°°
°°
Hypoxia-Inducible Factor 1¶Ń Induces Fibrosis and Insulin
Resistance in White Adipose Tissue
ABSTRACT
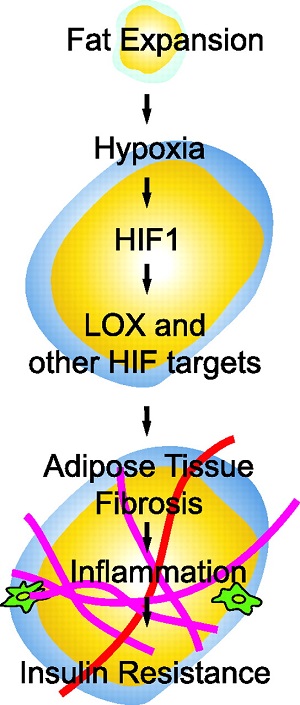
Adipose tissue can undergo rapid expansion during times of excess caloric
intake. Like a rapidly expanding tumor mass, obese adipose tissue becomes
hypoxic due to the inability of the vasculature to keep pace with tissue growth.
Consequently, during the early stages of obesity, hypoxic conditions cause an
increase in the level of hypoxia-inducible factor 1¶Ń (HIF1¶Ń) expression. Using a
transgenic model of overexpression of a constitutively active form of HIF1¶Ń, we
determined that HIF1¶Ń fails to induce the expected proangiogenic response. In
contrast, we observed that HIF1¶Ń initiates adipose tissue fibrosis, with an
associated increase in local inflammation. °įTrichrome- and picrosirius
red-positive streaks,°Ī enriched in fibrillar collagens, are a hallmark of
adipose tissue suffering from the early stages of hypoxia-induced fibrosis.
Lysyl oxidase (LOX) is a transcriptional target of HIF1¶Ń and acts by
cross-linking collagen I and III to form the fibrillar collagen fibers.
Inhibition of LOX activity by ¶¬-aminoproprionitrile treatment results in a
significant improvement in several metabolic parameters and further reduces
local adipose tissue inflammation. Collectively, our observations are consistent
with a model in which adipose tissue hypoxia serves as an early upstream
initiator for adipose tissue dysfunction by inducing a local state of fibrosis.
The dramatic rise in the prevalence of obesity has lead to increased efforts
aimed at gaining a better understanding of the physiology and pathophysiology of
adipose tissue and adipocytes. One of the more-surprising features of adipose
tissue described over the past 10 years is the realization that adipose tissue
in general and adipocytes in particular have the potential to be a rich source
of a vast array of secretory proteins. Since infiltrating immune cells, most
notably monocytes, are known to have a profound effect on adipocytes, interest
in the stromal fraction of adipose tissue has increased considerably. These
stromal components consist of fibroblastlike preadipocytes, endothelial cells,
vascular smooth muscle cells, neurons, and immune cells. It is currently not
established how these stromal components interact with adipocytes during adipose
tissue expansion. The nature of the local endothelium, a key constituent of the
vasculature, has received limited attention to date. Destruction of local
endothelial cells results in a reduction in fat mass during times of excess
caloric intake independent of food intake (2, 30, 38). Functioning through an as
yet unidentified mechanism, such a reduction in fat mass results in decreased
levels of steatosis in the liver and enhanced glucose tolerance. These metabolic
improvements are somewhat surprising, considering that the forced reduction of
fat mass in the context of lipodystrophies leads to a decrease rather than an
increase in systemic insulin sensitivity (30, 36). These observations highlight
the need for a better understanding of the adipose tissue vasculature.
During times of positive energy balance, adipose tissue absorbs the energy
surplus by increasing both cell size and number. The ability of adipose tissue
to expand critically depends on vascular outgrowth (4). At the same time, the
increased adipocyte size requires oxygen to diffuse over longer distances prior
to reaching adipocyte mitochondria; this is evident by a decreased partial
oxygen pressure (20 mmHg versus 40 mmHg) in obese versus lean mice, respectively
(20, 37, 53). Hypoxia in obese adipose tissue has been observed by several
groups and results in the induction of the key hypoxia regulator,
hypoxia-inducible factor 1 (HIF1) (20, 37, 49, 53). HIF1 is a heterodimer
consisting of the oxygen-regulated HIF1¶Ń subunit and the constitutively
expressed HIF1¶¬ (39). During normoxia, HIF1¶Ń is rapidly degraded by an
oxygen-dependent hydroxylation of two proline residues (P402/P564 in human
HIF1¶Ń), which enables binding to an E3 ligase complex, thus targeting the
protein for proteasomal degradation. Under hypoxic conditions, the level of
prolyl hydroxylation is reduced, and as a consequence, the protein accumulates
and translocates into the nucleus, where it binds to hypoxia response elements
in concert with HIF1¶¬ and p300. The stability of HIF1¶Ń can be uncoupled from the
local oxygen pressure by removal of the °įoxygen degradation domain°Ī (¶§ODD;
lacking amino acids 401 through 603) that comprises the two critical proline
residues. As a result, the half-life of HIF1¶Ń increases from 5 min to
approximately 60 min (23).
Here, our objectives were to address specifically the physiological consequences
of the local hypoxia in adipose tissue and the concomitant upregulation of
HIF1¶Ń. Taken together, we propose that HIF1¶Ń upregulation represents one of the
earliest events during adipose tissue expansion and an important step in the
sequential process of obesity-associated adipose tissue dysfunction.
Hypoxia-Inducible Factor 1¶Ń Induces Fibrosis and Insulin Resistance in White
Adipose Tissue | Molecular and Cellular Biology
https://mcb.asm.org/content/29/16/4467
°°
Adipose tissue hypoxia induces inflammatory M1 polarity of
macrophages in an HIF-1¶Ń-dependent and HIF-1¶Ń-independent manner in obese mice
°°
First Department of Internal MedicineUniversity of ToyamaToyamaJapan
Aims/hypothesis
As obesity progresses, adipose tissue exhibits a hypoxic and inflammatory
phenotype characterised by the infiltration of adipose tissue macrophages
(ATMs). In this study, we examined how adipose tissue hypoxia is involved in the
induction of the inflammatory M1 and anti-inflammatory M2 polarities of ATMs.
Methods
The hypoxic characteristics of ATMs were evaluated using flow cytometry after
the injection of pimonidazole, a hypoxia probe, in normal-chow-fed or
high-fat-fed mice. The expression of hypoxia-related and inflammation-related
genes was then examined in M1/M2 ATMs and cultured macrophages.
Results
Pimonidazole uptake was greater in M1 ATMs than in M2 ATMs. This uptake was
paralleled by the levels of inflammatory cytokines, such as TNF-¶Ń, IL-6 and
IL-1¶¬. The expression level of hypoxia-related genes, as well as
inflammation-related genes, was also higher in M1 ATMs than in M2 ATMs. The
expression of Il6, Il1¶¬ and Nos2 in cultured macrophages was increased by
exposure to hypoxia in vitro but was markedly decreased by the gene deletion of
Hif1a. In contrast, the expression of Tnf, another inflammatory cytokine gene,
was neither increased by exposure to hypoxia nor affected by Hif1a deficiency.
