ˇˇ
Reduction of O2 to H2O and Its Free Radical Intermediates
ˇˇ
Ë«ÇâÇŕÝďËŘŁ¨DATŁ©żÉŇÔŇÔ×ÔĘÉŇŔŔµĐÔ·˝Ę˝ÓŐµĽĚúµ°°×µÄČÜøĚ彵˝âŁ¬ÔöĽÓϸ°űÄÚÓÎŔëĚúˇŁ
H2O2 induces apoptosis via extrinsic and intrinsic pathway
Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells. Exposure of human lung epithelial cells to H2O2 induces apoptosis concomitant with cysteine oxidation and down-regulation of Bcl-2.
Sources of intracellular H2O2
ˇˇ
ˇˇ
ˇˇ
Diffusion of Oxygen from the Peripheral Capillaries into the Tissue Fluid
Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1‑phase arrest
https://www.spandidos-publications.com/or/40/3/1787In lung cancer cells, ROS formation is induced by various apoptosis-inducing agents, including cisplatin, chromium, and lipoic acid, and has been shown to be involved in the regulation of Bcl-2
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3596255/
ˇˇ
Ascorbic Acid and a Cytostatic Inhibitor of Glycolysis Synergistically Induce Apoptosis in Non-Small Cell Lung Cancer Cells
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0067081ˇˇ
Is Hydrogen Peroxide a Suitable Apoptosis Inducer for All Cell Types?
We speculated that highly proliferative cells would show the strongest apoptosis resistance, followed by primary cells and then highly differentiated cells. However, the exact opposite proved to be the case, with 293T cells showing the fastest apoptosis induction across H2O2 concentrations, as well as the shortest time to substantial induction, and the earliest end of induction compared to fibroblasts and cardiomyocytes. Moreover, the induction of apoptosis indices was accompanied by upregulation of RIP, a gene associated with necroptosis, at all H2O2 concentrations. Alternatively, terminally differentiated myocytes with no proliferative capacity showed minimal induction of apoptosis at concentrations inducing substantial apoptosis/necroptosis in 293T cells. In addition, internal ROS production may be different from one cell type to another and H2O2 produced during normal cell metabolism and production must be higher in rapidly proliferative cells. Therefore, H2O2 concentration used should be chosen carefully according to cell model in studies of apoptosis as the induction range differs markedly among cell types.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4993923/
ˇˇ
cytochromes of the c type that are found in eukaryotic mitochondria. They serve as redox intermediates that accept electrons from MITOCHONDRIAL electron transport complex iii and transfer them to MITOCHONDRIAL electron transport complex iv.
https://lookfordiagnosis.com/mesh_info.php?term=Cytochromes+C&lang=1
Ferricytochrome c protects mitochondrial cytochrome c oxidase against hydrogen peroxide-induced oxidative damage
https://pubmed.ncbi.nlm.nih.gov/20801213/
ˇˇ
ˇˇ
ˇˇ
https://courses.lumenlearning.com/wm-biology1/chapter/reading-electron-transport-chain/
When the arterial blood reaches the peripheral tissues, its Po2 in the capillaries is still 95 mm Hg.Yet, as shown in Figure 40-3, the Po2 in the interstitial fluid that surrounds the tissue cells averages only 40 mm Hg. Thus, there is a tremendous initial pressure difference that causes oxygen to diffuse rapidly from the capillaryDiffusion of oxygen from a tissue capillary to the cells. (Po2 in interstitial fluid = 40 mm Hg, and in tissue cells = 23 mm Hg.)
Reduction of oxygen. A single-electron transfer which converts molecular oxygen to the superoxide anion, creating an unstable molecule. The decomposition of hydrogen peroxide can be a source of the hydroxyl radical; this reaction requires both superoxide and hydrogen peroxide as precursors. These steps reduce oxygen to water by the addition of four electrons, yielding three reactive oxygen species: superoxide anion, hydrogen peroxide, and hydroxyl radical.
Selective visible-light-driven oxygen reduction to hydrogen peroxide using BODIPY photosensitizers†
Selective visible-light-driven O2 reduction to H2O2 was realized using BODIPY photosensitizers (PS) in the presence of ferrocene (Fc) as the reductant and acetic acid as the proton source. Mechanistic studies suggested that O2 could be activated by 3PS* through an energy transfer pathway to give singlet oxygen (1O2) in the absence of Fc. However, with Fc, 3PS* was first reductively quenched to PS¨B−, which was able to reduce O2 to the superoxide radical form in a subsequent electron transfer step.
https://pubs.rsc.org/en/content/articlelanding/2018/cc/c7cc09383g#!divAbstractˇˇ
Hydrogen peroxide oxidation and reduction reactions (HPORR) in the ORR(Oxgen Reduction Reaction)
Scheme 5: Reduction and oxidation of hydrogen peroxide.
Hydrogen peroxide oxidation and reduction reactions (HPORR) in the ORR
From theoretical calculations, the single soluble intermediate species that could participate in the ORR mechanism is H2O2. However, as discussed above, H2O2 is a stable intermediate in ORR only under some circumstances. It has not been detected in rotating ring-disc electrode (RRDE) experiments with either with massive Pt electrodes or Pt(111) electrodes in acidic solutions that contained moderately adsorbing anions provided that E > 0.35 V . Hydrogen peroxide can be reduced and oxidized by following two different irreversible reactions that lead to water and oxygen, respectively, as final products according to Scheme 5 .
Some reflections on the understanding of the oxygen reduction reaction at Pt(111)
https://www.beilstein-journals.org/bjnano/articles/4/108ˇˇ
Where do reactive oxygen species originate?
Cellular metabolism is a well established source of reactive oxygen species. These background cellular processes account for the baseline levels of oxidative DNA damage detected in normal tissues. Electron transport chains all are able to leak electrons to oxygen during normal function. Certain enzymes also release superoxides (including the oxidative burst released by phagocytic cells to destroy cells infected with viruses or bacteria). The formation of reactive oxygen species is described below (as well as physiological mechanisms to reduce their concentrations - superoxide dismutase and glutathione peroxidase/catalase):
ˇˇ
Reduction of O2 to H2O and its free radical intermediates (A) Lewis structures for molecular oxygen (O2) and its singlet electron derivatives superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (OH•). (B) The step wise reduction of O2 to H2O during aerobic respiration. The standard redox potential for reduction of each intermediate is also shown (reproduced and modified from [23]). Irradiation-mediated cleavage of H2O which produces OH• accounts for the damaging effects of radiation therapy.
ˇˇ
ROS have superoxide anion, hydrogen peroxide, and hydroxyl radical. Superoxide anion ( O 2 ∙ - ) generated from NADPH oxidation through NADPH oxidases. It reduces to hydrogen peroxide (H2O2) where superoxide dismutase (SOD) acts as catalyst. Hydrogen peroxide further reduces to H2O via catalase/oxidized iron (Fe2+) to highly reactive hydroxyl ion (OH−). During oxidative stress, when generation of reactive oxygen species spaces out their scavenging system, levels of oxidized reactive oxygen species accumulate and this damages many cellular factors.
ˇˇ
Selected examples of oxygen free radicals. (1) Superoxide anion is formed when molecular oxygen acquires an additional electron, (2) hydrogen peroxide can be generated by several metabolic reactions, (e.g., from superoxide), and (3) hydroxyl radicals can be formed from either the superoxide anion or from hydrogen per- oxide.
ˇˇ
http://cancer-research-frontiers.org/2018-4-101/
ˇˇ
ˇˇ
ˇˇ
Disulfide bridges in proteins
Disulfide (sulfur-sulfur) linkages between two cysteine residues are an integral component of the three-dimensional structure of many proteins. The interconversion between thiols and disulfide groups is a redox reaction: the thiol is the reduced state, and the disulfide is the oxidized state.
ˇˇ
Notice that in the oxidized (disulfide) state, each sulfur atom has lost a bond to hydrogen and gained a bond to a sulfur - this is why the disulfide state is considered to be oxidized relative to the thiol state.
The redox agent that mediates the formation and degradation of disulfide bridges in most proteins is glutathione, a versatile coenzyme that we have met before in a different context (section 14.2A). Recall that the important functional group in glutathione is the thiol, highlighted in blue in the figure below. In its reduced (free thiol) form, glutathione is abbreviated 'GSH'.
ˇˇ
In its oxidized form, glutathione exists as a dimer of two molecules linked by a disulfide group, and is abbreviated 'GSSG'.
A new disulfide in a protein forms via a 'disulfide exchange' reaction with GSSH, a process that can be described as a combination of two SN2-like attacks. The end result is that a new cysteine-cysteine disulfide forms at the expense of the disulfide in GSSG.
In its reduced (thiol) state, glutathione can reduce disulfides bridges in proteins through the reverse of the above reaction.
Disulfide bridges exist for the most part only in proteins that are located outside the cell. Inside the cell, cysteines are kept in their reduced (free thiol) state by a high intracellular concentration of GSH, which in turn is kept in a reduced state (ie. GSH rather than GSSG) by a flavin-dependent enzyme called glutathione reductase.
Disulfide bridges in proteins can also be directly reduced by another flavin-dependent enzyme called 'thioredoxin'. In both cases, NADPH is the ultimate electron donor, reducing FAD back to FADH2 in each catalytic cycle.ˇˇ
ˇˇ
ˇˇ
ˇˇ
ˇˇ
Chemical structures of a-lipoic acid and dihydrolipoic acid. a-Lipoic acid is an eight cabon disulfide compound containing a single chiral carbon. It is readily reduced in vivo to its dithiol form, dihydrolipoic acid. The R-enantiomer is the naturally occurring form of a-lipoic acid, while synthetic a-lipoic acid is a racemic mixture containing both the R-and S-enantiomers.
ˇˇ
ˇˇ
Association of free radicals, oxidative stress, and disease. The generation of free radicals including reactive oxygen and nitrogen species leads to increased oxidative stress. As portrayed in the diagram, this process is not tissue-specific , and could be a consequence of either increased production of free radicals, or reduced antioxidant defenses . Although causality has not been equivocally demonstrated, there is a large body of experimental evidence that shows the association of increased levels of oxidative stress and a variety of pathological conditions.
ˇˇ
ˇˇ
ˇˇ
ˇˇ
Potential areas of impact of oxidative stress in diabetes and the pre-diabetic state. Free radical generation leading to oxidative stress has the capacity to impact diabetes at multiple levels. The role of oxidative stress leading to microvascular complications has the most experimental support. Oxidative stress also plays a significant role in the development of macrovascular complications , including promoting atherosclerosis and the inhibition of nitric oxide-mediated vasodilation. 21 More recent evidence suggests an association of oxidative stress with both impaired insulin action (in vitro and in vivo) and the deterioration of b-cell function.
ˇˇ
ˇˇ
(a) Curcumin is a type of polyphenol. It has many anticancer activities. The source of curcumin is turmeric. It targets NF-¦ĘB as radiosensitizers. Curcumin has radioprotective effect and targets Nrf2. (b) Genistein, a natural radiosensitizer, is a type of polyphenol. Genistein is a derivative of soybean. It has the ability to inhibit the growth of cancer cell through apoptosis. It can elevate the efficacy of radiation therapy by combining with ionizing radiation. Genistein suppresses Akt and Erk, reduces survivin and cyclin B expression in cervical cancer, and inhibits NF-¦ĘB. It acts as radioprotector but the target is not yet determined. (c) Quercetin is the main flavonoid component. It can behave as antioxidant, anti-inflammatory, and antiviral. Quercetin causes apoptosis and arrests the cell cycle. Quercetin by inhibiting ATM elevates radiosensitization. It has radioprotective effect against radiation therapy.
ˇˇ
ˇˇ
ˇˇ
Pseudomonas aeruginosa (ÍÂ̼ٵĄ°űľúŁ©is an opportunistic Gram©\negative bacterium that is primarily responsible for infections related to cystic fibrosis (CF) airways, burn wounds, urinary tract infections, surgery©\associated infections, and HIV©\related illness. PyocyaninŁ¨ÂĚŧËŘŁ© and extracellular DNA (eDNA) are the major factors dictating the progression of biofilm formation and infection. Pyocyanin is a potent virulence factor causing cell death in infected CF patients and is associated with high mortality. eDNA is a key player in P. aeruginosa biofilm formation and is also responsible for the high viscosity of CF sputum that blocks the respiratory airway passages. In this chapter, we summarize our recent findings on the role of pyocyanin in facilitating P. aeruginosa biofilm formation. Pyocyanin promotes eDNA release in P. aeruginosa by inducing cell lysis mediated via hydrogen peroxide (H 2 O 2) production. Pyocyanin intercalates with the nitrogenous bases of DNA and creates structural perturbation on the double©\helix structure. Pyocyanin©\eDNA binding significantly influences P. aeruginosa cell surface hydropho©\ bicity and influences the physicochemical interactions facilitating bacterial cell©\to©\cell interaction (aggregation) and ultimately facilitates robust biofilm formation. A pyocyanin knockout (¦¤phzA©\G) mutant is shown to have significantly reduced eDNA release and biofilm formation in comparison to its wild©\type. To this end, we discover that antioxidant glutathione directly binds to pyocyanin and modulates pyocyanin structure and function, thus inhibiting pyocyanin©\eDNA binding and consequently hampering biofilm development.
ˇˇ
ˇˇ
Role of Pyocyanin and Extracellular DNA in Facilitating Pseudomonas aeruginosa Biofilm Formation
ˇˇ
Glutathione is the Protecter and Healer of the Body
Glutathione - You Are The Healer
https://youarethehealer.org/health-conditions/optizmize-your-health/detox-biotransformation-pathways/glutathione/ˇˇ
Our Protector
Glutathione is our protector, and friend. It tempers inflammation through its super hero antioxidant abilities. When it is depleted, inflammation can take over our body. Glutathione is found in nearly every compartment of our cells, including the nucleus. As the main antioxidant for mitochondria, glutathione is an absolute necessity for those little power-house mitochondrial factories who would otherwise become buried in reactive oxygen species.
What Glutathione Is, And Does - In A Nutshell
A tripeptide
Protects from reactive oxygen species
Protects from reactive nitrogen species
Cofactor for many antioxidant enzymes such as peroxidases and transferases
Storage form of cysteine
Storage form and transporter of nitric oxide
Metabolizes estrogens, leukotrienes, and prostaglandins
Involved in regulation of some transcription factors
Detoxifies many endogenous substances and xenobiotics
Gluatathione protects us from from free radicals through it's antioxidant activity. Free radicals are unstable molecules that cause damage in your body. They cause much of the inflammation that takes place in your body, and are associated with premature aging. Glutathione protects you from nasty free radicals such as reactive oxygen species, reactive nitrogen species and advanced glycation end products.
A Few Details
Glutathione (¦Ă-glutamyl-cysteinyl-glycine) is a tripeptide found in all cells where it is the major intracellular antioxidant. Composed of cysteine, glutamine and glycine, it is used as a major antioxidant, glutathione is the main nonprotein thiol (sulfhydryl/sulfur compound) responsible for the cellular redox balance. Glutathione is recycled to use it over, and over again. It is also synthesized anew by the body through combination of the three amino acids that is is composed of.
The organs that are highest in glutathione levels are the eye, the ears, the lung, the liver, the kidneys, and the skin. These organs have the highest concentrations of glutathione, for good reason, because these organs are subject to all of the toxins in the environment.ˇˇ
The Immune System and Glutathione
The central role of glutathione in a variety of cell functions related to immune defense has been demonstrated in studies.
A decrease in glutathione in antigen-presenting cells correlates with increased Th2 response.
Decreased glutathione found in macrophage cells from children with chronic asthma has been shown to be related to decreased bacterial phagocytosis.
Children with chronic asthma, had decreased glutathione related to post-translational modification of Nrf2.
Decreased glutathione and decreased macrophage defense againstˇˇ
ˇˇ
Glutathione as an Antioxidant and Redox Potential
Two Forms Of Glutathione
Glutathione exists in two forms. It is found in an oxidized form and a free or reduced form. Ony the free or reduced form has antioxidant activity. Once it is used and therefore oxidized, it needs to be recycled back to the reduced form again before it can continue with its antioxidant behavior. It is recycled back, and fourth, over, and, over again. The oxidized form is called glutathione disulfide (GSSG) and the reduced form is called glutathione (GSH). GSH is used by the body to neutralize reactive oxygen species which leads to its alteration into GSSG from GSH. (loses it's super hero antioxidant powers when it is oxidzed) The GSH needs the enzyme glutathione peroxidase to help it reduce free radicals. Glutathione is oxidized to GSSG in this process. Only GSH has the antioxidant activity. Think of the glutathione (GSH) as a super hero. It saves the body from damage by being oxidzed into (GSSG), rather than allowing free radicals to oxidize important cellular componants in our body (damaging mitochondria). Basically, it takes the hit to save our body from damage. However, to be able to continue functioning as an antioxidant, it has to change back into the reduced form and regaining it's super hero powers.ˇˇ
ˇˇ
Changing Glutathione Back To The Reduced Form GSH (Regaining Super Hero Antioxidant Powers)
GSSG can be recycled to GSH again and regain it's super hero antioxidant powers. This recycling back to GSH needs the enzyme glutathione reductase to facilitate the reduction of GSSG to GSH. It requires NADPH, and forms two GSH molecules from the one GSSG molecule.
Remember, we are examining reduced glutathione(GSH) and oxidized glutathione(GSSG). The ratio of GSH to GSSG controls the "redox potential" in the cells. It is the reduced glutathione that is guarding our body. GSH is a part of our antioxidant system and protects us from "free radicals". Free radicals are molecules that are unstable because they have unpaired electrons and are looking for another electron to steal in order to become stable again. They can steal electrons from the mitochondria, thereby damaging the mitochondria, causing inflammation and degeneration.
ˇˇ
Here is another way to examine this process: GSH will sacrifice themselves by giving an electron to a free radical. I mentioned that this happens with the help of an enzyme called glutathione peroxidase. This turns the glutathione (GSH) into glutathione disulfide (GSSG). The GSSG is now a free radical itself, but it can be turned back into its reduced state via an enzyme called glutathione reductase (GSR) using NADPH as an electron donor. This reinstates it back into its old GSH self again. Redox status (measure of reduced glutathione to oxidized glutathione) within the cells is reflected by the ratio of reduced GSH to oxidized GSSG (GSH/GSSG).This is used as a measure of cell toxicity. In healthy cells, more than 90% of the total glutathione pool is in the reduced form (GSH) and less than 10% in the disulfide form (GSSG).
You can see that these enzymes are a really important part of the process of glutathione's super hero abilities. We need enough glutathione, and we also need glutathione peroxidase, and glutathione reductase for this system to work effectively, and for us to stay healthy. Without this recycling process going smoothly we are in deep doo doo.ˇˇ
ˇˇ
ˇˇ
Antioxid Redox Signal. 2012 Jun 1; 16(11): 1323¨C1367.
doi: 10.1089/ars.2011.4123
PMCID: PMC3324814
PMID: 22146081
Redox Regulation of Mitochondrial Function
Diane E. Handy and Joseph Loscalzocorresponding author
Author information Article notes Copyright and License information Disclaimer
Cardiovascular Division, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts.ˇˇ
Redox-dependent processes influence most cellular functions, such as differentiation, proliferation, and apoptosis. Mitochondria are at the center of these processes, as mitochondria both generate reactive oxygen species (ROS) that drive redox-sensitive events and respond to ROS-mediated changes in the cellular redox state. In this review, we examine the regulation of cellular ROS, their modes of production and removal, and the redox-sensitive targets that are modified by their flux. In particular, we focus on the actions of redox-sensitive targets that alter mitochondrial function and the role of these redox modifications on metabolism, mitochondrial biogenesis, receptor-mediated signaling, and apoptotic pathways. We also consider the role of mitochondria in modulating these pathways, and discuss how redox-dependent events may contribute to pathobiology by altering mitochondrial function. Antioxid. Redox Signal. 16, 1323¨C1367.
ˇˇ
FIG. 2.
