íí
Peroxidase Activity of HemoglobiníñHaptoglobin Complexes
COVALENT AGGREGATION AND OXIDATIVE STRESS IN PLASMA AND MACROPHAGES*
the Departments of íýEnvironmental and Occupational Health and
**Critical Care Medicine, and
the í¼Safar Center for Resuscitation Research, University of Pittsburgh, Pittsburgh, Pennsylvania 15219 and
the ¶Research Institute of Physico-Chemical Medicine, Moscow 119992, Russia
Abstract
As a hemoprotein, hemoglobin (Hb) can, in the presence of H2O2, act as a peroxidase. In red blood cells, this activity is regulated by the reducing environment. For stroma-free Hb this regulation is lost, and the potential for Hb to become a peroxidase is high and further increased by inflammatory cells generating superoxide. The latter can be converted into H2O2 and feed Hb peroxidase activity. Haptoglobins (Hp) bind with extracellular Hb and reportedly weaken Hb peroxidase activity. Here we demonstrate that: (i) Hb peroxidase activity is retained upon binding with Hp; (ii) in the presence of H2O2, HbíñHp peroxidase complexes undergo covalent cross-linking; (iii) peroxidase activity of HbíñHp complexes and aggregates consumes reductants such as ascorbate and nitric oxide; (iv) cross-linked HbíñHp aggregates are taken up by macrophages at rates exceeding those for noncovalently cross-linked HbíñHp complexes; (v) the engulfed HbíñHp aggregates activate superoxide production and induce intracellular oxidative stress (deplete endogenous glutathione and stimulate lipid peroxidation); (vi) HbíñHp aggregates cause cytotoxicity to macrophages; and (vii) HbíñHp aggregates are present in septic plasma. Overall, our data suggest that under conditions of severe inflammation and oxidative stress, peroxidase activity of HbíñHp covalent aggregates may cause macrophage dysfunction and microvascular vasoconstriction, which are commonly seen in severe sepsis and hemolytic diseases.
As a hemoprotein, Hb,2 in the presence of oxidizing equivalents such as H2O2, can act as a peroxidase with very high oxidizing potential (1). In red blood cells, this potentially dangerous activity is strictly regulated by the reducing environment and the lack of oxidizing equivalents. The inadvertently appearing ferric forms of Hb are short-lived, and the hemoprotein is effectively converted into ferro-Hb (deoxy-Hb) by metHb reductase (2). Normally, less than 2% of total Hb exists in the form of MetHb because the rate of Hb reduction is far greater than its oxidation (2). For stroma-free Hb, however, this intracellular regulation is lost, and the likelihood for Hb to act as a peroxidase is high. This possibility is markedly increased in the course of severe inflammation (e.g. in sepsis) by the generation of superoxide radicals by immune cells. The latter can be spontaneously or catalytically (by extracellular superoxide dismutase) converted into H2O2, a fuel for Hb peroxidase activity. In line with this, several clinical and experimental investigations have established that lethality in sepsis is increased in the setting of hemolysis (3¿C5).
Circulating haptoglobin (Hp) provides an important endogenous defense against the toxic effects of Hb (6, 7). The major biological function of this abundant plasma protein is binding and recycling of stroma-free Hb via the macrophage CD163 receptor-mediated pathway (8, 9). It has been proposed that Hp possesses antioxidant activity and diminishes oxidative stress induced by stroma-free Hb (10¿C12). The antioxidant action of Hp toward Hb has been associated, at least in part, with weakening of its peroxidase activity (10) or preventing oxidation and cross-linking of Hb (7).
Previous work has demonstrated that peroxidase activity of different hemoproteins, including Hb, can induce protein self-oxidation leading to covalent cross-linking and aggregation (13¿C16). Whether these hetero-oligomeric covalent aggregates retain the peroxidase activity is unknown. If the aggregates retain peroxidase activity, they may continue to be a source of oxidative stress both in circulation as well as in phagocytizing cells involved in their clearance such as macrophages.
In the current work, we determined the extent to which: (i) Hb peroxidase activity is decreased by binding with Hp; (ii) peroxidase activity of HbíñHp complexes initiates cross-linking into covalent hetero-oligomers; (iii) peroxidase activity of HbíñHp complexes and aggregates utilizes nitric oxide (NO•); (iv) HbíñHp aggregates are taken up by macrophages as compared with noncovalent HbíñHp complexes; and (v) the engulfed HbíñHp aggregates induce oxidative stress and cytotoxicity. Here, we report that HbíñHp complexes and aggregates are potent peroxidases capable of inducing oxidative stress in both plasma and macrophages. We further demonstrate the presence of HbíñHp aggregates in septic plasma.
Go to:
EXPERIMENTAL PROCEDURES
Materials
RPMI 1640 medium, non-heat-inactivated fetal bovine serum were from the American Tissue Culture Collection (Manassas, VA). BioSep-SEC-S4000 column was purchased from Phenomenex, Inc. (Torrance, CA). Human Hb, Hp from pooled human plasma, haptoglobin phenotype 1-1 (Hp 1-1), haptoglobin phenotype 2-2 (Hp 2-2), diethylenetriaminepentaacetic acid (DTPA), 5,5-dimethyl-1-pyrroline N-oxide (DMPO), phorbol myristate acetate (PMA), dexamethasone, and H2O2 were acquired from Sigma-Aldrich. MultiTox-Glo Multiplex cytotoxicity assay kit was from Promega (Madison, WI). Rabbit DMPO nitrone adduct polyclonal antiserum was from Cayman Chemical Co. (Ann Arbor, MI), goat anti-human Hb antibody was from Bethyl Laboratories, Inc. (Montgomery, TX), rabbit anti-human Hp antibody was from Dako Cytomation (Carpinteria, CA), and anti-actin antibody was obtained from Calbiochem (Gibbstown, NJ). Anti-4-HNE antibody and phycoerythrin-conjugated anti-human CD163 antibody were from R & D Systems (Minneapolis, MN), goat anti-mouse horseradish peroxidase-conjugated antiserum, goat anti-rabbit alkaline phosphatase-conjugated antiserum, SilverSNAP stain kit, SuperSignal West Pico chemiluminescent substrate, and Lumi-Phos WB chemiluminescent substrate were purchased from Fisher.
Culture and Differentiation of THP-1 Cells
THP-1 cells (American Tissue Culture Collection) were cultured in the RPMI 1640 medium containing 10% fetal bovine serum, 0.05 mm 2-mercaptoethanol, and 5000 units of penicillin/streptomycin and maintained in suspension at 0.5¿C1.0 í┴ 106 cells/ml. The adherent, differentiated macrophages were obtained by the addition of PMA (20 nm) to culture medium for 4 days. The cells were then treated with fresh culture medium containing dexamethasone (250 nm) for 24 h to trigger CD163 expression (17).
Quantification of Cell Surface CD163 Expression
Cell surface CD163 expression was determined by flow cytometry using a phycoerythrin-conjugated anti-human CD163 antibody. A flow cytometer (Becton-Dickinson, Franklin Lakes, NJ) equipped with a 488-nm argon ion laser and supplied with the Cell Quest software was used to perform the analysis. Mean fluorescence intensity from 10,000 cells was acquired.
Uptake of Fluorescently Labeled HbíñHp Complexes and Aggregates
To quantify Hb or HbíñHp uptake, the cells were incubated with FITC-labeled respective ligands at a concentration of 10 ª╠g/ml (as the amount of Hb) in serum-free medium and analyzed using flow cytometry and confocal microscopy.
Flow Cytometry
After 2 h of incubation, the cells were washed with Ca2+-free phosphate-buffered saline (PBS) containing EDTA to dissociate extracellular HbíñHp from CD163 (18, 19), then collected, and quantified by mean fluorescence intensity from 10,000 acquired cells.
