��
Omega-3֬�����ά����C����NF-kB�ı���
1. vitamin C is kinase inhibitor,dehydroascorbic acid-DHA inhibits IKKalpha and IKKbeta,thus NFkB
2. Omega-3 is the ligand of PPAR-alpha/gamma that inhibit NF-kB signaling
3.NF-��B inhibitors and activators:
Inhibitors:vitamin c,curcumin, EGCG,ginger,vitamin K, omega-3,probiotics
4.chronic stress-induced cancer stem-like phenotype could be reversed by vitamin C
5. Vitamin K Inhibits NF-kappaB Activation
6.NFkB increases expression of ALDH1A, PD-L1,VEGF,polarizs macrophage to pro-tumor M2 subtype,
7.Ginger extract inhibits NFkB
8. Omega-3 Fatty Acids Inhibits NF-kB expression via PPAR��-dependent pathway.
9. fish oil diet significantly increased the level of PTEN protein in the breast tumors.
In addition, the fish oil diet attenuated the PI 3 kinase and Akt kinase activity in the tumors leading to significant inhibition of NF��B activation. Fish oil diet also prevented the expression of anti-apoptotic proteins Bcl-2 and Bcl-XL in the breast tumors with concomitant increase in caspase 3 activity.
10.Docosahexaenoic Acid Induces Cell Death in Human Non-Small Cell Lung Cancer Cells by Repressing mTOR via AMPK Activation and PI3K/Akt Inhibition.
ERBB is abbreviated from erythroblastic oncogene B, a gene isolated from avian genome. It is also frequently called HER2 (from human epidermal growth factor receptor 2) or HER2/neu.
HER2 is a member of the human epidermal growth factor receptor (HER/EGFR/ERBB) family. Amplification or over-expression of this oncogenehas been shown to play an important role ��
Wikipedia �� Text under CC-BY-SA licenseNAC has been shown to be a potent inhibitor of NF-��B activation in vascular endothelial cells (Schubert et al. 2002). NAC and other antioxidants have been reported to inhibit hydrogen peroxide-induced NF-��B activation (Gupta et al. 2010). The central role played by NF-��B signal pathway in physiological and pathological conditions has made it a potential target for pharmacological intervention
Omega-3 polyunsaturated fatty acid promotes the inhibition of glycolytic enzymes and mTOR signaling by regulating the tumor suppressor LKB1
��
Figure 4. Inflammation and NF-��B signaling cross-talk is an important link in brain cancer pathogenesis. The inflammatory cytokines (TNF-��, IL-1��, IL-6, and IL-8) released in the tumor microenvironment generate signals that lead to activation of I��B kinases (IKKs). IKKs induce phosphorylation and subsequent proteosomal degradation of NF-��B inhibitor, I��B��(inhibitor of Kappa B). The resulting free NF-��B (e.g., heterodimer of p65 and p50 subunits) then translocates to the nucleus and activate the transcription of various target genes that encode anti-apoptotic proteins (e.g., B-cell lymphoma 2/ Bcl-2), pro-inflammatory cytokines and chemokines, adhesion molecules (integrins and cadherins), proteases (MMPs), DNA repair proteins such as the MGMT (O6-methylguanine DNA methyltransferase). Many of these regulators contribute significantly to the oncogenesis of gliomas.https://www.mdpi.com/2218-273X/7/2/34/htm
��
��
A model illustrating the signaling pathway of NFkB activation. NFkB, shown here as a heterodimer consisting of p65 and p50 subunits, regulates expression of a number of inflammatory genes. IKK is activated following brain ischemia, causing the phosphorylation of its inhibitor protein, IkB. IkB is phosphorylated by its corresponding kinase (IKK) leading to ubiquitination (ubi) and degradation in proteasomes. This causes the dissociation of NFkB from IkB. Liberated NFkB then translocates into the nucleus, turning on the transcription of its downstream target genes, IKK 5 IkB kinase
��
Docosahexaenoic Acid Induces Cell Death in Human Non-Small Cell Lung Cancer Cells by Repressing mTOR via AMPK Activation and PI3K/Akt Inhibition.
Affiliation: Department of Biochemistry, School of Medicine, Chungnam National University, Daejeon 301-747, Republic of Korea ; Infection Signaling Network Research Center, School of Medicine, Chungnam National University, Daejeon 301-747, Republic of Korea.
��
��
The anticancer properties and mechanism of action of omega-3 polyunsaturated fatty acids (��3-PUFAs) have been demonstrated in several cancers; however, the mechanism in lung cancer remains unclear. Here, we show that docosahexaenoic acid (DHA), a ��3-PUFA, induced apoptosis and autophagy in non-small cell lung cancer (NSCLC) cells.
DHA-induced cell death was accompanied by AMP-activated protein kinase (AMPK) activation and inactivated phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling. Knocking down AMPK and overexpressing Akt increased mTOR activity and attenuated DHA-induced cell death, suggesting that DHA induces cell death via AMPK- and Akt-regulated mTOR inactivation. This was confirmed in Fat-1 transgenic mice, which produce ��3-PUFAs. Lewis lung cancer (LLC) tumor cells implanted into Fat-1 mice showed slower growth, lower phospho-Akt levels, and higher levels of apoptosis and autophagy than cells implanted into wild-type mice. Taken together, these data suggest that DHA-induced apoptosis and autophagy in NSCLC cells are associated with AMPK activation and PI3K/Akt inhibition, which in turn lead to suppression of mTOR; thus ��3-PUFAs may be utilized as potential therapeutic agents for NSCLC treatment.
��ʮ��̼��ϩ��ͨ������AMPK������PI3K/Akt������mTOR���Ӷ��յ��˷�Сϸ���ΰ�ϸ��������
���������Ժͦ�- 3�����֬��������û���(��3-PUFAs)�Ѿ�֤����һЩ��֢;Ȼ�����ΰ��Ļ����в������������,���DZ���,��ʮ��̼��ϩ��(DHA),��3-PUFA���յ�ϸ�������������ڷ�Сϸ���ΰ�(NSCLC)ϸ����
dhad�յ���ϸ������������amp��ĵ���ø(AMPK)�����ʧ�����֬������3-��ø(PI3K)/Akt/���鶯������ù�ذе���(mTOR)�ź�ͨ·���õ�AMPK������Akt������mTOR���ԣ�����DHA�յ���ϸ����������ʾDHA��ͨ��AMPK��Akt���ص�mTORʧ���յ�ϸ������������֬��1֤ʵת����С��,������3-PUFAs����Ұ����С����ȣ�Lewis lung cancer (LLC)����ϸ��ֲ��Fat-1С����������������ữaktˮƽ�ϵͣ�����������ˮƽ�ϸߡ�������������Щ���ݱ���dhad�յ���NSCLCϸ��������������AMPK�����PI3K/Akt�����йأ��Ӷ�����mTOR������;��˦�3-PUFAs���ܱ�������Ϊһ��DZ�ڵ�����ҩ���ڷ�Сϸ���ΰ������ơ�
https://openi.nlm.nih.gov/detailedresult.php?img=PMC4538321_BMRI2015-239764.006&req=4
https://openi.nlm.nih.gov/detailedresult.php?img=PMC4538321_BMRI2015-239764.006&req=4https://openi.nlm.nih.gov/detailedresult.php?img=PMC4538321_BMRI2015-239764.006&req=4
��
��
NF-��B activation by tumour necrosis factor requires the Akt serine�Cthreonine kinase | Nature
https://www.nature.com/articles/43466��
��
Cardiolipin oxidation and cytochrome c release in the mitochondrial... | Download Scientific Diagram
https://www.researchgate.net/figure/Cardiolipin-oxidation-and-cytochrome-c-release-in-the-mitochondrial-pathway-to-apoptosis_fig1_275060022��
��������ø����ֳ�D���������PPAR-alpha����Ҳ��ΪNR1C1���������Ǽ���1��C�飬��Ա1������һ������������PPARA�������ĺ����嵰�ס�[5]���������ø����ֳ�D������ĺ�������ø����ֳ�D�������һ��PPAR-���ǹ�������ø����ֳ�D�������Ǽ����һ���֡�����1990��˹�ٷҡ����֣�Stephen Green����¡��PPAR����ĵ�һ����Ա�����ѱ�ȷ��Ϊ�����������ø����ֳ�Ķ���������ΰ��°���ĺ����塣[6]
����
PPAR-���Ǹ�����֬�ʴ�л��ת¼���Ӻ���Ҫ���ڼ��� PPAR-��������ȱ���������±�������Ҷ�����ͪ�����DZ���ģ���ͪ�����ǶԳ��ڽ�ʳ�Ĺؼ���Ӧ�Է�Ӧ��[7] PPAR-���ļ���ͨ���ϵ���֬�������䣬֬�����Ϻͼ����Լ���������ø���������֬�����-�����йصĻ������ٽ�֬��������գ����úͷֽ��л��[8] PPAR-����Ҫͨ�������϶�������ϳɵ���������������Ƹ�Ѫ֢֬�ı�����ҩ��Լ����ֲ�ͬ��ɱ��������ݼ������ܼ����л��ܼ���ͳ��Ϊ��������ø����ֳ��������Դ���������֬���ᣬ���绨����ϩ���Լ����������֬���ᣬ�Լ�����֬���������Ļ�������绨����ϩ���л���15-�ǻ���ʮ��̼��ϩ������ijЩ��Ա�����硣 15��S��-HETE��15��R��-HETE��15��S��-HpETE��13-�ǻ�ʮ��̼��ϩ�ᣬһ���������л�����֯�ֲ�
PPAR-���ı�����Ѹ������֬�������֯����ߡ������ݶ����У��ڸ������ɫ֬����֯�з�����ߵ�PPAR-��mRNA����ˮƽ���������������ࡣ[9]��С���ʹ����������������з��ֽϵ͵�PPAR-������ˮƽ����PPAR-���ƺ��ڸ�����֯�и�ƽ�ȵر�����ڸΣ���������������и߱��
�ó��о�
ʹ��ȱ��������PPAR-����С����е��о�������PPAR-�����ڱ���Ϊ��������ø����ֳ���ĸ��ֺϳɻ������յ���������ø����ֳ������Ҫ��[10]ȱ��PPAR-����С��Խ�ʳ�ķ�ӦҲ��������������Ҫ�Ĵ�л���ң�����Ѫ��Ѫ����ͪ��ˮƽ�ͣ���Ѫ�Ǻ�֬���Ρ�[7]
ҩ��
PPAR-������������ҩ���ϸ�����壬������ҩ��������Ѫ֬�쳣��һ��ҩ�������ҩ����Ч����Ѫ����������������Ѫ��HDL-���̴�ˮƽ��[11]�����ѹ۲쵽������ҩ�����Ƶ��ٴ��洦��������͡��ҩ����ȣ��������û��ΰ룬�������ǶԱ�����ҩ���ڹ�״�������ಡ�������еĹ㷺Ӧ�óֱ���̬�ȡ� PPAR-�����������ܾ������ƷǾƾ���֬���ε����Ƽ�ֵ�� PPAR-��Ҳ������ijЩ������ҩ�����ò�λ��[12] [13]
���
PPAR-��ͨ���ı�����л���ı�������������ѧ���̡���ˣ�PPAR-���Ĺ�����������л��������ѧ����ֱ����ء�����������о�������PPAR-alpha�л����������֡�[8] PPAR-alpha�ľ���л������PDK4��ACOX1��CPT1����ͨ����ͨ�������������Ѿ�������������ͼ�ף���ͼ��˵����PPAR-��ͨ�������漰֬�ʴ�л����������ڶ�������Ϊ֬�ʴ�л����Ҫ���ڼ������á���Щ���С���������������ĵ�ͼ��PPAR-������Ӱ��֬������ȡ��ϸ���ڽ�ϣ��������-������������ø��֬������������ͪ���ã��������������£��������͵�֭�ϳɵĵ�������/���ڡ���
Omega-3֬���ᣨFAs���ǹ�������ø����ֳ�D������-����PPAR��������Ȼ���壬���Լ���PPAR��
Omega-3 fatty acids (FAs) are natural ligands of the peroxisome proliferator-activated receptor-�� (PPAR��)
Omega-3֬���ᣨFAs���ǹ�������ø����ֳ�D������-����PPAR��������Ȼ���壨Omega-3 fatty acids (FAs) are natural ligands of the peroxisome proliferator-activated receptor-�� (PPAR��)����������һ�ֺ����壬�ɵ��ڲ���֬�ʴ�л�Ļ���ı���ˮƽ�� PPAR�������L162V��̬�����л״�����йء����Ǽٶ���L162��������ȣ���O162-3 FA������Я��PPAR��-V162��λ�������������PPAR������л���ı����ϴ��ڲ��졣������Я�����������ָ����Ե�PPAR��-V162��λ����������L162��������Ե��������Ե�����Ѫ����ϸ���ֻ�Ϊ����ϸ�������ö�ʮ̼��ϩ�ᣨEPA������ʮ��̼��ϩ�ᣨDHA����EPA��DHA�Ļ���������ݱ�����������DHA��EPA��DHA�������L162��������ȣ�PPAR��-V162��λ����Я���ߵ�PPAR������֬����AI��APOA1���Ļ������ˮƽ���Ž��͡����⣬����EPA��DHA������֬����֬��ø��LPL�����������PPAR��L162V��̬�������б��ֳ��ϵ͵����ơ���ˣ�Я��PPAR��-V162��λ����ĸ����������Ӧomega-3 FA���������Ļ�������ʵĸı����ʾ��֬��ˮƽ�ĸ��ơ�Omega-3֬����(FAs)�������Ƕ�ʮ̼��ϩ��(EPA)�Ͷ�ʮ��̼��ϩ��(DHA)������Ѫ���кô���Omega-3֬�����֬�ʴ�л��Ӱ��������ɻ������ĸı�鵼�ġ��������˵,Omega-3֬���ἰ����������(PPAR������Ȼ���壬���ۻ�����ά����a������(R��R)֮ǰ����Ŀ�����ı���[13]���ض��İл������֬����֬��ø(LPL)[15](������������л������ø)����֬����AI (APOAI)[34](���ܶ�֬���Ĺؼ��ṹԪ��)���ܵ���˵,ŷ��٤- 3 FAs��PPAR������п��ܽ���Ѫ����������(TG)ˮƽ������֬�����̴�(���ܶ�֬�����̴�)��ˮƽ��
Omega-3 fatty acids regulate gene expression levels differently in subjects carrying the PPAR�� L162V polymorphism
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2745745/��
ͼ3��PPAR��ͨ��������Ҫ������ת¼����NF��B��AP-1����Ҫ�������ź�ͨ·�����⣬PPAR��ͨ���ϵ���ż������UCP2��UCP3����ROS�鵼����֢���йظ�����ϸ˵������μ��ı�����ͷ��ʾ����/�ϵ��������߱�ʾϸ��������̵�����/�µ��� Erk1 / 2-ϸ�����ź�Ӧ��ø1/2�� I��B-NF��B���Ƽ��� MAPK-�ٷ���ԭ�����ø�� NF��B�������Ӧ�B�� ROS-��������
NFKB mechanism of action - NF-��B - Wikipedia
https://en.wikipedia.org/wiki/NF-%CE%BAB#/media/File:NFKB_mechanism_of_action.pngNF-��B�����Bϸ���ĺ����Ӧ�������ǿ�ӣ���һ�ֵ����ʸ�����ɿ���DNA��ת¼��ϸ�����ӵIJ�����ϸ���� NF-��B�������������ж���ϸ�������У�������ϸ���Դ̼��ķ�Ӧ������Ӧ����ϸ�����ӣ����ɻ����ؽ��������������䣬������LDL�Լ�ϸ������ԭ��NF-��B�ڵ��ڶԸ�Ⱦ�����߷�Ӧ����ؼ����á� NF-��B�����ʵ��방֢�����Ժ����������Լ�������Ѫ֢���ݿˣ�������Ⱦ�����߷��������йء� NF-��BҲ��ͻ�������Ժͼ�������йء�
NF-��B���û��� �ڸ�ͼ�У���Rel��p50������ɵ�NF-��B�������Ϊ��������ʧ��״̬ʱ��NF-��Bλ�������Ƶ���I��B�����ϵİ����ܽ��С�ͨ������Ĥ����Ľ鵼������ϸ�����źſ��Լ���øI��B��ø��IKK���� IKK�����������ữI��B�����ף��Ӷ����·��ػ���I��B����NF-��B�ķ����Լ�����ø�����ս���I��B����Ȼ���NF-��B��λ��ϸ�����У������Ϊ��ӦԪ����RE�����ض�DNA���н�ϡ�Ȼ��DNA /NF-��B������ļ�����������ʣ����繲�������Ӻ�RNA�ۺ�ø�����ǽ�����DNAת¼��mRNA����������mRNA������ɵ����ʣ�����ϸ�����ܸı䡣��
ά����C�����ź���Ӧ��ʾ��ͼ��ά����C��ΪDHAͨ��������ת�˵�����ϸ������Ѹ�ٻ�ԭΪAA�� ROSͨ������IKK������NF-��B�źŴ���Ӧ�𣬶�AA���ROS������IKK�µļ������Щ�����У�AA������DHA����DHA����IKK�¡�
��
��
������Omega-3֬����ͨ��PPAR��������;������NF-��B�
Oxidized Omega-3 Fatty Acids Inhibit NF-��B Activation Via a PPAR��-Dependent Pathway
����Ӳ����Ѫ˨�γ���Ѫ������ѧ��2004;24:1621-1627
ժҪĿ�ġ�
�����о���Ŀ����ȷ�������ĺ���Ȼ��omega-3֬�������������MCP-1��IL-8��Ƥ�����Ӱ�죬���ң������Ч�����������ӱ����ȷ�������������ӱ���Ļ��ơ�
�����ͽ��-
ʹ��ø�����������ⶨ�������DZ���������EPA��DHA����δ������EPA��DHA����ϸ�������յ��ĵ���ϸ���������ף�MCP��-1����Ƥ������ڽ�С�̶�������IL-8���ڵ�ӾǨ���ʱ䶯�����У�������EPA����δ������EPA������Ч����ϸ�������յ�����Ƥϸ��������-��B��NF-��B���Ļ��ʹ�õ�����ӡ�����������DZ�������NF-��B�������Ԥ��I��B�������ữ����ģ���Ϊ������EPA��������ϸ�������յ���I��B�������ữ�ͷ��ػ������⣬������EPA������Ұ����С����Ƥϸ����NF-��B�ļ�����Թ�������ø����ֳ�D���������PPAR����ȱ�ݵ�С������Ƥϸ����NF-��B�ļ���û���������ã�����������EPA��ҪPPAR��������NF-��B��
���ۡ�
��Щ�о����������͵Ŀ������ÿ���������������omega-3֬����ͨ��PPAR��������;����NF-��B��������������¡�
����
��������ǰ���о��У�����ע�������EPA��PPAR������Ч���������������EPA��ϸ������Ƥ������õ�����������ҪPPAR����12��������Ʋ�������EPA��������ϸ�������յ���NF-��B��ͨ��PPAR��������;�������������DZ�������Ȼ������EPA������Ұ����ϸ����ϸ�������յ���NF-��B�Ļ������PPAR��ȱ������Ƥϸ��û���������ã������������EPAͨ��PPAR���鵼�˶�NF-��B���������á� PPAR���鵼������EPA��NF-��B����������ÿ�����PPAR����p50 / p65�ǻ���ֱ������á� Delerive���ˣ�1999�꣩ʹ�ù�����S-ת��ø����ʵ�����������ά���ף�PPAR����������������PPAR����������p65�������������[33]��
������֮ǰ���о�[11,12]һ�����Ƿ���omega-3֬��������������õ������ɾ���ǿ�������Ե������������Щ����������ƴ���Ӧ�����ϸ��ճ��������������ӵı��������omega-3֬����Ŀ������õ��������������ͨ��PPAR��������;������NF-��B�� ��-3֬����������ܿ��ܷ�����������ø������NADPH����ø�����������ø��������ø��֬����ø��������������Ļ����֢��������Щ�������������������PUFA����Щ��Ʒ�ļ����ɲ���������ЧPPAR���������Ϳ�NF-��B���Ե���Ч���Ͷ�������Ӧ���Ƽ��������������Լ�������
Oxidized Omega-3 Fatty Acids Inhibit NF-��B Activation Via a PPAR��-Dependent Pathway
Archana Mishra , Ashok Chaudhary , and Sanjeev Sethi Originally published1 Jul 2004https://doi.org/10.1161/01.ATV.0000137191.02577.86
Abstract
Objective�� The aim of this study was to determine the effects of oxidized versus native omega-3 fatty acids on the endothelial expression of chemokines MCP-1 and IL-8, and, if effective in inhibiting chemokine expression, to determine the mechanism for the inhibition of chemokine expression.
Methods and Results�� Using enzyme-linked immunosorbent assays, we show that oxidized EPA and DHA but not unoxidized EPA or DHA inhibit cytokine-induced endothelial expression of monocyte chemoattractant protein (MCP)-1 and, to a lesser extent, IL-8. In electrophoretic mobility shift assays, oxidized EPA but not unoxidized EPA potently inhibited cytokine-induced activation of endothelial nuclear factor-��B (NF-��B). Using Western blot analyses, we show that the inhibition of NF-��B activation was not caused by prevention of phosphorylation of I��B�� because oxidized EPA did not inhibit cytokine-induced phosphorylation and ubiquination of I��B��. Furthermore, oxidized EPA inhibited NF-��B activation in endothelial cells derived from wild-type mice but had no inhibitory effects on NF-��B activation in endothelial cells derived from peroxisome proliferator-activated receptor �� (PPAR��)-deficient mice, indicating that oxidized EPA requires PPAR�� for its inhibitory effects on NF-��B.
Conclusions�� These studies show that the antiinflammatory effects of fish oil may result from the inhibitory effects of oxidized omega-3 fatty acids on NF-��B activation via a PPAR��-dependent pathway.Discussion
Omega-3 fatty acids in fish oil has been reported to improve the prognosis of several chronic inflammatory diseases characterized by leukocyte accumulation, including atherosclerosis, inflammatory bowel disease, rheumatoid arthritis, psoriasis, etc.1�C4
Omega-3 fatty acids, such as EPA and DHA, are highly polyunsaturated and readily undergo oxidation at ambient and subambient temperatures, even in the absence of exogenous oxidizing reagents.5,6 In view of the ease with which omega-3 PUFA spontaneously oxidize and in vivo data suggesting extensive accumulation of oxidation products after fish oil consumption, we investigated the possibility that oxidized omega-3 fatty acids may be an important component of the observed antiinflammatory effects of fish oil. In our previous studies, we showed that oxidized EPA and not unoxidized EPA pretreatment of HUVEC inhibits leukocyte adhesion to cytokine-stimulated HUVEC, and this effect is mediated through a PPAR��-dependent pathway.11,12 In our present studies, we extend these observations and show that oxidized EPA is also effective in inhibiting cytokine-induced endothelial chemokine expression, particularly MCP-1, and propose a mechanism for these antiinflammatory effects.
The fact that oxidation of the omega-3 fatty acids is required for the aforementioned antiinflammatory effects is also pointed out by other studies. Nohe et al and De Caterina et al have shown that prolonged incubation of endothelial cells with native omega-3 fatty acids (26 hours of total exposure, 6 hours before and 20 hours after TNF�� stimulation) results in inhibition of cytokine and adhesion molecule expression, whereas shorter incubation periods (6 hours) has no effect.30�C32 This suggests that oxidation of omega-3 fatty acids takes place during the long incubation period, and that the oxidation product(s) and not the native omega-3 fatty acids are most likely responsible for the inhibition of leukocyte�Cendothelial interactions.
To ascertain that the inhibition of chemokine expression was not caused by the cytotoxic effects of oxidized EPA, we performed MTT assays, which showed that oxidized EPA in the doses used for our experiments had no effect on the viability of endothelial cells (data not shown). Also, oxidized EPA did not have any effect on the constitutively expressed surface proteins such as von Willebrand factor, endoglin, and human leukocyte antigen class I molecules.11,12
The expression of MCP-1 and IL-8 is regulated through activation of NF-��B. NF-��B activation appears to be necessary for the induction of chemokine genes, and deletion of NF-��B binding sites results in an inability to induce these genes.26 In these studies, we show that oxidized EPA and not unoxidized EPA inhibits cytokine-induced activation of NF-��B and promotes cytosolic retention of the p50 and p65 subunits. Hence, the oxidized EPA-mediated inhibition of MCP-1 and IL-8 in cytokine-stimulated endothelial cells could be explained by an inhibitory effect of oxidized EPA on NF-��B activity.
The difference in the extent of inhibition of MCP-1 and IL-8 expression by oxidized EPA is most likely caused by the differential effects of oxidized EPA on endothelial NF-��B and AP-1 activation. Oxidized EPA almost completely inhibits endothelial NF-��B activation, whereas it had minimal effects in preventing AP-1 activation. Because AP-1 activation alone can result in IL-8 expression,28 oxidized EPA had only a mild inhibitory effect on IL-8 expression.
What is the mechanism for the oxidized EPA-mediated inhibition of cytokine-induced NF-��B activation? We first hypothesized that oxidized EPA inhibits phosphorylation of I��B�� by inhibiting the IKK (kinase) complex. However, it is unlikely that oxidized EPA inhibits IKK (kinase) activity because phosphorylation and ubiquination of IkB�� is noted in oxidized EPA-treated endothelial cells. In fact, when normalized to actin controls, oxidized EPA pretreatment before TNF�� stimulation for 60 minutes resulted in 30% more p-IkB�� when compared with TNF��-treated cells. Also, it unlikely that oxidized EPA induces IkB�� expression because after 15 minutes of cytokine stimulation, no IkB�� is noted in the cytoplasm of cells pretreated with oxidized EPA. After 60 minutes, I��B�� starts to appear in the cytoplasm of oxidized EPA pretreated cells and its concentration is, in fact, somewhat less than the TNF��-treated cells. Thus, it is likely that oxidized EPA does not prevent NF-��B activation by increasing the expression of I��B��.DISCUSSION
In our previous studies we noted that oxidized EPA is a potent activator of PPAR�� and that PPAR�� is needed for the inhibitory effects of oxidized EPA on leukocyte�Cendothelial interactions,12 which lead us to hypothesize that oxidized EPA might inhibit cytokine-induced NF-��B activation through a PPAR��-dependent pathway. Here, we show that although oxidized EPA inhibits cytokine-induced NF-��B activation in wild-type cells, it has no inhibitory effects in PPAR��-deficient endothelial cells, suggesting that oxidized EPA mediates its inhibitory effects on NF-��B through PPAR��. The PPAR��-mediated inhibitory effects of oxidized EPA on NF-��B activation are possibly through direct interactions of PPAR�� with the p50/p65 subunits. Delerive et al (1999) have shown, using glutathione S-transferase pull-down experiments, that after fibrate (PPAR�� agonist) treatment, PPAR�� physically interacts with p65 in vitro.33
Taken together with our previous studies,11,12 we show that auto-oxidation of omega-3 fatty acids results in the generation of oxidized compounds with potent antiinflammatory properties that inhibit proinflammatory responses such as leukocyte adhesion receptor and chemokine expression. The central theme for the antiinflammatory effects of oxidized omega-3 fatty acids is likely through inhibition of NF-��B via a PPAR��-dependent pathway. The oxidation of omega-3 fatty acids is likely to occur in areas of active inflammation caused by increased expression of oxidative enzymes (eg, NADPH oxidase, myeloperoxidase, cyclooxygenase, lipoxygenase) and the generation of reactive oxygen species in these areas, which are capable of oxidizing PUFAs. The identification of these products could result in potent, low-toxicity, proinflammatory response inhibitors with potent PPAR�� agonist and anti-NF-��B properties for the treatment of inflammatory diseases.
��Oxidized Omega-3 Fatty Acids Inhibit NF-��B Activation Via a PPAR��-Dependent Pathway | Arteriosclerosis, Thrombosis, and Vascular Biology
https://www.ahajournals.org/doi/10.1161/01.atv.0000137191.02577.86��
��
�ڷ�����Omega3֬��������������ܰ�ϸ����Ѫ����CLL��ϸ���е�NFkB�
Oral Supplementation with Omega3 Fatty Acids Inhibits NFkB Activation In Chronic Lymphocytic Leukemia (CLL) Cells.
Oscar F. F Ballester
��飺�����Ӧ�B��NFkB���Dz���CLLϸ�������ʹ��Ĺؼ�ת¼���ӡ� NFkB����Ϊ�ǿ����������Ƹ��ֶ������������Ʒ�����ҪĿ�ꡣ�������ʵ�鶯��ģ���У���֤������OMEGA-3֬���ᣨO3FA��������NFkB���ԡ����ߺͷ�������������CLL��Rai 0-II�ڣ����������ƵĻ��ߣ��û���������I-II�����顣 O3FA������ܹ�����12���£�������ΧΪ2250 mg��EPA��DHA����ÿ�տ����ܵļ������ӵ�4500 mg��6750 mg����ͨ���ݶ����ķ��뵥����ϸ����������Ѫ��Ʒ�в���NFkB���ԣ����Է��ⵥλ/��g�����ʱ�ʾ�����о��ڼ��û��ߺͶ��ϵ����Ʒ��ʹ�ñ���LD50������ù�ؽ�������ϸ���������顣ͨ������ɫ������ϸ�����ܰ�ϸ��Ĥ֬���������������ԡ�
�����15�����߱���������飬����8��Ŀǰ�Ѿ�����˼ƻ����о���12���¡�û�з��ּ�����������ٴ��仯�� O3FA�����������á����䵼�º�ϸ�����ܰ�ϸ��Ĥ��O3FA�ɷֳʼ������������ӡ��ڻ���ʱ��CLL���ߵ�NFkB�������������߹۲쵽�ķ�Χ��2.05��104��2.32��105 NFkB������λ/�ˣ�������ʱCLL���ߵ���λ��Ϊ11.60��106
��Oral Supplementation with Omega3 Fatty Acids Inhibits NFkB Activation In Chronic Lymphocytic Leukemia (CLL) Cells.
Oscar F. F Ballester
Introduction: Nuclear factor kappa B (NFkB) is a critical transcription factor involved in the growth and survival of CLL cells. NFkB is recognized as an important target for the development of novel therapies for the treatment of various malignancies. In vitro and in experimental animal models, OMEGA-3 fatty acid (O3FA) supplementation has been shown to inhibit NFkB activity. Patients and Methods: Patients with early stage CLL (Rai stages 0-II) who required no therapy, where accrued to this phase I-II trial. O3FA supplements were given for a total of 12 months at doses ranging from 2250 mg (EPA plus DHA), escalated to 4500 mg and 6750 mg per day as tolerated. NFkB activity was measured in peripheral blood samples after separation of mononuclear cell by gradient centrifugation and expressed as luminescence units/�� g of protein. Baseline and multiple serial samples were obtained during the study period. In-vitro cytotoxicity assays to doxorubicin were conducted using standard LD50 methods. Compliance was monitored by analysis of red cell and lymphocyte membrane lipid composition by gas chromatography. Results: Fifteen patients have been accrued to the trial, 8 of them have currently completed the planned 12 months of the study period. No significant clinical changes in disease activity were noted. O3FA was well tolerated. Supplementation resulted in a dose-dependent increase of O3FA composition of red cell and lymphocyte membranes in a dose dependent manner. At baseline, CLL patients had NFkB above the range observed in normal controls (2.05 �� 104 to 2.32 �� 105 NFkB lum units/�� g). The median value in CLL patients at baseline was 11.60 �� 106
Oral Supplementation with Omega3 Fatty Acids Inhibits NFkB Activation In Chronic Lymphocytic Leukemia (CLL) Cells. | Blood | American Society of Hematology
https://ashpublications.org/blood/article/116/21/4607/66865/Oral-Supplementation-with-Omega3-Fatty-Acids��
NF-��B inhibitors and activators
Use this guide to find the right compound to activate or inhibit the NF-��B signaling pathway.
Multiple pharmacological compounds can activate or inhibit proteins involved in NF-kB signaling regulation. The table below highlights some of the small molecules that can be used to study NF-kB pathway.
��
Antioxidants
Antioxidants such as PDTC40 and NAC41 have shown potential to inhibit NF-��B activation either by exogeneous induction (e.g. LPS, TNF��) or hydrogen peroxide treatment. Antioxidants are likely inhibit NF-��B by scavenging reactive oxygen intermediates involved in the NF-��B pathway42.
Anti-inflammatory and immunosuppressant drugs
The commonly available NSAID, sodium salicylate was shown to bind IKK��43 and inhibit proteasome activity44 potentially reducing I��B degradation. A potent immunomodulatory glucocorticoid steroid, dexamethasone (DEX) exhibited interference with NF-��B activation and reduced TNF�� production45,46.
Investigations into immunosuppressant drugs revealed cyclosporin A (CsA) to inhibit NF-��B /RelA activation and block IL-2 and IL-8 gene expression47,48. FK506 (tacrolimus) another commercially available immunosuppressive drug blocks p50 nuclear translocation thereby reducing activation of its subsequent promoters and gene expression of inflammatory cytokines such as IL-249.��
NF-kB inhibitors and activators | Abcam
https://www.abcam.com/reagents/nf-kb-small-molecule-guide��
Two faces of PPAR��/NF��B signaling pathway in inflammatory responses to adipocytes lipolysis in grass carp Ctenopharyngodon idella
College of Animal Science and Technology, Northwest A&F University, Yangling, 712100, China
Highlights
•
Lipolysis has a dose-dependent differential impact on inflammatory response in adipocytes.
•
PPAR�� inhibited NF-��B signaling pathway in grass carp adipocytes.
•
Excess lipolysis produces ROS that contributes to adipocyte inflammation.
Abstract
Adipose tissue plays an important role in energy reservation, also be considered as vital immunological organ in animals. Adipocytes are the basic unit of adipose tissue, while little is known about the relationship between lipid metabolism and inflammatory response in fish adipocytes so far. In this study, forskolin was used to induce adipocyte lipolysis, and 5 ��M forskolin and 30 ��M forskolin both triggered lipolysis by increasing ATGL expression. Consequently, 30 ��M Forskolin instead of 5 ��M Forskolin induced the expression of NF-��B and its target pro-inflammatory cytokine genes including MCP-1, IL-6 and TNF-��. Further study found that low grade rate of lipolysis activated PPAR�� gene, and its inhibitory effect on the mRNA expression of NF-��B and its target genes inhibited the adipocyte inflammation. On the contrary, high grade rate of lipolysis increased the expression levels of NF-��B and its target genes, while their expression were attenuated by inhibition of reactive oxygen species (ROS) using ��-tocopherol, suggesting that ROS generated due to the PPAR��-mediated oxidation of released fatty acids from lipolysis may contribute to adipocyte inflammation. These results indicated that PPAR�� has dose effect in inflammatory responses to adipocyte lipolysis in grass carp. Taken together, grass carp adipocytes have immune activity. The inflammatory response is linked to the grade rate of adipocyte lipolysis in grass carp adipocytes, and excessive adipocyte lipolysis may promote a dynamic immune response in adipose tissue. This is the first study showing the regulatory effects of lipolysis on immune functions in fish adipocytes.