Mitochondrial hydrogen peroxide (H2O2) flux. Most of the reactive oxygen species (ROS) generated in mitochondria is in the form of equation M37 from the electron transport chain (ETC) (223, 349). Recent evidence suggests that NADPH-dependent oxidase 4 (NOX4) may be localized to mitochondria, where it may produce hydrogen peroxide and/or equation M38 (118). The TCA cycle enzyme ¦Á-ketoglutarate dehydrogenase (¦Á-KGDH) is also a potential source of equation M39 (423). Within the mitochondrial matrix, manganese-dependent superoxide dismutase (MnSOD) is the only SOD present (236); it reduces equation M40 to hydrogen peroxide. Mitochondrially located glutathione peroxidase-1 (GPx-1) utilizes glutathione (reduced glutathione [GSH]) as a cosubstrate in the reduction of hydrogen peroxide to water (126, 133). Similarly, the mitochondrially targeted peroxiredoxins (Prx3 or Prx5) reduce hydrogen peroxide using thioredoxin 2 (Trx2) to regenerate the active site (66, 73). Also important are glutathione reductase (GR) and Trx reductase (TrxR), which serve to reduce oxidized GSH and Trx, respectively (not represented in this figure). In the intermembrane space, it is thought that copper/zinc-dependent SOD (Cu/ZnSOD) may play a role in reducing equation M41 that is released from complex III. Within this space and in the cytosol, GPx-1 and Prx family members reduce hydrogen peroxide produced and released from the mitochondria or other sources.ˇˇ
Redox Regulation of Mitochondrial Function
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3324814/ˇˇ
ˇˇ
Stem cells and the impact of ROS signaling
Development 2014 141: 4206-4218; doi: 10.1242/dev.107086
Abstract
An appropriate balance between self-renewal and differentiation is crucial for stem cell function during both early development and tissue homeostasis throughout life. Recent evidence from both pluripotent embryonic and adult stem cell studies suggests that this balance is partly regulated by reactive oxygen species (ROS), which, in synchrony with metabolism, mediate the cellular redox state. In this Primer, we summarize what ROS are and how they are generated in the cell, as well as their downstream molecular targets. We then review recent findings that provide molecular insights into how ROS signaling can influence stem cell homeostasis and lineage commitment, and discuss the implications of this for reprogramming and stem cell ageing. We conclude that ROS signaling is an emerging key regulator of multiple stem cell populations.ˇˇ
Fig. 1.
ROS generation and scavenging. (A) Reactive oxygen species (ROS) include superoxide (O2.−), hydrogen peroxide (H2O2) and the highly reactive hydroxyl radical (OH.) (shown in red). O2.− can be generated from complexes I and III (shown in B) or through the oxidation of NADPH by NADPH oxidases. Subsequent reduction to H2O2 is catalyzed by superoxide dismutase (SOD). H2O2 can be further reduced to water (H2O) by catalase or can spontaneously oxidize iron (Fe2+) to form the highly reactive OH.. Under conditions of oxidative stress, when ROS generation outpaces the ROS scavenging system, accumulating levels of ROS oxidize and damage various cellular components. (B) The electron transport chain complexes I-IV harness electrons from NADH in a series of redox reactions, which are coupled to pumping protons (H+) into the mitochondrial intermembrane space. The proton motive force, a combination of the membrane potential (charge) and the concentration gradient (pH), powers ATP synthase (complex V). Normally, O2 acts as the final electron acceptor at complex IV, but aberrant reduction of O2 can occur at complexes I and III (red arrows), leading to the generation of O2.− (red).Stem cells and the impact of ROS signaling | Development
https://dev.biologists.org/content/141/22/4206ˇˇ
Mitochondria are the source of hydrogen peroxide for ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2892101
Jul 15, 2009 ˇ¤ Gyulkhandanyan AV, Pennefather PS. Shift in the localization of sites of hydrogen peroxide production in brain mitochondria by mitochondrial stress. J Neurochem. 2004; 90:405¨C21. Hersch SM, Yi H, Heilman CJ, Edwards RH, Levey AI. Subcellular localization and molecular topology of the dopamine transporter in the striatum and substantia nigra.
Cited by: 113
Publish Year: 2009
Author: Li Bao, Marat V. Avshalumov, Jyoti C. Patel, Christian R. Lee, Evan W. Miller, Christopher J. Chang,...
Redox Regulation of Mitochondrial Function
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3324814
Jun 01, 2012 ˇ¤ The outer membrane of the mitochondria is freely permeable to molecules <5000 kDa, in contrast to the inner mitochondrial membrane, which is highly impermeable.
Cited by: 331
Publish Year: 2012
Author: Diane E. Handy, Joseph Loscalzoˇˇ
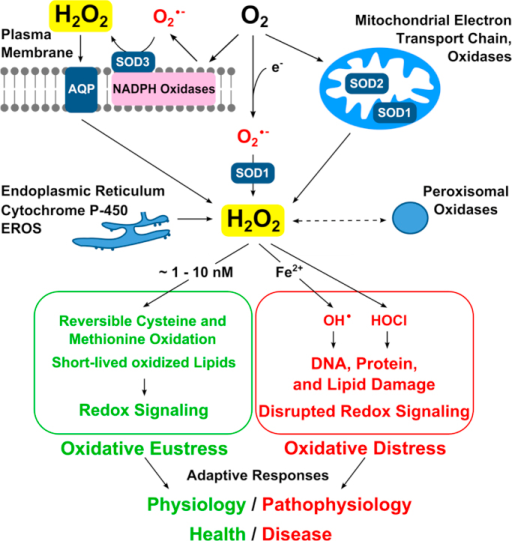
Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress ˇî
Hydrogen peroxide emerged as major redox metabolite operative in redox sensing, signaling and redox regulation. Generation, transport and capture of H2O2 in biological settings as well as their biological consequences can now be addressed. The present overview focuses on recent progress on metabolic sources and sinks of H2O2 and on the role of H2O2 in redox signaling under physiological conditions (1–10 nM), denoted as oxidative eustress. Higher concentrations lead to adaptive stress responses via master switches such as Nrf2/Keap1 or NF-κB. Supraphysiological concentrations of H2O2 (>100 nM) lead to damage of biomolecules, denoted as oxidative distress. Three questions are addressed: How can H2O2 be assayed in the biological setting? What are the metabolic sources and sinks of H2O2? What is the role of H2O2 in redox signaling and oxidative stress?
ˇˇˇˇ
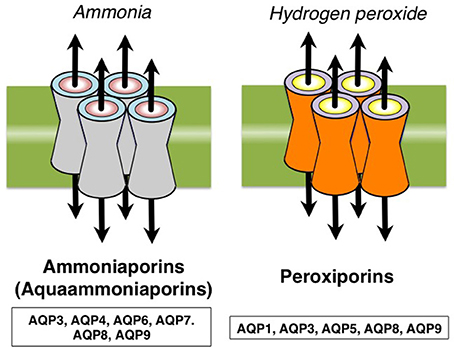
Role of hydrogen peroxide in oxidative stress. Top: Endogenous H2O2 sources include NADPH oxidases and other oxidases (membrane-bound or free) as well as the mitochondria. The superoxide anion radical is converted to hydrogen peroxide by the three superoxide dismutases (SODs 1,2,3). Hydrogen peroxide diffusion across membranes occurs by some aquaporins (AQP), known as peroxiporins. Bottom: In green, redox signaling comprises oxidative eustress (physiological oxidative stress). In red, excessive oxidative stress leads to oxidative damage of biomolecules and disrupted redox signaling, oxidative distress. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
ˇˇ
Role of hydrogen peroxide in oxidative stress. Top: End | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC5256672_gr3&req=4ˇˇ
ˇˇ
Frontiers | Aquaporins as Targets of Dietary Bioactive Phytocompounds | Molecular Biosciences
https://www.frontiersin.org/articles/10.3389/fmolb.2018.00030/fullˇˇ
ˇˇ
ˇˇ
Interpreting Intracellular Hydrogen Peroxide in Cancer Cells to Understand Cancer Susceptibility to Pharmacological Ascorbate Therapy
2017 Author(s): Erudaitius, DieaniraAdvisor(s): Rodgers, Victor G. J.
ˇˇ
The intravenous delivery of pharmacological ascorbate (P-AscH-) has recently been demonstrated to be a successful adjuvant in the treatment of some cancers. Administered as a series of infusions, P-AscH- generates high fluxes of extracellular hydrogen peroxide (H2O2), which is toxic to certain cancer cells while not affecting normal. In vitro studies indicate that cancer cells have a wide range in susceptibility to P-AscH- and subsequently to extracellular H2O2. The resulting intracellular H2O2 concentration is believed to accumulate differently in susceptible cancer cells as compared to non-susceptible cells. It is hypothesized that intracellular H2O2 concentration has a steady-state value that is significant for cell susceptibility and independent of cell type. Although this has been alluded to, this value has yet to be quantified. Further, the variations in cell parameters (i.e. membrane permeability via peroxiporins, catalase activity, etc.) for various cells are expected to be significant enough to alter intracellular H2O2 concentration, thereby impacting cell susceptibility. A steady-state model was developed which elucidates the parameter contribution to intracellular H2O2 accumulation. The intracellular H2O2 concentrations during P-AscH- therapy was quantified for pancreatic normal (H6c7; ascorbate non-responding), adenocarcinoma (MIA PaCa-2; ascorbate susceptible) and glioblastoma U-87 (non-responding), T98G (moderately susceptible) and LN-229 (highly susceptible) cell lines. Recognizing that MIA PaCa-2 has an enhanced expression of aquaporin-3 (AQP3) and the significance of AQP3 to plasma membrane permeability to H2O2, silenced AQP3 was also investigated. Interestingly, an increase in surviving fraction was observed for the silenced cells in clonogenic studies using therapeutic H2O2 concentrations. These results imply that cell-susceptibility to ascorbate therapy is significantly coupled to the plasma membrane permeability to H2O2, and in particular, elevated expressions of peroxiporins. Ultimately, this work provides insight to what targets are appropriate for improving P-AscH- therapy. Further, our mathematical results contradict the hypothesis that a unique intracellular H2O2 was sufficient for a specific clonogenic response. This aligns with recent work revealing that the combination of redox-active labile iron and high intracellular H2O2 concentration is the necessary and sufficient condition for cellular ascorbate-susceptibility. Quantifying the relationship of this combination to the clonogenic response is the subject of future research. Interpreting Intracellular Hydrogen Peroxide in Cancer Cells to Understand Cancer Susceptibility to Pharmacological Ascorbate Therapy
ˇˇ
https://escholarship.org/uc/item/1f50b0bh
ˇˇˇˇ
Overexpression of NOX4 has been found in diverse types of solid tumors, such as colorrectal cancer, prostate cancer [12], glioblastoma [13], liver cancer [14], and melanoma [15]. non-small cell lung cancer (NSCLC)
ˇˇ
ˇˇ
NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2©\amino©\1©\methyl©\6©\phenylimidazo[4,5©\b]pyridine (PhIP)
Abstract
NADPH oxidase/dual©\oxidase (Nox/Duox) family members have been implicated in nuclear factor kappa©\B (NF¦ĘB)©\mediated inflammation and inflammation©\associated pathologies. We sought to examine, for the first time, the role of Nox/Duox and NF¦ĘB in rats treated with the cooked meat heterocyclic amine carcinogen 2©\amino©\1©\methyl©\6©\phenylimidazo[4,5©\b]pyridine (PhIP). In the PhIP©\induced colon tumors obtained after 1 year, Nox1, Nox4, NF¦ĘB©\p50 and NF¦ĘB©\p65 were all highly overexpressed compared with their levels in adjacent normal©\looking colonic mucosa. Nox1 and Nox4 mRNA and protein levels also were markedly elevated in a panel of primary human colon cancers, compared with their matched controls. In HT29 human colon cancer cells, Nox1 knockdown induced G1 cell cycle arrest, whereas in Caco©\2 cells there was a strong apoptotic response, with increased levels of cleaved caspase©\3, ©\6, ©\7 and poly(ADP©\ribose)polymerase. Nox1 knockdown blocked lipopolysaccharide©\induced phosphorylation of I¦ĘB kinase, inhibited the nuclear translocation of NF¦ĘB (p50 and p65) proteins, and attenuated NF¦ĘB DNA binding activity. There was a corresponding reduction in the expression of downstream NF¦ĘB targets, such as MYC, CCND1 and IL1¦Â. The results provide the first evidence for a role of Nox1, Nox4 and NF¦ĘB in PhIP©\induced colon carcinogenesis, including during the early stages before tumor onset. Collectively, the findings from this investigation and others suggest that further work is warranted on the role of Nox/Duox family members and NF¦ĘB in colon cancer development.
NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2©\amino©\1©\methyl©\6©\phenylimidazo[4,5©\b]pyridine (PhIP) - Wang - 2011 - International Journal of Cancer - Wiley Online Library
https://onlinelibrary.wiley.com/doi/full/10.1002/ijc.25610ˇˇ
NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathwayˇˇ
ÉĎşŁ˝»Í¨´óѧ
highlights
•
Overexpression of NOX4 predicts prognosis of GC patients and tumor size.
•
NOX4 promotes GC cells growth via GLI1 pathway.
•
GLI1 expression is activated by ROS generated by NOX4.
•
GLI1 expression activated by ROS is in a dose-dependent way.
Abstract
NADPH Oxidase 4 (NOX4), a member of the NOX family, has emerged as a significant source of reactive oxygen species, playing an important role in tumor cell proliferation, apoptosis, and other physiological processes. However, the potential function of NOX4 in gastric cancer (GC) cell proliferation is yet unknown. The aim of this study was to illustrate whether NOX4 plays a role in regulating gastric cancer cell growth. First, the clinical information from 90 patients was utilized to explore the clinical value of NOX4 as a predictive tool for tumor size and prognosis. Results showed that NOX4 expression was correlated with tumor size and prognosis. In vitro assays confirmed that knockdown of NOX4 expression blocked cell proliferation and the expression of Cyclin D1, BAX, and so on. Interestingly, NOX4 promoted cell proliferation via activation of the GLI1 pathway. GLI1, a well-known transcription factor in the Hedgehog signaling pathway, was overexpressed to test whether NOX4 activates downstream signaling via GLI1. Overexpression of GLI1 reversed the inhibition of proliferation induced by NOX4 knockdown. In addition, overexpression of NOX4 increased GLI1 expression, and depletion of GLI1 expression decreased the effects induced by NOX4 overexpression. Further, ROS generated by NOX4 was required for GLI1 expression, as shown by use of the ROS inhibitor, diphenylene iodonium (DPI). In summary, the findings indicate that NOX4 plays an important role in gastric cancer cell growth and apoptosis through the generation of ROS and subsequent activation of GLI1 signaling. Hence, the targeting of NOX4 may be an attractive therapeutic strategy for blocking gastric cancer cell proliferation.
NOX4-driven ROS formation regulates proliferation and apoptosis of gastric cancer cells through the GLI1 pathway - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0898656818300469
ˇˇThe Expression and Prognostic Value of NOX4 in Gastric Cancer< Previous Article Next Article >
American Journal of Internal Medicine
Volume 6, Issue 6, November 2018, Pages: 144-151Î÷°˛˝»Í¨´óѧ
Abstract
Background: Gastric cancer (GC) is one of the most common malignant tumors worldwide which threaten the health of human. A lot of work has been done in tumor pathogenesis in recent years, while modest progress in diagnosis and treatment of gastric cancer have been made. Methods: Multiple databases including The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) were explored to identify the expression pattern of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 4 (NOX4), the main source of reactive oxygen species, in gastric cancer. We also analyzed the correlation between the expression level of NOX4 and gastric cancer patientsˇŻ clinical features. The prognostic value of NOX4 was explored in TCGA and K-M Plotter. Last, we utilized TCGA and DAVID databases to uncover the underlying molecular mechanisms of NOX4 with TCGA and DAVID databases. Results: We found that NOX4 was upregulated in tumors compared with adjacent non-tumor tissues and overexpression was correlated with tumor invasion and TNM stage in gastric cancer. Furthermore, NOX4 level could be an independent prognostic marker for GC, unacted on the choice of therapy or HER2 expression. Besides, we investigated the potential mechanisms of NOX4 in gastric cancer. Conclusions: Our findings proved that NOX4 may be a new prognostic factor or therapeutic marker for gastric cancer.
Keywords
The Expression and Prognostic Value of NOX4 in Gastric Cancer :: Science Publishing Group
http://www.sciencepublishinggroup.com/journal/paperinfo?journalid=252&doi=10.11648/j.ajim.20180606.11ˇˇ
NADPH Oxidase 1 Overexpression Enhances Invasion via Matrix Metalloproteinase-2 and Epithelial¨CMesenchymal Transition in Melanoma Cells
NADPH oxidase 1 (Nox1) is a member of the NADPH oxidase family that has not been well characterized in the melanocytic cell lineage. Here we demonstrated that Nox1 and Nox4 were detected in melanocytic lineage, with only Nox1 detected in normal human melanocytes and Nox4 in a subset of metastatic melanoma cell lines. The protein level and enzymatic activity of Nox1 was elevated in all melanoma cells as compared with normal melanocytes. Overexpression of GFP-Nox1 protein in Wm3211 primary melanoma cells increased invasion rate by 4- to 6-fold as measured by Matrigel invasion assay, whereas knocking down or inhibiting Nox1 decreased invasion by approximately 40¨C60% in Wm3211 and SK-Mel-28 cells. Matrix metalloproteinase-2 (MMP-2) was increased by Nox1 overexpression at the mRNA, protein, and activity levels, and decreased by Nox1 knockdown. MMP-2 promoter activity was also regulated by Nox1 knockdown. In addition, stable clones overexpressing Nox1 exhibited an epithelial¨Cmesenchymal transition (EMT) as examined by cell morphology and EMT markers; knockdown or inhibiting Nox1 led to a reversal of EMT. Supplementing MMP-2 to culture media did not induce EMT, suggesting that EMT induction by Nox1 was not through MMP-2 upregulation. In summary, Nox1 was overexpressed in all melanoma cell lines examined, and enhanced cell invasion by MMP-2 upregulation and EMT induction.
NADPH Oxidase 1 Overexpression Enhances Invasion via Matrix Metalloproteinase-2 and Epithelial¨CMesenchymal Transition in Melanoma Cells - Journal of Investigative Dermatology
https://www.jidonline.org/article/S0022-202X(15)35855-3/abstractˇˇ
ˇˇ
NADPH Oxidase Activation in Pancreatic Cancer Cells Is Mediated through Akt-dependent Up-regulation of p22phox*
the Veterans Affairs Greater Los Angeles Healthcare System and UCLA, Los Angeles, California 90073
Abstract
We recently showed that Nox4 NADPH oxidase is highly expressed in pancreatic ductal adenocarcinoma and that it is activated by growth factors and plays a pro-survival, anti-apoptotic role. Here we investigate the mechanisms through which insulin-like growth factor I and serum (FBS) activate NADPH oxidase in pancreatic cancer (PaCa) cells. We show that in PaCa cells, NADPH oxidase is composed of Nox4 and p22phox catalytic subunits, which are both required for NADPH oxidase activity. Insulin-like growth factor I and FBS activate NADPH oxidase through transcriptional up-regulation of p22phox. This involves activation of the transcription factor NF-¦ĘB mediated by Akt kinase. Up-regulation of p22phox by the growth factors results in increased Nox4-p22phox complex formation and activation of NADPH oxidase. This mechanism is different from that for receptor-induced activation of phagocytic NADPH oxidase, which is mediated by phosphorylation of its regulatory subunits. Up-regulation of p22phox represents a novel pro-survival mechanism through which growth factors and Akt inhibit apoptosis in PaCa cells.
Akt PKB Apoptosis NF-kB Transcription Factor Oxidase Pancreas
Previous Section
Next Section
Introduction
NADPH oxidases (1,¨C,3) are a major intracellular source of ROS.2 The phagocytic NADPH oxidase mediates oxidative burst, which kills the invading microorganisms. In non-phagocytic cells, NADPH oxidases play an important role in the regulation of various physiologic and patho-physiologic processes including proliferation, differentiation, and death.