Confocal Microscopy
After 1 h of incubation, the cells were treated with Trypan blue to quench extracellular fluorescence signal followed by fixation with paraformaldehyde. The images were obtained using Olympus Fluoview 1000 confocal microscope.
Assessment of Cytotoxicity
THP-1 cells were seeded in 96-well plate and differentiated with PMA followed by dexamethasone treatment. The cells were incubated with either Hb, HbíñHp complexes, or their aggregates at a concentration of 0.4 and 0.6 mg/ml (as the amount of Hb) for 1 h in serum-free medium, after rinsing with Ca2+-free PBS. The cells were further incubated for another 6 h (serum was added after 4 h of incubation to minimize the toxic effect of serum withdrawal). Cytotoxicity was determined using Promega MultiTox assay kit according to the manufacturer's manual by a Fusion ª┴ universal microplate analyzer (PerkinElmer Life Sciences). To make the data comparable, digitonin (30 ª╠g/ml) was used as 100% cytotoxicity standard.
Fluorescence Assay of GSH
Low molecular mass thiols (predominantly GSH) in cells were determined using ThioGloTM-1, a maleimide reagent that produces highly fluorescent adducts with SH groups. Briefly, after differentiated THP-1 cells were incubated with either Hb, Hb-aggregates, HbíñHp complexes, or HbíñHp aggregates in the same condition as in cytotoxicity assay, 0.2 í┴ 105 cells were suspended in PBS and lysed by freezing and thawing. GSH content was estimated by an immediate fluorescence response to ThioGloTM-1. The Fusion ª┴ microplate analyzer was employed for fluorescence assay using excitation at 388 nm and emission at 500 nm. A standard curve was established by the addition of GSH (0.04¿C4.0 ª╠m) to 100 mm phosphate buffer (pH 7.4) containing 10 ª╠m ThioGloTM-1. The total amount of protein was determined using the Bradford assay. The GSH level was expressed as nmol/mg protein.
Assessment of Superoxide Generation by Flow Cytometry
Oxidation-dependent fluorogenic dye, dihydroethidium (DHE, Molecular Probes) was used to evaluate intracellular production of superoxide radicals. Briefly, the cells were incubated with 5 ª╠m DHE for 10 min followed by washing with PBS. The cells were collected and resuspended in PBS after incubation with either Hb, Hb aggregates, HbíñHp complexes, or aggregates at concentrations of 0.4 mg/ml in cell culture medium without serum for 1 h. The fluorescence of ethidium was measured using a FACscan flow cytometer and Cell Quest software. Mean fluorescence intensity from 10,000 cells was acquired using a 585-nm band pass filter (FL-2 channel).
Formation of HbíñHp Aggregates
Hb aggregates and HbíñHp aggregates were prepared by incubation of Hb or HbíñHp (1:1) in the presence of H2O2 added in equal portions every 15 min (H2O2/Hb ratio 15:1) for 1 h. The reaction was stopped by the addition of catalase (1400 units/ml). In some cases oleic acid (oleic acid/Hb ratio 100:1) was added to increase the amount of HbíñHp aggregates. To produce FITC-labeled HbíñHp complexes, the HbíñHp mixture (1:1) was incubated in 50 mm phosphate buffer (pH 7.4) for 15 min, and then FITC was added at a ratio of 150 ª╠g of FITC/1 mg of protein and incubated for 1 h. Nonbound FITC was removed by spin column. To form labeled aggregates, FITC-labeled complexes were incubated with H2O2 (H2O2/Hb ratio 15:1) for 1 h. For EPR power saturation measurements, 200 ª╠m ascorbate was added to the formed aggregates and incubated for 15 min to reduce all of the protein immobilized radicals. The residual ascorbate was removed by ascorbate oxidase (1 unit/100 ª╠l; 5 min).
Peroxidase Activity of Hb, HbíñHp Complexes, and Aggregates
Formation of Ascorbate Radicals
Formation of ascorbate radicals was monitored by EPR spectroscopy using a JEOL-RE1X EPR spectrometer (Tokyo, Japan) at 25 íÒC. Ascorbate radical EPR spectra were recorded at the following settings: 3350 G, center field; 10 G, sweep width; 10 milliwatt, microwave power; 0.5 G, field modulation; 103, receiver gain; 0.03 s, time constant; 4 min, scan time. The time courses of ascorbate radical generation were obtained by repeated scanning of the EPR spectrum (5 G, sweep width; other instrumental conditions were the same).
UV Absorbance
UV absorbance measurements of ascorbate during its oxidation were performed by at 265 nm using UV160U spectrophotometer (Shimadzu Scientific Instruments).
Low Temperature EPR Measurements
To form heme-nitrosyl complexes, 10 ª╠m metHb was incubated with a nitroxyl donor, Angeli's salt (40 ª╠m), for 30 min at room temperature. The reaction was stopped by freezing the samples in liquid nitrogen. The spectra of nitrosylated Hb were recorded under the following instrumental settings: 3200 G, center field; 500 G, sweep width; 5 G, field modulation; 10 milliwatt, microwave power; 103, receiver gain; 0.1 s, time constant; 2 min, time scan. EPR spectra of oxoferryl-related tyrosyl radicals were measured at 77 K in 30 s after the addition of H2O2 (50 or 100 ª╠m) to the samples. EPR settings: center field, 3230 G; sweep width, 100 G; field modulation, 5 G; microwave power, 5 milliwatt; receiver gain, 103; time constant, 0.1 s; time scan, 2 min.
Plasma Samples
This study was approved by the Institutional Review Board of the Children's Hospital of Pittsburgh, and informed consent was obtained from parents for sample collection. Venous blood samples were collected in anticoagulant sodium citrate (3.2%)-containing tubes. The blood samples were centrifuged 10 min at 2400 rpm, and plasma was stored in −80 íÒC until further analysis.
Electrophoretic Techniques
PAGE
Hb, HbíñHp complexes, HbíñHp aggregates, and plasma samples were separated by native, SDS-PAGE, or SDS-DTT-PAGE in Tris-glycine buffer. Because of the high molecular mass of aggregates, gels with different porosity (12% acrylamide, 7.5% acrylamide, 2.5% acrylamide 0.5% agarose) were used. The gels were stained by SilverSNAP stain kit according to the manufacturer's manual.
Western Blotting Analysis
Proteins from SDS-PAGE gels were electrotransferred to a nitrocellulose membrane. After blocking with 5% nonfat milk dissolved in Tris-buffered saline/Tween 20 (0.05%) for 1 h, the membrane was incubated overnight with primary antibodies (at 4 íÒC). The membranes were washed several times followed by incubation with the secondary antibody conjugated with horseradish peroxidase or alkaline phosphatase for 60 min at room temperature.
For analysis of intracellular accumulation of HbíñHp aggregates and 4-hydroxynonenal (4-HNE) protein adducts control and HbíñHp aggregates treated THP-1 cells were washed three times with PBS containing EDTA by centrifugation, suspended in PBS, lysed by mild sonication, and centrifuged by 500 í┴ g for 10 min to remove cell debris. The supernatants were separated on 1% SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with West Pico (for peroxidase activity), anti-Hb or anti-Hp antibodies, monoclonal anti-4-HNE antibody, or anti-actin antibody for loading control.
The protein bands were visualized by using SuperSignal West Pico chemiluminescent substrate for the horseradish peroxidase-conjugated secondary antibody or Lumi-Phos WB chemiluminescent substrate for the alkaline phosphatase-conjugated antibody. The density of the bands was determined using EpiChemi II Darkroom (UVP BioImaging Systems, Upland, CA) or by exposure of Kodak scientific imaging film X-OMAT LS (Carestream Health Inc., Rochester, NY).