These results suggest that hypoxia induces the inflammatory phenotypes of
macrophages via Hif1a-dependent and -independent mechanisms. On the other hand,
the expression of inflammatory genes in cultured M2 macrophages treated with
IL-4 responded poorly to hypoxia.
Conclusions/interpretation
Adipose tissue hypoxia induces an inflammatory phenotype via Hif1a-dependent and
Hif1a-independent mechanisms in M1 ATMs but not in M2 ATMs.
Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an
HIF-1¶Ń-dependent and HIF-1¶Ń-independent manner in obese mice | SpringerLink
https://link.springer.com/article/10.1007/s00125-013-2885-1
°°
Oxygen deprivation and the cellular response to hypoxia in adipocytes ®C perspectives on white and brown adipose tissues in obesity
1Clore Laboratory, Buckingham Institute for Translational Medicine, University
of Buckingham, Buckingham, UK
2College of Science, King Saud University, Riyadh, Saudi Arabia
3Obesity Biology Unit, Institute of Ageing and Chronic Diseases, University of
Liverpool, Liverpool, UK
°°
°°
ĶĪ»Ľ£¨ĺ›Ī®Ķņ£¨īŐľ§°įļ÷Īš°ĪĶń“©őÔ‘Ĺņī‘Ĺ∂ŗ£®89£©£¨Ķę «”…”ŕ∆š÷ĪĹ”Ķń…ķņŪŌŗĻō–‘£¨»ťňŠ «ŐōĪūŃÓ»ňł––ň»§Ķń°£
į◊…ę÷¨∑ĺ£®WAT£© «≤ķ…ķ»ťňŠĶń÷ō“™≤ŅőĽ£¨∑ Ň÷◊ť÷Į÷–ĶńWATňģ∆Ĺ…żłŖ£®5£¨90£©°£Ņ…“‘ŐŠ≥Ų“ĽłŲń£–Õ£¨∆š÷–∑ Ň÷ňś◊Ň∑ Ň÷Ķń∑Ę’Ļ∂ÝĶľ÷¬WAT÷–»ťňŠĶń≤ķ…ķ‘Ųľ”£¨“Úő™īů–Õ÷¨∑ĺŌłįŻī””–—űīķ–Ľ◊™ĽĽő™—Š—űīķ–Ľ£®Õľ2£©°£»Ľļů£¨»ťňŠÕ®Ļż◊‘∑÷√ŕ/Ň‘∑÷√ŕ◊ų”√īŐľ§ŃŕĹŁĶńį◊…ę÷¨∑ĺŌłįŻ÷–ĶńUcp1ĪŪīÔ£¨ī”∂ÝĶľ÷¬briteŌłįŻĶńńľľĮļÕWAT÷–°įļ÷Īš°ĪĪŪ–ÕĶń≥ŲŌ÷°£’‚Ņ…ń‹∑ī”≥Ńňīů÷¨∑ĺŌłįŻĽżņŘĶń∑īĶųĹŕ∑ī”¶£¨’‚ «”…”ŕ»Ī—ű”’ĶľĶń»ťňŠ…żłŖĶľ÷¬≤ķ»»ŌłįŻĶń≤ķ…ķ£¨∂Ý≤ķ»»ŌłįŻĽŠ—űĽĮ÷¨÷ °£»Ľ∂Ý£¨Ļō”ŕbriteŌłįŻīŔ…ķ»»Ķń≥Ő∂»…–”–’ý“ť£¨Ļ„∑ļĶń—™Ļ‹–ő≥… «÷¨∑ĺŌłįŻīō∂‘ ”¶–‘≤ķ»»◊Ų≥Ų÷ō“™ĻĪŌ◊Ķń“Úňō÷ģ“Ľ°£
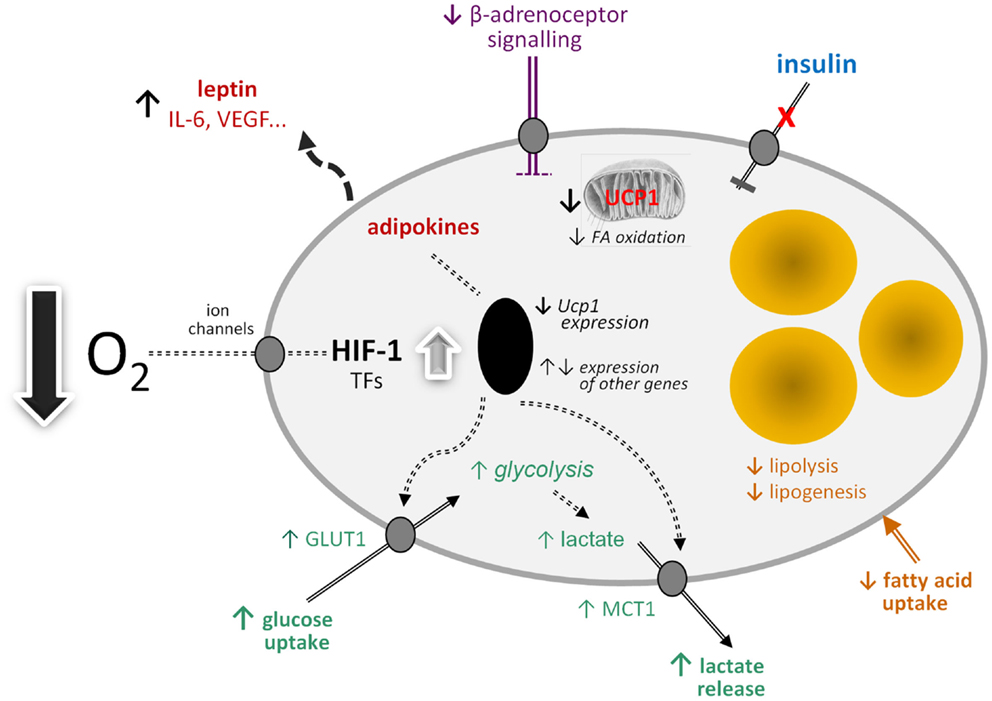
There are, of course, an increasing number of agents which have been reported to stimulate °įbrowning°Ī (89), but lactate is of particular interest because of its direct physiological relevance. WAT is an important site of lactate production and the levels in the tissue are increased in obesity (5, 90). A model can be proposed in which lactate production rises in WAT in obesity in response to developing hypoxia as large adipocytes switch from aerobic to anaerobic metabolism (Figure 2). The lactate then stimulates Ucp1 expression in neighboring white adipocytes, through an autocrine/paracrine action, leading to the recruitment of brite cells and the emergence of the °įbrowning°Ī phenotype in WAT. This could reflect a counter-regulatory response with the accumulation of large adipocytes resulting, via a hypoxia-induced elevation in lactate, in the production of thermogenic cells, which would oxidize lipid. However, the extent to which brite adipocytes may contribute to thermogenesis is debatable, with an extensive vascularization being one of the factors required for clusters of brite cells to make a significant contribution to adaptive heat production.
