Metabolic reprogramming: the emerging concept and associated therapeutic strategies
Oncogene-Driven Metabolic Alterations in Cancer
¡¡
Cancer metabolic reprogramming: importance, main features, and potentials for
precise targeted anti-cancer therapies | Phan | Cancer Biology & Medicine
http://www.cancerbiomed.org/index.php/cocr/article/view/629/655
Abstract
Cancer is the leading cause of human deaths worldwide. Understanding the biology
underlying the evolution of cancer is important for reducing the economic and
social burden of cancer. In addition to genetic aberrations, recent studies
demonstrate metabolic rewiring, such as aerobic glycolysis, glutamine
dependency, accumulation of intermediates of glycolysis, and upregulation of
lipid and amino acid synthesis, in several types of cancer to support their high
demands on nutrients for building blocks and energy production. Moreover,
oncogenic mutations are known to be associated with metabolic reprogramming in
cancer, and these overall changes collectively influence tumor-microenvironment
interactions and cancer progression. Accordingly, several agents targeting
metabolic alterations in cancer have been extensively evaluated in preclinical
and clinical settings. Additionally, metabolic reprogramming is considered a
novel target to control cancers harboring un-targetable oncogenic alterations
such as KRAS. Focusing on lung cancer, here, we highlight recent findings
regarding metabolic rewiring in cancer, its association with oncogenic
alterations, and therapeutic strategies to control deregulated metabolism in
cancer.
Keywords: Cancer, Non-small cell lung cancer, Cancer metabolism, Metabolic
reprogramming, Aerobic glycolysis, Oncogenic alteration
¡¡
INTRODUCTION
Despite numerous efforts for cancer treatment, cancer is the leading cause of
human deaths worldwide (Mathers and Loncar, 2006; Torre et al., 2015). Thus,
understanding the biology underlying the evolution of cancer is important for
reducing the economic and social burden of cancer. Recent investigations have
demonstrated the impact of metabolic reprogramming on the development and
progression of several types of human cancer, and deregulated metabolism is now
regarded as one of the hallmarks of cancer (Hanahan and Weinberg, 2011; Pavlova
and Thompson, 2016). Moreover, several findings demonstrate that mutations in
oncogenes and/or tumor suppressor genes can mediate metabolic rewiring in cancer
cells to support the high demands for building blocks and energy production in
these cells (Iurlaro et al., 2014; Nagarajan et al., 2016; Kerr and Martins,
2017). Because cancer cells are prone to several oncogenic mutations such as
RAS, EGFR, MYC, and BRAF mutations, these genes could also influence the
metabolic changes in cancer. Based on several studies on the association of
oncogenic alterations with the metabolic reprogramming (Kroemer and Pouyssegur,
2008; Hanahan and Weinberg, 2011; Iurlaro et al., 2014; Nagarajan et al., 2016;
Kerr and Martins, 2017), here, we summarize recent findings on the association
of oncogenic alterations with metabolic reprogramming in cancer, focusing on
lung cancer due to its great contribution to cancer incidence and mortality
rates. Further, we discuss the impact of metabolic alterations on the
tumor-microenvironment interaction and possible therapeutic options targeting
metabolic reprogramming.
GENERAL FEATURES OF METABOLIC REPROGRAMMING IN CANCER
Cancer cells have been known to possess markedly different metabolic features
compared with those of corresponding normal tissues (Tennant et al., 2010).
Unlike normal cells, cancer cells rearrange their cellular metabolic networks to
fulfill their high demands for building blocks and energy production to support
extensive proliferation and growth (Tennant et al., 2010; Kerr and Martins,
2017) (Fig. 1). The first defined cancer-specific metabolic alteration is the
Warburg effect, an aerobic glycolytic process discovered by Otto Warburg in 1926
(Warburg, 1956). In this process, cancer cells are dependent on glycolysis for
glucose metabolism even in the presence of oxygen, thereby producing high levels
of lactate and reducing the use of the tricarboxylic acid (TCA) cycle (Levine
and Puzio-Kuter, 2010). Because the TCA cycle and subsequent oxidative
phosphorylation produce cellular energy more efficiently than glycolysis, this
metabolic rewiring has been suggested as an alternative to compensate for
mitochondrial dysfunction in cancer cells (Warburg, 1956; Kerr and Martins,
2017). Indeed, mutations in the TCA cycle-associated enzymes, such as succinate
dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase
(IDH), have been found in several types of cancer including paraganglioma
(mutations in SDH), phaeochromocytoma (mutations in SDH), renal carcinoma
(mutations in FH), leiomyomatosis (mutations in FH), acute myeloid leukemia
(mutations in IDH), and glioblastoma (mutations in IDH), and these alterations
have been suggested to contribute to mitochondrial dysfunction in cancer and
tumorigenesis (King et al., 2006; Dang et al., 2010; Galluzzi et al., 2013;
Parker and Metallo, 2015). However, several recent findings have suggested the
essential role of functional mitochondria in cancer cells (Magda et al., 2008;
Whitaker-Menezes et al., 2011; Wallace, 2012). The upregulation of oxidative
phosphorylation has been noted in cancer cells (Whitaker-Menezes et al., 2011),
and the tumorigenic potential of cancer cells has also been shown to be
significantly reduced by depletion of mitochondrial DNA (Magda et al., 2008).
Therefore, in addition to ATP synthesis, metabolic switching to aerobic
glycolysis appears to be a means of supplying cancer cells with the precursors
of proteins, lipids, amino acids, and nucleic acids for building their cellular
structure and maintaining their upregulated proliferation. Thus, mitochondria
still play important roles in bioenergetics and biosynthesis in cancer cells
(Wallace, 2012).
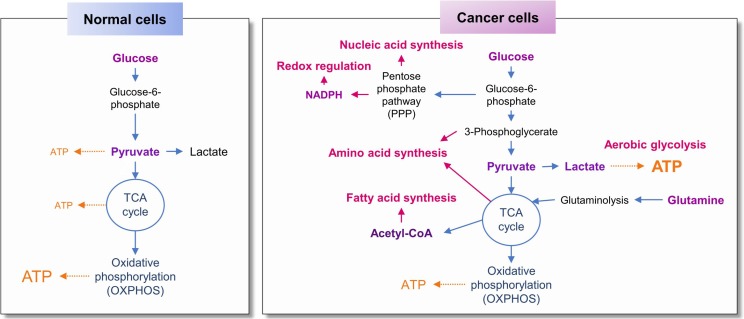
Fig. 1.
Metabolic reprogramming in cancer cells compared with normal cells.
Recent findings demonstrate the additional metabolic rewiring in cancer cells
and consequent alterations in cellular signaling pathways and the tumor
microenvironment, including changes in the metabolism of glucose, lipids, and
amino acids; regulation of the cellular redox state to tolerate reactive oxygen
species (ROS)-mediated damage in cellular compartments; and remodeling of the
extracellular matrix surrounding cancer cells. For instance, cancer cells
display elevated expression of the alternatively spliced form of pyruvate kinase
(PK), PK muscle isozyme M2 (PKM2) (Kroemer and Pouyssegur, 2008; Dong et al.,
2016). PK mediates the conversion of phosphoenolpyruvate (PEP) to pyruvate, the
rate-limiting step of glycolysis (Dong et al., 2016). Owing to the reduced
enzymatic activity of PKM2, the phosphorylated metabolites upstream of pyruvate
in the glycolytic pathway accumulate and are finally diverted into several
anabolic pathways to synthesize glycogen, triglycerides, phospholipids,
nucleotides, and amino acids (Gatenby and Gillies, 2004; Kroemer and Pouyssegur,
2008; Dong et al., 2016). In addition, cancer cells introduce acetyl-CoA into a
truncated TCA cycle, resulting in the export of acetyl-CoA into the cytosol,
where it serves as a precursor of fatty acids, cholesterol, and isoprenoids,
which are utilized for cell proliferation and growth (Kroemer and Pouyssegur,
2008). Fatty acid synthase and choline kinase, which mediate biosynthesis of
long-chain fatty acids and phosphatidylcholines, respectively, are also known to
be upregulated and activated in many types of cancer cells (Ramirez de Molina et
al., 2002; Menendez and Lupu, 2007; Kroemer and Pouyssegur, 2008). In the case
of amino acid metabolism, cancer cells express sensors of amino acid deficiency,
such as GATOR1, folliculin, and the Ras-like small GTPase Rag complex, to ensure
a sufficient supply of amino acids to activate rapamycin complex I (mTORC1)
(Bar-Peled and Sabatini, 2014; Tsun and Possemato, 2015). The upregulated uptake
of glutamine, a nonessential amino acid, through elevated expression of
glutamine transporters such as SLC1A5 and SLC38A2 has been thought to play
important roles in the supply of nitrogen, the uptake of essential amino acids,
and the maintenance of mTORC1 activation in cancer cells (Wise and Thompson,
2010). Consistent with these hypotheses, elevated expression of these glutamine
transporters is correlated with poor clinical outcomes in breast and lung
cancers (Hassanein et al., 2015; Jeon et al., 2015). Cancer cells also display
extensive conversion of glutamine to glutamate and upregulation of several
metabolic enzymes responsible for amino acid biosynthesis, including glutaminase
(GLS), phosphoglycerate dehydrogenase (PHG-DH), and asparagine synthetase (ASNS)
(Gao et al., 2009; Locasale et al., 2011; Possemato et al., 2011; Zhang et al.,
2014a; Tsun and Possemato, 2015). Moreover, the generation of nicotinamide
adenine dinucleotide phosphate (NADPH) by metabolizing glucose through the
pentose phosphate pathway (PPP) supports the defense of cancer cells against
oxidative or cellular stresses and the synthesis of fatty acids in cancer cells
(Gatenby and Gillies, 2004; Kroemer and Pouyssegur, 2008; Levine and
Puzio-Kuter, 2010). Further, the acidic tumor microenvironment is constructed
through the overproduction of lactate through aerobic glycolysis, facilitating
the invasion of tumor cells and blood vessels via matrix remodeling and
suppressing anticancer immunity (Fischer et al., 2007; Hunt et al., 2007;
Swietach et al., 2007; Kroemer and Pouyssegur, 2008; Levine and Puzio-Kuter,
2010). Collectively, these complex processes allow cancer cells to survive and
proliferate, but the details are known to be context dependent and
differentially regulated by various factors such as oncogenes/tumor suppressor
genes, microenvironments, and tissue of origin (Levine and Puzio-Kuter, 2010;
Yuneva et al., 2012; Hensley et al., 2016; Mayers et al., 2016; Kerr and
Martins, 2017). Thus, understanding the influence of cellular or environmental
factors, such as oncogene-induced metabolic switches, on cancer cell metabolism
is important for the development of better anticancer therapeutics targeting
altered metabolism in cancer cells.
¡¡
METABOLIC ALTERATIONS IN NON-SMALL CELL LUNG CANCER
Lung cancer is one of the main types of cancer due to its high prevalence and
poor survival rates (Mathers and Loncar, 2006; Torre et al., 2015).
Approximately 85% of all cases of lung cancer are non-small cell lung cancer
(NSCLC) (Molina et al., 2008). The three major types of NSCLC (adenocarcinoma
(ADC), squamous cell carcinoma (SQCC), and large cell carcinoma) are classified
based on histological and molecular/genetic features (Clinical Lung Cancer
Genome Project (CLCGP) and Network Genomic Medicine (NGM), 2013; Pikor et al.,
2013). Mutations in KRAS and EGFR as well as ALK rearrangements, among others,
are mainly found in lung ADC, which accounts for 30¨C40% of NSCLCs (Pikor et al.,
2013). Lung ADCs carrying these genetic alterations are addicted to the
associated signaling pathways for cell proliferation, growth, and survival and
thus can be vulnerable to the disruption of these signaling pathways
(Hrustanovic et al., 2015; Lin and Shaw, 2016). Indeed, several anticancer drugs
specifically targeting EGFR or ALK have been clinically used as a first-line
therapy for patients with lung ADC harboring these mutations (Saintigny and
Burger, 2012). However, none of these drugs have shown remarkable clinical
benefits, and drug resistance is still a large obstacle for efficient anticancer
treatment using these regimens (Lin and Shaw, 2016). Moreover, there is no
therapeutic option to control lung ADC carrying mutant KRAS. Although several
alternative approaches have been suggested, including targeting the functional
outputs of mutant KRAS or cellular addiction caused by mutant KRAS (Kerr and
Martins, 2017), it is important to develop novel therapeutic strategies to meet
clinical needs for the treatment of lung cancer, especially lung ADC carrying
mutations in oncogenes such as KRAS.
In line with the general metabolic reprogramming in cancer cells that has been
described previously, recent studies have demonstrated metabolic alterations in
NSCLC. Studies using NSCLC tumors surgically resected from patients after
radioisotope-labeled glucose (13C-glucose) infusion, NSCLC cells displayed
enhancements in glycolysis and the TCA cycle and subsequent enrichment of TCA
cycle intermediates compared with adjacent normal or benign lung tissues (Fan et
al., 2009; Hensley et al., 2016). In addition, the activity of pyruvate
carboxylase (PC), an enzyme mediating the irreversible carboxylation of pyruvate
to generate oxaloacetate (Gray et al., 2014), was elevated in NSCLC tumors
(Sellers et al., 2015; Hensley et al., 2016). Because upregulated PC activity
plays a role in the replenishment of TCA intermediates that have been utilized
in biosynthetic reactions (Kerr and Martins, 2017), this enhancement indicates
the rewiring of glucose metabolism to meet the high metabolic demands of cancer
cells. Moreover, silencing PC expression significantly reduced the
proliferative, colony-forming, and tumorigenic abilities of NSCLC cells,
suggesting that NSCLC cells are addicted to PC-mediated anaplerosis (the
reduction of TCA intermediates due to biosynthetic reactions). Thus, PC has the
potential to be a novel cellular target for anticancer drug development (Sellers
et al., 2015). A recent study shows that a subset of NSCLC cells utilizes
glycolysis for energy production and that these high glycolytic cells possess
elevated hexokinase 2 expression (Wu et al., 2015). Another recent study also
demonstrates the utilization of lactate as the main carbon source for the TCA
cycle in tumors from NSCLC patients and NSCLC tumor xenografts (Faubert et al.,
2017).
In addition to these changes in glucose metabolism, NSCLC cells exhibit
alterations in the metabolism of lipids, amino acids, and nucleic acids. For
example, the expression of acetyl-CoA carboxylase 1 (ACC1), one of the key
regulators of fatty acid synthesis, was elevated in NSCLC cells. Further,
pharmacological inhibition of ACC1 displayed significant antitumor effects in a
preclinical model of NSCLC (Svensson et al., 2016; Svensson and Shaw, 2016). The
expression and activity of ATP citrate lyase (ACLY), another key fatty acid
synthesis enzyme involved in the generation of cytosolic acetyl-CoA and
oxaloacetate, were also upregulated in NSCLC (Migita et al., 2008) and are
associated with poor clinical outcomes in NSCLC patients (Migita et al., 2008).
Consistent with the results of experiments targeting ACC1, siRNA-based ablation
of ACLY expression exhibited significant inhibitory effects on proliferation and
lipogenesis (Migita et al., 2008). Glycine decarboxylase (GLDC), a component of
a multienzyme complex responsible for glycine decarboxylation and serine
biosynthesis (Go et al., 2014) and involved in pyrimidine metabolism (Newman and
Maddocks, 2017), was also upregulated in lung tumor-initiating cells and
promoted cell transformation and tumorigenesis (Zhang et al., 2012). Elevated
GLDC expression was associated with poor survival in patients with NSCLC (Zhang
et al., 2012).
However, compared to altered glucose metabolism in NSCLC, the rewiring of other
metabolic pathways in NSCLC is still unclear and needs to be further elucidated.