Zymography
The samples were subjected to native or SDS-PAGE in Tris-glycine buffer in 7.5% polyacrylamide gel; peroxidase activity was determined by the incubation of gels or membranes containing electrotransferred proteins with SuperSignal West Pico Chemiluminescent Substrate, which is a typical substrate for peroxidases.
HPLC Chromatography
HPLC chromatography of oxidized HbíñHp was performed on BioSep-SEC-S4000 column in 50 mm phosphate buffer (pH 7.2). Collected fractions were concentrated by Ultracel YM-30 cut -off filters (Millipore, Bedford, MA). Assessment of peroxidase activity of fractions was performed by measuring the fluorescence of resorufin, an oxidation product of Amplex Red, or by assessment of ascorbate radical EPR signals. The reaction conditions for the Amplex Red assay were: 50 mm sodium phosphate buffer (pH 7.4), 100 ª╠m DTPA, 100 ª╠m Amplex Red, 50 ª╠m H2O2. The reaction conditions for EPR experiments were: 1 ª╠m ascorbate, 500 ª╠m etoposide, 200 ª╠m H2O2.
Statistical Analysis
HPLC ChromatographyData are expressed as means (standard deviation of at least triplicate determinations). The changes in variables were analyzed by a one-way analysis of variance for multiple comparisons. The differences were considered significant at p < 0.05.
Go to:
RESULTS
Effect of Hp on Peroxidase Activity of Hb
Initially, the involvement of Hb-driven peroxidase activity in its cross-linking with Hp and formation of HbíñHp aggregates was explored in model biochemical systems. To assess peroxidase activity of Hb, we utilized ascorbate as a well known substrate and monitored its oxidation by EPR spectroscopy. EPR is a specific and reliable method for detection of ascorbate radicals formed as one-electron intermediates during oxidation of ascorbate to dehydroascorbate (20). Incubation of Hb with ascorbate and H2O2 resulted in the formation of ascorbate radicals with a typical doublet signal in the spectrum with hyperfine splitting constants of 1.7 G (Fig. 1A, inset). Monitoring the time course of ascorbate radical EPR signals (Fig. 1A) revealed that Hp increased the lifespan of detectable ascorbate radicals, i.e. delayed its oxidation. This effect of Hp on the lifespan of ascorbate radicals generated by Hb/H2O2 was dependent on Hp/Hb ratios and saturated at ratios of 1:1 and higher (Fig. 1B). Without Hb (control), H2O2 did not cause the appearance of ascorbate radical signals (in the presence of the metal chelator DTPA, 100 ª╠m). Further, the magnitude of Hb-induced signal in the absence of H2O2 was ∼8-fold lower than that observed in its presence (data not shown). Thus, Hp suppressed oxidation of ascorbate by Hb/H2O2, resulting in delayed disappearance of its radical. In line with the EPR data, measurements of ascorbate absorbance (with a max at 265 nm) showed that Hb/H2O2-induced consumption of ascorbate was progressively inhibited by increasing concentrations of Hp (Fig. 1C). Similarly to EPR assessments, absorbance measurements demonstrated that increasing inhibition of Hb peroxidase activity by Hp reached saturation at HbíñHp ratio of 1:1. This is compatible with the known very high affinity of Hp for Hb (Kd is on the order of approximately í┴10−14 m) (21).
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480001.jpg
Open in a separate window
FIGURE 1.
H2O2-dependent oxidation of small molecular mass peroxidase substrates by free Hb and HbíñHp complexes monitored by EPR and by absorbance at 265 nm. A, typical EPR spectrum (inset) and time course of ascorbate radical EPR signals. B, lifespan of EPR signals from ascorbate radicals generated by HbíñHp complexes in the presence of H2O2. The reaction conditions were: 50 mm sodium phosphate buffer (pH 7.4), 100 ª╠m DTPA, 2.5 ª╠m Hb, 20 ª╠m ascorbate, 100 ª╠m H2O2, and Hp as indicated (means í└ S.D. from three independent experiments; *, p < 0.05 versus Hb). C, ascorbate oxidation by HbíñHp complexes. Inset, time course of characteristic ascorbate absorbance at 265 nm. The reaction conditions were: 50 mm sodium phosphate buffer (pH 7.4), 100 ª╠m DTPA, 1.25 ª╠m Hb, 20 ª╠m ascorbate, 100 ª╠m H2O2, and Hp as indicated. Line a, Hb; line b, HbíñHp −1:0.15; line c, HbíñHp −1:0.8; line d, HbíñHp-1:1 (means í└ S.D. from three independent experiments; *, p < 0.05 versus Hb). D, representative native PAGE stained for peroxidase activities of free Hb and its complexes with Hp 1-1, Hp 2-2 assessed by West Pico chemiluminescence substrate. E, averaged results of densitometric analysis of peroxidase activities of Hb complexes with Hp 1-1 and Hp 2-2 normalized to the activity of free Hb (*, p < 0.05 versus Hb). F, Lineweaver-Burk plots for peroxidase activity of Hb assayed by Amplex Red oxidation in the presence and absence of Hp. Hb (0.25 ª╠m) was preincubated for 10 min with Hp at a ratio of 1:1. The peroxidase activity was assessed by fluorescence of resorufin in PBS (pH 7.4) containing DTPA (100 ª╠m), H2O2 (50 ª╠m), and Amplex Red in the range of 5¿C150 ª╠m. Line 1, Hb; line 2, HbíñHp.
Peroxidase activity of free Hb and HbíñHp complexes was also assessed zymographically after native PAGE. By incubating gels with a typical chemiluminescent substrate for peroxidases, West Pico, we detected the peroxidase activity in bands, corresponding to free Hb as well as to its complexes with either Hp 1-1 or Hp 2-2. In agreement with the above results on ascorbate oxidation, Hp suppressed peroxidase activity of Hb toward West Pico (Fig. 1D). Quantification of zymograms revealed that the peroxidase activity of HbíñHp complexes was ∼30% of that for free Hb (Fig. 1E).
We showed that at Hp/Hb ratios exceeding 1:1, no free Hb could be detected on the gels, indicating that the complexes were the only species with the peroxidase activity. These results are consistent with data in the literature that stoichiometry of Hb interactions with Hp is 1:1 (22). Thus, kinetic estimates could be performed at this ratio of HbíñHp. The results obtained with Amplex Red as a typical peroxidase substrate demonstrated that Hp caused a mixed type of inhibition of Hb peroxidase activity, acting as a competitive inhibitor as well as a noncompetitive inhibitor (Fig. 1F). Apparently, Hp itself acted as a substrate of the peroxidase reaction catalyzed by Hb (see below). The noncompetitive mechanism of the inhibitory action may derive from covalent modification of HbíñHp complexes leading to the aggregation.
Hb-induced oxidation of several small molecule substrates: ascorbate, West Pico and Amplex Red, was partially and concentration-dependently inhibited upon binding of Hp, suggesting that oxidizing potential of Hb might be competitively redirected toward oxidation of Hp. Alternatively, binding of Hp could block access of small molecules (including H2O2) to the heme-catalytic site of Hb. To experimentally examine this, we studied interactions of Hb and its complexes with Hp with NO•, a small molecule that produces characteristic low temperature EPR spectra upon binding with the heme moiety in deoxy-Hb (23). When a source of nitroxyl radicals, Angeli's salt, was added to free Hb or HbíñHp complexes, a characteristic EPR spectrum of hexa-coordinate heme-nitrosyl complexes of Hb was readily detectable at 77 K (supplemental Fig. S1A, panels a and b). The magnitude of the signals was 15¿C20% lower in the presence of Hp (Fig. 2A). These results indicate that Hp does not preclude interactions of Hb with small molecules such as nitroxyl and NO•.