Both phagocytic and non-phagocytic NADPH oxidases involve proteins of the NOX family. The phagocytic NADPH oxidase (1) is composed of the Nox2 (gp91phox) and p22phox catalytic membrane-associated subunits and several regulatory cytosolic subunits (2,¨C,4). Non-phagocytic NADPH oxidases also require both a Nox isoform and p22phox to generate ROS. However, the non-phagocytic and phagocytic NADPH oxidases employ different regulatory subunits; furthermore, some non-phagocytic NADPH oxidases do not require cytosolic regulatory subunits for their activity (5,¨C,7).
Activation of the phagocytic NADPH oxidase occurs through assembly of a multicomponent complex comprised of the membrane-bound Nox2-p22phox subunits and the cytosolic subunits. Receptor-induced phosphorylation of the regulatory p47phox and p67phox subunits causes their recruitment from cytosol, resulting in conformational changes in the membrane-bound Nox2-p22phox complex and oxidase activation. In phagocytes, the Akt kinase mediates p47phox phosphorylation and thus NADPH oxidase activation (1, 8). The mechanisms of non-phagocytic NADPH oxidase activation remain poorly defined. Differently from the phagocytic NADPH oxidase, non-phagocytic oxidase activation may not involve the regulatory subunits. For example, various receptors including IGF-I (9), insulin, TGF¦Â, TNF¦Á, and TLR4 all activate Nox4 NADPH oxidase without engaging regulatory subunits (10,¨C,13).
We recently showed that Nox4 NADPH oxidase is highly expressed in pancreatic ductal adenocarcinoma (14, 15), and further, that it plays an important pro-survival role in pancreatic cancer (9, 14, 15). Growth factors (GFs) activate NADPH oxidase in PaCa cells, and the resulting ROS promote sustained activation of pro-survival kinases, such as JAK, thus suppressing apoptosis (15). Therefore, activation of NADPH oxidase is an important mechanism through which GFs protect PaCa cells from death. Of note, Nox4 NADPH oxidase was recently shown to promote endothelial tumor growth in mice (16).
The present study describes a novel mechanism whereby growth factors (in particular IGF-I) activate NADPH oxidase in PaCa cells, namely through transcriptional up-regulation of p22phox. We show that GFs stimulate Akt, which mediates activation of the transcription factor NF-¦ĘB to up-regulate p22phox expression. GF-induced p22phox up-regulation results in increased NADPH oxidase activity, leading to inhibition of apoptosis in PaCa cells.
NADPH Oxidase Activation in Pancreatic Cancer Cells Is Mediated through Akt-dependent Up-regulation of p22phox
http://www.jbc.org/content/286/10/7779.fullˇˇ
ˇˇ
NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart
Edited by Salvador Moncada, University College London, London, United Kingdom, and approved July 26, 2010
Abstract
NAD(P)H oxidases (Noxs) produce O2− and play an important role in cardiovascular pathophysiology. The Nox4 isoform is expressed primarily in the mitochondria in cardiac myocytes. To elucidate the function of endogenous Nox4 in the heart, we generated cardiac-specific Nox4−/− (c-Nox4−/−) mice. Nox4 expression was inhibited in c-Nox4−/− mice in a heart-specific manner, and there was no compensatory up-regulation in other Nox enzymes. These mice exhibited reduced levels of O2− in the heart, indicating that Nox4 is a significant source of O2− in cardiac myocytes. The baseline cardiac phenotype was normal in young c-Nox4−/− mice. In response to pressure overload (PO), however, increases in Nox4 expression and O2− production in mitochondria were abolished in c-Nox4−/− mice, and c-Nox4−/− mice exhibited significantly attenuated cardiac hypertrophy, interstitial fibrosis and apoptosis, and better cardiac function compared with WT mice. Mitochondrial swelling, cytochrome c release, and decreases in both mitochondrial DNA and aconitase activity in response to PO were attenuated in c-Nox4−/− mice. On the other hand, overexpression of Nox4 in mouse hearts exacerbated cardiac dysfunction, fibrosis, and apoptosis in response to PO. These results suggest that Nox4 in cardiac myocytes is a major source of mitochondrial oxidative stress, thereby mediating mitochondrial and cardiac dysfunction during PO.
ˇˇˇˇ
Our results suggest that Nox4 localized in mitochondria in cardiac myocytes is a major source of O2− production in the heart. In particular, up-regulation of Nox4 is primarily responsible for the increased mitochondrial O2− production in response to PO in the mouse heart. Furthermore, Nox4 plays a critical role in mediating mitochondrial dysfunction, apoptosis in cardiac myocytes, and eventual LV dysfunction in response to PO.
ˇˇ
O2− produced in mitochondria is rapidly converted into H2O2, which is readily diffusible in the intracellular space. However, increased production of O2− and/or H2O2 from mitochondrial Nox4 effectively oxidizes mitochondrial proteins, including aconitase (23).
ˇˇ
The biological toxicity of O2− is due to its capacity to inactivate the ˇ°iron-sulfur clusterˇ± containing enzymes (25), thereby liberating free iron in the cell, which can undergo Fenton chemistry and generate the highly reactive hydroxyl radical. TAC-induced inhibition of aconitase, an enzyme in the TCA cycle, was attenuated in c-Nox4−/− mice. Because Nox4 preferentially utilizes NADH as an electron donor (26), these results not only support our hypothesis that Nox4 is involved in mitochondrial oxidative stress but also raise an exciting possibility that Nox4 directly regulates the NADH/FADH2 generating enzymes in the TCA cycle, thereby initiating regulatory feedback mechanisms controlling its O2− producing activity.
ˇˇ
In summary, our results suggest that Nox4 in cardiac myocytes is a critical mediator of oxidative stress and cardiac dysfunction during PO. Because of its mitochondrial localization and up-regulation, Nox4 is a major source of O2− and/or H2O2 production in the heart, thereby playing an important role in mediating mitochondrial dysfunction during PO. We propose that Nox4 could be a target of future treatments for heart failure.
ˇˇ
NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart | PNAS
https://www.pnas.org/content/107/35/15565ˇˇ
Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons.
Department of Cellular and Integrative Physiology, University of Nebraska Medical Center, Omaha, NE 68198, USA.
Abstract
Angiotensin II (ANG II) plays an important role in the central regulation of systemic cardiovascular function. ANG II-mediated intraneuronal signaling has been shown to be predicated by an increase in mitochondrial superoxide (O₂∙-), yet the source of this reactive oxygen species (ROS) production remains unclear. NADPH oxidase 4 (Nox4), a member of the NADPH oxidase family, has been reported to be localized in mitochondria of various cell types and has been implicated in brain angiotensinergic signaling. However, the subcellular localization and function of Nox4 in neurons has not been fully elucidated. In this study, we hypothesized that Nox4 is expressed in neuron mitochondria and is involved in ANG II-dependent O₂∙--mediated intraneuronal signaling. To query this, Nox4 immunofluorescent staining and mitochondrial enrichment were performed in a mouse catecholaminergic neuronal cell model (CATH.a). Nox4 was shown to be present in neuron mitochondria as evidenced by colocalization with both the mitochondrial-localized protein manganese superoxide dismutase (MnSOD) and dye MitoTracker Red. Moreover, Nox4 expression was significantly increased in enriched mitochondrial fractions compared with whole cell lysates. Additionally, adenoviral-encoded small interfering RNA for Nox4 (AdsiNox4) caused a robust knockdown in Nox4 mRNA and protein levels, which led to the attenuation of ANG II-induced mitochondrial O₂∙- production. Finally, in the subfornical organ (SFO) of the brain, Nox4 not only demonstrated mitochondrial localization but was induced by chronic, peripheral infusion of ANG II. Collectively, these data suggest that Nox4 is a source of O₂∙- in neuron mitochondria that contributes to ANG II intraneuronal signaling.
www.ncbi.nlm.nih.gov/pubmed/23624625
Was this helpful?
NADPH oxidase 4 is an oncoprotein localized to mitochondria
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3040835
Aug 01, 2010 ˇ¤ We demonstrate that the NOX4 protein contains a 73 amino acid long mitochondrial localization signal at the N-terminus that is capable of transporting a passenger protein GFP into the mitochondria. Treatment of NOX4 overexpressing cells with catalase resulted in decreased tumorigenic characteristics. Together, this study provides evidence for an oncogenic function for NOX4 protein localized to mitochondria and suggests that NOX4 is a novel source of ROS produced in the mitochondria.
Cited by: 175
Publish Year: 2010
Author: Kelly A Graham, Mariola Kulawiec, Kjerstin M Owens, Xiurong Li, Mohamed Mokhtar Desouki, Dhyan Chand...
Subcellular localization of Nox4 and regulation in ...
https://www.pnas.org/content/106/34/14385
Aug 25, 2009 ˇ¤ ( i ) Immunoblot analysis in cultured mesangial cells and kidney cortex revealed that Nox4 is present in crude mitochondria, in mitochondria-enriched heavy fractions, and in purified mitochondria; ( ii ) immunofluorescence confocal microscopy also revealed that Nox4 localizes with the mitochondrial marker Mitotracker; and ( iii ) the mitochondrial localization prediction program MitoProt indicated that the probability score for Nox4 is identical to mitochondrial ˇ
Cited by: 460
Publish Year: 2009
Author: Karen L Block, Yves Gorin, Hanna E Abboudˇˇ
ˇˇ
Published: 19 October 2017
NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistanceˇˇ
ˇˇ
NADPH oxidases of the NOX family are membrane-bound enzymes, which use NADPH as a substrate to transfer electrons across membranes to molecular oxygen to generate superoxide, which is subsequently dismutated to hydrogen peroxide.
ˇˇ...the membrane bound NADPH oxidases of the NOX family are a major source of ROS in cancer. Seven membrane-bound NOX catalytic isoforms, referred to as NOX1 to NOX5, dual oxidase 1 (DUOX1) and DUOX2 have been identified, each of which displays similar but distinct structural, biochemical, and subcellular localization characteristics.
...NOX4 localizes to the mitochondrial compartment and is inhibited by AT
...Mitochondrial NOX4 is a novel energetic sensor
...Galatose enhances mitochondial metabolism-shunt cancer cells to mitochondrial respiration-increases ATP level by 40%,
cytotoxic drugs readily induced cell death in galactose-treated and VHL add back RCC cells compared to VHL-deficient cells grown in normal glucose (Fig. 4d, e).substituting galactose as the source of sugar in lieu of glucose enhances endogenous OXPHOS-driven ATP levels concomitant with a reduction of NOX activity within the mitochondrial compartment.
...NOX4 has been detected in other subcellular localizations of different cell types such as the endoplasmic reticulum, nucleus, and plasma membrane8, 27,28,29. T
... Acetylation of PKM2 mediates its destruction through a lysosomal mediated degradation pathway34. ..acetylation-dependent degradation of PKM2 is a mechanism that regulates drug-induced cell death in RCC; PKM2 is the critical NOX4 downstream target involved in RCC drug resistance, the first demonstration linking NOX4 and PKM2mitoTempol (a mitochondrial superoxide scavenger) is necessary and sufficient to sensitize VHL-deficient RCC to drug-induced cell death. IMPLICATIONS-CoQ10 also sensitizes cancer cells to -drug-induced cell death.
NOX4 is activated (loss of ATP levels within the mitochondria) during aerobic glycolysis.
Abstract
The molecular mechanisms that couple glycolysis to cancer drug resistance remain unclear. Here we identify an ATP-binding motif within the NADPH oxidase isoform, NOX4, and show that ATP directly binds and negatively regulates NOX4 activity. We find that NOX4 localizes to the inner mitochondria membrane and that subcellular redistribution of ATP levels from the mitochondria act as an allosteric switch to activate NOX4. We provide evidence that NOX4-derived reactive oxygen species (ROS) inhibits P300/CBP-associated factor (PCAF)-dependent acetylation and lysosomal degradation of the pyruvate kinase-M2 isoform (PKM2). Finally, we show that NOX4 silencing, through PKM2, sensitizes cultured and ex vivo freshly isolated human-renal carcinoma cells to drug-induced cell death in xenograft models and ex vivo cultures. These findings highlight yet unidentified insights into the molecular events driving cancer evasive resistance and suggest modulation of ATP levels together with cytotoxic drugs could overcome drug-resistance in glycolytic cancers.ˇˇ
Culturing mammalian cancer cells with galactose as the source of sugar in lieu of glucose blocks aerobic glycolysis and shunts energy reliability to mitochondrial respiration26. As an independent approach, we cultured 786-O cells with glucose or galactose containing media for 24 h in the presence or absence of rotenone and measured ATP levels. In parallel plates, mitochondrial fractions were prepared and NOX activity was examined. We find that ATP levels were increased ~40% in RCC cells cultured in galactose, which was inhibited by rotenone, whereas glucose had no effect (Supplementary Fig. 6a). The increase of ATP levels in the mitochondria from galactose-treated cells was concomitant with a paralleled reduction of NOX activity (Supplementary Fig. 6b).
ˇˇ
NOX4 expression is increased in the mitochondria of RCC tissue compared to normal control (Fig. 8a, b). Importantly, we find ATP efficiently inhibited NOX activity in isolated mitochondria from tumors (Fig. 8c). Figure 8d shows marked increase in PKM2 expression in RCC tumors compared to normal, whereas PKM1 expression appeared unchanged.
ˇˇ
PKM2 expression is higher in ex vivo patient cells compared to normal renal proximal tubular epithelial cells (Fig. 9d), whereas PKM1 expression was unchanged (Fig. 9e). Notably, NOX4 expression was higher in the total and mitochondrial fractions (Fig. 9f, g, respectively).
ˇˇ
We provide the first evidence that the NADPH oxidase isoform, NOX4, is a novel energetic sensor within the mitochondria, which serves as a checkpoint to couple mitochondrial energy metabolism to drug resistance in cancer cells. We suggest that during normal respiration, OXPHOS-driven ATP production in the mitochondria allosterically binds NOX4 through the Walker A, ATP-binding domain keeping NOX4-derived ROS low. Cellular events, such as cancer, which switch ATP production to aerobic glycolysis in the cytosol, thereby reducing ATP levels in the mitochondria relieves the breaks leading to NOX4 activation. Moreover, our data show that metabolic reprogramming-mediated activation of NOX4 inhibits PCAF-dependent acetylation- and lysosomal-mediated degradation of the oncogenic M2-PK (Fig. 9h).
ˇˇ
NOX4 expression is enhanced in the mitochondrial compartment in 80%+ of human RCC tumors that correlates with upregulation of PKM2 compared to normal adjacent tissue from the same individual.
ˇˇ
Together, we provide the first evidence that NOX4 functions as a mitochondrial energetic sensor and serves as a novel metabolic checkpoint, coupling the metabolic switch to cancer cell survival. We posit that targeted small molecules which mimic ATP binding to the NOX4 Walker A-binding site would be novel therapeutic approaches to reduce drug-resistance in renal cancer and likely other cancer cell types in which NOX4 drives drug resistance.ÔÚŇ»ĆđŁ¬ÎŇĂÇĚáą©Á˵ÚŇ»¸öÖ¤ľÝŁ¬Ö¤Ă÷NOX4łäµ±ĎßÁŁĚĺÄÜÁż´«¸ĐĆ÷Ł¬˛˘łäµ±ĐµĴúĐ»Ľě˛éµăŁ¬˝«´úлת»»ńîşĎµ˝°©Ď¸°ű´ć»îˇŁÎŇĂÇČĎÎŞŁ¬ÄŁÄâATPÓëNOX4 Walker A˝áşĎλµă˝áşĎµÄ°ĐĎňС·Ö×Ó˝«ĘÇĽőÉŮÉö°©ŇÔĽ°NOX4żÉÄÜÔÚĆäÖĐÇý¶ŻÄÍŇ©ĐÔµÄĆäËű°©Ď¸°űŔŕĐÍÖĐÄÍŇ©ĐÔµÄĐÂĐÍÖÎÁĆ·˝·¨ˇŁ
NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance | Nature Communications
https://www.nature.com/articles/s41467-017-01106-1ˇˇ
ˇˇ
Galactose Enhances Oxidative Metabolism and Reveals ...
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028536
Galactose is known to enhance mitochondrial metabolism and could be an excellent model to study mitochondrial dysfunction in human primary myotubes. The aim of the present study was to 1) characterize the effect of differentiating healthy human myoblasts in galactose on oxidative metabolism and 2) determine whether galactose can pinpoint a mitochondrial malfunction in post-diabetic ˇ
Cited by: 134
Publish Year: 2012
ˇˇGalactose Enhances Oxidative Metabolism and Reveals Mitochondrial Dysfunction in Human Primary Muscle Cells
Background
Human primary myotubes are highly glycolytic when cultured in high glucose medium rendering it difficult to study mitochondrial dysfunction. Galactose is known to enhance mitochondrial metabolism and could be an excellent model to study mitochondrial dysfunction in human primary myotubes. The aim of the present study was to 1) characterize the effect of differentiating healthy human myoblasts in galactose on oxidative metabolism and 2) determine whether galactose can pinpoint a mitochondrial malfunction in post-diabetic myotubes.
Methodology/Principal Findings
Oxygen consumption rate (OCR), lactate levels, mitochondrial content, citrate synthase and cytochrome C oxidase activities, and AMPK phosphorylation were determined in healthy myotubes differentiated in different sources/concentrations of carbohydrates: 25 mM glucose (high glucose (HG)), 5 mM glucose (low glucose (LG)) or 10 mM galactose (GAL). Effect of carbohydrates on OCR was also determined in myotubes derived from post-diabetic patients and matched obese non-diabetic subjects. OCR was significantly increased whereas anaerobic glycolysis was significantly decreased in GAL myotubes compared to LG or HG myotubes. This increased OCR in GAL myotubes occurred in conjunction with increased cytochrome C oxidase activity and expression, as well as increased AMPK phosphorylation. OCR of post-diabetic myotubes was not different than that of obese non-diabetic myotubes when differentiated in LG or HG. However, whereas GAL increased OCR in obese non-diabetic myotubes, it did not affect OCR in post-diabetic myotubes, leading to a significant difference in OCR between groups. The lack of an increase in OCR in post-diabetic myotubes differentiated in GAL was in relation with unaltered cytochrome C oxidase activity levels or AMPK phosphorylation.