Additionally, despite commonalities in metabolic reprogramming, the metabolic
alterations in individual NSCLC cells or tumors are highly heterogeneous
(Brunelli et al., 2014; Chen et al., 2014; Wu et al., 2015; Hensley et al.,
2016). Considering a high mutation burden in lung cancer, especially lung ADC
(Cancer Genome Atlas Research Network, 2014; Swanton and Govindan, 2016; Kerr
and Martins, 2017), and the association of alterations in oncogenes or tumor
suppressor genes with metabolic reprogramming (Levine and Puzio-Kuter, 2010;
Iurlaro et al., 2014; Nagarajan et al., 2016; Kerr and Martins, 2017), the
genetic heterogeneity of NSCLC appears to influence these metabolic diversities.
ROLE OF ONCOGENIC MUTATIONS IN METABOLIC REPROGRAMMING IN LUNG CANCER
Alterations in several oncogenes, such as MYC, RAS, and BRAF, have been known to
play a role in metabolic reprogramming (Iurlaro et al., 2014; Nagarajan et al.,
2016; Kerr and Martins, 2017). Briefly, MYC transcriptionally regulates some
metabolic enzymes involved in DNA synthesis and glycolysis, including
thymidylate kinase and lactate dehydrogenase A, respectively (Pusch et al.,
1997; Shim et al., 1997). MYC is also involved in the metabolic reprogramming of
fatty acids, glutamine, proline, and nucleic acids by direct transcriptional
regulation or indirect regulation utilizing microRNAs (Mannava et al., 2008; Gao
et al., 2009; Liu et al., 2012; Edmunds et al., 2014). In addition, increases in
the uptake and interconversion of a polyamine spermine, the metabolism of
inositol phospholipids, and aerobic glycolysis were observed in RAS-transformed
cells (Huang et al., 1988; Pakala et al., 1988; Chiaradonna et al., 2006).
Further, mutated RAS was found to mediate metabolic reprogramming in pancreatic
cancer by stimulating glucose uptake, channeling glycolytic intermediates into
the hexosamine biosynthesis pathway or pentose phosphate pathway, and directly
regulating aspartate transaminases (Ying et al., 2012; Son et al., 2013;
Nagarajan et al., 2016). BRAF is also known to regulate glucose and glutamine
metabolism in melanoma (Scott et al., 2011; Haq et al., 2013).
In the case of lung cancer, previous reports have suggested a link between
genetic mutations and metabolic rewiring in NSCLC, especially lung ADC. The
association of alterations in KRAS, EGFR, ALK, and STK11 genetic abnormalities
in lung ADC (Ji et al., 2007; Pikor et al., 2013) with metabolic changes is
described as follows (Fig. 2).
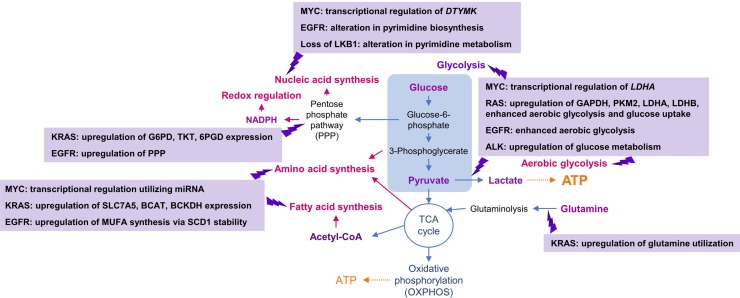
Fig. 2.
Contribution of genetic alterations to metabolic reprogramming in cancer.
Role of KRAS mutation in metabolic reprogramming in NSCLC
Mutations in the RAS oncogene are known to be a major driver of tumorigenesis
(Cox and Der, 2010; Pylayeva-Gupta et al., 2011; Hobbs et al., 2016). Three
isoforms of the RAS gene [Kirsten rat sarcoma viral oncogene homolog (KRAS),
neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) and Harvey rat sarcoma
viral oncogene homolog (HRAS)] encode four RAS proteins (KRAS4A, KRAS4B, NRAS,
and HRAS) (Pylayeva-Gupta et al., 2011; Hobbs et al., 2016). The two KRAS
isoforms arise from alternative RNA splicing of the KRAS gene (Pylayeva-Gupta et
al., 2011; Hobbs et al., 2016). Activating mutations have been identified at
three hotspots within the RAS protein (G12, G13, and Q61), but the mutation
frequency at each of the hotspots in the RAS isoform is known to be quite
different in each isoform (Pylayeva-Gupta et al., 2011; Hobbs et al., 2016). The
RAS protein is a small G protein whose activity is regulated by the GDP/GTP
cycle (Cox and Der, 2010; Pylayeva-Gupta et al., 2011; Hobbs et al., 2016). The
GTP-bound RAS, an activated form of the RAS protein, binds to downstream
effectors and triggers activation of signal transduction pathways, such as the
Raf-MEK-ERK pathway and the PI3K/Akt pathway, responsible for cell
proliferation, survival, and growth (Cox and Der, 2010; Pylayeva-Gupta et al.,
2011).
Mutations in the KRAS gene, including G12C, G12V, G12D, and G12A, are found in
approximately 30% of NSCLC patients with ADC histology (Kempf et al., 2016).
These mutations are found more frequently in smokers than in nonsmokers (25¨C35%
in smokers and 5% in nonsmokers) (Mao et al., 2010; Dearden et al., 2013; Kempf
et al., 2016). The KRAS mutation (G12D) is common in never-smokers, whereas the
KRAS mutation (G12C) is the most common mutation in NSCLC patients with a
history of smoking (Kempf et al., 2016). Mutations in KRAS and EGFR are mutually
exclusive (Kempf et al., 2016), but mutations in STK11 or TP53 are positively
correlated with KRAS mutations (Kempf et al., 2016). Although a recent report
describes the weak prognostic impact of the KRAS mutations in NSCLC (Roberts and
Stinchcombe, 2013), recent findings suggest a close association of the KRAS
mutations with poor prognosis of patients with NSCLC (Meng et al., 2013; Renaud
et al., 2015; Kempf et al., 2016). Accordingly, several anticancer approaches
targeting the RAS protein, including farnesyltransferase inhibitors, competitors
disrupting the RAS-chaperone interaction, and inhibitors of the RAS effector or
downstream signaling such as the MAPK pathway, mTOR, and Hsp90, have been
evaluated in preclinical and clinical settings. None, however, has shown
clinical benefits for anticancer treatment (Cox and Der, 2010; Kempf et al.,
2016), emphasizing the necessity of procuring alternative approaches to treat
cancer carrying RAS mutations.
Numerous findings demonstrate the involvement of mutant KRAS in the metabolic
rewiring of several types of human cancer (Pylayeva-Gupta et al., 2011;
Kimmelman, 2015; Lv et al., 2016; Kawada et al., 2017; Kerr and Martins, 2017),
including upregulation of glucose uptake, glutamine utilization, and aerobic
glycolysis (Onetti et al., 1997; Ying et al., 2012; Son et al., 2013). Using
patient-derived NSCLC tumors, cell lines, and animal models, several studies
have consistently identified the influence of mutant KRAS on metabolic
reprogramming in NSCLC. A recent study demonstrated the metabolism-related
proteomic profiles of NSCLC cell lines carrying intrinsic mutant KRAS (A549 and
H460) in comparison with those of normal bronchial epithelial cells
(Martin-Bernabe et al., 2014). These NSCLC cells expressed elevated levels of
enzymes involved in glycolysis (GAPDH, PKM2, LDHA, and LDHB) and PPP (G6PD, TKT,
and 6PGD) compared with normal cells, suggesting alterations in glucose
metabolism in NSCLC cells carrying mutant KRAS. It is known that these two cell
lines carry different KRAS mutations (G12S for A549; Q61H for H460) (Mahoney et
al., 2009; Acquaviva et al., 2012) and that the different amino acid
substitutions display distinct biological properties in terms of signaling
activation and sensitivity to anticancer agents (Garassino et al., 2011; Stolze
et al., 2015). Thus, cellular metabolism could be influenced by different KRAS
mutations. In line with this notion, a recent study demonstrated the impact of
different KRAS mutations on changes in metabolomic profiles (Brunelli et al.,
2014). In this study, different KRAS mutations at codon 12 (G12C, G12D, and
G12V) were evaluated. NSCLC cells carrying each of these mutations displayed
differential metabolic remodeling, including differences in redox buffering
systems and glutamine dependency (Brunelli et al., 2014). Among these mutations,
mutant KRAS (G12C) showed the most prominent metabolic changes in vitro. Of
note, these metabolic changes were maintained in a tumor xenograft model bearing
the same NSCLC cell line (Brunelli et al., 2014, 2016), suggesting that the in
vitro cell line model can be utilized to investigate metabolic alterations in
NSCLC patients. However, another independent study demonstrated discrepancies in
glucose metabolism using in vitro versus in vivo models (Davidson et al., 2016).
In this study, several mouse models, including two autochthonous mouse models
that develop spontaneous lung tumors (the KrasLA2/+ mouse model and the
KrasLSL-G12D/+;Trp53fl/fl (KP) mouse model with intratracheal delivery of
adenoviral Cre), a syngeneic xenograft model involving intratracheal inoculation
with lung tumor cells derived from the KP mouse model, and a tumor xenograft
model involving subcutaneous inoculation with human lung cancer cell lines, were
used for determining metabolic changes in vivo. Tumor cells arising in the KP
mouse model were used for in vitro determination of metabolic alterations
(Davidson et al., 2016). Both in vitro and in vivo models exhibited upregulated
lactate production. However, in contrast to a dependence on glutamine for TCA
cycle entry in vitro, lung tumors from these in vivo mouse models minimally
utilized glutamine as a carbon source for TCA cycle entry. Additionally, some
oxidative glucose metabolic enzymes, including pyruvate carboxylase and pyruvate
dehydrogenase (which generate oxaloacetate and acetyl-CoA, respectively), were
necessary for tumor formation and growth in these mouse models (Davidson et al.,
2016). Therefore, the environmental context needs to be taken into consideration
in the investigation of physiologically relevant metabolic alterations,
especially in the case of glucose metabolism.
¡¡
Additional studies also suggest that mutant KRAS mediates the changes in the
metabolism of amino acids, lipids, and folates. In a recent study using a mutant
Kras-driven model of spontaneous lung tumorigenesis (the KP mouse model), the
uptake and utilization of branched-chain amino acids (BCAAs), such as leucine
and valine, were elevated in KP mice possessing lung tumors (Mayers et al.,
2016). The expression of enzymes responsible for the catabolism of BCAAs,
including SLC7A5, BCAT, and BCKDH, was also upregulated in human NSCLC tumors,
and ablation of Bcat expression resulted in decreases in in vitro NSCLC cell
proliferation and in vivo NSCLC tumor growth (Mayers et al., 2016), indicating
the requirement of BCAA metabolism in NSCLC. In the same study, pancreatic
ductal adenocarcinoma (PDAC) carrying the same genetic alterations did not
utilize BCAA as a nitrogen source (Mayers et al., 2016), suggesting the
influence of tissue microenvironment-specific differences on metabolic
reprogramming over genetic mutations. In addition to amino acid metabolism,
mutant KRAS activated lipogenesis in lung ADC via induction of fatty acid
synthase through the ERK2-mediated pathway (Gouw et al., 2017). NSCLC cells
carrying mutant KRAS also showed a tendency to be dependent on the folate
metabolism pathway compared with those carrying wild-type KRAS (Moran et al.,
2014). Consistent with these findings, KRAS mutant NSCLC cells were sensitive to
antifolates such as methotrexate and pemetrexed, and the expression level of
enzymes related to folate metabolism, such as methylenetetrahydrofolate
dehydrogenase 2 (MTHFD2) was positively (Moran et al., 2014).
Moreover, despite the metabolic switch to aerobic glycolysis in cancer cells,
mitochondria are known to have a functional role in cell proliferation and
tumorigenesis (Magda et al., 2008; Whitaker-Menezes et al., 2011; Wallace,
2012). Likewise, deregulation of mitochondrial function through the ablation of
the expression of mitochondrial transcription factor A (TFAM) significantly
suppressed mutant Kras-driven lung tumor formation (Weinberg et al., 2010). In
this study, mitochondrial ROS generated through Complex III was essential for
mutant KRAS-induced anchorage-independent growth of cancer cells (Weinberg et
al., 2010). A previous report demonstrated the reduced expression of components
of Complex I in KRAS-transformed cells (Baracca et al., 2010). Considering that
both Complex I and Complex II mediate electron transfer to Complex III
(Mailloux, 2015), presumably, NSCLC cells expressing mutant KRAS might acquire
an alternative method (e.g., upregulation of Complex II) of compensating for the
KRAS-induced decrease in Complex I activity in order to maintain mitochondrial
function.
Role of EGFR mutations in metabolic reprogramming in NSCLC
Approximately 15¨C30% of NSCLC patients carry abnormalities in EGFR (Gridelli et
al., 2015). EGFR mutations are frequently observed in lung ADCs derived from
Asian patients with no smoking history (Gridelli et al., 2015). The most common
mutations in EGFR are a deletion at exon 19 (E746¨CA750) and substitutions at
exon 18 (G719C, G719S, G719A) and exon 21 (L858R), all of which are sensitive to
EGFR-targeted therapy (Pao and Miller, 2005; Gridelli et al., 2015). Aberrantly
activated EGFR activates signaling pathways driving the mitogenic, prosurvival,
and proinvasive phenotypes of the cancer cells (Zhang et al., 2010). In addition
to the direct modulation of signal transduction, aberrant EGFR mediates
metabolic reprogramming in NSCLC. For instance, global metabolic reprogramming,
such as enhanced aerobic glycolysis and upregulation of PPP, alters pyrimidine
biosynthesis and redox metabolism in EGFR mutant lung ADC cell lines
(Makinoshima et al., 2014). Combination treatment with erlotinib and a
glutaminase inhibitor (CB-839) drives EGFR mutant NSCLC cells to undergo
metabolic crisis, thereby leading to enhanced cell death, decreased cell
viability in vitro, and a rapid tumor regression in vivo (Momcilovic et al.,
2017), indicating the necessity of glutamine as a source for bioenergetics and
biosynthesis in NSCLCs carrying mutant EGFR. Moreover, EGFR increases
monounsaturated fatty acid (MUFA) synthesis by phosphorylating stearoyl-CoA
desaturase-1 (SCD1) via direct interaction and via maintaining the stability of
the SCD1 protein (Zhang et al., 2017). The level of phosphorylated SCD1
expression was found to be an independent prognostic factor for poor survival in
patients with NSCLC (Zhang et al., 2017). These results collectively indicate
that targeting alterations in glucose or lipid metabolism would be an
alternative combinatorial therapeutic approach for treatment of lung ADCs
harboring mutant EGFR.
Role of ALK rearrangement in metabolic reprogramming in NSCLC
ALK rearrangement accounts for approximately 3¨C7% of NSCLC cases (Katayama et
al., 2015). The most frequently observed ALK rearrangement is the EML4-ALK
fusion (Katayama et al., 2015). Several ALK inhibitors, including crizotinib and
ceritinib, have been clinically used for the treatment of patients with lung ADC
harboring alterations in ALK (Katayama et al., 2015). The impact of ALK
aberrations on metabolism in lung ADC has not been well characterized, but a
recent report indicates presence of upregulated glucose metabolism and highly
metastatic phenotypes in lung ADCs carrying ALK rearrangements (Choi et al.,
2013).
