different HMGB1 or metformin treatments on NET engulfment and efferocytosis. *: p<0.05; ns: nonsignificant.
Besides adverse effects mediated by extracellular HMGB1, inflammatory conditions are associated with metabolic reprogramming of immune and parenchymal cells that is associated with diminished activity of AMPK in macrophages [36]. Notably, AMPK activators, including metformin, have been shown to promote efferocytosis and also to reduce the severity of ALI [24�C26]. Thus, we examined whether AMPK activation can also recover the phagocytic capacity of HMDMs from ARDS patients. As shown in figure 5f, AMPK activation, i.e. phosphorylation of Thr172-AMPK, was significantly diminished in HMDMs treated with BAL fluid from ARDS patients. Moreover, we found that culture of ARDS HMDMs with metformin restored AMPK activation (figure 5g). This activation was associated with a significant increase in uptake of apoptotic neutrophils and NETs (figure 5h and i).
Discussion
Our study reveals major findings that could enhance or sustain lung inflammation during ARDS. First, although neutrophil lifespan is significantly increased, intra-alveolar neutrophils are releasing NETs through a pathway termed vital NETosis and BAL fluid of ARDS patients can increase the release of NETs, which could induce lung injury. Secondly, in ARDS conditions, the ability of macrophages to engulf NETs and apoptotic cells is significantly decreased. We also found that blocking HMGB1 and activating AMPK could enhance clearance of NETs and apoptotic cells.
Clinical and histological studies have suggested that the severity and outcome of ARDS were associated with the inflammatory process reflected in bronchoalveolar fluid [13]. A large number of clinical and animal studies have brought evidence that neutrophils have a direct influence on the onset and the persistence of ARDS. For instance, Steinberg et al. [12] found that alveolar macrophages increased in ARDS survivors compared to nonsurvivors, and reached the conclusion that sustained alveolar inflammation was associated with high mortality. Among factors that could sustain inflammatory conditions, increased NETosis and decreased ability of macrophages to engulf apoptotic cells and NETs appear to be critical.
We found that neutrophils could produce large amounts of NETs, spontaneously or when exposed to ARDS BAL fluid. NETs are composed of decondensed chromatin fibres coated with antimicrobial proteins. NETosis could require membrane rupture and the loss of neutrophil functions (so-called ��suicidal NETosis��) [17]. However, Yipp and Kubes [37] have demonstrated that, during the early phase of infection, NETosis involved neutrophils that did not undergo lysis and retained the ability to perform recruitment, chemotaxis and phagocytosis (so-called ��vital NETosis��). In our study, and probably in the ARDS setting, NETosis does not result in cell death, since we found that neutrophil lifespan was increased. Although the primary role of NETs is to avoid bacterial diffusion, NETs have been found to play deleterious effects on lung injury during ARDS and several studies have pointed out that NET formation during bacterial pneumonia only worsened lung injury, without any bactericidal activity [17, 38]. Moreover, Narasaraju et al. [15] demonstrated that, in mice challenged with influenza, NETs contributed to ALI by instigating alveolar capillary damage. Therefore, NETosis and clearance of NETs should be adequately regulated in vivo and defects in mechanisms responsible for NET clearance may contribute to perpetuated inflammation and worsening of tissue injury [15, 39]. Two mechanisms have been described in NET clearance: DNase I-dependent digestion and phagocytosis by macrophages [17]. The latter has been found to be diminished in our study. We also demonstrated that efferocytosis was decreased in ARDS. Clearance of cells undergoing apoptosis protects surrounding tissue from exposure to pro-inflammatory intracellular contents released from necrotic cells. Although there are convincing animal data showing that neutrophils secrete anti-inflammatory peptides while dying, failure to effectively remove apoptotic cells (particularly apoptotic neutrophils) perpetuates inflammation, exposing the lung to sustained inflammatory conditions that could increase neutrophil influx and NETosis and worsen lung injury [40, 41]. Thus, macrophages have a key position in the resolution of inflammation and initiation of tissue repair. Restoring the ability of macrophages to engulf both NETs and apoptotic cells could be of interest to decrease lung damage and pulmonary sequelae such as fibrosis.
We found that BAL fluid from ARDS patients could induce a decrease in efferocytosis in both healthy and ARDS macrophages, creating the possibility that therapeutic interventions could enhance efferocytosis and engulfment of NETs. Along these lines, the AMPK pathway appears to be a potential therapeutic target. We found in our study that the ability of macrophages to activate AMPK was decreased in inflammatory conditions, which could be associated with a defect in efferocytosis activity [24]. Notably, the ability of AMPK activation to enhance phagocytosis appears to be related to interaction with cytoskeletal organisation [24]. Lastly, we found that inhibition of HMGB1 could increase efferocytosis, as already reported in animal models of lung injury. HMGB1, originally described as a nuclear nonhistone DNA-binding protein, has subsequently been shown to be an alarmin involved in the inflammatory response playing a critical role in the recruitment of neutrophils, lung injury and suppressing bacterial clearance in the lung [31, 34, 42]. It is worth noting that metformin has been found to bind and inhibit the action of HMGB1, suggesting that the effects of metformin could be similar to those of HMGB1 inhibition [43]. High levels of HMGB1 in BAL fluid from ARDS patients can decrease efferocytosis, suggesting that inhibition of HMGB1, as well as activation of AMPK, could be of interest to restore efferocytosis, diminish lung injuries and ultimately improve lung function after ICU discharge.
Conclusion
Altogether, our results show that efferocytosis and NET engulfment, which could contribute to the persistence of lung inflammation, are diminished during ARDS. Restoration of AMPK activation with metformin and specific inhibition of HMGB1 appear to be promising targets to decrease lung inflammation and to limit alveolar damage and progression to lung fibrosis in patients with ARDS.Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS | European Respiratory Society
https://erj.ersjournals.com/content/52/2/1702590?ijkey=b20d11137eb04ca04e6e84c6eea829be31e36afa&keytype2=tf_ipsecsha��
High Mobility Group Box 1 (HMGB1) Phenotypic Role Revealed ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4153844
High mobility group box 1 (HMGB1) is an evolutionarily ancient protein that is present in one form or another in all eukaryotes. It fundamentally resides in the nucleus but translocates to the cytosol with stress and is subsequently released into the extracellular space.