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480002.jpg
Open in a separate window
FIGURE 2.
Detection of heme-nitrosyl complexes and protein-derived (Tyr•) radicals (peroxidase intermediates) by EPR spectroscopy. A, typical low temperature (77 K) EPR spectra of HbíñHp complexes and aggregates (insets) and magnitudes of EPR signals of heme-nitrosylated Hb (bar a), HbíñHp complexes (bar b), Hb aggregates (bar c), and HbíñHp aggregates (bar d). To form nitrosyl complexes, 40 ª╠m Angeli's salt was added to the solution containing 10 ª╠m Hb, and incubation was continued for 30 min, after which the samples were frozen (means í└ S.D. from 3¿C7 independent experiments; *, p < 0.05 versus Hb; **, p < 0.05 versus HbíñHp complexes). B, H2O2-dependent formation of protein-derived (Tyr•) radicals (peroxidase intermediates) by Hb (bar a), HbíñHp complexes (bar b), Hb aggregates (bar c), and HbíñHp aggregates (bar d). Shown are typical low temperature (77 K) EPR spectra of HbíñHp complexes and aggregates (insets) (measured at g = 2.005) and magnitudes of the respective protein-derived (Tyr) radicals (means í└ S.D. from three to six independent experiments; *, p < 0.05 versus Hb). C, power saturation curves for protein-derived radicals of Hb and HbíñHp complexes and aggregates. Hb aggregates and HbíñHp aggregates were obtained by incubating Hb (10 ª╠m) and HbíñHp in the presence of H2O2 (50 ª╠m added four times with a 15-min interval). 200 ª╠m ascorbate was added to the formed aggregates and incubated for 15 min to reduce all protein immobilized radicals, and the residual ascorbate was removed by ascorbate oxidase (1 unit/100 ª╠l; 5 min). 100 ª╠m H2O2 was added to Hb, Hb aggregates, HbíñHp complexes, and HbíñHp aggregates, then samples were frozen for 20 s, and EPR spectra of protein-derived radicals were measured. Catalase (10 ª╠g/ml) was added to stop the reaction.
Detection of H2O2-induced Hb Protein-immobilized Radicals; Intermediates of Peroxidase Reaction
In the absence of small molecule substrates, peroxidase activity of Hb can cause oxidation of protein itself yielding protein-immobilized radical intermediates, most commonly tyrosyl radicals (Tyr•) (13, 24). The production of these radical intermediates can be detected by low temperature EPR spectroscopy (25). Hb contains several potentially oxidizable tyrosine residues (Tyr42 and Tyr24 in ª┴-chain). A typical low temperature EPR spectrum obtained from an incubation system containing either Hb or HbíñHp complexes and H2O2 represents a characteristic signal of protein-derived (tyrosyl) radicals with a peak-to-trough width of 19 G and a g factor of 2.005 (25). (supplemental Fig. S1B, panels a and b). The magnitude of HbíñHp Tyr• radicals was significantly higher than that of Tyr• radicals of Hb (Fig. 2B, bars a and b). The proximity of Tyr• radical intermediates to the heme moiety of Hb can be probed by power saturation experiments. When these measurements were performed for Hb/H2O2 and HbíñHp/H2O2 incubations, no differences were found (Fig. 2C). This suggests that Hp did not significantly affect the structural organization of Hb as a peroxidase.
H2O2-dependent Aggregation of HbíñHp
Protein-immobilized (tyrosyl) radicals formed during the peroxidase reaction can realize their oxidizing potential to trigger the protein oligomerization process. In this reaction, the radicals recombine, resulting in carbon-carbon cross-linking that involves not only the initiating hemoprotein itself but surrounding proteins in close proximity yielding hetero-oligomers (13, 14, 16). This type of covalent cross-linking may be particularly effective between proteins in complexes. In the case of HbíñHp complexes, the formation of covalent hetero-oligomers is indicative of the recombination of radicals formed on both Hb and Hp and can be interpreted in terms of Hp being a substrate of the Hb-catalyzed peroxidase reaction. Both Hb and Hp contain several cysteines that can participate in disulfide bonding. Therefore, two different types of aggregates can be formed during co-oxidation of Hb with Hp: (i) S-S bridges (two coupled thiol groups that are dissociable by S-S reducing reagents) and (ii) Tyr-Tyr cross-links (nondissociable by S-S reducing reagents). PAGE analysis of fully denatured proteins in the presence of SDS and a disulfide reducing agent, DTT, revealed that incubation of Hb with Hp in the presence of H2O2 resulted in accumulation of aggregates with different molecular masses. Western blot analysis showed that these aggregates contained both Hb and Hp as evidenced by their positive staining with anti-Hb and anti-Hp antibodies (Fig. 3). Resistance of HbíñHp aggregates to DTT suggests that they are covalently linked via non S-S bonds. Although both Hp 1-1 and Hp 2-2 underwent oxidative cross-linking, a greater amount of very high molecular mass aggregates was formed after conjugation of Hb with Hp 2-2.
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480003.jpg
FIGURE 3.
PAGE of H2O2-induced covalent aggregation of Hb and Hp. SDS-PAGE (12% acrylamide) in the presence of DTT (40 ª╠m) stained with silver (panel a) and Western blots using anti-Hb antibody (panel b) and anti-Hp antibody (panel c). The arrows and brackets indicate positions of protein aggregates containing both Hb and Hp that were formed upon the addition of H2O2. Monomeric form of Hb (marked by a double-headed arrow) disappeared almost completely after incubation of Hb with H2O2 as a result of its oligomerization. Incubation system contained Hb (1 ª╠m) and Hp (equal weight amounts of Hp 1-1 and Hp 2-2 at a ratio of 1:1) in the presence or absence of H2O2 (50 ª╠m) in 50 mm phosphate buffer (pH 7.4). MW, molecular mass.
Quenching of HbíñHp Peroxidase-reactive Intermediates and Inhibition of HbíñHp Hetero-oligomerization by Reductants
The above results indicate that Hp acted as a substrate for Hb-driven peroxidase activity. Consequently, small molecule reducing substrates such as NO•, ascorbate, Amplex Red, or DMPO, known to interact with reactive intermediates of Hb peroxidase (15, 26), should prevent cross-linking of Hb with Hp. Increasing concentrations of these compounds can outcompete peroxidase activity of Hb toward Hpt and inhibit formation of aggregates. Indeed, we found that increasing concentrations of Amplex Red or DMPO progressively inhibited H2O2-dependent aggregation of Hb/Hpt as evidenced by PAGE (Fig. 4A and supplemental Fig. S2). NO• (generated by an NO donor, PAPANONate) and ascorbate acted in a similar way and inhibited cross-linking of Hb with Hpt (Fig. 4, B and C). Molecular proximity of Hp to Hb reactive intermediates, oxoferryl heme and/or protein immobilized (Tyr) radicals, makes Hp a preferred substrate of the peroxidase reaction. Indeed, Tyr-Tyr cross-linking of Hp with Hb was preventable by relatively high concentration of reducing substrates (for instance, 200-fold higher concentrations of ascorbate versus those of Hp were required to markedly inhibit the formation of aggregates).
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480004.jpg
FIGURE 4.
