癌症的光动力疗法 最新进展
PHOTODYNAMIC THERAPY OF CANCER: AN UPDATE
Abstract
Photodynamic therapy (PDT) is a clinically approved, minimally invasive therapeutic procedure that can exert a selective cytotoxic activity toward malignant cells. The procedure involves administration of a photosensitizing agent followed by irradiation at a wavelength corresponding to an absorbance band of the sensitizer. In the presence of oxygen, a series of events lead to direct tumor cell death, damage to the microvasculature and induction of a local inflammatory reaction. Clinical studies revealed that PDT can be curative particularly in early-stage tumors. It can prolong survival in inoperable cancers and significantly improve quality of life. Minimal normal tissue toxicity, negligible systemic effects, greatly reduced long-term morbidity, lack of intrinsic or acquired resistance mechanisms, and excellent cosmetic as well as organ function-sparing effects of this treatment make it a valuable therapeutic option for combination treatments. With a number of recent technological improvements, PDT has the potential to become integrated into the mainstream of cancer treatment.
Keywords: photodynamic therapy, laser, photosensitizer, cancer, singlet oxygen
Despite progress in basic research that has given us a better understanding of tumor biology and led to design of new generations of targeted drugs, recent large clinical trials for cancer, with some notable exceptions, have been able to detect only small differences in treatment outcomes.1-2 Moreover, the number of new clinically approved drugs is disappointingly low.3 These sobering facts indicate that to make further progress it is necessary to put an emphasis on other existing but still underappreciated therapeutic approaches. Photodynamic therapy (PDT) has the potential to meet many currently unmet medical needs. Although still emerging, it is already a successful and clinically approved therapeutic modality used for the management of neoplastic and non-malignant diseases. PDT was the first drug-device combination approved by the FDA almost two decades ago, but even so remains underutilized clinically.
PDT consists of three essential components - photosensitizer (PS, see Table 1 for the definitions of specialty terms), light and oxygen.4-5 None of these is individually toxic, but together they initiate a photochemical reaction that culminates in the generation of a highly-reactive product termed singlet oxygen (1O2, Table 1). The latter can rapidly cause significant toxicity leading to cell death via apoptosis or necrosis. Antitumor effects of PDT derive from three interrelated mechanisms - direct cytotoxic effects on tumor cells, damage to the tumor vasculature and induction of a robust inflammatory reaction that can lead to development of systemic immunity. The relative contribution of these mechanisms depends to a large extent on the type and dose of PS used, time between PS administration and light exposure, total light dose and its fluence rate (Table 1), tumor oxygen concentration and perhaps other still poorly recognized variables. Therefore, determination of optimal conditions for using PDT requires a coordinated interdisciplinary effort. This review will address the most important biological and physico-chemical aspects of PDT, summarize its clinical status and provide an outlook for its potential future development.
Table 1
Glossary of specialty terms
Specialty term Definition
Chaperone a protein that participates in the folding of newly synthesized or unfolded proteins into
a particular three-dimensional conformation
Damage-associated
molecular patterns
(DAMPs) intracellular proteins that, when released outside the cell following its injury, can
initiate or sustain an immune response in the noninfectious inflammatory response
Fluence rate the number of particles that intersect a unit area in a given amount of time (typically
measured in Watts per m2)
Fluorescence guided
resection a technique to enhance contrast of viable tumor borders that uses fluorescence emission
from tissue. Fluorescence can be enhanced by the addition of exogenous chromophores
(such as photosensitizers), with specific absorption and fluorescence properties
Ground state a state of elementary particles with the least possible energy in a physical system. This
is the usual (singlet) state of most molecules. One of the exceptions includes oxygen,
which in its ground state is a triplet and can be converted to a higher energy state of
singlet oxygen during PDT
Immunocompromised mice animals having an immune system that has been impaired by genetic modification,
disease or treatment
Immunocompetent mice animals having intact, i.e. normally functioning immune system
Intersystem crossing a radiationless process in which a singlet excited electronic state makes a transition to a
triplet excited state
Macromolecular
therapeutics proteins such as antibodies and growth factors for cell surface targeting, peptides and
mRNA for cancer vaccination, nucleotides for gene delivery and silencing as well as
drug moieties such as polymers and nanoparticles for delivery of therapeutics
Major histocompatibility
complex class I molecules transmembrane glycoproteins that bind short 8-11 amino-acid long peptides recognized
by T cell receptors
Naïve mice nonimmunized animals, i.e. those that were not previously exposed to a particular
antigen (such as tumor-associated antigen)
Pathogen-associated
molecular patterns
(PAMPs) evolutionary conserved microbial molecules that are not normally produced by
mammalian cells and are often common to whole classes of microorganisms. PAMPs
are recognized by pattern-recognition receptors
Pattern-recognition
receptors receptors that participate in the detection of pathogen-associated molecules and initiate
signaling cascades that triggering innate immune response
Photosensitizer a light-absorbing compound that initiates a photochemical or photophysical reaction
Singlet oxygen (1O2) an excited or energized form of molecular oxygen characterized by the opposite spin of
a pair of electrons, which is less stable and more reactive than the normal triplet oxygen
(O2)
Triplet state a state of a molecule or a free radical in which there are two unpaired electrons
Ubiquitin-proteasome
system the major intracellular pathway for protein degradation
Open in a separate window
Go to:
BASIC COMPONENTS OF PHOTODYNAMIC THERAPY
Photodynamic therapy is a two-stage procedure. Following administration of a light-sensitive PS tumor loci are irradiated with a light of appropriate wavelength. The latter can be delivered to virtually any organ in the body by means of flexible fiber-optic devices (Fig. 1). Selectivity is derived from both, the ability of useful photosensitizers to localize in neoplastic lesions and the precise delivery of light to the treated sites. Paradoxically, the highly localized nature of PDT is one of its current limitations, as the treatment is ineffective against metastatic lesions which are the most frequent cause of death in cancer patients. Ongoing research is focused on finding optimal PDT conditions to induce systemic immunity which might, at least to some extent, obviate this limitation in the future. PDT can be used either before or after chemotherapy, radiotherapy or surgery without compromising these therapeutic modalities. None of the clinically approved PSs accumulate in cells’ nuclei, limiting DNA damage that could be carcinogenic or lead to development of resistant clones. Moreover, the adverse effects of chemotherapy or radiation are absent. Radio- or chemoresistance do not affect sensitivity to PDT. Excellent cosmetic outcomes make PDT suitable for patients with skin cancers. There are no significant changes in tissue temperature and the preservation of connective tissue leads to minimal fibrosis, allowing retention of functional anatomy and mechanical integrity of hollow organs undergoing PDT. Selected patients with inoperable tumors, who have exhausted other treatment options, can also achieve improvement in quality of life with PDT. Finally, many PDT procedures can be performed in an out-patient or ambulatory setting thereby not only alleviating costs, but also making the treatment patient-friendly. The only adverse effects of PDT relate to pain during some treatment protocols and a persistent skin photosensitization that has been circumvented by the newer agents.
Figure 1
The principles of PDT
A photosensitizer (PS) is administered systemically or topically. After a period of systemic PS distribution it selectively accumulates in the tumor. Irradiation activates the PS and in the presence of molecular oxygen triggers a photochemical reaction that culminates in the production of 1O2. Irreparable damage to cellular macromolecules leads to tumor cell death via an apoptotic, necrotic or autophagic mechanism, accompanied by induction of an acute local inflammatory reaction that participates in the removal of dead cells, restoration of normal tissue homeostasis and sometimes in the development of systemic immunity.
Photosensitizers
Most of the photosensitizers used in cancer therapy are based on a tetrapyrrole structure, similar to that of the protoporphyrin contained in hemoglobin. An ideal photosensitizing agent should be a single pure compound to allow quality control analysis with low manufacturing costs and good stability in storage. It should have a high absorption peak between 600 and 800-nm (red to deep red) as absorption of photons with wavelengths longer than 800-nm does not provide enough energy to excite oxygen to its singlet state, and the capacity for forming a substantial yield of reactive oxygen species upon irradiation. Since the penetration of light into tissue increases with its wavelength, agents with strong absorbance in the deep red such as chlorins, bacteriochlorins and phthalocyanines offer improvement in tumor control. It should have no dark toxicity and relatively rapid clearance from normal tissues, thereby minimizing phototoxic side-effects. Other pertinent desirable properties of photosensitizing agents have been summarized elsewhere.6 While the interval between drug administration and irradiation is usually long, so that the sensitizer is given sufficient time to diffuse from normal tissues, reports now suggest that the tumor response may be sometimes better when light is delivered at a shorter drug-light interval when PS is still present in the blood vessels, thus producing marked vascular damage.7 Some reports suggest that a pronounced inflammatory response and necrotic cell death after illumination is important in the immune-stimulating function of PDT, while others suggest that PSs that produce more apoptosis and less inflammation are suitable for applications such as brain tumors where swelling is undesirable. Recent findings show that certain PDT-induced apoptotic cell death mechanisms are highly immunogenic and capable of driving antitumor immunity as well.8 Finally the light-mediated destruction of the PS known as photobleaching was previously thought to be undesirable, but some reports suggest that this property may make light dosimetry less critical as over-treatment is avoided when the PS is destroyed during the illumination.9
The first PS to be clinically employed for cancer therapy was a water-soluble mixture of porphyrins called hematoporphyrin derivative (HPD), a purified form of which later became known as Photofrin. Although Photofrin is still the most widely employed PS, the product has some disadvantages including a long-lasting skin photosensitivity and a relatively low absorbance at 630-nm.While a photodynamic effect can be produced with Photofrin, efficacy would be improved by red-shifting the red absorbance band and increasing the absorbance at the longer wavelengths. There has been a major effort among medicinal chemists to discover second-generation PSs and several hundred compounds have been proposed as potentially useful for anticancer PDT. Table 2 displays the most promising PSs that have been used clinically for cancer PDT (whether approved or in trials). The discovery that 5-aminolevulanic acid (ALA) was a biosynthetic precursor of the PS protoporphyrin IX10 has led to many applications in which ALA or ALA-esters can be topically applied, or administered orally. These are considered to be ‘pro-drugs’, needing to be converted to protoporphyrin to be active photosensitizers. Many hypotheses have been proposed to account for the tumor-localizing properties in PDT.11 These include the preponderance of leaky and tortuous tumor blood vessels due to neovascularization and absence of lymphatic drainage known as the enhanced permeability and retention effect.12 Some of the most effective compounds bind preferentially to low density lipoprotein (LDL) suggesting that upregulated LDL receptors found on tumor cells could be important.13
Table 2
Clinically applied photosensitizers.