��
��
Two faces of PPAR��/NF��B signaling pathway in inflammatory responses to adipocytes lipolysis in grass carp Ctenopharyngodon idella - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S1050464819303006��
��
NF-��B�źŴ����������������������е�����
NF-kappaB Signaling in Chronic Inflammatory Airway Disease
ͨ�����˶��������ӣ�Michael Schuliga��
ī������ѧ�β������о����ģ�LHRC����ī������ѧGrattan St.��Parkville 3010���Ĵ�����ά������
����
���������������Էμ�����COPD�����������������������������ͱ��Ͳ�ͬ����ҩ�����Ʒ�ʽ�ص�����������COPD�У�����Ӧ��ͨ���յ���֢������������������֢��������ԭ���е�ת¼���ӣ������ӣ�NF��-��B��NF-��B�����ǵ��ں�����������ϸ�����ӻ��Եĸ�����֢�������Ҫ�����ߡ���Ƥ�ʼ��أ�GCs������������Ҫ����ҩ��Ŀ��������漰����NF-��B�յ��Ļ���ת¼�������ϵ�GC���壨GRs����NF-��B���������NF-��B��Ӧ�����ת¼������ת¼����Ȼ�������ض�������COPD�У����ڷ����GR��Ⱦɫ�������鵰�ı仯��GC���µ�NF-��B�ķ�ʽ���ﱻ������NF-��B����Ʒ�������I��B��ø��IKKs�����Ƽ���������������COPD��DZ���Ʒ������⣬��תGR /�鵰����������ʾ������ͨ����ǿNF-��B������������̴�������������������ƪ���������������֢�е�NF-��B�źŴ���������Ϊ����������COPD�б��DZ����
��NF-kappaB Signaling in Chronic Inflammatory Airway Disease
by Michael Schuliga
Lung Health Research Centre (LHRC), Department Pharmacology and Therapeutics, University of Melbourne, Grattan St., Parkville 3010, Victoria, Australia
��
Abstract
Asthma and chronic obstructive pulmonary disease (COPD) are obstructive airway disorders which differ in their underlying causes and phenotypes but overlap in patterns of pharmacological treatments. In both asthma and COPD, oxidative stress contributes to airway inflammation by inducing inflammatory gene expression. The redox-sensitive transcription factor, nuclear factor (NF)-kappaB (NF-��B), is an important participant in a broad spectrum of inflammatory networks that regulate cytokine activity in airway pathology. The anti-inflammatory actions of glucocorticoids (GCs), a mainstay treatment for asthma, involve inhibition of NF-��B induced gene transcription. Ligand bound GC receptors (GRs) bind NF-��B to suppress the transcription of NF-��B responsive genes (i.e., transrepression). However, in severe asthma and COPD, the transrepression of NF-��B by GCs is negated as a consequence of post-translational changes to GR and histones involved in chromatin remodeling. Therapeutics which target NF-��B activation, including inhibitors of I��B kinases (IKKs) are potential treatments for asthma and COPD. Furthermore, reversing GR/histone acetylation shows promise as a strategy to treat steroid refractory airway disease by augmenting NF-��B transrepression. This review examines NF-��B signaling in airway inflammation and its potential as target for treatment of asthma and COPD.
Keywords: airway smooth muscle; allergic asthma; alveolar macrophages; cigarette smoke; eosinophils; epithelium; emphysema; histone deacetylase; phosphoinositide 3 kinase-delta (PI3K-��); sirtuins
��
Biomolecules | Free Full-Text | NF-kappaB Signaling in Chronic Inflammatory Airway Disease
https://www.mdpi.com/2218-273X/5/3/1266��
ά����C��һ�ּ�ø���Ƽ������⿹��Ѫ������I��B����ø�£�����NF-kB�ļ���
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits I��B�� Kinase ��
Department of Clinical Laboratories,1 Program in Molecular Pharmacology and Chemistry, Memorial Sloan-Kettering Cancer Center,2 Structural Biology Program, Department of Physiology and Biophysics, Mount Sinai School of Medicine, New York, New York3
*Corresponding author. Mailing address: Program in Molecular Pharmacology and Chemistry, Box 451, Memorial Sloan-Kettering Cancer Center, New York, NY 10021. Phone: (212) 639-8483. Fax: (212) 772-8589. E-mail: gro.ccksm.iks@edlog-d.
ABSTRACT
Reactive oxygen species (ROS) are key intermediates in cellular signal transduction pathways whose function may be counterbalanced by antioxidants. Acting as an antioxidant, ascorbic acid (AA) donates two electrons and becomes oxidized to dehydroascorbic acid (DHA). We discovered that DHA directly inhibits I��B�� kinase �� (IKK��) and IKK�� enzymatic activity in vitro, whereas AA did not have this effect. When cells were loaded with AA and induced to generate DHA by oxidative stress in cells expressing a constitutive active IKK��, NF-��B activation was inhibited. Our results identify a dual molecular action of vitamin C in signal transduction and provide a direct linkage between the redox state of vitamin C and NF-��B signaling events. AA quenches ROS intermediates involved in the activation of NF-��B and is oxidized to DHA, which directly inhibits IKK�� and IKK�� enzymatic activity. These findings define a function for vitamin C in signal transduction other than as an antioxidant and mechanistically illuminate how vitamin C down-modulates NF-��B signaling.ά����C��һ�ּ�ø���Ƽ������⿹��Ѫ������I��B����ø��
ժҪ
��������ROS����ϸ���ź�ת��ͨ·�еĹؼ��м��壬�书�ܿ��ܻᱻ������������������Ѫ�ᣨAA���䵱�����������ṩ�������Ӳ������������⿹��Ѫ�ᣨDHA�������Ƿ���DHA������ֱ������I��B����ø�£�IKK�£���IKK����ø���ԣ���AA��û���������á�����ϸ���м���AA��ͨ�����ﱾ������IKK�µ�ϸ���е�����Ӧ���յ�����DHAʱ��NF-��B�����ܵ����������ǵĽ��ȷ����ά����C���ź�ת���е�˫�ط������ã�����ʾ��ά����C��������ԭ״̬��NF-��B�ź��¼�֮���ֱ����ϵ�� AA�������NF-��B���ROS�м��壬����������DHA��ֱ������IKK�º�IKK����ø���ԡ���Щ���ֶ�����ά����C���źŴ����еĹ��ܣ�������Ϊ���������⣬���ӻ����ϲ�����ά����C����µ�NF-��B�źŴ�������
��ʳ�е�ά����C�����࣬�鳤�ද��������������ȱ��l-����ŵ-��-��������ø�������������Ǵ�����ϳ�;���е�����ø���Ķ��������������Ҫ��25���������������£�ά����C��Ҫ�Ի�ԭ��ʽ���ڣ�������Ѫ�ᣨAA����������������ʽ���⿹��Ѫ�ᣨDHA�������ڡ���������֪������ά����C�Ļ��ƣ�21��������������ϸ���е�ͨ��ϵͳͨ��������������ת�˵���ά����C��ΪDHAת�ˣ�34��������ϸ����DHA��Ѹ�ٻ�ԭ����AA����ʽ�ۻ���34��35�����ڶ���ת��ϵͳ��ר�ŵ�ϸ���������ã�����AAͨ����������AA��ת�˵��ף�SVCT1��/��SVCT2��ֱ��ת�˵�ϸ���У�33����
AA�ǽ�ԭ���ף�26������28����ȥ���������أ�18���Լ�������������8��������ϳ�ø�ĸ������ӡ���Ѫ����ϸ���У�AA��һ��ǿ��Ŀ����������ɴ����������ROS���ͻ��Ե���10��14����ϸ����ά����C����Ԥ��ϸ����������������Ӧ���յ���ͻ�䣨12��22��37�����ڴ������ɻ��Ĺ����У�����Ѫ����ṩһ�����ӣ���Ϊ���ȶ����м��忹��Ѫ�����ɻ�������ػ�ԭΪ����Ѫ�ᡣ����Ѫ����ſ����ṩ�ڶ������Ӳ�ת��ΪDHA��13��14���� DHA���ܻ�ԭΪAA�����ˮ��Ϊ2,3-��ͪ-�������ᣬȻ��лΪ������Ͳ��ᣨ14������װ��AA����¶�ڹ��������ϸ���У�AAת��ΪDHA������һЩͨ��������ת�˵��״�ϸ�����������Ӷ��ṩ��һ�ֽ�ά����Cѭ����ϸ�����������еĻ��ƣ�12�������ߣ��ɽ�ϸ����DHAת����ϸ�������Һ�ϸ������2��20���� DHA��Ҫ��ά����C����������ʽ�����ã�36����ROS��Ϊ��ѧ�ڶ���ʹ������ϸ����Ӧ����ؼ����ã���֮��������������ѡ�����źŷ�Ӧ��24�������磬ROS����ת¼���ӣ�����NF-��B����������������֢�͵����к���Ҫ��1��11��32��������ϸ�����ӣ����������������Ӧ���TNF-�����������������������ͨ���յ�I��B�������ữ������NF-��B��11��19�������ữ��I��B���ͷ�NF-��B����ͨ������ø��;���������⣨17������δ���ữ��I��B������е�NF-��B��ϣ���ֹ���Ǩ�ơ�����б���˵��AAͨ������p38˿��ԭ��ĵ���ø��MAPK��������Ƥϸ����TNF-���յ���NF-��B�Ļ��4�������ǣ������������AAͨ��������I��B�����ữ�йصļ�ø�ļ���������TNF-�������Ե�NF-��B���6����
�����о���ά����C��NF-��B����ĵ��ڣ�����DHA�������ϸ��������ֱ��������IKK�º�IKK���ļ�ø���ԡ���ˣ����ǵ����ݱ���ά����C����NF-��B���ܵ�˫�����û��ơ����ȣ���Ϊ�����������ROS��AA����ROS�鵼���ź�ת���¼�����Σ���������DHA��ά����Cֱ������IKK��ø���ԡ����Ϻͷ���
ά����C���ء�
��ǰ������6����HeLaϸ����װ��ά����C�������֮����ϸ�����������Һ��15 mM HEPES [pH 7.4]��135 mM NaCl��5 mM KCl��1.8 mM CaCl2��0.8 mM MgCl2����pH 7.4������30���ӣ�Ȼ���ò�ͬŨ�ȵ�DHA����30��������ͬ����Һ����37��C����3���ӡ� DHA����Sigma��������������ʥ·��˹����ͨ����AA�뿹��Ѫ������ø��Sigma������ø�ٲ�����
����ӡ��������
��ǰ�����Ʊ�ϸ����ȡ�6����ʹ�������ö��¡�������������ӡ�������������ữI��B������I��B������p38 MAPK�������ữp38 MAPK�������ữ��p44 / 42 MAPK��Cell Signaling Technology��Beverly��MA������p44 / 42 MAPKs��ŦԼ���������º���Upstate Biotech���Ϳ�FLAG���壨Sigma������Ĥ��������������øż���Ŀ���������G����һ����������ʹ����ǿ�Ļ�ѧ����ⶨ����Amersham Pharmacia Biotech��Piscataway��N.J������ʾ�����ʡ�
תȾ��ӫ����ø�ⶨ��
��pNF��B-luc��Clontech��Palo Alto��CA��˲ʱתȾHeLaϸ�������뺬IKK�£�SS / EE��������Ի���IKK�µ�˿����177��181�ֱ𱻹Ȱ����������������תȾͨ������Ʒ���Superfect��QIAGEN���������ǣ����������ǣ�����ϸ����ÿ����30 ngTNF-��һʽ���ݷ�����ʱ��Ϊͼ����ʾ��ʹ��ө����ø�������ⶨϵͳ��Promega���ⶨө����ø���ԡ���ø�ⶨ��
ͨ���ÿ�IKK�¿��壨Santa Cruz Biotechnology��Santa Cruz��CA���������߳�����TNF-��������ϸ����ȡ���з������Դ��IKK�¡��ӿ�FLAG�����飨Sigma����תȾHeLaϸ����ȡ���з����IKK�£�SS / EE������ͻ���塣��תȾ��293Tϸ���з��������IKK����IKK�¡���GenejammerתȾ�Լ���Stratagene��La Jolla��CA���ú���IKK����IKK�µ�����˲ʱתȾϸ���� 48Сʱ���ջ�ϸ������ͨ�����߳������뼤ø����30��C���ں���10��MATP��1��Ci[��-32P] ATP��1��g����Ļ���Һ����30��C��15���Ӻ�ⶨ��Դ��IKK�£�����IKK����IKK�º�IKK�£�SS / EE���ļ�ø���ԣ�������S-ת��ø[GST]-I��B������ͨ����������Ӱ������ữ�ĵ��6����ʹ�ô�����GST�ں�ø�����ữ���Ⱥ����ȶ�����1��PHAS-1����Ϊ���Calbiochem����������MAPK��p38���Ļ��ԣ������������̵�ָʾ���м�ø��Ӧ��Calbiochem ����
����DHA��ϲⶨ��
ͨ���ÿ�FLAG��������߳�������תȾ��HeLaϸ���з����IKK�£�SS / EE������ͻ���塣��Я��IKK�µ������ڼ�ø����Һ����6 mM [14C] DHA��6 mM [14C] AA��30��C����15���ӡ�ͨ����[14C] AA�뿹��Ѫ������øһ����������ڷ�������Һ��pH 5.5��������[14C] DHA����FLAG�Ĵ�������ϴ�Ѽ�ø������Beckman LS 6000LL����������صķ����Խ��м�����
ROS��⡣
��HeLaϸ���ں���0.2��ţѪ�������Ѫ���������б���16Сʱ��Ȼ����ϸ���м���ά����C��ϴ�ӣ�����ÿ����50 ngTNF-������10���ӡ�Ȼ��ϸ����20��M2'��7'-���ȶ���ӫ���ض���������DCF����37��C����10���ӣ���ʹ���䱸��Qimaging digital��Olympus BX60������40x��DP40��WIBA����ӫ����ӻ�ROS�������豸�����Retiga 1300C����QCapture������
�����ձ䡣ʹ��Stratagene��QuikChange XL�����ձ��Լ��а��������̵�˵�����ж����ձ䡣 ȥ�����ϸ����ά����C����TNF-���յ���NF-��B�������ʹ��HeLaϸ����Ϊģ��ϵͳ���о���ά����C����NF-��B��ķ��ӻ��ơ�ͨ����ϸ����¶��DHA��ʹ�主��ά����C��6�����ó���������֯����������AA�Ĵ�����������ص�αӰ��7������������ǰ�ķ���һ�£���NF-��B������ӫ����ø��������壨pNF��B-luc��תȾ��HeLaϸ������TNF-���������յ���ӫ����ø����������20����ͼ��ͼ1A��.1A���� ����װ��4.0 mM AA��ϸ����ʾө����ø���Ե��յ����ý�����ԼΪ�˱�����Ӧ��ө����ø���Ե�60�����ơ�ϸ����Ũ��Ϊ2.5 mM AA��TNF-���յ���ө����ø����û������Ӱ�죨P = 0.21��n = 3����βt���飩��ͼ��ͼ1A��.1A���� TNF-���յ���I��B�����ữ����װ��4 mM AA��ϸ����ʾI��B�������ữ���ͣ�ͼ��ͼ1B��.1B������Щ���������ϸ����ά����Cͨ����ֹI��B�������ữ������TNF-���յ���NF-��Bת¼��Ӧ��
ͼ�� 1��
ϸ����ά����C��iAA������TNF-���յ����źŴ����� ��A����תȾ��װ��ά����C��pNF��B-luc����������HeLaϸ����TNF-����+������6Сʱ��Ȼ��ⶨӫ����ø���ԡ����ݱ�ʾΪ����������ƽ��ֵ������Ǻű�ʾװ��ά����C��ϸ����δװ��ά����C��ϸ��֮���ͳ��ѧ���죨P = 0.001��n = 3����βѧ��t���飩 �� ��B����װ����4mM AA��δװ��AA��ϸ����TNF-��һ������ӡ����ָʾ��ʱ�䣨���ӣ����Ʊ�ϸ����ȡ���ͨ�������ữ-I��B�����������ӡ�����۲����ữ��I��B����p-I��B������δ���ữ��I��B����ʾ�������ӡ���С� ��C����TNF-����������ά����C��iAA����HeLaϸ��10���ӣ���ͨ������ӡ��������ữ��phos-p44 / 42���ͷ����ữp44 / 42 MAPK��p44 / 42���� ��D����TNF-������װ��AA��ϸ��������ʾ�����ữ��phos-p38���ͷ����ữp38 MAPK��p38��������ӡ���� ��E��������4 mMϸ����ά����C��+ iAA����δ����4 mMϸ����ά����C��-iAA����HeLaϸ����TNF-����+TNF��������������DCFͨ��ӫ�������ⶨROS��������壩����ʾ���ù�ѧ�����۲��ϸ������ͼ����TNF-���յ�MAPK;������p44 / 42 MAPK��p38 MAPK��16������ˣ����ǵ�����ϸ����AA�Ƿ�Ҳ����������Щ��ø�ļ��ͨ������ӡ������⣬TNF-���յ�HeLaϸ����p44 / 42 MAPK���ữ��ͼ��ͼ1C��.1C������AA����ϸ��������TNF-���յ���p44 / 42 MAPK���ữ��ͼ����ͼ1C��.1C������TNF-��������ϸ����ʾ���ữ��p38 MAPK�ʶ����ӣ�����AA����ϸ������p38 MAPK���ữ���Ž��ͣ�ͼ����ͼ1D��.1D��������TNF-������NF-��B�漰ROS��11����������Dz�����ά����C���ص�ϸ����TNF-���յ���ROS�����ˮƽ��ʹ��DCF��ӫ���������ϸ����ROS�� TNF-�����ƿ�����ϸ����ROS���ӣ�Ȼ��������û�м�װ��4 mMά����C��ϸ���е�ROSˮƽ��ͼ��ͼ1D��.1D������Щ���������ά����Cͨ���������IKK���ROS������I��B�������ữ���Ӷ�ͨ������I��B�������ữ����ֹ��TNF-�������Ե�NF-��B�Ļ�����һ��������źŴ����������ROS�Ļ��ۡ�
ϸ����ά����C��������ͻ���IKK�¡�
IKK�±�˿����177��181�������ữ�����ʹI��B�����ữ���ùȰ��������Щ˿����ɲ�������Ի��Ե�IKK�£�IKK�£�SS / EE����38����Ϊ���о�ά����C������IKK�»�����һ�����÷�ʽ�Ŀ����ԣ����Ƿ�����װ��ά����C��ϸ����IKK�£�SS / EE���Ļ��ԡ�pNF��B-luc�ͱ���IKK�£�SS / EE������ө����ø����������12����ͼ��ͼ2A��.2A����Ȼ��������4 mMά����C��ϸ��������IKK�£�SS / EE��������ӫ����ø���ԡ��ϵ�Ũ�ȵ�AA��2.5 mM��û������Ӱ�죨P = 0.11��n = 3����βt���飩��ͼ��ͼ2A��.2A����Ϊ��ȷ��ά����C�Ƿ�����IKK�£�SS / EE���Ļ��ԣ����Dz�������IKK�£�SS / EE��תȾ��ϸ�������ữI��B����ˮƽ����װ��ά����C��ϸ���У�IKK�£�SS / EE�������Ե�I��B�����ữ�ܵ����ƣ�ͼ��ͼ2B��.2B������IKK�£�SS / EE���ķ���������ϸ����ά����C�������û��Ӱ�죨����δ��ʾ������ˣ�������ά���ص�ϸ���У���IKK�£�SS / EE���ı����յ��Ķ�I��B�����ữ����������ֱ��ָʾ��ά����C��IKK�£�SS / EE�����Ե����ơ�
ͼ�� 2��
ϸ����ά����C��iAA����������ͻ���IKK�¡� ��A����ָʾŨ�ȵ�AA������pNF��B-luc��תȾ��HeLaϸ���ͱ�������ͻ���IKK��[IKK�£�SS / EE��]��������ӫ����ø���������ⵥλ����������ʾΪ������Ʒ��ƽ��ֵ����ƫ��Ǻű�ʾװ����ά����C��ϸ����δװ��ά����C��ϸ��֮���ͳ��ѧ���죨P = 0.006��n = 3����βѧ��t���飩�� ��B������IKK�£�SS / EE�����������������壨Ct��תȾ��HeLaϸ������2.5��4mMAAA����ʾ�����ữ��p-I��B��������I��B��������ӡ����
ά����C��������ʽDHA����������IKK�º�IKK���Ļ��ԡ�
����ϸ����ά����C������IKK�£�SS / EE���յ���I��B�����ữ����������о���AA�Ƿ��ֱ��������ϸ��ϵͳ��IKK�µļ�ø���ԡ�ͨ�����߳�����TNF-��������HeLaϸ���з���õ��Ļ��IKK�±��̶��ڵ���G-��֬�����ϣ�Ȼ��������⼤ø���ԵIJⶨ��6������ø��Ӧ�����������1��10 mM AA��IKK�»���û��Ӱ�죨ͼ��ͼ3A��.3A���������ƵIJⶨ�����£�100 mM��AA������IKK�»��ԣ����ǵ���ø��Ӧ������а����������Ǵ���DTT��ʱ�������������þ���ʧ�ˣ����������ƻ�����AA�������йأ�ͼ����ͼ3A��.3A �����ڷǻ�ԭ�����£�AA����ΪDHA��Ȼ�����ˮ��Ϊ������հ��ᣨ13��14����
ͼ�� 3�� ά����C��DHA����������ʽ����������IKK�º�IKK���� ��A���ⶨ���ÿ�IKK�¿����TNF-��������HeLaϸ����ȡ�������߳�����IKK�µ�IKK�»��ԡ�����ʾ������ø��Ӧ�������AAһ���������ijЩ����£�+ DTT����DTT���ӵ���ø��Ӧ������С�ͨ����������Ӱ�۲����ữ��GST-IkB����p-I��B��-GST���� ��B���ڻ���Һ�����գ���1mM DHA��AA�����ᣨOA���������ᣨTA�������²ⶨIKK�»��ԡ� ��C���ڻ���Һ������Ѫ������ø��AOx����AA��ø�ٲ�����DHA��AA + AOx����DHA�Ĵ����²ⶨIKK�»��ԡ��ֱ�ʹ����һ�ٻ�250 U����Ѫ������ø[AOx��1x����AOx��2.5x���� ��D����ӡ������ʾ���ڲ�ͬŨ�ȵ�DHA�����²ⶨIKK�»��ԣ���Լ�ø���Ա�ʾΪӡ�����¶��յİٷֱȡ�ͨ���ÿ�I��B�������IKK�¿����������ӡ�������Թ۲쵽�뻺��Һ��1 mM DHA����30���ӵļ�ø��Ӧ������е�GST-I��B����IKK�¡� ��E������ⶨ��תȾϸ�����߳����Ļ��IKK����IKK�µļ�ø���ԡ���DHA����������ⶨp38 MAPK��p38-GST���ļ�ø���ԡ� PHAS-1���������p-PHAS-1����ͨ����������Ӱȷ�����ữ�ĵ����ʣ�������Լ�ø������ʾΪӡ�����µİٷֱȡ�
Ϊ��ȷ��AA����Щ�������Ƿ��������IKK�µĻ��ԣ����ǽ���ø��Ӧ�������1 mM AA��DHA�������������һ���������1 mM DHAһ�����������IKK�µ����⼤ø���ԣ�ͼ��ͼ3B��.3B�����෴������ø��Ӧ�������1 mM AA�������������һ�����ʱ��δ����IKK�»��Ե�Ӱ�죨ͼ��ͼ3B��.3B����Ϊ�ų�DHA�Ƽ��м�ø��������Ⱦ��Ŀ����ԣ�ʹ�ÿ���Ѫ������ø��AAø������DHA�����ⶨ�����ƻ��ԡ�ø��������AA��DHAҲ����IKK�»��ԡ�������AA�Ϳ���Ѫ������øû���������ã�֤ʵ��DHA�Ļ��ԣ�ͼ����ͼ3C��.3C�������Ƿ������ƴ�Լ50����IKK�¼�ø������Ҫ��Լ100��MDHA��ͼ��ͼ3D��.3D���� DHA�����ƻ��Բ���DTT��Ӱ�죨����δ��ʾ������1 mM DHA����30���Ӳ�����������IKK�µĽ��⣨ͼ��ͼ3D����3D����֤ʵDHA������ֱ��������IKK�µ�ø���ԡ����ǻ��о���DHA�Ƿ�����IKK����p38 MAPK�Ļ��ԡ���תȾϸ���з����IKK����IKK�µļ�ø���Ա�DHA���ơ���500��M�£�DHA�������������м�ø���ԣ�ͼ����ͼ3E��.3E���� p38 MAPKҲ��DHA���ƣ���500��MDHA������Լ50���ļ�ø���ԣ�ͼ����ͼ3E3E����
Ϊ�˽�һ���о�DHA���Ƽ�ø�Ļ��������̶��ڵ���G�ϵ���Դ��IKK����DHA��AAһ�����������ڼ�ø��Ӧ֮ǰ������ϴ����ȥ��AA��DHA��ͼ����ͼ4A��.4A�� ����1 mM DHAԤ������IKK�±���ȫ���ƣ�����0.1 mM DHAԤ�����ı�����ԼΪ40������AAԤ�������ᵼ��IKK�»��Խ��ͣ�ͼ��ͼ4A��.4A������Щ���ֱ�����DHA����ͨ���ı���ø������������IKK�£�������ֱ�ӽ�ϼ�ø������ATP������ϻ����ϴ���DHA�������õ���ά�֣�����DHA����ATP������ϡ�ͨ��������ATP�����·���DHA�Ƿ������IKK�»������о���ATP��ϵĸ��š� ATP���־���DHA�����ƻ��ԣ�ͼ��ͼ4B����4B�����ⰵʾDHA���ܻ����ATP��IKK�µĽ�ϡ�
ͼ�� 4��
ATP����DHA�����ƻ��ԡ� ��A���ù��̵�ʾ��ͼ�� 15����15���ӣ��ң�����ͼ3,3��˵��ͨ����������Ӱȷ��IKK�µļ�ø���ԣ�����ʾ���ữ��I��B��-GST������ӡ���� ��B���ڻ���Һ��20��100��MATP�Ĵ����£���IKK������DHA��������ø������ʾΪ���յİٷֱȡ�����Ƭ����ʾ�����ữ��GST-I��B����pI��B��-GST����
�����⣬DHA���Ʋ��������ͻ���IKK�£�SS / EE����
Ϊ���о�DHA�Ƿ���IKK�£�����ʹ���˴�תȾϸ���з����IKK�£�SS / EE�����ڽ���о�֮ǰ������ȷ��DHA�Ƿ�����������IKK�£�SS / EE�����ԡ�������Ӧ�����������1 mM DHA������IKK�£�SS / EE���Ļ���40����ͼ��ͼ5A��.5A�����������������о���ά����C�Ƿ���IKK�£�SS / EE����ϡ�������IKK�£�SS / EE����������[14C] DHA��[14C] AA��������DHA�Ľ�ϻ�����ȣ�AA��ʾ�������ȡ�����IKK�£�SS / EE������Ԥ�������������ͣ�ͼ����ͼ5B��.5B���� DHA��IKK����Ľ�ϱ��������ȡ��Ԥ��������Ľ�ϴ�������ͼ��ͼ5B��.5B��������ATP������DHA�����ƻ��ԣ��������ȷ��DHA�Ƿ������IKK�£�SS / EE����ͻ���ATP���λ���ϡ������м�ø��ATP���λ���з��ֵĺ㶨������ͻ��ᵼ��ATP���ɥʧ���������¼�ø����ɥʧ��15������������IKK�£�SS / EE���о���������42��ͻ��ͻ���壬��Ϊ�����ᣨK42A�����ᣨK42R������������ⶨ�˼�ø���ԡ�����ͻ����ļ�ø���Ծ�����ȱ�ݣ�ͼ����ͼ5C��.5C��������Щ���һ�µ��ǣ�IKK�£�SS / EE��ͻ�����תȾ��ʾө����ø���Խ��ͣ�ͼ����ͼ5D��.5D���� DHA����IKK�£�SS / EE����ͬ��ˮƽ��ϵ�IKK�£�SS / EE��K42R��K42A��P = 0.14��n = 3��βt���飩��ͼ��ͼ3E��.3E�� ����Щ�������DHA��IKK�µĽ�϶����ڹ�����ATP���λ�㣬����ʾ����ATP��Ͽڴ���ͬ����ɢ���λ�㡣��
ͼ�� 5��
DHA��IKK�½�ϡ� ��A��������ⶨ��תȾϸ�������߳���������ͻ���IKK�£�IKK�£�SS / EE���ļ�ø���ԡ�ͨ����������Ӱȷ����ø���ԡ� ��B�����뿹FLAG���ϵ�IKK�£�SS / EE����[14C] DHA��[14C] AAһ������������FLAG��ϴ�ѣ���ͨ����˸�����ⶨ��IKK�£�SS / EE���йصķ����ԡ�ʹ����δתȾϸ�������գ���ȡ��һ����������ӣ����Թ����뿹FLAG���ӷ���������صķ����ԡ� ��C��������ⶨIKK�£�SS / EE����ATP���λ��ͻ����IKK�£�SS / EE��K42A��K42R�ļ�ø���ԡ��������ֻ�ͼ��ø������ʾΪӡ�����µ����ӱ����� ��D����pNF��B-luc����������IKK�£�SS / EE����K����K42A��K42R��תȾ��HeLaϸ����ө����ø���Ա��ⶨ��ө����ø������Westernӡ����WB��ǿ�ȵ����ⵥλ��ʾ�����ÿ����干תȾ��ϸ����ö�����ȡ�Ct���� ��E��������IKK�£�SS / EE����K����K42A��K42R��������[14C] DHAһ����������ϻ�����ʾΪ��IKK�£�SS / EE���Ķ��ս�ϵİٷֱȡ���
����Ӧ����AAת��ΪDHA������IKK�£�SS / EE���յ���ө����ø���ԡ�
Ϊ��ͨ������Ӧ��Դ������H2O2����AA����ϸ����DHA����������ȷ���˶�ϸ��������ӫ����ø����û��Ӱ���H2O2Ũ�ȡ���IKK�£�SS / EE��תȾ����50��MH2O2������δ��H2O2������ϸ����ʾ��ͬ��ө����ø���ԡ����ǣ�װ��2.5 mM AA����50��MH2O2������ϸ����ʾө����ø���Խ�����Լ50����P = 0.004��n = 3����βt���飩��ͼ����ͼ6A��.6A ����������ϸ����AA��2.5 mM��û������Ӱ�죨P = 0.5��n = 3����βt���飩����
ͼ�� 6��
ϸ����DHA����IKK�£�SS / EE���յ���ө����ø���ԡ� ��A����pNF��B-luc����������IKK�£�SS / EE����+����תȾ��ϸ��װ��2.5 mM iAA��ϸ����ά����C����δװ��iAA��-������50��MH2O2������δ��H2O2��-�����Ʊ�ϸ����ȡ��ⶨө����ø���Բ������ⵥλ��� ��B����5��MDMNQ������δ��DMNQ��-������תȾ��pNF��B-luc��IKK�£�SS / EE����+����2.5mM iAA��δװ��iAA��-����ϸ����תȾ��ϸ���� 4Сʱ���Ʊ�ϸ����ȡ�ӫ����ø���Ա�ʾΪ���ⵥλ��
���ǵ�����ROS��ϸ�������������������������������DMNQ�����Ƿ�����Ԥ������2.5 mMά����C��ϸ����IKK�£�SS / EE��������ө����ø���ԡ�DMNQ��Ũ�ȶ�ө����ø���Ի�����IKK�£�SS / EE��תȾ��ϸ����������Ϊ5��M��װ��2.5 mMά����C����5��MDMNQ������ϸ����ʾө����ø�������Ž��ͣ�P = 0.003��n = 3����βt���飩��ͼ��ͼ6B��.6B������Щ���������AA��DHA��ϸ����ת��������IKK�£�SS / EE���ļ�ø���ԣ�����DHA��������ø���Ƽ������á����ǽ��飬��װ��ά����C���������������µ�ϸ���У�AA��������ROS��ת��ΪDHA�����ɵ�DHA����ͨ���ٽ���������ת�˵����뿪ϸ��������Ϊֱ�Ӽ�ø���Ƽ��������ã���˴���ʾ������IKK����IKK�£�ͼ��ͼ7��.7������Щ����ͨ����������ά����C������Ӧ���뼤ø������ϵ��������
ͼ�� 7��
ά����C�����ź���Ӧ��ʾ��ͼ��ά����C��ΪDHAͨ��������ת�˵�����ϸ������Ѹ�ٻ�ԭΪAA�� ROSͨ������IKK���յ�NF-��B�źŴ���Ӧ�𣬶�AA���ROS������IKK�µļ������Щ�����У�AA������DHA����DHA����IKK�¡���
����
ROS����ϸ����л�����в����ģ�����Ҳ��Ϊ�ź�ת��;������Ҫ������롣��ˣ��������������ڵ���ϸ�����ⲿ�̼��ķ�Ӧ����ؼ����ã�24���� ROS�Ĵ�л������ֲ����弤���յ���ROS֮�����������ϸ����ά����C���ź�ת���е��������ã�5��27�������磬��֪��ϸ��-����ϸ������̼����ӣ�GM-CSF���Ͱ���3�ź��漰ROS��5��30����ά����C��Jak2����ˮƽ����GM-CSF��Ӧ�����Ʀ����ữGM-CSF���壨5��������֪NF-��BӦ�����ź�ת��������ROS�������Ѿ�������NF-��B�ļ����������ø�Ϳ���������������������������������N-������-1-���װ��ᣬ���������ף������ĵĹ����������ơ���ά����C��4��6��9��11��23�����������о��У�������ͼ�Ի�е��ʽ����ά����C������I��B�����ữ�е����á�
ά����C��Ϊϸ���ڿ�����������Ҫ���ѵõ����֤�����������ƹ��������յ���ϸ��������12��37����FAS���CD95�յ���ϸ��������27��������Ӧ���յ���DNA���ˣ�22����������Щ�����⣬ά����C�Ǹ���ø���ǻ�����Ӧ�ĸ������ӣ�13��14����ά����C��������Щ���ܣ���������Ϊ�����ӵĹ��ܣ������������ṩ���ӣ��ڴ˹����б�������DHA�������� DHA��Ҫ��ά����C�������������ã�36�������ݱ���Ҳ�����˼��Ǽ�ø���ԣ�8a����
���Ƿ���DHA���Գ䵱��ø���Ƽ�����ֱ������IKK�£�IKK����p38 MAPK�Ļ��ԣ��Ӷ���չ�����Ƕ�ά����C��ε���ROS������NF-��B�źŴ�������ʶ��4��6����ά����C����IKK��ø���Ե����֤����������ͻ���IKK��[IKK�£�SS / EE��]�յ��ķ�Ӧ���µ��������о�������ά����C��������ʽ���������ߴ�����IKK�º�IKK�£�SS / EE����ʹ�÷����Ա�ǵ�[14C] DHA�����Ƿ���DHA��IKK�½�ϣ���AA������Ȳ���ʾ���ƻ��ԣ�Ҳ����ʾ��IKK�µĽ�ϻ��ԡ�DHA��IKK���ͦ�ø���Ե��������ö�����������ά����C������NF-��B��Ӧ�Ļ��ơ����ȣ�AA������ϸ������-����������յ���ROS�źŴ������Ӷ���ֹROS�鵼�ķ�Ӧ�ļ�����������źŲ�����ROS����Σ�����AA������������DHA��������ROS����ļ�ø��������AA��ϸ����ͨ��ʵ��֤������һ�㣬��ͨ����H2O2��DMNQ����ʩ��ѹ�����յ�����DHA����Щ���ֱ���ά����C������IKK�������TNF-���ź�������ת��������п������á�
DHA���Ƽ�ø��ȷ�л����в�ȷ���������ǽ���DHA�������ø����λ�㸽���Ŀڴ��� ATP��ʼ����ʵ��Ľ����������������ATP���λ������ã����ǽ�ϲⶨ�Ľ��������������ATPλ�����DHA��ϲ��DZ���ġ���Щ���Ե�ì�ܽ������ͨ�����ڶ�����DHA���λ�������ڡ��Խ�ģ��ͼ������ģ������DHA�����ڴ�λ�㸽����϶�����ATP���λ���ϣ�����δ��ʾ����
�ݱ�����ά����C�����ü��������ἤø��ø���ԡ����ǣ����Ƚ�AA��DTT�������ɿ˷������������ã�29������Щ���������AA��ת��ΪDHA����DHA���������������ἤø�ķ��ӡ��ݱ������������һ�����øҲ��AA���ƣ�31����������ά����C����������Ӧ��״̬��ϸ����G2 / M�����յ��˶��ݵ�ϸ������ͣ�ͣ������Ʋ�DHAͨ���ӳ�ϸ�����ڵ���B-cdc2�Ļ���յ�ϸ������ͣ�ͣ�3��������ͨ������DHA����Ӱ����ϸ�����ڿ�������Ҫ�ļ�ø��Ӧ��ʹ��Щ��������������ǵ����ݱ���DHA��������IKK�£�IKK����p38 MAPK����T4������ἤø��DHA�����еֿ���������δ��ʾ����
NF-��B����Ƽ�������ˮ�����κͰ�˾ƥ�֣��ڼ�����֢�Ϳ��ܵ���������������Ҫ���á���Щҩ��ͨ����ATP��Ͼ���������IKK�µļ�ø���ԣ�38�������ǽ�ά����C��ΪNF-��B���Ƽ��ķ��ֱ�����ά����C����֢��ϸ�������з��������á�ά����C����ͨ�����ֲ�ͬ����صĻ���������NF-��B�ļ���µ�ROS�յ��ļ��ֱ������IKK����IKK�¡�Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC444845
Our findings with vitamin C as an NF-��B inhibitor point to a role for the vitamin in inflammation and apoptosis. Vitamin C can inhibit the activation of NF-��B by two distinct but related mechanisms: down-regulating ROS induced activation and directly inhibiting IKK�� and IKK��.
