¡¡
PD-1/PD-L1 Expression and Blockage in Human Tumors and Viral Infections
Summary
HIF-1alpha drives PD-L1 expression on immunes
Lactate via GPR81 drives PD-L1 expression on tumor cells
Endotoxin LPS drives PD-L1 expression on monocytes and macrophage,gastric cancer cells,thru NF-kB
Chemotherapy induces PD-L1 expression on tumor cells
IL-4/17,IFN-a/gamma,TNF-a,CM-GSF and inflammation induce PD-L1
NFkB increases expression of PD-L1 in ovarion cancer cells
Virus-infected cells THAT express high level of PD-L1 inhibits CTLs (mostly in chronic infections)aa
Expression PK M2 induces PD-L1 expression on immune cells and tumors
Lactate up-regulates the expression of PD-L1 in kidney and causes immunosuppression in septic Acute Renal Injury
Lactate activated PD-1/PD-L1 pathway can induce immunosuppression by inducing apoptosis in lymphocytes in septic AKI. Moreover, blocking GPR81 the receptor of lactate or PD-1/PD-L1 might be a new therapy for septic AKI.
Lactate up-regulates the expression of PD-L1 in kidney and causes immunosuppression in septic Acute Renal Injury - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1684118219301689¡¡
PD-L1 expression in human cancers and its association with clinical outcomes
1School of Medicine and Life Sciences, University of Jinan ¨C Shandong Academy of Medical Sciences, 2Department of Radiation Oncology, Shandong Cancer Hospital and Institute, 3School of Medicine, Shandong University, Jinan, People¡¯s Republic of China
*These authors contributed equally to this work
Abstract: PD-L1 is an immunoinhibitory molecule that suppresses the activation of T cells, leading to the progression of tumors. Overexpression of PD-L1 in cancers such as gastric cancer, hepatocellular carcinoma, renal cell carcinoma, esophageal cancer, pancreatic cancer, ovarian cancer, and bladder cancer is associated with poor clinical outcomes. In contrast, PD-L1 expression correlates with better clinical outcomes in breast cancer and merkel cell carcinoma. The prognostic value of PD-L1 expression in lung cancer, colorectal cancer, and melanoma is controversial. Blocking antibodies that target PD-1 and PD-L1 have achieved remarkable response rates in cancer patients who have PD-L1-overexpressing tumors. However, using PD-L1 as an exclusive predictive biomarker for cancer immunotherapy is questionable due to the low accuracy of PD-L1 immunohistochemistry staining. Factors that affect the accuracy of PD-L1 immunohistochemistry staining are as follows. First, antibodies used in different studies have different sensitivity. Second, in different studies, the cut-off value of PD-L1 staining positivity is different. Third, PD-L1 expression in tumors is not uniform, and sampling time and location may affect the results of PD-L1 staining. Therefore, better understanding of tumor microenvironment and use of other biomarkers such as gene marker and combined index are necessary to better identify patients who will benefit from PD-1/PD-L1 checkpoint blockade therapy.
Keywords: PD-L1, prognostic value, checkpoint blockade, immunotherapy, clinical outcome
[Full text] PD-L1 expression in human cancers and its association with clinical ou | OTT
https://www.dovepress.com/pd-l1-expression-in-human-cancers-and-its-association-with-clinical-ou-peer-reviewed-fulltext-article-OTT¡¡
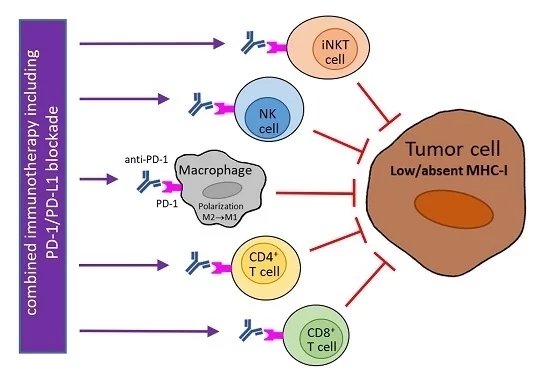
PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression
by Michal ŠmahelOrcID
Department of Genetics and Microbiology, Faculty of Science, Charles University, BIOCEV, Průmyslov¨¢ 595, 25250 Vestec, Czech Republic
Int. J. Mol. Sci. 2017, 18(6), 1331; https://doi.org/10.3390/ijms18061331
Abstract
The therapy of different advanced-stage malignancies with monoclonal antibodies blocking programmed cell death protein 1 (PD-1)/PD-1 ligand 1 (PD-L1) signaling has had an impressive long-lasting effect in a portion of patients, but in most cases, this therapy was not successful, or a secondary resistance developed. To enhance its efficacy in treated patients, predictive biomarkers are searched for and various combination treatments are intensively investigated. As the downregulation of major histocompatibility complex (MHC) class I molecules is one of the most frequent mechanisms of tumor escape from the host¡¯s immunity, it should be considered in PD-1/PD-L1 checkpoint inhibition. The potential for the use of a PD-1/PD-L1 blockade in the treatment of tumors with aberrant MHC class I expression is discussed, and some strategies of combination therapy are suggested.
Keywords: PD-1; PD-L1; checkpoint blockade; MHC class I; tumor escape; cancer immunotherapy; biomarker; interferon gamma
IJMS | Free Full-Text | PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression
https://www.mdpi.com/1422-0067/18/6/1331¡¡
The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance
G¨¹nther Schönrich* and Martin J. Raftery
Charit¨¦ ¨C Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Institute of Virology, Berlin, Germany
Programmed cell death protein (PD-1) and its ligands play a fundamental role in the evasion of tumor cells from antitumor immunity. Less well appreciated is the fact that the PD-1/PD-L1 axis also regulates antiviral immune responses and is therefore modulated by a number of viruses. Upregulation of PD-1 and its ligands PD-L1 and PD-L2 is observed during acute virus infection and after infection with persistent viruses including important human pathogens such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV). Experimental evidence suggests that insufficient signaling through the PD-1 pathway promotes immunopathology during acute infection by exaggerating primary T cell responses. If chronic infection is established, however, high levels of PD-1 expression can have unfavorable immunological consequences. Exhaustion and suppression of antiviral immune responses can result in viral immune evasion. The role of the PD-1/PD-L1 axis during viral infections is further complicated by evidence that PD-L1 also mediates inflammatory effects in the acute phase of an immune response.
In this review, we discuss the intricate interplay between viruses and the PD-1/PD-L1 axis.Concluding Remarks
It is a seductive proposition that a virus induces PD-1 ligands in order to inhibit and thus evade the host immune response. On the other hand, recent data on the regulation of PD-L1 expression during viral infection suggest that PD-L1 upregulation is rather a part of the normal innate response induced by IFNs and PRR signaling. The reason for this is still enigmatic. PD-L1 may have a yet not defined immunostimulatory role in the very early phase of viral infection. Later, it may adjust the quantity and quality of the antiviral CD8+ T cell response in such a way that virus is eliminated with minimal collateral tissue damage. The PD-1/PD-L1 axis may also be important to maintain antiviral Trm cells and Tex cells. Virus-induced PD-1 ligand expression as an immune evasion strategy should always be rigorously tested with this in mind.
Frontiers | The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance | Cellular and Infection Microbiology
https://www.frontiersin.org/articles/10.3389/fcimb.2019.00207/full¡¡
PD-L1 Expression on Retrovirus-Infected Cells Mediates Immune Escape from CD8+ T Cell Killing
Ilseyar Akhmetzyanova,# 1 Malgorzata Drabczyk,# 1 C. Preston Neff, 2 Kathrin Gibbert, 1 Kirsten K. Dietze, 1 Tanja Werner, 1 Jia Liu, 1 , 3 Lieping Chen, 4 Karl S. Lang, 5 Brent E. Palmer, 2 Ulf Dittmer, 1 and Gennadiy Zelinskyy 1 ,*
E. John Wherry, Editor
Author information Article notes Copyright and License information Disclaimer
1 Institute for Virology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany,
2 University of Colorado, Anschutz Medical Campus, Aurora, Colorado, United States of America,
3 Department of Infectious Diseases, Union Hospital of Tonji Medical College, Huazhong University of Science and Technology, Wuhan, P.R. China,
4 Department of Immunobiology, Yale School of Medicine, Yale University, New Haven, Connecticut, United States of America,
5 Institute for Immunology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany,
University of Pennsylvania, UNITED STATES,
#Contributed equally.
Abstract
Cytotoxic CD8+ T Lymphocytes (CTL) efficiently control acute virus infections but can become exhausted when a chronic infection develops. Signaling of the inhibitory receptor PD-1 is an important mechanism for the development of virus-specific CD8+ T cell dysfunction. However, it has recently been shown that during the initial phase of infection virus-specific CD8+ T cells express high levels of PD-1, but are fully competent in producing cytokines and killing virus-infected target cells. To better understand the role of the PD-1 signaling pathway in CD8+ T cell cytotoxicity during acute viral infections we analyzed the expression of the ligand on retrovirus-infected cells targeted by CTLs. We observed increased levels of PD-L1 expression after infection of cells with the murine Friend retrovirus (FV) or with HIV. In FV infected mice, virus-specific CTLs efficiently eliminated infected target cells that expressed low levels of PD-L1 or that were deficient for PD-L1 but the population of PD-L1high cells escaped elimination and formed a reservoir for chronic FV replication. Infected cells with high PD-L1 expression mediated a negative feedback on CD8+ T cells and inhibited their expansion and cytotoxic functions. These findings provide evidence for a novel immune escape mechanism during acute retroviral infection based on PD-L1 expression levels on virus infected target cells.
Author Summary
Virus-specific cytotoxic T cells can eliminate infected cells during acute viral infections, but in chronic infections these cells often become dysfunctional or ¡°exhausted.¡± The inhibitory receptor PD-1 is involved in the suppression of cytotoxic T cell responses in chronic infections. However, during many acute viral infections cytotoxic T cells up-regulate the PD-1 receptor but initially remain competent in killing virus infected target cells. Here we show that the ligand for PD-1, called PD-L1, can be induced on retrovirus infected cells and that the cells with the highest expression of PD-L1 escaped from cytotoxic T cell killing. Thus, PD-L1high infected target cells accumulated during the course of infection, formed the reservoir of virus persistence, and subsequently mediated a negative feedback on cytotoxic T cells via the PD-1 receptor that ultimately resulted in functional exhaustion of these cells. The current results provide evidence for a novel escape mechanism of viruses from cytotoxic T cell responses and may explain how viral reservoirs are established during chronic infections.
¡¡Retrovirus infection induces expression of PD-L1 on target cells in vitro
It has been demonstrated that PD-L1 expression can be induced by inflammatory cytokines [31], however if retrovirus infection of target cells is associated with enhanced levels of PD-L1 expression was less well established. The initial upregulation of PD-L1 on FV-infected target cell suggest that virus infection might influence PD-L1 expression. To test this we performed in vitro infection experiments of spleen cells with FV (Fig 3A). Splenocytes from naïve mice were incubated with F-MuLV infected mouse fibroblasts (M. Dunni). Infection of spleen cells was determined by detection of gp70 expression on the cell surface of the three different cell populations of interest. Gp70 positivity was associated with a significantly enhanced expression of PD-L1 on infected Ter119+, CD19+, and Gr1+ cells in comparison to populations of non-infected cells (Fig 3A). The overall expression levels of PD-L1 were similar on B cells and Gr-1+ cells but much lower on Ter119+ erythroblasts. The percentage of PD-L1high cells also significantly increased after FV infection (Fig 3B). Murine retroviruses have been reported to be sensed by TLR3 and TLR7 [32, 33] and this viral RNA recognition results in the induction of type I IFN responses [34]. Previous studies demonstrate that type I IFNs can stimulate the expression of PD-L1 on the surface of cells [35]. In order to determine whether FV infection induced transcription of IFN¦Á in vitro, we compared the levels of mRNA by RT-PCR in infected versus non-infected Ter119+ cells, CD19+ and Gr-1+ cells. FV infection induced IFN¦Á mRNA expression in CD19+ and Gr-1+ cells (Fig 3C). Thus, viral infection induces IFN¦Á production in vitro and in vivo [34], but does IFN¦Á influence the expression of PD-L1 on the surface of FV infected cells? To address this question spleen cells from wild type and IFN receptor deficient mice (IFNR1-/-) [36] were infected in vitro. FV infection more efficiently enhanced the PD-L1 expression level on the surface of gp70+ CD19+ cells and on gp70+ Gr-1+ cells from WT mice in comparison to IFNR1-/- mice (Fig 3D). The expression of PD-L1 on the surface of FV infected CD19+ and Gr-1+ cells was significantly enhanced after treatment with exogenous IFN¦Á (Fig 3E). This effect was not observed on FV infected cells from IFNR1-/- mice. Moreover, gp70+ B cells and gp70+ Gr-1+ cells from FV infected IFNR1-/- mice expressed significantly less PD-L1 than those cells from WT mice (Fig 3F). This data suggest that type I interferon signaling is involved in PD-L1 expression and that virus induced IFN¦Á is at least one important regulator of PD-L1 expression on infected cells during the early phase of FV infection.