Photosensitizer Structure Wavelength
(nm) Approved Trials Cancer types
Photofrin (HPD) porphyrin 630 World wide lung, esophagus, bile duct, bladder, brain,
ovarian
ALA porphyrin
precursor 635 World wide skin, bladder, brain, esophagus
ALA esters porphyrin
precursor 635 Europe skin, bladder
Foscan (mTHPC) Chlorine 652 Europe US head and neck, lung, brain, skin, bile duct
Verteporfin Chlorine 690 World wide
(AMD) UK ophthalmic, pancreatic, skin
HPPH chlorin 665 US head and neck, esophagus, lung
Purlytin (SnEt2) chlorin 660 US skin, breast
Taloporfin, LS11, MACE, Npe6 chlorin 660 US liver, colon, brain
Fotolon (PVP-Ce6), Radachlorin,
Photodithazine chlorin 660 Belarus,
Russia nasopharyngeal, sarcoma, brain
Silicon phthalocyanine (PC4) phthalocyanine 675 US cutaneous T cell lymphoma
Padoporfin (TOOKAD) bacteriochlorin 762 US prostate
Motexafin lutetium (LuTex) texaphyrin 732 US breast
Open in a separate window
There have been targeting studies in which PSs are covalently attached to various molecules that have some affinity for neoplasia or to receptors expressed on specific tumors.14 The intention is to rely on the ability of the targeting vehicle to control localization factors so that the PS can be chosen based on its photochemical properties. These vehicles include monoclonal antibodies, antibody fragments, peptides, proteins such as transferrin, epidermal growth factor and insulin, LDL, various carbohydrates, somatostatin, folic acid and many others.
Light sources
Blue light penetrates least efficiently through tissue while red and infrared radiations penetrate more deeply (Fig. 2). The region between 600 and 1200 nm is often called the optical window of tissue. However, light up to only about 800 nm can generate 1O2, since longer wavelengths have insufficient energy to initiate a photodynamic reaction.15 No single light source is ideal for all PDT indications, even with the same PS. Choice of light source should therefore be based on PS absorption (fluorescence excitation and action spectra), disease (location, size of lesions, accessibility, and tissue characteristics), cost and size. The clinical efficacy of PDT is dependent on complex dosimetry: total light dose, light exposure time, and light delivery mode (single vs. fractionated or even metronomic). The fluence rate also affects PDT response.16 Integrated systems that measure the light distribution and fluence rate either interstitially or on the surface of the tissues being treated are so far used only in experimental studies.
Figure 2
Light propagation through the tissues.
Both lasers and incandescent light sources have been used for PDT and show similar efficacies.17 Unlike the large and inefficient pumped dye lasers, diode lasers are small and cost-effective, are simple to install, have automated dosimetry and calibration features and a longer operational life. Such lasers are now being specifically designed for PDT. Light emitting diodes (LEDs) are alternative light sources with relatively narrow spectral bandwidths and high fluence rates.18-19 Lasers can be coupled into fibers with diffusing tips to treat tumors in the urinary bladder and the digestive tract. Inflatable balloons, covered on the inside with a strongly scattering material, formed to fit an organ, are also commercially available.20 It is quite feasible to implant a light source in solid organs deep in the body under image guidance. The choice of optimal combinations of PSs, light sources, and treatment parameters is crucial for successful PDT.21-22
Photophysics and photochemistry
Most PSs in their ground (i.e. singlet) state (Table 1) have two electrons with opposite spins located in an energetically most favorable molecular orbital. Absorption of light leads to a transfer of one electron to a higher-energy orbital (Fig. 3). This excited PS is very unstable and emits this excess energy as fluorescence and/or heat. Alternatively, an excited PS may undergo an intersystem crossing (Table 1) to form a more stable triplet state (Table 1) with inverted spin of one electron. The photosensitizer in triplet state can either decay radiationlessly to the ground state or transfer its energy to molecular oxygen (O2), which is unique in being a triplet in its ground state. This step leads to the formation of singlet oxygen (1O2), and the reaction is referred to as a Type II process.23 A Type I process can also occur whereby the PS reacts directly with an organic molecule in a cellular microenvironment, acquiring a hydrogen atom or electron to form a radical. Subsequent autoxidation of the reduced PS produces a superoxide anion radical (O2 •-). Dismutation or one-electron reduction of O2 •- gives hydrogen peroxide (H2O2), which in turn can undergo one-electron reduction to a powerful and virtually indiscriminate oxidant - hydroxyl radical (HO•). ROS generation via Type II chemistry is mechanistically much simpler than via Type I, and most PSs are believed to operate via Type II rather than Type I mechanism.
Figure 3
Photosensitization processes illustrated by a modified Jablonski diagram
Light exposure takes a photosensitizer molecule from the ground singlet state (S0) to an excited singlet state (S1). The molecule in S1 may undergo intersystem crossing to an excited triplet state (T1) and then either form radicals via a type 1 reaction or, more likely, transfers its energy to molecular oxygen (3O2) and form singlet oxygen (1O2), which is the major cytotoxic agent involved in PDT.
Mechanisms of PDT-mediated cytotoxicity
The lifetime of singlet oxygen (1O2) is very short (~10-320 ns), limiting its diffusion to only approximately 10-55 nm in cells.24 Thus, photodynamic damage will occur very close to the intracellular location of the PS.25 Photofrin is a complex mixture of porphyrin ethers with variable localization patterns mostly associated with lipid membranes. Of the other photosensitizing agents in current use, the chlorin NPe6 targets lysosomes, the benzoporphyrin derivative (BPD) targets mitochondria, m-tetrahydroxyphenylchlorin (mTHPC) has been reported to target mitochondria, ER or both, the phthalocyanine Pc 4 has a broad spectrum of affinity although mitochondria are reported to be a primary target.6 Other agents that have been developed can have multiple targets. Specific patterns of localization may vary also among different cell types.
PDT can evoke the three main cell death pathways: apoptotic, necrotic and autophagy-associated cell death (Fig. 4). Apoptosis is a generally major cell death modality in cells responding to PDT. Mitochondria outer membrane permeabilization (MOMP) after photodynamic injury is controlled by Bcl-2 family members and thought to be largely p53-independent.26 With mitochondria-associated PSs, photodamage to membrane bound Bcl-227-29 can be a permissive signal for MOMP and the subsequent release of caspase activators, such as cytochrome c and Smac/DIABLO, or other pro-apoptotic molecules, including apoptosis-inducing factor (AIF).26 Lysosomal membrane rupture and leakage of cathepsins from photo-oxidized lysosomes30-31 induces Bid cleavage and MOMP.31
Figure 4
Three major cell death morphotypes and their immunological profiles
Apoptosis is morphologically characterized by chromatin condensation, cleavage of chromosomal DNA into internucleosomal fragments, cell shrinkage, membrane blebbing and formation of apoptotic bodies without plasma membrane breakdown. Typically apoptotic cells release “find me” and “eat me” signals required for the clearance of the remaining corpses by phagocytic cells. At the biochemical level, apoptosis entails the activation of caspases, a highly conserved family of cysteine-dependent aspartate-specific proteases. Necrosis is morphologically characterized by vacuolization of the cytoplasm, swelling and breakdown of the plasma membrane resulting in an inflammatory reaction due to release of cellular contents and pro-inflammatory molecules. Classically, necrosis is thought to be the result of pathological insults or be caused by a bio-energetic catastrophe, ATP depletion to a level incompatible with cell survival. The biochemistry of necrosis is characterized mostly in negative terms by the absence of caspase activation, cytochrome c release and DNA oligonucleosomal fragmentation. Autophagy is characterized by a massive vacuolization of the cytoplasm. Autophagic cytoplasmic degradation requires the formation of a double-membrane structure called the autophagosome, which sequesters cytoplasmic components as well as organelles and traffics them to the lysosomes. Autophagosome-lysosome fusion results in the degradation of the cytoplasmic components by the lysosomal hydrolazes. In adult organisms, autophagy functions as a self-digestion pathway promoting cell survival in an adverse environment and as a quality control mechanism by removing damaged organelles, toxic metabolites or intracellular pathogens.
Phototoxicity is not propagated only through caspase-signaling but involves other proteases, such as calpains, as well as non-apoptotic pathways.26 Typically inhibition or genetic deficiency of caspases only delays phototoxicity or shifts the cell death modality towards necrotic cell death.32 Recent evidence suggests indeed that certain form of necrosis can be propagated through signal transduction pathways.33 The molecular mechanisms underlying programmed necrosis are still elusive, but certain events including activation of RIP1 (receptor interacting protein 1) kinase, excessive mitochondrial ROS production, lysosomal damage and intracellular Ca2+-overload, are recurrently involved.33-34 Severe inner mitochondria membrane photodamage or intracellular Ca2+-overload could promote mitochondrial permeability transition, an event that may favor necrotic rather than apoptotic phototoxicity.26,35
Photodamage of cells can also lead to the stimulation of macroautophagy (hereafter referred to as autophagy).36-37 This is a lysosomal pathway for the degradation and recycling of intracellular proteins and organelles. Autophagy can be stimulated by various stress signals including oxidative stress.38 This process can have both a cytoprotective and a pro-death role following cancer chemotherapy, including those involving ROS as primary damaging agents.38 Recent studies delineate autophagy as a mechanism to preserve cell viability following photodynamic injury.37 PSs that photodamage the lysosomal compartment may compromise completion of the autophagic process, causing incomplete clearance of the autophagic cargo. Accumulation of ROS-damaged cytoplasmic components may then potentiate phototoxicity in apoptosis competent cells.37 A better understanding of the interplay between autophagy, apoptosis and necrosis and how these processes lead to improved tumor response will be a requisite to devise better therapeutic strategies in PDT.
Cytoprotective mechanisms
Numerous publications have reported cytoprotective mechanisms that cancer cells exploit to avoid cytotoxic effect of PDT.26 The first mechanism identified was based on the large variation observed in the level of antioxidant molecules expressed in cancer cells.39 Both water-soluble antioxidants, e.g., some amino-acids, glutathione (GSH) or vitamin C and lipid-soluble antioxidants, e.g., vitamin E are present at variable levels in many cancer cell types explaining the large variation in PDT sensitivity.40 A second mechanism is associated with expression in cancer cells of enzymes that can detoxify ROS. Although there is no specific cellular enzyme that can directly detoxify 1O2, enzymes involved in other ROS metabolism can influence the cytotoxic effect of PDT. For example, superoxide dismutase (SOD) over-expression or treatment with SOD mimetics have been shown to counteract the cytotoxic effect of PDT.41 An increase of the SOD activity has been also observed in various cancer cell types following PDT, and this is associated with a decrease in glutathione peroxidase and catalase activities.42 The third cytoprotective mechanism involves proteins whose encoding genes are themselves induced by PDT. Many categories can be specified but most of them are part of signaling pathways that can regulate PDT-induced apoptosis [see ref 43 for a review] or participate in the repair of lesions induced by oxidative stress. NF-κB inhibition by over-expression of the IκBα super-repressor or by the use of pharmacological inhibitors strongly sensitizes cancer cells to apoptosis induced by PDT.44 Other stress-related transcription factors induced by PDT include AP-1, hypoxia inducible factor (HIF) or Nrf2. PDT was shown to up-regulate heme oxygenase-1 (HO-1) expression and the mechanism is dependent on Nrf2 nuclear accumulation and on p38MAPK and PI-3K activities. Because of the antioxidant activity of HO-1, it can be envisioned that Nrf2-dependent signal transduction can control cellular protection against PDT-mediated cytotoxic effect.