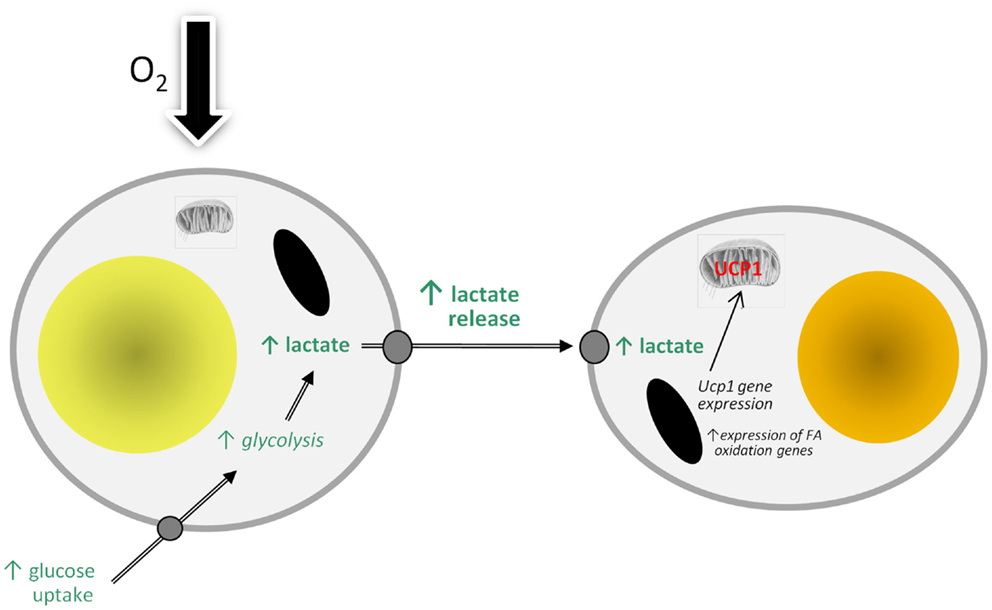
Figure 2. Model of how hypoxia may lead to the recruitment of brite adipocytes and the °įbrowning°Ī of white adipose tissue depots through stimulating the production and release of lactate. FA, fatty acid; UCP1, uncoupling protein-1.
°°
Frontiers | Oxygen Deprivation and the Cellular Response to Hypoxia in
Adipocytes ®C Perspectives on White and Brown Adipose Tissues in Obesity |
Endocrinology
https://www.frontiersin.org/articles/10.3389/fendo.2015.00019/full
Differences between white, brown and °įbrite°Ī fat tissue
https://www.gesundheitsindustrie-bw.de/en/article/news/differences-between-white-brown-and-brite-fat-tissue
°°
Adv Exp Med Biol. 2017;960:305-326. doi: 10.1007/978-3-319-48382-5_13.
Adipose Tissue Hypoxia in Obesity and Its Impact on
Preadipocytes and Macrophages: Hypoxia Hypothesis.
1 Faculty of Medicine, Department of General Surgery, Gazi University, Besevler,
Ankara, Turkey. dr.aengin@gmail.com.
2 Mustafa Kemal Mah. 2137. Sok. 8/14, 06520, Cankaya, Ankara, Turkey.
dr.aengin@gmail.com.
Abstract
Obese subjects exhibit lower adipose tissue oxygen consumption in accordance
with the lower adipose tissue blood flow. Thus, compared with lean subjects,
obese subjects have 44% lower capillary density and 58% lower vascular
endothelial growth factor (VEGF). The VEGF expression together with
hypoxia-inducible transcription factor-1 (HIF-1) activity also requires
phosphatidylinositol 3-kinase (PI3K)- and target of rapamycin (TOR)-mediated
signaling. HIF-1alpha is an important signaling molecule for hypoxia to induce
the inflammatory responses. Hypoxia affects a number of biological functions,
such as angiogenesis, cell proliferation, apoptosis, inflammation and insulin
resistance. Additionally, reactive oxygen radical (ROS) generation at
mitochondria is responsible for propagation of the hypoxic signal. Actually
mitochondrial ROS (mtROS) production, but not oxygen consumption is required for
hypoxic HIF-1alpha protein stabilization. Adipocyte mitochondrial oxidative
capacity is reduced in obese compared with non-obese adults. In this respect,
mitochondrial dysfunction of adipocyte is associated with the overall adiposity.
Furthermore, hypoxia also inhibits macrophage migration from the hypoxic adipose
tissue. Alterations in oxygen availability of adipose tissue directly affect the
macrophage polarization and are responsible from dysregulated adipocytokines
production in obesity. Hypoxia also inhibits adipocyte differentiation from
preadipocytes. In addition to stressed adipocytes, hypoxia contributes to immune
cell immigration and activation which further aggravates adipose tissue
fibrosis. Fibrosis is initiated in response to adipocyte hypertrophy in obesity.
Adipose Tissue Hypoxia in Obesity and Its Impact on Preadipocytes and
Macrophages: Hypoxia Hypothesis. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28585205
°°
Emerging Role of Adipose Tissue Hypoxia in Obesity and Insulin Resistance
Jianping Ye, Pennington Biomedical Research Center, Louisiana State University
System;
Abstract
Recent studies consistently support a hypoxia response in the adipose tissue in
obese animals. The observations have led to formation of an exciting concept,
adipose tissue hypoxia (ATH), in the understanding of major disorders associated
with obesity. ATH may provide cellular mechanisms for chronic inflammation,
macrophage infiltration, adiponectin reduction, leptin elevation, adipocyte
death, ER stress and mitochondrial dysfunction in white adipose tissue in
obesity. The concept suggests that inhibition of adipogenesis and triglyceride
synthesis by hypoxia may be a new mechanism for elevated free fatty acids in the
circulation in obesity. ATH may represent a unified cellular mechanism for
variety of metabolic disorders, and insulin resistance in patients with
metabolic syndrome. It suggests a new mechanism of pathogenesis of insulin
resistance and inflammation in obstructive sleep apnea. Additionally, it may
help us to understand the beneficial effects of caloric restriction, physical
exercise, and angiotensin II inhibitors in the improvement of insulin
sensitivity. In this review article, literatures are reviewed to summarize the
evidence and possible cellular mechanisms of ATH. The directions and road blocks
in the future studies are analyzed.