¡¡
Fig 3
PD-L1 expression on cells infected in vitro with FV or HIV.
Spleen cells were isolated from naive B6 mice and cultivated with F-MuLV infected Mus Dunni cells to infect mouse cells in vitro. Multi-parameter flow cytometry was used to determine PD-L1 expression (MFI) (A) and the percentage of PD-L1high cells (B) in different target cell populations of FV. C. Ter119+, CD19+, and Gr-1+ cells were isolated from naïve wild type mice and were infected with F-MuLV in vitro. mRNA from infected and non-infected cells was isolated for real time PCR quantification of the IFN¦Á mRNA expression. The numbers of IFN¦Á mRNA copies in relation to 105 copies of mRNA for ¦Â-actin is shown. Data was pooled from at least two independent experiments with similar results. Spleen cells were isolated from naїve wild type mice or from naïve IFNAR1-/- mice and cultivated with F-MuLV infected Mus Dunni cells to infect mouse cells in vitro. Multi-parameter flow cytometry was used to determine PD-L1 expression (MFI) on infected CD19+ and Gr-1+ cells (D) and in the presence of IFN¦Á (E) Data was pooled from at least two independent experiments with similar results. F. Multi-parameter flow cytometry was used to determine the expression of PD-L1 on the sur-face of gp70+Ter119+, gp70+CD19+, and gp70+Gr-1+ cells isolated from spleens of 6 day FV infected WT and IFNAR1-/- mice. Data was pooled from two independent experiments with similar results. Multi-parameter flow cytometry was used to determine the expression of PD-L1 on the surface of human CD4+ T cells after HIV-1 infection. Representative histograms of PD-L1 expression on human CD4+ T cells non-stimulated and non-infected, stimulated in vitro with PHA and infected with HIV-1 or cells only stimulated with PHA are shown. The data is shown for day three, seven and ten after infection (G). Expression of PD-L1 on human CD4+ T cells (H) and the percentage of PD-L1high CD4+ T cells (I) in populations of non-stimulated and non-infected, stimulated in vitro with PHA and infected with HIV-1 or cells only stimulated with PHA are shown at day ten after infection. Mean numbers plus SD from three independent experiments with similar results was shown. Differences between FV infected (gp70+) and FV non-infected (gp70-) mice cells were analyzed by an unpaired t-test. Differences between HIV infected (p24+) and HIV non-infected (p24-) CD4+ cells were analyzed by Mann-Whitney t test. Statistically significant differences between the groups are indicated in the figure (*p˂0.05, **p˂0.005).In order to show that human cells show enhanced expression of PD-L1 after infection with retrovirus as well, HIV infection of human CD4+ T cell was performed. It has previously been reported that the overall T cell compartment from HIV-infected patients expresses increased levels of PD-L1 [37], but this has not been attributed to virus infection on a single cell level. Therefore, we investigated the PD-L1 expression on CD4+ T cells from uninfected donors at day 3, 7 and 10 post HIV infection in vitro. Staining against intracellular p24 antigen was used to identify HIV infected cells. A representative histogram plot of a PD-L1 staining shows that PHA alone (which is needed to activate T cells so they become permissive to in vitro infection) increased PD-L1 expression but HIV infection further enhanced this expression during the course of infection (Fig 3G). Cumulative data of day 10 post infection from 8 donors shows that PD-L1 expression on HIV-infected CD4+ T cells (p24+) was significantly increased compared to p24- cells from the same cultures (Fig 3H). Similar results were achieved by determining the percentages of infected CD4+ cells expressing high levels of PD-L1 (Fig 3I). These data suggest that HIV infection of CD4+ T cells also causes increased expression of PD-L1 that may help the virus to evade antiviral CTL responses.
¡¡
Up-regulation of PD-L1 on FV infected cells protect them from CTL killing
Cytotoxic CD8+ T cells mediate elimination of infected target cells after day 6 of FV infection [26]. We hypothesize that cells with low-level expression of PD-L1 are more susceptible to CTL killing than cells expressing high levels of this molecule, which escape elimination by PD-1high CTL and subsequently get enriched (Fig 2). In order to confirm this theory an in vivo CTL killing assay was performed (Fig 4A). This assay allows to differentially detect the elimination of different donor cell populations in the same mouse. Cells from 5 day infected mice (low level of PD-L1) and cells from mice infected for 9 days (high level of PD-L1) (Fig 4B and 4C) were loaded with FV DbGagL peptide [38, 39], mixed 1:1 and adoptively transferred as target cells for virus-specific CD8+ T cells into FV infected mice (Fig 4A). The elimination of both populations of target cells was simultaneously analyzed in the same infected donor mouse (connected dots in Fig 4). This analysis was performed in the spleen (Fig 4D) and bone marrow (Fig 4E), because those are the organs with the highest FV loads and the strongest CTL activity [26]. In both organs elimination of target cells from donor mice was detected. However, the killing of splenocytes from 5 day infected mice (PD-L1low) was significantly higher in comparison to splenocytes from 9 day infected mice (PD-L1high). This confirms that enhanced expression of inhibitory ligands protected infected target cells from CTL killing.
¡¡In order to directly demonstrate the involvement of PD-L1 in the escape of target cells from CTL killing in vivo, two killing experiments with an impaired PD-L1 function on target cells were performed. Splenocytes from 9 day infected wild type mice or PD-L1 knockout mice were used as targets for CTL in vivo (Fig 4F). As expected, PD-L1 deficient target cells were more efficiently eliminated than wild type cells. Another modification of the in vivo CTL assay was performed to directly show the influence of PD-L1 on the efficacy of target cell killing. Half of the FV DbGagL peptide loaded spleen cells from 9 day infected mice (PD-L1high) were treated with blocking anti-PD-L1 antibodies and the other half with an isotype control antibody prior to their adoptive transfer into infected donor mice (Fig 4H). In all recipient mice splenocytes treated with anti-PD-L1 antibodies were killed significantly better than the cells with the control antibody. If the same experiment was performed with spleen cells from 5 day infected mice that express only low levels of PD-L1 either slight enhancement or reduction of target cell elimination after treatment with anti-PD-L1 antibodies was observed (Fig 4G). If a 10% increase of target cell killing in the population of anti-PD-L1 treated target cells was considered as significant enhancement compared to the isotype control, then only 2 out of 10 animals showed that in the group of mice receiving 5 day infected cells (Fig 5G), whereas all mice (5 out of 5) showed this in the 9 day group (Fig 5H). These different numbers were analyzed with the Mann-Whitney-Rank test, which indicates a significant difference between the groups with a p-value of 0,0014.
The data clearly indicate that PD-1 signaling is influencing the efficacy of CTL killing during acute FV infection.
To analyze whether the differences in PD-L1 expression between the Ter119+, CD19+ and Gr-1+ infected target cell populations and their differences in survival during acute FV infection correlated with CTL mediated elimination, we determined the killing of specific target cell subsets from 7 day infected mice in our in vivo CTL assay (Fig 4I). FV-specific CD8+ T cells most efficiently eliminated the Ter119+ cells (PD-L1low, Fig 2A), whereas killing of B cells (PD-L1high, Fig 2B) and Gr-1+ cells (PD-L1high, Fig 2C) was significantly reduced to half or even less than half of the Ter119+ cell killing. Thus, the PD-L1 expression levels on the different populations of infected target cells clearly correlated with the ability of virus-specific CTL to kill these cells during acute FV infection. In order to show the functional effect of the PD-L1 signaling on the CTL killing of different subpopulations, FV DbGagL peptide loaded spleen cells from 9 day infected wild type or PD-L1 knockout mice were used as targets for the killing assay (Fig 4J). When PD-L1 was absent the large difference in target cell elimination between Ter119+ cell and CD19+/Gr-1+ was gone as infected B cells and myeloid cells from PD-L1 knockout mice were significantly better eliminated than those from wild type mice. Thus, the lack of PD-L1 on the surface of infected target cells resulted in an enhanced susceptibility to CD8 T cell mediated killing.High expression of PD-L1 on infected target cells suppresses the functionality of CD8+ T cells
Virus-specific CD8+ T cells form tight contacts with infected target cells called cytotoxic synapses. Binding of virus-specific PD-1high CTLs to infected targets with high expression of PD-L1 may therefore have functional consequences for the effector CD8+ T cell. In order to analyze this, we infected PD-L1 knockout mice and compared their CTL response with wild type animals (Fig 6A and 6B). The absence of PD-L1 resulted in enhanced expansion of virus-specific CD8+ T cells and augmented production of the cytotoxic molecule granzyme B by FV-specific (tetramer+) CD8+ T cells. The expanded effector CD8+ T cells efficiently controlled FV infection in PD-L1 knockout mice (Fig 6C). Similar results were obtained from 10 day infected wild type mice treated once at day 7 with anti-PD-L1 blocking antibody (Fig 6D). When binding of PD-L1 to the PD-1 receptor was blocked by the antibody more granzyme B was produced by activated CD43+CD8+ T cells. These ex vivo data demonstrate the regulatory role of PD-L1 on the functionality of CD8+ T cells during acute FV infection.
High expression of PD-L1 on infected target cells suppresses the functionality of CD8+ T cells
Virus-specific CD8+ T cells form tight contacts with infected target cells called cytotoxic synapses. Binding of virus-specific PD-1high CTLs to infected targets with high expression of PD-L1 may therefore have functional consequences for the effector CD8+ T cell. In order to analyze this, we infected PD-L1 knockout mice and compared their CTL response with wild type animals (Fig 6A and 6B). The absence of PD-L1 resulted in enhanced expansion of virus-specific CD8+ T cells and augmented production of the cytotoxic molecule granzyme B by FV-specific (tetramer+) CD8+ T cells. The expanded effector CD8+ T cells efficiently controlled FV infection in PD-L1 knockout mice (Fig 6C). Similar results were obtained from 10 day infected wild type mice treated once at day 7 with anti-PD-L1 blocking antibody (Fig 6D). When binding of PD-L1 to the PD-1 receptor was blocked by the antibody more granzyme B was produced by activated CD43+CD8+ T cells. These ex vivo data demonstrate the regulatory role of PD-L1 on the functionality of CD8+ T cells during acute FV infection.
