ḂḂ
ÁṫẁŴĠḂ¶ẅ Influenza Virus Studies
Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failureḂî
ḂḂ
ḂḂ
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics
Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failureḂî
Author links open overlay panelHiroshiKidoYuushiOkumura1EtsuhisaTakahashiHai-YanPan2SiyeWangDengbingYao3MinYaoJunjiChidaMihiroYano
Division of Enzyme Chemistry, Institute for Enzyme Research, The University of Tokushima, Kuramoto-cho 3-18-15, Tokushima 770-8503, Japan
Received 31 March 2011, Revised 3 July 2011, Accepted 5 July 2011, Available online 23 July 2011.
Abstract
Influenza A virus (IAV) is one of the most common infectious pathogens in humans. Since the IVA genome does not have the processing protease for the viral hemagglutinin (HA) envelope glycoprotein precursors, entry of this virus into cells and infectious organ tropism of IAV are primarily determined by host cellular trypsin-type HA processing proteases. Several secretion-type HA processing proteases for seasonal IAV in the airway, and ubiquitously expressed furin and pro-protein convertases for highly pathogenic avian influenza (HPAI) virus, have been reported. Recently, other HA-processing proteases for seasonal IAV and HPAI have been identified in the membrane fraction. These proteases proteolytically activate viral multiplication at the time of viral entry and budding.In addition to the role of host cellular proteases in IAV pathogenicity, IAV infection results in marked upregulation of cellular trypsins and matrix metalloproteinase-9 in various organs and cells, particularly endothelial cells, through induced pro-inflammatory cytokines. These host cellular factors interact with each other as the influenza virusẀCcytokineẀCprotease cycle, which is the major mechanism that induces vascular hyperpermeability and multiorgan failure in severe influenza.
This mini-review discusses the roles of cellular proteases in the pathogenesis of IAV and highlights the molecular mechanisms of upregulation of trypsins as effective targets for the control of IAV infection. This article is part of a Special Issue entitled: Proteolysis 50 years after the discovery of lysosome.
Highlights
► Cellular proteases play key roles in virus entry and vascular hyperpermeability. ► Influenza A virus (IAV)-cytokine-protease cycle. ► IAV infection results in marked upregulation of cellular trypsins and MMP-9. ► Processing of IAV hemagglutinin (HA) is a pre-requisite for viral entry into cells. ► Identification of HA processing proteases for highly pathogenic avian IAV.
Fig. 10. The hypothesis of ḂḞInfluenzaẀCcytokineẀCprotease cycleḂḟ, involved in the pathogenesis of vascular hyperpermeability and tissue destruction in severe influenza. BBB, bloodẀCbrain barrier.
ḂḂ
5. Conclusions
It has been known for decades that the infectivity, infectious organ tropism and pathogenicity of IAV are primarily determined by host cellular trypsin-type proteases, which cleave the viral envelope fusion glycoprotein precursor HA0, because a HA-processing protease is not encoded in the viral genome. Although there are many host cellular proteases that recognize the carboxyl moiety of R residue as determined by synthetic substrates, only few proteases recognize the tertiary structure of the cleavage site of HA0, thus allowing IAV multiplication with proteolytic activation only in organs that express the HA-processing proteases [5], [17], [18]. As outlined in this review, the knowledge gained within the last few years has greatly improved our understanding on the role of host cellular proteases in the pathogenicity of IAV infection: (i) Several cell surface anchored trypsin-like serine proteases are candidates of HA-processing protease for seasonal human IAV, TMPRSS2, HAT and TMPRSS4. This is in addition to secreted HA-processing trypsin-type proteases in the airway, such as tryptase Clara, trypsin and mini-plasmin. In addition, MSPL and TMPRSS13, plasma membrane bound HA-processing proteases for HPAI viruses other than furin and PCs are found. Furin preferentially cleaves only HA with the R-K-K-R cleavage site motif with R at position P4, whereas MSPL/TMPRSS13 cleaves HA, including a wide range of strains of HPAI virus with the R/K-K-K-R motif with both K and R at position P4. (ii) IAV infection upregulates cellular trypsins, MMPs and cytokines, and also ATP depletion in various infected cells. The upregulated trypsins in various organs including endothelial cells changes IAV infectious organ tropism of seasonal IAV. Destruction of endothelial cell tight junctions by the induced trypsins causes viral invasion in various organs. Based on the results of studies on the relationship among upregulated factors, we propose the influenza virusẀCcytokineẀCprotease cycle hypothesis as one of the main mechanisms of vascular dysfunction in multiorgan failure, in association with cytokine storm in severe influenza (Fig. 10) [7]. (iii) Inhibitors of NF-ḊÊB and AP-1 effectively suppress the upregulation of proinflammatory cytokines and cellular trypsins, and improve survival rates of infected mice. In addition, administration of trypsin-type protease inhibitor aprotinin suppresses viral replication [7], upregulation of trypsins, MMPs and cytokines as well as ATP depletion and significantly improves cardiac function [8]. More selectively, trypsin-knockdown, particularly T2-knockdown, in cardiomyoblasts effectively suppresses IAV replication and ATP depletion accompanied by protection against mitochondrial membrane depolarization and apoptosis. These results suggest that upregulated cellular trypsins are one of the key pathogenic host factors in IAV-induced multiple organ failure. (iv) Since IAV infection induces impaired fuel utilization, patients with disorders of mitochondrial fatty acid oxidation are prone to severe multiple organ failure after IAV infection. (v) Upregulated cellular trypsins in various organs and endothelial cells might be effective drug targets for the control of multiple organ failure in severe influenza.