Role of LKB1 loss in metabolic reprogramming in NSCLC
LKB1, encoded by the STK11 gene, is a tumor suppressor gene which plays an
important role in the regulation of cellular growth and metabolism by
phosphorylation and activation of AMP-activated kinase (AMPK), an upstream
kinase controlling the mammalian target of rapamycin (mTOR) pathway, MARK/par-1,
and other AMPK-related kinases (Shackelford and Shaw, 2009). Approximately
15¨C35% of NSCLC patients harbor mutations in STK11 (Ji et al., 2007; Shackelford
and Shaw, 2009), which is more frequently observed in lung ADC than in lung SQCC
(Sanchez-Cespedes et al., 2002; Ji et al., 2007). According to its primary role
in the regulation of cellular metabolism, loss of LKB1 leads to deregulation of
cellular metabolism under conditions of energy stress (Carretero et al., 2007),
causing enhanced sensitivity to therapies targeting metabolism such as
phenformin (Shackelford et al., 2013) or therapies that induce energetic stress
such as erlotinib (Whang et al., 2016). In addition, metabolic reprogramming in
NSCLC harboring altered LKB1 has been demonstrated in a recently published
study. Using NSCLC cell lines carrying either KRAS mutations alone or both KRAS
mutations and loss of LKB1, this study identified that the additional loss of
LKB1 resulted in the accumulation of metabolites associated with the urea cycle
through upregulation of carbamoyl phosphate synthetase-1 (CPS1) (Kim et al.,
2017). Silencing of CPS1 expression suppressed the growth of tumor xenografts
derived from KRAS/STK11-mutant NSCLC cells through reduction of the pyrimidine
to purine ratio, thereby disrupting DNA replication (Kim et al., 2017). These
results indicate the existence of alterations in pyrimidine metabolism in
LKB1-deficient NSCLC cells and provides a novel therapeutic target for the
treatment of NSCLCs harboring loss of LKB1 expression.
TUMOR MICROENVIRONMENT-MEDIATED METABOLIC REPROGRAMMING IN CANCER
The interaction between tumors and the surrounding stromal cells that make up
the tumor microenvironment has been known to be implicated in cancer development
and progression (Quail and Joyce, 2013). Given the role of metabolic alterations
in cancer, the tumor-microenvironment interaction could be affected by metabolic
alterations in cancer cells and vice versa. For example, the differences in BCAA
metabolism between lung cancer and PDAC (Mayers et al., 2016) and in glutamine
dependent metabolism between in vitro and in vivo models (Davidson et al., 2016)
appear to be influenced by the environmental context. Nutrient sharing, nutrient
competition, and metabolite exchange between tumor and stromal cells are known
to influence and shape the tumor-microenvironment interaction (Lyssiotis and
Kimmelman, 2017). Indeed, lactate, amino acids, and fatty acids act as signaling
molecules that can be exchanged between tumor and stromal cells, resulting in
the regulation of signal transduction, gene expression, and characteristics of
neighboring cells (Lyssiotis and Kimmelman, 2017). Macromolecules or organelles
released from stromal cells can also support the biosynthetic and bioenergetic
needs of cancer cells (Spees et al., 2006; Chaudhri et al., 2013; Lyssiotis and
Kimmelman, 2017). Specifically, compared with normal fibroblasts, basal
autophagy was elevated in lung cancer-associated fibroblasts (CAFs) through the
influence of high glycolytic lung cancer cells, leading to the release of
dipeptides that could support surrounding cancer cells (Chaudhri et al., 2013).
Additionally, interactions with bone marrow-derived nonhematopoietic
stem/progenitor cells or skin fibroblasts rescued lung cancer cells with
mitochondrial defects and led to reactivation of their mitochondrial function
including electron transport chain activity (Spees et al., 2006). These
phenomena occurred through the transfer of mitochondria or mitochondrial DNA
from stem/progenitor cells or fibroblasts to lung cancer cells (Spees et al.,
2006). Collectively, these findings suggest a crucial association between
metabolic reprogramming and the tumor-microenvironment interaction. However,
details regarding mechanisms of action, the lung microenvironment-specific
consequences of these interactions, and their clinical impacts need to be
explored in further studies.
TARGETING METABOLIC REPROGRAMMING FOR THE TREATMENT OF CANCER
According to the importance of metabolic alterations in the development and
progression of cancer, several agents targeting cancer metabolism have been
developed and evaluated under preclinical and clinical studies (Kroemer and
Pouyssegur, 2008; Tennant et al., 2010; Nagarajan et al., 2016). Some
metabolism-targeting agents, such as mTOR inhibitors [rapamycin (sirolimus),
everolimus, and temsirolius] and metformin (AMPK activator and mitochondrial
Complex I inhibitor) are now approved for clinical use (Carracedo et al., 2013;
Nagarajan et al., 2016) (Table 1). Strategies targeting metabolic alterations
for anticancer therapy are detailed in the following sections (Nagarajan et al.,
2016).
Table 1.
Compounds targeting cancer metabolism in clinical studies
Name Target Clinical development stage Cancer types targeted
Agents targeting deregulated signaling pathways
Rapamycin (Sirolimus) mTOR Phase I/II Glioblastoma, Advanced cancer
Everolimus (RAD001) mTOR FDA approved Advanced renal cell carcinoma,
Pancreatic neuroendocrine tumors, Subependymal giant cell astrocytoma
Temsirolimus (CCI-779) mTOR FDA approved Advanced renal cell carcinoma
Ridaforolimus mTOR Phase I/II/III Advanced solid tumors
AZD8055 (MK-8669) mTOR Phase I Advanced solid tumors
Metformin AMPK Phase I/II/III Various advanced solid tumors
Agents targeting metabolic enzymes
2-Deoxygluose (2-DG) HK Phase I/II Various advanced solid tumors
TCD-717 CK Phase I Advanced solid tumors
Dichloroacetate PDK1 Phase I/II Advanced solid tumors, Head and neck
carcinoma, Brain tumor
Indoximod IDO Phase I/II Adult solid tumors, Advanced solid tumors, Acute
myeloid leukemia
Ivosidenib (AG-120) IDH1 Phase I/II Acute myeloid leukemia, Glioma, Advanced
cholangiocarcinoma, Advanced solid tumors
Enasidenib mesylate (AG-221) IDH2 Phase I/II Acute myeloid leukemia, Glioma,
Advanced solid tumors
AG-881 IDH1 or IDH2 Phase I Acute myeloid leukemia, Glioma
IDH1 peptide vaccine IDH1 Phase I Glioma
PEPIDH1M IDH1 Phase I Glioma
Agents depleting metabolites using recombinant enzymes (PEG-conjugated)
Arginase 1 Arginine Phase I/II Acute myeloid leukemia, Hepatocellular
carcinoma, Other solid tumors
Arginine deiminase Arginine Phase I/II/III Advanced solid tumors,
mesothelioma, small cell lung cancer, skin cancer
Asparaginase Asparagine Phase I/II/III Various types of leukemia and lymphoma
mTOR: mammalian target of rapamycin, AMPK: AMP activated protein kinase, HK:
hexokinase, CK: choline kinase, PDK1: pyruvate dehydrogenase kinase 1, IDO:
indoleamine 2,3-dioxygenase.
Targeting deregulated signaling pathways
Recent studies demonstrate the effectiveness of targeting the signaling pathways
downstream of oncogenes such as AMPK and mTOR, alone or in combination, in
several types of cancer. For example, metformin, an AMPK activator, inhibited
the biosynthesis of fatty acids and nucleic acids (Li et al., 2015), suppressed
the proliferation of lung cancer and the self-renewal capacity of hepatocellular
carcinoma stem cells by inducing apoptosis (Saito et al., 2013; Storozhuk et
al., 2013), and increased the radiosensitivity of lung and breast cancer cells
(Storozhuk et al., 2013; Zhang et al., 2014b). The mTOR inhibitor rapamycin also
inhibited the cell proliferation in several types of cancer including colorectal
cancer, glioma, pancreatic cancer, and recurrent glioblastoma (Houchens et al.,
1983; Eng et al., 1984; Grewe et al., 1999; Cloughesy et al., 2008). In a phase
I clinical trial, rapamycin showed anticancer activity in PTEN-deficient
glioblastoma (Cloughesy et al., 2008). Rapamycin analogs with improved water
solubility, such as everolimus and temsirolimus, also exhibited potent
anticancer effects on several types of cancer alone or in combination with other
anticancer agents (Vignot et al., 2005) and have been clinically used for the
treatment of advanced renal cell carcinoma, pancreatic neuroendocrine tumors,
and subependymal giant cell astrocytoma (Benjamin et al., 2011).
Targeting metabolic enzymes
2-Deoxyglucose (2-DG) has a similar structure to glucose and is unable to be
metabolized in mammals (Nagarajan et al., 2016). Thus, 2-DG can inhibit multiple
glycolytic steps by competitively acting with glucose (Nagarajan et al., 2016).
2-DG is phosphorylated by HK2 and phosphorylated 2-DG acts an inhibitor of HK2
(Wick et al., 1957). In addition, various inhibitors targeting metabolic
enzymes, including lonidamine and 3-bromopyruvate (hexokinase inhibitors),
TLN-232 (a pyruvate kinase inhibitor), orlistat and cerulenin (fatty acid
synthase inhibitors), dichloroacetate (a PDK1 inhibitor), MN58b and TCD-717
(choline kinase inhibitors), soraphen A (an acetyl-CoA carboxylase inhibitor),
indoximod [an indoleamine 2,3-dioxygenase (IDO) inhibitor], ivosidenib (AG-120),
enasidenib mesylate (AG-221 mesylate), AG-881, IDH305, PEPIDH1M
(IDH1R132H-specific peptide vaccine) (inhibitors targeting mutated IDH1 or
IDH2), and SB-2049990 (an ATP citrate lyase inhibitor), have been evaluated in
preclinical and clinical studies (Table 1) (Hatzivassiliou et al., 2005; Wang et
al., 2005; Al-Saffar et al., 2006; Beckers et al., 2007; Kroemer and Pouyssegur,
2008; Tennant et al., 2010; Mondesir et al., 2016; Nagarajan et al., 2016).
Depleting metabolites using recombinant enzymes
Strategies to inhibit a specific metabolic pathway using recombinant enzymes to
reduce a specific oncogenic metabolite have been developed recently (Nagarajan
et al., 2016). For instance, recombinant arginine deiminase and arginase I
(which degrade and deplete arginine) conjugated with polyethylene glycol (PEG)
(pegylated arginine deiminase and pegylated arginase 1, respectively) have been
evaluated in phase I/II clinical trials for the treatment of advanced melanoma
and advanced hepatocellular carcinoma (Izzo et al., 2004; Glazer et al., 2010;
Yang et al., 2010; Ott et al., 2013; Yau et al., 2013; Nagarajan et al., 2016).
Recombinant l-asparaginase (which degrades and depletes asparagine) conjugated
with PEG (PEG-asparaginase) is also in clinical trials for the treatment of
pediatric and adult acute lymphoblastic leukemia, multiple myeloma, and advanced
solid tumors (Taylor et al., 2001; Agrawal et al., 2003; Fu and Sakamoto, 2007;
Kurtzberg et al., 2011).
Specifically, in lung cancer, despite the various anticancer approaches
targeting cancer metabolism described above, no metabolism-targeted drugs have
been approved for lung cancer treatment. Currently, most metabolism-targeting
agents for lung cancer are still under preclinical evaluation (Nagarajan et al.,
2016). Of note, agents targeting unique oncogene-driven metabolic rewiring have
been relatively poorly developed and should be investigated in further studies.
For lung cancer treatment, cellular markers specifically elevated in NSCLC cells
harboring oncogenic alterations, including BCAT (Mayers et al., 2016), SCD1
(Zhang et al., 2017), and CPS1 (Kim et al., 2017), could be potential candidates
for developing novel anticancer agents specifically disrupting oncogene-driven
metabolic reprogramming in NSCLC. In addition, metabolic synthetic lethality can
be a valuable therapeutic approach considering the metabolic vulnerabilities of
NSCLC carrying oncogenic mutations (Bensaad and Harris, 2013; Megchelenbrink et
al., 2015; Kerr and Martins, 2017).
CONCLUSION
Cancer cells demand large nutrient supplies and thus reprogram their metabolic
pathways to ensure metabolic flexibility, cellular homeostasis, energy
production, cell proliferation, and survival. In addition to direct modulation
of signal transduction pathways causing oncogenic addiction, alterations in
oncogenes also contribute to metabolic rewiring in cancer cells, resulting in
the promotion of cancer cell proliferation, survival, and metastatic
dissemination. Accordingly, metabolic reprogramming is now considered an
important characteristic of several types of cancer, including NSCLC. Despite
several ongoing approaches to target cancer metabolism, metabolic reprogramming
should be therapeutically explored in additional studies. In addition, the
influence of metabolic rewiring on the interaction between cancer cells and the
tumor microenvironment needs to be extensively investigated to comprehensively
understand the course of cancer development and progression, providing
mechanistic insights on several anticancer therapies targeting metabolism,
microenvironmental interactions, and evasion of anticancer immunity. However,
metabolic heterogeneity may reduce the responsiveness of metabolism-targeting
anticancer drugs; thus, an in-depth exploration of metabolic status in cancer
cells will be necessary to determine detailed metabolic changes at the cellular
and molecular levels. Further, the clinical impact of metabolic alterations on
cancer and the relevant biomarkers to predict or diagnose metabolic
reprogramming should also be identified to develop tailored precision medicine
targeting metabolic rewiring for the treatment of cancer.
Oncogene-Driven Metabolic Alterations in Cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5746037/
¡¡
Cancer metabolic reprogramming: importance, main features,
and potentials for precise targeted anti-cancer therapies
1Department of Molecular and Cellular Oncology, 2Department of General Internal
Medicine, Ambulatory Treatment and Emergency Care, 3Department of Endocrine
Neoplasia and Hormonal Disorders, The University of Texas MD Anderson Cancer
Center, 1515 Holcombe Blvd, Houston, Texas, TX77030, USA
Abstract
Cancer cells are well documented to rewire their metabolism and energy
production networks to support and enable rapid proliferation, continuous
growth, survival in harsh conditions, invasion, metastasis, and resistance to
cancer treatments. Since Dr. Otto Warburg¡¯s discovery about altered cancer cell
metabolism in 1930, thousands of studies have shed light on various aspects of
cancer metabolism with a common goal to find new ways for effectively
eliminating tumor cells by targeting their energy metabolism. This review
highlights the importance of the main features of cancer metabolism, summarizes
recent remarkable advances in this field, and points out the potentials to
translate these scientific findings into life-saving diagnosis and therapies to
help cancer patients.
Keywords: Cell cycle; energy metabolism; glycolysis; glutaminolysis;
mitochondria biogenesis
¡¡
Cancer metabolism: major remodeling of cellular energy production and metabolic pathways in tumors
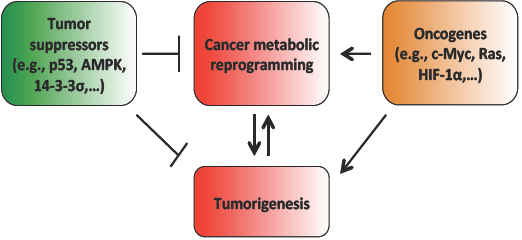
Cancer metabolic reprogramming has been recognized as one of the ten cancer
hallmarks by Drs. Hanahan and Weinberg in their seminal review paper published
in 20111. Some of the most striking changes of tumor cellular bioenergetics
include elevation of glycolysis, increase in glutaminolytic flux, upregulation
of amino acid and lipid metabolism, enhancement of mitochondrial biogenesis,
induction of pentose phosphate pathway and macromolecule biosynthesis1-17.
Glycolysis
Compared to normal cells, cancer cells prefer using glycolysis even in normoxic
condition18-20. This phenomenon is often referred as the Warburg effect because
Dr. Otto Warburg discovered and reported these metabolic alterations in tumors
in 1930 and 195618-20. Many decades later, numerous studies have provided
additional insights into the abnormality of cancer metabolism.