PDT was found to induce expression of various HSPs for which a protective role in PDT has been described. For example, transfection of tumor cells with HSP27 gene increased survival of tumor cells after PDT.45 Similarly, increased HSP60 and HSP70 levels are inversely correlated with sensitivity to the photodynamic treatment.46-47 The simplest explanation for these observations is the ability of HSPs to bind to oxidatively damaged proteins. Moreover, intracellular function of HSPs is not only restricted to protein refolding. Many HSPs “client” proteins play a critical role in the regulation of prosurvival pathways. PDT also leads to increased ubiquitination of carbonylated proteins thereby tagging them for degradation in proteasomes, which prevents formation of toxic protein aggregates.48
Go to:
ANTIVASCULAR EFFECTS OF PDT
Photodynamic perturbation of tissue microcirculation was first reported in 1963.49 A study by W.M. Star et al.50 utilized a window chamber to make direct observations of implanted mammary tumor and in adjacent normal tissue microcirculation in rats before, during, and at various times after PDT sensitized with HPD. An initial blanching and vasoconstriction of the tumor vessels was followed by heterogeneous responses including eventual complete blood flow stasis, hemorrhage, and in some larger vessels, the formation of platelet aggregates. Observations performed on excised tissues from murine models, demonstrated a wide range of vascular responses including disruption of blood flow to subcutaneous urothelial tumors and to normal rat jejunum, breakdown of the blood brain barrier in the normal brain of mice, and endothelial cell and organelle damage in subcutaneous tumors and normal tissue.51-52
Other studies demonstrated that tumor cells treated with a potentially curative photodynamic dose in vivo were clonogenic if removed immediately from the host.53-54 Progressive loss in clonogenicity was seen when tumors were left in the host for increasing durations; this corresponded to progression of PDT-induced hypoxia as determined radio-biologically. Hypoxia sufficient to preclude direct tumor cell killing was identified at sub-curative PDT doses. These studies suggested a central role for vascular damage in governing the tumor response to PDT in mouse models.
Many reports cited above directly implicate the endothelium as a primary target for PDT in vivo; this stimulated research into the relative sensitivity of endothelial cells to PDT and the responses of endothelial cells that could initiate the various phenomena at the vessel level. Gomer et al.55 showed that bovine endothelial cells were significantly more sensitive to Photofrin-PDT than smooth muscle cells or fibroblasts from the same species. This increased sensitivity, assessed by clonogenic assay, was not a result of increased Photofrin accumulation. Sensitivity to HPD-mediated PDT of bovine aorta endothelial cells and human colon adenocarcinoma cells was investigated by West et al.56 Exponentially growing endothelial cells were significantly more sensitive than similarly proliferating tumor cells, and the difference in sensitivity was accompanied by greater PS accumulation in the endothelial cells. Endothelial cell responses to sub-lethal doses of PDT may also contribute to vascular changes observed in tissue.
Increased vessel permeability to albumin in the rat cremaster muscle during and after Photofrin-PDT was reported by Fingar et al.57 More recently, intravital fluorescence imaging has been used to demonstrate treatment-induced increases in tumor vessel permeability for verteporfin- and NPe6-PDT.58-59 In a pioneering study, Synder et al.60 showed that HPPH-PDT induction of increased tumor vascular permeability resulted in enhanced accumulation of Doxil, a liposome-encapsulated formulation of doxorubicin. When Doxil was administered immediately after PDT, tumor control and selectivity were potentiated significantly relative to either modality alone. In a study motivated by the need to deliver chemotherapeutic agents to the brain adjacent to a tumor, ALA-PDT was used successfully to transiently disrupt the blood brain barrier in normal rat brain in vivo.61 These and other aspects of vascular-targeted PDT represent important current research directions.
Go to:
PDT AND THE IMMUNE RESPONSE
Inflammation and innate immunity
PDT frequently provokes a strong acute inflammatory reaction observed as localized edema at the targeted site.4 This reaction is a consequence of PDT-induced oxidative stress. Thus, PDT can be ranked among cancer therapies (including cryotherapy, hyperthermia and focused ultrasound ablation) producing chemical/physical insult in tumor tissue perceived by the host as localized acute trauma. This prompts the host to launch protective actions evolved for dealing with threat to tissue integrity and homeostasis at the affected site.62 The acute inflammatory response is the principal protective effector process engaged in this context. Its main task is containing the disruption of homeostasis, ensure removal of damaged cells, and then promote local healing with restoration of normal tissue function.
The inflammation elicited by PDT is a tumor antigen non-specific process orchestrated by the innate immune system.62 The recognition arm of this system, in particular pattern-recognition receptors (PRRs, Table 1), is responsible for detecting the presence of PDT-inflicted tumor-localized insult revealed to its sensors as the appearance of “altered-self”.62 PDT appears particularly effective in generating rapidly an abundance of alarm/danger signals, also called damage-associated molecular patterns (DAMPs, Table 1) or cell death-associated molecular patterns (CDAMPs), at the treated site that can be detected by the innate immunity alert elements.62
The onset of PDT-induced inflammation is marked by dramatic changes in the tumor vasculature that becomes permeable for blood proteins and proadhesive for inflammatory cells.62 This occurs even with those PSs that mainly target tumor rather than vascular cells, where the inflammatory process is predominantly initiated by signals originating from photooxidative damage produced in perivascular regions with chemotactic gradients reaching the vascular endothelium. The inflammatory cells, led by neutrophils and followed by mast cells and monocytes/macrophages, rapidly and massively invade tumors undergoing PDT (Fig. 5).4,63 Their primary task is to neutralize the source of DAMPs/CDAMPs by eliminating debris containing compromised tissue elements including injured and dead cells. Damage and dysfunction of photodynamically-treated tumor vasculature frequently ends up with vascular occlusion that serves to “wall off” the damaged tumor tissue until it is removed by phagocytosis thereby preventing the spreading of the disrupted homeostasis.62 Depletion of these inflammatory cells or inhibition of their activity after PDT was shown to diminish therapeutic effect.64-67 Among cytokines involved in the regulation of the inflammatory process, the most critical role in tumor PDT response is played by IL-1β and IL-6.68-69 Blocking the function of various adhesion molecules was proven also detrimental to PDT response.68-69 On the other hand, blocking anti-inflammatory cytokines such as IL-10 and TGF-β can markedly improve the cure rates after PDT.62
Figure 5
PDT-induced effects
Light-mediated excitation of photosensitizer-loaded tumor cells leads to production of reactive oxygen species (ROS) within these cells, leading to cell death (predominantly apoptotic and necrotic). Tumor cell kill is further potentiated by damage to the microvasculature (not shown), which further restricts oxygen and nutrient supply. Tumor cell death is accompanied by activation of the complement cascade, secretion of proinflammatory cytokines, rapid recruitment of neutrophils, macrophages and dendritic cells (DCs). Dying tumor cells and tumor cell debris is phagocytosed by phagocytic cells, including DCs, which migrate to the local lymph nodes and differentiate into professional antigen-presenting cells. Tumor antigen presentation within the lymph nodes is followed by clonal expansion of tumor-sensitized lymphocytes that home to the tumor and eliminate residual tumor cells.
PDT and adaptive immunity
Numerous pre-clinical and clinical studies have demonstrated that PDT can influence the adaptive immune response in disparate ways; some regimens result in potentiation of adaptive immunity, while others lead to immunosuppression. The precise mechanism leading to potentiation vs. suppression is unclear; however it appears as though the effect of PDT on the immune system is dependent upon the treatment regimen, the area treated and the photosensitizer type.66,70 PDT induced immune suppression is largely confined to cutaneous and transdermal PDT regimens involving large surface areas.70-71
PDT efficacy appears to be dependent upon the induction of anti-tumor immunity. Long-term tumor response is diminished or absent in immunocompromized mice (Table 1).64,72 Reconstitution of these animals with bone marrow or T cells from immunocompetent mice (Table 1) results in increased PDT efficacy. Clinical PDT efficacy also appears to depend on anti-tumor immunity. Patients with vulval intraepithelial neoplasia (VIN) who did not respond to ALA-PDT were more likely to have tumors that lacked major histocompatibility complex class I molecules (MHC-I, Table 1) than patients who responded to ALA-PDT.73 MHC-I recognition is critical for activation of CD8+ T cells and tumors that lack MHC-I are resistant to cell-mediated anti-tumor immune reactions.74 VIN patients who responded to PDT had increased CD8+ T cell infiltration into the treated tumors as compared to non-responders. Immunosuppressed and immunocompetent actinic keratoses and Bowen’s disease patients had similar initial response rates to PDT; however, immunosuppressed patients exhibited greater persistence of disease or appearance of new lesions.75
Canti et al.76 were the first to show PDT-induced immune potentiation, demonstrating that cells isolated from tumor-draining lymph nodes of PDT-treated mice were able to confer tumor resistance to naïve mice (Table 1). Subsequent studies demonstrated that PDT directed against murine tumors resulted in the generation of immune memory.77 Recent reports have shown that clinical anti-tumor PDT also increases anti-tumor immunity. PDT of multifocal angiosarcoma of the head and neck resulted in increased immune cell infiltration into distant untreated tumors that was accompanied by tumor regression.78 PDT of basal cell carcinoma (BCC) increased immune cell reactivity against a BCC-associated antigen.79
The mechanism whereby PDT enhances anti-tumor immunity has been examined for the past several decades. PDT activates both humoral and cell-mediated anti-tumor immunity, although the importance of the humoral response is unclear. PDT efficacy in mice and humans is reduced in the absence of CD8+ T cell activation and/or tumor infiltration.64,73,80 Therefore most mechanistic studies have focused on the means by which PDT potentiates CD8+ T cell activation. It is clear that induction of anti-tumor immunity following PDT is dependent upon induction of inflammation.81 PDT-induced acute local and systemic inflammation is postulated to culminate in the maturation and activation of dendritic cells (DCs). Mature DCs are critical for activation of tumor specific CD8+ T cells and induction of anti-tumor immunity.82 DCs are activated in response to PDT69 and migrate to tumor draining lymph nodes where they are thought to stimulate T cell activation.69,83 Generation of CD8+ effector and memory T cells is frequently, but not always dependent upon the presence and activation of CD4+ T cells.84 PDT induced anti-tumor immunity may64 or may not depend on CD4+ T cells80 and may be augmented by natural killer (NK) cells.80
PDT-mediated enhancement of anti-tumor immunity is believed to be due, at least in part, to stimulation of DCs by dead and dying tumor cells, suggesting that in vitro PDT-treated tumor cells may act as effective anti-tumor vaccines.85 This hypothesis has been proven by several studies using a wide variety of photosensitizers and tumor models in both preventative and therapeutic settings.67,85-87
Mechanistic studies showed that incubation of immature DCs with PDT-treated tumor cells leads to enhanced DC maturation, activation and increased ability to stimulate T cells.85,88 PDT of tumor cells causes both cell death and cell stress4,89-90 and it is hypothesized that the activation of DCs by PDT-treated cells is the result of recognition of DAMPs/CDAMPs released/secreted/exposed by PDT from dying cells.91-93 HSP70 is a well-characterized DAMP that interacts with the danger signal receptors, TLRs (Toll-like receptors) 2 and 494 and is induced by PDT.95 The level of expression of HSP70 in PDT-treated tumor cells appears to correlate with an ability to stimulate DC maturation96 and initiation of inflammation.92,97 Furthermore, opsonization of photodynamically-treated tumor cells by complement proteins increases the efficacy of PDT-generated vaccines.86 PDT therefore induces multiple danger signals capable of triggering antigen-presenting cell activation and anti-tumor immunity.