Cited by: 106
Publish Year: 2004
Author: JuDepartment of Clinical Laboratories,1 Program in Molecular Pharmacology and Chemistry, Memorial Sloan-Kettering Cancer Center,2 Structural Biology Program, Department of Physiology and Biophysics, Mount Sinai School of Medicine, New York, New York3
*Corresponding author. Mailing address: Program in Molecular Pharmacology and Chemistry, Box 451, Memorial Sloan-Kettering Cancer Center, New York, NY 10021. Phone: (212) 639-8483. Fax: (212) 772-8589. E-mail: gro.ccksm.iks@edlog-d.��
ά����Cͨ������I��B�����ữ����TNF���յ���NF��B�,Ӱ����֢�������͵�������
Vitamin C Suppresses TNF��-Induced NF��B Activation by Inhibiting I��B�� Phosphorylation†
ժҪ
ϸ����̼��źż���ת¼����NF��B�������漰����Ӧ����֢��ϸ�����Ļ��������ڹ��̡������������Ӧ���TNF����ͨ���漰NF��B�յ���ø��NIK�����伤�����ζ��ǻ�I��B��ø��IKK����ȷ����ļ�ø;������NF��B�� IKK�������ữI��B��NF��B���ܵ�������ڼ��������Ƿ���ϸ����ά����C�Լ��������Է�ʽ������ϸ��ϵ��HeLa������U937��������Ѫ��HL-60������MCF7����ԭ����Ƥϸ����HUVEC����TNF���յ���NF��B���ά����C��һ����Ҫ�Ŀ��������������ϸ��ͨ������������ʽ��ά����C-���⿹��Ѫ�ᣨDHA����ϸ���ڻ��ۿ���Ѫ��(AA)����Ϊ����Ѫ����������ڹ��ɽ������������ǿ����������������ͨ����ϸ����DHAһ�����Ϊϸ��������ά����C��װ��ά����C��ϸ����ʾ��TNF���յ���NF��B����λ��NF��B�����ı������ת¼��I��B�����ữ���Ž��͡����ǵ�����ָ����ά����Cͨ������TNF���յ��Ķ�����p38 MAP��ø��NIK��IKK�¼�ø����������NF��B����Ļ��ơ���Щ���������ϸ���ڵ�ά����C����ͨ������NF��B���Ӱ����֢���������͵������̡�
Extracellular stimuli signal for activation of the transcription factor NF��B, leading to gene expression regulating processes involved in immune responses, inflammation, and cell survival. Tumor necrosis factor-�� (TNF��) activates NF��B via a well-defined kinase pathways involving NF��B-inducing kinase (NIK), which activates downstream multisubunit I��B kinases (IKK). IKK in turn phosphorylates I��B, the central regulator of NF��B function. We found that intracellular vitamin C inhibits TNF��-induced activation of NF��B in human cell lines (HeLa, monocytic U937, myeloid leukemia HL-60, and breast MCF7) and primary endothelial cells (HUVEC) in a dose-dependent manner. Vitamin C is an important antioxidant, and most cells accumulate ascorbic acid (AA) intracellularly by transporting the oxidized form of the vitamin, dehydroascorbic acid (DHA). Because ascorbic acid is a strong pro-oxidant in the presence of transition metals in vitro, we loaded cells with vitamin C by incubating them with DHA. Vitamin C-loaded cells showed significantly decreased TNF��-induced nuclear translocation of NF��B, NF��B-dependent reporter transcription, and I��B�� phosphorylation. Our data point to a mechanism of vitamin C suppression of NF��B activation by inhibiting TNF��-induced activation of NIK and IKK�� kinases independent of p38 MAP kinase. These results suggest that intracellular vitamin C can influence inflammatory, neoplastic, and apoptotic processes via inhibition of NF��B activation.
Cited by: 259
Publish Year: 2002
Author: Juan M. C��rcamo, Alicia Pedraza, Oriana B��Program in Molecular Pharmacology and Chemistry and Departments of Clinical Chemistry and Medicine, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, Box 451, New York, New York 10021
��
PPARalpha��ϸ����л����֢�Ŀ������ԡ����ߣ���ǰ��PPAR������������֬Ѫ֢����Ѫ�ܼ����йأ���Ϊ���ǵ��ڵ��������Ǻ�֬�ʴ�л�Ļ���ı���.PPARalpha����Ŀ���ֳ���ٵ���������DZ�ڵĿ�ת�����Ե�����֤����ʾ��������PPARalpha�����������еĿ������á����������������ʵ�״̬�£��������������������У���PPARalpha��AMP�����ĵ���ø(AMPK)Эͬ���ã���i�������°�Akt���ԣ���ii������ϸ����ֳ��iii����ʹ�ǽͽ�������ϸ�����롰��л���� Metabolic catastrophy���� PPARalpha������DZ�ڿ������ð���������֢����ż�����ף�UCP�����ϵ����Ӷ�����������������IJ�����ϸ����ֳ��
ժҪ
��������ø����ֳ�D�����壨PPARs����Ϊ���ưе���������˹㷺�Ĺ�ע����ǰ��PPAR������������֬Ѫ֢����Ѫ�ܼ����йأ���Ϊ���ǿɵ��ڵ��������Ǻ�֬�ʴ�л�Ļ���ı�������PPAR����Ҳ����Ϊ��DZ�ڵĿ���ҩ�����нϵ͵�ȫ�����ԡ� PPARalpha����Ŀ���ֳ���ٵ���������DZ�ڵĿ�ת�����Ե�����֤�ݴ�ʹ��������PPARalpha�����������еĿ������á� PPARalpha�����ͨ�����֬����ϳɲ��ٽ�֬�����-����������ϸ��������ƽ�⡣���������������ʵ�״̬�£�ͨ�����������������У���PPARalpha��AMP�����ĵ���ø��AMPK)Эͬ���ã���i�������°���Akt���ԣ���ii������ϸ����ֳ���Լ���iii��ǿ���ǽͽ�������ϸ�����롰��л���ѡ��� PPARalpha������DZ�ڿ������ð���������֢����ż�����ף�UCP�����ϵ����Ӷ�����������������IJ�����ϸ����ֳ��
��֮���к�ǿ��ǰ�ᣬ���Ͷ��Ժ����������õ�PPAR����Ӧ����Ϊ�Կ���ɢ��֢�������Ƽ�����������ִ�ҽѧ�ͻ����о�����Ҫ��ս��
��
ͼ3��PPAR��ͨ��������Ҫ������ת¼����NF��B��AP-1����Ҫ�������ź�ͨ·�����⣬PPAR��ͨ���ϵ���ż������UCP2��UCP3����ROS�鵼����֢���йظ�����ϸ˵������μ��ı�����ͷ��ʾ����/�ϵ��������߱�ʾϸ��������̵�����/�µ��� Erk1 / 2-ϸ�����ź�Ӧ��ø1/2�� I��B-NF��B���Ƽ��� MAPK-�ٷ���ԭ�����ø�� NF��B�������Ӧ�B�� ROS-��������
Fig3: PPAR�� antagonizes main inflammatory signaling pathways throughrepression of the main inflammatory transcription factors: NF��B and AP-1.Additionally, PPAR�� reduces ROS-mediated inflammation by upregulation ofuncoupling proteins UCP2 and UCP3. See the text for more detailedexplanation. Arrowheads represent activation/upregulation, and blunted linesindicate inhibition/downregulation of the cellular proteins or processes.AP-1��activating protein-1; Erk1/2��extracellular signal response kinase 1/2; I��B��inhibitor of NF��B; MAPK��mitogen activated protein kinase; NF��B��nuclear factor ��B; ROS��reactive oxygen species.
Affiliation: Department of Food Biotechnology, Faculty of Food Technology, Agricultural University of Krakow, ul. Balicka 122, 31149 Krakow, Poland.
��
EPA��DHA����HK-2ϸ����LPS�յ�����֢��Ӧ��PPAR-�������Ի��Ƶ�֤��
EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: Evidence for a PPAR-�èCdependent mechanism
����
������о������������к��Ц�-3�����֬���ᣨ��-3PUFAs����ʮ̼��ϩ�ᣨEPA����C20��5��3���Ͷ�ʮ��̼��ϩ�ᣨDHA����C22��6��3�����ӻ��������༲���ķ������ر�����IgA������IgAN���С���������Խ��Խ�˽����͵��������ã����Ԧ�-3PUFA�����û���֪֮���١��ݱ�������������ø����ֳ�D�����壨PPAR���ļ��������˴���ϸ�����ӵIJ����� EPA��DHA����ʾ�ɼ���PPAR�������о���Ŀ���Ǽ���-3PUFA�Ƿ�ͨ����������С��ϸ���е�PPARs���п������á�
����
������ʵ���о�ʹ�������������˽�����С��ϸ��ϵ[����2��HK-2��ϸ��]���Ӧ�-3PUFA��������ϸ�����ռ�������������������ø�����������ⶨ��ELISA����������ϸ���з������ϸ��RNA������ʵʱ�����ۺ�ø����Ӧ��PCR������HK-2ϸ���Ʊ�����ȡ������ת¼���Ӽ���ⶨ��
���
EPA��DHAŨ�ȷֱ�Ϊ10��mol/ L��100��mol/ L����Ч������֬���ǣ�LPS���յ��ĺ�����-��B��NF-��B����͵���ϸ����������1��MCP-1���ı�� EPA��DHA��������HK-2ϸ����PPAR-��mRNA�͵����ʻ��ԣ�2��3����������Ϊ100��mol/ L��˫��A����ˮ�����ѣ�BADGE��������EPA��DHA�յ���PPAR-�û����������EPA��DHA��LPS�յ���HK-2ϸ����NF-��B����������á������EPA��DHA�Ķ���ϸ����ȣ�PPAR-�õĹ������һ��������NF-��B�Ļ��
����
���ǵ����ݱ�����EPA��DHA����ͨ��HK-2ϸ����PPAR-��������;���µ�LPS�յ���NF-��B�����Щ���������EPA��DHA����PPAR-�ÿ����������������õ�DZ�ڻ���֮һ��EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: Evidence for a PPAR-�èCdependent mechanism - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0085253815505322��
The Inflammation Link: NF-κB Remains a Difficult but Intriguing Target
While anticancer therapies aimed at particular pathways have mushroomed in recent years, one crucial target has remained elusive: nuclear factor kappa-light-chain-enhancer of activated B cells (NF-��B). The pathway is constitutively active in the majority of cancers and provides a mechanistic link between chronic inflammation and tumorigenesis. As such, it represents an important target for anticancer therapy. The complex regulation of NF-��B activation has presented significant challenges for the development of such agents, but researchers are now beginning to better understand and embrace this complexity to drive development of a variety of novel NF-��B-targeting strategies.
The NF-��B Family
NF-��B was discovered in late 1980s as a nuclear factor that binds to the enhancer region of the kappa-light chain of immunoglobulin in B cells. It was subsequently shown to be a ubiquitous family of transcription factors that are present in all cells and control the transcription of more than 500 target genes involved in critical cellular pathways including proliferation and apoptosis. The NF-��B pathway is activated by a wide range of stimuli, including stress, cytokines, free radicals, ultraviolet irradiation, and bacterial or viral antigens.
NF-��B is activated by a number of upstream signaling pathways, including the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor-2 (HER2), platelet-derived growth factor receptor (PDGFR), and c-KIT pathways. Another key activator of NF-��B is the receptor-activator of NF-��B (RANK), which is a type of tumor necrosis factor (TNF) receptor. Further adding to the complex regulation of NF-��B signaling, it undergoes numerous posttranslational modifications, including methylation and acetylation, and it interacts with several other transcription factors, such as signal transducer and activator of transcription (STAT)-3 and hypoxia inducible factor (HIF)-1��, both of which regulate NF-��B activity.
The Many Roles of NF-��B
Roles of NF-kB EBV, indicates Epstein-Barr virus; EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; HBV, hepatitis B virus; HCV, hepatitis C virus; HHV-8, human herpesvirus 8; HTLV, human T-lymphotropic virus; IL, interleukin; PDGFR, platelet-derived growth factor receptor; TNF, tumor necrosis factor. Adapted from Sethi G, Sung B, Aggarwal BB. Nuclear factor-��B activation: from bench to bedside. Exp Biol Med. 2008; 233(1): 21-31.
Linking Inflammation and Cancer The activation of NF-��B plays a role in the suppression of apoptosis, stimulation of proliferation, and promotion of migration and invasion, all of which are hallmarks of cancer. Unsurprisingly, therefore, NF-��B has been implicated in tumorigenesis. What is surprising is that oncogenic mutations in the NF-��B genes are rare and largely limited to lymphoid malignancies, yet NF-��B activation is observed in almost all tumors.
... to read the full story��
��֢���ӣ�NF-��B��Ȼ��һ�����ѵ���Ȥ��Ŀ��
The Inflammation Link: NF-κB Remains a Difficult but Intriguing Target
ͼʾ��EBV��ʾ����˹̹-�Ͷ������� EGFR����Ƥ�����������壻 HER2���˱�Ƥ������������2�� HBV���Ҹβ����� HCV�������ײ����� HHV-8�����������8�� HTLV������T�ܰͲ����� IL�����أ� PDGFR��ѪС�������������������壻 TNF�������������ӡ�
���ܽ���������ض�;���Ŀ����Ʒ���������ӿ�֣���һ���ؼ���Ŀ����Ȼ�����������Bϸ�������Ӧ�-������ǿ����NF-��B������;���ڴ������֢�о�������Ի��ԣ����ṩ��������֢����������֮��Ļ�����ϵ����ˣ��������˿������Ƶ���ҪĿ�ꡣ NF-��B����ĸ��ӵ���Ϊ����ҩ��Ŀ���������ش���ս�������о���Ա���ڿ�ʼ���õ�����ͽ������ָ����ԣ����ƶ���������NF-��B������Եķ�չ��
NF-��B����
NF-��B��1980���ĩ��������һ�ֺ����ӣ���Bϸ���������Ħ���������ǿ�������ϡ����֤������һ������������ϸ���е��ձ���ڵ�ת¼���Ӽ��壬�ɿ����漰�ؼ�ϸ��;����������ֳ�͵�������500����л����ת¼�� NF-��B;�������ִ̼��������Ӧ����ϸ�����ӣ����ɻ��������������Լ�ϸ������ԭ��
NF-��B�����������ź�ͨ·���������Ƥ�����������壨EGFR�����˱�Ƥ������������2��HER2����ѪС��Դ�������������壨PDGFR����c-KIT;���� NF-��B����һ���ؼ��������NF-��B���弤�����RANK��������һ�������������ӣ�TNF�����塣��һ��������NF-��B�źŵĸ��ӵ��أ������������෭������Σ���������������������������������ת¼���ӣ������ź�ת���Ӻ�ת¼�����ӣ�STAT��-3�͵����յ����ӣ�����ã� HIF��-1�������߾�����NF-��B���ԡ�
NF-��B���������� NF-kB EBV�����ñ�������˹̹-�Ͷ������� EGFR����Ƥ�����������壻 HER2���˱�Ƥ������������2�� HBV���Ҹβ����� HCV�������ײ����� HHV-8�����������8�� HTLV������T�ܰͲ����� IL�����أ� PDGFR��ѪС�������������������壻 TNF�������������ӡ��ı���Aghiwal BB��Sethi G��SungB��������-��B����ӹ���̨�����ߡ� Exp Biol Med�� 2008�ꣻ 233��1����21-31�� ����֢�방֢��ϵ����NF-��B�ļ���������ϸ���������̼���ֳ�Լ��ٽ�Ǩ�ƺ���Ϯ�з������ã�������Щ���ǰ�֢�ı�־����ˣ�����������ǣ�NF-��B�����������йء����˾��ȵ��ǣ�NF-��B�����е��°�ͻ����ټ������Һܴ�̶��������ܰ����������������Ǽ��������������ж��۲쵽��NF-��B��� ...�Ķ�ȫ����
The Inflammation Link: NF-κB Remains a Difficult but Intriguing Target
https://www.onclive.com/publications/oncology-live/2013/june-2013/the-inflammation-link-nf-b-remains-a-difficult-but-intriguing-target��
��֢101����֢������֢��ȼ��2012��11��11�գ�ҽѧ��ʿBrian D. Lawenda
����Ϊ���Ǵ������ͨ������֢��Ϊ������˺���Ⱦ�ı����Է�Ӧ��û���������ǽ��������ص���������������������һ���ܷ����ĭ�С��� ���ֻ����ô��... ��֢��Ϊ���࣬�����ԡ��͡����ԡ���
���Է��ף������ǵ�������Ϊ����ʱ������ϸ������ϸ��������ϸ���ȣ��ڼ���֯�е�����Ӧ�������˻��Ⱦʱ�ͻᷢ�����á���Щϸ������Χ��֯��Ѫ�����ͷų����������Ի�ѧ���ʺ͵����ʣ��Ӷ��� �����κ����������ߣ���ϸ��������������������������ϸ���ź�����Ѫ�������Ӹ������Ѫ����ʹ����ϸ����������Ӫ���������룩������֯����Ѫ�ܵ�������������֯��������Щ��֯�ṩӪ�����˹��̳�Ϊ�����ԡ���֢�������ڸ�Ⱦ�õ����ƻ��˿����������Сʱ�������ڽ������������ʵ������ˡ���
��
������֢��
������֢���Ա����������ܵжԻ������ֺ����������ԡ���֢�����ڴ��ڵ�ˮƽ�����ȵ���֢״̬���������������꣩���˺�����ɱ�����ǡ����ֲ������ִ�ҽѧ����Ҫ�ķ���֮һ������������ʶ����������֢�뼸���������Լ�����������֢���ķ����ͷ�չ�йء�
������֢���������������ߺ����Ը�Ⱦ�Լ�������Ϊ���ԣ�����Щ�����У��漰����֯�����˳����Ļ�ѧ�͵������źŴ�����������������
������֢������������ȵ��ص������ԣ�����Ѫ��ˮƽ�������ߣ��ȵ��غ��ȵ�������������1��IGF-1���IJ������ӣ���л�ۺ�����2�����ͷ���֢��������Щ���ؾ�����ʾ�����Ӱ�֢��չ����չ�����ķ��ա�
�����Է���Inflammasome��
���Ǵ��������ǰ��δ��˵������֢С�塱����Ϊ����������ŷ��ֵģ�2002�꣩����Щ�����ʸ���������������ϵͳ���ɻ�ȱ��һ���֡�����������ġ�Σ�ո�Ӧ���ס���ɣ���Щ���ױ�������ϸ�������ɻ���ѧ���ʼ��
һ���������֢С��ͻ������ǵ�����ϸ��������ϸ���������źţ���ʼ������������֢���ס������о���������֢С���ںܴ�̶��Ϲ����ڷ��ֵ��µ�������֢״̬��
�������Ӧ�B��NF-kB���ڰ�֢�е�����
The Role of Nuclear Factor Kappa-B (NF-kB) in Cancer
��
������ϸ���״μ��κ���֯���˻�ѹ���ļ���ʱ�����Ǿͻἤ��һ�ֳ�Ϊ�����Ӧ�B��NF-kB����ϸ�����ס��õ����ǿ�����֯��֢����Ҫ�����ء������NF-kB�ź�֪ͨϸ����ͨ������;��������ͨ����������С�壩��ʼ����������֢����֯���Ķ��ֵ����ʡ�
��ĿǰΪֹ��ֻҪ�ڰ�ǰ����֯��û�г��������źţ��Ϳ����ˡ���
��������֢�����£�������֯�ȷǷ�����֯���������ͻ���DNA���ˡ��������ڷ�����֯�д������ɻ��IJ�����ʵ���ϣ��������ɻ������˺��ж��ı�¶��ÿ�������ǵ�ϸ���в�����ʮ�ڸ�DNAͻ�䡣������Щ�߷�Ӧ�Ի�ѧ������DNA�����ʱ�����ǿ��Բ���DNAͻ�䡣ֻҪϸ���ܹ�����ЩDNAͻ�䣨�����ϸ���������õ�DNA�����ƣ�����ϸ�������ź�ʹ�侭��ϸ����ɱ���̣���������������ǰϸ���Ͳ��ᷢ����
���ǣ��ڳ�����¶��NF-kB�����ź��ڼ䣨��������֢�з�������ϸ��������DNA�����ƾ����رա���¾����Ŵ�ͻ���ϸ�����ۡ������Щϸ�������㹻��ͻ�䣬���ǿ���ת��Ϊ��ǰϸ��������ת��Ϊ��ϸ����
���ǣ��ڳ�����¶��NF-kB�����ź��ڼ䣨��������֢�з�������ϸ��������DNA�����ƾ����رա���¾����Ŵ�ͻ���ϸ�����ۡ������Щϸ�������㹻��ͻ�䣬���ǿ���ת��Ϊ��ǰϸ��������ת��Ϊ��ϸ����
�ڴ���������֢������£���ǰ�Ͱ�����֯�ᷢ���źţ�ָʾ����ѣ���������������Χ��֯������ѪҺ���ܰܣ��Ӷ�ʹ���ǿ��Խ��������������λ�����ⱻ��Ϊ��ת���ԡ���ɢ������Щ��֢�źŻ���ʹ�����������ϸ��ʧ��Ӷ�ʹ����ʶ��������γɵİ�ϸ����
��ϸ��ѧ��ȷ���Լ��������ʹ���͵͵����������֮һ�����ǿ��Լ����Լ���NF-kB����ʹ�����ܹ�����Χ��֯��������ͬ����֢���ף��Ӷ��ٳ����������֯�����������ơ�
ʵ�����������С�壨inflammasome)����ͨ���ಽ�����漰NF-kB��������ϸ��������ϸ����ʶ����κ���֯���˻�����Ӧ��ʱ���������Ȼἤ��NF-kB��Ȼ���NF-kB��ϸ�������ź����γ���֢С�嵰���������1��2�������������������������������Խ�һ����Ӧ��֯���˻�����Ӧ�����ڴ˲��裨��3�����У��������֢С����ϸ�������ź��Բ������ͷ���֢���ף���IL-1�ȣ���
��Inflammation 101: The Fuel That Feeds Cancer
Nov 11, 2012 Brian D. Lawenda, M.D.
I think most of us typically think of inflammation as our body��s protective response to injury and infection. Without it, we��d be in serious trouble (unless we lived in a sealed bubble.)
If it were only that simple��
Inflammation comes in two categories, ��acute�� and ��chronic.��
Acute Inflammation:
When our body is behaving properly, immune cells (white blood cells, macrophages, etc.) kick into gear when they detect oxidative stress, injury or infection in the tissues. These cells release a storm of inflammatory chemicals and proteins into the surrounding tissues and into the blood stream to:
Destroy any foreign invaders (i.e. bacteria, viruses)
Signal more immune cells to the area
Dilate blood vessels to increase blood flow to the area (to enable the influx of immune cells, oxygen and nutrients)
Trigger the growth of tissues and new blood vessels (to aid in tissue repair and supply nutrients to these tissues)
This process is called ��acute�� inflammation, and will resolve within hours-to-weeks after the infection is controlled or the injury is healed��unless it doesn��t��
Chronic Inflammation:
Whereas acute inflammation protects us from our hostile environment, ��chronic�� inflammation (a prolonged state of low-level, smoldering inflammation, lasting months-to-years) can hurt or even kill us. This difference is one of the most important discoveries of modern medicine. We now recognize that chronic inflammation is involved in the development and progression of almost every chronic disease��including cancer.
The role of chronic inflammation is particularly poignant in the autoimmune and chronic infectious diseases, where the involved tissues are flooded with persistent chemical and protein signaling to repair and grow.
Chronic inflammation can also significantly reduce your body��s sensitivity to insulin, leading to persistently elevated levels of blood sugar, increased production of insulin and insulin-like growth factor 1 (IGF-1), metabolic syndrome, type 2 diabetes and obesity. All of these factors have been shown to increase the risk of cancer development, progression and recurrence.
The ��Inflammasome��
Most of us have never heard of ��inflammasomes�� before, as they were discovered relatively recently (in 2002.) These protein complexes are an integral part of our body��s immune system. They are comprised of special, ��danger sensing proteins�� that are activated by viruses, bacteria, and free radical chemicals.
Once activated, the inflammasomes signal our immune cells (macrophages) to begin to produce a storm of inflammatory proteins. The latest research indicates that inflammasomes are responsible in large part for the chronic inflammatory state that results from obesity.The Role of Nuclear Factor Kappa-B (NF-kB) in Cancer
When immune cells first detect any signs of tissue injury or stress, they activate a cellular protein, called nuclear factor kappa-B (or NF-kB.) This protein is the main ��switch�� that turns on the inflammation in the tissues. Activated NF-kB signals the cell (via many pathways, including through the activation of inflammasomes) to begin to produce numerous proteins involved in inflammation and tissue repair.
So far so good, as long as the growth signals are not happening in precancerous or cancerous tissues��
During chronic inflammatory conditions, inflamed tissues are predisposed to developing and accumulating DNA damage at a greater rate than non-inflamed tissues. This is due to the production of large amounts of free radicals in inflamed tissues [in fact, billions of DNA mutations develop in our cells each day as a result of free radical injury and toxic exposures.] When these highly-reactive chemicals interact with DNA, they can create DNA mutations. As long as the cell is able to either repair these DNA mutations (most cells have built-in DNA repair mechanisms) or signal the cell to undergo a process of cell suicide (��apoptosis��), precancerous cells will not develop.
However, during constant exposure to NF-kB directed signals (which happens during chronic inflammation), both apoptosis and DNA repair mechanisms are shut down. This leads to an accumulation of cells with genetic mutations. If these cells develop enough mutations, they can transform into precancerous cells and, eventually, cancer cells.
In the presence of chronic inflammation, precancerous and cancerous tissues are signaled to divide, grow and even invade into surrounding tissues (including blood and lymph vessels, which gives them access to the rest of the body; this is referred to as ��metastatic�� spread.) These inflammatory signals also inactivate immune cells in the area, preventing them from being able to identify and attack the newly formed cancer cells.
One of the sneaky things that cancer cells learn to do to ensure their own growth and survival, is that they can activate their own NF-kB. This enables them to turn on all of the same inflammatory proteins in the surrounding tissues, thereby hijacking the body��s tissue repair and growth mechanisms.
Experiments have shown that inflammasome activation involves NF-kB through a multi-step process. When the immune cells (macrophages) identify any tissue injury or oxidative stress, they first activate NF-kB. Activated NF-kB then signals the cells to form inflammasome protein complexes (steps 1 and 2), which are now ready (or ��primed) to further respond to the tissue injury or oxidative stress. At this step (step 3), the activated inflammasome signals the cell to produce and release inflammatory protein (i.e. IL-1, etc.)Inflammation 101: The Fuel That Feeds Cancer - Integrative Oncology Essentials
https://integrativeoncology-essentials.com/2012/11/inflammation-101-the-fuel-that-feeds-cancer��
Institute of General Pathology, 2Institute of Pathology, 3Institute of Pharmacology and 4Institute of Histology, Catholic University, L.go F. Vito, 1, 00168 Rome, Italy
Omega-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE 2 induced ERK-1 and -2 and HIF-1�� induction pathwayn-3 Polyunsaturated fatty acids (PUFAs) inhibit the development of microvessels in mammary tumors growing in mice. Human colorectal tumors produce vascular endothelial growth factor (VEGF) whose expression is up-regulated in tumor cells by both cyclooxygenase-2 (COX-2) and PGE 2 and directly correlated to neoangiogenesis and clinical outcome.
The goal of this study was to examine the capability of n-3 PUFAs to regulate VEGF expression in HT-29 human colorectal cells in vitro and in vivo . Constitutive VEGF expression was augmented in cultured HT-29 cells by serum starvation and the effects of eicosapentaenoic (EPA) or docosahexaenoic acid (DHA) on VEGF, COX-2, phosphorylated extracellular signal-regulated kinase (ERK)-1 and -2 and hypoxia-inducible-factor 1-�� (HIF-1��) expression and PGE 2 levels were assessed.
Tumor growth, VEGF, COX and PGE 2 analysis were carried out in tumors derived from HT-29 cells transplanted in nude mice fed with either EPA or DHA. Both EPA and DHA reduced VEGF and COX-2 expression and PGE 2 levels in HT-29 cells cultured in vitro . Moreover, they inhibited ERK-1 and -2 phosphorylation and HIF-1�� protein over-expression, critical steps in the PGE 2 -induced signaling pathway leading to the augmented expression of VEGF in colon cancer cells.
EPA always showed higher efficacy than DHA in vitro . Both fatty acids decreased the growth of the tumors obtained by inoculating HT-29 cells in nude mice, microvessel formation and the levels of VEGF, COX-2 and PGE 2 in tumors.
The data provide evidence that these n-3 PUFAs are able to inhibit VEGF expression in colon cancer cells and suggest that one possible mechanism involved may be the negative regulation of the COX-2/PGE 2 pathway. Their potential clinical application as anti-angiogenic compounds in colon cancer therapy is proposed.