Cited by: 21
Publish Year: 2014
Author: Daolin Tang, Rui Kang, Bennett Van Houten, Herbert J. Zeh, Timothy R. Billiar, Michael T. Lotze
High mobility group box 1 (HMGB1) as a novel frontier in ...
https://onlinelibrary.wiley.com/doi/full/10.1111/jnc.14663
The emerging research suggests that high�\mobility group box protein 1 (HMGB1), a DNA�\binding protein that is both actively secreted by inflammatory cells and released by necrotic cells, might contribute to the pathogenesis of epilepsy.
Cited by: 10
Publish Year: 2019
A��
High-mobility group box 1 ( HMGB1 ) is actively and/or passively released leading to multiple signaling pathways. Actively released HMGB1 from brain slice cultures found histochemical evidence of release from neurons by ethanol (Zou and Crews 2014), although HMGB1 release likely occurs from most brain cell types. Neurons and glia release HMGB1 during glutamate excitation (Maroso et al. 2010, 2011). HMGB1 is also released during necrotic cell death activating innate immune signaling. HMGB1 has multiple signaling mechanisms regulated by oxidation of cysteines. Fully oxidized HMGB1 ( blue , left ) does not activate proinflammatory signaling, although it may contribute to resolution of the proinflammatory state. HMGB1 in the all-thiol form ( yellow , middle ) is an agonist at the receptor for advanced glycation end- products ( RAGE ; Allette et al. 2014). All-thiol-HMGB1 also forms heterodimers with proinflammatory molecules such as interleukin-1beta ( IL-1 �� ; Wahamaa et al. 2011) with the HMGB1-IL1 heterodimer synergistically enhancing stimulation of the IL-1 �� receptor leading to proinflammatory gene induction through activation of NF- �� B transcription (Venereau et al. 2012). Disulfide-HMGB1 ( red , right ) activates Toll-like receptor 4 ( TLR4 ) also leading to nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells ( NF- �� B ) and induction of proinflammatory cytokines. Adapted from Tang et al. (2012)
��
Neuronal excitability releases HMGB1, which increases cytokine secretion by microglia that activate astrocytes that increase glutamate increasing neuronal excitability through glutamate, HMGB1, and other signals. Glutamate, alcohol, and other factors release HMGB1 from neurons causing microglia to become hyper-ramified, resulting in their release of high-mobility group box 1 ( HMGB1 ) and other proinflammatory cytokines. These innate immune signaling molecules stimulate astrocytes reducing glutamate uptake (Zou and Crews 2005) and increasing astrocyte release of glutamate that induce neuronal excitability causing further increases in HMGB1 release. Increased neuronal hyperexcitability can contribute to potentiation of specific neuronal connectivity and/or to excitotoxic neuronal cell death. Figure adapted from Kettenmann et al. (2013)
��
Ethanol in the gut causes leakage of bacterial products into the portal vein increasing hepatic TNF �� release into the blood which induces neuroimmune gene expression in the brain. High doses of consumed alcohol in the gut (i.e., at least 2 �C 3 g/kg ETOH intragastric doses [Ferrier et al. 2006]) increases permeability allowing bacterial products such as endotoxin-lipopolysaccharide ( LPS ) to enter portal circulation. Alcohol and LPS enter portal circulation leading to induction of liver tumor necrosis factor-alpha ( TNF �� ) and other proinflammatory cytokines that are released into the blood and enter the brain through cytokine-specific receptor transport (e.g., the TNF �� receptor) (See Qin et al. 2007 for details). This activates positive loops of proinflammatory
��... constitutively express Toll-like receptor (TLR) 4 and other innate immune receptors that are responsive to proinflammatory signals like high-mobility group box 1 (HMGB1) but are also responsive to neurotransmitters (Kettenmann et al. 2013). Innate immune gene upregulation with rapid monocyte responses to infection was first characterized in blood as acute phase response proteins that today are known to include multiple cytokines, chemokines, proteases, cellular oxidases, and cytokine receptors. Acute phase responses and monocyte activation involve amplification in the expression of a large number of genes through kinase signaling pathways that converge on two distinct transcription factors: nuclear factor kappa-light-chain-enhancer of activated B cells (NF- �� B) and activator protein-1 (AP-1). Both NF- �� B and AP-1 induction promote the expression of innate immune cytokines (Li and Verma 2002; Valles et al. 2004), including tumor necrosis factor-alpha (TNF �� ) and interleukin-1beta (IL-1 �� ) as well as upregulation of TLRs and other cytokine receptors. In addition, innate immune proteases and oxidases are induced, particularly cyclooxygenase (COX-2) and nicotinamide ade- nine dinucleotide phosphate oxidase (NADPH oxidase) as well as major histocompatibility (MHC) signaling molecules, such as beta-2 microglobulin. These monocyte-microglial- expressed proteins and their receptors are innate immune signaling molecules (ISMs) that are expressed in the brain (Blanco and Guerri 2007; Guerri and Pascual 2010; Valles et al. 2004). This review will refer to these brain signaling molecules as B neuroimmune ^ due to their characterization in the immune system of the brain, while recognizing that signaling across multiple unique brain cells differs from immune inflammation in response to infection. Brain neuroimmune signaling primarily involves monocyte-microglial innate immune signals and not adaptive immune antibodies. Although microglia are unique tissue- specific brain monocyte-like cells, similar to all monocytes, microglia undergo morphological changes that characterize stages of activation (Graeber 2010) (Fig. 1). Resting ramified microglia likely contribute trophic and other signals similar to the wound healing monocyte phenotype termed M2 that upon activation can become hyper-ramified, with secretion of proinflammatory cytokine signals (Beynon and Walker 2012). However, activated microglia do not necessarily al- ways adopt an M1 phenotype as Marshall et al. (2013) found that young adult rats subjected to a 4-day binge model of alcohol led to partial microglial activation as evidenced by increased expression of OX-42 but not a fully activated phenotype characterized by expression of OX-6 or ED-1. This partial microglial action was accompanied by an increase in the anti-inflammatory cytokine IL-10 and no increase in proinflammatory cytokines IL-6 or TNF �� . Further proinflammatory activation, known as M1 monocyte phenotype, involves expansion of processes to a B bushy morphology ^ and finally a B phagocytic ^ rounded morphology (Colton 2009). The relationship between morphological changes in monocyte-like cells including microglia and the secretion of ISM