��
Natural Cures for H.Pylori Infection
���б�ĩ��������ʼ��֪���Ⱥ�������ӡ���������ѧ��
��
��
contents
H.pylori infection is infectious
Stomach acid at pH 2.5 or lower kills H.pylori
chemical formula of urea: CH4N2O
urease: account for 10~15% of total protein,intracellular neutral pH optimum urease,surface urease is vital for H.p survival
Arginase:the source of urea comes from both urease and arginine converted from arginase
location of urase: mostly intrabacterial, few on surface(outer urease)
urease is activated at acidic environment lower than pH 5.0
H. pylori urease has a neutral pH optimum between 7.5 and 8.5 but essentially no activity below a pH of 4.5, and activity is lost irreversibly at this pH. The activity of cytoplasmic urease is low at neutral pH but increases 10- to 20-fold as the external pH falls between 6.5 and 5.5, and its activity remains high down to pH ∼2.5 . Thus, cytoplasmic, not surface, urease is ...
induction of Arg-1 is type 2 cytokine dependent.
https://www.jimmunol.org/content/167/11/6533
��
�쳣�����ƻ��������ݸ˾�
����ͻ��ϸ�����������ϵ������⣬���������˾��������˱������������������߷�Ӧ����Ӧ�����߷�Ӧ�ķ�����
��ĿǰΪֹ���Ѿ������˼���ϸ������(����һЩ������ı���ͼ�н�����˵��)�����磬
���������˾���ë���ķ�չʹ���Dz���toll������(һ��ʶ����������ϸ����ë����������������)��Ӱ�졣
���������˾���LPS�²��Աȸ��������Գ���ϸ���ĵ�1000�������Աȸ��������Գ���ϸ���ĵ�500����
���������˾�����ֱ�Ӵ���Ƥϸ��Ĥ��õ��̴������������ϵ�������֬�ʲ��С�
���Ѿ���ϸ����������ͨ������glucosyl�Ļ�õ��̴���(cholesterol-����Ӧ�������ǻ�ת��ø)��
���⣬�����ݸ˾����ܼ��پ���ϸ��������ɱ����һ��������NO)��
���ľ�����ø�����ϸ���յ���һ�������ϳ�ø��������L-�����ᣬ���߱�ϸ�������ϳ�����(����ø�ĵ���)��
���⣬��¶�����������˾��IJ�����ϵ�����ϸ���о�����øII��Arg-2)��ת¼���Ӷ���L -������ֽ�Ϊ���غ�L -���ᣬ�������л�ɶష���Ӷ���������ϸ���ĵ�����
��
�����ݸ˾�����֢������ڵĹ�ϵ�������ݸ˾���Ⱦ��ͨ�������м������θ����ڣ�������֢��Ӧ����֢ϸ��������IL-1������ԭø��TNF-ϸ�������˱�ϸ�������Է��ڡ���vaca�յ�ezrin����ˮ���⣬����H+K+ atpøa -�ǻ������ӻ���Ҳ���������Է��ڡ�
���������ݸ˾���Ⱦ�ɵ���θ����θ����ࣨhypochlorhydria or hyperchlorhydria������ȡ����θ�����س̶Ⱥͷֲ����������Ⱦ�Ļ��߳��ڷ�չΪθ��������������θ�ף�pangastritis�����ɽ�չΪθ�����/���ٰ����෴���ڴ�Լ12%�����Ը�Ⱦ�����з���θ���θ�ף���������θ����࣬����ܵ���ʮ��ָ������(23)����
��
Relationships between H. pylori, inflammation, and acid secretion. H. pylori infection can reduce acid secretion and increase inflammation via multiple intermediates. Increased production of IL-1�� and TNF-�� from inflammatory cells inhibits acid secretion from parietal cells. Acid secretion is also inhibited by repression of H+K+ ATPase ��-subunit promoter activity, in addition to VacA-induced proteolysis of ezrin.
Chronic H. pylori infection may result in hypochlorhydria or hyperchlorhydria, depending on the severity and distribution of gastritis. Most patients infected long-term develop pangastritis associated with hypochlorydria, which may progress to gastric ulceration and/or adenocarcinoma. Conversely, antral predominant gastritis occurs in approximately 12% of chronically infected patients and is characterized by hyperchlorhydria, which may lead to duodenal ulcer disease (23).Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk | Clinical Microbiology Reviews
https://cmr.asm.org/content/23/4/713��
https://www.wjgnet.com/1007-9327/full/v17/i28/3300.htm
��
Activation begins at a medium pH of 6.5 and reaches its maximum value at pH 5.5. Urease activity remains constant down to a pH between 2.5 and 3.0 and is detectable even at a pH of 2.0.
The bacteria are bioenergetically neutralophiles, meaning they are able to survive between pH 4 and 8 and grow between pH 6 and 8.
gastric epithelial cells renew every 3~7 days
Distribution of Heme oxygenase ��HO-1) in the body: KCs, macrophages, endothelial cells, neuro and glial cells, epithelial cells
HO-1 is expressed primarily in myeloid cells and that HO-1 levels in these cells were directly proportional to cytoprotection
cagA + strains of H. pyloriactivate the transcription factor NF-��B in gastric epithelial cells
The elimination of pathogenic Gram-positive and Gram-negative bacterial strains is an important medical problem and can be achieved by bacteria lysis, inhibition of bacterial adhesion, and increasing the permeability of bacterial cell walls [33�C35].
Highlights
1. inhibitors of urease: quercetin, linoseed oil��onion,garlic/allicin,EGCG/green tea��curcumin/turmeric��vitamin C, Manuka honey,ginger,saponins/licorice glycyrrhizin, sulforaphane and other isothiocyanates
2. Blocking of NFkB signaling pathway��quercetin, curcumin,lactobaccillus,HO-1, EPA,DHA, Vitamin C
3. inhibitors of h.p adherence to mucus/Protectors of thiol groups on gastric epithelial cells: ECGC,vitamin C
4. Kills H.pylori:omega-3,flaxseed oil, DHA,ginger/gingerols/honey��curcumin��vitamin C/NAC? saponins/licorice, astralagus, ginseng
5. enhances healing:ginger,honey��curcumin/turmuric, banana
6.promotors of H.p: baking soda,soda drinks,Urea, arginase2,
7. inhibitor of acid secrection:ginger, upripe green banana,
7b.inhibitors of arginase 2: all alpha-amino acids(non-competitive, di-aminoacids, competitive), especially orthinine(strongly, competitive and non-competiitive), lysine(competitive inhibitor,0.6 times as effective as orthinine), Nitric oxide, CB-1158, borate, l-hydroxyarginine, the most potent physiologic inhibitor of arginase,
7c. breaking h.p membrane: saponins-selectively release cholesterol/making pores on membrane,
7d.activators of iNOS:saponins/glycyrrhizin,norvaline,HO-1?
7e.inducers of HO-1: nitric oxide, heme(decrease the level of phosphorylated CagA (p-CagA)), nitrates,vitamin c, arginine,oxygen��lansoprazole,statins, aspirin, niacin, certain prostaglandins, eicosanoids such as epoxyeicosatrienoic (EETs) and free and complexed metals ( anti-inflammatory effect on the dependent of HO-1 expression, activation of the pro-inflammatory response mediated by p-CagA are inhibited in HO-1-expressing cells, H. pylori inhibits in gastric epithelial cells the expression of hmox-1 (Gobert et al., 2011), the gene encoding heme oxygenase-1 (HO-1), a potent anti-inflammatory and antioxidant enzyme (Ryter et al., 2006). Moreover, we showed that when HO-1 was upregulated in epithelial cells prior to H. pylori infection, there was a reduction of IL-8 release (Gobert et al., 2011). HO-1 downregulates H. pylori-induced NF-��B, p38, and ERK1/2 signaling in gastric epithelial cells)
7f. Inducers of ariginases: lactic acid (tumor cells, T cells, muscle cells), gliadin(���ף���candida albicans chitin, H.pylori,
8. H.pylori infection induces apoptosis in gastric epithelial cells(and macrophages), injury by components of H.p may causes proliferation and neoplasia, studied by Lodz univ, Poland
9. H.pylori infection and activated T cells and interferon gamma and TNF-alpha induce enhanced expression of PD-L1 in gastric epithelial cells,dentritic cells, leading to apoptosis of cytotoxic T cells.
10.CagA is associated with an increased risk of developing gastric cancer. Shh signaling mediates H. pylori-induced PD-L1 expression. Consistent with our findings, we have shown that H. pylori infection induces an increase in Shh secretion and signaling via a CagA dependent pathway
11.PPIs enhance hypochlorihydria thus increases gastric cancer risk
12.VacA targets mitochondria,infection caused a time-dependent increase in mitochondrial-membrane permeability and release of cytochrome c into the cytosol. triggered apoptosis. makes channel
13. curcumin inhibits shikimate dehydrogenase
14. banana: increases mucus resistance
15. H0-1 protects stomach thru vitamin c. gastric epithelial cells require vitamin C to translate HO-1 mRNA into active protein, which then may exert gastroprotection by its antioxidant and vasodilative properties.
16. The Immune Battle by macrophage against Helicobacter pylori Infection: NO Offense. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages.
17.cagA or vacA secreted by Helicobacter pylori Induces Apoptosis of Macrophages in Association with Alterations in the Mitochondrial Pathway.infection caused a time-dependent increase in mitochondrial-membrane permeability and release of cytochrome c into the cytosol. triggered apoptosis.
18.Ginger:anti-ulcer,inhibitor of PPIs,anti-H.p
20. Urease from Helicobacter pylori is inactivated by sulforaphane and other isothiocyanates
21. ulcer protective activity of ginger aqueous extract: cinnamic acid, gallic acid, cinnamic acid inhibits H/K pump, gallic acid antioxidant
22. Muc1 Mucin Limits Both Helicobacter pylori Colonization of the Murine Gastric Mucosa and Associated Gastritis
23.Helicobacter pylori Modulates Lymphoepithelial Cell Interactions Leading to Epithelial Cell Damage through Fas/Fas Ligand Interactions
24. Docosahexaenoic Acid Inhibits Helicobacter pylori Growth In Vitro and Mice Gastric Mucosa Colonization
25. H. pylori is sensitive to unsaturated free fatty acids through their incorporation into phospholipids and membrane destruction.alpha-lenolenic acid at concentrations of 10(-3) M killed virtually all organisms via membrane fraction. ��saponins+ALA+Vc work like a chained bomb)
26. 20% of h.pylori conolize in gastric epithelial cells, 80% in mucusa
H. pylori VacA toxin. VacA is a secreted channel-forming toxin unrelated to other known bacterial toxins. Most H. pylor istrains contain a vacA gene, but there is marked variation among strains in VacA toxin activity.https://my.vanderbilt.edu/coverlab/our-research/
��
27.Most gastric MALT lymphomas are characterized by their association with the Helicobacter pylori (HP) infection and are cured by first-line HP eradication therapy (HPE).
28.A natural flavonoid leucocyanidin present in unripe plantain banana pulp (Musa sapientum L. var. paradisiaca) protects the gastric mucosa from aspirin-induced erosions
29. 7-dehydrocholesterol proved to be fatally toxic to H. pylori. phosphatidylcholines (PC) is the most prevalent glycerophospholipid in mammals and exists in sufficient abundance to kill H. pylori in gastric mucosa and gastric juice of humans [29,30]. In sum, our study demonstrated that H. pylori aggressively incorporates exogenous non-esterified cholesterol into the cell membrane, in order to survive in the presence of PCs.
30.Muc1 provides a protective barrier, which limits both acute and chronic colonization by H pylori, as well as playing a major role in limiting the inflammation induced by Helicobacter infection.
31.H. pylori induces apoptosis of infected macrophages. the activation of c-Myc and induction of ODC in response to H. pylori is dependent on phosphorylation of extracellular signalregulated kinase (ERK). knockdown of ODC by siRNA increased iNOS protein expression and NO production in response to H. pylori. This enhanced killing of the bacterium, whereas spermine inhibited killing. a pathway of macrophage apoptosis in which the activation of arginase, ODC, and SMO(PAO1) is required, with the latter resulting in generation of H2O2 that activates the mitochondr-ial pathway of apoptosis.
ODC:Ornithine decarboxylase
https://www.europeanmedical.info/cancer-research/a-1.html
��
http://www.cell.com/trends/microbiology/fulltext/S0966-842X(16)00041-X
��
31.b.Curcumin as tyrosine kinase inhibitor in cancer treatment
32.In addition to CagA, we found that VacA was also involved in the induction of apoptosis. In epithelial cells, VacA has been shown to target the mitochondria, cause cytochrome c release, activate caspase 3, and lead to cleavage of downstream apoptosis-inducing substrates, such as poly(ADP-ribose) polymerase (21). Although H. pylori can induce distinct signal transduction pathways in macrophages versus epithelial cells (44), since we observed activation of the mitochondrial pathway, we speculate that VacA may also target mitochondria in macrophages.
33. H. pylori induces epithelial-cell apoptosis through a type II pathway in vitro through activation of caspases 8, 9, 6, and 3, as well as a time-dependent activation of Bid and release of cytochrome c
34. H. pylori infection of mice induced a rapid infiltration of macrophages into the stomach. Treatment of mice with an inhibitor of ERK phosphorylation attenuated phosphorylation of c-Fos, expression of ODC, and apoptosis in gastric macrophages.
35. omega-3 fatty acids have a role in transcriptional regulation by phosphorylation inhibition of JNK, ERK, and MAPK proteins, which downregulates protein-1 expression. 26 Furthermore, omega-3 fatty acids are proposed to have a role in restraining nuclear factor-��B nuclear translocation secondary to I��B phosphorylation
35.reactivate macrophage for ulcer healing via beta-glucan
36.chitin-chitosan
37.VITAMIN C, OXYGEN, nitric oxide, HO-1,CO, MACROPHAGE,ULCER HEALING
gastric epithelial cells require vitamin C to translate HO-1 mRNA into active protein, which then may exert gastroprotection by its antioxidant and vasodilative properties.
inducers of HO-1: nitric oxide and hemin
http://dmm.biologists.org/content/9/12/1473
��
38. Heme oxygenase-1 inhibits phosphorylation of the Helicobacter pylorioncoprotein CagA in gastric epithelial cells
hmox-1 expression and HO-1 protein levels were diminished in the epithelial cells of the stomach of cagA+ H. pylori-infected patients
enhance HO-1 expression in gastric epithelial cells
38b.Curcumin as tyrosine kinase inhibitor in cancer treatment
��
38c. �ʲ��� "when taken orally, glycyrrhizin converts of aglycone metabolite 18beta-glycyrrhetinic acid (GRA)" that induces nitrous oxide synthase, the stimulate Th1 response.
39. Inhibitors of arginase: L-norvaline, polyphenols, borate, NOHA hydroxy-arginine, flavonoids (2S)-5,2��,5��-trihydroxy-7,8-dimethoxy flavanone (TDF)
��
http://www.sigmaaldrich.com/technical-documents/articles/biofiles/how-antibiotics-work.html
Immune Evasion by Helicobacter pylori is Mediated by Induction of Macrophage Arginase II1
H. pylori upregulates macrophage Arg2, thereby restricting iNOS protein levels and NO production, and enhancing macrophage apoptosisArg2, but not Arg1, is upregulated in H. pylori-stimulated macrophages, induction of macrophage Arg2 by H. pylori inhibits iNOS translation, NO production, and bacterial killing in vitro, inhibition of arginase blocks H. pylori-induced macrophage apoptosis in our in vitro studies, staining demonstrates enhanced Arg2 levels in H. pylori-infected macrophages, Arg2−/− macrophages are more abundant, express more iNOS, and have increased nitrotyrosine staining as compared to WT macrophages during H. pylori infection, H. pylori stimulation induces Arg2 expression in macrophages, H. pylori infection increases macrophage apoptosis that is abolished in Arg2−/− mice,
induction of Arg2 by H. pylori is a mechanism by which the pathogen escapes the host innate immune response and contributes to the immunopathogenesis of the infection.
both Arg-I and Arg-II are to hydrolyze l-arginine to produce urea and l-ornithineThe primary function of Arg-I is to remove excessive nitrogen produced from amino acid metabolism through hepatic urea cycle, which is otherwise toxic for our body
The gastric cancer-causing pathogen Helicobacter pylori up-regulates spermine oxidase (SMOX) in gastric epithelial cells, causing oxidative stress-induced apoptosis and DNA damage.
The Expression of Heme Oxygenase-1 Induced by Lansoprazole
endothelial NO reveals important anti-oxidative and anti-inflammatory functions and suppresses expression of the adhesion molecules40.Human Gastric Epithelium Produces IL-4 and IL-4��2 Isoform Only upon Helicobacter Pylori Infection
https://journals.sagepub.com/doi/abs/10.1177/03946320070200041741.Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection.
42. gastric epithelial cells recognize Cag pathogenicity island (PAI)-positive bacteria via the NOD1 receptor (81), and NOD1 regulates the direct killing of the bacteria by the antimicrobial peptides, human ��-defensins (33).
��
At 10-5 mol/L, chemically synthesized hBD-2 (30 ��g/mL) was reported to completely inhibit growth of H pylori, while recombinant hBD-3 began to inhibit growth of H pylori at a concentration of 10-7 mol/L[5,11]. In the present study, we demonstrated that synthesized hBD-3 as well as hBD-2 inhibited H pylori growth at concentrations of 50 ��g/mL or more.
hBD-3 is frequently expressed in gastric mucosa with H pylori infection showing gastritis, but not in inflamed mucosa without H pylori infection.
43. ��������Astragaloside IV inhibits TGF-��1-induced epithelial-mesenchymal transition through inhibition of the PI3K/Akt/NF-��B pathway in gastric cancer cells.
The influence of saponins on cell membrane cholesterol.
saponin-induced cholesterol liberation as well as the saponin-induced inhibition of cholesterol uptake with the membrane toxicity
The results suggested that the general cytotoxicity of saponins is mainly dependent on their membrane toxicity and that the membrane toxicity might be caused by the loss of cholesterol from the cell membrane.
The experiments revealed a correlation between the membrane toxicity and the reduction in surface tension.
Cholesterol is Believed to Play a Role in Bacterial Selectivity of AMPs
One of the major differences between bacterial and eukaryotic cell membranes is the presence of a large amount of cholesterol in eukaryotic cell membranes and a complete absence in bacterial cell membranes . Cholesterol has been shown to protect human erythrocytes from attack by magainin 2 . Similar studies on model membranes have demonstrated that the presence of cholesterol reduces AMP binding and suppresses the disruption of lipid bilayer structure by AMPS.
Composition of Membranes is Key to Amp Selectivity
Bacteria have difficulty in developing resistance to AMPs because the toxicity of AMP is mostly mediated by a non-specific process rather than by an interaction with a specific protein target. Most AMPs lyse bacteria by directly interacting with the lipid bilayer of the bacterial cell membrane and disrupting the lipid bilayer structurenote:cholesterol protects h.p from attack by AMP; h.p vaciously uptake cholesterol; intracellular h.p decrease AMP production; H. pylori uptakes cholesterol from epithelial cells; cholesterol enhances the resistance of H. pylori.
Cholesterol Enhances Helicobacter pylori Resistance to Antibiotics and LL-37
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3101455/Helicobacter pylori's cholesterol uptake impacts resistance to docosahexaenoic acid - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S1438422113001914
Metabolic labelling of cholesteryl glucosides in Helicobacter pylori reveals how the uptake of human lipids enhances bacterial virulenceMetabolic labelling of cholesteryl glucosides in Helicobacter pylori revealshow the uptake of human lipids enhances bacterial virulence - Chemical Science (RSC Publishing)
https://pubs.rsc.org/en/content/articlelanding/2016/sc/c6sc00889e#!divAbstract
Helicobacter pylori, the most common etiologic agent of gastric diseases including gastric cancer, is auxotrophic for cholesterol and has to hijack it from gastric epithelia. Upon uptake, the bacteria convert cholesterol to cholesteryl 6'-O-acyl-��-D-glucopyranoside (CAG) to promote lipid raft clustering in the host cell membranes. However, how CAG appears in the host to exert the pathogenesis still remains ambiguous. Herein we identified hp0499 to be the gene of cholesteryl ��-D-glucopyranoside acyltransferase (CGAT). Together with cholesteryl glucosyltransferase (catalyzing the prior step), CGAT is secreted via outer membrane vesicles to the host cells for direct synthesis of CAG. This significantly enhances lipid rafts clustering, gathers adhesion molecules (including Lewis antigens and integrins ��5, ��1), and promotes more bacterial adhesion. Furthermore, the clinically used drug amiodarone was shown as a potent inhibitor of CGAT to effectively reduce the bacterial adhesion, indicating that CGAT is a potential target of therapeutic intervention.��
H. pylori, Hormones and Your Gut Health �� TRUE NATURE ...
https://www.truenaturehealthconsulting.com/blog/...
Oct 17, 2018 �� The reinforced barrier creates resistance of H. pylori to phosphatidylcholines, a group of phospholipids that includes linoleic and/or arachidonic acid. These are polyunsaturated fatty acids (PUFAs) that confer antibacterial actions which are fatal to the typically steroid-free H. pylori membrane. Despite its low abundance, phosphatidyl serine in the cell membrane plays key roles in various ��Human ��-defensin-3 induction in H pylori-infected gastric mucosal tissues https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4100659/
��
https://www.superfoodly.com/dgl-licorice/
��
��
��
within colonies of H. pylori, the circular, or coccoid forms were found at the centre of the colony while the spiral forms were found at the edges and were actively dividing
Helicobacter pylori
http://tolweb.org/treehouses/?treehouse_id=4��
https://vmicro.iusm.iu.edu/hs_vm/docs/lab12_7.htm
��
https://en.wikibooks.org/wiki/Medical_Physiology/Gastrointestinal_Physiology/Secretions
��
Figure 2. - Pathogenesis of Helicobacter pylori (H. pylori) infection. Several virulence factors, such as CagA and VacA, encoded by H. pylori genes, interact with gastric epithelial cells and the immune system, resulting in an inflammatory response, mucosal damage and, eventually, gastric cancerogenesis (see description in the text).
https://www.spandidos-publications.com/10.3892/ijo.2012.1701
��
��
��
��
��
Bavituximab �C Novel Checkpoint Inhibitor in Phase 3 | Cancer Biology
https://blogs.shu.edu/cancer/2015/02/04/bavituximab-novel-checkpoint-inhibitor-in-phase-3/Arginase expression is the default mode of tissue macrophages, but can also be amplified by signals, such as IL-4/13 or transforming growth factor-�� (TGF-��) that accelerates wound healing and tissue repair. In macrophages, basal polyamine (putrescine, spermidine, and spermine) levels are relatively low but are increased upon IL�\4 stimulation.
��
��
https://www.nature.com/articles/s41419-020-2391-6
��
ODC:Ornithine decarboxylase SM0: spermine oxidase
��
ODC:Ornithine decarboxylase
https://www.europeanmedical.info/cancer-research/a-1.html
��
��
Schematic diagram of Helicobacter pylori infection��adhesion and invasion) and pathogenesis. The urease activity and flagella-mediated motility of H. pylori facilitate its survival and movement toward the lower mucus gel above the epithelium, followed by several adhesins, including blood-antigen binding protein A, sialic acid-binding adhesin, and other outer membrane proteins interacting with receptors on the host epithelium cells. After successful colonization, toxins, including cytotoxin-associated gene A, and vacuolating cytotoxin A, are involved in damage of host tissue and intracellular replication. Source publication
��Schematic diagram of Helicobacter pylori infection and pathogenesis.... | Download Scientific Diagram
https://www.researchgate.net/figure/Schematic-diagram-of-Helicobacter-pylori-infection-and-pathogenesis-The-urease-activity_fig1_301234135��
http://www.labpedia.net/test/244 �����ݸ˾���������ԭ��
��
A scholarly paper published in 2011 by Hirofumi Shimomura, following extensive research, showed that pregnenolone, dehydroepiandrosterone (DHEA), epiandrosterone (EA) and estrone (E1) are all absorbed into the lipid membrane of H. pylori. As steroids, they have the capacity to strengthen and reinforce the membrane barrier and increase its resistance to treatment/eradication. The reinforced barrier creates resistance of H. pylori to phosphatidylcholines, a group of phospholipids that includes linoleic and/or arachidonic acid. These are polyunsaturated fatty acids (PUFAs) that confer antibacterial actions which are fatal to the typically steroid-free H. pylori membrane.
H. pylori, Hormones and Your Gut Health �� TRUE NATURE FUNCTIONAL HEALTH CONSULTING
https://www.truenaturehealthconsulting.com/blog/2018/9/10/h-pylori-hormones-and-your-gut-health��
��
The well-studied catalytic role of urease, the Ni-dependent conversion of urea into carbon dioxide and ammonia, has been shown to protect Helicobacter pylori against the low pH environment of the stomach lumen.
Noncatalytic Antioxidant Role for Helicobacter pylori Urease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6088170/��
��
��
Frontiers | The Role of microRNAs in Helicobacter pylori Pathogenesis and Gastric Carcinogenesis | Cellular and Infection Microbiology
https://www.frontiersin.org/articles/10.3389/fcimb.2011.00021/full
Induction of Cyclooxygenase-2 Overexpression in Human Gastric Epithelial Cells by Helicobacter pylori Involves TLR2/TLR9 and c-Src-Dependent Nuclear Factor-��B Activation | Molecular Pharmacology
http://molpharm.aspetjournals.org/content/66/6/1465��
The schematic gastric carcinogenesis triggered by a vicious cycle within the CagL-integrin priming-intragastric pH elevation during chronic H. pylori infection.
Fig. 2: Schematic representation of the pro-oncogenic actions of the H. pylori CagA oncoprotein.
figure2
When delivered into gastric epithelial cells, the CagA oncoprotein perturbs multiple intracellular signaling pathways and then promotes malignant transformation of the host cells by providing cancer-hallmark capabilities. There is a mutual feedforward stimulatory mechanism between the pro-oncogenic activities of CagA and pro-inflammatory responses.
Fig 13 Proposed model of H. pylori induced PD-L1 expression in the gastric epithelium. (A) Shh secretion is induced from the acid secreting parietal cells in response to H. pylori infection that is driven by a CagA dependent mechanism. (B) We propose that Shh induces the expression of PD-L1 on GSII/PgA transdifferentiated cells. PD-L1 then interacts with PD-1 on the surface of CTLs and shuts down the CTL effector function which may lead to the survival of these transdifferentiated metaplastic cells. (C) The addition of PD-1Inh blocks the interaction between PD-1 on CTLs and PD-L1 on transdifferentiated/SPEM cells allowing the CTLs to destroy these cells.
��
��
H. pylori Virulence Factors: Toxins (CagA, VacA, DupA, OipA, IceA) | SpringerLink
https://link.springer.com/chapter/10.1007/978-981-287-706-2_5
��
��
https://en.wikibooks.org/wiki/Medical_Physiology/Gastrointestinal_Physiology/Secretions
��
Gastric mucosal gland showing heavy colonization of H. pylori (arrows) (Giemsa stain x1000). 20% of H.pylori conolize in epithelial cells.
Gastric mucosal gland showing heavy colonization of H. pylori (arrows)... | Download Scientific Diagram
https://www.researchgate.net/figure/Gastric-mucosal-gland-showing-heavy-colonization-of-H-pylori-arrows-Giemsa-stain_fig3_317588143
H. pylori colonizes the primate stomach, a harsh environment. The stomach lumen ranges between pH 1�C5 [8], conditions at which H. pylori is viable for only ~30min [9]. Additionally, stomach contents are cleared regularly, and the gastric mucosa undergoes constant turnover [10]. Accordingly, H. pylori must rapidly initiate colonization and localize where the environment is more hospitable: within 15��m from the gastric epithelial cells [11], and deep within gastric glands [12].��
Colonization, localization, and inflammation: The roles of H. pylori chemotaxis in vivo
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5862749/��
https://openi.nlm.nih.gov/detailedresult?img=PMC3615616_acb-46-8-g001&req=4
��
��
Team:Slovenia/Background/The problem - 2008.igem.org
http://2008.igem.org/Team:Slovenia/Background/The_problem
http://myplace.frontier.com/~dffix/medmicro/helic.htm
��
The Helicobacter pylori Genome
Genome size: 1.7 million base pairs
Number of Genes: approximately 1600
% Genes unique to H. pylori: 17%
Two strains of Helicobacter pylori have genomes which have been completely sequenced (Tomb et al., 1997; Alm et al. 1999). The first genome sequenced was isolated from a gastritis patient in the United Kingdom and was sequenced using the whole genome random sequencing method (Tomb et al., 1997). The second strain of H. pylori was isolated from a patient in the United States in 1994 and was sequenced in 1996 (Alm et al., 1999). The two Helicobacter pylori genomes were compared by Alm et al. (1999) and were found to differ in only six to seven percent of genes. These genes appeared to be clustered within a single region of the genome (Alm et al., 1999).
Morphology
H. pylori is a Gram-negative bacterium which is enclosed within two membranes (Curry & Jones, 1990). When first observed by Warren and Marshall in biopsy samples from gastritis patients, H. pylori was found to have a spiral shape; however this bacterium has also been observed to take on curved, rod (bacillary) or circular (coccoid) forms (Meyers, 2007; Chalmers et al., 2004). H. pylori typically has between four to seven flagella localized at one pole of the bacterium (Salyers & Whitt, 2002). The combination of its curved shape and unipolar flagella allows H. pylori to move easily through the thick mucus layer of the human stomach (Salyers & Whitt, 2002).
Studies conducted by Ng et al. (1985) found that within colonies of H. pylori, the circular, or coccoid forms were found at the centre of the colony while the spiral forms were found at the edges and were actively dividing. It is theorized that the circular form of H. pylori is inactive and represents a survival adaptation which allows the organism to survive unfavourable conditions (Curry & Jones 1990). It is also theorized that it is the coccal forms that are involved in the transmission of H. pylori.
Physiology
Helicobacter pylori has a microaerophilic physiology meaning that it thrives best in low oxygen enviroments (Helicobacter Foundation, 2006). Analysis of the Helicobacter pylori genome sequenced by Tomb et al. (1997) indicated that glucose was the primary carbon source used for energy production.
Another interesting aspect of H. pylori physiology is that it thrives at a neutral pH of 7.0 (Helicobacter Asssociation, 2006). In order to protect itself from the acidic environment of the stomach, H. pylori burrows its way into the stomach's mucus lining (Helicobacter Asssociation, 2006). There H. pylori use the powerful enzyme urease to maintain a favourable pH by breaking down urea in the stomach into ammonia and bicarbonate�Cstrong bases which counteract the acid in the stomach (Chalmers et al., 2004). Urease enzyme activity has been shown to be essential to the colonization of H. pylori and as a result, H. pylori has developed a unique 2 subunit urease enzyme that is exceptionally powerful (Chalmers et al., 2004).
H. pylori obtains its nutrients by taking advantage of the human inflammatory reponse. The human body will send extra nutrients to the area colonized by H. pylori in order to help white blood cells attack the bacteria (Helicobacter Foundation, 2006). H. pylori however, are inaccessible to these cells because of their location within the mucus and are therefore able to use the excess nutrients provided by their host with no risk to themselves (Helicobacter Foundation, 2006).Helicobacter pylori
http://tolweb.org/treehouses/?treehouse_id=4722��
H. pylori, Hormones and Your Gut Health
What is H. pylori?
Helicobacter pylori, previously known as Campylobacter pylori, is a Gram-negative, microaerophilic bacterium usually found in the stomach. It was identified in 1982 by Australian scientists Barry Marshall and Robin Warren, who made the link between H. pylori and gastric ulcers, a condition not previously associated with GI microbial disease.
H. pylori and Food
The fermentation of malabsorbed carbohydrates in the gut produces hydrogen gas. Hydrogen gas is the preferred energy source for H. pylori. Elevated hydrogen gases are also associated with other nasty bugs such as Salmonella, E. coli and Campylobacter jejuni. Sugars (including fructose), starches, and some fibrous carbohydrates contribute to this fermentation and gas production.
Other important considerations are food sensitivities and leaky gut syndrome. A mucosal barrier that is damaged by autoimmune reactions to foods is a very vulnerable place for pathogens to enter and set up a colony. The stomach is designed to be a closed system, ready and able to attack pathogens as they enter, with sufficient healthy bacteria as well as stomach acid. When this environment is opened in leaky gut conditions, illness risks increase dramatically.
When a person has H. pylori, the strain diversity increases over time. The presence of any virulence factor increases the potential for symptoms and disease. The average person with HP has 2.5 different strains. Each strain will have its own set (or not) of virulence factors. A bacteria like H. pylori can double in amount every 1-2 hours. So over a short period of time, levels can increase dramatically.
H. pylori, Hormones and Neurotransmission
Another area of great interest surrounding H. pylori is the advancement in understanding of negative influences of steroidal hormones on the infection. A scholarly paper published in 2011 by Hirofumi Shimomura, following extensive research, showed that pregnenolone, dehydroepiandrosterone (DHEA), epiandrosterone (EA) and estrone (E1) are all absorbed into the lipid membrane of H. pylori. As steroids, they have the capacity to strengthen and reinforce the membrane barrier and increase its resistance to treatment/eradication. The reinforced barrier creates resistance of H. pylori to phosphatidylcholines, a group of phospholipids that includes linoleic and/or arachidonic acid. These are polyunsaturated fatty acids (PUFAs) that confer antibacterial actions which are fatal to the typically steroid-free H. pylori membrane. Despite its low abundance, phosphatidyl serine in the cell membrane plays key roles in various phenomena such as the coagulation cascade, clearance of apoptotic cells, and recruitment of signaling molecules. Phosphatidyl serine also localizes in endocytic organelles which are responsible for ��cell drinking�� and ��cell eating��, two forms of moving nutrients into cells by the creation of vacuoles. Also important to note is that phospholipid molecules such as phosphatidyl serine have both polar and non-polar lipid regions, and because of this, can have difficulty finding stability to begin with. The phosphate portion of the molecule seeks water, while the lipid portion seeks oil. Between the innate instability and the blockage of phosphatidyl serine by steroidal hormones, these endocytic cellular nutrition functions become impaired.
In opposition to DHEA, EA and pregnenolone, estradiol (E2), androstenedione, and progesterone are harmful for the survival of H. pylori. Progesterone exhibits the most effective antibacterial action against H pylori. Progesterone inhibits the absorption of free-cholesterol by H. pylori and conversely, high levels of free-cholesterol inhibit the antibacterial actions of progesterone. These two hormones appear to bind to identical sites on the bacteria and therefore can obstruct each other��s effects.
One study indicates that licorice root may positively impact the treatment of H. pylori. Licorice is an adaptogenic herb which supports adrenal function and hormone balance. This is more proof that hormones and our guts are intimately connected. Further information indicates that there are likely effects on the brain-gut axis by H. pylori. These may arise from a direct neurotoxic effect, activation of inflammatory processes in nerves and micronutrient deficiency. Intermediate effects of chronic H. pylori infection on brain-gut axis function have been clinically observed as: (1) the alteration of feeding patterns; (2) cognitive and memory dysfunction, increased vulnerability to stress and anxiety- and depressive-like behaviors.
What all of this means for a person infected with H. pylori is that the infection does not exist in isolation of the body��s complex metabolic functions. It is not a simple scenario of a gut infection that is easily eradicated with conventional therapies. The infection burrows deep into the lining of the gut where antibiotics may not reach it, and is impacted by adrenal and liver health/balance. Other factors such as poor cholesterol synthesis and hormone toxicity can alter the natural balances and strategies employed by a healthy body in attacking an infection such as H. pylori.
H. pylori and Health Management
In summation, H. pylori is a complex pathogen capable of derailing proper nourishment and increasing other disease risks including GI cancers. A comprehensive understanding and investigation of the body��s metabolic processes and needs is highly advised. Checks and cross-checks with influences and counter-influences are necessary. Some examples of this are:
Is the diet of the infected person appropriate for their needs? Are there sufficient foods to stimulate acid production? Is the liver being nourished by the individual��s diet?
Are there dietary and/or toxin stressors adding to pathogen hosting and/or poor gut membrane integrity?
Is the liver producing adequate enzymes for adrenal hormone production?
Are cholesterol and other steroid hormone levels normal and balanced? Are there excesses and imbalances that could be altering the cell membrane integrity? What is the source of the inflammation causing such imbalances?
What role is chronic stress playing in the expression of immune and detoxification processes in the body?
H. pylori, Hormones and Your Gut Health �� TRUE NATURE FUNCTIONAL HEALTH CONSULTING
https://www.truenaturehealthconsulting.com/blog/2018/9/10/h-pylori-hormones-and-your-gut-health��
Molecular Hydrogen as an Energy Source for Helicobacter pylori | Science
https://science.sciencemag.org/content/298/5599/1788.fullMolecular Hydrogen as an Energy Source for Helicobacter pylori
Olson, Jonathan W.; Maier, Robert J.
Abstract
The gastric pathogen Helicobacter pylori is known to be able to use molecular hydrogen as a respiratory substrate when grown in the laboratory. We found that hydrogen is available in the gastric mucosa of mice and that its use greatly increased the stomach colonization by H. pylori. Hydrogenase activity in H. pylori is constitutive but increased fivefold upon incubation with hydrogen. Hydrogen concentrations measured in the stomachs of live mice were found to be 10 to 50 times as high as the H. pylori affinity for hydrogen. A hydrogenase mutant strain is much less efficient in its colonization of mice. Therefore, hydrogen present in animals as a consequence of normal colonic flora is an energy-yielding substrate that can facilitate the maintenance of a pathogenic bacterium.Molecular Hydrogen as an Energy Source for Helicobacter pylori - NASA/ADS
https://ui.adsabs.harvard.edu/abs/2002Sci...298.1788O/abstractHelicobacter pylori Urease Suppresses Bactericidal Activity of Peroxynitrite via Carbon Dioxide Production
Kumamoto University School of Medicine
ABSTRACT
Helicobacter pylori can produce a persistent infection in the human stomach, where chronic and active inflammation, including the infiltration of phagocytes such as neutrophils and monocytes, is induced. H. pylori may have a defense system against the antimicrobial actions of phagocytes. We studied the defense mechanism of H. pylori against host-derived peroxynitrite (ONOO−), a bactericidal metabolite of nitric oxide, focusing on the role of H. pylori urease, which produces CO2 and NH3 from urea and is known to be an essential factor for colonization. The viability of H. pylori decreased in a time-dependent manner with continuous exposure to 1 ��M ONOO−, i.e., 0.2% of the initial bacteria remained after a 5-min treatment without urea. The bactericidal action of ONOO− against H. pylori was significantly attenuated by the addition of 10 mM urea, the substrate for urease, whereas ONOO−-induced killing of a urease-deficient mutant of H. pylori or Campylobacter jejuni, another microaerophilic bacterium lacking urease, was not affected by the addition of urea. Such a protective effect of urea was potentiated by supplementation with exogenous urease, and it was almost completely nullified by 10 ��M flurofamide, a specific inhibitor of urease. The bactericidal action of ONOO− was also suppressed by the addition of 20 mM NaHCO3 but not by the addition of 20 mM NH3. In addition, the nitration of l-tyrosine of H. pylori after treatment with ONOO− was significantly reduced by the addition of urea or NaHCO3, as assessed by high-performance liquid chromatography with electrochemical detection. These results suggest that H. pylori-associated urease functions to produce a potent ONOO− scavenger, CO2/HCO3−, that defends the bacteria from ONOO− cytotoxicity. The protective effect of urease may thus facilitate sustained bacterial colonization in the infected gastric mucosa.
Helicobacter pylori Urease Suppresses Bactericidal Activity of Peroxynitrite via Carbon Dioxide Production
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC98327/��
[37] Peroxynitrite reactions with carbon dioxide ...
https://www.sciencedirect.com/science/article/pii/S007668799901099X
Peroxynitrite decomposition was studied in 100 mM phosphate buffer containing 0.1 mM dtpa and was followed spectro- scopically at 302 nm in the presence of CO2-HCO3- as previously de- scribed. 17 The disappearance of ONOO- in the presence of excess COz-HCO3- follows pseudo first-order kinetics (Fig. 1).
Cited by: 126
Publish Year: 1999
Author: Rafael Radi, Ana Denicola, Bruce A. Freeman
Nitric Oxide Cycle, Superoxide and Peroxynitrite
www.medicalinsider.com/cardiac3.html
Increasing bodily CO2 levels - by rebreathing or by going below sea level (or presumably breathing a higher partial pressure of O2 - causing one to breathe less and thus build up more CO2); CO2 being a scavenger of Peroxynitrite. CO2 reacts with Peroxynitrite (ONOO-) ����
Analysis of 75 onion cultivars showed the following: Total quercetin content in a kilogram of yellow, pink and red onions varied from 54 mg to 286 mg in onions grown in 1992. A kilogram is just over 35 ounces. The best yellow onion they tested was the Sweet Savannah, which accounted for the 286 mg result. White onions showed only trace amounts of quercetin. Onions stored for 5 months showed some change in quercetin content. Variation in Quercetin Content in Different Colored Onions
Onions and Quercetin ��
https://www.grow-your-vitamins.com/walking-onions-and-quercetin/��
��
Utilization of quercetin and quercetin glycosides from onion (Allium cepa L.) solid waste as an antioxidant, urease and xanthine oxidase inhibitors
Highlights
•
Onion solid waste (OSW) used as cheap and valuable source for production of value-added products.
•
HPLC revealed significant amount of quercetin and quercetin glycosides in OSW.
•
OSW extracts inhibited the urease and xanthine oxidase activity in vitro.
•
OSW showed potent antioxidant activity with in vitro scavenging assays.
Abstract
This study aimed to determine the flavonol glycosides from onion solid waste (OSW) using HPLC analysis, with antioxidant and enzyme inhibitory activities. We found considerable amount of quercetin-4��-O-monoglucoside (QMG: 254.85), quercetin-3,4��-O-diglucoside (QDG: 162.34), quercetin (Q: 60.44), and isorhamnetin-3-glucoside (IMG: 23.92) (mg/100 g) dry weight (DW) of OSW. For OSW, the methanol and ethanol showed the strongest antioxidant activities, followed by ethyl acetate, chloroform, and n-hexane extracts. Among the flavonols, Q and QDG possessed higher antioxidant activities. OSW and flavonol glycosides displayed significant enzyme inhibitory activity, with IC50 values ranging from 12.5 �� 0.11 to 32.5 �� 0.28 for OSW, 8.2 �� 0.07 to 16.8 �� 0.02 for flavonol glycosides, and 4.2 �� 0.05 ��g/mL for thiourea (positive control) towards urease; while 15.2 �� 0.8 to 35.8 �� 0.2 (��g/mL) for OSW, 10.5 �� 0.06 to 20.8 �� 0.05 (��g/mL) for flavonol glycosides, and 6.5 �� 0.05 ��g/mL for allopurinol (positive control) towards xanthine oxidase, respectively. The OSW and flavonol glycosides may thus be considered as potential antioxidant and antigout agents.
Graphical abstract
Utilization of quercetin and quercetin glycosides from onion (Allium cepa L.) solid waste as an antioxidant, urease and xanthine oxidase inhibitors - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S030881461730821X��
Inhibition of Helicobacter pylori and Associated Urease by Oregano and Cranberry Phytochemical Synergies
Research has indicated that urease of H. pylori is located in the cytoplasm in freshly prepared cultures and in the outer membrane in older cultures (15). In addition to pathogenicity from H. pylori, evidence indicates that ammonia generated by urease can cause injury to the gastroduodenal mucosa (33, 42). Specific inhibition of urease activity has been proposed as a possible strategy to inhibit this microorganism (25). It has been demonstrated that a urease-negative mutant does not cause gastritis in nude mice due to difficulty in colonization (40). These results suggest the important role of urease in bacterial colonization.
...
Previous research also indicated that host antioxidant stimulation is related to enhanced H. pylori inhibition (2). Therefore, we have proposed to develop a specific phenolic antioxidant profile to inhibit H. pylori. Our strategy couples the benefits of antioxidant activity with specific phenolic profiles to inhibit H. pylori. Further, we have previously investigated whether botanical phytochemical mixtures contribute to antioxidant functionality and antimicrobial effects through synergy (41).
Previous studies have indicated the antimicrobial potential of phytochemical extracts (6, 7, 11). Oregano and cranberry are useful botanicals which are generally recognized as safe for food flavoring and as potential functional ingredients, which are known for their antimicrobial activity linked to the phenolic moiety. Phenolic phytochemicals such as ellagic acid and rosmarinic acid have the potential to interact with proteins and alter their conformation. These phytochemicals can directly interact with the receptors on the cell membrane and could affect normal functioning of ion pumps (17, 27, 28, 32, 39, 41). Also, the partially hydrophobic nature of phenolic constituents allows for accumulation and attachment in the bacterial cytoplasmic membrane, where inhibitory effects may eventually lead to cell death. Recent evidence has also indicated that altering multidrug resistance pumps on the cytoplasmic membrane of bacteria by inhibitors or genetic knockout can enhance the antimicrobial function of phytochemicals (36). Therefore, in oregano and cranberry phenolic profiles, specific phenolics may inhibit multidrug resistance pumps, allowing other phenolics in a synergistic profile to inhibit the bacterium.
Plate assay results indicated that the oregano and cranberry extract mixture was superior in inhibiting H. pylori than individual extracts at the same phenolic concentration. When different extract ratios based on phenolic content were used, a larger inhibition zone was observed, indicating higher susceptibility to a specific ratio (25% oregano and 75% cranberry) of extract mixture. This may be due to one (or more than one) specific phenolic present in the extract that damages the membrane first, making cells more sensitive to the other phenolics (36). As a consequence, impairment of proton pumps and loss of H+-ATPase in damaged membranes can cause disruption in the normal cellular function of the microorganism and therefore lead to cell death (Fig. 6). Further, the acidic nature of phenolic-containing extracts themselves at higher concentrations may create a low-pH microenvironment due to proton donation and cell membrane disruption due to stacking (32), which is likely more effective than low pH alone.
Our results indicated that the activity of urease was inhibited in the presence of phytochemical extracts.
Inhibition of Helicobacter pylori and Associated Urease by Oregano and Cranberry Phytochemical Synergies | Applied and Environmental Microbiology
https://aem.asm.org/content/71/12/8558��
An overview on the potential of natural products as ureases inhibitors: A review��
a
Departamento de Botânica, Instituto de Ci��ncias Biol��gicas, Universidade Federal de Minas Gerais, Av. Pres. Antônio Carlos, 6627, Pampulha, Belo Horizonte, MG 31270-901, Brazil
b
Departamento de Qu��mica, Instituto de Ci��ncias Exatas, Universidade Federal de Minas Gerais, Av. Pres. Antônio Carlos, 6627, Pampulha, Belo Horizonte, MG 31270-901, Brazil
Received 14 July 2014, Revised 21 September 2014, Accepted 22 September 2014, Available online 13 October 2014.
Abstract
Ureases, enzymes that catalyze urea hydrolysis, have received considerable attention for their impact on living organisms�� health and life quality. On the one hand, the persistence of urease activity in human and animal cells can be the cause of some diseases and pathogen infections. On the other hand, food production can be negatively affected by ureases of soil microbiota that, in turn, lead to losses of nitrogenous nutrients in fields supplemented with urea as fertilizer. In this context, nature has proven to be a rich resource of natural products bearing a variety of scaffolds that decrease the ureolytic activity of ureases from different organisms. Therefore, this work compiles the state-of-the-art researches focused on the potential of plant natural products (present in extracts or as pure compounds) as urease inhibitors of clinical and/or agricultural interests. Emphasis is given to ureases of Helicobacter pylori, Canavalia ensiformis and soil microbiota although the active site of this class of hydrolases is conserved among living organisms.An overview on the potential of natural products as ureases inhibitors: A review - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S2090123214001088��
Introduction
Urease (EC 3.5.1.5) is a key enzyme for the global nitrogen cycle, occurring in plants, fungi and bacteria. This type of hydrolase speeds up by one-hundred-trillion-fold the urea hydrolysis rate to ammonia (NH3) and carbon dioxide [1], [2], [3].
Since its discovery in plants [4], Canavalia ensiformis (Fabaceae) urease has been exhaustively investigated and became the milestone in Biochemistry science as the first enzyme to be crystallized [5] and also proven to be strictly dependent on nickel ions (Ni2+) [6]. The dependence on nickel ions for catalytic activity is a unique feature of urease among hydrolytic enzymes [1], [2]. The first three-dimensional structure of a urease was fully reported by Jabri and coworkers in 1995 from Crystallography studies performed with urease from Klebsiella aerogenes [7]. Later on, other structures were disclosed for ureases from Bacillus pasteurii [8], Helicobacter pylori [9] and most recently C. ensiformis [10]. Indeed, the elucidation of the urease structure from a legume was crucial to better understand the requirements for ureolytic activity of this class of enzymes in different organisms [10]. The great similarity of amino acid sequence among ureases from multiple origins [11] suggests a common ancestral for this enzyme. Ureases share a basic trimeric array with 1, 2 or 3 subunits that can fuse forming hexameric or dodecameric architecture.Each active site contains two Ni2+ ions apart from each other in 3.5�C3.7 Å, bridged by oxygen atoms of a lysine carbamate residue and a hydroxide ion [3], [12]. Plants and fungi ureases exhibit a single polypeptide chain while bacteria have two or three different subunits (��, �� and ��) [1], [13]. The incorporation of Ni2+ in protein structure is assisted by accessory proteins, believed to be urease-specific chaperones [11].
INTRODUCTION
��ø��EC 3.5.1.5����ֲ������ϸ���з�����ȫ��ѭ���Ĺؼ�ø���������͵�ˮ��ø������ˮ��ɰ���NH3���Ͷ�����̼���ٶ�����˰��ڱ���[1]��[2]��[3]��
�Դ���ֲ���з���[4]���������ô�滨��Fabaceae������ø�;����꾡���о�������Ϊ���ﻯѧ��ѧ�������̱�����Ϊ��һ�����ᾧ��ø[5]�����ұ�֤���ϸ������������ӣ�Ni2 +��[ 6]����ˮ��ø�У������������ӵĴ���������ø�Ķ�������[1]��[2]��Urease (EC 3.5.1.5) is a key enzyme for the global nitrogen cycle, occurring in plants, fungi and bacteria. This type of hydrolase speeds up by one-hundred-trillion-fold the urea hydrolysis rate to ammonia (NH3) and carbon dioxide [1], [2], [3].
Since its discovery in plants [4], Canavalia ensiformis (Fabaceae) urease has been exhaustively investigated and became the milestone in Biochemistry science as the first enzyme to be crystallized [5] and also proven to be strictly dependent on nickel ions (Ni2+) [6]. The dependence on nickel ions for catalytic activity is a unique feature of urease among hydrolytic enzymes [1], [2]. The first three-dimensional structure of a urease was fully reported by Jabri and coworkers in 1995 from Crystallography studies performed with urease from Klebsiella aerogenes [7]. Later on, other structures were disclosed for ureases from Bacillus pasteurii [8], Helicobacter pylori [9] and most recently C. ensiformis [10]. Indeed, the elucidation of the urease structure from a legume was crucial to better understand the requirements for ureolytic activity of this class of enzymes in different organisms [10]. The great similarity of amino acid sequence among ureases from multiple origins [11] suggests a common ancestral for this enzyme. Ureases share a basic trimeric array with 1, 2 or 3 subunits that can fuse forming hexameric or dodecameric architecture. Each active site contains two Ni2+ ions apart from each other in 3.5�C3.7 Å, bridged by oxygen atoms of a lysine carbamate residue and a hydroxide ion [3], [12]. Plants and fungi ureases exhibit a single polypeptide chain while bacteria have two or three different subunits (��, �� and ��) [1], [13]. The incorporation of Ni2+ in protein structure is assisted by accessory proteins, believed to be urease-specific chaperones [11].