°°
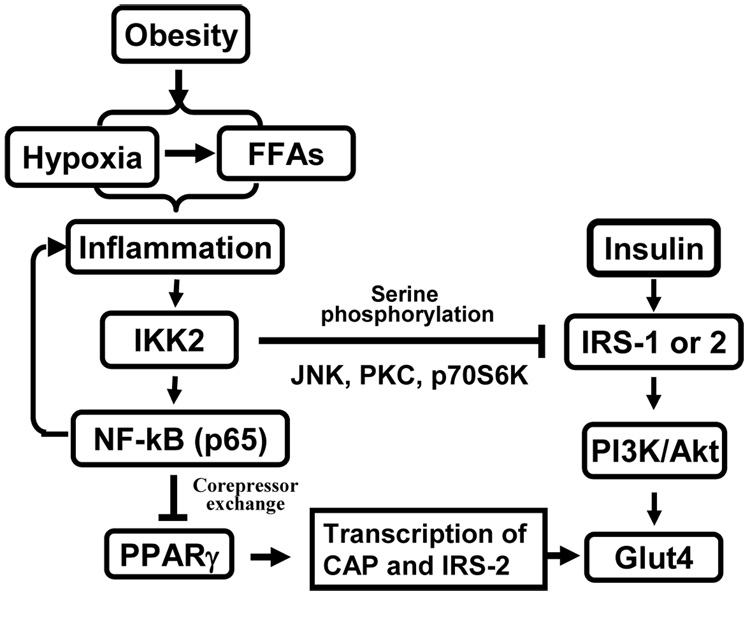
Adipose tissue hypoxia and inflammation response
ATH may provide an answer to the question about the cause of chronic
inflammation in adipose tissue in obesity. It may also explain the impact of
ischemia/reperfusion in the adipose tissue (71). The hypoxia is able to induce
inflammation in adipose tissue by induction of gene expression in adipocytes and
macrophages. This possibility was demonstrated using primary cells and cell
lines (20, 21). The induced genes include TNF-¶Ń, IL-1, IL-6, MCP-1 (Monocyte
chemoattractant protein-1), PAI-1 (plasminogen activator inhibitor-1), MIF
(macrophage migration inhibition factor), iNOS (inducible nitric oxide
synthase), MMP9 (matrix metalloproteinases 9), and MMP2. The molecular mechanism
of gene expression is related to activation of NF-kB and HIF-1¶Ń. All of these
genes are targets of NF-kB, and some of them are also targets of HIF-1 (PAI-1,
MIF, and iNOS) (63).
Activation of NF-kB by hypoxia is well-established in the fields of cancer
biology, immunology, and cardiovascular research as being reviewed (72®C77).
In cancer research, it is proposed that activation of NF-kB is responsible for
tumor resistance to radiotherapy or chemotherapy in cancer patients (75). NF-kB
increases tumor survival through anti-apoptosis effects. In cardiovascular
study, activation of NF-kB by hypoxia is proposed to mediate inflammation for
lung injury, and tissue damage in ischemia-reperfusion (76). It is known that
hypoxia activates NF-kB through IKK-independent pathway as being reviewed
(72, 78). In response to hypoxia, NF-kB is disassociated from IkBa in the
absence of IkBa degradation. The disassociation leads to nuclear translocation
and transcriptional activation of inflammatory cytokines.
Transcription factor NF-kB is a master regulator of inflammation response
(78®C80). It is formed by two proteins of the Rel family, p65 and p50 (78). NF-kB
controls transcription of many pro-inflammatory cytokines (such as TNF-¶Ń, IL-1,
and IL-6) or inflammation mediator (such as iNOS, and VACM). The signaling
pathway for NF-kB activation has been well documented in reviews (81, 82). In
the absence of activation, NF-kB is associated with IkB¶Ń (inhibitor) and
retained in the cytoplasm. When cells are stimulated by extracellular signal,
such as TNF-¶Ń or LPS, activation of IKK2 leads to phosphorylation,
ubiquitination and degradation of IkB¶Ń protein in proteasome. In the absence of
IkB¶Ń, NF-kB will be activated and translocated into the nucleus to initiate
transcription of target genes, which include IkBa, TNF-¶Ń, IL-1 and IL-6. After
the IkB¶Ń protein level is restored from this transcription-based process, NF-kB
will be associated with IkB¶Ń and then excluded from the nucleus. In this
classical signaling pathway, activation of NF-kB is dependent on activation of
IKK2. In general, inhibition of IKK2 leads to suppression of the transcriptional
activity of NF-kB.
°°
ATH in adipocyte cell death and plasma FFA elevation
Hypoxia may be a potential risk factor for adipocyte death in adipose tissue of
obese subjects. An increase in adipocyte death was reported in adipose tissue of
obese subjects, and was proposed to induce macrophage infiltration (18). In
dietary obese mice, adipocyte death was increased with growth of fat pads (19).
However, the reason of adipocyte death is not clear. Our study suggests that
hypoxia induces necrosis in 3T3-L1 adipocytes (99). This observation provides an
underlying mechanism of cell death in adipose tissue. The cell death may promote
lipolysis and release of FFA into blood stream under insulin resistance. This
will explain the increase in plasma FFA in obesity. In adult rats, plasma FFA in
the vein blood is induced by acute hypoxia in an ischemia model (99). In the
newborn mice, plasma FFA is induced by systemic hypoxia (128). In ischemia
research, hypoxia has been well-documented in the induction of cell death in
heart and brain (129). In adipose tissue, ischemia induces damages in several
forms, such as edema congestion and bleeding (71).
Possible causes of adipose tissue hypoxia
The physiological basis of ATH might be related to reduction in adipose tissue
blood flow (ATBF) (ml/min/100g tissue) and capillary density. Reduction in ATBF
has been reported in obesity in both humans (149®C151) and animals (152®C154). The
reduction means that blood perfusion is reduced in each unit of adipose tissue
in obesity. In obese people, the ATBF rate was 30®C40% lower (P < 0.02®C0.05) than
that of non-obese subjects (153). Although an earlier study suggests that ATBF
was not reduced in obesity (155), all of later studies consistently support the
reduction of ATBF (149®C154). The ATBF reduction was observed only in the obese
diabetic rats (obese Zucker rat), but not in the non-obese diabetic GK rats
(154), suggesting a role of adipose tissue mass in the control of blood flow.
Insulin resistance may not lead to the ATBF reduction since it occurs in both
obese Zucker rats and non-obese GK rats. The ATBF reduction is associated with
insulin resistance in obesity (150, 151). Although the association has been
known in obesity for years, the intermediate events linking the two conditions
remains unknown. The adipose tissue hypoxia may be a potential link.
A reduction in capillary density may contribute to the adipose tissue hypoxia
(156). Capillary density is determined by angiogenesis that requires
proliferation and tube formation of endothelial cells. Endothelial proliferation
was driven by growth factors including VEGF, and FGF2. The tube formation and
capillary maturation are controlled by a different set of cytokines including
PDGF, TGF-¶¬ and Angiopoietin. We observed that VEGF expression was not increased
in response to hypoxia in the adipose tissue of ob/ob mice although expression
of other hypoxia response genes was up-regulated (20). This defect was
associated with a reduced endothelial density in the tissue (156). The evidence
suggests that angiogenesis is deficient in the adipose tissue of obese mice, and
this defect may account for the reduction in adipose tissue blood flow in
obesity. The detail molecular events underlying the angiogenic defect remain to
be investigated in obese condition.
Blood perfusion is reduced from a decrease in vasodilation or increase in
vasoconstriction. An increase in vasoconstriction in obesity is supported by
literature. Angiotensin II (Ang II) is a serum peptide with known function to
increase vasoconstriction. Ang II is a component in the renin-angiotensin system
(RAS), and produced after hydrolysis of Ang I by angiotensin-converting enzyme
(ACE). Ang II acts on both the type 1 (AT1) and type 2 (AT2) receptors (157,
158). In obesity, the Ang II activity is increased in adipose tissue and in
circulation (159®C161). This may contribute to the ATBF reduction through an
increase in vasoconstriction (162). Additionally, the Ang II inhibitors are
known to enhance blood perfusion in adipose tissue (161). The inhibitors also
decrease inflammation in adipose tissue, and increase systemic insulin
sensitivity (163, 164). It remains to be tested if the pharmacological
inhibitors for Ang II improve oxygen supply in the adipose tissue in obesity.