The implications of PDT-induced anti-tumor immunity and efficacious PDT-generated vaccines are significant and provide an exciting possibility for using PDT in the treatment of metastatic disease and as an adjuvant in combination with other cancer modalities. Several pre-clinical studies demonstrated that PDT is able to control the growth of tumors present outside the treatment field80,98 although others have failed to demonstrate control of distant disease following PDT.99 PDT was also shown to be in an effective surgical adjuvant in non-small-cell lung cancer patients with pleural spread.101
Go to:
COMBINATIONS OF PDT WITH OTHER THERAPIES
Combinations of various therapeutic modalities with non-overlapping toxicities are among the commonly-used strategies to improve the therapeutic index of treatments in modern oncology. Two general approaches may increase antitumor effectiveness of PDT: (i) sensitization of tumor cells to PDT; and (ii) interference with cytoprotective molecular responses triggered by PDT in surviving tumor or stromal cells. Any interactions between PDT and PDT-sensitizing agents will be confined to the illuminated area. Therefore, the potentiated toxicity of the combinations is not systemic. This should be of special importance in elderly or debilitated patients who tolerate more intensive therapeutic regimes poorly. Moreover, considering its unique 1O2-dependent cytotoxic effects, PDT can be safely combined with other antitumor treatments without the risk of inducing cross-resistance.102
There have been few studies on combinations of PDT with standard antitumor regimens. PDT can be used in combination with surgery as a neoadjuvant, adjuvant or repetitive adjuvant treatment, preferably fluorescence image-guided to confine illumination to the most suspicious lesions. PDT has also been successfully combined with radiotherapy and chemotherapy (Table 3).
Table 3
Combinations of PDT and various therapeutic modalities in cancer treatment – a comprehensive summary.
Drug or treatment modality Outcome / Results
Chemotherapeutics and novel anticancer drugs
Anthracyclines Doxorubicin improves PDT-mediated tumor growth control in mice257
Platinum compounds Cisplatin potentiates antitumor activity of PDT in mice257
Antimetabolites Methotrexate enhances in vitro cytotoxicity of ALA-PDT by up-regulation of protoporphyrin IX production258
Microtubule inhibitors Vincristine administered prior or immediately after PDT improves its antitumor activity in mice259
DNA methyltransferase inhibitors 5-azadeoxycytidine prolongs survival of PDT-treated animals and improves tumor growth control260
Proteasome inhibitors Bortezomib enhances PDT-mediated ER-stress in cancer cells in vitro and significantly delays post-PDT tumor
re-growth in mice48
Radiotherapy
Two-way enhancement of antitumor effects: PDT sensitizes cancer cells to radiotherapy261 and radiotherapy increases anticancer efficacy of PDT,262
prolonged tumor growth control induced by combined treatment212
Drugs modulating arachidonic acid cascade
Cyclooxygenase-2 (COX-2) inhibitors COX-2 inhibitors (such as NS-398109, nimesulid263 or celecoxib264) potentiate antitumor effects of PDT, possibly
through indirect antiangiogenic effects
Lipoxygenase (LOX) inhibitors MK-886, that also serves as a FLAP inhibitor, sensitizes tumor cells to PDT-mediated killing265
Agents increasing photosensitizer accumulation in tumor cells
Vitamin D Increases 5-ALA-induced protoporphyrin IX accumulation and thus potentiates PDT cytotoxicity in vitro266
Imatinib Increases intracellular accumulation of 2nd generation PSs and thus potentiates PDT cytotoxicity in vitro and in
vivo103
Lipid lowering drugs Lovastatin – a HMG-CoA reductase inhibitor improves in vitro LDL binding and Photofrin uptake by cancer
cells267
Salicylate and related drugs Enhancement of PDT efficacy in vitro via increased PS uptake by tumor cells268
Approaches increasing oxygen delivery to tumor cells
Erythropoietin (EPO) EPO improves chemotherapy-induced anemia and restores antitumor efficacy of PDT in mice269, however, EPO
might also inhibit direct PDT-mediated cytotoxicity towards certain cancer cells270
Hyperbaric oxygen Increased antitumor effects of PDT in mice271 and in advanced pleural tumors in humans272
Hyperthermia In various treatment regimens, hyperthermia potentiates antitumor efficacy of PDT in vitro and in animal
models.273 Short time interval between these two treatment modalities might increase normal tissue injury via
vascular effects274
Targeting cytoprotective mechanisms and increasing of radical formation in cancer cells
Disruption of heme degradation pathway Targeting of HO-1 with selective inhibitors107, siRNA275 as well as a siRNA-mediated knockdown of
ferrochelatase275 or chelatation of iron ions276 potentiate antitumor effects of PDT
Inhibition of superoxide dismutase 2-methoxyestradiol, a natural SOD inhibitor enhances PDT cytotoxicity in vitro and improves antitumor effects of
PDT in mice41
NO synthase inhibition Improved tumor response to PDT in mice108
HSP90 modulation Interference with HSP90 client proteins binding using a geldanamycin derivative improves responsiveness to
PDT both in vitro and in vivo106
Lowering cellular glutathione content Depleting GSH levels in tumor cells using buthionine sulfoximine significantly enhances PDT efficacy in vitro and
in vivo277
Vitamin E and its analogues α-tocopherol-mediated radical production enhances PDT toxicity in vitro and in vivo278
Targeting of tumor vasculature
Antiangiogenic treatment Anti-VEGF279 or anti-VEGFR280 monoclonal antibodies, matrix metalloproteinase inhibitor (prinomastat)281, TNP-
470282 and other anti-angiogenic agents110,283 as well as adenovirus-driven IL-12 expression284 potentiate
antitumor effects of PDT in mice
Apoptosis promotion or G1 cell cycle inhibition in PDT-treated cells
Bcl-2 antagonist synergizes with PDT in in vitro cytotoxicity285
Ursodeoxycholic acid sensitizes mitochondrial membranes in tumor cells to PDT-mediated damage286
A ceramide analogue delays tumor re-growth post PDT in mice287
Rapamycin (a mTOR inhibitor) delivered post PDT enhances its in vitro cytotoxicity288
Other approaches
Combinations of two different
photosensitizers 5-ALA- and low dose Photofrin-PDT show enhanced antitumor efficacy in vitro and in vivo with no risk of
prolonged skin photosensitivity113
BPD- and benzothiazine-PDT synergize in antitumor activity in vitro and in vivo289
Hypoxia-activated bioreductive drugs Improved tumor response to PDT in mice exposed to mitomycin C290
Open in a separate window
Abbreviations used: 5-ALA, 5-aminolaevulinic acid; BPD, benzoporphyrin derivative; COX, cyclooxygenase; EPO, erythropoietin; FLAP, 5-lipoxygenase activating protein; GSH, glutathione; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; HO-1, heme oxygenase-1; HSP, heat shock protein; LOX, lipoxygenase; mTOR, mammalian target of rapamycin; PS, photosensitizer; SOD, superoxide dismutase; VEGFR, vascular endothelial growth factor receptor.
Another approach to promote PDT efficacy involves increased PS delivery or impaired loss from tumor cells. The first approach involves conjugation of PS to various tumor-targeting molecules as is described above. This may be important in the treatment of tumors where large surface areas are illuminated and hence increased tumor selectivity is desired14, e.g. superficial spreading bladder cancer or metastases to peritoneum and pleural cavity. The use of compounds that impair PS efflux has also been demonstrated to effectively sensitize tumor cells to PDT, although such approaches seem to be limited to those PSs that are the substrates of outward transport systems such as ABCG2.103 Another approach involves increased conversion of ALA or its esters into protoporphyrin IX by iron-chelating agents.104
Development of novel target-specific antitumor drugs has enabled examination of a number of concept-based combinations that in various molecular mechanisms sensitize tumor cells to cytotoxic effects of PDT. Proteins are major targets for oxidative reactions as they constitute nearly 70% of the dry weight of cells. Oxidized proteins can be re-folded by molecular chaperones (Table 1) such as HSPs. Inefficient restoration of their structure leads to accumulation of misfolded proteins and their aggregation that precipitates cell death. Accumulation of damaged or misfolded proteins within ER triggers a process called ER-stress, which can be ameliorated by unfolded protein response (UPR) or can lead to cell death 105. Therapeutic approaches that interfere with re-folding or removal of oxidized proteins can be used to sensitize tumor cells to PDT. For example, modulation of HSP function with geldanamycin, a HSP90 inhibitor, sensitizes tumor cells to PDT.106 Bortezomib, a proteasome inhibitor successfully used in the treatment of hematological disorders potentiates cytotoxic effects of PDT by aggravation of ER-stress.48 Moreover, several apoptosis-modulating factors such as rapamycin, Bcl-2 antagonists, ursodeoxycholic acid or ceramide analogues have been shown to increase PDT-mediated cancer cell death (Table 3).
Transformed cells deeply seated within the tumor mass receive suboptimal light doses and survive due to induction of numerous cytoprotective mechanisms. Targeting enzymes participating in ROS scavenging (such as superoxide dismutase, heme oxygenase-1 or nitric oxide synthase) with selective inhibitors has been shown to improve antitumor activity of PDT.41,107-108 Antivascular effects of PDT can be further potentiated by COX inhibitors109, antiangiogenic or antivascular drugs110 or monoclonal antibodies targeting factors promoting neovascularization (such as VEGF)111 significantly improving tumor growth control after PDT. Finally, combining PDT with agents that target signal transduction pathways such as the anti-EGFR agent, cetuximab may also improve the efficacy of PDT.112 Moreover, combining two different PSs in one treatment regimen leads to simultaneous targeting of tumor as well as vascular cells.113 The use of agents that enhance the efficacy without increasing the normal tissue effects of PDT thereby improving the therapeutic index will represent a major focus of clinical research going forward.
Go to:
CLINICAL PDT
The clinical use of PDT for cancer dates to the late 1970s when there was a study on the effects of HPD + light in five patients with bladder cancer.114 In 1978, Dougherty reported the first large series of patients successfully treated with PDT with HPD.115 Complete or partial responses were observed in 111 of 113 malignant lesions. Of the large variety of tumors examined, none was found to be unresponsive. Since this early work, there have been over 200 clinical trials for PDT.
Recent systematic reviews116-117 revealed that PDT can be considered a reasonable option in the treatment of malignant and pre-malignant non-melanoma skin lesions. It is also useful in the treatment of Barrett’s esophagus and unresectable cholangiocarcinoma. However, its effectiveness in the management of other types of tumors has not yet been unequivocally proven. The major reason for this is that only few adequately powered randomized controlled trials were performed so far. Systematic analysis of the literature is limited due to lack of optimal PDT parameters (illumination conditions or PS dose) that could be comparable among these studies.
PDT produces mostly superficial effects. Due to a limited light penetration through tissues the depth of tumor destruction ranges from few millimeters up to one centimeter. This apparent disadvantage can be favorably exploited in the treatment of superficial diseases, such as premalignant conditions (mucous dysplasia, actinic keratosis), carcinoma in situ or superficial tumors (such as malignant pleural mesothelioma118 or intraperitoneal disseminated carcinomatosis119-120). Moreover, PDT can be used supplemental to surgery, to irradiate tumor bed and increase the probability of long-term local disease control.