is poorly understood, although increased severity of pathology is associated with greater ISM induction and activated morphology. Like all monocytes, microglial activation can lead to NF- �� B transcription of proinflammatory genes, which signal to other microglia as well as astroglia, oligodendroglia, and neurons, amplifying neuroimmune gene induction within and across cells by induction of TLRs and cytokine receptors, many belonging to the IL-1 �� receptor family that activate kinase cascades that converge on NF- �� B (Blanco and Guerri 2007; Blanco et al. 2004, 2005; Fernandez-Lizarbe et al. 2009, 2013; Pascual et al. 2011b; Valles et al. 2004). The amplification of ISMs across cells and tissue can lead to pathology, and understanding the processes of monocyte signaling provides insight into microglial signaling in brain. The most severe acute example of monocyte activation during infection is sepsis. Sepsis and the systemic inflammatory response syndrome refer to a B cytokine storm, ^ which involves a pronounced increase in multiple proinflammatory cytokines and other ISMs that cause a potentially fatal innate immune reaction consisting of positive feed-forward loops between cytokines and immune and tissue cells that result in highly elevated cytokine blood levels, multi-organ failure, and death (Osterholm 2005). Models of sepsis that involve activation of an acute phase-like response lead to increased expression of multiple cytokines that are induced in distinct phases. During the initial phase, TNF �� and IL-1 �� expression is increased during the first several hours after innate immune activation and then subside. The second phase involves HMGB1, which is an agonist at multiple receptors that contribute to further activation of proinflammatory cascades (Fig. 2). Disulfide-HMGB1 is a TLR4 agonist (Tang et al. 2012), and thiol-HMGB1 is an agonist at the receptor for advanced glycation end products (RAGE; Allette et al. 2014) and also dimerizes with proinflammatory molecules (Tang et al. 2012; Venereau et al. 2012), such as IL-1 �� (Wahamaa et al. 2011) that enhances IL-1 �� receptor induction of proinflammatory molecules through NF- �� B. HMGB1 increases in blood approximately 16 h after infection in models of sepsis and persists for several days during which mice die. Mortality is prevented by anti-HMGB1 antibody treatment (Wang et al. 2001) consistent with HMGB1 induction of a B cytokine storm ^ by acting through multiple receptors that converge on proinflammatory NF- �� B signaling. Survivors of sepsis models show prolonged increases in serum HMGB1 and cognitive deficits that are blunted with HMGB1 antibody treatment (Chavan et al. 2012). To model alcoholic hepatitis and alcohol-induced release of gut endotoxin, we systemically administer lipopolysaccharide (LPS) and polyinosinic:polycytudylic acid (poly I:C). Administration of these endotoxins systemically after ethanol treatment exacerbates the innate immune response. Acute binge drinking also increases serum endotoxin levels albeit at a lower level observed under septic conditions. High binge drinking doses cause the gut to become permeable or B leaky ^ (Ferrier et al. 2006). Only high doses of ethanol, e.g., at least 2 �C 3 g/kg ETOH intragastric doses (Ferrier et al. 2006), potentiate gut innate immune signaling, disrupting gut tight junctions, and opening sites that allow the gut biome bacteria and their endotoxins to enter the portal circulation leading to the liver where they can initiate a proinflammatory response (Sims et al. 2010). Released LPS potentiates alcohol-induced liver inflammation and secretion of proinflammatory cytokines, including the proinflammatory cytokine TNF �� , which is released into the blood. Proinflammatory cytokines in the blood are transported across the blood �C brain barrier (Banks and Erickson 2010; Qin et al. 2007) such that both cytokines and alcohol enter the brain where they induce neuroimmune activation. Innate immune signaling molecules in the brain appear to contribute to both brain health and pathology. Indeed, recent studies find that MHC molecules contribute not only to most neurodegenerative diseases (Gage 2002; Glass et al. 2010) as well as alcohol and drug dependence (Crews 2012) but are also critically involved in brain development (Huh et al. 2000). Within the brain microglia, innate immune cytokines, such as TNF �� , IL-1 �� , and HMGB1 as well as TLRs, purinergic receptors (e.g., P2X7), various cytokine receptors, and innate immune proteases and oxidases, all amplify through NF- B and AP-1 loops that confound studies that are focused on studying a single neuroimmune signaling molecule (Fig. 3). NF- �� B regulates the transcription of proinflammatory innate immune genes as well as many other genes (Perkins 2007). Ethanol increases NF- �� B �C DNA binding and expression of multiple innate immune genes including proinflammatory cytokines, TNF �� , IL-1 �� , and MCP-1, the proinflammatory oxidase, iNOS, and proteases TACE and tPA (Zou and Crews 2010). Previously, we found that ethanol increased NF- �� B p65 nuclear immunohistochemistry consistent with NF- �� B p50 ⁄ p65 subunit nuclear translocation and transcription activation. Similarly, we found that antibodies to p50 or p65 super-shifted EMSA gels, suggesting that ethanol increased brain NF- �� B p65 �C p50 heterodimer �C DNA binding (Zou and Crews 2006). Taken together, ethanol-induced NF- �� B �C DNA binding and target gene expression support ethanol activation of NF- �� B transcription of proinflammatory genes. However, using an ELISA-based DNA binding analysis, we found large increases in NF- �� B subunit p50 protein but not NF- �� B p65 protein. A similar finding has been reported for prefrontal cortex gene expression in the post-mortem human alcoholic brain (Okvist et al. 2007). Array analysis of gene expression in post-mortem alcoholic prefrontal cortex found 479 transcripts with NF- �� B �C DNA binding sites that were generally upregulated, with analysis of NF- �� B subunit proteins indicating NF- �� B p50 was the dominant subunit expressed in human alcoholic brain (Okvist et al. 2007). Although homodimers of NF- �� B p50 inhibit transcription (Perkins 2007), increases in NF- �� B p50 protein are often associated with increased transcription. Several mechanisms are involved in increased NF- �� B p50 activation of transcription including protease processing of inhibitory NF- �� B p105 to transcriptionally active NF- �� B p50 (Hoffmann et al. 2006) or through NF- �� B p50 homodimer association with BCL3, atypical I �� B, and other proteins that activate gene transcription involving NF- �� B p50 (Ghosh and Hayden 2008). Thus, an increase in NF- �� B p50 is consistent with increased NF- �� B gene transcription. Although ...