��n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE 2 induced ERK-1 and -2 and HIF-1�� induction pathway | Carcinogenesis | Oxford Academic
https://academic.oup.com/carcin/article/25/12/2303/2475914��
Omega-3֬�������ư��װ�����ģ���е���������
Omega-3 Fatty Acids Inhibit Tumor Growth in a Rat Model of Bladder Cancer
ժҪ
Omega-3����-3��֬�������ڶ��ְ�֢���͵�Ԥ���������н����˲��ԣ�����δ�ԡ����ڡ����װ�����Ч���з����������о�ּ��������ʮ̼��ϩ�ᣨEPA���Ͷ�ʮ��̼��ϩ�ᣨDHA��������ڰ��װ�����ģ���еĻ�ѧԤ����Ч����20�ܵ�ʵ�鷽���У���44ֻ����Wistar�����Ϊ4�飺�°���-N-����-N-��4-�Ƕ�������������BBN���� ��-3��DHA + EPA�����ͦ�-3+ BBN��
�������8���ڸ���BBN�ͦ�-3���ڵ�20��ʱ���ռ�ѪҺ�Ͱ��ײ������·��Ƥ����������Ĵ��ڣ���֢����ֳ��������ԭ״̬�ı�־�����װ��ķ�����Ϊ���գ�0��������-3��0������BBN��65�����ͦ�-3+ BBN��62.5������ ��-3+ BBN��������������ԭλ������BBN��ȣ�����������Լ��٣�0.9��0.1µmm3��112.5��6.4µmm3����ͬ��������ʾ��MDA / TAS���ʽ��ͣ�����ֹ��BBN�յ���Ѫ��CRP��TGF-��1��CD31��
��֮��omega-3֬�����ڰ��װ�����ģ�������ƶ��ǰ�Ͷ��Բ���ķ�չ������������ڿ��ף�������������ֳ�Ϳ�Ѫ�������������¡�
ͼ1
���ĺ����֯��̬ѧ���������գ�a���ͦ�-3-�����Ĵ���b�������а�����ʾ������ۡ���BBN�飨c���У�65.0���Ĵ�����ֳ���ʢ�İ��װ�������ռ���˰�����ǻ������һ���У����ױ��ֳ����ŵ�Ѫ�ܹ�����������ʾ�����γɵ���Ѫ�����ɡ���Ԥ���Ԧ�-3+ BBN�飨d���У����������ٷ�����ͬ��������������Լ��١�
N-����-N-��4-�Ƕ�������������BBN���յ��Ĵ�����װ�ʵ��ģ�����о����֢��չ�ĺ����Ҿ�����֤��ģ�͡�ʵ���ϣ�������������װ�����֯ѧ�����ԣ����ѳ�Ϊ�о��������������Լ��������Ʋ�����Ч�����ģ��[10-12]����·��Ƥ������һ�������������Ĺ��̣��������˴����Ե���Ϯ�Բ���ķ��Ӻ���̬�仯�������������������������Ƥ�쳣������ǰ�ı仯�Լ����Բ��䣨��ͷ״���Ͱ���[12-14]����ˣ����������Щ;�����������ƿ���Ԥ�����װ��ķ�չ����������ǰ�ı����Ѿ�֤����Ԥ�����Ե�����Ч�������������Լ�ʹ�ÿ��Ϳ���ֳ�����е��о�[15-19]��
�����֬���ᣨPUFA�������ھ��������������ã������䵱ϸ����������Դ�������ʺϳɵĵ��ڼ���ϸ��Ĥ�ṹ�������֬����֬�Ļ�����ɲ��֣��Լ����������ԣ����ԣ�ϸ��Ĥ�Ķ���ѧ�Ͷ�̬�仯���Լ����༤�أ�ϸ�����ϸ���ڵ���ʹ���������ǰ��[20��21]��
�ж����������ԣ������Ժ���ֳ�Լ�����ʹϸ�����ڷ�������ת���� ��-3֬���������俹�����Ϳ������ԣ��ѱ�������Ϊ��ѧԤ������/�����Ƽ������˲��ԣ�������ҩ���/����ƽ�����ڶ��ְ�֢[27-30]�������ڰ��װ����ƶ���Ч���д���������ˣ����о���Ŀ���������������շ��İ��װ�����ģ����������-3֬���ᣬ�ر��Ƕ�ʮ̼��ϩ�ᣨEPA���Ͷ�ʮ��̼��ϩ�ᣨDHA���Ļ�ѧԤ��Ч����Omega-3 Fatty Acids Inhibit Tumor Growth in a Rat Model of Bladder Cancer
��EXTRACT
Omega-3 (��-3) fatty acids have been tested on prevention and treatment of several cancer types, but the efficacy on ��in vivo�� bladder cancer has not been analyzed yet. This study aimed at evaluating the chemopreventive efficacy of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) mixture in an animal model of bladder cancer. Forty-four male Wistar rats were divided into 4 groups during a 20-week protocol: control; carcinogen��N-butyl-N-(4-hydroxybutyl) nitrosamine (BBN); ��-3 (DHA + EPA); and ��-3 + BBN. BBN and ��-3 were given during the initial 8 weeks. At week 20 blood and bladder were collected and checked for the presence of urothelium lesions and tumors, markers of inflammation, proliferation, and redox status. Incidence of bladder carcinoma was, control (0%), ��-3 (0%), BBN (65%), and ��-3 + BBN (62.5%). The ��-3 + BBN group had no infiltrative tumors or carcinoma in situ, and tumor volume was significantly reduced compared to the BBN (0.9 �� 0.1 mm3 versus 112.5 �� 6.4 mm3). Also, it showed a reduced MDA/TAS ratio and BBN-induced serum CRP, TGF-��1, and CD31 were prevented. In conclusion, omega-3 fatty acids inhibit the development of premalignant and malignant lesions in a rat model of bladder cancer, which might be due to anti-inflammatory, antioxidant, anti-proliferative, and anti-angiogenic properties.
The experimental model of rat bladder cancer induced by N-butyl-N-(4-hydroxybutyl) nitrosamine (BBN) is an appropriate and validated model to study human cancer development. In fact, due to the histological similarities with the human bladder cancer, it has been the most used model for the study of tumor pathophysiology, as well as for the evaluation the efficacy of therapeutic strategies [10�C12]. The urothelial carcinogenesis is a continuous and slow process that goes through molecular and morphological changes, from benign to aggressive lesions, including initial dysplastic and proliferative epithelial abnormalities, preneoplastic changes, and malignant lesions (papilloma and carcinoma) [12�C14]. Thus, an early treatment targeting these pathways could hypothetically prevent bladder cancer development and growth. Previous reports have demonstrated beneficial effects of preventive strategies, including our own studies using anti-inflammatory and anti-proliferative agents [15�C19].
Polyunsaturated fatty acids (PUFAs) have many physiological roles in the body, including acting as sources of cellular energy, regulators of protein synthesis, building blocks of phospholipids and glycolipids required for cell membrane structure, and components of membranes that regulate the fluidity, permeability, and dynamics of cell membranes, as well as precursors for many hormones, inter- and intracellular messengers as well as their receptors [20, 21].
There is a wide range of chronic inflammatory, oxidative, and proliferative conditions that make cells susceptible to neoplastic transformation. The omega-3 fatty acids, due to their antioxidant and anti-inflammatory properties have been tested as chemopreventive and/or therapeutic agents, per se and in combination with drugs and/or radiotherapy, in several cancer types [27�C30], but the putative efficacy on bladder cancer remains to be elucidated. The purpose of this study was, thus, to evaluate the chemopreventive efficacy of ��-3 fatty acids, in particular eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), in a rat model of bladder cancer induced by a nitrosamine.Omega-3 Fatty Acids Inhibit Tumor Growth in a Rat Model of Bladder Cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3705844/��
n-3 PUFA�����˽᳦��ϸ����VEGF�ı���Ӷ�����COX-2 / PGE 2�յ���ERK-1��-2�Լ�HIF-1�����յ�;��
n-3�����֬���ᣨPUFA������С������������Ѫ�ܵķ�����
�˽᳦ֱ����������Ѫ����Ƥ�������ӣ�VEGF���������������ϸ���б�������ø2��COX-2����PGE 2�ϵ�����������Ѫ�����ɺ��ٴ����ֱ����ء�
�����о���Ŀ����������������о�n-3 PUFA����HT-29�˽᳦ֱ��ϸ����VEGF�����������HT-29�˽᳦ֱ��ϸ���ڼ���Ѫ�幩Ӧ�DZ���VEGF�������˶�ʮ̼��ϩ�ᣨEPA�����ʮ��̼��ϩ�ᣨDHA����VEGF��COX-2�����ữϸ�����źŵ��ڼ�ø��ERK��-1��ERK-2��ȱ���յ�����1-����HIF-1�����ı����PGE 2��ˮƽ��Ӱ�졣
EPA��DHAι������������ֲ��HT-29ϸ����������������VEGF��COX��PGE 2�� EPA��DHA���ɽ�������������HT-29ϸ��VEGF��COX-2���PPGE 2ˮƽ�����⣬��������ERK-1��-2���ữ�Լ�HIF-1�����Ĺ��ȱ������PGE 2�յ����źŴ���;���еĹؼ����裬���½᳦��ϸ����VEGF�ı������ӡ�
EPA���������DZ��ֳ���DHA���ߵĹ�Ч������֬�����������ͨ���������н���HT-29ϸ����õ�������������Ѫ���γ��Լ�������VEGF��COX-2��PGE 2��ˮƽ��
�����ṩ����Щn-3 PUFA�ܹ����ƽ᳦��ϸ����VEGF�����֤�ݣ��������漰��һ�ֿ��ܵĻ��ƿ�����COX-2 / PGE 2;���ĸ����ء�
���������Ϊ��Ѫ�����ɻ������ڽ᳦�������е�DZ���ٴ�Ӧ�á�n-3 Polyunsaturated fatty acids (PUFAs) inhibit the development of microvessels in mammary tumors growing in mice.
Institute of General Pathology, 2Institute of Pathology, 3Institute of Pharmacology and 4Institute of Histology, Catholic University, L.go F. Vito, 1, 00168 Rome, Italy
��
Human colorectal tumors produce vascular endothelial growth factor (VEGF) whose expression is up-regulated in tumor cells by both cyclooxygenase-2 (COX-2) and PGE 2 and directly correlated to neoangiogenesis and clinical outcome.
The goal of this study was to examine the capability of n-3 PUFAs to regulate VEGF expression in HT-29 human colorectal cells in vitro and in vivo .
Constitutive VEGF expression was augmented in cultured HT-29 cells by serum starvation and the effects of eicosapentaenoic (EPA) or docosahexaenoic acid (DHA) on VEGF, COX-2, phosphorylated extracellular signal-regulated kinase (ERK)-1 and -2 and hypoxia-inducible-factor 1-�� (HIF-1��) expression and PGE 2 levels were assessed.
Tumor growth, VEGF, COX and PGE 2 analysis were carried out in tumors derived from HT-29 cells transplanted in nude mice fed with either EPA or DHA. Both EPA and DHA reduced VEGF and COX-2 expression and PGE 2 levels in HT-29 cells cultured in vitro . Moreover, they inhibited ERK-1 and -2 phosphorylation and HIF-1�� protein over-expression, critical steps in the PGE 2 -induced signaling pathway leading to the augmented expression of VEGF in colon cancer cells.
EPA always showed higher efficacy than DHA in vitro . Both fatty acids decreased the growth of the tumors obtained by inoculating HT-29 cells in nude mice, microvessel formation and the levels of VEGF, COX-2 and PGE 2 in tumors.
The data provide evidence that these n-3 PUFAs are able to inhibit VEGF expression in colon cancer cells and suggest that one possible mechanism involved may be the negative regulation of the COX-2/PGE 2 pathway.
Their potential clinical application as anti-angiogenic compounds in colon cancer therapy is proposed.
n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE 2 induced ERK-1 and -2 and HIF-1�� induction pathway | Carcinogenesis | Oxford Academic
https://academic.oup.com/carcin/article/25/12/2303/2475914#authorNotesSectionTitle��
Omega-3 Fatty Acids Inhibit Tumor Growth in a Rat Model of ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3705844
Jun 20, 2013 �� Omega-3 (��-3) fatty acids have been tested on prevention and treatment of several cancer types, but the efficacy on ��in vivo�� bladder cancer has not been analyzed yet.This study aimed at evaluating the chemopreventive efficacy of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) mixture in an animal model of bladder cancer.
Cited by: 14
Publish Year: 2013
Author: Belmiro Parada, Fl��vio Reis, Raquel Cerejo, Patr��cia Garrido, Jos�� Sereno, Maria Xavier-Cunha, Paula...
Academic Editor: Achille Cittadini
Omega-3 fatty acid compound found to halt cancerous tumor ...
https://www.naturalnews.com/040931_omega-3_fatty_acids_cancer_tumors.html
Jun 25, 2013 �� Omega-3 DHA metabolite stops cancerous metastasis by halting blood supply to tumors The bioactive long-chain omega-3 fat, docosahexaenoic acid (DHA), found in its free form in fatty fish and fish oil supplements, is broken down by the body into a metabolite form known as epoxy docosapentaenoic acid (EDP).
UC Davis Researchers: Key Discovery Involving Omega-3 ...
entomology.ucdavis.edu/News/UC_Davis_Researchers...
Apr 01, 2013 �� UC Davis Researchers: Key Discovery Involving Omega-3 Fatty Acids and Cancer ... discovered cytochrome P450 epoxygenase metabolites of omega-3 fatty acid DHA or epoxy docosapentaenoic acids (EDPs) block blood supply to the tumor and ... ��The study by Zhang and colleagues has uncovered a previously unrecognized anti-cancer effect of omega-3 ...UC Davis Researchers: Key Discovery Involving Omega-3 Fatty Acids and Cancer - UC Davis Department of Entomology and Nematology
http://entomology.ucdavis.edu/News/UC_Davis_Researchers__Key_Discovery_Involving_Omega-3_Fatty_Acids_and_Cancer/��
GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects
1
Department of Medicine, Division of Endocrinology and Metabolism, University of California, San Diego, La Jolla, CA 92093, USA
2
Division of Pharmacology, Shiga University of Medical Science, Tsukinowa, Seta, Otsu-city, Shiga, 520-2192 Japan
3
Tethys Bioscience, 3410 Industrial Boulevard, Suite 103, West Sacramento, CA 95691, USA
Summary
Omega-3 fatty acids (��-3 FAs), DHA and EPA, exert anti-inflammatory effects, but the mechanisms are poorly understood. Here, we show that the G protein-coupled receptor 120 (GPR120) functions as an ��-3 FA receptor/sensor. Stimulation of GPR120 with ��-3 FAs or a chemical agonist causes broad anti-inflammatory effects in monocytic RAW 264.7 cells and in primary intraperitoneal macrophages. All of these effects are abrogated by GPR120 knockdown. Since chronic macrophage-mediated tissue inflammation is a key mechanism for insulin resistance in obesity, we fed obese WT and GPR120 knockout mice a high-fat diet with or without ��-3 FA supplementation. The ��-3 FA treatment inhibited inflammation and enhanced systemic insulin sensitivity in WT mice, but was without effect in GPR120 knockout mice. In conclusion, GPR120 is a functional ��-3 FA receptor/sensor and mediates potent insulin sensitizing and antidiabetic effects in vivo by repressing macrophage-induced tissue inflammation.GPR120 Is an Omega-3 Fatty Acid Receptor Mediating Potent Anti-inflammatory and Insulin-Sensitizing Effects - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0092867410008883��
Akt-mediated regulation of NF��B and the essentialness of ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2767458
Dec 15, 2009 �� The serine/threonine kinase Akt (cellular homolog of murine thymoma virus akt8 oncogene), also known as PKB (protein kinase B), is activated by lipid products of phosphatidylinositol 3-kinase (PI3K). Akt phosphorylates numerous protein targets ��
Cited by: 281
Publish Year: 2009
Author: Dong Bai, Lynn Ueno, Peter K. Vogt
��NF-��B activation by tumour necrosis factor requires the Akt serine�Cthreonine kinase | Nature
Publish Year: 1999
NF-��B activation by tumour necrosis factor requires the Akt serine�Cthreonine kinase
Abstract
Activation of the nuclear transcription factor NF-��B by inflammatory cytokines requires the successive action of NF-��B-inducing kinase (NIK) and an I��B-kinase (IKK) complex composed of IKK�� and IKK��1,2,3,4,5. Here we show that the Akt serine�Cthreonine kinase6 is involved in the activation of NF-��B by tumour necrosis factor (TNF). TNF activates phosphatidylinositol-3-OH kinase (PI(3)K) and its downstream target Akt (protein kinase B). Wortmannin (a PI(3)K inhibitor), dominant-negative PI(3)K or kinase-dead Akt inhibits TNF-mediated NF-��B activation. Constitutively active Akt induces NF-��B activity and this effect is blocked by dominant-negative NIK. Conversely, NIK activates NF-��B and this is blocked by kinase-dead Akt. Thus, both Akt and NIK are necessary for TNF activation of NF-��B. Akt mediates IKK�� phosphorylation at threonine 23. Mutation of this amino acid blocks phosphorylation by Akt or TNF and activation of NF-��B. These findings indicate that Akt is part of a signalling pathway that is necessary for inducing key immune and inflammatory responses.
NF-��B activation by tumour necrosis factor requires the Akt serine�Cthreonine kinase | Nature
https://www.nature.com/articles/43466
��
NF-��B activation by tumour necrosis factor requires the Akt serine�Cthreonine kinase | Nature
https://www.nature.com/articles/43466
Cross-Talk between NFkB and the PI3-Kinase/AKT Pathway Can ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3386924
Jun 29, 2012 �� Cross-talk between NFkB pathway and PI3-kinase/AKT pathway has been shown in various cancers , , however, the relationship between these two pathways have not been fully explored in PEL cells. We found that inhibition of NFkB pathway by Bay11-7085 not only inactivates the NFkB pathway, it also inactivates AKT as well as down-stream targets of ...
Cited by: 116
Publish Year: 2012
A��
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC444845
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits I��B�� Kinase �� Juan M. C��rcamo , 1, 2 Alicia Pedraza , 2 Oriana B��rquez-Ojeda , 2 Bing Zhang , 3 Roberto Sanchez , 3 and David W. Golde 2, *
Cited by: 106
Publish Year: 2004
Author: Juan M. C��rcamo, Alicia Pedraza, Oriana B��rquez-Ojeda, Bing Zhang, Roberto Sanchez, David W. Golde
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid ...
europepmc.org/articles/PMC444845
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits I��B�� Kinase �� Juan M. C��rcamo , 1, 2 Alicia Pedraza , 2 Oriana B��rquez-Ojeda , 2 Bing Zhang , 3 Roberto Sanchez , 3 and David W. Golde 2, *
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid ...
https://www.researchgate.net/publication/8455763_Vitamin_C_Is_a_Kinase_Inhibitor...
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid Inhibits I B Kinase Article in Molecular and Cellular Biology 24(15):6645-52 �� September 2004 with 103 Reads How we measure 'reads'
��
https://content.iospress.com/articles/journal-of-alzheimers-disease/jad170589
��
Pharmacological Ascorbate-Induced HIF-1�� Degradation and VEGF Suppression in Pancreatic Cancer
Justin Wilkes, MD, Juan Du, PhD, Zita Sibenaller, PhD, Adrienne Klinger, Joseph J. Cullen, MD, FACS
University of Iowa, Carver College of Medicine, Iowa City, IAHypoxia-inducible factor-1�� (HIF-1��) mediates cellular responses to hypoxia by up-regulating vital homeostatic processes, such as vascular endothelial growth factor (VEGF). In pancreatic adenocarcinoma, HIF-1�� strongly affects prognosis and correlates with metastatic disease. Pharmacologic doses of ascorbate (P-AscH-; vitamin C) inhibit pancreatic cancer cell growth by an auto-oxidation reaction resulting in generation of extracellular hydrogen peroxide. We hypothesized that in hypoxic conditions, P-AscH- induces HIF-1�� degradation, VEGF suppression, and cytotoxicity.
��Pharmacological Ascorbate-Induced HIF-1�� Degradation and VEGF Suppression in Pancreatic Cancer - Journal of the American College of Surgeons
https://www.journalacs.org/article/S1072-7515(16)30403-3/abstract��
Expression of hypoxia inducible factor 1�� and 2�� and its ...
https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-017-0388-y
Oct 30, 2017 �� We found an inverse correlation between vitamin C level and HIF-1�� but not HIF-2�� expression in thyroid lesions. These results agree with our in vitro study showing that vitamin C induced a dose - dependent decrease of HIF-1�� but not HIF-2�� protein level in ��
Cited by: 4
Publish Year: 2017
Author: Paweł J��źwiak, Piotr Ciesielski, Agnieszka Z��
Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury.
Department of Pharmacology, Weill Medical College, Cornell University, New York, New York 10021, USA.
Abstract
Reactive oxygen species (ROS)-induced mitochondrial abnormalities may have important consequences in the pathogenesis of degenerative diseases and cancer. Vitamin C is an important antioxidant known to quench ROS, but its mitochondrial transport and functions are poorly understood. We found that the oxidized form of vitamin C, dehydroascorbic acid (DHA), enters mitochondria via facilitative glucose transporter 1 (Glut1) and accumulates mitochondrially as ascorbic acid (mtAA). The stereo-selective mitochondrial uptake of D-glucose, with its ability to inhibit mitochondrial DHA uptake, indicated the presence of mitochondrial Glut. Computational analysis of N-termini of human Glut isoforms indicated that Glut1 had the highest probability of mitochondrial localization, which was experimentally verified via mitochondrial expression of Glut1-EGFP. In vitro mitochondrial import of Glut1, immunoblot analysis of mitochondrial proteins, and cellular immunolocalization studies indicated that Glut1 localizes to mitochondria. Loading mitochondria with AA quenched mitochondrial ROS and inhibited oxidative mitochondrial DNA damage. mtAA inhibited oxidative stress resulting from rotenone-induced disruption of the mitochondrial respiratory chain and prevented mitochondrial membrane depolarization in response to a protonophore, CCCP. Our results show that analogous to the cellular uptake, vitamin C enters mitochondria in its oxidized form via Glut1 and protects mitochondria from oxidative injury. Since mitochondria contribute significantly to intracellular ROS, protection of the mitochondrial genome and membrane may have pharmacological implications against a variety of ROS-mediated disorders.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC444845��
Hum Mol Genet. 2010 Oct 1;19(19):3721-33. doi: 10.1093/hmg/ddq286. Epub 2010 Jul 16.
Mitochondrial GLUT10 facilitates dehydroascorbic acid import and protects cells against oxidative stress: mechanistic insight into arterial tortuosity syndrome.
Lee YC1, Huang HY, Chang CJ, Cheng CH, Chen YT.
Author information
1
Institute of Biomedical Sciences, Academia Sinica, Taipei 11529, Taiwan, Republic of China.
Abstract
Mutations in glucose transporter 10 (GLUT10) alter angiogenesis and cause arterial tortuosity syndrome (ATS); however, the mechanisms by which these mutations cause disease remain unclear. It has been reported that in most cells, mitochondria are the major source of reactive oxygen species (ROS). Moreover, mitochondria are known to incorporate as well as recycle vitamin C, which plays a critical role in redox homeostasis, although the molecular mechanism(s) underlying mitochondrial vitamin C uptake are poorly understood. We report here that GLUT10 localizes predominantly to the mitochondria of smooth muscle cells and insulin-stimulated adipocytes, where GLUT10 is highly expressed. We further demonstrate that GLUT10 facilitates transport of l-dehydroascorbic acid (DHA), the oxidized form of vitamin C, into mitochondria, and also increases cellular uptake of DHA, which in turn protects cells against oxidative stress. This protection is compromised when GLUT10 expression in mitochondria is inhibited. In addition, we found that aortic smooth muscle cells from GLUT10-mutant mice have higher ROS levels than those from wild-type mice. Our results identify the physiological role of GLUT10 as the mitochondrial DHA transporter, and demonstrate that GLUT10 protects cells from oxidative injury. Furthermore, our findings provide a mechanism to explain the ascorbate in mitochondria and show how loss-of-function GLUT10 mutations may lead to arterial abnormalities in ATS. These results also reinforce the importance of vitamin C and ROS in degenerative diseases.
Mitochondrial GLUT10 facilitates dehydroascorbic acid import and protects cells against oxidative stress: mechanistic insight into arterial tortuos... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/20639396��
Vitamin K Inhibits NF-kappaB Activation
https://www.wellnessresources.com/studies/vitamin-k-inhibits-nf-kappab-activation
Furthermore, menaquinone-4, a form of vitamin K2, reduced the activation of nuclear factor ��B (NF��B) and inhibited the phosphorylation of IKK��/�� after treatment of cells with LPS. These results clearly show that the anti-inflammatory activity of vitamin K is mediated via the inactivation of the NF��B signaling ��
Vitamin C blocks TNF-��-induced NF-kB activation and ICAM-1 expression in human neuroblastoma cells
Abstract
Interactions of the cell adhesion molecules are known to play important roles in mediating inflammation. The proinflammatory cytokine, tumor necrosis factor-�� (TNF-��), activates the NF-kB signaling pathway, which induces the expression of various genes, such as intercellular adhesion molecule-1 (ICAM-1). In this study, the effect of vitamin C on the ICAM-1 expression induced by TNF-�� in a human neuroblastoma cell line, SK-N-SH was investigated. Treatment with vitamin C resulted in the downregulation of the TNF-��-induced surface expression and ICAM-1 mRNA levels in a concentration-dependent manner. Moreover, a gel shift analysis indicated that vitamin C dose-dependently inhibited the NF-kB activation and lkB�� degradation induced by TNF-��. Taken together, these results suggest that vitamin C downregulates TNF-��-induced ICAM-1 expression via the inhibition of NF-kB activation.
Vitamin C bloc ks TNF-��-induced NF- kB activation and ICAM-1 expression in human neuroblastoma cellsSpringerLink
https://link.springer.com/article/10.1007/BF02975434
Vitamin C Inhibits NF-��B Activation by TNF Via the Activation of p38 Mitogen-Activated Protein Kinase
Abstract
The transcription factor NF-��B is a central mediator of altered gene expression during inflammation, and is implicated in a number of pathologies, including cancer, atherosclerosis, and viral infection. We report in this study that vitamin C inhibits the activation of NF-��B by multiple stimuli, including IL-1 and TNF in the endothelial cell line ECV304 and in primary HUVECs. The induction of a NF-��B-dependent gene, IL-8, by TNF was also inhibited. The effect requires millimolar concentrations of vitamin C, which occur intracellularly in vivo, particularly during inflammation. Vitamin C was not toxic to cells, did not inhibit another inducible transcription factor, STAT1, and had no effect on the DNA binding of NF-��B. Inhibition by vitamin C was not simply an antioxidant effect, because redox-insensitive pathways to NF-��B were also blocked. Vitamin C was shown to block IL-1- and TNF-mediated degradation and phosphorylation of I-��B�� (inhibitory protein that dissociates from NF-��B), due to inhibition of I-��B kinase (IKK) activation. Inhibition of TNF-driven IKK activation was mediated by p38 mitogen-activated protein kinase, because treatment of cells with vitamin C led to a rapid and sustained activation of p38, and the specific p38 inhibitor SB203580 reversed the inhibitory effect of vitamin C on IKK activity, I-��B�� phosphorylation, and NF-��B activation. The results identify p38 as an intracellular target for high dose vitamin C.
Vitamin C Inhibits NF-��B Activation by TNF Via the Activation of p38 Mitogen-Activated Protein Kinase | The Journal of Immunology
https://www.jimmunol.org/content/165/12/7180
Quantum chemical study of the autoxidation of ascorbate ...
https://www.researchgate.net/publication/304104302_Quantum_chemical_study_of_the...Ascorbate readily undergoes pH-dependent autoxidation producing hydrogen peroxide (H(2)O(2)). In the presence of catalytic metals this oxidation is accelerated.
��
Vitamin K Inhibits NF-kappaB Activation
Abstract
Vitamin K is essential for blood coagulation and bone metabolism in mammals. This vitamin functions as a cofactor in the posttranslational synthesis of ��-carboxyglutamic acid (Gla) from glutamic acid residues. However, other functions of vitamin K have been reported recently. We previously found that vitamin K suppresses the inflammatory reaction induced by lipopolysaccharide (LPS) in rats and human macrophage-like THP-1 cells. In this study, we further investigated the mechanism underlying the anti-inflammatory effect of vitamin K by using cultures of LPS-treated human- and mouse-derived cells. All the vitamin K analogues analyzed in our study exhibited varied levels of anti-inflammatory activity. The isoprenyl side chain structures, except geranylgeraniol, of these analogues did not show such activity; warfarin did not interfere with this activity. The results of our study suggest that the 2-methyl-1,4-naphtoquinone ring structure contributes to express the anti-inflammatory activity, which is independent of the Gla formation activity of vitamin K. Furthermore, menaquinone-4, a form of vitamin K2, reduced the activation of nuclear factor ��B (NF��B) and inhibited the phosphorylation of IKK��/�� after treatment of cells with LPS. These results clearly show that the anti-inflammatory activity of vitamin K is mediated via the inactivation of the NF��B signaling pathway.
Vitamin K Inhibits NF-kappaB Activation
https://www.wellnessresources.com/studies/vitamin-k-inhibits-nf-kappab-activation��
The NF-��B Pathway and Cancer Stem Cells
1Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, NC 27599, USA; ude.cnu.liame@knira
Abstract
The NF-��B transcription factor pathway is a crucial regulator of inflammation and immune responses. Additionally, aberrant NF-��B signaling has been identified in many types of cancer. Downstream of key oncogenic pathways, such as RAS, BCR-ABL, and Her2, NF-��B regulates transcription of target genes that promote cell survival and proliferation, inhibit apoptosis, and mediate invasion and metastasis. The cancer stem cell model posits that a subset of tumor cells (cancer stem cells) drive tumor initiation, exhibit resistance to treatment, and promote recurrence and metastasis. This review examines the evidence for a role for NF-��B signaling in cancer stem cell biology.
Keywords: NF-��B, cancer, cancer stem cells, tumor-initiating cells, self-renewal
The NF-��B Pathway and Cancer Stem Cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4931665/��
Ovarian cancer: New drug may prevent recurrence
New research in mice identifies a compound that prevents ovarian cancer recurrence by eradicating the cancer stem-like cells that conventional chemotherapy leaves behind.
New findings could change the face of ovarian cancer treatment.
According to the National Cancer Institute, there were over 22,000 new cases of ovarian cancer in the United States in 2018. More than 14,000 of these resulted in death.
Ovarian cancer is not very common, but its recurrence rate is notoriously high. According to previous estimates, "between 70 and 90 percent of all women with ovarian cancer" will have a recurrence at some point after their diagnosis, depending on how advanced the disease was.
While a person's outlook depends on various factors, just under 50 percent of those who develop ovarian cancer go on to survive for 5 years, according to more recent data.
New research, however, may have found a way to prevent ovarian cancer from coming back. A team of scientists, led by Dr. Ronald Buckanovich, who is also a professor of medicine at the University of Pittsburgh in Pennsylvania, discovered a drug that targets stem-like ovarian cancer cells and stops the cancer from returning.
Dr. Buckanovich and his colleagues published their findings in the journal Cell Reports.
Plucking the root of ovarian cancer
Dr. Buckanovich explains that, even though chemotherapy can destroy up to 99 percent of ovarian cancer cells, the treatment still "misses" stem-like cancer cells.
"You can think of stem-like cells as seeds," explains Dr. Buckanovich, who is also the co-director of the Women's Cancer Research Center at Pittsburgh University. "They put down roots and grow into a plant," he says.
"I especially like the dandelion analogy. When we treat cancer, we're essentially mowing the lawn. But the problem is that dandelions always come back."
The researcher also explains that 11 stem-like cancer cells are enough to form a tumor. So, the researchers set out to find a compound that can eradicate these stem-like cells.
To do so, the scientists carried out a range of chemical experiments. Specifically, they aimed to find a compound that inhibits a pathway known as ALDHA. The cancer cells rely on this pathway to get rid of the toxins that they produce when they replicate quickly.
Study co-author Edward Grimley, Ph.D., who is a Postdoctoral Research Associate in the lab of Dr. Buckanovich, spoke to Medical News Today about the methods the researchers used to find the ALDHA-inhibitor.
Grimley explained that the team screened for a variety of chemical analogs to a "small-molecule known to inhibit the ALDH1A family of enzymes."
"From these experiments, we identified 673A, a potent inhibitor of the ALDH1A family," said Grimley. Then, the researchers showed that this drug efficiently killed cancer stem-like cells in ovarian cancer cell lines.Ovarian cancer: New drug may prevent recurrence
https://www.medicalnewstoday.com/articles/324706.php#1��
Surgery for Recurrent Ovarian Cancer Does Not Help Survival - National Cancer Institute
Median overall survival was 50.6 months in the surgery group and 64.7 months in the no-surgery group.
��
NF-kB promotes ovarian tumorigenesis via classical pathways supporting proliferative cancer cells and alternative pathways supporting ALDH+ cancer stem-like cells
Carrie D House, Elizabeth Jordan, Lidia Hernandez, Michelle Ozaki, Jana M James, Marianne Kim, Michael J Kruhlak, Eric Batchelor, Fathi Elloumi, Margaret C. Cam and Christina M. Annunziata1Women's Malignancies Branch, NCI/NIH
2Medical Oncology Branch, NCI/NIH
3Women's Malignancies Branch, NIH/NCI
4Medical Oncology Branch, National Cancer Institute
5Experimental Immunology Branch, National Cancer Institute
6Laboratory of Pathology, National Cancer Institute
7Developmental Therapeutics Branch, National Institutes of Health
8Neuro-Oncology Branch, National Institutes of Health
Abstract
Understanding the mechanisms supporting tumor-initiating cells (TIC) is vital to combat advanced stage recurrent cancers. Here we show that in advanced ovarian cancers NF-kB signaling via the RelB transcription factor supports TIC populations by directly regulating the cancer stem-like associated enzyme aldehyde dehydrogenase (ALDH). Loss of RelB significantly inhibited spheroid formation, ALDH expression and activity, chemoresistance, and tumorigenesis in subcutaneous and intrabursal mouse xenograft models of human ovarian cancer. RelB also affected expression of the ALDH gene ALDH1A2. Interestingly, classical NF-kB signaling through the RelA transcription factor was equally important for tumorigenesis in the intrabursal model, but had no effect on ALDH. In this case, classical signaling via RelA was essential for proliferating cells, whereas the alternative signaling pathway was not. Our results show how NF-kB sustains diverse cancer phenotypes via distinct classical and alternative signaling pathways, with implications for improved understanding of disease recurrence and therapeutic response.
Received February 3, 2017.
Revision received February 13, 2017.
Accepted October 20, 2017.
Copyright ©2017, American Association for Cancer Research.NF-kB promotes ovarian tumorigenesis via classical pathways supporting proliferative cancer cells and alternative pathways supporting ALDH+ cancer stem-like cells | Cancer Research
https://cancerres.aacrjournals.org/content/early/2017/10/25/0008-5472.CAN-17-0366��
��
Interactions of NF-��B signaling pathway in pancreatic cancer (PC). As indicated in the diagram, both classical and non-classical NF-��B pathways interact with a number of signaling pathways to remain constitutively activated in pancreatic cancer cells and promotes tumorigenesis and metastasis.
��
��
��
NF-��B in pancreatic cancer: Its key role in chemoresistance
Author links open overlay panelQuanxiaoLiab1GangYanga1MengyuFengaSuliZhengaZheCaoaJiangdongQiuaLeiYouaLianfangZhengcYaHuaTaipingZhangadYupeiZhaoa
a
Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100730, China
b
Second Affiliated Hospital, Harbin Medical University, Harbin, 150086, China
c
Department of Nuclear Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100730, China
d
Clinical Immunology Center, Chinese Academy of Medical Sciences, Beijing, 100730, China
Highlights
•
NF-��B activation influences biological behaviors and chemoresistance in PC cells.