Skin tumors
PDT using Photofrin and ALA and its derivatives has been extensively studied in the treatment of both premalignant and malignant skin tumors.121-122 In the definitive setting, PDT is currently approved in the United States, Canada and the European Union for the treatment of actinic keratosis and approved in the EU and Canada for treatment of basal cell carcinoma (BCC). PDT has demonstrated efficacy in treating squamous cell carcinoma in situ/Bowen’s disease and has also been used with some success to treat extramammary Paget’s disease. However, the results of PDT for squamous cell carcinoma (SCC) of the skin using topical photosensitizers have been disappointing, with recurrence rates of >50%.121-122
PDT for actinic keratosis and PDT for SCC in situ/Bowen’s disease
Successful results for PDT of nonhyperkeratotic actinic keratosis have been achieved with systemically administered Photofrin as well as topically applied ALA and methyl-ALA (MAL). Twenty randomized controlled trials that reported the use of PDT in the treatment of actinic keratosis (AK) have been identified. Kennedy et al.123 introduced topically applied ALA for the treatment of nonhyperkeratotic AK with complete response rates for AK lesions exceeding 75%. In a placebo controlled trial, ALA-PDT showed a significantly superior complete response rate as compared to sham PDT using vehicle + light of 89% vs 13%, P<0.001.124 Similar results were obtained using MAL-PDT.125-126 Other studies have compared PDT for AK to cryotherapy or topical 5-fluorouracil (5-FU) cream. In one study, 119 subjects with 1501 AK lesions of the scalp and face were randomly assigned to receive MAL-PDT to either the left or right sided lesions with cryotherapy used to treat the contralateral side.127 Twenty-four weeks after therapy, both treatment groups showed a high response rate 89% for MAL-PDT vs. 86% for cryotherapy; p = 0.2, but MAL-PDT showed superior cosmesis and patient preference. Similar results have been found in other large randomized trials of MAL-PDT vs cryotherapy, with complete response rates for both ranging from 68-81% for cryotherapy and 69-92% for MAL-PDT.19,125-126,128 In conclusion, multiple trials have demonstrated complete response rates of 70% to 90% with good to excellent cosmetic outcomes in >90% of patients for PDT treatment of AK. In a randomized study comparing 5-FU cream to either of ALA-PDT or MAL-PDT in treatment of AK, equivalent complete response rates were found with comparable or superior tolerability for PDT.129-130 Current studies focus on novel photosensitizer drugs and re-formulations of ALA, such as nanoemulsion or patch based applicators, that may increase the complete response rate for AK at 12 months to >95%.131 The results of ALA-PDT in the treatment of Bowen’s disease (squamous cell carcinoma in situ) have been equally positive and so far were reported in 6 randomized clinical trials. Randomized, controlled trials comparing ALA-PDT or MAL-PDT to cryotherapy or 5-FU cream reveal complete response rates of 82-100% for PDT vs 67-100% for cryotherapy or 79-94% for 5-FU at 12-24 months.132-134
PDT for basal cell cancer
Other indications for ALA-PDT include superficial and nodular basal cell carcinoma.135-137 Six randomized clinical trials have reported the results of PDT for nodular BCC, 5 evaluated PDT efficacy in the treatment of superficial BCC and two were done in patients with mixed superficial and nodular BCC. In the largest single institution experience with 1440 nodular and superficial BCCs, PDT using systemically administered Photofrin shows an initial (6 month) complete response rate of 92%, with a recurrence rate of less than 10% at 4 years.138 At this same institution, a 92% complete response rate was achieved with topical ALA-PDT in 330 patients with superficial BCC, but the response rate dropped to 71% in 75 patients with nodular BCC.138 In a multicenter randomized trial of MAL-PDT vs cryotherapy for superficial BCC, complete response rates at 3 months were 97% and 95%, with 22% and 20% 5 year recurrence rates for MAL-PDT and cryotherapy, respectively.139 In this study, the excellent to good cosmetic outcome was 89% for MAL-PDT and 50% for cryotherapy. However, when topical PDT is compared to surgery for BCC, topical ALA or MAL-PDT consistently shows an increase in recurrence rate as compared to surgery for both superficial and nodular BCC. A randomized controlled trial of MAL-PDT vs surgical excision in 196 patients with superficial BCC showed a 9.3% recurrence rate for PDT vs 0% recurrence rate for surgery at 12 months.140 However, the good to excellent cosmetic outcome was 94% and 60% for the PDT and surgical excision patients, respectively. Similarly, in trials of PDT vs surgery for nodular BCC, recurrence rates are less than 5% for surgery versus 14-30% with ALA-PDT.141-144 As with superficial BCC, cosmetic effects are consistently shown to be more favorable with ALA-PDT. In summary, PDT can be appropriate and effective treatment alternative to cryosurgery or surgical excision for selected BCC.
Head and neck tumors
PDT has been successfully employed to treat early carcinomas of the oral cavity, pharynx, and larynx preserving normal tissue and vital functions of speech and swallowing.145 Multiple institutions have published small series of results demonstrating the efficacy of PDT for head and neck cancer.146 Only one small clinical trial was randomized and compared Photofrin-PDT with chemotherapy (5-FU and cisplatin) in the treatment of nasopharyngeal carcinoma.147 Although no details on randomization procedures or blinding was provided the clinical response was better with PDT (p=0.001), and there was improvement in Karnofsky score. Biel et al. reported the largest series of over 300 patients accrued over a 15-year clinical time period and treated with Photofrin-mediated PDT.148 Among the treated lesions there were predominantly squamous cell carcinomas of the oral cavity, pharynx or larynx, but also Kaposi’s sarcoma, melanoma and squamous cell carcinoma in head and neck area. The treatment protocol most commonly involved the administration of 2.0 mg/kg of Photofrin 48h prior to irradiation with 630 nm light from Nd:YAG pumped dye laser. The light fluences delivered ranged between 50 and 75 J/cm2 for oral cavity, nosopharyngeal and skin lesions and at 80 J/cm2 for laryngeal tumors.149
Among the reported group there were 133 patients that presented with recurrent or primary CIS, T1N0 and T2N0 laryngeal carcinomas and were treated with PDT with curative intent. After a single PDT procedure the patients were followed-up on average for 96 months and at 5 years demonstrated 90% cure rate. The second group of patients subjected to PDT treatment consisted of 138 patients with CIS and T1N0 squamous cell carcinomas of the oral cavity. Similarly, one PDT treatment was delivered and the patients were followed up for up to 211 months. All patients were reported to achieve complete pathological and clinical response and the cure rate at 5 years remained at 100%. PDT was also used for more advanced stages of oral cavity lesions. 52 patients with T2N0 as well as T3N0 SCC also received single PDT treatment that led to complete pathological and clinical response affording for 100% cure rate at 3 years.
Overall over 500 patients with early stage oral cavity, larynx, pharynx and nasopharynx lesions were treated with Photofrin-based PDT worldwide with similar success.149-152 The small number of recurrences were usually salvaged with either repeated PDT or surgical resection. Complications seen in these series were limited to cutaneous photosensitivity, and local pain following therapy was usually controlled by oral analgesics.
The intense development of second generation of photosensitizers has seen them entering the clinical application in head and neck lesions as well. Several series have reported on the use of the second-generation photosensitizers such as ALA and mTHPC.153,154 The large multi-center phase II trials evaluated the application of Foscan-mediated PDT in the treatment of primary oropharyngeal cancers. The study by Hopper et al.153 of early oral cancer, where the tumors were up to 2.5 cm in diameter, reported the complete response rate of 85% (97/114) at twelve weeks and disease free survival at 75% at 2 years. In another study by Copper et al.155 PDT was used in the treatment of a total of 27 patients with 42 second or multiple primary head and neck tumors. Cure rates for stage I or in situ disease were 85% versus 38% for stage II/III.
Perhaps the most interesting study reported the application of Foscan-mediated PDT for advanced disease. 128 patients with advanced head and neck cancer were treated with a single PDT session.156 The patients included in this study had failed conventional therapy or were unsuitable for such treatment. PDT delivered at 96 h after Foscan administration allowed for 100% tumor mass reduction in 43% of lesions and the remaining lesions were reduced by at least 50%. In this trial tumor mass reduction was measured for each lesion by multiplying lesion’s length by width. The 100% tumor mass reduction represented a complete local tumor clearance. Over half of the treated patients also achieved substantial quality-of-life benefit. Overall the complete response rates as determined for every patient according to the World Health Organization (WHO) criteria were 13%, but interestingly, this figure rose to 30% when the total surface area of the tumor could be illuminated and the depth estimate was less than 1 cm. A relatively limited work that has been done with 5-aminolevulinic acid for head and neck lesions reported results that were slightly inferior to the once observed with Photofrin and Foscan.154,157-158
Taken together, the data from phase I/II trials strongly suggest that PDT could be an effective primary and alternative treatment modality for patients presenting with early head and neck tumors and that further research in this area, including randomized trials, is needed.
Digestive system tumors
The application of PDT in gastrointestinal (GI) tract has been divided into two groups: PDT of the esophagus and beyond. Barrett’s esophagus and various grades of dysplasia and early esophageal cancer are the best-studied PDT applications in the GI tract.159-160 Premalignant conditions such as Barrett’s esophagus with high grade dysplasia are theoretically ideal for treatment with PDT.161 These are superficial and large mucosal areas easily accessible for light. Barrett’s esophagus is the development of intestinal-type metaplasia in the esophagus and is associated with gastroesophageal reflux disease. Dysplasia may arise in the setting of Barrett’s esophagus and can lead to the development of adenocarcinoma. Although historically, the standard treatment was distal esophagectomy, this treatment is associated with significant morbidity and a 3-5% mortality rate. Therefore, endoscopic ablative therapies have become attractive alternatives for patients with Barrett’s esophagus, including argon plasma coagulation and PDT.
Seven randomized clinical trials have been reported to evaluate PDT in Barrett’s esophagus patients with high-grade dysplasia or superficial carcinoma. Most were relatively small, included less than 50 patients, and did not clearly report on study methods. Therefore, it is premature to state whether PDT is superior, equivalent, or inferior to other ablative treatments. The most frequent adverse effects included prolonged skin photosensitivity and esophageal strictures, especially when using Photofrin. However, the frequency of the latter does not appear to be higher as compared with argon plasma coagulation. There is insufficient information on the clinical factors that might be useful in predicting the likelihood of strictures after PDT.