PD-L1 Expression on Retrovirus-Infected Cells Mediates Immune Escape from CD8+ T Cell Killing
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4617866/¡¡
Programmed death 1 expression in cancer and infection: tissue versus periphery
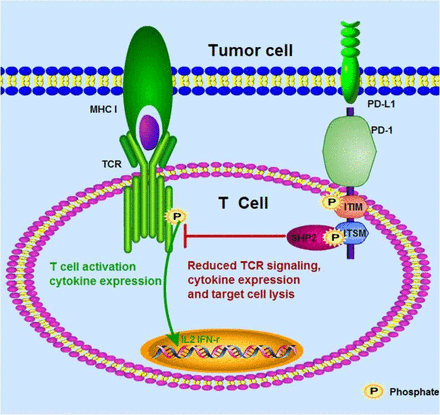
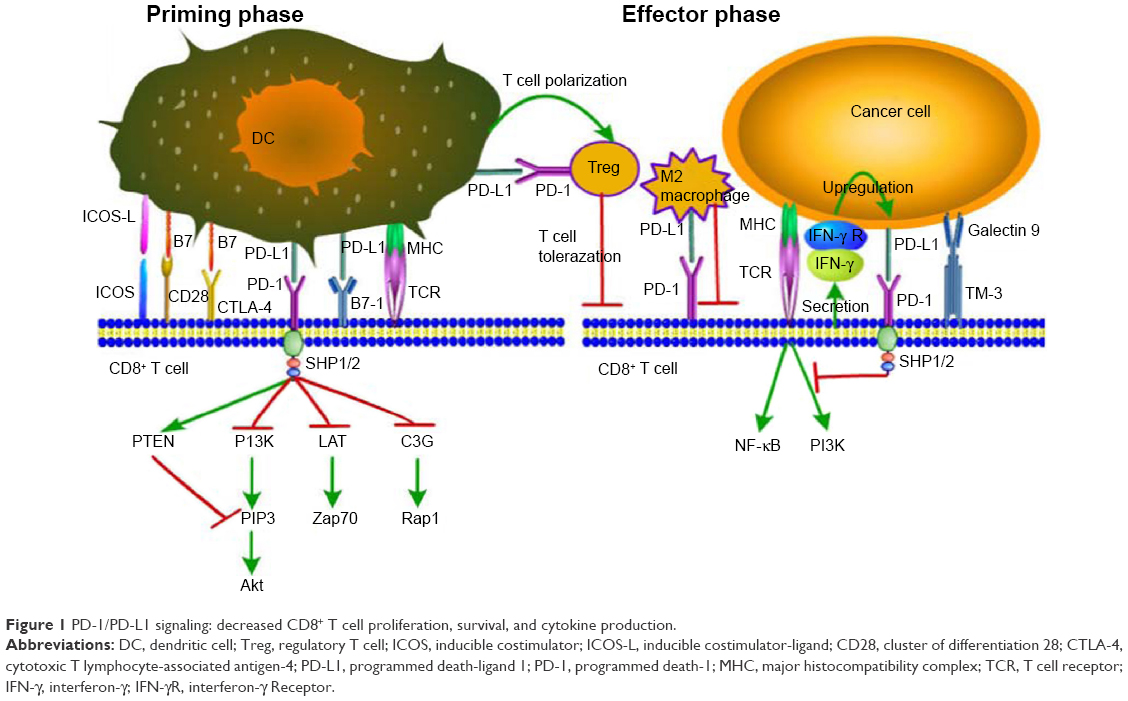
The chronicity of cancer and persistent infections provides constant antigen exposure to antigen-reactive T-cells, leading to cellular exhaustion and abrogation of effector functions.1 The expression of cell surface-bound molecules such as programmed cell death protein 1 (PD-1/CD279) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4/CD152) on antigen-specific T-cells are markers of exposure to immunogenic stimuli.2 PD-1 and CTLA-4 identify as immune checkpoints due to their crucial role in regulating the magnitude and quality of T-cell responses. Other immune checkpoint molecules of clinical relevance are the T-cell immunoglobulin and mucin-domain containing 3 (TIM-3), with its nominal ligand galectin 9, and to a lesser extent high-mobility group Box 1 (HMGB1)3 and the lymphocyte-activation gene 3 (LAG-3), which binds to major histocompatibility complex (MHC) class II molecules with higher affinity than the CD4 receptor.4
This review focuses on PD-1 and its ligands. PD-1 expression on T-cells is rapidly induced after antigen exposure, following engagement between the T-cell receptor (TCR) and its cognate epitope-loaded MHC molecule in the draining lymph nodes.5 PD-1 RNA is detectable in T-cells as rapidly as 2 h post TCR-specific stimulation.6 In addition to TCR-dependence, the presence of interleukin 2 (IL-2), IL-7, IL-15, vascular endothelial growth factor (VEGF), IL-6, and transforming growth factor beta (TGF-¦Â) in the local cytokine milieu in lymph nodes and diseased tissue additionally contribute to PD-1 up-regulation on T-cells.7, 8, 9, 10 PD-1 interacts with its ligands, namely PD-L1 (B7-H1/CD274) and PD-L2 (B7-DC/CD273). The PD-1 ligands are expressed on transformed cells, professional antigen-presenting cells (pAPCs), and epithelial cells, as well as T-cells.11 This interaction provides signals that tolerize T-cells to their antigenic targets, disarming their effector functions.1, 7 Type 1 interferons (IFN-¦Á/¦Â) and tumour necrosis factor alpha (TNF-¦Á) can up-regulate PD-L1 expression on T-cells, B-cells, natural killer (NK) cells, myeloid cells, and epithelial cells,5, 7 while PD-L2 expression is inducible via interferon gamma (IFN-¦Ã), granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-4 signalling.7 Figure 1 is a schematic representation of the factors driving PD-1 and PD-L1/2 expression by T-cells and transformed cells, respectively.
Figure 1
Factors that induce PD-1, PD-L1, and PD-L2 expression in host cells. Although the major trigger for PD-1 expression on T-cells is TCR engagement with the epitope-loaded MHC molecule on transformed cells (infected or cancerous cells), a cytokine milieu consisting of proinflammatory mediators such as IFN-¦Ã, type 1 interferons, IL-6, VEGF, and IL-2 augment the activation of this immune checkpoint pathway. In addition, cytokines important for T-cell maintenance, i.e., IL-7 and IL-15, also contribute to PD-1 expression during chronic disease. TGF-¦Â is another physiologically important cytokine that induces PD-1 expression and potentiates regulatory T-cell activity. Antigenic molecules such as Mycobacterium tuberculosis LAM can activate the PD-1 pathway in T-cells via TLR2 signalling. PD-L1/2 expression on transformed host cells is inducible by exposure to IFN-¦Ã, type 1 interferons, TNF-¦Á, and GM-CSF − thus indicative of intense inflammation. Bacterial LPS can also induce PD-L1 expression, via TLR4 involvement and NF-¦ÊB activation. The anti-inflammatory cytokine IL-4 can also trigger PD-L2 expression. Abbreviations: TCR, T-cell receptor; LAM, lipoarabinomannan; LPS, lipopolysaccharide; TLR, toll-like receptor; NF-¦ÊB, nuclear factor kappa B.
A summary of PD-1 expression by various human cell types is presented in Table 1.88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 Biologically, PD-1 expression may play a beneficial role in ameliorating and curbing the development of autoimmune disease − although this is not true for cancer and infections.12 PD-1 expression on T-cells identifies immune cells that specifically recognize and react to transformed as well as to pathogen-derived antigens. More recent studies have shown that PD-1+ tumour-infiltrating T-cells (TIL) contain a distinct repertoire of tumour-reactive CD8 T-cells that express oligoclonal TCRs, specific for mutated tumour epitopes, termed neoantigens.13 PD-1+ CD8 T-cells recognizing the patient¡¯s cancer cells are also found in peripheral blood mononuclear cells (PBMCs), albeit in significantly lower numbers compared to the tumour microenvironment.13 PBMC-derived neoantigen-specific CD8 T-cells can also be expanded ex vivo for cellular therapy.14 These PD-1+ CD8 TILs co-express additional surface markers associated with activation and potential tolerance, such as TIM-3, LAG-3, and the cytotoxic lymphocyte (CTL) activation marker 4-1BB.13
¡¡
Table 1Summary of PD-1, PD-L1, and PD-L2 in a variety of human cell types. Cell type PD-1 expression level PD-L1 expression level PD-L2 expression level Ref. ¡¡ Constitutive Cytokine-induced Constitutive Cytokine-induced Constitutive Cytokine-induced ¡¡ Naive T-cells Nil Yes Nil Yes Nil Yes 5,7,18,31, Activated T-cells Yes Increased Yes Increased Nil Yes 61,88,89, 90,91, 92,93, 94,95, 96,97, 98,99 Exhausted T-cells Yes Increased Yes Increased Nil Yes ¡¡ Regulatory T-cells Yes Increased Yes Increased Nil Yes ¡¡ NK cells Nil Yes Nil Yes Nil Yes ¡¡ Naive B-cells Yes Increased Yes Increased Yes Increased ¡¡ Plasma cells Yes Increased Yes Increased Yes Increased ¡¡ Plasmacytoid DCs Nil Nil Yes Increased Yes Increased ¡¡ Myeloid DCs Nil Yes Yes Increased Yes Increased ¡¡ Macrophages Nil Yes Yes Increased Yes Increased ¡¡ Monocytes Nil Yes Yes Increased Nil Yes ¡¡ Airway and lung epithelia Nil Nil Nil Yes Nil Yes ¡¡ Liver non-parenchymal cells Nil Yes Yes Increased Yes Increased ¡¡ Mesenchymal stromal cells Nil Nil Yes Increased Yes Increased ¡¡ Pancreatic islets Nil Nil Yes Increased Yes Increased ¡¡ Fibroblasts Nil Nil Yes Increased Yes Increased ¡¡ HUVEC Nil Nil Nil Yes Yes Increased
¡¡¡¡
Anti-PD-1/PD-L1 therapy for infectious diseases: learning from the cancer paradigm - International Journal of Infectious Diseases
https://www.ijidonline.com/article/S1201-9712(17)30031-0/fulltext¡¡
The effect of chemotherapy on PD-1(programmed cell death 1)/PD-L1( programmed cell death 1 ligand) axis: Some chemotherapeutical drugs may finally work through immune response
Most tumors are immunogenic which would trigger some immune response. Chemotherapy also has immune potentiating mechanisms of action. But it is unknown whether the immune response is associated with the efficacy of chemotherapy and the development of chemoresistance. Recently, there is a growing interest in immunotherapy, among which the co-inhibitory molecules, programmed cell death 1/programmed cell death 1 ligand (PD-1/PD-L1) leads to immune evasion. Since some reports showed that conventional chemotherapeutics can induce the expression of PD-L1, we try to summarize the effect of chemotherapy on PD-1/PD-L1 axis and some potential molecules relevant to PD-1/PD-L1 in chemoresistance in this review.