•
Activation of NF-��B responding to chemotherapy results in inducible chemoresistance.
•
Side effects need be considered when inhibiting NF-��B in PC adjuvant therapy.
Abstract
Pancreatic cancer is considered a lethal disease with a high mortality and an extremely low five-year survival rate. Chemotherapy plays a pivotal role in pancreatic cancer treatment both in an adjuvant setting after complete resection and in the case of unresectable metastatic disease. However, none of the available combination chemotherapy regimens has resulted in satisfactory survival outcomes. Recent studies have revealed that both constitutive and induced activation of nuclear factor kappa B (NF-��B) in pancreatic cancer cells are closely associated with cell proliferation, invasion, anti-apoptosis, inflammation, angiogenesis and chemotherapeutic resistance. Therefore, NF-��B inhibitors in combination with cytotoxic compounds have been reported as novel agents that improve chemotherapy sensitivity in pancreatic cancer. In this review, we outline recent developments in the understanding of the role of the NF-��B signaling pathway and its associated genes in the progression of pancreatic cancer and highlight some potentially effective strategies for pancreatic cancer treatment.��
NF-��B in pancreatic cancer: Its key role in chemoresistance - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0304383518301381��
Cancers (Basel). 2019 Aug; 11(8): 1182.
Published online 2019 Aug 15. doi: 10.3390/cancers11081182
NF-��B Signaling in Ovarian Cancer
Brittney S. Harrington and Christina M. Annunziata*
Women��s Malignancies Branch, National Cancer Institute, Bethesda, MD 20892, USA
Abstract
The NF-��B signaling pathway is a master and commander in ovarian cancer (OC) that promotes chemoresistance, cancer stem cell maintenance, metastasis and immune evasion. Many signaling pathways are dysregulated in OC and can activate NF-��B signaling through canonical or non-canonical pathways which have both overlapping and distinct roles in tumor progression. The activation of canonical NF-��B signaling has been well established for anti-apoptotic and immunomodulatory functions in response to the tumor microenvironment and the non-canonical pathway in cancer stem cell maintenance and tumor re-initiation. NF-��B activity in OC cells helps to create an immune-evasive environment and to attract infiltrating immune cells with tumor-promoting phenotypes, which in turn, drive constitutive NF-��B activation in OC cells to promote cell survival and metastasis. For these reasons, NF-��B is an attractive target in OC, but current strategies are limited and broad inhibition of this major signaling pathway in normal physiological and immunological functions may produce unwanted side effects. There are some promising pre-clinical outcomes from developing research to target and inhibit NF-��B only in the tumor-reinitiating cancer cell population of OC and concurrently activate canonical NF-��B signaling in immune cells to promote anti-tumor immunity.
Keywords: nuclear factor-kappa B, RelA, RelB, stemness, chemoresistance, ovarian cancer��
1. Introduction
Ovarian cancer (OC) is the most lethal gynecologic malignancy [1]. The low survival rate of approximately 40% owes to the intra- and inter-tumor heterogeneity, diagnosis at an advanced stage and high rate of recurrence and chemoresistance that classifies this disease [2]. There is increasing evidence that the high rate of disease recurrence after chemotherapy is due to residual populations of cancer stem cells (CSCs) that escaped therapy and re-establish the disease, leading to progression [3]. Populations of CSCs have been identified by certain markers in OC such as CD133, CD44, and ALDH and efforts are focused on strategies to target these populations to prevent recurrent disease [3,4]. The inflammatory but immunosuppressive tumor microenvironment also contributes to OC progression by the production of cytokines that promote survival signaling in OC cells and suppress the infiltration of anti-tumor immune cells [5].
The increased expression of NF-��B signaling molecules had been associated with poor prognosis in many cancers including bladder cancer [6], non-small-cell lung cancer [7] and renal cell carcinoma [8,9]. In OC, the expression levels of NF-��B proteins, p65 and p50, are elevated in malignant and borderline serous ovarian tumors [10,11] and the elevated expression of subunits of the NF-��B pathway, such as the activating I��B kinases (IKKs) �� and ��, have been associated with worse overall survival in OC [12,13,14,15]. Constitutive activation of NF-��B was found to be a feature of cell lines derived from granulosa cell tumors of the ovary, thought to be driven by IKK��; however, the upstream drivers of this pathway were not fully elucidated [16]. In terms of prognostic value, the elevated expression of NF-��B subunits, p65 and p50, has been reported as being greater than 90% of OC cases examined [17,18] and is, therefore, not useful to distinguish patients based on expression of these markers alone. Correlative studies investigating NF-��B pathway proteins and other markers have been reported. The inhibitory proteins that sequester NF-��B in the cytoplasm I��B inversely correlate with p65 expression in OC with poor prognosis, where loss of I��B and increased total p65 expression was associated with worse overall survival [19]. Similarly, the high expression of NF-��B p65 combined with low expression of tumor suppressor PTEN were found to be independent risk factors for chemoresistant ovarian cancer [18]. Another study, focusing on upstream activators of canonical NF-��B signaling in OC, found that high expression of both toll-like receptor 4 (TLR4) and Myd-88 was associated with a poorer outcome and a more aggressive tumor phenotype and was significantly correlated with the expression of NF-��B pathway proteins IKK�� and matrix metalloprotease 9 (MMP9) [17]. The prognostic significance of NF-��B activity in OC is related to its roles in promoting properties of aggressive OC such as chemoresistance [15,20,21,22,23,24] and maintenance of cancer stem cell populations that contribute to recurrence [3,25].
The NF-��B signaling pathway has been implicated in the progression of many cancers, in both blood and solid malignancies [26]. The NF-��B pathway controls multiple cellular processes that can be exploited by cancer cells such as the inhibition of apoptosis in response to chemotherapy, transcription of self-renewal genes in CSCs and the production of anti-tumor cytokines for immune evasion [27]. Due to its role in many normal cellular functions, targeting NF-��B in tumor cells presents a challenge in OC [28]. In this review, we examine the role of the NF-��B signaling pathway in OC chemo-resistance and disease recurrence, inflammation and immunosuppression, and the strategies for targeting this pathway clinically in OC.
��2. Canonical and Non-Canonical NF-��B Signaling in OC
The activation of NF-��B signaling in OC has been shown to promote invasiveness, stemness and chemoresistance. NF-��B is a group of transcription factors consisting of five proteins; RelA/p65, c-Rel, RelB, p50 (NF-��B 1) and p52 (NF-��B 2), which are generated by selective degradation in the proteasome of p105 and p100, respectively [28,29]. The composition of the subunits driving NF-��B transcriptional activity directs the signaling towards the canonical or non-canonical pathway. The canonical pathway activates c-Rel-p50 or RelA-p50/p52, while the non-canonical pathway typically activates RelB-p52 [28,29]. Under basal conditions when NF-��B signaling is inactive, the subunits are sequestered in the cytoplasm bound to inhibitory proteins that must be degraded to allow nuclear translocation for transcriptional activity. Inactive c-Rel or RelA/p65 is bound to an inhibitor of ��B (I��B) or p105, both of which can be proteasomally degraded after phosphorylation by I��B kinases (IKK) to generate the active RelA/p50 dimer for nuclear translocation and activation of canonical signaling [29,30]. Similarly, RelB is bound to p100 in an inactive state, which can also be phosphorylated by an IKK, but this results in ubiquitination and partial degradation of p100 which generates p52 in the proteasome for nuclear translocation and non-canonical transcriptional activity [29,30].
There are overlapping and distinct activators of the canonical and non-canonical NF-��B signaling pathways and these can differ depending on the cell type [28]. Proinflammatory signaling can activate canonical NF-��B signaling in the innate immune response to pathogens and mitogen-activated protein kinase (MAPK) signaling can enhance cell survival through cellular inhibitors of apoptosis proteins (cIAPs) and canonical NF-��B signaling [29]. Non-canonical NF-��B signaling can be activated in response to adaptive immunity, for B cell activation and expansion and in bone remodeling [31]. Recently, research efforts have focused on delineation of the canonical and non-canonical pathways for their roles in disease progression.
2.1. Canonical NF-��B Signaling in Proliferation and Chemoresistance
The canonical pathway of NF-��B activation has been well established for its anti-apoptotic, pro-angiogenic function in many cancers [32,33]. NF-��B transcription is activated after the degradation of inhibitory I��B proteins by phosphorylation by IKKs, allowing translocation to the nucleus with either RelA/p65 (canonical) or RelB (non-canonical) signaling pathway subunits [25,27,34]. Due to their critical function in the activation of NF-��B signaling, IKKs have been investigated as a target for inhibitors to prevent the nuclear translocation of NF-��B subunits for transcriptional activity. In OC, IKK�� was shown to promote the anchorage-independent growth, proliferation and invasion of OC cells [13]. The pro-angiogenic chemokines IL-8 and VEGF are direct targets of NF-��B and are upregulated by activation of canonical NF-��B signaling through IKKs [13,35,36].
Canonical NF-��B signaling through p65/RelA can be activated in response to inflammatory chemokines and cytokines in the tumor microenvironment. Increasing M2 (pro-tumor) macrophage infiltration and increased p65 phosphorylation and NF-��B activity during tumor progression could be observed intra-vitally in a syngeneic mouse model of OC, using an NF-��B-dependent GFP/luciferase reporter cell line [37]. Inhibition of NF-��B activity in this model was associated with decreased expression of M2 markers and increasing expression of M1 (anti-tumor) markers [37]. This has been confirmed in vitro, where increased expression of p65 is observed in OC cells co-cultured with macrophages, which also showed increased invasiveness and migration compared to OC cells alone [38,39]. Inhibition of NF-��B signaling by inhibitors or RNA interference significantly reduced the invasive phenotype of the co-cultured OC cells [39].
NF-��B activity mediates resistance to stressors of OC cells that occurs during disease progression and metastasis such as oxidative stress and anchorage-independent survival [40]. Inhibition of NF-��B activity in cultured OC cell lines significantly inhibited anchorage-independent growth [40]. Canonical NF-��B signaling regulates the expression of several antioxidant response genes to alleviate elevated cellular reactive oxygen species levels in OC cells [40]. Similarly, numerous studies have shown that the NF-��B pathway is activated in platinum-resistant OC cells to modulate apoptosis and promote cell survival [20,22,41,42,43]. In one example, OC cell lines with resistance to multiple therapies (platinum, paclitaxel and erlotinib) had high NF-��B activity, as measured by p65 phosphorylation, which increased with increasing doses of chemotherapy drugs [21]. Treatment with the NF-��B inhibitor BAY 11-7082 significantly reduced the viability, clonogenicity and anoikis resistance of OC cell lines as a monotherapy in vitro and sensitized cells to multiple therapies [21]. The proposed mechanism of action of BAY 11-7082 appeared to be inhibition of anti-apoptotic genes cIAP1, Bcl-2, and Bcl-xl, direct targets of RelA, and enhanced the expression of p21, a regulator of cell proliferation in the resistant cell lines [21].
DNA damage can also activate NF-��B signaling in a retrograde nuclear to cytoplasmic signaling cascade that is dependent on ataxia telangiectasia-mutated (ATM) [44]. Chemotherapeutic agents that cause genotoxic stress, such as etoposide and doxorubicin, activate NF-��B pro-survival signaling [45,46]. In response to DNA damage, the IKK�� subunit��also known as NF-��B essential modulator (NEMO)��translocates into the nucleus for post-translational modification, phosphorylation by ATM and monoubiquitination for nuclear export [44,47]. In the cytoplasm, the modified NEMO-ATM complex can activate IKKs to phosphorylate and degrade inhibitory I��B�� subunits and activate canonical NF-��B survival signaling [44].
Both pro- and anti-apoptotic signaling can be activated downstream of NF-��B in OC cell lines, suggesting a biphasic role for NF-��B in OC progression [24]. In a comparison of chemoresistant cell lines with high NF-��B transcriptional activity with their isogenic, chemosensitive parental cells, it was found that inhibition of the NF-��B pathway using a dominant negative phosphorylation-deficient mutant of I��B�� had opposing effects on the chemoresistant and chemosensitive cells of the same origin [24]. The I��B�� mutation caused reduced tumor growth of the aggressive, chemoresistant cells in vivo but this same mutation acted to promote tumor growth in the isogenic chemosensitive cells [24]. Apoptosis could be induced in the aggressive chemoresistant cell lines by introduction of this mutation, whereas the chemosensitive cells showed reduced apoptosis [41]. This polar difference in activity and cell viability was related to the ability of NF-��B to differentially regulate mitogen-activated protein kinase (MAPK) phosphorylation to produce pro- or anti- apoptotic signaling in OC cells [24]. This study highlighted the canonical pathway activity and its potential to contribute to chemoresistance and OC cell survival.
Canonical NF-��B signaling may be important in maintaining the proliferative cell populations of tumors, whereas non-canonical NF-��B signaling maintains the CSC populations. In a comparative study of shRNAs targeting RelA or RelB, OC cells grown in tumor-initiating cell (TIC) culture conditions had significantly fewer Ki67 positive, proliferative cell populations and reduced viability with loss of RelA, compared with the loss of RelB [4].
2.2. Non-Canonical NF-��B Signaling and OC Persistence
Activation of the non-canonical NF-��B signaling pathway has been associated with CSC maintenance and self-renewal in OC [4]. A number of genes have been identified as markers of CSCs in OC including CD133, CD117, aldehyde dehydrogenases (ALDH) and CD44 [48,49]. In CD44+ SKOV3 cells there was increased expression of RelA but the expression of non-canonical NF-��B proteins RelB and IKK�� was greater, as was the expression of stem cell markers NANOG, SOX2 and OCT4/3 compared to CD44− cells [25]. The CD44+ population showed greater colony-forming capacity in vitro and tumorigenicity in vivo which could be reversed by knocking down IKK�� or by expressing a dominant negative mutant in the CD44+ population [25]. In other cancer types, IKK�� can be activated and function separately from NF-��B pathway signaling [50,51], but no direct evidence of this has been shown in OC as yet. Thus, the importance of IKK�� in this model suggests that the CD44+ CSC population was likely maintained by the non-canonical NF-��B signaling pathway via RelB.
Similar results were reported in the CSC population in other OC cell lines, where both RelA and RelB expression increased in OC cells grown under TIC culture conditions but RelB was more highly expressed in the ALDH+/CD133+ CSC population of OV90 and ACI23 cell lines compared to RelA [4]. Depletion of RelA or RelB in cell line xenografts had differing effects based on the site of tumorigenesis. In subcutaneous and intrabursal xenografts of OC cell lines with inducible knockdown of RelA or RelB with shRNA, loss of either RelA or RelB significantly reduced tumor burden compared to controls [4]. However, intraperitoneal xenografts of shRelA and shRelB cells showed dramatically fewer numbers of tumor cells with shRelB compared to shRelA and control, suggesting there is a greater requirement of non-canonical NF-��B signaling in intraperitoneal, anchorage-independent metastasis in this model [4].
Further evidence of differing roles for canonical and non-canonical NF-��B signaling in OC progression was demonstrated in the context of ECM-interacting protein Transglutaminase (TG2). TG2 can promote OC metastasis in vitro and in vivo and can activate both canonical and non-canonical NF-��B signaling pathways [52,53]. TG2 signaling intracellularly activates canonical NF-��B and a secreted form of TG2 can activate non-canonical NF-��B [52]. In the latter case, RelB activation by secreted TG2 induced CD44 expression, downregulated E-cadherin, and increased invasiveness and peritoneal metastasis in vivo, but interestingly, did not affect cell proliferation in vitro [52]. This is consistent with previous findings about the functions of RelB in OC proliferative and CSC populations [4].
Non-canonical NF-��B signaling is also important for CSC maintenance in endometrioid endometrial cancer (EEC). RelB but not RelA, was significantly upregulated in EEC, and silencing RelB in EEC cell lines affected colony formation, tumor growth in vivo and cell cycle progression [54]. The cells silenced for RelB accumulated in G0/G1, upregulated cell cycle arresting proteins p27 and p21, and activated apoptosis [46]. In comparison, cells with enhanced RelB expression showed advanced cell cycle progression and upregulation of proliferative, apoptosis and cell-cycle-transition-regulating genes such as c-Myc, cyclins D1 and E, and Bcl-2 and Bcl-xl [54].
3. NF-��B Activity in the Inflammatory Ovarian Cancer Microenvironment
The OC microenvironment is unique in that multiple cell types other than the tumor cells are coopted to promote metastasis and inhibit immune activity [5]. There are both inflammatory and immunosuppressive components of the milieu that are controlled by NF-��B activity in the cancer cells, as well as infiltrating immune cells. OC cells secrete immunosuppressive cytokines to promote pro-tumor phenotypes in tumor-associated macrophages (TAMs), which in turn, secrete immunoinhibitory cytokines such as IL-10 and contribute to constitutive NF-��B activation in OC cells by secreting TNF��, promoting survival, CSC expansion and metastasis [59,60].
3.1. Modulation of NF-��B Activity in Infiltrating Immune Cells
TAMs are typically polarized in the pro-tumor ��M2�� subtype which secrete high levels of IL-10 and TNF��, and decreased IL-12 to promote the immunosuppressive environment and increase invasiveness in cancer cells [61,62]. In a co-culture experiment of mouse bone-marrow-derived macrophages (BMDMs) with mouse ovarian cancer cell line ID-8, the importance of upstream activators of canonical NF-��B signaling was demonstrated by specific knockouts of Myd-88, IL1R, TLR2 and TLR4 in the BMDMs, which caused decreased invasion of ID-8 cells in vitro and decreased tumor growth in vivo [62]. Specific inhibition of the NF-��B classical pathway activating kinase IKK�� in BMDMs caused a shift in the polarization from the pro-tumor M2 subtype to anti-tumor M1 subtype in vitro and in vivo [62]. IKK��-inhibited BMDMs had decreased production of immunosuppressive cytokines and increased nitric oxide synthesis to promote apoptosis and increased IL-12 production for cytotoxic immune activation [62].
The OC microenvironment contains both pro-inflammatory and immunosuppressive cells so there is a contribution of cytokines by both anti-tumor M1- and pro-tumor M2-polarized macrophages. While M2 macrophages may be recruited locally to the tumor cells to be immunosuppressive, M1 macrophages are also recruited and promote chronic inflammation and high levels of cytokines that are intended to promote cytotoxic T cell recruitment, but these can also act on OC cells to promote survival and metastasis [60,63,64]. For example, treatment with the conditioned medium (CM) of M1-polarized macrophages caused nuclear translocation RelA/p65 and activation of NF-��B transcription, increased invasion and migration in OC cell lines [65]. It was determined that the CM produced by M1-polarized macrophages contained high levels of TNF�� and IL-6 and that TNF�� activated NF-��B in the OC cells [65].
Dendritic cells are also able to infiltrate the OC microenvironment and can be switched from immunostimulatory to immunosuppressive [66]. A target for cancer immunotherapy is programmed cell death-1 (PD-1), which is a negative regulator of immune cytotoxicity that is expressed on immune cells, particularly T cells [67]. Regulation of immune cytotoxicity through PD-1 is directed by interaction with its ligand (PD-L1), which is expressed on normal cells but has also been detected on cancer cells as a strategy to avoid immune cytotoxicity [67]. DCs from OC tumors and ascites that expressed PD-1 had decreased anti-tumor activity due to inhibition of canonical, immunogenic NF-��B activity [68]. Blockade of PD-1 on the DCs restored their pro-inflammatory functions by activation of canonical NF-��B with degradation of I��B��, allowing the nuclear translocation of RelA/p65 [68]. In the PD-1-blocked DCs, the upregulation of co-stimulatory molecules and secretion of immunogenic and proinflammatory cytokines TNF��, Il-6 and G-CSF was found to be dependent on NF-��B [68]. Unfortunately, the co-activation of NF-��B activation in OC cells in response to these cytokines can be utilized to further promote the immunosuppressive pro-tumor environment.
3.2. NF-��B Activation in Ovarian Cancer Cells to Alter the Microenvironment
OC cells contribute to the maintenance of the tumor microenvironment by secreting immunosuppessive cytokines to prevent migration of anti-tumor immune cells and to polarize macrophages to a pro-tumor M2 subtype [61,69]. This phenomenon triggers the expression of multiple cytokines including TNF�� from tumor-infiltrating macrophages exposed to OC cells [61]. Accordingly, constitutive NF-��B signaling is activated in OC cells in response to co-culture with M2 macrophages and in response to high TNF�� [38,69]. In addition, constitutively activated NF-��B signaling in OC cells also leads to increased immunosuppressive cytokines to further promote tumor growth [70]. The activation of canonical NF-��B signaling in OC cells is a major component of the immunosuppressive cytokine secretion, and inhibition of the pathway in OC cells caused the immune cell infiltrates to switch to an anti-tumor phenotype in vitro and in vivo [70,71,72].
Persistent inflammation has been associated with ovarian cancer development, in women with pelvic inflammatory disease or in response to pathogenic infections of Chlamydia trachomatis or Mycoplasma genitalium [73]. Recognition of pathogen-associated molecular patterns (PAMPs) by cell receptors such as TLRs initiates the innate immune response by activating NF-��B, which stimulates the production of pro-inflammatory cytokines including TNF��, IL-6, IL-8, and MCP-1 [74]. Persistent, chronic inflammation following infection and constitutive NF-��B activity may contribute to ovarian carcinogenesis by producing pro-inflammatory and pro-angiogenic cytokines and through anti-apoptotic signaling [73].
Another way OC cells use NF-��B signaling for immune evasion is through PD-L1 upregulation [75]. In response to chemotherapy, the expression of p65/RelA and PD-L1 increased in OC cell lines in vitro and in vivo [75]. Silencing p65/RelA prevented PD-L1 upregulation, confirming its dependence on NF-��B activity [75]. Thus, the OC microenvironment activates NF-��B signaling in a variety of cell types to promote tumor cell survival, immune evasion and metastasis.��
��
Figure 1
Strategies to target NF-��B in ovarian cancer. NF-��B signaling in cancer cells can be activated by interacting proteins in the tumor microenvironment, such as lysophosphatidic acid (LPA) signaling through CD44 to promote NF-��B activity and cytokine secretion. NF-��B signaling can also be activated by TNF�� binding to cell surface receptors Myd-88 and TLR4, or through TNF�� independent receptors such as TG2. NF-��B activity promotes cytokine secretion to dampen immune responses and switch macrophages from inflammatory M1 phenotypes to M2 immunoinhibitory phenotypes. Selective activation of NF-��B signaling in dendritic cells and macrophages with toll-like receptor (TLR) agonists or blockade of PD-1 could promote anti-tumor immunity and recruitment of additional anti-tumor immune cells. Alternatively, selective inhibition of NF-��B in tumor cells by I��B kinase (IKK) inhibitors could attenuate pro-tumor survival signaling in the tumor cells without disrupting inflammatory NF-��B activity in immune cells.NF-��B Signaling in Ovarian Cancer
5. Conclusions
The NF-��B signaling pathway has been implicated in OC tumorigenesis through its roles in mediating chemoresistance, maintenance of CSC populations, metastasis and immune evasion. The NF-��B signaling proteins and IKKs are overexpressed in OC tumors and correlate with poorer patient outcomes. Both canonical and non-canonical NF-��B signaling contribute to OC progression with overlapping and distinct roles in promoting chemoresistance and CSC maintenance. The inflammatory tumor microenvironment is also heavily influenced by NF-��B signaling, from the inflammatory cytokines that activate NF-��B signaling in OC cells and the immunosuppressive phenotype of TAMs and other immune cells that have diminished NF-��B activity. Targeting NF-��B in OC to eliminate the CSC population is desirable, but current strategies are limited to inhibition of IKKs. More research is needed to dissect the pathways and identify strategies to modulate NF-��B to reduce metastasis and tumor recurrence. There is a need to strike a balance between the inhibition of the non-canonical NF-��B in OC cells which enables long-term resistance and tumor re-initiation from CSC and the activation of canonical NF-��B in immune cells to generate an effective anti-tumor immune response.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6721592/��
BMC Complement Altern Med. 2007; 7: 44.
Published online 2007 Dec 20. doi: 10.1186/1472-6882-7-44
Ginger inhibits cell growth and modulates angiogenic factors in ovarian cancer cells
Jennifer Rhode,#1 Sarah Fogoros,#2 Suzanna Zick,3 Heather Wahl,4 Kent A Griffith,5 Jennifer Huang,4 and J Rebecca Liucorresponding author4
188th Medical Group, Wright-Patterson AFB, Ohio, USA
2National Institutes of Health, National Human Genome Research Institute, Cancer Genetics Branch, USA
3Department of Family Medicine, University of Michigan, Ann Arbor, MI, USA
4Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, University of Michigan, Ann Arbor, MI, USA
5Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA
corresponding authorCorresponding author.
#Contributed equally.
Jennifer Rhode: moc.loa@edohRMnJ; Sarah Fogoros: vog.hin.liam@sorogof; Suzanna Zick: ude.hcimu.dem@kcizs; Heather Wahl: ude.hcimu@lhawh; Kent A Griffith: ude.hcimu@gtnek; Jennifer Huang: ude.hcimu@hynej; J Rebecca Liu: ude.hcimu@uilr
This article has been cited by other articles in PMC.
Go to:
Abstract
Background
Ginger (Zingiber officinale Rosc) is a natural dietary component with antioxidant and anticarcinogenic properties. The ginger component [6]-gingerol has been shown to exert anti-inflammatory effects through mediation of NF-��B. NF-��B can be constitutively activated in epithelial ovarian cancer cells and may contribute towards increased transcription and translation of angiogenic factors. In the present study, we investigated the effect of ginger on tumor cell growth and modulation of angiogenic factors in ovarian cancer cells in vitro.
Methods
The effect of ginger and the major ginger components on cell growth was determined in a panel of epithelial ovarian cancer cell lines. Activation of NF-��B and and production of VEGF and IL-8 was determined in the presence or absence of ginger.
Results
Ginger treatment of cultured ovarian cancer cells induced profound growth inhibition in all cell lines tested. We found that in vitro, 6-shogaol is the most active of the individual ginger components tested. Ginger treatment resulted in inhibition of NF-kB activation as well as diminished secretion of VEGF and IL-8.
Conclusion
Ginger inhibits growth and modulates secretion of angiogenic factors in ovarian cancer cells. The use of dietary agents such as ginger may have potential in the treatment and prevention of ovarian cancer.��
Results
Ginger inhibits growth in ovarian cancer cells as compared to non-transformed ovarian epithelial cells
Continuous exposure to ginger extract resulted in a marked reduction in cell growth after 1�C5 days of exposure in A2780 ovarian cancer cells (Figure (Figure1A,1A, p < .0001 at all doses and time points). We tested additional ovarian cancer cell lines to determine if this was an effect unique to the A2780 ovarian cancer cells. Ginger treatment resulted in similar effects in all cell lines tested, including the chemoresistant cell lines SKOV3 and ES-2[39] (Figure 1B,C, p < .05 for all doses and time points). Untransformed human ovarian surface epithelial cells (HOSE) were minimally affected by ginger extract exposure at days 1 and 3, and showed some inhibition in growth by day 5 (Figure (Figure1D,1D, p > .05 for days 1 and 3, p < .05 for day 5). To confirm that ginger treatment inhibited cell growth, treated cells were analyzed by trypan blue exclusion as well. As expected, ginger treatment resulted in a profound inhibition of cell proliferation and growth at doses of 50 ��g/ul and higher (Figure (Figure22).
Continuous ginger exposure inhibits growth in ovarian cancer cells in vitro. A, B, C: The effect of ginger on growth of A2780, SKOV3 and ES2 ovarian cancer cell lines was assessed by using sulforhodamine B assays. Cells were incubated continuously with media containing ginger at the indicated concentrations and growth was assayed at Days 1, 3 and 5 of exposure. Ginger treated cells displayed significant growth inhibition as compared to control treated cells (p < .05 for all cell lines, all ginger concentrations). D: Human ovarian surface epithelial cells were treated with the indicated concentrations of ginger with minimal effect seen following days 1�C3 of culture (p > .05). HOSE demonstrated some inhibition of growth by day 5 of treatment (p < .05). Data are presented as means �� S.D, and are representative of at least 3 independent experiments.
Figure 2
Continuous ginger exposure inhibits growth in ovarian cancer cells in vitro. A, B, C: The effect of ginger on growth of A2780, SKOV3 and ES2 ovarian cancer cell lines was assessed using the trypan blue exclusion assay. Cells were incubated continuously with media containing ginger at the indicated concentrations and viable cells were counted on days Days 1, 3 and 5 of exposure. Ginger treated cells displayed significant growth inhibition as compared to control treated cells.��
To determine whether lower doses of ginger could also inhibit cell growth, an extended range of concentrations was tested. In the A2780 and ES-2 cell lines, ginger concentrations of less than 50 ��g/ml did not significantly impact cell growth, whereas in the SKOV3 cell line, some inhibition of cell growth was seen with ginger concentrations as low as 30 ��g/ml (Figure (Figure33 and data not shown).
��
6-Shogaol is the most active of the individual ginger components tested in ovarian cancer cells
Previous investigators have shown bio-activity of various individual ginger components in several tumor types [30,40-43]. To determine the relative bio-activity in ovarian cancer, A2780 ovarian cancer cells were treated with 6-, 8- and 10-gingerol as well as 6-shogaol. In contrast to other published findings, we found that 6-, 8- and 10-gingerol had no effect on the growth or viability of ovarian cancer cells (p > .05 at all time points). Treating cells with whole ginger extract or 6-shogaol resulted in profound growth inhibition (Figure (Figure4A,4A, p < .05 at all time points for both ginger and 6-shogaol treated cells). Morphologically, cells treated with ginger appeared markedly growth inhibited, similar to cisplatin treated cells (Figure (Figure5).5). Cells cultured with vehicle control (DMSO) continued to proliferate.��
Figure 5
Morphologic appearance of ginger treated ovarian cancer cells. A2780 ovarian cancer cells were incubated with DMSO solvent, ginger (75 ��g/ml, replenished on day 3), or Cisplatin (2.5 ��g/ml). Cells were examined by light microscopy at 1, 3, and 5 days of treatment.��
We next determined if continuous exposure to individual ginger components was necessary to cause the growth inhibitory effect seen in ovarian cancer cells. Similar to the use of whole ginger root extract, continuous exposure to 6-shogaol was necessary to cause the growth inhibitory effect seen in ovarian cancer cells. We treated cells with ginger and individual ginger components for 24 hours, after which the cells were washed and media was changed. Once ginger or 6-shogaol was removed from the media, cell growth resumed (Figure (Figure4B4B).
Ginger inhibits NF-��B in ovarian cancer cells
Because we found that ginger markedly suppressed ovarian cancer cell proliferation in vitro, and several genes that regulate proliferation are regulated by NF-��B, we hypothesized that ginger may mediate its anti-neoplastic activity in ovarian cancer cells though modulation of this pathway. Constitutive activation of NF-��B has been described in many tumor types including ovarian cancer[9], suggesting that targeting NF-��B may have an anti-neoplastic effect in this tumor type. Natural products such as ginger, or ginger components such as zerumbone can inhibit NF-��B in other cell types [16,44,45]. We chose two chemoresistant ovarian cancer cell lines (CaOV3 and SKOV3) to evaluate the effect of ginger treatment on activation of NF-��B. As shown in Figure Figure6,6, treatment with ginger extract resulted in a significant inhibition of NF-��B activation in CaOV3 and SKOV3 cell lines��
Figure 6
Ginger inhibits NF-kB activation in ovarian cancer cells. Representative ovarian cancer cell lines with high endogenous NF-kB activation, (A) CaOV3 and (B) SKOV3 were transfected with an NF-��B-dependent reporter plasmid (pBVIx-Luc). Cells were treated with DMSO (vehicle control) or ginger (75 ��g/ml). NF-��B activation was determined by measuring relative luciferase activity 48 hours after treatment. Luciferase activity is reported as arbitrary relative light units (mean +/- S.D.) Ginger treatment resulted in inhibition of NF-��B activation (p < .05 for both cell lines). Representative data is shown.
Ginger Inhibits IL-8 and VEGF Secretion in Ovarian Cancer Cells
IL-8 can function as a paracrine and/or autocrine growth factor in some tumor types, and the secretion of IL-8 protein from tumor cells themselves is thought to be crucial for these effects[46,47]. In ovarian cancer patients, elevated IL-8 expression has been found in ascites as well as in serum[33]. Furthermore, IL-8 has been shown to stimulate proliferative growth in ovarian cancer cells in vitro[35]. Because IL-8 secretion is thought to be regulated in part by NF-��B, and ginger can clearly inhibit NF-��B in ovarian cancer cells, we hypothesized that ginger could also inhibit IL-8 secretion. Using a representative panel of ovarian cancer cell lines, we found that A2780 and CaOV3 cells produced negligible amounts of IL-8 (<0.05 pg/ml), whereas the cell lines ES-2 and SKOV3 had high constitutive expression of IL-8 (Figure (Figure7A).7A). Treatment with ginger resulted in significant inhibition of IL-8 production in the ES-2 and SKOV3 cell lines (p < .05 for both cell lines).��
Figure 7
Ginger inhibits VEGF and IL-8 in ovarian cancer cells. Ovarian cancer cells were cultured with DMSO (vehicle control) or Ginger (75 ��g/ml) for 48 hours. Production of the angiogenic factor s IL-8 (A) and VEGF (B) were assayed using ELISA assays. (A.) Only ES-2 and SKOV3 cells expressed high IL-8 levels at Baseline, and ginger treatment resulted in a significant decrease in IL-8 production (p < .05 for both cell lines). (B.) VEGF production was reduced in all cell lines following ginger treatment (p = .19, .18, .007, and .07 for A2780, CaOV3, ES-2 and SKOV3 cells respectively).
VEGF, the most important inducer of angiogenesis, is also under transcriptional control of NF-��B[9]. Serum VEGF levels as well as tumor expression of VEGF are associated with poor prognosis in ovarian cancer patients [31], and inhibition of VEGF function using Avastin™ has shown promise in the treatment of ovarian cancer patients[48]. Because ginger treatment resulted in inhibition of NF-��B, we next sought to determine whether ginger could similarly inhibit VEGF in ovarian cancer cells. In all cell lines tested, there was high endogenous production of VEGF, and ginger treatment resulted in inhibition of VEGF secretion. Inhibition of VEGF secretion was most evident in the ES-2 cell line (p = .007), as compared to the other cell lines tested (p = .19, .18, and .07 for A2780, CaOV3, and SKOV3 respectively, Figure Figure7B7B).��
Discussion
The analysis of epidemiologic data and disparities in global incidence of ovarian cancer may provide clues to uncover environmental and biologic factors that contribute towards the development of ovarian cancer. Dietary prevalence of foods such as ginger, garlic, soy, curcumin, chilies and green tea are thought to contribute to the decreased incidence of colon, gastrointestinal, prostate, breast and other cancers in South East Asian countries [49]. Accumulating evidence suggests that many dietary factors may be used alone or in combination with traditional chemotherapeutic agents to prevent or treat cancer. The potential advantage of many natural or dietary compounds seems to focus on their potent anticancer activity combined with low toxicity and very few adverse side effects.