Inhibition of HbíñHp peroxidase-reactive intermediates and hetero-oligomerization and immuno-spin trapping of H2O2-induced protein-immobilized radicals as evidenced by PAGE. A¿CC, protection against H2O2-induced HbíñHp aggregation by Amplex Red, PAPANONOate, and ascorbate as evidenced by SDS-DTT-PAGE (7.5% acrylamide). The arrows and brackets indicate positions of protein aggregates containing both Hb and Hp formed upon the addition of H2O2. Staining was performed using SilverSNAP stain kit (A), anti-Hb antibody (B and C, panels a) and anti-Hp antibody (B and C, panels b). D, detection of protein-immobilized radical intermediates using antibody against a spin trap DMPO after SDS-PAGE (7.5% acrylamide) followed by Western blot analysis. The arrows and brackets indicate positions of protein aggregates reacting with DMPO. Staining was performed using anti-DMPO antibody (panel a), anti-Hb antibody (panel b), and anti-Hp antibody (panel c). E, inhibition of Hb/Hpt hetero-oligomerization by DMPO as evidenced by SDS-DTT-PAGE (7.5% acrylamide) stained with silver. The arrow indicates the position of covalent protein aggregates formed upon the addition of H2O2. The incubation system contained Hb (1 ª╠m) and Hp1¿C1 in the presence or absence of H2O2 (50 ª╠m) and the indicated concentrations of Amplex Red, PAPANONOate, ascorbate, and DMPO in 50 mm phosphate buffer (pH 7.4). MW, molecular mass.
Recently, a new technique, immuno-spin trapping, has been developed for specific and sensitive detection of protein-immobilized radicals (24, 27, 28). It is based on the use of antibodies to protein radical-DMPO adducts. When HbíñHp complexes were incubated with H2O2 in the presence of DMPO, protein-DMPO adducts were observed in HbíñHp aggregates on Western blots using anti-DMPO antibody (Fig. 4D). As expected, the spin trap, DMPO, suppressed covalent hetero-oligomerization of Hb with Hp (Fig. 4E).
Peroxidase Activity Is Retained in HbíñHp Aggregates
To characterize peroxidase activity of nondenatured HbíñHp complexes, we employed several experimental approaches to comparatively evaluate peroxidase activity of HbíñHp aggregates versus their complexes as follows.
Native PAGE
Using nondenaturing conditions of native gels, we found that peroxidase activity of Hb decreased in the order Hb > HbíñHp complexes > HbíñHp aggregates. In fact normalized to the unit of Hb content, the ratios of peroxidase activity were 100:50:10 for Hb, HbíñHp complexes, and HbíñHp aggregates, respectively (Fig. 5A). Importantly, however, the aggregates still displayed significant and measurable activity.
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480005.jpg
Open in a separate window
FIGURE 5.
Comparison of the peroxidase activity of free Hb, HbíñHp complexes, and aggregates. A, assessment of peroxidase activity of free Hb, HbíñHp complexes, and HbíñHp aggregates after native PAGE with subsequent Western blotting using West Pico chemiluminescence substrate. Panel a, representative gel stained with West Pico for peroxidase activity. Panel b, representative gel stained with SilverSNAP stain kit for detection of protein. Panel c, averaged peroxidase activity assessments based on the densitometry of bands (normalized to the amount of Hb) (*, p < 0.05 versus Hb). B, HPLC separation of HbíñHp complexes and HbíñHp aggregates and assessments of peroxidase activity in the fractions. Panel a, HPLC chromatogram of HbíñHp complexes and HbíñHp aggregates separated on BioSep-SEC-S4000 column; insets show native PAGE of two fractions corresponding to HbíñHp complexes and HbíñHp aggregates. Aggregation of HbíñHp was induced by incubation of Hb and Hp with H2O2 (H2O2/Hb ratio 15:1, 60 min). Panels b and c, peroxidase activity of HbíñHp complexes and HbíñHp aggregates measured by fluorescence of Amplex Red oxidation product, resorufin (panel b) and by EPR spectroscopy of ascorbate radical signals (presented as lifespan of ascorbate radical signals) (panel c) (*, p < 0.05 versus HbíñHp complexes). C, H2O2-dependent peroxidase activity of HbíñHp aggregates revealed on SDS-PAGE. The incubation system contained Hb (1 ª╠m) and Hp (equal weight amounts of Hp 1-1 and Hp 2-2 at a ratio of 1:1) in the presence or absence of H2O2 (25 ª╠m) in 50 mm phosphate buffer (pH 7.4). After PAGE in 2.5% acrylamide, 0.5% agarose, zymography was performed using West Pico chemiluminescence substrate. The arrows and brackets indicate positions of HbíñHp aggregates.
HPLC Separation
To more accurately examine the peroxidase activity of the aggregates, we performed their HPLC separation and assessed the activity in the fractions obtained (Fig. 5B, panel a). Based on assessments of Amplex Red oxidation, we confirmed that HbíñHp aggregates exerted considerable peroxidase activity, which was, however, lower than that of HbíñHp complexes (Fig. 5B, panel b). In concordance with this, EPR measurements of ascorbate radicals demonstrated an increased lifespan of ascorbate radical signals in the spectra (corresponding to lower magnitude of EPR signals, i.e. lower peroxidase activity toward ascorbate) (Fig. 5B, panel c).
SDS-PAGE
To separate high molecular mass aggregates of HbíñHp, we used high porosity mixed SDS gel (2.5% acrylamide, 0.5% agarose). Using West Pico as a peroxidase substrate, we found that peroxidase activity was preserved in HbíñHp aggregates (both HbíñHp 1-1 and HbíñHp 2-2) (Fig. 5C). As expected, SDS did not dissociate covalently cross-link HbíñHp aggregates. However, quantitative comparisons of peroxidase activities of HbíñHp aggregates versus those of HbíñHp complexes or free Hb were impossible because of the strong inhibitory effect of denaturing conditions of SDS gels on the activity of free Hb (data not shown). Indeed, it has been shown that treatment with SDS dissociates Hb into subunits with lower (versus nondenatured Hb) peroxidase activity, likely because of partial release of noncovalently bound heme (29).
EPR Spectroscopy
Assessments of peroxidase-reactive intermediates by low temperature EPR spectroscopy confirmed the formation of protein-derived (tyrosyl) radicals when covalently aggregated HbíñHp were incubated in the presence of H2O2 (Fig. 2B, bars c and d, and supplemental Fig. S1B, panels c and d). No differences between HbíñHp aggregates and Hb were found in power saturation experiments, suggesting that no changes in the structural organization of the catalytic heme environment occurred after the formation of HbíñHp aggregates (Fig. 2C).
Further, accessibility of the heme-catalytic site to small molecules and its reactivity toward NO• was directly demonstrated in HbíñHp aggregates by the formation of heme-nitrosyl complexes. When the aggregates were incubated with the nitroxyl radical generator Angeli's salt, characteristic signals of NO•-heme complexes were detectable in low temperature EPR spectra (supplemental Fig. S1A, panels c and d and Fig. 2, A and B, bars c and d). Overall, these results indicate that peroxidase activity is retained by HbíñHp aggregates that, if taken up by macrophages, may cause intracellular damage.
Phagocytosis of HbíñHp Aggregates by THP-1 Macrophages
Endocytosis of HbíñHp complexes is a known pathway for their elimination mediated by CD163 receptors in macrophages (8, 30, 31). To explore the uptake of HbíñHp aggregates, we employed differentiated THP-1 human monocytes. In THP-1 cells treated with PMA and dexamethasone, we used flow cytometry and confirmed a significant increase in surface expression of CD163 (Fig. 6A). Fluorescently labeled HbíñHp complexes were readily taken up by CD163 expressing THP-1 cells (Fig. 6, B and C). Most notably, the uptake was 2-fold higher when fluorescently labeled HbíñHp covalently cross-linked aggregates were incubated with differentiated CD163 expressing THP-1 cells. In contrast uptake of cross-linked Hb aggregates was lower than that of intact nonaggregated Hb. As expected, we did not see a significant uptake of Hb, HbíñHp complexes, or HbíñHp aggregates by undifferentiated THP-1 cells. The intracellular accumulation of the HbíñHp aggregates after incubation of THP-1 cells in the presence of HbíñHp aggregates was evidenced also by the presence of anti-Hb- and anti-Hp-positive high molecular mass material with peroxidase activity in cell lysates (Fig. 6D). This material was not detected in lysates obtained from naïve cells.
