烟曲霉侵袭性曲霉病的发病机制
Pathogenesis of Aspergillus fumigatus in Invasive Aspergillosis
Taylor R. T. Dagenais, Nancy P. Keller
摘要:曲霉是一种全球性的广泛存在于各种生态位的腐生植物。已经鉴定出近200种曲霉,其中不到20种已知会导致人类疾病。其中,烟曲霉是最常见的,在免疫功能低下的患者群体中,其侵袭性曲霉病(IA)的发生率增加在很大程度上是由烟曲霉引起的。IA是一种毁灭性的疾病,在某些患者群体中死亡率高达90%。传统上,鉴定和评估烟曲霉特异性因子在IA发病机制中的作用的研究主要集中在单基因缺失和突变特性的研究上。结合最近分析全球真菌对不同环境或宿主条件的反应的大规模方法,这些研究已经确定了许多因素,有助于烟曲霉的总体致病潜力。在这里,我们提供了一个关于烟曲霉属侵袭性疾病发病机制的重要发现的概述。
图1所示。
烟曲霉的感染生活史。曲霉菌在环境中普遍存在,无性繁殖导致空气分生孢子的产生。特定免疫抑制患者组吸入可导致肺分生孢子的形成、萌发,以及PMN介导的真菌控制(皮质类固醇治疗)或菌丝生长不受控制(PMN缺乏浸润,在严重情况下传播(中性粒细胞减少)。
介绍
曲霉是一种普遍存在的腐生真菌,在全球碳氮循环中发挥着重要作用。虽然曲霉的主要生态位是土壤或腐烂的植被,但它产生的疏水分生孢子很小,很容易分散到空气中,并能在多种环境条件下存活。曲霉属,包括近200种,对公共健康有巨大的影响,有利的是作为工业应用的主要工具,不利的是作为植物和人类病原体(71)。几种曲霉菌因其丰富的酶谱在食品和药品工业生产中得到了应用。例如,黑曲霉被用于柠檬酸、淀粉酶、果胶酶、植酸酶和蛋白酶的工业生产;terreus用于降胆固醇药物洛伐他汀;将大豆和大米分别发酵成酱油和清酒。曲霉在农业中也有不太知名的一面。黄曲霉科植物,尤其是黄曲霉和寄生黄曲霉,可以用黄曲霉毒素污染几种常见的农作物,黄曲霉毒素是一种具有免疫抑制特性的剧毒致癌物(228,230)。食用受污染的作物可导致严重疾病或死亡,这是发展中国家的一个常见问题。
人类病原体烟曲霉
在人类曲霉的致病种类中,烟曲霉是人类感染的主要病原体,其次是黄曲霉、土曲霉、黑曲霉和模式生物尼都兰(54,135)。曲霉会根据宿主的免疫状况引发一系列人类疾病(54,107)。在肺功能改变的个体,如哮喘和囊性纤维化患者中,曲霉可引起过敏性支气管肺曲霉病,这是一种对真菌成分的过敏反应。非侵袭性曲菌瘤可在反复暴露于分生孢子后形成,并以已存在的肺部空洞为目标,如结核病患者已愈合的病灶。侵袭性曲霉菌病(IA)可能是曲霉菌相关疾病中最具破坏性的一种,主要针对免疫功能严重受损的患者。这种致命疾病的高危人群是血液学恶性肿瘤患者,如白血病;实体器官和造血干细胞移植患者;长期接受皮质类固醇治疗的病人,通常用于预防和/或治疗移植病人的移植物抗宿主病;具有遗传免疫缺陷的个体,如慢性肉芽肿病(CGD);以及感染人类免疫缺陷病毒的个体(54 97 126 133 148 162 227)。在高危人群中,死亡率从40%到90%不等,取决于宿主免疫状况、感染地点和采用的治疗方案等因素(114)。IA的严重程度和发病率的增加需要更好地理解宿主和真菌之间的相互作用,这有助于烟曲霉病的发病机制(130)。发病机制和毒力是在宿主免疫功能改变的背景下使用的术语,因为这种生物体本质上是一种机会性病原体,疾病的病理和进展是真菌生长和宿主反应的结果。在这篇综述中,我们将在这些免疫缺陷的背景下讨论烟曲霉作为感染生命周期进程的致病潜力。侵袭性曲霉病
传染性生命周期。曲霉主要是腐生植物,生长在环境中死亡或腐烂的物质上。曲霉菌的感染生命周期始于分生孢子(无性孢子)的产生,这些孢子很容易分散到空气中,确保在室内和室外环境中无处不在(图1)(65,137)。人类感染的主要途径是吸入这些空气中的分生孢子,然后在细支气管或肺泡腔中沉积分生孢子。在健康个体中,未被粘液纤毛清除的分生孢子遇到上皮细胞或肺泡巨噬细胞,后者是肺内的主要吞噬细胞。肺泡巨噬细胞主要负责吞噬和杀死分生孢子曲霉菌,并启动促炎反应,将中性粒细胞(一种多形核细胞[PMN])招募到感染部位。逃避巨噬细胞杀伤并萌发的分生孢子成为浸润中性粒细胞的目标,中性粒细胞能够破坏菌丝。发生IA的风险主要是由于这些宿主防御功能的紊乱,以及真菌的特性导致烟曲霉在这种肺环境中存活和生长(176)。尽管其他宿主的反应与抗病性有关,但在这篇综述中,我们将重点讨论真菌与真菌防御最重要的主要先天成分之间的相互作用。危险因素和病理。引起IA风险增加的主要宿主免疫缺陷是中性粒细胞减少和皮质类固醇诱导的免疫抑制,而IA在这些免疫抑制条件下的病理结果不同,如前所述,对患者和动物模型(9,17,53,192)。长期的中性粒细胞减少是IA最主要的危险因素,通常是高细胞毒性治疗的结果,如环磷酰胺,这是用于移植病人或血液疾病。环磷酰胺是一种DNA烷基化剂,它与DNA结合,干扰细胞复制,破坏包括中性粒细胞在内的循环白细胞。在中性粒细胞减少症患者和化疗诱导的中性粒细胞减少症动物模型中,IA的特征是快速和广泛的菌丝生长导致血栓形成和出血(41,192)。缺乏炎症浸润,尽管肿瘤坏死因子α(TNF-α)的生产,导致低水平的炎症。没有中性粒细胞的恢复,血管侵犯和通过血液传播到其他器官的结果。
许多非中性粒细胞减少症患者,最常见的是那些接受皮质类固醇治疗的患者,如接受皮质类固醇预防或治疗移植物抗宿主病的同种异体移植患者,都易患IA,尽管疾病的病理有很大的不同。IA在这些患者和非嗜中性粒细胞动物模型中是非血管侵袭性的,其特征是真菌发育受限,伴有化脓性肉芽肿浸润、组织坏死和过度炎症。皮质类固醇对吞噬细胞功能有重要影响,包括但不限于吞噬细胞功能受损、吞噬细胞氧化爆发、细胞因子和趋化因子的产生以及细胞迁移(见文献116)。多项研究表明,皮质类固醇损害吞噬细胞杀死烟曲霉分生孢子和菌丝的功能能力(37,92,132,171,172,214)。尽管类固醇对先天免疫细胞功能有影响,但中性粒细胞被吸收到肺中,防止菌丝入侵,但会造成炎症环境,导致组织损伤。这种加重的炎症反应通常被认为是死亡的原因,而在嗜中性粒细胞宿主中观察到的真菌生长不受控制。在每种免疫抑制方案下,真菌发育和宿主反应的巨大差异突出了在宿主免疫状态和随后对真菌感染反应的背景下研究曲霉菌发病机制的重要性。烟曲霉生物学
对来自不同宿主源和地理位置的临床和环境分离株的基因组比较表明,如果给予适当的宿主,熏蒸A.的任何环境菌株都可能具有致病性(50)。与其他物种相比,烟曲霉表现出独特的组合基本特征,有助于致病性。曲霉菌感染的主要途径是吸入空气中的分生孢子并沉积在细支气管或肺泡腔。答:来自烟的平均大小分生孢子(2到3μm)是理想的浸润深度进入肺泡,而更大的其他人类病原体包括a . flavus和分生孢子a尼日尔可能更容易被删除的上呼吸道黏膜纤毛的清除。此外,烟羽a .比其他致病物种更耐高温,在37°C生长良好,能够承受50°C以上的温度,例如在经常有人居住的生态位――腐烂的植被中遇到的温度。据推测,生长在高温下可能会诱导独特的应激反应基因的表达,从而带来额外的毒性效益,尽管这一理论缺乏证据。
多项研究表明,曲霉在37℃下的径向生长和萌发率与致病性有关。烟曲霉a .的形态发生相关基因的缺失,包括环amp依赖蛋白激酶信号通路基因pkaR的调控亚基和ras家族亚同源基因rasB的缺失,导致其在体外发芽率和生长速度降低,这与IA小鼠模型的毒力降低有关(67,235)。calcineurin通路的突变体参与细胞应激反应和形态发生,在37℃下生长明显受损,分生孢子萌发和极化菌丝生长缺陷,在IA的多个动物模型中致病能力显著受损(48)。此外,对烟曲霉、黄曲霉和黑曲霉的生长进行了比较,发现发芽率与病原菌的流行率存在相关性。这些菌种在30℃以下的发芽率相似,但在37℃和42℃时发芽率有所不同(8)。因此,在体内生长速率,特别是37℃时,可能是造成烟曲霉致病性的一个因素。确实,在烟羽a .中缺失一个参与核糖体生物发生的基因cgrA,对果蝇昆虫模型(25℃)的生长和毒力没有影响,但在37℃时径向生长较慢,在动物模型(37℃)中毒力降低(19,23)。这些研究将37℃下的生长速度与毒性联系起来。
呼吸道定殖
吸入分生孢子曲霉是一种常见的现象,因为它们在环境中无处不在;据估计,平均每人每天可吸入200个分生孢子。在易感染ia的患者群体中,肺粘膜防御功能受损,导致真菌定植和生长。
曲霉与可溶性肺成分的相互作用