Conclusions/Significance
Our results indicate that differentiating human primary myoblasts in GAL enhances aerobic metabolism. Because this cell culture model elicited an abnormal response in cells from post-diabetic patients, it may be useful in further studies of the molecular mechanisms of mitochondrial dysfunction.ˇˇ
°ëČéĚÇÔöÇżČËĚĺłőĽ¶ĽˇČâϸ°űµÄŃő»Ż´úĐ»˛˘˝ŇĘľĎßÁŁĚ幦ÄÜŐĎ°
±łľ°
µ±ÔÚ¸ßĆĎĚŃĚÇĹŕŃř»ůÖĐĹŕŃřʱŁ¬ČËŔŕÔ´úĽˇąÜ(Human primary myotubes)ľßÓи߶ȵÄĚǽͽâ×÷ÓĂŁ¬Ňň´ËşÜÄŃŃĐľżĎßÁŁĚ幦ÄÜŐĎ°ˇŁŇŃÖŞ°ëČéĚÇżÉÔöÇżĎßÁŁĚĺµÄ´úĐ»Ł¬˛˘ÇŇżÉÄÜĘÇŃĐľżČËŔŕÔ´úĽˇąÜÖĐĎßÁŁĚ幦ÄÜŐĎ°µÄľřĽŃÄŁĐ͡Ł±ľŃĐľżµÄÄżµÄĘÇŁş1Ł©±íŐ÷°ëČéĚÇÖĐ˝ˇżµµÄČËŔŕłÉĽˇĎ¸°ű(health human myoblasts)·Ö»Ż¶ÔŃő»Ż´úĐ»µÄÓ°Ď죬ŇÔĽ°2Ł©Č·¶¨°ëČéĚÇĘÇ·ńżÉŇÔČ·¶¨ĚÇÄň˛ˇşóĽˇąÜ(post-diabetic myotubes.)ÖеÄĎßÁŁĚ幦ÄÜŇ쳣ˇŁ
·˝·¨/Ö÷ŇŞ·˘ĎÖ
Č·¶¨ÁË˝ˇżµµÄĽˇąÜÖеĺÄŃőÂĘŁ¨OCRŁ©Ł¬ČéËáˮƽŁ¬ĎßÁŁĚ庬ÁżŁ¬ÄűĂĘËáşĎøşÍϸ°űÉ«ËŘCŃő»ŻĂ¸»îĐÔŇÔĽ°AMPKÁ×ËữŁ¬ŐâĐ©ĽˇąÜµÄĚĽË®»ŻşĎÎďŔ´Ô´/Ũ¶Č˛»Í¬Łş25 mMĆĎĚŃĚÇŁ¨¸ßĆĎĚŃĚÇŁ¨HGŁ©Ł©Ł¬5 mMĆĎĚŃĚÇŁ¨µÍĆĎĚŃĚÇŁ¨LGŁ©Ł©»ň10 mM°ëČéĚÇŁ¨GALŁ©ˇŁ»ą˛â¶¨ÁËŔ´×ÔĚÇÄň˛ˇşó»ĽŐßşÍĆĄĹäµÄ·ĘĹÖ·ÇĚÇÄň˛ˇĘÜĘÔŐߵļˇąÜÖĐĚĽË®»ŻşĎÎď¶ÔOCRµÄÓ°Ď졣ÓëLG»ňHGĽˇąÜĎŕ±ČŁ¬GALĽˇąÜµÄOCRĎÔ×ĹÔöĽÓŁ¬¶řÎŢŃőĚǽͽâĎÔ×ĹĽőÉ١Ł GALĽˇąÜÖеÄŐâÖÖOCRÔöĽÓÓëϸ°űÉ«ËŘCŃő»ŻĂ¸»îĐԺͱí´ďÔöĽÓŇÔĽ°AMPKÁ×ËữÔöĽÓÓйءŁÔÚLG»ňHGµÄ·Ö»ŻĚÇÄň˛ˇşóĽˇąÜµÄOCRÓë·ĘĹÖ·ÇĚÇÄň˛ˇĽˇąÜÎ޲îŇ졣µ«ĘÇŁ¬ľˇąÜGALÔöĽÓÁË·ĘĹÖ·ÇĚÇÄň˛ˇĽˇąÜÖеÄOCRŁ¬µ«Ëü˛˘Î´Ó°ĎěĚÇÄň˛ˇşóĽˇąÜÖеÄOCRŁ¬´Ó¶řµĽÖÂÁ˝×éÖ®ĽäµÄOCRĎÔ×Ų»Í¬ˇŁÔÚGALÖĐ·Ö»ŻµÄĚÇÄň˛ˇşóĽˇąÜÖĐŁ¬OCRȱ·¦ÔöĽÓÓëϸ°űÉ«ËŘCŃő»ŻĂ¸»îĐÔˮƽδ¸Ä±ä»ňAMPKÁ×ËữÓйءŁ
˝áÂŰ/ŇâŇĺ
ÎŇĂǵĽáąű±íĂ÷Ł¬ÔÚGALÖĐ·Ö»ŻČËŔŕÔ´úłÉĽˇĎ¸°űżÉÔöÇżÓĐŃő´úĐ»ˇŁŇňÎŞ´Ëϸ°űĹŕŃřÄŁĐÍÔÚĚÇÄň˛ˇ»ĽŐßµÄϸ°űÖĐŇýĆđŇ쳣·´Ó¦Ł¬ËůŇÔËüżÉÄÜÔÚ˝řŇ»˛˝ŃĐľżĎßÁŁĚ幦ÄÜŐĎ°µÄ·Ö×Ó»úÖĆÖĐÓĐÓáŁ
ˇˇGalactose Enhances Oxidative Metabolism and Reveals Mitochondrial Dysfunction in Human Primary Muscle Cells
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028536ˇˇ
ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis
ˇˇNorthwestern University
Reactive oxygen species (ROS) derived from NADPH oxidase (NOX) and mitochondria play a critical role in growth factor-induced switch from a quiescent to an angiogenic phenotype in endothelial cells (ECs).ˇˇ
However, how highly diffusible ROS produced from different sources can coordinate to stimulate VEGF signaling and drive the angiogenic process remains unknown.
ˇˇ
Using the cytosol-and mitochondria-targeted redox-sensitive RoGFP biosensors with real-time imaging, here we show that VEGF stimulation in human ECs rapidly increases cytosolic RoGFP oxidation within 1 min, followed by mitochondrial RoGFP oxidation within 5 min, which continues at least for 60 min.
ˇˇ
Silencing of Nox4 or Nox2 or overexpression of mitochondria-targeted catalase significantly inhibits VEGF-induced tyrosine phosphorylation of VEGF receptor type 2 (VEGFR2-pY), EC migration and proliferation at the similar extent.
ˇˇ
Exogenous hydrogen peroxide (H2O2) or overexpression of Nox4, which produces H2O2, increases mitochondrial ROS (mtROS), which is prevented by Nox2 siRNA, suggesting that Nox2 senses Nox4-derived H2O2to promote mtROS production. Mechanistically, H2O2increases S36 phosphorylation of p66Shc, a key mtROS regulator, which is inhibited by siNox2, but not by siNox4. Moreover, Nox2 or Nox4 knockdown or overexpression of S36 phosphorylation-defective mutant p66Shc(S36A) inhibits VEGF-induced mtROS, VEGFR2-pY, EC migration, and proliferation.
ˇˇ
In summary, Nox4-derived H2O2in part activates Nox2 to increase mtROS via pSer36-p66Shc, thereby enhancing VEGFR2 signaling and angiogenesis in ECs. This may represent a novel feed-forward mechanism of ROS-induced ROS release orchestrated by the Nox4/ Nox2/pSer36-p66Shc/mtROS axis, which drives sustained activation of angiogenesis signaling program.
ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis ˇŞ Northwestern Scholars
https://www.scholars.northwestern.edu/en/publications/ros-induced-ros-release-orchestrated-by-nox4-nox2-and-mitochondri
NOX4Ö§łÖĚǽͽⲢ´Ů˝ř·ÇСϸ°ű·Î°©Ď¸°űÖеĹȰ±őŁ°·´úĐ»
NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells
ąă¶«ĘˇŇ©żĆ´óѧ ÖĐÉ˝´óѧ°©Ö˘ÖĐĐÄ
Highlights
• NOX4 directes glucose metabolism to both glycolysis and PPP in NSCLC.
• NOX4 promotes glutaminolysis into total GSH synthesis for oxidative resistance.
• The selective NOX4 inhibitor GKT137831 may be potentially used against NSCLC.
Abstract
Our previous studies have confirmed that NADPH oxidase 4 (NOX4) is abundantly expressed in non-small cell lung cancer (NSCLC) and contributes to cancer progression.ˇˇ
Nevertheless, the comprehensive mechanisms for NOX4-mediated malignant progression and oxidative resistance of cancer cells remain largely unknown.
ˇˇ
This study found that NOX4 directed glucose metabolism not only to the glycolysis but also to pentose phosphate pathway (PPP) pathway for production of NADPH in NSCLC cell lines. Besides, we also found that NOX4 promoted glutaminolysis into total GSH synthesis. Specifically, the data showed that ectopic NOX4 expression did not induce apoptosis of NSCLC cells; however, inhibition of GSH production resulted in obvious apoptotic death of NOX4-overexpressed NSCLC cells.
ˇˇ
Furthermore, we demonstrated that NOX4-induced glycolysis probably via ROS/PI3K/Akt signaling-dependent c-Myc upregulation. The selective NOX4 inhibitor, GKT137831, significantly inhibited glucose and glutamine metabolic phenotypes both in vitro and in vivo, and itself or combination with 2-DG, a synthetic glycolytic inhibitor, suppressed cancer cell growth both in vivo and in vitro.
ˇˇ
Elimination of NOX4-derived H2O2 effectively reversed NOX4 overexpression-mediated metabolic effects in NSCLC cells. NOX4 levels were significantly correlated with increased glucose and glutamine metabolism-related genes, as well as Akt phosphorylation and c-Myc expression in primary NSCLC specimens.
ˇˇ
In conclusion, these results reveal that NOX4 promotes glycolysis, contributing to NSCLC growth, and supports glutaminolysis for oxidative resistance. Therefore, NOX4 may be a promising target to reverse malignant progression of NSCLC.
ˇˇˇˇ
Model: NOX4 stimulates glycolysis and supports glutaminolysis for resistance to apoptosis in NSCLC cells. The red, purple and green lines represented glycolysis, PPP and glutamine metabolism pathway, respectively.
ˇˇ
NOX4Ö§łÖĚǽͽⲢ´Ů˝ř·ÇСϸ°ű·Î°©Ď¸°űÖеĹȰ±őŁ°·´úĐ»
ąă¶«ĘˇŇ©żĆ´óѧ ÖĐÉ˝´óѧ°©Ö˘ÖĐĐÄFree Radical Biology and Medicine
Volume 101, December 2016, Pages 236-24
ŇŞµă
• ÔÚ·ÇСϸ°ű·Î°©ÖĐŁ¬NOX4˝«ĆĎĚŃĚÇ´úĐ»ŇýµĽÖÁĚǽͽâşÍPPPˇŁ
• NOX4´Ů˝řąČ°±őŁ°··Ö˝âÎŞ×ÜGSHşĎłÉŁ¬ľßÓĐżąŃő»ŻĐÔˇŁ
• ѡÔńĐÔNOX4ŇÖÖĆĽÁGKT137831żÉÄÜÓĂÓÚżąNSCLCˇŁ
ŐŞŇŞ
ÎŇĂÇŇÔÇ°µÄŃĐľżŇѾ֤ʵŁ¬NADPHŃő»ŻĂ¸4Ł¨NOX4Ł©ÔÚ·ÇСϸ°ű·Î°©Ł¨NSCLCŁ©ÖĐ´óÁż±í´ďŁ¬˛˘ÓĐÖúÓÚ°©Ö˘µÄ·˘ŐąˇŁ
Č»¶řŁ¬NOX4˝éµĽµÄ¶ńĐÔ˝řŐąşÍ°©Ď¸°űµÄżąŃő»ŻĐÔµÄČ«Ăć»úÖĆČÔĘÇδ֪֮ĘýˇŁ
ŐâĎîŃĐľż·˘ĎÖŁ¬NOX4˛»˝ö˝«ĚÇ´úлָµĽĚǽͽ⣬¶řÇŇ»ąÖ¸µĽÁËÔÚNSCLCϸ°űϵÖвúÉúNADPHµÄÎěĚÇÁ×ËáÍľľ¶Ł¨PPPŁ©Íľľ¶ˇŁ´ËÍ⣬ÎŇĂÇ»ą·˘ĎÖŁ¬NOX4´Ů˝řąČ°±őŁ°··Ö˝âÎŞ×ÜGSHşĎłÉˇŁľßĚĺ¶řŃÔŁ¬ĘýľÝĎÔĘľŇěλNOX4±í´ď˛»ÓŐµĽNSCLCϸ°űµňÍöˇŁČ»¶řŁ¬ŇÖÖĆGSH˛úÉúµĽÖÂNOX4ąý±í´ďµÄNSCLCϸ°űĂ÷ĎÔµňÍöËŔÍöˇŁ
´ËÍ⣬ÎŇĂÇÖ¤Ă÷ÁËNOX4ÓŐµĽµÄĚǽͽâżÉÄÜĘÇͨąýROS / PI3K / AktĐĹşĹŇŔŔµµÄc-MycÉϵ÷ˇŁŃˇÔńĐÔNOX4ŇÖÖĆĽÁGKT137831ÔÚĚĺÍâşÍĚĺÄÚľůĎÔ×ĹŇÖÖĆĆĎĚŃĚǺ͹Ȱ±őŁ°·´úĐ»±íĐÍŁ¬¶řĆä×ÔÉí»ňÓë2-DGŁ¨Ň»ÖֺϳɵÄĚǽͽâŇÖÖĆĽÁŁ©ÁŞşĎĘąÓĂŁ¬ÔňÔÚĚĺÄÚşÍĚĺÍâľůŇÖÖĆÁË°©Ď¸°űµÄÉúł¤ˇŁ
ĎűłýNOX4ŃÜÉúµÄH2O2żÉÓĐЧÄćתNSCLCϸ°űÖĐNOX4ąý±í´ď˝éµĽµÄ´úĐ»×÷ÓᣠNOX4ˮƽÓëÔöĽÓµÄĆĎĚŃĚǺ͹Ȱ±őŁ°·´úĐ»ĎŕąŘ»ůŇňŁ¬ŇÔĽ°Ô·˘ĐÔNSCLC±ę±ľÖеÄAktÁ×ËữşÍc-Myc±í´ďĎÔ×ĹĎŕąŘˇŁ
×ÜÖ®Ł¬ŐâĐ©˝áąű±íĂ÷Ł¬NOX4´Ů˝řĚǽͽ⣬´Ů˝řNSCLCµÄÉúł¤Ł¬˛˘Ö§łÖąČ°±őŁ°··Ö˝âŇÔ˛úÉúżąŃő»ŻĐÔˇŁŇň´ËŁ¬NOX4żÉÄÜĘÇÄćתNSCLC¶ńĐÔ˝řŐąµÄÓĐĎŁÍűµÄÄż±ęˇŁ
ÍĽĐθĹŇŞ
ÄŁĐÍŁşNOX4´ĚĽ¤ĚǽͽⲢ֧łÖąČ°±őŁ°··Ö˝âŇÔµÖżąNSCLCϸ°űµÄµňÍöˇŁşěÉ«Ł¬×ĎÉ«şÍÂĚÉ«Ďß·Ö±đ´ú±íĚǽͽ⣬PPPşÍąČ°±őŁ°·´úл;ľ¶ˇŁNOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0891584916309881ˇˇ
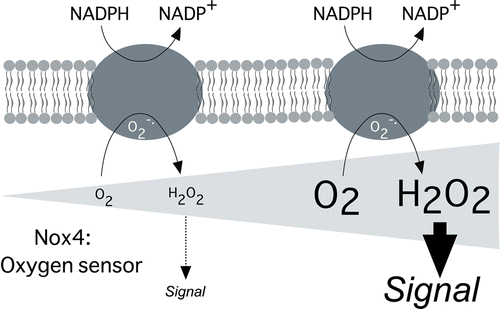
Nox4: A Hydrogen Peroxide-Generating Oxygen Sensor
published 2014
Abstract
Nox4 is an oddity among members of the Nox family of NADPH oxidases [seven isoenzymes that generate reactive oxygen species (ROS) from molecular oxygen] in that it is constitutively active. All other Nox enzymes except for Nox4 require upstream activators, either calcium or organizer/activator subunits (p47phox, NOXO1/p67phox, and NOXA1). Nox4 may also be unusual as it reportedly releases hydrogen peroxide (H2O2) in contrast to Nox1¨CNox3 and Nox5, which release superoxide, although this result is controversial in part because of possible membrane compartmentalization of superoxide, which may prevent detection. Our studies were undertaken (1) to identify the Nox4 ROS product using a membrane-free, partially purified preparation of Nox4 and (2) to test the hypothesis that Nox4 activity is acutely regulated not by activator proteins or calcium, but by cellular pO2, allowing it to function as an O2 sensor, the output of which is signaling H2O2. We find that approximately 90% of the electron flux through isolated Nox4 produces H2O2 and 10% forms superoxide. The kinetic mechanism of H2O2 formation is consistent with a mechanism involving binding of one oxygen molecule, which is then sequentially reduced by the heme in two one-electron reduction steps first to form a bound superoxide intermediate and then H2O2; kinetics are not consistent with a previously proposed internal superoxide dismutation mechanism involving two oxygen binding/reduction steps for each H2O2 formed. Critically, Nox4 has an unusually high Km for oxygen (∼18%), similar to the values of known oxygen-sensing enzymes, compared with a Km of 2¨C3% for Nox2, the phagocyte NADPH oxidase. This allows Nox4 to generate H2O2 as a function of oxygen concentration throughout a physiological range of pO2 values and to respond rapidly to changes in pO2.ˇˇ
Oxygen Dependence of Nox4
Herein, we explore the hypothesis that Nox4 activity is regulated not only by its expression level but also by oxygen availability and that it therefore functions as an oxygen sensor. For an enzyme to function as an oxygen sensor, it must fulfill two criteria. First, a sine qua non of an oxygen-sensing enzyme is an unusually high Km for oxygen that allows it to respond to physiological ranges of oxygen concentrations, which in tissues can range from 2¨C5% to around 20% in the lung, with intermediate concentrations in the circulatory system.(52) Second, its enzymatic activity must be linked to an effect or signal that can be translated into a cellular response. While Nox4 has been speculated to function in this manner,(26, 53, 54) the first of these criteria has not been previously evaluated. At least two oxygen-dependent enzymes, the HIF1-¦Á prolyl hydroxylase PHD and the HIF1-¦Á asparagine hydroxylase FIH-1, fit this paradigm and function as bona fide oxygen sensors, linked to the HIF1-¦Á-dependent transcriptional response to hypoxia.(55, 56) In both cases, the enzymes have high Km values for oxygen (10¨C20% O2 for PHD and ∼8% O2 for FIH-1) that render the enzyme activity proportional to physiological ranges of tissue pO2 values. Because hydroxylation by PHD targets HIF1-¦Á for degradation and that by FIH-1 is inhibitory, the net effect of lowering pO2 is to activate HIF1-¦Á-dependent transcription. In contrast, oxygen-dependent enzymes (collagen prolyl hydroxylase, P450 enzymes, and cytochrome c oxidase) that participate in cellular functions unrelated to oxygen sensing show low Km values for O2 (ranging from 0.2 to 3%), allowing them to function even at low to moderately low pO2 values. While an enzyme with a low Km for oxygen will be nearly saturated at tissue levels of oxygen, a high Km for oxygen allows oxygen-sensing enzymes to respond in a nearly linear manner with respect to oxygen concentration. For example, if the oxygen concentration were to increase from 3 to 12%, Nox4 activity would increase ∼300%. In contrast, an enzyme with an oxygen Km of 2% would increase its activity only ∼25%. Thus, while a low Km allows an enzyme to function nearly optimally at tissue pO2 values, it cannot respond dynamically to changes in pO2; in contrast, a high Km enzyme operating at a subsaturating pO2 will change its activity dynamically with changes in pO2.
The oxygen dependence for Nox2- versus Nox4-dependent ROS generation is compared in Figure 2. Nox2-dependent superoxide generation in either intact human neutrophils or a cell-free system shows a Km for oxygen of 3.1 or 2.3%, respectively, corresponding to the range of Km values seen in enzymes that participate in metabolic or cellular housekeeping functions. Because inflamed or infected tissues are often moderately hypoxic, this would allow the Nox2 system to continue to function at a significant rate under these conditions. On the other hand, Nox4 in both intact cells and lysates shows an oxygen Km value for H2O2 generation of 16¨C20%, corresponding to the Km range seen for other known oxygen-sensing enzymes. Under our assay conditions, the rate of Nox4-dependent H2O2 generation at both 21 and 1% oxygen was approximately linear for up to 3 h and was inhibited by DPI (data not shown). Because tissue oxygen concentrations are often on the order of 2¨C3%, this means that Nox4 will generate H2O2 approximately in direct proportion to oxygen at concentrations below ∼10%, making it a sensitive reporter of tissue oxygenation.