Fig. 4. Schematic diagram of host cellular responses in the mouse airway after IAV infection.
In this review, we discuss host cellular HA processing proteases for seasonal and highly pathogenic avian influenza (HPAI) viruses and the relationship among IAV, cytokines and host cellular proteases that induce vascular hyperpermeability and multi-organ failure in severe influenza. We also discuss the genetic background of influenza high risk patients and effective targets for the control of IAV infection.
2. Host cellular viral envelope processing proteases of seasonal human and highly pathogenic avian IAV
Limited proteolysis of envelope glycoprotein HA, is an important step for IAV entry into cells. This cleavage induces the maturation of HA protein and leads to the fusion between virions and host cell membranes. However, IAV cannot process HA by itself, because no HA-processing protease is encoded in its genome. Therefore, viral entry into the cells is determined by the host cellular processing proteases [3], [17], [18], [19]. Proteolytic activation of seasonal human IAV HA occurs extracellularly by airway proteases, such as tryptase Clara [18], mini-plasmin [20], pancreatic trypsin [3], cellular trypsins [4], [7], [8], [21], porcine lung tryptase [22] and TC30 [23]. Fig. 1A shows that tryptase Clara purified as a single protein band on SDS-PAGE under reducing conditions from rat lung and cationic trypsin from bovine pancreas cleave [3H]glucosamine-labeled influenza virus HA into subunits HA1 and HA2. Direct amino acid sequencing of the amino-terminus of the HA2 subunit revealed residues G-L-F-G-A-I-A-G-, indicating that the cleavage sites by tryptase Clara and trypsin was between R325 and G326, the amino-terminal site of membrane fusion domain of HA [18]. Fig. 1B shows that tryptase Clara and trypsin triggered the IAV infectivity in a dose-dependent manner. Although the induction of infectivity by tryptase Clara was less efficient than that induced by trypsin, in contrast, tryptase Clara cleaved selectively even at higher concentrations and did not decrease viral infectivity.
Download : Download high-res image (78KB)Download : Download full-size image
Fig. 1. Proteolytic activation of IAV Aichi/2/68(H3N2) in vitro by tryptase Clara and trypsin. A. SDS-PAGE of the viral glycoproteins. [3H]Glucosamine-labeled non-activated virus isolated from culture media of MadinẀCDarby canine kidney (MDCK) cells (lane 1) was treated with bovine pancreatic cationic trypsin (10 ḊÌg/ml) for 15 min (lane 2), tryptase Clara purified from rat lung (10 ḊÌg/ml) for 15 min (lane 3), tryptase Clara (50 ḊÌg/ml) for 15 min (lane 4), or tryptase Clara (50 ḊÌg/ml) for 30 min (lane 5) at 37 ḂãC. SDS-PAGE proceeded on a 13% acrylamide gel under reducing conditions. B. Activation of virus infectivity. Non-activated virus propagated in MDCK cells was suspended in PBS (pH 7.2) and digested with trypsin (Ḃŵ) or with tryptase Clara (Ḃñ) for 15 min at 37 ḂãC. The infectivity of the activated virus was assayed by immunofluorescent cell-counting method [18]. CIU, cell-infecting unit.
In addition to these secreted-type serine proteases, transmembrane serine protease 2 (TMPRSS2) and human airway trypsin-like protease are also candidates for the processing proteases in the human airway membrane [24]. In addition, it was reported recently that TMPRSS2 and TMPRSS4 proteolytically activate the 1918 Spanish flu influenza virus HA [25]. These trypsin-type endoproteases recognize the carboxyl moiety of a single R residue within the consensus cleavage motif Q/E-X-R (where X is any amino acid except C and basic amino acids) of most of the seasonal human IAV known to date, including pneumotropic and neurotropic IAV strains [26], [27].
These proteases show different tissue distribution in the airway [17]. Mini-plasmin, a truncated kringle 1ẀC4 domains derivative of plasmin, is found in folded rat epithelial cells; tryptase Clara is located in Clara cells of rat inferior bronchiolar divisions, such as the terminal and respiratory bronchioles; and lung anionic trypsin is distributed in stromal cells of rat peri-bronchiolar regions. Under physiologic conditions, the distribution of mini-plasmin is limited to rat superior bronchiolar divisions but amounts as high as ~ 2 ḊÌM may be produced from blood plasminogen at inflammatory loci by the processing proteases, such as elastase and cathepsin G from granulocytes and cathepsin D from monocytes, during clot formation in the lung [17], [20]. In addition, IAV infection induces marked upregulation of cellular trypsins in the lung, heart, brain and vascular endothelial cells [4], [7], [8]. These proteases may play critical roles in the spread of IAV with vascular hyperpermeability and tissue damage. We also found mast cell tryptase and tryptase TC30 as HA processing enzymes in porcine lung, although human mast cell tryptase did not proteolytically activate HA0 of IAV in humans [22], [23].