High-mobility group box 1 ( HMGB1 ) is actively and/or passively... | Download Scientific Diagram
https://www.researchgate.net/figure/High-mobility-group-box-1-HMGB1-is-actively-and-or-passively-released-leading-to_fig2_273782517��
��
The innate immune architecture of lung tumors and its ...
https://onlinelibrary.wiley.com/doi/10.1002/path.5241
Jan 25, 2019 �� In contrast, IM��s reside between the alveoli and capillaries. Unlike AM��s, they express CD11b and are thought to be maintained from monocytic progenitors that are recruited from the periphery 22. Relative to AM��s, less is known about the physiological role of IM��s.
Cited by: 1
Publish Year: 2019
Author: Simon Milette, Pierre O Fiset, Logan A Walsh, Jonathan D Spicer, Daniela F Quail
(PDF) The role of macrophages in influenza A virus infection
https://www.researchgate.net/publication/277675881_The_role_of_macrophages_in...
The role of macrophages in influenza A virus infection. ... Surface marker AM��s IM��s EM��s Comments R ef. C ... of ep ithelial cells with virus initiates in ammation and activ ates AM��s, ...
Lung Interstitial Macrophages Redefined: It Is Not That ...
https://www.atsjournals.org/doi/full/10.1165/rcmb.2017-0158ED
Aug 01, 2017 �� Strategies for depleting IM��s or AM��s using clodronate or F4/80 antibody (a pan monocytic/macrophage marker), respectively, have produced varied results. For example, depletion of IM��s, but not AM��s, exaggerated endotoxin/antigen-induced airway allergic responses , whereas depletion of sessile AM��s augmented LPS-induced lung injury . Thus ...
Cited by: 2
Publish Year: 2017
Author: Sekhar P. Reddy, Dolly Mehta��
The innate immune architecture of lung tumors and its implication in disease progression - Milette - 2019 - The Journal of Pathology
AECs mediate innate immunity
AECs act as a first line of defense against antigens that have escaped mucociliary clearance. Single cell�\RNA sequencing studies have shown that AECs can be subdivided into multiple subtypes 3, 5. The best characterized subtypes include squamous type I and cuboidal type II cells, which are responsible for gas exchange with the endothelium and production of pulmonary surfactant, respectively 11. Being endowed with an arsenal of pattern recognition receptors (PRRs) such as Toll�\like receptors (TLRs), Nod�\like receptors and retinoic acid�\inducible gene (RIG)�\I�\like receptors, AECs promptly sense pathogen�\associated molecules and initiate NF�\��B�\dependent inflammatory cascades through the release of antimicrobial peptides, cytokines and chemokines 11. Of particular importance are microbial ssRNA�\ and CpGDNA�\sensing TLR�\3, TLR�\7, TLR�\9, RIG�\I and melanoma differentiation�\associated protein�\5, which have been implicated in the rapid release of interferon (IFN)�\��, granulocyte�\macrophage colony�\stimulating factor (GM�\CSF), interleukin (IL)�\6 and IL�\8, a potent neutrophil�\recruiting chemokine 12. This robust cytokine response is critical to subsequently stimulate adaptive immunity, and neutralize the harmful effects of foreign pathogens.
However, owing to the hypersensitivity of AEC PRRs, these receptors can sometimes become diverted to mediate autoimmunity. For example, in mouse models of allergic asthma, activation of TLR�\4 by dust mites on AECs is associated with local production of IL�\25, IL�\33, GM�\CSF and thymic stromal lymphopoietin (TSLP) 13. These cytokines work in concert to stimulate activation and pulmonary infiltration of dendritic cells (DCs), lymphocytes, neutrophils and eosinophils 13. In addition, studies of pulmonary alveolar proteinosis have shown that GM�\CSF is specifically necessary for lung homeostasis, such that GM�\CSF autoantibodies or genetic deletion causes abnormal lung development and surfactant accumulation, despite normal peripheral hematopoiesis 14-16. Together these studies highlight a unique ability of AECs to undergo ��innate immune mimicry��, and orchestrate inflammation and autoimmunity. Therefore, the contribution of AECs in the pulmonary microenvironment is essential to understanding both normal and pathologic lung function.��
Figure 1
Open in figure viewerPowerPoint
The premetastatic niche and early metastatic colonization in the lungs. Schematic depiction of the various components of early metastasis, including the premetastatic niche, circulating tumor cells, colonization, and dormancy. Most metastases form adjacent to the lung capillaries as they exit from the vasculature. (A) Integrin ��6��1�\bearing tumor�\derived exosomes accumulate in the premetastatic lung, where they promote the recruitment of pro�\inflammatory myeloid cells and deposition of fibronectin 76. Exosomal snRNA activate AECs, which in turn secrete neutrophil�\recruiting S100 proteins 77. (B) C5a activates AM��s, which in turn secrete TGF�\�� and participate in ECM remodeling 78. (C) Inflammatory stimuli such as tobacco smoke or microbial infection can induce the formation of NETs in the lung, which cleave laminin 79. Laminin cleavage generates epitopes that activate integrin signaling in dormant cancer cells to stimulate their awakening. (D) Circulating tumor cells in the vasculature are captured by NETs, which can be induced by inflammation or soluble G�\CSF 80, 81.��
Figure 2
Cell�Ccell interactions in the TIME of primary lung tumors. Schematic representation of the interactions between cancer cells and various pulmonary cell types that regulate tumor progression, as well as the soluble factors involved. Most primary lung tumors develop from bronchial or alveolar epithelial cells, and are therefore often found adjacent to the airspaces. (A) Malignant transformation of AECs can be triggered by activated myeloid�\cell derived ROS 82. (B) Cancer cells expanding in the lung parenchyma (alveolus or bronchial epithelium) can be attacked by the immune system and undergo apoptosis through several surveillance mechanisms. Pro�\inflammatory neutrophils and AM�� can secrete ROS and TNF�\��. Neutrophils can also act as antigen presenters to activate tumor�\infiltrating lymphocytes 83, 84. (C) V��9V��2 T cells can promote DC maturation and activate macrophages via TNF�\�� and IFN�\�� secretion 85-87. Cancer cell death is also induced by NK cell�\ and CTL�\derived GrzB 88. The cytotoxicity of these cells can be further enhanced by the activity of ILC2s and ILC3s 89, 90. (D) The formation of TLSs promotes the generation of anti�\tumor plasma cells. (E) Cancer cells can evade immune attack through the activity of tolerogenic lymphoid cells including Tregs, �æ� T17 and Foxp3+ �æ� T cells 91, 92. These cells shut down cell�\mediated responses through the expression of immune checkpoints, including PD�\L1 and CTLA�\4. Macrophages and neutrophils can also be reprogrammed to acquire an immunosuppressive phenotype (e.g. in response to hypoxia). This promotes tumor expansion through the release of immunosuppressive and pro�\angiogenic factors such as IL�\10, TGF�\��, Arg1 and VEGF. The tolerogenic activity of these cells can be further enhanced under hypoxic conditions 93-95.The innate immune architecture of lung tumors and its implication in disease progression - Milette - 2019 - The Journal of Pathology - Wiley Online Library
https://onlinelibrary.wiley.com/doi/10.1002/path.5241��
��
Cell Death Dis. 2019 Jun 5;10(6):442. doi: 10.1038/s41419-019-1684-0.