In addition to the ATBF reduction, the increase in adipocyte size may contribute
to the interstitial hypoxia. In tissue, oxygen can only defuse about 120 micron
(165, 166). When adipocyte diameter increases to (or above) 120 micron, oxygen
will not be able to reach the cells beyond 120 micron from the capillary. The
diameter of a large adipocyte can be over 150 micron (167). This distance effect
remains to be tested in the adipose tissue in obesity.
Application of ATH
In addition to the role in pathogenesis of insulin resistance, ATH may provide
an alternative mechanism for insulin sensitization by several factors, such as
physical exercise, fasting, weight loss and Ang II inhibitors. ATBF is increased
in response to stress such as exercise (198®C200), mental stress (201), fasting
(202) and nutrient intake (149, 203®C205). ATBF is increased by the Ang II
inhibitor (161), epinephrine (206®C208), insulin and NO (nitric oxide) (209).
Insulin sensitivity is improved by physical exercise, fasting, and the Ang II
inhibitors, and ATBF is increased in all of these conditions. An improvement in
oxygen supply may contribute to the mechanism of insulin sensitization under
these conditions. This possibility needs to be tested in experiment.
Emerging Role of Adipose Tissue Hypoxia in Obesity and Insulin Resistance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2650750/
Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an
HIF-1¶Ń-dependent and HIF-1¶Ń-independent manner in obese mice | SpringerLink
https://link.springer.com/article/10.1007/s00125-013-2885-1
°°
∑«ĺ∆ĺę–‘÷¨∑ĺłő—◊ļÕ—◊–‘ĺř …ŌłįŻM1‘ŕ÷¨∑ĺ◊ť÷ĮļÕłő‘ŗĽżĺŘŌŗĻō
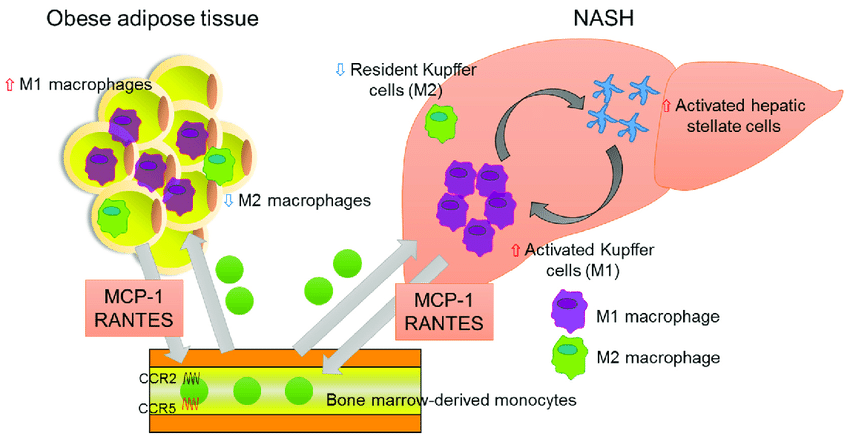
Association of chemokines and NASH. In adipose tissue of the obese, bone-marrow-derived monocytes are recruited from the bloodstream, predominantly via MCP-1-CCR2 signaling. The RANTES-CCR5 pathway also plays an important role in monocyte recruitment in adipose tissue. Infiltrated macrophages in obese adipose tissue undergo a phenotypic switch from alternative M2 macrophages to classical M1 macrophages. The latter secrete pro-inflammatory cytokines, which result in insulin resistance, adipokine dysfunction, and excess lipid accumulation in the liver. In the fatty liver, the recruitment and activation of immune cells, including Kupffer cells, contribute to hepatic inflammation, which is involved in hepatic stellate cell activation.
Association of chemokines and NASH. In adipose tissue of the obese,... |
Download Scientific Diagram
https://www.researchgate.net/figure/Association-of-chemokines-and-NASH-In-adipose-tissue-of-the-obese-bone-marrow-derived_fig2_316676050
°°
Annu Rev Physiol. 2010;72:219-46. doi: 10.1146/annurev-physiol-021909-135846.
Macrophages, inflammation, and insulin resistance.
Olefsky JM1, Glass CK.
Author information
1
Department of Medicine, University of California-San Diego, La Jolla, CA
92093-0651, USA. jolefsky@ucsd.edu
Abstract
Obesity induces an insulin-resistant state in adipose tissue, liver, and muscle
and is a strong risk factor for the development of type 2 diabetes mellitus.
Insulin resistance in the setting of obesity results from a combination of
altered functions of insulin target cells and the accumulation of macrophages
that secrete proinflammatory mediators. At the molecular level, insulin
resistance is promoted by a transition in macrophage polarization from an
alternative M2 activation state maintained by STAT6 and PPARs to a classical M1
activation state driven by NF-kappaB, AP1, and other signal-dependent
transcription factors that play crucial roles in innate immunity. Strategies
focused on inhibiting the inflammation/insulin resistance axis that otherwise
preserve essential innate immune functions may hold promise for therapeutic
intervention.
Macrophages, inflammation, and insulin resistance. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/20148674/
°°
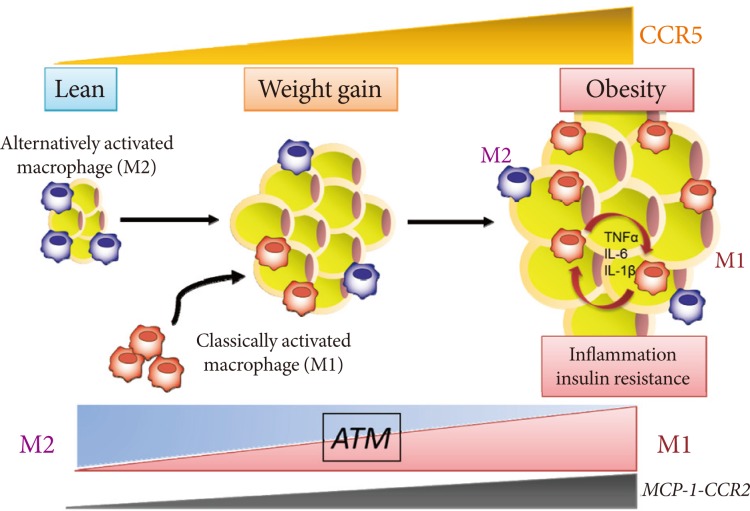
Chemokine Systems Link Obesity to Insulin Resistance
Tsuguhito Ota
Department of Cell Metabolism and Nutrition, Brain/Liver Interface Medicine
Research Center, Kanazawa University School of Medicine, Kanazawa, Japan.
Abstract
Obesity is a state of chronic low-grade systemic inflammation. This chronic
inflammation is deeply involved in insulin resistance, which is the underlying
condition of type 2 diabetes and metabolic syndrome. A significant advance in
our understanding of obesity-associated inflammation and insulin resistance has
been recognition of the critical role of adipose tissue macrophages (ATMs).