ˇˇMechanism of H2O2 Generation
Two mechanisms are possible for the generation of H2O2 by Nox4. Because the FAD domain does not conduct this reaction directly,(30) both possible mechanisms require single electron transfers from heme to oxygen. According to a ˇ°superoxide dismutaseˇ± mechanism that was previously suggested,(33) two superoxide molecules are formed in sequence and retained at the active site, and their sequestered dismutation before release from the enzyme results in H2O2 formation. Such a mechanism is shown in Figure 3 (bottom), along with its rate equation. The oxygen dependence for such a mechanism involves two oxygen binding events and predicts a sigmoidal oxygen dependence, as shown by the dashed line in Figure 3. According to an internal superoxide reduction mechanism (Figure 3, top mechanism and rate equation), a single oxygen binds and is reduced in two sequential electron transfer steps from the heme, using a retained superoxide intermediate. Because there is a single oxygen binding event, such a mechanism predicts a simple hyperbolic Michaelis¨CMenten curve as shown by the solid black line in the top panel of Figure 3. Data replotted from Figure 2 show an excellent fit to the theoretical line predicted by a mechanism involving a single oxygen binding event for each H2O2 formed (i.e., a mechanism in which sequential electrons are introduced into the oxygen from the heme), while they do not conform to the internal superoxide dismutation mechanism that would require two distinct oxygen binding events for each H2O2 formed. Therefore, while either mechanism could account for a small production of superoxide (depending on the relative rate of dissociation compared with those of subsequent steps), kinetics are consistent only with a mechanism involving a single bound superoxide intermediate (termed a ˇ°sequential one-electron reduction mechanismˇ± in Figure 3). Because such a mechanism has certain features in common with a superoxide reductase enzymatic activity, we also investigated the effects of azide and cyanide on H2O2 and superoxide generation in Nox4-expressing cells. A bacterial superoxide reductase shows inhibition of the production of H2O2 by these agents,(57) with accumulation of superoxide. The H2O2 electrode system was used for hydrogen peroxide detection, because these agents inhibit the HRP that is needed in the Amplex Red assay. For superoxide, the DHE assay was used to allow for high sensitivity. While it was not possible to use cyanide using the electrode system because of signal instability, 0.6 mM sodium azide produced 70% inhibition of Nox4-dependent H2O2 generation but did not cause any increase in superoxide production, suggesting that azide inhibits the overall Nox4 enzyme activity rather than a superoxide reduction step per se. Likewise, 1 mM KCN failed to increase superoxide generation in Nox4-expressing cells compared with that in control cells. Inhibition of Nox4 by azide is of interest in that Nox2 is not inhibited by azide or cyanide. Thus, azide inhibition may point to differentiating features of the oxygen-binding heme site in Nox2 versus Nox4.
Nox4: A Hydrogen Peroxide-Generating Oxygen Sensor | Biochemistry
https://pubs.acs.org/doi/10.1021/bi500331y#ˇˇ
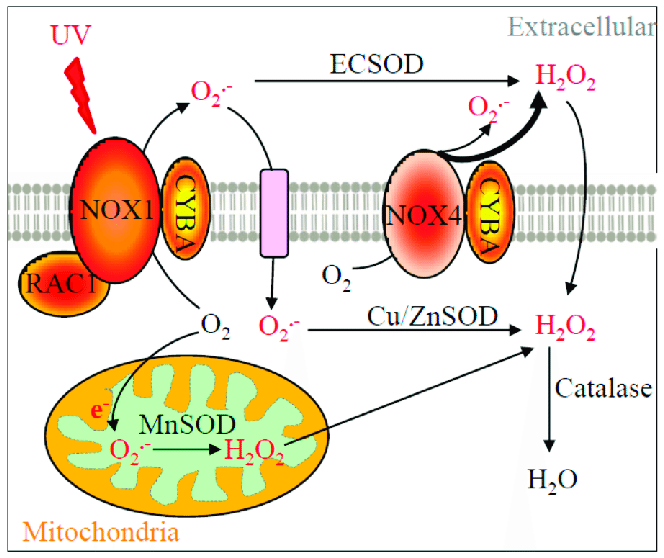
Diagram of the relevant reactive oxygen species (ROS) production pathway. NOX1, NOX4, CYBA, RAC1, SOD enzymes, catalase, their subcellular locations, and their functions in ROS production and metabolism are depicted in this diagram. NOX1 enzyme complex utilizes CYBA as one of its subunits and is activated by RAC1-GTPase to produce superoxide. On the other hand, NOX4 only couples with CYBA to generate hydrogen peroxide and superoxide. Of particular note, only plasma membrane NOX4 is shown in this diagram but mitochondrial or nuclear NOX4 has also been reported [16]. NOX1 is activated by UV to enhance its superoxide production, which requires the GTPase activity of RAC1. Superoxide is further metabolized into hydrogen peroxide at various subcellular locations by different SOD isozymes. Hydrogen peroxide is then converted into water molecules by catalase. Other additional redox enzymes (e.g., glutathione peroxidases, which also convert hydrogen peroxide into water) are not the focus in this study and therefore not included. Black arrows indicate the cellular movement of oxygen, ROS, and enzymatic metabolisms. A bold arrow represents a greater relative amount of ROS produced.
Diagram of the relevant reactive oxygen species (ROS) production... | Download Scientific Diagram
https://www.researchgate.net/figure/Diagram-of-the-relevant-reactive-oxygen-species-ROS-production-pathway-NOX1-NOX4_fig1_322512783ˇˇ
ˇˇ
World J Gastroenterol. 2003 Mar 15; 9(3): 562¨C567.
Published online 2003 Mar 15. doi: 10.3748/wjg.v9.i3.562
PMCID: PMC4621583
PMID: 12632519
Role of mitochondrial dysfunction in hydrogen peroxide-induced apoptosis of intestinal epithelial cells
Third Military Medical University,
Abstract
AIM: To study the role of mitochondrial dysfunction in hydrogen peroxide-induced apoptosis of intestinal epithelial cells.
METHODS: Hydrogen peroxide-induced apoptosis of human intestinal epithelial cell line SW-480 was established. Cell apoptosis was determined by Annexin-V and PI double-stained flow cytometry and DNA gel electrophoresis. Morphological changes were examined with light and electron microscopy. For other observations, mitochondrial function, cytochrome c release, mitochondrial translocation and membrane potential were determined simultaneously.
RESULTS: Percentage of apoptotic cells induced with 400 ¦Ěmol/L hydrogen peroxide increased significantly at l h or 3 h after stimulation and recovered rapidly. Meanwhile percentage of apoptotic cells induced with 4 mmol/L hydrogen peroxide increased with time. In accordance with these changes, we observed decreased mitochondrial function in 400 ¦Ěmol/L H2O2-stimualted cells at 1 h or 3 h and in 4 mmol/L H2O2-stimualted cells at times examined. Correspondingly, swelling cristae and vacuole-like mitochondria were noted. Release of cytochrome c, decreased mitochondrial membrane potential and mitochondrial translocation were also found to be the early signs of apoptosis.
CONCLUSION: Dysfunctional mitochondria play a role in the apoptosis of SW-480 cell line induced by hydrogen peroxide.
Role of mitochondrial dysfunction in hydrogen peroxide-induced apoptosis of intestinal epithelial cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4621583/ˇˇ
ˇˇ
Role of the Mitochondrial Permeability Transition and Cytochrome c Release in Hydrogen Peroxide-Induced Apoptosis
We investigated the role of the mitochondrial inner membrane permeability transition and subsequent release of cytochrome c into the cytosol during oxidative stress-evoked apoptosis. Sublethal oxidative stress was applied by treating L929 cells with 0.5 mM H2O2 for 90 min. Then the cellular localization of cytochrome c was examined by immunofluorescent staining and Western blotting. H2O2 treatment caused the permeability transition and pore formation, resulting in membrane depolarization and translocation of cytochrome c from the mitochondria into the cytosol. Pretreatment with cyclosporin A and aristolochic acid (to inhibit pore formation) significantly attenuated a reduction of the mitochondrial membrane potential, as well as signs of apoptosis such as DNA fragmentation, increased plasma membrane permeability, and chromatin condensation. Therefore, exposure to H2O2 caused the opening of permeability transition pores in the inner mitochondrial membrane. An essential role of cytosolic cytochrome c in the execution of apoptosis was demonstrated by its direct microinjection into the cytosol, thus bypassing the need for cytochrome c release from the mitochondrial intermembrane space. Microinjection of cytochrome c caused caspase-dependent apoptosis.
Role of the Mitochondrial Permeability Transition and Cytochrome c Release in Hydrogen Peroxide-Induced Apoptosis | Request PDF
https://www.researchgate.net/publication/11505540_Role_of_the_Mitochondrial_Permeability_Transition_and_Cytochrome_c_Release_in_Hydrogen_Peroxide-Induced_Apoptosisˇˇ
ĎßÁŁĚĺͨ͸ĐÔת±äşÍϸ°űÉ«ËŘcĘÍ·ĹÔÚąýŃő»ŻÇâÓŐµĽµÄϸ°űµňÍöÖеÄ×÷ÓĂ
ÎŇĂǵ÷˛éÁËŃő»ŻÓ¦Ľ¤ÓŐ·˘µÄϸ°űµňÍöąýłĚÖĐĎßÁŁĚĺÄÚĤͨ͸ĐÔת±äşÍϸ°űÉ«ËŘcĘͷŵ˝Ď¸°űÖĘÖеÄ×÷ÓáŁÍ¨ąýÓĂ0.5 mM H2O2´¦ŔíL929ϸ°ű90·ÖÖÓŔ´Ę©ĽÓŃÇÖÂËŔĐÔŃő»ŻÓ¦Ľ¤ˇŁČ»şóͨąýĂâŇßÓ«ąâȾɫşÍµ°°×ÖĘÓˇĽŁĽě˛éϸ°űÉ«ËŘcµÄϸ°ű¶¨Î»ˇŁ H2O2´¦ŔíµĽÖÂÉř͸ĐÔת±äşÍż×Đγɣ¬µĽÖÂϸ°űÉ«ËŘc´ÓĎßÁŁĚĺ˝řČëĤµÄČĄĽ«»ŻşÍŇĆλˇŁÓĂ»·ćßľúËŘAşÍÂí¶µÁĺËá˝řĐĐÔ¤´¦ŔíŁ¨ŇÔŇÖÖĆż×Đγɣ©żÉĎÔ×ĹĽőČőĎßÁŁĚĺĤµçλµÄ˝µµÍŁ¬ŇÔĽ°µňÍöĽŁĎóŁ¬ŔýČçDNAƬ¶Î»ŻŁ¬ÖĘĤͨ͸ĐÔÔöĽÓşÍȾɫÖĘŨËőˇŁŇň´ËŁ¬±©Â¶ÓÚH 2 O 2µĽÖÂĎßÁŁĚĺÄÚĤµÄͨ͸ĐÔąý¶Éż×µÄżŞ·ĹˇŁÍ¨ąý˝«ĆäÖ±˝ÓĎÔ΢עÉäµ˝°űÖĘČÜ˝şÖĐŁ¬Ö¤Ă÷ÁË°űÖĘϸ°űÉ«ËŘcÔÚÖ´ĐĐϸ°űµňÍöÖеÄÖŘŇŞ×÷ÓĂŁ¬´Ó¶řČĆąýÁË´ÓĎßÁŁĚĺĤĽäżŐĽäĘÍ·Ĺϸ°űÉ«ËŘcµÄĐčŇŞˇŁĎ¸°űÉ«ËŘcµÄĎÔ΢עÉäŇýĆđë×Ěěµ°°×øŇŔŔµĐÔµňÍöˇŁ
ĎßÁŁĚĺͨ͸ĐÔת±äşÍϸ°űÉ«ËŘcĘÍ·ĹÔÚąýŃő»ŻÇâÓŐµĽµÄϸ°űµňÍöÖеÄ×÷ÓĂË÷ȡPDF
https://www.researchgate.net/publication/11505540_Role_of_the_Mitochondrial_Permeability_Transition_and_Cytochrome_c_Release_in_Hydrogen_Peroxide-Induced_ApoptosisCytochrome c release and caspase activation in hydrogen ...
https://febs.onlinelibrary.wiley.com/doi/full/10.1016/S0014-5793(98)00630-9
The present study was to determine whether hydrogen peroxide or tributyltin, agents known to disrupt mitochondrial function and induce apoptosis, stimulates mitochondrial cytochrome c release and activation of caspase©\3. Hydrogen peroxide induces apoptosis [ 11 ], and is also known to interfere with mitochondrial functions.
Cited by: 293
Publish Year: 1998
Author: H¨¦l¨¨ne Stridh, Monica Kimland, Dean P. Jones, Sten Orrenius, Mark B. Hampton
Sulfite Oxidase Activity of Cytochrome C: Role of Hydrogen ...
https://www.researchgate.net/publication/285651701_Sulfite_Oxidase_Activity_of...
Cytochrome c (Cyt c) plays a vital role in the mitochondrial electron transport chain (ETC). In addition, it is a key regulator of apoptosis.ˇˇ
Cytochrome c release and caspase activation in hydrogen peroxide©\ and tributyltin©\induced apoptosis
The ability of H2O2 and tributyltin (TBT) to trigger pro©\caspase activation via export of cytochrome c from mitochondria to the cytoplasm was investigated. Treatment of Jurkat T lymphocytes with H2O2 resulted in the appearance of cytochrome c in the cytosol within 2 h. This was at least 1 h before caspase activation was observed. TBT caused cytochrome c release already after 5 min, followed by caspase activation within 1 h. Measurement of mitochondrial membrane potential (¦¤¦·m) showed that both H2O2 and TBT dissipated ¦¤¦·m, but with different time courses. TBT caused a concomitant loss of ¦¤¦·m and release of cytochrome c, whereas cytochrome c release and caspase activation preceded any apparent ¦¤¦·m loss in H2O2©\treated cells. Thus, our results suggest that different mechanisms are involved in triggering cytochrome c release with these apoptosis©\inducing agents.
Cytochrome c release and caspase activation in hydrogen peroxide©\ and tributyltin©\induced apoptosis - Stridh - 1998 - FEBS Letters - Wiley Online Library
https://febs.onlinelibrary.wiley.com/doi/full/10.1016/S0014-5793%2898%2900630-9ˇˇ
ąýŃő»ŻÇâşÍČý¶ˇ»ůÎýÓŐµĽµÄϸ°űµňÍöÖĐϸ°űÉ«ËŘcµÄĘÍ·ĹşÍë×Ěěµ°°×øµÄ»î»Ż
ŃĐľżÁËąýŃő»ŻÇâşÍČý¶ˇ»ůÎýŁ¨TBTŁ©Í¨ąýϸ°űÉ«ËŘc´ÓĎßÁŁĚĺĘäłöµ˝Ď¸°űÖĘ´Ą·˘Ç°ë×Ěěµ°°×øŁ¨pro-caspase)Ľ¤»îµÄÄÜÁ¦ˇŁÓĂH2O2´¦ŔíJurkat TÁÜ°Íϸ°űµĽÖÂ2СʱÄÚϸ°űÖĘÖĐłöĎÖϸ°űÉ«ËŘcˇŁŐâÖÁÉŮÔڹ۲쵽ë×Ěěµ°°×øĽ¤»î֮ǰ1СʱˇŁ TBTÔÚ5·ÖÖÓşóŇŃľŇýĆđϸ°űÉ«ËŘcĘÍ·ĹŁ¬Č»şóÔÚ1СʱÄÚĽ¤»îcaspaseˇŁĎßÁŁĚĺĤµçλŁ¨¦¤¦·mŁ©µÄ˛âÁż˝áąű±íĂ÷Ł¬H2O2şÍTBTľůşÄɢÁ˦¤¦·mŁ¬µ«Ę±łĚ˛»Í¬ˇŁ TBTµĽÖ¦¤¦·mµÄËđʧşÍϸ°űÉ«ËŘcµÄĘÍ·ĹŁ¬¶řÔÚH2O2´¦ŔíµÄϸ°űÖĐŁ¬Ď¸°űÉ«ËŘcµÄĘÍ·ĹşÍcaspase»î»ŻĎČÓÚČÎşÎĂ÷ĎԵĦ¤¦·mËđʧˇŁŇň´ËŁ¬ÎŇĂǵĽáąű±íĂ÷Ł¬ŐâĐ©µňÍöÓŐµĽĽÁ´Ą·˘Ď¸°űÉ«ËŘcĘͷŵĻúÖƲ»Í¬ˇŁ
ąýŃő»ŻÇâşÍČý¶ˇ»ůÎýÓŐµĽµÄϸ°űµňÍöÖĐϸ°űÉ«ËŘcµÄĘÍ·ĹşÍë×Ěěµ°°×øµÄ»î»Ż-Stridh-1998-FEBS Letters-WileyÔÚĎßÍĽĘéąÝ
https://febs.onlinelibrary.wiley.com/doi/full/10.1016/S0014-5793%2898%2900630-9ˇˇ
ˇˇ
oxidation of ascorbic acid generates hydrogen peroxide
ˇˇ
cited from Ascorbic acid: Chemistry, biology and the treatment of cancerˇî
Department of Radiation Oncology, University of Iowa College of Medicine, Iowa City, IA, USA
Abstract
Since the discovery of vitamin C, the number of its known biological functions is continually expanding. Both the names ascorbic acid and vitamin C reflect its antiscorbutic properties due to its role in the synthesis of collagen in connective tissues. Ascorbate acts as an electron-donor keeping iron in the ferrous state thereby maintaining the full activity of collagen hydroxylases; parallel reactions with a variety of dioxygenases affect the expression of a wide array of genes, for example via the HIF system, as well as via the epigenetic landscape of cells and tissues. In fact, all known physiological and biochemical functions of ascorbate are due to its action as an electron donor. The ability to donate one or two electrons makes AscH− an excellent reducing agent and antioxidant. Ascorbate readily undergoes pH-dependent autoxidation producing hydrogen peroxide (H2O2). In the presence of catalytic metals this oxidation is accelerated. In this review, we show that the chemical and biochemical nature of ascorbate contribute to its antioxidant as well as its prooxidant properties. Recent pharmacokinetic data indicate that intravenous (i.v.) administration of ascorbate bypasses the tight control of the gut producing highly elevated plasma levels; ascorbate at very high levels can act as prodrug to deliver a significant flux of H2O2 to tumors. This new knowledge has rekindled interest and spurred new research into the clinical potential of pharmacological ascorbate. Knowledge and understanding of the mechanisms of action of pharmacological ascorbate bring a rationale to its use to treat disease especially the use of i.v. delivery of pharmacological ascorbate as an adjuvant in the treatment of cancer.
Keywords: Ascorbate, Oxidative stress, Hydrogen peroxide, Cancer, Pharmacology, Nutritionˇˇ
2. Chemistry and biochemistry
Ascorbic acid (AscH2, vitamin C) is a water-soluble ketolactone with two ionizable hydroxyl groups. It has two pKaˇŻs, pK1 is 4.2 and pK2 is 11.6; thus, the ascorbate monoanion, AscH−, is the dominant form at physiological pH (Fig. 1). Ascorbate is an excellent reducing agent and readily undergoes two consecutive, one-electron oxidations to form ascorbate radical (Asc•−) and dehydroascorbic acid (DHA). The ascorbate radical is relatively unreactive due to resonance stabilization of the unpaired electron; it readily dismutes to ascorbate and DHA (kobs =2ˇÁ105 M−1 s−1, pH 7.0) [4].
2Asc•- + H+ ↔ AscH- + DHA. (1)
These properties make ascorbate an effective donor antioxidant [13].
Pure ascorbic acid is a white crystalline powder, extremely soluble in water making a colorless solution. Sodium ascorbate often contains significant quantities of oxidation products departing a yellow color. At pH values between 6 and 7.8, the purity and concentration of an ascorbate solution can be easily determined by its absorbance, ¦Ĺ265 =14,500 M−1 s−1 [14].
Ascorbate oxidizes readily. The rate of oxidation is dependent on pH and is accelerated by catalytic metals [14]. In the absence of catalytic metals, the spontaneous oxidation of ascorbate is quite slow at pH 7.0 [15]. This autoxidation, i.e. oxidation in the absence of catalytic metals, occurs via the ascorbate dianion, Asc2− [16]. At pH 7.0 the dominant species for vitamin C is AscH− (99.9%) with low concentrations of AscH2 (0.1%) and Asc2− (0.005%). The amount of Asc2− will increase by a factor of ten with a one unit increase in pH; this will increase the rate of autoxidation by a factor of 10. In an air-saturated phosphate buffer (pH 7.0), it has been estimated that the observed pseudo-first-order rate constant for the autoxidation of ascorbate is on the order of 10−7¨C10−6 s−1 at pH 7.0 [15], consistent with a rate constant of ˇÖ300 M−1 s−1 for the true autoxidation of Asc2−, Reaction 2 [17].
k=300M−1s−1Asc2−+O2ˇúAsc∙−+O2∙−.