Ureases in the context of Helicobacter pylori
The increase of medium pH by the accumulation of NH3 is a urease trait of tremendous medical importance [3]. Urine and/or gastrointestinal infections by ureolytic bacteria can cause health complications in humans and animals, which include kidney stone formation, pyelonephritis, hepatic encephalopathy and ultimately hepatic coma [3], [12]. Therefore, major public health issues are related with H. pylori, gram-negative bacteria that are able to survive in an environment as acidic as that of the stomach (pH 2). As a consequence, H. pylori infection can induce gastric inflammation and increase the risk for the development of duodenal and gastric ulcers, gastric adenocarcinoma and gastric lymphoma [3], [14]. About 50% of global population is committed by H. pylori. This bacteria species can persist in the stomach for the whole life of infected individuals without causing disease symptoms. The high prevalence of H. pylori in human population indicates that such microorganism has developed mechanisms for resistance against host defenses [14]. Urease enzyme in cytoplasm and/or bound to H. pylori surface is the main virulence factor of such human pathogen [15]. It is postulated that the lyses of some pathogen cells leads to the release of cytosolic ureases that bind to the surface of intact bacterial cells and cause the hydrolysis of urea present in human guts at a concentration of 3 mM. The NH3 formed increases the medium pH, which creates a friendly environment for H. pylori survival [15], [16].
During the past 20 years, the recommended first-line therapy for H. pylori eradication consisted of the combination of the antibiotics amoxicillin and clarithromycin with omeprazole, a proton pump cell inhibitor. However, the increase of H. pylori resistance to these antibiotics (particularly to clarithromycin) made this therapy a non-attractive option in recent years [2], [17], [18]. Indeed, other treatment strategies have emerged to fight H. pylori infection, which include the use of bismuth salts combined with a proton pump cell inhibitor or the combination of other classes of antibiotics (e.g. fluoroquinolones, aminopenicillins, tetracyclines, etc.) [2], [18], [19].
Additionally, urease inhibitors may be effective therapies for the treatment of diseases caused by urease-dependent pathogenic microorganisms. However, the commercially available urease inhibitors, such as phosphorodiamidates, hydroxamic acid derivatives and imidazoles are toxic and of low stability, features that prevent their clinical use [20], [21]. Then, the search for novel urease inhibitors with improved stability and low toxicity is mandatory to improve life quality of human beings and animals.Potential of plant extracts as urease inhibitors
Studies with focus on urease of clinical interest
The ethnomedicinal use of plants to treat chronic gastritis, ulcers and related gastroduodenal disorders, diseases that can be caused by H. pylori, is widely reported [37], [38], [39]. Studies carried out with several plant extracts allowed for the identification of urease inhibitors that may be useful for the control of H. pylori strains growth [40], [41], [42], [43].
Alk(en)yl thiosulfinates (TS) are the main constituents of many foodstuffs, for example diallyl thiosulfinate (allicin) corresponds to around 70% of TS content in fresh aqueous garlic extract [44], [45]. Commonly used as a flavoring, garlic (Allium sativum; Liliaceae) is recognized as an antimicrobial and anti-urease food due to allicin levels [44], [46], [47]. The urease inhibition by garlic extract is an irreversible time- and TS-concentration dependent; 18-min incubation of urease with garlic extract is sufficient to cause total loss of enzyme activity [44]. The inhibitory effect of TS-enriched garlic extract was attributed to the ability of TS to oxidize the �CSH group of a cysteine residue present in the enzyme active site [44].
Plant juices obtained from A. sativum (garlic), Allium cepa (yellow and white onions), Allium porrum (leek), Brassica oleraceae var. capitata (cabbage; Brassicaceae) and Brassica oleraceae var. gemmifera (Brussels sprouts) were also effective urease inhibitors [45]. It was found that the higher the TS content, the better the juice was concerning the inhibition of ureolytic activity of urease. Thus, the best inhibitory effects were achieved when garlic juice was used, followed by the employment of Brussels sprouts one. With exception of cabbage juice, all foodstuffs juice tested lost the effect after heating at 95 ��C [45]. Therefore, authors recommend the ingestion of raw garlic, onion, cabbage and Brussels sprout so that the urease inhibitory properties can be preserved and still work in the treatment of H. pylori infection [45]. The in vitro anti-H. pylori activity of methanolic leaf extracts (50 mg/mL) of Allium ascalonicum (Liliaceae) was found to be due to the ability of such extract to decrease urease activity [48]. The methanolic extracts were determined to contain alkaloids, cardiac glycosides, saponins and traces of flavonoids.��
Isolated plant natural products as urease inhibitors
Polyphenols, specially flavonoids, have been pointed out as notable H. pylori urease inhibitors [58], [59], [60]. Therefore, genistein, an isoflavone widely produced by plants of Fabaceae family, was found to inhibit H. pylori urease by 50% when used at 430 ��g/mL while its 7-O-glucoside derivative exhibited no effect on the enzyme activity (Fig. 1) [58].
The therapeutic potential of Lonicera japonica (Caprifoliaceae) against H. pylori is well known [61]. A pool of flavonoids extracted from flowers of this plant exhibited an IC50 value of 946 ��M on H. pylori urease [62]. By testing pure compounds, the flavonols quercetin, rutin, myricetin and myricitrin and the flavones luteolin and luteolin 7-O-glucoside were found the most potent against H. pylori urease, presenting IC50 values of 11.2 ��M, 67.6 ��M, 77.2 ��M, 98.7 ��M, 35.5 ��M, and 55.8 ��M, respectively [62]. Quercetin-4��-O-d-glucoside (Fig. 1) isolated from A. cepa (Liliaceae) showed an IC50 of 190 ��M against C. ensiformis urease [63]. Other, quercetin glucoderivatives (Fig. 1) isolated from Psidium guajava fruits (guava; Myrtaceae) negatively affected the activity of C. ensiformis urease, such as isoquercitrin (IC50 = 160 ��M), quercitrin (IC50 = 200 ��M), avicularin (IC50 = 140 ��M) and guaijaverin (IC50 = 120 ��M). The IC50 for quercetin aglycone toward C. ensiformis urease was determined to be 80 ��M [63].
A study carried out with seven natural products isolated from a butanolic subfraction of the ethanolic extract of Celtis africana (Celtidaceae) revealed the remarkable antiureolitic property of four flavone C-glucosides with IC50 lower than 50 ��M (Table 1) [64]....
Methyl gallate and 1,2,3,4,6-penta-O-galloyl-d-glucoside (PGG) (Fig. 2), widely produced by Paeonia lactiflora (Paeoniaceae) roots, were tested as pure compounds against H. pylori urease [67]. It was observed that PGG (IC50 = 72 ��M) is roughly as potent as the reference inhibitor acetohydroxamic acid. Methyl gallate presented an IC50 of 1.3 mM [67].
An overview on the potential of natural products as ureases inhibitors: A review - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S2090123214001088��
Valorization of onion solid waste and their flavonols for assessment of cytotoxicity, enzyme inhibitory and antioxidant activities
Abstract
Onion (Allium cepa L.) is rich with flavonols which perceived benefits to human health. Flavonols like quercetin and quercetin glycosides from onion solid waste (OSW) have been extracted and tested against enzymes of clinical importance in Alzheimer's disease and diabetes and be shown to have cytotoxic and antioxidant effects. A simple high-performance liquid chromatography-diode array detector method using a Zorbax Eclipse XDB C18 column was developed to separate quercetin-3, 4��-O-diglucoside, quercetin-4��-O-monoglucoside, and quercetin from OSW. These compounds were identified using infrared, ultra-violet, 1H, and 13C nuclear magnetic resonance spectroscopic techniques. The OSW solvent fractions and flavonols showed significant antioxidant activities using DPPH (1, 1-diphenyl-2-picrylhydrazyl), FRAP (ferric reducing antioxidant power), and ABTS (2,2��-azino-bis-3-ethylbenzthiazoline-6-sulphonic acid) radical scavenging assays. The samples exhibited significant in vitro anti-cholinesterase activity with strong antidiabetic effects. OSW extracted with methanol and ethanol showed greater in vitro anti-cholinesterase and hypoglycemic effects than QDG, QMG, and Q possibly due to interactions between multiple compounds and/or complex multivariate interactions with other factors in OSW. In addition, cytotoxicity assays showed that OSW and QDG, QMG, and Q could inhibit the proliferation of selected cancer cell lines. Results indicate that OSW and flavonol glycosides are potential antioxidant, antidiabetic, anticancer, and sedative agents.
Valorization of onion solid waste and their flavonols for assessment of cytotoxicity, enzyme inhibitory and antioxidant activities - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0278691518301303��
Saudi J Biol Sci. 2015 Jul; 22(4): 398�C403.
Effect of different exposed lights on quercetin and quercetin glucoside content in onion (Allium cepa L.)
Eun Young Ko,a,1 Shivraj Hariram Nile,a,1 Kavita Sharma,a Guan Hao Li,b,⁎ and Se Won Parka,⁎
aDepartment of Bioresources and Food Science, College of Life and Environmental Sciences, Konkuk University, Seoul 143-701, Republic of Korea
bDepartment of Food Science, Agricultural College, Yanbian University, Park Road 977, Yanji City, Jilin Province 133002, China
Guan Hao Li: nc.ude.uby@ilhg; Se Won Park: rk.ca.kuknok@krapwes
⁎Corresponding authors. nc.ude.uby@ilhg, rk.ca.kuknok@krapwes
1E.Y. Ko and S.H. Nile, have equal contribution and considered as first authors.
Abstract
Quercetin and quercetin glucosides are the major flavonols present in onion (Allium cepa L.) and are predominantly present as quercetin, quercetin-3,4��-diglucoside and quercetin-4��-glucoside. Effect of different light wavelengths on onion after harvest and storage, with fluorescent, blue, red and ultra violet light influenced the quercetin and quercetin glucosides profile. In a peeled onion, all the light treatments elevated quercetin content in bulb. Among them, particularly fluorescent light effect was more eminent which stimulates the maximum synthesis of quercetin in onion. In case of whole onion bulb, skin and pulp showed different responses to light treatment, respectively. The pulp had the highest quercetin glucosides under blue light, whereas the lowest under fluorescent light. Onion skin showed nearly opposite pattern as compared to the pulp. In particular, light treatment proved to be a better way to increase the level of quercetin content in onions which might be utilized for industrial production of bioactive compounds from onion and onion waste products.
Onion is rich in two groups of phytochemicals (flavonoids and the alk(en)yl cysteine sulphoxides) that are beneficial to human health. The former is divided into two major groups, the flavonols and the anthocyanins which have attracted interest in recent years.��
��ͬ���ն��������Ƥ�غ���Ƥ���������պ�����Ӱ��
��и��������彡�����������ֲ�ﻯѧ���ʣ����ͪ���飨ϩ�������װ�����������ǰ�߷�Ϊ�����࣬��ͪ���ͻ����أ����������������ǵ���Ȥ��
��Ƥ�غ���Ƥ����������У��������е���Ҫ��ͪ������Ҫ����Ƥ�أ���Ƥ��-3,4'-���������պ���Ƥ��-4'-�������յ���ʽ���ڡ��ջ�ʹ����ͬ�����Ĺ����е�Ӱ�죬ӫ�⣬���⣬����������Ӱ����Ƥ�غ���Ƥ�ص����������ס���ȥƤ����У����еĹ��մ�����������۾�����Ƥ�صĺ��������У�������ӫ��Ч����Ϊ���ţ���̼����������Ƥ�ص����ϳɡ�����������۾�������£�Ƥ������Թ���ķ�Ӧ�ֱ�ͬ�������������¾�����ߵ���Ƥ���������գ�����ӫ����¾�����͵���Ƥ���������ա��������ȣ����Ƥ��ʾ�������෴�ķ�Ӧ���ر�أ���ʵ֤��������������������Ƥ�غ���ˮƽ�ĽϺ÷��������������Ƥ�غ��������ڴ���к���з����й�ҵ����������Ի����Keywords: Onion, Light effect, Quercetin, Quercetin glucosides
Effect of different exposed lights on quercetin and quercetin glucoside content in onion (Allium cepa L.)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4486465/��
Front. Cell. Infect. Microbiol., 03 January 2012 | https://doi.org/10.3389/fcimb.2011.00021
The role of microRNAs in Helicobacter pylori pathogenesis and gastric carcinogenesis
Jennifer M. Noto1* and Richard M. Peek1,21 Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA2 Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, USAGastric carcinogenesis is a multistep process orchestrated by aberrancies in the genetic and epigenetic regulation of oncogenes and tumor suppressor genes. Chronic infection with Helicobacter pylori is the strongest known risk factor for the development of gastric cancer. H. pylori expresses a spectrum of virulence factors that dysregulate host intracellular signaling pathways that lower the threshold for neoplastic transformation. In addition to bacterial determinants, numerous host and environmental factors increase the risk of gastric carcinogenesis. Recent discoveries have shed new light on the involvement of microRNAs (miRNAs) in gastric carcinogenesis. miRNAs represent an abundant class of small, non-coding RNAs involved in global post-transcriptional regulation and, consequently, play an integral role at multiple steps in carcinogenesis, including cell cycle progression, proliferation, apoptosis, invasion, and metastasis. Expression levels of miRNAs are frequently altered in malignancies, where they function as either oncogenic miRNAs or tumor suppressor miRNAs. This review focuses on miRNAs dysregulated by H. pylori and potential etiologic roles they play in H. pylori-mediated gastric carcinogenesis.
��Frontiers | The Role of microRNAs in Helicobacter pylori Pathogenesis and Gastric Carcinogenesis | Cellular and Infection Microbiology
https://www.frontiersin.org/articles/10.3389/fcimb.2011.00021/full��
Induction of Cyclooxygenase-2 Overexpression in Human Gastric Epithelial Cells by Helicobacter pylori Involves TLR2/TLR9 and c-Src-Dependent Nuclear Factor-��B Activation
Ya Jen Chang, Ming Shiang Wu, Jaw Town Lin, Bor Shyang Sheu, Tatsushi Muta, Hiroyasu Inoue and Ching-Chow Chen
Molecular Pharmacology December 2004, 66 (6) 1465-1477; DOI: https://doi.org/10.1124/mol.104.005199
Abstract
Gastric epithelial cells were incubated with a panel of clinical isolates of Helicobacter pylori, including nonulcer dyspepsia with gastritis (HS, n = 20), gastric ulcer (HU, n = 20), duodenal ulcer (HD, n = 21), and gastric cancer (HC, n = 20). HC strains induced a higher cyclooxygenase-2 (COX-2) expression than those from HS, HD, and HU. The bacterial virulence factors and the host cellular pathways were investigated. Virulence genes of iceA, vacA, babA2, cagA 3�� repeat region, and hrgA failed to show any association with the disease status and COX-2 expression. Methylation-specific polymerase chain reaction revealed HC strains not affecting the methylation status of COX-2 promoter. Nuclear factor (NF)-��B, NF-interleukin 6, and cAMP response element were found to be involved in COX-2 induction. We explored a novel NF-��B activation pathway. The mutants of TLR2 and TLR9, but not TLR4, inhibited H. pylori-induced COX-2 promoter activity, and neutralizing antibodies for TLR2 and TLR9 abolished H. pylori-induced COX-2 expression. Phosphatidylinositol-specific phospholipase C (PI-PLC), protein kinase C (PKC), and Src inhibitors inhibited COX-2 induction. The dominant-negative mutants of NIK and various I��B kinase complexes, including IKK�� (Y188F), IKK�� (Y199F), and IKK�� (FF), inhibited the COX-2 promoter activity. Phosphorylation of GST-IKK�� (132-206) at Tyr188 and Tyr199 by c-Src was found after H. pylori infection.In summary, H. pylori induces COX-2 expression via activations of NF-��B, NF-interleukin 6, the cAMP response element. In NF-��B activation, H. pylori acts through TLR2/TLR9 to activate both the cascade of PI-PLC��/PKC��/c-Src/IKK��/�� and the cascade of NIK/IKK��/��, resulting in the I��B�� degradation and the expression of COX-2 gene. The COX-2 overexpression may contribute to the carcinogenesis in patients colonized with these strains.
Received July 21, 2004.
Accepted September 20, 2004.
The American Society for Pharmacology and Experimental TherapeuticsInduction of Cyclooxygenase-2 Overexpression in Human Gastric Epithelial Cells by Helicobacter pylori Involves TLR2/TLR9 and c-Src-Dependent Nuclear Factor-��B Activation | Molecular Pharmacology
http://molpharm.aspetjournals.org/content/66/6/1465��
Gastroenterology. 2000 Jul;119(1):97-108.
H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells.
Maeda S1, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, Akanuma M, Shiratori Y, Omata M.
Author information
1
Department of Gastroenterology, University of Tokyo, Tokyo, Japan. maeda-2IM@h.u-tokyo.ac.jp
Abstract
BACKGROUND & AIMS:
H. pylori infection on gastric epithelial cells has been shown to induce NF-kappaB activation, but the mechanism of intracellular signal conduction that leads to NF-kappaB activation is not clear. The aim of this study was to analyze the molecular mechanism responsible for H. pylori-mediated NF-kappaB activation on gastric cancer cells.
METHODS:
NF-kappaB activation by H. pylori was tested by using luciferase reporter assay. IkappaBalpha degradation by H. pylori infection was assessed by immunoblotting. IKKalpha and IKKbeta activation was analyzed by kinase assay. In transfection experiments, effects of dominant negative IkappaBalpha, IKKalpha, IKKbeta, NF-kappaB-inducing kinase (NIK), TRAF2, and TRAF6 mutants were investigated. The effects of an IKKbeta-specific inhibitor, aspirin, on NF-kappaB activation and IL-8 secretion were also analyzed.
RESULTS:
H. pylori promotes degradation of IkappaBalpha, a cytoplasmic inhibitor of NF-kappaB. In kinase assay, H. pylori induced IKKalpha and IKKbeta catalytic activity in gastric cancer cells. Transfection of kinase-deficient mutant of either IKK inhibited H. pylori-mediated NF-kappaB activation dose-dependently. Aspirin inhibited both NF-kappaB activation and IL-8 secretion induced by H. pylori. NF-kappaB activation was also inhibited by transfection of kinase-deficient NIK or a dominant negative mutant of upstream adapter protein TRAF2 or TRAF6.
CONCLUSIONS:
H. pylori induces NF-kappaB activation through an intracellular signaling pathway that involves IKKalpha, IKKbeta, NIK, TRAF2, and TRAF6.H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer ... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/10889159��
Published: 19 March 2012
Lactobacillus acidophilus ameliorates H. pylori-induced gastric inflammation by inactivating the Smad7 and NF��B pathways
Yao-Jong Yang, Ching-Chun Chuang, Hsiao-Bai Yang, Cheng-Chan Lu & Bor-Shyang Sheu
BMC Microbiology volume 12, Article number: 38 (2012) Cite this article
Abstract
Background
H. pylori infection may trigger Smad7 and NF��B expression in the stomach, whereas probiotics promote gastrointestinal health and improve intestinal inflammation caused by pathogens. This study examines if probiotics can improve H. pylori-induced gastric inflammation by inactivating the Smad7 and NF��B pathways.
Results
Challenge with H. pylori increased IL-8 and TNF-�� expressions but not TGF-��1 in MKN45 cells. The RNA levels of Smad7 in AGS cells increased after H. pylori infection in a dose-dependent manner. A higher dose (MOI 100) of L. acidophilus pre-treatment attenuated the H. pylori-induced IL-8 expressions, but not TGF-��1. Such anti-inflammatory effect was mediated via increased cytoplasmic I��B�� and depletion of nuclear NF��B. L. acidophilus also inhibited H. pylori-induced Smad7 transcription by inactivating the Jak1 and Stat1 pathways, which might activate the TGF-��1/Smad pathway. L. acidophilus pre-treatment ameliorated IFN-��-induced Smad7 translation level and subsequently reduced nuclear NF-��B production, as detected by western blotting.
Conclusions
H. pylori infection induces Smad7, NF��B, IL-8, and TNF-�� production in vitro. Higher doses of L. acidophilus pre-treatment reduce H. pylori-induced inflammation through the inactivation of the Smad7 and NF��B pathways.
Background
Helicobacter pylori infection is considered a major factor inducing chronic gastritis, peptic ulcer, and even gastric cancer in humans [1�C3]. In mice and human studies, the gastric mucosa of H. pylori-infected subjects show up-regulated NF-��B pathway and Th1 type cytokine responses [4�C9], which may disturb the integrity of the gut epithelial barrier [10]. Accordingly, the inactivation of the NF-��B pathway and its downstream immune cascades may be helpful in preventing serious H. pylori-induced complications.
������˾�ͨ�����Smad7��NF��B;�����������ݸ˾��յ���θ��֢
������Ŀ�ģ�
θ��Ƥϸ���ϵ������ݸ˾���Ⱦ����ʾ���յ�NF-��B������ã�������NF-��B���ϸ�����źŴ��������в�����������о���Ŀ���Ƿ�������θ��ϸ���������ݸ˾��鵼��NF-��B��ķ��ӻ��ơ�
������
�����ݸ˾���NF-kappaB������ͨ��ʹ��ӫ����ø��������������Եġ�ͨ������ӡ�����������ݸ˾���Ⱦ�����IkappaBalpha���⡣ͨ����ø�ⶨ������IKKalpha��IKKbeta�����תȾʵ���У��о������Ը�IkappaBalpha��IKKalpha��IKKbeta��NF-��B�յ���ø��NIK����TRAF2��TRAF6ͻ��������á���������IKKbeta���������Ƽ���˾ƥ�ֶ�NF-��B�����IL-8���ڵ�Ӱ�졣
���
�����ݸ˾��Ĺ���������MKN45ϸ����IL-8��TNF-���ı����������TGF-��1�ı�������ݸ˾���Ⱦ��AGSϸ����Smad7��RNAˮƽ�Լ��������Է�ʽ���ӡ��ϸ�����MOI 100����������˾�Ԥ�������Լ��������ݸ˾��յ���IL-8��������ܽ���TGF-��1�����ֿ���������ͨ��ϸ����I��B�������Ӻͺ�NF��B�ĺľ����鵼�ġ�������˾���ͨ�����Jak1��Stat1;�������������ݸ˾��յ���Smad7ת¼������ܻἤ��TGF-��1/ Smad;����������˾�Ԥ�����ɸ���IFN-���յ���Smad7����ˮƽ���������ٺ�NF-��B�IJ������絰��ӡ��������⡣
����
�����ݸ˾���Ⱦ���������յ�Smad7��NF��B��IL-8��TNF-���IJ������ϸ�����������˾�Ԥ������ͨ��Smad7��NF��B;����ʧ����������ݸ˾��յ�����֢��
����
�����ݸ˾���Ⱦ����Ϊ�ǵ�����������θ�ף���������������θ������Ҫ����[1-3]����С��������о��У������ݸ˾���Ⱦ�����θ�Ĥ��ʾ���ϵ���NF-��Bͨ·��Th1��ϸ�����ӷ�Ӧ[4-9]������ܻ���ų���Ƥ���ϵ�������[10]����ˣ�NF-��Bͨ·����������������ʧ�����������Ԥ�����ص������ݸ˾�����IJ���֢��Lactobacillus acidophilus ameliorates H. pylori -induced gastric inflammation by inactivating the Smad7 and NF��B pathways | BMC Microbiology | Full Text
https://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-12-38��
A Tale of Two Toxins: Helicobacter Pylori CagA and VacA ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3109773
Nov 23, 2010 �� Cytotoxin-Associated Gene A. CagA is arguably the most well studied virulence factor of H. pylori.It is encoded on the cag pathogenicity island, which is a horizontally acquired 40 kb DNA segment that encodes for a type IV secretion system, and is the only known effector protein to be injected into host cells (Censini et al., 1996; Akopyants et al., 1998).
����
The Role of CagA in the Gastric Biology of Helicobacter pylori
Steffen Backert and Martin J. Blaser
DOI: 10.1158/0008-5472.CAN-16-1680 Published July 2016
See related article by Blaser et al., Cancer Res 1995;55:2111�C5.
Visit the Cancer Research 75th Anniversary timeline.
Helicobacter pylori (H. pylori) was discovered in 1983 by the Australian scientists Warren and Marshall as a gastric pathogen, causing peptic ulcer disease. In 2005, they received the Nobel Prize in Medicine or Physiology for their discoveries, especially because the use of antibiotics to treat ulcers changed the practice of medicine. In the early 1990s, our group and others showed that H. pylori is also involved in causing gastric cancer. Between ulcers and cancer, H. pylori became firmly established as a major human pathogen.
Yet, colonization by H. pylori is extremely common worldwide, affecting about half of the world's population, and most carriers develop neither ulcers nor cancer. As such, we had been investigating whether some strains might be more pathogenic than others. In 1988 to 1989, my laboratory was creating libraries of H. pylori genes to identify and clone important antigens. A screen of these libraries using serum from an H. pylori carrier (M.J. Blaser) identified a strongly immunoreactive clone, which we ultimately characterized. Concurrently, we were asking whether carriers developed antibody responses to specific bacterial proteins, including the secreted vacuolating cytotoxin (VacA). In 1990, we reported that people carrying VacA+ strains produced antibodies to a band at about 128 kDa that was absent in carriers of strains that did not have toxin activity (1). Both of our approaches identified the very same protein, which we and colleagues in Siena later jointly named as cytotoxin-associated gene A (cagA and the resulting protein CagA; refs. 2, 3). Studies reported in 1991 by Jean Crabtree and her colleagues in England confirmed our earlier association of a high molecular weight protein (they measured it at 120 kDa) with peptic ulcer disease (4). That independent confirmation by another team, across an ocean, using parallel but different investigative methods, showing essentially the exact same result, confirmed for us that we were on the track of something important.
In 1995, we reported in Cancer Research that carriage of a CagA+ strain was associated with increased risk of gastric cancer (5), that is, the article we are remembering today, but it was one link in a long chain of studies involving multiple scientists around the world. In 1995, we also reported that adjacent to cagA on the bacterial chromosome was a homolog to a type IV secretion system (T4SS) protein (cagE or virB4), and mutating that gene eliminated major CagA effects on epithelial cells (6), but at this stage we did not understand all of the steps in the process. After these early studies, a breathtaking expansion of research on CagA occurred with more than 3,000 citations in PubMed until today. A major question asked by many researchers was: Why are cagA+ H. pylori strains especially associated with ulcers and cancer?
A major breakthrough in the study of CagA came in 2000 when five groups independently reported that H. pylori may encode a functioning membrane-spanning T4SS and that the major substrate that it translocates into host cells is the CagA protein itself (7). All these genes and others are present on a bacterial chromosomal locus that has been called the cag island. The island, a locus of about 40 kb carrying up to 32 genes, is typically flanked by 31-bp direct repeats, was acquired horizontally from a yet unknown ancestor and integrated into the H. pylori chromosome (6, 7). In later years, we have learned that the cag T4SS also translocates other H. pylori substrates, including cell wall components (peptidoglycan) driving much of the proinflammatory response. However, H. pylori also possesses other T4SSs with different activities.
The CagA protein has been recognized as a marker for the entire cag island and has remarkable functions. During infection, CagA localizes to the host plasma membrane and undergoes tyrosine phosphorylation. This was surprising at the time, as the previously published H. pylori genome sequences do not encode tyrosine kinases. These data suggested that host tyrosine kinases may target CagA upon delivery (7). After this discovery, we and others showed that phosphorylation occurs at specific Glu-Pro-Ile-Tyr-Ala (EPIYA)-sequence motifs by c-SRC and c-ABL family kinases (8). The action is precise: the bacteria inject CagA molecules that mimic host proteins and thereby act as molecular ��Trojan horses�� containing a bacterial hidden core message that allows the microbe to substantially influence the host cell (7). In fact, intracellular CagA has since been shown to interact with multiple host cell proteins, either depending on its phosphorylation or not (Fig. 1). A common variant of the cagA gene, encoding an EPIYT-motif, has an altered function (9).
Figure 1.
Model for the translocation of CagA into host cells and role in subverting multiple host signaling cascades. A, CagA phosphorylation-independent and phosphorylation-dependent signal transduction events (B) are displayed. Black arrows, activated signaling events; red lines, inhibitory pathways. Using a type IV secretion system (Cag-pilus), H. pylori delivers CagA across both bacterial and host cell membranes into gastric epithelial cells. Translocation requires integrin ��5��1 and phosphatidylserine. Upon delivery, members of the oncogenic c-SRC and c-ABL tyrosine kinases can phosphorylate CagA at EPIYA and EPIYT sequences. Intracellular CagA then modulates multiple signaling cascades associated with cell polarity, cell cycle, cell proliferation, disruption of tight and adherent junctions, cytoskeletal rearrangements, cell elongation, proinflammatory responses, and suppression of apoptosis, as indicated. The phospho-EPIYT-motif differs from phospho-EPIYA as it leads to enhanced Akt kinase activation (*) and suppression of IL8 secretion and AGS cell elongation (9). Panels A and B were updated from Backert and colleagues (Helicobacter 2010;15:163�C176) with kind permission from Wiley.
The first identified binding partner of phospho-CagA was the phosphatase SHP-2, stimulating gastric epithelial (AGS) cell elongation through an ERK signaling pathway (10). CagA can also recruit the tight junction proteins JAM and ZO-1 (11), as well as E-cadherin and PAR1, resulting in altered epithelial cell-to-cell junctions (Fig. 1). Subsequently, the various forms of CagA have been discovered to interact with >25 host signaling factors promoting proinflammatory, proliferative, cell cycle�Crelated, and antiapoptotic gene alterations (Fig. 1). Thus, CagA is a remarkably multifunctional effector protein of H. pylori, acting as a signaling ��master key�� to subvert normal host cell functions, presumably to directions favorable for H. pylori survival. Accordingly, alterations in cellular growth and of epithelial integrity by CagA can trigger precancerous alterations in the stomach, which is one explanation for the high frequency of gastric cancer in patients carrying CagA+H. pylori (5). On the basis of these findings, the study of CagA biology has evolved in recent years as a paradigm for a group of bacterial effector proteins, which are identified by the host as ��self�� and are being phosphorylated in the same fashion. Like EPIYA-motif phosphorylation in H. pylori, similar tyrosine-phosphorylated effector proteins have been also identified in other bacteria. In enteropathogenic Escherichia coli (EPEC) as well as in Anaplasma, Bartonella, Chlamydia, Ehrlichia, and Haemophilus species, these effectors also may impact host signaling cascades and disease development.The H. pylori T4SS forms an extracellular, needle-like pilus, and its assembly requires multiple protein�Cprotein interactions and several pilus-associated factors. Upon host cell contact and H. pylori adherence, the pilus is induced and then exports CagA across both the bacterial and epithelial membranes into the host cell cytoplasm (12). Studies of the injection mechanism of CagA showed that the T4SS pilus with exposed CagL proteins binds to the epithelial cell integrin member ��5��1; this was the first described host cell receptor for a bacterial T4SS (12). Thus, CagA appears to not be randomly delivered across the host cell membrane, but rather in a tightly receptor-controlled fashion. On the basis of the crystal structure of an approximately 100-kDa amino-terminal CagA fragment, a possible mechanism has been proposed for its cell-surface binding (13). Mapping of preserved areas in the CagA crystal structure detected four conserved surface-exposed patches (CSP1-4) that represent putative hotspots for protein�Cprotein interactions. The proximal part of a single-layer ��-sheet (covering CSP4) mediates the specific interaction of CagA with ��1 integrin to trigger its own translocation, possibly by an endocytic import pathway (13). Taken together, these data provide comprehensive evidence for a unique receptor-dependent internalization mechanism of CagA into host cells.
Animal models for H. pylori infection have been developed in the last 20 years, which highlight the significance of CagA in pathogenesis. For example, in Mongolian gerbils, CagA+H. pylori may trigger premalignant and malignant pathologies. Four weeks after experimental challenge, nearly every animal developed gastric dysplasia, and by 8 weeks, about two-thirds developed adenocarcinoma (14). A host transcription factor that is aberrantly activated in gastric premalignant lesions is ��-catenin, which upregulates cancer-related genes (14). In addition to this infection model, a fundamental experimental connection between CagA and gastric carcinogenesis in vivo has been described by the generation of transgenic C57BL/6J mice expressing CagA (13). These transgenic mice developed gastric epithelial hyperplasia even without H. pylori infection, and some developed polyps and adenocarcinomas in the stomach (and small intestine) after 72 weeks. In addition, systemic expression of CagA in mice resulted in the development of leukocytosis with IL3/GM-CSF hypersensitivity, and some animals developed B-cell lymphomas and myeloid leukemias (15). In that model, the CagA transgene that could be phosphorylated intracellularly by SRC family kinases was critical to the development of pathology. These observations were further supported by two other transgenic model systems, in Drosophila and zebrafish (16). Transgenic expression of CagA in these model organisms revealed significantly elevated rates of JNK activation and Wnt target gene upregulation, resulting in intestinal epithelial cell proliferation as well as the appearance of adenocarcinoma and small cell carcinoma (16). Taken together, these studies establish that H. pylori can induce the expansion of gastric adenocarcinoma in gerbils and other model systems. Oncogenic transformation clearly proceeds in a fashion dependent on a functional T4SS during infection, and expression of CagA alone is sufficient to cause severe malignant lesions in transgenic animals. It is reasonable to propose that gastric epithelial cells carrying translocated CagA could acquire cancer stem cell�Clike properties, making them candidates from which gastric cancer develops; future studies should investigate this hypothesis.
On the basis of the above findings, cagA was described as the first bacterial oncogene (15), but as with many other oncogenes in humans, the story became even more complex. The disease-associated factor CagA is also associated with health, and its lack can lead to disease. Although cagA+H. pylori strains were preferentially associated with both ulcers and gastric cancer, we and others found that these same strains had an inverse (protective) relationship with premalignant and malignant conditions of the esophagus, specifically Barrett esophagus and esophageal adenocarcinoma (17). More recently, numerous studies have shown an inverse association of H. pylori with asthma and other manifestations of allergy, including rhinitis and atopy, especially for those conditions that begin in childhood, and especially with cagA+ strains (18). Thus, these microbes and their genes do not exist in a vacuum. The context in the stomach, and perhaps elsewhere, is important in terms of the phenotypes conveyed to their hosts. The enhanced host interactions of cagA+ strains drive proliferative, inflammatory, and immune phenomena not only in vitro but also in vivo, which is a double-edged sword. In short, the CagA molecular interactions create a state of chronic inflammation that injures the stomach, which worsens across the decades, leading to atrophy and metaplasia, and in some cases, to cancer. But a similar (if not identical) process leads to the recruitment of immunocytes to the stomach wall. Some of these, for example, FOXP3+-regulatory T cells, may have favorable actions to prevent childhood-onset asthma, as shown in experimental asthma models in mice (19).
In aggregate, new data suggest that carrying a CagA+H. pylori strain may not lead to enhanced mortality, compared with a CagA− strain, or to no strain at all (20). It may just be that the causes of death vary for all three groups, with H. pylori inducing gastric cancer cost, but possibly vascular disease benefits. This is in line with the longstanding coevolution of humans with H. pylori. Although progress is being made, especially in the experimental models of asthma, the key mechanisms and its switches are not identified to the degree needed to intervene, so as to prevent or treat disease. One future for CagA is to understand these interactions at the cellular and tissue levels to optimize health. Another future is to harness CagA and its parts, as drugs to force cells to respond according to our medical goals, not the organism's biological needs. By comparison, as Clostridium botulinum has yielded ��botox�� for therapeutic (and other) purposes, an ancient molecule so well evolved for interacting with host cells as CagA likely will have future utility. Perhaps, it might be an antioncogene to reduce cell growth, unbridled proliferation, or to trigger apoptosis via particular pathways. At this interface of host and coevolved microbe, with continued investigation nearly 30 years after its discovery, we predict a bright future. It appears that H. pylori and CagA will continue as rewarding research topics.
The Role of CagA in the Gastric Biology of Helicobacter pylori | Cancer Research
https://cancerres.aacrjournals.org/content/76/14/4028
Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis
Song-Ze Ding,†,1,2 Joanna B Goldberg,1 and Masanori Hatakeyama21Department of Microbiology, University of Virginia Health System, Charlottesville, VA 22908, USA
2Division of Molecular Oncology, Institute for Genetic Medicine, Hokkaido University, Sapporo, 060-0817, Japan
Abstract
Chronic colonization of the human stomach by Helicobacter pylori, a Gram-negative bacterium, is the major cause of chronic gastritis, peptic ulcers and gastric cancer. Recent progress has elucidated important bacterial and host factors that are responsible for H. pylori-induced gastric inflammation and gastric malignancy. H. pylori cytotoxin-associated antigen A is the major oncogenic factor injected into host cells from bacteria and it disrupts epithelial cell functions. Together with H. pylori cag pathogenicity island, it causes general inflammatory stress within gastric mucosa and activates multiple oncogenic pathways in epithelial cells. A growing list of these pathways includes NF-��B, activator protein-1, PI3K, signal transducers and activators of transcription 3, Wnt/��-catenin and cyclooxygenase 2. H. pylori induces epigenetic alterations, such as DNA methylation and histone modification, which play critical roles in oncogenic transformation. In addition, investigations into gastric stem cell or progenitor cell biology have shed light on the mechanisms through which gastric cancer may originate. Continued investigation in these areas will yield novel insights and help to elucidate the mechanisms of bacteria-induced carcinogenesis.Executive summary
�� Chronic infection with Helicobater pylori in the human stomach lasts decades and causes persistent inflammation. Most>H. pylori-infected patients have no clinical symptoms. Approximately 10�C20% of H. pylori-infected patients will develop peptic ulcers and 1% will develop gastric cancer.
�� Upon infection, H. pylori activates multiple oncogenic pathways, such as activator protein-1, NF-kB, Wnt/��-catenin, signal transducers and activators of transcription 3, PI3K and cyclooxygenase-2, all of which have been shown to contribute to the oncogenic transformation processes.
�� Bacterial virulence factors, such as cytotoxin-associated antigen A and cag pathogenecity island, are critical for causing inflammation and the activation of oncogenic pathways. In addition, cytotoxin-associated antigen A protein also disrupts the epithelial cell normal contact and cell polarity, which confer the epithelial cell oncogenic potential.
�� H. pylori-induced epigenetic alternation, including DNA methylation and histone modification, as well as its interaction with gastric stem cells or progenitor cells are important topics that will greatly enhance our understanding of the bacteria-induced carcinogenesis; therefore, they are currently the target of extensive investigation.
Keywords: cancer, chromatin, epigenetics, gastric epithelial cell, Helicobacter pylori, histone, oncogeneHelicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2882595/
Cited by: 140
Publish Year: 2010
Author: Song-Ze Ding, Joanna B Goldberg, Masanori Hatakeyama
Helicobacter pylori CagA promotes epithelial mesenchymal ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6251132
Nov 22, 2018 �� Background. Helicobacter pylori (H. pylori) delivers oncoprotein CagA into gastric epithelial cells via the T4SS and drives activation of multiple oncogenic signalling pathways.YAP, a core effector of the Hippo tumour suppressor pathway, is frequently overexpressed in human cancers, suggesting its potential tumor-promoting role.
Cited by: 8
Publish Year: 2018
Author: Nianshuang Li, Nianshuang Li, Yan Feng, Yi Hu��
J Exp Clin Cancer Res. 2018; 37: 280.
Published online 2018 Nov 22. doi: 10.1186/s13046-018-0962-5
Pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway
Nianshuang Li,1,2 Yan Feng,2 Yi Hu,1 Cong He,1 Chuan Xie,1 Yaobin Ouyang,1 Stephen C. Artim,2 Deqiang Huang,1 Yin Zhu,1 Zhijun Luo,3 Zhongming Ge,corresponding author#2 and Nonghua Lucorresponding author#1
Author information Article notes Copyright and License information Disclaimer
1Department of Gastroenterology, The First Affiliated Hospital of Nanchang University, 17 Yong Waizheng Street, Donghu District, Nanchang, 330006 Jiangxi Province China
2Division of Comparative Medicine, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 USA
3Department of Biochemistry, Boston University School of Medicine, 72 East Concord Street, Boston, MA 02118 USAAbstract
Background
Helicobacter pylori (H. pylori) delivers oncoprotein CagA into gastric epithelial cells via the T4SS and drives activation of multiple oncogenic signalling pathways. YAP, a core effector of the Hippo tumour suppressor pathway, is frequently overexpressed in human cancers, suggesting its potential tumor-promoting role. Although CagA is a casual factor in H. pylori induced gastric carcinogenesis, the link between CagA and YAP pathway has not been identified. In this work, we investigated the regulation of oncogenic YAP pathway by H. pylori CagA.
Methods
Expression of YAP and E-cadherin protein in human gastric biopsies were assessed by immunohistochemistry. H. pylori PMSS1 cagA− isogenic mutant strains were generated. Gastric epithelial cells were co-cultured with H. pylori wild-type cagA+ strains or isogenic mutants and were also treated by recombinant CagA expression. Immunofluorescence was performed for YAP localization. Immunoblot and quantitative PCR were performed for examining levels of YAP, downstream effectors and markers of epithelial-mesenchymal transition. Verteporfin and siRNA silencing were used to inhibit YAP activity.
Results
YAP is significantly upregulated in human gastric carcinogenesis. We generated PMSS1 CagA isogenic mutant strains with chloramphenicol resistance successfully. Our analysis indicated that H. pylori infection induced YAP and downstream effectors in gastric epithelial cells. Importantly, knockout of CagA in 7.13 and PMSS1 strains reduced the expression of YAP by H. pylori infection. Moreover, Inhibition of YAP suppressed H. pylori infection-induced Epithelial-mesenchymal transition (EMT).
Conclusion
Our results indicated that H. pylori CagA as a pathogenic protein promotes oncogenic YAP pathway, which contributes to EMT and gastric tumorigenesis. This study provided a novel mechanistic insight into why cagA+ H. pylori infection is associated with a higher risk for the development of gastric cancer.Helicobacter pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6251132/��
Toxins (Basel). 2019 Nov; 11(11): 677.
Published online 2019 Nov 19. doi: 10.3390/toxins11110677
Helicobacter pylori Virulence Factors Exploiting Gastric Colonization and its Pathogenicity
Shamshul Ansari1 and Yoshio Yamaoka2,3,4,5,*
1Department of Microbiology, Chitwan Medical College and Teaching Hospital, Bharatpur 44200, Chitwan, Nepal; moc.oohay@384irasnaluhsmahs
2Department of Environmental and Preventive Medicine, Oita University Faculty of Medicine, Idaigaoka, Hasama-machi, Yufu, Oita 879-5593, Japan
3Global Oita Medical Advanced Research Center for Health, Idaigaoka, Hasama-machi, Yufu, Oita 879-5593, Japan
4Department of Medicine, Gastroenterology and Hepatology Section, Baylor College of Medicine, 2002 Holcombe Blvd., Houston, TX 77030, USA
5Borneo Medical and Health Research Centre, Universiti Malaysia Sabah, Kota Kinabaru, Sabah 88400, Malaysia
*Correspondence: pj.ca.u-atio@akoamayy
Abstract
Helicobacter pylori colonizes the gastric epithelial cells of at least half of the world��s population, and it is the strongest risk factor for developing gastric complications like chronic gastritis, ulcer diseases, and gastric cancer. To successfully colonize and establish a persistent infection, the bacteria must overcome harsh gastric conditions. H. pylori has a well-developed mechanism by which it can survive in a very acidic niche. Despite bacterial factors, gastric environmental factors and host genetic constituents together play a co-operative role for gastric pathogenicity. The virulence factors include bacterial colonization factors BabA, SabA, OipA, and HopQ, and the virulence factors necessary for gastric pathogenicity include the effector proteins like CagA, VacA, HtrA, and the outer membrane vesicles. Bacterial factors are considered more important. Here, we summarize the recent information to better understand several bacterial virulence factors and their role in the pathogenic mechanism.
��2. Virulence Factors Associated with Escape to High Acidic Environment
After transit to the gastric lumen, the H. pylori encounters extremely harsh conditions of pH around 2.0. However, H. pylori possesses several factors like urease, bacterial shape and flagella mediating motility to interact with the harsh gastric environment (Table 1). The acidic conditions help the bacteria to express some genetic determinants that neutralize the acidic environment (reviewed by Ansari and Yamaoka [7]).Table 1
Virulence factors necessary for H. pylori mediated pathogenicity.
Virulence Factors Mechanism Effects References Acid escape virulence factors Urease Production of NH3 and CO2 Neutralizes the gastric acidity [14,15,16] NH3 damages the gastric epithelium [18,19] CO2 protects the bacteria from killing by metabolic products [20] Angiogenesis Causes gastric cancer progression [21] Activation of PI3K-AKT-mTOR pathway Enhances the progression of cancers [22] Bacterial shape Helical bacterial shape Enhances the bacterial penetration into the mucous layer protecting the bacteria [28] Flagella Motility Helps bacteria to move away from the acidic environment [30] Flagellin activation Displays higher motility that protects the bacteria [31] Epithelial cells colonizing factors BabA Binds with the epithelial cell receptor Leb Mediates bacterial attachment and colonization [34] Enhances CagA translocation [35] Induces double strand breaks in the host cells [36] SabA Binds with sialyl-Lex antigen Mediates bacterial attachment and colonization [37] OipA Bacterial adherence to the gastric epithelium Damages mucosal layer [38] Induces interleukin (IL)-8 expression [38] Causes host cell apoptosis [39] HopQ Bacterial adherence to the gastric epithelium Inhibits immune cell activities [40] Epithelial cells pathogenicity factors cagPAI Encodes syringe like T4SS Translocation of CagA and peptidoglycan [41,42,43,44] CagT Acts as core complex protein in T4SS Helps in the translocation of CagA [45] CagY Binds with integrin Modulates the immune response to promote the bacterial persistence [46] Alters T4SS functions [47] Cag�� Unknown Associates with T4SS function and mediates CagA delivery [48] CagL Acts as core complex protein in T4SS and binds with integrin Helps in the translocation of CagA [42,49] Induces IL-8 expression [50] CagA Phosphorylation of tyrosine Causes cellular proliferation [51] Causes IL-8 expression [52] Causes cell elongation [53] Down regulates the heat shock protein 1 [54,55] VacA Vacuolization of epithelial cells Causes cell vacuolization [56] Causes cell necrosis [57] Causes cellular apoptosis [58,59,60] Endoplasmic reticulum stress Enhances activation of autophagy and increased cellular death [61] HtrA Acts as protease Degrades mis-folded proteins [62,63] Enables delivery of CagA [64,65,66,67,68] Cleaves the tight junction proteins (occluding, claudin-8, and E-cadherin) [64,65,66,67,68,69,70,71] Outer membrane vesicles Clathrin dependent and independent internalization Protects pathogen from the toxic effects of reactive oxygen species [72] Impairs cellular functions [73] Induces dendritic cell functions [74,75] ��-glutamyl transpeptidase Transpeptidation and amino acid synthesis Enhances cell apoptosis [76] Inhibits cellular proliferation [77] Arrests cell cycle [78] VacA dependent vacuolation of epithelial cells Epithelial cell destruction [79] ��
2.1. Urease
A large amount of intracellular urease is produced by H. pylori, constituting around 10% of the total bacterial protein production. In addition to intracellular urease, H. pylori also contains extracellular urease on the bacterial surface due to the lysis of some bacteria in the stomach [12,13].
Urease-catalyzed urea hydrolysis (endogenous and exogenous) results in ammonia (NH3) and carbamate production, which is spontaneously decomposed to yield another ammonia (NH3) and carbonic acid (H2CO3). The carbonic acid is broken down to CO2 and water (H2O) molecules. Ammonia in its protonated form (NH4+) neutralizes stomach acidity and plays an important role in providing a favorable nearly neutral micro-environment around H. pylori [14]. CO2 is converted to bicarbonate (HCO3−) and H+ in the periplasmic space by periplasmic ��-carbonic anhydrase, maintaining the periplasmic pH close to 6.1 via an acid acclimation mechanism. In this way, NH3 and CO2 production provides the necessary environment for H. pylori��s gastric survival [15,16]. However, continuous urease expression is required by bacteria for successful colonization even after its survival adaptation in the stomach [17].
In addition to its role in acid neutralization, several in vitro studies uncovered urease��s role and its catalytic products in pathogenicity establishment. Ammonia production disrupts the tight cell junctions, breaches the cellular integrity, and damages the gastric epithelium [18,19]. CO2 protects the bacterium from the bactericidal activity of metabolic products like nitric oxide and intracellular killing by phagocytes [20]. Urease was recently reported to possibly contribute to tumor growth and metastatic dissemination by inducing angiogenesis, or new blood vessel formation from pre-existing vasculature, and playing a key role in gastric cancer progression [21]. Chronic H. pylori infection has been found to induce hypoxia-induced factor (HIF), which contributes to the development and progression of several cancers. A recent study showed that the H. pylori urease activated the PI3K-AKT-mTOR pathway in gastric cells. The activation of this pathway increases HIF-�� expression [22]. Moreover, H. pylori urease was found to drive the differentiation of endothelial cells by producing reactive oxygen species and activating the lipoxygenase pathway via pro-inflammatory properties, contributing to H. pylori infection progression to gastric carcinogenesis [23]. Furthermore, H. pylori urease was shown to bind to major histocompatibility complex (MHC) class II molecules and induce cell apoptosis [24].
Keywords: Helicobacter pylori, gastritis, peptic ulcer, gastric cancer, CagA, cagPAI
Helicobacter pylori Virulence Factors Exploiting Gastric Colonization and its Pathogenicity
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6891454/��
World J Gastrointest Pharmacol Ther. 2016 May 6; 7(2): 171�C178.
Non-pharmacological treatment of Helicobacter pylori
Haim Shmuely, Noam Domniz, and Jacob Yahav
Author information Article notes Copyright and License information Disclaimer
Haim Shmuely, Jacob Yahav, Department Medicine D, Kaplan Medical Center, Rehovot 76100, Israel
Noam Domniz, Sheba Medical Center, Tel-Hashomer, Ramat Gan 52506, Israel��
GINGER
Several preclinical studies suggest that ginger, an agent linked to gastric and colon carcinogenesis, generates a protective effect against H. pylori[57,58]. Ginger phenolic fractions provide inhibitory effects on the growth of H. pylori, scavenge free radicals, reduce power abilities, protect DNA and inhibit lipid peroxidation[59,60].
Mahady et al[58] reported on the chemo-preventative effects of ginger which directly impede H. pylori growth, particularly CagA+ strains. The authors showed that gingerols and ginger extracts inhibit the development of H. pylori in vitro of 19 clinical strains. In addition, the fraction comprising the gingerols and 6-shogoal was very successful in inhibiting the growth of H. pylori CagA+ strains. This documentation suggests that specific ginger extracts containing gingerols may assist in treating or preventing H. pylori CagA and strains in vivo.