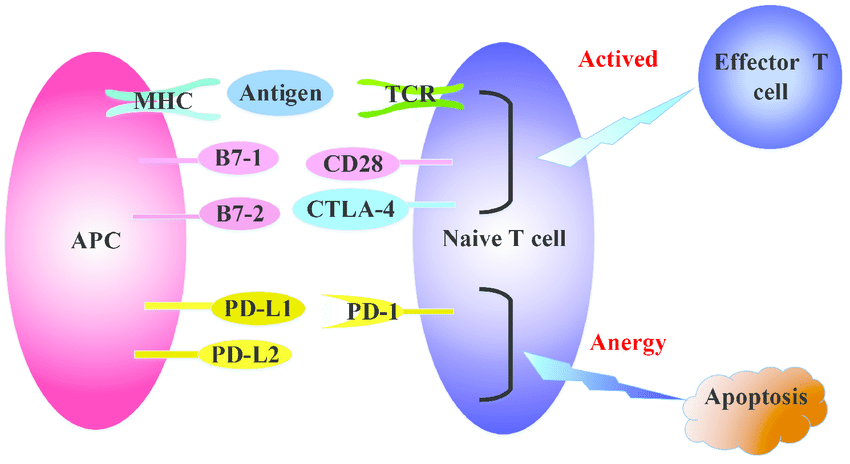
The regulation of T cell activation. T cell receptor (TCR) recognizes the tumor antigens in the context of major histocompatibility complex (MHC) expressed on professional antigen presenting cells (APCs). Then APCs deliver a second signal by positive co-stimulatory molecules CD28/B7-1/B7-2 to fully activate naive T cells. CTLA-4 can bind with B7-1/B7-2 competitively to inhibit the activation of CD28/B7-1/B7-2, resulting in inactivation of T cells. PD-1 binds with its ligands, PD-L1 and PD-L2 to attenuate lymphocyte activation.¡¡
figure
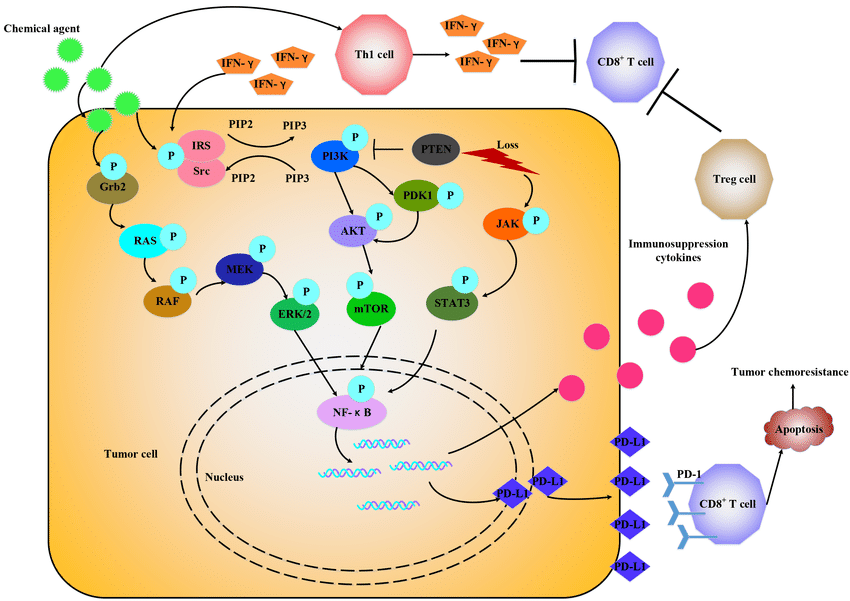
Chemotherapeutic agents promote PD-1/PD-L1 expression through various signals. Chemotherapeutic agents via IFN-¦Ã-dependent and IFN-¦Ã-independent way to upregulate PD-L1 expression by activating different signals, like RAS/RAF, PI3K/ AKT, JAK/STAT3 and release some immune suppression cytokines to attenuate antitumor immune response.
(PDF) The effect of chemotherapy on programmed cell death 1/ programmed cell death 1 ligand axis: Some chemotherapeutical drugs may finally work through immune response
https://www.researchgate.net/publication/296055196_The_effect_of_chemotherapy_on_programmed_cell_death_1_programmed_cell_death_1_ligand_axis_Some_chemotherapeutical_drugs_may_finally_work_through_immune_response
¡¡Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells.
https://www.nature.com/articles/onc2017188
Jun 12, 2017 ¡¤
Feng J1, Yang H2, Zhang Y3, Wei H4, Zhu Z5, Zhu B6, Yang M7, Cao W7, Wang L1, Wu Z3,8.
Author information
1
Medical Oncology, The First Affiliated Hospital of Wannan Medical College, Wuhu, China.
2
The Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Tianjin Lung Cancer Center and Institute, Tianjin Medical University General Hospital, Tianjin, China.
3
Anhui Province Key laboratory of Active Biological Macro-molecules Research, Wannan Medical College, Wuhu, China.
4
Department of Central Laboratory, Wannan Medical College, Wuhu, China.
5
School of Pharmacy, Wannan Medical College, Wuhu, China.
6
School of Forensic Medicine, Wannan Medical College, Wuhu, China.
7
School of Anesthesiology, Wannan Medical College, Wuhu, China.
8
School of Preclinical Medicine, Wannan Medical College, Wuhu, China.
Abstract
The clinical success of immunotherapy that inhibits the negative immune regulatory pathway programmed cell death protein 1/PD-1 ligand (PD-1/PD-L1) has initiated a new era in the treatment of metastatic cancer. PD-L1 expression is upregulated in many solid tumors including lung cancer and functions predominantly in lactate-enriched tumor microenvironments. Here, we provided evidence for PD-L1 induction in response to lactate stimulation in lung cancer cells. Lactate-induced PD-L1 induction was mediated by its receptor GPR81. The silencing of GPR81 signaling in lung cancer cells resulted in a decrease in PD-L1 protein levels and functional inactivation of PD-L1 promoter activity. In addition, GPR81-mediated upregulation of PD-L1 in glucose-stimulated lung cancer cells that recapitulates the enhanced glycolysis in vivo was dependent on lactate dehydrogenase A (LDHA). We also demonstrated that activation of GPR81 decreases intracellular cAMP levels and inhibits protein kinase A (PKA) activity, leading to activation of the transcriptional coactivator TAZ. Interaction of TAZ with the transcription factor TEAD was essential for TAZ activation of PD-L1 and induction of its expression. Furthermore, we found that lactate-induced activation of PD-L1 in tumor cells led to reduced production of interferon-¦Ã and induction of apoptosis of cocultured Jurkat T-cell leukemia cells. Our findings reveal an unexpected role of lactate in contributing to tumor cell protection from cytotoxic T-cell targeting and establishes a direct connection between tumor cell metabolic reprograming and tumor evasion from the immune response.¡¡
°©»ùÒò¡£ 2017Äê10ÔÂ19ÈÕ; 36£¨42£©£º5829-5839¡£ doi£º10.1038 / onc.2017.188¡£ EPUB 2017Äê6ÔÂ12ÈÕ¡£
Ö×Áöϸ°ûÀ´Ô´µÄÈéËáÑÎͨ¹ýGPR81ÓÕµ¼È˷ΰ©Ï¸°ûÖÐTAZÒÀÀµÐÔµÄPD-L1Éϵ÷¡£
·ëJ1£¬ÑîH2£¬ÕÅY3£¬ÎºH4£¬ÖìZ5£¬ÖìB6£¬ÑîM7£¬²ÜW7£¬ÍõL1£¬ÎâZ3,8¡£
×÷ÕßÐÅÏ¢
1¸ö
ÎߺþÊÐÍîÄÏҽѧԺ¸½ÊôµÚһҽԺҽѧÖ×Áö¿Æ¡£
2
Ìì½òÒ½¿Æ´óѧ×ÜÒ½ÔºÌì½ò·Î°©ÖÐÐĺÍÑо¿Ëù£¬Ìì½òÊзΰ©×ªÒÆÓëÖ×Áö΢»·¾³Ìì½òÊÐÖصãʵÑéÊÒ£¬Ìì½ò¡£
3
ÍîÄÏҽѧԺ£¬°²»ÕÊ¡»îÐÔÉúÎï´ó·Ö×ÓÑо¿ÖصãʵÑéÊÒ£¬Îߺþ¡£
4
ÍîÄÏҽѧԺÖÐÑëʵÑéÊÒ£¬Îߺþ¡£
5
ÍîÄÏҽѧԺҩѧԺ£¬Îߺþ¡£
6
ÍîÄÏҽѧԺ·¨Ò½Ñ§Ôº£¬Îߺþ¡£
7
ÍîÄÏҽѧԺÂé×íѧԺ£¬Îߺþ¡£
8
ÍîÄÏҽѧԺ»ù´¡Ò½Ñ§Ôº£¬Îߺþ¡£
¡¡ÕªÒª
ÒÖÖÆÒõÐÔÃâÒßµ÷½Ú;¾¶³ÌÐòÐÔϸ°ûËÀÍöµ°°×1 / PD-1ÅäÌ壨PD-1 / PD-L1£©µÄÃâÒßÁÆ·¨µÄÁÙ´²³É¹¦¿ª´´ÁËתÒÆ°©ÖÎÁƵÄмÍÔª¡£ PD-L1±í´ïÔÚ°üÀ¨·Î°©ÔÚÄÚµÄÐí¶àʵÌåÁöÖÐÉϵ÷£¬²¢ÇÒÖ÷ÒªÔÚ¸»º¬ÈéËáµÄÖ×Áö΢»·¾³Öз¢»Ó×÷Óá£ÔÚÕâÀÎÒÃÇÌṩÁ˶Էΰ©Ï¸°ûÖÐÈéËá´Ì¼¤µÄPD-L1ÓÕµ¼µÄÖ¤¾Ý¡£ÈéËáÓÕµ¼µÄPD-L1ÓÕµ¼ÊÇÓÉÆäÊÜÌåGPR81½éµ¼µÄ¡£·Î°©Ï¸°ûÖÐGPR81ÐźÅתµ¼µÄ³ÁĬµ¼ÖÂPD-L1µ°°×ˮƽ½µµÍºÍPD-L1Æô¶¯×Ó»îÐÔ¹¦ÄÜʧ»î¡£´ËÍ⣬GPR81½éµ¼µÄÆÏÌÑÌǴ̼¤µÄ·Î°©Ï¸°ûÖÐPD-L1µÄÉϵ÷¸ÅÀ¨ÁËÌåÄÚÔöÇ¿µÄÌǽͽ⣬ÕâÒÀÀµÓÚÈéËáÍÑÇâøA£¨LDHA£©¡£ÎÒÃÇ»¹Ö¤Ã÷£¬GPR81µÄ¼¤»î»á½µµÍϸ°ûÄÚcAMPµÄˮƽ£¬²¢ÒÖÖƵ°°×¼¤Ã¸A£¨PKA£©µÄ»îÐÔ£¬´Ó¶øµ¼ÖÂת¼¹²¼¤»îÒò×ÓTAZµÄ¼¤»î¡£ TAZÓëת¼Òò×ÓTEADµÄÏ໥×÷ÓöÔÓÚTAZ¼¤»îPD-L1ºÍÓÕµ¼Æä±í´ïÖÁ¹ØÖØÒª¡£´ËÍ⣬ÎÒÃÇ·¢ÏÖ£¬ÈéËáÓÕµ¼µÄÖ×Áöϸ°ûÖÐPD-L1µÄ¼¤»îµ¼Ö¸ÉÈÅËØ-¦ÃµÄ²úÉú¼õÉÙÒÔ¼°ÓÕµ¼¹²ÅàÑøµÄJurkat Tϸ°û°×Ѫ²¡Ï¸°ûµòÍö¡£ÎÒÃǵķ¢ÏÖ½ÒʾÁËÈéËáÔÚ´Ù½øÖ×Áöϸ°ûÃâÊÜϸ°û¶¾ÐÔTϸ°û°ÐÏòµÄ×÷ÓÃÖз¢»ÓÁ˳öºõÒâÁϵÄ×÷Ó㬲¢ÔÚÖ×Áöϸ°û´úлÖرà³ÌÓëÃâÒßÓ¦´ðÌÓ±ÜÖ×ÁöÖ®¼ä½¨Á¢ÁËÖ±½ÓÁªÏµ¡£Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28604752¡¡
PD-L1 Overexpression During Endotoxin Tolerance Impairs the Adaptive Immune Response in Septic Patients via HIF1¦Á
Innate Immunity Group
Tumor Immunology Lab, IdiPAZ, La Paz University Hospital, Madrid, Spain
The Journal of Infectious Diseases, Volume 217, Issue 3, 1 February 2018, Pages 393¨C404, https://doi.org/10.1093/infdis/jix279
Published: 29 July 2017 Article history
Abstract
Sepsis, among other pathologies, is an endotoxin tolerance (ET)¨Crelated disease. On admission, we classified 48 patients with sepsis into 3 subgroups according to the ex vivo response to lipopolysaccharide. This response correlates with the Acute Physiology and Chronic Health Evaluation (APACHE) II score and the ET degree. Moreover, the ET-related classification determines the outcome of these patients. Programmed cell death¨Cligand 1 (PD-L1) expression on septic monocytes is also linked with ET status. In addition to the regulation of cytokine production, one of the hallmarks of ET that significantly affects patients with sepsis is T-cell proliferation impairment or a poor switch to the adaptive response. PD-L1/programmed cell death-1 (PD-1) blocking and knockdown assays on tolerant monocytes from both patients with sepsis and the in vitro model reverted the impaired adaptive response. Mechanistically, the transcription factor hypoxia-inducible factor-1¦Á (HIF1¦Á) has been translocated into the nucleus and drives PD-L1 expression during ET in human monocytes. This fact, together with patient classification according to the ex vivo lipopolysaccharide response, opens an interesting field of study and potential personalized clinical applications, not only for sepsis but also for all ET-associated pathologies.RESULTS
Some studies of patients with sepsis have reported the upregulation of both PD-L1 and PD-1 on monocytes and T cells, respectively
PD-L1 Expression in Patients With Sepsis Correlates With Their ET Status
PD-L1/PD-1 Crosstalk Controls the Impairment of T-Lymphocyte Proliferation During ET
HIF1¦Á Drives PD-L1 Expression on ET Monocytes
HIF1¦Á Modulates T-Cell Response Through PD-L1 Overexpression in ET Patients With Sepsis
Graphical resume. Tolerant monocytes are characterized by a deregulated inflammatory response, increased phagocytosis, and impaired lymphocyte proliferation. The immunosuppressive phenotype is mediated by IRAK-M expression and the reduction of lymphocyte proliferation is mediated by PD-L1 expression. However, the master regulator of IRAK-M and PD-L1 is transcription factor HIF1¦Á, which also increases phagocytosis capacity in monocytes under ET. Abbreviations: ET, endotoxin tolerance; HIF1¦Á, hypoxia-inducible factor-1¦Á; IRAK-M, interleukin-1 receptor-associated kinase¨CM; PD-1, programmed cell death-1; PD-L1, programmed cell death¨Cligand 1; TLR-4, Toll-like receptor 4.The reported low absolute lymphocyte count during sepsis [48] could be explained by a potential PD-L1¨Cinduced apoptosis (Supplementary Figure 1).