吸入后,分生孢子虫立即接触到由呼吸道粘膜和呼吸道上皮组成的气道粘膜。这种肺液由粘液、蛋白质、脂质、离子、水和其他细胞分泌物组成,有助于清除吸入的微粒或病原体。在这种复杂的液体中还有opsonic PRRs,它覆盖在吸入的病原体上,有助于宿主防御。在这些蛋白质中有集合素,一组由II型细胞和Clara细胞分泌的c型凝集素受体,它们以钙依赖的方式结合碳水化合物分子。许多致病真菌,包括烟曲霉,有一个碳水化合物丰富的细胞壁,可以被最常见的集合素,甘露糖结合凝集素(MBL)和表面活性蛋白SP-A和SP-D识别。体外实验表明,MBL、SP-A和SP-D能够结合和凝集烟曲霉分生孢子,并通过巨噬细胞和中性粒细胞增强烟曲霉的吞噬和杀灭作用(2,3,120,144)。MBL、SP-A和SP-D最近被发现可以激活补体(59)。在体内,集合体已被证明是重要的:MBL - /−和SP-D - /−小鼠对IA表现出更高的敏感性,重组MBL、SP-D和SP-A已被用于增强宿主对动物模型中曲霉菌病的防御(94,121)。因此,通过增强补体激活、吞噬、杀死分生孢子或聚集分生孢子用于其他宿主防御,集合体可能有助于分生孢子的清除。宿主对微生物最早的反应之一是激活补体,补体是一种血清蛋白的集合,它识别并结合保守的微生物成分,导致调理作用或破坏。虽然补体成分主要存在于血清中,但也存在于支气管肺泡液中,虽然含量较低,但有可能参与宿主对曲霉菌的防御。存在三种补体途径,在C3与微生物表面结合时收敛。早期研究表明,烟曲霉分生孢子与菌丝结合C3。与其他人类致病物种相比,烟曲霉和黄曲霉每单位分生孢子表面结合的C3分子更少(75)。大多数结合C3被裂解成iC3b, iC3b是吞噬补体受体的配体;因此,烟曲霉和黄曲霉可能不太容易受到互补介导的吞噬作用或吞噬细胞识别(193,194)。几种曲霉菌的分生孢子和菌丝也能结合补体抑制因子H、其剪接产物FHR-1和因子H家族蛋白FFHL-1,阻止补体级联的激活(13,213)。与经典的凝集素通路抑制剂c4b结合蛋白结合的烟曲霉已被观察到(213)。最后,烟曲霉和黄曲霉(而非黑曲霉)被发现产生一种可溶性的互补抑制因子,可能是脂质衍生的,可以阻止选择性通路激活(219,221)。由此看来,烟曲霉和黄曲霉都具有抑制或减少补体激活的防御机制,而黄曲霉的这种防御机制鲜为人知。因此,烟曲霉抑制补体活化的能力可能有助于该生物体的整体发病机制。
参与曲霉防御的其他可溶性成分包括五羟色胺PTX3和纤溶酶原。PTX3是一种可溶性的opsonin,由吞噬细胞产生,有助于微生物识别(28)。缺乏PTX3基因的小鼠易感染IA,这与吞噬细胞对烟曲霉分生孢子a .的识别能力降低有关(69)。最近的一项研究表明纤溶酶原(纤溶酶原是纤维分解途径的组成部分)的遗传变异与IA易感性有关(233)。与烟曲霉结合的纤溶酶原也被检测到,当其裂解为活性纤溶酶时,可通过其在细胞外基质降解中的作用增强传播(13)。
曲霉与呼吸道上皮细胞的相互作用
尽管呼吸道上皮细胞在启动对许多吸入病原体的抗菌先天免疫反应方面具有重要作用,但很少有研究探讨呼吸道上皮细胞在宿主对曲霉的防御中的作用(12,127)。作为吸入分生孢子的第一个细胞, 呼吸道上皮细胞可能参与了烟曲霉的整体免疫反应。上皮细胞可能分泌可溶性抗菌化合物,在呼吸道防御中发挥直接作用。defensin家族成员对多种微生物和抗菌肽具有广谱活性是由上皮细胞体外孵化后与来自烟a(1)体外,曲霉菌发芽分生孢子和菌丝,但不是休息分生孢子,被主持人PRRs上皮细胞和诱导细胞因子和趋化因子的产生如白介素、TNF-α,引发(10,14)。皮质类固醇可以消除这种炎症反应,质疑皮质类固醇治疗的IA高危患者上皮细胞的功能(14)。上皮细胞可能协助启动对烟熏A.的促炎反应,尽管它们的作用可能远不如肺泡巨噬细胞那么强大。烟曲霉分生孢子被多种上皮细胞(包括气管上皮细胞、肺泡II型细胞、人鼻上皮细胞和A549肺上皮细胞系)结合并吞噬(66,155,223)。被A549上皮细胞吞噬的分生孢子进入酸性吞噬溶酶体并可被杀死,尽管一些分生孢子能够萌发并同时离开吞噬溶酶体和肺细胞,但没有证据表明肺细胞受到损伤(222,223)。答:来自烟分生孢子还能抑制药物――或者TNF-α-induced细胞凋亡在初级上皮细胞和上皮细胞株体外,虽然体内的影响是未知的(18)。
一些真菌来源的因素可能有助于烟曲霉与气道上皮细胞结合和调节的能力(图2)。有趣的是,包括烟曲霉在内的致病性曲霉比非致病性曲霉表现出更多的分生孢子唾液酸(224)。粘附于纤维蛋白原、基底膜糖蛋白层粘连蛋白和细胞外基质成分纤连蛋白也部分由唾液酸残基和分生孢子表面的其他蛋白介导(7,30,31,34,46,202,203)。分生孢子与这些成分结合的重要性与肺损伤或窘迫是IA的危险因素有关。纤维蛋白原(加工后的纤维蛋白)和纤维连接蛋白附着在受伤表面,如受损上皮表面,层粘连蛋白暴露于上皮损伤或剥离。因此,唾液酸,以及可能与牙槽骨成分结合的其他分生孢子因子,可以通过增强对上皮细胞和损伤组织成分的粘附和定植,促进致病性。
图2所示。
烟曲霉与呼吸道上皮细胞的相互作用。吸入后,烟曲霉遇到呼吸道上皮(气管内壁、支气管和细支气管)、上呼吸道的粘液和液体内壁,最终到达肺泡腔。真菌产物(如红色所示)可能通过组织损伤(交叉发线)和上皮细胞或受损基底膜的附着增强定植。分生孢子也可能萌发并通过基底膜或上皮细胞摄取后侵入周围的肺组织。烟曲霉可能通过改变上皮功能和生存能力的分泌产物促进在其他健康肺组织中的定植。从烟曲霉菌株中提取的培养滤液在A549细胞中诱导细胞收缩、脱屑和肌动蛋白细胞骨架重排(93,98)。丝氨酸蛋白酶抑制剂和半胱氨酸蛋白酶抑制剂均能抑制滤液的活性,说明其具有一定的蛋白酶活性。事实上,当特定的丝氨酸蛋白酶AF-ALF被删除时,培养滤液不能诱导肌动蛋白细胞骨架损伤(98)。在熏蒸菌培养滤液中的蛋白酶活性也与人类鼻上皮细胞脱离和局部接触的丧失有关,后者可能有助于萌发菌丝侵入肺组织(98,169,201)。
从烟曲霉病患者身上提取的烟曲霉培养滤液和痰标本中提取的人呼吸上皮细胞损伤和纤毛节律减慢与次级代谢产物有关,尤其是神经胶质毒素,在浓度较高的情况下,还与福马西林和海伏酸有关(5,6,43)。在气道上皮气液界面模型中,许多烟曲霉菌株的分生孢子和菌丝滤液中产生的致震代谢物疣状菌素也参与了改变鼻上皮细胞的跨上皮耐药、超极化和细胞质空泡化(29,96)。疣状菌素与分生孢子和菌丝成分有关,在早期感染时可影响气道上皮细胞,但在体内的产生尚未观察到(96)。因此,熏鱼能够干扰粘膜纤毛清除,结合呼吸上皮细胞和基底膜蛋白,侵入或破坏上皮细胞,建立感染,并可能逃避其他宿主的防御。曲霉和肺泡巨噬细胞
巨噬细胞对曲霉菌的反应。肺泡巨噬细胞是呼吸道的主要吞噬细胞,是宿主防御分生孢子曲霉菌的重要组成部分。肺泡巨噬细胞以一种依赖于放线菌素的方式吞噬分生孢子曲霉,这一过程由宿主细胞PRRs识别病原体相关的分子模式介导。来自烟PRR订婚的配体形成了一种促炎反应的特点是重要的细胞因子和趋化因子的产生对于主机防御这种生物,包括TNF-αIL-1β,il - 6,引发,巨噬细胞炎症蛋白1α,单核细胞化学引诱物蛋白1(39岁,131,138,158)。TLR2、TLR4和c型凝集素受体dectin-1是参与烟曲霉识别和宿主细胞活化的最具特征的PRRs。体外研究表明,分生孢子和菌丝通过TLR2和TLR4激活巨噬细胞,TLR2可以识别分生孢子和菌丝形态,而TLR4只能识别菌丝形态(143)。使用TLR−/−小鼠进行的研究也表明,TLR4可能在体内发挥重要作用,TLR2也可能在体内发挥潜在作用。在中性粒细胞减少模型中,TLR2−/−和TLR4−/−小鼠比野生型小鼠表现出更高的真菌负担(11,15)。尽管在这些研究中,TLR4−/−小鼠的存活率低于野生型小鼠,但TLR2−/−小鼠的结果却与之相反。TLRs在非嗜中性粒细胞模型中的作用尚未得到很好的研究,尽管TLR4多态性在异基因造血干细胞移植患者中与IA风险增加有关(22)。
与TLRs不同,dectin-1在免疫抑制和免疫能力强的宿主中都对宿主防御曲霉菌至关重要(226)。Dectin-1特定的真菌碳水化合物β葡聚糖(1,3),通常掩盖在休息a来自烟由蛋白质的分生孢子hydrophobin层。分生孢子的肿胀后,β(1、3)葡聚糖变得暴露和存在肿胀的分生孢子,germlings,菌丝形态类型(70、77)。Dectin-1-β(1、3)葡聚糖接触导致吞噬,巨噬细胞激活,强烈的促炎反应诱导(35、70、77、119、189、210)。因此,在TLRs的辅助作用下,dectin-1能够使先天免疫细胞吞噬和杀死分生孢子,并引发炎症反应。肺泡巨噬细胞通过活性氧(ROS)和吞噬溶酶体酸化杀死吞噬溶酶体内膨胀的分生孢子(83,159,220)。Philippe等人首次证明了ROS在巨噬细胞介导的分生孢子杀伤中的作用。在这些实验中,来自p47phox−/−CGD小鼠的肺泡巨噬细胞,或经NADPH氧化酶化学抑制剂处理的野生型肺泡巨噬细胞,其杀死烟孢子A.的能力显著受损(159)。虽然其他研究表明nadph介导的氧化反应不会导致肺泡巨噬细胞杀死分生孢子,但有几个因素(巨噬细胞与分生孢子的比例、共孵育时间、动物菌株和/或检测的细胞类型)可能导致相互矛盾的结果(136,177)。非氧化机制确实有助于分生孢子的杀灭。活性氮种类是无效的,尽管其他候选品种(如抗菌肽)尚未经过测试(159)。总的来说,免疫抑制的IA易感患者肺泡巨噬细胞效应功能降低,可能是皮质类固醇介导的抑制,也可能是化疗诱导的衰竭,导致烟曲霉逃避巨噬细胞杀伤的能力降低。
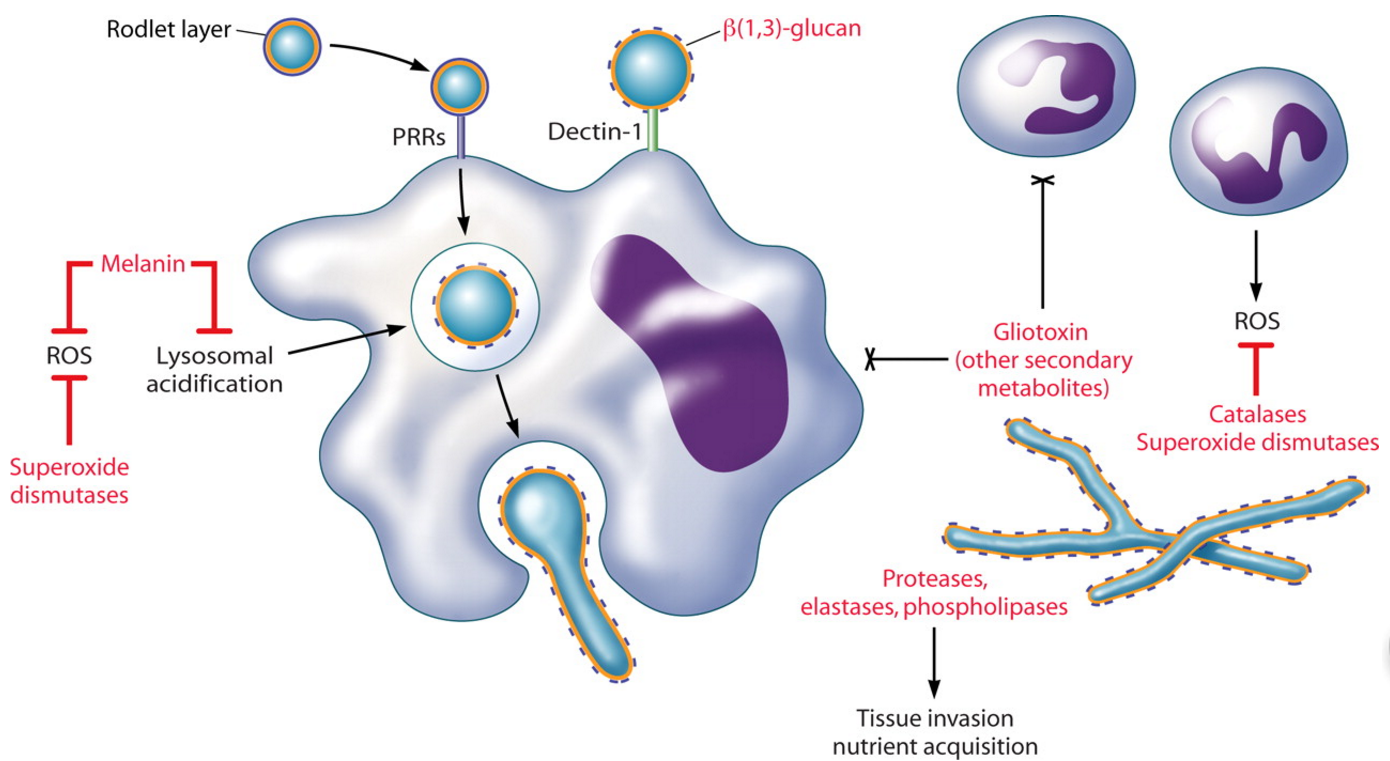
答:来自烟防御。(我)黑色素。除了屏蔽β(1、3)葡聚糖和延迟巨噬细胞激活,休息a来自烟分生孢子对巨噬细胞死亡(图3)。对宿主防御的黑色素的保护作用,特别是通过清除ROS,被描述为许多致病真菌(87、106)。在曲霉中,黑色素提供分生孢子色素,用于区分某些种类。在熏蒸a . fumigatus中,Jahn等人在UV诱变后首次描述了一种白色的无色素菌株(88)。利用烟曲霉的cosmid文库对野生型表型进行互补,发现聚酮合成酶基因pksP是色素产生的来源(105)。与野生分生孢子相比,白色突变体表现出超微结构的细胞壁差异,对氧化剂H2O2和NaOCl的敏感性增加(88)。此外,由于野生型黑色素清除培养基中的ROS,白色分生孢子比野生型诱导更大的单核细胞和中性粒细胞产生ROS。单核细胞比野生型分生孢子能杀死更多被摄取的突变分生孢子,可能是通过ros介导的机制。在系统性IA动物模型中,白色分生孢子毒性较弱,首次表明黑色素在烟曲霉病发病机制中起直接作用。这些研究表明黑色素作为活性氧清除剂是一个重要的致病因素。值得注意的是,IA的全身动物模型不能完全概括IA的感染过程,因为分生孢子是通过尾巴直接注入血液的,而不是鼻内或气管内。显然,这种管理可能导致宿主对烟曲霉的反应非常不同。然而,在鼻内模型中对另一种黑色素突变体的研究(见下文)支持黑色素在烟曲霉病发病机制中的作用。一项研究还表明氰化物不敏感的替代氧化酶参与了保护分生孢子不受巨噬细胞ros介导的杀伤(hyphal成分尚未探索)(123)。然而,在那项研究中,RNA干扰被用来降低黑色素突变体中aox的表达,因此只有在黑色素缺乏的情况下,替代氧化酶才可能在保护巨噬细胞不被杀死方面发挥重要作用。其他研究表明,人类巨噬细胞吞噬pksP突变分生孢子导致吞噬体酸化增加,这是吞噬溶酶体融合的结果(89)。氯喹的加入增加了吞噬溶酶体融合和pH值,导致与突变体相似的野生型杀伤。因此,功能性pksP可以阻止一定程度的吞噬溶酶体融合,提高分生孢子的存活率。此外,该研究表明吞噬溶酶体酸化是一种分生孢子杀灭机制。