We found different potencies for these cellular proteases in the proteolytic potentiation of various virus strains. As shown in Fig. 2, among the proteases examined in our studies, pancreatic trypsin efficiently activated the infectivity of almost all strains except IAV WSN/33(H1N1), while small size derivatives of plasmin, such as mini-plasmin and micro-plasmin, a truncated kringle 1ẀC5 domains derivative of plasmin, also activated all strains though less efficiently than trypsin [20]. The proteolytic potentiating activity of plasmin was the highest for IAV WSN strain, weak for IAV Aichi/2/68(H3N2) strain, and nonexistent for IAV seal/Massachusetts/1/81(H7N7). Mutational evolution of IAV HA0 to adapt to these host cellular processing proteases in the airway allows efficient multiplication of the virus in vivo and ultimately causing epidemic of HA infection. In addition to the host cellular proteases, microbial proteases also proteolytically activate influenza virus HA0 in bacterial infection of the airways and play a role in the spread of the virus [28], [29].
Download : Download high-res image (116KB)Download : Download full-size image
Fig. 2. Potentiation by plasmin, mini-plasmin, microplasmin, and trypsin of IAV WSN(H1N1) (A), IAV seal/Massachusetts/1/81(H7N7) (B), and IAV Aichi/2/68(H3N2) (C). MDCK cell-grown non-activated IAV strains were treated for 30 min at 37 ḂãC with trypsin (Ḃñ), plasmin (Ḃŵ), mini-plasmin (Ḃø), or microplasmin (Ḃö) at the indicated concentrations. The enzyme reaction was stopped by the addition of 100 ḊÌM aprotinin. Infectivity is shown as cell-infecting unit (CIU).
As is the case for all HPAI viruses of subtypes H5 and H7 known to date, the cleavage of HA occurs at the C-terminal R residue in the consensus multi-basic motifs, such as R-X-K/R-R with R at position P4 and K-K/R-K/T-R with K at P4, and leads to systemic infection. The trypsin type serine proteases for seasonal human IAV described above are not susceptible for proteolytic cleavage of HA of HPAI virus. Early studies demonstrated that the ubiquitously expressed furin and proprotein convertases (PCs) are activating proteases of HPAI viruses [30], [31], [32], [33], [34]. Furin and PCs are calcium-dependent subtilisin-like serine proteases in the trans-Golgi network that cleave the consensus multi-basic motif R-X-K/R/X-R with R at position P4 [35], [36], [37], [38], [39]. However, replacement of P4 R by K and a nonbasic amino acid significantly suppresses the processing activities of furin and PCs [37], [39], [40]. These findings suggest the possible involvement of host cellular protease(s), other than furin and PCs, in the processing of HA of HPAI viruses with multi-basic cleavage motifs with K at position P4.
We recently found that ubiquitous type II transmembrane serine proteases (TTSPs), MSPL and its splice variant TMPRSS13, are novel candidates of HA processing proteases of all HPAI viruses known to date [41], [42], [43], [44]. The TTSPs have a common structure; a short cytoplasmic domain and a transmembrane domain at the N-terminus side, and a serine protease domain at the extracellular C-terminus. Most of the TTSPs identified so far recognize a single R at position P1, but the newly isolated MSPL/TMPRSS13 has unique substrate specificities of the preferential recognition of a paired basic residue at the cleavage site [41], [43], [44]. Thus, MSPL/TMPRSS13, which are ubiquitously expressed in the plasma membranes, particularly highly expressed in the lungs, brain and vascular epithelial cells of human and chicken [42], [43], can activate various fusogenic viral envelope glycoproteins of HPAI viral strains. To clarify the roles of MSPL and TMPRSS13 in the proteolytic activation of HA of HPAI viruses, we searched for cells that express MSPL/TMPRSS13 at below detection levels and found human endothelial cell line ECV304 as the corresponding cell line. We then established MSPL/TMPRSS13 stably expressing ECV304 cells.
Fig. 3 shows the role of MSPL/TMPRSS13 in HAPI virus infection and multiplication. Wild type (WT) ECV304 cells, ECV304-WT, and stable transfectant ECV304 cells expressing full-length human MSPL, ECV304-MSPL, were infected with the HPAI virus A/Crow/Kyoto/53/2004 (H5N1) (HA: NḂä-R-K-K-RḂýG-CḂä) [41] and the genetically modified mutant recombinant virus (HA: NḂä-K-K-K-RḂýG-CḂä). Although processing of HA from the mutant recombinant virus (with the motif NḂä-K-K-K-R-CḂä) in ECV-WT cells was hardly detected, conversion of HA0 to the mature form was detected in ECV-MSPL cells. In addition, this conversion was suppressed by BowmanẀCBirk trypsin inhibitor (BBI), a membrane non-permeable high-molecular mass inhibitor against MSPL/TMPRSS13. To test for the generation of infective virus, the conditioned media from 1-day cultures of ECV304-WT and ECV304-MSPL cells infected with WT and mutant HPAI H5N1 viruses were inoculated into newly prepared cells and cultured for 24 h. Although infection with WT virus carrying the HA cleavage motif of R-K-K-R spread from the conditioned media of both ECV304-WT and ECV304-MSPL cells, infection with mutant virus carrying the HA cleavage motif K-K-K-R spread only from the condition medium of ECV304-MSPL cells. These results strongly suggest that the expression of MSPL, but not furin, potentiates multicycle replication of HPAI virus with the K-K-K-R HA cleavage motif. The cleavability of HA peptides of 14-residues synthetic peptides derived from HA cleavage sites of HPAI strains, such as A/chick/Penn/1370/83 (H5N2) [45] and A/FPV/Rostock/34 (H7N1) [31], by MSPL/TMPRSS13 was also studied. MSPL cleaved both the H5 HA peptide with the K-K-K-R motif and the H7 HA peptide with the R-K-K-R motif at the correct positions, while furin cleaved only at a single site of R with R at position P4, NḂä-K-K-R-K-K-RḂý-G-CḂä and hardly cleaved the H5 HA peptide with K at position P4. These findings suggest that MSPL/TMPRSS13 covers diverse cleavage specificities including non-susceptible specificity to furin.