Epithelial ovarian carcinoma is the leading cause of death among patients with gynecologic cancers. Despite multiple modalities of treatment including surgery and chemotherapy, ovarian cancer patients continue to have one of the lowest 5-year survival rates [1]. The significant morbidity and limited success of surgery and chemotherapy for ovarian cancer has led to searches for alternative therapies. Recently, ginger root and its main poly-phenolic constituents (gingerols and zerumbone) been shown to exhibit anti-inflammatory [16-19], and anti-neoplastic activity [20-24] in several cell types through inhibition of the transcription factor NF-��B [25-28]. NF-��B plays an important role in tumorigenesis, given its ability to control the expression and function of numerous genes involved in cell proliferation, sustained angiogenesis, and evasion of apoptosis. Different tumor types, including ovarian cancer, have been shown to express high constitutive NF-��B activity[9]. In this study we show that ginger blocks NF-��B activation in ovarian cancer cells, resulting in inhibition of NF-��B regulated gene products involved in cellular proliferation and angiogenesis.
Many of the pathways that mediate adaptive survival strategies in cancer cells are under the transcriptional control of NF-��B [9]. We have shown here that in ovarian cancer cells, NF-��B is constitutively activated, and blocking NF-��B activation with ginger results in suppressed production of NF ��B regulated angiogenic factors and selectively inhibits ovarian cancer cell growth. We have found that ginger selectively inhibits ovarian cancer cell growth, as compared to non-transformed ovarian epithelial cells. Previous reports indicate that the ginger component 6-shogaol induces cell death in chemoresistant hepatoma cells [50], yet inhibits cell death in non-neoplastic spinal cord cells [51], suggesting that ginger and ginger components' effects are cell type specific. The apparent contradictory findings may be due to a differential effect of ginger on transformed cells (i.e. cancer cells) vs. untransformed cells. Phytochemicals such as ginger, generally have multiple molecular targets. This pleiotropism may constitute an advantage in the treatment of ovarian cancer, where multiple factors contribute towards the carcinogenic process.
Conclusion
The results of this study indicate that ginger may exhibit anti-neoplastic effects through the inhibition of NF-��B. Further studies utilizing ginger in an in vivo model of ovarian cancer will provide a platform for the development of ginger as a therapeutic tool in this disease.Ginger inhibits cell growth and modulates angiogenic factors in ovarian cancer cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2241638/��
Zingiber officinale attenuates retinal microvascular changes in diabetic rats via anti-inflammatory and antiangiogenic mechanisms. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/27293376��
��
The NF��B luciferase mouse: a new tool for real time ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1773458
The NF��B luciferase mouse: a new tool for real time measurement of NF��B activation in the whole animal D A Mann Liver Research Group, University of Southampton, Southampton, UK; Email: ku.ca.notos@2mad��
Gut. 2002 Dec; 51(6): 769�C770.
The NF��B luciferase mouse: a new tool for real time measurement of NF��B activation in the whole animal
D A Mann
Liver Research Group, University of Southampton, Southampton, UK; ku.ca.notos@2mad
Of the many different transcription factors found in mammalian cells by far the best known among scientists and clinicians alike is nuclear factor ��B (NF��B). In addition to its pivotal role as a regulator of innate and adaptive immune responses, NF��B has also been linked as a contributory factor in many human disorders.1 NF��B stimulates transcription of over 150 different genes encoding cytokines, adhesion molecules, immunoreceptors, acute phase proteins, enzymes, and regulators of the cell cycle and apoptosis.2 Transcription of these genes is stimulated in response to binding of NF��B to a specific 10 base pair DNA sequence (GGGRNNYYCC, where R corresponds to a purine, Y represents a pyrimidine, and N can be any base) located in either the enhancer or upstream promoter regions of a gene. Binding of NF��B to these sequences is tightly regulated by the inhibitory function of the I��B-�� protein which forms a complex with NF��B that masks functional domains of the transcription factor required for its nuclear localisation and DNA binding activities. The mechanism by which I��B-�� bound NF��B is activated is now well understood (fig 1 ▶).3 Following stimulation of the cell by an extracellular signal, such as the interaction of tumour necrosis factor �� with its receptor, the recently discovered I��B kinase (IKK) complex is activated. The IKK complex then catalyses phosphorylation of I��B-�� at two N terminal serine residues (ser 32 and 36) which then renders I��B-�� a substrate for ubiquitination and finally for degradation by the 26S proteasome. These events are rapid, and within minutes of the initial stimulation event result in the nuclear translocation of active NF��B. A remarkable number of different inducers of the NF��B pathway have been described, including a plethora of cytokines and a surprising variety of different viral and bacterial gene products.2 Given the ability of NF��B to be activated in response to this diverse range of extracellular stimuli and its subsequent transcriptional activation of a vast number of target genes, it is not surprising that the transcription factor has been implicated in so many disease processes. Gastrointestinal diseases in which NF��B has been linked as a contributor to pathogenesis include fibrogenesis, inflammatory bowel disease, acute pancreatitis, responses to infections by bacteria and viruses, and last but not least cancer.4��
Figure 1
Events leading to activation of nuclear factor ��B (NF��B) dependent gene transcription. Following the interaction of a cell surface receptor with its ligand (for example, tumour necrosis factor �� (TNF-��)), the I��B kinase (IKK) complex is activated resulting in phosphorylation (P), ubiquitination (Ub), and proteasome mediated degradation of I��B-��. Free NF��B (p65:p50) dimers then translocate to the nucleus where additional phosphorylation events on p65 and recruitment of the coactivator and histone acetyltransferase CBP/p300 leads to transcriptional activation of up to 150 different genes. INOS, inducible nitric oxide synthase; COX2, cyclooxygenase 2.��
Carlsen H, Moskaug JØ, Fromm SH, et al. In vivo imaging of NF-kappa B activity. J Immunol 2002;168:1441�C6.
A wide range of human disorders involves inappropriate regulation of NF-kappaB, including cancers and numerous inflammatory conditions. Toward our goal to define mechanisms through which NF-kappaB leads to the development of disease, we have developed transgenic mice that express luciferase under the control of NF-kappaB, enabling real-time in vivo imaging of NF-kappaB activity in intact animals. We show that in the absence of extrinsic stimulation, strong luminescence is evident in lymph nodes in the neck region, thymus, and Peyer��s patches. Treating mice with TNF-alpha, IL-1alpha, or LPS increased the luminescence in a tissue-specific manner, with the strongest activity observed in skin, lungs, spleen, Peyer��s patches, and the wall of the small intestine. Liver, kidney, heart, muscle, and adipose tissue displayed less intense activities. Also, exposure of skin to a low dose of UV radiation increased luminescence in the exposed areas. Furthermore, induction of chronic inflammation resembling rheumatoid arthritis produced strong NF-kappaB activity in the affected joints, as revealed by in vivo imaging. Thus, we have developed a versatile model for monitoring NF-kappaB activation in vivo.
To date, most of what has been inferred about the potential role played by NF��B in gastrointestinal disease has been derived from a combination of in vitro and ex vivo studies measuring changes in NF��B nuclear localisation or DNA binding activity. The problem with these studies is that they are based on the often incorrect assumption that the transcriptional activity (or trans-activation) of NF��B can be directly correlated with its degree of nuclear location and DNA binding. Similarly, the suggestion that many commonly used anti-inflammatory drugs including aspirin, sulphasalazine, and glucocorticoids influence the activation of NF��B is largely based on this assumption. In fact, the degree of trans-activation by NF��B is also influenced by additional regulatory mechanisms including post-translational modifications (for example, phosphorylation) of NF��B and its interaction with transcriptional coactivators such as CBP/p300.5 The paper by Carlsen et al describes the production of a new experimental tool that provides a significant advance in our ability to quantify changes in NF��B dependent transcription in real time, in a living animal and in a relatively non-invasive manner. What Carlson et al have produced is a transgenic mouse that carries in its genome a firefly luciferase gene under the control of three tandem NF��B DNA binding sites. Consequently, this mouse will express luciferase in any cell in which active NF��B is present; moreover, the level of luciferase expressed is proportional to the level of NF��B trans-activation in a cell or tissue. Measurement of luciferase expression is achieved by intravenous injection of the animal with a suitable substrate such as d-luciferin which in the presence of the enzyme leads to a bioluminescent reaction that can be detected in the whole animal using an image intensifier coupled to a CCD camera. As firefly luciferase has only a short biological half-life (two hours), the system is capable of measuring temporal fluctuations in NF��B activity which is a tremendous advance over other systems such as DNA binding assays that only provide a static snapshot of NF��B activity. But perhaps the most impressive advantage is that the system is sufficiently sensitive to image and quantify NF��B activity within the internal organs (including the small intestine and the liver) of the intact animal.
How does the availability of the NF��B luciferase mouse impact on gastroenterology? The answer is that for the first time we can now properly monitor the influence of exogenous agents and pathological conditions on NF��B activity in the gut and liver. This includes the potential to screen for new drugs that target aberrant NF��B activity. It should also be possible to accurately determine the effective dose and administration regimen for a given anti-NF��B drug in an organ specific manner. Rather than simply determining the delivery and uptake of these drugs into the gut and liver, the NF��B luciferase mouse will now tell us the biological efficacy of the drug within the tissue and over what timeframe the drug can be expected to remain effective. As well as impacting on clinical research the NF��B reporter mouse has tremendous potential in basic research, especially when combined with experimental in vivo models of disease. One of the major problems associated with studying NF��B is its amazing sensitivity to changes in the cellular environment; the very process of isolating tissues and organs from an animal may itself alter NF��B activity. To date it has therefore been virtually impossible to determine changes in the transcriptional activity of NF��B during a disease process. Carrying out in vivo models of gastrointestinal disease in the NF��B luciferase mouse will enable us to accurately quantify the degree to which NF��B dependent transcription is increased throughout the entire course of a disease. Such studies will significantly improve our understanding of the role played by NF��B in gastrointestinal diseases.��
The NF��B luciferase mouse: a new tool for real time measurement of NF��B activation in the whole animal
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1773458/��
��
NFkB Luciferase Reporter HeLa Stable Cell Line
Description:
NFkB Responsive Luciferase Reporter Hela Stable Cell Line is derived from human cervical cancer, and stably express firefly luciferase reporter gene under the control of the NFkB response element. This cell line is an ideal cellular model for monitoring the activation of NFkB Pathway triggered by stimuli treatment, enforced gene expression and gene knockdown.
Principle
NFkB plays an important role in controlling many biological processes including immune and inflammatory responses, developmental processes, cellular growth, and apoptosis. In response to the various stimuli, such as stress, cytokines, free radicals, ultraviolet irradiation, and bacterial or viral antigens, NFkB is activated and translocate from cytoplasm to nucleus, where NFkB binds to its response element on the promoter region and regulates a wide spectrum of gene expression. Dysfunction of NFkB activity is associated with cancer, inflammatory and autoimmune disease, and viral infection. Monitoring the NFkB activity is essential to unveil the mechanism of these diseases and conduct drug discovery. Signosis has established NFkB luciferase reporter Hela stable cell line that has been stably transfected with pTA-NFkB-luciferase reporter vector, which contains 4 repeats of NFkB binding sites, a minimal promoter upstream of the firefly luciferase coding region. Therefore, the cell line can be used as a reporter system for monitoring the activation of NFkB triggered by stimuli treatment, enforced gene expression and gene knockdown. The stable cell line can be used for studying NFkB signaling pathways activated by different cytokines, such as TNFa, and many other stimuli.
The cell line was established by transfection using a pTA-NFkB-luciferase reporter vector, which contains 4 repeats of NFkB binding sites, a minimal promoter upstream of the firefly luciferase coding region, along with hygromycin expression vector followed by hygromycin selection. The hygromycin resistant clones were subsequently screened for TNFa-induced (or IL-1) luciferase activity.
����
Analysis of TNFa/NFkB Pathway Reporter Cell Line in response to TNFa treatment. The cells were seeded on a 96-well plate for overnight with DMEM including 10% FBS. The cell then were treated with 20ng/ml TNFa in DMEM + 0.1% FBS for 6 hours.
��
Principle behind TF luciferase reporter. TF luciferase reporter stable cell line utilizes artificial promoter constructs to drive luciferase expression. The promoter region can consists of multiple repeats of a cis-element TF binding site, a DNA fragment from the promoter region of a known TF downstream gene, or a DNA fragment containing putative/known TF binding sites. There are several ways that a TF can be activated, such as through extracellular stimuli or through intracellular signaling pathways. Once activated, the TF translocates to the nucleus and often interacts with relevant co-factors to drive gene expression. Once luciferase is expressed, it can generate light in an enzymatic assay and the amount of light measured is positively correlated with the level of TF activation.
��
Signosis
https://www.signosisinc.com/product/nfkb-luciferase-reporter-hela-stable-cell-line-sl-0001-np��
Published: 14 September 2017
Endogenous ��-3 Fatty Acid Production by fat-1 Transgene and Topically Applied Docosahexaenoic Acid Protect against UVB-induced Mouse Skin Carcinogenesis
Hye-Won Yum, Jin Park, Hyun-Jung Park, Jun Wan Shin, Yong-Yeon Cho, Su-Jung Kim, Jing X. Kang & Young-Joon Surh
Scientific Reports volume 7, Article number: 11658 (2017) Cite this article
Abstract
The present study was intended to explore the effects of endogenously produced ��-3 polyunsaturated fatty acids (PUFAs) on ultraviolet B (UVB)-induced skin inflammation and photocarcinogenesis using hairless fat-1 transgenic mice harboring ��-3 desaturase gene capable of converting ��-6 to ��-3 PUFAs. Upon exposure to UVB irradiation, fat-1 transgenic mice exhibited a significantly reduced epidermal hyperplasia, oxidative skin damage, and photocarcinogenesis as compared to wild type mice. The transcription factor, Nrf2 is a master regulator of anti-inflammatory and antioxidant gene expression. While the protein expression of Nrf2 was markedly enhanced, the level of its mRNA transcript was barely changed in the fat-1 transgenic mouse skin. Topical application of docosahexaenoic acid (DHA), a representative ��-3 PUFA, in wild type hairless mice induced expression of the Nrf2 target protein, heme oxygenase-1 in the skin and protected against UVB-induced oxidative stress, inflammation and papillomagenesis. Furthermore, transient overexpression of fat-1 gene in mouse epidermal JB6 cells resulted in the enhanced accumulation of Nrf2 protein. Likewise, DHA treated to JB6 cells inhibited Nrf2 ubiquitination and stabilized it. Taken together, our results indicate that functional fat-1 and topically applied DHA potentiate cellular defense against UVB-induced skin inflammation and photocarcinogenesis through elevated activation of Nrf2 and upregulation of cytoprotective gene expression.
Introduction
Skin is repeatedly exposed to various external stressors, such as solar radiation and microbes1,2. In response to changing external and internal environment, the skin produces cytokines, neurotransmitters, neuroendocrine hormones and their corresponding receptors3,4,5. These capabilities of skin allow it to preserve the homeostasis which is critical for organism fitness and survival.
Exposure of skin to excessive ultraviolet B (UVB) causes the production of reactive oxygen species (ROS), which results in massive infiltration of inflammatory cells, especially, neutrophils at the site of tissue injury6. ROS play important roles in the modulation of intracellular signal transduction involved in each stage of photocarcinogenesis, i.e., initiation, promotion, and progression7. One of the key enzymes mediating inflammatory signal transduction is cyclooxygenase-2 (COX-2) that catalyzes the rate-limiting step in the biosynthesis of prostaglandins. Nuclear factor kappa B (NF-��B) and signal transducer and activator of transcription 3 (STAT3) are two representative redox-sensitive transcription factors responsible for regulating COX-2 expression8,9,10,11,12,13,14. A battery of antioxidant enzymes play a central role in counteracting excessive ROS accumulation, thereby maintaining the cellular redox balance. Examples are heme oxygenase-1 (HO-1) and NAD(P)H:quinone oxidoreductase 1 (NQO1). The proximal promoter regions of genes encoding HO-1 and NQO1 harbor a consensus sequence known as antioxidant response element (ARE) which is a preferred binding site of nuclear factor-erythroid related factor-2 (Nrf2)15.
Long chain ��-3 polyunsaturated fatty acids (��-3 PUFAs) abundant in fish oil have been reported to retain marked anti-inflammatory and anti-oxidative properties, which contribute to their chemopreventive potential16,17. In contrast, ��-6 PUFAs have pro-inflammatory activities. The ��-6/��-3 PUFA ratio has been considered essential for determining the risk for some human malignancies18,19,20,21. Kang et al. developed genetically engineered mice of C57BL6 background carrying a fat-1 gene which encodes an ��-3 fatty acid desaturase derived from Caenorhabditis elegans 22,23. These transgenic mice are capable of converting ��-3 PUFAs from ��-6 PUFAs, thereby maintaining a significantly elevated ��-3/��-6 PUFA ratio in their tissues and organs.
In the present study, hairless fat-1 transgenic mice were generated by cross-breeding of male fat-1 +/− mice with female SKH-1 hairless mice. By utilizing these hairless fat-1 transgenic mice maintained on ��-6 PUFA containing diet, we investigated the effect of endogenously formed ��-3 PUFAs on the development of UVB-induced tumors as well as oxidative stress and inflammation in the skin.��
Endogenous ��-3 Fatty Acid Production by fat-1 Transgene and Topically Applied Docosahexaenoic Acid Protect against UVB-induced Mouse Skin Carcinogenesis | Scientific Reports
https://www.nature.com/articles/s41598-017-11443-2��
Published: 29 November 2019
Inhibition of lung cancer growth and metastasis by DHA and its metabolite, RvD1, through miR-138-5p/FOXC1 pathway
Xiaoming Bai, Jiaofang Shao, Sujin Zhou, Zhenggang Zhao, Fanghong Li, Rong Xiang, Allan Z. Zhao & Jinshun Pan
Journal of Experimental & Clinical Cancer Research volume 38, Article number: 479 (2019) Cite this article�Ͼ�ҽ�ƴ�ѧ
Abstract
Background
Non small cell lung cancer (NSCLC) is one of the most common cancers in the world. DHA is known to be capable of suppressing NSCLC cell proliferation and metastasis. However, the mechanisms by which DHA exhibits its antitumor effects are unknown. Here we aimed to identify the effects and mechanisms of DHA and its metabolites on lung cancer cell growth and invasion.
Methods
As measures of cell proliferation and invasion ability, the cell viability and transwell assays were used in vitro. Transgenic mfat-1 mice, which convert ��-6 PUFAs to ��-3 PUFAs, were used to detect the effect of endogenous DHA on tumor transplantation. An LC − MS/MS analysis identified the elevation of several eicosanoid metabolites of DHA. By using qPCR miRNA microarray, online prediction software, luciferase reporter assays and Western blot analysis, we further elucidated the mechanisms.
Results
Addition of exogenous DHA inhibited the growth and invasion in NSCLC cells in vitro. Endogenously produced DHA attenuated LLC-derived tumor growth and metastasis in the transgenic mfat-1 mice. Among the elevation of DHA metabolites, resolvin D1 (RvD1) significantly contributed to the inhibition in cell growth and invasion. MiRNA microarray revealed that the level of miR-138-5p was significantly increased after RvD1 treatment. MiR-138-5p mimics decreased cell viability and invasion; while miR-138-5p inhibitor abolished RvD1-mediated suppression of cell viability and invasion. The expression of FOXC1 was significantly reduced upon overexpression of miR-138-5p while luciferase reporter assay showed that FOXC1 was a direct target of miR-138-5p. In vivo, endogenous DHA by the mfat-1 transgene enhanced miR-138-5p expression and decreased FOXC1 expression. Furthermore, overexpression of FOXC1 reversed the inhibition in cell viability and invasion induced by RvD1 treatment.
Conclusions
These data identified the RvD1/miR-138-5p/FOXC1 pathway as a novel mechanism by DHA and its metabolite, RvD1, and the potential of targeting such pathway as a therapeutic strategy in treating NSCLC.��
Background
Lung cancer is the most common cancer in both incidence and death rate, with non-small cell lung cancer (NSCLC) and small-cell lung cancer accounting for 85 and 15% of the reported cases, respectively [1,2,3]. Despite advances in detection and improvements to standard of care, NSCLC is often diagnosed at an advanced stage and bears poor prognosis [1]. Although a combination of resection and chemotherapy as well as immunotherapy can improve survival, the prognosis of lung cancer is still poor [4].
Docosahexaenoic acid (DHA), an ��-3 polyunsaturated fatty acid (PUFA), is an essential fatty acid [5]. Epidemiological literatures, including cross-sectional and migrational studies, have revealed a protective effect of DHA against the development of various cancers [6,7,8]. In NSCLC, DHA not only inhibits cancer cell proliferation and migration [9], but also suppresses angiogenesis [10]. Several DHA metabolites have been found to participate in the control of carcinogenesis [10]. For example, resolvin D1 (RvD1), one of DHA metabolites, has been reported to suppress tumor growth in several murine cancer cell lines at the doses 10,000 times lower than DHA [11]. The potential effects and underlying mechanisms of RvD1 on NSCLC cell growth and invasion, however, are completely unknown, apart from inhibiting TGF-��1-induced epithelial mesenchymal transition in lung cancer [12, 13].
MiRNAs are small, endogenous noncoding RNAs (18�C23 nucleotides) that post-transcriptionally regulate gene expression by targeting the 3��-untranslated regions (3��-UTR) of mRNAs [14]. Aberrant expression of some miRNAs has been reported to be associated with malignant phenotypes in various cancers, participating in not only cell proliferation, but also invasion and metastasis [15, 16]. In addition, several miRNAs were involved in the control of cancer cell growth and invasion by DHA metabolites [9, 13]. Among them, miR-138-5p is postulated as a tumor suppressor. Accordingly, the expression of miR-138-5p is decreased in many cancer tissues, compared with adjacent noncancerous tissues; and reduced expression of miR-138-5p is significantly correlated with patients�� clinicopathological factors and poor survival [17, 18]. Among NSCLC cases, miR-138-5p is involved in the elevation of GADD45A and inhibits cell growth [19]. However, little is known about the relationship between DHA and miR-138-5p.
��
A series of recent studies involving the usage of a transgenic genetic model, fat-1 (or mfat-1) transgenic mice, have shed new light into the mechanisms underlying the protective effects of ��-3 PUFAs [20]. The fat-1 transgenic mice, initially generated in Kang��s lab [21, 22] (and later the mfat-1 mouse model independently designed by our group [23]) carry a globally expressed C. elegans fat-1 transgene that encodes an ��-3 fatty acid desaturase. The FAT-1 enzyme can convert ��-6 PUFAs to the corresponding ��-3 forms by adding a double bond to the ��-3 position, thus allowing endogenous production of ��-3 PUFAs while reducing the ��-6: ��-3 ratio in tissues without special dietary adjustment [23, 24]. The fat-1 transgenic model allows researchers to bypass the traditional lengthy dietary approach of feeding fish-oil to the animals, and have been widely applied to the studies related to autoimmune diseases, tumorigenesis, metabolic and cardiovascular diseases, as well as neurological diseases.
In current studies, we found that endogenous DHA produced by mfat-1 gene significantly inhibited cell viability and metastasis, and increased RvD1 production in lung cancer cells. Further, miR-138-5p was upregulated in RvD1-treated lung cancer cells. Herein, we intend to advance the role and mechanisms of DHA and RvD1 on NSCLC growth and metastasis, with the primary focus on miR-138-5p-mediated pathway.��
Inhibition of lung cancer growth and metastasis by DHA and its metabolite, RvD1, through miR-138-5p/FOXC1 pathway | Journal of Experimental & Clinical Cancer Research |
https://jeccr.biomedcentral.com/articles/10.1186/s13046-019-1478-3��
��
��
f1-ijms-14-05501: Resolvin signaling pathways in different cell types. (A) In polymorphonuclear neutrophils (PMNs), RvE1 binds to ChemR23, activates G��i/o, which activates extracellular signal-regulated kinase (ERK), and eventually blocks TNF-�� signaling. Alternatively, RvE1 binds to BLT1 and blocks the activation of adenylate cyclase and NF��B signaling; (B) In Macrophages, RvE1 binds to the ChemR23 receptor and activates Akt via mTOR and alternatively blocks TNF-�� signaling via ERK. Also, RvD1 binds to GPR32 to enhance miRNA expression and activate transcription factors leading to increased phagocytosis; (C) In acinar cells, RvD1 binds to the ALX/FPR2 receptor leading to Akt activation and blocking TNF-�� signaling.
��
Understanding resolvin signaling pathways to improve oral health.
��
Affiliation: Department of Periodontics and Endodontics, School of Dental Medicine, University at Buffalo, the State University of New York, Buffalo, NY 14214-3932, USA. olgabake@buffalo.edu.
The discovery of resolvins has been a major breakthrough for understanding the processes involved in resolution of inflammation. Resolvins belong to a family of novel lipid mediators that possess dual anti-inflammatory and pro-resolution actions. Specifically, they protect healthy tissue during immune-inflammatory responses to infection or injury, thereby aiding inflammation resolution and promoting tissue healing. One of the major concerns in modern medicine is the management and treatment of oral diseases, as they are related to systemic outcomes impacting the quality of life of many patients. This review summarizes known signaling pathways utilized by resolvins to regulate inflammatory responses associated with the oral cavity.
��
Resolvin signaling pathways in different cell types. (A | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3634469_ijms-14-05501f1b&req=4��
��
DHA is a more potent inhibitor of breast cancer metastasis to bone and related osteolysis than EPA
Md M. Rahman, Maria Veigas, Paul J. Williams, and Gabriel Fernandes
Breast Cancer Res Treat. Author manuscript; available in PMC 2014 Oct 1
Abstract
Breast cancer patients often develop bone metastasis evidenced by osteolytic lesions, leading to severe pain and bone fracture. Attenuation of breast cancer metastasis to bone and associated osteolysis by fish oil (FO), rich in EPA and DHA, has been demonstrated previously. However, it was not known whether EPA and DHA differentially or similarly affect breast cancer bone metastasis and associated osteolysis. In vitro culture of parental and luciferase gene encoded MDA-MB-231 human breast cancer cell lines treated with EPA and DHA revealed that DHA inhibits proliferation and invasion of breast cancer cells more potently than EPA. Intra-cardiac injection of parental and luciferase gene encoded MDA-MB-231 cells to athymic NCr nu/nu mice demonstrated that DHA treated mice had significantly less breast cancer cell burden in bone, and also significantly less osteolytic lesions than EPA treated mice. In vivo cell migration assay as measured by luciferase intensity revealed that DHA attenuated cell migration specifically to the bone. Moreover, the DHA treated group showed reduced levels of CD44 and TRAP positive area in bone compared to EPA treated group. Breast cancer cell burden and osteolytic lesions were also examined in intra-tibially breast cancer cell injected mice and found less breast cancer cell growth and associated osteolysis in DHA treated mice as compared to EPA treated mice. Finally, doxorubicin resistant MCF-7 (MCF-7dox) human breast cancer cell line was used to examine if DHA can improve sensitization of MCF-7dox cells to doxorubicin. DHA improved the inhibitory effect of doxorubicin on proliferation and invasion of MCF-7dox cells. Interestingly, drug resistance gene P-gp was also down-regulated in DHA plus doxorubicin treated cells. In conclusion, DHA attenuates breast cancer bone metastasis and associated osteolysis more potently than EPA, possibly by inhibiting migration of breast cancer cell to the bone as well as by inhibiting osteoclastic bone resorption.
Keywords: Breast cancer, bone metastasis, omega-3 fatty acids, docosahexaenoic acid, osteolysis��
had significantly less breast cancer cell burden in bone, and also significantly less osteolytic lesions than EPA treated mice. In vivo cell migration assay as measured by luciferase intensity revealed that DHA attenuated cell migration specifically to the bone. Moreover, the DHA treated group showed reduced levels of CD44 and TRAP positive area in bone compared to EPA treated group. Breast cancer cell burden and osteolytic lesions were also examined in intra-tibially breast cancer cell injected mice and found less breast cancer cell growth and associated osteolysis in DHA treated mice as compared to EPA treated mice. Finally, doxorubicin resistant MCF-7 (MCF-7dox) human breast cancer cell line was used to examine if DHA can improve sensitization of MCF-7dox cells to doxorubicin. DHA improved the inhibitory effect of doxorubicin on proliferation and invasion of MCF-7dox cells. Interestingly, drug resistance gene P-gp was also down-regulated in DHA plus doxorubicin treated cells. In conclusion, DHA attenuates breast cancer bone metastasis and associated osteolysis more potently than EPA, possibly by inhibiting migration of breast cancer cell to the bone as well as by inhibiting osteoclastic bone resorption.
Keywords: Breast cancer, bone metastasis, omega-3 fatty acids, docosahexaenoic acid, osteolysisInhibition of lung cancer growth and metastasis by DHA and its metabolite, RvD1, through miR-138-5p/FOXC1 pathway | Journal of Experimental & Clinical Cancer Research | Full Text
https://jeccr.biomedcentral.com/articles/10.1186/s13046-019-1478-3��
Docosahexaenoic Acid Induces Cell Death in Human Non-Small Cell Lung Cancer Cells by Repressing mTOR via AMPK Activation and PI3K/Akt Inhibition
��
The anticancer properties and mechanism of action of omega-3 polyunsaturated fatty acids ( �� 3-PUFAs) have been demonstrated in several cancers; however, the mechanism in lung cancer remains unclear. Here, we show that docosahexaenoic acid (DHA), a �� 3-PUFA, induced apoptosis and autophagy in non-small cell lung cancer (NSCLC) cells. DHA-induced cell death was accompanied by AMP-activated protein kinase (AMPK) activation and inactivated phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling. Knocking down AMPK and overexpressing Akt increased mTOR activity and attenuated DHA-induced cell death, suggesting that DHA induces cell death via AMPK- and Akt-regulated mTOR inactivation. This was confirmed in Fat-1 transgenic mice, which produce �� 3-PUFAs. Lewis lung cancer (LLC) tumor cells implanted into Fat-1 mice showed slower growth, lower phospho-Akt levels, and higher levels of apoptosis and autophagy than cells implanted into wild-type mice. Taken togetuher, these data suggest that DHA-induced apoptosis and autophagy in NSCLC cells are associated with AMPK activation and PI3K/Akt inhibition, which in turn lead to suppression of mTOR; thus �� 3-PUFAs may be utilized as potential therapeutic agents for NSCLC treatment.
��
��
https://openi.nlm.nih.gov/detailedresult.php?img=PMC4538321_BMRI2015-239764.006&req=4
��
��
www.tara.tcd.ie/bitstream/handle/2262/33699/Vitamin C inhibits NF-kappa B activation by...Vitamin C Inhibits NF-kB Activation by TNF Via the Activation of p38 Mitogen-Activated Protein Kinase1 Andrew G. Bowie2 and Luke A. J. O��Neill The transcription factor NF-kB is a central mediator of altered gene expression during inflammation, and is implicated in a
IJMS | Free Full-Text | Mollugin Has an Anti-Cancer Therapeutic Effect by Inhibiting TNF-��-Induced NF-��B Activation | HTML
https://www.mdpi.com/1422-0067/18/8/1619/htm��
Mol Cell Biol. 1998 Sep;18(9):5157-65.
Human T-cell leukemia virus type 1 Tax induction of NF-kappaB involves activation of the IkappaB kinase alpha (IKKalpha) and IKKbeta cellular kinases.
Geleziunas R1, Ferrell S, Lin X, Mu Y, Cunningham ET Jr, Grant M, Connelly MA, Hambor JE, Marcu KB, Greene WC.
Author information
1
Gladstone Institute of Virology and Immunology, Microbiology, and Immunology University of California, San Francisco, San Francisco, California 94141-9100, USA. Romas_Geleziunas@quickmail.ucsf.edu
Abstract
Tax corresponds to a 40-kDa transforming protein from the pathogenic retrovirus human T-cell leukemia virus type 1 (HTLV-1) that activates nuclear expression of the NF-kappaB/Rel family of transcription factors by an unknown mechanism. Tax expression promotes N-terminal phosphorylation and degradation of IkappaB alpha, a principal cytoplasmic inhibitor of NF-kappaB. Our studies now demonstrate that HTLV-1 Tax activates the recently identified cellular kinases IkappaB kinase alpha (IKKalpha) and IKKbeta, which normally phosphorylate IkappaB alpha on both of its N-terminal regulatory serines in response to tumor necrosis factor alpha (TNF-alpha) and interleukin-1 (IL-1) stimulation. In contrast, a mutant of Tax termed M22, which does not induce NF-kappaB, fails to activate either IKKalpha or IKKbeta. Furthermore, endogenous IKK enzymatic activity was significantly elevated in HTLV-1-infected and Tax-expressing T-cell lines. Transfection of kinase-deficient mutants of IKKalpha and IKKbeta into either human Jurkat T or 293 cells also inhibits NF-kappaB-dependent reporter gene expression induced by Tax. Similarly, a kinase-deficient mutant of NIK (NF-kappaB-inducing kinase), which represents an upstream kinase in the TNF-alpha and IL-1 signaling1����Tϸ����Ѫ������NF-��B��˰���յ��漰IkappaB��ø����IKKalpha����IKKbetaϸ����ø�ļ��
���������Ǵ�ѧ�ɽ�ɽ��У������˹ͨ����������ѧ������ѧ������ѧ�о�����
Tax��Ӧ��һ�������²�����ת¼��������Tϸ����Ѫ������1�ͣ�HTLV-1����40 kDaת�����ף��õ���ͨ��δ֪���Ƽ���NF-kappaB / Rel����ת¼���ӵĺ˱��TAX����ٽ�IkappaB alpha��һ����Ҫ��NF-kappaBϸ�������Ƽ�����N�����ữ�ͽ��⡣
���ǵ��о����ڱ���HTLV-1TAX�����������������ϸ����øIkappaB��øalpha��IKKalpha����IKKbeta������ͨ��������Nĩ�˵���˿���������ữIkappaB alpha������Ӧ�����������Ӧ���TNF-alpha���Ͱ���1��IL-1���̼���
���֮�£����յ�NF-��B�ij�ΪTax��M22ͻ���岻�ܼ���IKKalpha��IKKbeta��
ͬ����NIK��øȱ����ͻ���壨NF-kappaB�յ��ļ�ø����������IKKalpha��IKKbeta�����TNF-alpha��IL-1�ź�ͨ·�е����μ�ø����ֹ��NF-kappaB��Tax�յ������ǣ���Ȼ���漰��Щ������ϸ������;���е���Ĥ����Ԫ������ΪTRAF2��TRAF6�ν��ӵ����Ը�ͻ���������Ч����Ϸֱ�ͨ��TNF-����IL-1�����ϸ����β�ͷ����ź�,����NF-��B��TAX�յ���
��������о���ʾ��NF-��B;���ڳ���Tϸ����Ѫ����һ������T�Ͱ�Ѫ������1�ͣ�HTLV-1���������Ϯ�Զ����������б�����Լ������������TAXͨ���븻�����ص�IKK�������������������������Լ��
��֮����Щ�о�������HTLV-1 Tax���ô���ϸ�������źŴ���������ĩ�ˣ��Ӷ�������NF-��B���յ�����ϸ������;������ͨ��Tax����NF-��B�IJ����ı������HTLV-1�鵼������Tϸ��ת���з��Ӻ������ã��ٴ�����Ϊ����Tϸ����Ѫ������
Human T-cell leukemia virus type 1 Tax induction of NF-kappaB involves activation of the IkappaB kinase alpha (IKKalpha) and IKKbeta cellular kinases. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/9710600��
Sci Rep. 2019 Nov 5;9(1):16014. doi: 10.1038/s41598-019-52408-x.