One hundred and two patients with Barrett’s esophagus and high-grade dysplasia (69 patients) or mucosal adenocarcinoma (33 patients) were treated with photodynamic therapy using Photofrin as an alternative to esophagectomy (median series follow-up time of 1.6 years). After treatment with PDT, there was complete ablation of glandular epithelium with one course of photodynamic therapy in 56% of patients. Strictures requiring dilation occurred in 20 patients (20%) and was the most common serious adverse event. Photodynamic therapy failed to ablate dysplasia or carcinoma in four patients and subsequent esophagectomy was curative in three of these patients. The authors concluded that PDT is a highly effective, safe and minimally invasive first-line treatment for patients with Barrett’s dysplasia and mucosal adenocarcinoma.162 Corti et al. followed 62 patients with esophageal cancer who were treated with HPD-mediated PDT.163 Eighteen of these patients had in situ cancer (Tis), 30 had T1 tumors, 7 had T2 tumors, and 7 had recurrence of tumors at the anastomotic site from prior surgery. Radiation was delivered to selected patients. The complete response rate after PDT alone was 37% (23 out of 62 patients) and 82% (51 out of 62 patients) after PDT and radiation. The complete response rate to PDT alone was the highest in Tis/T1 patients (44%) compared to T2 patients (28%). Patients with recurrence at the anastomotic site did not respond to PDT. Median local progression-free survival was 49 months for patients with Tis/T1 lesions, 30 months for patients with T2 lesions, and 14 months for patients with recurrent tumors. Of those who had a CR, 48% remained disease free through the follow-up period (range 3 to 90 months). Three cases (7%) of esophageal stricture and 1 case (3%) of tracheoesophageal fistula were reported. Based upon these data, the authors concluded that PDT was effective therapy for early stage esophageal cancer and also demonstrated that radiotherapy could be used in those patients who did not respond completely to PDT. What is also clear from these studies is that in tumors with greater depth of penetration (T2 or greater) PDT is not an optimal treatment option. A randomized, Phase III trial of Photofrin-mediated PDT for Barrett’s esophagus and high grade dysplasia has been performed by the International Photodynamic Group for High-Grade Dysplasia in Barrett’s Esophagus.164 Patients were randomized to treatment with omeprazole (37 patients) or omeprazole with PDT (128 patients). At 5 years, PDT was significantly more effective than omeprazole alone in eliminating high grade dysplasia (77% [106/138] vs 39% [27/70], P<0.0001). A secondary endpoint of preventing progression to cancer showed a significant difference (P=0.027) with about half the likelihood of cancer occurring in the PDT arm (21/138 [15%] versus 20/70 [29%]). There was also a significantly (P=0.004) longer time to progression to cancer favoring PDT. It is based upon these data that the United States FDA approved Photofrin-mediated PDT for patients with Barrett’s esophagus and high grade dysplasia who do not undergo surgery. It should be noted that a recent Cochrane review concluded that radiofrequency ablation has significantly fewer complications than PDT and is efficacious at eradicating both dysplasia and Barrett’s esophagus. Long-term follow-up data are still needed before radiofrequency ablation should be used in routine clinical care.165 These Phase II and III trials of PDT for high-grade dysplasia demonstrate that this therapy prevents the development of invasive carcinoma and is a safe and reliable treatment option.166-168 Despite this positive assessment there are certain challenges. Stricture formation, potential skin phototoxicity, severe chest pain and nausea are quite problematic. It is believed however that with improved dosimetry and new PSs those limitations could be overcome.
PDT has been applied to a variety of tumor types in the GI tract beyond the esophagus.169 Early clinical studies from Japan of PDT in the stomach suggested great promise170-171, but regrettably were not followed by randomized clinical trials so far. PDT for early duodenal and ampullary cancers and advanced adenomas has been also investigated in pilot studies that indicated promising results, but further work is required to optimize the treatment conditions.172-173 The most promising results have been achieved in cholangiocarcinoma (CC). The case reports of PDT for CC began to emerge in the 1990s,174 but it was not until Ortner et al. published an uncontrolled, observational pilot study of 9 patients with inoperable CC treated with Photofrin-mediated PDT.175 In a follow up study 70 patients were treated including 20 who were randomized to PDT followed by bilateral plastic stenting.176 The median survival in the PDT + stenting group was a remarkable 493 days compared to only 98 days in the stenting alone group. Patients quality of life also improved significantly. Other studies have shown similar results.177-179 Although only two clinical trials for CC176,178 were randomized both reached a similar conclusion that PDT has a therapeutic effect on nonresectable CC. The most common complication was cholangitis that developed in every fourth patient undergoing PDT + stenting, which was higher than the rates observed in control patients treated with stenting alone. Other rare adverse effects reported include cholecystitis, abscess formation, pancreatitis, biliary leakage, and biloma. Consequently, a multicenter clinical trial has been recently initiated to obtain regulatory approval in the USA and Canada.169
Among other applications for PDT in the GI tract there are studies of PDT for unresectable pancreatic cancers180 and numerous reports that have looked at using PDT to eradicate colon polyps as well as to palliate bulky colon and rectal cancers.181-184 The use of PDT in these tumors is still considered experimental as there are not high level data to support the routine use of PDT for these indications at this time. In addition, PDT may have efficacy in treating hepatocellular carcinoma, which remains one of the most common form of cancer worldwide. Early results from clinical trials have been quite promising and a phase III study is currently underway to evaluate the efficacy of Talaporfin-mediated PDT using interstitial LEDs as compared to institution-specific standard treatment.185
PDT for intraperitoneal malignancies
As with pleurally disseminated malignancies, the treatment of patients with peritoneal carcinomatosis or sarcomatosis is typically palliative in nature. PDT has the potential to combine selective destruction of cancerous tissue compared to normal tissue with the ability to treat and conform to relatively large surface areas. Moreover, the intrinsic, physical limitation in the depth of visible light penetration through tissue limits PDT damage to deeper structures, thereby providing additional potential for tumor cell selectivity. This is especially true after surgical debulking (cytoreduction) where the residual tumor is microscopic or less than 5 mm in depth. A phase I trial of intraoperative PDT following maximal surgical debulking that was performed with 70 patients, mostly with recurrent ovarian cancer carcinomatosis or peritoneal sarcomatosis, resulted in a 76% complete cytologic response rate with tolerable toxicity.186 In the follow-up phase II study, patients were enrolled, stratified according to cancer type (ovarian, gastrointestinal, or sarcoma), and given doses of Photofrin and light at the maximally tolerated dose that was defined in the phase I trial.119,187 As in the phase I trial, intraperitoneal PDT was associated with a postoperative capillary leak syndrome that necessitated fluid resuscitation in the immediate postoperative period that was in excess of the typical fluid needs of patients who receive surgery alone.188 Other than the capillary leak syndrome188 and the skin photosensitivity, the complication rates were similar to the complication rates typically observed after similarly extensive surgery in the absence of PDT. With a 51-month median follow-up, the median failure-free survival and overall survival rates for the patients who received PDT were 3 months and 22 months in ovarian cancer patients; 3.3 months and 13.2 months in gastrointestinal cancer patients and 4 months and 21.9 months in sarcoma patients, respectively. Six months after therapy, the pathologic complete response rate was three of 33 (9.1%), two of 37 (5.4%), and four of 30 (13.3%) for the patients with ovarian cancer, gastrointestinal cancer, and sarcoma, respectively. The median survival of almost 2 years in the ovarian patients and over 1 year in the gastrointestinal patients suggested some benefit from this treatment compared to historical controls. In the patients with sarcoma the prolonged overall survival was primarily due to patients with sarcomatosis from gastrointestinal stromal tumors who were treated with imatinib when it became available. Given the narrow therapeutic index of PDT in the treatment of peritoneal carcinomatosis, this therapy has potential to benefit patients but requires further study.
Urinary system tumors
Prostate Cancer
Patients with prostate cancer who elect definitive radiotherapy have limited options for salvage therapy for isolated local failure. Moreover, first line, definitive management of early stage prostate cancer with either surgery or ionizing radiation therapy has significant associated morbidities due to the proximity of normal structures such as nerves, bladder and rectum. The intrinsic limitation in the range of PDT-mediated damage imposed by visible light has the potential to selectively treat the prostate while sparing the surrounding normal tissues. By adapting the techniques developed for interstitial brachytherapy with radioactive seeds, light can be delivered to the entire prostate gland using interstitial cylindrically diffusing optical fibers. Unlike chemotherapy or radiation therapy, the mechanism of cell killing by PDT is not dependent on DNA damage or cell cycle effects, decreasing the chances of therapy cross resistance and eliminating late normal tissue effects such as second malignancy. All of these factors combine to make prostate cancer an attractive target for clinical trial development.
Several groups have published clinical trial results for prostate PDT using second generation PS. In a pilot study of mTHPC-mediated PDT, 14 patients who experience biopsy confirmed local failure following definitive radiotherapy for early stage prostate cancer were treated using up to 8 implanted interstitial cylindrically diffusing optical fibers.189 Of these patients, 13 were considered to have received a high light dose (≥50 J/cm2). Response of prostate specific antigen to therapy was observed in 9 patients and a complete pathologic response was observed in 5 patients. One patient developed a urorectal fistula after a rectal biopsy was performed 1 month following PDT. Four patients developed stress incontinence and four patients developed decreased erectile function. In a follow-up report of definitive mTHPC-mediated PDT as first line therapy, six patients with organ confined, Gleason 6 adenocarcinoma of the prostate, were treated with 4-8 interstitial fibers with implants designed to cover only the areas of the prostate with biopsy proven disease.190 Four of these patients had a second PDT treatment due to biopsy confirmed persistent disease at 3 month follow-up. While the treatment was relatively well tolerated, and all patients showed evidence of necrosis on post-procedure imaging or biopsy, all 6 patients had biopsy confirmed residual disease after PDT.
Another group has studied Motexafin Lutetium (MLu) as a photosensitizer for prostate PDT.191-192 In the Phase I trial, 17 patients with biopsy confirmed, locally recurrent prostate adenocarcinoma following definitive radiotherapy were treated with increasing doses of 732 nm (red) light using interstitial fibers. The primary goal of this trial was to determine the maximally tolerated dose and dose limiting toxicities of MLu-mediated prostate PDT, and one important secondary goal was to begin to develop the capability to perform real-time measurements of tissue optical properties, tissue levels of oxygen and photosensitizer to eventually allow real-time light fluence modulation that would provide a more homogenous dose of PDT to the entire prostate gland. As in the mTHPC study, one patient developed a urorectal fistula that was attributed to inhomogeneity of light dose. The remainder of toxicities observed in these patients was mild to moderate and consisted of urinary toxicities, including stress incontinence. Although not designed to measure efficacy, a significant difference was found in time to biochemical failure (prostate specific antigen recurrence) between the low and high PDT dose cohorts, providing some evidence of biochemical and pathologic disease response to PDT.
Another group has investigated vascular-targeted PDT using Pd-bacteriopheophorbide (Padoporfin, Tookad) mediated PDT and a short drug-light interval. In the phase I trial, 24 patients with biopsy confirmed local failure following definitive radiotherapy for prostate adenocarcinoma were treated with Padoporfin-mediated PDT using 2 interstitial fibers.193-194 This study demonstrated that vascular-targeted PDT could be safely performed in this patient population. In the follow-up phase II study, 28 patients were treated with increasing light doses.195 After 6 months of follow-up, less residual cancer was noted on biopsy as the light dose increased. All had negative biopsies at follow-up if >60% of the prostate was determined to be avascular by post-PDT magnetic resonance imaging (MRI). Toxicities were significant, with 2 patients developing urethrorectal fisulas. This study demonstrated the potential for pathologic complete response over a short-term follow up. Together, these studies suggest that while prostate PDT is feasible, comprehensive treatment of the entire gland will be necessary and improved techniques and dosimetry will be critical in providing an acceptable toxicity profile.
Bladder Cancer
Bladder cancers, which are often superficial and multifocal, can be assessed and debulked endoscopically. In addition, the geometry of the bladder should allow for improved and homogeneous delivery of light. These factors make superficial bladder cancer an attractive target for PDT. In general, early response rates (2 to 3 months) to PDT have been about 50% to 80% of patients with longer-term (1 to 2 years) durable responses in 20% to 60% of patients. It should be noted that many of the patients treated in these studies had recurrent disease that developed after standard therapies such as BCG.