Download : Download high-res image (214KB)Download : Download full-size image
Fig. 3. Cleavage of HA proteins in WT HPAI virus A/Crow/Kyoto/53/2004 (H5N1) and the mutant (Mut) virus by MSPL and multiple infections with the viruses. (A) ECV304-MSPL and ECV304-WT cells were infected with WT HPAI virus A/Crow/Kyoto/53/2004 (H5N1) (H5N1-WT; carrying the HA motif R-K-K-R) and genetically modified mutant virus (H5N1-Mut; carrying the HA motif K-K-K-R) for 24 h. The cell lysates were subjected to SDS-PAGE and analyzed by western immunoblotting with anti-H5N1 monoclonal antibody (4 C12). BowmanẀCBirk trypsin inhibitor (BBI), an inhibitor of MSPL [41]. Molecular mass markers are on the right. (B) Immunofluorescence assay was performed as described previously [41]. The cells were fixed with 4% paraformaldehyde in PBS containing 0.1% Triton-X-100. The binding of polyclonal antibodies to the viral protein was detected with an AlexaFluor 488-conjugated secondary antibody (1:500). Bar, 50 ḊÌm.
3. Influenza virusẀCcytokineẀCprotease cycle in the pathogenesis of vascular hyperpermeability and multiple organ failure in severe influenza
IAV infection increases the levels of proinflammatory cytokines such as TNF-ḊÁ, IL-6, and IL-1ḊÂ in the airway and blood. These cytokines upregulate cellular trypsins and MMP-9 through activation of nuclear factor-kappa B (NF-ḊÊB) and activator protein 1 (AP-1) [7]. Fig. 4 is a schematic illustration of a typical series of biological responses in the airway fluid of mice after IAV infection. The initial response before the peak of viral proliferation at days 4ẀC5 post-infection is significant increases in proinflammatory cytokine levels (ḂḞcytokine stormḂḟ). Immediately after that, ectopic trypsin and MMP-9 are markedly upregulated, and viral titers increase in the airway. Just after the peak of viral proliferation, the innate and adaptive immune responses of protective immunity are induced for defense and recovery, or oppositely on rare occasions, multi-organ failure.
Download : Download high-res image (195KB)Download : Download full-size image
Fig. 4. Schematic diagram of host cellular responses in the mouse airway after IAV infection.
Fig. 5 shows upregulation of cytokines and cellular trypsins, increase in viral RNA after IAV infection, and effects of NF-ḊÊB and AP-1 inhibitors on the upregulation and survival of infected mice. The levels of TNF-ḊÁ and IL-6 in the lungs, the site of initial virus infection, were increased persistently for 6 days, and that of IL-1ḊÂ peaked at days 4ẀC6 post-infection (Fig. 5A). Since these cytokine responses are associated with activation of transcription factors, NF-ḊÊB and AP-1 [15], [46], [47], [48], we treated mice once daily for 4 days with NF-ḊÊB inhibitors: pyrrolidine dithiocarbamate (PDTC) and N-acetyl-l-cysteine (NAC), and AP-1 inhibitors: nordihydroguaiaretic acid (NDGA). PDTC and NDGA significantly suppressed the upregulation of TNF-ḊÁ and IL-1ḊÂ (P < 0.001), and NAC suppressed TNF-ḊÁ (P < 0.001) and IL-6 (P < 0.01) at day 4 post-infection. Trypsin activities analyzed by gelatin zymography showed upregulation of cellular trypsins in mice lung, brain, and heart during infection for 6 days (Fig. 5B). Trypsin induction was inhibited also by treatment with PDTC, NAC and NDGA, probably via blockade of NF-ḊÊB and AP-1 binding in the promoter region of the gene (S.R. Talukder et al., manuscript in preparation). Viral RNA replication in various organs at day 4 post-infection was suppressed by more than one order of magnitude by PDTC, NAC and NDGA (Fig. 5C). Suppression of viral multiplication and induction of cytokines and trypsins by treatment with these inhibitors significantly improved the survival of mice at day 14 post-infection (Fig. 5D).
Download : Download high-res image (339KB)Download : Download full-size image
Fig. 5. Upregulation of cytokines and trypsins, increase in viral RNA after IAV infection in mice, and effects of NF-ḊÊB and AP-1 inhibitors on the upregulation and survival of infected mice. (A) Mice were infected with 250 plaque-forming units (PFU) of IAV WSN/33(H1N1) virus (WSN) with and without treatment with pyrrolidine dithiocarbamate (PDTC), N-acetyl-l-cysteine (NAC), and nordihydroguaiaretic acid (NDGA). Levels of tumor necrosis factor (TNF)-ḊÁ, interleukin (IL)-6, and IL-1ḊÂ in lung homogenates (n = 3) were analyzed before (Control) and at day 2 (WSN-D2), day 4 (WSN-D4) and day 6 (WSN-D6) after infection. Cytokine levels in lungs of animals treated once daily for 4 days with PDTC (PDTC-D4), NAC (NAC-D4), and NDGA (NDGA-D4) were also measured. Data are mean ḂÀ SEM. **P < 0.01, #P < 0.001, versus WSN-D4. (B) Trypsin activities analyzed by gelatin zymography of infected mice for 0ẀC6 days. Animals were treated with PDTC, NAC, and NDGA once daily for 4 days. Each lane represents the same experimental conditions as in (A). (C) Mice were infected with WSN and also treated with PDTC, NAC, and NDGA. Quantitative analysis of viral NS1 RNA copies normalized by ḊÂ-actin at day 4 after infection was conducted by real-time PCR (n = 3). Data are mean ḂÀ SEM. #P < 0.001 versus without drug treatment. (D) Survival rates. Mice of each group (n = 10) were infected with WSN at 250 PFU and 500 PFU. Animals infected with WSN at 250 PFU were treated with PDTC, NAC, and NDGA once daily for 4 days, and the survival rates of the different groups were compared. *P < 0.05, #P < 0.001 versus without drug treatment.