pksP的首次发现与Tsai等和Watanabe等人对色素生物合成涉及的另一个基因arp1的鉴定相一致,arp1是一种萘环酮合成酶,与scytalone脱水酶同源(206,225)。arp1突变体呈红粉色,与野生分生孢子相比,其结合的C3分子更少,这意味着arp1合成的黑色素或色素中间体可以防御宿主补体。对烟曲霉分生孢子色素生物合成的进一步研究发现,包括pksP(也称为alb1)在内的6个基因簇参与了二羟基萘-黑色素生物合成(207)。与Jahn等人的观察相似,alb1的缺失导致了小鼠的白色表型和毒性降低(205)。alb1突变体也更容易与C3结合,这强化了黑色素或黑色素中间体可能阻止补体激活的观点。另一种黑色素,pyomelanin,由酪氨酸降解途径产生,也可能有助于致病性,因为突变体对ROS的敏感性增强(178)。总的来说,黑色素通过保护分生孢子免受多种宿主防御,尤其是肺泡巨噬细胞的防御,似乎是烟曲霉病发病的一个重要决定因素。
(二)ROS防御中介。作为有毒活性氧的清除剂,其他与致病性有关的分子包括小棒和超氧化物歧化酶(SODs)。分生孢子被疏水层包围,疏水层由杆状蛋白组成。在烟曲霉的两个杆状体基因中,rodA只负责杆状体的产生,而rodA突变体对肺泡巨噬细胞的杀伤表现出更高的易感性(156)。无棒分生孢子在IA大鼠模型中也诱导了微弱的炎症反应(183)。考虑到分生孢子的膨胀对巨噬细胞的活化至关重要,无棒的分生孢子可以诱导更快速、更强的巨噬细胞活化和分生孢子的清除。两种真核超氧化物歧化酶(Cu/Zn-SOD)和锰超氧化物歧化酶(Mn-SOD)在烟毒蛾发病机制中的作用尚未得到很好的研究。在分生孢子和菌丝的细胞壁中检测到Cu/Zn-SOD,在培养滤液中检测到SOD活性(73,78)。虽然铜/锌- sod在体外低铁条件下上调,一些烟曲霉感染患者的血清与铜/锌- sod发生反应,但在体内的具体作用尚未确定(38,73,79)。McDonagh等人在体外和感染IA小鼠模型时发现,在中性粒细胞的作用下,Mn-SOD转录产物水平升高,说明这种SOD在氧化应激防御中起作用(129)。对罕见的高毒性突变体的研究证实了氧化应激对致病性的影响。中断的α(1、3)葡聚糖合酶基因ags3导致毒性增加IA的动物模型,与体外抗氧化应激相关,可能增加黑色素相关内容或增加发芽率观察到在这个突变体(128)。另一个与氧脂素生物合成无关的突变体,在该突变体中,烟曲霉的三个环氧化酶基因被RNA干扰沉默,也显示出与对H2O2抗性增加相关的毒力增强(208)。环氧化酶基因的个体缺失并没有导致毒性的增加,尽管一个基因ppoC的缺失导致了对H2O2更强的抵抗力,以及异常的分生孢子形态和巨噬细胞吞噬能力的增加(49)。最后,糖基磷脂酰肌醇锚定蛋白ECM33的缺失导致一个高毒性菌株的萌发和对细胞壁不稳定剂的耐药性增加(174)。有趣的是,编码C2H2锌指转录因子的ace2基因的缺失导致了分生孢子色素沉着、细胞壁组织的改变以及对H2O2的抗性(63)。ace2突变体在非嗜中性粒细胞小鼠模型中具有高毒性,但在嗜中性粒细胞模型中则没有,这表明该突变体对嗜中性粒细胞的氧化防御具有抵抗力。该突变体中ags3、ecm33和ppoC的转录本也被检测,发现其转录本低于野生型,这表明这些基因的ACE2调控可能导致了这些突变体之间的一些表型相似性。
图3所示。
烟曲霉与吞噬细胞的相互作用。肺泡巨噬细胞吞噬通过PRRs吸入分生孢子。分生孢子的肿胀(巨噬细胞)的内部或外部释放保护小棒层,暴露β(1、3)葡聚糖供dectin-1识别。Dectin-1-β(1、3)葡聚糖相互作用是主要负责激活的巨噬细胞炎性反应,包括分生孢子的杀害。中性粒细胞附着在菌丝上并脱颗粒,通过氧化和非氧化机制破坏菌丝。中性粒细胞也可能聚集分生孢子,阻止萌发。吞噬细胞功能受损是IA的主要危险因素。真菌产物(红色显示)可能通过逃避或调节宿主的防御而导致这些免疫缺陷宿主的真菌致病性。适应哺乳动物的肺部环境
在哺乳类动物的肺中,熏鱼的萌发和菌丝的生长,在宿主肺防御系统存活后,需要激活营养感知、获取和生物合成途径,从宿主环境中获取营养。代谢途径的研究对于理解烟曲霉的发病机制至关重要,因为它可以识别宿主的营养限制以及烟曲霉为适应这种环境生态位而专门利用的相互反应途径。
哺乳动物组织退化
烟曲霉的基因组编码了一个广泛的降解酶库,支持这种真菌在环境植物物质上的无处不在的生长(56)。虽然许多这些酶特定的植物细胞壁成分,很有可能一些真菌酶可能参与发病机理,由于丝状哺乳动物宿主的增长需要宿主组织的分解营养收购和入侵(图3)。当需要蛋白质分泌了内质网的蛋白质折叠能力,展开的蛋白质反应(UPR)发起的。这可能发生在特定的环境压力下,如细胞壁扰动、热应力和营养限制。Richie等人最近的一项研究表明,通过缺失hacA基因导致UPR的破坏,导致了一个突变体,该突变体对各种压力敏感,包括哺乳动物肺组织的生长(168)。此外,该突变体在三种IA小鼠模型中明显受损,这意味着UPR需要调节体内组织降解和获取营养所需的蛋白分泌通路。
由于弹性蛋白是肺组织的重要组成部分,大多数关于蛋白酶参与IA的研究都集中在曲霉弹性酶的作用上。Kothary等人首先描述了弹性蛋白酶的产生与侵袭性疾病之间的相关性(100)。在该研究中,我们选择了几株烟曲霉产弹酶和非产弹酶环境菌株来感染用可的松治疗的(非中性粒细胞)小鼠。所有产生弹性体的菌株都是致命的,而感染非产生弹性体菌株的小鼠中有近三分之二存活了下来。此外,还观察到感染了产生弹性蛋白酶菌株的小鼠肺部真菌负担和组织坏死程度较高,这表明弹性酶活性与真菌侵袭和致病性有关。另一项临床和环境A. fumigatus菌株的研究发现,IA患者的所有菌株都表现出弹性酶活性,但超过三分之一的环境菌株缺乏弹性酶活性(21)。IA患者菌株的平均弹性酶活性也高于曲霉瘤患者或烟曲霉a菌群的菌株,这表明弹性酶活性与侵袭性疾病的致病性有关。
蛋白水解酶的数量和功能冗余使得很难确定弹性酶活性或单个蛋白酶在致病性中的重要性。丝氨酸蛋白酶alp最初被认为是弹性水解活性的来源之一(165)。在曲霉病患者血清中产生抗Alp抗体,在受感染的人肺组织中检测到Alp,将这种蛋白酶的产生与感染联系起来。然而,几项研究表明,烟曲霉alp (Afalp)突变体在可的松处理或中性粒细胞减少小鼠体内的毒力没有显著差异(134,199)。此外,对IA患者肺血管组织的组织学检查发现血管壁缺乏弹性溶解,这使弹性酶活性对IA的贡献受到质疑(55)。另一方面,Kolattukudy等人发现了另一种与Afalp具有很强同源性的弹性蛋白酶,并且该蛋白酶的A. fumigatus突变体在辐照(中性粒细胞减少)小鼠中产生的毒力显著低于野生型的总弹性酶活性的10%(99)。该丝氨酸蛋白酶通过免疫金电镜定位于中性粒细胞减数分裂动物肺内萌发分生孢子和菌丝(99,125)。以类似的方式,一种具有弹性水解活性的金属蛋白酶被定位于中性粒细胞感染动物肺部侵入菌丝。本文还描述了从烟曲霉中纯化的一种天球蛋白酶,它能降解人的弹性蛋白、I型和III型胶原蛋白以及纤维连接蛋白,并通过在中性粒细胞模型中穿透菌丝分泌(81,108)。这些研究表明,在感染过程中会产生一种或多种具有弹性水解活性的蛋白酶,尽管目前还不清楚弹性酶活性是否与发病机制有关。我们怀疑,烟曲霉蛋白酶对弹性酶活性的影响只是导致营养物质降解和真菌入侵宿主组织的一个因素。研究各种曲霉菌sp.临床分离株,a .来自烟临床分离株被发现不仅弹性蛋白酶活性,而且酸性蛋白酶和磷脂酶活动,而株a flavus只有弹性蛋白酶活性,在尼日尔,只检测到(4)磷脂酶活动。因此,各种蛋白水解酶的结合可能导致的能力来自烟a降解宿主组织营养收购和入侵。
营养物质的合成和获取
通过研究代谢途径突变体的表型,经典地确定了烟曲霉在感染过程中的营养需求。例如,使用生长营养pyrG、pabaA和lysF突变株进行的感染研究表明,真菌需要分别合成尿嘧啶/尿苷、叶酸和赖氨酸,以维持在体内的生存和毒性(36,52,113,175)。到目前为止,这些是对生长营养菌株的唯一研究,证明了真菌对宿主中不可获得的营养物质的生物合成的迫切需要。
氮代谢对烟曲霉的致病性也有一定的影响。曲霉可以利用广泛的氮源,并通过硝酸盐同化对全球氮素循环做出重要贡献,其中环境硝酸盐被吸收并转化为铵,随后转化为谷氨酰胺和谷氨酸。参与硝酸盐转运和加工的蛋白质受区域基因位点的转录调控。Hensel等人发现,在低氮源条件下生长的区域突变体与野生型熏蒸A.之间存在明显的生长差异(74)。虽然在嗜中性粒细胞减少的IA模型中,区域突变体表现出与野生型菌株相似的毒性,但在肺组织中观察到该突变体的延迟生长表型。区域- hygr转化体在体内的还原速率也提示了区域调控氮代谢的有益作用。在另一项研究中,Panepinto等人基于烟熏a .与人内皮细胞接触时的转录上调,鉴定了ras相关蛋白RhbA(153)。在贫氮条件下,rhbA突变体生长较慢,在嗜中性粒细胞减少的IA模型中表现出明显的致病性降低。肺部病变的检查进一步证实了体内rhbA突变体的生长减少。从这些研究中可以明显看出,氮代谢的多样性对烟曲霉的总体致病性有贡献。