In normal cells, glucose is catabolized to pyruvate, which can be later
converted to acetyl-CoA to fuel the tricarboxylic acid cycle (TCA cycle, or
Krebs cycle). TCA cycle generates NADH and FADH2 to provide mitochondrial
respiratory chain with electrons for energy production. This is an effective
energy production mode since each glucose molecule can produce up to 36 ATP,
largely thanks to mitochondrial respiration. In normal cells, glycolysis is
prioritized only when oxygen supply is limited. In contrast, cancer cells
preferentially use glycolysis even in the abundance of oxygen2,3,5,7,18-21. This
is why tumor glycolysis is often called ¡°aerobic glycolysis¡±, or the Warburg
effect, to distinguish from the normal anaerobic glycolysis of healthy cells.
However, cancer cells have to compensate for the 18-fold lower efficacy of
energy generation (glycolysis only makes 2 ATP per glucose molecule consumed
while mitochondrial respiration can produce up to 36 ATP for each glucose
molecule catabolized). Part of the solution is to upregulate glucose
transporters, especially Glut1, Glut2, Glut3, and Glut4, to uptake more
glucose5,22-24. In fact, the increase in glucose uptake is a major feature
distinguishing tumor cells from normal cells. This difference has been widely
exploited in Positron Emission Tomography (PET) imaging modality using
radiolabeled analogs of glucose such as 18F-fluorodeoxyglucose as a tracer to
visualize tumors.
In addition, tumors remarkably elevate the expression of the majority of
glycolytic enzymes. Major oncogenes such as Ras, Myc, and HIF-1¦Á are reported to
be master inducers of cancer glycolysis3,5,24. Many glycolytic enzymes are also
upregulated in tumors because of elevated c-Myc and HIF-1¦Á transcriptional
activity and insufficient p53-mediated control. Indeed, c-Myc and HIF-1¦Á are
well recognized as two master inducers of glycolysis through direct or indirect
transactivation of cancer glycolytic genes. These two transcription factors
coordinate to promote the expression of key glycolytic enzymes such as HK2,
PFK1, TPI1, LDHA, among others, in tumors2,3,5,7,21,25,26. In fact, most of
glycolytic gene promoter areas contain consensus Myc and HIF-1¦Á binding motifs.
While HIF-1¦Á is mainly functional in hypoxia, c-Myc is well known to promote its
glycolytic target genes¡¯ expression in normoxia. This coordination allows tumors
to continuously drive glycolysis for supporting their rapid proliferation and
accelerated biosynthesis2,3,7,11,15,16,21.
In contrast, p53 is known to suppress glucose uptake by directly inhibiting the
transcription of glucose transporter Glut1 and Glut427,28 and suppressing the
expression of Glut328. Glut3 is an NF-¦ÊB target gene and p53 is found to block
NF-¦ÊB activation, thereby considerably reducing Glut3 transcription and
expression28. p53 also induces the expression of TIGAR to slow down cancer
glycolytic flux29,30. Fructose 2,6-bisphosphate is an important allosteric
activator of PFK1, a major glycolytic enzyme. Fructose 2,6-bisphosphate is
produced by PFK2 from fructose 1-phosphate. By converting fructose
2,6-bisphosphate back to fructose 1-phosphate, TIGAR significantly slows down
tumor glycolysis29,30.
The interaction among p53, c-Myc and HIF-1¦Á has a decisive impact on the status
of cancer glycolysis2,5,7,16,21,30. Many studies have characterized the
communication between these three master regulators of cancer glycolysis and how
the balance among these factors control the status of cancer metabolism.
On the other hand, the way tumor cells process pyruvate, the end product of
glycolysis, is also different from normal cells. In normal cells, most of
pyruvate is converted to acetyl-CoA to fuel the TCA cycle. Some pyruvate is used
to produce alanine or lactate. In contrast, pyruvate-to-lactate is a preferred
reaction in tumor cells due to the upregulation of lactate dehydrogenase A
(LDHA). This reaction is beneficial for cancer cells as it helps regenerate NADH
to accelerate glycolysis2,3,5,11,25. Furthermore, lactate is secreted into tumor
microenvironment via MCT4 transporter to fuel other cancer cells that do not
have frequent access to nutrient supplies from blood stream. Lactate could be
uptaken by MCT1 transporter and used by the TCA cycle for metabolism. The
symbiosis of lactate-producing cancer cells and lactate-consuming tumor cells is
an effective way for tumors¡¯ adaptation to the diverse and constantly changing
conditions in tumors, which is caused by the leaky and poorly formed tumor blood
vessel network7,31-33. Furthermore, converting pyruvate to lactate also reduces
reactive oxygen species¡¯ levels, thereby diminishing the intracellular oxidative
stress in cancer cells and promoting tumors¡¯ survival2,7. Moreover, lactate also
lowers the pH of extracellular microenvironment and facilitates the activity of
metalloproteases for breaking down extracellular matrix. Thus, lactate is an
inducer of cancer invasion and metastasis34,35.
Importantly, glycolysis provides cancer cells with not only energy but also
necessary precursors for biosynthesis, which is similar to stem cells¡¯ metabolic
profiles. Several glycolytic metabolites such as glucose-6-phosphate,
dihydroxyacetone phosphate, among others, could be diverted into other metabolic
pathways. For instance, glucose-6-phosphate is often consumed by pentose
phosphate pathway to synthesize nucleotides and NADPH (a major reducing agent
important for redox homeostasis and drug detoxifying reactions).
Dihydroxyacetone phosphate could be used for lipid synthesis, which is important
for assembling new organelles and cells to promote tumor growth and
proliferation. Metabolites from glycolysis are also important materials for
amino acid production and macromolecules synthesis, which is required for active
cell division and large-scale biosynthetic programs2,3,5,7,16,36,37. In addition
to their metabolic function, glycolytic enzymes play active roles in promoting
cancer survival, metastasis, invasion, chromatin remodeling, gene expression
regulation, and other essential cellular processes2,38. Thus targeting
glycolytic enzymes¡¯ activities could be useful strategies for cancer therapy.
Glutaminolysis
In addition to glycolysis, many tumors also rely on glutaminolysis to fuel their
cellular bioenergetics and metabolism. Glutaminolysis is a series of biochemical
reactions catabolizing glutamine into downstream metabolites such as glutamate,
¦Á-ketoglutarate. The products of glutaminolysis are very important to fuel the
TCA cycle of tumors. The intermediates of TCA cycles could be used for the
synthesis of lipid, cholesterol, amino acids and other essential metabolites.
Moreover, NADH and FADH2 from the TCA cycle provide electrons for the electron
transport chain of mitochondria to generate ATP. Thus, similar to glycolysis,
glutaminolysis supplies cancer cells with not only ATP but also crucial
precursors for continuous biosynthesis and accelerated
proliferation3,5,13,15,16,22,25.
Glutaminolysis upregulation in tumors is mediated by c-Myc4,9,13,39. Multiple
studies demonstrate that c-Myc promotes both glutamine uptake and the catabolic
process of glutamine. In fact, c-Myc transactivates ASCT2 and SN2, two important
glutamine transporters on cellular membrane9,40. c-Myc also suppresses miR-23a/b
to upregulate GLS1 expression41,42. GLS1 is a major enzyme for glutaminolysis.
Therefore, c-Myc is an important inducer of glutaminolysis in tumors.
Interestingly, while promoting cancer metabolic reprogramming, c-Myc also
renders cancer cells addicted to glutaminolysis, which opens a new therapeutic
window to selectively suppress and eliminate cancer cells9,13-15,39,43.
Therefore, targeting tumor glutaminolysis and c-Myc-induced-glutamine addiction
is a promising anti-cancer metabolism therapy.
Pentose phosphate pathway
Pentose phosphate pathway (PPP) is a classical metabolic pathway consisting of
two branches. In the oxidative arm, PPP converts glucose-6-phosphate, a
glycolytic intermediate, into ribulose-5-phosphate and generates NADPH. NADPH is
then used for glutathione production, detoxification reactions, and biosynthesis
of lipids as well as other macromolecules. The non-oxidative PPP branch involves
reversible carbon-exchanging reactions with the final products as
fructose-6-phosphate and glyceraldehyde-3-phosphate. These metabolites can
participate in glycolysis and downstream metabolic pathways44. PPP is commonly
viewed as a line of defense counteracting reactive oxidative stress and
producing ribose-5-phosphate for nucleotide synthesis. However, new studies
suggest that PPP has important impacts on various aspects of cancer, including
proliferation, apoptosis, invasion, drug resistance, and metastasis44. These
exciting findings unveil PPP as a potential target for anti-cancer metabolism
therapies.
Rapidlyproliferating cancer cells constantly demand nucleotides and other
materials for biosynthesis. Therefore, by providing NAPDH and pentose phosphate
for nucleotide synthesis, PPP is important and frequently upregulated in many
types of tumors5,44. In fact, the activity of glucose-6-phosphate dehydrogenase
(G6PD), a major PPP enzyme, increases in proliferating cancer cells45. G6PD,
transketolase (TK) and other PPP enzymes are elevated in multiple types of
cancer and facilitated tumors¡¯ accelerated proliferation44,46,47. In addition,
G6PD also promotes cancer survival by producing NADPH, a key tool for tumor
cells to defend against oxidative stress, chemotherapy-induced cytotoxic damage,
as well as for promoting biosynthesis44. Hence, G6PD function is tightly
controlled by the tumor suppressor p53. Indeed, p53 associates with G6PD and
prevents this enzyme from forming active dimer complexes48. It is noteworthy
that G6PD is directly transactivated by HIF-1¦Á49. The function of G6PD is
strictly regulated in normal cells but highly activated in cancer cells, making
G6PD a strong oncogene candidate44. Interestingly, G6PD and TK functions are
both suppressed by resveratrol50, suggesting the usage of this natural product
in cancer treatment and prevention.
While normal cells frequently rely on the oxidative branch of PPP for
ribose-5-phosphate production; cancer cells use both arms, e.g., oxidative and
non-oxidative, of PPP to generate ribose-5-phosphate for nucleic acid
synthesis51-53. Furthermore, cancer cells can use ribose-5-phosphate in both de
novo and salvage pathways to synthesize nucleotides. These flexible metabolic
programs help cancer cells effectively adapt to constantly changing nutritional
conditions of tumor microenvironment.
In addition, PPP also protects tumor cells from apoptosis by counteracting
oxidative stress and facilitating DNA damage repair. In fact, nonsteroidal
anti-inflammatory medications induce apoptosis and shrinkage of colon carcinoma
and polyps by regulating PPP54. Moreover, G6PD inhibitors, e.g., DHEA and 6-AN,
promote apoptosis in mouse fibroblasts and PC-12 neural cells while
overexpression of G6PD protects cells from H2O2-induced cell death55. Knocking
down of G6PD also increases oxidative stress-mediated toxicity in melanoma
cells56. The vital role of PPP in protecting cells from programmed cell death is
additionally proven in vivo such as in stem cells and peripheral blood
mononuclear cells of patients lacking G6PD55,57,58. Interestingly, the
cytoprotective function of PPP is not limited to defending against reactive
oxygen species but also expands to helping DNA damage repair. Indeed, upon DNA
damage, ATM quickly activates G6PD functions to accelerate PPP for quenching
reactive oxygen species, increasing nucleotide synthesis and enabling effective
DNA repair. Therefore, knocking down G6PD significantly impairs DNA damage
repair ability59,60. Some other studies describe the impact of PPP on regulation
of autophagy61, but the molecular mechanism is still not completely understood.
Surprisingly, PPP also induces tumor angiogenesis. Leopold et al.62 and Pan et
al.63 reported the crosstalk between G6PD and VEGF and tight association between
G6PD and angiogenesis. These studies show that VEGF stimulate G6PD expression
via Src signaling and G6PD is important for VEGF-induced-endothelial cell
migration by increasing the phosphorylation of VEGR receptor Flk-1/KDR. G6PD
also increases the proangiogenic activity of endothelial NO by providing NADPH
and stimulates Akt-induced activation of endothelial nitric oxide synthase
(eNOS)62.
PPP additionally promotes tumor resistance to chemotherapy and radiation by
multiple mechanisms. First, PPP provides cancer cells with NAPDH, a potent
anti-oxidative agent that protects cancer cells from reactive oxygen
species-induced cell death caused by chemotherapy and radiation44; Second, PPP
facilitates DNA damage repair by providing material for nucleotide synthesis;
Third, by shifting cancer metabolism away from mitochondrial respiration, PPP
lowers the intracellular concentrations of reactive oxygen species, thereby
increasing tumor endurance and survival during chemotherapy and radiation
treatment; Fourth, NAPDH derived from PPP, is an important element for
glutathione (GSH) generation. GSH is frequently used in detoxification
reactions, enabling cancer resistance to a variety of chemotherapeutic agents.
GSH conjugation to these xenobiotics also facilitates the activity of MDR1 and
MDR2 to discard cytotoxic substances. Therefore, increase in G6PD expression and
PPP flux increase intracellular GSH levels and reduce drug accumulation in
cancer cells64. However, there are still many exceptions where PPP neither
significantly contributes to drug resistance nor promotes the effect of certain
chemotherapeutic agents in several cancer cell lines. This complexity requires
more study to fully elucidate the contribution of PPP in protecting cells from
anti-cancer treatments44.
In short, PPP is an important metabolic pathway providing cancer cells with
NADPH, ribose-5-phosphate and other essential intermediates. NAPDH is crucial
for counteracting oxidative stress and biosynthesis reactions.
Ribose-5-phosphate is a major element for nucleotide synthesis. Interestingly,
the impact of PPP on cancer cells is well beyond oxidative defense. Indeed, PPP
upregulation promotes cancer cell survival, angiogenesis, proliferation,
invasion, metastasis, and resistance to radiation and chemotherapies. Therefore,
elevated and active PPP enzymes, for instance, TKTL or G6PD, are frequently
observed in malignant, aggressive, proliferative and drug-resistant cancer
cells44. The new exciting discoveries about PPP open new therapeutic windows but
also require more study to refine rational approaches for precise and effective
targeting of this vital metabolic pathway in cancer cells.
Mitochondrial biogenesis
Another major change in cancer metabolism is the enhancement of mitochondrial
biogenesis. In contrast to conventional concepts, mitochondria play very
important roles in cancer because these vital organelles are the nexus of many
essential metabolic pathways65. Mitochondria are not only the energy generators
but also the factories synthesizing many indispensable molecules for cellular
biosynthesis, growth and proliferation. Moreover, mitochondria additionally
control the redox balance and Ca2+ concentration, which is essential for
cellular homeostasis65. Therefore, impairment of mitochondrial function or lack
of mitochondrial biogenesis seriously suppresses tumorigenesis, tumor formation
and growth65-71. Furthermore, in comparison with healthy and well differentiated
cells, cancer cells frequently rewire their mitochondria to switch from a
maximal energy production by mitochondrial electron transport chain to a
well-adjusted balance among constant energy requirement, large-scale biogenesis
programs and rapid cell proliferation65. Therefore, mitochondrial biogenesis and
mitochondria are truly essential for tumor cells65. Hence, increase in
mitochondria biogenesis is a significant advantage for cancer.
It is well established that c-Myc is a strong promoter of mitochondrial
synthesis. In fact, c-Myc induces the expression of many nuclear-encoded
mitochondrial genes. More importantly, c-Myc directly transactivates
mitochondrial transcription factor A (TFAM). TFAM is a transcription factor that
is indispensable for mitochondrial genes transcription and mitochondrial DNA
replication72. In reality, TFAM promotes the right formation of mitochondrial
transcription and replication complexes and facilitates the correct positioning
of mitochondrial DNA for optimal gene transcription and proper mitochondrial DNA
duplication65. As the synthesis of new mitochondrial components and replication
of mitochondrial DNA are vital for de novo mitochondrial formation, c-Myc,
indeed, plays a crucial role in elevating the number of mitochondria. As a
consequence, lack of Myc expression and transactivational activity remarkably
reduces mitochondrial mass as well as mitochondrial biogenesis, resulting in a
severely suppressive impact on many metabolic pathways of cancer cells and
tumorigenesis ultimately72.