In most animal species, trypsin has three major isoforms: cationic trypsin (T1), anionic trypsin (T2), and mesotrypsin (T3) [49], [50]. To analyze the linkage between these cytokines and cellular trypsins in human endothelial cells, changes in the expression of human trypsin (hPRSS) genes (T1ẀC3) were analyzed after 6-h exposure to 10 ng/mL TNF-ḊÁ, IL-6, and IL-1ḊÂ (Fig. 6). All tested cytokines tended to upregulate hPRSS expression levels, especially TNF-ḊÁ (##P < 0.01) and IL-1ḊÂ (#P < 0.05), and the upregulation was inhibited by simultaneous treatment with the respective neutralizing antibodies (100 ng/ml) to these cytokines (*P < 0.05; **P < 0.01). Cellular trypsins mediate the post-translational proteolytic cleavage of viral envelope HA and subsequent tissue damage in various organs. These results suggest close relationship among factors, influenza virus, cytokines and host cellular proteases cycle in the pathogenesis of influenza.
Download : Download high-res image (88KB)Download : Download full-size image
Fig. 6. Increase in human trypsin (hPRSS) expression in endothelial cells after WSN infection and cytokine treatment, and its suppression by anti-cytokine antibodies. (A) hPRSS mRNA levels in the cells measured by RT-PCR after viral infection for 0ẀC12 h and the percentage change in the expression. (B) Increase in hPRSS mRNA levels in the cells after treatment with cytokine (TNF-ḊÁ, IL-6, and IL-1ḊÂ) for 6 h and their suppression by anti-cytokine antibodies. Data are mean ḂÀ SEM (n = 3). ##P < 0.01, #P < 0.05 versus the control. **P < 0.01, *P < 0.05, versus treatment with each antibody.
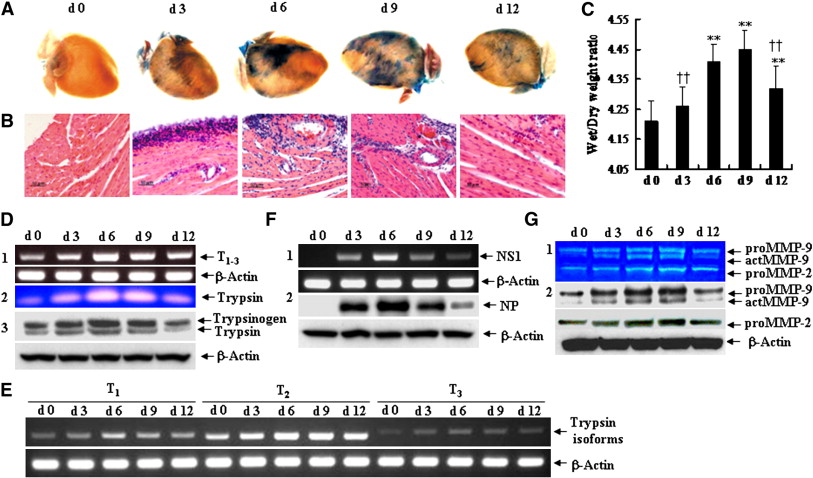
Fig. 7 shows the results of studies on upregulation of trypsin and MMP-9 in the myocardium associated with increased vascular hyperpermeability, tissue edema, and inflammatory cell infiltration after IAV infection in mice [8]. In these studies, mice intra-nasally inoculated with IAV exhibited acute myocarditis, as confirmed by histological examination of hematoxylin and eosin-stained sections. Inflammatory infiltrates appeared in the subepicardium at day 3 post-infection, followed by extensive infiltration across the interstitium and perivascular areas deep into the myocardium, accompanied by extracellular matrix destruction during days 6 and 9, and then alleviated at day 12 (Fig. 7AẀCC).Coronary vascular permeability, monitored by extravasation (using Evans blue dye) and tissue edema (by wet/dry ratio), started to increase at day 3 after infection, reaching a peak during days 6 and 9, and then decreased significantly at day 12.