SQSTM-1/p62 potentiates HTLV-1 Tax-mediated NF-��B activation through its ubiquitin binding function.
Schwob A1,2, Teruel E1,2, Dubuisson L1,2, Lormi��res F1,2, Verlhac P3,4, Abudu YP5, Gauthier J1,2, Naoumenko M1,2, Cloarec-Ung FM1,2,6, Faure M3, Johansen T5, Dutartre H1,2, Mahieux R1,2, Journo C7,8.
Author information
1
International Center for Research in Infectiology, Retroviral Oncogenesis Laboratory, INSERM U1111 - Universit�� Claude Bernard Lyon 1, CNRS, UMR5308, École Normale Sup��rieure de Lyon, Universit�� Lyon, Lyon, France.
2
Equipe labellis��e "Fondation pour la Recherche M��dicale", Paris, France.
3
International Center for Research in Infectiology, Autophagy, Infection, Immunity Laboratory, INSERM U1111 - Universit�� Claude Bernard Lyon 1, CNRS, UMR5308, École Normale Sup��rieure de Lyon, Universit�� Lyon, Lyon, France.
4
Department of Biomedical Sciences of Cells and Systems, Section Molecular Cell Biology, University Medical Center Groningen, Groningen, The Netherlands.
5
Molecular Cancer Research Group, Institute of Medical Biology, University of Tromsø - The Arctic University of Norway, Tromsø, Norway.
6
École Normale Sup��rieure Paris-Saclay, Universit�� Paris-Saclay, Cachan, France.
7
International Center for Research in Infectiology, Retroviral Oncogenesis Laboratory, INSERM U1111 - Universit�� Claude Bernard Lyon 1, CNRS, UMR5308, École Normale Sup��rieure de Lyon, Universit�� Lyon, Lyon, France. chloe.journo@ens-lyon.fr.
8
Equipe labellis��e "Fondation pour la Recherche M��dicale", Paris, France. chloe.journo@ens-lyon.fr.
Abstract
The NF-��B pathway is constitutively activated in adult T cell leukemia, an aggressive malignancy caused by Human T Leukemia Virus type 1 (HTLV-1). The viral oncoprotein Tax triggers this constitutive activation by interacting with the ubiquitin-rich IKK complex. We previously demonstrated that Optineurin and TAX1BP1, two members of the ubiquitin-binding, Sequestosome-1 (SQSTM-1/p62)-like selective autophagy receptor family, are involved in Tax-mediated NF-��B signaling. Here, using a proximity-dependent biotinylation approach (BioID), we identify p62 as a new candidate partner of Tax and confirm the interaction in infected T cells. We then demonstrate that p62 knock-out in MEF cells as well as p62 knock-down in HEK293T cells significantly reduces Tax-mediated NF-��B activity. We further show that although p62 knock-down does not alter NF-��B activation in Jurkat T cells nor in infected T cells, p62 does potentiate Tax-mediated NF-��B activity upon over-expression in Jurkat T cells. We next show that p62 associates with the Tax/IKK signalosome in cells, and identify the 170-206 domain of p62 as sufficient for the direct, ubiquitin-independent interaction with Tax. However, we observe that this domain is dispensable for modulating Tax activity in cells, and functional analysis of p62 mutants indicates that p62 could potentiate Tax activity in cells by facilitating the association of ubiquitin chains with the Tax/IKK signalosome. Altogether, our results identify p62 as a new ubiquitin-dependent modulator of Tax activity on NF-��B, further highlighting the importance of ubiquitin in the signaling activity of the viral Tax oncoprotein.
PMID: 31690813 PMCID: PMC6831704 DOI: 10.1038/s41598-019-52408-xSQSTM-1/p62 potentiates HTLV-1 Tax-mediated NF-��B activation through its ubiquitin binding function. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31690813��
J Biol Chem. 2007 Feb 9;282(6):4185-92. Epub 2006 Dec 4.
Activation of NF-kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin-dependent relocalization of I kappa B kinase.
Harhaj NS1, Sun SC, Harhaj EW.
Author information
1
Department of Microbiology and Immunology, Sylvester Comprehensive Cancer Center, The University of Miami, Miller School of Medicine, Miami, Florida 33136, USA.
Abstract
Human T cell leukemia virus type 1 (HTLV-1) is the etiological agent of adult T cell leukemia. HTLV-1 encodes a trans-activating protein, Tax, which is largely responsible for the oncogenic properties of the virus. Tax promotes T cell transformation by deregulating the activity of various cellular factors, including the transcription factor NF-kappaB. Tax activates the IkappaB kinase (IKK) via physical interaction with the regulatory subunit, IKKgamma, although it is unknown precisely how Tax activates the IKK complex. Here we show that Tax modulates the cellular localization of the IKK complex. The IKKs relocalize from a broad distribution in the cytoplasm to concentrated perinuclear "hot spots" in both HTLV-1-transformed lines and in Tax-expressing Jurkat cells. Relocalization of IKK is not observed with Tax mutants unable to activate NF-kappaB, suggesting that only activated forms of IKK are relocalized. However, relocalization of IKK is strictly dependent on Tax expression because it does not occur in ATL cell lines that lack Tax expression or in Jurkat cells treated with phorbol 12-myristate 13-acetate and ionomycin. Furthermore, IKKgamma is required for redistribution because cells lacking IKKgamma were unable to relocalize IKKalpha upon expression of Tax. We also find that Tax ubiquitination likely regulates IKK relocalization because mutation of three critical lysine residues in Tax renders it unable to relocalize IKK and activate the canonical and noncanonical NF-kappaB pathways. Finally, we have observed that the perinuclear IKK in Tax-expressing cells colocalizes with the Golgi, and disruption of Golgi with either nocodazole or brefeldin A leads to a redistribution of IKK to the cytoplasm. Together, these results demonstrate that Tax induces relocalization of the IKK complex in a ubiquitin-dependent manner, and dynamic changes in the subcellular localization of the IKK complex may be critical for Tax function.Activation of NF-kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin-dependent relocalization of I kappa... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/17145747��
Abnormal activation of transcription factor NF-kB by oncoprotein of Human T cell leukemia Virus 1 (HTLV-1)
��Wednesday, February 24, 2016
Speaker: Prof. Jun-ichiro Inoue
Division of Cellular and Molecular Biology, Department of Cancer Biology
Institute of Medical Science, University of TokyoAbstract: Activation of NF-kB by human T cell leukemia virus type 1 Tax is thought to be crucial in T cell transformation and the onset of adult T cell leukemia. Tax constitutively activates NF-kB through activation of the IkB kinase (IKK) complex, which is only transiently activated in response to physiological stimulation in a polyubiquitination-dependent manner. Although polyubiquitination of Tax was reported to be required for IKK activation, most studies have been performed using intact cells, in which secondary NF-kB activation can be induced by various cytokines that are secreted due to Tax-mediated primary NF-kB activation. Therefore, a cell-free assay system, in which IKK can be activated by adding highly purified recombinant Tax to cytosolic extract, was used to analyze Tax-induced IKK activation. In contrast to the cytosolic extract, the purified IKK complex was not activated by Tax, whereas it was efficiently activated by MEKK1, which does not require polyubiquitination to activate IKK. Critical roles of various polyubiquitin chains in the Tax-induced NF-kB activation will be discussed.
"Abnormal activation of transcription factor NF-kB by oncoprotein of Human T cell leukemia Virus 1 (HTLV-1)" | OIST Groups
https://groups.oist.jp/csu/event/abnormal-activation-transcription-factor-nf-kb-oncoprotein-human-t-cell-leukemia-virus-1��
Curcumin Treatment Suppresses IKKb Kinase Activity of ...
https://clincancerres.aacrjournals.org/content/clincanres/17/18/5953.full.pdf
Curcumin Treatment Suppresses IKKb Kinase Activity of Salivary Cells of Patients with Head and Neck Cancer: A Pilot Study ... Curcumin inhibited IKKb kinase activity in the saliva of HNSCC patients, and this ... (IKK) kinase, a complex consisting of 3 proteins: IKKa, IKKb, and IKKg [also known as the NF-kB essential mod-ulator (NEMO)], is ...��
��Linking JNK signaling to NF-��B: a key to survival
Salvatore Papa, Francesca Zazzeroni, Can G. Pham, Concetta Bubici, Guido Franzoso Journal of Cell Science 2004 117: 5197-5208; doi: 10.1242/jcs.01483 Summary In addition to marshalling immune and inflammatory responses, transcription factors of the NF-��B family control cell survival. This control is crucial to a wide range of biological processes, including B and T lymphopoiesis, adaptive immunity, oncogenesis and cancer chemoresistance. During an inflammatory response, NF-��B activation antagonizes apoptosis induced by tumor necrosis factor (TNF)-��, a protective activity that involves suppression of the Jun N-terminal kinase (JNK) cascade. This suppression can involve upregulation of the Gadd45-family member Gadd45��/Myd118, which associates with the JNK kinase MKK7/JNKK2 and blocks its catalytic activity. Upregulation of XIAP, A20 and blockers of reactive oxygen species (ROS) appear to be important additional means by which NF-��B blunts JNK signaling. These recent findings might open up entirely new avenues for therapeutic intervention in chronic inflammatory diseases and certain cancers; indeed, the Gadd45��-MKK7 interaction might be a key target for such intervention. Linking JNK signaling to NF-��B: a key to survival |
��
Viral pathogenesis
The pro-survival activity of NF-��B also plays a crucial role in viral pathogenesis (reviewed by Kucharzack et al., 2003). Indeed, the need for an inducible gene expression program to maintain cell survival might have originally evolved as a mechanism for disposing of infected cells that, because of viral takeover, exhibit grossly defective transcription. Not surprisingly, many viruses have adapted to this host defense mechanism by developing their own anti-apoptotic strategies or acquiring genes that either induce or mimic NF-��B. Examples of such genes, many of which are implicated in viral oncogenesis, include: v-FLIP of human herpesvirus 8 (HHV-8), which is linked to Kaposi's sarcoma and lymphoma; Tax of HTLV-1, which causes adult T-cell leukemia (ATL); and of course v-Rel, which is encoded by the avian retrovirus REV-T (Kucharczak et al., 2003; Benedict et al., 2002).
��
Journal of Cell Science https://jcs.biologists.org/content/117/22/5197
��
Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and JNK activation.
Background: Glioblastoma multiforme (GBM) is the most aggressive of the primary brain tumors, with a grim prognosis despite intensive treatment. In the past decades, progress in research has not significantly increased overall survival rate.
Methods: The in vitro antineoplastic effect and mechanism of action of Casiopeina III-ia (Cas III-ia), a copper compound, on rat malignant glioma C6 cells was investigated.
Results: Cas III-ia significantly inhibited cell proliferation, inducing autophagy and apoptosis, which correlated with the formation of autophagic vacuoles, overexpression of LC3, Beclin 1, Atg 7, Bax and Bid proteins. A decrease was detected in the mitochondrial membrane potential and in the activity of caspase 3 and 8, together with the generation of intracellular reactive oxygen species (ROS) and increased activity of c-jun NH(2)-terminal kinase (JNK). The presence of 3-methyladenine (as selective autophagy inhibitor) increased the antineoplastic effect of Cas III-ia, while Z-VAD-FMK only showed partial protection from the antineoplastic effect induced by Cas III-ia, and ROS antioxidants (N-acetylcysteine) decreased apoptosis, autophagy and JNK activity. Moreover, the JNK -specific inhibitor SP600125 prevented Cas III-ia-induced cell death.
Conclusions: Our data suggest that Cas III-ia induces cell death by autophagy and apoptosis, in part due to the activation of ROS -dependent JNK signaling. These findings support further studies of Cas III-ia as candidate for treatment of human malignant glioma.F9: Suggested pathway initiated by Cas III-ia leading to autophagy and apoptosis in C6 glioma cells. Cas III-ia may cause oxidative stress by JNK activation. JNK can phosphorylate Bcl-2 and release Beclin 1 enhancing autophagy. Alternatively, JNK can induce phosphorylation of c-jun; phospho-c-jun favors the production of AP-1 which, in turn, increases the expression of many genes among which are c-jun itself, Beclin, Atg7, Bax and FasL. Beclin 1 and Atg 7 may induce autophagy and FasL can stimulate Fas-R inducing the activation of caspase 8, which then degrades Bid into Bidt inducing Bax oligomerization and the depolarization of the mitochondrial membrane with release of cyt c and the consequent activation of caspase 3. Autophagy and apoptosis can thus induce cell death.
Suggested pathway initiated by Cas III-ia leading to au | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3404907_1471-2407-12-156-9&req=4��
J Neurosci. 2014 Jan 29; 34(5): 1903�C1915.
Omega-3 Fatty Acids Protect the Brain against Ischemic Injury by Activating Nrf2 and Upregulating Heme Oxygenase 1
Meijuan Zhang,1,2,3,* Suping Wang,2,* Leilei Mao,1,2,* Rehana K. Leak,4 Yejie Shi,2 Wenting Zhang,1 Xiaoming Hu,1,2,6 Baoliang Sun,5 Guodong Cao,2,6 Yanqin Gao,1 Yun Xu,3 Jun Chen,corresponding author1,2,6 and Feng Zhangcorresponding author1,2,5,6
Abstract
Ischemic stroke is a debilitating clinical disorder that affects millions of people, yet lacks effective neuroprotective treatments. Fish oil is known to exert beneficial effects against cerebral ischemia. However, the underlying protective mechanisms are not fully understood. The present study tests the hypothesis that omega-3 polyunsaturated fatty acids (n-3 PUFAs) attenuate ischemic neuronal injury by activating nuclear factor E2-related factor 2 (Nrf2) and upregulating heme oxygenase-1 (HO-1) in both in vitro and in vivo models. We observed that pretreatment of rat primary neurons with docosahexaenoic acid (DHA) significantly reduced neuronal death following oxygen-glucose deprivation. This protection was associated with increased Nrf2 activation and HO-1 upregulation. Inhibition of HO-1 activity with tin protoporphyrin IX attenuated the protective effects of DHA. Further studies showed that 4-hydroxy-2E-hexenal (4-HHE), an end-product of peroxidation of n-3 PUFAs, was a more potent Nrf2 inducer than 4-hydroxy-2E-nonenal derived from n-6 PUFAs. In an in vivo setting, transgenic mice overexpressing fatty acid metabolism-1, an enzyme that converts n-6 PUFAs to n-3 PUFAs, were remarkably resistant to focal cerebral ischemia compared with their wild-type littermates. Regular mice fed with a fish oil-enhanced diet also demonstrated significant resistance to ischemia compared with mice fed with a regular diet. As expected, the protection was associated with HO-1 upregulation, Nrf2 activation, and 4-HHE generation. Together, our data demonstrate that n-3 PUFAs are highly effective in protecting the brain, and that the protective mechanisms involve Nrf2 activation and HO-1 upregulation by 4-HHE. Further investigation of n-3 PUFA neuroprotective mechanisms may accelerate the development of stroke therapies.Omega-3 Fatty Acids Protect the Brain against Ischemic Injury by Activating Nrf2 and Upregulating Heme Oxygenase 1
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3905150/��
molecular Cancer, 2015
Activation of the NF-��B pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma
Fei Wang, Jin-Lian Yang, Ke-ke Yu, Mei Xu, You-zhi Xu, Li Chen, Yan-min Lu, Hao-shu Fang, Xin-yi Wang, Zhong-qian Hu, Fei-fei Li, Lixin Kan, Jia Luo & Si-Ying Wang
Molecular Cancer volume 14, Article number: 10 (2015) Cite this article
Abstract
Background
Hepatocellular carcinoma (HCC), the most common form of primary liver cancer, is the third leading cause of cancer-related death in human. Alcohol is a known risk factor for HCC. However it is still unclear whether and how alcohol enhances the progression and metastasis of existing HCC.
Methods and results
We first retrospectively investigated 52 HCC patients (24 alcohol-drinkers and 28 non-drinkers), and found a positive correlation between alcohol consumption and advanced Tumor-Node-Metastasis (TNM) stages, higher vessel invasion and poorer prognosis. In vitro and in vivo experiments further indicated that alcohol promoted the progression and migration/invasion of HCC. Specifically, in a 3-D tumor/endothelial co-culture system, we found that alcohol enhanced the migration/invasion of HepG2 cells and increased tumor angiogenesis. Consistently, higher expression of VEGF, MCP-1 and NF-��B was observed in HCC tissues of alcohol-drinkers. Alcohol induced the accumulation of intracellular reactive oxygen species (ROS) and the activation of NF-��B signaling in HepG2 cells. Conversely, blockage of alcohol-mediated ROS accumulation and NF-��B signaling inhibited alcohol-induced expression of VEGF and MCP-1, the tumor growth, angiogenesis and metastasis.
Conclusion
This study suggested that chronic moderate alcohol consumption may promote the progression and metastasis of HCC; the oncogenic effect may be at least partially mediated by the ROS accumulation and NF-ĸB-dependent VEGF and MCP-1 up-regulation.Activation of the NF-��B pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma | Molecular Cancer | Full Text
https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-014-0274-0��
The omega-3 polyunsaturated fatty acid docosahexaenoic acid inhibits proliferation and progression of non-small cell lung cancer cells through the reactive oxygen species-mediated inactivation of the PI3K /Akt pathway
Yuanqin Yin, Chengguang Sui, Fandong Meng, Ping Ma & Youhong Jiang
Cancer Institute, First Affiliated Hospital, China Medical University, Shenyang, Liaoning, 110001, China
Abstract
Background
Docosahexaenoic acid(DHA) inhibits tumor growth and progression in various cancers, including lung cancer. However, the mechanisms involved remain unclear. The aim of this study was to identify the mechanism of DHA in inhibiting progression of non-small cell lung cancer (NSCLC) in vitro.
Methods
The proliferation of A549 was tested by MTT, and cell apoptosis was analysed using flow cytometer. The migration and invasion were examined respectively by wound healing assay and Transwell invasion assay. The level of ROS (reactive oxygen species, ROS) was checked by DCF (dichlorodihydrofluorescein, DCF) production in cells. The apoptosis associated protein (caspase-3, PARP,Bax,Bcl-2 and survivin) and metastases associated proteins including HEF1, MMP9 and VEGF were detected by Western blot, and the same method was used in the expression of PI3K and Akt.
Results
DHA inhibited proliferation and induced apoptosis of A549 cells. Moreover, it suppressed the invasion and metastasis of A549 cells, while downregulating the levels of metastasis-associated proteins, including HEF1, matrix metallopeptidase (MMP9), and vascular endothelial growth factor (VEGF), in a dose -dependent manner. In addition, DHA inactivated Akt phosphorylation. All of these responses were associated with the accumulation of intracellular ROS. DHA downregulated the level of antioxidant enzymes such as catalase, while the antioxidant N-acetyl-cysteine (NAC) reversed the effect of DHA, which further validated our findings.
Conclusions
The present study demonstrates that DHA inhibits the development of non-small lung tumors through an ROS-mediated inactivation of the PI3K/Akt signaling pathway.
The omega-3 polyunsaturated fatty acid docosahexaenoic acid inhibits proliferation and progression of non-small cell lung cancer cells through the reactive oxygen species-mediated inactivation of the PI3K /Akt pathway | Lipids in Health and Disease | Full Text
https://lipidworld.biomedcentral.com/articles/10.1186/s12944-017-0474-x��
The Journal of Nutritional Biochemistry
Anti-inflammatory effects of EPA and DHA are dependent upon time and dose-response elements associated with LPS stimulation in THP-1-derived macrophages
Abstract
The long-chain n-3 polyunsaturated fatty acids (LC n-3 PUFA) of fish oil, eicosapentanoic (EPA) and docosahexanoic (DHA) acids are considered cardioprotective. Inflammation elicited by macrophages is increasingly recognised in the aetiology of metabolic syndrome. This study investigated the differential anti-inflammatory potential of EPA and DHA through cytokine production and nuclear factor (NF)-��B signalling in a human macrophage model. We investigated the dependency of LC n-3 PUFA immune-modulation on concentration and duration of lipopolysaccharide (LPS) activation.
Interleukin (IL)-1��, IL-6 and tumor necrosis factor-�� secretion from EPA, DHA and control cells were differentially limited by LPS concentration. In all cases, there was no benefit in activating cells with >0.1 ��g/ml LPS. LC n-3 PUFA decreased proinflammatory cytokines production, an effect modulated by LPS concentration. Expression of the transcription factor NF-��B p65 was significantly reduced in the nucleus and retained in the cytoplasm of EPA- and/or DHA-treated macrophages during 5-h activation with 0.1 ��g/ml LPS. Nuclear binding of p65 was significantly reduced in EPA- and DHA-treated cells at 2-h LPS activation. Over the time course, expression of nuclear I��B�� was significantly reduced, cytoplasmic NF-��B p50 significantly increased and cytoplasmic cleaving enzyme I��B inhibitor complex significantly reduced in LC n-3 PUFA-treated cells.
EPA and DHA down-regulated the production of proinflammatory cytokines associated with the aetiology of metabolic syndrome, NF-��B transcriptional activity and upstream cytoplasmic signalling events. Immune responses are dynamic, and the present study suggests a nutrient sensitive window of LPS activation at which EPA and DHA are strongly anti-inflammatory.��
EPA��DHA�Ŀ���������������THP -1��Դ�ľ���ϸ����LPS�̼���ص�ʱ��ͼ�����Ӧ����
ժҪ
�����еij���n-3�����֬����(LC n-3 PUFA)����ʮ̼��ϩ��(EPA)�Ͷ�ʮ��̼��ϩ��(DHA)����Ϊ�������ౣ�����á�����ϸ���������֢�ڴ�л�ۺ����IJ���ѧ�б�Խ��Խ�����ʶ�������о�������EPA��DHA����DZ�ڿ���ϸ�����ӵ������ͺ�ת¼����(NF) -��B�ź����������ϸ��ģ�͡������о���LC n-3 PUFA���ߵ��ڶ�֬����(LPS)����Ũ�Ⱥͳ���ʱ���������ϵ��
����(IL) 1��,���غ���������factor-������EPA��DHA�Ϳ���ϸ����ͬLPSŨ�ȵ����ơ������������,û������> 0.1��g /����LPS����ϸ����LC n-3 PUFA���ʹ���ϸ�����ӵIJ�������������LPSŨ�ȵ��ڡ������ת¼����NF-��B p65����������ϸ���˺�ϸ�����еı���EPA - 5 h�ڼ��/��DHA-treated����ϸ�����0.1��g /�������ϻ��ˡ��ڼ���2Сʱ��EPA��dha������ϸ���У�p65�ĺ˽���������١��˵�ʱ�����,����I��B�����Լ���,ϸ����NF-��B p50��������,�����ѿ�I��Bø���Ƽ�����LC n - 3 PUFA-treatedϸ���������١�
EPA��DHA���ƴ���ϸ�����ӵ��������л�ۺ����IJ���ѧ�й�,NF-��Bת¼���λ�Ͱ����ź��¼������߷�Ӧ�Ƕ�̬�ģ�Ŀǰ���о�����LPS�����һ��Ӫ�����д��ڣ���������ڣ�EPA��DHA���к�ǿ�Ŀ������á���
Anti-inflammatory effects of EPA and DHA are dependent upon time and dose-response elements associated with LPS stimulation in THP-1-derived macrophages - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0955286309000370��
Fish oil targets PTEN to regulate NF��B for downregulation of anti-apoptotic genes in breast tumor growth
Abstract
The molecular mechanism for the beneficial effect of fish oil on breast tumor growth is largely undefined. Using the xenograft model in nude mice, we for the first time report that the fish oil diet significantly increased the level of PTEN protein in the breast tumors.In addition, the fish oil diet attenuated the PI 3 kinase and Akt kinase activity in the tumors leading to significant inhibition of NF��B activation. Fish oil diet also prevented the expression of anti-apoptotic proteins Bcl-2 and Bcl-XL in the breast tumors with concomitant increase in caspase 3 activity.
To extend these findings we tested the functional effects of DHA and EPA, the two active ��-3 fatty acids of fish oil, on cultured MDA MB-231 cells. In agreement with our in vivo data, DHA and EPA treatment increased PTEN mRNA and protein expression and inhibited the phosphorylation of p65 subunit of NF��B in MDA MB-231 cells. Furthermore, DHA and EPA reduced expression of Bcl-2 and Bcl-XL. NF��B DNA binding activity and NF��B-dependent transcription of Bcl-2 and Bcl-XL genes were also prevented by DHA and EPA treatment. Finally, we showed that PTEN expression significantly inhibited NF��B-dependent transcription of Bcl-2 and Bcl-XL genes.
Taken together, our data reveals a novel signaling pathway linking the fish oil diet to increased PTEN expression that attenuates the growth promoting signals and augments the apoptotic signals, resulting in breast tumor regression.
Keywords: PTEN, NF��B, DHA, EPA, Breast tumor growth, Apoptotic signal
Fish oil targets PTEN to regulate NF��B for downregulation of anti-apoptotic genes in breast tumor growth
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2847452/��
Mol Cell Biol. 2003 Nov; 23(22): 8070�C8083.
I��B Kinase-Independent I��B�� Degradation Pathway: Functional NF-��B Activity and Implications for Cancer Therapy
Vinay Tergaonkar, Virginie Bottero, Masahito Ikawa,† Qiutang Li, and Inder M. Verma* Laboratory of Genetics, The Salk Institute for Biological Studies, La Jolla, California 92037
ABSTRACT
Antiapoptotic activity of NF-��B in tumors contributes to acquisition of resistance to chemotherapy. Degradation of I��B is a seminal step in activation of NF-��B. The I��B kinases, IKK1 and IKK2, have been implicated in both I��B degradation and subsequent modifications of NF��B.
Using mouse embryo fibroblasts (MEFs) devoid of both IKK1 and IKK2 genes (IKK1/2−/−), we document a novel I��B degradation mechanism. We show that this degradation induced by a chemotherapeutic agent, doxorubicin (DoxR), does not require the classical serine 32 and 36 phosphorylation or the PEST domain of I��B��. Degradation of I��B�� is partially blocked by phosphatidylinositol 3-kinase inhibitor LY294002 and is mediated by the proteasome. Free NF-��B generated by DoxR-induced I��B degradation in IKK1/2−/− cells is able to activate chromatin based NF-��B reporter gene and expression of the endogenous target gene, I��B��. These results also imply that modification of NF-��B by IKK1 or IKK2 either prior or subsequent to its release from I��B is not essential for NF-��B-mediated gene expression at least in response to DNA damage. In addition, DoxR-induced cell death in IKK1/2−/− MEFs is enhanced by simultaneous inhibition of NF-��B activation by blocking the proteasome activity. These results reveal an additional pathway of activating NF-��B during the course of anticancer therapy and provide a mechanistic basis for the observation that proteasome inhibitors could be used as adjuvants in chemotherapy.
��
The transcription factor NF-��B has been documented to play roles in innate and adaptive immune responses (19). Deregulated activity of NF-��B pathway has also been observed and linked to the progression of several human malignancies (27). It is becoming increasingly clear that NF-��B can modulate multiple aspects of cell growth and death (10). Understanding the regulation of NF-��B activity is likely to shed insight into cancer progression and treatment. NF-��B, a dimer of two similar or heterologous subunits, is predominantly found in the cytoplasm complexed with the I��B family of inhibitory molecules (37). Degradation of I��B proteins in response to stimuli is an obligatory step in NF-��B activation (14). This degradation is mediated by the proteasome and requires that the I��B molecules are phosphorylated at specific serine residues (4). Hence, the role of the I��B kinases (IKKs), IKK1 (also called IKK��) and IKK2 (also called IKK��), which induce signal-dependent phosphorylation and degradation of I��B molecules, is extensively investigated. The cytoplasmic >700-kDa complex containing IKK1 and IKK2 receives and transmits various stimuli that lead to I��B�� degradation and NF-��B DNA binding. Apart from the two IKK enzymes, the IKK complex is believed to consist of other known and unknown proteins which might have regulatory or chaperone-like functions (5, 39). Mice which were homozygous null for both IKK1 and IKK2 die at embryonic day 9.5 (18). Mouse embryo fibroblasts (MEFs) derived from IKK1/2−/− double knockout embryos are severely defective in I��B degradation and NF-��B activation, in response to ��classical�� stimuli such as cytokines and bacterial products recognized by pattern recognition receptors (19). These results emphasize that IKK1 and IKK2 are absolutely essential for I��B degradation and NF-��B activation (18). The rate-limiting step in the activation of NF-��B is believed to be its release from the inhibitor I��B molecules, which retain NF-��B in the cytoplasm and additionally inhibit its DNA-binding ability. Defective I��B�� activity has been mechanistically linked to the persistent nuclear NF-��B seen in many human tumors (27). However, other genetic modifications of NF-��B subsequent to its release from I��B are now thought to be necessary for generating functional NF-��B activity in the context of transcription of target genes. These changes, which are mainly initiated by selective phosphorylation, increase its interaction with p300/CBP and also the chromatin remodeling factors. Several kinases, including GSK3, NIK, CKII, PKA, PKC��, TBK/t2k/NAK, and IKK themselves have been thought to be required for modifying NF-��B after its release from I��B�� (10). Since the same catalytic activity of IKK is required both for I��B�� degradation and NF-��B modification, it has been difficult to dissect the contribution of IKKs in processes subsequent to the release of NF-��B from I��B. In the present study, using IKK1/2−/− MEFs, we decipher a novel mechanism of I��B�� degradation in response to the anticancer agent doxorubicin (DoxR), independent of IKK1 and IKK2. Further, treatment of IKK1/2−/− MEFs with DoxR leads to NF-��B transcriptional activity and, more importantly, this activity correlates with the protection of these MEFs from apoptosis. Our results demonstrate that certain nonclassical stimuli can activate NF-��B upon prolonged treatment and that this ��sneak�� NF-��B activity might have clinical relevance during the course of long-term chemotherapy.
I��B Kinase-Independent I��B�� Degradation Pathway: Functional NF-��B Activity and Implications for Cancer Therapy
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC262380/
��Post-ischemic inflammation: molecular mechanisms and therapeutic implications
Zhen Zheng & Midori A. Yenari
Pages 884-892 | Published online: 19 Jul 2013Department of Neuroscience, School of Medicine, Standord University
Abstract
Ischemic stroke is characterized by the disruption of cerebral blood flow (CBF). This reduction of CBF results in energy failure and secondary biochemical disturbances, eliciting a robust in situ inflammatory response.Post-ischemic inflammation is a dynamic process involving a complicated set of interactions among various inflammatory cells and molecules. The resident inflammatory brain cells, microglia, are especially activated in response to ischemic insults, many of which are regulated by nuclear transcription factor, kappa B (NF-kB).
As a result, several inflammatory genes are expressed, leading to local generation of various cytokines, which in turn promulgate inflammatory signals. Meanwhile, endothelial cells lining the local cerebral blood vessels are stimulated to produce adhesion molecules, causing the migration of peripheral circulating leukocytes into the compromised brain tissue, an event that amplifies inflammatory signaling cascades.
Post-ischemic inflammation appears to serve multiple purposes, depending on its timing and magnitude, as well as the topographic distribution of various inflammatory molecules. Data from experimental manipulations of some inflammatory molecules are yielding insight into therapeutic strategies for ischemic stroke.
This review focuses on some recent advances regarding the regulation of inflammatory signaling pathways, the detrimental effects of post-ischemic inflammation and the potential molecular targets for ischemic stroke therapy.
Post-ischemic inflammation: molecular mechanisms and therapeutic implications: Neurological Research: Vol 26, No 8
https://www.tandfonline.com/doi/abs/10.1179/016164104X2357��
Cancer Biol Ther. 2013 Nov 1
Omega-3 polyunsaturated fatty acid promotes the inhibition of glycolytic enzymes and mTOR signaling by regulating the tumor suppressor LKB1
Rafaela Andrade-Vieira, Jae H Han, and Paola A Marignani*
The omega-3 polyunsaturated fatty acids (��3PUFAs) are a class of lipids biologically effective for the treatment of inflammatory disorders, cardiovascular disease and cancer. Patients consuming a high dietary intake of ��3PUFAs have shown a low incidence of metabolic disorders, including cancer. Although the effects of ��3PUFAs intake was shown to be involved in the prevention and treatment of these diseases, the underlying molecular mechanisms involved are not well understood. Here, we show that ��3PUFA, docosahexaenoic acid (DHA) enhanced the tumor suppressor function of LKB1. We observed that when LKB1 expressing cells are treated with DHA, there is an increase in LKB1 activity leading to phosphorylation of AMPK and inhibition of mTOR signaling. Abrogation of LKB1 in MCF-7 cells by siRNA reversed this phenotype. Furthermore, cellular metabolism was altered and ATP levels were reduced in response to DHA treatment, which was further attenuated in cells expressing LKB1. More importantly, in mammary epithelial cells expressing LKB1, the rate of glycolysis was decreased as a result of diminished expression of glycolytic enzymes. Functionally, these events lead to a decrease in the migration potential of these cells. Overall, our discovery shows for the first time that LKB1 function is enhanced in response to ��3PUFA treatment, thereby resulting in the regulation of cell metabolism.