在某些情况下,在IA动物模型中没有表型的代谢途径突变体可以提供有关感染期间营养有效性的有用信息。对于烟曲霉来说,C2化合物和脂肪酸作为碳和能量的唯一来源,其生长需要异柠檬酸裂解酶或苹果酸合成酶。然而,在IA动物模型中,这两种酶的突变体都具有完全的毒性,这表明烟曲霉利用了其他碳源(147,179)。其他人类致病性真菌上调异柠檬酸裂解酶、苹果酸合成酶,和β-oxidation酶由宿主巨噬细胞吞噬作用,暗示glucose-poor C2 /脂肪acid-rich环境。据推测,脂质生长的能力,也被认为是丰富的宿主细胞内,解释了他们的完全毒性表型。然而,这些突变体也在碳水化合物和氨基酸的碳源上生长良好。考虑到一些分生孢子可能不会遇到肺泡巨噬细胞,很可能多种碳同化途径可以弥补乙醛酸循环的损失。一个候选途径是柠檬酸甲酯循环,这是真菌生存和致病性所必需的(82,122)。在降解氨基酸进行碳同化的过程中,有毒的丙酰辅酶A在细胞内积累,并被柠檬酸甲酯合酶所阻止。烟曲霉体内柠檬酸甲酯合成酶的缺失导致了培养过程中生长和次生代谢产物的减少,增加了体内外巨噬细胞杀伤的敏感性,降低了IA虫鼠模型的毒力(82,122)。目前尚不清楚分生孢子的死亡是由于毒性丙酰辅酶A的积累而独立于或除了对宿主防御的敏感性之外。然而,烟曲霉体内的碳同化需要柠檬酸甲酯循环。这些研究还表明,烟曲霉利用原生或寄主蛋白降解来获得氨基酸和其他营养物质。氨基酸可以从降解的蛋白质中获得,也可以由含碳和含氮的前体合成。在氨基酸饥饿条件下,氨基酸生物合成相关基因的表达受cpcA基因座调控的保守通路控制。cpcA的缺失导致IA动物吸入模型中毒性较弱的菌株,尽管肺组织在真菌生长或传播方面,突变体与野生型之间没有显著差异(101)。由于cpcA突变体是原养型的,因此推测在体内,导致cpcA活化的不是缺乏氨基酸而是不平衡。因此,cpcA调控氨基酸的生物合成途径,为哺乳动物肺的生长提供理想的营养条件。
铁获取
铁是许多生物合成途径的必要成分,在酶促反应中起辅助作用,在电子传递系统中起催化作用。然而,体内游离铁的不稳定性和宿主防御机制的固着严重限制了铁的有效性。对于包括烟曲霉在内的许多人类病原体,从宿主获取铁的能力是一个必要的毒力决定因素(图3)。烟曲霉利用两个系统获取铁:铁离子介导的铁吸收和还原性铁同化(180)。烟炱产生四种已知的铁铁特异性螯合剂。曲霉菌分泌褐藻碱C和三乙酰褐藻碱C螯合细胞外铁,而铁蛋白和羟基铁蛋白分别参与菌丝和分生孢子细胞内铁的储存(62,72)。sidA是第一个在siderophore生物合成途径中发现的基因,催化所有siderophore生物合成的第一步,即l-鸟氨酸的羟基化(76)。缺乏细胞外和细胞内铁储存的sidA突变体在低铁条件下或人血清中不能生长或生长不良,对H2O2的敏感性增加,生长速度降低(76,180)。此外,sidA突变体在中性粒细胞减少和非中性粒细胞减少的IA小鼠模型中是无毒的。
Schrettl等人最近发现了参与特异性铁包膜合成的铁调控基因:sidC、sidD、sidF和sidG(181)。在嗜中性粒细胞减少的IA模型中,这些基因中任何一个的缺失都减弱了毒力,这表明在感染期间,细胞外和细胞内的铁粒在铁的获取和储存方面都需要。对单个突变体的鉴定表明,细胞内由sidC和sidG调控的铁粒为萌发和抗氧化胁迫储存铁,这两者对发病机制都很重要。此外,sidD和sidF调控细胞外铁素体的产生,促进菌丝生长,可能通过体外实验从人转铁蛋白中获得铁。虽然烟曲霉利用与铁离子吸收无关的机制在体外获得铁,但它们对毒力似乎没有必要。ftrA的一个突变体,参与还原性铁同化的铁渗透酶,在体外表现为野生型表型,在体内保持完全的毒力(180)。因此,铁离子介导的铁离子捕获和细胞内储存对烟曲霉的毒力至关重要。
为了全面了解熏蒸a .在早期感染期间的适应性,McDonagh等人在嗜中性粒细胞减少的IA模型中对受感染小鼠肺部的熏蒸a .进行了转录分析(129)。与迄今为止的体外和体内研究一致,与富含铁的实验室培养基相比,在肺初始适应过程中上调的基因包括铁团和铁转运基因,以及参与金属获取、氮分解代谢、脂质和氨基酸分解代谢以及碳水化合物转运的基因。从整体上看,这些研究证明了营养物质感知、获取和生物合成的多样性,有助于烟曲霉的整体致病潜力。
额外的环境压力
除了获取营养外,烟曲霉还必须适应环境压力,如温度(如上所述)、pH值和氧气限制。McDonagh等人在分析真菌对哺乳动物肺在IA期间的适应性时发现,体内差异表达的基因(与实验室培养基相比)与体外碱性条件下表达的基因(pH 7)相关,如pH调节剂PacC(129)。PacC最早在a . nidulans中发现,是一种转录因子,当生长在生理pH值等碱性条件下时,PacC激活碱性表达基因,抑制酸性表达基因(200)。在IA的中性粒细胞减少模型中,pacC是a . nidulans完全毒力所必需的,这意味着pH适应是曲霉的致病特性之一(20)。对低氧环境的适应也被认为是烟曲霉致病性的重要因素。炎症部位,如那些在曲霉感染的肺组织中发现的,聚集了免疫细胞,它们代谢可用的氧气并阻塞血管,创造了一个缺氧的环境。在真核病原体中,参与低氧适应的基因知之甚少。一类转录因子,甾醇调节元件结合蛋白,首先在裂殖酵母pombe中被发现,它在低氧条件下介导依赖氧的甾醇合成和生长(80)。SrbA是pombe甾醇调节元件结合蛋白Sre1的同源物,最近被鉴定为烟曲霉属(229)。srbA的缺失导致甾醇生物合成和细胞壁形态的改变。尽管srbA突变体的生长和分生孢子在富氧条件下不受影响,但在低氧条件下,srbA突变体的菌丝生长和形态发生改变,在中性粒细胞减少的IA小鼠模型中是无毒的。在CGD小鼠中,srbA突变体的毒性明显低于野生型。此外,感染后嗜中性粒细胞肺的组织学分析显示宿主清除了srbA突变体,这导致了烟曲霉需要适应低氧条件才能建立侵袭性感染的假设。相比之下,McDonagh等人未能在体外厌氧条件下证明哺乳动物肺中差异表达的基因与厌氧条件之间存在联系,尽管厌氧条件完全缺氧,不适合与缺氧进行比较(129)。虽然还没有人明确指出曲霉菌感染部位的氧供应情况,但缺氧适应似乎是包括白色念珠菌和新型隐球菌在内的其他真菌病原体发病机制的一个关键组成部分,值得在烟曲霉菌中进行进一步研究(64)。
结论
烟曲霉是人类曲霉病最常见的病原。免疫功能严重受损的患者,尤其是那些患有血液病恶性肿瘤或接受过移植的患者,有感染最严重曲霉菌感染IA的风险。尽管对曲霉菌发病机制的研究非常广泛,但很少有明确的因素导致与烟熏相关的IA。也许最重要的是这个物种的基本生物学特征。小的、容易通过空气传播的分生孢子进入下呼吸道,烟曲霉在37℃下生长良好,发芽率超过其他种类。除了这些特征,烟曲霉还能适应哺乳动物肺部的环境条件。在有IA风险的免疫抑制个体中,分生孢子定植受损的肺组织或肺上皮细胞,逃避巨噬细胞的杀伤,降解周围组织以获得或合成生长所需的营养物质。根据潜在的宿主免疫状态,烟曲霉可能不受控制地生长和传播(中性粒细胞减少),或被中性粒细胞控制,导致过度炎症(类固醇诱导的免疫抑制)。烟曲霉的发病机制是多因素的,本综述中描述的许多真菌特性可能是导致烟曲霉在IA中流行的原因之一。突变体的描述和对体内条件的整体分析为了解烟曲霉的总体发病机制提供了重要的线索。结合这些方法,跨物种比较和结合宿主对曲霉菌感染的反应可以进一步了解烟曲霉IA发病机制的复杂性。
美国微生物学会Pathogenesis of Aspergillus fumigatus in Invasive Aspergillosis
Taylor R. T. Dagenais, Nancy P. KellerPathogenesis of Aspergillus fumigatus in Invasive Aspergillosis Summary: Aspergillus species are globally ubiquitous saprophytes found in a variety of ecological niches. Almost 200 species of aspergilli have been identified, less than 20 of which are known to cause human disease. Among them, Aspergillus fumigatus is the most prevalent and is largely responsible for the increased incidence of invasive aspergillosis (IA) in the immunocompromised patient population. IA is a devastating illness, with mortality rates in some patient groups reaching as high as 90%. Studies identifying and assessing the roles of specific factors of A. fumigatus that contribute to the pathogenesis of IA have traditionally focused on single-gene deletion and mutant characterization. In combination with recent large-scale approaches analyzing global fungal responses to distinct environmental or host conditions, these studies have identified many factors that contribute to the overall pathogenic potential of A. fumigatus. Here, we provide an overview of the significant findings regarding A. fumigatus pathogenesis as it pertains to invasive disease.