Download : Download high-res image (451KB)Download : Download full-size image
Fig. 7. Increased vascular hyperpermeability, tissue edema and inflammatory cell infiltration in acute myocarditis, and kinetics of upregulation of trypsins in the myocardium after IAV infection. (A) Vascular hyperpermeability monitored by extravasation (Evans blue staining) during the course of infection from day 0 (d 0) to 12 (d 12). (B) Hematoxylin and eosin staining. Bar = 50 ḊÌm. (C) Heart edema determined by wet/dry weight ratio. Data are mean ḂÀ SD of 10 mice in each group. ⁎⁎P < 0.01 versus d 0; ††P < 0.01 versus d 9. (D1, E and F1) RT-PCR-based detection of trypsin1ẀC3, trypsin isoforms T1, T2 and T3 and IAV NS1 gene in the hearts from day 0 to 12 after infection. (D2) Detection of trypsin activity by zymography, (D3) trypsinogen and trypsin by western immunoblotting, and (F2) viral nucleoprotein (NP). (G) Kinetics of up-regulation of proMMP-9, proMMP-2, and activated (act) MMP-9 in the hearts determined by zymography (G1) and western immunoblotting (G2) from day 0 to 12 post-infection. Each result is representative of three experiments.
Trypsin in mice has also three major isoforms (T1ẀC3) [50]. In the above studies, infection of IAV first upregulated trypsins (T1ẀC3) in the heart at day 3, reached a peak at day 6, remained at high levels until day 9, and then started to decrease at day 12 (Fig. 7D) [8]. The majorities of upregulated T1ẀC3 were trypsinogens, as determined by western immunoblotting, and were partially converted to active trypsins with a peak at day 6 monitored by zymography. In uninfected mice, T2 was the predominant isoform in the heart, while the level of T1 was less than that of T2, and T3 was barely detected. After infection, T2 was predominantly induced with a peak during days 6 and 9 (Fig. 7E). The viral levels in the heart monitored by viral NS1 gene and nucleoprotein (NP) showed that it also peaked at day 6 and started to decrease at day 9 (Fig. 7F). Analysis of the distribution of the increased levels of trypsins, viral protein and Evans' blue dye (a marker of hyperpermeability and tissue injury) showed their overlapped localization, suggesting their close relationship in the infected foci [8].
Limited amounts of trypsin(s) effectively converts various proMMPs, particularly proMMP-9, into active MMPs [51], [52] and the latter in turn convert proinflammatory cytokines to active cytokines [53], resulting in cardiomyocyte injury and extracellular matrix degradation. Along with an increase in trypsin levels, upregulated proMMP-9 was readily converted to active MMP-9 (actMMP-9) from day 3 postinfection, whereas upregulated proMMP-2 was not (Fig. 7G). The actMMP-9 levels increased to a peak value during day 6 and 9, followed by a decrease in the levels at day 12.
4. Trypsin knockdown inhibits influenza virusẀCcytokineẀCprotease cycle and mitochondrial ATP depletion
Since host cellular trypsins facilitate IAV entry and viral replication in target cells and trypsin efficiently converts proMMPs to active MMPs in tissues, we analyzed the effects of silencing major trypsin genes, T1 and T2 in mice heart, on viral replication and production of actMMP-9 and cytokines in cardiomyoblast H9c2 cells. The expression levels of T1 and T2 genes determined by RT-PCR were selectively and significantly suppressed in trypsin1-short hairpin (sh) RNA gene silencing plasmid (T1-sh) and trypsin2-shRNA gene silencing plasmid (T2-sh) cells, respectively, compared with their levels in the control (C-sh) cells (Fig. 8A) [8]. Viral replication monitored by NS1 gene and the levels of viral NP showed significant suppression of IAV replication in T1- and T2-knockdown cells; the magnitude of suppression in T2-knockdown was significantly higher than in T1-knockdown (Fig. 8B1ẀC2). Furthermore, a significant reduction in the number of immunofluorescence-labeled viral antigen in the infected cells (red) was detected under T1- and T2-knockdown conditions, particularly T2-knockdown condition, compared to those in C-sh cells (Fig. 8C1ẀC2).
Download : Download high-res image (356KB)Download : Download full-size image
Fig. 8. Effects of trypsin knockdown on IAV replication, MMPs upregulation and actMMP-9 production in H9c2 cardiomyoblasts. (A) Silence efficiency of T1 and T2 genes was determined by RT-PCR in T1-sh, T2-sh and C-sh cell lines. (B1) Effects of T1- and T2-knockdown on viral replication monitored by NS1 gene in cell lysates by RT-PCR, NP in culture media by western immunoblotting, and (B2) relative values of NS1 and NP versus infection control (C-sh). (C1) Detection of viral antigen by immunofluorescence (red) in H9c2 cells at 24 h after infection. Nuclei were stained by 4,6-diamidino-2-phenylindole (DAPI). Bar = 25 ḊÌm. (C2) Percentage of infected cells. Data are mean ḂÀ SD of three independent experiments. (D) Effects of T1- and T2-knockdown on the expression of proMMP-9, proMMP-2 and actMMP-9 in cell lysates and culture media of T1-sh, T2-sh, and C-sh cells infected with IAV or mock monitored by western immunoblotting. ⁎P < 0.05, ⁎⁎P < 0.01 versus mock infection; ††P < 0.01 versus infection control; ##P < 0.01 versus T1-knockdown.
Upregulation of both proMMP-9 and proMMP-2 was detected in C-sh cells after infection and the upregulation was significantly suppressed by T1- and T2-knockdown; the magnitude of suppression was higher in T2-knockdown than in T1-knockdown (Fig. 8D). Furthermore, a significant suppression of actMMP-9 production monitored by the ratio of actMMP-9/proMMP-9 was observed in culture media of T1- and T2-knockdown cells, the efficiency of T2-knockdown was higher than in T1-knockdown.