Lipid synthesis
Increase in lipid metabolism is another remarkable feature of cancer metabolism.
Lipids are important building blocks of new organelles and cells. Lipid
synthesis is a multiple step process involving several enzymes such as ATP
citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN),
and stearoyl-CoA desaturase (SCD). This procedure starts with converting
acetyl-CoA to malonyl-CoA by ACC. A series of condensation reactions by FASN
results in saturated fatty acids. Fatty acids could be desaturated by SCD.
Cancer cells frequently upregulate de novo fatty acid synthesis to satisfy their
demands for lipids73-75. FASN elevation is observed in breast, prostate and
other types of cancer73,76-79. FASN is a target gene of HIF-1¦Á and frequently
overexpressed in an Akt and SREBP1-dependent manner80. ACLY, often activated by
Akt81, is indispensable for tumor transformation and formation both in vitro and
in vivo81,82. ACC is also very important for tumorigenesis as inhibition of ACC
stops cancer growth and induces apoptosis of prostate cancer cells83.
Furthermore, cancer cells often have higher lipid accumulation in form of lipid
droplets in relative to normal cells84.
Cholesterol synthesis, or the mevalonate pathway, is also an important aspect of
lipid biosynthesis because cholesterol is a major component of membranes
controlling the membrane fluidity and formation of lipid rafts. Cholesterol is
vital for activation of Ras-Raf signaling pathway85 and deregulation of
cholesterol synthesis is correlated with tumorigenic transformation86.
Interestingly, statin-mediated inhibition of HMGCR, an important enzyme of the
mevalonate pathway, considerably ameliorates the effectiveness of chemotherapies
in acute myeloid leukemia87, hepatocellular carcinoma88, and other types of
cancer through epigenetic pattern modification89.
The sterol regulatory element-binding proteins (SREBPs) are the main
transcription factors controlling the expression of most of enzymes involved in
fatty acid and cholesterol synthesis. SREBPs are helix-loop-helix 125 kDa
proteins that require a protein cleavage at the endoplasmic reticulum for
activation73. While SREBP1 controls fatty acid, triacylglycerol and phospholipid
synthesis, SREBP2 regulates cholesterol generation90. SREBPs are controlled by
tumor suppressors and oncogenes. AMPK, for instance, inhibits SREBP activation91
and suppresses ACC91, thereby keeping lipid synthesis in check. Loss of pRb
upregulates SREBP1 and SREBP2, thereby activating Ras signaling92. p53 mutants,
on the other hand, coordinates with SREBP to transactivate
cholesterol-synthesizing enzymes93. Of note, SREBP1 and SREBP2 are often
overexpressed in cancer76 and play an important role in cancer cell survival94.
At the organism level, excessive lipid synthesis contributes to tumorigenesis.
It has been well documented that obesity increases the risk of cancer73. In
fact, excessive lipid concentrations in liver and muscle cells induce insulin
resistance by impairing insulin signaling and reducing glucose uptake. Insulin
resistance forces pancreatic cells to secrete more insulin and insulin-like
growth factors, which is very beneficial for cancer proliferation and
survival95-97. Obesity also increases inflammation, which contributes to insulin
resistance and tumorigenesis98. Dietary restriction may reverse these
tumorigenic trends but in certain scenarios, especially when PI3K/Akt signaling
is overactivated, the tumor-suppressing impact of dietary limitation
decreased99. A possible explanation is that nutrient restriction may reduce the
levels of circulating insulin and insulin-like growth factors. However, the
constitutive activation of PI3K/Akt may compensate for that insulin signaling
decrease100.
Fatty acid oxidation
While glycolysis, glutaminolysis, fatty acid synthesis have been well
characterized during the past few decades; fatty acid oxidation (FAO) still
remains a little known metabolic pathway. However, recent studies have
demonstrated the important contribution of FAO to tumorigenesis101.
Fatty acids are a rich energy source that can yield to up to two times more ATP
than carbohydrates when needed. Fatty acids could be oxidized in mitochondria or
by cytoplasmic lipophagy, a new fatty acid catabolic process102. FAO is a
repeated multi-round process leading to the production of acetyl CoA, NADH, and
FADH2 in each cycle. Acetyl-CoA can be imported into TCA cycle to generate more
NADH and FADH2, which subsequently fuel mitochondrial respiration chain for ATP
production. Acetyl-CoA can also fuel TCA cycle for synthesis of citrate.
Citrate-derived isocitrate and malate can be respectively converted to
¦Á-ketoglutarate by IDH1 or pyruvate by malic enzyme (ME1)102. Both reactions
generate NADPH, which plays a very important role in maintaining redox
homeostasis, inducing cell survival, enabling xenobiotics detoxification and
promoting biosynthesis for cell growth and division103. Of note, NAPDH is
crucial for the function of many anabolic enzymes to sustain large-scale
biosynthetic programs in many cancer cells.
NAPDH derived from FAO is very important for cancer cells to quench reactive
oxidative stress. For instance, blocking glioma tumor¡¯s FAO leads to rapid
depletion of NADPH, surge of reactive oxidative species¡¯ concentrations and
increase in apoptosis104. NADPH produced by FAO is also relevant to the
maintenance of hematopoietic stem cells because these cells are very sensitive
and vulnerable to reactive oxidative stress. In fact, increased reactive oxygen
species levels inhibit hematopoietic stem cells¡¯ self-renewal and leads to cell
differentiation105-107. Jeon et al.108 reported that LKB1-APMK regulates the
balance between NADPH consumption by fatty acid synthesis and NAPDH production
by FAO. In fact, AMPK blocks fatty acid synthesis in tumors by phosphorylating
and inactivating acetyl-CoA carboxylase (ACC)109, antagonizing PPAR signal
transduction110 and regulating CTP1C expression111. Therefore, AMPK is a potent
inhibitor of fatty acid synthesis in cancer cells.
Needless for further emphasis, ATP is by large one of the most important
molecules for cancer cells. Due to its rapid proliferation and accelerated
activities, tumors are almost constantly in high demand for ATP. ATP is the most
frequently used energy currency and a major material for phosphorylation
reactions, an essential mode of cellular signal transduction and protein
modification. ATP is also an indispensable element for DNA and RNA replication
and repair. The function of MDR1 and other ABC pumps on cellular membrane, a
major tumors¡¯ line of defense against chemotherapy, absolutely requires ATP.
Recently, ATP production by FAO has been shown to prevent anoikis, a type of
cell death due to loss of attachment to extracellular matrix although the
molecular mechanism still remains unclear and warrants more study103,112. The
Pandolfi group113 also reported that the promyelocytic leukemia (PML) protein
induced FAO by activating peroxisome-proliferator-activated receptors (PPARs),
leading to poor survival and clinical outcomes of breast cancer patients.
Moreover, Tak Mak¡¯s lab111 additionally found that carnitine
palmitoyl-transferase 1 isoform C (CPT1C) is an oncogene that induces cancer
growth, ATP production, FAO and confers resistance to mTORC1 inhibitors. CPT1
proteins mediate the import of fatty acids into mitochondria for FAO reactions.
CPT1 links carnitine to fatty acids and transports the conjugated products
(acyl-carnitines) into mitochondria. Therefore, the oncogenic property of CPT1C
is a good example illustrating the potential of FAO in tumorigenesis.
FAO is also important in ensuring cancer cell survival in a manner that is
independent of ATP production101. In fact, CPT1 proteins suppress the
pro-apoptotic function of Bax and Bak by modulating the formation of
mitochondrial permeability transition pores and reducing cytochrome c
release114,115. The results from Samudio et al.116 and Vickers group117
additionally indicate that FAO can promote cancer cell survival by preventing a
cytotoxic intracellular surge of fatty acid concentrations. On the other hand,
several groups show that the increase in reactive oxygen species due to
FAO-induced mitochondrial respiration could be harmful for leukemia cells.
However, this toxicity could be resolved by upregulating uncoupling protein 2
and 3 (UCP2, UCP3) that effectively dissipate the gradient proton in
mitochondria and decrease mitochondrial oxidative phosphorylation efficiency118.
Thus, fatty acid oxidation promotes cancer cell survival, and provides tumors
with necessary energy and precursors. The new findings about FAO reveal
fascinating understandings about cancer metabolic reprogramming and unveil very
promising opportunities for anti-cancer therapeutic approaches. However,
additional knowledge is needed to successfully develop effective therapies
targeting this important catabolic process in cancer.
Interestingly, Hu et al.119 has recently completed a massive meta-analysis of
over 2,500 microarrays including 22 types of cancer to compare the metabolic
gene expression landscape of tumors relative to that of corresponding normal
tissues. From this comprehensive transcriptomics analysis, three important
observations have been reported: (1) despite the process of tumor evolution,
there is still a significant degree of similarity in the gene expression
metabolic profiles of tumors in comparison with those of the normal tissues
where tumors originate; (2) the metabolic gene expression landscape across
different types of tumors is heterogeneous. However, glycolysis, nucleotide
synthesis, aminoacyl-tRNA synthesis, and pentose phosphate pathway are
consistently upregulated and increasingly important in actively proliferating
cancer cells; (3) hundreds of metabolic isoenzymes demonstrate remarkable and
cancer-specific expression alterations, representing new significant therapeutic
opportunities for anti-cancer metabolism therapies. These isoenzymes are
important for cancer. Some enzymes such as isocitrate dehydrogenase and fumarate
dehydratase, may even imitate or aggravate the impact of tumorigenic genetic
mutations119.
In short, metabolic reprogramming is an important cancer hallmark
characterized by the upregulation of glycolysis, glutaminolysis, lipid
metabolism, mitochondrial biogenesis, pentose phosphate pathway as well as other
biosynthetic and bioenergetic pathways. These cancer metabolic programs provide
tumor cells with not only necessary energy but also crucial materials to support
large-scale biosynthesis, rapid proliferation, survival, invasion, metastasis
and resistance to anti-cancer therapies. Therefore, exploiting the unique
features of cancer metabolism for cancer detection, treatment and monitoring is
a very promising trend in cancer therapeutics, diagnosis and prevention.
Cancer metabolism and diagnostic imaging
The distinguished features of cancer metabolism have been extensively exploited
for initial diagnosis, staging disease, monitoring tumor responses to therapies,
and detecting cancer recurrence120. Therefore, nowadays, metabolic molecular
imaging plays an indispensable role in clinical oncology. These diagnostic
methods are non-invasive and can accurately detect the changes in selective
biologic processes of tumors compared to normal surrounding tissues both at the
initial tumor sites and metastatic locations over an extended period of time.
The information provided by advanced imaging modalities such as PET, magnetic
resonance spectroscopy imaging (MRSI), magnetic resonance imaging (MRI), is very
valuable for cancer detection, prevention, and treatment120.
Positron emission tomography(PET)
PET is frequently combined with X-ray computed tomography (CT) to provide
detailed information about cancer and anatomic locations of tumors. PET measures
the signals of radiolabeled tracers taken up by cancer cells. PET is safe and
widely used in clinics because the small amount of imaging probes doesn¡¯t
interfere with normal physiological processes. 18F-fluoro-2-deoxyglucose (FDG)
is the most commonly used PET imaging material. Since most of tumors have a high
glycolytic flux, elevated glucose uptake and increased hexokinase function, they
will often have higher FDG signals relative to normal tissues. After being
imported into tumor cells, FDG is phosphorylated by hexokinase but
phosphorylated FDG cannot be further catabolized by glycolytic pathway.
Therefore, phosphorylated FDG molecules are accumulated in tumors and can be
detected by PET scanners. In clinics, FDG-PET scan is commonly used for
determining cancer stages, identifying cancer recurrence and assessing tumor
response to anti-cancer therapies121,122.
In addition to upregulated glycolysis, other patterns of cancer metabolism are
also used for molecular oncology imaging using PET scan. Choline, for example,
is frequently absorbed by tumor cells and used for new cellular membrane
biosynthesis, an important process for cell division. Therefore 11C and 18F
radiolabeled choline tracers have been successfully applied in hepatocellular
carcinoma, lung, brain, and prostate cancer diagnosis123-126. Similarly,
3'-deoxy-3'-18F-fluorothymidine is often used to monitor cancer cell
proliferation in vivo. 3'-deoxy-3'-18F-fluorothymidine is a thymidine analog and
frequently phosphorylated by thymidine kinase 1. This enzyme is highly active in
rapidly dividing cells, e.g., tumor cells, especially in S phase. Thus,
3'-deoxy-3'-18F-fluorothymidine PET can identify and measure tumor malignancy,
tracking the efficacy of anti-cancer therapies127. Many other tracers are also
used in PET imaging modality to monitor specific biological processes of tumors.
For instance, 68Ga-DOTATOC, a high-affinity ligand for somatostatin receptor 2,
is used to detect neuroendocrine cancer masses128. 16-¦Á-18F-fluoro-17¦Â-estradiol
is used to quantify ER¦Á and ER¦Â expression129. Tumor angiogenesis and the
effectiveness of anti-angiogenic therapeutic agents are measured by tracers
containing arginine-glycine-aspartic acid-peptide ligands. These ligands
associate with ¦Áv¦Â3 integrin whose expression is elevated on newly formed blood
vessels130. Nitroimidazole is also exploited to image hypoxic areas where tumors
are frequently located131.
In summary, PET with radiolabeled metabolic tracers is certainly a valuable and
powerful imaging method with vast applications in clinical oncology. This
diagnostic modality is continuously improved and more advanced tracers are in
development. However, radiation is still a major concern for PET and its
tracers. The radiation containment and safety are also other significant issues
for PET application in clinics120. In addition, a complete understanding about
cancer metabolic patterns and bioenergetics programs is crucial to continuously
innovate metabolic tracers-based PET scan imaging.
The combination of MRI and MRSI
MRI and MRSI are often combined in clinical oncology diagnostics because 1H MRSI
is easily compatible with currently available MRI scanners in clinics132-134. 1H
MRSI has a high sensitivity and could be applied on a number of tracers120.
During the past few years, MRSI has made significant advances and rapidly become
a reliable imaging modality. A number of 1H tracers have been successfully
developed. For instance, 1H choline-containing metabolites are employed to
measure tumor malignancy. Choline is an important component of cellular
membrane. Higher choline concentrations are detected in aggressive and malignant
tumors in comparison with benign and normal tissues135,136. In fact, many breast
tumors contain a large amount of choline while benign tumor masses often have
low levels of choline135,136. Since the accumulation of choline is associated
with increased cell proliferation in brain, breast, cervical and prostate
cancers133,137-139, choline availability could be used as a marker for
predicting tumor histologic grade, aggressiveness, and even response to
anti-cancer therapies with low unspecific detection rates120,139. Moreover, as
brain tumors often have increased choline concentrations and diminished levels
of N-acetyl aspartate, the ratio of choline/N-acetyl aspartate has been used to
evaluate the aggressiveness of several types of brain tumors140-142.
Choline/creatinine ratio measurement is also a valuable indicator of
oligodendroglial cancer grade143.
13C tracers are emerging important diagnostic probes although their application
is still at early stages. Recently, Nelson et al.144 reported a successful
preclinical study and phase I clinical trial results with 31 prostate cancer
patients. This is a pioneer project examining the applicability and safety of
hyperpolarized 13C pyruvate tracers to monitor and evaluate the metabolic
changes, especially 13C pyruvate-to-13C lactate flux, of prostate tumors in
patients. This technique enabled a 10,000-fold increase in signals compared to
regular MRI. Results were very promising with excellent safety profiles and
accurate detection of 13C pyruvate-to-13C lactate flux in tumor areas that were
subsequently proven by biopsy-based pathological and histological analyses. The
success of this pioneer study paves a new way for non-invasive, safe, precise,
and sensitive cancer diagnosis as well as tumor monitoring. A number of new
types of 13C metabolic tracers are under development and will certainly play a
major role in cancer detection and imaging in future.