(2)
Thus, true autoxidation is indeed very slow. In most laboratory settings, oxidation of ascorbate is catalyzed by adventitious catalytic metals in the buffers as well as metals that enter the buffer for laboratory equipment [15,18]. In near-neutral buffers with contaminating metals, the oxidation and subsequent loss of ascorbate can be very rapid.Ascorbic acid: Chemistry, biology and the treatment of cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3608474/ˇˇ
»ŻŃ§ÓëÉúÎﻯѧ
żą»µŃŞËᣨAscH2Ł¬Î¬ÉúËŘCŁ©ĘÇľßÓĐÁ˝¸öżÉµçŔëôÇ»ůµÄË®ČÜĐÔÍŞÄÚőĄˇŁËüÓĐÁ˝¸öpKaŁ¬pK1ÎŞ4.2Ł¬pK2ÎŞ11.6Ł»Ňň´ËŁ¬żą»µŃŞËᵥŇőŔë×ÓAscH-ĘÇÉúŔípHµÄÖ÷ŇŞĐÎĘ˝Ł¨ÍĽ1Ł©ˇŁżą»µŃŞËáĘÇŇ»ÖÖĽ«şĂµÄ»ąÔĽÁŁ¬ČÝŇ׾ŔúÁ˝´ÎÁ¬ĐřµÄµĄµç×ÓŃő»ŻŁ¬Đγɿą»µŃŞËá×ÔÓÉ»ůŁ¨Asc•-Ł©şÍÍŃÇâżą»µŃŞËᣨDHAŁ©ˇŁÓÉÓÚδĹä¶Ôµç×ӵŲŐńÎȶ¨Ł¬żą»µŃŞËá×ÔÓÉ»ůĎŕ¶ÔĂ»ÓĐ·´Ó¦ĐÔˇŁËüşÜČÝŇ×±äłÉżą»µŃŞËáşÍDHAŁ¨kobs = 2ˇÁ105 M-1 s-1Ł¬pH 7.0Ł©[4]ˇŁ
2Asc•-+ H +↔AscH- + DHAˇŁ Ł¨1Ł©
ŐâĐ©ĚŘĐÔĘążą»µŃŞËáłÉÎŞÓĐЧµÄą©ĚĺżąŃő»ŻĽÁ[13]ˇŁ
´żµÄżą»µŃŞËáĘÇ°×É«˝áľ§ĐÔ·ŰÄ©Ł¬Ľ«Ň×ČÜÓÚË®Ł¬ĐÎłÉÎŢÉ«ČÜŇşˇŁżą»µŃŞËáÄĆͨłŁ°üş¬´óÁżµÄŃő»Ż˛úÎłĘ»ĆÉ«ˇŁÔÚpHÖµ˝éÓÚ6şÍ7.8Ö®ĽäʱŁ¬żą»µŃŞËáČÜŇşµÄ´ż¶ČşÍŨ¶ČżÉŇÔͨąýĆäÎüĘŐÂʦĹ265= 14,500 M-1 s-1ÇáËÉČ·¶¨[14]ˇŁ
żą»µŃŞËáČÝŇ×Ńő»ŻˇŁŃő»ŻËŮÂĘȡľöÓÚpHÖµŁ¬˛˘ÓÉ´ß»Ż˝đĘôĽÓËŮ[14]ˇŁÔÚĂ»ÓĐ´ß»Ż˝đĘôµÄÇéżöĎÂŁ¬żą»µŃŞËáµÄ×Ô·˘Ńő»ŻÔÚpH 7.0ʱ·ÇłŁÂý[15]ˇŁŐâÖÖ×ÔŃő»ŻŁ¬Ľ´ÔÚÎŢ´ß»Ż˝đĘôµÄÇéżöϵÄŃő»ŻŁ¬ĘÇͨąýżą»µŃŞËáµÄ¶ţĽŰŇőŔë×ÓAsc2- [16]·˘ÉúµÄˇŁÔÚpH 7.0ʱŁ¬Î¬ÉúËŘCµÄÖ÷ŇŞÖÖŔŕĘÇAscH-Ł¨99.9ŁĄŁ©Ł¬AscH2Ł¨0.1ŁĄŁ©şÍAsc2-Ł¨0.005ŁĄŁ©µÄŨ¶ČµÍˇŁ pHֵÿÔöĽÓ1µĄÎ»Ł¬Asc2-µÄÁż˝«ÔöĽÓ10±¶ˇŁŐ⽫ʹ×ÔŃő»ŻµÄËŮÂĘÔöĽÓ10±¶ˇŁÔÚżŐĆř±ĄşÍµÄÁ×ËáŃλşłĺŇşŁ¨pH 7.0Ł©ÖĐŁ¬ľÝąŔĽĆŁ¬ËůąŰ˛ěµ˝µÄżą»µŃŞËá×ÔŃő»ŻµÄÄâŇ»Ľ¶·´Ó¦ËŮÂĘłŁĘýÔĽÎŞ10ˇŁ pH 7.0ʱΪ-7-10-6 s-1 [15]Ł¬ÓëAsc2-µÄŐćʵ×ÔŃő»Ż·´Ó¦2µÄËŮÂĘłŁĘýˇÖ300M-1 s-1Ň»ÖÂ[17]ˇŁ
k = 300M-1s-1Asc2- + O2ˇúAsc∙-+ O2∙-
Ł¨2Ł©
Ňň´ËŁ¬ŐćŐýµÄ×ÔŃő»ŻČ·Ęµ·ÇłŁ»şÂýˇŁÔÚ´ó¶ŕĘýʵŃéĘŇ»·ľłÖĐŁ¬»şłĺŇşÖеIJ»¶¨´ß»Ż˝đĘôŇÔĽ°˝řČëʵŃéĘŇÉ豸»şłĺŇşµÄ˝đĘô»á´ß»Żżą»µŃŞËáµÄŃő»Ż[15,18]ˇŁÔÚľßÓĐÎŰČľĐÔ˝đĘôµÄ˝Ó˝üÖĐĐԵĻşłĺŇşÖĐŁ¬żą»µŃŞËáŃεÄŃő»ŻşÍËćşóµÄËđʧ»á·ÇłŁŃ¸Ë١Łˇˇ
3. Biosynthesis and uptake
Human erythrocytes express a high number of GLUT1, but have no SVCT proteins [38]. The ascorbate level in erythrocytes is similar to the plasma from which they were taken [39,40]. It is speculated that erythrocytes recycle and release ascorbate to help maintain plasma levels of ascorbate. With the exception of erythrocytes, intracellular ascorbate concentrations are higher than extracellular fluids. Studies have shown that ascorbate accumulates to millimolar concentrations in neutrophils, lymphocytes, monocytes, and platelets (Table 1). In circulating lymphocytes, 4 mM can be achieved in healthy young women with oral ascorbate supplementation [41]. Intracellular ascorbate not only contributes to the intracellular reducing environment, it can also quench extracellular oxidants by transferring electrons across the plasma membrane [42]. In addition, there appears to be regulated ascorbate efflux from astrocytes to neurons, thereby bolstering antioxidant defense [43]. Furthermore, the release of ascorbate from cells may also reduce non-transferrin-bound iron, thereby stimulating the uptake of iron by cells [44].Table 1
Ascorbate content of human tissues.
Tissue Ascorbate (mmol/kg wet tissue) Ascorbate (mM) Ref. Adrenal glands 1.7¨C2.3 ˇˇ [45] Pituitary gland 2.3¨C2.8 ˇˇ [45] Liver 0.8¨C1 ˇˇ [45] Spleen 0.8¨C1 ˇˇ [45] Pancreas 0.8¨C1 ˇˇ [45] Kidneys 0.28¨C0.85 ˇˇ [45] Skeletal muscle 0.17¨C0.23 ˇˇ [45] Brain 0.74¨C0.85 ˇˇ [45,226,227,234] Placenta 0.23¨C0.72 ˇˇ [48] Plasma (healthy) ˇˇ 0.04¨C0.08 [41,45,51] Red blood cell ˇˇ 0.04¨C0.06 [39] Neutrophil ˇˇ 1.2 [41] Lymphocyte ˇˇ 4.0 [41] Monocyte ˇˇ 3.2 [41] Platelet ˇˇ 3.7 [41,56] Cerebral spinal fluid ˇˇ 0.15¨C0.25a [235¨C237] Neuron ˇˇ 10 [238] Glial cells ˇˇ 1 [238] Lens ˇˇ 2.5¨C3.4 [46] Corneal epithelium ˇˇ 12.5 [49] Aqueous humor ˇˇ 0.4¨C1.1 [46] Alveolar macrophage ˇˇ 0.32 [50] Bronchoalveolar lavage ˇˇ 0.04¨C0.06 [51] Saliva ˇˇ 0.04¨C0.05 [45] aAscorbate levels in cerebral spinal fluid are approximately three to four times that of plasma.
Ascorbic acid: Chemistry, biology and the treatment of cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3608474/ˇˇ
Oxidative stress has been shown to increase the levels of catalytic iron in tissues. A series of electron paramagnetic resonance (EPR) studies demonstrated that ultraviolet light increased the levels of labile iron in skin [186]; heat stress-induced oxidative events increased the level of labile iron in liver [187]; and ischemia reperfusion increased the level of catalytic iron in the heart [188]. In addition, ionizing radiation and some chemotherapeutic drugs have been shown to increase catalytic free iron levels [189¨C191]. Since it takes only very low concentrations of catalytic metals to bring about the rapid oxidation of ascorbate [192,193], approaches that increase catalytic iron could potentially enhance the cytotoxicity of pharmacologic ascorbate in vivo [194].
ˇˇ
...almost thirty years ago, Linus Pauling and his colleagues [194] designed a copper:glycylglycylhistidine complex that mimics the binding of albumin to copper; it enhanced the antitumor activity of ascorbate against Ehrlich ascites tumor cells. In addition, extracellular fluids contain little or no catalase activity, and low levels of SOD and GPx [208]. Thus, the H2O2 generated by ascorbate oxidation in the extracellular space could accumulate to a concentration greater than the intracellular level resulting in net rate of diffusion across the cell membrane into cells, resulting in toxicity to cancer cells [10].
ˇˇ
Pharmacological ascorbate inhibits tumor growth in mice. In mice bearing tumor xenografts of pancreatic cancer, treatment with 4 g kg−1 ascorbate (i.p., twice daily) significantly decreased the rate of tumor growth [11,12,164] (Table 4). Upon cessation of treatment with pharmacological ascorbate, the rate of tumor growth increased. This rate was similar to the rate observed in the control group, i.e. no ascorbate (Du et al., unpublished results). In this tumor model pharmacologic ascorbate as a single agent was cytostatic, not cytotoxic. Obviously, the use of intravenous high dose ascorbate as a single agent for cancer was not curative. To test the combination effect of standard chemotherapy with ascorbate, Espey et al. [209] have shown that synergistic cytotoxic effects can be achieved with gemcitabine and ascorbate in pancreatic cancer both in vitro and in a nude-mouse model. These data provide support for investigating the use of pharmacologic ascorbate as an adjuvant for conventional cancer chemotherapies.
Three phase I clinical trials of intravenous ascorbate in patients with advanced cancer measured plasma concentrations of ascorbate and evaluated clinical consequences [162,220,221]. In the study by Riordan and colleagues, patients were given continuous infusion of 0.15 to 0.7 g kg−1 day−1 for up to eight weeks; in the Hoffer study, ascorbate was administrated three times per week at fixed doses of 0.4, 0.6, 0.9 and 1.5 g kg−1; and in the most recent study by Monti et al. [221], patients received 50, 75 and 100 g per infusion (three infusions per week) for 8 weeks with concomitant i.v. gemcitabine and oral erlotinib. In the Riordan study, ascorbate concentrations in serum during therapy ranged from 0.28 to 3.8 mM. In the Hoffer study, peak plasma ascorbate concentrations ranged from 2.4 to 26 mM. When 1.5 g kg−1 was administered, plasma ascorbate concentrations exceeded 10 mM for ˇÖ4.5 h [220], levels that have been shown to induce cell killing in a variety of cancer cells [9,11,12,164]. Finally in the Monti study, patients that received 100 g of ascorbate achieved peak plasma concentrations between 25 and 32 mM. All three trials demonstrated that high-dose intravenous ascorbate was well tolerated in cancer patients with normal renal function; the combination of ascorbate infusion with standard of care chemotherapies did not increase toxicity.
ˇˇ
ˇˇ
Elevated heme synthesis and uptake underpin intensified ...
cancerres.aacrjournals.org/content/early/2019/03/22/0008-5472.CAN-18-2156
Jan 01, 2019 ˇ¤ Notably, tumors formed by cells with increased heme synthesis or uptake also displayed elevated oxygen consumption rates and ATP levels. These data show that elevated heme flux and function underlie enhanced OXPHOS and tumorigenicity of NSCLC cells. Targeting heme flux and function offers a potential strategy for developing therapies for lung cancer.
Cited by: 3
Publish Year: 2019ˇˇ
Enhanced Heme Function and Mitochondrial Respiration ... https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3660535 Evidently, in cancer cells, the levels of the rate-limiting heme biosynthetic enzyme ALAS and heme uptake proteins HCP1 and HRG1 are strongly enhanced (Figure 8). Increasing the availability of a key component, heme, can lead to enhanced levels of an array of oxygen-utilizing hemoproteins in mitochondria, as well as in other cellular compartments ( Figure 8 ). Cited by: 57 Publish Year: 2013 A
ˇˇEnhanced Heme Function and Mitochondrial Respiration ... https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3660535 Evidently, in cancer cells, the levels of the rate-limiting heme biosynthetic enzyme ALAS and heme uptake proteins HCP1 and HRG1 are strongly enhanced (Figure 8). Increasing the availability of a key component, heme, can lead to enhanced levels of an array of oxygen-utilizing hemoproteins in mitochondria, as well as in other cellular compartments ( Figure 8 ). Cited by: 57 Publish Year: 2013 A
ˇˇ
ˇˇ
PLoS One. 2013; 8(5): e63402.
Published online 2013 May 21. doi: 10.1371/journal.pone.0063402
Enhanced Heme Function and Mitochondrial Respiration Promote the Progression of Lung Cancer Cells
Abstract
Lung cancer is the leading cause of cancer-related mortality, and about 85% of the cases are non-small-cell lung cancer (NSCLC). Importantly, recent advance in cancer research suggests that altering cancer cell bioenergetics can provide an effective way to target such advanced cancer cells that have acquired mutations in multiple cellular regulators. This study aims to identify bioenergetic alterations in lung cancer cells by directly measuring and comparing key metabolic activities in a pair of cell lines representing normal and NSCLC cells developed from the same patient. We found that the rates of oxygen consumption and heme biosynthesis were intensified in NSCLC cells. Additionally, the NSCLC cells exhibited substantially increased levels in an array of proteins promoting heme synthesis, uptake and function. These proteins include the rate-limiting heme biosynthetic enzyme ALAS, transporter proteins HRG1 and HCP1 that are involved in heme uptake, and various types of oxygen-utilizing hemoproteins such as cytoglobin and cytochromes. Several types of human tumor xenografts also displayed increased levels of such proteins. Furthermore, we found that lowering heme biosynthesis and uptake, like lowering mitochondrial respiration, effectively reduced oxygen consumption, cancer cell proliferation, migration and colony formation. In contrast, lowering heme degradation does not have an effect on lung cancer cells. These results show that increased heme flux and function are a key feature of NSCLC cells. Further, increased generation and supply of heme and oxygen-utilizing hemoproteins in cancer cells will lead to intensified oxygen consumption and cellular energy production by mitochondrial respiration, which would fuel cancer cell proliferation and progression. The results show that inhibiting heme and respiratory function can effectively arrest the progression of lung cancer cells. Hence, understanding heme function can positively impact on research in lung cancer biology and therapeutics.
Enhanced Heme Function and Mitochondrial Respiration Promote the Progression of Lung Cancer Cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3660535/ˇˇ
Iron Induces Death In Tumor Cells
Date:
March 13, 2009
Source:
Helmholtz Association of German Research Centres
Summary:
Tumor cells and healthy cells differ considerably in metabolism intensity. Scientists have now taken advantage of this difference; by releasing cellular iron, they were able to induce death selectively in tumor cells.
Share:
FULL STORY
Rapid growth of cancer cells and their frequent divisions have their price: Cancer cells need considerably more energy than healthy cells. Their metabolism runs at full speed and requires large amounts of micronutrients, particularly iron. However, high levels of iron in the cell lead to the production of extremely harmful free radicals.
To protect itself from these, the cell inactivates free iron by binding it to what are called iron storage proteins.
Collaborating with physicians of the Dermatology Department of Mannheim University Hospitals, Dr. Karsten G¨ąlow and Professor Dr. Peter Krammer, head of the Division of Immunogenetics at DKFZ, investigated S¨¦zary's disease (also called S¨¦zary syndrome), an extremely aggressive type of cutaneous T cell lymphoma. The majority of currently available treatments are not really effective against this fatal type of cancer.
Using a molecular-biological trick, G¨ąlow and colleagues succeeded in blocking the production of one of the iron storage proteins in lymphoma cells. This leads to a rise in the level of free, non-bound iron in these cells. The iron boosts the production of free oxygen radicals which cause oxidative stress and, thus, cause damage to the cancer cells and induce their death. Healthy cells with their low iron level, however, survive the treatment unharmed.
The DKFZ researchers have already found evidence that this iron effect also works in other lymphomas. They are now investigating whether selective release of iron may be a suitable approach for developing a novel cancer treatment.
Michael K. Kiessling, Claus D. Klemke, Marcin M. Kami¨˝ski, Ioanna E. Galani, Peter H. Krammer, and Karsten G¨ąlow: Inhibition of constitutively activated NF-¦ĘB induces ROS- and iron dependent cell death in cutaneous T cell lymphoma. Cancer Research 2009; DOI:10.1158/0008-5472.CAN-08-3221
The German Cancer Research Center is funded by the German Federal Ministry of Education and Research (90%) and the State of Baden-W¨ąrttemberg (10%).
Iron Induces Death In Tumor Cells -- ScienceDaily
https://www.sciencedaily.com/releases/2009/03/090311103607.htmˇˇ
Article
Published: 21 May 2019
Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis
Guo-Qing Chen, Fahad A. Benthani, Jiao Wu, Deguang Liang, Zhao-Xiang Bian & Xuejun Jiang
Cell Death & Differentiation (2019)Cite this articleˇˇ
School of Chinese Medicine, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China
Cell Biology Program, Memorial Sloan Kettering Cancer Center, New York City, NY, 10065, USA
Abstract
The antimalarial drug artemisinin and its derivatives have been explored as potential anticancer agents, but their underlying mechanisms are controversial. In this study, we found that artemisinin compounds can sensitize cancer cells to ferroptosis, a new form of programmed cell death driven by iron-dependent lipid peroxidation. Mechanistically, dihydroartemisinin (DAT) can induce lysosomal degradation of ferritin in an autophagy-independent manner, increasing the cellular free iron level and causing cells to become more sensitive to ferroptosis. Further, by associating with cellular free iron and thus stimulating the binding of iron-regulatory proteins (IRPs) with mRNA molecules containing iron-responsive element (IRE) sequences, DAT impinges on IRP/IRE-controlled iron homeostasis to further increase cellular free iron. Importantly, in both in vitro and a mouse xenograft model in which ferroptosis was triggered in cancer cells by the inducible knockout of GPX4, we found that DAT can augment GPX4 inhibition-induced ferroptosis in a cohort of cancer cells that are otherwise highly resistant to ferroptosis. Collectively, artemisinin compounds can sensitize cells to ferroptosis by regulating cellular iron homeostasis. Our findings can be exploited clinically to enhance the effect of future ferroptosis-inducing cancer therapies.