Induction of proinflammatory cytokines (IL-6, IL-1ḊÂ and TNF-ḊÁ) by IAV infection in culture media, was suppressed under T1- and T2-knockdown conditions, particularly IL-6 in T2-knockdown (Fig. 9A) [8]. Since influenza-associated encephalopathy is a sign of impaired mitochondrial fuel utilization in severe influenza with multiple organ failure [54], we further analyzed the effects of trypsin knockdown on mitochondrial membrane potential (∆ḊṖm), intracellular ATP level and apoptosis after IAV infection for 24 h. Active mitochondria with high ∆ḊṖm appeared red by cationic lipophilic probe JC-1[54], [55], as shown in the cells with mock infection (Fig. 9B, C-sh, mock). Viral infection markedly reduced the red fluorescence indicating mitochondrial depolarization, and reduction of ∆ḊṖm. T1- and T2-knockdown, particularly T2-knockdown, improved the infection-induced mitochondrial depolarization (Fig. 9B, T2-sh, infection) [8]. The numbers of apoptotic cells analyzed by TUNEL staining were significantly reduced under T1- and T2-knockdown conditions, particularly T2-knockdown condition. Furthermore, ATP levels were reduced in H9c2 cells to about 61% after viral infection and this reduction was abrogated by trypsin-knockdown, particularly T2-knockdown (Fig. 9C).
Download : Download high-res image (323KB)Download : Download full-size image
Fig. 9. IAV infection-induced cytokine upregulation, mitochondrial membrane depolarization, ATP depletion and apoptosis, and abrogation by T1- and T2-knockdown in H9c2 cardiomyoblasts. (A) Effects of T1- and T2-knockdown on IL-6, IL-1ḊÂ, and TNF-ḊÁ levels in culture media were analyzed by ELISA at 24 h after infection. (B) ∆ḊṖm of C-sh, T1-sh and T2-sh cells infected with mock or IAV was analyzed after IAV infection for 24 h. Top: ∆ḊṖm was monitored by treatment for 15 min with JC-1 (red). Bottom: Apoptotic cells detected by TUNEL staining (light blue). Nuclei were stained by DAPI. Bar = 25 ḊÌm. Right: Percentage of apoptotic cells among total cells. (C) ATP levels under the same experimental conditions in B. Data are mean ḂÀ SD of three independent experiments. ⁎P < 0.05, ⁎⁎P < 0.01 versus mock infection; †P < 0.05, ††P < 0.01 versus infection control; #P < 0.05, ##P < 0.01 versus T1-knockdown.
IAV infection induces impaired fuel utilization as described above. A large proportion of patients suffering from disabling or fatal influenza-associated encephalopathy exhibits a thermolabile phenotype of compound homo/heterozygous variants of carnitine palmitoyltransferase II (CPT II) [54], [56], [57]. CPT Ḋ© and II are important factors in ATP generation through mitochondrial long-chain fatty acid oxidation [58], [59], [60]. ATP generation in endothelial cells, in particular, is derived as much as 70% from fatty acid oxidation versus 30% from glycolysis [61]. Considered together, these results suggest the involvement of influenza virusẀCcytokineẀCprotease cycle in endothelial cells of patients with disordered mitochondrial fatty acid oxidation, in the enhanced development of severe vascular dysfunction, resulting in multiple organ failure.
5. Conclusions
It has been known for decades that the infectivity, infectious organ tropism and pathogenicity of IAV are primarily determined by host cellular trypsin-type proteases, which cleave the viral envelope fusion glycoprotein precursor HA0, because a HA-processing protease is not encoded in the viral genome. Although there are many host cellular proteases that recognize the carboxyl moiety of R residue as determined by synthetic substrates, only few proteases recognize the tertiary structure of the cleavage site of HA0, thus allowing IAV multiplication with proteolytic activation only in organs that express the HA-processing proteases [5], [17], [18]. As outlined in this review, the knowledge gained within the last few years has greatly improved our understanding on the role of host cellular proteases in the pathogenicity of IAV infection: (i) Several cell surface anchored trypsin-like serine proteases are candidates of HA-processing protease for seasonal human IAV, TMPRSS2, HAT and TMPRSS4. This is in addition to secreted HA-processing trypsin-type proteases in the airway, such as tryptase Clara, trypsin and mini-plasmin. In addition, MSPL and TMPRSS13, plasma membrane bound HA-processing proteases for HPAI viruses other than furin and PCs are found. Furin preferentially cleaves only HA with the R-K-K-R cleavage site motif with R at position P4, whereas MSPL/TMPRSS13 cleaves HA, including a wide range of strains of HPAI virus with the R/K-K-K-R motif with both K and R at position P4. (ii) IAV infection upregulates cellular trypsins, MMPs and cytokines, and also ATP depletion in various infected cells. The upregulated trypsins in various organs including endothelial cells changes IAV infectious organ tropism of seasonal IAV. Destruction of endothelial cell tight junctions by the induced trypsins causes viral invasion in various organs. Based on the results of studies on the relationship among upregulated factors, we propose the influenza virusẀCcytokineẀCprotease cycle hypothesis as one of the main mechanisms of vascular dysfunction in multiorgan failure, in association with cytokine storm in severe influenza (Fig. 10) [7]. (iii) Inhibitors of NF-ḊÊB and AP-1 effectively suppress the upregulation of proinflammatory cytokines and cellular trypsins, and improve survival rates of infected mice. In addition, administration of trypsin-type protease inhibitor aprotinin