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480006.jpg
FIGURE 6.
Uptake of HbíñHp complexes and aggregates by THP-1 cells. A, CD163 expression in differentiated and nondifferentiated THP-1 cells. In THP-1 cells treated with PMA and dexamethasone (Dex), flow cytometry analysis showed an increase in surface expression of CD163. Expression of CD163 was detected by flow cytometry using phycoerythrin (PE)-conjugated antibody. Inset, typical histogram of nondifferentiated (gray) or differentiated (unfilled) THP-1 cells (means í└ S.D. from three independent experiments; ***, p < 0.001 versus nondifferentiated cells). B, uptake of Hb, HbíñHp complexes and HbíñHp aggregates by THP-1 cells as evidenced by flow cytometry. Note that the uptake of fluorescently labeled HbíñHp aggregates was higher than that of HbíñHp complexes. Inset, shown is a typical histogram of differentiated THP-1 cells incubated with HbíñHp complexes (filled purple) or HbíñHp aggregates (green unfilled) (means í└ S.D. from four independent experiments; *, p < 0.05; **, p < 0.01 versus Hb; ##, p < 0.01 versus HbíñHp). C, confocal microscopy of THP-1 cells incubated with fluorescently labeled Hb, HbíñHp complexes, or HbíñHp aggregates. Inset, shown is a typical photomicrograph of THP-1 cells with engulfed HbíñHp aggregates (green). Blue, staining of nuclei with Hoechst 33342. The images were obtained using an Olympus Fluoview 1000 confocal microscope with DP25 digital camera. Magnification was 600í┴, and the aperture was 150 ª╠m. For quantification, three fields were randomly chosen (150 cells in each field) (means í└ S.D. from three independent experiments; ***, p < 0.001 versus Hb; ###, p < 0.001 versus HbíñHp complexes.) D, detection of HbíñHp aggregates in THP-1 cells by SDS-PAGE. Lane i, naïve THP-1 cells; lane ii, THP-1 cells incubated with HbíñHp aggregates.
Uptake of HbíñHp Aggregates Is Accompanied by Oxidative Stress and Cytotoxicity in THP-1 Cells
To assess macrophage responses to engulfed HbíñHp aggregates, we measured the production of superoxide radicals and biomarkers of oxidative stress: GSH and lipid peroxidation products. We found that accumulation of HbíñHp aggregates was accompanied by a markedly enhanced intracellular production of superoxide radicals assessed by flow cytometry using DHE as a probe (Fig. 7A). Neither Hb nor HbíñHp complexes elicited increased superoxide production in THP-1 cells. Dismutation of superoxide radicals yields H2O2 that can fuel the peroxidase activity and hence induce oxidative stress. Indeed, we found that incubation of THP-1 cells with HbíñHp aggregates, but not with Hb or HbíñHp complexes, caused a significant loss of intracellular GSH and accumulation of a typical end product of lipid peroxidation, 4-HNE (detected as its protein adduct) (Fig. 7, B and C).
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480007.jpg
Open in a separate window
FIGURE 7.
Uptake of HbíñHp aggregates by THP-1 cells is accompanied by increased production of superoxide radicals and oxidative stress. A, superoxide generation by THP-1 cells incubated with free Hb, Hb aggregates, HbíñHp complexes, or aggregates measured by flow cytometry with dihydroethidium. Inset, shown is a typical histogram of ethidium fluorescence response from control (filled) and HbíñHp aggregate-loaded (unfilled) THP-1 cells (means í└ S.D. from three independent experiments; *, p < 0.05 versus control). B, intracellular GSH levels in THP-1 cells incubated with free Hb, Hb aggregates, HbíñHp complexes, or aggregates (means í└ S.D. from three independent experiments; *, p < 0.05 versus control). C, Western blot analysis of 4-HNE protein adducts in THP-1 cells treated with HbíñHp aggregates (panel a). Densitometric analysis of anti-4-HNE antibody-positive bands (normalized to amount of actin) (panel b) (*, p < 0.05 versus control). D, cytotoxicity of Hb, HbíñHp complexes and HbíñHp aggregates in differentiated THP-1 cells (means í└ S.D. from three independent experiments; **, p < 0.001 versus control). MW, molecular mass.
Combined, these results indicate that activation of THP-1 cells by phagocytized HbíñHp aggregates caused enhanced production of oxygen radicals, and the peroxidase activity of accumulating aggregates was likely responsible for the induction of oxidative stress. Therefore, we tested whether uptake of HbíñHp aggregates was associated with cytotoxicity. Indeed, engulfment of HbíñHp covalent aggregates induced a dose-dependent cytotoxicity; neither Hb nor HbíñHp complexes showed significant toxic effects under the same incubation conditions (Fig. 7D).
HbíñHp Aggregates in Septic Plasma
We further reasoned that HbíñHp aggregates can be present in plasma during sepsis, in which oxidative stress and hemolysis commonly occur (32¿C34). Using native PAGE, we analyzed septic plasma and revealed peroxidase activity in the bands corresponding to complexes of Hb with Hp. Fig. 8 presents an example of detection of HbíñHp aggregates with peroxidase activity in septic plasma. On native gel, distinctive bands positively staining for peroxidase activity were observed for free Hb, HbíñHp 1-1 complexes, and aggregated material with low electrophoretic mobility. Staining with anti-Hb and anti-Hp antibodies confirmed the presence of both proteins in aggregates (Fig. 8A). We were also able to ascertain the presence of HbíñHp aggregates on PAGE after SDS-DTT treatment (Fig. 8B). Under these denaturing and reducing PAGE conditions, HbíñHp complexes were dissociated and observed as subunits with molecular masses of <50 kDa. The presence of high molecular mass bands is compatible with covalent hetero-oligomerization of HbíñHp in septic plasma (Fig. 8B).
An external file that holds a picture, illustration, etc.
Object name is zbc0490994480008.jpg
FIGURE 8.
Detection of HbíñHp aggregates in septic plasma. A, gels after native PAGE (7.5% acrylamide) were stained with West Pico chemiluminescence substrate for peroxidase activity (panel a), Western blot analyses with anti-Hb (panel b), and anti-Hp antibodies (panel c). B, Western blot analyses with anti-Hb (panel a) and anti-Hp (panel b) antibodies after SDS-DTT-PAGE (7.5% acrylamide). The arrows and brackets indicate the positions of high molecular mass aggregates.
Go to:
DISCUSSION
Peroxidase Activity of HbíñHp Complexes and Aggregate
Release of Hb from ruptured red blood cells, where its concentrations are as high as 2.5 mm, can lead to micromolar Hb levels in whole plasma, particularly as a result of microangiopathic shear stress (35). The findings of this manuscript support a novel pathogenic mechanism, which could be an unrecognized mediator of injury and death in patients with hemolysis and conditions associated with systemic inflammation, including sepsis. Normally, the HbíñHp complex is rapidly eliminated via CD163-mediated pathway, thus preventing peroxidase activity of free Hb. However, under conditions of severe inflammation and oxidative stress, the otherwise innocuous HbíñHp complexes are themselves covalently cross-linked into hetero-oligomeric HbíñHp aggregates with peroxidase function. The latter scavenge nitric oxide in plasma and kill macrophages after phagocytosis. This new insight may be useful for new therapeutic approaches in patients with the commonly lethal combination of hemolysis and sepsis.