Chemokines are small proteins that direct the trafficking of immune cells to
sites of inflammation. In addition, chemokines activate the production and
secretion of inflammatory cytokines through specific G protein-coupled
receptors. ATM accumulation through C-C motif chemokine receptor 2 and its
ligand monocyte chemoattractant protein-1 is considered pivotal in the
development of insulin resistance. However, chemokine systems appear to exhibit
a high degree of functional redundancy. Currently, more than 50 chemokines and
18 chemokine receptors exhibiting various physiological and pathological
properties have been discovered. Therefore, additional, unidentified
chemokine/chemokine receptor pathways that may play significant roles in ATM
recruitment and insulin sensitivity remain to be fully identified. This review
focuses on some of the latest findings on chemokine systems linking obesity to
inflammation and subsequent development of insulin resistance.
Chemokine Systems Link Obesity to Insulin Resistance - ScienceCentral
https://www.e-sciencecentral.org/articles/SC000002363
°°
Exercise and Adipose Tissue Macrophages: New Frontiers in Obesity Research?
1Combat Protection and Performance Program, DSO National Laboratories,
Defence Medical and Environmental Research Institute, Singapore
2Department of Physiology, Yong Loo Lin School of Medicine, National University
of Singapore, Singapore
3Division of Endocrinology, Department of Medicine, Khoo Teck Puat Hospital,
Singapore
4Institute for Memory Impairments and Neurological Disorders (MIND Institute),
University of California Irvine, Irvine, CA, USA
°°
ABSTRACT
Obesity is a major public health problem in the twenty-first century.
Mutations in genes that regulate substrate metabolism, subsequent dysfunction in
their protein products, and other factors, such as increased adipose tissue
inflammation, are some underlying etiologies of this disease. Increased
inflammation in the adipose tissue microenvironment is partly mediated by the
presence of cells from the innate and adaptive immune system. A subset of the
innate immune population in adipose tissue include macrophages, termed adipose
tissue macrophages (ATMs), which are central players in adipose tissue
inflammation. Being extremely plastic, their responses to diverse molecular
signals in the microenvironment dictate their identity and functional
properties, where they become either pro-inflammatory (M1) or anti-inflammatory
(M2). Endurance exercise training exerts global anti-inflammatory responses in
multiple organs, including skeletal muscle, liver, and adipose tissue. The
purpose of this review is to discuss the different mechanisms that drive
ATM-mediated inflammation in obesity and present current evidence of how
exercise training, specifically endurance exercise training, modulates the
polarization of ATMs from an M1 to an M2 anti-inflammatory phenotype.
°°
Introduction
The immune system is instrumental in mediating a number of physiological
processes in the mammalian species, including pathogen surveillance, wound
repair, and metabolic regulation. Accumulating evidence shows that the immune
system interacts with other organ systems, including the adipose tissue.
Traditionally, adipose tissue is known for its role in energy homeostasis,
especially as a storage depot for lipids. The scientific paradigm of this
once-neglected organ shifted, when researchers discovered novel secretory
functions of adipose tissue in the mid-1990s. Seminal studies demonstrated that
gene and protein products of hormones that regulate satiety, such as leptin,
were found to be overexpressed in, and secreted from adipocytes (1, 2).
Adipose tissue was further recognized as an endocrine-like organ, particularly
after adipose tissue per se was shown to express and secrete cytokines that can
exert effects in distant organs, such as tumor necrosis factor (TNF)-¶Ń and
interleukin (IL)-6, among other cytokines previously thought to originate only
from immune cells (3, 4). Cytokines, hormones, and other protein factors
secreted from adipose tissue were subsequently termed °įadipokines,°Ī and they can
exhibit autocrine, paracrine, and endocrine functions (5). Since then, adipose
tissue has also been recognized to be in a chronic inflammatory state in an
obese host, wherein immune cells, such as macrophages, were found in greater
abundance ®C 40% of all cells within white adipose tissue (WAT) of obese mice,
relative to 10% in lean mice (6). In addition, this increased macrophage density
correlated with glucose and insulin resistance (7). In an obese state, more than
90% of all adipose tissue macrophages (ATMs) were found at sites of adipocyte
death, where they form a crown-like structure (8) and participate in tissue
remodeling, such as scavenging lipids from necrotic adipocytes (9) or inducing
vessel growth (10).
Resident macrophages are phenotypically heterogeneous, where two distinct forms
of macrophages, M1 and M2 phenotypes, are found within adipose tissue of both
obese and lean individuals (or mice), with the extent of each phenotype
dependent on local signals from the adipose microenvironment. In general, murine
studies have demonstrated that excess adiposity increases the proportion of M1
to M2 macrophages in WAT (8). Lumeng°Įs group reported that after diet-induced
obesity, murine ATMs had high gene expressions of cluster of differentiation
(CD)11c+, TNF-¶Ń, and inducible nitric oxide synthase (iNOS), which are markers
characteristic of M1 macrophages, whereas ATMs from lean mice expressed many
genes characteristic of M2 macrophages, including arginase 1 (Arg1), Ym1, and
IL-10 (8).phenotype.
°°
Obesity-Related Metabolic Stress, Inflammation, and Macrophage
Polarization
Obesity- and exercise-induced macrophage polarization requires an integration of
metabolic and immune crosstalk. First, excess adipocyte lipid availability
during obesity increases (i) the availability of fatty acids for activation of
immune and metabolic mediators of inflammatory response, including toll-like
receptor (TLR)-4, nuclear factor-kappa B (NF-¶ B), IkB kinase (IKK)-¶¬, Jun kinase
(JNK)-1, fatty acid binding proteins (FABPs), PPAR(s), (ii) cellular stress
[unfolded protein response (UPR) in the endoplasmic reticulum], (iii)
mitochondrial reactive oxygen species (ROS) production and mitochondrial
dysfunction, and (iv) protein synthesis (mammalian target of rapamycin (mTOR)
hyperactivation) (15®C17). Each of these factors will be discussed in turn.
Excess Availability of Fatty Acids In the obese state, increased
concentrations of saturated fatty acids activate (i) TLR4 in both adipocytes and
macrophages (18), (ii) NF-¶ B in adipocytes (19), (iii) IKK¶¬ in myeloid cells
(20), and (iv) JNK in adipocytes (21), all of which activate downstream
inflammatory cytokines and proteins, such as TNF-¶Ń, IL-6, and iNOS (16). These
pro-inflammatory cytokines and proteins participate in a feedback loop between
adipocytes and circulating monocytes, culminating in the M1 polarization of
macrophages that infiltrate the adipose tissue. Although well characterized as a
cellular lipid chaperone, ligand-bound FABP4 also demonstrates novel roles in
inflammation and in its interaction with nuclear receptors, such as PPARs (15).