INTRODUCTION
Aspergillus species are ubiquitous, saprophytic fungi that play a significant role in global carbon and nitrogen recycling. Although their primary ecological niche is soil or decaying vegetation, aspergilli produce small, hydrophobic conidia that disperse easily into the air and can survive a broad range of environmental conditions. The genus Aspergillus, which includes almost 200 species, has a tremendous impact on public health both beneficially as the workhorse of industrial applications and negatively as plant and human pathogens (71). Several Aspergillus species are utilized for their rich enzymatic profile in the industrial production of foods and pharmaceuticals. For example, Aspergillus niger is used for the industrial production of citric acid, amylases, pectinases, phytases, and proteases; A. terreus is used for the cholesterol-lowering drug lovastatin; and A. oryzae is used for the fermentation of soybeans and rice into soy sauce and sake, respectively. Aspergilli also have a less reputable side in the agricultural industry. Aspergillus section Flavi, particularly A. flavus and A. parasiticus, can contaminate several common crops with aflatoxin, a highly toxic carcinogen with immunosuppressive properties (228, 230). The consumption of contaminated crops can cause serious illness or death and is a common problem in developing countries.
The Human Pathogen A. fumigatus
Among the human pathogenic species of Aspergillus, A. fumigatus is the primary causative agent of human infections, followed by A. flavus, A. terreus, A. niger, and the model organism, A. nidulans (54, 135). Aspergilli cause a wide range of human ailments depending on the immune status of the host (54, 107). In individuals with altered lung function such as asthma and cystic fibrosis patients, aspergilli can cause allergic bronchopulmonary aspergillosis, a hypersensitive response to fungal components. Noninvasive aspergillomas may form following repeated exposure to conidia and target preexisting lung cavities such as the healed lesions in tuberculosis patients. Invasive aspergillosis (IA) is perhaps the most devastating of Aspergillus-related diseases, targeting severely immunocompromised patients. Those most at risk for this life-threatening disease are individuals with hematological malignancies such as leukemia; solid-organ and hematopoietic stem cell transplant patients; patients on prolonged corticosteroid therapy, which is commonly utilized for the prevention and/or treatment of graft-versus-host disease in transplant patients; individuals with genetic immunodeficiencies such as chronic granulomatous disease (CGD); and individuals infected with human immunodeficiency virus (54, 97, 126, 133, 148, 162, 227). Mortality rates range from 40% to 90% in high-risk populations and are dependent on factors such as host immune status, the site of infection, and the treatment regimen applied (114). The severity and increased incidence of IA necessitate a better understanding of the interplay between host and fungus that contributes to A. fumigatus pathogenesis (130). Pathogenesis and virulence are terms used here in the context of altered host immune function, as this organism is inherently an opportunistic pathogen, and disease pathology and progression are the result of both fungal growth and the host response. In this review, we will thus discuss the pathogenic potential of A. fumigatus as a progression of the infectious life cycle within the context of these immunodeficiencies.Invasive Aspergillosis