We provide several lines of evidence confirming the preservation of peroxidase activity both in HbíñHp complexes and aggregates. These include direct assessments of the activity in isolated HPLC fractions, low temperature EPR spectroscopy of characteristic intermediates, immuno-spin trapping, PAGE zymography using prototypical peroxidase substrates, and formation of heme-nitrosylated species. The Western blot analyses confirmed the hetero-oligomeric nature of aggregates and involvement of both Hb and Hp in the cross-links.
Different Clearance of HbíñHp Complexes and Aggregates
Recently, aggregation of Hb via its peroxidase function has been demonstrated. This resulted in blunted interaction of Hb with Hp and consequently decreased CD163-dependent uptake of the complexes (14). Similarly, we report decreased uptake of Hb aggregates by THP-1 macrophages differentiated to express high levels of CD163 receptors. Although these results are very important, circulating Hp has been considered as a significant endogenous defense against Hb peroxidase (6) because of its very high affinity binding (>1014 m−1) to Hb (21). Hp is an acute phase protein, and its normal plasma levels (ranging from 0.45 to 3 mg/ml) (8) increase 2¿C5-fold during inflammation (36). These amounts of Hp are sufficient to bind micromolar levels of free Hb. The binding and elimination of HbíñHp complexes would be expected to decrease circulating Hp levels, particularly under conditions of hemolysis (37). However, complete depletion of Hp and accumulation of free Hb aggregates may be typical of extremely severe conditions with hemolysis, for example sickle cell disease. More commonly, released free Hb is scavenged by Hp, resulting in the formation of HbíñHp complexes. The role of this major function of Hp is underscored by its significant power as a predictor of disease severity and clinical outcomes (38¿C40).
Buehler et al. (7) demonstrated that binding of Hb with Hp yields complexes that protect Hb from oxidative and structural modification; as a result of this protection, CD163-mediated uptake of HbíñHp complexes is preserved (7). These findings are in agreement with the results of the current study, which was focused on covalent cross-linking between Hb and Hp mediated by the peroxidase activity of the former in the complexes and realized through the recombination of protein immobilized radicals, most likely Tyr• radicals. However, our studies on the uptake and elimination of oxidatively cross-linked HbíñHp aggregates yielded results that were not completely concordant with those of Buehler et al. (7).
Buehler et al. reported similar engulfment of nonoxidized HbíñHp complexes and oxidized HbíñHp aggregates by CD163 expressing HEK293 cells; our results indicate that oxidatively cross-linked HbíñHp aggregates were taken up more effectively by THP-1 macrophages than nonoxidized complexes. There are several explanations for these apparent discrepancies: (i) The uptake of HbíñHp aggregates likely depends on their molecular mass. We used Hp isolated from pooled plasma that contained all three major phenotypes of Hp: Hp 1-1, Hp 2-1, and Hp 2-2. Polymeric forms of Hp 2-1 and Hp 2-2 have multiple molecular species with very high molecular masses: up to 300 and 900 kDa, respectively, and the molecular mass of HbíñHp complexes in this case can be as high as 1.5 MDa. Cross-linking of several such complexes would yield large aggregates with very high molecular masses: up to 3¿C5 MDa. In contrast, in the study by Buehler et al. (7), Hp 1-1 has been employed with a molecular mass of 86 kDa; Hp 1-1 forms relatively small complexes with Hb, ∼150 kDa (6). Consequently, cross-linking of these complexes yielded aggregates of smaller molecular masses (∼300 kDa). (ii) It is possible that both CD163-dependent as well as CD-163-independent pathways could be involved in engulfment of HbíñHp aggregates by THP-1 cells in our experiments. As professional phagocytes, THP-1 cells used in our study express not only CD163 but also other receptors potentially involved in the engulfment and uptake of large aggregates. In particular, a scavenger receptor of type B family, CD36, is known to be effective in the uptake of oxidatively modified high molecular mass aggregates, including oxidatively modified lipoproteins (41). Moreover, CD91 receptor (that belongs to the low density lipoprotein receptor superfamily) recognizes more than 40 different ligands, among which are heme-hemopexin, lipoproteins, viruses, and complexes of proteases with protease inhibitors (42). Neither of these receptors are expressed in HEK293 cells that have epithelial origin (43, 44). (iii) Greater uptake of oxidatively cross-linked HbíñHp aggregates versus nonoxidized HbíñHp complexes in our experiments could be also due to the fact that our í░oxidizedí▒ samples were highly enriched with cross-linked aggregates as evidenced by the percentage of aggregates versus nonaggregated forms of HbíñHp on silver-stained gels (as illustrated by Fig. 5A). Notably, the content of cross-linked HbíñHp aggregates used in experiments reported by Buehler et al. (7) was significantly lower than that used in the current study.
Binding and Aggregation of Hb with Different Phenotypes of Hp
In humans, unlike other mammals, there are two alleles for the Hp gene (Hp 1 and Hp 2). Accordingly two encoded proteins Hp 1-1 and Hp 2-2 represent an 86-kDa dimeric molecule and a polymeric form up to 900 kDa (6). Thus, HbíñHp cross-linking can produce high molecular mass aggregates (especially in the case of Hp 2-2), readily taken up by macrophages. Binding of Hb with Hp is believed to neutralize free the pro-oxidant effects of Hb (7, 10, 11). Based on structural considerations, however, this antioxidant function of Hp is not immediately apparent. ª┴/ª┬ dimers of Hb participating in the formation of HbíñHp complexes contain three Hp-binding sites that include amino acid residues 11¿C25 and 131¿C146 of the ª┬-globin chain and residues 121-l27 of the ª┴-chain in human Hb. Although two of these sites participate in interactions of the polypeptide chains with the heme (residues 100¿C140), neither of the Hb-binding regions of Hp seems to be in direct contact with the heme crevice (21, 45, 46). As a result, binding of Hp to Hb leaves the heme iron ready to interact with small molecules, such as O2 (47), NO, or H2O2. In line with these earlier reports (23, 24, 25), our EPR findings showed heme-nitrosylated complexes and protein-immobilized (Tyr•) radicals in HbíñHp complexes and aggregates. However, the specific location and proximity to heme of Hp-binding sites on the Hb molecule is sufficient for the role of Hp as a competitive (versus low molecular mass reductants) substrate for Hb-dependent peroxidase reactions.
In the circulation, cross-linked HbíñHp aggregates retained the peroxidase activity and hence continued to act as a source of oxidative stress and damage. In the presence of oxidizing equivalents, such as H2O2 and organic hydroperoxides including lipid hydroperoxides, heme-peroxidases are activated to highly reactive intermediates. The cleavage of the O-O bond of hydroperoxide is associated with the formation of oxoferryl (Fe4+=O) species and a cation radical of porphyrin-compound I (48, 49). In the case of Hb, the highly reactive radical on the porphyrin ring is unstable, causing one-electron oxidation of one of amino acid residues in the immediate proximity, most commonly tyrosine, tryptophan, or histidine, thus producing protein-immobilized radicals (27, 50, 51). Either of the two reactive radical species, porphyrin radical and/or protein-immobilized (Tyr•) radical, can act as a pro-oxidant depleting the major intracellular antioxidants such as GSH, ascorbate (52), and oxidizing biomolecules, such as proteins and lipids (51, 53, 54). In line with this, we observed that uptake of HbíñHp aggregates by THP-1 cells resulted in enhanced superoxide radical production, most likely by NADPH oxidase-dependent pathways known to be activated during phagocytosis (55, 56). Dismutation of superoxide radicals yields H2O2 required for maintenance of peroxidase activity of HbíñHp complexes. The latter catalyzed oxidative attack on thiols and lipids as evidenced by depletion of GSH and accumulation of protein adducts with 4-hydroxy-nonenal, one of the typical end products of lipid peroxidation (57, 58).