FABP4−/− macrophages demonstrated impaired IKK and NF-¶ B activity, concomitant
with a reduction in protein expressions of cyclooxygenase (COX)-2 and iNOS, as
well as lower LPS-stimulated secretions of monocyte chemotactic protein (MCP)-1,
TNF-¶Ń, and IL-6 (22). Obesity-associated hyperlipidemia presents excess fatty
acids that bind to FABP4 in adipocytes or stromal macrophages in the adipose
tissue microenvironment, inducing the secretion of pro-inflammatory cytokines
that recruit greater numbers of M1 macrophages. This view is supported by
co-culture experiments where deletion of FABP4 in adipocytes resulted in MCP-1
gene expression in macrophage, and deletion of FABP4 in macrophages improved
insulin signaling and glucose uptake in adipocytes (23). Peroxisome
proliferator-activated receptors are transcription factors that can be activated
by fatty acids, and belong to the nuclear receptor family (24). They are
expressed in adipose tissue, skeletal muscle, liver, macrophages, and other
organs, although the three isoforms (¶Ń, ¶ń, and ¶√) are tissue specific (25). Both
PPAR¶√ and PPAR¶ń expressions are negatively associated with obesity, where they
modulate adipogenesis and lipid oxidation, respectively (26). In addition, PPAR¶ń
is required for inducing macrophage M2 polarization, as PPAR¶ń−/− macrophages
demonstrated a decrease in M2 macrophage profile expression, as gene expressions
of IL-13, IL-4, and macrophage galactose N-acetyl-galactosamine were all
reduced, compared with wild-type macrophages (27). In addition, myeloid-specific
PPAR¶ń−/− mice demonstrated an increase in gene expression of M1 macrophage
markers (MCP-1, TNF-¶Ń, IL-6) (27). Similarly, PPAR¶√−/− macrophages are polarized
toward the M1 phenotype (28), suggesting that PPARs play a role in mediating
macrophage polarization. Mitochondrial ROS, Cellular Stress, and Aberrant
Protein Synthesis Obesity augments metabolic stress in the organism. At the
cellular level, this is demonstrated through mitochondrial dysfunction and mTOR
hyperactivation, whereby the integrated cellular signaling of these two
pathological conditions can contribute to ER stress (15). Excess lipids in the
adipocyte increase the substrate load for inefficient mitochondrial oxidative
phosphorylation, leading to generation of ROS that can damage mitochondrial
constituents, and perpetuate a cycle of ROS-induced damage and further
mitochondrial dysfunction. Adipocyte mitochondrial dysfunction permits further
excess cellular lipid build-up, leading to the production of the
pro-inflammatory cytokines described earlier and recruitment and polarization of
M1 macrophages. Overabundance of lipids in the adipocyte is sensed by mTOR as a
high intracellular energy state, leading to hyperactivation of mTOR, which
chronically can lead to uncontrolled protein synthesis, increased ER stress,
UPR, and JNK activation (15). Lipid overload can also directly mediate the
inflammatory response by inducing ER stress, via the initiation of UPR (15). The
UPR activates cyclic-AMP-responsive element-binding protein H (CREBH), which
induces C-reactive protein (CRP) and serum amyloid P-component (SAP) production,
both of which are mediators of the acute phase response that contributes to
pro-inflammatory cytokine production. Lipid overload can indirectly simulate the
pro-inflammation state, as the ER responds to both mitochondrial dysfunction and
mTOR hyperactivation and integrates their respective signals to generate ROS
(15) and activate the JNK- (29) and NF-¶ B-mediated (30) inflammation pathways.
Thus, the ER, mTOR, and mitochondria can both directly and indirectly induce M1
macrophage activation via production of pro-inflammatory cytokines directly, and
through inter-organelle crosstalk as described. In addition to lipid overload,
glucose intolerance and insulin resistance are associated with obesity, which
may be partly explained by reduced serum concentrations of adiponectin (31), an
important adipokine that promotes glucose uptake, insulin sensitivity, and
¶¬-oxidation in peripheral tissues. Adiponectin is an anti-inflammatory and M2
macrophage polarizing molecule, as it attenuates TLR4-mediated NF-¶ B activation
(32) and upregulates IL-10 production in macrophages (33). In addition,
adenoviral delivery of adiponectin in wild-type mice increased Arg1 expression
in peritoneal macrophages, whereas peritoneal macrophages isolated from
adiponectin−/− mice showed increased M1 macrophage polarization (34). Thus,
adiponectin deficiency in obesity could alter macrophage polarization fates to
favor M1 activation.
°°
Effects of Exercise on Metabolic Stress, Inflammation, and Macrophage Polarization
Attenuation of Metabolic Stress
Exercise training increases adipocyte-specific gene and protein expression of
AMPK and PGC-1¶Ń (35), which in turn enhances ¶¬-oxidation and mitochondrial
biogenesis, allowing for greater lipid oxidation per mitochondrion. Improved
mitochondrial ¶¬-oxidation reduces oxidative stress and mitochondrial
dysfunction, thus limiting pro-inflammatory cytokine production and reducing
signals for ER stress-mediated inflammation. Improved function and reduced
stress in both organelles as direct or indirect consequences of exercise
training can, thus, attenuate macrophage M1 polarization or recruitment via
reduced stress signaling. Improved exercise-induced lipid oxidation attenuates
the need to transport excess fatty acids. For instance, FABP4 concentrations in
circulation were reduced with aerobic training in obese women (36). This
attenuation in serum FABP4 may be mediated by improved AMPK signaling, since
metformin, an AMPK agonist, reduced macrophage Forkhead box O1 (FOXO1)-mediated
transcription of FABP4 protein expression (37).
Peroxisome proliferator-activated receptors also respond favorably to exercise
training in that protein expression of PPAR¶ń increased by 53% in adipose tissue
of exercise-trained rats fed a high-fat diet, compared with sedentary rats on
the same diet (38). Similar outcomes in PPAR¶√ were attained with (i)
exercise-trained rats, with increased DNA-binding activity in adipocytes (39)
and in (ii) exercise-trained humans, with increased gene expression in
adipocytes (35). Such outcomes translate to improved adipocyte lipogenesis and
oxidation, reducing free fatty acids in the adipose tissue microenvironment for
PPAR¶ń/¶√-mediated M1 macrophage activation.
Exercise training improves glucose and insulin sensitivity, mediated partly by
improved AMPK/insulin receptor substrate (IRS)-1/phosphoinositol-3 kinase (PI3K)
signaling. Such exercise-induced improvements are associated with increased gene
(40®C42) and protein (43) expression of adiponectin and gene expression of
adiponectin receptor (40) in adipose tissues of rats and humans.
Reduction of Inflammation and Modulating Phenotypes of Macrophages
Physical inactivity has been associated with several chronic metabolic and
inflammatory diseases, such as type 2 diabetes mellitus (T2D) (44®C46).
Furthermore, a sedentary lifestyle is accompanied by the accumulation of
visceral fat, which predisposes adipose tissue to infiltration by
pro-inflammatory immune cells, increases adipokine secretion and the development
of a low-grade, systemic inflammatory state (47). Low-grade systemic
inflammation is associated with the pathology of several diseases, including
neurodegenerative diseases and insulin resistance. Chronic moderate exercise, in
contrast, has been shown to exert anti-inflammatory effects and, therefore,
protects against chronic inflammation-associated diseases (44®C46, 48). This
protective effect of regular exercise may be mediated through both the reduction
of visceral fat mass and the induction of an anti-inflammatory environment with
each bout of exercise (48, 49).