H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses.
Lee ACY1, Zhang AJX1,2,3,4, Chu H1,2,3,4, Li C1, Zhu H1, Mak WWN1, Chen Y1, Kok KH1,2,3,4, To KKW1,2,3,4, Yuen KY5,6,7,8.
Author information1
Department of Microbiology, The University of Hong Kong, Hong Kong, China.
2
State Key Laboratory of Emerging Infectious Diseases, Hong Kong, China.
3
Carol Yu Centre for infection, Hong Kong, China.
4
Research Centre of Infection and Immunology, The University of Hong Kong, Hong Kong, China.
5
Department of Microbiology, The University of Hong Kong, Hong Kong, China. kyyuen@hku.hk.
6
State Key Laboratory of Emerging Infectious Diseases, Hong Kong, China. kyyuen@hku.hk.
7
Carol Yu Centre for infection, Hong Kong, China. kyyuen@hku.hk.
8
Research Centre of Infection and Immunology, The University of Hong Kong, Hong Kong, China. kyyuen@hku.hk.
Abstract
We previously demonstrated that avian influenza A H7N9 virus preferentially infected CD14+ monocyte in human peripheral blood mononuclear cells (PBMCs), which led to apoptosis. To better understand H7N9 pathogenesis in relation to monocyte cell death, we showed here that extensive phosphorylation of mixed lineage kinase domain-like (MLKL) protein occurred concurrently with the activation of caspases-8, -9 and -3 in H7N9-infected monocytes at 6 h post infection (hpi), indicating that apoptosis and necroptosis pathways were simultaneously activated. The apoptotic morphology was readily observed in H7N9-infected monocytes with transmission electron microscopy (TEM), while the pan-caspase inhibitor, IDN6556 (IDN), accelerated cell death through necroptosis as evidenced by the increased level of pMLKL accompanied with cell swelling and plasma membrane rupture. Most importantly, H7N9-induced cell death could only be stopped by the combined treatment of IDN and necrosulfonamide (NSA), a pMLKL membrane translocation inhibitor, but not by individual inhibition of caspase or RIPK3. Our data further showed that activation of apoptosis and necroptosis pathways in monocytes differentially contributed to the immune response of monocytes upon H7N9 infection. Specifically, caspase inhibition significantly enhanced, while RIPK3 inhibition reduced the early expression of type I interferons and cytokine/chemokines in H7N9-infected monocytes. Moreover, culture supernatants from IDN-treated H7N9-infected monocyte promoted the expression of co-stimulatory molecule CD80, CD83 and CD86 on freshly isolated monocytes and monocyte-derived dendritic cells (MDCs) and enhanced the capacity of MDCs to induce CD3+ T-cell proliferation in vitro. In contrast, these immune stimulatory effects were abrogated by using culture supernatants from H7N9-infected monocyte with RIPK3 inhibition. In conclusion, our findings indicated that H7N9 infection activated both apoptosis and necroptosis in monocytes. An intact RIPK3 activity is required for upregulation of innate immune responses, while caspase activation suppresses the immune response.
PMID: 31165725 PMCID: PMC6549191 DOI: 10.1038/s41419-019-1684-0H7N9 influenza A virus activation of necroptosis in human monocytes links innate and adaptive immune responses. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31165725��
Research Highlight
Published: 09 July 2013
ROS sets the stage for macrophage differentiation
Anthony Covarrubias, Vanessa Byles & Tiffany Horng
Cell Research volume 23, pages984�C985(2013)Cite this article
Department of Genetics and Complex Disease, Harvard School of Public Health, Harvard University
While M1 macrophages are highly pro-inflammatory and microbicidal, M2 macrophages and the related tumor associated macrophages (TAMs) regulate tissue remodeling and angiogenesis and can display immunomodulatory activity. In July issue of Cell Research, Zhang et al. show that ROS production, critical for the activation and functions of M1 macrophages, is necessary for the differentiation of M2 macrophages and TAMs, and that antioxidant therapy blocks TAM differentiation and tumorigenesis in mouse models of cancer.
Macrophages are key orchestrators in both the initiation and resolution stages of inflammation, and function as sentinel cells that maintain homeostasis and protect against infection. They are activated by many stimuli including pathogen-associated molecular patterns (PAMPs), endogenous danger-associated molecular patterns (DAMPs), and cytokines found in the tissue microenvironment1. During their activation, macrophages can polarize to pro-inflammatory or anti-inflammatory states with distinct phenotypes and physiological responses �� the classical pro-inflammatory M1 state induced by LPS and interferon-�� (IFN-��) and the ��alternative�� M2 state triggered by IL-4 and IL-132. The M1 state is characterized by increased expression of pro-inflammatory cytokines as well as microbicidal activity, while M2 macrophages upregulate the anti-inflammatory cytokine IL-10 and participate in tissue remodeling, wound repair, and host defense against large parasites.