Poor spatial resolution used to be a challenge for MRSI133,138,145,146, but new
advances and ongoing technological improvements are addressing this limiting
factor, making MRSI a promising adjunct to MRI. Combining conventional MRI with
MRSI will enable accurate, safe and non-invasive characterization of tumors.
This new diagnostic strategy is especially important when collecting lesion
biopsies is risky, painful and difficult. Thus, in future, this new combinatory
imaging modality will reduce patients¡¯ discomfort, concern, risk, pain, and
avoid unnecessary invasive diagnostic procedures while increasing the accuracy,
reliability and sensitivity of diagnosis120.
In summary, diagnostic imaging plays a crucial role in cancer detection and
treatment. Exploiting the unique features of cancer metabolism is a very
promising direction for developing novel diagnosis methods to accurately detect
cancer lesions even at early stages and precisely monitor tumors¡¯ responses to
therapies.
Therapeutic implications
Given the vital role of metabolic reprogramming for tumorigenesis, targeting
cancer bioenergetics is a very promising and rapidly rising direction for
anti-cancer therapy development nowadays. Many compounds have been developed to
selectively and effectively inhibit metabolic enzymes that are important for
tumors. These inhibitors are currently at various stages of clinical trial
process and we expect to see them in clinics within five to ten years from now.
One of the most common trends in anti-cancer metabolism therapies is to inhibit
enzymes that are exclusively or mostly expressed or used in tumor cells. This
therapeutic strategy would effectively eliminate tumors while minimizing damage
to normal cells. Several groups have successfully developed inhibitors for
Glutaminase 1 (GLS1), a glutaminase isoform that is highly upregulated in cancer
cells, and proved the efficacy of blocking GLS1 in cancer treatment147,148. This
tactic bases on previous studies showing a significant dependence of
c-Myc-overexpressing cancer cells on glutaminolysis9,11-15,25,149. Similarly,
modulating the activity of PKM2, a glycolytic enzyme that is frequently elevated
in tumors, is also a promising therapy150,151. Fatty synthase (FASN) is
important for palmitate synthesis and this enzyme¡¯s expression is elevated in
many tumors. Therefore, several groups have developed FASN inhibitors to target
tumorigenesis75,152. Many inhibitors for HIF and HIF targets, for instance
monocarboxylate transporter MCT4 and carbonic anhydrase IX (CAIX) are also
potential anti-cancer drugs in future153-156. Similarly, MCT1 and carbonic
anhydrase XII are targets of great potential153,154. MCT1 and MCT4 suppressors
inhibit cancer growth in vitro and in vivo and invasion in vitro157-160. In
fact, interfering with lactate transport by MCT1 and MCT4 inhibitors has been
shown to induce tumor cell starvation and subsequent apoptosis158.
Blocking lactate production using dichloroacetate (DCA) shows promising results
with minor side effects in early phase clinical trials, especially in
glioblastoma patients161,162. DCA is found to promote pyruvate-to-acetyl-CoA
flux and reduce pyruvate-to-lactate conversion, thereby inducing tumor shrinkage
and apoptosis in vivo161-163. Clinical trial data show that DCA also suppresses
tumor angiogenesis, blocks HIF1-¦Á signaling and activates p53 in glioblastoma
multiforme patients161. Initial studies additionally find that DCA inhibits
pyruvate dehydrogenase kinase 1 (PDK1) activity and thereby activating the
function of pyruvate dehydrogenase 1 (PDH1), an important enzyme catalyzing the
pyruvate-to-acetyl-CoA biochemical reaction162,163. However, more and larger
clinical studies are needed to fully elucidate the mechanism of action of this
interesting compound and further evaluate its efficacy in cancer patients.
Glycolysis inhibitors are also of interest for many groups and pharmaceutical
companies. For instance, FX11, a selective suppressor of lactate dehydrogenase A
(LDHA) activity, was tested by Le et al.164 and is currently studied by National
Cancer Institute¡¯s Experimental Therapeutics Program (NExT). 2-deoxyglucose
(2-DG) is among the most advanced cancer metabolism inhibitors in clinical
trials (Phase II). 2-DG reversibly inhibits hexokinase to block glycolysis. 2-DG
usage in combination with radiation demonstrates a good safety profile and
slightly improves survival of glioblastoma multiforme patients165,166. However,
the effects of 2-DG may be limited by high concentration of glucose because
2-DG-mediated inhibition of hexokinase is reversible.
Inhibiting mutant isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase
2 (IDH2) is a remarkable therapeutic approach because these mutant enzymes have
distinct activities compared to normal IDH1 and IDH2 in the healthy cells. On
the other hand, metformin, a common anti-diabetics medication, has demonstrated
very promising impact in cancer treatment. It is well known that metformin
inhibits mitochondrial complex I of liver cells, thereby decreasing ATP
production. Lack of ATP subsequently stimulates LKB1-AMPK pathway and blocks
gluconeogenesis, leading to lower blood glucose concentrations, improved
sensitivity to insulin and diminished insulin production167. It is currently
unclear whether metformin improves cancer patient clinical outcomes by lowering
blood glucose levels and insulin/insulin-like growth factors generation or by
directly targeting cancer cells. Nevertheless, the usage of metformin has been
well documented to ameliorate cancer patient survival168,169 and metformin are
harmful for cancer stem cells170. Clinical trials testing the impact of
metformin on cancer in patients are ongoing (Table 1).
Table 1
Table 1 List of several potential anti-cancer metabolism compounds
Full table
Importantly, there is also an urgent need to develop effective inhibitors to
target the key inducers of cancer metabolic reprogramming such as c-Myc and Ras.
Ras mutations and c-Myc upregulation are frequent in many common types of cancer
and these dysregulations are major drivers of tumorigenesis and resistance to
therapies171,172. However, despite our relentless efforts, effectively and
directly inhibiting Ras and c-Myc still requires a lot more study because these
two proteins are currently undruggable targets. Interestingly, several
preclinical research projects show that targeting metabolic enzymes
significantly inhibits tumors carrying Ras mutation and c-Myc
overexpression9,173. In fact, suppressing glycolysis and glutaminolysis
remarkably antagonizes the growth of tumors bearing those genetic
alterations9,164,174,175. These observations imply a new way to treat tumors
carrying genetic mutations that can¡¯t be directly targeted.
Another striking example of successful anti-cancer metabolism therapies is
L-asparaginase. L-asparaginase mediates deamination reactions to degrade
asparagine into aspartic acid176, thereby reducing asparagine availability to
cancer cells and suppressing their growth177,178. This therapy is very effective
for acute lymphoblastic leukemia (ALL) and related leukemia subtypes because ALL
cells are unable to synthesize asparagine179. Therefore, these cancer cells have
to rely on extracellular asparagine sources and become very vulnerable when
asparagine supplies are limited.
However, lymphocytes, especially T cells, have similar metabolic programs as
those in tumor cells. For instance, lymphocytes also depends on glutamine
metabolism180, suggesting that systematically targeting glutaminolysis for
cancer treatment may severely affect adaptive immune responses and also innate
immunity to a certain degree. These metabolic similarities between cancer cells
and lymphocytes explain why many agents targeting cancer metabolism are also
strong immunosuppressants. For instance, cyclosporine, a potent anti-cancer drug
that inhibits mTOR, significantly suppresses immune system. Suppressor of
nicotinamide phosphoribosyltransferase (NAMPT), an enzyme responsible for
nicotinamide adenine dinucleotide (NAD+) regeneration, is poisonous to
lymphocytes181. In fact, early clinical trials data show that FK866, a
NAD+synthesis inhibitor, leads to mild lymphopenia and severe
thrombocytopenia182.
These findings suggest that immunosuppression could be a challenge for therapies
designed to target cancer cells¡¯ bioenergetics as the Achilles¡¯ heel of tumors.
Nevertheless, there is still a significant therapeutic window for anti-cancer
metabolic therapies. We just need to identify the key differences in the
bioenergetics patterns of tumors and those of healthy cells in order to optimize
our therapies for precisely inhibiting the unique metabolic targets in cancer
cells. A significant example is to use BPTES to selectively block GLS1, a
glutaminase enzyme isoform that is crucial for cancer cells and specifically
upregulated in tumors147,148.
Conclusion
Metabolic reprogramming is a major hallmark of cancer, which is characterized by
upregulated glycolysis, glutaminolysis, lipid metabolism, pentose phosphate
pathway, mitochondrial biogenesis, among others. These metabolic programs
provide cancer cells with not only energy but also vital metabolites to support
large-scale biosynthesis, continuous proliferation and other major processes of
tumorigenesis. Potent oncogenes as c-Myc, HIF1¦Á, Ras and PI3K/Akt are important
promoters of cancer metabolic alterations. In contrast, major tumor suppressors
such as p53 and LKB1/AMPK antagonize those changes and keep cellular metabolism
in check (Figure 1 and Figure 2). Rfewiring metabolism is very beneficial for
tumor survival, invasion, metastasis, growth, angiogenesis, proliferation and
resistance to anti-cancer therapies. Although there is still much to study and
discover, recent remarkable advances in this field have unveiled exciting
therapeutic windows to precisely and effectively target cancer metabolism and
bioenergetics (Figure 3). It is expected that anti-cancer metabolism therapies
will play an important role in clinical oncology within five or ten years.
Figure 1 The impacts of tumor suppressors and oncogenes on cancer metabolic
reprogramming, an important cancer hallmark. Cancer metabolic alterations are
the results of oncogene activation and mutant metabolic enzymes. Cancer
metabolic reprogramming promotes tumorigenesis by facilitating and enabling
rapid proliferation, survival, invasion, metastasis, resistance to therapies and
other central cellular processes of tumorigenesis. On the other hand, as
tumorigenesis advances, cancer cells acquire more mutations and changes that
further enhance metabolic reprogramming and, in turn, accelerate tumor growth,
proliferation and progression. Tumor suppressors, for instance, p53, and AMPK,
exert their suppressive regulation on cancer metabolic alterations by blocking
the function, activation and expression of essential cancer metabolic genes. Our
recent results also show that 14-3-3¦Ò, a downstream target gene of p53,
effectively opposes and reverses cancer metabolic reprogramming. Our data
indicate that 14-3-3¦Ò accelerates the degradation of c-Myc, an important
transcription factor promoting cancer metabolic reprogramming183. In contrast,
oncogenes such as c-Myc, HIF-1¦Á, Ras, and Akt are major inducers of tumor
bioenergetics alterations by upregulating the expression or activation of key
metabolic enzymes such as HK2, GLS1, LDHA, among others. The balance between
tumor suppressors and oncogenes has a decisive impact on the status of cancer
metabolism.
Figure 2 Summary of key changes in cancer metabolic reprogramming. Cancer
metabolic reprogramming is characterized by enhanced glycolysis, PPP, lipid
metabolism, glutaminolysis, mitochondrial biogenesis, among others. These
pathways provide cancer cells with not only essential energy but also important
precursors to support large-scale biosynthesis, rapid proliferation, continuous
growth, tissue invasion, metastasis, survival and resistance to anti-cancer
therapies. For instance, glycolysis generates 2 ATP per glucose consumed and
provides materials for PPP and other biosynthetic programs. Similarly, PPP
supplies tumors with ribose-5-phosphate and NADPH. Ribose-5-phosphate is a major
element for nucleotide synthesis, which is used in DNA replication, RNA
synthesis, and DNA damage repair, among others. NADPH is a key line of defense
counteracting oxidative stress and a crucial metabolite for a number of
biosynthesis reactions. NADPH is produced by 4 biochemical reactions mediated by
G6PD, 6PLGD, ME1 and IDH1. In addition, fatty acid synthesis is indispensable
for formation of new cellular membranes and proliferation. A number of fatty
acid synthesis enzymes such as ACC, ACLY and FASN are upregulated or activated
by oncogenes such as c-Myc, HIF-1¦Á, Akt, among others. On the other hand, FAO is
also important for cancer cells because it generates energy, NADPH and other
necessary metabolites. Fatty acids are imported into mitochondria by CPT1 and
oxidized to generate acetyl-CoA. Acetyl-CoA fuels the TCA cycle to generate NADH
and FADH2. The latter metabolites donate electrons to mitochondrial ETC for ATP
generation. CPT1 also antagonizes Bax and Bad-mediated apoptosis by preventing
the formation of mitochondrial membrane transition pores and reducing cytochrome
c release. Citrate produced by the TCA cycle can be transported from
mitochondria to cytosol. Cytosolic citrate is used in a number of reactions to
produce acetyl-CoA, oxaloacetate and isocitrate. These metabolites are important
for lipid synthesis, NAPDH production, and many other central cellular
processes. Mitochondrial biogenesis is also a striking feature of cancer
metabolic reprogramming. Mitochondria are not only the energy generators but
also the factories for synthesizing many essential metabolites for cancer
growth, proliferation and metastasis. In addition, the metabolic lactate-based
symbiosis is another remarkable characteristic of cancer metabolism. Cancer
cells frequently upregulate LDHA to facilitate the conversion of pyruvate to
lactate. Lactate is then secreted to tumor microenvironment via MCT4
transporters and can be taken by neighboring cancer cell thanks to MCT1
importers. Lactate is thereafter used for other metabolic pathways in tumors.
This metabolic symbiosis facilitates the survival of cancer cells in harsh
conditions. Thus, metabolic reprogramming is a major cancer hallmark. It is
characterized by the upregulation of a number of inter-connected metabolic
pathways providing cancer cells with vital energy and metabolites. This
metabolic plasticity is essentially important because it allows cancer cells to
effectively and rapidly adapt to the rapidly changing conditions of tumor
microenvironment. In addition, the flexibility of cancer bioenergetics also
enables rapid proliferation, continuous growth, invasion, metastasis and
resistance to anti-cancer therapies. Therefore, further knowledge about cancer
metabolic reprogramming is very important for successful development of precise
and efficacious anti-cancer metabolism therapies. Dashed arrows indicate
indirect effects or multi-step processes. Abbreviations: HK2, hexokinase 2;
LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6PGLD,
6-phosphogluconate dehydrogenase; ACC, acetyl-CoA carboxylase; ACLY, ATP citrate
lyase; FASN: fatty acid synthase, SCD, stearoyl-CoA desaturase; CPT, carnitine
palmitoyltransferase; CPT1C, carnitine palmitoyltransferase 1C; PDH, pyruvate
dehydrogenase; PDK, pyruvate dehydrogenase kinase; UCP, uncoupling proteins;
MCT, monocarboxylic acid transporter; ME1, malic enzyme; IDH1, isocitrate
dehydrogenase1; GLS1, glutaminase; GLUD, glutamate dehydrogenase; FAO, fatty
acid oxidation; ETC, electron transport chain; PPP, pentose phosphate pathway;
TCA, tricarboxylic acid cycle; ¦Á-KG, alpha-ketoglutarate.
Figure 3 Summary of the mechanism of several important drug candidates for
anti-cancer metabolism therapies. Phloretin inhibits the import of glucose, a
major source of nutrient for cancer cells. 2DG, 3BrPA, and Lonidamine inhibit
HK2, a rate-limiting step of glycolytic pathway. 3PO blocks PFK1 activation by
inhibiting PFKFB3 (PFK2). FX11 selectively inhibits LDHA, a major metabolic
enzyme of cancer. BPTES and 968 suppress the function of GLS1. GLS1 is a
glutaminolytic enzyme that is highly and selectively upregulated in cancer. DCA
inactivates PDH kinase (PDK), thereby increasing PDH activity and enhances the
conversion of pyruvate to acetyl-CoA and decreases cancer glycolysis. Metformin
blocks energy production of cancer cells by inhibiting mitochondrial complex I,
suppresses lipid and protein synthesis, modulates glycolysis. At the organism
level, by lowering blood glucose concentration, metformin decreases glucose
supply, as well as insulin and insulin-like growth factor signaling availability
for tumor cells. MCT inhibitors impair the metabolic lactate-based symbiosis of
cancer cells. Many other anti-cancer metabolism compounds are under development.