Keywords: LKB1, aerobic glycolysis, cell metabolism, omega-3 PUFA, cancer, mTOR signaling, DHA, glycolytic enzymes, hexokinase, LDH-A
Omega-3 polyunsaturated fatty acid promotes the inhibition of glycolytic enzymes and mTOR signaling by regulating the tumor suppressor LKB1
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3925660/��
��
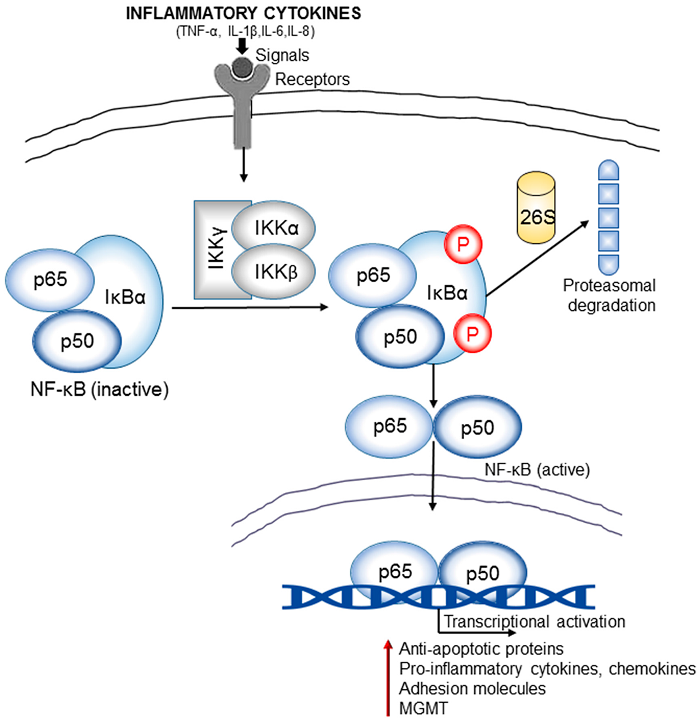
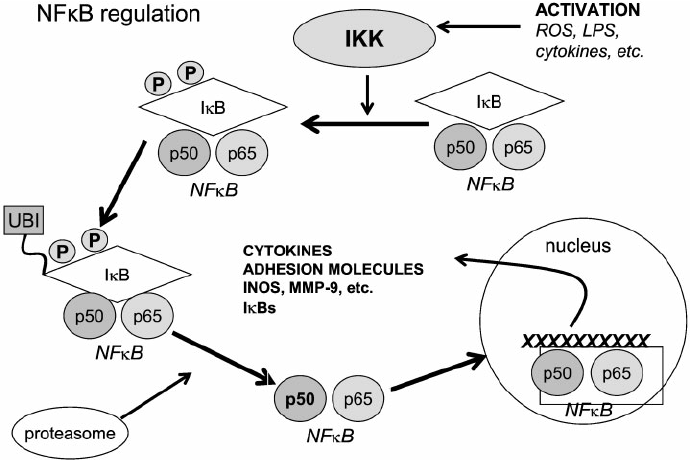
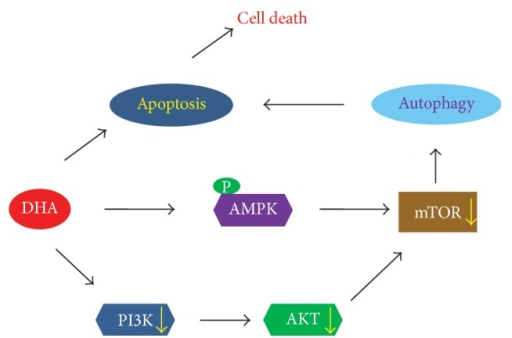
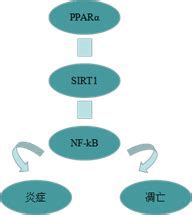
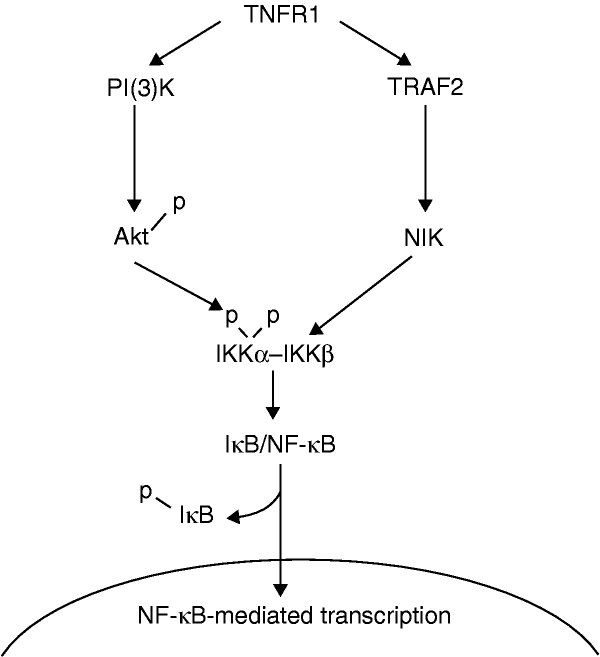
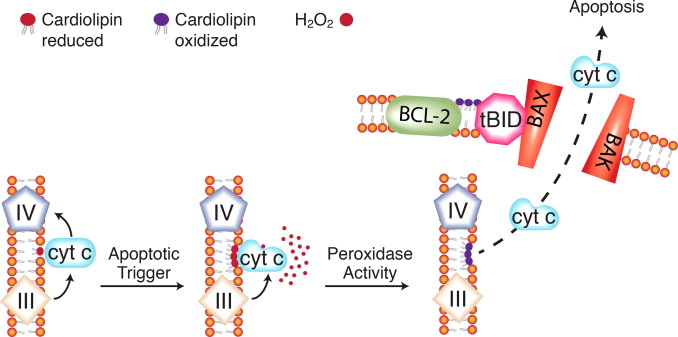

.jpg)
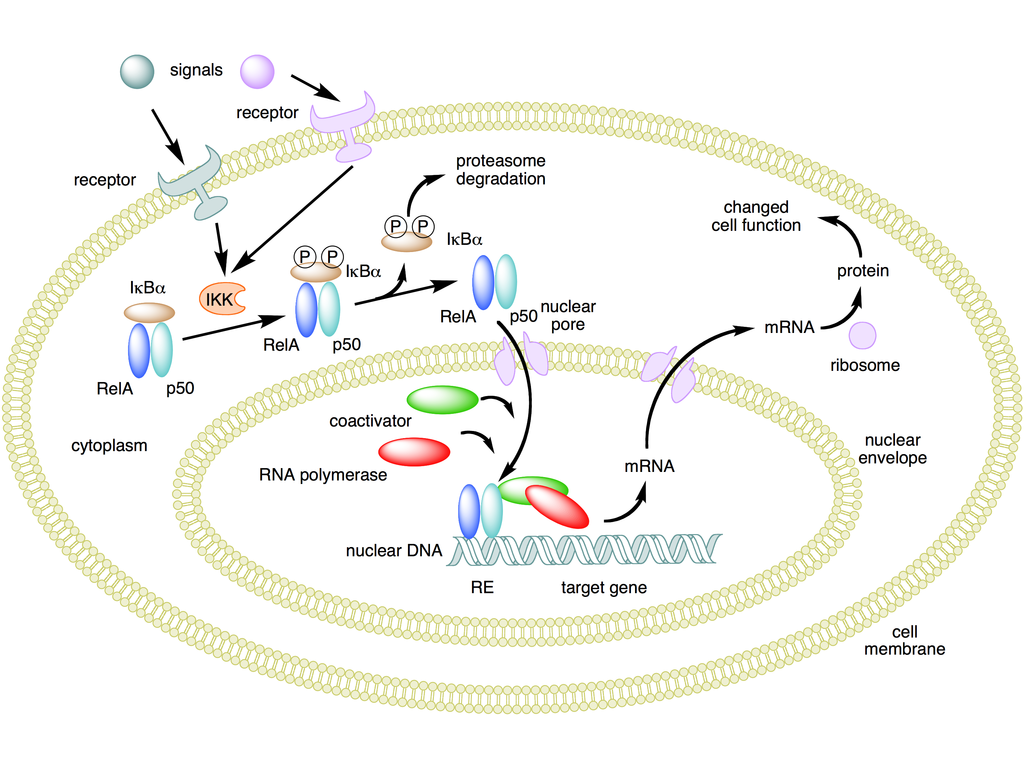
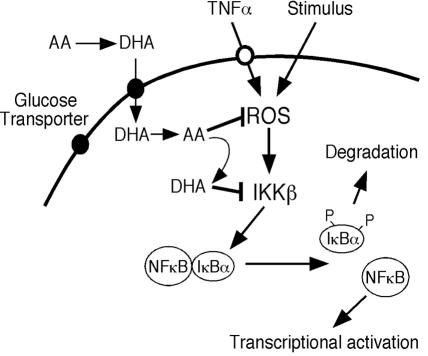
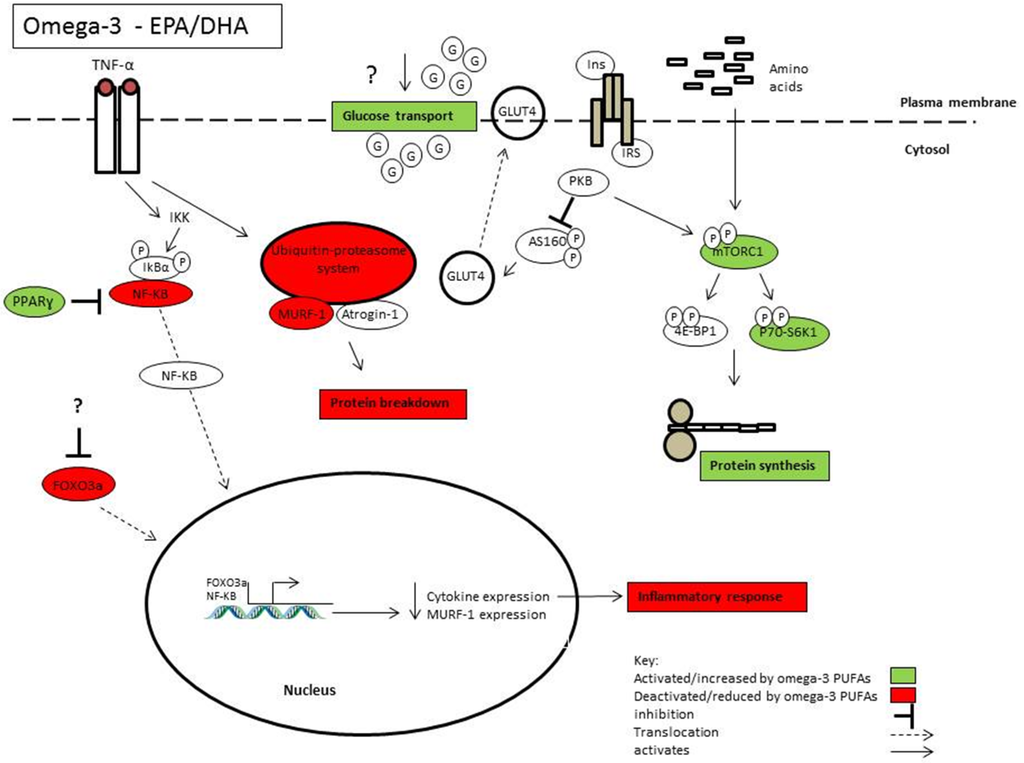
.jpg)

.jpg)
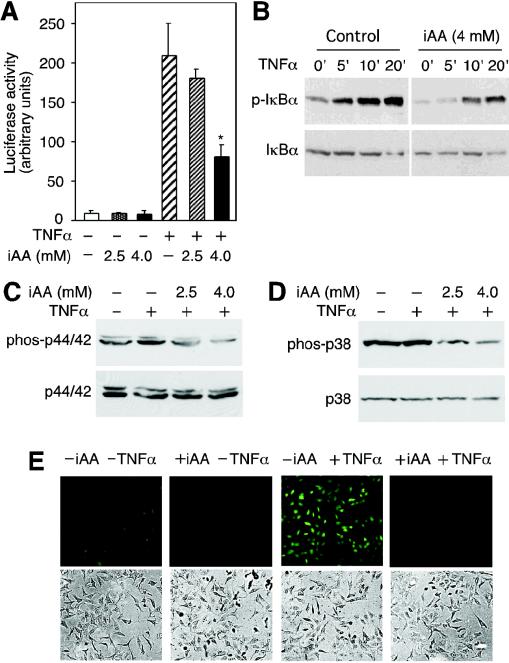
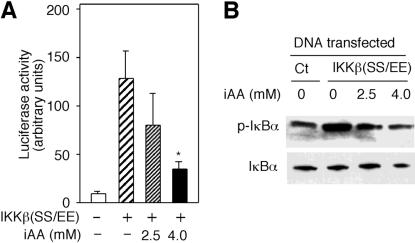
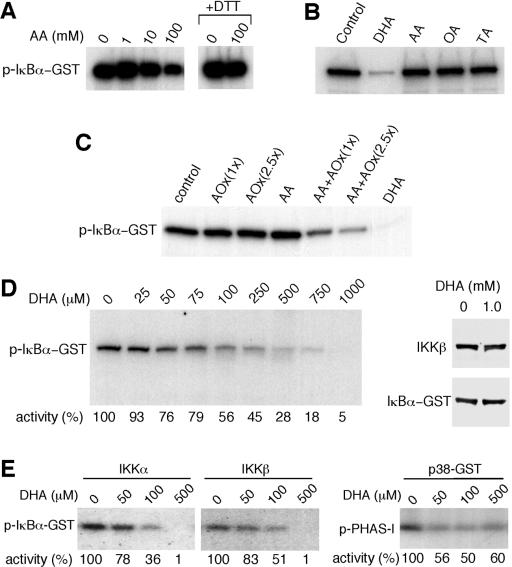
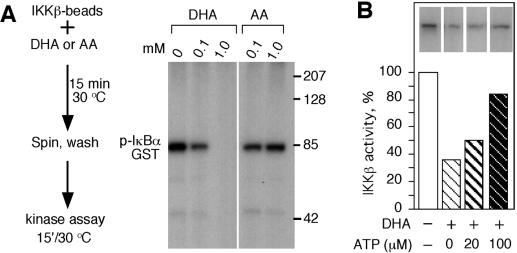
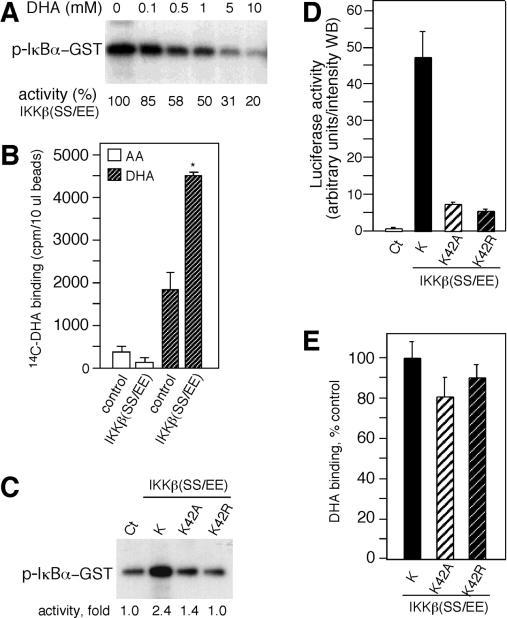
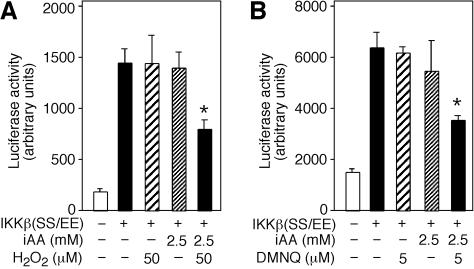
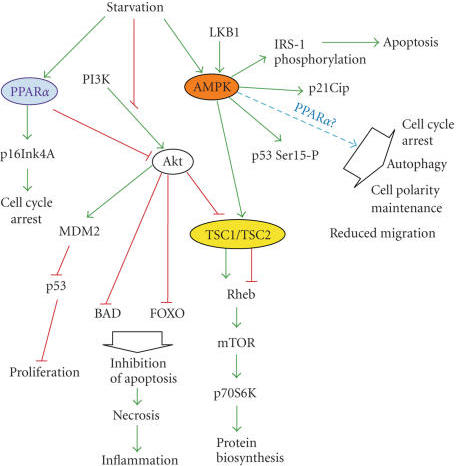
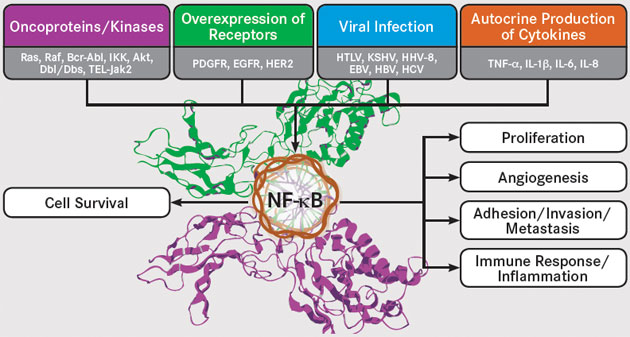
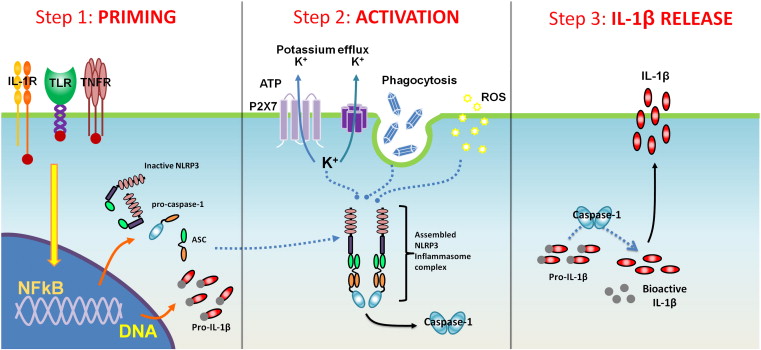
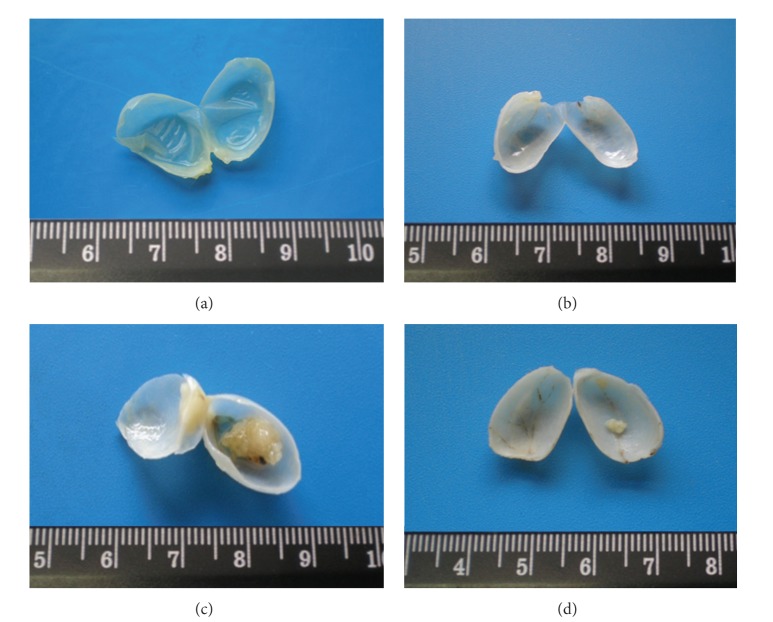
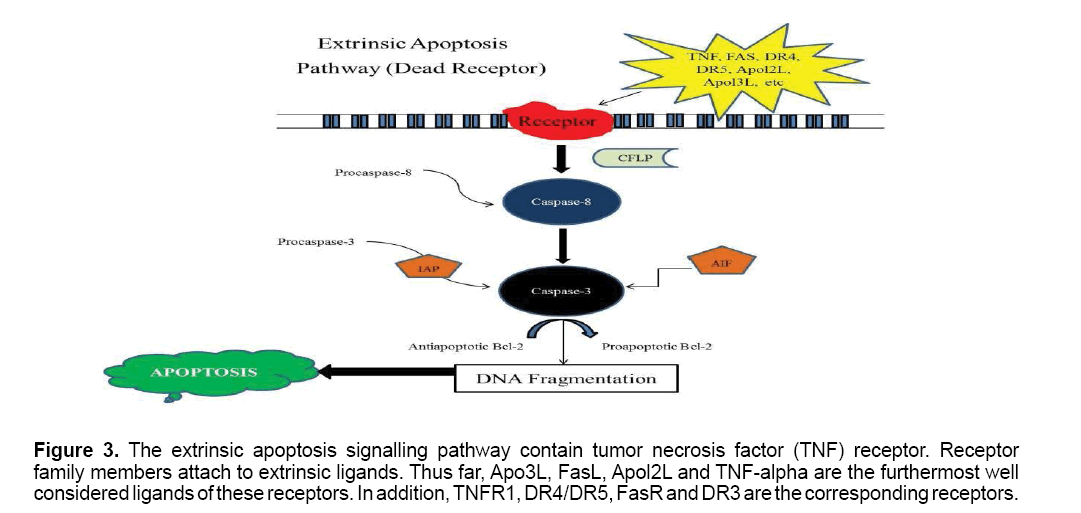
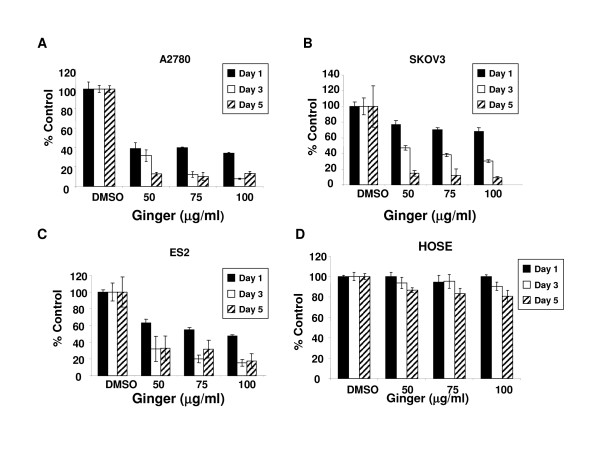
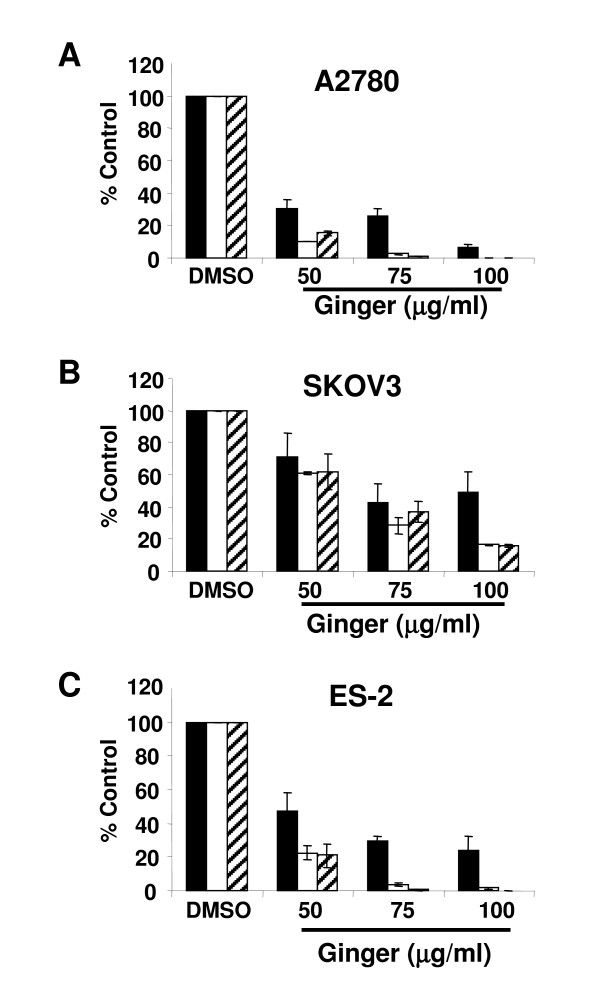
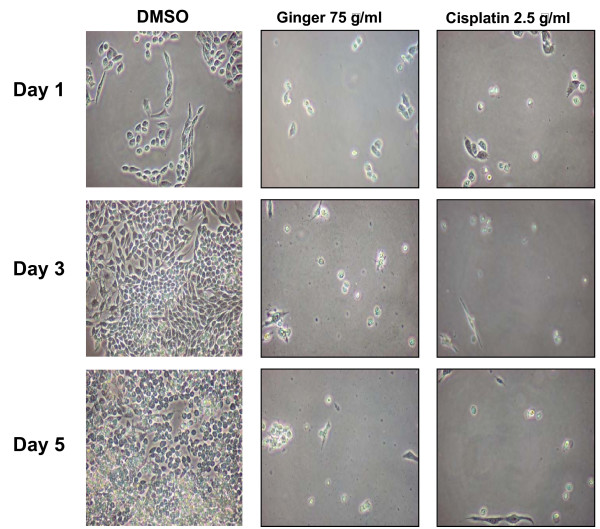
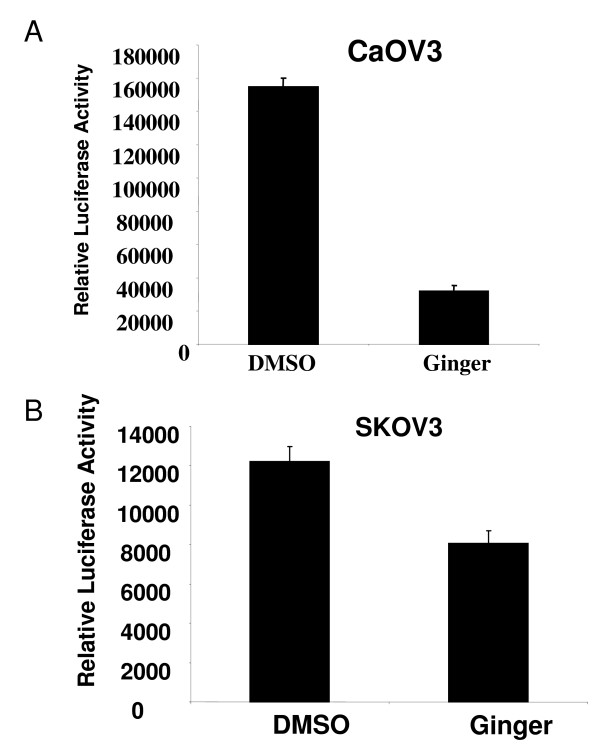
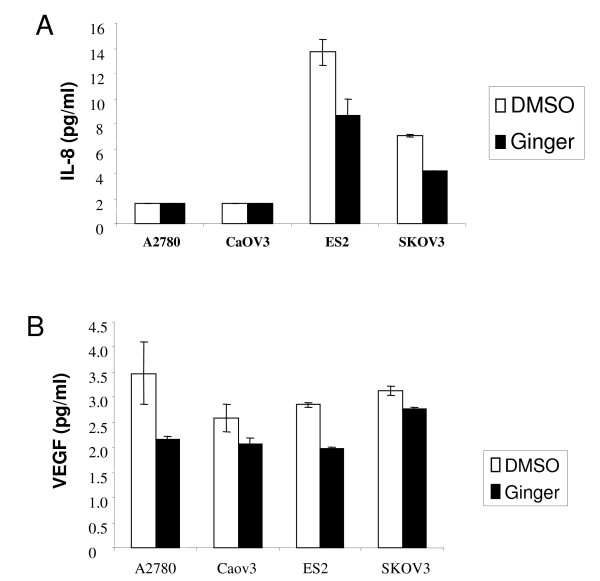
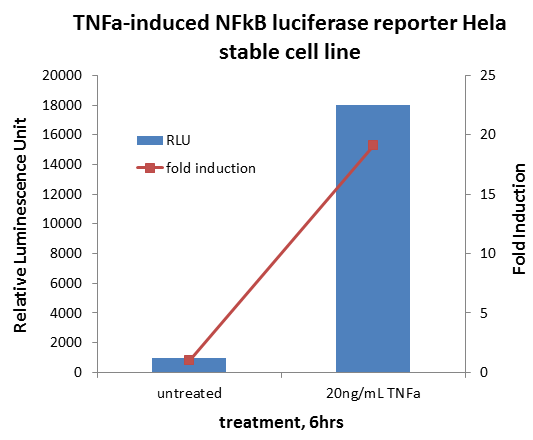
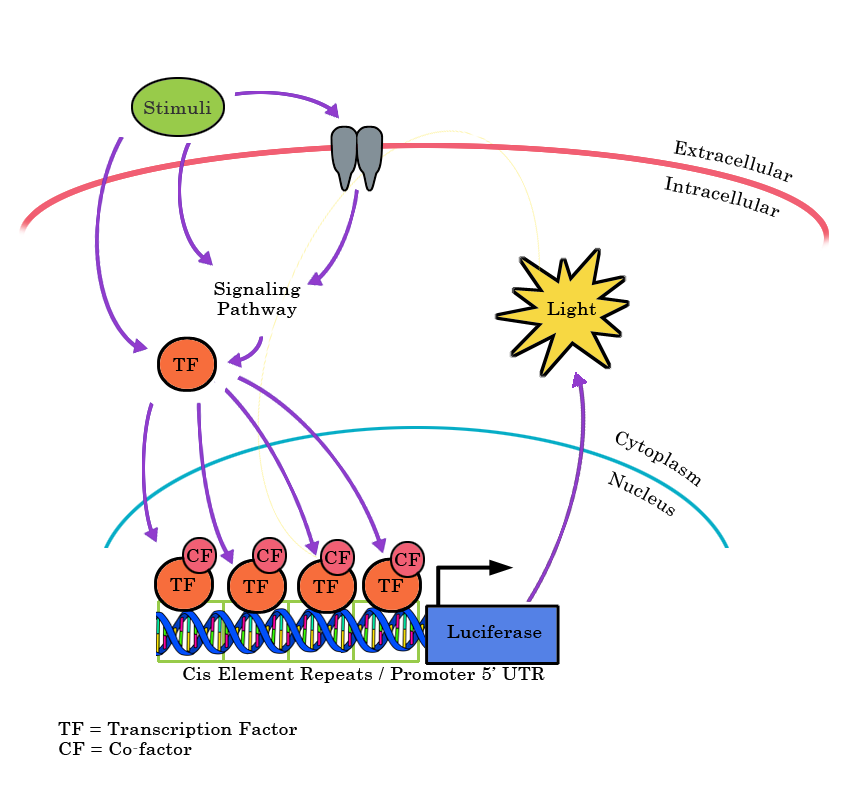
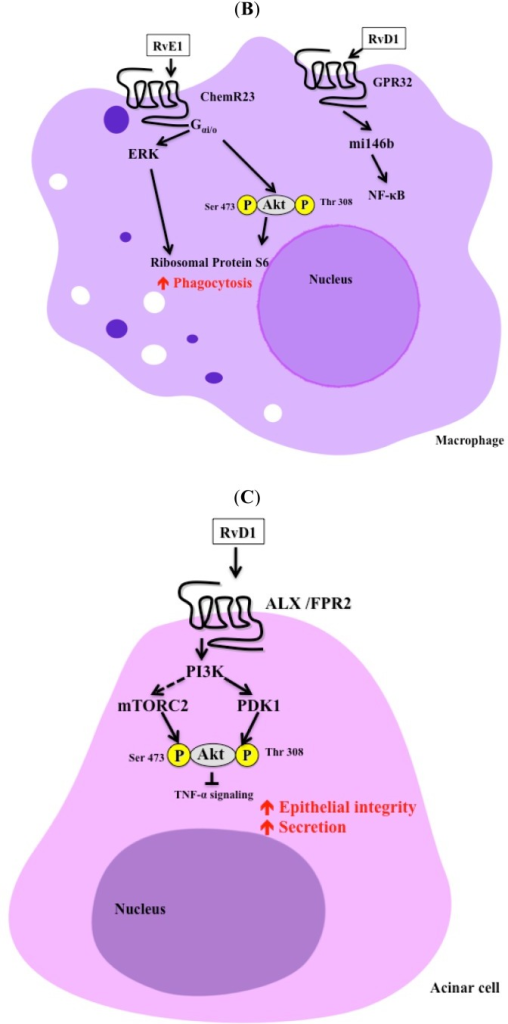
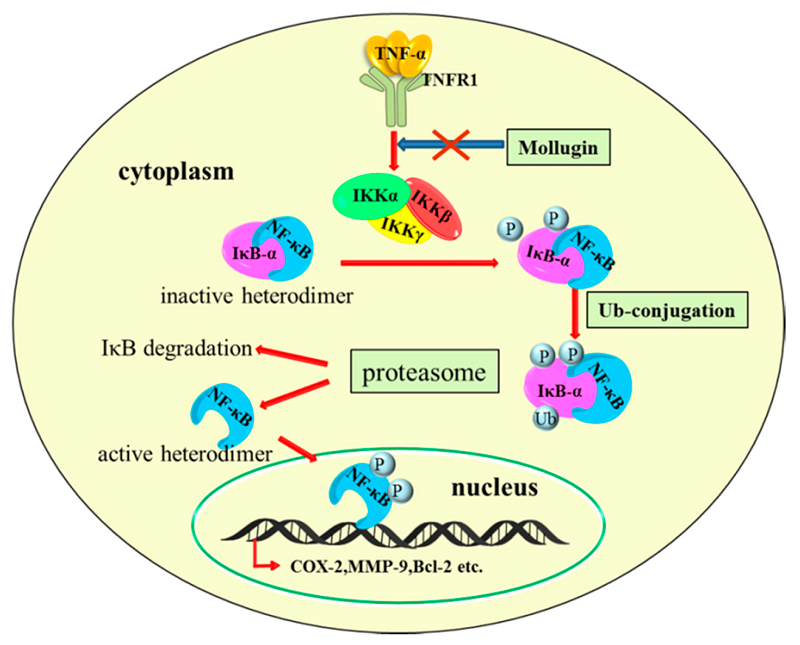


{kind=link}