Early studies used hematoporphyrin derivative (HPD)-mediated PDT. In one study, focal HPD-mediated PDT was used to treat 50 superficial bladder transitional cell carcinomas (TCC) in 37 patients and achieved a 74% complete response rate.196 Another study used HPD-mediated PDT to treat the entire bladder wall for 34 patients with refractory carcinoma in situ (CIS) of the bladder and achieved a 73.5% complete response rate at 3 months.197 However, by 2 years, 77.8% of these patients experienced disease recurrence. In these studies, treatment of superficial bladder cancer with PDT is generally well tolerated, with dysuria, hematuria, and skin photosensitivity being the most common acute toxicities. However, bladder wall fibrosis/diminished bladder capacity has been and continues to be a problem in some treated patients. With improved dosimetry and the use of Photofrin as a photosensitizer, other investigators have achieved durable complete response rates as high as 60% for refractory bladder CIS or superficial TCC.198-199 Studies of locally applied (intravesical) ALA demonstrate that similar durable complete response rates of 52-60% at 2-3 years can be achieved for patients with treatment refractory bladder CIS without the prolonged skin photosensitivity experienced using systemic Photofrin.200-201
While most of the patients treated with bladder PDT are refractory to BCG, one randomized controlled study has compared a single Photofrin-mediated PDT to multiple BCG treatments (induction + maintenance) and found that these therapies are equivalent in durable treatment response.202 Studies combining intravesical immunotherapies such as BCG or chemotherapies such as mitomycin C with PDT showed that these therapies may significantly enhance the PDT responsiveness of bladder tumors.203-204 Despite these promising results, PDT for bladder cancer remains largely investigational with limited use. PDT for bladder cancer is approved in Canada and some EU nations, but has not been approved by the US FDA.
Non-small cell lung cancer and mesothelioma
PDT for non-small cell lung cancer (NSCLC) was first used in 1982 by Hayata and colleagues to achieve tumor necrosis and reopening of the airway.205 PDT for lung cancer is particularly useful for (i) patients with advanced disease where PDT is used as a palliation strategy206-208 and (ii) patients with early central lung cancer when patients are unable to undergo surgery.209-210 PDT is considered to be more specific and lesion-oriented compared to other available modalities, produces less collateral damage and therefore fewer complications. Indeed, a randomized trial of PDT versus Nd:YAG laser therapy for obstructing NSCLC lesions showed equal initial efficacy for these two treatments, with a longer duration of response for PDT.208 PDT + palliative radiation also appears to increase the time to bronchus re-occlusion when combined as compared to radiation alone.211-212
In patients with early stage lung cancer, PDT has been used to successfully treat patients for whom surgery is not feasible. In one phase II study, 54 patients with 64 lung carcinoma lesions underwent Photofrin-mediated PDT and showed an 85% complete response rate with a 6.5% local failure rate at 20.2 months.210 Other studies have supported these excellent results, with complete response rates averaging 73% in studies totaling 359 patients.211,213-214 For radiographically occult lung cancers, results are equally good, with one typical study showing a complete response rate of 94% with 80% local control at 5 years.215 Second generation photosensitizers have also been used in early stage lung cancer treatment. Recently Usuda et al.216 reported a series of seventy cancer lesions < or =1.0 cm in diameter and 21 lesions >1.0 cm in diameter treated with NPe6-PDT. The complete response rates were 94.0% (66 of 70) and 90.4% (19 of 21), respectively. NPe6-PDT was capable of destroying the residual cancer lesions observed after the mass of large tumors had been reduced by electrocautery. Another report217 described the results of 529 PDT procedures performed on 133 patients that presented with non-small cell lung cancer (89 patients), metastatic airway lesions (31 patients), small cell lung cancer (4 patients), benign tumors (7 patients), and other (unspecified) lung conditions (2 patients). The lesions were most commonly located in the main stem bronchi (71 patients). Most patients received two treatments during a 3-day hospitalization and returned in 2 weeks for two more PDTs. The authors concluded that PDT can be safely and effectively used in the described setting leading to improved dyspnea in selected patients. Small number of randomized clinical trials in NSCLC and insufficient reporting on study methods and treatment outcomes do not enable to draw firm conclusions on PDT efficacy and safety. PDT remains a very promising therapeutic approach in the treatment of NSCLC.
NSCLC with pleural spread is incurable with standard treatment modalities such as surgery, chemotherapy or ionizing radiotherapy and median survival rates in these patients typically range from 6 to 9 months. Surgery alone has been unsuccessful in obtaining local control and does not extend survival beyond palliative chemotherapy, which remains the standard of care for treatment of this disease. Based on promising phase I results, a pilot phase II trial of Photofrin-mediated PDT was performed to investigate the efficacy of combined surgery and PDT for patients with either recurrent or primary NSCLC with pleural spread, the majority of whom had N2 nodal involvement and bulky pleural disease.101,218 In this study, local control of pleural disease at 6 months was achieved in 11 of 15 (73%) of evaluable patients and median overall survival for all 22 patients was 21.7 months. These results are highly encouraging in this population of patients and suggest that additional investigation in this area is warranted.
Malignant pleural mesothelioma (MPM) is a cancer of the pleura that, like NSCLC with pleural spread, has no currently available curative options. In a phase II study of Photofrin-mediated PDT following extrapleural pneumonectomy for MPM, patients with stage I and II disease experienced a median survival of 36 months with a 2-year survival rate of 61% while patients with stage III and IV disease experienced a median survival time of 10 months.219 Both of these were significantly improved compared to historical series of surgery alone. However, in a single randomized phase III study of surgery versus surgery with PDT, patients received similar treatment as described above, but did not appear to benefit from the addition of PDT to surgery.220 This trial was potentially underpowered and also involved surgical debulking that could leave disease of up to 5 mm thickness as opposed to a marcoscopically complete resection. Trials of intraoperative PDT using mTHPC showed that mTHPC PDT is feasible and has potentially acceptable toxicity.221-222 One important finding in these studies of resection with PDT for MPM is that a lung-sparing, tumor debulking surgery can be combined with PDT to achieve local control rates similar to those observed with extrapleural pneumonectomy. Indeed, a more recent study of macroscopically complete, lung sparing surgical debulking followed by intraoperative Photofrin-mediated PDT for patients with locally advanced MPM found a median survival that had not been reached with a 2.1 year median follow-up in patients following radical pleurectomy with PDT.223 Thus, PDT for MPM needs to be further evaluated in clinical trials of lung sparing surgery.
Brain tumors
PDT is currently undergoing intensive clinical investigation as an adjunctive treatment for brain tumors.224 The major tumor lesions particularly suitable for PDT treatment are newly diagnosed and recurrent brain tumors due to their high uptake of photosensitizers. Since early 1980s close to one thousand patients have received PDT for brain lesions worldwide. Perria et al.225 reported one of the earliest attempts to use PDT to treat the post-resection glioma cavity in humans and Kaye et al.226 reported a phase I/II trial involving 23 patients with glioblastoma multiforme (GBM) and anaplastic astrocytoma (AA). Other brain lesions treated with PDT included malignant ependymomas,227-228 malignant meningiomas,229 melanoma and lung cancer brain metastasis,226,229 and recurrent pituitary adenomas.230 The initial trails provided encouraging results and the authors concluded that PDT can be used as an adjuvant therapy in brain tumors patients. The photosensitizers employed so far were various formulations of hematoporphyrin derivatives (HPD, Photofrin), ALA as well as mTHPC. The light sources used to activate those photosensitizers included lamps, dye lasers, gold vapor potassium titanyl phosphate (KTP) dye lasers and diode lasers.
Currently photosensitizers are being evaluated both as intraoperative diagnostic tools by means of photodetection (PD) and fluorescence guided resection (FGR, Table 1) as well as during PDT as an adjunctive therapeutic modality.229,231-233 All three approaches take advantage from the higher uptake of PS by the malignant cells and are used intraoperatively. The most recently published trials that employed PD, FGR and PDT provided additional encouraging results but the initial delay in tumor progression did not translate to extended overall survival.234-237
Stylli et al. reported the results of a total of 375 patients treated at the Royal Melbourne Hospital.234 Among the 375 patients the lion’s share consisted of newly diagnosed (138 patients) and recurrent (140 patients) glioblastomas multiforme. Additional histological types included newly diagnosed (41 patients) and recurrent (46 patients) anaplastic astrocytomas. Patients received 5 mg/kg of hematoprophyrin derivative 24 hours prior to surgery and the light dose was 70-260 J/cm2. In the follow-up the mean survival of both types of GBM was between 14.3-14.9 months and about 28-41% of patients survived more than two years. For AA the mean survival was between 66.6 and 76.5 months and 57-73% survived more than 3 years.
Muller et al. reported the results of a prospective randomized controlled trial using adjuvant Photofrin-mediated PDT in the study group.236 The 96 patients treated for supratentorial gliomas with Photofrin-PDT at St. Michael’s Hospital in Toronto, Canada were randomized to two groups that received either 40 J/cm2 or 120 J/cm2. The patients that received the higher dose (48 patients) survived on average for 10 months while the 49 patients in the low dose group survived on average 9 months and the difference between both groups was not statistically significant at 0.05 level.
Stummer et al. reported the results of the ALA study group, a multicenter prospective randomized controlled trial in Germany.235 This trial compared the effectiveness of ALA based FGR to conventional surgery. The 322 patients with suspected malignant gliomas were followed-up for 35.4 months. Patients randomized to FGR group demonstrated much better time to progression (5.1 months) compared to 3.6 months in the controls, which translated in greater survival of 16.7 versus 11.8 months respectively. However, the difference in overall survival was not statistically significant.
Eljamel et al. reported a single center prospective randomized controlled study that employed the techniques of ALA based FGR, protoporphyrin IX (PpIX) spectroscopy and fractionated Photofrin-mediated PDT in GBM patients.237 The PDT was delivered up to 500 J/cm2 in five fractions. Among the 27 recruited patients 13 received FGR and PDT and demonstrated the mean survival of 52.8 weeks compared to 24.6 weeks of the control group. The mean time to tumor progression was 8.6 months in the FGR and PDT group compared to 4.8 months in the control group.
The current standard therapies that include surgery, radiation therapy and chemotherapy afford for median survival of about 15 months and although there is limited data comparing PD, FGR and photodiagnosis with those standard therapies the initial results from randomized trials are encouraging. It remains to be seen whether PDT for brain tumors remains a palliative or at most an alternative treatment modality. The new classes of PSs, the better understanding of dosimetry and further improvement in technology may significantly change the currently achieved clinical outcome. Additionally, pre-clinical data indicating that protracted light delivery may increase the therapeutic index of PDT in the brain combined with newer technologies such as implantable, LED-based light delivery systems could lead to significant improvements in treatment outcomes.224
Barriers for adoption of PDT into routine clinical practice
Despite being first described in the early 1900s238, the use of PDT to treat cancer patients has been relatively slow to enter mainstream clinical practice. Even when used clinically, PDT for cancer remains in many cases an alternative or palliative treatment or is used within the context of a clinical trial. For the PDT novice, the array of associated technologies such as lasers, applicators/fiber optics and power meters along with the need to perform manual calculations for dosimetry can be daunting. When performed with the assistance of a radiation oncologist or medical physicist with some training in optical methods and dosimetry, this difficulty can be overcome more easily. Another potential problem is the scarcity of phase III clinical trials that could demonstrate the superiority of PDT over other modalities.116 While more randomized trials of PDT are needed, other technologies and therapies with a similar deficiency in phase III data have been much more readily adopted by clinicians. Finally, the first generation PSs exhibited a prolonged skin sensitivity to visible light and this likely limited the use of these drugs in the palliative setting, especially for patients with a life expectancy of less than 6-12 months. However, better understanding of dosimetry, light emitting diode (LED) and diode-based laser technologies with simplified user interfaces and new PSs with decreased duration of skin photosensitivity, combined with mechanistic studies that may allow patient or tumor specific selection of therapy suggest that PDT has the potential to finally make the transition to obtain widespread clinical use in the oncologic community.