Infectious life cycle.Aspergilli are predominantly saprophytes, growing on dead or decaying matter in the environment. The infectious life cycle of Aspergillus begins with the production of conidia (asexual spores) that are easily dispersed into the air, ensuring ubiquity in both indoor and outdoor environments (Fig. 1) (65, 137). The primary route of human infection is via the inhalation of these airborne conidia, followed by conidial deposition in the bronchioles or alveolar spaces. In healthy individuals, conidia that are not removed by mucociliary clearance encounter epithelial cells or alveolar macrophages, the primary resident phagocytes of the lung. Alveolar macrophages are primarily responsible for the phagocytosis and killing of Aspergillus conidia as well as the initiation of a proinflammatory response that recruits neutrophils (one type of polymorphonuclear cell [PMN]) to the site of infection. Conidia that evade macrophage killing and germinate become the target of infiltrating neutrophils that are able to destroy hyphae. The risk of developing IA results primarily from a dysfunction in these host defenses in combination with fungal attributes that permit A. fumigatus survival and growth in this pulmonary environment (176). Although other host responses have been associated with disease resistance, for this review, we will focus on fungal interactions with the primary innate components that are most important for fungal defense.Risk factors and pathology.The primary host immunodeficiencies that are responsible for the increased risk of IA are neutropenia and corticosteroid-induced immunosuppression, and the pathological consequences of IA under these immunosuppressive conditions differ, as described previously for patients and animal models (9, 17, 53, 192). Prolonged neutropenia is classically defined as the most dominant risk factor for IA and is often the result of highly cytotoxic therapies such as cyclophosphamide, which is used for transplant patients or those with hematological diseases. Cyclophosphamide, a DNA-alkylating agent, binds to DNA and interferes with cellular replication, depleting circulating white blood cells including neutrophils. In neutropenic patients and animal models of chemotherapy-induced neutropenia, IA is characterized by thrombosis and hemorrhage from rapid and extensive hyphal growth (41, 192). The lack of inflammatory infiltrates, despite the production of tumor necrosis factor alpha (TNF-α), results in low levels of inflammation. Without neutrophil recovery, angioinvasion and dissemination to other organs via the blood result.
A variety of nonneutropenic patients, most commonly those on corticosteroid therapy such as allogeneic transplant patients receiving corticosteroids for prophylaxis or treatment of graft-versus-host disease, are susceptible to IA, although the pathology of the disease is quite different. IA in these patients and nonneutropenic animal models is nonangioinvasive, characterized by limited fungal development with pyogranulomatous infiltrates, tissue necrosis, and excessive inflammation. Corticosteroids have significant consequences for phagocyte function, including but not limited to the impairment of phagocytosis, phagocyte oxidative burst, production of cytokines and chemokines, and cellular migration (reviewed in reference 116). Several studies have shown that corticosteroids impair the functional ability of phagocytes to kill A. fumigatus conidia and hyphae (37, 92, 132, 171, 172, 214). Despite the effects of steroids on innate immune cell function, neutrophils are recruited to the lung and prevent hyphal invasion but create an inflammatory environment that results in tissue injury. This exacerbated inflammatory response is generally regarded as being the cause of death, in contrast to the uncontrolled fungal growth observed in neutropenic hosts. The dramatic differences in both fungal development and host responses under each immunosuppressive regimen highlight the importance of studying Aspergillus pathogenesis within the context of host immune status and subsequent response to fungal infection.