M2-like macrophage polarization is of particular pathophysiological consequence in the setting of cancer. Early in tumor development, monocytes are recruited by tumor and stromal cell-derived chemokines to take up residence at the tumor site, where they differentiate into macrophages in response to MCSF produced by tumor cells. Such tumor-associated macrophages (TAMs) facilitate multiple steps in tumorigenesis, including promotion of tumor cell proliferation and resistance to apoptosis as well as secretion of pro-angiogenic factors and proteolytic enzymes that aid tumor cell metastasis. TAMs also display some immunosuppressive features, such as IL-10 and TGF-�� production and poor antigen presentation, which conspire to prevent tumor cell killing by infiltrating T cells. Thus, the characteristics most critical for the tumor-promoting profile of TAMs bear semblance to the M2 phenotype. Although the details of such M2 polarization are not well characterized, IL-4 produced by T-cells in the tumor, as well as other tumor-derived factors, may be critical3.
In July issue of Cell Research, a study by Zhang et al.4 provides new insights into control of macrophage differentiation and activation. In particular, the authors show that ROS production is important in M2 but not M1 macrophage differentiation. Their experimental protocol is to treat monocytes for 6 days with M-CSF or GM-CSF to induce differentiation to macrophages, followed by polarization with IL-4 (M2 state) or LPS and IFN-�� (M1 state). Interestingly, pre-treating monocytes with the antioxidant butylated hydroxyanisole (BHA) prior to differentiation inhibits M2 but not M1 polarization, as indicated by analysis of macrophage differentiation markers and M1/M2 polarization markers. The authors attribute this to the effects of BHA, i.e., block of ROS production, in inhibiting ERK activation during macrophage differentiation, consistent with previous reports implicating a role for ROS as well as MAP kinases in macrophage differentiation5. Furthermore, LPS and IFN-�� but not IL-4 stimulation can ��rescue�� ERK activation, perhaps in a manner dependent on ROS production, thus explaining why M2 but not M1 polarization is impaired by antioxidant treatment (Figure 1).
Figure 1
figure1
M1 macrophages are highly pro-inflammatory and microbicidal and are polarized by treatment with LPS+IFN��, while M2 macrophages mediate tissue repair, angiogenesis and immunomodulation. Tumor associated macrophages (TAMs), which are M2-like, are associated with worsened clinical prognosis in many cancers and are thought to be skewed by a combination of tumor-derived factors and other cytokines present in the tumor microenvironment. ROS production increases during M-CSF- or GM-CSF-induced macrophage differentiation from monocytes, and the antioxidant BHA specifically inhibits M2 and TAM polarization. LPS+IFN�� treatment is able to overcome the effects of BHA to induce normal M1 polarization, revealing a specific role for ROS in macrophage polarization.
Full size image
As the M2-like properties of TAMs are thought to promote tumorigenesis, Zhang et al. go on to investigate the consequences of BHA administration in mouse models of cancer. They demonstrate that in vivo treatment of BHA can attenuate cancer initiation, progression, and metastasis in multiple models. As ROS can promote tumor cell proliferation, survival, and DNA damage, BHA could be acting directly on the tumor cells to prevent growth and metastasis6. However, BHA had no effects on the proliferation of three tumor cell lines in vitro. The authors propose that TAM differentiation may be a critical target, as BHA administration reduced TAM numbers as well as levels of TAM markers. Moreover, in at least one of the models, BHA administration was ineffective when macrophages were depleted by clodronate injection.
Collectively, the findings of Zhang et al. are intriguing for several reasons. First, ROS production is usually associated with the activation and functions of M1 rather than M2 macrophages. ROS production downstream of LPS signaling mediates production of pro-inflammatory cytokines (in part through MAP kinase activation). ROS and nitric oxide (NO) production by NADPH oxidase and iNOS, respectively, as well as mROS upregulation are key to the antimicrobial activity of M1 macrophages7. Indeed NO production can inhibit oxidative metabolism, pivotal to the survival and function of M2 macrophages8. Thus ROS production may be important in M1 activation and function while the requirement for ROS in M2 differentiation may be most critical during MCSF-mediated differentiation rather than IL-4-triggered polarization. Future studies to better understand the role of ROS production in macrophage differentiation and activation may be informative. Second, it would be interesting to further probe the effects of BHA in inhibiting tumorigenesis. The authors' in vitro studies suggest inhibition of TAM differentiation as one underlying mechanism, but one can envision additional possibilities. At least in some cancers, tumor cells and other immune cells in the microenvironment produce ROS that promote inflammation9, thus contributing to tumorigenesis. mROS has been linked to activation of HIF1��, which can facilitate angiogenesis and metastasis. Indeed, it is worth pointing out that ROS can regulate many cellular processes, some of which have already been alluded to, including signal transduction (e.g., downstream of growth factor receptors and innate immune signaling pathways as well as MAP kinase activation), redox signaling, autophagy, and respiratory burst and other antimicrobial activities10. Thus it is likely that other cellular processes perturbed by antioxidant treatment contribute to the effects of BHA in reducing tumorigenesis.
Finally, the study by Zhang et al. suggests that treatment with BHA or perhaps other antioxidants could be considered in therapeutic control of cancer. Indeed, there is tremendous interest in the clinical use of antioxidants for treating many diseases. Given the pleiotropic activities of ROS mentioned above, it would be important to better understand the molecular pathways by which antioxidants exert their effects.ROS sets the stage for macrophage differentiation | Cell Research
https://www.nature.com/articles/cr201388��
Immunobiology. 2011 Jan-Feb;216(1-2):164-72. doi: 10.1016/j.imbio.2010.06.003. Epub 2010 Jun 15.
Pro- and anti-inflammatory control of M-CSF-mediated macrophage differentiation.
Popova A1, Kzhyshkowska J, Nurgazieva D, Goerdt S, Gratchev A.