Go to:
NOVEL STRATEGIES IN PHOTODYNAMIC THERAPY
Two-photon PDT
The standard method in PDT is to use an organic PS, activated by continuous light, administered as an acute, high-dose single treatment. There are several fundamentally different approaches that are under pre-clinical investigation, involving different photophysics, chemistry and/or photobiological mechanisms. In 2-photon PDT short (~100 fs) laser pulses with very high peak power are used, so that two light photons are absorbed simultaneously by the PS. Since each photon only contributes half the excitation energy, near-infrared light can be used to achieve deeper tissue penetration. The subsequent photochemistry and photobiological effects are the same as in 1-photon PDT. Starkey et al. reported 2 cm effective treatment depth in tumor xenografts; this is considerably greater than what would typically be achieved by 1-photon activation.239 Alternatively, if the laser beam is strongly focused, then the activation volume may be extremely small. This may be exploited to target individual blood vessels240, reducing damage to adjacent tissues. Both approaches have used novel PSs designed to have very high 2-photon cross sections.239-240 Potentially, either strategy could overcome light attenuation limitations, particularly in pigmented tumors such as melanoma.
Metronomic PDT
In metronomic PDT (mPDT) both the drug and light are delivered at very low dose rates over an extended period (hours-days). This can result in tumor cell-specific apoptosis, with minimal tissue necrosis.241 To date, the main focus has been in glioma to minimize direct photodynamic damage to adjacent normal brain and secondary damage from the inflammatory response to PDT-induced tumor necrosis. Dose-dependent tumor responses have been demonstrated in vitro242 and in an intracranial model using ALA and an implanted optical fibre source.243 It is not known if this concept applies to other PSs or organ sites. There is evidence that the molecular pathways in mPDT may be different from those of acute, high-dose PDT.244
PDT molecular beacons
The concept of PDT molecular beacons (MBs) derives from the use of MBs as fluorescent probes with high target specificity. The PS is linked to a quenching molecule, so that it is inactive until the linker is cleaved by a target-specific enzyme (Fig. 6). Alternatively, the linker may be an antisense oligonucleotide (hairpin) loop, which is opened by hybridization to complementary mRNA. PDT beacons were first demonstrated using a caspase-3 linker between pyropheophorbide and a carotenoid quencher, achieving 8-fold and 4-fold quenching and unquenching, respectively, as demonstrated by the singlet oxygen yield.245 Subsequently, matrix metalloproteinase (MMP)-based beacons were reported in vitro and in vivo, with high selectivity between MMP+/− tumors.246 Hairpin-type beacons targeted to c-raf-1 mRNA had even higher tumor-to-non tumor specificity and almost complete restoration of the PDT efficacy upon hybridization in human breast cancer cells in vitro.247 The most important characteristic of MB is that tumor selectivity no longer depends solely the PS delivery, but also on the tumor specificity of the unquenching interaction and selectivity of the beacon to this interaction. Recently, asymmetric hairpin beacons were described to balance high quenching efficiency with 2-step activation (cleavage and dissociation) to enhance tumor cell uptake.248
Figure 6
PDT molecular beacons
A peptide linker that is a substrate of a cancer-associated enzyme (e.g. a protease) is conjugated to a photosensitizer (PS) and a singlet oxygen (1O2) quencher. Proximity of the PS and quencher ensures inhibition of 1O2 generation during irradiation of normal cells. In the presence of an enzyme the substrate sequence is cleaved and the PS and quencher are separated thereby enabling photoactivation of the PS.
Nanotechnology in PDT
Nanoparticles (NP) have several potential roles in PDT: for PS delivery, as PSs per se, and as energy transducers.249 Liposomal NPs are used clinically for delivery of the water-insoluble photosensitizer BPD.250 The potential advantage of NPs is that a high ‘payload’ can be delivered and they can be ‘decorated’ with multiple targeting moieties such as antibodies or peptides. Other approaches251 include: biodegradable polymers, ceramic (silica) and metallic (gold, iron oxide) NPs; magnetic NPs, in which an applied magnetic field enhances localization to the tumor; and hybrid NPs that allow both PDT and either another therapeutic strategy such as hyperthermia or an imaging technique such as magnetic resonance imaging. NP delivery of 2-photon PSs has also been reported, since these typically have very poor water solubility.252 Materials that themselves generate 1O2 upon photoexcitation include silicon NPs and quantum dots. The latter may also be linked to organic PSs, where they absorb the light energy with high efficiency and transfer it to the PS. Upconverting NPs have been investigated, in which relatively long wavelength light (near infrared) is absorbed and converted to shorter wavelength light that activates the attached PS.251 These concepts illustrate a general advantage of NP-based PDT in that the photophysical and photochemical properties of the PS can be uncoupled from the delivery and activation processes. A final recent approach is the encapsulation of a PS inside polymeric NPs that in turn are incorporated into liposomes containing a second drug such as an antiangiogenic agent (or vice versa).253 This co-delivery increases the therapeutic synergy of the two modalities.
Photochemical internalization
A large number of technologies have been developed to enhance translocation of macromolecular therapeutics (Table 1) into the cytosol. These technologies are mainly designed to enhance cellular uptake of macromolecules via endocytosis and stimulate their endosome-to-cytosol translocation. Photochemical internalization (PCI) was specifically designed to enhance the release of endocytosed macromolecules into the cytosol. It is based on the use of PSs located in endocytic vesicles as shown in Fig. 7.30 PDT-generated 1O2 induces a release of macromolecules from the endocytic vesicles into the cytosol.254 The physico-chemical requirements of the PSs utilized in PCI are strong amphiphilicity hindering their penetration through membranes and the presence of hydrophobic region necessary for penetration sufficiently deep into cell membranes in order to efficiently produce singlet oxygen in a membranous environment.255 The unique properties of the PCI process may be used to activate the therapeutics only in the light exposed area while unexposed normal tissues are spared. PCI has been shown to increase the biological activity of several molecules that do not readily penetrate the plasma membrane, including type I ribosome-inactivating proteins (RIPs), immunotoxins, plasmids, adenoviruses, various oligonucleotides, dendrimer-based delivery of chemotherapeutica and unconjugated chemotherapeutics such as bleomycin and doxorubicin.255 In addition, PCI allows for utilizing therapeutics without intrinsic properties for endosome-to-cytosol translocation. An example is the use of the highly toxic ribosome-inactivating protein - diphtheria toxin (DT). In a PCI-based treatment regimen DT may be replaced with type I RIPs such as gelonin and saporin exerting low translocation efficiency and thereby reducing the side-effects from the toxins.256 The clinical documentation of therapeutic effects of macromolecular therapeutics for intracellular targets on solid tumors is, however, limited. An ongoing phase I/II clinical trial evaluating PCI of bleomycin has been reported to result in encouraging tumor responses. Out of 14 patients treated so far (squamous cell carcinoma of the head & neck, adenocarcinoma of the breast, chondroblastic osteosarcoma and skin adnexal tumor) complete clinical regression was observed in all evaluable tumors within a few weeks after treatment, although two recurrences were seen at the 3 month follow up (Berg, unpublished). The treatment has left the healthy tissue underneath the tumor largely unaffected, indicating high specificity for the tumor tissue. These promising properties of the PCI technology have the potential to enhance the antitumor efficacy and to exert a high grade of specificity due to the combination of targeted therapeutics with light-activated cytosolic delivery induced by PSs preferentially accumulating in solid tumors.
Figure 7
The principles of the PCI technology
The photosensitizer (PS) and the therapeutic compound (D) in this example linked to a monoclonal antibody as a targeting moiety are delivered to the target cells. The photosensitizer and the therapeutic compound are both unable to penetrate the plasma membrane and both are thus endocytosed reaching initially the endocytic compartments (endosome). The photsensitizers used in PCI are integrated into the membranes of the endocytic vesicles. Upon light exposure the photosensitizer becomes activated and form singlet oxygen oxidizing membrane constituents resulting in rupture of the endocytic membranes, allowing the therapeutic compound to reach cellular compartments where its therapeutic targets are located (T1 or T2 (nucleus)). In the absence of light the therapeutic compound may be degraded in the lysosomes.
Go to:
CONCLUSIONS
PDT is still considered to be a new and promising antitumor strategy. Its full potential has yet to be shown and its range of applications alone or in combination with other approved or experimental therapeutic approaches is definitely not exhausted. The advantages of PDT compared with surgery, chemotherapy or radiotherapy are reduced long-term morbidity and the fact that PDT does not compromise future treatment options for residual or recurrent disease. Due to a lack of natural mechanisms of 1O2 elimination and a unique mechanism of cytotoxicity mutations that confer resistance to radiotherapy or chemotherapy do not compromise antitumor efficacy. Moreover, PDT can be repeated without compromising its efficacy. These are significant limiting factors for chemotherapeutics and radiotherapy. Finally, many conventional antitumor treatments carry risk of inducing immunosuppression. PDT-induced immunogenic cell death associated with induction of a potent local inflammatory reaction offers the possibility to flourish into a therapeutic procedure with excellent local antitumor activity and capable of boosting the immune response for effective destruction of metastases. Interdisciplinary uniqueness of PDT inspires specialists in physics, chemistry, biology and medicine and its further development and novel applications can only be limited by their enormous imagination.
CA Cancer J Clin. Author manuscript; available in PMC 2012 Jul 1.
Published in final edited form as:
CA Cancer J Clin. 2011 Jul-Aug; 61(4): 250–281.
Published online 2011 May 26. doi: [10.3322/caac.20114]
Patrizia Agostinis,1 Kristian Berg,2 Keith A. Cengel,3 Thomas H. Foster,4 Albert W. Girotti,5 Sandra O. Gollnick,6 Stephen M. Hahn,3 Michael R. Hamblin,7,8,9 Asta Juzeniene,2 David Kessel,10 Mladen Korbelik,11 Johan Moan,2,12 Pawel Mroz,7,8 Dominika Nowis,13 Jacques Piette,14 Brian C. Wilson,15 and Jakub Golab13,16,*
PHOTODYNAMIC THERAPY OF CANCER: AN UPDATE https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3209659/
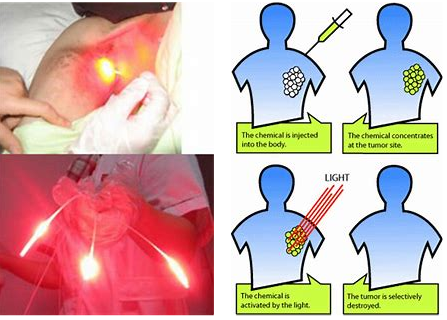
.png)
.png)
.png)
.png)
.png)
.png)
.png)