Several possible mechanisms have been described regarding the beneficial
anti-inflammatory effects of regular physical activity, including: (i) reduction
in visceral fat mass (with a subsequent decreased production and release of
pro-inflammatory adipokines), (ii) reduction in the expression of TLRs on
monocyte and macrophages (50), and (iii) induction of several anti-inflammatory
molecules from leukocytes and skeletal muscle (51, 52). In addition, the
inhibition of monocyte/macrophage infiltration into adipose tissue and the
phenotypic switching of macrophages within adipose tissue (52, 53) have been
proposed recently. The last two mechanisms are of great importance, since
obesity is accompanied by ATMs infiltration into adipose tissue, and induces a
phenotypic switch in ATM polarization from an anti-inflammatory M2 phenotype to
a pro-inflammatory M1 phenotype and, hence, contributing to insulin resistance
(27, 54®C56).
The anti-inflammatory function of exercise might prevent chronic inflammatory
diseases through the induction of phenotypic switching from M1 to M2
macrophages, as well as inhibit macrophage infiltration into adipose tissue.
There are few studies investigating the role of exercise training on macrophage
phenotype switching in adipose tissues. In an earlier study by Kawanishi and
colleagues (53), these investigators showed, for the first time, that treadmill
running (16 weeks) significantly decreased CD11c (M1 macrophage-specific marker)
mRNA expression and increased CD163 (M2 macrophage marker) mRNA expression in
adipose tissues of obese mice. In a recent study, these authors showed that
exercise training decreased TNF-¶Ń mRNA and CD11c levels in the adipose tissues
of high-fat-diet obese mice (57).
Similar observations were also reported in other pre-clinical studies (58, 59).
First, Oliveira et al. (58) reported that two single bouts of swim exercise of 3
h each and separated by 45 min of rest, induced a M1-to-M2 phenotype switch in
WAT and stromal vascular fraction (SVF) of rats on a high-fat diet, as evidenced
by the increased protein expression of macrophage galactose-type C-type lectin1
(MGL1), a M2 macrophage marker. By contrast, protein expression of TNF-¶Ń and
iNOS were downregulated in the SVF. Likewise, chronic treadmill running (up to
12 weeks) resulted in the attenuation of CD11c in the adipose tissue of mice on
high-fat diets, although surprisingly, the gene expression of two M2-macrophage
markers, Arg1 and CD206, was increased in sedentary mice on the high-fat diet,
and decreased in chronically trained mice, also on the high-fat diet (59) The
authors suggest that the improvements in inflammatory profiles in these mice may
involve an attenuation of both M1 and M2 macrophages in adipose tissue.
In humans, only a single study (60) to date had investigated outcomes involving
macrophage polarization after exercise training. In this randomized controlled
trial that spanned 12 weeks, overweight (BMI: 25®C30 kg/m2, body fat >25%), young
men were stratified into one of four experimental groups: (i) endurance training
group, (ii) dietary control group, (iii) endurance training and increased diet
without weight loss group, or (iv) control group. Protein expression of CD68, a
pan-macrophage marker, was not significantly different among the four
conditions; however, CD163, a M2 macrophage marker was increased in subcutaneous
adipose tissue of both exercise groups, but not in either the dietary control
group or the control group.
Given the paucity of studies that investigated exercise and macrophage phenotype
switching, it is clear that more research is needed to determine the proximal
signaling pathways that guide M1 vs. M2 macrophage modification in adipose
tissues by exercise. It is possible that exercise may indirectly regulate
macrophage phenotype through the enhancement of blood-derived or contracting
muscles-derived M2-type marker production. We and others have clearly shown that
exercise strongly induced the expression and release of IL-10, Arginase-1,
CD163, and IL-6 from human leukocytes and skeletal muscles (49®C51).
The mechanism by which exercise induces macrophages polarization toward an M2
phenotype is likely to be related to the induction of PPAR¶√ and its co-factors
(PGC-1¶Ń/¶¬). PPAR¶√ and PGC-1a are known for their important roles in the
regulation of efficient energy utilization and oxidative phosphorylation, both
of which are reduced in obesity and insulin resistance (61, 62). In particular,
PPAR¶√ plays an important role in controlling adipose tissue inflammation and
insulin resistance through the activation and infiltration of alternatively
activated (M2) macrophages (63, 64). A previous study by Odegaard and colleagues
(28) showed that PPAR¶√ deficiency in macrophages impairs M2 macrophage
activation and predisposes the animals to development of diet-induced obesity,
insulin resistance, and glucose intolerance. Further evidence for the role of
PPAR-¶√ in macrophage activation stems from studies by Stienstra et al. (64) who
showed a repolarization of adipose tissues macrophages to an M2 phenotype
following treatment of mice with PPAR¶√ agonist. The authors speculated that M2
macrophages might play a role in PPAR¶√-dependent expansion and remodeling of
adipose tissue.
The PPAR¶√ hypothesis becomes more convincing, given that participation in
exercise programs activates PPAR¶√ and PPAR¶√-mediated signaling events in adipose
tissue and monocytes/macrophages (35, 65®C68), and that, exercise-induced
activation of M2 macrophages is mediated via PPAR¶√ and its co-factors (PGC-1¶Ń/¶¬)
(69). The beneficial role of exercise-induced PPAR¶√ in serum lipid profiles
(increased HDL-cholesterol and decreased total cholesterol, LDL-cholesterol, and
triglycerides) has also been reported previously (67, 70). Thus,
exercise-triggered adipocyte- and/or monocyte/macrophage-specific PPAR¶√
activation may constitute an additional rationale for prescribing exercise in
obesity and type 2 diabetes. Our working hypothesis of how exercise modulates
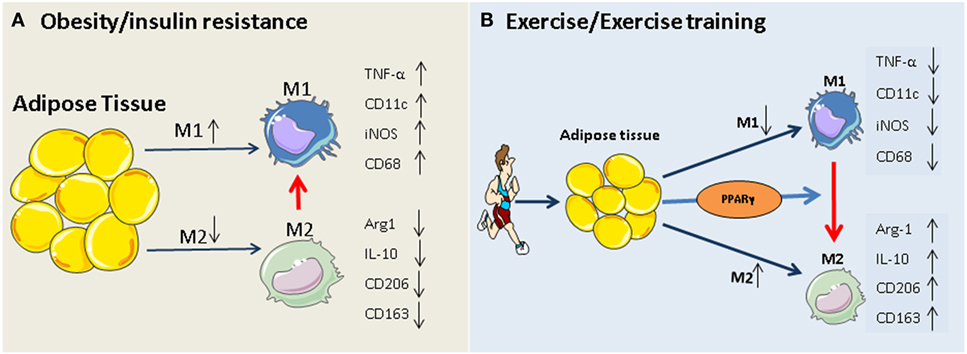
macrophage polarization is summarized in Figure 1.
Frontiers | Exercise and Adipose Tissue Macrophages: New Frontiers in Obesity
Research? | Endocrinology
https://www.frontiersin.org/articles/10.3389/fendo.2016.00065/full
°°
°°
°°