suppresses viral replication [7], upregulation of trypsins, MMPs and cytokines as well as ATP depletion and significantly improves cardiac function [8]. More selectively, trypsin-knockdown, particularly T2-knockdown, in cardiomyoblasts effectively suppresses IAV replication and ATP depletion accompanied by protection against mitochondrial membrane depolarization and apoptosis. These results suggest that upregulated cellular trypsins are one of the key pathogenic host factors in IAV-induced multiple organ failure. (iv) Since IAV infection induces impaired fuel utilization, patients with disorders of mitochondrial fatty acid oxidation are prone to severe multiple organ failure after IAV infection. (v) Upregulated cellular trypsins in various organs and endothelial cells might be effective drug targets for the control of multiple organ failure in severe influenza.Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failure - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1570963911001968ḂḂ
Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions
Kirsty R. Short1,2, Jennifer Kasper3
, Stijn van der Aa1
, Arno C. Andeweg1
,
Fatiha Zaaraoui-Boutahar1
, Marco Goeijenbier1
, Mathilde Richard1
,
Susanne Herold4
, Christin Becker4
, Dana P. Scott5
, Ronald W.A.L. Limpens6
,
Abraham J. Koster6
, Montserrat BẀḃrcena6
, Ron A.M. Fouchier1
,
Charles James Kirkpatrick3 and Thijs Kuiken1
Affiliations: 1
Dept of Viroscience, Erasmus Medical Center, Rotterdam, The Netherlands. 2
School of
Biomedical Sciences, University of Queensland, Brisbane, Australia. 3
Institute of Pathology, University Medical
Center, Johannes Gutenberg University, Mainz, Germany. 4
University of Giessen and Marburg Lung Center
(UGMLC), Justus-Liebig-University of Giessen, Member of the German Center for Lung Research (DZL),
Giessen, Germany. 5
Rocky Mountain Veterinary Branch, Division of Intramural Research, National Institute of
Allergy and Infectious Diseases, National Institutes of Health, Hamilton, MT, USA. 6
Dept of Molecular Cell
Biology, Section Electron Microscopy, Leiden University Medical Centre, Leiden, The Netherlands.
Correspondence: Thijs Kuiken, Dept of Viroscience, Erasmus Medical Center, Dr Molewaterplein 50, 3015GE
Rotterdam, The Netherlands. E-mail: t.kuiken@erasmusmc.nl
ABSTRACTA major cause of respiratory failure during influenza A virus (IAV) infection is damage to
the epithelialẀCendothelial barrier of the pulmonary alveolus. Damage to this barrier results in flooding of
the alveolar lumen with proteinaceous oedema fluid, erythrocytes and inflammatory cells. To date, the
exact roles of pulmonary epithelial and endothelial cells in this process remain unclear.
Here, we used an in vitro co-culture model to understand how IAV damages the pulmonary epithelialẀC
endothelial barrier. Human epithelial cells were seeded on the upper half of a transwell membrane while
human endothelial cells were seeded on the lower half. These cells were then grown in co-culture and IAV
was added to the upper chamber.
We showed that the addition of IAV (H1N1 and H5N1 subtypes) resulted in significant barrier damage.
Interestingly, we found that, while endothelial cells mounted a pro-inflammatory/pro-coagulant response
to a viral infection in the adjacent epithelial cells, damage to the alveolar epithelialẀCendothelial barrier
occurred independently of endothelial cells. Rather, barrier damage was associated with disruption of tight
junctions amongst epithelial cells, and specifically with loss of tight junction protein claudin-4.
Taken together, these data suggest that maintaining epithelial cell integrity is key in reducing pulmonary
oedema during IAV infection.
Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions
https://erj.ersjournals.com/content/erj/47/3/954.full.pdfḂḂ
Potential contribution of alveolar epithelial type I cells ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5696455
Dec 22, 2017 ḂĊ The epithelial barrier function of TJs in the lungs is mainly established by claudins. Claudin-5 is expressed in pulmonary epithelial as well as in endothelial cells . Claudins-4 and -18 are present in AECIẀCAECI juctions, whereas AECIẀCAECII junctions additionally contain claudin-3 [45,46].
Cited by: 13
Publish Year: 2017
Author: Michael Kasper, Kathrin BarthḂḂ
Reactive oxygen and nitrogen species during viral infections
https://www.researchgate.net/publication/264090810_Reactive_oxygen_and_nitrogen...
Reactive oxygen species (ROS) are frequently produced during viral infections. Generation of these ROS can be both beneficial and detrimental for many cellular functions.
The Role of Reactive-Oxygen-Species ... - PubMed Central (PMC)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3039955
Oxidative stress occurs when oxygen radical formation and levels exceed those of antioxidants, potentiating cell responses such as apoptosis, tumorgenesis, and immune-response [1ẀC3]. However, the necessity of ROS for immune function could be exploited by ḂḞpotential pathogensḂḟ to reduce host responses in order to enhance survival and ...
Cited by: 146
Publish Year: 2011
Author: Ralee Spooner, Özlem Yilmaz
Role of free radicals in viral pathogenesis and mutation ...
https://www.researchgate.net/publication/12069418_Role_of_free_radicals_in_viral...
Oxygen radicals and nitric oxide (NO) are generated in excess in a diverse array of microbial infections. Emerging concepts in free radical biology are now shedding light on the pathogenesis of ...
Author: Takaaki AkaikeḂḂ
ḂḂ
ḂḂ