Targeting cancer metabolism is a very promising direction for anti-cancer
therapies. It is expected that inhibitors of tumor metabolism will play an
important role in clinical oncology within five or ten years. These medications
could be used alone or in combination with other current anti-cancer therapies
to increase efficacy. Abbreviations: 2DG, 2-deoxyglucose; 3BrPA,
3-bromopyruvate; HK2, hexokinase 2; PFK1, phosphofructose kinase 1; LDHA,
lactate dehydrogenase A; GLS1, glutaminase 1; DCA, dicholoroacetate; PDH,
pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; MCT, monocarboxylic
acid transporter.
However, the efficacy of anti-cancer metabolism therapies will need to be
carefully evaluated because cancer cells are well known for their metabolic
plasticity and heterogeneity1,2,11,21,119,184. That may enable tumors to bypass
certain inhibition mediated by therapeutic agents. Furthermore, as we have seen
during the past decades, inhibiting individual enzymes or blocking single
pathways seldom leads to effective cancer treatment. Therefore, it is highly
likely that anti-cancer metabolism approaches need to be combined with other
therapies to improve therapeutic effects and clinical outcomes. Further
understanding about cancer metabolic reprogramming is certainly needed for
effective therapy development. Nevertheless, exploiting the unique features and
weakness of tumor metabolism for cancer treatment, detection and monitoring is
clearly a very promising direction.
Cancer metabolic reprogramming: importance, main features, and potentials for
precise targeted anti-cancer therapies | Phan | Cancer Biology & Medicine
http://www.cancerbiomed.org/index.php/cocr/article/view/629/655
¡¡
Metabolic reprogramming: the emerging concept and
associated therapeutic strategies
Go J. Yoshida
Journal of Experimental & Clinical Cancer Research volume 34, Article number:
111 (2015) Cite this article
Abstract
Tumor tissue is composed of cancer cells and surrounding stromal cells with
diverse genetic/epigenetic backgrounds, a situation known as intra-tumoral
heterogeneity. Cancer cells are surrounded by a totally different
microenvironment than that of normal cells; consequently, tumor cells must
exhibit rapidly adaptive responses to hypoxia and hypo-nutrient conditions. This
phenomenon of changes of tumor cellular bioenergetics, called ¡°metabolic
reprogramming¡±, has been recognized as one of 10 hallmarks of cancer. Metabolic
reprogramming is required for both malignant transformation and tumor
development, including invasion and metastasis. Although the Warburg effect has
been widely accepted as a common feature of metabolic reprogramming,
accumulating evidence has revealed that tumor cells depend on mitochondrial
metabolism as well as aerobic glycolysis. Remarkably, cancer-associated
fibroblasts in tumor stroma tend to activate both glycolysis and autophagy in
contrast to neighboring cancer cells, which leads to a reverse Warburg effect.
Heterogeneity of monocarboxylate transporter expression reflects cellular
metabolic heterogeneity with respect to the production and uptake of lactate. In
tumor tissue, metabolic heterogeneity induces metabolic symbiosis, which is
responsible for adaptation to drastic changes in the nutrient microenvironment
resulting from chemotherapy. In addition, metabolic heterogeneity is responsible
for the failure to induce the same therapeutic effect against cancer cells as a
whole. In particular, cancer stem cells exhibit several biological features
responsible for resistance to conventional anti-tumor therapies. Consequently,
cancer stem cells tend to form minimal residual disease after chemotherapy and
exhibit metastatic potential with additional metabolic reprogramming. This type
of altered metabolic reprogramming leads to adaptive/acquired resistance to
anti-tumor therapy. Collectively, complex and dynamic metabolic reprogramming
should be regarded as a reflection of the ¡°robustness¡± of tumor cells against
unfavorable conditions. This review focuses on the concept of metabolic
reprogramming in heterogeneous tumor tissue, and further emphasizes the
importance of developing novel therapeutic strategies based on drug
repositioning.
Introduction
Tumor tissue consists of a heterogeneous cellular population. Stromal cells such
as neurons, vascular endothelial cells, fibroblasts, and macrophages in cancer
tissue drive chemotherapy resistance [1] as well as tumor survival and
progression [2, 3]. Even in pure populations of tumor cells, heterogeneity is
present as a result of genetic mutation and epigenetic modulations. This
cellular heterogeneity can be explained by a hierarchical model, in which cancer
stem-like cells (CSCs) can provide transient amplifying cells and differentiated
non-CSCs involved in establishing the tumor tissue [4, 5]. CSCs possess several
biological features of ¡°stemness¡±, a combination of phenotypes including
plasticity in the transition between quiescent (G0 phase) and proliferative
states [6] and resistance to redox stress and chemotherapeutic agents [7, 8].
Importantly, accumulating evidence suggests that metabolic reprogramming is
crucial in order for CSCs to maintain unlimited self-renewal potential and
hyper-adaptation to drastic changes in the tumor microenvironment [9¨C11].
Intra-tumoral heterogeneity due to the presence of CSCs is primarily responsible
for our inability to induce the same therapeutic effect among cancer cells as a
whole [12, 13]. CSCs are very likely to contribute to the formation of minimal
residual disease (MRD) [1]. The term ¡®MRD¡¯ is most often used in the context of
hematological malignant disorders [14], but the underlying concept is quite
convenient in discussion of clinically undetectable resistant clones after
conventional anti-tumor therapies [1]. Thus, MRD is expected to contribute
prominently to latent relapse and distant metastasis (Fig. 1).
Fig. 1
figure1
Cancer stem cells and MRD formation. Heterogeneous tumor tissue with
combined-modality therapy leads to the formation of MRD, which is clinically
undetectable. Transiently reduced heterogeneity is observed in MRD, which is
enriched in CSCs. Relapse or metastasis results in re-acquisition of a
heterogeneous population that is more potentially aggressive in terms of its
degree of ¡°stemness¡±
Full size image
Aberrant proliferation of cancer cells is supported by enhanced adaptation to
nutrient microenvironment mediated by alterations in energy metabolism.
Consequently, metabolic reprogramming is believed to be one of the hallmarks of
tumor cells in parallel with genomic instability, tumor-provoking chronic
inflammation, escape from the immune system, etc. [5]. Although aerobic
glycolysis, termed the Warburg effect, is a characteristic metabolic feature of
cancer cells [15, 16], recent investigations revealed that other metabolic
features, in particular, the reverse Warburg effect [17, 18], metabolic
symbiosis [19, 20], and addiction to glutamine metabolism [21, 22], create
challenges for anti-cancer treatment due to adaptive or acquired
chemoresistance. This review article focuses on the relationship between
metabolic reprogramming and tumor heterogeneity, as well as on the development
of promising therapeutic strategies by drug repositioning targeting metabolic
reprogramming.
Conventional Warburg effect and emerging concepts
In 1924, Otto Warburg discovered that tumor cells tend to produce large amounts
of lactate from glucose, regardless of the available oxygen level [15, 16]. This
situation is similar to anaerobic glycolysis, implying that oxidative
phosphorylation (OXPHOS) is replaced by glycolysis in normal differentiated
cells under hypoxia [23, 24]. However, cancer cells appear to engage in
glycolytic metabolism before they are exposed to hypoxic conditions [15, 16].
OXPHOS in mitochondria generates as many as 36 mol ATP from 1 mol glucose,
whereas the conversion of glucose to pyruvate or lactate produces only 2 or 4
mol ATP, respectively [25, 26]. It remains unclear why cancer cells largely
depend on this ¡°inefficient¡± metabolic pathway, even when enough oxygen is
available [27, 28]. In striking contrast to normal cells, cancer cells
preferentially uptake and convert glucose into lactate even in the presence of
sufficient oxygen [29]. This seemingly ¡°inefficient¡± metabolic characteristic
relies largely on aberrant upregulation of GLUT1, a glucose transporters
abundantly expressed in cancer cells [30, 31], although one contradictory study
reported that GLUT1 is not necessarily involved in the Warburg effect depending
on the degree of tumor invasiveness [32]. Inefficient ATP synthesis becomes an
obstacle for cancer cells only when their energy resources are scarce. However,
this is not the case in proliferating cancer cells with aberrant angiogenesis
[29]. Tumor cells finely regulate ATP synthesis by regulating substrate uptake,
as well as enzymes related to glycolysis, which enables them adapt to the
nutrient microenvironment [33]. Moreover, the regulation of adenosine
monophosphate-activated protein kinase (AMPK) signal transduction, a sensor of
energy status, is intimately connected to the Warburg effect, one form of
metabolic reprogramming of cancer cells [34, 35]. Indeed, genetic ablation of
AMPK activates mammalian target of rapamycin (mTOR) signal with ectopic
expression of hypoxia-inducible factor-1 alpha (HIF-1 alpha), resulting in rapid
cellular proliferation accompanied by activation of aerobic glycolysis [35].
This strongly suggests the importance of cancer metabolic reprogramming in
maintaining the interaction between the oxygen-sensing transcription factor and
the nutrient-sensing signal pathway.
Metabolic reprogramming in response to chemotherapy
Tumor heterogeneity in regard to mitochondrial metabolism, in seeming
contradiction to the Warburg effect, is considered to induce the diversity in
activated metabolic pathways [36] (Fig. 2). Notably, MRD in several kinds of
cancers is enriched in CSCs, leading to intra-tumoral heterogeneity and poor
prognosis [1, 9, 10, 37]. Non-CSCs of bladder cancer, for instance, release
prostaglandin E2 (PGE2) when they undergo apoptosis during the course of
chemotherapy. PGE2 promotes the awakening of dormant G0-phased CSCs into the
proliferative state [9]. Given that PGE2-mediated metabolic activation in
mitochondria has been demonstrated in non-malignant cells [38], it is possible
that activated CSCs undergo altered metabolic reprogramming (Fig. 3). Similarly,
the survivors after transient depletion of a driver oncogene (i.e., activated
mutant KRAS G12D in pancreatic cancer) tend to depend heavily on OXPHOS in
mitochondria rather than aerobic glycolysis. Comprehensive analysis of metabolic
pathways of survivors after chemotherapy revealed the prominent expression of
genes that regulate mitochondrial function, autophagy and lysosome degradation
activity, as well as a strong reliance on mitochondrial respiration and
diminished dependence on the Warburg effect [10]. Autophagy is a
metabolic-recycling pathway involving proteasome-independent degradation of
cellular components (e.g., old and dysfunctional mitochondria), which is
partially responsible for cancer chemoresistance [39].
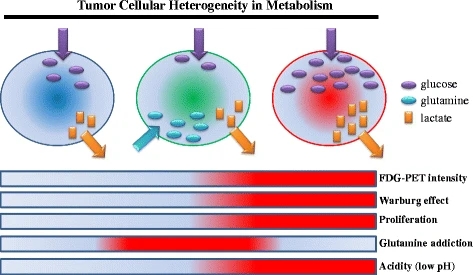
figure2
Tumor heterogeneity in metabolism. The degree of addiction to glucose or
glutamate differs among various types of cancer cells. Tumor cells robustly
importing glucose via the GLUT1 transporter are responsible for the high
intensity of FDG-PET in the clinical settings. Cancer cells that express high
levels of GLUT1 also induce a low-pH acidic tumor microenvironment, thereby
increasing the invasive potential of tumors
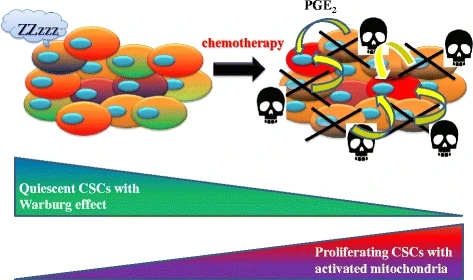
figure3
Iatrogenic activation of CSCs with altered metabolic reprogramming. Non-CSCs are
susceptible to chemotherapy and undergo apoptosis. Released PGE2 awakens the
dormant CSCs localized in the niche. Proliferating CSCs are likely to exhibit
additional metabolic reprogramming, concomitant with up-regulation of
OXPHOS-related molecules
Full size image
Furthermore, malignant melanoma cells that survive and proliferate after
treatment with mutant BRAF (V600E) inhibitor tend to exhibit relative dependence
on mitochondrial metabolism [11]. Because BRAF suppresses oxidative
phosphorylation (OXPHOS), MRD cells up-regulate proliferator-activated
receptor-gamma coactivator-1 (PGC1-alpha). The BRAF (V600E)-MITF-PGC1-alpha axis
promotes the biogenesis of mitochondria and causes BRAF-mutant melanoma cells to
become addicted to mitochondrial metabolism [11]. Because histone H3 lysine 4
(H3K4)-demethylase JARID1B-highly expressing melanoma cells proliferate slowly
and are highly dependent on mitochondrial metabolism [11, 40],
chemotherapy-induced metabolic reprogramming in tumor tissue is likely to be
responsible for the enrichment of CSCs in MRD.
Metabolic interaction driven by tumor heterogeneity
Initially, the concept of Warburg effect was believed to be confined to cancer
cells. More recently, the emerging concept of the ¡°reverse Warburg effect¡±,
however, has attracted considerable attention. Tumor cell-derived reactive
oxygen species (ROS) decrease the expression of caveolin-1 in cancer-associated
fibroblasts (CAFs). CAFs are the major component of tumor stroma, and as such
they express alpha-smooth muscle actin (alpha-SMA) and are widely recognized to
drive tumor progression and metastasis [41]. Loss of caveolin-1 in CAFs results
in elevated ROS levels, which in turn stabilize HIF-1 alpha [17, 42]. In brief,
cancer cells create ¡°pseudo-hypoxic¡± conditions for fibroblasts. Because the
transcription factor HIF-1 alpha promotes glycolysis and provides tumor cells
with lactate and glutamate, elevated production of ROS in cancer cells
indirectly induces uptake of intermediate metabolites of the tricarboxylic acid
(TCA) cycle in mitochondria. CAFs consume more glucose and secrete more lactate
than normal fibroblasts. Furthermore, CAFs depend significantly on autophagy,
and the activation of autophagy in tumor stroma leads to chemoresistance [18,
42] (Fig. 4).
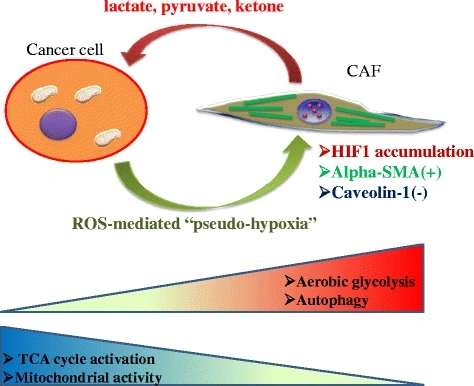
figure4
Interaction of caveolin 1-deficient CAFs with tumor cells. Cancer cells induce a
pseudo-hypoxic microenvironment rich in ROS derived from metabolic
reprogramming. By contrast, CAFs negative for caveolin 1 provide tumor cells
with lactate, pyruvate, and ketone bodies. Notably, although cancer cells depend
heavily on mitochondrial metabolism, CAFs exhibit the Warburg effect and
activation of the autophagic pathway
Full size image
As mentioned above, fibroblasts surrounding epithelial cancer cells undergo
metabolic reprogramming resembling the phenotype associated with the Warburg
effect. Metabolic symbiosis between epithelial cancer cells and CAFs requires
that each cell express a different subtype of monocarboxylate transporter (MCT).