·˘˛ĽĘ±ĽäŁş2019Äę5ÔÂ21ČŐ
ÇŕÝďËŘ»ŻşĎÎďͨąýµ÷˝ÚĚúÎČ̬Ŕ´Ęą°©Ď¸°ű¶ÔĚúËŔÍöĂô¸Đ
łÂąúÇ죬Fahad A.BenthaniŁ¬Î⽿Ł¬ÁşµÂąâŁ¬BŐ×ĎéşÍ˝ŞŃ§ľü
ϸ°űËŔÍöÓë·Ö»ŻŁ¨2019Ł©Ô®Ňý±ľÎÄ
Ďă¸Ű˝ţ»á´óѧÖĐҽѧԺŁ¬Ďă¸ŰľĹÁú
ϸ°űÉúÎďѧĽĆ»®Ł¬ĽÍÄî˹¡ˇ¤żĚŘÁŐ°©Ö˘ÖĐĐÄŁ¬Ĺ¦ÔĽŁ¬Ĺ¦ÔĽŁ¬10065Ł¬ĂŔąú
łéĎó
ŇŃľŃĐľżÁËżąĹ±Ň©ÇŕÝďËŘĽ°ĆäŃÜÉúÎď×÷ΪDZÔڵĿą°©Ň©Ł¬µ«ĆäDZÔÚ»úÖĆÉĐ´ćŐůŇ顣ÔÚŐâĎîŃĐľżÖĐŁ¬ÎŇĂÇ·˘ĎÖÇŕÝďËŘŔ໯şĎÎďżÉŇÔĘą°©Ď¸°ű¶ÔĚúËŔÍöĂô¸ĐŁ¬¶řĚúËŔÍöĘÇĚúŇŔŔµĐÔÖ¬ÖĘąýŃő»ŻÇý¶ŻµÄłĚĐňĐÔϸ°űËŔÍöµÄŇ»ÖÖĐÂĐÎĘ˝ˇŁ´Ó»úÖĆÉĎ˝˛Ł¬Ë«ÇâÇŕÝďËŘŁ¨DATŁ©żÉŇÔŇÔ×ÔĘÉŇŔŔµĐÔ·˝Ę˝ÓŐµĽĚúµ°°×µÄČÜøĚ彵˝âŁ¬´Ó¶řÔöĽÓϸ°űÖеÄÓÎŔëĚúˮƽŁ¬˛˘ĘąĎ¸°ű¶ÔĚúĐⲡ¸üĽÓĂô¸ĐˇŁ´ËÍ⣬ͨąýÓëϸ°űÓÎŔëĚúµŢşĎŁ¬´Ó¶ř´ĚĽ¤Ěúµ÷˝Úµ°°×Ł¨IRPŁ©Óë°üş¬ĚúĎěÓ¦ÔŞĽţŁ¨IREŁ©ĐňÁеÄmRNA·Ö×ӵĽáşĎŁ¬DATײ»÷IRP / IREżŘÖƵÄĚúÎČ̬Ł¬´Ó¶ř˝řŇ»˛˝ÔöĽÓϸ°űÓÎŔëĚúˇŁÖŘŇŞµÄĘÇŁ¬ÔÚĚĺÍâşÍСĘóŇěÖÖŇĆֲģĐÍÖĐŁ¬Í¨ąýÓŐµĽĐÔÇĂłýGPX4ÔÚ°©Ď¸°űÖĐŇý·˘ÁËĚúËŔÍöŁ¬ÎŇĂÇ·˘ĎÖDATżÉŇÔÔöÇżGPX4ŇÖÖĆÓŐµĽµÄĚúËŔÍöŁ¬·ńÔň¸ĂČş°©Ď¸°ű¶ÔĚúËŔÍöľßÓи߶ȿąĐÔˇŁ×ÜĚĺ¶řŃÔŁ¬ÇŕÝďËŘ»ŻşĎÎďżÉͨąýµ÷˝Úϸ°űĚúÎČ̬Ŕ´ĘąĎ¸°ű¶ÔĚúËŔÍöĂô¸ĐˇŁÎŇĂǵķ˘ĎÖżÉÔÚÁŮ´˛ÉĎĽÓŇÔŔűÓĂŁ¬ŇÔÔöǿδŔ´ÓŐµĽĚúËŔÍöµÄ°©Ö˘ÖÎÁƵÄЧąűˇŁ
ÇŕÝďËŘ»ŻşĎÎďͨąýµ÷˝ÚĚúÎČ̬Ŕ´Ęą°©Ď¸°ű¶ÔĚúËŔÍöĂô¸Đ
Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis | Cell Death & Differentiation
https://www.nature.com/articles/s41418-019-0352-3ˇˇ
Open Access
Int. J. Mol. Sci. 2019, 20(1), 39; https://doi.org/10.3390/ijms20010039
Review
A Dual Role of Heme Oxygenase-1 in Cancer Cells
by Shih-Kai Chiang 1, Shuen-Ei Chen 1,2,3,4 and Ling-Chu Chang 5,*OrcID
1
Department of Animal Science, National Chung Hsing University, Taichung 40227, Taiwan
Received: 9 November 2018 / Accepted: 19 December 2018 / Published: 21 December 2018
Abstract: Heme oxygenase (HO)-1 is known to metabolize heme into biliverdin/bilirubin, carbon monoxide, and ferrous iron, and it has been suggested to demonstrate cytoprotective effects against various stress-related conditions. HO-1 is commonly regarded as a survival molecule, exerting an important role in cancer progression and its inhibition is considered beneficial in a number of cancers. However, increasing studies have shown a dark side of HO-1, in which HO-1 acts as a critical mediator in ferroptosis induction and plays a causative factor for the progression of several diseases. Ferroptosis is a newly identified iron- and lipid peroxidation-dependent cell death. The critical role of HO-1 in heme metabolism makes it an important candidate to mediate protective or detrimental effects via ferroptosis induction. This review summarizes the current understanding on the regulatory mechanisms of HO-1 in ferroptosis. The amount of cellular iron and reactive oxygen species (ROS) is the determinative momentum for the role of HO-1, in which excessive cellular iron and ROS tend to enforce HO-1 from a protective role to a perpetrator. Despite the dark side that is related to cell death, there is a prospective application of HO-1 to mediate ferroptosis for cancer therapy as a chemotherapeutic strategy against tumors.
Keywords: ferroptosis; heme oxygenase-1; iron; reactive oxygen species; glutathione; chemotherapyˇˇ
Figure 3. Model of HO-1-mediated ferroptosis. HO-1 exerts a cytoprotective effect by scavenging ROS during moderate activation. By contrast, excessive activation of HO-1 increases labile Fe2+, leading to ROS overload and death of cancer cells.ˇˇ
Ferroptosis and Cancer
Ferroptosis is a recently identified type of cell death that is morphologically, genetically, and mechanistically distinct from regulated cell death, including apoptosis, necroptosis, and autophagy [6]. Ferroptotic cells are morphologically characterized by small mitochondria, collapsed mitochondrial cristae, and increased mitochondrial membrane density [6]. Mechanistically, ferroptosis is induced by iron accumulation and lipid peroxidation, accompanied by glutathione depletion. Excessive lipid peroxidation impairs the cellular membrane fluidity, permeability, and cellular integrity, eventually leading to cell death [6,18,19,20]. Ferroptosis can be induced by the overloading of iron (ferric ammonium citrate), glutathione/glutamine antiporter system Xc− inhibition (e.g., erastin, sorafenib, sulfasalazine, and lanperisone), and glutathione peroxidase 4 (GPx4) inactivation (RSL3, DPI7). Pharmacological manipulations, such as iron chelators (deferoxamine, ciclipirox, and deferiprone), glutathione replenishment (N-acetyl-l-cysteine, ¦Â-mercaptoethanol, cysteine/cystine, intracellular glutathione), and inhibitors of ROS production and lipid peroxidation (liporxstatin-1, ferrostatin-1, zileuton) that modulate the ferroptotic process have been shown to function against diseases, including cancer, neurotoxicity, and ischemia/reperfusion-induced injury [18,19,20].
The interrelationship between ferroptosis and cancer progression has been validated using ferroptotic agents. Some small molecules (e.g., erastin and RSL3) and clinical cancer drugs (e.g., sorafenib, sulfasalazine, and artesunate) induced cell death via the inhibition of system Xc− and GPx4 in various types of cancer cells [6,7,19]. A delay of ferroptosis protects cancer cells from metabolic oxidative stress, and thus increases their survival and distal metastasis [21]. Besides, the induction of ferroptosis was shown to overcome artesunate-induced resistance in head and neck cancer cells [22], and the induction of ferroptosis that contributes to anticancer activity has been identified in different cancer types [23]. Diffuse large B-cell lymphomas and renal cell carcinomas strongly rely on GPx4 availability to maintain redox status, and thus may suggest a high sensitivity to ferroptosis [7]. Certain cancer cells, such as pancreatic cancer cell lines (MIA PaCa-2, PANC-1, and BxPC-3) and a subset of triple-negative cancer cells also greatly depend on system Xc− to mediate cysteine uptake for growth, as well as survival under oxidative stress conditions [24,25], suggesting that system Xc− might serve as a good chemotherapeutic target. These results delineate the potential of ferroptotic process in clinicalˇˇ
ˇˇ
Few irons in the cytosol are deposited in the labile iron pool where the redox-active iron (Fe2+) is oxidized by hydrogen peroxide to Fe3+ (ferric) and therefore generates ROS, including soluble radical (HO⋅), lipid alkoxy (RO⋅), and hydroxide ion (OH−) via the Fenton reaction. Free iron in the redox-active form is easily accessed as a pro-oxidant [49]. Thus, the NPBI system is crucial for iron overload to induce the lipid peroxidation [52,53]. Overloading of iron can promote the Fenton reaction and ROS generation [49]. Excessive ROS production consequently results in the peroxidation of adjacent lipid and oxidative damage of DNA and proteins, eventually inducing ferroptosis [6,7,18,19,20].
Cellular iron homeostasis and distribution are regulated by specific iron-regulating proteins. At a low iron level, these regulatory proteins can bind to the iron-response element of target genes to inhibit the expression of iron-binding proteins, such as ferritin and ferroportin, but increase the expression of transferrin receptor [56]. Enhanced HO-1 activity was shown to increase the cellular iron level and also promote ferritin production to sequester iron and following pro-oxidant effects in the meanwhile [33,57]. However, the iron-binding ability of ferritin is disturbed by oxidative stress, causing an uncontrolled release of iron, finally resulting in excessive accumulation of iron in the cytosol and enhancement of lipid peroxidation. The importance of iron in ferroptosis was thoroughly confirmed by increased iron uptake and the presence of iron chelators, such as deferoxamine and ciclopirox, and deferiprone [6,18,19,20]. Similarly, supplementation with an exogenous source of iron, such as ferric ammonium citrate, ferric citrate, and iron chloride hexahydrate enhanced ferroptosis [6]. Activation of iron metabolism-related proteins also contributes to ferroptosis. Upregulation of transferrin receptor 1 resulted in a higher sensitivity to ferroptosis induction [58,59]. The iron-response element binding protein 2 (IREB2) is essential for erastin-induced ferroptosis [6]. Moreover, a decrease in ferritin for iron storage may cause a free iron overload [58], thereby enhancing ferroptosis induction by stimulatory agents [6]. Increased degradation of NCOA4, a receptor cargo protein of ferritin, resulted in ferritinophagy and ferritin degradation, leading to an increase in free iron and ROS generation [60].
Ferroptosis induction by HO-1 is mediated by iron augmentation and lipid peroxidation [15,16,17]. The induction is tightly associated with oxidative stress, due to its predominant sensitivity to oxidative inducers, such as redox-active iron, heme, hemoglobin, and heme-containing proteins [61,62]. The upregulation of HO-1 can enhance heme degradation and ferritin synthesis and change the intracellular iron distribution [57].ˇˇ
IJMS | Free Full-Text | A Dual Role of Heme Oxygenase-1 in Cancer Cells | HTML
https://www.mdpi.com/1422-0067/20/1/39/htmˇˇ
Proc Natl Acad Sci U S A. 1999 Jun 8; 96(12): 6751¨C6756.
Cell Biology
Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: The role of ferritin
ABSTRACT
In mammalian cells, the level of the iron-storage protein ferritin (Ft) is tightly controlled by the iron-regulatory protein-1 (IRP-1) at the posttranscriptional level. This regulation prevents iron acting as a catalyst in reactions between reactive oxygen species and biomolecules. The ultraviolet A (UVA) radiation component of sunlight (320¨C400 nm) has been shown to be a source of oxidative stress to skin via generation of reactive oxygen species. We report here that the exposure of human primary skin fibroblasts, FEK4, to UVA radiation causes an immediate release of ˇ°freeˇ± iron in the cells via proteolysis of Ft. Within minutes of exposure to a range of doses of UVA at natural exposure levels, the binding activity of IRP-1, as well as Ft levels, decreases in a dose-dependent manner. This decrease coincides with a significant leakage of the lysosomal components into the cytosol. Stabilization of Ft molecules occurs only when cells are pretreated with lysosomal protease inhibitors after UVA treatment. We propose that the oxidative damage to lysosomes that leads to Ft degradation and the consequent rapid release of potentially harmful ˇ°freeˇ± iron to the cytosol might be a major factor in UVA-induced damage to the skin.
Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: The role of ferritin
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC21987/ˇˇ
ˇˇ
RESEARCH ARTICLE| MAY 24 2001
Intracellular ascorbic acid enhances the DNA single-strand breakage and toxicity induced by peroxynitrite in U937 cells
Biochem J (2001) 356 (2): 509-513.
A well-established protocol to increase the intracellular content of ascorbic acid was used to investigate the effects of the vitamin on DNA single-strand breakage and toxicity mediated by authentic peroxynitrite (ONOO−) in U937 cells. This protocol involved exposure for 60min to 100¦ĚM dehydroascorbic acid, which was taken up by the cells and converted into ascorbic acid via a GSH-independent mechanism. At the time of exposure to ONOO−, which was performed in fresh saline immediately after loading with dehydroascorbic acid, the vitamin present in the cells was all in its reduced form. It was found that, in cells that are otherwise ascorbate-deficient, an increase in their ascorbic acid content does not prevent, but rather enhances, the DNA-damaging and lethal responses mediated by exogenous ONOO−. These results therefore suggest that acute supplementation of ascorbic acid can be detrimental for individuals with pathologies associated with a decrease in ascorbic acid and in which ONOO− is known to promote deleterious effects.
Intracellular ascorbic acid enhances the DNA single-strand breakage and toxicity induced by peroxynitrite in U937 cells | Biochemical Journal | Portland Press
https://portlandpress.com/biochemj/article-abstract/356/2/509/39253/Intracellular-ascorbic-acid-enhances-the-DNA?redirectedFrom=fulltextˇˇ
J Neurochem. 2014 May;129(4):663-71. doi: 10.1111/jnc.12663. Epub 2014 Feb 19.
The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism.
Cisternas P1, Silva-Alvarez C, Mart¨Şnez F, Fernandez E, Ferrada L, Oyarce K, Salazar K, Bolaños JP, Nualart F.
Author information
1
Laboratorio de Neurobiolog¨Şa, Departamento de Biolog¨Şa Celular, Centro de Microscop¨Şa Avanzada CMA BIOBIO, Facultad de Ciencias Biol¨®gicas, Universidad de Concepci¨®n, Concepci¨®n, Chile.
Abstract
Vitamin C is an essential factor for neuronal function and survival, existing in two redox states, ascorbic acid (AA), and its oxidized form, dehydroascorbic acid (DHA). Here, we show uptake of both AA and DHA by primary cultures of rat brain cortical neurons. Moreover, we show that most intracellular AA was rapidly oxidized to DHA. Intracellular DHA induced a rapid and dramatic decrease in reduced glutathione that was immediately followed by a spontaneous recovery. This transient decrease in glutathione oxidation was preceded by an increase in the rate of glucose oxidation through the pentose phosphate pathway (PPP), and a concomitant decrease in glucose oxidation through glycolysis. DHA stimulated the activity of glucose-6-phosphate dehydrogenase, the rate-limiting enzyme of the PPP. Furthermore, we found that DHA stimulated the rate of lactate uptake by neurons in a time- and dose-dependent manner. Thus, DHA is a novel modulator of neuronal energy metabolism by facilitating the utilization of glucose through the PPP for antioxidant purposes.
© 2014 International Society for Neurochemistry.
The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/24460956ˇˇ
Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1‑phase arrest
Authors: Woo Hyun Park
Affiliations: Department of Physiology, Medical School, Research Institute for Endocrine Sciences, Chonbuk National University, Jeonju, Jeollabuk 54907, Republic of Korea
Abstract
Hydrogen peroxide (H2O2) is frequently applied to cultured cells to induce oxidative stress. The present study investigated the molecular and cellular effects of exogenous H2O2 on Calu‑6 and A549 lung cancer cells. Based on MTT assays, H2O2 inhibited the growth of Calu‑6 and A549 cells with IC50 values of ~50 and 100 µM at 24 h, respectively. Cells treated with H2O2 demonstrated a considerable G1‑phase arrest of the cell cycle. H2O2 dose‑dependently augmented the numbers of dead (trypan blue‑positive) and Annexin V‑FITC‑stained cells in these cells, which was accompanied by the reduction of Bcl‑2 and pro‑caspase‑3 levels, as well as the upregulation of caspase‑3 and ‑8 activities. In addition, H2O2 triggered the failure of mitochondrial membrane potential (MMP; ¦¤¦·m). However, relatively higher doses of H2O2 did not raise the percentages of sub‑G1 cells in these cell lines. All the tested caspase inhibitors (Z‑VAD for pan‑caspases, Z‑DEVD for caspase‑3, Z‑IETD for caspase‑8 and Z‑LEHD for caspase‑9) decreased the percentages of sub‑G1 and Annexin V‑FITC‑stained cells in the H2O2‑treated Calu‑6 and A549 cells. However, caspase inhibitors did not significantly prevent the loss of MMP (¦¤¦·m) in H2O2‑treated lung cancer cells. In conclusion, H2O2 inhibited the growth of Calu‑6 and A549 lung cancer cells through cell death and G1‑phase arrest. H2O2‑induced cell death resulted from necrosis, as well as caspase‑dependent apoptosis.
ˇˇ
Figure 7. - Schematic diagram representing the proposed cellular effects of H2O2 in lung cancer cells. H2O2, hydrogen peroxide; MMP (¦¤¦·m), mitochondrial membrane potential.Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1‑phase arrest
https://www.spandidos-publications.com/or/40/3/1787ˇˇ
ˇˇ
Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo
Qi Chen, Michael Graham Espey, Andrew Y. Sun, Je-Hyuk Lee, Murali C. Krishna, Emily Shacter, Peter L. Choyke, Chaya Pooput, Kenneth L. Kirk, Garry R. Buettner, and Mark Levine
PNAS May 22, 2007 104 (21) 8749-8754; https://doi.org/10.1073/pnas.0702854104
Communicated by E. R. Stadtman, National Institutes of Health, Bethesda, MD, March 27, 2007 (received for review January 31, 2007)H2O2 Formation in Extracellular Fluid with Parenteral Administration of Ascorbate.