In conclusion, the present study has demonstrated that human blood sepsis monocytes under ET expressed high levels of PD-L1 on their cell surface via the induction of HIF1¦Á, and governed the impaired induction of T-cell proliferation during ET. The mechanism described completes the picture of the ET hallmarks and the factors involved. In addition, our findings open a potential clinical application, not only for patients with sepsis but also for those ET-related pathologies.
ÄÚ¶¾ËØÄÍÊÜÆÚ¼äPD-L1¹ý±í´ïÏ÷ÈõÁËŧ¶¾Ö¢»¼Õßͨ¹ýHIF1¦ÁµÄÊÊÓ¦ÐÔÃâÒß·´Ó¦
ÏÈÌìÃâÒßС×é
Î÷°àÑÀÂíµÂÀïÀ°Í˹´óѧҽԺIdiPAZÖ×ÁöÃâÒßѧʵÑéÊÒ
´«È¾²¡ÔÓÖ¾£¬µÚ217¾í£¬µÚ3ÆÚ£¬2018Äê2ÔÂ1ÈÕ£¬µÚ393-404Ò³£¬https£º//doi.org/10.1093/infdis/jix279
·¢²¼£º2017Äê7ÔÂ29ÈÕÎÄÕÂÀúÊ·
ÕªÒª
ŧ¶¾Ö¢ÊÇÆäËûÓëÄÚ¶¾ËØÄÍÊÜÐÔ£¨ET£©Ïà¹ØµÄ¼²²¡¡£ÈëԺʱ£¬ÎÒÃǸù¾Ý¶ÔÖ¬¶àÌÇ£¨LPS)µÄÀëÌå·´Ó¦½«48Ãûŧ¶¾Ö¢»¼Õß·ÖΪ3¸öÑÇ×é¡£¸Ã·´Ó¦Óë¼±ÐÔÉúÀíºÍÂýÐÔ½¡¿µÆÀ¹À£¨APACHE£©II·ÖÊýºÍET³Ì¶ÈÏà¹Ø¡£´ËÍ⣬ÓëETÏà¹ØµÄ·ÖÀà¾ö¶¨ÁËÕâЩ»¼ÕßµÄÔ¤ºó¡£»¯Å§ÐÔµ¥ºËϸ°ûÉϵijÌÐòÐÔϸ°ûËÀÍöÅäÌå1£¨PD-L1£©±í´ïÒ²ÓëET״̬Óйء£³ýÁ˵÷½Úϸ°ûÒò×ӵIJúÉúÍ⣬ET¶Ôŧ¶¾Ö¢»¼ÕßµÄÏÔ×ÅÓ°ÏìÖ®Ò»ÊÇTϸ°ûÔöÖ³ÕÏ°»ò¶ÔÊÊÓ¦ÐÔ·´Ó¦µÄ²»Á¼×ª»»¡£¶Ôŧ¶¾Ö¢»¼ÕߺÍÌåÍâÄ£Ð͵ÄÄÍÊÜÐÔµ¥ºËϸ°û½øÐÐPD-L1 /³ÌÐòÐÔϸ°ûËÀÍö1£¨PD-1£©×è¶ÏºÍÇõÍÊÔÑé¿ÉÄæתÊÜËðµÄÊÊÓ¦ÐÔ·´Ó¦¡£´Ó»úÖÆÉϽ²£¬×ªÂ¼Òò×ÓµÍÑõÓÕµ¼Òò×Ó-1¦Á£¨HIF1¦Á£©ÒÑתÒƵ½ºËÖУ¬²¢ÔÚÈ˵¥ºËϸ°ûµÄETÆÚ¼äÇý¶¯PD-L1±í´ï¡£ÕâÒ»ÊÂʵÓë¸ù¾ÝÀëÌåÖ¬¶àÌÇ·´Ó¦½øÐеĻ¼Õß·ÖÀàÒ»Æð£¬ÎªÅ§¶¾Ö¢ÒÔ¼°ËùÓÐÓëETÏà¹ØµÄ²¡Àíѧ¿ª±ÙÁËÓÐȤµÄÑо¿ÁìÓòºÍDZÔڵĸöÐÔ»¯ÁÙ´²Ó¦Ó᣽á¹û
һЩŧ¶¾Ö¢»¼ÕßµÄÑо¿±¨¸æÁËPD-L1ºÍPD-1·Ö±ðÔÚµ¥ºËϸ°ûºÍTϸ°ûÉϵ÷
ŧ¶¾Ö¢»¼ÕßPD-L1µÄ±í´ïÓëÆäET״̬Ïà¹Ø
PD-L1 / PD-1´®ÈÅ¿ØÖÆETÆÚ¼äTÁÜ°Íϸ°ûÔöÖ³µÄËðº¦
HIF1¦ÁÇý¶¯ETµ¥ºËϸ°ûÉÏPD-L1±í´ï
HIF1¦Áͨ¹ýPD-L1¹ý±í´ïµ÷½Úŧ¶¾Ö¢ET»¼ÕßµÄTϸ°û·´Ó¦
ͼÐλ¯¼òÀú¡£ÄÍÊÜÐÔµ¥ºËϸ°ûµÄÌØÕ÷ÊÇÑ×Ö¢·´Ó¦Ê§µ÷£¬ÍÌÊÉ×÷ÓÃÔö¼ÓºÍÁÜ°Íϸ°ûÔöÖ³ÊÜËð¡£ÃâÒßÒÖÖƱíÐÍÓÉIRAK-M±í´ï½éµ¼£¬ÁÜ°Íϸ°ûÔöÖ³µÄ¼õÉÙÓÉPD-L1±í´ï½éµ¼¡£µ«ÊÇ£¬IRAK-MºÍPD-L1µÄÖ÷Òªµ÷¿ØÒò×ÓÊÇת¼Òò×ÓHIF1¦Á£¬ËüÔÚETÏÂÒ²Ôö¼ÓÁ˵¥ºËϸ°ûµÄÍÌÊÉÄÜÁ¦¡£Ëõд£ºET£¬ÄÚ¶¾ËØÄÍÊÜÐÔ£» HIF1¦Á£¬È±ÑõÓÕµ¼Òò×Ó-1¦Á£» IRAK-M£¬°×½éËØ1ÊÜÌåÏà¹Ø¼¤Ã¸¨CM£» PD-1£¬³ÌÐòÐÔϸ°ûËÀÍö-1£» PD-L1£¬³ÌÐòÐÔϸ°ûËÀÍö-ÅäÌå1£» TLR-4£¬TollÑùÊÜÌå4¡£
ŧ¶¾Ö¢Æڼ䱨µÀµÄ¾ø¶ÔÁÜ°Íϸ°û¼ÆÊýµÍ[48]¿ÉÒÔÓÃDZÔÚµÄPD-L1ÓÕµ¼µÄϸ°ûµòÍöÀ´½âÊÍ£¨²¹³äͼ1£©¡£
×ÜÖ®£¬±¾Ñо¿±íÃ÷£¬ETϵÄÈËѪ°ÜѪ֢µ¥ºËϸ°ûͨ¹ýÓÕµ¼HIF1¦ÁÔÚÆäϸ°û±íÃæ±í´ï¸ßˮƽµÄPD-L1£¬²¢¿ØÖÆÁËETÆÚ¼äTϸ°ûÔöÖ³µÄÓÕµ¼ÊÜËð¡£ËùÃèÊöµÄ»úÖÆÍêÉÆÁËET±êÖ¾µÄͼÐκÍËùÉæ¼°µÄÒòËØ¡£´ËÍ⣬ÎÒÃǵķ¢ÏÖΪŧ¶¾Ö¢»¼ÕßÒÔ¼°ÓëETÏà¹ØµÄ²¡Àíѧ´ò¿ªÁËDZÔÚµÄÁÙ´²Ó¦Óá£
¡¡PD-L1 Overexpression During Endotoxin Tolerance Impairs the Adaptive Immune Response in Septic Patients via HIF1¦Á | The Journal of Infectious Diseases | Oxford Academic
https://academic.oup.com/jid/article/217/3/393/4056038¡¡
Front Cell Infect Microbiol. 2019; 9: 207.
Published online 2019 Jun 13. doi: 10.3389/fcimb.2019.00207
PMCID: PMC6584848
PMID: 31263684
The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance
G¨¹nther Schönrich* and Martin J. Raftery
Abstract
Programmed cell death protein (PD-1) and its ligands play a fundamental role in the evasion of tumor cells from antitumor immunity. Less well appreciated is the fact that the PD-1/PD-L1 axis also regulates antiviral immune responses and is therefore modulated by a number of viruses. Upregulation of PD-1 and its ligands PD-L1 and PD-L2 is observed during acute virus infection and after infection with persistent viruses including important human pathogens such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV). Experimental evidence suggests that insufficient signaling through the PD-1 pathway promotes immunopathology during acute infection by exaggerating primary T cell responses. If chronic infection is established, however, high levels of PD-1 expression can have unfavorable immunological consequences. Exhaustion and suppression of antiviral immune responses can result in viral immune evasion. The role of the PD-1/PD-L1 axis during viral infections is further complicated by evidence that PD-L1 also mediates inflammatory effects in the acute phase of an immune response. In this review, we discuss the intricate interplay between viruses and the PD-1/PD-L1 axis.
¡¡Virus-induced upregulation of PD-1 ligands on hematopoietic cells.