Researchers studying Mongolian gerbils noted that ginger extract prevented and treated H. pylori-induced infection and inflammation[61]. Moreover, additional research was implemented to clarify the in vitro mechanism of the ginger extract. These results confirm the medicinal properties of ginger in Ayurveda and folklore medicines and further advocate that ginger be considered a new therapeutic approach in the treatment of gastric disorders.Non-pharmacological treatment of Helicobacter pylori
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4848239/��
Anticancer Res. 2003 Sep-Oct;23(5A):3699-702.
Ginger (Zingiber officinale Roscoe) and the gingerols inhibit the growth of Cag A+ strains of Helicobacter pylori.
Mahady GB1, Pendland SL, Yun GS, Lu ZZ, Stoia A.
Department of Pharmacy Practice, UIC/NIH Center for Botanical Dietary Supplements Research, PAHO/WHO Collaborating Centre for Traditional Medicine, University of Illinois at Chicago, 833 S. Wood St, MC 877, Chicago, IL 60612, USA. mahady@uic.edu
Abstract
BACKGROUND:
Ginger root (Zingiber officinale) has been used traditionally for the treatment of gastrointestinal ailments such as motion sickness, dyspepsia and hyperemesis gravidarum, and is also reported to have chemopreventative activity in animal models. The gingerols are a group of structurally related polyphenolic compounds isolated from ginger and known to be the active constituents. Since Helicobacter pylori (HP) is the primary etiological agent associated with dyspepsia, peptic ulcer disease and the development of gastric and colon cancer, the anti-HP effects of ginger and its constituents were tested in vitro.
MATERIALS AND METHODS:
A methanol extract of the dried powdered ginger rhizome, fractions of the extract and the isolated constituents, 6-,8-,10-gingerol and 6-shogoal, were tested against 19 strains of HP, including 5 CagA+ strains.
RESULTS:
The methanol extract of ginger rhizome inhibited the growth of all 19 strains in vitro with a minimum inhibitory concentration range of 6.25-50 micrograms/ml. One fraction of the crude extract, containing the gingerols, was active and inhibited the growth of all HP strains with an MIC range of 0.78 to 12.5 micrograms/ml and with significant activity against the CagA+ strains.
CONCLUSION:
These data demonstrate that ginger root extracts containing the gingerols inhibit the growth of H. pylori CagA+ strains in vitro and this activity may contribute to its chemopreventative effects.Ginger (Zingiber officinale Roscoe) and the gingerols inhibit the growth of Cag A+ strains of Helicobacter pylori.
Mahady GB, Pendland SL, Yun GS, Lu ZZ, Stoia A
Anticancer Res. 2003 Sep-Oct; 23(5A):3699-702.Ginger (Zingiber officinale Roscoe) and the gingerols inhibit the growth of Cag A+ strains of Helicobacter pylori. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/14666666/��
Ginger and Cinnamon for Helicobacter-Induced Dyspepsia
by | Aug 9, 2016 | 2012, Cinnamon, Digestive Health, Ginger |
Written by Joyce Smith, Staff Writer. Treatments with ginger/omeprazole and cinnamon/ginger/omeprazole were significantly more effective with higher rates of eradicating H. pylori than the Cinnamon/omeprazole regimen (70% & 80% versus 50%).
botanicals - cinnamonHelicobacter pylori (H. pylori or HP) is now recognized as the primary cause of gastritis and peptic ulcer disease. According to the CDC, more than two thirds of the world��s population is infected with H. pylori 1. These gram negative bacteria are responsible for numerous gastrointestinal disorders including dyspepsia, duodenal and gastric ulcer, gastric cancer and even mucosa-associated lymphoid tissue lymphoma 2-4. It causes an inflammatory reaction that leads to gastritis that ultimately results in the clinical symptoms of the infection 4.
Because of increased antibiotic resistance and adverse reactions to antibiotic treatment, antibiotics for treating H. pylori are less effective now and successful treatment is greatly compromised. Also, reinfections of H. pylori are very common 5 and alternative treatment options are desperately needed. Herbal medicines have been used for many years to treat GI disorders such as gastritis, peptic ulcer, and dyspepsia 6. In fact, a number of in vitro studies have shown that ginger and cinnamon extracts are effective against H. pylori 7,8.
In this study, researchers tested the effectiveness of ginger and cinnamon in treating H. pylori induced gastritis when these herbs were used in combination with omeprazole (commonly known as Prilosec) to treat H. pylori- associated functional dyspepsia. This was an open-label efficacy study of 60 H. pylori-positive patients (52 men, 8 women) aged 24-56 years with functional dyspepsia but no history of peptic ulcers, gastric reflux, irritable bowel syndrome, or treatment with proton pump inhibitors.
Patients were randomized into 3 groups of 20 per group and given a 14 day treatment of the following ginger, cinnamon, and omeprazole combinations:
Group 1: Ginger (400 mg twice daily) plus omeprazole (20 mg twice daily)
Group 2: Cinnamon (850 mg twice daily) plus omeprazole (20 mg twice daily)
Group 3: Ginger (400 mg twice daily), Cinnamon (850 mg twice daily) plus omeprazole (20 mg twice daily)
In addition, all study participants were advised of the following dietary and lifestyle changes:
Avoid high-fat meals, alcohol, caffeine, and foods that trigger dyspeptic symptoms such as coffee, onions, peppers, citrus fruits, spices and carbonated vegetables
Eat frequent small meals throughout the day and no late evening meals
Adopt relaxation and stress management techniques
Stop smoking and lose weight
Stool specimens were collected before and 4 weeks after eradication treatment and tested for the presence of H.pylori 9 using the non-invasive Helicobacter Pylori Stool Antigen (HpSA) Test Kit, Code # 53850 10.
F1 30 mg L. acidophilus (5��109 CFU), 30 mg L. reuteri (5 �� 109 CFU), 330 mg inulin, 5 mg silica, 5 mg talc
F2 12 mg L. plantarum (5��109 CFU), 20 mg L. rhamnosus (5��109 CFU) and 60mg B. animalis subsp. L. lactis (5 �� 109 CFU), 298 mg inulin, 5 mg silica, 5 mg talc
F3 390 mg inulin, 5 mg silica, 5 mg talc
Although all treatment regimens dramatically reduced dyspeptic symptoms and were well tolerated with negligible side effects, stool specimen analyses revealed that the ginger/omeprazole and cinnamon/ginger/omeprazole treatments had significantly higher rates of eradicating H. pylori than the Cinnamon/omeprazole regimen (70% & 80% versus 50%, P �� 0.05). Also, dyspeptic symptom were dramatically reduced within hours following the administration of treatment regimens, particularly with the triple eradication (cinnamon/ginger/omeprazole) regimen.
Conclusion: This study demonstrates that ginger and to a lesser extent cinnamon preparations traditionally used for the treatment of gastrointestinal disorders are effective as components of H. pylori eradication regimens and have little or no adverse reactions. In fact, they could potentially become an inexpensive anti-H. pylori alternative for antibiotic-resistant therapies.
Limitations of this study: Open Label studies or clinical trials do not attempt to disguise the new drug or treatment, meaning that no standard treatment or placebo is used. This leans towards bias, as both the patient and the physician are aware of which groups are receiving what type of treatment.
Source: Ali AM. Efficacy of ginger and cinnamon pharmaceutical preparations in patients with Helicobacter pylori�Cassociated functional Dyspepsia. Prime Journal of Microbiology Research. 2012;2(1):67-72.Ginger and Cinnamon for Helicobacter-Induced Dyspepsia | NHRI
http://www.naturalhealthresearch.org/ginger-cinnamon-helicobacter-induced-dyspepsia/��
Treating H. pylori��blocking acid secretion results in other problems
When it comes to calming down the burning pain of an inflamed stomach lining or ulcer, reducing the amount of acid in the stomach may seem like a good idea. But two new studies with laboratory mice, conducted by Howard Hughes Medical Institute scientists at the University of Michigan Medical School, indicate it could be exactly the wrong thing to do.
Mice treated with prescription drugs called Proton Pump Inhibitors or PPIs, which block acid production, acquired more bacteria and developed more inflammatory changes in their stomach linings than untreated mice did. The study concluded that Proton Pump Inhibitors (including drugs marketed under the trade names Prilosec® and Prevacid®) dissipate stomach acid, which serves an important anti-microbial function and protects the body from ingested microorganisms.
And when acid suppression is removed, the majority of ulcers, particularly those caused by H. pylori, recur.2224Are antibiotics a good idea?
Physicians typically prescribe a triple antibiotic regimen using metronidazole, clarithromycin, and amoxicillin to treat H. pylori-caused ulcers. The problem is, according to Dr. Manfred Kist, a leading German microbiologist, the bacterium is becoming resistant to the standard antibiotics used to treat the infection. Antibiotics can also upset the delicate balance of helpful bacteria in your gastrointestinal tract, paving the way for additional problems.
Kist, director of Germany��s National Reference Center for Helicobacter pylori, claimed in a Reuter��s article that many gastroenterologists don��t gather enough information before treating H. pylori infected patients with antibiotics. ��This is a trend that will lead to increased drug resistance of H. pylori,�� he said. Kist added that 40 to 50% of patients are now resistant to metronidazole, one of the three standard antibiotics used in Germany to treat H. pylori infection. This number rose from 30 percent in the 1980s.24
New sources of anti-H. pylori drugs are needed��which is why the new research showing that herbal extracts can eradicate the H. pylori bacteria is so exciting.Eradicate H. pylori Infection and Protect Yourself from Ulcers Naturally! - Us Smart Publications
https://smart-publications.com/articles/eradicate-h-pylori-infection-and-protect-yourself-from-ulcers-naturally/��
7 Herbs shown to kill H. pylori bacteria �� and reduce risk of peptic ulcer disease
Ginger Extract
Most people are familiar with ginger as a seasoning, but it actually has been used for thousands of years as a medicinal herb. Interestingly, almost all of its benefits have been validated by modern science, and it is the pharmacopoeias of many countries.
Ginger is reported to help prevent cardiovascular disease by inhibiting platelet aggregation and promoting healthy cholesterol absorption.
It helps promote healthy digestion, and is particularly helpful as an anti-nausea and vomiting remedy, both prophylactically and acutely, from a variety of causes, including motion sickness, post-operative recovery, morning sickness, flu and even drug side effects. Ginger has also been shown to prevent ulcer formation.37
A study at the University of Illinois, Chicago, found that ginger root extracts inhibit the growth of H. pylori in vitro.38
Thyme Extract
A popular herbal remedy in ancient Egypt, Greece and Rome, thyme was mainly used for headaches, digestive problems, respiratory illness, and as a mood-enhancer.
Researchers who investigated the anti-microbial properties of 21 essential oils against five important food-borne pathogens, including E. coli, noted that thyme was among the top three at inhibiting the bacteria.25
Also, food and engineering researchers at Technion (the Israel Institute of Technology) found that when thyme extract was compared to several antibacterials, it had a significant inhibitory effect on H. pylori. They suggested that since thyme extract has significant therapeutic potential, it should be further studied in clinical trials.26
Terminalia Chebula Extract
Commonly called haritaki in Ayurvedic Medicine, this herb is from a tree that grows in India. The plant is used extensively in the preparation of many ayurvedic formulations for a variety of health concerns including chronic ulcers, fungal skin infections, digestive problems, constipation, heart disease, tumors, nervous disorders, asthma, and more.
Its principle constituents contain chebulagic, chebulinic acid and corilagin, and its fruit is used as an antiviral, coagulant, and laxative. Terminalia also has a wide antibacterial and antifungal spectrum, and is also known as an adaptogen and hepatoprotective (liver protectant).2728
More recently, in a study comparing with three other ayurvedic herbs, terminalia was found to be the best antioxidant among the four extracts.29
A study at the University of Tehran, Iran, tested extracts of terminalia chebula on H. pylori, which showed significant antibacterial activity on H. pylori and other bacterium.30
The H. pylori bacterium has been estimated to be present in 30-40% of the U.S. population, and is considered the world��s most common chronic infection.
Evodia Extract
Evodia is a seasonal tree, native to northern China and Korea. Its reddish-brown fruit has been used for thousands of years to treat gastrointestinal disorders such as nausea, vomiting, and diarrhea. Evodia has also been traditionally used as a painkiller to treat headaches that accompany nausea and vomiting, and relieve pain associated with abdominal hernias.34
Research done in Japan and Korea has found that evodia extracts strongly inhibit the growth of H. pylori, which reinforces its traditional use for digestive ailments.3536
Licorice Extract
Licorice has been used for thousands of years in Traditional Chinese Medicine as a tonic to rejuvenate the heart and spleen, and as a treatment for ulcers, cold symptoms, and skin disorders. It also has a long history throughout the world as a medicinal herb for numerous health complaints.
Modern herbalists commonly utilize licorice in treating adrenal insufficiencies such as hypoglycemia and Addison��s disease, counteracting stress, and in purifying the liver and bloodstream. In combination with other herbs, it is recommended as a demulcent to soothe mucous membranes, and as an expectorant useful in treating flu, colds, respiratory disorders, and bronchitis.
Licorice is commonly used in Europe to treat ulcers, and research is proving that it does, indeed, have very potent therapeutic effects. Licorice extracts protect the esophagus, stomach, and intestinal lining from stomach acids. In cases of heartburn, licorice also helps repair the stomach��s protective mucous lining. In the following studies, two of the standard anti-ulcer medications failed to perform as well as licorice extracts.
In a recent study at the Institute of Medical Microbiology and Virology, Kiel, Germany, researchers found that licorice extract produced a potent effect against strains of H. pylori that are resistant against clarithromycin, one of the antibiotics typically used in the three antibiotic treatment regimen. The authors concluded that this study provides hope that licorice extract can form the basis for an alternative therapeutic agent against H. pylori.32
A study at Toho University, Chiba, Japan, found that licorice extracts are also effective against H. pylori strains that are resistant to both amoxicillin and clarithromycin, making them viable as chemopreventive agents for peptic ulcer or gastric cancer in H. pylori-infected individuals.33Berberine ������
Berberine is a plant alkaloid isolated from the roots and bark of several plants including golden seal, barberry, coptis chinensis and Yerba mansa. Berberine-containing plants have been used medicinally in ayurvedic and Chinese medicine, and are known to have antimicrobial activity against a variety of organisms including bacteria, viruses, fungi, protozoans, helminths, and chlamydia. Currently, the predominant clinical uses of berberine include bacterial diarrhea, intestinal parasite infections, and ocular trachoma infections.
More recently, berberine has been demonstrated to be effective against H. pylori.42
Curcumin
Curcumin is the substance that gives the spice turmeric its yellow color. Curry powder, which is used extensively in Indian cuisine, is largely made of turmeric and other spices. Curcumin contains many powerful antioxidants and anti-inflammatory compounds, which have been shown to support colon health, a healthy cardiovascular system, and most recently brain health. Dozens of studies have shown that it is a chemopreventative, and more recently it has been shown to exert a strong antibacterial effect against H. pylori.
Studies carried out in the U.S.39, Italy40, and Germany41, produced results showing a significant in vitro effect of its extracts against H. pylori, leading researchers to conclude that curcumin could be considered a valuable support in the treatment of the infection.
Should you take these herbs without knowing for sure if you have H. pylori?
That��s a good question. Although you can get tested for H. pylori, the tests are very expensive. All of these herbs can be taken both therapeutically and prophylactically. In other words, they are perfectly safe to take whether you know you have H. pylori, or whether you just want to protect yourself from its devastating effects.
Thyme, licorice, evodia, terminalia chebula, berberine, curcumin and ginger have a long history of providing numerous health benefits without risk of side effects. And it��s important to remember that H. pylori has been implicated as a culprit in several diseases, not just peptic ulcer disease (PUD). It could be residing in your arteries or liver, in which case, you could end up with cardiovascular disease or worse.
So play it safe. If you know that you have an H. pylori infection, take these herbs at a high dose as a therapeutic treatment for 10 days, several times a year. And if you aren��t sure you have H. pylori but want to protect yourself, it��s perfectly safe to take these herbs year round at a low dose.
Caution: If you are pregnant or breastfeeding, you should consult with your health care professional before taking herbs.Conclusion
We now know that nine out of ten peptic ulcers are caused by an infection from the bacterium Helicobacter pylori and not by stress or spicy foods as previously thought. We also know that H. pylori may be a culprit in numerous other diseases, including cardiovascular and autoimmune, and various skin disorders and stroke.
Several H. pylori strains have become resistant to two of the three antibiotics used in the triple antibiotic drug therapy prescribed to eradicate it, and researchers are vigorously studying a number of herbal extracts to find an alternative treatment.Eradicate H. pylori Infection and Protect Yourself from Ulcers Naturally! - Us Smart Publications
https://smart-publications.com/articles/eradicate-h-pylori-infection-and-protect-yourself-from-ulcers-naturally/��
Nutr Res Pract. 2016 Dec; 10(6): 623�C628.
Published online 2016 Sep 9. doi: 10.4162/nrp.2016.10.6.623
PMCID: PMC5126412
PMID: 27909560
Induction of heme oxygenase-1 with dietary quercetin reduces obesity-induced hepatic inflammation through macrophage phenotype switching
Chu-Sook Kim,1 Hye-Seon Choi,2 Yeonsoo Joe,2 Hun Taeg Chung,2 and Rina Yucorresponding author1
Abstract
BACKGROUND/OBJECTIVES
Obesity-induced steatohepatitis accompanied by activated hepatic macrophages/Kupffer cells facilitates the progression of hepatic fibrinogenesis and exacerbates metabolic derangements such as insulin resistance. Heme oxyganase-1 (HO-1) modulates tissue macrophage phenotypes and thus is implicated in protection against inflammatory diseases. Here, we show that the flavonoid quercetin reduces obesity-induced hepatic inflammation by inducing HO-1, which promotes hepatic macrophage polarization in favor of the M2 phenotype.
MATERIALS/METHODS
Male C57BL/6 mice were fed a regular diet (RD), high-fat diet (HFD), or HFD supplemented with quercetin (HF+Que, 0.5g/kg diet) for nine weeks. Inflammatory cytokines and macrophage markers were measured by ELISA and RT-PCR, respectively. HO-1 protein was measured by Western blotting.
RESULTS
Quercetin supplementation decreased levels of inflammatory cytokines (TNF��, IL-6) and increased that of the anti-inflammatory cytokine (IL-10) in the livers of HFD-fed mice. This was accompanied by upregulation of M2 macrophage marker genes (Arg-1, Mrc1) and downregulation of M1 macrophage marker genes (TNF��, NOS2). In co-cultures of lipid-laden hepatocytes and macrophages, treatment with quercetin induced HO-1 in the macrophages, markedly suppressed expression of M1 macrophage marker genes, and reduced release of MCP-1. Moreover, these effects of quercetin were blunted by an HO-1 inhibitor and deficiency of nuclear factor E2-related factor 2 (Nrf2) in macrophages.
CONCLUSIONS
Quercetin reduces obesity-induced hepatic inflammation by promoting macrophage phenotype switching. The beneficial effect of quercetin is associated with Nrf2-mediated HO-1 induction. Quercetin may be a useful dietary factor for protecting against obesity-induced steatohepatitis.
Keywords: Obesity, quercetin, inflammation, macrophageInduction of heme oxygenase-1 with dietary quercetin reduces obesity-induced hepatic inflammation through macrophage phenotype switching
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5126412/��
Arch Biochem Biophys. 2014 Dec 15;564:83-8. doi: 10.1016/j.abb.2014.09.005. Epub 2014 Sep 18.
Heme oxygenase-1 and anti-inflammatory M2 macrophages.
Naito Y1, Takagi T2, Higashimura Y3.
Author information
1
Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Kyoto 602-8566, Japan. Electronic address: ynaito@koto.kpu-m.ac.jp.
2
Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Kyoto 602-8566, Japan.
3
Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Kyoto 602-8566, Japan; Department of Food Factor Science, Kyoto Prefectural University of Medicine, Kyoto, Kyoto 602-8566, Japan.
Abstract
Heme oxygenase-1 (HO-1) catalyzes the first and rate-limiting enzymatic step of heme degradation and produces carbon monoxide, free iron, and biliverdin. HO-1, a stress-inducible protein, is induced by various oxidative and inflammatory signals. Consequently, HO-1 expression has been regarded as an adaptive cellular response against inflammatory response and oxidative injury. Although several transcriptional factors and signaling cascades are involved in HO-1 regulation, the two main pathways of Nrf2/Bach1 system and IL-10/HO-1 axis exist in monocyte/macrophage. Macrophages are broadly divisible into two groups: pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages. More recently, several novel macrophage subsets have been identified including Mhem, Mox, and M4 macrophages. Of these, M2 macrophages, Mhem, and Mox are HO-1 highly expressing macrophages. HO-1 has been recognized as having major immunomodulatory and anti-inflammatory properties, which have been demonstrated in HO-1 deficient mice and human cases of genetic HO-1 deficiency. However, the mechanism underlying the immunomodulatory actions of HO-1 remains poorly defined. This review specifically addresses macrophage polarization. The present current evidence indicates that HO-1 induction mediated by multiple pathways can drive the phenotypic shift to M2 macrophages and suggests that HO-1 induction in macrophages is a potential therapeutic approach to immunomodulation in widely diverse human diseases.
��
Copyright © 2014 Elsevier Inc. All rights reserved.
KEYWORDS:
Heme oxygenase; Inflammation; Macrophage; PolarizationHeme oxygenase-1 and anti-inflammatory M2 macrophages. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25241054��
Sultan Qaboos Univ Med J. 2006 Dec; 6(2): 71�C76.
PMCID: PMC3074916
PMID: 21748138
The Antibacterial Activity of Honey on Helicobacter Pylori
Basil C Nzeako* and Faiza Al-Namaani
Author information Copyright and License information Disclaimer
Abstract
Objective:
This project aimed to assess the antibacterial potential of various brands of honey sold in Muscat area on some isolates of H. pylori and to determine if there is any synergy between honey and amoxycillin or clarithromycin used in the treatment of H. pylori gastritis and duodenal ulcer.
Methods:
Eight samples of commercial honey were used in the experiment after they were checked for purity by sub-culturing on blood agar and incubating for 48 hours at 37��c. Honey samples showing gross contamination were discarded. Purified culture isolates of H. pylori from our laboratory stock cultures were swabbed on chocolate plate using 1x 104 cfu/ml. One hundred microlitres (100��l) of various honey samples were placed on each plate which was subsequently incubated microaerophilically at 37ºc for 3 days. The presence or absence of growth inhibition zones on each plate was noted and an average zone size of each honey was taken. Honey samples with high zone sizes were further diluted from 1:2�C1:8 to find the end-points of their growth inhibition concentrations and the experiment was repeated in triplicates. The synergistic effect between honey, amoxycillin and clarithromycin was done in triplicates by placing honey at various distances between each antibiotic after swabbing chocolate agar with 1x 104 cfu/ml of H. pylori. The plates were incubated as before.
Results:
All honey samples produced growth inhibition zones with H. pylori no at dilution of honey but had different zone sizes at 1:2�C1:8 dilutions. Black Forest honey had the highest antibacterial activity followed by Langnese honey. None of the honeys had a synergistic effect with either clarithromycin or amoxycillin.
Conclusion:
We conclude that, in vitro, some honey brands possess antibacterial activity against H. pylori and that no synergy or antagonism was observed between honey and clarithromycin or honey and amoxicillin using H. pylori as a test organism. Though no synergy or antagonism was observed between honey, amoxicillin or clarithromycin, it has been suggested that the use of honey with triple therapy regimen may help shorten the time required to eliminate H. pylori from stomach lining of patients with gastritis or duodenal ulcer caused by H. pylori infection
Keywords: Antibacterial, Amoxicillin, Clarithromycin, Honey, H. pylori, Triple therapy, Synergyhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC3074916/
��
Cellular & Molecular Immunology volume
Published: 05 December 2019
Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis
Atsushi Takahashi-Kanemitsu, Christopher T. Knight & Masanori Hatakeyama
Department of Microbiology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
Abstract
Chronic infection with Helicobacter pylori cagA-positive strains is the strongest risk factor for gastric cancer. The cagA gene product, CagA, is delivered into gastric epithelial cells via the bacterial type IV secretion system. Delivered CagA then undergoes tyrosine phosphorylation at the Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs in its C-terminal region and acts as an oncogenic scaffold protein that physically interacts with multiple host signaling proteins in both tyrosine phosphorylation-dependent and -independent manners. Analysis of CagA using in vitro cultured gastric epithelial cells has indicated that the nonphysiological scaffolding actions of CagA cell-autonomously promote the malignant transformation of the cells by endowing the cells with multiple phenotypic cancer hallmarks: sustained proliferation, evasion of growth suppressors, invasiveness, resistance to cell death, and genomic instability. Transgenic expression of CagA in mice leads to in vivo oncogenic action of CagA without any overt inflammation. The in vivo oncogenic activity of CagA is further potentiated in the presence of chronic inflammation. Since Helicobacter pylori infection triggers a proinflammatory response in host cells, a feedforward stimulation loop that augments the oncogenic actions of CagA and inflammation is created in CagA-injected gastric mucosa. Given that Helicobacter pylori is no longer colonized in established gastric cancer lesions, the multistep nature of gastric cancer development should include a ��hit-and-run�� process of CagA action. Thus, acquisition of genetic and epigenetic alterations that compensate for CagA-directed cancer hallmarks may be required for completion of the ��hit-and-run�� process of gastric carcinogenesis.Introduction
Gastric cancer is the fifth most common malignancy and the third leading cause of cancer death, accounting for 819,000 deaths worldwide in 2018.1 While gastric cancer is diagnosed most frequently in developed nations, there exist large geographic variations in gastric cancer incidence.1 In Eastern Asian countries such as Japan, China, and Korea, the incidence is 8-fold greater than that in North America, accounting for more than half of the total gastric cancer incidence.1 Gastric cancer is also primarily a male-dominant cancer, where the incidence in men is 2.2 times greater than that in women.1 The majority of gastric cancers are adenocarcinomas, which are categorized according to Lauren��s criteria into two main histologically based types of gastric cancer: intestinal-type and diffuse-type gastric carcinomas.2 Intestinal-type gastric carcinomas are characterized by the formation of a well-differentiated glandular structure and a relatively well-defined multistep, multifactorial process of histological progression: normal gastric mucosa, chronic atrophic gastritis, intestinal metaplasia, dysplasia and gastric cancer.2,3 Intestinal-type gastric carcinomas are commonly attributed to environmental factors and occur primarily in elderly male patients.4 Diffuse-type gastric carcinomas are usually not well differentiated, lack adhesion, and spread aggressively throughout the stomach.2 These gastric carcinomas are predominantly observed in younger individuals and affect men and women equally.5,6,7 Due to their frequent nodal and distant metastases, they have a worse prognosis than intestinal-type gastric carcinomas.8,9 In addition, gastric cancer has been divided into four distinct subtypes by comprehensive molecular classification: genomically stable, chromosomally unstable, microsatellite unstable, and Epstein-Barr virus (EBV)-positive.10
Helicobacter pylori (H. pylori) is a gram-negative, microaerophilic bacterium that was discovered by Marshall and Warren to infect the epithelium of the stomach.11 It has been found to have infected roughly half of the world��s population, making it one of the most common human infectious agents worldwide.12 While other microorganisms and viruses are unable to survive in the harsh acidic conditions (pH < 2) of the stomach, H. pylori colonizes the stomach by penetrating the gastric mucous layer to reach the epithelial cell layer (pH 5�C6).13,14 Additionally, H. pylori can neutralize surrounding acid through the secretion of urease, an enzyme responsible for converting urea into bicarbonate and ammonia.15 In this newly acquired niche, H. pylori continues to thrive as a monoculture and continuously elicits the host's cellular and humoral immune responses to the site of infection.16,17,18,19 Consequently, the death of immune and epithelial cells at the site of the immune response provides nutrients to the gastric pathogen, allowing for continued colonization of the stomach over the lifespan of the host.20 H. pylori infection is transmitted from host to host through the fecal-oral or oral-oral route and is primarily acquired due to poor hygiene and crowded conditions that facilitate transmission of infection mainly among family members.21
While the development of gastric cancer is variably influenced by both environmental factors and host genetics, there is undoubtedly a significant impact of H. pylori in the development of gastric cancer.22,23,24 In epidemiological studies, H. pylori has been identified as an agent of peptic ulcers (gastric ulcers and duodenal ulcers).25 Clinico-epidemiological studies have also provided a strong relationship between H. pylori infection and the development of mucosa-associated lymphoid tissue (MALT) lymphoma and adenocarcinomas,26,27,28,29,30,31 and the results of subsequent large-scale prospective cohort studies have further supported this association.32,33 H. pylori infection in Mongolian gerbils has also provided evidence for its role in gastric carcinogenesis.34,35,36 In 1994, the International Agency for Research on Cancer, World Health Organization (IARC/WHO) classified H. pylori as a class 1 carcinogen, the only bacterium given this classification.37 It is now well accepted that H. pylori is the strongest risk factor for the development of both intestinal-type and diffuse-type gastric adenocarcinomas, accounting for ~75% of all human gastric cancer cases.38,39
This review summarizes the recent advances in research aimed at the elucidation of the molecular mechanism of gastric carcinogenesis actively driven by the Helicobacter pylori-derived CagA oncoprotein, focusing on the CagA-induced pro-oncogenic perturbation of multiple cell signals that coordinately generate functional interplays not only amongst themselves but also with the inflammatory responses of host cells.CagA, An H. Pylori virulent protein delivered into gastric epithelial cells
H. pylori can be divided into two major subpopulations based on the presence or absence of the cagA gene that encodes the CagA protein: cagA-positive and cagA-negative strains.40 The cagA gene is one of the 27�C31 putative genes that are present in a 40-kilobase genomic DNA segment known as the cag pathogenicity island (cag PAI).40,41 This DNA segment is thought to have been introduced via horizontal transfer from an unknown organism.40 Approximately 20 genes found in the cag PAI encode components of the type IV secretion system (T4SS), a syringe-like structure that is capable of delivering CagA into the cytoplasm of gastric epithelial cells.40,41 CagA is the only effector protein that is known to be secreted by the T4SS.42,43,44,45 Worldwide, cagA-positive strains are responsible for ~60% of the H. pylori infections in individuals. Strains isolated in East Asian countries such as Japan, China, and Korea, however, are almost all cagA-positive strains.46 cagA-positive H. pylori strains are associated with acute gastritis, peptic ulceration, and gastric cancer.40,47 It was first reported in 1995 that infection with cagA-positive strains increased the risk of gastric cancer, with a risk that was at least one order of magnitude higher risk than that of cagA-negative strains.48
H. pylori contains several adhesins, including BabA/B, SabA, AlpA/B, HopQ, HopZ, and OipA, that mediate the tight adherence of the bacteria to gastric epithelial cells and consequently initiate and facilitate the formation of the T4SS.49,50,51 The assembled T4SS is composed of three subassemblies, an outer membrane core complex (OMCC), periplasmic ring complex (PRC), and central stalk.52,53,54 The H. pylori T4SS is composed of additional components that are not present in the prototypical T4SSs present in other species.53 Structural analysis by cryogenic microscopy revealed that the H. pylori T4SS contains an expanded OMCC and a symmetry mismatch between the OMCC and the PRC,55 and its importance in CagA injection warrants further investigation. Injection of CagA requires recognition by the H. pylori T4SS of an Arg-rich CagA-secretion signal sequence (a 20-amino-acid sequence) in the carboxyl-terminal (C-terminal).56 Interaction of the C-terminal region of CagA (an 100-amino-acid sequence found near the CagA-secretion signal sequence) with CagF, a secretion chaperone-like protein, of the T4SS is required for injection of CagA.57,58 However, the C-terminal region is not sufficient for injection of the CagA protein because both the amino-terminal (N-terminal) region and the C-terminal region are required for this process.56 Therefore, the recognition and injection of CagA may be a two-step process requiring first either the C-terminal or N-terminal region followed by the recognition of the second region or a simultaneous recognition of the two distinct regions.
Translocation of CagA into host gastric epithelial cells by H. pylori is achieved via a specific interaction between surface adhesins of the bacteria and receptors for the bacterial component on the host cells. In recent studies, carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) have been identified as a set of protein receptors on epithelial cells that are essential for CagA delivery, which is mediated by specific binding of CEACAMs with the outer membrane adhesin HopQ of the bacteria.50,51,59,60 The CagL protein, a pilus surface component of the T4SS, has also been reported to be an adhesin that mediates CagA delivery by interacting with and thereby activating ��5��1 integrin on target gastric epithelial cells in an arginine-glycine-aspartate (RGD) motif-dependent manner.61 CagL stimulates SRC family kinase (SFK) activity, which in turn phosphorylates the delivered CagA.61 The ��5��1 integrin also interacts with other components of the T4SS, including CagY and CagI, which may cooperatively stabilize the T4SS-host cell interaction.62 To further facilitate the delivery of CagA, H. pylori induces the membrane phospholipid phosphatidylserine (PS) found in the inner membrane leaflet of epithelial cells to be exposed on the outer membrane.63 CagA interacts with the exposed PS to initiate its secretion into the host epithelial cell.63Molecular structure of the CagA protein
CagA is a 128�C145-kilodalton (kDa) protein that is composed of a structured N-terminal region and an intrinsically disordered/unstructured C-terminal tail.64,65,66 The N-terminus of CagA, which comprises ~70% of the entire CagA, further consists of three structured domains: domain I, domain II, and domain III. Variations in the molecular weight of CagA are due to the structural polymorphisms in its C-terminal region, which exist in distinct strains of H. pylori64 (Fig. 1). Once injected into the host gastric epithelial cells, CagA is localized to the inner leaflet of the plasma membrane. The mechanism for intracellular localization of CagA is strongly dependent on the status of apicobasal polarity of the epithelial cells in which CagA is delivered. In polarized epithelial cells, the positively charged phosphatidylserine (PS)-binding K-Xn-R-X-R motif located in domain II of the N-terminal region of CagA interacts with PS, which plays an important role in tethering CagA to the membrane63 (Fig. 1). However, in nonpolarized epithelial cells, the intrinsically disordered C-terminal tail plays an essential role in the localization of CagA to the membrane.67
Fig. 1: Schematic structure of the H. pylori CagA protein.
figure1
The CagA protein is composed of a structural N-terminal region and an intrinsically disordered C-terminal region. The K-Xn-R-X-R motif is required for CagA to physically associate with the membrane phospholipid phosphatidylserine (PS) in cells. The EPIYA (Glu-Pro-Ile-Tyr-Ala) motifs in the C-terminal region are the tyrosine phosphorylation sites of CagA. The EPIYA-repeat region of CagA includes the common EPIYA segments, EPIYA-A, EPIYA-B, and an East Asian CagA-specific EPIYA-D segment, or a variable number of Western CagA-specific EPIYA-C segments, which are a results of the sequence polymorphisms of the CagA protein. The CM motif, which is composed of 16 amino acid residues, serves as a PAR1b binding site, promoting the multimerization of the CagA protein. Based on sequence polymorphisms, the CM motif is subdivided into 2 groups: a canonical CM motif, which has conserved PAR1b-binding ability, and a noncanonical CM motif, which lacks the binding ability. East Asian CagA possesses a single East Asian CagA-specific CM motif (CME), whereas Western CagA possesses multiple Western CagA-specific CM motifs (CMW). The structure of CagA with noncanonical CM motifs, which lack binding ability to PAR1b/MARK2, is shown (bottom panel). The Amerindian CagA, which is derived from the H. pylori v225 strain, possesses an internally deleted noncanonical CM motif. The ABC��-type Western CagA, which was cloned from the H. pylori TH2099 strain that had colonized housed macaques, possesses a derivative of the CMW motif with amino acid substitutions (CMW��) as well as the atypical EPIYA-C segment that contains the ELIYA sequence.
Full size image
While the bacterial CagA protein has no significant homology in its primary structure with known proteins in humans and other species, it undergoes tyrosine phosphorylation by host tyrosine kinases specifically on the Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs in the C-terminal polymorphic region (EPIYA-repeat region)45,68,69 (Fig. 1). From the sequence flanking the EPIYA motif, 4 distinct EPIYA segments have been identified in the CagA protein: EPIYA-A, EPIYA-B, EPIYA-C, and EPIYA-D.70,71 H. pylori strains in regions excluding East Asian countries contain the CagA protein with EPIYA segments arranged as EPIYA-A (32 amino acids), EPIYA-B (40 amino acids), and EPIYA-C (34 amino acids) and is thereby referred to as Western CagA or ABC-type CagA.72 The EPIYA-C segment is present in variable numbers of copies among distinct Western CagA variants, typically represented in tandem between 1 to 3 times.72 The EPIYA-repeat region of CagA found in East Asian countries also possesses EPIYA-A and EPIYA-B segments but, instead of the tandem EPIYA-C segment, contains a distinct EPIYA-containing segment termed EPIYA-D (47 amino acids), and the CagA protein is referred to as East Asian CagA or ABD-type CagA70 (Fig. 1).
Due to the variation of the sequence flanking the tyrosine (Y) residue, the distinct EPIYA segments are tyrosine-phosphorylated selectively by different kinases. SFKs, including c-Src, Fyn, Lyn, and YES, specifically phosphorylate the EPIYA-C and EPIYA-D segments, while the c-Abl tyrosine kinase phosphorylates all of the segments.73,74,75 Interestingly, CagA delivered into host cells has been found to only be phosphorylated on 1 or 2 EPIYA segments, but never 3 or more, in experiments utilizing H. pylori strains with ABC-type CagA.75 EPIYA-A and EPIYA-C or EPIYA-B and EPIYA-D are preferably phosphorylated in combination in Western CagA and East Asian CagA, respectively.75 Therefore, there may be a stepwise event in which EPIYA-C or EPIYA-D is phosphorylated by SFKs at the start of an infection followed by phosphorylation of EPIYA-A or EPIYA-B by c-Abl at a subsequent time point.75 Dephosphorylation of the given SRC/c-Abl-mediated phosphotyrosyl residues of the EPIYA motifs is mediated by SH2 domain-containing protein tyrosine phosphatase (PTPase) 1 (SHP1) in cells.76 Since the binding/scaffolding capability of EPIYA motifs to intracellular proteins is tyrosine phosphorylation-dependent, the phosphorylation status of EPIYA motifs strongly influences the pathobiological activity of the CagA protein.
At the plasma membrane, the delivered CagA is present as a multimerized complex, which requires the conserved 16-amino-acid sequence [CagA-multimerization (CM) motif] in the C-terminal region of CagA.77,78 The CM motif is located immediately distal to either the EPIYA-C segment in Western CagA or the EPIYA-D segment in East Asian CagA77 (Fig. 1). There are alterations in 5 amino acid residues in the CM motif between Western CagA and East Asian CagA. The Western CagA CM motif is therefore termed CMW, and East Asian CagA is termed CME.79 The 16 amino acids at the N-terminal sequence of EPIYA-C are identical to those in CMW, and therefore, CagA containing multiple EPIYA-C segments also contains increased numbers of CM motifs77 (Fig. 1). In addition to CagA multimerization, the CM motif is also involved in the stability of delivered CagA, the degradation of which is mediated by an autophagy-dependent mechanism.80,81,82,83Pro-oncogenic scaffolding functions of CagA that perturb host cell signaling
Deregulation of SHP2, the pro-oncogenic PTPase involved in the regulation of cell growth, motility, and morphology
When the CagA protein is expressed in human gastric epithelial cells (e.g., the AGS cell line), the hummingbird phenotype, a morphological change characterized by an elongated cell shape with elevated cell motility, is observed in a CagA-tyrosine-phosphorylation-dependent manner.84,85 Since the unique morphology of the hummingbird cell is similar to that of cells treated with hepatocyte growth factor (HGF),84,86 it was suggested that the tyrosine-phosphorylated CagA is capable of aberrantly emitting a mitogenic cue, which is reminiscent of that generated upon HGF stimulation. Posttranslational tyrosine phosphorylation of proteins is known to function as a switch that mediates intracellular signal transduction to an SH2 domain-containing protein through a phospho-tyrosine/SH2-domain interaction, which is utilized in mitogenic signaling pathways triggered by growth factors.86
SH2 domain-containing PTPase 2 (SHP2) was the first identified SH2 domain-containing protein that could bind to the tyrosine-phosphorylated EPIYA-C (EPIpYA-C) or EPIYA-D (EPIpYA-D) site of the CagA protein.85 SHP2 is a nonreceptor type PTPase encoded by the PTPN11 gene that possesses two regulatory SH2 domains, an enzymatic PTPase domain and a C-terminal tail region. In physiological conditions, SHP2 is in an enzymatically inactive conformation, in which catalytic reaction to its substrates is interfered with due to the autoinhibitory intramolecular interaction of its SH2 domains with the PTPase domain. Binding of a tyrosine-phosphorylated peptidyl ligand to the SH2 domains changes the structure of the PTPase from the autoinhibitory/closed conformation to an enzymatically active/opened conformation.87 Genetic analysis has provided evidence that cytoplasmic SHP2 is activated by growth factor stimuli and that SHP2 activity is required for full activation of the RAS-RAF-MEK-ERK pathway, a pro-oncogenic signaling pathway involved in the proliferation and differentiation of cells (e.g., neuroblastoma cells).88,89 The binding of EPIpYA-C or EPIpYA-D of CagA protein to SHP2 aberrantly maintains the active/opened conformation of the PTPase, thereby constitutively activating the downstream MEK-ERK kinases90 (Fig. 2). SHP2 has also been reported to regulate the signaling pathways associated with cell morphology and motility.88 For instance, SHP2 dephosphorylates and inactivates focal adhesion kinase (FAK), a key regulator of the dynamics of focal adhesions.91 Therefore, the tyrosine-phosphorylated CagA-induced hummingbird phenotype is due to the perturbation of both SHP2-MEK-ERK signaling and SHP2-FAK signaling through EPIpYA-C-mediated or EPIpYA-D-mediated aberrant activation of the SHP2 phosphatase70,85,91,92 (Fig. 2).
Fig. 2: Schematic representation of the pro-oncogenic actions of the H. pylori CagA oncoprotein.
figure2
When delivered into gastric epithelial cells, the CagA oncoprotein perturbs multiple intracellular signaling pathways and then promotes malignant transformation of the host cells by providing cancer-hallmark capabilities. There is a mutual feedforward stimulatory mechanism between the pro-oncogenic activities of CagA and pro-inflammatory responses.
Full size image
Since gain-of-function mutations of the PTPN11 gene, which encodes SHP2, have been found in patients with hematopoietic malignancies [juvenile myelomonocytic leukemia (JMML), childhood myelodysplastic syndrome, B-cell acute lymphoblastic leukemia and acute myelocytic leukemia] and solid tumors, SHP2 is thought to be a bona fide oncoprotein.93,94,95 In addition to the SHP2 substrates associated with RAS-ERK signaling and FAK, Parafibromin [also known as cell division cycle 73 (CDC73)], a core component of the RNA polymerase II-associated factor (PAF) complex, which is localized in the nucleus of cells, has also been reported as a substrate of SHP2.96 Parafibromin is a transcriptional scaffold protein that promotes transcription of target genes transactivated by transcriptional coactivators/effectors, such as ��-catenin, Gli1, NICD (Notch intracellular domain) and YAP/TAZ, through the formation of complexes dependent on the status of tyrosine phosphorylation.96,97,98,99 Regarding the SHP2-parafibromin axis, an immunohistochemical study of H. pylori-infected gastric epithelia has shown that a small fraction of CagA is present in the nucleus.100 Although further investigation is required, Parafibromin and its downstream transcriptional circuit could be a potential target of the CagA/SHP2 complex in the nucleus.
Unlike EPIpYA-C and EPIpYA-D, the EPIpYA-A and EPIpYA-B motifs are utilized as docking sites for another SH2-containing protein, C-terminal SRC kinase (CSK). CSK is a protein tyrosine kinase that is responsible for the phosphorylation of an inhibitory phosphorylation site of SFKs, which phosphorylate EPIYA-C and EPIYA-D of CagA.101 Therefore, CagA/CSK complex formation contributes to the attenuation of CagA-SHP2 signaling by downregulating phosphorylation on EPIYA-C and EPIYA-D via inhibition of SFKs.102 In addition to SHP2 and CSK, proteomic screening of intracellular host proteins that physically associate with tyrosine-phosphorylated CagA peptides has revealed five additional SH2-domain-containing proteins as potential CagA-binding partners (SHP1, PI3K, GRB2, GRB7 and RAS-GAP), indicating that the pathobiological roles of CagA in hijacking multiple signaling pathways in the host cells are also mediated through these tyrosine-phosphorylated proteins.103Inhibition of PAR1b, the serine/threonine kinase that regulates cell polarity and microtubule dynamics
Tyrosine phosphorylation of the EPIYA motifs is not necessary for all of the pathobiological actions of the CagA protein. Proteomic analysis of the coprecipitates of the CagA protein revealed that partitioning defective-1 (PAR1) family serine/threonine kinases interact with CagA, irrespective of the tyrosine phosphorylation status of the CagA protein.104 The CagA-PAR1 interaction requires the CM motif of CagA, which directly binds to the catalytic cleft of the PAR1 kinase, thereby inhibiting its kinase activity.104,105 Due to the high sequence homology in the kinase domain among the isoforms, CagA is capable of inhibiting all four mammalian PAR1 isoforms, including PAR1b, a predominant isoform in epithelial cells.79,106 In polarized epithelial cells, PAR1b is localized to the basolateral membrane domain, where it is distinguished from that in the apical membrane domain by the tight junctions present between them. Along with the other PAR proteins, PAR1b plays an indispensable role in maintaining tight junctions, which control paracellular permeability across the epithelial cell monolayer, a fundamental role in the establishment and maintenance of apicobasal polarity in epithelial cells.107 CagA expression in a polarized epithelial cell monolayer (e.g., MDCK cells) causes polarity defects characterized by nonpolarized distribution of tight junctional proteins and basolateral proteins, such as ZO-1 and E-cadherin, respectively, which impairs tight junctional barriers of the monolayer via the CM motif-dependent inhibition of PAR1b (Fig. 2).104 Additionally, PAR1 kinases have an alternative name, MARKs (microtubule affinity-regulating kinases), and these were independently cloned and identified as kinases crucial for maintaining microtubule dynamics via the phosphorylation of microtubule-associated substrates.108 Furthermore, it was found that CagA expression in mitotic gastric epithelial cells leads to a delay in the prophase/metaphase transition, which is concomitantly associated with spindle misorientation at the onset of anaphase, followed by chromosomal segregation with an abnormal division axis. The effects of CagA in cell division are abolished by the coexpression of PAR1b, indicating that CagA-mediated inhibition of PAR1b is required for the prophase/metaphase delay and the subsequent spindle misorientation, those of which may be caused by perturbed microtubule stability as well as microtubule-based spindle dysfunction109 (Fig. 2). In addition, PAR1b is known to phosphorylate the RhoA-specific guanine exchange factor H1 (GEF-H1) to inhibit GEF activity, which is required for the induction of RhoA-dependent stress fiber formation.110 Therefore, CagA alters not only the microtubule-based cytoskeletal system but also the actin-based cytoskeletal system, the coordination of which may contribute to the induction of abnormal cell polarity, cell morphology and cell movement in CagA-expressing cells (Fig. 2). Moreover, it has recently been reported that MST1/2 (mammalian Ste20-like kinase 1/2), a component of the tumor-suppressing Hippo signaling pathway, is a novel substrate of PAR1b.111 Therefore, further investigation may reveal the involvement of perturbed MST1/2-LATS1/2-YAP/TAZ signaling induced by the CagA-PAR1b complex in the pathobiological action of the CagA protein.
Adherence junctional defects and Wnt activation, which induce aberrant cell-fate reprograming
In addition to the tight junctional defects, CagA has been reported to target an adherence junctional component, which is functionally associated with Wnt signaling as well as epithelial-mesenchymal transition (EMT) (Fig. 2). CagA has the ability to physically interact with the cytoplasmic domain of E-cadherin in a CM motif-dependent manner. The CagA/E-cadherin interaction impairs and destabilizes E-cadherin/��-catenin complex formation and aberrantly translocates the membranous fraction of ��-catenin to the nucleus, where it subsequently transactivates Wnt target genes in a ��-catenin/TCF-dependent manner.112,113 The following alternative mechanisms of CagA-mediated Wnt activation have been reported: CagA potentiates Wnt signaling through activation of phosphoatidylinositol-3-kinase (PI3K)-AKT signaling by physically interacting with the HGF-stimulated c-Met receptor via the CM motif in a tyrosine phosphorylation-independent manner;114 CagA physically associates with glycogen synthase kinase (GSK)-3�� via its C-terminal region, thereby sequestering GSK-3�� and subsequently potentiating Wnt activation;115 and CagA promotes the proteasome-mediated degradation of the RUNX3 tumor suppressor, which attenuates ��-catenin/TCF-dependent transcription.116,117 It has been reported that the aberrant activation of Wnt signaling by CagA induces ectopic expression of intestine-specific caudal-related homeobox transcription factors (CDX1 and CDX2), and this transcriptionally activates the stemness-associated reprograming factors Sal-like protein 4 (SALL4) and Kr��ppel-like factor 5 (KLF5).118,119 These findings suggest that ectopic expression of these transdifferentiation-associated transcriptional factors by CagA in gastric epithelial cells may play a key role in the development of intestinal metaplasia. Regarding the pathobiological action of CagA in adherens junctions, it has been reported that tyrosine-phosphorylated CagA has the ability to disrupt adherens junctions by forming a complex with CRK adaptor proteins (CRK-I, CRK-II and CRK-L)120 (Fig. 2). Impairment of the adherens junctions and tight junctions in polarized epithelial cells by CagA is an important cue that triggers EMT-like dedifferentiation, which is characterized by morphological change, invasive motility, and induction of mesenchymal-specific proteins such as snail.121,122,123��
Induction of genomic/chromosomal instability concomitant with inactivation of tumor suppressor p53
CagA was originally identified as a product of the ��cytotoxin-associated gene A (cagA)��. However, the CagA protein itself does not show cytotoxic activity against host epithelial cells. Along with the degradation of the tumor suppressor RUNX3, CagA subverts the tumor suppressor function of apoptosis-stimulating protein of p53 2 (ASPP2), also known as Bcl2-binding protein (Bbp)/tumor suppressor p53-binding protein 2 (p53BP2), by physical interacting with the N-terminal region of CagA (Fig. 2). Through the interaction with ASPP2, CagA counteracts the p53-mediated apoptotic response upon cellular stress by promoting the degradation of the p53 tumor suppressor.124,125,126 CagA has also been reported to enhance p53 degradation by activating E3 ubiquitin ligases, HDM2 (human double minute 2) and ARF-BP1 (ARF-binding protein 1), both of which are negatively regulated by p14ARF.127,128 A recent study revealed that CagA aberrantly promoted the induction of TRIP12 (thyroid hormone receptor-interacting protein 12), an E3 ubiquitin ligase for the p14ARF protein.129 Therefore, loss of the ARF gene locus as well as the CagA-mediated degradation of the p14ARF protein in gastric epithelial cells may potentiate the p53 degradation induced by CagA.