ˇˇ
With in vivo validation of ascorbate as a prodrug for selective H2O2 formation, we can now suggest mechanisms to account for selective ascorbate action. In vitro data indicate that external pharmacologic ascorbate concentrations are required for external H2O2 formation (14). We propose that external H2O2 formed from pharmacologic ascorbate concentrations diffuses into cells (31) and mediates toxicity in sensitive cells by ATP depletion (23) via one or more of three pathways (Fig. 5). First, H2O2 may cause DNA single-strand breaks, repaired by polyADP-ribose polymerase (PARP). Enhanced PARP activity may deplete NAD+, resulting in ATP depletion (27, 29). Second, H2O2 removal within cells may be mediated in part by glutathione (GSH) peroxidase. GSH peroxidase has an essential requirement for GSH, which, upon enzyme activity, is oxidized to GSH disulfide (GSSG). GSSG is regenerated to GSH with reducing equivalents from NADPH, which in turn is regenerated from glucose via the pentose shunt. Glucose used to reduce NADP+ to NADPH is not available for ATP generation (26). In cancer cells that depend on anaerobic metabolism for ATP generation (the Warburg effect), loss of glucose to the pentose shunt may result in decreased ATP, leading to cell death (32¨C35). Third, mitochondria in some cancer cells may have increased sensitivity to H2O2 (28, 34, 36). Mitochondria in such cells may be less efficient at baseline in generating ATP compared with normal cells. Enhanced mitochondrial sensitivity to H2O2, with or without inefficient generation of ATP at baseline, may result in decreased ATP production. These pathways for ATP depletion induced by H2O2 are independent, and more than one could be responsible for cell death in sensitive cells (28, 34). Pharmacologic ascorbate concentrations should not impair normal cells because their primary ATP generation is via aerobic metabolism and because their mitochondria may not be as sensitive to H2O2 as those in some cancer cells.
ˇˇ
Fig. 5.
Pharmacologic ascorbic acid concentrations: mechanisms for selective cell death. Pharmacologic ascorbic acid concentrations produce extracellular H2O2, which can diffuse into cells, deplete ATP in sensitive cells, and thereby cause cell death. ATP may be depleted by three mechanisms. (i) DNA damage induced by H2O2 activates PARP. Activated PARP catabolizes NAD+, thereby depleting substrate for NADH formation and consequent ATP synthesis. (ii) H2O2 is catabolized by concurrent oxidation of GSH to GSSG. To reduce GSSG back to GSH, GSH reductase utilizes NADPH, which is provided by the pentose shunt from glucose. Glucose used to reduce NADP+ to NADPH cannot be used for glycolysis or NADH production so that ATP generation is decreased. (iii) H2O2 may directly damage mitochondria, especially ATP synthase, so that ATP production falls. Some cancer cells rely primarily on glycolysis rather than on oxidative phosphorylation respiration for ATP production (the Warburg effect). Compared with oxidative phosphorylation, ATP generation by glycolysis is inefficient. In glycolysis-dependent cancer cells, decreased glycolysis may lower intracellular ATP. Cancer cells that are glycolysis-dependent may be particularly sensitive to pharmacologic ascorbic acid concentrations, compared with cells that use oxidative phosphorylation. See text for additional details.ˇˇ
Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo | PNAS
https://www.pnas.org/content/104/21/8749ˇˇ
Ascorbic Acid and a Cytostatic Inhibitor of Glycolysis Synergistically Induce Apoptosis in Non-Small Cell Lung Cancer Cells
Published: June 11, 2013
Owensboro Cancer Research Program, Owensboro, Kentucky, United States of America
University of Louisville, Louisville, Kentucky
Ascorbic acid (AA) exhibits significant anticancer activity at pharmacologic doses achievable by parenteral administration that have minimal effects on normal cells. Thus, AA has potential uses as a chemotherapeutic agent alone or in combination with other therapeutics that specifically target cancer-cell metabolism. We compared the effects of AA and combinations of AA with the glycolysis inhibitor 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3-PO) on the viability of three non-small cell lung cancer (NSCLC) cell lines to the effects on an immortalized lung epithelial cell line. AA concentrations of 0.5 to 5 mM caused a complete loss of viability in all NSCLC lines compared to a <10% loss of viability in the lung epithelial cell line. Combinations of AA and 3-PO synergistically enhanced cell death in all NSCLC cell lines at concentrations well below the IC50 concentrations for each compound alone. A synergistic interaction was not observed in combination treatments of lung epithelial cells and combination treatments that caused a complete loss of viability in NSCLC cells had modest effects on normal lung cell viability and reactive oxygen species (ROS) levels. Combination treatments induced dramatically higher ROS levels compared to treatment with AA and 3-PO alone in NSCLC cells and combination-induced cell death was inhibited by addition of catalase to the medium. Analyses of DNA fragmentation, poly (ADP-ribose) polymerase cleavage, annexin V-binding, and caspase activity demonstrated that AA-induced cell death is caused via the activation of apoptosis and that the combination treatments caused a synergistic induction of apoptosis. These results demonstrate the effectiveness of AA against NSCLC cells and that combinations of AA with 3-PO synergistically induce apoptosis via a ROS-dependent mechanism. These results support further evaluation of pharmacologic concentrations of AA as an adjuvant treatment for NSCLC and that combination of AA with glycolysis inhibitors may be a promising therapy for the treatment of NSCLC.
Ascorbic Acid and a Cytostatic Inhibitor of Glycolysis Synergistically Induce Apoptosis in Non-Small Cell Lung Cancer Cells
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0067081ˇˇ
ˇˇ
Mol Biol Cell. 2013 Mar 15; 24(6): 858¨C869.
Regulation of apoptosis by Bcl-2 cysteine oxidation in human lung epithelial cells
Sudjit Luanpitpong,a Pithi Chanvorachote,b Christian Stehlik,c William Tse,d Patrick S. Callery,a Liying Wang,e and Yon Rojanasakula,d,1
Kunxin Luo, Monitoring Editor
University of California, Berkeley
ABSTRACT
Hydrogen peroxide is a key mediator of oxidative stress known to be important in various cellular processes, including apoptosis. B-cell lymphoma-2 (Bcl-2) is an oxidative stress¨Cresponsive protein and a key regulator of apoptosis; however, the underlying mechanisms of oxidative regulation of Bcl-2 are not well understood. The present study investigates the direct effect of H2O2 on Bcl-2 cysteine oxidation as a potential mechanism of apoptosis regulation. Exposure of human lung epithelial cells to H2O2 induces apoptosis concomitant with cysteine oxidation and down-regulation of Bcl-2. Inhibition of Bcl-2 oxidation by antioxidants or by site-directed mutagenesis of Bcl-2 at Cys-158 and Cys-229 abrogates the effects of H2O2 on Bcl-2 and apoptosis. Immunoprecipitation and confocal microscopic studies show that Bcl-2 interacts with mitogen-activated protein kinase (extracellular signal-regulated kinase 1/2 [ERK1/2]) to suppress apoptosis and that this interaction is modulated by cysteine oxidation of Bcl-2. The H2O2-induced Bcl-2 cysteine oxidation interferes with Bcl-2 and ERK1/2 interaction. Mutation of the cysteine residues inhibits the disruption of Bcl-2¨CERK complex, as well as the induction of apoptosis by H2O2. Taken together, these results demonstrate the critical role of Bcl-2 cysteine oxidation in the regulation of apoptosis through ERK signaling. This new finding reveals crucial redox regulatory mechanisms that control the antiapoptotic function of Bcl-2.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3596255/
ˇˇ
NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4692134
Dec 20, 2015 ˇ¤ NADPH oxidases (NOXs) and mitochondria are the major source of ROS in vascular cells in both normal and pathological conditions. Depending on the cell type, NOX1, NOX2, and NOX4 homologs together with p22phox make up the membrane-bound catalytic core .
Mitochondrial dysfunction and oxidative stress in heart ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6923355
Dec 19, 2019 ˇ¤ NOXs are a family of proteins involved in intracellular ROS production and catalyze the transfer of NADPH to molecular oxygen to generate superoxide and hydrogen peroxide. Of particular interest is NOX4, which partially localizes to the mitochondrial inner membrane in cardiomyocytes and renal cells 20¨C22.ˇˇ
ˇˇ
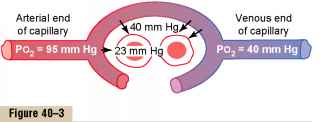

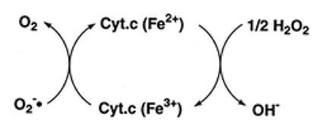
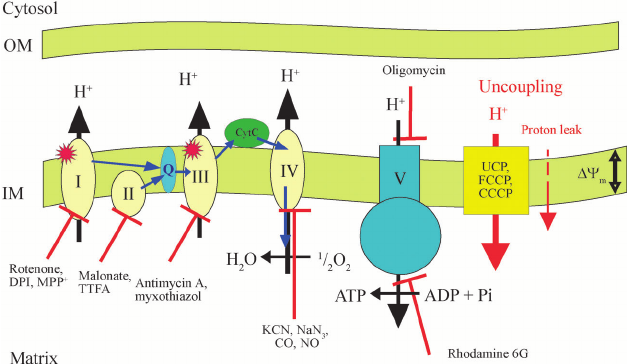
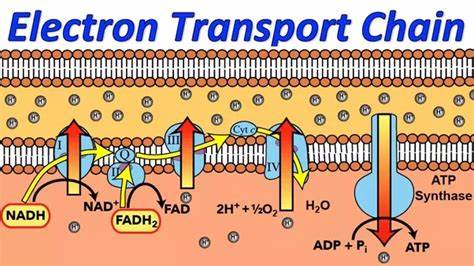

.png)
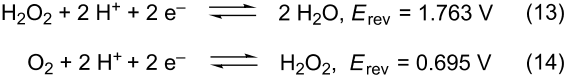

.png)

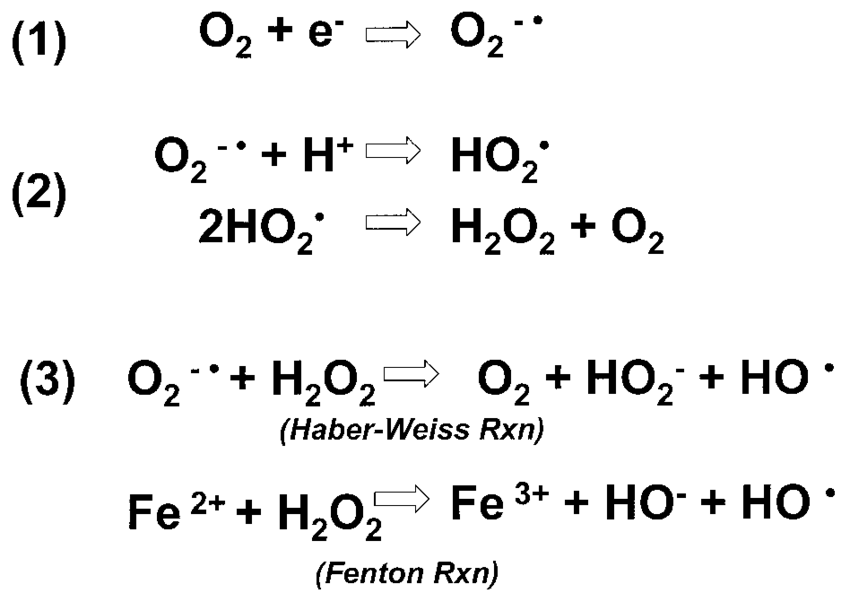
.jpg)
.png)
.png)
.png)

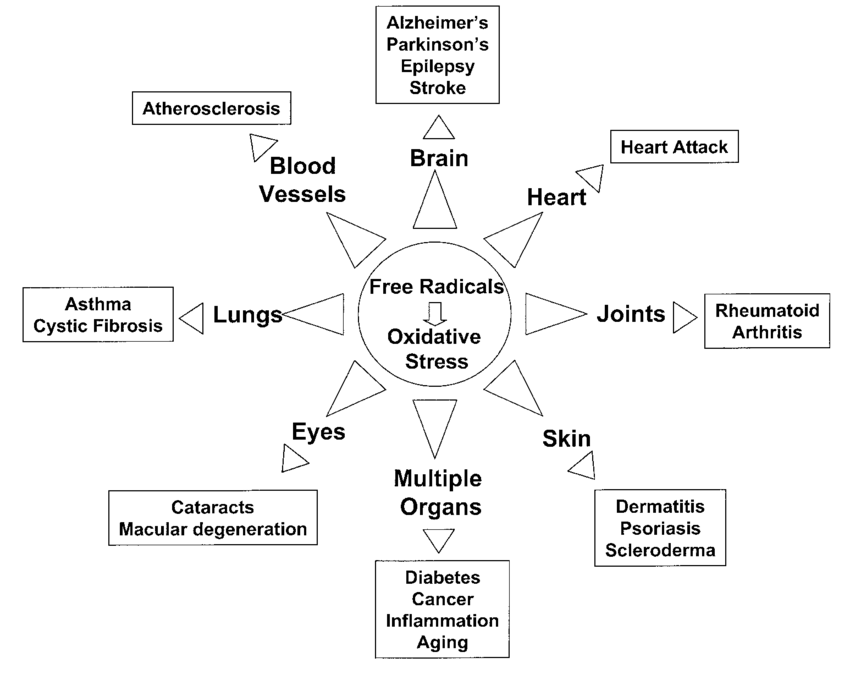
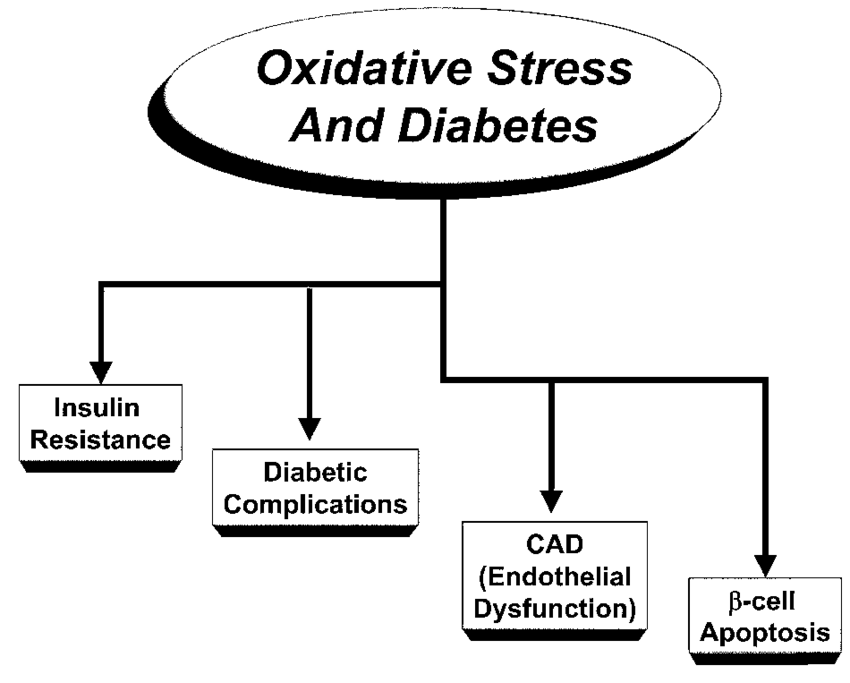

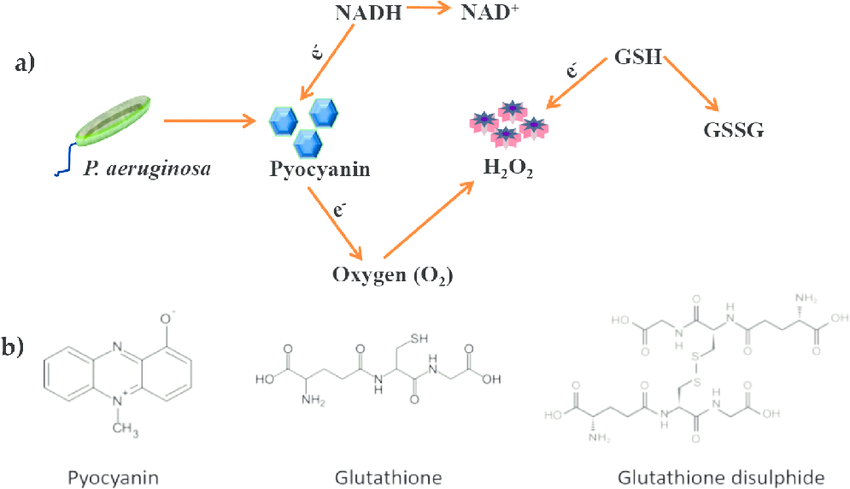
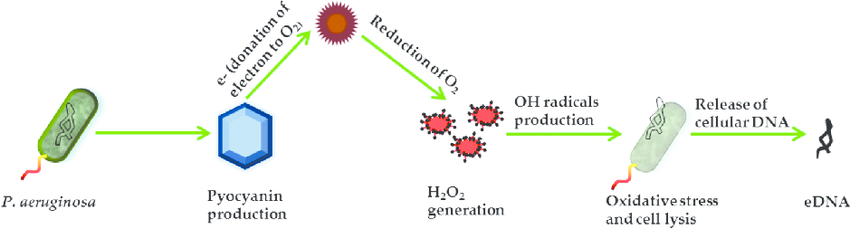

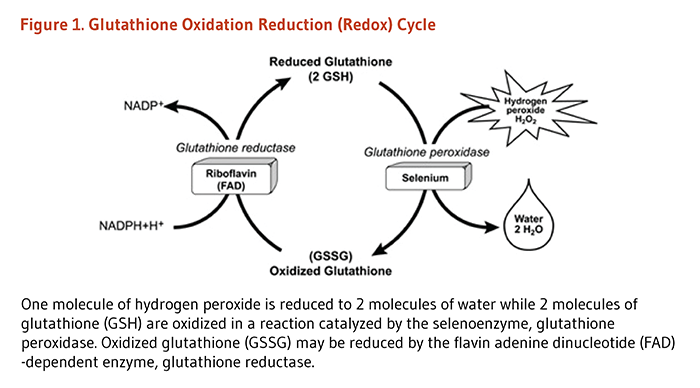
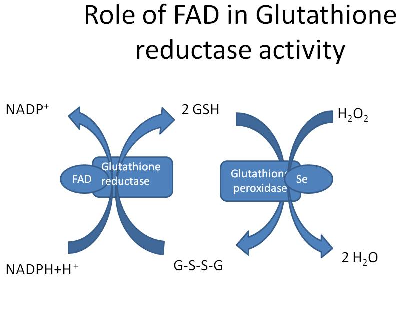
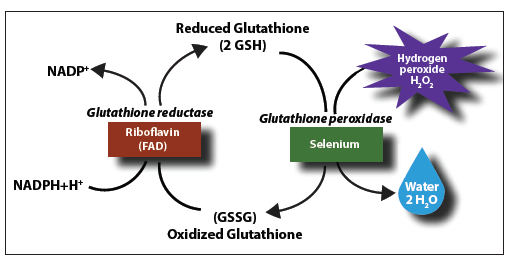
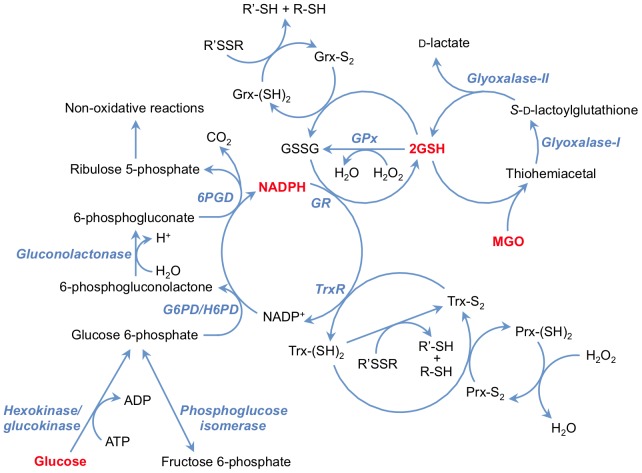
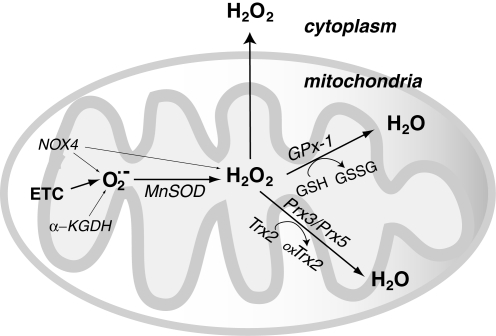
.jpg)