Virus Findings References LCMV Arm and clone13 Increased PD-L1 expression on myeloid DCs and marginal zone macrophages; decreased T cell motility in the marginal zone of the spleen due to PD-L1 Zinselmeyer et al., 2013 LCMV High PD-L1 expression on Kupffer cells in the liver Shaabani et al., 2016 IAV Type I IFN induced PD-L1 expression on virus-infected professional APCs in the airways Erickson et al., 2012; Valero-Pacheco et al., 2013; Rutigliano et al., 2014; Staples et al., 2015; McKendry et al., 2016 JEV PD-L1 upregulation on virus-infected DCs in vitro and decreased expansion of Treg cells by virus-infected DCs after PD-L1 blockade Gupta et al., 2014 EOBV Increased numbers of PD-L1 transcripts during EOBV infection of monocytes derived from macaques Menicucci et al., 2017 HV PD-L1/2 upregulation on DCs; high amounts of soluble PD-1 and PD-L2 in the circulation of HV-infected patients Raftery et al., 2018
Virus Findings References LCMV Arm and clone13 Increased PD-L1 expression on myeloid DCs and marginal zone macrophages; decreased T cell motility in the marginal zone of the spleen due to PD-L1 Zinselmeyer et al., 2013 LCMV High PD-L1 expression on Kupffer cells in the liver Shaabani et al., 2016 IAV Type I IFN induced PD-L1 expression on virus-infected professional APCs in the airways Erickson et al., 2012; Valero-Pacheco et al., 2013; Rutigliano et al., 2014; Staples et al., 2015; McKendry et al., 2016 JEV PD-L1 upregulation on virus-infected DCs in vitro and decreased expansion of Treg cells by virus-infected DCs after PD-L1 blockade Gupta et al., 2014 EOBV Increased numbers of PD-L1 transcripts during EOBV infection of monocytes derived from macaques Menicucci et al., 2017 HV PD-L1/2 upregulation on DCs; high amounts of soluble PD-1 and PD-L2 in the circulation of HV-infected patients Raftery et al., 2018 FV PD-L1 expression on erythroid precursor cells and CD4+ T lymphocytes Akhmetzyanova et al., 2015 HIV PD-L1/2 upregulation on monocytes, DCs and macrophages; Correlation between level of PD-L1 expression and disease progression Boasso et al., 2008; Meier et al., 2008; Wang et al., 2008; Rodriguez-Garcia et al., 2011 SIV Upregulation of PD-L1 on DCs; correlation between level of PD-L1 expression and disease progression; improved function of antiviral T cells function after PD-L1 blockade Xu et al., 2010 HSV-1 Increased PD-L1 expression on DCs in the draining lymph nodes after virus inoculation into foot pads of mice Channappanavar et al., 2012 VZV PD-L1/2 upregulation on human monocytes, B cells, NK cells, and NKT cells Jones et al., 2019 KSHV Increased PD-L1 expression on monocytes Host et al., 2017 Ad, adenovirus; EOBV, Ebola virus; FV, Friend retrovirus; HIV, human immunodeficiency virus; HSV-1, herpes simplex virus type 1; HV, hantavirus; IAV, Influenza A virus; JEV, Japanese encephalitis virus; KSHV, Kaposi's sarcoma-associated herpesvirus; LCMV, lymphocytic choriomeningitis virus; LCMV Arm, LCMV strain Armstrong; LCMV clone13, LCMV strain clone13; RSV, respiratory syncytial virus; Treg cells, regulatory T cells; VZV, varicella zoster virus.
Table 2
Virus-induced PD-L1 upregulation on non-hematopoietic cells.
Viruses Findings References LCMV PD-L1 upregulation on fibroblastic reticular cells Zinselmeyer et al., 2013 Ad Increased PD-L1 expression on primary human hepatocytes Grakoui et al., 2006; Muhlbauer et al., 2006 HBV Upregulated PD-L1 expression on hepatocytes derived from a transgenic mouse model of BV infection Maier et al., 2007 IAV, MHPV, PIV-3, RSV Increased levels of PD-L1 on alveolar and bronchiolar epithelial cells after virus infection in vitro and in patients with viral acute lower tract infections Stanciu et al., 2006; Telcian et al., 2011; Erickson et al., 2012; McNally et al., 2013 RABV Type I IFN-dependent PD-L1 upregulation on virus-infected mouse and human neuronal cells in vitro and on neuronal cells in virus-infected mice Lafon et al., 2008 HSV-1 PD-L1 upregulation on mouse neuroblastoma cells Chentoufi et al., 2011 HSV-1 PD-L1 upregulation on virus-infected neurons in ganglia Jeon et al., 2013 HSV-1 PD-L1 upregulation on epithelial cells in the virus-infected cornea Jeon et al., 2018 Ad, adenovirus; EOBV, Ebola virus; HMPV, human metapneumovirus; HBV, hepatitis B virus; HSV-1, herpes simplex virus type 1; HV, hantavirus; IAV, Influenza A virus; JEV, Japanese encephalitis virus; LCMV, lymphocytic choriomeningitis virus; LCMV Arm, LCMV strain Armstrong; LCMV WE, LCMV strain WE; PIV-3, parainfluenza virus type 3; RABV, rabies virus; RSV, respiratory syncytial virus; Treg cells, regulatory T cells; VHF, viral hemorrhagic fever.
Figure 1
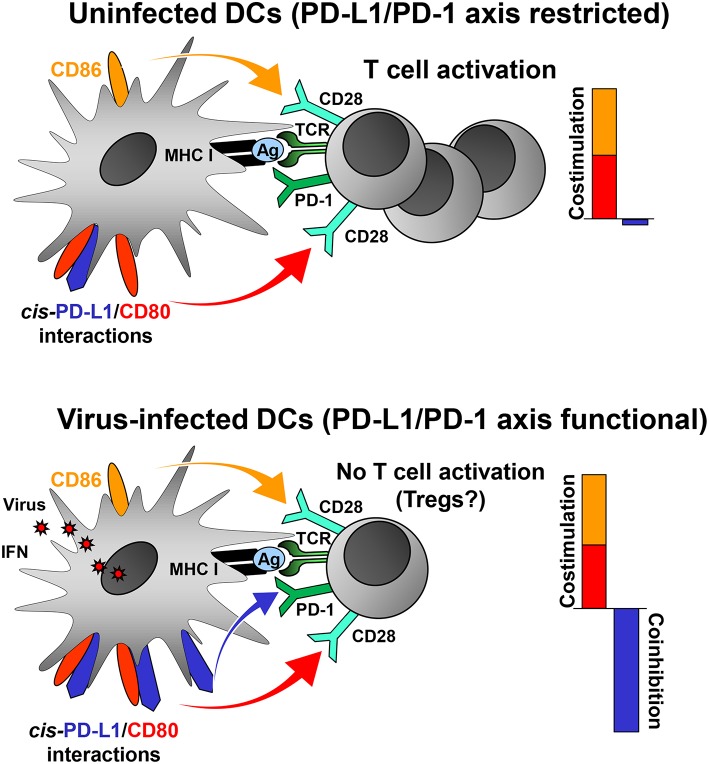
PD-L1 mediated viral regulation of T cell activation. Upper graph: In the absence of viral infection mature dendritic cells (DCs) express relatively low levels of PD-L1. Recognition of cognate antigen (Ag) bound to MHC class I molecules by T cell receptor (TCR) results in upregulation of PD-1 on T cells. DCs express co-stimulatory molecules CD80 and CD86 allowing efficient co-stimulation of T cells via CD28. The PD-1/PD-L1 axis is not co-inhibitory due to restriction by cis-PD-L1/CD80 interactions, and thus T cells are activated. Lower graph: In the context of viral infection DCs upregulate PD-L1 due to exposure to viral PAMPs and high levels of type I IFN. The restricting cis-PD-L1/CD80 interactions are most likely overwhelmed by virus-induced PD-L1 resulting in PD-1 signaling and prevention of T cell activation. The consequences of this for the generation of Tregs is as of yet unknown.¡¡
3
Figure 2
The PD-1/PD-L1 checkpoint in acute virus infection. Early phase: The infected tissue produces type I IFNs and possibly type III IFNs, which strongly induce antiviral IFN-stimulated genes (ISGs) but only moderate PD-L1 levels. Antiviral CD8+ T cells eliminate virus-infected cells. At this stage, the PD-1/PD-L1 checkpoint activity is low and does not restrict the antiviral immune response. Late Phase: Type II IFN and TNF-¦Á is secreted by CD8+ T cells and other immune cells. In addition, hematopoietic cells such as plasmacytoid DCs (pDCs) produce large amounts of type I IFN. This results not only in virus elimination but also increases PD-L1 expression. The high checkpoint activity downregulates terminal differentiation of antiviral CD8+ T cells. Ideally, the strength and quality of the CD8+ T cell response is balanced out in such a way that the viral intruder is eliminated without causing immunopathology.
Keywords: PD-1, PD-L1, PD-L2, antiviral immune responses, viral immune evasion, virus-induced immunopathogenesis, virusesThe PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6584848/¡¡
J Cell Biochem. 2018 Dec;119(12):9997-10004. doi: 10.1002/jcb.27329. Epub 2018 Aug 26.
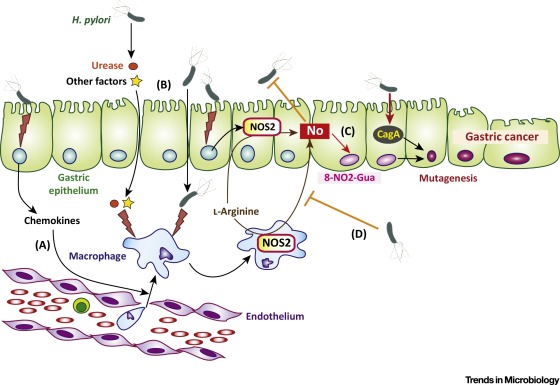
LPS promotes the expression of PD-L1 in gastric cancer cells through NF-¦ÊB activation.
Li H1, Xia JQ1, Zhu FS1, Xi ZH1, Pan CY1, Gu LM1, Tian YZ1.
Author information
1
Department of Gastroenterology, Affiliated Hospital of Integrated Traditional Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, China.
Abstract
Gastric cancers are a group of highly aggressive malignancies with a huge disease burden worldwide. Gastric infections, such as helicobacter pylori, can induce the occurrence of gastric cancers. However, the role of gastric infection in gastric cancer development is unclear. Programmed death-ligand 1 (PD-L1, B7-H1) is a member of the B7 family of cell surface ligands, which binds the PD-1 transmembrane receptor and inhibits T-cell activation within cancer tissues. It has been reported that the expression of PD-L1 is inversely related to the prognosis of patients with gastric cancers. Therefore, the regulation of PD-L1 expression in gastric cancers needs to be studied. In the current study, we explored the possible effects of lipopolysaccharide (LPS) on PD-L1 expression in gastric cancer cells. We observed that LPS stimulation could markedly increase PD-L1 expression in gastric cancer cells. Furthermore, we found that nuclear factor-¦ÊB (NF-¦ÊB) activation was involved in PD-L1 expression in gastric cancer cells exposed to LPS stimulation through p65-binding to the PD-L1 promoter. Taken together, these data indicate that gastric infection might promote the development of gastric cancers thought the LPS-NF-¦ÊB-PD-L1 axis.
¡¡¡¡
Jϸ°ûÉúÎﻯѧ¡£2018Äê12ÔÂ;119(12):9997-10004¡£doi: 10.1002 / jcb.27329¡£Epub 2018 8ÔÂ26ÈÕ¡£
LPS´Ù½øPD-L1µÄ±í´ïÔÚθ°©Ï¸°ûͨ¹ýNF-¦ÊB¼¤»î
Li H1, Xia JQ1, Zhu FS1, Xi ZH1, Pan CY1, Gu LM1, Tian YZ1¡£
×÷ÕßÐÅÏ¢
1 ÄϾ©ÖÐÒ½Ò©´óѧÖÐÎ÷Ò½½áºÏÒ½ÔºÏû»¯ÄÚ¿Æ£¬½ËÕÄϾ©¡£
ÕªÒª
θ°©ÊÇÒ»×é¶ñÐԳ̶ȼ«¸ßµÄ¶ñÐÔÖ×Áö£¬ÔÚÊÀ½ç·¶Î§ÄÚ¾ßÓо޴óµÄ¼²²¡¸ºµ£¡£Î¸¸ÐȾ£¬ÈçÓÄÃÅÂݸ˾ú¸ÐȾ£¬¿ÉÒýÆðθ°©µÄ·¢Éú¡£È»¶ø£¬Î¸¸ÐȾÔÚθ°©·¢Éú·¢Õ¹ÖеÄ×÷ÓÃÉв»Ã÷È·¡£³ÌÐòÐÔËÀÍöÅäÌå1 (PD-L1, B7- h1)ÊÇϸ°û±íÃæÅäÌåB7¼Ò×åµÄ³ÉÔ±Ö®Ò»£¬ËüÓëPD-1¿çĤÊÜÌå½áºÏ£¬ÒÖÖÆ°©Ï¸°û×éÖ¯ÄÚTϸ°ûµÄ»î»¯¡£Óб¨µÀ³ÆPD-L1µÄ±í´ïÓëθ°©»¼ÕßµÄÔ¤ºó³Ê¸ºÏà¹Ø¡£Òò´Ë£¬PD-L1ÔÚθ°©Öеıí´ïµ÷¿ØÐèÒª½øÒ»²½Ñо¿¡£ÔÚ±¾Ñо¿ÖУ¬ÎÒÃÇ̽ÌÖÁËÖ¬¶àÌÇ(LPS)¶Ôθ°©Ï¸°ûÖÐPD-L1±í´ïµÄ¿ÉÄÜÓ°Ïì¡£ÎÒÃǹ۲쵽LPS´Ì¼¤¿ÉÒÔÏÔÖøÔö¼Óθ°©Ï¸°ûÖÐPD-L1µÄ±í´ï¡£´ËÍâ,ÎÒÃÇ·¢ÏÖºËfactor-¦ÊB (NF-¦ÊB)¼¤»î²ÎÓëPD-L1±í´ïÔÚθ°©Ï¸°û±©Â¶ÓÚLPS´Ì¼¤Í¨¹ýp65-binding PD-L1Æô¶¯×Ó¡£×ÛÉÏËùÊö,ÕâЩÊý¾Ý±íÃ÷θ¸ÐȾ¿ÉÄÜ´Ù½øθ°©Ö¢µÄ·¢Õ¹ÈÏΪLPS-NF-¦ÊB-PD-L1Öá¡£
©2018ÍþÀûÆÚ¿¯ÓÐÏÞ¹«Ë¾
© 2018 Wiley Periodicals, Inc.LPS promotes the expression of PD-L1 in gastric cancer cells through NF-¦ÊB activation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/30145830¡¡
expression of PD-L1 was strongly enhanced by LPS.
Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10
*Ragon Institute of Massachusetts General Hospital, Massachusetts Institute of Technology and Harvard University,
¡¬Howard Hughes Medical Institute, Charlestown, Massachusetts, USA;
†AIDS and Cancer Virus Program, SAIC Frederick Inc., National Cancer Institute at Frederick, Frederick, Maryland, USA;
‡Department of Medical Oncology, Dana Farber Cancer Center, Department of Medicine, Harvard Medical School, Boston, Massachusetts, USA; and
¡ìDivision of Infectious Diseases, Massachusetts General Hospital, Boston, Massachusetts, USA
1.Correspondence: Ragon Institute of MGH, MIT and Harvard University, 149 13th St., Charlestown, MA 02129, USA., E-mail: gro.srentrap@hganavakd
Thus, basal expression of PD-L1 was low, and expression of PD-L1 was strongly enhanced by LPS. In contrast, macrophages showed higher constitutive levels of PD-L2 expression that were only modestly increased with LPS stimulation.
¡¡...After stimulation with LPS, PD-L1 expression was clearly up-regulated for all donors (Fig. 1A, upper) in terms of fluorescence intensity (Fig. 1B) and in the percent of positive cells (Fig. 1C). The difference between nonstimulated and LPS-stimulated cells was statistically significant (P<0.0001; Fig. 1B), and macrophages showed a 20-fold increase in the integrated intensity of PD-L1 after LPS treatment.
Cited by: 93
Publish Year: 2011
Author: Marta Rodr¨ªguez-Garc¨ªa, Filippos Porichis, Olivier G. d
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3058820¡¡
Immunol Lett. 2017 Apr;184:7-14. doi: 10.1016/j.imlet.2017.02.006. Epub 2017 Feb 14.
Inflammatory cytokines IL-17 and TNF-¦Á up-regulate PD-L1 expression in human prostate and colon cancer cells.
Abstract
Programmed cell death protein 1 (PD-1) acts on PD-1 ligands (PD-L1 and PD-L2) to suppress activation of cytotoxic T lymphocytes. Interleukin-17 (IL-17) and tumor necrosis factor-¦Á (TNF-¦Á) are co-expressed by T helper 17 (TH17) cells in many tumors. The purpose of this study was to test if IL-17 and TNF-¦Á may synergistically induce PD-L1 expression in human prostate cancer LNCaP and human colon cancer HCT116 cell lines. We found that IL-17 did not induce PD-L1 mRNA expression, but up-regulated PD-L1 protein expression in HCT116 and LNCaP cells. TNF-¦Á induced PD-L1 mRNA and protein expression in both cell lines. Neither IL-17 nor TNF-¦Á induced PD-L2 mRNA or protein expression. IL-17 and TNF-¦Á acted individually rather than cooperatively in induction of PD-L1 expression. IL-17 and/or TNF-¦Á activated AKT, nuclear factor-¦ÊB (NF-¦ÊB), and extracellular signal-regulated kinases 1/2 (ERK1/2) signaling pathways in HCT116 cells, whereas only NF-¦ÊB signaling was activated in LNCaP cells. NF-¦ÊB inhibitor could diminish PD-L1 protein expression induced by IL-17 and/or TNF-¦Á in both HCT116 and LNCaP cell lines. ERK1/2 inhibitor could also reduce PD-L1 protein expression induced by IL-17 and/or TNF-¦Á in HCT116 cells, while AKT inhibitor could abolish PD-L1 protein expression induced by IL-17 and/or TNF-¦Á in LNCaP cells. These results suggest that IL-17 and TNF-¦Á act individually rather than cooperatively through activation of NF-¦ÊB and ERK1/2 signaling to up-regulate PD-L1 expression in HCT116 cells, while the two inflammatory cytokines act through activation of NF-¦ÊB signaling, in the presence of AKT activity, to up-regulate PD-L1 expression in LNCaP cells.
Copyright © 2017 European Federation of Immunological Societies. Published by Elsevier B.V. All rights reserved.
KEYWORDS:
Colon cancer; Interleukin-17; Programmed cell death protein 1 ligand 1; Prostate cancer; Tumor necrosis factor-¦Á
PMID: 28223102 PMCID: PMC5362328 DOI: 10.1016/j.imlet.2017.02.006Inflammatory cytokines IL-17 and TNF-¦Á up-regulate PD-L1 expression in human prostate and colon cancer cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28223102¡¡
¡¡
Pyruvate Kinase M2 Is Required for the Expression of the ...
https://www.frontiersin.org/articles/10.3389/fimmu.2017.01300
Oct 13, 2017 ¡¤ Furthermore, RNA silencing of PKM2 inhibited LPS-induced PD-L1 expression. This regulation occurs through direct binding of PKM2 and Hif-1¦Á to HRE sites on the PD-L1 promoter. Moreover, TEPP-46 inhibited expression of PD-L1 on macrophages, DCs and T cells as well as tumour cells in a mouse CT26 cancer model.
Cited by: 18
Publish Year: 2017
Author: Eva M. Palsson-McDermott, Lydia Dyck, Zbigni¡¡
Front. Immunol., 13 October 2017 | https://doi.org/10.3389/fimmu.2017.01300
Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors
imageEva M. Palsson-McDermott*, imageLydia Dyck, imageZbigniew Zasłona, imageDeepthi Menon, imageAnne F. McGettrick, imageKingston H. G. Mills and imageLuke A. O¡¯Neill
School of Biochemistry and Immunology, Trinity College Dublin, Trinity Biomedical Science Institute, Dublin, Ireland
Blocking interaction of the immune checkpoint receptor PD-1 with its ligand PD-L1 is associated with good clinical outcomes in a broad variety of malignancies. High levels of PD-L1 promote tumor growth by restraining CD8+ T-cell responses against tumors. Limiting PD-L1 expression and function is therefore critical for allowing the development of antitumor immune responses and effective tumor clearance. Pyruvate kinase isoform M2 (PKM2) is also a key player in regulating cancer as well as immune responses. PKM2 catalyzes the final rate-limiting step of glycolysis. Furthermore, PKM2 as a dimer translocates to the nucleus, where it stimulates hypoxia-inducible factor 1¦Á (Hif-1¦Á) transactivation domain function and recruitment of p300 to the hypoxia response elements (HRE) of Hif-1¦Á target genes. Here, we provide the first evidence of a role for PKM2 in regulating the expression of PD-L1 on macrophages, dendritic cells (DCs), T cells, and tumor cells. LPS-induced expression of PD-L1 in primary macrophages was inhibited by the PKM2 targeting compound TEPP-46. Furthermore, RNA silencing of PKM2 inhibited LPS-induced PD-L1 expression. This regulation occurs through direct binding of PKM2 and Hif-1¦Á to HRE sites on the PD-L1 promoter. Moreover, TEPP-46 inhibited expression of PD-L1 on macrophages, DCs, and T cells as well as tumor cells in a mouse CT26 cancer model. These findings broaden our understanding of how PKM2 may contribute to tumor progression and may explain the upregulation of PD-L1 in the tumor microenvironment.Results
PKM2 Expression and Function Play a Key Role in LPS-Induced PD-L1 Expression of Activated Macrophages and Dendritic Cells (DCs)
Since PD-L1 is important in maintaining immune homeostasis by regulating an overactive immune response, we first examined expression of PD-L1 in activated macrophages and DCs. Stimulation of RAW 264.7 macrophages (Figure 1A), murine bone marrow-derived macrophages (BMDM, Figure 1B), or murine BMDC (Figure 1C) with LPS for 24 h (100 ng/ml), induced a robust increase in expression of PD-L1 surface protein (top panels), and pdl1 RNA transcript (bottom panels).
PD-L1 was recently shown to be regulated by Hif-1¦Á during hypoxia.
As a consequence of reduced PKM2 expression, LPS-induced PD-L1 expression was decreased (Figure 1D, middle panel, compare lane 3 and 4, ¦Á-PKM2, to lanes 1 and 2, SC) further implicating a role for PKM2 in PD-L1 expression in macrophages.PKM2 and Hif-1¦Á Bind Directly and Simultaneously to Two HRE Promoter Regions of the PD-L1 Promoter
We next designed biotinylated oligonucleotides spanning two of the hypoxia response elements (HRE) present in the PD-L1 promoter, HRE1 and HRE4, which are known to bind Hif-1¦Á during hypoxia (7). As seen in Figure 2A, endogenous PKM2 (left panels) as well as Hif-1¦Á (right panels) bind to the HRE1 and HRE4 elements of the PD-L1 promoter in primary macrophages. PKM2 binds basally to the promoter in resting BMDM cells (2A, lanes 1 and 4 top panel), however, binding of Hif-1¦Á and of PKM2 to the two oligonucleotide sequences are significantly increased in response to LPS (compare lanes 1¨C2, 4¨C5, 7¨C8, and 10¨C11, top panel). Furthermore, modifying PKM2 into a tetrameric configuration using TEPP-46 reduces binding of both PKM2 itself as well as Hif-1¦Á to the promoter sequences (Figure 2A, top panels, compare lanes 2¨C3, 5¨C6, 8¨C9, and 11¨C12).PKM2 Regulates Expression of PD-L1 in Immune as well as Murine Tumor Cells
PD-1 and its ligand PD-L1 play an important role in the subversion of antitumor immune responses. The murine CT26 colon carcinoma model was used to study the role of PKM2 in the regulation of PD-L1 expression in an in vivo setting. Flow cytometric analysis of CT26 tumor-bearing mice revealed that immune cells (CD45+) expressed increased levels of PD-L1 in dLN compared with naïve LN (Figure 3C). The expression level of PD-L1 was more dramatically increased on tumor-infiltrating immune cells compared to LN cells (Figures 3A,C), suggesting that the tumor microenvironment promotes PD-L1 expression. Tumor and stromal cells (CD45−) expressed the highest levels of PD-L1 compared with immune cells (Figures 3A,C). Of the tumor-infiltrating immune populations, CD3+ T cells expressed the lowest levels of PD-L1, while CD11c+ DCs or CD11b+F4/80+ monocytes/macrophages expressed the highest levels of PD-L1 (Figure 3B). To test whether PD-L1 expression in the CT26 tumor model was regulated by PKM2, mice were injected with TEPP-46 every 2¨C3 days. TEPP-46 effectively reduced the expression of PD-L1 in CD45+ leukocytes, CD11c+ DCs, CD3+ T cells, or CD19+ B cells in the dLN (Figure 3D). Moreover, of the cells occupying the tumor microenvironment, TEPP-46 significantly downregulated PD-L1 expression on leukocytes in general (CD45+) and specifically on CD11c+ DCs, CD3+ T cells, or F4/80+CD11b+ monocytes/macrophages (Figure 3E). Interestingly, TEPP-46 also downregulated PD-L1 expression on CD45− cells, which comprise tumor and stromal cells (Figures 3F,G). In addition, overall PD-L1 mRNA expression levels measured by RT-PCR in dLN and tumor samples were reduced (Figure 3H).Frontiers | Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2017.01300/full
LPS-induced expression of PKM2 causes concurrent phosphorylation of Tyrosine 105 to an extent comparable or greater to the increased expression levels. This phosphorylation is evident after just 1 hr, with the strongest induction after 48 hr (Figure 1 A, middle, 10.3-fold increase in relative band intensity).