AIRWAY COLONIZATION
Inhalation of Aspergillus conidia is a common occurrence due to their ubiquitous presence in the environment; estimates suggest that the average person may inhale up to 200 conidia per day. In IA-susceptible patient populations, the mucosal defenses of the lung are compromised, leading to fungal colonization and growth.
Aspergillus Interactions with Soluble Lung Components
Following inhalation, A. fumigatus conidia immediately encounter the airway mucosa comprised of the fluid lining the respiratory tract and airway epithelia. This pulmonary fluid is comprised of mucus, proteins, lipids, ions, water, and other cellular secretions that contribute to the mucociliary clearance of inhaled particles or pathogens. Also within this complex fluid are opsonic PRRs that coat inhaled pathogens and contribute to host defense. Among these proteins are the collectins, a group of C-type lectin receptors secreted by type II cells and Clara cells that bind carbohydrate moieties in a calcium-dependent manner. Many pathogenic fungi, including A. fumigatus, have a carbohydrate-rich cell wall that can be recognized by the most common collectins, mannose-binding lectin (MBL) and the surfactant proteins SP-A and SP-D. In vitro, MBL, SP-A, and SP-D have been shown to bind and agglutinate A. fumigatus conidia as well as enhance the phagocytosis and killing of A. fumigatus conidia by macrophages and neutrophils (2, 3, 120, 144). MBL, SP-A, and SP-D were more recently found to activate complement (59). Collectins have been demonstrated to be important in vivo: MBL−/− and SP-D−/− mice exhibit increased susceptibility to IA, and recombinant MBL, SP-D, and SP-A have been used to enhance host defenses against aspergillosis in animal models (94, 121). Collectins may thus contribute to conidial clearance by enhancing complement activation, phagocytosis, and killing of conidia or aggregating conidia for other host defenses.One of the earliest host responses to microorganisms is the activation of complement, a collection of serum proteins that recognize and bind conserved microbial constituents, resulting in opsonization or destruction. Although found predominantly in serum, complement components are present, albeit at lower levels, in bronchoalveolar fluid and have the potential to be involved in host defense against Aspergillus. Three complement pathways exist, converging at binding of C3 to the microbial surface. Early studies demonstrated that A. fumigatus conidia and hyphae bind C3. In comparison to other human pathogenic species, A. fumigatus as well as A. flavus bind fewer C3 molecules per unit of conidial surface (75). The majority of bound C3 is cleaved to iC3b, a ligand for phagocytic complement receptors; thus, A. fumigatus and A. flavus may be less susceptible to complement-mediated phagocytosis or phagocyte recognition (193, 194). Conidia and hyphae from several Aspergillus species also bind the alternative complement inhibitor factor H, its splice product FHR-1, and factor H family protein FFHL-1, preventing the activation of complement cascades (13, 213). Binding to the classical and lectin pathway inhibitor C4b-binding protein has been observed for A. fumigatus (213). Finally, A. fumigatus and A. flavus, but not A. niger, were found to produce a soluble complement-inhibitory factor, potentially lipid derived, that prevented alternative pathway activation (219, 221). It would thus appear that A. fumigatus and, to a lesser-known extent, A. flavus have defense mechanisms to inhibit or reduce complement activation. Thus, the ability of A. fumigatus to inhibit complement activation may contribute to the overall pathogenesis of this organism.
Other soluble components involved in Aspergillus defense include the pentraxin PTX3 and plasminogen. PTX3 is a soluble opsonin produced by phagocytes that facilitates microbial recognition (28). Mice deficient in PTX3 are susceptible to IA, which correlated with a reduced recognition of A. fumigatus conidia by phagocytes (69). A recent study implicated genetic variation in plasminogen, a component of the fibrolytic pathway, in susceptibility to IA (233). Plasminogen bound to A. fumigatus has also been detected, which, when cleaved into active plasmin, could enhance dissemination via its role in the degradation of the extracellular matrix (13).
Aspergillus Interaction with Respiratory Epithelia
Despite the importance of respiratory epithelia in initiating antimicrobial innate immune responses against many inhaled pathogens, few studies have examined the role of the airway epithelia in the host defense against Aspergillus (12, 127). As the first cells encountered by inhaled conidia, airway epithelia likely contribute to the overall immune response to A. fumigatus. Epithelial cells may secrete soluble antimicrobial compounds that play a direct role in airway defense. Members of the defensin family of antimicrobial peptides have broad-spectrum activity against multiple microbes and are produced by epithelial cells in vitro following incubation with A. fumigatus (1). In vitro, Aspergillus germinating conidia and hyphae, but not resting conidia, are recognized by host PRRs on epithelial cells and induce the production of cytokines and chemokines such as IL-6, TNF-α, and IL-8 (10, 14). Corticosteroid administration can eliminate this inflammatory response, questioning the function of epithelial cells in corticosteroid-treated patients at risk for IA (14). Epithelial cells likely assist in initiating proinflammatory responses against A. fumigatus, although their contribution is likely far less robust than that of the alveolar macrophage.. fumigatus conidia have been shown to bind and be engulfed by a variety of epithelial cells including tracheal epithelial cells, alveolar type II cells, human nasal epithelial cells, and the A549 lung epithelial cell line (66, 155, 223). Conidia engulfed by A549 epithelia enter acidic phagolysosomes and can be killed, although some conidia are able to germinate and exit both the phagolysosome and pneumocyte without evidence of pneumocyte damage (222, 223). A. fumigatus conidia are also able to inhibit drug- or TNF-α-induced apoptosis in primary epithelial cells and epithelial cell lines in vitro, although the in vivo implications of this are unknown (18). Several fungally derived factors may contribute to the ability of A. fumigatus to bind and modulate the airway epithelium (Fig. 2). One factor contributing to A. fumigatus binding and uptake by epithelial cells is the presence of sialic acid residues on conidia (34, 51). Interestingly, pathogenic species of aspergilli, including A. fumigatus, display more conidial sialic acid than do nonpathogenic aspergilli (224). Adhesion to fibrinogen, the basement membrane glycoprotein laminin, and the extracellular matrix component fibronectin is also partially mediated by sialic acid residues and other proteins on the conidial surface (7, 30, 31, 34, 46, 202, 203). The significance of conidial binding to these components is linked to the fact that lung injury or distress is a risk factor for IA. Fibrinogen (fibrin after processing) and fibronectin attach to wounded surfaces such as the surface of damaged epithelia, and laminin is exposed upon epithelial injury or detachment. Sialic acid, and perhaps other conidial factors that bind to alveolar components, could thus contribute to pathogenicity by enhancing adhesion to and colonization of epithelia and components of injured tissue.
A. fumigatus may facilitate colonization in otherwise healthy lung tissue via secreted products that alter epithelial function and viability. Culture filtrates from A. fumigatus strains have been shown to induce cell shrinkage, desquamation, and actin cytoskeleton rearrangement in A549 cells (93, 98). The activity of filtrates could be inhibited with serine and cysteine protease inhibitors, implicating protease activity. Indeed, when a specific serine protease, AF-ALF, was deleted, culture filtrates failed to induce actin cytoskeleton damage (98). Protease activity in A. fumigatus culture filtrates has also been linked to human nasal epithelial cell detachment and loss of focal contacts that may assist germinating hyphae in invading the lung tissue (98, 169, 201).
Human respiratory epithelial cell damage and slowed ciliary beat frequency from A. fumigatus culture filtrates and sputum samples obtained from patients with pulmonary aspergillosis have been linked to secondary metabolites, specifically gliotoxin and, at higher concentrations, fumagillin and helvolic acid (5, 6, 43). The tremorigenic metabolite verruculogen, produced in conidial and hyphal filtrates of many A. fumigatus strains, has also been implicated in modifying transepithelial resistance, hyperpolarization, and cytoplasmic vacuolization of human nasal epithelial cells in an air-liquid interface model of the airway epithelium (29, 96). Associated with conidia and hyphal elements, verruculogen could impact the airway epithelium during early infection, although production in vivo has yet to be observed (96). Thus, A. fumigatus is able to interfere with mucociliary clearance, bind respiratory epithelia and basement membrane proteins, and invade or damage epithelial cells to establish infection and potentially evade other host defenses.Aspergillus and the Alveolar Macrophage Macrophage responses to Aspergillus.Alveolar macrophages are the primary resident phagocytic cells of the respiratory tract and a critical component of the host defense against Aspergillus conidia. Alveolar macrophages phagocytose Aspergillus conidia in an actin-dependent manner, a process mediated by the recognition of pathogen-associated molecular patterns by host cell PRRs. PRR engagement of A. fumigatus ligands generates a proinflammatory response characterized by the production of cytokines and chemokines that are important for host defense against this organism, including TNF-α, IL-1β, IL-6, IL-8, macrophage inflammatory protein 1α, and monocyte chemoattractant protein 1 (39, 131, 138, 158). TLR2 and TLR4 and the C-type lectin receptor dectin-1 are the most well-characterized PRRs involved in the recognition of A. fumigatus and the activation of host cells. In vitro studies have demonstrated that conidia and hyphae activate macrophages through TLR2 and TLR4, and TLR2 recognizes both conidial and hyphal morphologies, whereas TLR4 recognizes only the hypha form (143). Studies using TLR−/− mice also suggest an essential role for TLR4, and potentially TLR2, in vivo. In neutropenic models, TLR2−/− and TLR4−/− mice exhibit higher fungal burdens than wild-type mice (11, 15). Although TLR4−/− mice have lower survival rates than wild-type mice in these studies, contradicting results were demonstrated for TLR2−/− mice. The role of TLRs in nonneutropenic models has not been well studied, although TLR4 polymorphisms in allogeneic hematopoietic stem cell transplant patients have been associated with an increased risk for IA (22). Unlike TLRs, dectin-1 is essential for the host defense against Aspergillus in both immunosuppressed and immunocompetent hosts (226). Dectin-1 is specific for the fungal carbohydrate β(1,3)-glucan, which is normally masked on resting A. fumigatus conidia by the proteinaceous hydrophobin layer. Following conidial swelling, β(1,3)-glucan becomes exposed and is present on swollen conidia, germlings, and hypha morphotypes (70, 77). Dectin-1-β(1,3)-glucan engagement results in phagocytosis, macrophage activation, and a strong induction of proinflammatory responses (35, 70, 77, 119, 189, 210). Thus, dectin-1, with additional contributions by TLRs, enables innate immune cells to phagocytose and kill conidia as well as elicit proinflammatory responses.