In addition to stimulating proteasomal degradation of p53, CagA has been reported to promote the acquisition of spontaneous loss-of-function mutations in the p53 gene. Chronic infection with cagA-positive H. pylori in the gastric epithelium induces ectopic expression of AID (activation-induced cytidine deaminase), a DNA/RNA editing enzyme essential for the somatic hypermutation and class-switch recombination of immunoglobulin genes, via nuclear factor-��B (NF-��B) activation in a T4SS-dependent manner. The ectopic expression of AID results in the accumulation of nucleotide alterations, including spontaneous loss-of-function mutations in the TP53 tumor suppressor gene130 (Fig. 2). Furthermore, chronic exposure to the CagA protein induces aneuploidy/polyploidy in gastric epithelial cells, which may be due to the CagA/PAR1b-mediated defects in microtubule-based mitotic spindle formation109 (Fig. 2). Although the molecular mechanism is still under investigation, it was recently reported that CagA-mediated PAR1b inhibition induced DNA double-strand breaks (DSBs).131 Consistently, infection of gastric epithelial cells with cag PAI-positive H. pylori strains has been shown to induce DSBs, which are concomitantly associated with reduced expression of the DSB repair factor RAD51.132 Notably, H. pylori-induced gastritis, particularly that induced by H. pylori cagA-positive strains, is associated with CpG hypermethylation of MGMT, the gene encoding the DNA repair protein O6-methylguanine DNA methyltransferase (MGMT).133 CagA also upregulates expression of the anti-apoptotic protein MCL1 (myeloid cell leukemia sequence-1) through activation of pro-survival MEK-ERK signaling134 (Fig. 2). Collectively, these observations indicate that upon delivery of CagA, host gastric epithelial cells not only acquire resistance to cell death via inactivation of the p53 tumor suppressor but also induce chromosomal/genomic instability, both of which are hallmarks of cancer cells135 (Fig. 2)��
When delivered into gastric epithelial cells, the CagA oncoprotein perturbs multiple intracellular signaling pathways and then promotes malignant transformation of the host cells by providing cancer-hallmark capabilities. There is a mutual feedforward stimulatory mechanism between the pro-oncogenic activities of CagA and pro-inflammatory responses.
Sequence Polymorphisms of CagA that Confer Differential Oncogenic Potential
Since the CagA protein has an H. pylori strain-specific polymorphism in its primary structure, especially in its C-terminal region, which contains distinct EPIYA-A/B/C/D segments and CM motifs, the pathobiological activity of CagA differs among the distinct CagA species. The amino acid sequence flanking the phospho-tyrosine (pY) residue of the EPIYA-D segment is perfectly matched to the consensus motif of the phospho-tyrosyl peptide that can bind to the SH2 domain of SHP2, while a single amino acid mismatch is present at the pY + 5 position in the pY-flanking sequence of the EPIYA-C segment70,71 (Fig. 3). A quantitative biochemical study revealed that the East Asian CagA (ABD)-specific EPIpYA-D peptide binds to the N-SH2 domain of SHP2 ~100-fold more strongly than the Western CagA (ABC)-specific EPIpYA-C peptide. The phenylalanine (F) residue at the pY + 5 position of the EPIpYA-D peptide was found to form a high-affinity monovalent bond with the hollow pocket on the N-SH2 phosphopeptide-binding floor of SHP2136 (Fig. 3). Accordingly, East Asian CagA exhibits a stronger ability to bind/deregulate SHP2 and a greater capability to induce SHP2-dependent morphological changes in gastric epithelial cells than Western CagA.70,136 Collectively, the findings reveal that the East Asian CagA-specific EPIpYA-D motif is qualitatively very different from the Western CagA-specific EPIpYA-C motif in terms of the biological activity required for deregulation of the SHP2 oncoprotein, which may causatively account for the higher incidence of gastric cancers in East Asian countries than in Western countries. Notably, clinical evidence supports that gastric cancer is more closely associated with H. pylori strains carrying East Asian CagA than with H. pylori strains carrying Western CagA in geographical regions where two distinct strains cocirculate.137,138,139Fig. 3: Polymorphism-dependent differential binding of the EPIYA motif of CagA to SHP2.
The consensus sequence of the phosphotyrosyl peptide for binding to the N-SH2 domain of SHP2 is shown. The sequence of the amino acid residues following the EPIpYA-D motif in East Asian CagA (magenta) is perfectly matched to the N-SH2 domain-binding consensus sequence (black). In contrast, a single mismatched amino acid residue is shown at the pY + 5 position in the EPIpYA-C peptide derived from Western CagA (light green). The EPIpYA/N-SH2 physical interactions are visualized by the crystal structures of the SHP2 tandem SH2 domains (surface representation, white) complexed with the EPIpYA-D peptide (magenta) and EPIpYA-C peptide (light green). The phospho-tyrosine residues (pY) in the EPIYA peptides are observed to interface with a deep pocket of the SH2 groove. The phenylalanine residue (F) at the pY + 5 position of the EPIpYA-D peptide contributes to a binding interface with high affinity for the SH2 domain.
figure3
The consensus sequence of the phosphotyrosyl peptide for binding to the N-SH2 domain of SHP2 is shown. The sequence of the amino acid residues following the EPIpYA-D motif in East Asian CagA (magenta) is perfectly matched to the N-SH2 domain-binding consensus sequence (black). In contrast, a single mismatched amino acid residue is shown at the pY + 5 position in the EPIpYA-C peptide derived from Western CagA (light green). The EPIpYA/N-SH2 physical interactions are visualized by the crystal structures of the SHP2 tandem SH2 domains (surface representation, white) complexed with the EPIpYA-D peptide (magenta) and EPIpYA-C peptide (light green). The phospho-tyrosine residues (pY) in the EPIYA peptides are observed to interface with a deep pocket of the SH2 groove. The phenylalanine residue (F) at the pY + 5 position of the EPIpYA-D peptide contributes to a binding interface with high affinity for the SH2 domain.
Full size image
In contrast to the East Asian CagA species, the great majority of which is composed of CagA with a single EPIYA-D segment for SHP2-binding, Western CagA species can be subdivided into two major groups: CagA with a single EPIYA-C segment for SHP2-binding (type I Western CagA) and CagA with multiple EPIYA-C segments (type II Western CagA), which account for ~60�C70% and 30�C40% of all Western CagA species, respectively.140,141 Type II Western CagA is characterized by tandemly arranged EPIYA-C duplication or triplication, which is thought to be causally due to genetic recombination that occurred in the EPIYA-repeat region141,142 (Fig. 1). Biochemical analysis revealed that type II Western CagA exhibits stronger binding to SHP2 than does type I Western CagA. Moreover, even among the type II Western CagA species, a CagA with a larger number of EPIYA-C segments displays stronger binding to SHP2 than a CagA with a smaller number of EPIYA-C segments.140,143,144 Based on the fact that the two SH2 domains (N-SH2 and C-SH2) are present in a single SHP2 molecule, type II Western CagA is thought to acquire the ability to bind to SHP2 with high affinity via divalent interaction of the multiplicated EPIpYA-C segments with N-SH2 and C-SH2 domains. In contrast, type I Western CagA is thought to bind to SHP2 via the monovalent low-affinity interaction of EPIpYA-C with N-SH2 or C-SH2.136 These quantitative differences in the SHP2-binding ability between the two types of Western CagA account for the differential degrees of SHP2-dependent pathobiological actions of type I and type II Western CagA in gastric epithelial cells, since the levels of the induced hummingbird and invasive phenotypes are positively correlated with the number of EPIYA-C segments in distinct Western CagA types.70,140,145 Consistently, another polymorphism in the EPIYA repeat region of CagA that confers multiplication of EPIYA-A and EPIYA-B segments also proportionally attenuates the CagA/SHP2-dependent induction of the hummingbird phenotype through EPIpYA-A- or EPIpYA-B-mediated activation of CSK, which in turn inhibits SRC-dependent tyrosine phosphorylation of CagA on EPIYA-C or EPIYA-D motifs.143 Notably, several clinico-epidemiological studies have shown that infection with H. pylori strains carrying type II Western CagA is strongly associated with the risk of gastric cancer when compared to infection with H. pylori carrying type I Western CagA.146,147 This further suggests that the quantitative difference in SHP2 deregulation is a crucial factor that determines the distinct level of oncogenic activity of Western CagA species.
Comprehensive analysis of sequence polymorphisms in EPIYA motifs of the CagA protein showed that the EPIYA-B site was the most polymorphic among the 4 distinct EPIYA-A/B/C/D sites. While almost all (>98%) of the EPIYA-C sites are composed of the authentic EPIYA sequence, the authentic EPIYA-B sequence accounts for only 55.5% of the entire population of EPIYA-B polymorphisms and is also shared by the EPIYT-B sequence (32.9%) and others. CagA physically interacts with the SH2 domain of PI3K via the tyrosine-phosphorylated EPIYA-B site, which thereby potentiates PI3K-AKT signaling. A coimmunoprecipitation experiment and structural modeling of the EPIpYA-B or EPIpYT-B peptide bound to the SH2 domain of PI3K indicated that the threonine (T) residue at the pY + 1 position of EPIpYT-B gives higher affinity to the SH2 domain for binding compared to that of the authentic EPIpYA-B sequence.148 Since clinical evidence shows a weaker association of CagA carrying the EPIYT-B polymorphism with gastric cancer than CagA carrying the EPIYA-B motif, involvement of the EPIYA-B polymorphism-dependent differential activation of PI3K-AKT requires further investigation.148
H. pylori strain-specific polymorphisms of the CagA protein are also observed in the CM motif. Among CagA species isolated so far, the CM motif can be classified into two groups according to the PAR1b-binding capability: a canonical CM motif, which conserves the binding ability, and a noncanonical CM motif, which does not. The canonical CM motif is further subdivided into two groups, the Western CagA-specific CMW motif and the East Asian CagA-specific CME motif. Most of the Western CagA species possess 2-4 repeated CMW motifs, with some exceptions. On the other hand, East Asian CagA species possess a single CME motif77,79,149 (Fig. 1). In a similar fashion to the case of the EPIYA-C/D polymorphism, the single CME motif exhibits approximately 2-fold stronger PAR1b-binding ability than a single CMW motif. An increased number of CMW motifs in a single Western CagA protein exhibits an enhanced PAR1b-binding ability in a synergistic manner. The differential PAR1b-binding activity among the distinct CagA species is proportional to the degree of pathobiological activity of the CagA/PAR1b complex such as tight junctional disruption and stress fiber formation in gastric epithelial cells.150 The biological activities of CagA possessing the noncanonical CM motif have also been investigated. The CagA species with an Amerindian-type CM motif (CMAm motif), which is thought to be an internally deleted derivative of the canonical CM motif, showed neither PAR1b-binding nor alterations in epithelial cell polarity, while EPIpYA-mediated SHP2-binding was retained150,151 (Fig. 1). Related to this, it has been reported that the cagA gene isolated from an H. pylori strain that colonized housed macaque encodes an ABC-type Western CagA derivative (ABC��-type CagA), which is characterized by variations in residues in both the EPIYA-C segment and the CM motifs (Fig. 1). Due to the amino acid substitutions present in the noncanonical CM motifs, the ABC��-type CagA lacks binding ability to PAR1b. Additionally, the atypical EPIYA-C segment, which is characterized by amino acid substitutions in the EPIYA-C motif (ELIYA), causes impaired SHP2-binding activity of ABC��-type CagA. In contrast to the standard H. pylori strain with ABC-type cagA, infection of a gastric organoid with H. pylori carrying the ABC��-type cagA failed to elicit CagA-dependent epithelial disruption, suggesting that binding to SHP2 and PAR1b may play a key role in the virulence of distinct CagA species.152��
Oncogenic potential of CagA in vivo
The pathobiological actions of CagA observed in gastric epithelial cells in vitro suggest that CagA perturbs multiple signaling pathways highly associated with carcinogenesis. Consistently, a number of epidemiological studies have shown an intimate association of CagA with the development of H. pylori-associated gastric cancer.153,154,155 However, at that time, no in vivo evidence convincingly supported a crucial role of CagA in gastric carcinogenesis, although a key role of cag PAI, encoding the T4SS for CagA delivery, was indicated.36,156 To clarify the causal link between CagA and in vivo oncogenesis, transgenic mice that express wild-type CagA protein (East Asian CagA) ubiquitously throughout the body were generated (WT cagAE-Tg mice).157 Transgenic expression of the CagA protein caused hyperplasia of the gastric epithelium. Most importantly, in some of the WT cagAE-Tg mice, adenocarcinoma spontaneously developed in the stomach and small intestine. Since the CagA protein produced in the transgenic system indeed underwent tyrosine phosphorylation on EPIYA motifs in cells, abnormal signal transduction perturbed by tyrosine-phosphorylated CagA was thought to be involved in the in vivo oncogenicity of CagA. To investigate the role of the tyrosine phosphorylation of CagA, cagA transgenic mice that expressed tyrosine phosphorylation-resistant CagA (PR CagA), a CagA derivative with tyrosine-to-phenylalanine substitutions in all EPIYA-A/B/D motifs, were also established (PR cagAE-Tg mice). In contrast to the WT cagAE-Tg mice, the PR cagAE-Tg mice did not show gastrointestinal abnormalities, suggesting the requirement of tyrosine phosphorylation on EPIYA motifs for the in vivo pathogenicity of CagA protein in the gastrointestinal tract in mice.157 In addition, the WT cagAE-Tg mice showed leukocytosis associated with hypersensitivity to hematopoietic cytokines, including interleukin-3 (IL-3) and granulocyte-macrophage colony-stimulating factor (GM-CSF), in bone marrow cells. Some of those WT cagAE-Tg mice developed myeloid leukemias as well as B-cell lymphomas.157 The development of such hematological abnormalities, including hematological malignancies, occurred in mice expressing a gain-of-function mutant of SHP2.158,159 In contrast, the PR cagAE-Tg mice, which systemically expressed a CagA protein lacking SHP2-binding ability, did not show any hematological abnormalities, even though the expression levels of PR CagA in PR cagAE-Tg mice were comparable to or even higher than those of wild-type CagA in WT cagAE-Tg mice, suggesting an indispensable role of the tyrosine phosphorylation on EPIYA motifs in the hematological diseases observed in WT cagAE-Tg mice.157 The involvement of SHP2 deregulation by CagA was supported by further in vivo evidence obtained from transgenic mice systemically expressing wild-type Western CagA (WT cagAW-Tg mice) at levels comparable to those of East Asian CagA in WT cagAE-Tg mice. The WT cagAW-Tg mice developed gastric epithelial hypertrophy and gastrointestinal tumors in addition to lymphoid malignancies but did not develop myeloid abnormalities, which were found in WT cagAE-Tg mice. Furthermore, the incidence of tumors in WT cagAW-Tg mice was significantly lower than that in WT cagAE-Tg mice, indicating that Western CagA is qualitatively less oncogenic than East Asian CagA in vivo.160
Another cagA-transgenic (cagA-Tg) animal with either systemic CagA expression or tissue-specific CagA expression in the distal esophagus and anterior intestine was generated using zebrafish. The cagA-Tg zebrafish showed hyperplasia in the adult intestinal epithelium. Moreover, transgenic expression of the cagA gene in p53-null zebrafish induced intestinal small-cell carcinoma and adenocarcinoma.161 These results collectively suggest a key role of tyrosine phosphorylation in the in vivo pathobiological/oncogenic activity of the CagA protein, which may cooperate with alterations of cancer-associated genes in host cells.
Although cagA-positive H. pylori-associated human gastric cancers frequently coexist with precancerous chronic atrophic gastritis,162 the gastrointestinal neoplasms that developed in the WT cagA-Tg mice, as well as the hyperplasia shown in the cagA-Tg zebrafish did not show any overt inflammation.157,160,161 These observations are consistent with the notion that the CagA protein per se is not a potent inflammogen163,164,165 and indicate that inflammation is not necessarily required for the oncogenesis evoked by CagA in vivo. However, chronic inflammation, which is associated with cagA-positive H. pylori infection, has been reported to promote the oncogenesis of cancer-predisposed gastric epithelial cells.165 To experimentally investigate whether the inflammatory response can enhance the pro-oncogenic action of CagA in vivo, WT cagAE-Tg mice, which systemically expressed CagA, were treated with a colitis inducer, dextran sulfate sodium (DSS). The DSS-treated WT cagAE-Tg mice showed a higher incidence of colonic dysplasia than did non-DSS-treated WT cagAE-Tg mice and DSS-treated control mice, suggesting that colonic inflammation accelerated the pro-oncogenic potential of CagA in this experimental model in the colon.166
Induction of proinflammatory host responses by cagA-Positive H. pylori
Clinico-pathological observations strongly indicated proinflammatory actions of the CagA protein in gastric pathogenesis.167 Animal infection studies showed that cagA-positive H. pylori strains elicited a greater severity of mucosal inflammation than cagA-negative strains.137,156 DSS-treated WT cagAE-Tg mice, which systemically expressed CagA, also exhibited deterioration of DSS-induced colitis compared to DSS-treated wild-type control mice.166 Immunoblot analysis of the colon revealed a substantial decrease in the level of I��B (inhibitor of NF-��B), which prevented nuclear translocation of NF-��B, in a CagA/PAR1b-dependent manner.166 Although the CagA-mediated I��B reduction per se was insufficient to activate NF-��B signaling, a primary mediator of the inflammatory response, it made cells hypersensitive to stimuli that activate NF-��B.166,168 It was reported that CM motif-dependent NF-��B activation by CagA is also induced by activation of IKK (I��B kinase), which promotes I��B degradation, through the CagA-c-Met-PI3K-AKT axis.114 IKK activation by CagA was also reported to be mediated by the activation of TGF-��-activated kinase 1(TAK1). CagA physically interacts with TNF receptor-associated factor 6 (TRAF6)-TAK1 complex and promotes aberrant poly-ubiquitination/activation of TAK1, leading to activation of IKK.169 Independent of NF-��B signaling, CagA induces the proinflammatory cytokine interleukin-1�� (IL-1��), which is cleaved/matured by the inflammasome complex in epithelial cells166 (Fig. 2). It has also been reported that CagA gives rise to the induction of the pro-inflammatory cytokine interleukin-8 (IL-8), probably due to EPIpYA-B-dependent PI3K activation, which may be influenced by polymorphisms at the EPIYA-B site.148 In addition, CagA is reported to activate STAT3 (signal transducer and activator of transcription 3), which is a proinflammatory transcription factor, via the IL-6/gp130 signaling pathway by a mechanism independent of the tyrosine phosphorylation of CagA170 (Fig. 2).
H. pylori-evoked proinflammatory responses of host cells are caused not only by CagA but also by other factors of the bacterium. It has been reported that bacterial peptidoglycan delivered through the T4SS from H. pylori stimulates Nod1 (nucleotide-binding oligomerization domain 1), a cytoplasmic pathogen recognition receptor, in epithelial cells and activates NF-��B, which induces inflammatory cytokines including IL-1, IL-6, IL-8, TNF�� (tumor necrosis factor ��) and/or IFN-�� (interferon-��).171,172 The involvement of H. pylori lipopolysaccharide (LPS) in NF-��B activation in gastric epithelial cells has been reported; H. pylori LPS itself may stimulate NF-��B through TLR2 (toll-like receptor 2) or TLR4-mediated signaling.173 However, recent studies have revealed that T4SS-delivered ADP-glycero-��-D-manno-heptose (ADP heptose), an LPS metabolite that is a novel pathogen-associated molecular pattern (PAMP) of gram-negative bacteria, including H. pylori, plays a central role in NF-��B activation by H. pylori via ALPK1 (��-kinase 1)-TIFA (TRAF-interacting protein with forkhead-associated domain) signaling.174,175,176 In addition to the H. pylori-epithelial cell interaction, H. pylori also infects immune cells in the gastric epithelial niche and thereby activates the NLRP3 inflammasome to induce IL-1�� in a cag PAI-dependent manner.177,178,179 It is thus thought that cagA-positive H. pylori induces a host inflammatory response, which enhances the oncogenic potential of CagA, by perturbing immune signaling in both gastric epithelial cells and infiltrating immune cells.��
Gastric carcinogenesis evoked by the H. Pylori CagA oncoprotein
It is generally thought that normal cells evolve progressively to a neoplastic state by successively acquiring the following ten hallmark capabilities: sustained proliferative signaling, evasion of growth suppressors, activation of invasion/metastasis, replicative immortality, induction of angiogenesis, resistance to cell death, genome instability/mutability, induction of tumor-promoting inflammation, evasion of immune destruction, and deregulation of cellular energetics.135 Based on this concept, the multistep process of human tumor pathogenesis can be accounted for by the stepwise acquisition of these characteristics, which subsequently enable normal cells to become tumorigenic and malignant.135 cagA-positive H. pylori-associated gastric carcinogenesis is also thought to progress in this stepwise fashion.2,3 In vitro and in vivo analyses of the CagA protein thus far have collectively characterized CagA as a bona fide oncoprotein that confers six of the ten cancer hallmark capabilities to host cells. Five of these hallmarks are induced cell-autonomously: (1) sustained proliferative signaling (SHP2-ERK and Wnt signaling), (2) evasion of growth suppressors (inactivation of RUNX3 and p53), (3) activation of invasion and metastasis (hummingbird phenotype, EMT-like morphological changes, loss of epithelial cell polarity, and junctional defects), (4) resistance to cell death (p53 inactivation and MCL1 induction), and (5) genome instability and mutability (AID induction and mitotic defects). The remaining hallmark, (6) induction of tumor-promoting inflammation (NF-��B/inflammasome activation), is induced non-cell-autonomously (Fig. 2) (Table 1).
Table 1 Pro-oncogenic effects of cagA-positive H. pylori infection.
Full size table
Spontaneous development of gastrointestinal neoplasms in cagA-Tg mice in the absence of infiltrating immune cells indicates that prolonged CagA expression is sufficient for carcinogenesis in this particular transgenic system.157 This suggests that inflammatory responses are not indispensable for CagA-induced gastrointestinal carcinogenesis, which may depend on mutual feedforward augmentation of the five malfunctional biological processes in CagA-expressing cells. This notion is consistent with the observations in claudin18-null (Cldn18−/−) mice, a mouse model of chronic atrophic gastritis that develops neither dysplasia nor carcinoma in the stomach.180 However, along with the well-recognized notion that chronic inflammation fosters carcinogenesis, inflammation indeed strengthens the oncogenic potential of the CagA protein.165,166 Although it remains unclear to what extent CagA per se is involved in H. pylori-induced gastritis, the oncogenic actions of CagA and cagA-positive H. pylori-induced inflammation reinforce each other in the progression of gastric carcinogenesis (Fig. 4).
Fig. 4: Schematic representation of H. pylori CagA-directed ��hit-and-run�� gastric carcinogenesis.
When delivered into gastric epithelial cells, the H. pylori-derived CagA oncoprotein perturbs multiple intracellular signaling pathways, which promotes malignant transformation of the host cells. There is a mutual feedforward stimulatory mechanism between the oncogenic activities of CagA and the pro-inflammatory responses against cagA-positive H. pylori infection. The direct priming of pro-oncogenic signaling by CagA in the precancerous stage promotes the acquisition of genetic and epigenetic alterations that can compensate for the perturbed cell signaling by CagA. Therefore, the established gastric cancer cells no longer require CagA protein to maintain their malignant phenotypes.
When delivered into gastric epithelial cells, the H. pylori-derived CagA oncoprotein perturbs multiple intracellular signaling pathways, which promotes malignant transformation of the host cells. There is a mutual feedforward stimulatory mechanism between the oncogenic activities of CagA and the pro-inflammatory responses against cagA-positive H. pylori infection. The direct priming of pro-oncogenic signaling by CagA in the precancerous stage promotes the acquisition of genetic and epigenetic alterations that can compensate for the perturbed cell signaling by CagA. Therefore, the established gastric cancer cells no longer require CagA protein to maintain their malignant phenotypes.
Full size image
Inconsistent with the well-recognized pivotal roles of H. pylori infection in gastric carcinogenesis, established gastric cancer cells maintain their transformed phenotypes in the absence of H. pylori colonization. Therefore, during the process of multistep gastric carcinogenesis, CagA-injected gastric epithelial cells must acquire genomic and epigenomic alterations that can compensate for the pro-oncogenic functions of the CagA protein. This ��hit-and-run�� hypothetical process of cagA-positive H. pylori-induced gastric carcinogenesis is thought to be achieved by the induction of genomic instability due to the ectopic expression of AID and/or the mitotic defects caused by CagA. The CagA-induced genetic mutability, which occurs concomitantly with the acquisition of resistance to cell death, enables the cells to accumulate genetic alterations. Aberrantly regulated cell cycle progression occurring via the tyrosine-phosphorylated CagA-SHP2-ERK axis, which is influenced by CagA polymorphisms, may accelerate the process of AID-dependent and/or mitosis-dependent acquisition of genetic alterations181 (Fig. 4). Genetic and epigenetic alterations frequently found in gastric cancers, including amplification of genes involved in RAS-ERK signaling (KRAS, FGFR2, EGFR, ERBB2, and MET),182 gain of function of the CTNNB1 gene (encoding ��-catenin),183 and loss of function of the TP53 gene (encoding p53) and the CDH1 gene (encoding E-cadherin),184,185,186,187 may play key roles in the functional compensation of CagA-directed cancer hallmarks.
An in vivo lineage tracing study in a mouse stomach indicated that the adult pyloric epithelium self-renews within two weeks.188 Therefore, to contribute to the initiation and progression of the multistep ��hit-and-run�� carcinogenesis in the gastric epithelium, H. pylori must adhere to and subsequently deliver CagA into long-lived stem/progenitor cells. Regarding this issue, direct contact of H. pylori with the stem/progenitor cell population, which is defined as either Lgr5+/Axin2+ cells or Lgr5−/Axin2+ cells, has been shown in human/mouse gastric glands. Furthermore, H. pylori infection of stem/progenitor cells causes glandular hyperplasia associated with an expansion of the cells with Lgr5 expression and/or Axin2 expression upon the function of T4SS, which delivers CagA into the cells.189,190 Additionally, since both Lgr5 and Axin2 are known to be specific target genes of Wnt signaling, which plays an indispensable role in the maintenance and self-renewal of epithelial stem cells, deregulation of Wnt signaling by CagA may promote the expansion of stem/progenitor cells, which may accelerate the progression of ��hit-and-run�� gastric carcinogenesis.Conclusions and future perspectives
H. pylori CagA is the first identified bacterial oncoprotein that hijacks multiple cellular processes associated with carcinogenesis. The functional interplay that mutually occurs and accelerates the CagA-induced cell-autonomous pro-oncogenic actions is thought to be fundamentally required for the generation of the ��hit-and-run�� circuit that drives the acquisition of genetic alterations for multistep carcinogenesis. Additionally, mutual reinforcement between the pro-oncogenic actions of CagA and H. pylori-induced inflammation adds another layer of oncogenic interplay by further accelerating the acquisition of genetic alterations that eventually generate cells with the cancer hallmark phenotypes in a CagA-independent manner (Fig. 4). In several previous experiments, oncogenic stress induced by CagA provoked premature cell senescence [oncogene-induced senescence (OIS)] through aberrant induction of the cell cycle inhibitor p21.112,123,191 Therefore, the acquisition of genetic alterations that can overcome the CagA-induced OIS may also be a key step in cagA-positive H. pylori-induced ��hit-and-run�� gastric carcinogenesis.
In addition to the interaction between H. pylori and host cells, a functional interplay between CagA and other virulent proteins of H. pylori [e.g., vacuolating cytotoxin A (VacA)] may further modulate the oncogenic actions of CagA.82,191 Moreover, since sustained tyrosine phosphorylation of CagA is induced via SHP1 suppression in EBV-coinfected gastric epithelial cells, colonized cagA-positive H. pylori may modulate and benefit from the composition of the gastric mucosal microbiota, suggesting a yet-to-be-known extra layer of interplay: mutual modulation between the oncogenic actions of CagA and the gastric mucosal microbiota.76,192,193 Although the biological relevance of bacteria other than H. pylori in the stomach needs to be further investigated, a functional interplay between cagA-positive H. pylori and the gastric microbiota, which includes EBV, may provide better insight into understanding the mechanism of CagA-induced ��hit-and-run�� gastric carcinogenesis.
H. pylori infection is crucial for gastric carcinogenesis, whereas H. pylori is no longer required in established gastric cancer. The concept of CagA-directed ��hit-and-run�� carcinogenesis provides insights for a better understanding of this paradoxical phenomenon by suggesting a dual oncogenic action of the CagA protein: priming of pro-oncogenic signaling pathways and promotion of genomic instability. Further investigation is needed to evaluate the relevance of the hypothetical concept.Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis | Cellular & Molecular Immunology
https://www.nature.com/articles/s41423-019-0339-5��
Shp2: Indoline Scaffold Protein Tyrosine ... - | Moffitt
https://moffitt.org/research-science/innovation/office-of-innovation/available...
Apr 09, 2007 �� Shp2 is a protein tyrosine phosphatase (PTPase) that mediates growth factor and cytokine signaling. It is also involved in cell transformation by oncogenic growth factor receptor kinases and by the oncogenic bacterium H. pylori. Mutations of Shp2 are associated with several types of leukemias as well as the developmental disorder Noonan Syndrome.
[PDF]Inhibitor of Shp2 Protein Tyrosine Phosphatase
labpages2.moffitt.org/media/3149/06a038-inhibitors-of-shp2-tomv1.pdf
Opportunity Summary: Shp2 is a protein tyrosine phosphatase (PTPase) that mediates growth factor and cytokine signaling. Aberrant Shp2 expression is associated with several types of leukemias and developmental disorders. Technology Abstract • Shp2 is a protein tyrosine phosphatase (PTPase) that mediates growth factor and cytokine signaling.
SHP2 regulates proliferation and tumorigenicity of glioma ...
https://www.ncbi.nlm.nih.gov/pubmed/28852935
SHP2 is a cytoplasmic protein tyrosine phosphatase (PTPase) involved in multiple signaling pathways and was the first identified proto-oncogene PTPase. Previous work in glioblastoma (GBM) has demonstrated the role of SHP2 PTPase activity in modulating the oncogenic phenotype of ��
Cited by: 4
Publish Year: 2017
Author: Laura Roccograndi, Zev A. Binder, Logan Zhang, Nicola Aceto, Zhuo Zhang, Mohamed Bentires-Alj, Ichir...
The phosphotyrosine phosphatase SHP2 is a critical ...
https://www.nature.com/articles/1206798
Oct 09, 2003 �� The Src homology 2 domain-containing phosphotyrosine phosphatase (SHP2) is a critical mediator of RTK signaling, but its role in oncogenic RTK-induced cell ��
Cited by: 110
Publish Year: 2003
Auth��
Scand J Gastroenterol. 2006 Sep;41(9):1013-8.
Gastric expression of inducible nitric oxide synthase and myeloperoxidase in relation to nitrotyrosine in Helicobacter pylori-infected Mongolian gerbils.
Elfvin A1, Bölin I, Lönroth H, Fändriks L.
Author information
1
Department of Gastrosurgical Research, Göteborg Vaccine Research Institute (GUVAX), Sahlgrenska Academy, Göteborg University, Gothenburg, Sweden. anders.elfvin@vgregion.se
Abstract
OBJECTIVE:
For obscure reasons Helicobacter pylori infection of the gastric mucosa is maintained despite a pronounced host defence response. The present study elucidates possible H. pylori-related interference in the oxy- and nitro-radical formation pathways.
MATERIAL AND METHODS:
Male Mongolian gerbils were infected with two different H. pylori strains, TN2GF4 and SS1. At 3, 6, 12 or 18 months after inoculation, gastric expressions of myeloperoxidase (MPO), inducible nitric oxide synthase (iNOS) and nitrotyrosine were assessed by Western blotting.
RESULTS:
Expression of both iNOS and MPO was markedly up-regulated in the H. pylori-infected animals compared with non-infected controls. The TN2GF4-infected animals initially (at 3 and 6 months) demonstrated pronounced expression of both iNOS and MPO. The SSI-infected animals exhibited a slower onset with significantly increased iNOS after 12 and 18 months. Nitrotyrosine expression was slightly elevated in the infected groups at 3 and 6 months compared with that in the controls. Nitrotyrosine levels then decreased and were no longer significantly different from those of controls (TN2GF4-infected animals) or were lower (SS1-infected animals) than in the controls.
CONCLUSIONS:
The results indicate that peroxynitrite formation as reflected by nitrotyrosine expression is low or even inhibited in chronic H. pylori infection despite pronounced expression of enzymes representing both the oxy- and nitro-radical formation pathways. The results support the theory that H. pylori survival is related to functional inhibition of mucosal enzymatic NO and/or oxy-radical formation.Gastric expression of inducible nitric oxide synthase and myeloperoxidase in relation to nitrotyrosine in Helicobacter pylori-infected Mongolian ge... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/16938713��
The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts
Weronika Gonciarz, Agnieszka Krupa, Krzysztof Hinc, Michał Obuchowski, Anthony P Moran †, Adrian Gajewski, Magdalena Chmiela
Published: August 7, 2019
Abstract
Background
Helicobacter pylori colonizes the human gastric mucosa, causing chronic inflammation, peptic ulcers and gastric cancer. A cascade of harmful processes results from the interaction of these bacteria with the gastric epithelium.
Aim
To investigate these processes in terms of upregulation of oxidative stress and cell apoptosis and downregulation of the pro-regenerative activity of cells.
Methods
We employed an in vivo guinea pig model at 7 or 28 days postinoculation with H. pylori, corresponding to an acute or chronic stage of infection, respectively, and an in vitro model of guinea pig primary gastric epithelial cells and fibroblasts treated with bacterial components: glycine acid extract (GE), urease subunit A (UreA), cytotoxin-associated gene A protein (CagA) and lipopolysaccharide (LPS). Cells were evaluated for metabolic activity (MTT reduction), myeloperoxidase (MPO) and metalloproteinase (MMP-9) secretion, lipid peroxidation (4-hydroxynonenal (4HNE)), migration (wound healing), proliferation (Ki-67 antigen) and cell apoptosis (TUNEL assay; Bcl-xL, Bax, Bcl-2 expression; caspase 3 cleavage).
Results
Significant infiltration of the gastric mucosa by inflammatory cells in vivo in response to H. pylori was accompanied by oxidative stress and cell apoptosis, which were more intense 7 than 28 days after inoculation. The increase in cell proliferation was more intense in chronic than acute infection. H. pylori components GE, CagA, UreA, and LPS upregulated oxidative stress and apoptosis. Only H. pylori LPS inhibited cell migration and proliferation, which was accompanied by the upregulation of MMP-9.
Conclusions
H. pylori infection induces cell apoptosis in conjunction with increased oxidative stress. Elevated apoptosis protects against deleterious inflammation and neoplasia; however, it reduces cell integrity. Upregulation of cell migration and proliferation in response to injury in the milieu of GE, CagA or UreA facilitates tissue regeneration but increases the risk of neoplasia. By comparison, downregulation of cell regeneration by H. pylori LPS may promote chronic inflammation.The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts
https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0220636��
Clin Exp Immunol. 2010 Sep; 161(3): 551�C559.
Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection
Y-Y Wu,*† C-W Lin,* K-S Cheng,‡ C Lin,†�� Y-M Wang,* I-T Lin,* Y-H Chou,* and P-N Hsu¶††
Author information Article notes Copyright and License information
*Department of Medical Laboratory Science and Biotechnology, China Medical University and Hospital, Taichung, Taiwan
ABSTRACT
B7-H1 [programmed death-ligand-1 (PD-L1)] is a B7-family member that binds to programmed death-1 (PD-1). Recently, deficiency of PD-L1 has been demonstrated to result in accelerated gastric epithelial cell damage in gastritis, and PD-L1 is suggested to play a critical role in regulating T cell homeostasis. Here, we aimed to gain more insight into gastric PD-L1 expression, regulation and function during Helicobacter pylori infection. PD-L1 expression in human gastric epithelial cells was analysed using Western blotting, quantitative polymerase chain reaction and fluorescence activated cell sorter analysis. Furthermore, co-culture experiments of human gastric epithelial cells with primary human T cells or Jurkat T cells were conducted. PD-L1 expression in primary human gastric epithelial cells was strongly enhanced by H. pylori infection and activated T cells, and augmented markedly by further stimulation with interferon-�� or tumour necrosis factor-��. Moreover, PD-L1 expression in gastric epithelial cells significantly induced apoptosis of T cells. Our results indicate that a novel bidirectional interaction between human gastric epithelial cells and lymphocytes modulates PD-L1 expression in human gastric epithelial cells, contributing to the unique immunological properties of the stomach.
Fig. 2
Programmed death-ligand-1 (PD-L1) expression of human gastric epithelial cells after Helicobacter pylori infection. (a) Human gastric adenocarcinoma (AGS) cells were infected with H. pylori and RNA was isolated 24 and 48 h after infection. Non-infected AGS cells served as controls. (b) Primary human gastric epithelial cells were infected with H. pylori and RNA was isolated 24 and 48 h after infection. Non-infected primary human gastric epithelial cells served as controls. PD-L1 mRNA expression was quantified in relation to ��-actin expression by real-time polymerase chain reaction. *P < 0��05 compared to control.Expression of PD-L1 in gastric epithelial cell-mediated T cell apoptosis
It has been demonstrated that PD-L1 expressed by tumour cells is able to induce apoptosis in T cells. With regard to these observations, we wanted to evaluate whether T cell apoptosis might be regulated by PD-L1 expressed in gastric epithelial cells. Therefore, AGS cells were cultured in the presence or absence of IFN-�� or TNF-�� and blocking antibodies to PD-L1, respectively. Unbound antibody was removed prior to establishment of co-cultures with Jurkat T cells to ensure that the effect of the antibodies was due to blockade of PD-L1 on the gastric epithelial cells and not on T cells, as PD-L1 can be expressed on activated human T cells [23]. After 8 h of co-culture, apoptosis in Jurkat T cells was examined by FACS analysis. Numbers of apoptotic Jurkat T cells were increased significantly in co-cultures with AGS cells activated by IFN-�� or TNF-�� compared to co-cultures with untreated control cells (2��39 �� 0��9-fold, P = 0��013 and 2��05 �� 0��7-fold, P = 0��006, respectively). In the presence of blocking antibodies to PD-L1, apoptosis in Jurkat T cells was reduced significantly in co-cultures with IFN-��- and TNF-��-activated AGS cells (0��59 �� 0��2, P = 0��0003 and 0��65 �� 0��1, P = 0��004, respectively), demonstrating that PD-L1 expression on the surface of gastric epithelial cells mediates T cell apoptosis. A representative FACS result is depicted in Fig. 6. Similar results were obtained in co-culture experiments with primary human gastric epithelial cells and T cells, respectively (data not shown).DISCUSSION
We have demonstrated for the first time that primary human gastric epithelial cells express constitutively low levels of PD-L1, while its expression is enhanced strongly by activated T cells and H. pylori infection, and augmented markedly by further stimulation with IFN-�� or TNF-��. Moreover, PD-L1 expression on primary human gastric epithelial cells induces apoptosis in T cells.
H. pylori infection is one of the most common infections of humans because 50% of the global population carries this bacterium in the stomach, and in some individuals infection leads to serious diseases. Despite the inflammatory and immune responses that are elicited by H. pylori infection, it remains chronic. Various studies have suggested that T cell functions are affected during infection with H. pylori. Early studies to assess T cell function in response to H. pylori suggested that T cells in infected individuals were hyporesponsive compared with non-infected individuals. Gastric T cells from infected individuals were suppressed in their proliferation and IFN-�� production [24]. The mechanisms responsible for the low response were not clear.
Basic expression but even more inducible induction of PD-L1 on gastric epithelial cells may have several implications. IFN-�� is a key proinflammatory molecule released by activated T cells, up-regulating MHC expression and antigen presentation pathways. However, IFN-�� can also dampen the extent of T cell activation and limit T cell-mediated tissue damage [17,25�C27]. The observed profound up-regulation of PD-L1 expression on gastric epithelial cells may serve as a part of a tissue protective negative feedback mechanism in T cell-mediated inflammatory events. PD-L1 delivers an inhibitory signal to T cell activity; our finding of induced PD-L1 expression on human gastric epithelial cells in response to TNF-�� is of particular interest. One might speculate that TNF-�� therapy affects both anti-bacterial but, via PD-L1, also immunosuppressive and anti-inflammatory mechanisms, eventually favouring bacterial persistence. We would like to hypothesize that TNF-��-induced PD-L1 on human gastric epithelial cells accounts in part for some of these reported gastric protective effects.
Interestingly, we observed increased levels of PD-L1 in human primary gastric epithelial cells after H. pylori infection. Similarly, previous studies found an up-regulation of PD-L1 expression in gastric epithelial cell lines in response to H. pylori[28] and in lymphocytes in response to H. pylori[29]. Taking into account that PD-L1 may have receptors other than PD-1 on activated T cells [30�C32], we used anti-PD-L1 mAb instead of anti-PD-1 mAb or fusion protein and could partially reverse the proapoptotic effect of human gastric epithelial cells on lymphocytes. At this time, however, we do not know whether this is mediated through ligation of PD-1. Conversely, it may be speculated that the observed PD-L1 up-regulation in human gastric epithelial cells by activated lymphocytes is mediated exclusively via substantial IFN-�� or TNF-�� expression compared to resting cells. In any case, PD-L1 up-regulation by activated lymphocytes has been observed in activated hepatic stellate cells [32]. Furthermore, IFN-�� has been shown to up-regulate PD-L1 expression in dermal fibroblasts [33]. Interestingly, also in vivo substantially increased expression of B7-H1 on keratinocytes was found close to numerous T cell infiltrates in the subepithelium of patients with chronic inflammatory mucocutaneous disease [12]. In addition, in Transwell experiments, the level of PD-L1 expression was even higher in the Transwell cultures, indicating that perhaps cell�Ccell contact reduced the soluble factors released by activated T cells.
Our results suggest a bidirectional interaction between human gastric epithelial cells and lymphocytes. Primary human gastric epithelial cells isolated from human stomach express low levels of PD-L1. Exposure to IFN-�� or TNF-��, an inflammatory cytokine released by activated T cells, results in up-regulation of PD-L1 on gastric epithelial cells that in turn induces apoptosis in lymphocytes. Herewith, gastric epithelial cells may contribute to gastric tolerance induction by deletion of activated T cells through induction of apoptosis. Currently, the cellular mechanism by which, in certain in vivo settings, PD-L1 co-stimulates T cell responses, and in other circumstances these responses are inhibited, remains elusive. Our study may provide a first step for the elucidation of this mechanism in the stomach, an organ with unique immunological properties.
Keywords: apoptosis, B7-H1, gastric epithelial cell, Helicobacter pylori, PD-1, PD-L1Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2962974/��
��
Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection
1 Department of Molecular Genetics, Biochemistry, and Microbiology, Cincinnati OH, United States of America
2 Department of Pharmacology and Systems Physiology, University of Cincinnati, Cincinnati OH, United States of America
3 Division of Developmental Biology, Cincinnati Children��s Hospital Medical Center, Cincinnati OH, United States of America
4 Center for Stem Cell and Organoid Medicine, Cincinnati Children��s Hospital Medical Center, Cincinnati OH, United States of America
5 Department of Biomedical Engineering, University of Cincinnati, Cincinnati OH, United States of America
6 Department of Pediatric Surgery, Cincinnati Children��s Hospital Medical Center, Cincinnati OH, United States of America
7 Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States of America
8 Department of Pathology and Lab Medicine, University of Cincinnati College of Medicine, Cincinnati OH, United States of America
University of Illinois, UNITED STATESAbstract
Helicobacter pylori (H. pylori) is the major risk factor for the development of gastric cancer. Our laboratory has reported that the Sonic Hedgehog (Shh) signaling pathway is an early response to infection that is fundamental to the initiation of H. pylori-induced gastritis. H. pylori also induces programmed death ligand 1 (PD-L1) expression on gastric epithelial cells, yet the mechanism is unknown. We hypothesize that H. pylori-induced PD-L1 expression within the gastric epithelium is mediated by the Shh signaling pathway during infection. To identify the role of Shh signaling as a mediator of H. pylori-induced PD-L1 expression, human gastric organoids generated from either induced pluripotent stem cells (HGOs) or tissue (huFGOs) were microinjected with bacteria and treated with Hedgehog/Gli inhibitor GANT61. Gastric epithelial monolayers generated from the huFGOs were also infected with H. pylori and treated with GANT61 to study the role of Hedgehog signaling as a mediator of induced PD-1 expression. A patient-derived organoid/autologous immune cell co-culture system infected with H. pylori and treated with PD-1 inhibitor (PD-1Inh) was developed to study the protective mechanism of PD-L1 in response to bacterial infection. H. pylori significantly increased PD-L1 expression in organoid cultures 48 hours post-infection when compared to uninfected controls. The mechanism was cytotoxic associated gene A (CagA) dependent. This response was blocked by pretreatment with GANT61. Anti-PD-L1 treatment of H. pylori infected huFGOs, co-cultured with autologous patient cytotoxic T lymphocytes and dendritic cells, induced organoid death. H. pylori-induced PD-L1 expression is mediated by the Shh signaling pathway within the gastric epithelium. Cells infected with H. pylori that express PD-L1 may be protected from the immune response, creating premalignant lesions progressing to gastric cancer.
Author summary
Gastric cancer is the 5th most common cancer worldwide and the 3rd most common cause of cancer-related death. Helicobacter pylori (H. pylori) infection is the major risk factor for the development of gastric cancer. Our laboratory has reported that the Sonic Hedgehog (Shh) signaling pathway is an early response of the gastric epithelium to infection and fundamental to the initiation of H. pylori-induced gastritis. H. pylori also induces programmed death ligand 1 (PD-L1) expression on gastric epithelial cells, yet the mechanism is unknown. PD-L1 is a protective ligand that is known to suppress the immune system by shutting down T cell effector function. We hypothesized that H. pylori-induced PD-L1 expression within the gastric epithelium is mediated by the Shh signaling pathway during infection. Moreover, we showed that metaplastic cells may survive chronic inflammation by expressing the immunosuppressive ligand PD-L1 for the persistence of infection and progression of disease to cancer.Introduction
Helicobacter pylori (H. pylori) infects nearly 50% of the world's population and is the number one risk factor for gastric cancer [1]. Albeit a controversial issue, it may be that although H. pylori infection treated with antibiotics is cleared, once a patient has progressed to a metaplastic phenotype, elimination of the bacteria does not reduce the risk of developing gastric cancer [2]. H. pylori induces pathogenesis by injecting one key virulence factor cytotoxic associated gene A (CagA) into the gastric epithelial cells [3]. Importantly, CagA stimulates a drastic increase in Sonic Hedgehog (Shh) signaling from parietal cells, a response that is mediated by NF��B signaling [4, 5]. Shh is a gastric morphogen known to initiate gastritis in response to H. pylori infection [4]. Upon infection H. pylori induces the secretion of Shh from the acid-secreting parietal cells [4]. Following a sustained increase in Shh secretion and signaling, macrophages are recruited to the infection site [4]. These macrophages secrete IL-1�� which inhibits acid secretion causing atrophic gastritis and the atrophy of parietal cells [4, 6]. Overall, Shh signaling plays a fundamental role in the initiation of H. pylori-induced gastritis [4, 5]. It has also been observed that following H. pylori infection programmed death ligand 1 (PD-L1) expression on the gastric epithelium is drastically increased [7]. The expression of PD-L1 in human gastric biopsies of infected patients has never been investigated. PD-L1 interacts with programmed death 1 (PD1) on the surface of cytotoxic T lymphocytes (CTLs) rendering CTLs unable to induce apoptosis [8, 9]. Thus, PD-L1 signaling induces cellular proliferation and survival [10, 11].
H. pylori infection combined with the atrophy of the acid secreting parietal cells leads to the development of spasmolytic polypeptide/Trefoil Factor (TFF) 2-expressing metaplasia (SPEM) [12, 13]. SPEM is the first step in a series of neoplastic changes that occur in the gastric epithelium prior to the development of gastric cancer [14, 15]. In the setting of chronic inflammation and persistent bacterial infection there is the progression of SPEM to intestinal metaplasia and gastric cancer [15]. PD-L1 is a protective ligand that is known to suppress the immune system by shutting down T cell effector function [8, 9]. Here we demonstrate that H. pylori-induced PD-L1 expression is mediated by Shh signaling as an early epithelial response to infection and a mechanism by which the bacteria evades the immune response. We also demonstrate here that SPEM cells may survive chronic inflammation by expressing the immunosuppressive ligand PD-L1 for the persistence of infection and progression of disease to cancer.
Fig 13 Proposed model of H. pylori induced PD-L1 expression in the gastric epithelium. (A) Shh secretion is induced from the acid secreting parietal cells in response to H. pylori infection that is driven by a CagA dependent mechanism. (B) We propose that Shh induces the expression of PD-L1 on GSII/PgA transdifferentiated cells. PD-L1 then interacts with PD-1 on the surface of CTLs and shuts down the CTL effector function which may lead to the survival of these transdifferentiated metaplastic cells. (C) The addition of PD-1Inh blocks the interaction between PD-1 on CTLs and PD-L1 on transdifferentiated/SPEM cells allowing the CTLs to destroy these cells.Discussion
In the current study we show that PD-L1 is induced following initial H. pylori infection. Previous studies using primary gastric epithelial cells collected from biopsies of patients diagnosed with dyspepsia and gastric cancer cell lines showed an increase in PD-L1 expression following H. pylori infection [7, 27]. The novelty of this study is the use of gastric organoids derived from patients. To the best of our knowledge we are the first to show induction of PD-L1 expression in human tissue and human derived-organoid models as an early response to bacterial infection. Importantly, induction of PD-L1 expression in gastric organoids and epithelial monolayers was not observed in response to infection with G27 H. pylori strain bearing a CagA deletion (��CagA). The involvement of CagA in H. pylori-induced PD-L1 expression is significant because CagA is associated with an increased risk of developing gastric cancer [3, 28]. Our data suggests a role of PD-L1 as a potential mechanism by which virulent strains of H. pylori allow for the persistence of infected gastric epithelial cells.
Shh signaling mediates H. pylori-induced PD-L1 expression. Consistent with our findings, we have shown that H. pylori infection induces an increase in Shh secretion and signaling via a CagA dependent pathway [5]. We further demonstrate that canonical Shh downstream effectors were drastically increased specifically in the fundus/corpus of the stomach following H. pylori infection. Shh signaling in the epithelium of infected antral differentiated organoids was not observed. An explanation for this observation is data demonstrating that Shh is secreted from the acid-secreting parietal cells within the fundic region of the stomach [29, 30]. Indeed by Acridine Orange accumulation we demonstrate here functional acid-secreting parietal cells with FHGOs, huFGOs and gastric epithelial monolayers. In the presence of GANT 61, a Gli/Hedgehog signaling inhibitor, and vismodegib, that targets the Hedgehog signaling pathway by blocking Ptch and SMO, the expression of canonical SHH downstream effectors decreased. Interestingly, PD-L1 and Shh expression robustly increase in fundic organoids following infection with H. pylori, a response that was ablated with GANT 61 and vismodegib treatment. Parietal cells in this region secrete Shh which subsequently induces the secretion of acid, normal epithelial cell function and regeneration [29, 31�C33]. We advance our initial studies by demonstrating that the release of Shh within the corpus mediates the early induction of PD-L1 expression in response to bacterial infection. In support of our findings, it has been documented that Mycobacteria-responsive Shh signaling within human dendritic cells also mediates PD-L1 expression [34].