Pyruvate Kinase M2 Regulates Hif-1¦Á ... - ScienceDirect
www.sciencedirect.com/science/article/pii/S1550413114005567¡¡
LPS are among toxic substances produced by H. pylori exhibiting low endotoxic activity compared to typical bacterial LPS. The aim of this study was to investigate bioactivity of LPS produced by different serotypes of Helicobacter pylori compared to Escherichia coli and Brucella abortus LPS.
Bioactivity and immunological evaluation of LPS from ...
www.ncbi.nlm.nih.gov/pmc/articles/PMC3696850/¡¡
Helicobacter pylori Lipopolysaccharides Upregulate Toll-Like Receptor 4 Expression and Proliferation of Gastric Epithelial Cells via the MEK1/2-ERK1/2 Mitogen-Activated Protein Kinase Pathway
Helicobacter pylori is recognized as an etiological agent of gastroduodenal diseases. H. pylori produces various toxic substances, including lipopolysaccharide (LPS). However, H. pylori LPS exhibits extremely weakly endotoxic activity compared to the typical LPS, such as that produced by Escherichia coli, which acts through Toll-like receptor 4 (TLR4) to induce inflammatory molecules. The gastric epithelial cell lines MKN28 and MKN45 express TLR4 at very low levels, so they show very weak interleukin-8 (IL-8) production in response to E. coli LPS, but pretreatment with H. pylori LPS markedly enhanced IL-8 production induced by E. coli LPS by upregulating TLR4 via TLR2 and the MEK1/2-ERK1/2 pathway. The transcription factor NF-Y was activated by this signal and promoted transcription of the tlr4 gene. These MEK1/2-ERK1/2 signal-mediated activities were more potently activated by LPS carrying a weakly antigenic epitope, which is frequently found in gastric cancers, than by LPS carrying a highly antigenic epitope, which is associated with chronic gastritis. H. pylori LPS also augmented the proliferation rate of gastric epithelial cells via the MEK1/2-ERK1/2 pathway. H. pylori LPS may be a pathogenic factor causing gastric tumors by enhancing cell proliferation and inflammation via the MEK1/2-ERK1/2 mitogen-activated protein kinase cascade in gastric epithelial cells.
Helicobacter pylori is a microaerophilic, spiral gram-negative bacterium that can colonize gastric epithelial cells or gastric mucin. H. pylori is recognized as a causative factor of chronic gastritis, gastroduodenal ulcers, gastric cancer, and mucosa-associated lymphatic tissue lymphoma. The cell injury and inflammation caused by chronic H. pylori infection is believed to underlie these diseases (5). H. pylori produces several factors that induce inflammatory reactions. For example, vacuolating toxin (VacA), an ammonium ion produced by H. pylori urease, and monochloramines are cytotoxic (9, 33). The monochloramines form from an ammonium ion produced by urease and hypochlorite produced by phagocytic cells. H. pylori activates NF-¦ÊB through the type IV secretion system, which consists of proteins encoded by cag pathogenicity island (8). H. pylori 60-kDa heat shock protein (HSP60) induces the production of inflammatory cytokines via the mitogen-activated protein (MAP) kinase pathway (40). The chronic inflammation induced by these substances may indirectly cause gastroduodenal diseases, including gastric cancer. In addition, direct mechanisms for carcinogenesis have been reported. For example, H. pylori CagA is inserted into host cells via the type IV secretion system, and it activates various host intracellular signaling pathway, which leads to the promotion of cell proliferation and motility, inhibition of apoptosis, and increased inflammatory cytokine production (reviewed in reference 22). Another direct mechanism is that H. pylori induces host activation-induced cytidine deaminase, which promotes genetic mutations in tumor suppressors, such as p53 (20).
In general, gram-negative bacterial lipopolysaccharides (LPSs) are key inducers of inflammation through their role as agonists of Toll-like receptors (TLRs). However, H. pylori LPSs show extremely low endotoxic activity compared to typical gram-negative bacterial LPSs, such as those from Escherichia coli (11, 23, 25, 34). The weak endotoxic activity allows H. pylori to establish chronic colonization or infection, rather than causing a systemic inflammatory response, such as septic shock. The typical LPSs are recognized by the TLR4 complex expressed on host cells, which consists of TLR4, CD14, and MD2. However, there is controversy over which TLR contributes to signal transduction by H. pylori LPS. Some reports suggest that H. pylori LPS transduces signals via the TLR4 system (12, 13, 24), whereas others (15, 30), including our recent report (37), suggest that TLR2 is required for the signal transduction induced by H. pylori LPS. Notably, Triantafilou et al. (32) suggested that LPS derived from some H. pylori strains antagonizes the TLR4 signaling activated by a typical LPS.
We have proposed that H. pylori LPSs are divided into three types: a smooth LPS that carries a highly antigenic epitope, a smooth LPS that carries a weakly antigenic epitope, and a rough LPS that lacks an O-polysaccharide chain (34, 39). The highly antigenic epitope and the weakly antigenic epitope are located on the O-polysaccharide chain adjacent to the core region of the LPS. Most H. pylori-infected individuals have high titer antibodies against the highly antigenic epitope in sera, whereas about half of infected individuals have low titers of antibodies against the weakly antigenic epitope (35, 39). The LPS carrying the weakly antigenic epitope is frequently found in strains obtained from patients with gastric cancer, whereas the LPS with the highly antigenic epitope is preferentially found in strains associated with chronic gastritis (34, 39), suggesting that the antigenicity of LPS is related to the pathogenicity of H. pylori strains. However, the relationship is still unclear. In the present study, we found that H. pylori LPS upregulates TLR4, thereby potentiating the effects of LPS from other organisms, such as E. coli. H. pylori LPS also increased the proliferation of gastric epithelial cells. These properties might contribute to gastric inflammation and carcinogenesis.Helicobacter pylori Lipopolysaccharides Upregulate Toll-Like Receptor 4 Expression and Proliferation of Gastric Epithelial Cells via the MEK1/2-ERK1/2 Mitogen-Activated Protein Kinase Pathway | Infection and Immunity
https://iai.asm.org/content/78/1/468¡¡
Vet Pathol. Author manuscript; available in PMC 2015 Dec 2.
Published in final edited form as:
Vet Pathol. 2013 Sep; 50(5): 895¨C902.
Lipopolysaccharide enhances mouse lung tumorigenesis: A model for inflammation-driven lung cancer
Tamene Melkamu,1,* Xuemin Qian,2,* Pramod Upadhyaya,2 M Gerard O'Sullivan,2,3 and Fekadu Kassie2,3
1Department of Animal Science, University of Minnesota, Minneapolis Minnesota, USA
2Masonic Cancer Center, University of Minnesota, Minneapolis Minnesota, USA
3College of Veterinary Medicine, University of Minnesota, Minneapolis Minnesota, USA
Abstract
The association between pulmonary inflammation and lung cancer is well-established. However, currently there are no appropriate models that recapitulate inflammation-related lung cancer in humans. In the present study, we examined, in two tumor bioassays, enhancement by bacterial lipopolysaccharide (LPS) of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in A/J mice.¡¡
Mice that were treated with NNK alone developed 29.6 ¡À 9.8 and 36.2 ¡À 4.1 lung tumors/mouse in experiment 1 and 2, respectively. Chronic intranasal instillation of LPS to NNK-treated mice increased the multiplicity of lung tumors to 47.3 ¡À 16.1 and 51.2 ¡À 4.8 lung tumors/mouse in experiment 1 and 2, corresponding to a significant increase by 60% and 41%, respectively. Moreover, administration of LPS to NNK-pretreated mice significantly increased the multiplicity of larger tumors and histopathologically more advanced lesions (adenoma with dysplasia and adenocarcinoma), macrophage recruitment to the peritumoral area and expression of inflammation-, cell proliferation- and survival-related proteins. Overall, our findings demonstrated the promise of the NNK-LPS-A/J mice model to better understand inflammation-driven lung cancer, dissect the molecular pathways involved and identify more effective preventive and therapeutic agents against lung cancer.
Keywords: 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, animal model, lipopolysaccharide, lung tumor, macrophageLipopolysaccharide enhances mouse lung tumorigenesis: A model for inflammation-driven lung cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4667800/¡¡
Anti-PD-1/PD-L1 therapy for infectious diseases: learning from the cancer paradigm
Martin Raoa, Davide Valentinia,b, Ernest Dodooa,c, Alimuddin Zumlad, Markus Maeurera,b,low asterisk,'Correspondence information about the author Markus MaeurerEmail the author Markus MaeurerEmail the author Markus Maeurer
Corresponding Editor: Eskild Petersen, Aarhus, Denmark.
Highlights
•T cells are constantly exposed to antigens in chronic diseases, succumbing to cellular exhaustion and reduced immunoreactivity Programmed cell death.
•(PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) expression on antigen-specific T cells marks immunogenic stimuli exposure.
•PD-1 and CTLA-4 identify as immune checkpoints due to their crucial role in regulating the magnitude and quality of T-cell responses.
•The success of PD-1/PD-L1 blockade in cancer prompts extensive clinical evaluation in chronic infections in light of increasing drug resistance.
Summary
Objectives
Immune checkpoint pathways regulate optimal host immune responses against transformed cells, induce immunological memory, and limit tissue pathology. Conversely, aberrant immune checkpoint activity signifies a poor prognosis in cancer and infectious diseases. Host-directed therapy (HDT) via immune checkpoint blockade has revolutionized cancer treatment with therapeutic implications for chronic infections, thus laying the foundation for this review.
Methods
Online literature searches were performed via PubMed, PubMed Central, and Google using the keywords ¡°immune checkpoint inhibition¡±; ¡°host-directed therapy¡±; ¡°T cell exhaustion¡±; ¡°cancer immunotherapy¡±; ¡°anti-PD-1 therapy¡±; ¡°anti-PD-L1 therapy¡±; ¡°chronic infections¡±; ¡°antigen-specific cells¡±; ¡°tuberculosis¡±; ¡°malaria¡±; ¡°viral infections¡±; ¡°human immunodeficiency virus¡±; ¡°hepatitis B virus¡±; ¡°hepatitis C virus¡±; ¡°cytomegalovirus¡± and ¡°Epstein¨CBarr virus¡±. Search results were filtered based on relevance to the topics covered in this review.
Results
The use of monoclonal antibodies directed against the antigen-experienced T-cell marker programmed cell death 1 (PD-1) and its ligand PD-L1 in the context of chronic infectious diseases is reviewed. The potential pitfalls and precautions, based on clinical experience from treating patients with cancer with PD-1/PD-L1 pathway inhibitors, are also described.
Conclusions
Anti-PD-1/PD-L1 therapy holds promise as adjunctive therapy for chronic infectious diseases such as tuberculosis and HIV, and must therefore be tested in randomized clinical trials.Anti-PD-1/PD-L1 therapy for infectious diseases: learning from the cancer paradigm - International Journal of Infectious Diseases
https://www.ijidonline.com/article/S1201-9712(17)30031-0/fulltext¡¡
¡¡

.jpg)