Epithelial cancer cells express MCT1, which contributes to uptake of lactate
provided by caveolin1-null CAFs expressing MCT4 [17, 43]. Tumor cells synthesize
pyruvate from lactate, providing the TCA cycle with an intermediate metabolite.
Notably, an extracellular space rich in lactate reflects acidic conditions,
which in turn lead to the formation of pseudo-hypoxic conditions.
It should be emphasized, however, that this reverse Warburg effect is not
necessarily present in all tumor types. Tumors expressing high levels of MCT4 or
mesenchymal phenotype do not tend to exhibit the reverse Warburg phenomenon.
Instead, cancer cells exhibit hierarchical metabolic heterogeneity:
MCT4-expressing tumor cells perform glycolysis and secrete lactate via MCT4,
whereas MCT1-expressing cells import lactate via MCT1 and perform OXPHOS. In
addition, the amount of glucose uptake is lower in MCT1-positive cancer cells
than in MCT4-positive cells [19, 20] (Fig. 5). This metabolic heterogeneity is
referred to as metabolic symbiosis, and this kind of lactate shuttle is also
observed between neurons and astrocytes in the normal brain tissue [44]. It is
notable that normal and cancerous tissues share finely regulated mechanisms of
metabolic symbiosis.
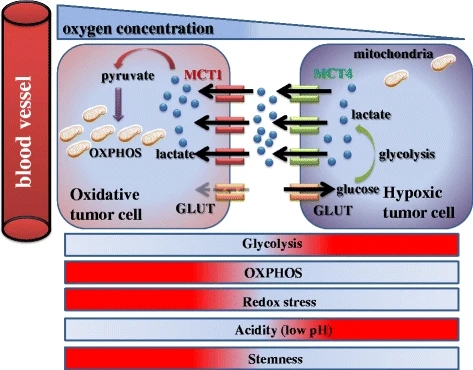
figure5
Metabolic symbiosis between oxidative/aerobic tumor cells and
hypoxic/glycolytic cells. Tumor heterogeneity induces a lactate shuttle between
hypoxic and oxidative cancer cells. While MCT4-positive hypoxic cells contribute
to formation of an acidic microenvironment by aerobic glycolysis and secretion
of lactate, MCT1-expressing oxidative cells utilize lactate as a substrate of
the TCA cycle, and consequently exhibit stem-like characteristics. Notably, in
contrast with MCT1-positive cancer cells, glucose uptake is robust in
MCT4-expressing cells
Cancer stem-like cells in metabolic symbiosis
Importantly, well-oxygenated/aerobic cancer cells expressing high levels of MCT1
efficiently produce metabolic intermediates, as well as ATP, by utilizing
lactate derived from hypoxic/glycolytic cells expressing high levels of MCT4.
Redox stress is a major hallmark of cancer tissues that drives robust metabolism
in adjacent proliferating MCT1-positive cancer cells, which are rich in
mitochondria, mediated by the paracrine transfer of mitochondrial fuels such as
lactate, pyruvate, and ketone bodies [19, 20] (Figs. 4 and 5).
Most importantly, genotoxic stress due to chemotherapy or irradiation, which
increase ROS levels, promotes a CSC-like phenotype [45¨C47]. Because CSCs exhibit
a rapidly proliferating and poorly differentiated phenotype, MCT1-positive
cancer cells are likely to harbor stem-like phenotypes in heterogeneous
populations of tumor cells. After all, activated mitochondrial metabolism
produces enough energy not only for self-renewal by proliferation but also for
invasion/distant metastasis, both of which are activated in CSCs.
Thus, the pharmacological blockage of MCT1 is useful for the treatment of
cancer. MCT1 inhibition disrupts metabolic symbiosis, and MCT1-positive aerobic
cancer cells can no longer uptake lactate [20], which suggests that
MCT1-positive CSCs play a fundamental role in maintaining the hierarchy in tumor
cellular society, in contrast to MCT4-positive cells (Fig. 5).
Acquisition of stem-like and malignant phenotypes with metabolic reprogramming
The cooperation of amino acid transporters is necessary for cancer cells to
undergo metabolic reprogramming and maintain stem-like phenotypes. For example,
triple-negative breast cancer (TNBC) cells, which lack estrogen receptor,
progesterone receptor, and the tyrosine kinase receptor HER2, exhibit addiction
to glutamine metabolism due to coordination between the xCT and ASCT2 amino acid
transporters [48, 49]: xCT uptakes cystine in exchange for glutamine, for use in
GSH synthesis [7], whereas ASCT2 uptakes glutamine in a collaborative manner
[50]. Glutamine is simultaneously imported via ASCT2 transporter and exported in
exchange for leucine via the LAT1/4F2 (CD98 heavy chain) antiporter [48]. The
glutamine uptake pathway contributes to the synthesis of alpha-KG, promoting the
TCA cycle in mitochondria, as well as glutamate, thereby promoting synthesis of
nucleotides required for cellular proliferation [48] (Fig. 6). Thus, metabolic
reprogramming, which is orchestrated by the elevated expression and interaction
of amino acid transporters, contributes to the activation of glutamine metabolic
reprogramming and protects tumor cells against accumulation of oxidative stress
mediated by cystine metabolic reprogramming.
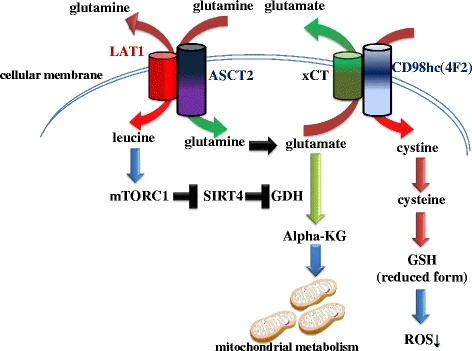
figure6
Metabolic reprogramming of amino acids due to coordinated transporters.
ASCT2/LAT1 and xCT/CD98hc transporter complexes in tumor cells activate the
mTORC1-SIRT4-GDH axis and glutathione synthesis, respectively. The former
pathway promotes conversion of glutamate into alpha-KG, a substrate of the TCA
cycle, whereas the latter pathway maintains redox status
Remarkably, circulating tumor cells (CTCs) that have undergone metabolic
reprogramming provide themselves with a microenvironment that is favorable for
colonization and distant metastasis. Recent work showed that CTCs derived from
colon adenocarcinoma and positive for CD110, the thrombopoietin receptor, can
home to the pre-metastatic niche and colonize metastatic hepatic tissue due to
elevated lysine catabolism [51, 52]. Lysine degradation provides CD110-positive
CTCs with glutamate and acetyl-CoA, which contributes to the synthesis of
anti-oxidant GSH and p300-dependent LRP6 acetylation, respectively [52, 53].
This metabolic reprogramming promotes the metastatic potential of CTCs via a
reduction in ROS levels, elevation of self-renewal potential, and activation of
the Wnt/beta-catenin signal pathway [52]. Thus, CTCs resemble CSCs during the
process of metastasis, at least in terms of the ¡®education¡¯ of the
pre-metastatic niche. Most importantly, this metastatic phenotype is supported
by lysine metabolic reprogramming.
A subpopulation of cancer cells that depend heavily on aerobic glycolysis
robustly uptakes and consumes glucose, whereas another subpopulation engages in
OXPHOS and glutaminolysis with activated mitochondrial metabolism. The
efficiency of lactate production in the former (MCT4-positive) subpopulation is
much higher than in the latter (MCT1-positive) subpopulation, which relies on
OXPHOS and glutamine-derived TCA cycle in the mitochondria [54] (Fig. 5). Thus,
tumor cells tend to decrease microenvironmental pH via elevated lactate
secretion. The acidic tumor microenvironment induces expression of matrix
metalloproteinases (MMPs), especially MMP-2 and MMP-9 [55]. Thus, metabolic
reprogramming remarkably enhances the invasion and metastatic potentials of
cancer cells.
Activation of glutamine metabolism driven by oncogene addiction
Mitochondria plays a much more important role in cancer metabolism than
previously expected, and glutaminolysis is the most common metabolic pathway
regulated in this organelle [56]. Glutaminolysis is the series of biochemical
reactions by which glutamine is catabolized into downstream metabolites, e.g.,
alpha-ketoglutarate (alpha-KG) and glutamate. Via the TCA cycle, alpha-KG
undergoes catabolism to malate, which is transported into the cytoplasm and
converted to pyruvate, and then ultimately to lactate [22]. Mechanistically,
mTORC1 signaling promotes glutamine anaplerosis via upregulation of glutamate
dehydrogenase (GDH) [57]. SIRT4 is a mitochondrial-localized member of the
sirtuin family of NAD-dependent enzymes that play fundamental roles in
metabolism, stress response and longevity [58]. In regard to glutaminolysis,
SIRT4 is a critical negative regulator for glutamine metabolism in mitochondria
[58], which is down-regulated at the transcriptional level when the mTOR
signaling pathway is activated [57]. Thus, mTOR inhibitors such as rapamycin are
expected to block mTORC1-SIRT4-GDH axis, which is essential for glutaminolysis
[57] (Fig. 6).
As mentioned above, tumor tissue consists of a cellular population that is
heterogeneous in terms of dependency on the Warburg effect and mitochondrial
metabolism. Relative to slow-cycling CSCs, proliferative cancer cells tend to
take up a great deal of glutamine, as well as glucose, for the generation of
metabolites [54]. Both aerobic glycolysis and glutaminolysis are frequently
simultaneously activated in malignant cancer cells [36, 59]. Seemingly
paradoxically, however, some cancer cell lines cannot survive and proliferate in
the absence of glutamine, despite the fact that glutamine is a non-essential
amino acids that can be synthesized from glucose [60]. Glutamine is a primary
substrate for the TCA cycle and is required to maintain the redox state via the
production of nicotinamide adenine dinucleotide phosphate (NADPH).
Glutaminolysis enables cancer cells to reduce NADP+ to NADPH, a reaction that is
catalyzed by malic enzymes. NADPH is a required electron donor for reductive
steps in lipid synthesis, nucleotide metabolism, and maintenance of reduced GSH
[21]. In this way, metabolic reprogramming of glutaminolysis enables cancer
cells to regulate redox state.
Oncogenic c-Myc mediates elevation of glutaminolysis in cancer cells. c-Myc
promotes both glutamine uptake and glutamine catabolism [61]. Because of
c-Myc-mediated metabolic reprogramming, cancer cells tend to exhibit ¡°glutamine
addiction¡± [48, 61]. This is a typical example of metabolic reprogramming in
cancer cells with oncogene-addiction [62, 63], suggesting a potential ¡°Achilles¡¯
heel¡± of tumor cells that are addicted to glutamine metabolism in manner that is
mediated by c-Myc.
Therapeutic strategies targeting metabolic reprogramming
Drug repositioning (DR), screening for anti-cancer therapeutic effects of
conventionally administered medications for non-malignant disorders, has
attracted a great deal of attention because the safety and frequency of side
effects of these medicines have been already proven [64]. Proton pump inhibitor
(PPIs), for instance, are acid-activated pro-drugs that inhibit H/K-ATPase
expressed in gastric parietal cells and are conventionally used for the
treatment of gastric ulcer [65]. PPIs have exert synergistic effects on
chemotherapy [66] by modulating the acidic microenvironment [67] or
down-regulating microRNAs involved in chemotherapy resistance [68]. Other
typical examples of DR include sulfasalazine [7, 8, 69], itraconazole [70, 71],
terfenadine [72, 73], and simvastatin [74, 75] are described in Table 1. To
address their anti-tumor therapeutic effects in clinical settings, all of those
drugs are being tested in clinical trials or xenograft experiments.
Table 1 Typical examples of conventional drugs as anti-tumor agents
Full size table
Here, we will describe in detail the potential effects of metformin as an
anti-cancer drug. DR has revealed, for example, that metformin, an oral drug
widely used to treat type 2 diabetes mellitus (DM) [76], prevents tumor growth
and development. A large number of retrospective clinical studies also show that
metformin prevents carcinogenesis and improves clinical prognosis [77¨C79].
Metformin activates AMPK signal transduction, which not only decreases insulin
resistance in type 2 DM [76] but also blocks AMPK-mediated mTOR activation even
in CSCs [77]. mTOR signals are regulated by amino-acid transporters,
characterized by the L-type amino acid transporter 1 (LAT1; SLC7A5) and the
glutamine/amino acid transporter (ASCT2; SLC1A5) [80, 81], which is why the
AMPK-mTOR axis functions as a sensor of dynamic change in the nutrient/growth
factor microenvironment. In particular, leucine uptake via LAT1 activates the
mTOR signal pathway [81, 82] leading to poor prognosis [83, 84]. Because EpCAM
is a functional CSC marker that forms a complex with amino-acid transporters
such as LAT1 [82, 85], it is reasonable that the LAT1 expression level would be
positively correlated with poor prognosis [83, 84]. Therefore, the
LKB1-AMPK-mTOR axis is orchestrated by amino-acid concentration in the tumor
microenvironment, and this axis promotes metabolic reprogramming of cancer cells
in response to the microenvironment.
Remarkably, recent investigations have revealed that this anti-type 2 DM drug
suppresses ectonucleotide pyrophosphatase/phosphodiesterase family member 1
(ENPP1). Consequently, metformin can inhibit the generation of the subpopulation
of cancer cells that express high levels of ABCG2, an ATP-binding cassette (ABC)
transporter responsible for active drug efflux. Mechanistically, the cytosolic
domain of ENPP1 is crucial for interaction with ABCG2 at the cellular membrane;
thus ENPP1 contributes to drug resistance by promoting the stabilization of
ABCG2 [86, 87]. In addition, metformin induces microRNA-27b-mediated suppression
of ENPP1, which reduces chemoresistance and tumor seeding potential [86]. ENPP1
is widely accepted as a cause of insulin resistance in type 2 DM [88],
emphasizing the significance of drug repositioning. Collectively, these
observations indicate that this anti-DM agent is a promising means to attenuate
the malignant behavior of cancer cells, much like other drugs conventionally
administered for non-cancerous diseases.
Conclusions
The complex and dynamic metabolic reprogramming should be regarded as a
reflection of the ¡°robustness¡± of tumor cells against unfavorable conditions.
Hyper-adaptation due to metabolic reprogramming of cancer cells is likely to
give us a great opportunity to attack the ¡°shatter point¡± in heterogeneous tumor
tissue. DR enables us to identify ¡°silver bullets¡± for the treatment of tumor
tissues in metabolically heterogeneous cell populations. To facilitate
development of novel therapeutic strategies, the synergistic effects of
repositioned drugs with conventional anti-cancer agents should be evaluated in
clinical trials in the near future.
Abbreviations
alpha-KG:
Alpha-ketoglutarate
AMPK:
Adenosine monophosphate-activated protein kinase
CAFs:
Cancer-associated fibroblasts
CSC:
Cancer stem-like cell
CTC:
Circulating tumor cells
DM:
Diabetes mellitus
DR:
Drug-repositioning
ECM:
Extracellular matrix
ENPP1:
Ectonucleotide pyrophosphatase/phosphodiesterase family member 1
GDH:
Glutamate dehydrogenase
HIF-1 alpha:
Hypoxic inducible factor-1 alpha
LAT1:
L-type amino acid transporter 1
MCT:
Monocarboxylate transporter
MMP:
Matrix metalloproteinases
MRD:
Minimal residual disease
mTOR:
Mammalian target of rapamycin
NADPH:
Nicotinamide adenine dinucleotide phosphate
OXPHOS:
Oxidative phosphorylation
ROS:
Reactive oxygen species
TCA:
Tricarboxylic acid
Metabolic reprogramming: the emerging concept and associated therapeutic
strategies | Journal of Experimental & Clinical Cancer Research | Full Text
https://jeccr.biomedcentral.com/articles/10.1186/s13046-015-0221-y
¡¡