Alveolar macrophages kill conidia that have swollen within the phagolysosome with reactive oxygen species (ROS) and phagolysosomal acidification (83, 159, 220). Philippe et al. first demonstrated the role of ROS in macrophage-mediated conidial killing. In these experiments, alveolar macrophages from p47phox−/− CGD mice, or wild-type alveolar macrophages treated with chemical inhibitors of NADPH oxidase, were significantly impaired in their ability to kill A. fumigatus conidia (159). Although other studies suggested that NADPH-mediated oxidative responses do not contribute to alveolar macrophage killing of conidia, several factors (macrophage-to-conidium ratios, coincubation times, and animal strains and/or cell types tested) can lead to conflicting results (136, 177). It does appear that nonoxidative mechanisms contribute to conidial killing. Reactive nitrogen species are ineffective, although other candidates (antimicrobial peptides, for example) have yet to be tested (159). Overall, immunosuppressed patients who are susceptible to IA have reduced alveolar macrophage effector functions, either from corticosteroid-mediated suppression or from chemotherapy-induced depletion, resulting in the ability of A. fumigatus to escape macrophage killing.
A. fumigatus defenses. (i) Melanin.In addition to masking β(1,3)-glucan and delaying macrophage activation, resting A. fumigatus conidia are resistant to macrophage killing (Fig. 3). The protective role of the pigment melanin against host defenses, specifically via scavenging ROS, has been described for many pathogenic fungi (87, 106). In Aspergillus, melanin provides the conidial pigment that has been used to distinguish between some species. In A. fumigatus, a white, pigmentless strain was first described by Jahn et al. following UV mutagenesis (88). Complementation of the wild-type phenotype using an A. fumigatus cosmid library identified a polyketide synthase gene, pksP, as being the source of pigment production (105). The white mutant displayed ultrastructural cell wall differences and increased susceptibility to the oxidants H2O2 and NaOCl in comparison to wild-type conidia (88). Additionally, white conidia induced greater monocyte and neutrophil production of ROS than did the wild type as a result of wild-type melanin scavenging ROS from the culture medium. Monocytes were able to kill more ingested mutant conidia than wild-type conidia, presumably via ROS-mediated mechanisms. In an animal model of systemic IA, the white conidia were less virulent, demonstrating for the first time the direct role of melanin in A. fumigatus pathogenesis. These studies implicate melanin as being an important contributor to pathogenesis as an ROS scavenger. It should be noted that the systemic animal model of IA does not fully recapitulate the infectious process of IA, as conidia are instilled directly into the blood via the tail vain as opposed to intranasally or intratracheally. Clearly, this administration could lead to a very different host response to A. fumigatus. Studies of another melanin mutant in an intranasal model (see below), however, support the role for melanin in A. fumigatus pathogenesis. One study also implicated the involvement of the cyanide-insensitive alternative oxidase in protecting conidia from macrophage ROS-mediated killing (the hyphal component not being explored) (123). In that study, however, RNA interference was used to knock down aox expression in a melanin mutant such that alternative oxidase may be important for protection against macrophage killing only in the context of a melanin deficiency.dditional studies indicated that human macrophage engulfment of pksP mutant conidia resulted in an increased acidification of the phagosome as a result of phagolysosomal fusion (89). The addition of chloroquine, which increases phagolysosomal fusion and pH, resulted in wild-type killing similar to that of the mutant. A functional pksP therefore prevented some level of phagolysosomal fusion, increasing conidial survival. Furthermore, that study implicated phagolysosome acidification as being a mechanism of conidial killing. The initial discovery of pksP coincided with the identification by Tsai et al. and Watanabe et al. of another gene involved in pigment biosynthesis, arp1, a naphthopyrone synthase and homologue of scytalone dehydratase (206, 225). Mutants of arp1 were reddish-pink and bound fewer C3 molecules than wild-type conidia, implicating melanin or a pigmented intermediate synthesized by arp1 in the defense against host complement. Further study of conidial pigment biosynthesis in A. fumigatus identified a six-gene cluster involved in dihydroxynaphthalene-melanin biosynthesis, including pksP (also called alb1) (207). The deletion of alb1, similar to the observations by Jahn et al., led to a white phenotype and reduced virulence in mice (205). The alb1 mutants were also more susceptible to C3 binding, reinforcing the notion that melanin or melanin intermediates may prevent complement activation. Another melanin, pyomelanin, produced by the tyrosine degradation pathway, may also contribute to pathogenicity, as mutants showed an enhanced sensitivity to ROS (178). Overall, melanin appears to be a significant determinant of A. fumigatus pathogenesis by protecting conidia against multiple host defenses, particularly those of the alveolar macrophage. (ii) Mediators of ROS defense.Other molecules implicated in pathogenicity as scavengers of toxic ROS include rodlets and superoxide dismutases (SODs). Conidia are surrounded by a hydrophobic layer comprised of rodlet proteins. Of the two rodlet genes in A. fumigatus, rodA is solely responsible for rodlet production, and rodA mutants display increased susceptibility to alveolar macrophage killing (156). Rodletless conidia also induced a weak inflammatory response in a rat model of IA (183). Given that conidial swelling is essential for macrophage activation, the rodletless conidia may induce more rapid and robust macrophage activation and conidial elimination. The two eukaryotic SOD enzymes, Cu/Zn-SOD and Mn-SOD, have not been well studied for their role in A. fumigatus pathogenesis. Cu/Zn-SOD has been detected in the cell wall of conidia and hyphae, and SOD activity in culture filtrates has been demonstrated (73, 78). Although Cu/Zn-SOD is upregulated under low-iron conditions in vitro and sera from some patients with A. fumigatus infections react with Cu/Zn-SOD, a specific role in vivo has yet to be identified (38, 73, 79). McDonagh et al. found increased levels of Mn-SOD transcripts in response to neutrophils in vitro and during infection in a murine model of IA, implicating this SOD in oxidative stress defense (129).
Studies of rare hypervirulent mutants substantiate the contribution of oxidative stress resistance to pathogenicity. A disruption of the α(1,3)-glucan synthase gene ags3 led to increased virulence in an animal model of IA, correlating with resistance to oxidative stress in vitro, perhaps related to the increased melanin content or increased germination rate observed in this mutant (128). An unrelated mutant of oxylipin biosynthesis, in which the three cyclooxygenase genes in A. fumigatus were silenced by RNA interference, also demonstrated increased virulence correlating with increased resistance to H2O2 (208). The individual deletion of the cyclooxygenase genes did not yield an increase in virulence, although a loss of one gene, ppoC, resulted in greater resistance to H2O2 as well as aberrant conidium morphology and increased phagocytosis by macrophages (49). Finally, the deletion of the glycosylphosphatidylinositol-anchored protein ECM33 resulted in a hypervirulent strain with increased germination and resistance to cell wall-destabilizing agents (174). Intriguingly, the deletion of the ace2 gene, encoding a C2H2 zinc finger transcription factor, resulted in altered conidial pigmentation, cell wall organization, and resistance to H2O2 (63). The ace2 mutant was hypervirulent in a nonneutropenic mouse model but not in a neutropenic model, implicating resistance to neutrophil oxidative defenses. Transcripts of ags3, ecm33, and ppoC in this mutant were also examined and found to be lower than those of the wild type, suggesting that ACE2 regulation of these genes may contribute to some of the phenotypic similarities among these mutants.
参考文献:Pathogenesis of Aspergillus fumigatus in Invasive Aspergillosis | Clinical Microbiology Reviews https://cmr.asm.org/content/22/3/447
.png)
.png)