Author information
Abstract
Macrophages play a key role in inflammation, tissue regeneration and tolerance. Their differentiation is regulated by tissue cells derived CSF-1 (M-CSF). The ability of macrophages to use autocrine M-CSF to control their differentiation and function remained controversial. In this study we investigated the regulation of M-CSF production by Th1 and Th2 cytokines (IFN-�� and IL-4) and tolerogenic stimuli - glucocorticoid dexamethasone in primary human monocyte derived macrophages. We show that IFN-�� and IL-4 efficiently induce production of M-CSF while glucocorticoid inhibited it in a dose dependent manner. Since glucocorticoid inhibits production of inflammatory cytokines we tested whether this effect is a result of inhibited M-CSF production. We showed that exogenous M-CSF rescues the ability of glucocorticoid-treated macrophages to produce TNF and IL-6 in response to LPS. These data indicate that glucocorticoid-treated macrophages retain the ability to respond to M-CSF. Analyzing the mechanism of this responsiveness, we showed that dexamethasone up-regulates surface expression of M-CSF receptor - CSF-1R. We conclude that the ability of macrophages to produce M-CSF secures macrophage differentiation under Th1 and Th2 conditions if tissue cells are unable to supply enough M-CSF. Increased surface expression of CSF-1R in tolerogenic conditions guarantees response to minute amounts of exogenous M-CSF.
Copyright © 2010 Elsevier GmbH. All rights reserved.
PMID: 20619482 DOI: 10.1016/j.imbio.2010.06.003Pro- and anti-inflammatory control of M-CSF-mediated macrophage differentiation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/20619482��
Cancer Res. 2003 Jul 1;63(13):3632-6.
Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways.
Liu W1, Reinmuth N, Stoeltzing O, Parikh AA, Tellez C, Williams S, Jung YD, Fan F, Takeda A, Akagi M, Bar-Eli M, Gallick GE, Ellis LM.
Author information
1
Department of Cancer Biology, The University of Texas M. D. Anderson Cancer Center, Houston, Texas 77030-4009, USA.
Abstract
Overexpression of cyclooxygenase-2 (COX-2) has been observed in human colorectal cancer. COX-2 expression in human tumors can be induced by growth factors, cytokines, oncogenes, and other factors. The mechanisms regulating COX-2 expression in human colon cancer have not been completely elucidated. We hypothesized that the proinflammatory cytokine interleukin-1 beta (IL-1 beta) mediates COX-2 expression in HT-29 human colon cancer cells. Treatment of HT-29 cells with IL-1 beta induced expression of COX-2 mRNA and protein in a time- and dose-dependent manner. Inhibitors of the extracellular signal-regulated kinase 1/2, c-Jun NH(2)-terminal kinase, P38 mitogen-activated protein kinase, and nuclear factor-kappa B (NF-kappa B) signaling pathways blocked the ability of IL-1 beta to induce COX-2 mRNA. In contrast, Wortmannin, a phosphoinositide 3-kinase inhibitor upstream of protein kinase B/Akt, led to a slight increase in COX-2 mRNA expression after IL-1 beta treatment. Electrophoretic mobility shift assay on nuclear extracts demonstrated that IL-1 beta induced NF-kappa B DNA binding activity in HT-29 cells, and the activated NF-kappa B complex was eliminated after treatment with an inhibitor of NF-kappa B. Supershift assay indicated that the two NF-kappa B subunits, p65 and p50, were involved in activation of NF-kappa B complex by IL-1 beta stimulation. The stability of COX-2 mRNA was not altered by IL-1 beta treatment. These data demonstrate that IL-1 beta induces COX-2 expression in HT-29 cells through multiple signaling pathways and NF-kappa B.Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/12839952��
Published: 18 October 2018
Macrophages promote epithelial proliferation following infectious and non-infectious lung injury through a Trefoil factor 2-dependent mechanism
Li-Yin Hung, Debasish Sen, Taylor K. Oniskey, Jeremey Katzen, Noam A. Cohen, Andrew E. Vaughan, Wildaliz Nieves, Anatoly Urisman, Michael F. Beers, Matthew F. Krummel & De��Broski R. Herbert
Mucosal Immunology volume 12, pages64�C76(2019)Cite this article
Abstract
Coordinated efforts between macrophages and epithelia are considered essential for wound healing, but the macrophage-derived molecules responsible for repair are poorly defined.This work demonstrates that lung macrophages rely upon Trefoil factor 2 to promote epithelial proliferation following damage caused by sterile wounding, Nippostrongylus brasiliensis or Bleomycin sulfate. Unexpectedly, the presence of T, B, or ILC populations was not essential for macrophage-driven repair. Instead, conditional deletion of TFF2 in myeloid-restricted CD11cCre TFF2 flox mice exacerbated lung pathology and reduced the proliferative expansion of CD45− EpCAM+ pro-SPC+ alveolar type 2 cells. TFF2 deficient macrophages had reduced expression of the Wnt genes Wnt4 and Wnt16 and reconstitution of hookworm-infected CD11cCre TFF2flox mice with rWnt4 and rWnt16 restored the proliferative defect in lung epithelia post-injury.
These data reveal a previously unrecognized mechanism wherein lung myeloid phagocytes utilize a TFF2/Wnt axis as a mechanism that drives epithelial proliferation following lung injury.
Macrophages promote epithelial proliferation following infectious and non-infectious lung injury through a Trefoil factor 2-dependent mechanism | Mucosal Immunology
https://www.nature.com/articles/s41385-018-0096-2��
Int J Mol Sci. 2018 Sep; 19(9): 2821.
Published online 2018 Sep 18. doi: 10.3390/ijms19092821
Monocytes and Macrophages as Viral Targets and Reservoirs
Ekaterina Nikitina,1,2,3,* Irina Larionova,3,4 Evgeniy Choinzonov,5 and Julia Kzhyshkowska3,6
Author information Article notes Copyright and License information Disclaimer
1Department of Episomal-Persistent DNA in Cancer- and Chronic Diseases, German Cancer Research Center, 69120 Heidelberg, Germany
2Department of Oncovirology, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, Tomsk 634050, Russia
3Department of Translational Cellular and Molecular Biomedicine, Tomsk State University, Tomsk 634050, Russia; ur.liam@_fortim (I.L.); ed.grebledieh-inu.amdem@akswokhsyhzk.ailuJ (J.K.)