In biopsies collected from H. pylori infected patients, PD-L1 expression co-localized with proteins that classically mark SPEM cells including TFF2 and CD44v9 [16, 17]. Different regions of the stomach respond differently to early transforming factors. For example, individuals most at risk of developing gastric cancer are those in whom the bacteria colonize the corpus (or fundus) of the stomach, when acid secretion is impaired. In contrast, bacterial colonization of the antrum is associated with low levels of inflammation in the corpus, high acid secretion and the development of duodenal ulcer disease [35�C37]. Differences in the regional response to H. pylori infection is evident from our studies. The use of human PSC-derived antral and fundic gastric organoids has allowed us to identify how these unique regions of the human stomach differentially respond to H. pylori infection.
To identify whether PD-L1 expression protects the epithelium from chronic inflammation, we developed an organoid/autologous immune cell co-culture system. Organoids infected with H. pylori highly expressed PD-L1 and suppressed CTL proliferation. CTLs are the main pro-apoptotic cell within the gastric cancer microenvironment [38]. When H. pylori infected organoids were co-cultured with CTLs and treated with a PD-1 inhibitor (PD-1Inh) there was an increase in proliferating CTLs and a decrease in live PD-L1 expressing gastric epithelial cells. Therefore, this suggests that PD-L1 expression was protective to the infected cells. These results are significant because once a patient progresses to a metaplastic state, the eradication of H. pylori does not decrease the risk of developing gastric cancer [2].
PD-L1 expression lasts through to the development of gastric cancer. Up to 69% of all gastric cancers express PD-L1 [39]. Here we present an organoid/immune cell co-culture to model infection with H. pylori and treatment with immune checkpoint inhibitors. From this study, we proposed that PD-L1, that is induced by parietal cell-derived Shh, may be protective to SPEM cells in the presence of bacterial infection (Fig 13A and 13B). When the interaction between PD-1 and PD-L1 is inhibited, activated CTLs may target the SPEM glands (Fig 13C). These models could be used to devise a therapy for patients that have progressed to a metaplastic state and would therefore not benefit from eradication of H. pylori. In addition, this co-culture system could possibly be used to discover new therapies for gastric cancer.Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6380601/��
Sonic hedgehog is one of three proteins in the mammalian signaling pathway family called hedgehog, the others being desert hedgehog (DHH) and Indian hedgehog (IHH). SHH is the best studied ligand of the hedgehog signaling pathway. It plays a key role in regulating vertebrate organogenesis,...
Sonic hedgehog - Wikipedia
en.wikipedia.org/wiki/Sonic_Hedgehog_Pathway
Sonic hedgehog signalling pathway: a complex network
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4117150
Sonic Hedgehog (Shh) signalling cascade is one of the intricate signal transduction mechanisms that govern the precisely regulated developmental processes of multicellular organisms. Along with establishing the patterns of cellular differentiation to direct complex organ formation, it also has an important role in post-embryonic tissue regeneration and repair processes.
Cited by: 100
Publish Year: 2014
Author: Zia Choudhry, Azadeh A. Rikani, Adnan Maqsood Choudhry, Sadaf Tariq, Fozia Zakaria, Muhammad Waheed ...
A highlight on Sonic hedgehog pathway | Cell Communication ...
https://biosignaling.biomedcentral.com/articles/10.1186/s12964-018-0220-7
Mar 20, 2018 �� Hedgehog (Hh) signaling pathway plays an essential role during vertebrate embryonic development and tumorigenesis. It is already known that Sonic hedgehog (Shh) pathway is important for the evolution of radio and chemo-resistance of several types of tumors.
Cited by: 21
Publish Year: 2018
Author: Gabriela Basile Carballo, J��ssica Ribeiro Honorato, Giselle Pinto Farias de Lopes, Tania Cristina Le...
Targeting the Sonic Hedgehog Signaling Pathway: Review of ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4773745
Feb 15, 2016 �� The sonic hedgehog (Shh) signaling pathway is a major regulator of cell differentiation, cell proliferation, and tissue polarity. Aberrant activation of the Shh pathway has been shown in a variety of human cancers, including, basal cell carcinoma, malignant gliomas, medulloblastoma, leukemias, and cancers of the breast, lung, pancreas, and prostate.
Cited by: 279
Publish Year: 2016
Author: Tadas K. Rimkus, Richard L. Carpenter, Shadi Qasem��
Cancers (Basel). 2016 Feb; 8(2): 22.
Published online 2016 Feb 15.
Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors
Tadas K. Rimkus,1 Richard L. Carpenter,1 Shadi Qasem,2,3 Michael Chan,3,4 and Hui-Wen Lo1,3,*
William Chi-shing Cho, Academic Editor
Author information Article notes Copyright and License information Disclaimer
1Department of Cancer Biology, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA; ude.htlaehekaw@SUKMIRT (T.K.R.); ude.htlaehekaw@tnepracr (R.L.C.)
2Department of Pathology, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA; ude.htlaehekaw@mesaqs
3Comprehensive Cancer Center, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA; ude.htlaehekaw@nahcm
4Department of Radiation Oncology, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA
*Correspondence: ude.htlaehekaw@olh; Tel.: +1-336-716-0695; Fax: +1-336-716-0255
Abstract
The sonic hedgehog (Shh) signaling pathway is a major regulator of cell differentiation, cell proliferation, and tissue polarity. Aberrant activation of the Shh pathway has been shown in a variety of human cancers, including, basal cell carcinoma, malignant gliomas, medulloblastoma, leukemias, and cancers of the breast, lung, pancreas, and prostate. Tumorigenesis, tumor progression and therapeutic response have all been shown to be impacted by the Shh signaling pathway. Downstream effectors of the Shh pathway include smoothened (SMO) and glioma-associated oncogene homolog (GLI) family of zinc finger transcription factors. Both are regarded as important targets for cancer therapeutics. While most efforts have been devoted towards pharmacologically targeting SMO, developing GLI-targeted approach has its merit because of the fact that GLI proteins can be activated by both Shh ligand-dependent and -independent mechanisms. To date, two SMO inhibitors (LDE225/Sonidegib and GDC-0449/Vismodegib) have received FDA approval for treating basal cell carcinoma while many clinical trials are being conducted to evaluate the efficacy of this exciting class of targeted therapy in a variety of cancers. In this review, we provide an overview of the biology of the Shh pathway and then detail the current landscape of the Shh-SMO-GLI pathway inhibitors including those in preclinical studies and clinical trials.
Keywords: sonic hedgehog pathway, smoothened, GLI, tGLI1, inhibitors, PTCH, targeted therapy
1. Introduction
The Hedgehog (HH) gene was discovered in 1980 by Nusslein-Volhard and Wieschaus through genetic analysis of the fruit fly Drosophila melanogaster [1]. In the early 1990s, three HH gene homologs were discovered in vertebrates; Sonic Hedgehog (SHH), Indian Hedgehog (IHH), and Desert Hedgehog (DHH) [2,3,4]. DHH and IHH have been shown to play important roles in normal tissue development, including pancreas and testis organogenesis and bone formation [5,6,7,8]. Shh is the most potent of these ligands and is the most widely expressed in adult tissues [9,10]. Shh signaling plays an essential role in embryonic development and is critical for maintenance of tissue polarity. It has been shown that Shh is the dominant oncogenic HH ligand, as ectopic expression of Shh was sufficient to induce basal cell carcinoma in mice [11,12]. The Shh pathway is tightly regulated in most adult tissues but hyperactivation of this pathway is found in many solid tumors [13,14,15,16,17,18,19,20]. Aberrant Shh signaling has been implicated in many human cancers that account for up to 25% of human cancer deaths [21]. Greater understanding of the role of Shh signaling in human cancers has clearly indicated the need for development of anti-cancer therapies targeting the Shh pathway.
1.1. Shh Signaling Pathway Overview��
Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4773745/��
H. pylori CagL-Y58/E59 prime higher integrin ��5��1 in adverse pH condition to enhance hypochlorhydria vicious cycle for gastric carcinogenesis.Affiliation: Institute of Clinical Medicine, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan.
Abstract
Background/aims: H. pylori CagL amino acid polymorphisms such as Y58/E59 can increase integrin ��5��1 expression and gastric cancer risk. Hypochlorhydria during chronic H. pylori infection promotes gastric carcinogenesis. The study test whether CagL-Y58/E59 isolates may regulate integrin ��5��1 to translocate CagA via the type IV secretory system even under adverse pH conditions, and whether the integrin ��5��1 expression primed by H. pylori is a pH-dependent process involving hypochlorhydria in a vicious cycle to promote gastric carcinogenesis.
Methods: The expressions of integrin ��5 and ��1, CagA phosphorylation, IL-8, FAK, EGFR, and AKT activation of AGS cells exposed to CagL-Y58/E59 H. pylori, isogenic mutants, and different H. pylori CagL amino acid replacement mutants under different pH values were determined. Differences in the pepsinogen I/II ratio (indirectly indicating gastric acidity) and gastric integrin ��5��1 expression were compared among the 172 H. pylori-infected patients with different cancer risks.
Results: Even under adversely low pH condition, H. pylori CagL-Y58/E59 still keep active integrin ��1 with stronger binding affinity, CagA translocation, IL-8, FAK, EGFR, and AKT activation than the other mutants (p<0.05). The in vitro assay revealed higher priming of integrin ��5��1 by H. pylori under elevated pH as hypochlorhydria (p<0.05). In the H. pylori-infected patients, the gastric integrin ��5��1 expressions were higher in those with pepsinogen I/II ratio <6 than in those without (p<0.05).
Conclusions: H. pylori CagL-Y58/E59 prime higher integrin under adverse pH and may involve to enhance hypochlorhydria vicious cycle for gastric carcinogenesis, and thus require an early eradication.
Bottom Line: H. pylori CagL amino acid polymorphisms such as Y58/E59 can increase integrin ��5��1 expression and gastric cancer risk.Differences in the pepsinogen I/II ratio (indirectly indicating gastric acidity) and gastric integrin ��5��1 expression were compared among the 172 H. pylori-infected patients with different cancer risks.In the H. pylori-infected patients, the gastric integrin ��5��1 expressions were higher in those with pepsinogen I/II ratio <6 than in those without (p<0.05).
The schematic gastric carcinogenesis triggered by a vicious cycle within the CagL-integrin priming-intragastric pH elevation during chronic H. pylori infection.
(1) The H. pylori with CagL-Y58/E59 can prime and preserve more integrin ��5��1, even under adverse pH conditions in the stomach, to address efficient T4SS with more CagA translocation, IL-8 secretion to have more severe gastric mucosa destruction;
(2) Such damage of gastric mucosa is linked with loss of parietal cells to elevate the intragastric pH;
(3) the H. pylori CagL (especially the CagL-Y58/E59) contributes to hypochlorhydria via the dissociation of ADAM17 from integrin ��5��1 (Saha etal. 2008 [44], 2010 & this study);
(4) Under the increase of intragastric pH value or with a drop of pepsinogen I/II ratio <6, this study showed that the integrin ��5��1 expression can be triggered up, and thus result in a positive vicious cycle that makes efficient T4SS deliver more CagA translocation to contribute the gastric carcinogenesis.
��
The schematic gastric carcinogenesis triggered by a vic | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3757014_pone.0072735.g005&req=4��
Hypochlorhydria induced by a proton pump inhibitor leads to intragastric microbial production of acetaldehyde from ethanol
S. Väkeväinen J. Tillonen M. Salaspuro H. Jousimies�\Somer H. Nuutinen M. Färkkilä
First published: 24 December 2001 https://doi.org/10.1046/j.1365-2036.2000.00858.x Citations: 34
Professor M.Salaspuro Research Unit of Alcohol Diseases, Helsinki University Central Hospital, PL 345, Tukholmankatu 8 F, 00029 HUS, Finland. E�\mail: mikko.salaspuro@hus.fi
Abstract
Background:
Acetaldehyde, produced locally in the digestive tract, has recently been shown to be carcinogenic in humans.
Aim:
To examine the effect of iatrogenic hypochlorhydria on intragastric acetaldehyde production from ethanol after a moderate dose of alcohol, and to relate the findings to the changes in gastric flora.
Methods:
Eight male volunteers ingested ethanol 0.6 g/kg b.w. The pH, acetaldehyde level and microbial counts of the gastric juice were then determined. The experiment was repeated after 7 days of lansoprazole 30 mg b.d.
Results:
The mean (�� S.E.M.) pH of the gastric juice was 1.3 �� 0.06 and 6.1 �� 0.5 (P < 0.001) before and after lansoprazole, respectively. This was associated with a marked overgrowth of gastric aerobic and anaerobic bacteria (P < 0.001), by a 2.5�\fold (P=0.003) increase in gastric juice acetaldehyde level after ethanol ingestion, and with a positive correlation (r=0.90, P < 0.001) between gastric juice acetaldehyde concentration and the count of aerobic bacteria.
Conclusions:
Treatment with proton pump inhibitors leads to hypochlorhydria, which associates with intragastric overgrowth of aerobic bacteria and microbially�\mediated acetaldehyde production from ethanol. Since acetaldehyde is a local carcinogen in the concentrations found in this study, long�\term use of gastric acid secretory inhibitors is a potential risk�\factor for gastric and cardiac cancers.
INTRODUCTION
Due to its acidity, the stomach is usually free of microbes. However, oral microbes may survive in the stomach with a pH above 4.0 and bacterial proliferation can be expected when the pH rises over 5.0.1, 2 Therefore, gastric and duodenal microbial overgrowth is a common finding during long�\term use of gastric proton pump inhibitors or H2�\receptor antagonists, as well as in certain other similar conditions, e.g. achlorhydria caused by atrophic gastritis.345678
Via alcoholic fermentation mediated by microbial alcohol dehydrogenases normal gut flora, bacterial overgrowth and yeast infection can be responsible for endogenous ethanol production in the digestive tract of both experimental animals and humans.9101112 Small amounts of endogenous ethanol have also been found in the gastric juice of patients receiving cimetidine.13 Under aerobic and microaerobic conditions and in the presence of excess alcohol the microbial alcohol dehydrogenase reaction runs in the opposite direction resulting in marked acetaldehyde production both in vitro and in vivo.141516
Many recent epidemiological studies have shown that the risk of ethanol�\associated digestive tract cancers is greatly increased in Asian subjects with a genetically deficient ability to metabolize acetaldehyde.17181920212223 After adjustment for age, daily alcohol consumption, and amount of cigarette smoking, significantly increased risks (odds ratios) in the presence of mutant aldehyde dehydrogenase�\2 have been found for oropharyngolaryngeal (11.1), oesophageal (12.5), stomach (3.5), colon (3.4), lung (8.2) and oesophageal cancer concomitant with oropharyngolaryngeal and/or stomach cancer (54.2).20 Very recently, we found that individuals with this gene deficiency have two to three times higher in vivo salivary acetaldehyde levels after a moderate dose of alcohol than those with normal aldehyde dehydrogenase�\2.24 Accordingly, aldehyde dehydrogenase�\2�\deficient heavy drinkers are exposed for years or decades to markedly higher local acetaldehyde levels than those with the normal aldehyde dehydrogenase�\2 genotype. When this information is combined with the above mentioned epidemiological data, our finding provides strong evidence for the local carcinogenic action of acetaldehyde in humans. Therefore, all conditions possibly associating with enhanced local production of acetaldehyde can be considered as potential risk�\factors for upper digestive tract cancers.
The aim of this study was to examine in humans the effect of 1 week of treatment with a proton pump inhibitor on gastric flora and its capacity to produce acetaldehyde from ethanol in vivo after ingestion of a moderate dose of alcohol.RESULTS
Lansoprazole treatment raised the mean (�� S.E.M.) pH level of the gastric juice from 1.3 �� 0.06 to 6.1 �� 0.5, P < 0.001 ( Figure 1). This was associated with a significant increase in the mean intragastric acetaldehyde level from 22.1 �� 2.3 ��M to 55.4 �� 8.0 ��M, P=0.003. The intragastric acetaldehyde levels increased in all volunteers during the medication; the highest measured level was 100.5 ��M ( Figure 2). The mean ethanol concentration of the gastric juice was 1.7% (range 0.7�C3.4%) and 2.6% (range 1.2�C4.1%), P=0.054, non�\significant, before and during the treatment, respectively. Lansoprazole did not change the mean salivary acetaldehyde level (44.7 �� 6.8 ��M before the medication and 36.1 �� 6.4 ��M during it, P=0.21, non�\significant).
image
Figure ure 1
Open in figure viewerPowerPoint
The effect of lansoprazole treatment (30 mg b.d.) for 7 days on the pH of the gastric juice in eight volunteers after ethanol (0.6 g/kg b.w.) ingestion.
image
Figure ure 2
Open in figure viewerPowerPoint
The effect of lansoprazole treatment (30 mg b.d.) for 7 days on the acetaldehyde level of the gastric juice in eight volunteers after ethanol (0.6 g/kg b.w.) ingestion.
Before the medication, minor growth of aerobic bacteria was detected in the gastric juice of two volunteers, 1 �� 101 and 2 �� 101 colony forming units(cfu)/mL, the other aerobic cultures were negative. The anaerobic bacterial cultures of the gastric juices before the treatment were all negative. During lansoprazole, the total aerobic counts were 1.3 �� 0.7 �� 106 cfu/mL and the total anaerobic counts were 1.5 �� 0.8 �� 106 cfu/mL. The increase in both total counts was highly significant (P < 0.001). The cultures of yeasts were negative both before and during lansoprazole treatment. A vast selection of oral bacterial species were present; Table 1 summarizes the bacteriological results. There was also a highly significant positive correlation (r=0.90, P < 0.001) between the individual total aerobic bacterial counts and the individual gastric juice acetaldehyde levels during the medication ( Figure 3A). The correlation between individual acetaldehyde levels and anaerobic bacterial counts was also positive (r=0.76, P=0.021) ( Figure 3B).
Table 1. The effect of lansoprazole on the mean gastric bacterial counts (cfu/mL) in eight volunteers
image
image
Figure ure 3
Open in figure viewerPowerPoint
(A, B) Correlation between individual total aerobic and anaerobic bacterial counts and individual gastric juice acetaldehyde levels in eight volunteers after ethanol (0.6 g/kg b.w.) ingestion and during lansoprazole (30 mg b.d.) treatment.
Histologically gastric mucosal biopsies were within normal limits in all of the volunteers, and all of them were Helicobacter pylori�\negative.
DISCUSSION
It has been shown that acetaldehyde can be formed from ethanol in vitro by incubating human bronchopulmonary washings, and in vivo in mouthwashes of ethanol�\rinsing volunteers, both thought to be of bacterial origin.28, 29 Moreover, we have shown the production of marked amounts of acetaldehyde in the saliva after moderate alcohol ingestion.14 This acetaldehyde production can be significantly reduced by the use of antiseptic chlorhexidine mouthwash, indicating a microbial background for acetaldehyde production from ethanol by oral microbes.14 In this study salivary acetaldehyde levels after alcohol ingestion were comparable to those reported earlier.14
Diminished gastric acid secretion is associated with an increased number of microbes in the gastric juice.8, 30 This was confirmed in this study. The baseline aerobic and anaerobic cultures of bacteria and yeasts from the acidic gastric juices were nearly negative, which also confirms earlier findings.8, 3031 During the induction of hypochlorhydria with the proton pump inhibitor, both aerobic and anaerobic bacteria were found more frequently and in higher loads from the gastric juices. Stomatococcus, Streptococcus and Neisseria species were the most prevalent and numerous aerobic species. Since these bacteria are common species colonizing the oropharynx, the microbes that are able to survive in the neutral gastric milieu originate from the oral cavity. This is in accordance with earlier findings.31
The characteristics of microbially mediated acetaldehyde production from ethanol both in vitro and in vivo have been established in several studies.141516, 25, 32333435 This is the first study to show the in vivo acetaldehyde production from ethanol by the microbial overgrowth in the hypochlorhydric stomach. Acetaldehyde production was enhanced in all our volunteers after lansoprazole treatment and this paralleled with the increased number of microbes in the gastric juice. In fact, we were able to show a highly significant positive correlation between the individual total aerobic bacterial counts and the individual acetaldehyde levels during the medication. This correlation was more obvious than the one between acetaldehyde levels and the number of total anaerobic bacteria. This is in line with our earlier findings, which show that, by and large, only aerobes representing normal human gut flora possess alcohol dehydrogenase activity.15, 34
The subjects, who had the lowest gastric juice pH levels and therefore low bacterial counts after the medication, had only slight increases in their gastric juice acetaldehyde levels. These findings suggest that acetaldehyde in the stomach is produced by oral aerobic microbes which are able to grow in the neutral gastric milieu. However, small amounts of acetaldehyde were also detected from the acidic gastric juices. Part of the acetaldehyde that is formed from ethanol in the oral cavity is evidently distributed with saliva via the oesophagus to the stomach. Since no microbes were able to grow in the acidic gastric juice, it can be assumed that acetaldehyde in the acidic stomach is most probably originated from the oral cavity. This concept is strongly supported by our earlier findings indicating that microbial acetaldehyde production is almost totally abolished at pH 4.0 or lower.25
Alcohol consumption is a well�\known risk�\factor for upper digestive tract cancers.36373839 However, the tumour�\promoting effect of alcohol has so far been obscure, since ethanol itself is not carcinogenic. In contrast, acetaldehyde, the first metabolite of ethanol, has been shown to have many mutagenic and carcinogenic effects in cell cultures and animal models.40414243 As a highly reactive compound it can, for example, form adducts with macromolecules, proteins and DNA.444546 Acetaldehyde adducts can interfere with normal cellular functions and lead to cellular destruction and promote carcinogenesis. Moreover, acetaldehyde can cleave folate, which has been shown to lead to local folate deficiency and, thereby, to the hypomethylation of DNA and the promotion of carcinogenesis.47 Acetaldehyde is also known to be one of the major components of tobacco smoke.48
Stronger evidence for acetaldehyde as the major factor responsible for ethanol�\associated carcinogenesis is derived from recent studies linking the genotypes of ethanol�\metabolizing enzymes to markedly enhanced tumour risk.17181920212223, 49, 50 In Asian ��flushers��, single point mutation in the gene that codes low Km aldehyde dehydrogenase�\2 leads to nearly total inactivation of this enzyme. Thereby, aldehyde dehydrogenase�\2�\deficient individuals are exposed after alcohol ingestion to markedly higher local acetaldehyde levels than those with the normal aldehyde dehydrogenase�\2 enzyme.24 Alcoholics with cancer of oropharynx, larynx, oesophagus, stomach, colon or lungs had markedly higher frequencies of this mutant aldehyde dehydrogenase�\2*1/*2 genotype than cancer�\free alcoholics.20 Interestingly, microbial acetaldehyde production from ethanol has been described in all these organs.14, 28, 29, 33 This human ��knockout model�� for deficient acetaldehyde removal provides strong evidence for the local carcinogenic action of acetaldehyde in humans.
It should be noted that the gastric juice acetaldehyde levels found in this study were equal to the salivary acetaldehyde levels found in aldehyde dehydrogenase�\2�\deficient subjects after a moderate dose of alcohol.24 Furthermore, many of the in vitro and animal studies dealing with the carcinogenic action of acetaldehyde have been carried out with acetaldehyde concentrations less than 500 ��M.43, 51, 52 Accordingly, acetaldehyde levels of the gastric juice found in this study can be considered to be comparable to those suggested to be carcinogenic in other studies.
It has also been shown that Caucasians with rapid metabolizing alcohol dehydrogenase (alcohol dehydrogenase�\3*1) allele, which leads to higher and quicker production of acetaldehyde, have greater risk for upper digestive tract cancers than those with slow metabolizing allele if high amounts of alcohol are consumed.49, 50 These studies also provide convincing evidence for the carcinogenicity of acetaldehyde in humans. Aldehyde dehydrogenase�\2�\deficient genotype does not exist amongst Finns. The possible role of alcohol dehydrogenase�\3*1 allelle on our results remains to be established in future studies.
In conclusion, the use of gastric proton pump inhibitors leads to hypochlorhydria and overgrowth of bacteria in the gastric juice. This associates with enhanced intragastric production of acetaldehyde via alcohol dehydrogenase mediated ethanol oxidation carried out by the aerobic bacteria representing normal oral microflora. Since acetaldehyde is a local carcinogen, long�\term use of the inhibitors of gastric acid secretion may increase the risk for upper digestive tract cancers, especially amongst frequently drinking subjects with aldehyde dehydrogenase�\2�\deficient or alcohol dehydrogenase�\3*1 genotype.��
Hypochlorhydria induced by a proton pump inhibitor leads to intragastric microbial production of acetaldehyde from ethanol - Väkeväinen - 2000 - Alimentary Pharmacology & Therapeutics - Wiley Online Library
https://onlinelibrary.wiley.com/doi/full/10.1046/j.1365-2036.2000.00858.x��
J Dig Dis. 2011 Apr;12(2):51-9. doi: 10.1111/j.1751-2980.2011.00480.x.
Acetaldehyde and gastric cancer.
Salaspuro M1.
Author informationResearch Unit on Acetaldehyde and Cancer, University of Helsinki, Helsinki, Finland. mikko.salaspuro@helsinki.fi
Abstract
Aldehyde dehydrogenase (ALDH2) and alcohol dehydrogenase (ADH) gene polymorphisms associating with enhanced acetaldehyde exposure and markedly increased cancer risk in alcohol drinkers provide undisputable evidence for acetaldehyde being a local carcinogen not only in esophageal but also in gastric cancer. Accordingly, acetaldehyde associated with alcoholic beverages has recently been classified as a Group 1 carcinogen to humans.Microbes are responsible for the bulk of acetaldehyde production from ethanol both in saliva and Helicobacter pylori-infected and achlorhydric stomach.
Acetaldehyde is the most abundant carcinogen in tobacco smoke and it readily dissolves into saliva during smoking. Many foodstuffs and 'non-alcoholic' beverages are important but unrecognized sources of local acetaldehyde exposure.
The cumulative cancer risk associated with increasing acetaldehyde exposure suggests the need for worldwide screening of the acetaldehyde levels of alcoholic beverages and as well of the ethanol and acetaldehyde of food produced by fermentation. The generally regarded as safe status of acetaldehyde should be re-evaluated. The as low as reasonably achievable principle should be applied to the acetaldehyde of alcoholic and non-alcoholic beverages and food. Risk groups with ADH-and ALDH2 gene polymorphisms, H. pylori infection or achlorhydric atrophic gastritis, or both, should be screened and educated in this health issue.
L-cysteine formulations binding carcinogenic acetaldehyde locally in the stomach provide new means for intervention studies.
© 2011 The Author. Journal of Digestive Diseases © 2011 Chinese Medical Association Shanghai Branch, Chinese Society of Gastroenterology, Renji Hospital Affiliated to Shanghai Jiaotong University School of Medicine and Blackwell Publishing Asia Pty Ltd.Acetaldehyde and gastric cancer. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/21401890Formulation and in�\vivo evaluation of l�\cysteine chewing gums for binding carcinogenic acetaldehyde in the saliva during smoking
Alma Kartal Jaana Hietala Into Laakso Pertti Kaihovaara Ville Salaspuro Mia Säkkinen Mikko Salaspuro Martti Marvola
First published: 18 February 2010 https://doi.org/10.1211/jpp.59.10.0004 Citations: 13
Abstract
Cigarette smoke contains toxic amounts of acetaldehyde that dissolves in saliva, posing a significant risk of developing oral, laryngeal and pharyngeal carcinomas. l�\Cysteine, a non�\essential amino acid, can react covalently with carcinogenic acetaldehyde to form a stable, non�\toxic 2�\methylthiazolidine�\4�\carboxylic acid...Formulation and in�\vivo evaluation of l�\cysteine chewing gums for binding carcinogenic acetaldehyde in the saliva during smoking - Kartal - 2007 - Journal of Pharmacy and Pharmacology - Wiley Online Library
https://onlinelibrary.wiley.com/doi/abs/10.1211/jpp.59.10.0004��
���Ѿ������������ȩ�Ĵ�л����
FEMS����ѧ��־����191��1����51-5��2000��11��
24.87���ո�������ѧ
Ram��nMira deOrduña
28.22hang
G��J��Ƥ¡
��ʤǿ
����
��ȩ�������ʳƷ�д��ڵĻӷ��Է�ζ������������������ƺͰ����Ѿƺ���Ҫ������˾�����Oenococcus����9�������ܹ��ھ�ֹϸ��ϵͳ�д�л��ȩ��������Pediococcus�������ܡ�������Ҵ������併������ġ�����˾���OenococcusҲ�ܹ�����SO��2������ȩ����ĸ��BayanusPremi��reCuv��e��Oenococcus oeni Lo111�ľ�Ϣϸ����ͬ��������������Lo111��л�˽�ĸ��������ȩ��ƻ����������������SO��2�������ȩ�������Ըйٺ���ɫ�����Լ������Ѿ���ʹ��SO��2������Ӱ�졣Acetaldehyde metabolism by wine lactic acid bacteria
Article in FEMS Microbiology Letters 191(1):51-5 �� November 2000
24.87Oregon State University
Ram��n Mira de Orduña
28.22Changins
G J Pilone
S Q Liu
Abstract
Acetaldehyde is a volatile flavor compound present in many fermented foods and is important in the production of red and white wines. Nine strains of the genera Lactobacillus and Oenococcus were able to metabolize acetaldehyde in a resting cell system, whereas two Pediococcus strains were not. Acetic acid and ethanol were produced from its degradation. A Lactobacillus and an Oenococcus were able to degrade SO(2)-bound acetaldehyde, as well. A coincubation of resting cells of Saccharomyces bayanus Premi��re Cuv��e and Oenococcus oeni Lo111 showed that strain Lo111 metabolized acetaldehyde produced by the yeast. The ability of malolactic bacteria to degrade free and SO(2)-bound acetaldehyde has implications for sensory and color qualities and the use of SO(2) in wine.Acetaldehyde metabolism by wine lactic acid bacteria | Request PDF
https://www.researchgate.net/publication/12319971_Acetaldehyde_metabolism_by_wine_lactic_acid_bacteria��
�����ض�ӡ�������ݸ˾������꼰С���Ⱦ�Ŀ�������
Ronita De��1 Parag Kundu��2 Snehasikta Swarnakar��2 T. Ramamurthy��1 Abhijit Chowdhury��3 G.Balakrish Nair��1��Asish K.Mukhopadhyay1��*
������Ϣ����ע�Ͱ�Ȩ��������Ϣ��������
���������볦�������о�����ӡ�ȼӶ�����700010��1ӡ�Ȼ�ѧ����ѧ�о�����ӡ�ȼӶ�����700032��2����ϵͳ����ѧԺ���о���ҽѧ�������о�ѧԺ��ӡ�ȼӶ�����3
ժҪ
����ʧ�����������ݸ˾���ص�θʮ��ָ����������θ�ף������������θ���������ע����Ҫԭ��
�����أ����Խ��ƵĶ��������飬�����֤ʵ������ֹ�����ݸ˾�������������˽�������������ڵֿ����������ݸ˾���Ⱦ�����ж�65�������ݸ˾��ٴ�������Ŀ������ԡ������ص�MIC��ΧΪ5��g/ ml��50��g/ ml����ʾ�������������������ݸ˾���������Ч�ԣ��������Ļ�������ء�
���ֳ���ͬ������MICs�������ݸ˾������б��뽪��������ø��aroE����ĺ��������У��������ƺ�������Ǿ��������Ƽ������ã����������ؽ鵼�Ķ�ӡ�������ݸ˾�������������ƿ��ܲ���������������the��·�ϡ���֯ѧ��齪�����������ݸ˾���Ⱦ��C57BL / 6С���еĿ������ü����ڼ������Ⱦ�����θ�����еĹ�Ч��
��������ʾ���������ݸ˾���Ⱦ�ľ�����DZ������Ϊ���ڸ�����ȾС��������ݸ˾��Լ��ָ������ݸ˾������θ���˷���dz���Ч��
�����о�Ϊ�����ض������ݸ˾���Ⱦ������Ч���ṩ����ӱ�ļ��⣬����������Ϊ����Ʒ���DZ������Ϊ��һ���о����������ص����Ϳ����б꿪���˵�·����
Antimicrobial Activity of Curcumin against Helicobacter pylori Isolates from India and during Infections in Mice▿
Ronita De,1 Parag Kundu,2 Snehasikta Swarnakar,2 T. Ramamurthy,1 Abhijit Chowdhury,3 G. Balakrish Nair,1 and Asish K. Mukhopadhyay1,*
Author information Article notes Copyright and License information Disclaimer
National Institute of Cholera and Enteric Diseases, Kolkata 700010, India,1 Indian Institute of Chemical Biology, Kolkata 700032, India,2 School of Digestive Diseases, Institute of Post Graduate Medical Education and Research, Kolkata, India3
ABSTRACT
Treatment failure is a major cause of concern for the Helicobacter pylori-related gastroduodenal diseases like gastritis, peptic ulcer, and gastric cancer.Curcumin, diferuloylmethane from turmeric, has recently been shown to arrest H. pylori growth. The antibacterial activity of curcumin against 65 clinical isolates of H. pylori in vitro and during protection against H. pylori infection in vivo was examined. The MIC of curcumin ranges from 5 ��g/ml to 50 ��g/ml, showing its effectiveness in inhibiting H. pylori growth in vitro irrespective of the genetic makeup of the strains.
The nucleotide sequences of the aroE genes, encoding shikimate dehydrogenase, against which curcumin seems to act as a noncompetitive inhibitor, from H. pylori strains presenting differential curcumin MICs showed that curcumin-mediated growth inhibition of Indian H. pylori strains may not be always dependent on the shikimate pathway. The antimicrobial effect of curcumin in H. pylori-infected C57BL/6 mice and its efficacy in reducing the gastric damage due to infection were examined histologically.
Curcumin showed immense therapeutic potential against H. pylori infection as it was highly effective in eradication of H. pylori from infected mice as well as in restoration of H. pylori-induced gastric damage.
This study provides novel insights into the therapeutic effect of curcumin against H. pylori infection, suggesting its potential as an alternative therapy, and opens the way for further studies on identification of novel antimicrobial targets of curcumin.
RESULTS
Differential inhibition of H. pylori growth in vitro by curcumin.
Among 65 Helicobacter pylori strains tested against curcumin, 47 strains were isolated from patients with duodenal ulcers, whereas 12 and 6 cases were isolated from patients with antral gastritis and nonulcer dyspepsia, respectively. Among these strains, 64 strains, including ATCC 43504, showed a positive amplicon for cagA and the other 2 provided an empty site amplicon, indicating absence of the total cag PAI. MICs were determined by the agar dilution method. Curcumin inhibited the growth of all 65 clinical isolates as well as the reference strain ATCC 43504. The MIC of curcumin ranged from 5 ��g/ml to 50 ��g/ml, and the majority of the strains (81%) showed a MIC of either 10 ��g/ml (23%) or 15 ��g/ml (58%) (Fig. (Fig.2).2). A few strains showed higher MICs (7.7% and 1.5% of the strains yielded MICs of 30 ��g/ml and 50 ��g/ml, respectively). These results clearly confirm that curcumin acts as a potent growth inhibitor for Indian H. pylori strains irrespective of the disease status. However, the roles of polymorphism in target genes and strain-specific differences in the MICs of curcumin need further investigation. The MICs of amoxicillin and clarithromycin ranged from 0.03 to 2 ��g/ml and 0.02 to 0.12 ��g/ml, respectively. The MICs of amoxicillin, curcumin, and clarithromycin against reference strain ATCC 43504 were 0.03, 15, and 0.02 ��g/ml, respectively.Curcumin eradicates H. pylori from H. pylori-infected mouse stomach.
Since an in vitro assay showed that curcumin had antibacterial effects against H. pylori, we tested the effect of curcumin on H. pylori colonization in mice. A group of mice were infected with AM1 and SS1 separately. Three weeks after the infection, they were sacrificed and gastric tissues were checked by a urease test, which showed positive results indicating H. pylori colonization. Establishment of H. pylori infection was also confirmed by PCR assay of the H. pylori-specific gene vacA using DNA isolated from H. pylori-infected stomach tissues. Colonization was further reconfirmed by quantitative culture in blood agar plates. Two weeks after infection, a group of mice were treated with curcumin for 7 days. The effect of curcumin was assessed by a urease test with the respective mouse gastric tissues. It was observed from quantitative culture that curcumin, irrespective of strains, eradicated H. pylori from infected mouse stomach. To further confirm curcumin's anti-H. pylori potential, the H. pylori-specific gene vacA was amplified by PCR using DNA isolated from stomach tissues of H. pylori-infected and treated mice, while the mouse-specific GAPDH gene served as a control. Figure Figure44 shows that curcumin treatment completely eliminated H. pylori from mouse stomach. Thus, these in vivo data are in accordance with the in vitro results, confirming curcumin's anti-H. pylori potential.DISCUSSION
Eradication of H. pylori with a proton pump inhibitor-based triple therapy is presently used to treat H. pylori infection (21). Though it has a success rate of 80 to 90%, problems like treatment failure and contraindications for some patients are common. Furthermore, rapidly emerging drug resistance in H. pylori strains during treatment with various antibiotics is a major obstacle for successful eradication therapies (1). Because of the prevalence of antibiotic-resistant H. pylori strains, there is an increasing search for safe and effective nonantibiotic compounds that inhibit H. pylori growth. In the Indian traditional medical system, a number of plants and plant products are known to possess potent medicinal properties, suggesting their usefulness in treatment. Recent studies have shown that curcumin possesses anti-H. pylori potential (11, 15). This prompted us to explore its antimicrobial potential against Indian H. pylori strains that are geographically distinct from east Asian and Western strains. Moreover, a majority of the Indian population harbors H. pylori and quite a number of them suffer from H. pylori-associated gastrointestinal diseases.
In the present study, we have primarily shown that curcumin potentially inhibited the growth of all the 65 H. pylori strains in vitro that were isolated from infected patients suffering from gastrointestinal disorders. It is noteworthy that a majority of these strains were metronidazole resistant, with MICs ranging from 16 ��g/ml to >64 ��g/ml (7). So, our results suggest that curcumin acts through mechanisms distinctly different from the mode of action of these antibiotics for inhibition of H. pylori growth. Han et al. (11) demonstrated that the growth-inhibitory activity of curcumin against H. pylori is due to inhibition of the shikimate pathway, necessary for synthesis of aromatic amino acids in bacteria. Thus, the aroE genes, encoding SDH, from H. pylori strains showing different MICs were analyzed for nucleotide and amino acid sequence variations. Striking differences in the nucleotide sequences of the genes from these strains corroborated the fact that curcumin targets the shikimate pathway and indicate that polymorphism in aroE may lead to differential inhibition of H. pylori growth. However, strains having identical sequences also had different curcumin MICs. This may be due to the strain-to-strain variations in the rate of curcumin uptake/efflux, or it may be that the antimicrobial effect of curcumin on H. pylori follows distinct mechanisms that may not be solely dependent on inhibition of the shikimate pathway. A recent study by Rai et al. suggests that curcumin may inhibit bacterial cell proliferation by inhibiting the assembly dynamics of FtsZ (a bacterial protofilament), which polymerizes to form a Z ring at the midcell that orchestrates bacterial cell division (22).
The mouse model of H. pylori infection has been widely used in investigations of host responses to H. pylori infection as well as in eradication studies. In the present study, we have shown that curcumin treatment completely eradicated H. pylori from infected mouse stomach. This eradication by curcumin was irrespective of the bacterial genotype, which is independent of the presence of the cag PAI. These data are of immense importance for the development of alternative therapy against H. pylori infection since studies on high doses of curcumin in animals and humans have confirmed a lack of any toxic side effects (9, 25). Histological analysis clearly showed that curcumin is highly effective in repairing damaged tissue. All these observations not only indicate the therapeutic potential of curcumin against H. pylori infections but also highlight the anti-inflammatory effect of curcumin. Various clinical trials suggest a therapeutic potential for curcumin in diseases such as familial adenomatous polyposis, inflammatory bowel disease, ulcerative colitis, colon cancer, pancreatic cancer, hypercholesteremia, atherosclerosis, pancreatitis, psoriasis, and arthritis (9). It has been claimed that curcumin and related compounds have anti-human immunodeficiency virus type 1 and 2 activity in a recent patent application (19).
In conclusion, our study highlights the potential antibacterial activity of curcumin against H. pylori in vitro, as curcumin was highly effective in inhibiting H. pylori growth irrespective of the genetic makeup of the strains, although its MIC is relatively high; this may be due to the poor bioavailability of curcumin (22). Our studies also showed that curcumin-mediated inhibition of H. pylori growth involved mechanisms that may not always be dependent on the shikimate pathway, which opens the way for further studies directed toward determination of novel antimicrobial targets of curcumin. Curcumin showed immense therapeutic potential against H. pylori infections and H. pylori-associated gastroduodenal diseases, as it was equally effective in eradicating H. pylori from infected mouse stomach. Moreover, the gastric damage induced by H. pylori infection was almost completely restored by curcumin, thus highlighting its potential as an alternative therapy against H. pylori infection. Overall, this study provides novel insights in the therapeutic potential of curcumin against H. pylori infections, although further studies are required to extrapolate its effect on humans.
��Antimicrobial Activity of Curcumin against Helicobacter pylori Isolates from India and during Infections in Mice
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2663130/J Biol Chem. 2006 Nov 10;281(45):34651-62. Epub 2006 Sep 11.
Cag pathogenicity island-independent up-regulation of matrix metalloproteinases-9 and -2 secretion and expression in mice by Helicobacter pylori infection.
Kundu P1, Mukhopadhyay AK, Patra R, Banerjee A, Berg DE, Swarnakar S.
Author information
Indian Institute of Chemical Biology, Kolkata 700032, India.
Abstract
Helicobacter pylori cag pathogenicity island (PAI) is a major determinant of gastric injury via induction of several matrix metalloproteinases (MMPs). In the present study, we examined the influence of the cag PAI on gastric infection and MMP-9 production in mice and in cultured cells. A new mouse colonizing Indian H. pylori strain (AM1) that lacks the cag PAI was used to study the cag PAI importance in inflammation. Groups of C57BL/6 mice were inoculated separately with H. pylori strains AM1 and SS1 (cag+), gastric tissues were histologically examined, and bacterial colonization was scored by quantitative culture. Mice infected with either cag+ or cag- H. pylori strains showed gastric inflammation and elevated MMP-3 production. Significant up-regulation of pro-MMP-9 secretion and gene expression in H. pylori infected gastric tissues indicate dispensability of cag PAI for increased pro-MMP-9 secretion and synthesis in mice. In agreement, cell culture studies revealed that both AM1 and SS1 were equipotent in pro-MMP-9 induction in human gastric epithelial cells. Both strains showed moderate increase in MMP-2 activity in vivo and in vitro. In addition, increased secretion of tumor necrosis factor (TNF)-alpha, interleukin (IL)-1beta, and IL-6 induced pro-MMP-9 secretion and synthesis in AM1 or SS1 strain-infected mice suggesting elicitation of pro-inflammatory cytokines by both cag- and cag+ genotype. Moreover, tissue inhibitors of metalloproteinase-1 expression were decreased with increase in pro-MMP-9 induction. These data show that H. pylori may act through different pathways other than cag PAI-mediated for gastric inflammation and contribute to up-regulation of MMP-9 via pro-inflammatory cytokines.Cag pathogenicity island-independent up-regulation of matrix metalloproteinases-9 and -2 secretion and expression in mice by Helicobacter pylori in... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/16966323/��
Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori
Steffen Backert, Marguerite Clyne & Nicole TegtmeyerUniversity College Dublin, School of Biomolecular and Biomedical Sciences, Science Center West, Belfield Campus, Dublin-4, Ireland
Cell Communication and Signaling volume 9, Article number: 28 (2011)
Abstract
Helicobacter pylori is a highly successful pathogen uniquely adapted to colonize humans. Gastric infections with this bacterium can induce pathology ranging from chronic gastritis and peptic ulcers to gastric cancer. More virulent H. pylori isolates harbour numerous well-known adhesins (BabA/B, SabA, AlpA/B, OipA and HopZ) and the cag (cytotoxin-associated genes) pathogenicity island encoding a type IV secretion system (T4SS). The adhesins establish tight bacterial contact with host target cells and the T4SS represents a needle-like pilus device for the delivery of effector proteins into host target cells such as CagA. BabA and SabA bind to blood group antigen and sialylated proteins respectively, and a series of T4SS components including CagI, CagL, CagY and CagA have been shown to target the integrin ��1 receptor followed by injection of CagA across the host cell membrane.The interaction of CagA with membrane-anchored phosphatidylserine may also play a role in the delivery process. While substantial progress has been made in our current understanding of many of the above factors, the host cell receptors for OipA, HopZ and AlpA/B during infection are still unknown. Here we review the recent progress in characterizing the interactions of the various adhesins and structural T4SS proteins with host cell factors. The contribution of these interactions to H. pylori colonization and pathogenesis is discussed.
��
Introduction
H. pylori colonises the stomach of about half of the human world population, which is associated with chronic, often asymptomatic gastritis in all infected individuals. Depending on various criteria, more severe gastric diseases including peptic ulcer disease can occur in up to 10-15% of infected persons [1�C3]. H. pylori infections are commonly diagnosed with a strong inflammatory response, but the bacteria evolved numerous mechanisms during evolution to avoid recognition and clearance by the host defence machineries, and if not treated with antibiotics, they can persist for life. H. pylori-induced gastritis is the strongest singular risk factor for developing cancers of the stomach; however, only a small proportion of infected individuals develop malignancy such as mucosa-associated lymphoid tissue (MALT) lymphoma and even gastric adenocarcinoma [1�C3]. Gastric adenocarcinoma constitutes the second leading cause of cancer-associated death worldwide, and about 700,000 people die from this malignancy annually [3]. The clinical outcome of infection with H. pylori is dependent on a very complex scenario of host-pathogen crosstalk. Disease progression is determined by various factors including the genetic predisposition of the host, the bacterial genotype and environmental parameters [1�C3]. The cellular and molecular mechanisms developed by H. pylori to undermine host defence strategies have been under intense investigation worldwide.
Clinical H. pylori strains are highly diverse both in their genetic information and potential to induce pathogenicity. Myriads of bacterial factors have been reported to influence the pathogenesis of H. pylori infections. There are two classical virulence determinants expressed by H. pylori, the CagA protein encoded by the cytotoxin-associated genes pathogenicity island (cag PAI) and the vacuolating cytotoxin (VacA). Secreted VacA can trigger various responses including pore formation in the host cell membrane, modification of endo-lysosomal trafficking, cellular vacuolation, immune cell inhibition and apoptosis. VacA's activities are highlighted in several review articles [1�C4] and will not be discussed here. In the mid nineties, the cag PAI was entirely sequenced from various H. pylori strains and found to represent a 40-kb DNA insertion element in the chromosome, which is flanked by 31-bp direct repeats and carrying up to 32 genes [5, 6]. Large scientific interest concentrates on the CagA protein which is present in more virulent isolates, but is typically absent in less virulent H. pylori strains. Thus, CagA serves as a virulence marker for the cag PAI [7, 8]. Work in the last ten years has shown that the cag PAI encodes type-IV secretion system (T4SS) which injects CagA into target cells where it interferes with multiple host cell signaling cascades [9, 10]. Other well-described pathogenicity-associated mechanisms include flagella-driven bacterial motility, urease-mediated neutralization of pH, HtrA-mediated cleavage of E-cadherin, modification of host cell cholesterol, shedding of outer-membrane vesicles and peptidoglycan-dependent immune responses [1�C3, 11�C13]. In addition, H. pylori carries several classical surface adhesins permitting tight adherence of the bacteria to gastric epithelial cells. Here we review the various molecular adhesion strategies of H. pylori to gastric epithelial target cells which facilitate bacterial binding. We also discuss the structure and function of the T4SS, and how it makes contact with host cell surface factors to inject the CagA effector protein.
H. pylori is one of the most successful human pathogens. Studies of host-bacterial interactions using their fundamental adhesins and the virulence factors CagA and T4SS have provided us with many detailed insights in processes ultimately connected to H. pylori colonisation and pathogenesis.
in one in vivo study, in which phosphorylated CagA was isolated from biopsies of atrophic gastritis and in noncancerous tissues from H. pylori-positive patients using immunoprecipitation and Western blotting approaches [90].Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori | Cell Communication and Signaling | Full Text
https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-9-28��
H. pylori Virulence Factors: Toxins (CagA, VacA, DupA, OipA, IceA)
Authors and affiliations
Jung Mogg Kim
Department of MicrobiologyHanyang University College of MedicineSeoulSouth Korea
First Online: 17 June 2016
Abstract
Helicobacter pylori (H. pylori) specifically colonizes the human stomach. The majority of bacteria live in the mucus layer overlying gastric epithelial cells and only a small proportion of bacteria are found interacting with the epithelial cells. To succeed in the long-term colonization, H. pylori has developed a unique set of virulence factors, which allow survival in a harsh ecological niche. Clinical outcomes associated with H. pylori infections are largely mediated by a complex interaction between bacterial, host, and environmental factors. Over the past year, a variety of studies focusing on both host and bacterial factors have proceeded. Among the bacterial factors that contribute to the pathophysiology associated with H. pylori infections, it is remarkable that the cytotoxin-associated gene A (CagA) protein is delivered into gastric epithelial cells via bacterial type IV secretion system, leading to induction of diverse immune responses and gastroduodenal diseases. The vacuolating cytotoxin (VacA) is also one of the most important virulence factors and functions as an intracellular-acting protein exotoxin, in which one of the most important target sites for VacA is the mitochondrial inner membrane in the host. In addition, the outer membrane inflammatory protein (OipA), the induced by contact with epithelium (IceA), and duodenal ulcer promoting (dupA) gene have emerged as virulence factors, although the specific genes involved in virulence are still being determined. This chapter summarizes the results of the most relevant studies regarding H. pylori virulence factors such as CagA, VacA, OipA, IceA, and dupA gene and discusses their molecular mechanism for generating gastrointestinal diseases.
Keywords
CagA Helicobacter pylori VacA Virulence factorH. pylori Virulence Factors: Toxins (CagA, VacA, DupA, OipA, IceA) | SpringerLink
https://link.springer.com/chapter/10.1007/978-981-287-706-2_5��
A new strategy used by Helicobacter pylori to target mitochondria
��November 29, 2017
by Institut Pasteur
Gastric cancer: A new strategy used by Helicobacter pylori to target mitochondria
Gastric tissue infected by H. pylori. Nuclei are in blue (Hoechst) and mitochondria in red (Mitotracker labelling). Credit: Laurent Chatre - CNRS/Institut Pasteur
Scientists from the Institut Pasteur and CNRS have recently identified new strategies used by Helicobacter pylori bacteria to infect cells. By specifically targeting mitochondria, these bacteria, despite being extracellular, can optimize infection in the host. These findings pave the way for new strategies to combat H. pylori infection, which is associated with most cases of gastric cancer and several other gastric disorders. The results were published on Nov. 21 in the journal Scientific Reports.
Helicobacter pylori is a bacterial pathogen that colonizes the stomach of approximately half of the world's population. Infection with H. pylori is acquired in childhood and lasts for decades. H. pylori is the main risk factor for gastric cancer and is linked to more than 80 percent of cases. Gastric cancer, the third most common cause of cancer-related death, often has poor prognosis because it tends to be diagnosed at an advanced stage. It is responsible for about 800,000 deaths each year worldwide.