HbíñHp Aggregation in Severe Inflammation
Under conditions of severe inflammation and massive recruitment of macrophages, generated steady state concentrations of superoxide radicals and H2O2 (in the high micromolar range) (59, 60) may be sufficient for the realization of peroxidase activity of HbíñHp complexes. Accordingly we were able to detect the presence of HbíñHp aggregates in plasma samples from septic patients. Hemolytic anemia complicated by sepsis remains a major health problem in all age groups in both developing and industrialized countries. The release and presence of stroma-free Hb in circulation, robust inflammatory response accompanied by oxidative stress (with production of reactive oxygen and nitrogen species and depletion of antioxidants), microvascular thrombosis and vasoconstriction, and macrophage deactivation leading to immune paralysis are the major hallmarks of severe hemolytic disease and sepsis. Although these critical pathogenesis factors have been identified, the links between them and the mechanisms leading to their appearance are not completely understood.
The primary water-soluble antioxidant of plasma is ascorbate that readily interacts with reactive peroxidase intermediates (52). Decreased levels of plasma ascorbate have been documented during sepsis and displayed strong correlation with the survival (61, 62). Administration of ascorbate was found to protect against circulatory dysfunction in sepsis, reducing the incidence of organ failure and the duration of patient hospitalization (63, 64).
Nitric oxide (NO•) is also utilized as one of the reducing substrates for peroxidases, resulting in its oxidative depletion and possibly dysregulation of vascular tone. This may be enhanced by a loss of control over ascorbate-dependent decomposition of S-nitrosothiols (accumulating in plasma during sepsis), resulting in further apparent deficiency of releasable NO (65). If uninterrupted, the vicious peroxidase cycle can consume the majority of essential antioxidants and cause their depletion and deficiency. Based on the proposed mechanism, targeted strategies can be developed to eliminate HbíñHp aggregates (e.g. via affinity binding), thus breaking the viscous peroxidase cycle.Peroxidase Activity of HemoglobiníñHaptoglobin Complexes
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2781594/íí
Blood. 2013 Feb 21; 121(8): 1276¿C1284.
Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins
Dominik J. Schaer,corresponding author1 Paul W. Buehler,2 Abdu I. Alayash,2 John D. Belcher,3 and Gregory M. Vercellotticorresponding
1Division of Internal Medicine, University Hospital, Zurich, Switzerland;
2Laboratory of Biochemistry and Vascular Biology, Division of Hematology, Center for Biologics Evaluation and Research, Food and Drug Administration, Bethesda, MD; and
3Division of Hematology, Oncology, and Transplantation, University of Minnesota Medical School, Minneapolis, MN
Abstract
Hemolysis occurs in many hematologic and nonhematologic diseases. Extracellular hemoglobin (Hb) has been found to trigger specific pathophysiologies that are associated with adverse clinical outcomes in patients with hemolysis, such as acute and chronic vascular disease, inflammation, thrombosis, and renal impairment. Among the molecular characteristics of extracellular Hb, translocation of the molecule into the extravascular space, oxidative and nitric oxide reactions, hemin release, and molecular signaling effects of hemin appear to be the most critical. Limited clinical experience with a plasma-derived haptoglobin (Hp) product in Japan and more recent preclinical animal studies suggest that the natural Hb and the hemin-scavenger proteins Hp and hemopexin have a strong potential to neutralize the adverse physiologic effects of Hb and hemin. This includes conditions that are as diverse as RBC transfusion, sickle cell disease, sepsis, and extracorporeal circulation. This perspective reviews the principal mechanisms of Hb and hemin toxicity in different disease states, updates how the natural scavengers efficiently control these toxic moieties, and explores critical issues in the development of human plasma¿Cderived Hp and hemopexin as therapeutics for patients with excessive intravascular hemolysis.
Introduction
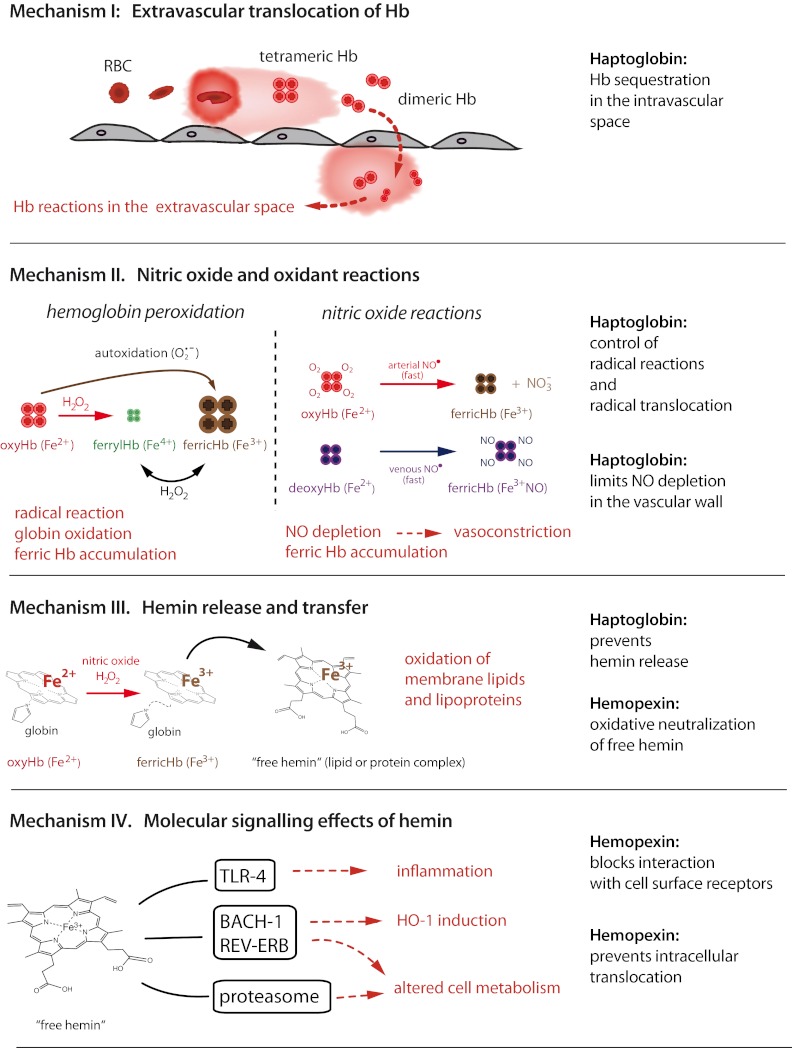
When hemoglobin (Hb) bursts from RBCs because of hemolysis, the naked Hb, devoid of its antioxidant sentries that are normally available within the RBC, can wreak oxidative havoc in the vasculature and in exposed tissues.1 To neutralize Hb and its reactive ferric protoporphyrin-IX group (hemin), specialized plasma scavenger proteins sequester the toxic moieties and transit them to compartments where heme-oxygenases can break down hemin into less toxic metabolites. Other molecules and reducing substances contribute to this protective physiology. However, when these clearance and detoxifying systems are overwhelmed by intravascular hemolysis, such as during sickle cell disease, blood transfusion, malaria, or sepsis, Hb and hemin trigger vascular and organ dysfunction that leads to adverse clinical effects (Figure 1).
Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3578950/íí