4Department of Molecular Oncology and Immunology, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, Tomsk 634050, Russia
5Head and Neck Department, Cancer Research Institute, Tomsk National Research Medical Center, Russian Academy of Sciences, Tomsk 634050, Russia; ur.cmint@vonoznyohc
6Institute of Transfusion Medicine and Immunology, Medical Faculty Mannheim, Heidelberg University, 68167 Heidelberg, Germany
Abstract
Viruses manipulate cell biology to utilize monocytes/macrophages as vessels for dissemination, long-term persistence within tissues and virus replication.Viruses enter cells through endocytosis, phagocytosis, macropinocytosis or membrane fusion. These processes play important roles in the mechanisms contributing to the pathogenesis of these agents and in establishing viral genome persistence and latency.
Upon viral infection, monocytes respond with an elevated expression of proinflammatory signalling molecules and antiviral responses, as is shown in the case of the influenza, Chikungunya, human herpes and Zika viruses. Human immunodeficiency virus initiates acute inflammation on site during the early stages of infection but there is a shift of M1 to M2 at the later stages of infection. Cytomegalovirus creates a balance between pro- and anti-inflammatory processes by inducing a specific phenotype within the M1/M2 continuum.
Despite facilitating inflammation, infected macrophages generally display abolished apoptosis and restricted cytopathic effect, which sustains the virus production.
The majority of viruses discussed in this review employ monocytes/macrophages as a repository but certain viruses use these cells for productive replication. This review focuses on viral adaptations to enter monocytes/macrophages, immune escape, reprogramming of infected cells and the response of the host cells.
Keywords: monocyte/macrophage, virus, persistence, reservoir, cell response, inflammation, cancerMonocytes and Macrophages as Viral Targets and Reservoirs
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6163364/��
PLoS One. 2012;7(1):e29443. doi: 10.1371/journal.pone.0029443. Epub 2012 Jan 4.
Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation.
Hoeve MA1, Nash AA, Jackson D, Randall RE, Dransfield I.
1 MRC Centre for Inflammation and Research, Queen's Medical Research Institute, University of Edinburgh, Edinburgh, United Kingdom. m.hoeve@ed.ac.uk
Abstract
Influenza virus infection accounts for significant morbidity and mortality world-wide. Interactions of the virus with host cells, particularly those of the macrophage lineage, are thought to contribute to various pathological changes associated with poor patient outcome.Development of new strategies to treat disease therefore requires a detailed understanding of the impact of virus infection upon cellular responses.
Here we report that human blood-derived monocytes could be readily infected with the H3N2 influenza virus A/Udorn/72 (Udorn), irrespective of their phenotype (CD14(++)/CD16(-), CD14(++)/CD16(+) or CD14(dim)CD16(++)), as determined by multi-colour flow cytometry for viral haemagglutinin (HA) expression and cell surface markers 8-16 hours post infection.
Monocytes are relatively resistant to influenza-induced cell death early in infection, as approximately 20% of cells showed influenza-induced caspase-dependent apoptosis. Infection of monocytes with Udorn also induced the release of IL-6, IL-8, TNF�� and IP-10, suggesting that NS1 protein of Udorn does not (effectively) inhibit this host defence response in human monocytes.
Comparative analysis of human monocyte-derived macrophages (Mph) demonstrated greater susceptibility to human influenza virus than monocytes, with the majority of both pro-inflammatory Mph1 and anti-inflammatory/regulatory Mph2 cells expressing viral HA after infection with Udorn. Influenza infection of macrophages also induced cytokine and chemokine production. However, both Mph1 and Mph2 phenotypes released comparable amounts of TNF��, IL-12p40 and IP-10 after infection with H3N2, in marked contrast to differential responses to LPS-stimulation.
In addition, we found that influenza virus infection augmented the capacity of poorly phagocytic Mph1 cells to phagocytose apoptotic cells by a mechanism that was independent of either IL-10 or the Mer receptor tyrosine kinase/Protein S pathway.
In summary, our data reveal that influenza virus infection of human macrophages causes functional alterations that may impact on the process of resolution of inflammation, with implications for viral clearance and lung pathology.
PMID: 22238612 PMCID: PMC3251590 DOI: 10.1371/journal.pone.0029443
[Indexed for MEDLINE] Free PMC ArticleInfluenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/22238612��
J Immunol. 1999 Jun 15;162(12):7322-9.
Virus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway.
Pirhonen J1, Sareneva T, Kurimoto M, Julkunen I, Matikainen S.
Author information
1
Department of Virology, National Public Health Institute, Helsinki, Finland. jaana.pirhonen@ktl.fi
Abstract
Monocytes and macrophages play a significant role in host's defense system, since they produce a number of cytokines in response to microbial infections. We have studied IL-1 beta, IL-18, IFN-alpha/beta, and TNF-alpha gene expression and protein production in human primary monocytes and GM-CSF-differentiated macrophages during influenza A and Sendai virus infections.Virus-infected monocytes released only small amounts of IL-1 beta or IL-18 protein, whereas 7- and 14-day-old GM-CSF-differentiated macrophages readily produced these cytokines. Constitutive expression of proIL-18 was seen in monocytes and macrophages, and the expression of it was enhanced during monocyte/macrophage differentiation.
Expression of IL-18 mRNA was clearly induced only by Sendai virus, whereas both influenza A and Sendai viruses induced IL-1 beta mRNA expression. Since caspase-1 is known to cleave proIL-1 beta and proIL-18 into their mature, active forms, we analyzed the effect of a specific caspase-1 inhibitor on virus-induced IL-1 beta and IL-18 production.
The release of IL-1 beta and IL-18, but not that of IFN-alpha/beta or TNF-alpha, was clearly blocked by the inhibitor.
Our results suggest that the cellular differentiation is a crucial factor that affects the capacity of monocytes/macrophages to produce IL-1 beta and IL-18 in response to virus infections. Furthermore, the virus-induced activation of caspase-1 is required for the efficient production of biologically active IL-1 beta and IL-18.
PMID: 10358182
[Indexed for MEDLINE] Free full textVirus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/10358182��
��