H. pylori has several virulence factors that interact with specific targets in the cell and directly affect the severity of gastric disease. Vacuolating cytotoxin A (VacA) was previously the only main H. pylori factor known to act on mitochondria, causing cellular membrane and organelle dysfunction, ultimately leading to cell death.
Scientists from the Institut Pasteur and the CNRS have discovered that H. pylori uses at least two additional strategies to target mitochondria. These strategies do not lead to cell death, but maintain an environment that is conducive to bacterial proliferation.
Their results show that H. pylori affects both mitochondrial transport systems (used to transfer proteins into mitochondria) and the machinery for the replication and maintenance of the mitochondrial genome. The scientists also discovered that, contrary to what was previously believed, VacA is not the only H. pylori component capable of affecting mitochondria. This suggests that the bacteria may produce other mitochondria-interacting factors that have not been yet identified.
Co-author Miria Ricchetti of the Institut Pasteur says, "The damage to mitochondria caused by H. pylori bacteria is temporary and disappears once the infection has been eliminated. Despite remarkably high levels of stress, mitochondria, like cells, can remain functional and withstand infection for longer than previously thought. It is important for us to bear this in mind when looking for strategies to inhibit the bacterium's pathogenic potential."
Co-author Eliette Touati of the Institut Pasteur, says, "We have observed in a mouse model that this type of damage is associated with a worsening of gastric lesions. The damage may therefore affect the chronicity and severity of infection by H. pylori. Understanding these new interactions between pathogen and host cells (via mitochondria) is vital for the development of effective strategies to combat H. pylori infection. The aim is to reduce the persistence of the bacteria in the stomach and curb associated conditions, especially cancer."A new strategy used by Helicobacter pylori to target mitochondria
https://phys.org/news/2017-11-strategy-helicobacter-pylori-mitochondria.html��
Gut Microbes. 2010 Nov-Dec; 1(6): 392�C395.
Targeting of Helicobacter pylori VacA to mitochondria
Antoine Galmiche1 and Joachim Rassowcorresponding author2
1Laboratoire de Biochimie Inserm ERI12; Hopital Nord; CHU Amiens Picardie; Amiens Cedex 1, France
2Institut f��r Physiologische Chemie; Ruhr-Universität Bochum; Bochum, Germany
corresponding authorCorresponding author.
Correspondence to: Joachim Rassow; Email: ed.bur@wossar.mihcaoj
Abstract
One of the major virulence factors of Helicobacter pylori is the vacuolating toxin vaca. It has been known for a long time that the toxin enters host cells by endocytosis. On the other hand there is ample evidence that vaca is able to trigger apoptosis and this effect has been attributed in part to interactions with mitochondria. However, for 10 years it was difficult to reconcile the obvious accumulation of vaca in endosomes with mitochondrial targeting. The accessibility of the mitochondria to the toxin was enigmatic.In our new study, we investigated the activities of p34, the toxic subunit of vaca, in more detail. We found that the p34 N-terminus carries a unique targeting sequence for import into mitochondria and for insertion into the mitochondrial inner membrane. By forming an anion channel in this membrane, the toxin has the ability to interfere directly with mitochondrial functions. Taking into account additional results from independent studies, we discuss the implications of our findings with respect to intracellular traffic, the remarkable possibility of a direct transfer of VacA from endosomes to mitochondria and vaca-dependent cell death.
Key words: Helicobacter pylori, VacA, endosome, mitochondria, protein targeting, protein transfer, protein import, Bax, Bak, apoptosisSeveral important virulence factors of H. pylori were already identified many years ago.1 Most prominent are the urease of the bacteria and the toxins CagA and VacA: the urease helps the bacteria in keeping the environment at a neutral pH.2 CagA is a protein which is injected into host cells by a type IV secretion system and interferes with signalling pathways.3 The vacuolating toxin VacA is released by the bacteria using a C-terminal autotransporter. VacA binds to the outer surface of host cells and is internalized by endocytosis.4,5 Inside the cells, VacA can target mitochondria and at least in some cases, it triggers the release of cytochrome c and apoptosis.6,7 Until recently, it was completely unclear how and why VacA produces these effects.
Targeting of Helicobacter pylori VacA to mitochondria
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3056105/��
Sci Rep. 2017 Nov 21;.
Helicobacter pylori targets mitochondrial import and components of mitochondrial DNA replication machinery through an alternative VacA-dependent and a VacA-independent mechanisms.
Chatre L1,2, Fernandes J3,4,5, Michel V3,4, Fiette L6,7, Av�� P6,7, Arena G1,8,9, Jain U10,11, Haas R10,12, Wang TC13, Ricchetti M14,15, Touati E16,17.
1
Stem Cells and Development, Team Stability of Nuclear and Mitochondrial DNA, Institut Pasteur, 25-28 Rue du Dr. Roux, Paris, France.
2
CNRS UMR3738, Paris, France.
3
Unit of Helicobacter Pathogenesis, Team Genotoxicity, Infection and Cancer, Institut Pasteur, 25-28 Rue du Dr. Roux, Paris, France.
4
CNRS ERL3526, Paris, France.
Abstract
Targeting mitochondria is a powerful strategy for pathogens to subvert cell physiology and establish infection. Helicobacter pylori is a bacterial pathogen associated with gastric cancer development that is known to target mitochondria directly and exclusively through its pro-apoptotic and vacuolating cytotoxin VacA.By in vitro infection of gastric epithelial cells with wild-type and VacA-deficient H. pylori strains, treatment of cells with purified VacA proteins and infection of a mouse model, we show that H. pylori deregulates mitochondria by two novel mechanisms, both rather associated with host cell survival. First, early upon infection VacA induces transient increase of mitochondrial translocases and a dramatic accumulation of the mitochondrial DNA replication and maintenance factors POLG and TFAM. These events occur when VacA is not detected intracellularly, therefore do not require the direct interaction of the cytotoxin with the organelle, and are independent of the toxin vacuolating activity. In vivo, these alterations coincide with the evolution of gastric lesions towards severity. Second, H. pylori also induces VacA-independent alteration of mitochondrial replication and import components, suggesting the involvement of additional H. pylori activities in mitochondria-mediated effects. These data unveil two novel mitochondrial effectors in H. pylori-host interaction with links on gastric pathogenesis.
Helicobacter pylori targets mitochondrial import and components of mitochondrial DNA replication machinery through an alternative VacA-dependent an... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29162845��
VacA Genotype in Helicobacter pylori
By Elnaz Saeidi, Amirhossein Sheikhshahrokh, Abbas Doosti and Reza Ranjbar
Submitted: March 7th 2018Reviewed: August 29th 2018Published: September 18th 2019
DOI: 10.5772/intechopen.81203
Helicobacter pylori infection has been recognized as a worldwide problem. H. pylori infection is the most prevalent cause of chronic gastritis and has been related to peptic ulcer disease and gastric cancer. It is considered that H. pylori infects half of the world��s population. Several virulence factors are produced by H. pylori in which each of them is related to an increase in the risk of disease development. The vacuolating cytotoxin (VacA) is one of these virulence factors. The first defined action of VacA was induction of intracellular vacuolation. VacA uses a variation in other effects on target cells, such as disruption of mitochondrial functions, stimulation of apoptosis, and blockade of T-cell proliferation, for the induction of vacuolation. In addition, VacA has an important role for colonization of H. pylori in vivo.
Keywords
Helicobacter pylori disease vacuolating cytotoxin (VacA) vacuolation
1. Introduction
Helicobacter pylori (H. pylori) is a gram-negative and microaerophilic bacterium, which usually colonizes in the human stomach. H. pylori affects about half of the human population worldwide, which exists in their upper gastrointestinal tract. Though all the factors have not been known, we could say that the infection is most likely to happen at a young age and happens more common in developing countries [1]. Prevalence of infection is through human contact mainly via the gastric-oral way [2]. H. pylori is related to some diseases such as peptic ulcer disease, gastric ulcers, mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric cancer [3].
Study on this microaerophilic spiral-shaped bacterium is interesting. H. pylori is a part of a quickly growing genus. New species are being derived from numerous vertebrate hosts. In addition, other Helicobacter species are being derived from nongastric parts in humans and might have a role in diseases that formerly had no certain etiologic factor.
H. pylori has polar-sheathed flagella, which helps in motility. In addition, these structures also have a terminal bulb that could make it more adapted to swimming through mucus. Moreover, on the surface, there are special biological characteristics in the lipopolysaccharide, and in order to escape from the host responses, genes that control addition of the O-side chains can phase vary. Moreover, H. pylori has a special peptidoglycan structure, which is different from other gram-negative bacteria. Also H. pylori releases an autotransported vacuolating cytotoxin that makes the abnormal phenotype of vacuolation in host cells.
For the first time in 1982, two Australian scientists Barry Marshall and Robin Warren identified H. pylori in a patient with chronic gastritis and gastric ulcers. Before that, it was not believed to have a microbial reason. By the successful culture of H. pylori, a large number of researchers investigated the epidemiology of transmission of the organism. In addition, it is connected with the development of duodenal ulcers and stomach cancer. More than 80% of infected population with the bacterium are asymptomatic, and it might have an important impress in the natural stomach ecology [4]. However, almost 10�C15% of people infected with H. pylori shown severe gastric disorders containing peptic ulcers, gastric lymphoma, mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric adenocarcinoma [5].
In 1983, H. pylori was cultured from human gastric tissue for the first time [5]. After many years, a proteinaceous component known as ��vacuolating cytotoxin�� was found in H. pylori broth culture supernatants. By adding vacuolating cytotoxin to cultured eukaryotic cells, the cells became vacuolated [6]. Formerly, bacterial toxins had not been reported by this function. In the further studies, the identity of the vacuolating toxin was shown [7, 8] and revealed that vacuolating cytotoxin (VacA) has different characteristics and activities compared to other bacterial toxins.
Multiple virulence factors are produced by H. pylori in which each of them is related to an increase in the risk of disease extension. Cytotoxin-associated gene A (CagA) and the vacuolating cytotoxin (VacA) are the virulence factors [9].
Infection by H. pylori strains including the toxigenic allelic s1 form of VacA increased the risk of peptic ulceration and gastric cancer [10]. VacA was termed because of its ability to cause ��vacuole��-like membrane vesicles in the cytoplasm of gastric cells [11]. However, its function in H. pylori pathogenesis has not been clear yet. VacA is a pore-forming toxin (PFT). VacA uses a variety of other effects on target cells, such as disruption of mitochondrial functions, stimulation of apoptosis, and blockade of T-cell proliferation, for the induction of vacuolation [12]. In addition, VacA has an important role for colonization of H. pylori in vivo [13].
A type IV secretion system is encoded in the cag pathogenicity island (cagPAI), and it replaces CagA into gastric epithelial cells. It causes morphological changes and proinflammatory cytokine secretion [14].
10. Conclusion
Helicobacter pylori has been investigated since its first culture in 1982 from a gastric biopsy. Cytotoxin-associated gene A (CagA) and the vacuolating cytotoxin (VacA) are the virulence factors which are produced by H. pylori and are related to an increase in the risk of disease extension [9].
VacA has a significant role in the pathogenesis of H. pylori-associated diseases, especially in peptic ulceration and distal gastric adenocarcinoma. For this reason, VacA has been studied widely. VacA is still under examination in order to find out its accurate role in these diseases. However, VacA makes H. pylori able to colonize in the human gastric mucosa. Moreover, VacA could have a role in gastric epithelial damage. For this reason, VacA is a target for therapeutic intervention and also is considered for usage in a vaccine against H. pylori.��
VacA Genotype in Helicobacter pylori | IntechOpen
https://www.intechopen.com/books/helicobacter-pylori-new-approaches-of-an-old-human-microorganism/vaca-genotype-in-em-helicobacter-pylori-em-��
J Ethnopharmacol. 1987 Sep-Oct;21(1):11-9.
Effect of banana powder (Musa sapientum var. paradisiaca) on gastric mucosal shedding.Department of Pharmacology, Banaras Hindu University, Varanasi, India
Mukhopadhyaya K1, Bhattacharya D, Chakraborty A, Goel RK, Sanyal AK.
Author information
Abstract
Banana pulp powder (Musa sapientum Linn. var. paradisiaca) was studied for its effects on gastric mucosal resistance. Banana-treated (0.5 g/kg orally, twice daily for 3 days) rats of either sex showed: (i) a significant increase in the [3H]thymidine incorporation into mucosal cell DNA; (ii) a significant increase in the total carbohydrate (sum of total hexoses, hexosamine, fucose and sialic acid) content of gastric mucosa; (iii) a significant decrease in gastric juice DNA and protein; (iv) a significant increase in the total carbohydrates and carbohydrate/protein ratio of gastric juice. Aspirin treatment to rats caused similar effects as banana on the [3H]thymidine incorporation into mucosal cell DNA but showed opposite effects on the other parameters. These results suggest that banana treatment increased and aspirin decreased the gastric mucosal resistance as evidenced by a respective decrease and increase in gastric juice DNA, the latter serving as an index of the rate of mucosal shedding. Increased cellular mucus may be the factor for increased mucosal resistance. The results of the present study tend to confirm that plantain banana powder strengthens mucosal resistance and promotes the healing of ulcers.�㽶�۶�θճĤ�����Ӱ�졣
������˹ӡ�ȴ�ѧҩ��ѧϵ������������ӡ��
Mukhopadhyaya K1, Bhattacharya D, Chakraborty A, Goel RK, Sanyal AK��
ժҪ
�㽶������о����θճĤ������Ӱ�졣���㽶����(0.5 g/kg�ڷ���ÿ�����Σ�����3��)�������������ֳ�:(i)ճĤϸ��DNA��[3H]������ऽ����������;(��)θճĤ��̼ˮ������(���ǡ����ǰ��������ǡ���Һ��)������������;(��)θҺDNA���������Լ���;(��)θҺ����̼ˮ�������̼ˮ������/�����ʱ����������ߡ���˾ƥ�ֶԴ�����������㽶��[3H]������ल���ճĤϸ��DNA���������ƣ���������������Ӱ���෴����Щ����������㽶����������θճĤ�ֿ�������˾ƥ�ֽ�����θճĤ�ֿ�����θҺDNA�ֱ���ٺ����ӣ�θҺDNA��ΪθճĤ�����ʵ�һ��ָ�ꡣϸ���Һ���ӿ�����ճĤ�ֿ������ӵ����ء����о����������֤ʵ�Ž�����ǿճĤ�ֿ������ٽ��������ϡ�
PMID: 2447444 DOI: 10.1016/0378-8741(87)90089-4Effect of banana powder (Musa sapientum var. paradisiaca) on gastric mucosal shedding. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/2447444��
Banana not only is delicious, but also has a lot of medical value.
This fruit is made of water (75%), carbohydrate (23%), proteins (1%), fatty acids (0,3%) and alimentary fiber (2,3%) and their proportions can change according to the grade of maturation and species. Moreover, pulp contains lots of vitamins such as vitamin A, B2, B1, C, PP, B6 (it promotes protein��s metabolism) and E, and it is full of minerals as calcium, phosphorus, iron and potassium.
However, most important and studied effects of bananas are:
• inhibition of hiv replication
• improving physical performances
• prevention from hypertension and stroke
• cure for depression
• regulation of bowel��s motility
• cross reaction in allergies.
• antiacid action on stomachNow let��s focus on the effect antiacid, various researchers have reported that bananas pulp protect the gastric mucosa in the laboratory animals against NSAIDs and other ulcerogens. Also, it appears to stimulate growth of the gastric lining after damage has occurred.
What is the peptic ulcer?
Peptic ulcer is a sore that forms in the lining of the stomach or the duodenum. A peptic ulcer results from an imbalance between some endogenous aggressive factor(s) [hydrochloric acid, pepsin, refluxed bile, leukotrienes, reactive oxygen species ROS] and cytoprotective factors, which include the function of the mucus-bicarbonate barrier, surface active phospholipids, prostaglandins (PGs), mucosal blood flow, cell renewal and migration, non-enzymatic and enzymatic antioxidants and some growth factors.The pathogenesis of ulcers is multifactorial and includes diverse factors such as a stressful lifestyle, alcohol consumption, use of steroidal and nonsteroidal anti-inflammatory drugs (NSAIDs) and drugs which stimulate gastric acid and pepsin secretion, Helicobacter pylori infections, smoking, lower socioeconomic status and family history.
The pattern and distribution of gastritis strongly correlate with the risk of clinical duodenal or gastric ulcers, mucosal atrophy, gastric carcinoma or gastric lymphoma.
Therapeutic target
The main therapeutic target is the control of gastric secretion using antacids, H2 receptor blockers like ranitidine, famotidine, anticholinergics like pirenzepin, telezipine or proton pump blockers like omeprazole, lansoprazole, etc. The prevention or cure of peptic ulcers is one of the most challenging problem in medicine because gastric ulcer therapy faces drawbacks and most of the drugs currently available in the market show limited efficacy against gastric diseases and are often associated with severe side-effects.
The development of new anti-ulcer drugs from medicinal plants is an attractive proposition, because diverse chemical compounds with anti-ulcer activities have been isolated from these plants, and they have shown to produce promising results in the treatment of gastric ulcers. The bioactive molecules (generally alkaloids, glycosides, lupeols, essential oils, e.t.c) isolated from crude extracts have been used directly as therapeutic agents or as starting materials for the synthesis of useful drugs or serve as a model for pharmacologically active compounds in the process of drugs in synthesis.
Banana as a drug
The use of Musa sapientum pulp in peptic ulcer as a component of herbal medicine has been evaluated and found effective. Some studies reported that pectin and phosphatidylcholine in green banana strengthens the mucous-phospholipid layer that protects the gastric mucosa. Other studies highlight reported that leucocyanidin, a natural flavonoid from the unripe banana (Musa sapientum) pulp, protects the gastric mucosa from erosions. Leucocyanidin and the synthetic analogues, hydroxyethylated leucocyanidin and tetra-allyl leucocyanidin were found to protect the gastric mucosa in aspirin-induced erosions in rat by increasing gastric mucus thickness. In addition to what other studies provide evidence on anti-ulcerogenic activity of banana pulp powder in aspirin-, indomethacin-, phenylbutazone-, prednisolone-induced gastric ulcers, and cysteamine- and histamine-induced duodenal ulcers in rats and guinea-pigs, respectively.
Furthermore, people from the South-Western Nigeria do blend the dried Musa sapientumpeels with the yam flour, which is one of their stable foods. Folklore has it that this meal has ameliorative effect on the patients with gastric pain and ulcer.The Leucocyanidin
The Leucocyanidin is a colorless chemical compound, belonging to the group of leucoantociani idrossiflavani, which is extracted from many plants including banana (Musa sapientum) pulp; increases capillary resistance , antagonizes the loss fraction of blood elements and normalizes capillary permeability .
��
The mechanism of action
The mechanism of action of Musa sapientum is by stimulating the growth of the gastric mucosa by increasing mucosal protein i.e. sialic acid and hexosamine, which in turn increase the production of mucus and thus prevent erosion by the ulcer. These significant increased levels of sialic acid and hexosamine correlated with the increased mass of mucosa in the stomach of animals treated with banana. The mode of action of the banana appears to be unlike that of conventional anti-ulcerogenic drugs in that it promotes mucus secretion by stimulating the growth of mucosal cells. The regenerated mucosa cells would rapidly seal damaged areas with a secretory layer of mucus and prevent further erosions due to gastric HCl and pepsin. The active components such as leucocyanidin may be responsible for the antiulcer properties and protect the mucosa by stimulation of cell proliferation, promoting mucus secretion, increasing mucus resistance, inhibiting the Hcl secretion and thus healing the ulcer.
Conclusion
We can conclude by saying that the positive effects of the banana are quite comparable, if not better than the antacid drugs still on the market.�㽶��Ϊҩ��
�Ѿ�����������������������Ϊ��ҩʹ�õİŽ�����ʹ�ã�����������Ч��һЩ�о���������ɫ�㽶�еĹ�������֬���������ǿ����θճĤ��ճҺ��֬�㡣�����о�ǿ��ָ��������������һ������δ�����㽶��Musa sapientum���������Ȼ���ͪ���ɱ���θճĤ������ʴ�����ְ����غͺϳ����������һ����Ļ�ɫ�պ���ϩ�����Ļ�ɫ��ͨ������θճҺ�����������˾ƥ���շ��Ĵ��������е�θճĤ���������о��⣬�㽶����˾ƥ�֣�������������������ͪ���������������θ�����Լ����װ����鰷�����ʮ��ָ������Ŀ��������Ҳ��֤�ݡ�
���⣬���������������ϲ������ǵ�ȷ������İŽ���ľ����ɽҩ�ۻ����һ���������ǵ��ȶ�ʳƷ֮һ������ѧ��Ϊ��ٷ���θʹ���������и������á�
������
��������һ����ɫ��������ڰ״��ӣ�������ֲ������ȡ�������㽶��Musa sapientum��ֽ��������ëϸѪ�ֿܵ�������ѪҺԪ�ص���ʧ�����淶ëϸѪ�����ԡ����û���
�㽶�����û�����ͨ������ճĤ������Һ��ͼ��ǰ��̼�θճĤ���������Ӷ�����ճҺ�IJ������Ӷ���ֹ�������ʴ����Һ��ͼ��ǰ�����Щ�������ӵ�ˮƽ�����㽶�����Ķ����θ��ճĤ������������ء��㽶�����÷�ʽ�ƺ��볣�濹����ҩ��ͬ����Ϊ��ͨ���̼�ճĤϸ�����������ٽ�ճҺ���ڡ�������ճĤϸ����Ѹ����ճҺ�ķ��ڲ��ܷ�����������ֹ����θ���θ����ø������Ľ�һ����ʴ�����Գɷ������������ؿ��ܸ����������ʲ�ͨ���̼�ϸ����ֳ���ٽ�ճҺ���ڣ�����ճҺ���ԣ���������ڴӶ���������������ճĤ��The effects of banana against gastric ulcers
http://flipper.diff.org/app/items/info/7316��
Helicobacter pylori VacA, acting through receptor protein tyrosine phosphatase ��, is crucial for CagA phosphorylation in human duodenum carcinoma cell line AZ-521
Masayuki Nakano, Kinnosuke Yahiro, Eiki Yamasaki, Hisao Kurazono, Junko Akada, Yoshio Yamaoka, Takuro Niidome, Masanori Hatakeyama, Hidekazu Suzuki, Taro Yamamoto, Joel Moss, Hajime Isomoto, Toshiya Hirayama
Disease Models & Mechanisms 2016 9: 1473-1481; doi:1Department of Bacteriology, Institute of Tropical Medicine, Nagasaki University, 1-12-4, Sakamoto, Nagasaki 852-8523, Japan
2Department of International Health, Institute of Tropical Medicine, Nagasaki University, 1-12-4, Sakamoto, Nagasaki 852-8523, Japan
3Department of Molecular Infectiology, Graduate School of Medicine, Chiba University, 1-8-1, Inohana, Chuo-ku, Chiba 260-8670, Japan
4Division of Food Hygiene, Department of Animal and Food Hygiene, Obihiro University of Agriculture and Veterinary Medicine, Nishi 2-11, Inada-cho, Obihiro, Hokkaido 080-8555, Japan
5Department of Environmental and Preventive Medicine, Oita University Faculty of Medicine, Idaigaoka 1-1, Yufu, Oita 879-5593, Japan
6Department of Medicine, Gastroenterology and Hepatology Section, Baylor College of Medicine, Houston, TX 77030, USA
7Department of Applied Chemistry and Biochemistry, Graduate School of Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
8Division of Microbiology, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-Ku, Tokyo 113-0033, Japan
9Medical Education Center, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan
10Cardiovascular and Pulmonary Branch, NHLBI, National Institutes of Health, Bethesda, MD 20892-1590, USA
11Division of Medicine and Clinical Science, Tottori University Faculty of Medicine, 86 Nishi-cho, Yonago, Tottori 683-8503, Japan
ABSTRACT
Helicobacter pylori, a major cause of gastroduodenal diseases, produces vacuolating cytotoxin (VacA) and cytotoxin-associated gene A (CagA), which seem to be involved in virulence. VacA exhibits pleiotropic actions in gastroduodenal disorders via its specific receptors. Recently, we found that VacA induced the phosphorylation of cellular Src kinase (Src) at Tyr418 in AZ-521 cells. Silencing of receptor protein tyrosine phosphatase (RPTP)��, a VacA receptor, reduced VacA-induced Src phosphorylation. Src is responsible for tyrosine phosphorylation of CagA at its Glu-Pro-Ile-Tyr-Ala (EPIYA) variant C (EPIYA-C) motif in Helicobacter pylori-infected gastric epithelial cells, resulting in binding of CagA to SHP-2 phosphatase. Challenging AZ-521 cells with wild-type H. pylori induced phosphorylation of CagA, but this did not occur when challenged with a vacA gene-disrupted mutant strain. CagA phosphorylation was observed in cells infected with a vacA gene-disrupted mutant strain after addition of purified VacA, suggesting that VacA is required for H. pylori-induced CagA phosphorylation. Following siRNA-mediated RPTP�� knockdown in AZ-521 cells, infection with wild-type H. pylori and treatment with VacA did not induce CagA phosphorylation. Taken together, these results support our conclusion that VacA mediates CagA phosphorylation through RPTP�� in AZ-521 cells. These data indicate the possibility that Src phosphorylation induced by VacA is mediated through RPTP��, resulting in activation of Src, leading to CagA phosphorylation at Tyr972 in AZ-521 cells.
Putative model showing VacA-induced CagA phosphorylation during H. pylori infection in AZ-521 cells. H. pylori injects CagA into gastric epithelial cells by a type-IV secretion system and secretes VacA into the extracellular space (step 1). Secreted VacA binds to the extracellular domain of RPTP�� (step 2). VacA induces Src phosphorylation at Tyr418 through RPTP��, resulting in activation of Src (step 3). It has been shown that activated Src promotes tyrosine phosphorylation at Tyr972 of an EPIYA-C motif in translocated CagA and pCagA then inactivates Src (Selbach et al., 2003; Mueller et al., 2012) (step 4). Finally, pCagA interacts with other host molecules, including SHP2 phosphatase (step 5).
INTRODUCTION
Helicobacter pylori is a major causative agent for the development of gastroduodenal diseases, including chronic gastritis, peptic ulcer and gastric cancers (Blaser and Atherton, 2004; Peek and Blaser, 2002). It has been proposed that about 50% of the world's population is infected with H. pylori, but only a small number of the infected individuals develop severe clinical manifestations such as gastric adenocarcinoma (Wroblewski et al., 2010). Although a number of virulence factors have been found in H. pylori, vacuolating cytotoxin (VacA) and cytotoxin-associated gene A (CagA) are considered to be the major factors in H. pylori-induced diseases (Blaser and Atherton, 2004; Peek and Blaser, 2002).
VacA is a potent cytotoxin secreted by most clinical isolates of H. pylori, and shows pleiotropic actions in cultured gastric epithelial cells, including generation of vacuoles in the cytoplasm, mitochondrial damage leading to apoptosis, and modulation of signal transduction pathways associated with immune responses (Boncristiano et al., 2003; Hisatsune et al., 2007; Isomoto et al., 2010; Nakayama et al., 2004, 2009; Yamasaki et al., 2006). To facilitate their biological actions in host cells, VacA binds to specific surface receptors. We have identified three different cell surface proteins as VacA receptors: receptor protein tyrosine phosphatase �� and �� (RPTP�� and RPTP��) and low-density lipoprotein receptor-related protein-1 (LRP1) (Yahiro et al., 1999, 2003, 2012). In addition, other molecules, including sphingomyelin, have been reported to serve as VacA receptors (Gupta et al., 2010). Of these VacA receptors, during H. pylori infection, RPTP�� is associated with the development of gastric ulcers in experimental animal models and LRP1 is involved in VacA-dependent autophagy, followed by CagA degradation in infected host cells (Fujikawa et al., 2003; Tsugawa et al., 2012; Yahiro et al., 2012). These data suggest that both receptors are involved in intoxication by VacA. Therefore, we speculate that both receptors, RPTP�� and LRP1, are associated with the development of gastric disorders in H. pylori infection. However, the role of RPTP�� in intoxication with VacA is unclear.
Previous studies have shown that RPTP�� contributes to activation of cellular Src kinase (Src) and other Src family kinase (Su et al., 1999). It has been shown that Src activity is elevated in RPTP��-overexpressing cultured cells, whereas the opposite was observed in RPTP��-deficient cells (den Hertog et al., 1993; Harder et al., 1998; Su et al., 1999; Zeng et al., 2003; Zheng et al., 1992). Furthermore, it has been reported that Src kinase activity is reduced in RPTP��-knockout mice (Harder et al., 1998; Ponniah et al., 1999). Therefore, RPTP�� is an important physiological regulator of Src. RPTP�� can dephosphorylate both phosphorylated tyrosine residues, pTyr530 and pTyr418 (human Src numbering throughout; the inhibitory phosphorylation site and active site of Src, respectively), thereby causing Src activation following autophosphorylation of Tyr418 (Boggon and Eck, 2004; Vacaru and den Hertog, 2010; Zheng et al., 2000). In addition, based on immunohistochemistry using human gastric cancer tissues, it has been suggested that RPTP�� is associated with the progression of gastric cancer (Wu et al., 2006).
In the present study, we show the role of RPTP�� in VacA intoxication and also demonstrate that VacA is associated with CagA phosphorylation in AZ-521 cells during H. pylori infection. We propose the possibility that VacA induces CagA phosphorylation through RPTP�� in AZ-521 cells.
RESULTS
VacA induces Src phosphorylation in vitro
Previous studies have shown that VacA modulates signal transduction pathways in AZ-521 cells (Hisatsune et al., 2007; Nakayama et al., 2009). In this study, we found that VacA enhanced phosphorylation at Tyr418 in Src, but heat-inactivated VacA (iVacA) did not have similar effects (Fig. 1A). We also examined the effect of VacA on Src phosphorylation at Tyr418 using other gastric epithelial cells: AGS cells (a human gastric adenocarcinoma cell line) and NUGC3 cells (a gastric cancer cell line). Although VacA stimulated Src phosphorylation at Tyr418 in NUGC3 cells as well as in AZ-521 cells, we did not find any effects of VacA in AGS cells (Fig. 1B), suggesting that Src phosphorylation induced by VacA is dependent on cell type.VacA-induced Src phosphorylation is mediated by RPTP��
VacA intoxicates host cells through specific surface receptors. We previously identified three molecules (RPTP��, RPTP�� and LRP1) as VacA receptors (Yahiro et al., 1999, 2003, 2012). To evaluate which of these VacA receptors contributes to Src phosphorylation, we used small-interfering RNA (siRNA)-mediated knockdowns, which targeted each of the VacA receptors, in AZ-521 cells. Although VacA induced phosphorylation at Tyr418 of Src in the presence of control siRNA, VacA did not induce phosphorylation at Tyr418 of Src in siRNA-mediated RPTP�� knockdown AZ-521 cells (Fig. 2A). On the other hand, in AZ-521 cells, VacA enhanced phosphorylation at Tyr418 in Src after RPTP�� or LRP1 silencing (Fig. 2B,C). To verify these results, we also examined Src phosphorylation induced by VacA using the RPTP�� constitutive-knockdown AZ-521 cells constructed by a shRNA lentiviral expression system (Yahiro et al., 2012). We found that VacA did not enhance phosphorylation at Tyr418 in Src in RPTP�� constitutive-knockdown AZ-521 cells (Fig. 3), consistent with the results using the siRNA-mediated RPTP�� knockdown AZ-521 cells (Fig. 2A). Taken together, we speculate that RPTP��, but not RPTP�� or LRP1, is involved in VacA-dependent phosphorylation at Tyr418 in Src.
VacA production by H. pylori induces CagA phosphorylation and co-immunoprecipitation with SHP2 phosphatase
Previous studies have shown that CagA is delivered into gastric epithelial cells directly via a type-IV secretion system and then translocated CagA is tyrosine-phosphorylated by Src family kinases, including Src (Odenbreit et al., 2000; Selbach et al., 2002; Stein et al., 2002). In this study, we have shown that VacA induces phosphorylation at Tyr418 in Src in AZ-521 cells (Figs 1 and 2). We therefore hypothesized that Src phosphorylation induced by VacA is important in CagA phosphorylation during H. pylori infection. To evaluate this hypothesis, we examined whether VacA production of H. pylori stimulates CagA phosphorylation by challenging cells with wild-type and vacA mutant strains in AZ-521 cells. Although total CagA protein, precipitated with anti-CagA antibody, was similar in both wild-type and vacA mutant strains, the amount of phosphorylated CagA (pCagA) seen following infection with wild-type H. pylori was significantly greater than that seen following infection with a vacA mutant strain (Fig. 4A). We next verified the effect of VacA on SHP2 phosphatase, because it has been demonstrated that SHP2 phosphatase specifically binds to pCagA in H. pylori-infected host cells (Higashi et al., 2002a). When we carried out immunoprecipitation using anti-CagA antibody following H. pylori infection of AZ-521 cells, precipitated SHP2 phosphatase following infection with a vacA mutant strain was decreased compared with that seen with wild-type H. pylori infection (Fig. 4A). In addition, we also analyzed the effect of VacA on CagA phosphorylation in NUGC3 cells. We found that CagA phosphorylation was stimulated in the early phase (2 h) of infection with wild-type H. pylori more than was seen following infection with a vacA mutant strain. This was not the case in the late phase of infection (8 h) (Fig. S1).VacA induces CagA phosphorylation in cagA gene-transfected cells in an RPTP��-dependent manner
Next, we examined the effect of RPTP�� silencing on VacA-induced CagA phosphorylation in cells infected with wild-type H. pylori. We found that silencing of the RPTP�� gene with siRNA resulted in a significant reduction of VacA-induced CagA phosphorylation, compared with that in control siRNA-transfected cells. A prior study showed that ��1 integrin is associated with CagA translocation in H. pylori-infected cells through its interaction with CagL, a component of the type IV secretion apparatus of H. pylori, resulting in induction of phosphorylation in both of FAK and Src (Kwok et al., 2007). In addition, they speculated that interaction between CagL and the ��1-integrin�CFAK�CSrc signaling pathway was associated with CagA phosphorylation (Kwok et al., 2007). When we examined the effect of ��1-integrin gene silencing on VacA-induced CagA phosphorylation in cells infected with wild-type H. pylori, we found that silencing of the ��1-integrin gene mediated by siRNA did not result in differences of VacA-induced CagA phosphorylation compared to control siRNA-transfected cells (Fig. 6A), suggesting that RPTP�� is responsible for CagA phosphorylation induced by VacA in AZ-521 cells. In addition, we assessed whether RPTP�� is crucial for CagA phosphorylation by VacA using AZ-521 cells co-transfected with RPTP�� siRNA and plasmid harboring an HA-tagged cagA gene. We found that VacA did not induce CagA phosphorylation in AZ-521 cells co-transfected with RPTP�� siRNA and plasmid harboring an HA-tagged cagA gene (Fig. 6B). Taken together, these results indicate that RPTP�� contributes to the VacA-induced CagA phosphorylation in AZ-521 cells during H. pylori infection.
In summary, we have shown that VacA promotes CagA phosphorylation through RPTP�� in AZ-521 cells (Fig. 7). Our findings demonstrate the possibility that VacA induces CagA phosphorylation in AZ-521 cells during H. pylori infection through the VacA-RPTP��-Src signaling pathway. Activation of CagA by a VacA-dependent pathway in AZ-521 cells seems to be important in the pathogenesis seen in H. pylori infection.Helicobacter pylori VacA, acting through receptor protein tyrosine phosphatase ��, is crucial for CagA phosphorylation in human duodenum carcinoma cell line AZ-521 | Disease Models & Mechanisms
https://dmm.biologists.org/content/9/12/1473��
Avicenna J Phytomed. 2016 Sep-Oct; 6(5): 495�C501.
PMCID: PMC5052411
PMID: 27761418
Assessment of antibacterial effect of garlic in patients infected with Helicobacter pylori using urease breath test
Mahmoud Zardast,1 Kokab Namakin,2,* Jamil Esmaelian Kaho,3 and Sarira Sadat Hashemi3
Author information Article notes Copyright and License information
1Birjand Atherosclerosis AndCoronary Artery Research Center, Department of pathology, Birjand University of Medical Sciences (BUMS), Birjand, Iran
2Department of pediatrics, Birjand University of Medical Sciences (BUMS), Birjand, Iran
3Birjand University of Medical Sciences, Birjand, Iran (BUMS)
*Corresponding Author: Tel: +98 915 561 2001, Fax: +985632220505, d_namakin@yahoo.com
Abstract
Objective:
Helicobacter pylori (H. pylori) is the most common pathogenic bacteria in the stomach. The aim of the current study was to explore the effect of oral garlic administration on bacterial urease activity inside the stomach and its contribution to the treatment of H. pylori infection.
Materials and Methods:
In this clinical trial, 15 patients were studied quantitatively with Urease Breath Test (UBT). The patients with gastrointestinal symptoms and a positive serum H. pylori IgG were enrolled. UBT was performed for each patient in three sessions as follows: at the beginning of the study, an initial UBT was performed based on which, the positive cases entered the study and the negative ones were excluded. Second UBT was done three days later in patients who were not receiving any treatment and were considered as the control, whereas the third UBT was performed three days after prescribing two medium-sized cloves of garlic (3 g) with their meal, twice a day (at noon and in the evening). The collected data were analyzed using ANOVA and Bonferroni tests and the significance level was set at p<0.05.
Results:
the mean UBT significantly differed before and after treatment with garlic cloves, being significantly lower after garlic consumption. No meaningful difference was observed in the mean UBT without garlic consumption between the first and second steps.
Conclusion:
Raw garlic has anti-bacterial effects against H. pylori residing in the stomach and may be prescribed along with routine drugs for the treatment of gastric H. pylori infection.Materials and Methods
H. pylori detection techniques
H. pylori can be detected by several methods. Among them, few methods are able to quantitatively evaluate the activity of intra-mucosal live bacteria. Anti-H.pylori antibodies do not immediately decrease following treatment initiation and require eat least a few months. Therefore, the immediate level of antibodies in serum does not show the success of treatment. Endoscopy and biopsy are relatively aggressive techniques which cannot assess the early effects of treatment easily. Fecal antigens also show a lot of diversity. Quantitative UBT, on the other hand, can assess the early bacteriostatic effects of bacteria. By this method, the urease activity level of bacteria can be measured in a short time period (Count Per Minutes (CPM)).
Study design
In this pre-post-clinical trial, 15 patients with H. pylori infection were studied. The patients were enrolled by simple sampling. All patients who referred to the Gastroenterology clinic of Vali-Asr Hospital, Birjand, Iran from May 2013 to Jan 2014 with complaints of dyspepsia and gastrointestinal problems such as abdominal pain, loss of appetite, nausea and flatulence with a positive H. pylori IgG test were asked to participate in the study after fully explaining the study protocol. A questionnaire consisting of demographic data was completed for each participant and they were subsequently referred to Shafa Laboratory of Immunology, Birjand, Iran in order to completecarbon-14 urease breath test (UBT) for the diagnosis of active H. pylori infection.
The study protocol was approved by the Ethics Committee of Birjand University of Medical Sciences, Birjand, Iran and an informed consent was obtained from each participant prior to study entrance.
UBT conditions and performance
The conditions required for performing a quantitative UBT were as follows: fasting for at least 6 hours, no antibiotics and bismuth consumption during the last4 weeks, not having used any proton pump inhibitors, antacids, and histamine receptor blockers (e.g. ranitidine, famotidine and cimetidine) at least for a week and not smoking since one hour before the test. Each patient swallowed a C*14 urea capsule with a glass of water in the seated position and after 10 minutes they blew into a special breathing card; the respiratory card was placed in a plastic bag with aluminum coverage and was counted by a Gamma counter in the least possible time frame. In case of positivity of the gamma counter, the patient was diagnosed with active H. pylori infection and entered the study.
For three days, the patients were banned from consuming garlic or any other preparations of the alliums family, antibiotics and acid-reducing drugs. At the end of the three treatment-free days, the quantitative UBT count was performed once again and recorded as the control. Two medium cloves of raw garlic (3 g) were then administered toeach patient twice a day for 3 days with their daily meals. At the end of the 3-day treatment course, quantitative UBT counting was repeated for the patients, this time regarded as the cases.
Statistical analysis
Gamma counter results, expressed as Count Per Minute (CPM), were analyzed by SPSS statistical software ver. 16 and the Kolmogorov-Smirnov test was applied to assess the normal distribution of the collected data. Since the data were normally distributed, intra- group ANOVA and Bonferroni tests were used to analyze the data at the significance level of p<0.05.Discussion
The results of this study showed that consumption of fresh garlic inhibits H. pylori activity inside the stomach mucosa to a great extent which indicates a bacteriostatic effect for garlic; bactericidal effects may take place at higher concentrations of garlic or following longer treatment courses. Various studies support the inhibitory effect of garlic on the growth of H. pylori (Jonkers et al., 1999 ▶).
In this context, Cutler and Wilson (2004) ▶ showed that garlic has a wide range of antibacterial activities and is also effective against H. pylori infection. Also, Ghobeh et al. (2010) ▶ reported that consumption of 4 gr of garlic powder leads to eradication of bacteria in 87% of H. pylori positive individuals. However, McNulty et al. (2008) ▶ study did not reveal any association between consumption of garlic powder and eradication of H. pylori infection.
Numerous studies have investigated the effect of garlic on various bacteria. Tsao et al. (2001) ▶ studied the effect of garlic oil and diallyltrisulfide and diallyltetrasulfide on Pseudomonas aeruginosa and showed that these elements potentially prevent or treat nosocomial infections and infections due to antibiotic-resistant strains of bacteria. Kazemizadeh et al. (2011) ▶ studied the effect of garlic extract on Enterococcus faecalis whereas Hosseini Jazini et al. (2007) ▶ studied the effect of garlic extract on multidrug-resistant Acinetobacter strains. These studies have pointed out the broad range of antibacterial effects of garlic.
In vitro administration of garlic extract at room temperature prevented the activity of gram negative bacteria such as Salmonella and E. coli and gram-positive bacteria including streptococcus type A and anthracis type B. Such inhibitory effects of garlic are even more marked than those of penicillin as 1mg of allicin is as effective as 15 standard units of penicillin (Farkhondeh and Aliporyegane., 2012).
The main components of garlicareorganosulfur (aleinandallicin), organic acids, carbohydrates and vitamins and the most important property of garlic, which is its antimicrobial effect, is attributed to allicin or garlic oil with 162.3 KD molecular weight.
Allicin is not naturally present in garlic cloves, but it is produced after hydrolysis and oxidation of alein. The mechanism(s) for the antimicrobial activity of allicin and garlic extract have not been yet fully investigated; however, a number of mechanisms have been suggested in this respect. Interference with the function of enzymes and proteins containing the sulfhydryl group (SH) is one of such mechanisms. Allicin irreversibly inhibits the SH proteases and NADP-dependent alcohol dehydrogenase (Shapoury et al., 2004 ▶). According to Inder and Qiutang (2002) ▶ study, garlic blocks the activity of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-��B ).This factor increases the expression of inflammatory cytokines and is one of the key molecules in inflammation and cancer. Activation of this nuclear factor is mediated byTLR4 receptor (Toll-like receptors). TLR4 are involved in induction of immune responses. Many of these receptors contain cysteine in their extracellular and cytoplasmic sectors (Iwalokun et al., 2004 ▶).
In addition, allicin in garlic contains compounds called thiosulfinate that can interact with cysteine. As a result, allicin can react with the cysteine which is in the structure of these receptors resulting in the inhibition of signaling pathways associated with TLR4 on the surface of cellular receptors. Allicin prevents the activation of NF-��B by inhibiting TLR4 signaling. This inhibition is considered as one of the main mechanisms via which garlic induced itsanti-inflammatory effects (Ghobeh et al., 2010 ▶).
Furthermore, H. pylori produces Heat Shock Proteins (HSP), urease and lipopolysaccharidase which are absorbed by the stomach epithelial cells. Following absorption, they pass through the mucous and synthesize inflammatory factors such as CRP, IL-8 and TNF-a (Hekmatdoosta et al., 2015 ▶).
Iwalokun et al. (2004) ▶ findings suggest that garlic extract is effective on the pathogenesis of toxic bacteria by preventing the toxin production. Moreover, it has been noted that allicin can reduce H. pylori infection by blocking nitrous synthesis and scavenging bnitrates and free radicals from the body.
This study had certain limitations; the study population was small and the effect of garlic was not compared with the routine anti-H. pylori regimens.
Key Words: Helicobacter pylori, Urease Breath Test (UBT), Garlic, StomachAssessment of antibacterial effect of garlic in patients infected with Helicobacter pylori using urease breath test
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5052411/��
Gastrointestinal - Garlic and helicobacter pylori revisited
medherb.com/Therapeutics/Gaastrointestinal...
In-vitro-research indicates that concentrations of allicin such as would be found in 2-6 cloves of garlic blended in a quart of water are effective against H. pylori (Sivam et al), a dose is in the same range that Dharmananda reports effective clinically.
SHP1 inactivates H. pylori oncoprotein | Technology Org
https://www.technology.org/2016/04/01/shp1-inactivates-h-pylori-oncoprotein
A University of Tokyo research group found that the enzyme SHP1 is the long-sought protein tyrosine phosphatase that inactivates the oncogenic (cancer inducing) potential of the CagA protein ...
allicin vs Helicobacter - Helicobacter Forums
https://www.helico.com/forums/viewtopic.php?t=202959
Apr 16, 2012 �� We should use as well some 1g of allicin... 1 g is probably much to much but when some dose of 100mg allicin show no effect it will be convincing (I mean real allicin not powder not sliced garlic cloves of unknown origin, no garlic oil with low allicin content) ... ↳ Ideas for Resistant H.pylori Infections ↳ Yoghurt, Lactobacilli ...��
Therap Adv Gastroenterol. 2016 Nov; 9(6): 836�C844.
Helicobacter pylori and gastric acid: an intimate and reciprocal relationship
Helge L. Waldum, Per M. Kleveland, and Øystein F. Sørdal
Helge L. Waldum, Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Trondheim, Norway;
Abstract
Helicobacter pylori (Hp) is the main cause of gastritis, peptic ulcer disease and gastric cancer. There are still unanswered questions related to the interaction between Hp and man, like what determines the susceptibility for the initial infection and the mechanisms for the carcinogenic effect. The initial infection seems to require a temporal gastric hypoacidity. For Hp to survive in the gastric mucous layer, some acidity is necessary. Hp itself is probably not directly carcinogenic. Only when inducing oxyntic mucosal inflammation and atrophy with hypoacidity, Hp predisposes for gastric cancer. Gastrin most likely plays a central role in the Hp pathogenesis of duodenal ulcer and gastric cancer.
Keywords: gastric acid, gastric cancer, gastrin, gastritis, Helicobacter pylori, peptic ulcer
Production of acid in the upper gastrointestinal tract has been preserved during phylogenesis [Johnsen, 1998], reflecting the importance of the main function of gastric juice; that is killing of swallowed microorganisms [Wilder-Smith et al. 1992]. The normal gastric juice creates a hostile milieu for microorganisms, making the luminal content of the stomach as well as the small intestine relatively sterile. Inflammation of the gastric mucosa was some decades ago so prevalent [Siurala et al. 1968] that gastritis even was considered a natural consequence of aging. However, it was rather early recognized that gastritis was related to gastric cancer since gastric cancer only developed in stomachs with gastritis [Morson, 1955]. It was, therefore, a great breakthrough when it was shown that Helicobacter pylori (Hp) infection was the major cause of gastritis [Marshall and Warren, 1984]. Although it is more than 25 years since the central role of Hp in the pathogenesis of upper gastrointestinal disease was realized, there are still unresolved questions related to the interaction between Hp and the host, like the mechanism for the carcinogenic effect and the susceptibility for Hp infection. This paper aims to make a concise review of the interactions between Hp and humans.
Acute infection by Helicobacter pylori
Whereas it is well known how Helicobacter pylori (Hp) can survive in the superficial mucous layer by its urease activity causing a livable pH in its vicinity, the mechanisms by which Hp can survive and proliferate when infecting a normal stomach are not so well understood. Nevertheless, the two known voluntary infections were successful only after inhibition of gastric acidity [Marshall et al. 1985; Morris and Nicholson, 1987], suggesting the role of gastric juice in the defense against the initial infection. Both these subjects developed self-limited symptoms from the epigastric area with fullness, nausea and vomiting some days after the infection and lasting for about a week. Similarly, in an outbreak of gastritis due to contamination of equipment used in a study where gastric acid secretion was determined multiple times in healthy subjects, the participants developed hypoacidity, gastritis and similar symptoms [Ramsey et al. 1979] as those voluntarily infected with Hp. Hp has retrospectively been presumed to be the causative agent. Also in these subjects, Hp may have been introduced to the stomach without gastric juice, which was continuously aspirated or as part of studying meal-stimulated acid secretion where the luminal content in vivo was titrated to pH 5.0. Thus, during the two voluntary infections [Marshall et al. 1985; Morris and Nicholson, 1987], as well as the transmission by the nasogastric tube [Ramsey et al. 1979], Hp entered a stomach without acid [Figure 1(a)], allowing the bacterium to bury into the mucous layer before normal gastric acidity was reestablished. In most cases of chronic Hp gastritis, there is no information of any symptomatic episode. This may indicate that most of the acute infections are asymptomatic, or alternatively, a gastroenteritis due to Hp infection has been misdiagnosed as viral. Moreover, childhood Hp infection seems to be prevalent [Thomas et al. 1999], which may be explained by reduced gastric acidity during early life [Agunod et al. 1969; Rodbro et al. 1967]. Alternatively, gastroenteritis by other causes may make the gastric content hypoacidic and thus give time and possibility for Hp to proliferate and infect the stomach. The higher frequency of Hp infection in underdeveloped countries [Weaver, 1995] may perhaps be explained by a higher frequency of gastroenteritis in these countries. The initial infection affects both oxyntic and antral mucosa [Ramsey et al. 1979; Morris and Nicholson, 1987]. We do not know the mechanisms behind the resolution of the initial infection resulting in loss of symptoms and restoration of acid secretion [Morris and Nicholson, 1987; Ramsey et al. 1979]. Nevertheless, it seems that the infection persists in all infected subjects, with some having the ability to limit the infection to the antral mucosa whereas others develop a chronic pan-gastric infection at an early stage.
Figure 1.
The different phases of the relationship between Helicobacter and gastric acid.
(a) The initial infection is facilitated by reduced gastric acidity. (b) The initial infection causes reduction in gastric acidity. (c) Although the infection persists, gastric acidity is by some reason or the other restored. (d) When the Hp infection causes atrophy of the oxyntic glands, gastric acid secretion declines. (e) When the oxyntic atrophy is so pronounced that sufficient gastric acidity is not reached, hypergastrinemia develops and the patient is concomitantly predisposed for gastric cancer.
Thus, gastric acidity probably has a protective role in the defense against the initial Hp infection. The use of inhibitors of gastric acid secretion in small children, which recently has been reported to predispose for Clostridium infection [Nylund et al. 2014], may also make these children more susceptible to Hp infection.
Mechanisms of hypoacidity during the initial infection
The course of acute Hp gastritis or epidemic hypochlorhydria, which has been used synonymously, may last for weeks or months [Marshall et al. 1985; Morris and Nicholson, 1987; Ramsey et al. 1979]. The mechanism behind the reduced gastric acidity in the acute phase of Hp infection is not known, but properties both by Hp itself and the inflammation this infection provokes have been implicated [Calam, 1995] [Figure 1(b)]. Thus, Hp infection may induce the production of cytokines like interleukin (IL)-1�� [Noach et al. 1994] having an inhibitory effect on gastric acid secretion [Wallace et al. 1991]. A direct effect on acid secretion by Hp itself is supported by studies in isolated parietal cells [Cave and Vargas, 1989]. Among the Hp-derived factors involved in the reduction of gastric acidity during acute infection are NH3, fatty acids or a substance having inhibitory effects on the H+/K+-ATP-ase [Calam, 1995]. For more than 20 years it has been shown that Hp and fatty acids produced by Hp block H+/K+-ATP-ase [Beil et al. 1994] and more recently that Hp represses proton-pump expression as well [Saha et al. 2010]. Some cytokines liberated by the inflammation may also have a profound effect on acid secretion [Saperas et al. 1990].
Transition to chronic infection
We do not know for sure, but the prevailing hypothesis is that, once infected, Hp gastritis becomes chronic; if not, Hp is eradicated by treatment. The transition from acute to chronic gastritis is accompanied by restoration of gastric acid secretion [Ramsey et al. 1979; Morris and Nicholson, 1987]. However, what happens in the stomach in this phase is not known. Moreover, why the infection is confined to the antral mucosa in some patients whereas pan-gastritis occurs in others is unknown, although differences in mucosal acidity may play a role. The oral spread of infection caused by inhibition of gastric acid secretion [Logan et al. 1995] also suggests that local acidity plays a role in the distribution of Hp infection.
The spiral shape and flagella help Hp to bore into the mucous layer [Calam, 1995] where there is a pH gradient due to H+ diffusing from the luminal side and HCO3− from the epithelial cells. Hp is dependent on a near neutral pH to thrive; the urease activity of Hp [Marshall et al. 1990] increases pH in the vicinity, thus making it possible for Hp to survive at a more acidic place. In many ways, a slightly acidic milieu is ideal for Hp growth since its NH3 production otherwise could induce too alkaline milieu for the bacterium [Scott et al. 1998].
When Hp infection is mainly confined to the antral mucosa, there may be increased gastrin release leading to augmented gastric acid secretion predisposing to duodenal ulcer [Levi et al. 1989]. The mechanism for the increased gastrin release is possibly local alkalization by NH3 produced by Hp urease. The urease theory for the stimulation of gastrin release is attractive, but is admittedly not well supported experimentally. Thus an infusion of urea into the stomach of seven patients with duodenal ulcer infected with Hp increased intragastric ammonium concentration threefold, but did not affect plasma gastrin [Chittajallu et al. 1991]. However, it is not clear whether ammonium produced intragastrically will reach and affect the function of the G cell directly or the somatostatin D cell, both being localized deep in the glands. Interestingly, when Chittajallu and colleagues determined the ammonium concentration 1 month after Hp eradication, it was significantly reduced compared with the baseline value before eradication [Chittajallu et al. 1991]. Thus, it may be that the ammonium concentration before starting the urea infusion had maximal effect? More problematic for the urease theory with respect to stimulation of gastrin release by Hp is the lack of a reduction in gastrin in six patients with Hp-positive duodenal ulcer dosed with the urease inhibitor acetohydroxamic acid [El Nujumi et al. 1991]. The efficacy of the urease inhibitor was controlled by the urease breath test [El Nujumi et al. 1991]. However, since the degree of hypergastrinemia in patients with duodenal ulcer secondary to Hp is very small [Lanzon-Miller et al. 1987] and well within the normal range, gastrin has to be determined with a sensitive and accurate method to detect any difference, and only six subjects may be too few to detect a significant change [El Nujumi et al. 1991]. Moreover, the ammonium concentration after acetohydroxamic acid intake was about half of that measured the placebo day. Since we do not know the concentration relationship between intragastric ammonium and gastrin release, this single point study does not exclude that the urease activity of Hp is responsible for the hypergastrinemic effect. In another study from the same group [El-Omar et al. 1993] gastrin and acid secretion in Hp-positive patients with duodenal ulcer, Hp-positive healthy individuals and Hp-negative healthy individuals were determined at the basal state and during stimulation with gastrin-releasing peptide (GRP). They found that basal and GRP induced gastrin release as well as acid secretion were highest in patients with duodenal ulcer and Hp, but also augmented compared with Hp-positive healthy controls. At reexamination 1 month after Hp eradication, gastrin was at the level of healthy Hp negative controls whereas gastric acid secretion, although markedly reduced, apparently still was higher than in controls [El-Omar et al. 1993]. From these results it may be interpreted that Hp is responsible for inappropriate gastrin release, whereas trophic effects by this slight hypergastrinemia, particularly on the enterochrommafine like (ECL) cell [Brenna and Waldum, 1992], still results in elevated gastric acid secretion. It is also tempting to suggest that the difference in gastric acid secretion between Hp-positive persons with or without duodenal ulcer [El-Omar et al. 1993] may reflect different degrees of trophic effect on the cells regulating acid secretion.
There have been reports indicating that antral Hp infection could have trophic (positive or negative) effects on G and especially D cells [Moss et al. 1992], which would be expected to influence gastrin release even at neutral pH. In this context it should be recalled that neuroendocrine (NE) cells have a long lifespan [Fossmark et al. 2005], and thus any effect could persist for a long time after Hp eradication. The trophic effects could be caused by chronic effects of alkalization by ammonium. Therefore, it is difficult to exclude from the present literature that the effect of Hp on gastrin release is not caused by urease activity. Whether H+ directly affects the G cell or the antral D cell is not settled, but is not important from a functional point of view. Both the G cell and the antral D cell are of the open type and thus influenced by the gastric content. From this it is probable the gastrin release from the G cell is affected both directly from the gastric content and indirectly via somatostatin from D cells. However, there is an indication for an increase in antral gastrin and a fall in antral somatostatin in patients infected with Hp [Odum et al. 1994]
Previously it was hypothesized that duodenal ulcers developed at spots of gastric metaplasia [Carrick et al. 1989]. However, patients with gastrinoma develop ulcers without Hp infection [Weber et al. 1997], showing that increased acid secretion is sufficient to induce peptic ulcers. It should be added that the degree of gastrin increase in blood in patients with duodenal ulcer is small [Lanzon-Miller et al. 1987] due to the restraint on gastrin release by the increase in gastric acidity [Walsh et al. 1975] provoked by the small gastrin elevation. The sensitivity for gastrin with respect to its main physiological action, that is stimulation of histamine release from the ECL cell, is very high [Sandvik and Waldum, 1990]. Therefore, in most patients with duodenal ulcer there is no hypergastrinemia, but nevertheless a certain inappropriate hypergastrinemia in relation to gastric acidity [Smith et al. 1990]. There is a case report describing increased gastric acid secretion and a moderate hypergastrinemia in two patients with peptic ulcer where both gastrin and gastric acid secretion fell to normal levels after eradication of Hp [Metz et al. 1995]. The mechanism for the higher gastrin values than expected from gastric acidity in these two patients was not explained.
Mechanism for the hypersecretion of acid in patients with Hp-related duodenal ulcer
Patients with duodenal ulcer have for a long time been known to have increased gastric acid secretion [Wormsley and Grossman, 1965] and inappropriate gastrin release induced by antral Hp infection is now the accepted cause since gastrin as well as acid secretion (both basal and stimulated GRP and pentagastrin) are reduced 6 months after eradication of Hp [Harris et al. 1996] [Figure 1(c)]. The reduction in pentagastrin-stimulated acid secretion after Hp eradication probably reflects a reduction in the trophic effect on the ECL cell by gastrin, which is evident even at rather low gastrin concentrations which affect ECL cell proliferation [Brenna and Waldum, 1992]. At the time when many of the studies on the role and mechanism for Hp-induced gastric acid hypersecretion were done, many of the researchers apparently did not accept the central role of the ECL cell in the regulation of gastric acid secretion [Waldum et al. 1991]. Meals and GRP would by an increase in gastrin release be expected to augment their maximal acid secretion in people infected with Hp. In common with pentagastrin stimulation, meal and GRP will also increase acid secretion due to the trophic effects of gastrin on the ECL cell mass. Histamine release is the restrictive factor in maximal gastrin-stimulated acid secretion [Kleveland et al. 1987]. The fall in pentagastrin-stimulated acid secretion after Hp eradication seen in patients with duodenal ulcer [El-Omar et al. 1993] most probably reflects a reduction in ECL cells secondary to a reduction in gastrin.
With time, Hp-induced inflammation spreads to the oxyntic mucosa. What makes the oxyntic mucosa more resistant to Hp infection compared with the antral mucosa is not known, although acidity has been proposed to play a role. Naturally, the acid-producing oxyntic glands are located in the oral part of the stomach where they empty their content to the lumen. It is, however, hard to imagine how this should affect the acidity in the mucous layer. However, the local luminal acidity is higher in the oxyntic area, and this could be of importance in the defense against Hp infection and also explain the oral spread of the infection from the antral mucosa. In any way, also the oxyntic mucosa is infected, starting as a superficial gastritis to begin with and developing into a gastritis affecting the deeper layer of the mucosa leading to hypoacidity and marked atrophy of most elements but not the target cell of gastrin, the ECL cell. At the early phases of oxyntic gastritis, gastric acidity is only slightly reduced and may be restored by Hp eradication [Tari et al. 2007]. As the oxyntic glands are destroyed, the capacity to produce acid is reduced, leading to hypoacidity and marked secondary hypergastrinemia. At this stage the capacity to restore normal acid secretion is limited [Iijima et al. 2004]. In this context we will also mention our previous study on Mongolian gerbils where we showed that the gastrin antagonist netazepide prevented the oxyntic inflammation provoked by Hp infection [Sordal et al. 2013]. The functions of the superficial cells (production of HCO3− and mucous) are also reduced by the gastritis and thus predisposing for gastric peptic ulcer [Byrd et al. 2000] [Figure 1(d)].
It may be concluded that the confusion related to the mechanisms for Hp-induced inappropriate hypergastrinemia and acid hypersecretion are mainly due to the trophic changes induced on long-living NE cells. Furthermore, an incorrect view on the regulation of gastric acid secretion at the time of most of these studies has also contributed to misunderstandings.
Hp and gastric cancer
As previously stated, gastric cancer seldom develops in a stomach without gastritis [Morson, 1955]. Similarly, Hp is an accepted cause of gastric cancer [Parsonnet et al. 1991]. It is also well known that Hp gastritis, when affecting the antrum only, predisposes to duodenal ulcer [Levi et al. 1989], which rarely occurs together with gastric cancer [Hansson et al. 1996]. First when Hp has induced an atrophic gastritis in the oxyntic mucosa, there is an increased risk of gastric cancer [Fossmark et al. 2015]. The central role of oxyntic mucosal atrophy in gastric carcinogenesis has previously been shown [Iijima et al. 2004]. All together, these facts suggest that the carcinogenic effect of Hp infection is related to the oxyntic atrophy and not directly to the inflammation or the agent itself [Figure 1(e)]. This view is also supported by the long unsuccessful search for a carcinogenic factor in Hp. Hp strains differ in their expression of certain genes; cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA). Particularly, CagA has been implicated in gastric carcinogenesis [Parsonnet et al. 1997; Gwack et al. 2006; Amieva and Peek, 2016]. However, even the carcinogenic effect of CagA cannot be separated from its effect on inflammation, gastric acidity and gastrin in blood [Konturek et al. 2002]. In general, the role of virulence factors associated to Hp were reviewed some years ago [Wen and Moss, 2009]. Oxyntic mucosal atrophy leads to hypoacidity with secondary microbiological intragastric changes. If changes in gastric milieu should be the cause of Hp-induced gastric cancer, it is strange that patients with so-called ��autoimmune�� gastritis affecting only the oxyntic mucosa develop cancers in the oxyntic area [Walker et al. 1971]. Oxyntic atrophy also leads to hypergastrinemia as a consequence of gastric hypoacidity, and we have previously implicated gastrin in Hp-induced gastric carcinogenesis [Waldum et al. 2015]. The target cell of gastrin, the ECL cell, is regulated functionally and trophically by gastrin [Waldum et al. 2014a]. The role of the ECL cell in gastric carcinogenesis seems hitherto to have been greatly underestimated [Waldum et al. 2014a]. The two types of gastric cancer according to Lauren [Lauren, 1965], the intestinal and diffuse types, seem to represent separate entities since they do not transform into each other. The ECL cell may be the cell of origin for the diffuse carcinomas [Waldum et al. 1998]. Lack of E-cadherin is an important factor in the pathogenesis of diffuse gastric carcinomas [Becker et al. 1994], and interestingly, we did not find E-cadherin expression even in normal ECL cells [Waldum et al. 2014b]. Hp infection plays a central role in the pathogenesis of both types [Parsonnet et al. 1997], although metaplasia is mainly associated to the intestinal type [Solcia et al. 1996]. In any ways, hypergastrinemia has been shown to be implicated in Hp-associated gastric carcinogenesis [Konturek et al. 2002; Sun et al. 2014]. A central role of gastrin in Hp-induced gastric carcinogenesis is also supported by animal studies [Sordal et al. 2013; Takaishi et al. 2009]. Very recently it was reported that Hp could infect deep into the glands and reach the stem cell area [Sigal et al. 2015]. Such deep localization of Hp was mainly found in the antral area, whereas the oxyntic area seems to be mostly involved in Hp-induced gastric carcinogenesis [Fossmark et al. 2005]. Moreover, in contrast to virus bacteria, it has hitherto not been shown to have a direct carcinogenic effect.
Hp and gastrinoma
Most patients with peptic ulcer due to gastrinoma are not infected with Hp [Weber et al. 1997], demonstrating that increased gastric acid secretion alone is sufficient to provoke peptic ulcers. Gastrinomas seldom manifest themselves before adulthood [Soga and Yakuwa, 1998], whereas Hp infection most often occurs during childhood [Thomas et al. 1999]. The lower incidence of Hp infections in patients with gastrinoma compared with age-matched controls indicates that increased gastric acidity may eradicate Hp.
Hp and hypoacidity/anacidity
During the early phases of oxyntic gastritis caused by Hp, gastric acid secretion may be only moderately reduced and Hp eradication even in patients with some degree of atrophy can augment acid secretion [Tari et al. 2007]. In patients with pan-gastritis and oxyntic atrophy, Hp may not be detectable [Karnes et al. 1991]. It is presumed that Hp cannot live under these conditions. Hp may have been replaced by other microorganisms which can live in this situation in the stomach, or alternatively, NH3 production by Hp urease creates a local milieu too alkaline for the agent itself in a stomach without acid [Marshall et al. 1990]. The role of acid for Hp to thrive is also demonstrated by the effects of acid inhibition in combination with antibiotics in eradication of Hp [Unge et al. 1989], as well as the possible oral spread in the stomach during treatment with inhibitors of gastric acid secretion [Logan et al. 1995]. Interestingly, the new inhibitor of gastric acid secretion, vonoprazan, belonging to potassium-competitive acid blockers, and probably more efficient in inhibiting acid secretion than proton-pump blockers, seems to be more efficient in combination with antibiotics in eradicating Hp compared with proton-pump inhibitors [Murakami et al. 2016].
Conclusion
It may be concluded that Hp is dependent on temporal hypoacidity or anacidity for its primary infection, but acidity to survive for a long time. Hp infection in the antral mucosa causes duodenal ulcers induced by increased gastric acid secretion secondary to slight increased gastrin release from the G cells, probably due to NH3 production provoked by urease. When infecting the oxyntic mucosa causing inflammation, the functions (mucous and HCO3− production) of the superficial cells are reduced, predisposing for gastric peptic ulcer. Long-term infection of the oxyntic mucosa causes atrophy and marked reduced gastric acid secretion, leading to gastric hypoacidity and marked hypergastrinemia that probably predisposes for gastric cancer. HP does not survive in a too acidic (patients with gastrinoma) or in an anacidic stomach.
The interactions between Hp and the stomach are very complex, but we now understand the pathogenesis of most of the diseases in the stomach and duodenum, since Hp plays a central role in most of these conditions.Helicobacter pylori and gastric acid: an intimate and reciprocal relationship
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5076771/��
��
Dig Dis Sci. 2015 Jun
Gastrin May Mediate the Carcinogenic Effect of Helicobacter pylori Infection of the Stomach.
Waldum HL1, Hauso Ø, Sørdal ØF, Fossmark R.
Author information
Department of Cancer Research and Molecular Medicine, Faculty of Medicine, Norwegian University of Science and Technology, Prinsesse Kristinas Gate 1, 7006, Trondheim, Norway, helge.waldum@ntnu.no.
Abstract
Gastric cancer occurs almost exclusively in patients with gastritis. Since Helicobacter pylori (Hp) was proved to cause gastritis, Hp was also expected to play a role in gastric carcinogenesis. Despite extensive studies, the mechanisms by which Hp cause gastric cancer are still poorly understood.However, there is evidence that the anatomical site of Hp infection is of major importance. Infection confined to the antral mucosa protects against gastric cancer but predisposes to duodenal ulcer, whereas Hp infection of the oxyntic mucosa increases the risk of gastric cancer. Hp infection does not predispose to cancers in the gastric cardia.
In patients with atrophic gastritis of the oxyntic mucosa, the intragastric pH is elevated and the concentration of microorganisms in the stomach is increased. This does not lead to increased risk of gastric cancer at all anatomical sites. The site specificity of Hp infection in relation to cancer risk indicates that neither Hp nor the changes in gastric microflora due to gastric hypoacidity are carcinogenic per se. However, reduced gastric acidity also leads to hypergastrinemia, which stimulates the function and proliferation of enterochromaffin-like (ECL) cells located in the oxyntic mucosa. The ECL cell may be more important in human gastric carcinogenesis than previously realized, as every condition causing long-term hypergastrinemia in animals results in the development of neoplasia in the oxyntic mucosa. Patients with hypergastrinemia will far more often develop carcinomas in the gastric corpus. In conclusion, hypergastrinemia may explain the carcinogenic effect of Hp.
Dig Dis Sci�� 2015��6��
θ���ؿ��ܽ鵼θ�����ݸ˾���Ⱦ���°�����
Waldum HL1��HausoØ��SørdalØF��Fossmark R.
Ų����ѧ������ѧҽѧԺ��֢�о������ҽѧϵ
θ������ֻ������θ�����С�������ʵ֤�������ݸ˾���Hp��������θ�ף����HpҲ������θ�������з������á����ܽ����˹㷺���о�������Hp����θ���Ļ�����֪֮���١����ǣ���֤�ݱ�����Hp��Ⱦ�Ľ��ʲ�λ�dz���Ҫ��������θ�ճĤ�ĸ�Ⱦ��Ԥ��θ��������ʮ��ָ�������߲���ճĤ(oxyntic mucosa)��Hp��Ⱦ�����ӻ�θ���ķ��ա� Hp��Ⱦ��������Gastric cardia��
��������ճĤ(oxyntic mucosa)ή����θ�����У�θ��pH���ߣ�θ������Ũ�����ߡ��ⲻ�ᵼ�����н��ʲ�λ��θ���������ӡ� Hp��Ⱦ�방֢�����йص�λ�������Ա�����Hp������θ��������θ��Ⱥ�仯�������������°��ԡ����ǣ����͵�θ���Ҳ�ᵼ�¸�θ����Ѫ֢(hypergastrinemia)���Ӷ��̼�λ��θ�Ĥ�ij��ȸ������ף�ECL��ϸ���Ĺ��ܺ���ֳ�� ECLϸ������θ�����������п��ܱ���ǰ��ʶ���ĸ�Ϊ��Ҫ����Ϊ�����ﳤ�ڸ�θ����Ѫ֢��ÿ��������ᵼ�´߲���ճĤ���γɡ���θ����Ѫ֢�Ļ����������θ�巢�����䡣��֮����θ����Ѫ֢���Խ���Hp���°����á�
Gastrin May Mediate the Carcinogenic Effect of Helicobacter pylori Infection of the Stomach. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25480404��
J Gastroenterol Hepatol. 2004 Sep;19(9):988-93.
Hypergastrinemia after Helicobacter pylori infection is associated with bacterial load and related inflammation of the oxyntic corpus mucosa.
Chuang CH1, Sheu BS, Yang HB, Kao AW, Cheng HC, Yao WJ.
Department of Internal Medicine, Medical College, National Cheng Kung University, Tainan, Taiwan.
Abstract
BACKGROUND AND AIM:
Helicobacter pylori infection causes hypergastrinemia. This study aimed to determine the association between serum gastrin and the severity of H. pylori-related gastric histology.
METHODS:
A total of 458 dyspeptic patients were included in this study after the absence of gastric malignancy was confirmed using endoscopy. The gastric specimens of each patient were collected from the antrum and corpus for the analysis of H. pylori-related histology changes by updated Sydney's system. Before endoscopy, the fasting blood samples were collected for gastrin analysis.
RESULTS:
The H. pylori-infected patients had higher gastrin levels than those without infection (P = 0.01). Gastrin levels were related to H. pylori density and acute and chronic inflammation scores in the corpus mucosa (P < 0.05), but not in the antral mucosa (P = NS). Gastrin levels were also not related to the presence of gastric atrophy. Multivariate regression showed that the gastrin level was only related to acute corpus inflammation. However, in the patients without infection, the gastrin level was also associated with acute corpus inflammation. Nevertheless, the patients with denser H. pylori infection were more likely to have acute corpus gastritis than those with lighter H. pylori infection, and thus presented with higher gastrin levels (P < 0.05).
CONCLUSIONS:
The increased level of gastrin of serum after H. pylori infection was associated with acute inflammation in the gastric corpus mucosa, but not in the antral mucosa. Denser H. pylori infection causes more severe corpus gastritis and thus may lead to a higher fasting level of gastrin of serum.��
Team:ATOMS-Turkiye/Project/Ulcer - 2015.igem.org
http://2015.igem.org/Team:ATOMS-Turkiye/Project/Ulcer��
Anti-Helicobacter Effects of Vitamin C
In an in vitro study, 10 to 20 mg vitamin C per ml could effectively inhibit Helicobacter pylori growth under microaerobic conditions, whereas in an aerobic milieu, vitamin C even promoted H. pylori survival in concentrations ranging from 2 to 20 mg/mL [105]. These observations might be explained by the antioxidant properties of vitamin C, protecting microaerophilic bacteria against toxic effects of ROS. Following one-week treatment of H. pylori-infected Mongolian gerbils with 10 mg vitamin C daily, gastric pathogen loads could be significantly lowered [106]. In support, several clinical studies reported more effective H. pylori eradication upon vitamin C application to infected humans [107�C109]. In addition, oxidative stress, apoptotic responses, and decreased cellular viability that had been induced in an H. pylori-infected human gastric adenocarcinoma cell line could be counteracted by vitamin C application in its L-ascorbic acid-2-glucoside form [110].
Immunomodulatory and Antimicrobial Effects of Vitamin C
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6798581/��
Including results for vitamin c enhance activity of ho-1.
Do you want results only for vitaminc enhance sactivity of HO-1?
Vitamin C, a Multi-Tasking Molecule, Finds a Molecular ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5959041
While the mechanism involved in the induction of HO-1 remains to be determined, it is likely that vitamin C provokes a pro-oxidant state in cells that subsequently activates HO-1 gene expression. Indeed, HO-1 is known as an oxidative stress-responsive gene, and as described next, vitamin C at pharmacological doses behaves as a pro-oxidant. 4.3.
Cited by: 4
Publish Year: 2016
Author: Y. Robert Li
Gastroprotection by vitamin C��a heme oxygenase-1-dependent mechanism?
Author links open overlay panelJan CBeckeraNinaGrosserbPeterBoknikcHenningSchröderbWolframDomschkeaThorstenPohlea
a
Department of Medicine B, University of M��nster, M��nster, Germany
b
Department of Pharmacology and Toxicology, School of Pharmacy, University of Halle, Halle, Germany
c
Institute of Pharmacology und Toxicology, University of M��nster, M��nster, Germany
Received 21 October 2003, Available online 13 November 2003.
Abstract
Free oxygen radicals contribute to gastric mucosal damage induced by acetylic�Csalicylic acid (ASA). Vitamin C has been shown to reduce gastric toxicity of ASA in humans.We intended to assess the role of heme oxygenase-1 (HO-1) in this process by application of these substances to AGS and KATO III cells. HO-1 expression was monitored by real-time RT-PCR, Western blot, and HO activity measurement. HO-1 mRNA was significantly elevated by either ASA or vitamin C in gastric epithelial cells, combination of both substances further increased expression.
HO-1 protein and enzyme activity rose in cells exposed to vitamin C alone or combined with ASA, but not after stimulation with ASA alone.
In contrast to endothelia, in which ASA simultaneously induces HO-1 mRNA and protein expression, gastric epithelial cells require vitamin C to translate HO-1 mRNA into active protein, which then may exert gastroprotection by its antioxidant and vasodilative properties.
Keywords
Acetylic�Csalicylic acidGastric epithelial cellsGastroprotectionHeme oxygenaseProtein expressionReal-time RT-PCRVitamin CGastroprotection by vitamin C��a heme oxygenase-1-dependent mechanism? - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0006291X03022460��
Crit Rev Immunol. 2001;21(5):399-425.
Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue.
Mills CD1.
Author information
1
Department of Surgery and Diabetes Institute for Immunology and Transplantation, University of Minnesota Hospitals and Clinics, Minneapolis 55455, USA. mills002@tc.umn.edu
Abstract
Macrophages can metabolize arginine to nitric oxide in quantities that inhibit pathogens or nearby host cells. They can instead metabolize arginine to ornithine (a precursor of polyamines and collagen) in quantities that stimulate pathogens or nearby host cells.Macrophages are essentially the only circulating cells that can make these life or death decisions with arginine. Macrophages expressing these destructive or constructive phenotypes have been termed M-1 or M-2 because they also stimulate TH1 or TH2 responses, respectively.
Factors that influence whether a macrophage expresses the M-1 or M-2 phenotype and the real or potential impact on immune responses and other host processes are discussed.
PMID: 11942557
[Indexed for MEDLINE]https://www.ncbi.nlm.nih.gov/pubmed/11942557
��
J Immunol. 2010 Mar 1;184(5):2572-82. doi: 10.4049/jimmunol.0902436. Epub 2010 Jan 22.
Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages.
Lewis ND1, Asim M, Barry DP, Singh K, de Sablet T, Boucher JL, Gobert AP, Wilson KT.
Author information
Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37240, USA.
Abstract
Helicobacter pylori infection of the stomach causes peptic ulcer disease and gastric cancer. Despite eliciting a vigorous immune response, the bacterium persists for the life of the host.An important antimicrobial mechanism is the production of NO derived from inducible NO synthase (iNOS). We have reported that macrophages can kill H. pylori in vitro by an NO-dependent mechanism, but supraphysiologic levels of the iNOS substrate l-arginine are required. Because H. pylori induces arginase activity in macrophages, we determined if this restricts NO generation by reducing l-arginine availability.
Inhibition of arginase with S-(2-boronoethyl)-l-cysteine (BEC) significantly enhanced NO generation in H. pylori-stimulated RAW 264.7 macrophages by enhancing iNOS protein translation but not iNOS mRNA levels. This effect resulted in increased killing of H. pylori that was attenuated with an NO scavenger.
In contrast, inhibition of arginase in macrophages activated by the colitis-inducing bacterium Citrobacter rodentium increased NO without affecting iNOS levels. H. pylori upregulated levels of arginase II (Arg2) mRNA and Arg2 protein, which localized to mitochondria, whereas arginase I was not induced.
Increased iNOS protein and NO levels were also demonstrated by small interfering RNA knockdown of Arg2 and in peritoneal macrophages from C57BL/6 Arg2(-/-) mice.
In H. pylori-infected mice, treatment with BEC or deletion of Arg2 increased iNOS protein levels and NO generation in gastric macrophages, but treatment of Arg2(-/-) mice with BEC had no additional effect.
These studies implicate Arg2 in the immune evasion of H. pylori by causing intracellular depletion of l-arginine and thus reduction of NO-dependent bactericidal activity.
PMID: 20097867 PMCID: PMC2841360 DOI: 10.4049/jimmunol.0902436
[Indexed for MEDLINE] Free PMC ArticleArginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/20097867��
Microb Pathog. 2018 Oct;123:1-8. doi: 10.1016/j.micpath.2018.06.033. Epub 2018 Jun 21.
The role of T helper 1-cell response in Helicobacter pylori-infection.
Bagheri N1, Salimzadeh L2, Shirzad H3.
Author information
Abstract
Helicobacter pylori (H. pylori) is a human pathogen affecting over 50% of the world population. This pathogen is usually associated with chronic inflammation of the gastric mucosa that can lead to peptic ulcer disease (PUD) and gastric cancer (GC), especially in susceptible individuals.These outcomes have been attributed to the interaction of several factors, including host genetic susceptibility, local innate and adaptive immune responses, virulence factors of H. pylori, and environmental factors.
T helper (Th) cell subsets and their signature cytokines especially IFN-��, contribute to anti-bacterial response, but at the mean time sustaining chronic inflammatory responses in the site of infection.
It has been acknowledged that H. pylori-infection results in a Th1-dominant response and that inflammation of the gastric mucosa depends mainly on Th1 cell responses. But, the mechanism of the role of Th1 cell responses in H. pylori-infection has not yet been clearly explained. In this review, we will focus on the role of Th1 involved in H. pylori-infection, its interaction with Th17/Treg cells and its association with the clinical consequences of the infection.
Copyright © 2018. Published by Elsevier Ltd.
KEYWORDS:
Gastric cancer; Gastritis; Helicobacter pylori; Peptic ulcer disease; Th1
PMID: 29936093 DOI: 10.1016/j.micpath.2018.06.033The role of T helper 1-cell response in Helicobacter pylori-infection. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29936093��
Oncogene. 2015 Apr 2;34(14):1865-71. doi: 10.1038/onc.2014.135. Epub 2014 May 19.
Matrix metalloproteinase 7 restrains Helicobacter pylori-induced gastric inflammation and premalignant lesions in the stomach by altering macrophage polarization.
Krakowiak MS1, Noto JM2, Piazuelo MB2, Hardbower DM3, Romero-Gallo J2, Delgado A2, Chaturvedi R2, Correa P2, Wilson KT4, Peek RM Jr5.
Author information
1
Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, USA.
2
Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA.
3
1] Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA [2] Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, USA.
4
1] Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, USA [2] Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA [3] Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN, USA [4] Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN, USA.
5
1] Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, USA [2] Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA.
Abstract
Helicobacter pylori is the strongest risk factor for the development of gastric cancer. Although the specific mechanisms by which this pathogen induces carcinogenesis have not been fully elucidated, high-expression interleukin (IL)-1�� alleles are associated with increased gastric cancer risk among H. pylori-infected persons.In addition, loss of matrix metalloproteinase 7 (MMP7) increases mucosal inflammation in mouse models of epithelial injury, and we have shown that gastric inflammation is increased in H. pylori-infected MMP7(-/-) C57BL/6 mice.
In this report, we define mechanisms that underpin such responses and extend these results into a genetic model of MMP7 deficiency and gastric cancer. Wild-type (WT) or MMP7(-/-) C57BL/6 mice were challenged with broth alone as an uninfected control or the H. pylori strain PMSS1.
All H. pylori-challenged mice were successfully colonized. As expected, H. pylori-infected MMP7(-/-) C57BL/6 mice exhibited a significant increase in gastric inflammation compared with uninfected or infected WT C57BL/6 animals. Loss of MMP7 resulted in M1 macrophage polarization within H. pylori-infected stomachs, as assessed by Luminex technology and immunohistochemistry, and macrophages isolated from infected MMP7-deficient mice expressed significantly higher levels of the M1 macrophage marker IL-1�� compared with macrophages isolated from WT mice.
To extend these findings into a model of gastric cancer, hypergastrinemic WT INS-GAS or MMP7(-/-) INS-GAS mice were challenged with H. pylori strain PMSS1. Consistent with findings in the C57BL/6 model, H. pylori-infected MMP7-deficient INS-GAS mice exhibited a significant increase in gastric inflammation compared with either uninfected or infected WT INS-GAS mice.
In addition, the incidence of gastric hyperplasia and dysplasia was significantly increased in H. pylori-infected MMP7(-/-) INS-GAS mice compared with infected WT INS-GAS mice, and loss of MMP7 promoted M1 macrophage polarization.
These results suggest that MMP7 exerts a restrictive role on H. pylori-induced gastric injury and the development of premalignant lesions by suppressing M1 macrophage polarization.
PMID: 24837365 PMCID: PMC4237684 DOI: 10.1038/onc.2014.135��
CELL SCIENCES RESEARCH ARTICLE
Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in human gastric epithelial cells: role in epithelial cell migration
Lydia E. Wroblewski, P.-J. M. Noble, Adelina Pagliocca, D. Mark Pritchard, C. Anthony Hart, Fiona Campbell, Andrew R. Dodson, Graham J. Dockray, Andrea Varro
Journal of Cell Science 2003 116: 3017-3026; doi: 10.1242/jcs.00518
Summary
Epithelial cell responses to bacterial infection include induction of matrix metalloproteinase 7 (MMP-7). Here, we identify increased MMP-7 expression in the gastric epithelium in response to the oncogenic bacterium Helicobacter pylori, and report on the mechanisms and consequences for gastric epithelial cell migration.In patients infected with H. pylori, there was increased MMP-7 in gastric biopsies detected by western blot. MMP-7 was localized to the advancing edge of migrating gastric epithelial cell colonies, including lamellipodia. Rates of spreading of gastric gland cells were higher in H. pylori-infected cultures compared with control, and this was inhibited by antisense oligonucleotides to MMP-7.
Complementary data were obtained in a gastric cancer cell line (AGS cells). In the latter, H. pylori induced expression of an MMP-7-luciferase promoter/reporter vector through mechanisms that involved activation of Rho and Rac. RhoA acted through activation of both NF-��B and AP-1, whereas Rac activated NF-��B but not AP-1.
MMP-7 is commonly upregulated in gastric cancer; since H. pylori is a recognized gastric carcinogen, the data suggest a new mechanism by which the bacterium might predispose towards gastric neoplasia.
Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in human gastric epithelial cells: role in epithelial cell migration | Journal of Cell Science
https://jcs.biologists.org/content/116/14/3017��
Free Radic Biol Med. 2017 Apr;105:16-27. doi: 10.1016/j.freeradbiomed.2016.09.024. Epub 2016 Sep 25.
Polyamine- and NADPH-dependent generation of ROS during Helicobacter pylori infection: A blessing in disguise.
Gobert AP1, Wilson KT2.
Author information
1
Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, United States; Center for Mucosal Inflammation and Cancer, United States.
2
Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, United States; Department of Pathology, Microbiology, and Immunology, United States; Department of Cancer Biology, United States; Center for Mucosal Inflammation and Cancer, United States; Vanderbilt Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, TN 37232, United States; Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN 37212, United States. Electronic address: keith.wilson@vanderbilt.edu.
Abstract
Helicobacter pylori is a Gram-negative bacterium that specifically colonizes the gastric ecological niche. During the infectious process, which results in diseases ranging from chronic gastritis to gastric cancer, the host response is characterized by the activation of the innate immunity of gastric epithelial cells and macrophages. These cells thus produce effector molecules such as reactive oxygen species (ROS) to counteract the infection.The generation of ROS in response to H. pylori involves two canonical pathways: 1) the NADPH-dependent reduction of molecular oxygen to generate O2•-, which can dismute to generate ROS; and 2) the back-conversion of the polyamine spermine into spermidine through the enzyme spermine oxidase, leading to H2O2 production. Although these products have the potential to affect the survival of bacteria, H. pylori has acquired numerous strategies to counteract their deleterious effects.
Nonetheless, ROS-mediated oxidative DNA damage and mutations may participate in the adaptation of H. pylori to its ecological niche. Lastly, ROS have been shown to play a major role in the development of the inflammation and carcinogenesis.
It is the purpose of this review to summarize the literature about the production of ROS during H. pylori infection and their role in this infectious gastric disease.
Published by Elsevier Inc.
KEYWORDS:
Gastric cancer; Helicobacter pylori; NADPH oxidase; Polyamines; Reactive oxygen species
PMID: 27682363 PMCID: PMC5366100 DOI: 10.1016/j.freeradbiomed.2016.09.024
[Indexed for MEDLINE] Free PMC ArticlePolyamine- and NADPH-dependent generation of ROS during Helicobacter pylori infection: A blessing in disguise. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/27682363��
Trends Microbiol. 2016 May;24(5):366-376. doi: 10.1016/j.tim.2016.02.005. Epub 2016 Feb 22.
The Immune Battle against Helicobacter pylori Infection: NO Offense.
Gobert AP1, Wilson KT2.
Author information
1
Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Center for Mucosal Inflammation and Cancer, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
2
Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Center for Mucosal Inflammation and Cancer, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN 37212, USA. Electronic address: keith.wilson@vanderbilt.edu.
Abstract
Helicobacter pylori is a successful pathogen of the human stomach. Despite a vigorous immune response by the gastric mucosa, the bacterium survives in its ecological niche, thus favoring diseases ranging from chronic gastritis to adenocarcinoma.The current literature demonstrates that high-output of nitric oxide (NO) production by the inducible enzyme NO synthase-2 (NOS2) plays major functions in host defense against bacterial infections.
However, pathogens have elaborated several strategies to counteract the deleterious effects of NO; this includes inhibition of host NO synthesis and transcriptional regulation in response to reactive nitrogen species, allowing the bacteria to face the nitrosative stress. Moreover, NO is also a critical mediator of inflammation and carcinogenesis.
In this context, we review the recent findings on the expression of NOS2 in H. pylori-infected gastric tissues and epithelial cells, the role of NO in H. pylori-related diseases and H. pylori gene expression, and the mechanisms whereby H. pylori regulates NO synthesis by host cells.
Published by Elsevier Ltd.
KEYWORDS:
Helicobacter pylori; gastric cancer; nitric oxide; polyamines
Comment in
Helicobacter pylori, Its Urease and Carbonic Anhydrases, and Macrophage Nitric Oxide Synthase. [Trends Microbiol. 2017]
Effect of CO2 on Peroxynitrite-Mediated Bacteria Killing: Response to Tsikas et al. [Trends Microbiol. 2017]
PMID: 26916789 PMCID: PMC4841705 DOI: 10.1016/j.tim.2016.02.005
[Indexed for MEDLINE] Free PMC ArticleThe Immune Battle against Helicobacter pylori Infection: NO Offense. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/26916789��
Role of urease in megasome formation and Helicobacter pylori survival in macrophages.
Schwartz JT1, Allen LA
Author information
Journal of Leukocyte Biology, 16 Mar 2006, 79(6):1214-1225
Abstract
Previous studies have demonstrated that Helicobacter pylori (Hp) delays its entry into macrophages and persists inside megasomes, which are poorly acidified and accumulate early endosome autoantigen 1.Herein, we explored the role of Hp urease in bacterial survival in murine peritoneal macrophages and J774 cells.
Plasmid-free mutagenesis was used to replace ureA and ureB with chloramphenicol acetyltransferase in Hp Strains 11637 and 11916. ureAB null Hp lacked detectable urease activity and did not express UreA or UreB as judged by immunoblotting. Deletion of ureAB had no effect on Hp binding to macrophages or the rate or extent of phagocytosis. However, intracellular survival of mutant organisms was impaired significantly.
Immunofluorescence microscopy demonstrated that (in contrast to parental organisms) mutant Hp resided in single phagosomes, which were acidic and accumulated the lysosome marker lysosome-associated membrane protein-1 but not early endosome autoantigen 1. A similar phenotype was observed for spontaneous urease mutants derived from Hp Strain 60190.
Treatment of macrophages with bafilomycin A1, NH4Cl, or chloroquine prevented acidification of phagosomes containing mutant Hp. However, only ammonium chloride enhanced bacterial viability significantly. Rescue of ureAB null organisms was also achieved by surface adsorption of active urease.
Altogether, our data indicate a role for urease and urease-derived ammonia in megasome formation and Hp survival.
Role of urease in megasome formation and Helicobacter pylori survival in macrophages. - Abstract - Europe PMC
https://europepmc.org/article/PMC/1868427��
Infect Immun. 2004 May; 72(5): 2889�C2898.
Helicobacter pylori Induces Apoptosis of Macrophages in Association with Alterations in the Mitochondrial Pathway
Rena J. Menaker,1,2 Peter J. M. Ceponis,1,3 and Nicola L. Jones1,2,4,*Research Institute, Hospital for Sick Children,1 Department of Physiology,2 Department of Laboratory Medicine and Pathobiology,3 Department of Paediatrics, University of Toronto, Toronto, Canada4
ABSTRACT
Helicobacter pylori is a gastric bacterial pathogen that evades host immune responses in vivo and is associated with the development of gastritis, peptic ulcer disease, and gastric cancers.Induction of macrophage apoptosis is a method employed by multiple pathogens to escape host immune responses. Therefore, we hypothesized that H. pylori induces apoptosis of infected macrophages.
RAW 264.7 cells were infected with H. pylori strain 60190, and apoptosis was assessed. Transmission electron microscopy and fluorescence microscopy showed that infected macrophages displayed morphological features characteristic of apoptosis.
Quantification by acridine orange-ethidium bromide fluorescent-dye staining showed that apoptosis was dose and time dependent, and apoptosis was further confirmed by increased binding of annexin V-fluorescein isothiocyanate (FITC) to externalized phosphatidylserine of infected but not of control macrophages.
Macrophages infected with isogenic mutants of H. pylori strain 60190 deficient in either cagA or vacA induced significantly less apoptosis than the parental strain, as assessed by increased binding of annexin V-FITC. Western blot analysis of whole-cell protein lysates revealed that infection with strain 60190 induced a time-dependent increase in cleavage of procaspase 8 and disappearance of full-length Bid compared with uninfected cells. Furthermore, pharmacological inhibition of caspase 8 caused a decrease in levels of apoptosis. Finally, infection caused a time-dependent increase in mitochondrial-membrane permeability and release of cytochrome c into the cytosol.
These results suggest that H. pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Elimination of this key immunomodulatory cell may represent a mechanism employed by the bacterium to evade host immune responses.
Helicobacter pylori Induces Apoptosis of Macrophages in Association with Alterations in the Mitochondrial Pathway
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC387848/��
Infect Immun. 2008 May; 76(5): 2235�C2239.
Macrophages Are Mediators of Gastritis in Acute Helicobacter pylori Infection in C57BL/6 Mice▿
Maria Kaparakis,1,† Anna K. Walduck,1 Jason D. Price,1,2 John S. Pedersen,3 Nico van Rooijen,4 Martin J. Pearse,2,5 Odilia L. C. Wijburg,1,2,‡* and Richard A. Strugnell1,2,‡
Department of Microbiology and Immunology and the NHMRC Bacterial Pathogenesis Group, The University of Melbourne, Parkville, Victoria 3010, Australia,1 CRC for Vaccine Technology,2 Department of Anatomical Pathology, Alfred Hospital, Prahran, Victoria 3181, Australia,3 Department of Molecular Cell Biology, Vrije Universiteit VUMC, Amsterdam, The Netherlands,4 CSL Limited, Poplar Rd., Parkville, Victoria 3052, Australia5
*Corresponding author. Mailing address: Department of Microbiology and Immunology, The University of Melbourne, Melbourne, VIC 3010, Australia. Phone: 61 3 8344 9919. Fax: 61 3 9347 1540. E-mail: ua.ude.bleminu@ailido
†Present address: Department of Microbiology, Monash University, Melbourne, Australia.
‡Contributed equally as senior authors.
This article has been cited by other articles in PMC.
ABSTRACT
Helicobacter pylori is the etiological agent of human chronic gastritis, a condition seen as a precursor to the development of gastrointestinal ulcers or gastric cancer. This study utilized the murine model of chronic H. pylori infection to characterize the role of macrophages in the induction of specific immune responses and gastritis and in the control of the bacterial burden following H. pylori infection and vaccination. Drug-loaded liposomes were injected intravenously to deplete macrophages from C57BL/6 mice, and effective removal of CD11b+ cells from the spleens and stomachs of mice was confirmed by immunofluorescence microscopy. Transient elimination of macrophages from C57BL/6 mice during the early period of infection reduced the gastric pathology induced by H. pylori SS1 but did not affect the bacterial load in the stomach. These data suggest that macrophages are important to the severity of gastric inflammation during H. pylori infection.
Helicobacter pylori is the causative agent of human gastroduodenal disease, resulting in acute gastric inflammation (16), ulcers, gastric adenocarcinoma, or mucosa-associated lymphoid tissue lymphoma (18, 24). H. pylori resides extracellularly within the gastric mucus layer of the human stomach, with only 1% of the organisms attached to host epithelial cells (11). The normal gastric mucosa of H. pylori-negative adults and children is populated by very few macrophages (12). Macrophages and neutrophils enter gastric tissue in response to H. pylori infection (20) and increase in number with the severity of gastritis and the duration of infection (12). H. pylori can activate macrophages and elicit interleukin-1 (IL-1), tumor necrosis factor alpha, IL-6, IL-8, MIP-1��, and GRO-�� production (5, 6, 13), which induce the recruitment and activation of inflammatory cells, including macrophages and T cells.Macrophages are bone marrow-derived mononuclear phagocytes that reside within tissues, act as scavengers of immunogenic debris, and eliminate foreign particles by phagocytosis (7). Macrophages are thought to have a central function in controlling and regulating the immune response; once activated, they produce proinflammatory cytokines and chemokines (7). Activated macrophages also express major histocompatibility complex class II and can therefore activate antigen-specific CD4+ T cells (4).
Macrophages Are Mediators of Gastritis in Acute Helicobacter pylori Infection in C57BL/6 Mice
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2346689/��
Gastroenterology. Author manuscript; available in PMC 2013 Jul 11.
Published in final edited form as:
Gastroenterology. 2011 Feb; 140(2): 412�C424.
Gastric Epithelial Stem Cells
JASON C. MILLS* and RAMESH A. SHIVDASANI‡��
Author information Copyright and License information Disclaimer
*Division of Gastroenterology, Departments of Medicine, Pathology & Immunology, and Developmental Biology, Washington University School of Medicine, St. Louis, Missouri
‡Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts
��Department of Medicine, Brigham & Women��s Hospital and Harvard Medical School, Boston, Massachusetts
Address requests for reprints to: Jason C. Mills, MD, PhD, Washington University School of Medicine, Box 8124, 660 South Euclid Avenue, St Louis, Missouri 63110. ude.ltsuw@sllimj; fax: (314) 362-7487. Ramesh A. Shivdasani, MD, PhD, Dana-Farber Cancer Institute, 44 Binney Street, Boston, Massachusetts 02115. ude.dravrah.icfd@inasadvihs_hsemar; fax: (617) 582-7198
Abstract
Advances in our understanding of stem cells in the gastrointestinal tract include the identification of molecular markers of stem and early progenitor cells in the small intestine. Although gastric epithelial stem cells have been localized, little is known about their molecular biology. Recent reports describe the use of inducible Cre recombinase activity to indelibly label candidate stem cells and their progeny in the distal stomach, (ie, the antrum and pylorus).No such lineage labeling of epithelial stem cells has been reported in the gastric body (corpus). Among stem cells in the alimentary canal, those of the adult corpus are unique in that they lie close to the lumen and increase proliferation following loss of a single mature progeny lineage, the acid-secreting parietal cell. They are also unique in that they neither depend on Wnt signaling nor express the surface marker Lgr5.
Because pathogenesis of gastric adenocarcinoma has been associated with abnormal patterns of gastric differentiation and with chronic tissue injury, there has been much research on the response of stomach epithelial stem cells to inflammation.
Chronic inflammation, as induced by infection with Helicobacter pylori, affects differentiation and promotes metaplasias. Several studies have identified cellular and molecular mechanisms in spasmolytic polypeptide�Cexpressing (pseudopyloric) metaplasia.
Researchers have also begun to identify signaling pathways and events that take place during embryonic development that eventually establish the adult stem cells to maintain the specific features and functions of the stomach mucosa.
We review the cytologic, molecular, functional, and developmental properties of gastric epithelial stem cells.
Figure 3
Origins of principal corpus epithelial lineages. The self-renewing stem cell gives rise to each of the principal epithelial lineages of the corpus. There is ultrastructural evidence for the transient intermediates for each lineage (eg, presurface cells depicted in Figure 2); however, available evidence indicates greater complexity in the zymogenic lineage, which arises from a long-lived (��1 week in mice) intermediate, the mucous neck cell, with its own distinct ultrastructure and probable function.
Keywords: Stomach Stem Cells, Epithelial Self-Renewal, Tissue MetaplasiaGastric Epithelial Stem Cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3708552/��
Br J Pharmacol. 2000 Aug; 130(7): 1531�C1538.
Cytotoxicity associated with induction of nitric oxide synthase in rat duodenal epithelial cells in vivo by lipopolysaccharide of Helicobacter pylori: inhibition by superoxide dismutase
Dominique Lamarque,1 Anthony P Moran,2 Zoltan Szepes,1 Jean-Charles Delchier,1 and Brendan J R Whittle3,*
11Institut National de la Sant�� et de la Recherche M��dicale (INSERM U.99) et Service d'H��patologie et de Gastroent��rologie, Hôpital Henri Mondor, F-94010 Cr��teil, France
22Department of Microbiology, National University of Ireland, Galway, Ireland
33The William Harvey Research Institute, St. Bartholomew' & Royal London School of Medicine & Dentistry, Charterhouse Square, London EC1M 6BQAbstract
The products released by Helicobacter pylori (H. pylori) in the gastric antral and duodenal mucosa may be involved in mucosal ulceration by stimulating the local formation of cytotoxic factors such as nitric oxide (NO), superoxide or peroxynitrite.
The present study investigates the ability of purified H. pylori lipopolysaccharide (LPS) to induce nitric oxide synthase (iNOS) in rat duodenal epithelial cells following in vivo challenge and its interaction with superoxide in promoting cellular damage and apoptosis.
H. pylori LPS (0.75�C3 mg kg−1 i.v. or 3�C12 mg kg−1 p.o.) induced a dose�Cdependent expression of iNOS activity after 5 h in the duodenal epithelial cells, determined by [14C] arginine conversion to citrulline.
The epithelial cell viability, as assessed by Trypan Blue exclusion and MTT conversion, was reduced 5 h after challenge with H. pylori LPS, while the incidence of apoptosis was increased.
The iNOS activity and reduction in cell viability following H. pylori LPS challenge i.v. was inhibited by the selective iNOS inhibitor, 1400 W (0.2�C5 mg kg−1 i.v.).
Concurrent administration of superoxide dismutase conjugated with polyethylene glycol (250�C500 i.u. kg−1, i.v.), which did not modify the cellular iNOS activity, reduced the epithelial cell damage provoked by i.v. H. pylori LPS, and abolished the increased incidence of apoptosis.
These results suggest that expression of iNOS following challenge with H. pylori LPS provokes duodenal epithelial cell injury and apoptosis by a process involving superoxide, implicating peroxynitrite involvement. These events may contribute to the pathogenic mechanisms of H. pylori in promoting peptic ulcer disease.
Keywords: Helicobacter pylori, nitric oxide, inducible nitric oxide synthase, duodenal epithelial cells, duodenum, lipopolysaccharideCytotoxicity associated with induction of nitric oxide synthase in rat duodenal epithelial cells in vivo by lipopolysaccharide of Helicobacter pylori: inhibition by superoxide dismutase
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1572225/��
The importance of the surface urease of Helicobacter pylori: fact or fiction?
University of Carlifornia, Los Ageles
��
The urease produced by the gastric denizen, Helicobacter pylori, is an essential component of its adaptation to life in the acidic environment of the stomach. H. pylori synthesizes larger quantities of this enzyme than any other known microorganism and experiments in a variety of animal models (e.g. piglet, mouse and ferret) have shown that mutants lacking urease activity are unable to infect the stomach 1 . The reaction catalyzed by urease produces 2 mole ammonia and 1 mole carbon dioxide for every mole of urea hydrolyzed. As the p K a of the ammonia/ammonium couple is 9.2, urease activity results in alkalization of the medium to this pH, rapidly in unbuffered solutions and more slowly in buffered solutions given an infinite amount of urea. The urease of H. pylori has a pH optimum of ∼8.0 and, moreover, is irreversibly inactivated at pH <4.0�C4.5 (Ref. 2 ).
Keywords
Helicobacter pylori
urease
surface urease
intracellular urease
gastric juice
KlebsiellaThe importance of the surface urease of Helicobacter pylori: fact or fiction?: Trends in Microbiology
https://www.cell.com/trends/microbiology/fulltext/S0966-842X(01)02226-0#%20��
Chemodetection and Destruction of Host Urea Allows Helicobacter pylori to Locate the Epithelium
Julie Y. Huang
Emily Goers Sweeney
Michael Sigal
Calvin J. Kuo
Karen Guillemin
Manuel R. Amieva
Show all authorsHighlights
•
H. pylori is attracted to metabolites emanating from the human gastric epithelium
•
Urea is the primary host metabolite that attracts H. pylori
•
H. pylori��s high-affinity chemoreceptor TlpB detects nanomolar amounts of urea
•
Urea degradation by bacterial urease facilitates sensing through TlpB
Summary
The gastric pathogen Helicobacter pylori interacts intimately with the gastric mucosa to avoid the microbicidal acid in the stomach lumen. The cues H. pylori senses to locate and colonize the gastric epithelium have not been well defined. We show that metabolites emanating from human gastric organoids rapidly attract H. pylori. This response is largely controlled by the bacterial chemoreceptor TlpB, and the main attractant emanating from epithelia is urea. Our previous structural analyses show that TlpB binds urea with high affinity. Here we demonstrate that this tight binding controls highly sensitive responses, allowing detection of urea concentrations as low as 50 nM. Attraction to urea requires that H. pylori urease simultaneously destroys the signal. We propose that H. pylori has evolved a sensitive urea chemodetection and destruction system that allows the bacterium to dynamically and locally modify the host environment to locate the epithelium.
Chemodetection and Destruction of Host Urea Allows Helicobacter pylori to Locate the Epithelium: Cell Host & Microbe
https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(15)00294-2#%20��
Helicobacter pylori Makes a Molecular Incision to Gain Epithelial Entry: Cell Host & Microbe
https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(17)30403-1��
Acute and chronic gastric ulcer gastritis: treatment with drugs and alternative means
Alexey Portnov, medical expert
Last reviewed: 17.06.2019Ulcerative gastritis: symptoms, treatment, diet and nutrition, prevention | Competently about health on iLive
https://iliveok.com/health/acute-and-chronic-gastric-ulcer-gastritis-treatment-drugs-and-alternative-means_128275i15938.htmlMucus Production - Regulation - TeachMePhysiologyTeachMePhysiology
https://teachmephysiology.com/gastrointestinal-system/stomach/mucus-production/��
Scand J Gastroenterol Suppl. 1991;180:88-94.
Histamine-producing cells in the stomach and their role in the regulation of acid secretion.
Håkanson R1, Sundler F.
Author information
1
Dept. of Pharmacology, University of Lund, Sweden.
Abstract
Histamine H2-receptor antagonists inhibit basal and all kinds of stimulated acid secretion with about the same effectiveness, and hence, local release of histamine is thought to be necessary for the stimulation of parietal cells.A local histamine pool is physiologically relevant only if it is within the diffusion distance of the parietal cells, if it is effectively mobilized by gastrin, and if it is endowed with the machinery for a rapid replenishment of the histamine that has been released. Histamine in the stomach occurs in endocrine cells (so-called enterochromaffin-like (ECL) cells), mast cells, and neurons.
The ECL cells are peptide hormone-producing cells. In mammals they are located basally in the oxyntic gland area, in the chief-cell-rich region. Parietal cells predominate in the mid-region. ECL cells respond readily to gastrin with histamine release and histamine resynthesis. Thus, the ECL cells fulfil important prerequisites of a physiologically relevant histamine pool. however, it is not apparent how the preferential localization of ECL cells at the base of the glands may be conducive to a direct effect of released histamine on the parietal cells, unless histamine reaches the parietal cells via capillary transport from the glandular base. Mast cells are numerous in the stomach of, for example, man, pigs, dogs, and cats. They occur scattered throughout the stomach wall without any obvious relation to the parietal cells.(ABSTRACT TRUNCATED AT 250 WORDS).
Histamine-producing cells in the stomach and their role in the regulation of acid secretion. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/2042038��
Mucus Production 10Litle Each Day
Gastrointestinal
The Stomach
Mucus ProductionMucus production
The production of mucus is conducted by stomach surface epithelial cells and foveolar cells. The mucus itself is around 95 per cent water, with the remaining five per cent made up of polymers that give the mucus its gel-like viscosity. The viscosity of mucus is dynamic and can be altered by the rate of secretion from glandular cells or rate of breakdown by proteolytic enzymes within the stomach lumen.
The bicarbonate element of the mucus is important as it allows an increased pH local to the epithelial cells, protecting them from the highly acidic stomach environment. These bicarbonate ions are formed in the mucus-secreting cells by reacting carbon dioxide with water, using the enzyme carbonic anhydrase. The bicarbonate ions are then pumped into the mucus layer by exchanging them with chloride ions.
Mucus is secreted by the stomach epithelial cells, but the mucus is mainly secreted from foveolar cells, found in the necks of the gastric pits. Mucus-secreting cells are the most abundant cell type in the stomach, giving indications of how important mucus is to the functioning stomach.
��
Control of Secretion
Much like the control of salivary secretions, the gastric secretions (including mucus secretion) is largely controlled by neural influences. An increase in mucus production is signalled by a stimulation of the Vagus nerve and is mediated by prostaglandins. The cells respond to external factors such as mechanical stress and elements of the cephalic and gastric digestion phases by increasing mucus productions as required. In healthy patients, there is always a thick mucus layer to protect the stomach from auto-digestion.Clinical Relevance - Peptic Ulcer Disease
If for some reason the mucus layer is breached, the epithelial cells are exposed to concentrated stomach acid and the digestive enzymes contained within the gastric juices. As the stomach wall is made up of the same proteins and lipids as many foods we eat, if this mucosal barrier is broken the stomach will begin to digest itself, forming a peptic ulcer.
There are certain precipitations to this condition, including gastric colonisation of Helicobacter pylori, exposure to nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen, and excess acid secretion as seen in Zollinger-Ellison syndrome.
The effect of NSAIDs on gastric mucus secretion has been known for around 40 years. Drugs such as ibuprofen and aspirin have revolutionised the immediate management of pain and inflammation, but can have adverse effects on the gastrointestinal mucosa. NSAIDs work by blocking the synthesis of prostaglandins, which play an important role in both pain sensation and mucosal maintenance.
When prostaglandins are blocked, less gastric mucus is secreted, cellular junctions become less tight and mucosal blood flow is less adequately maintained. This makes the mucus layer less effective at protecting the stomach epithelium as well as less effective at managing any subsequent damage to the epithelium. This is why patients on chronic intakes of NSAIDs are at a much higher risk of peptic ulcers.Mucus Production - Regulation - TeachMePhysiologyTeachMePhysiology
https://teachmephysiology.com/gastrointestinal-system/stomach/mucus-production/��
Developmental Anatomy and Physiology of the Stomach
Steven J. Czinn, Samra Sarigol Blanchard, in Pediatric Gastrointestinal and Liver Disease (Fourth Edition), 2011
Pepsinogen
Pepsinogen is a powerful and abundant protein digestive enzyme secreted by the gastric chief cells as a proenzyme and then converted by gastric acid in the gastric lumen to the active enzyme pepsin. The role of pepsin and its precursor in protein digestion was first described in the 19th century. Pepsinogens consist of a single polypeptide chain with a molecular weight of approximately 42,000 Da. Pepsinogens are synthesized and secreted primarily by the gastric chief cells of the human stomach before being converted into the proteolytic enzyme pepsin, which is crucial for digestive processes in the stomach. Furthermore, pepsin can activate additional pepsinogen autocatalytically. Pepsinogens belong to the endopeptidase family of aspartic proteinases. The aspartic proteinases are also called acid proteinases because they act between pH 1.5 and 5.0. The mucosal lining of human gastric mucosa produces four types of pepsinogen: pepsinogen I (PGA or PGI), pepsinogen II (PGC or PGII), cathepsin E, and cathepsin D. It has been reported that the moment of the first appearance of measurable pepsinogen in the fetal stomach varies considerably between species. In the human fetus, granules appear in the peptic cells at weeks 32 to 36. Cephalic vagal stimulation strongly stimulates pepsinogen secretion. Acetylcholine, cholecystokinin, gastrin, secretin, VIP, epidermal growth factor, and nitric oxide can stimulate pepsinogen.21
Anticholinergics, histamine H2-receptor antagonists, and vagotomy decrease pepsinogen secretion. Raised serum levels of type 1 pepsinogen have been associated with duodenal ulcer and gastrinoma, whereas atrophic gastritis (with or without pernicious anemia) has been associated with low levels of type 1 pepsinogen.Pepsinogen - an overview | ScienceDirect Topics
https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/pepsinogen��
Ginger Among Herbs Touted As Safer, Natural Alternatives To PPIs For Heartburn Relief
Amanda Burbank
August 8, 2016
An article published by The Alternative Daily shared the details of a recent study published in the Journal of the American Society of Nephrology that indicates users of proton pump inhibitors, or PPIs, such as well-known name-brands Nexium, Prevacid, and Prilosec, were found to have a 28 percent increased risk of kidney damage, and a 98 percent greater risk of kidney failure, compared to those who took H2 blockers, which include Tagamet, Zantac and Pepcid.
Kidney damage isn��t the only dangerous side-effect that has been linked to PPIs. The Alternative Daily lists the loss of bone density, nutritional deficiencies and an increased risk of heart attacks as other adverse effects. These drugs, which are prescribed to more than 15 million Americans, are also available over-the-counter.
The publication strives to share ��innovative ideas in alternative health�� with its readers, and suggests heartburn can be prevented and treated naturally, without consumers having to resort to taking medications.
��If you��re someone who is reliant on heartburn medications, it��s important to explore alternative treatments,�� The Alternative Daily reported.
The three ingredients suggested by the site for fighting heartburn safely are turmeric, dill and ginger root. Turmeric is listed as being beneficial at reducing symptoms of indigestion, inflammation, pain and ulcers as well as providing antioxidants. Dill is noted to improve digestion and prevent gas.
The third ingredient, ginger root, is a logical natural substitute for antacid drugs. The Alternative Daily reports that a 2007 study in Molecular Research and Food Nutrition found that ginger performed six to eight times better than PPI lansoprazole, an example of which is Prevacid.
��It was found to not only inhibit acid reflux but actually promote the production of proteolytic enzymes, which break down proteins �� something which conventional drugs deactivate,�� the article read.
But keep in mind that while fresh herbs are generally safe, consumers should stay away from processed herbal supplements, which are not regulated by the U.S. Food and Drug Administration (FDA). These products often boast unsubstantiated health claims, and may contain undeclared ingredients including dangerous drugs and contaminants.
It��s always a good idea to consult your doctor before following any advice about dietary changes.
The Alternative Daily offers its readers a recipe for an herbal tea with ingredients chosen to reduce heartburn and boost overall immunity. The recipe can be found by following the publication link provided below.Ginger among herbs touted as safer, natural alternatives to PPIs for heartburn relief | Righting Injustice
http://www.rightinginjustice.com/news/2016/08/08/ginger-among-herbs-touted-as-safer-natural-alternatives-to-ppis-for-heartburn-relief/��
NATURAL ALTERNATIVES TO HEARTBURN MEDICATION
Much like antibiotics, heartburn drugs of all kinds are grossly over-prescribed. According to most health care professionals, perhaps one in a hundred people actually need these drugs, while 99 out of 100 really need to change their diets and/or use harmless alternatives.
There are probably as many natural remedies for hyperacidity as there are prescription drugs, so one has many choices. If you are not sure what to do, a natural health care professional will be able to help.
Since stress, poor lifestyle choices such as cigarette smoking, and physical inactivity can cause hyperacidity as well as just about any disorder, efforts should definitely be made to make major changes in these areas.
Helicobacter pylori bacterial infections are usually thought to be the cause of peptic ulcer disease and the symptoms that usually trigger doctors to prescribe either proton pump inhibitors or other drugs. There are a variety of tests your doctor can order for you to make the diagnosis. These include blood tests, breath tests, x-rays, ultrasounds, scopes and even biopsies. Medical doctors treat the H. pylori infection with antibiotics and a variety of antacids and/or acid suppressing drugs.
With respect to diet, the best thing to do is eat frequent small meals throughout the day instead of the usual three large meals. Drink more spring or purified water to help dilute acid excess. Therapeutic vegetable juices include carrot, spinach, beet, cucumber, parsley, celery, cabbage and potato. These should be used liberally throughout the day (2 quarts daily). Raw potato juice just before breakfast can help reduce acid regurgitation. Avoid red meat, alcohol, hot sauces, spicy and fried foods, added salt, caffeine products, sugar and refined carbohydrate products.
If you use aspirin, replace it with white willow bark capsules. At least this will not further aggravate the discomfort.
A large number of people suffer from hyperacidity because of an allergy to milk protein (casein) or gluten found in most grains. Food allergy or sensitivity testing might be a good idea in the more stubborn cases failing to respond to other diet and supplement changes.
Supplements that have a healing effect against H. pylori include garlic, wild mountain oregano oil, berberine, lactobacillus acidophilus and other probiotics, bovine colostrum, essential fatty acids (flaxseed oil, cod liver oil, salmon oil, evening primrose oil, borage oil), licorice root tincture or herbal tea, choline, lecithin, PABA, bismuth, bentonite, goldenseal, slippery elm, burdock, manuka honey, ginger root, mastic gum and aloe vera juice.
It should be noted here that the long-term use of licorice can elevate blood pressure in some sensitive individuals. The glycyrrhetinic acid component of licorice is what is responsible for this potential side effect. The best way of getting around this problem while still taking advantage of licorice��s ability to protect the gastrointestinal lining from acid irritation is to use deglycyrrhizinated licorice (DGL). Many herbal brands manufacture DGL, a supplement widely available at most health food stores and pharmacies specializing in natural remedies.
The four herb combination of burdock, slippery elm, Turkish rhubarb, and sheep sorrel (a.k.a. Essiac) is effective for a wide range of gut problems including hiatus hernia, duodenal ulcers, gastritis, colitis, Crohn��s disease, non-specific indigestion, irritable bowel syndrome, hemorrhoids and bowel infections. It can be used alone or in combination with DGL, aloe vera juice or prescription medications in more resistant cases. Check with your doctor or naturopath for a personalized treatment regime.
��Drugs You Don��t Need And the Best Natural Alternatives
https://vitalitymagazine.com/article/drugs-you-dont-need-and-the-best-natural-alternatives/��
Ginger as Treatment for Heartburn �C Outperforms Acid-Blockers
Big Pharma profits when people treat symptoms with pills and syringes rather than removing the source of the problem and treating the condition naturally. People across cultures and centuries, however, have treated gastric discomfort and other ailments with ginger, and it seems we still don��t need Big Pharma��s help.
In a 2007 study published in the journal Molecular Research and Food Nutrition, researchers compared the anti-ulcer and anti-Helicobacter plyori (a bacteria linked to ulcers) properties of ginger and conventional acid-blockers like Lansoprazole, or Prevacid. Remarkably, ginger performed six to eight times better than did the drug. Rather than interfering with or removing stomach acid barrier (and thereby deactivating proteolytic enzymes and increasing risk of infection), ginger inhibits acid reflux and contains potent proteolytic enzymes comparable to the celebrated papaya. Researchers cite the ginger root as ��potential in-expensive multistep blockers against ulcer.��
In addition to being a treatment for heartburn, the health benefits of ginger are more than abundant, with the spice also being antibacterial, antiviral, and has antiparasitic properties. It��s been found to kill cancer cells more effectively than often harmful cancer drugs and even ease the common cough.
Read more: https://naturalsociety.com/ginger-powerful-treatment-for-heartburn/#ixzz6Gxz1LpDG
Follow us: @naturalsociety on Twitter | NaturalSociety on FacebookGinger More Powerful than Drugs as Treatment for Heartburn
https://naturalsociety.com/ginger-powerful-treatment-for-heartburn/��
Send to
Mol Nutr Food Res. 2007 Mar;51(3):324-32.
Inhibition of gastric H+, K+-ATPase and Helicobacter pylori growth by phenolic antioxidants of Zingiber officinale.
Siddaraju MN1, Dharmesh SM.Department of Biochemistry and Nutrition, Central Food Technological Research Institute, Mysore 570-020, Karnataka, India.
Abstract
Ulcer is a common global problem characterized by acute gastric irritability, bleeding, etc. due to either increased gastric cell proton potassium ATPase activity (PPA) or perturbation of mucosal defence. Helicobacter pylori has been identified as a major ulcerogen in addition to oxidative stress and nonsteroidal anti-inflammatory drugs. In this paper, we report ginger-free phenolic (GRFP) and ginger hydrolysed phenolic (GRHP) fractions of ginger (Zingiber officinale) as potent inhibitors of PPA and H. pylori growth. GRFP and GRHP inhibited PPA at an IC(50) of 2.9 +/- 0.18 and 1.5 +/- 0.12 microg/mL, exhibiting six- to eight-fold better potency over lansoprazole. GRFP is constituted by syringic (38%), gallic (18%) and cinnamic (14%) acids and GRHP by cinnamic (48%), p-coumaric (34%) and caffeic (6%) acids as major phenolic acids. GRFP and GRHP further exhibited free radical scavenging (IC(50) 1.7 +/- 0.07 and 2.5 +/- 0.16), inhibition of lipid peroxidation (IC(50) 3.6 +/- 0.21 and 5.2 +/- 0.46), DNA protection (80% at 4 microg) and reducing power abilities (80-338 U/g) indicating strong antioxidative properties. GRFP and GRHP may thus be potential in-expensive multistep blockers against ulcer.��
���������������θH+��K+- atpø�������ݸ˾��������������á�
Siddaraju MN1, Dharmesh SM��
ӡ�ȿ������˰�������570-020����ʳƷ�����о������ﻯѧ��Ӫ��ѧϵ��
ժҪ
������һ��������ȫ�������⣬�������Ǽ���θӦ���ԡ���Ѫ�ȣ���ԭ�������θϸ�������Ӽ�atpø����(PPA)���ӣ�Ҳ������ճĤ�����ܵ����š������ݸ˾��dz�����Ӧ���ͷ��������ҩ�������Ҫ����ԭ������ƪ�����У����DZ����������� �����(GRFP)�ͽ�ˮ���(GRHP)������ΪPPA��h.p ylori��������Ч���Ƽ���GRFP��GRHP��2.9��0.18��1.5��0.12g/mL��IC(50)����PPA����ʾ�����������������˱���Ч����GRFP��Ҫ�ɶ�����(38%)��ûʳ����(18%)�������(14%)��ɣ���GRHP��Ҫ�������(48%)�����㶹����(34%)�Ϳ�����(6%)��ɡ�GRFP��GRHP��һ�����ֳ�������ɻ�(IC(50) 1.7 +/- 0.07��2.5 +/- 0.16)������֬�ʹ�����(IC(50) 3.6 +/- 0.21��5.2 +/- 0.46)��DNA����(4gʱ80%)�ͻ�ԭ����(80-338 U/g)�����н�ǿ�Ŀ��������ܡ���ˣ�GRFP��GRHP���������������DZ�ڵİ���Ķಽ��ϼ���
PMID: 17295419 DOI: 10.1002/mnfr.200600202Inhibition of gastric H+, K+-ATPase and Helicobacter pylori growth by phenolic antioxidants of Zingiber officinale. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/17295419��
Evid Based Complement Alternat Med. 2011; 2011: 249487.
Gastroprotective Effect of Ginger Rhizome (Zingiber officinale) Extract: Role of Gallic Acid and Cinnamic Acid in H+, K+-ATPase/H. pylori Inhibition and Anti-Oxidative Mechanism
Siddaraju M. Nanjundaiah, Harish Nayaka Mysore Annaiah, and Shylaja M. Dharmesh*
Author information Article notes Copyright and License information Disclaimer
Department of Biochemistry and Nutrition, Central Food Technological Research Institute, CSIR, Mysore 570 020, Karnataka, India
Abstract
Zinger officinale has been used as a traditional source against gastric disturbances from time immemorial. The ulcer-preventive properties of aqueous extract of ginger rhizome (GRAE) belonging to the family Zingiberaceae is reported in the present study. GRAE at 200 mg kg−1 b.w. protected up to 86% and 77% for the swim stress-/ethanol stress-induced ulcers with an ulcer index (UI) of 50 �� 4.0/46 �� 4.0, respectively, similar to that of lansoprazole (80%) at 30 mg kg−1 b.w. Increased H+, K+-ATPase activity and thiobarbituric acid reactive substances (TBARS) were observed in ulcer-induced rats, while GRAE fed rats showed normalized levels and GRAE also normalized depleted/amplified anti-oxidant enzymes in swim stress and ethanol stress-induced animals. Gastric mucin damage was recovered up to 77% and 74% in swim stress and ethanol stress, respectively after GRAE treatment. GRAE also inhibited the growth of H. pylori with MIC of 300 �� 38 ��g and also possessed reducing power, free radical scavenging ability with an IC50 of 6.8 �� 0.4 ��g mL−1 gallic acid equivalent (GAE). DNA protection up to 90% at 0.4 ��g was also observed. Toxicity studies indicated no lethal effects in rats fed up to 5 g kg−1 b.w. Compositional analysis favored by determination of the efficacy of individual phenolic acids towards their potential ulcer-preventive ability revealed that between cinnamic (50%) and gallic (46%) phenolic acids, cinnamic acid appear to contribute to better H+, K+-ATPase and Helicobacter pylori inhibitory activity, while gallic acid contributes significantly to anti-oxidant activity.��������ȡ���θ�ı�������:ûʳ������������H+��K+-ATPase/H�е����á������ݸ˾������ƺͿ���������
ӡ�ȿ������˰�������570 020����ʳƷ�����о������ﻯѧ��Ӫ��ѧϵ
ժҪ
��Զ��ʱ���������ͱ�������ͳ�Ŀ�θ��ҩ����ı����˽���ֲ�ェ��ˮ���GRAE���Ŀ��������á�GRAE��200 mg kg - 1 b.w����ӾӦ��/�Ҵ�Ӧ��������ı����ʷֱ�Ϊ86%��77%������ָ��(UI)Ϊ50��4.0/46��4.0����30 mg kg - 1 b.wʱ��������(80%)�ı��������ơ������յ�����H+��K+-ATPase���Ժ�����ͱ����ᷴӦ����(TBARS)�������ߣ�GRAE����������ӾӦ�����Ҵ�Ӧ���յ�����GRAEˮƽ����������GRAE����/����������øҲ������������ӾӦ�����Ҵ�Ӧ����θ�Һ���˵Ļָ��ֱ�Ϊ77%��74%����Ҳ������������������������˷�300��38��gҲ���л�ԭ����,���ɻ����������IC50 6.8��0.4��g����−1ûʳ���ᵱ��(GAE)��DNA����90%��0.4��gҲ�۲쵽�������о�������������Ϊ5����- 1�����صĴ�����û���������á��ɷַ����IJⶨ�����������DZ�ڵĹ�Чulcer-preventive����֮��¶,���(50%)�ͷ�����(46%)���ᡢ������ƺ������ڸ��õ�H +, K + atpø�����������˾����ƻ,ûʳ�������������ڿ��������ԡ�
Scheme 1
Ulcerogens generate oxidative stress (OS) leading to susceptibility for ulcer formation by activating H+, K+-ATPase, enabling H. pylori colonization and invasion, mucosal damage, and so forth, ginger downregulates these events.3.5. Anti-Helicobacter pylori Activity of GRAE
The bacteria isolated from endoscopic samples were Gram-negative, motile and showed positive for urease, catalase and oxidase tests (Table 5). Further, it was confirmed by the response to antibiotics as it was resistant to antibiotics like erythromycin, nalidixic acid, polymixin B, penicillin and vancomycin and was susceptible to amoxicillin, clarithriomycin and metronidazole. The appearance of a characteristic white mucilaginous colony confirms the identity of bacteria as H. pylori.In the agar diffusion method, GRAE showed a clear inhibition zone around the disc at 50 ��g mL−1 concentration equivalent to that of a susceptible antibiotic amoxicillin at 10 ��g mL−1 (Figure 2(a)). MIC values determined by broth dilution method indicated significant anti-H. pylori activity at 300 �� 38 ��g mL−1 at P ~ .003.
SEM observation revealed the efficacy of GRAE action in inhibiting the H. pylori growth. Figure 2(b) shows the uniform rod-shaped normal H. pylori cells, whereas the cells treated with amoxicillin, GRAE, gallic and cinnamic acid changed from helical form to coccoid and became necrotic (showed with arrows in Figures 2(c)�C2(f)). Coccoid form with blebs in the bacterial surface, appearance of vacuoles, granules and an area of low electron density in the cytoplasm (showed with arrow marks) were observed in GRAE-treated sample indicating the lysis of H. pylori. Results were also substantiated by viability test, which indicated the loss of 85% viability upon treatment with GRAE supporting anti-microbial nature of GRAE.4. Discussion
Ulcer results from an imbalance between aggressive factors and the maintenance of mucosal integrity through the endogenous defense mechanisms. To regain the balance, different therapeutics including spice and plant extracts have been used. In the previous paper, we had shown that free and bound phenolics of ginger possessed potential ulcer preventive activity in vitro, including inhibition of H+, K+-ATPase and H. pylori growth [9]. However, in view of addressing a question whether the traditional practice of using crude ginger extract in either boiled water or cold water extract can yield compounds which are gastroprotective in nature; we evaluated in vitro and in vivo ulcer-preventive properties of GRAE and determined whether it also contained phenolic acids that favors gastroprotection as reported in our previous papers [9, 17].
ROS are implicated in the pathogenesis of several diseases. Free radicals are continuously produced during normal physiologic events and removed by anti-oxidant defense mechanisms, including enzymes such as SOD, CAT and enzymes involved in the glutathione redox cycle. Free radicals cause lipid peroxidation and production of highly toxic lipid derivatives, which in turn can modify cell functions and even may lead to cell death. Oxidative modification of proteins may result in structural impairment and also change their functional properties such as their involvement in signaling, critical for numerous cellular functions. They affect the vasomotor function of vasculature throughout the body via alterations in the activity of the autonomic nervous system, thus changing the blood flow to involve tissues such as mucosa. Oxidative stress (OS) being major source in causing ROS-mediated ulceration, up/down regulation of anti-oxidant/anti-oxidative enzymes reveal the ability of GRAE to counteract the OS condition and hence protection to ulcer.
GRAE at 200 mg kg−1 b.w. protected swim stress/ethanol-induced ulcer lesions up to 86% similar to that of lansoprazole (80%), a known antiulcer drug at 30 mg kg−1 b.w. Bloody streaks, inflammations, oozing of blood into the lumen of the stomach, and so forth, observed in ulcerous animals were not found in GRAE ingested animals, similar to those of healthy rats indicating the gastroprotective effect of GRAE. Further, we followed the protective effect investigating the biochemical parameters such as alterations in the gastric mucin, oxidants, GSH, H+, K+-ATPase and anti-oxidant enzymes level including CAT, SOD, peroxidase, and so forth, in the ulcerated organ��stomach as well in the metabolizing organ��liver in all groups of rats��healthy, ulcerated and GRAE/lansoprazole treated. Preventive anti-oxidant enzymes such as SOD and CAT are the first line of defense against ROS. Administration of GRAE resulted in a significant increase in the SOD, CAT and reduced GSH levels (Tables (Tables11 and and2)2) similar to those of control animals, suggesting the efficacy of GRAE in preventing free radical-induced damage during ulceration. SOD, GPx, CAT and GSH all contribute to maintain anti-oxidant status during oxidative stress condition such as ulcers. SOD and GPx levels were increased during ulceration to scavenge superoxide and peroxy radicals generated during ulceration. Depletion in CAT and reduced GSH suggest the utilization of these components towards the neutralization of increased free radicals.
In our experimental model, ∼3.4-fold reduction in gastric mucin and 2.4-fold reduced glutathione as well as 2.6-fold increased oxidative product��TBARS in the stomach were normalized by GRAE (Tables (Tables11�C3) treatment. Gastric ulcers are often a chronic disease and it may persist for 10�C20 years as characterized by repeated episodes of healing and re-exacerbations [23]. Stress-induced ulcer better resembles clinical ulcers in chronicity, severity and practicality of experiencing stress due to varietal patterns of lifestyle in day to day life and serves the most reliable model to study ulcer healing process [24, 25]. The incidence of swim stress-induced ulcer is predominant in the glandular part of the stomach leading to gastric mucosal/mucin damage. GRAE significantly prevented ulcers both by reducing the oxidative stress as well as boosting the mucosal defense.
Further, during our study, we evaluated the possible mechanism of protection to gastric ulcer apart from up-regulation of anti-oxidant and anti-oxidant enzyme levels. Gastric H+, K+-ATPase located in the apical membrane of parietal cells, pumps protons into the gastric lumen, using energy derived from the hydrolysis of ATP, and is thus involved in gastric acid secretion. Accordingly, the activity of gastric H+, K+-ATPase was measured in the stomach homogenate, which showed 3-fold up-regulation of the enzyme in ulcer condition and was normalized by treatment with GRAE (Table 3). Results were further substantiated by sheep H+, K+-ATPase inhibition by GRAE with an IC50 of 16.5 �� 1.2 ��g mL−1 on par or better than lansoprazole (19.3 �� 2.2 ��g w/w), indicating the potential multi-targeted effect of GRAE in preventing swim stress-induced ulcers in experimental rats. GRAE may find itself more useful as a H+, K+-ATPase (proton pump) inhibitor than the existing pump inhibitors, since they have adverse effects as reported particularly under conditions of pregnancy/lactation and alcohol or any other drug consumption. Least toxicity of GRAE may also find GRAE as useful alternative source for ulcer healing therapeutics.
Besides, GRAE also exhibited the anti-H. pylori activity. The MIC values obtained confirmed the significant (P = .003) anti-H. pylori activity, and as reported in previous papers results were supported by an observation of Vattem et al. [18], where phenolic phytochemicals such as cinnamic acid, cinnamaldehyde, coumarins and flavonoids have been suggested to exhibit high anti-H. pylori activity. GRAE with higher content of cinnamic acid showed better inhibition of H. pylori. As mentioned in earlier studies, phenolics were thought to exert their anti-microbial effect by causing (i) hyperacidification at the plasma membrane interface of the microorganism [26] or (ii) intracellular acidification, resulting in disruption of H+, K+-ATPase required for ATP synthesis of microbes [27] or (iii) may be related to inactivation of cellular enzymes causing membrane permeability changes [27].
Also, it is intriguing to observe that cinnamic acid is acting as a potent inhibitor of H+, K+-ATPase, and also H. pylori probably by stronger binding of cinnamic acid to membrane domains of H+, K+-ATPase and H. pylori than gallic acid, which is a poor inhibitor of H+, K+-ATPase and H. pylori (Figure 6). Lack of correlation between fold inhibitory activity versus binding between gallic/cinnamic acid still do not rule out the proposed mechanism. Because being hydrophobic cinnamic acid binding ability may be higher with H+, K+-ATPase and H. pylori which carry membrane domains than HSA alone which is devoid of this hydrophobic domain.
GRAE also exhibited reducing power and prevented free radical-induced lipid and DNA peroxidation. This anti-oxidative property also contributes significantly to reduce ulcer condition and justifies the ethno medical claims. Table 6 shows the relative concentration of individual phenolic acids towards the antiulcer activity. Our data in the present and previous papers indicate that cinnamic acid, caffeic acid and p-coumaric acids contribute significantly to inhibition of H+, K+-ATPase and H. pylori growth in the free as well as in the bound form. Recently, we also reported the role of cinnamic acid in cross-linking property of pectic polysaccharide and its contribution hence to the structural stability (manuscript communicated).
Table 6
Relative percentage contribution of individual phenolic acids towards anti-oxidant, anti-H. pylori and H+, K+-ATPase inhibition.
GRAE Phenolics (mg g−1) AOX (% contribution) PPI (% contribution)H pylori-inhibition (% contribution)
Gallic acid 3.4 93 9 66
Cinnamic acid 3.8 1 88 30
Other phenolics 0.4 6 3 4
Gallic acid significantly contributed to anti-oxidant activity than cinnamic acid where as cinnamic acid contributed to H+, K+-ATPase inhibition.
5. Conclusions
The present study clearly demonstrated that aqueous extract of ginger was able to protect the gastric mucosa from stress-induced mucosal lesions and inhibits gastric acid secretion probably by blocking H+, K+-ATPase action, inhibiting growth of H. pylori and offering anti-oxidant protection against oxidative stress-induced gastric damage The results confirm the popular use of ginger for its medicinal properties in Ayurveda and folklore medicines. These results further suggest the use of ginger for gastric disorders that needs to be considered as possibilities for new therapeutic approaches.Gastroprotective Effect of Ginger Rhizome (Zingiber officinale) Extract: Role of Gallic Acid and Cinnamic Acid in H+, K+-ATPase/H. pylori Inhibition and Anti-Oxidative Mechanism
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3136331/��
Trans-Cinnamic acid ��ʽ�����
Cinnamic acid exists as trans and cis isomers, but the trans form is the one most often found in nature and is the article of commerce. It is obtained from cinnamon bark and balsam resins such as storax. It was first isolated in 1872 by F. Beilstein (of Handbook of Organic Chemistry fame) and A. Kuhlberg. It is synthesized by the Perkin reaction between Ac2O and PhCHO.
trans-Cinnamic acid is used in the manufacture of flavors, dyes, and pharmaceuticals; but its major use is for the production of its methyl, ethyl, and benzyl esters. These esters are important components of perfumes. The acid is also a precursor to the sweetener aspartame.trans-Cinnamic acid - American Chemical Society
https://www.acs.org/content/acs/en/molecule-of-the-week/archive/c/trans-cinnamicacid.html��
Gastroprotective [6]-Gingerol Aspirinate as a Novel Chemopreventive Prodrug of Aspirin for Colon Cancer
Published: 09 January 2017
Gastroprotective [6]-Gingerol Aspirinate as a Novel Chemopreventive Prodrug of Aspirin for Colon Cancer
Yingdong Zhu, Fang Wang, Yantao Zhao, Pei Wang & Shengmin Sang
Abstract
A growing body of research suggests daily low-dose aspirin (ASA) reduces heart diseases and colorectal cancers. However, the major limitation to the use of aspirin is its side effect to cause ulceration and bleeding in the gastrointestinal tract. Preclinical studies have shown that ginger constituents ameliorate ASA-induced gastric ulceration.We here report the design and synthesis of a novel prodrug of aspirin, [6]-gingerol aspirinate (GAS). Our data show that GAS exerts enhanced anti-cancer properties in vitro and superior gastroprotective effects in mice. GAS was also able to survive stomach acid and decomposed in intestinal linings or after absorption to simultaneously release ASA and [6]-gingerol. We further present that GAS inactivates both COX-1 and COX-2 equally. Our results demonstrate the enhanced anticancer properties along with gastroprotective effects of GAS, suggesting that GAS can be a therapeutic equivalent for ASA in inflammatory and proliferative diseases without the deleterious effects on stomach mucosa.
Gastroprotective [6]-Gingerol Aspirinate as a Novel Chemopreventive Prodrug of Aspirin for Colon Cancer | Scientific Reports
https://www.nature.com/articles/srep40119��
��
THE BENEFITS OF THE USE OF GINGER
IN HERBAL PREPARATIONS
CHEMICAL CONSTITUENTS OF GINGER
by Martha WhitneyThis site brought to you by The School of Natural Healing & Christopher Publications
Zingiber officinale is the botanical name of ginger. It is botanically correct to refer to ginger as a rhizome rather than a root. Ginger can therefore be propagated from budded sections. Ginger is one of the world��s top ten favored spices. Yet surprisingly, its tremendous medicinal value is virtually unknown.
Ginger can be divided into four principle parts: taste or pungency, essential oil or fragrance, macro/micro-nutrients, and synergists. An oily-resinous substance dissected from the plant comprising 5 to 10 percent of the plant is called ginerol.4
This oleoresin was then broken down into close to thirty elements. Gingerols or zingiberene may be responsible for taste and scent respectively. In fact, within ginger there are hundreds of ingredients that are referred to as synergists. These interact to make the plant as a whole the powerful healer that it is.
There may be dozens of different complex chemical interactions allowing ginger to prevent or benefit conditions like heart attacks, arthritis and ulcers. There are more than four hundred taste, fragrance, nutrient and synergistic constituents interacting to create an endless number of medicinal benefits.
One gram of one of ginger��s principle constituent, zingibain, can actually tenderize as much as twenty pounds of meat. The obvious impact or effect is improved digestion. This enzyme can enhance the effectiveness of other antibacterial elements by as much as 50%. The enzyme zingibain can aid immunity to the effect of digesting parasites and their eggs, and is associated with anti-inflammatory activity. This is due in part to the fact that ginger acts as an antioxidant with more than twelve constituents superior to vitamin E.5 This action empowers ginger to help neutralize free radicals which are widely recognized as participation or being responsible for the inflammation process.
Each of ginger��s 477 constituents could be listed. This impressive list includes the well known ascorbic acid, caffeic acid, capsaicin, beta-sitosterol, beta-carotene, curcumin, lecithin, limonene, selenium and tryptophan. It is nothing less than an exercise in complete and utter futility to try and isolate the ��active�� element from ginger.
4 Felter, H. W., and Lloyd, J. U. King��s American Dispensatory. Portland, Ore.: Eclectic Medical, 1983, 2110.
5. a. Govindarajan, V.S. ��Ginger �CChemistry, technology, and quality evaluation: Part 2.: Critical Reviews in Food Science and Nutrition 17, no. 8 (1082): 189-258 (p. 230), citing Hirahara, F. ��Antioxidative activity of various spices on oils and fatrs.Chemical Constituents of Ginger
https://www.herballegacy.com/Whitney_Chemical.html��
Zingibain, zingipain, or ginger protease (EC 3.4.22.67) is a cysteine protease enzyme found in ginger (Zingiber officinale) rhizomes. It catalyses the preferential cleavage of peptides with a proline residue at the P2 position.
Zingibain - Wikipedia
en.wikipedia.org/wiki/Zingibain
����
GINGER RHIZOME: A NEW SOURCE OF PROTEOLYTIC ENZYME
E. H. THOMPSON I. D. WOLF C. E. ALLEN
First published: May 1973 https://doi.org/10.1111/j.1365-2621.1973.tb02836.x Citations: 64
Paper No. 8158. Scientific Journal Series, Minnesota Agricultural Experiment Station, St. Paul.
Abstract
The proteolytic activity of ginger rhizome was studied with bovine serum albumin (BSA), collagen and actomyosin as substrates. A semipurified, powdered enzyme preparation was prepared by buffer extraction of an acetone powder of ginger rhizome and subsequent acetone precipitation of the proteolytic principle from the buffer extract. With 3% BSA as substrate, a relatively high proteolytic activity occurred over a pH range of 4.5�C6.0, with an optimum pH of 5.0. The optimum temperature for proteolysis of BSA was 60��C during a 10 min reaction time, with rapid denaturation of the enzyme occurring at 70�� C. NaCl in cone up to 10% produced about a 20 and 50% reduction in proteolysis of collagen and BSA, respectively. The ginger protease was protected by dithiothreitol during extraction and reaction, indicating the involvement of −SH groups at the active site. The analyses of soluble peptide amino acids or terminal amino acids suggest that the proteolysis of collagen is many fold greater than that of actomyosin. The combined proteolysis of these two muscle protein fractions by the ginger protease resulted in significantly more tender meat. According to conventional nomenclature, ��Zingibain�� is the proper name for this proteolytically active principle in Zingiber officinale roscoe or ginger rhizome which is commonly referred to as gingerroot. For meat applications, a possible advantage of zingibain over papain and ficin is the greater proteolysis of collagen in comparison to actomyosin. When compared to reported values for bromelain, zingibain has a higher optimum activity temperature, which is desirable in some applicationsGINGER RHIZOME: A NEW SOURCE OF PROTEOLYTIC ENZYME ...
https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1365-2621.1973.tb02836.x��
Yonago Acta Med. 2011 Mar; 54(1): 11�C19.
Protective Effects of Ginger against Aspirin-Induced Gastric Ulcers in Rats
Zhongzhi Wang, Junichi Hasegawa, Xinhui Wang, Akiko Matsuda, Takahiro Tokuda, Norimasa Miura, and Tatsuo Watanabe*
Author information Article notes Copyright and License information Disclaimer
Division of Pharmacotherapeutics, Department of Pathophysiological and Therapeutic Science, School of Medicine, Tottori University Faculty of Medicine, Yonago 683-8503, Japan
*Division of Integrative Physiology, Department of Functional, Morphological and Regulatory Science, School of Medicine, Tottori University Faculty of Medicine, Yonago 683-8503, Japan
Abstract
We investigated the mechanism underlying the protective effects of ginger against gastric damage induced by aspirin in rats. Gastric mucosal lesions were produced by orally administering 200 mg/kg aspirin suspended in 1% carboxymethylcellulose solution to pyloric-ligated male Wistar rats.Ginger powder (200 mg/kg) markedly reduced the aspirin-induced gastric hemorrhagic ulcer area. The total acidity of gastric juice was not significantly influenced by aspirin or ginger. Ginger powder did not affect the aspirin-induced reduction in mucosal prostaglandin E2 (PGE2) content; however, it did ameliorate the aspirin-induced increases in mucosal activity of the inducible form of NO synthase (iNOS) and plasma tumor necrosis factor (TNF)-�� and interleukin (IL)-1�� levels.
In the next experiment, high and low doses of 6-gingerol and 6-shogaol were used instead of ginger powder in the same experimental model to examine their roles in the anti-ulcer mechanism of ginger. Both 6-gingerol and 6-shogaol reduced aspirin induced ulcer formation, mucosal iNOS and plasma TNF-�� and IL-1�� levels.
In conclusion, ginger powder prevents the aspirin induced gastric ulcer formation by reducing mucosal iNOS activity and the plasma levels of inflammatory cytokines but does not affect gastric juice or acid production or mucosal PGE2 content. This protective effect of ginger powder against gastric ulcers may be attributable to both gingerol and shogaol.
��
������˾ƥ���´���θ����ı�������
����־�����ȴ���һ�����»ԡ��������ӡ�����߹㡢���ֺͶɱߴ���*
��ȡ��ѧҽѧԺ�������������ƿ�ѧϵҩ������ѧ�����ձ�Yonago 683-8503
*��ȡ��ѧҽѧԺ���ܡ���̬�͵��ڿ�ѧϵ�ۺ�����ѧ����Yonago 683-8503
ժҪ
�����о��˽���˾ƥ�����´���θ���˵ı������û��ơ���1%�ȼ���ά����Һ����Һ�ڷ�200 mg/kg��˾ƥ�֣��Խ������Ž���������Wistar�������θճĤ���ˡ�
������(200 mg/kg)�����Լ��ٰ�˾ƥ������θ�����Ѫ�������˾ƥ�ֺ�������θҺ����ȵ�Ӱ�첻�����������۲�Ӱ�찢˾ƥ���յ���ճĤǰ������E2 (PGE2)��������;Ȼ��,�������˰�˾ƥ���յ���iNOSø�ϳɺ�Ѫ��������������(TNF) -���Ͱ���(IL) 1��ˮƽ�����ӡ�
�ڽ�������ʵ���У�����ͬ��ʵ��ģ���У��ֱ��øߡ��ͼ����Ľ����غͽ�ϩ�Ӵ��������ۣ��о���������������������е����á������غͽ�ϩ�ӽ��Ͱ�˾ƥ���յ������γ�,ճĤiNOS��Ѫ��TNF-����IL-1��ˮƽ��
����������������ͨ������ճĤiNOS���Ժ�Ѫ������ϸ������ˮƽ����ֹ��˾ƥ���յ���θ�����γɣ�����Ӱ��θҺ��θ��IJ�����ճĤPGE2�����������۶�θ��������ֱ������ÿ��������ڽ����غͽ�ϩ�ӹ�ͬ���õĽ����Gastric juice and acid production
Both ginger and aspirin reduced gastric juice production /100 g body weight. However this effects disappeared by coadministration of ginger and aspirin. The total acidity showed no significant differences among these conditions (Table 1).Mucosal iNOS expression
Aspirin administration led to a significant increase in gastric mucosal iNOS activity in the ulcerated rats as compared to that measured in the control or ginger administered rats (Fig. 4). The coadministration of ginger powder resulted in a significant decrease in gastric mucosal iNOS activity compared with that observed in the aspirin alone group (P < 0.05). No differences were seen in iNOS levels between the ginger powder treated Group 2 and Group 1 (control).
One of the mechanisms by which aspirin damages the gastric mucosa is the increased production of NO due to the overexpression of iNOS (Kontureck et al., 2006). NO is a mediator not only of gastrointestinal mucosal defense (Calatayud et al., 2001), but also of its damage (Muscara and Wallace, 1999). It has been shown that different concentrations of NO have completely opposite effects in the same tissue (Wallace and Millor, 2000). In general, the mucosal and endothelial NOS isoforms produce low amounts of NO. However, the high quantity of NO produced by iNOS damages the epithelium (Piotrowski et al., 1999; Wallace and Miller, 2000). The excessive release of NO from gastric epithelial cells induced by aspirin has been reported to exert detrimental effects (Whittle, 2003; Hsu and Liu, 2004). Inhibiting aspirin-induced increases in iNOS expression in the gastric mucosa leads to a reduction in gastric mucosal damage (Konturec et al., 2006). In the present study, ginger powder reduced iNOS activity and inhibited the production of gastric ulcers, even in the presence of aspirin.Effects of 6-gingerol and 6-shogaol in the presence of aspirin
High and low doses of 6-gingerol and 6-shogaol, the major constituents of ginger powder, had no adverse effects on the stomach and inhibited the ulcer formation induced by aspirin (Fig. 6). 6-Gingerol and 6-shogaol exerted inhibitory effects on the aspirin induced increases in mucosal iNOS activity (Fig. 7) and plasma TNF-�� and IL-1�� levels (Fig. 8) in the same manner as ginger powder.
��
From these results, ginger powder is suggested to protect the stomach against the ulcer formation induced by aspirin by reducing iNOS activity in the gastric mucosa and inflammatory cytokine (TNF-�� and IL-1��) expression. These effects of ginger powder seem to be derived from the actions of gingerol and shogaol, the main ingredients of ginger.
Keywords: aspirin, gastric damage, ginger powder, inflammatory cytokine, inducible form of NO synthase activityProtective Effects of Ginger against Aspirin-Induced Gastric Ulcers in Rats
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3763798/��
Medicina (Kaunas). 2019 Mar; 55(3): 64.
Gastroprotective Effect of Zingerone on Ethanol-Induced Gastric Ulcers in Rats
Neda Sistani Karampour,1 Ardeshir Arzi,1,* Anahita Rezaie,2 Marzieh Pashmforoosh,3 and Fatemeh Kordi3
1Department of Pharmacology, Jundishapur University of Medical Sciences, Ahvaz 6135715794, Iran; moc.oohay@6831inatsis.n
Abstract
Background and objectives: Zingerone is an ingredient of ginger (Zingiber officinale) with different pharmacological activities. Several studies have investigated the effect of zingerone on various gastrointestinal diseases, including irritable bowel syndrome and diarrhea. This study is aimed to evaluate the effect of zingerone on ethanol-induced gastric ulcers in rats.Materials and Methods: Gastric ulcers were induced by ethanol (96%, 5 mL/kg, po) in male wistar rats and zingerone (50, 100, and 200 mg/kg) was administrated orally. Normal saline and ranitidine were used as negative and positive control, respectively. In this study, the number and length of ulcers, and malondialdehyde (MDA) and nitric oxide (NO) levels in stomach tissues were determined.
Results: The findings showed that the mean number and length of gastric ulcers were significantly lower in zingerone-received groups than ethanol group (P < 0.05). The level of malondialdehyde was decreased in the stomach of zingerone groups (P < 0.05) compared to the ethanol group. In addition, zingerone treatment prevented the decrease of nitric oxide level by ethanol in the stomach tissue.
Conclusions: The present study showed that zingerone has a protective effect on the ethanol-induced gastric ulcer, which may be due to its free radical scavenging activity.
����ͪ���Ҵ���θ��������θ��������
Neda Sistani Karampour,1 Ardeshir Arzi,1 Anahita Rezaie,2 Marzieh Pashmforoosh,3 Fatemeh Kordi3
1 ����Jundishapurҽ�ƴ�ѧҩ��ѧϵ��Ahvaz 6135715794������;moc.oohay@6831inatsis.n
ժҪ
������Ŀ��:����ͪ������(Zingiber officinale)��һ�ֳɷ֣����в�ͬ��ҩ�����ԡ������о��Ѿ�����������ͪ�Ը���θ��������Ӱ�죬���������ۺ�֢��к�����о�ּ����������ͪ���Ҵ���θ��������Ӱ�졣
�����뷽��:���Ҵ�(96%��5 mL/kg, po)�յ�wistar���Դ���θ���ڷ�����ͪ(50��100��200 mg/kg)�����Զ�����Ϊ������ˮ�飬���Զ�����Ϊ�����涡�顣���о��ⶨ��θ����������ͳ��ȣ��Լ�θ��֯�б���ȩ(MDA)��һ������(NO)��ˮƽ��
���:����ͪ��θ�����ƽ�������ͳ������Ե����Ҵ���(P < 0.05)�����Ҵ���Ƚϣ�����ͪ��θ��֯����ȩˮƽ����(P < 0.05)�����⣬����ͪ������ֹ���Ҵ�����θ��֯��һ������ˮƽ��
����:����ͪ���Ҵ���θ������һ���ı������ã���������������ɻ��йء�
Keywords: gastric ulcer, zingerone, ethanol, malondialdehyde, nitric oxide, ratGastroprotective Effect of Zingerone on Ethanol-Induced Gastric Ulcers in Rats
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6473471/��
Helicobacter pylori Containing Only Cytoplasmic Urease Is Susceptible to Acid
Partha Krishnamurthy, Mary Parlow, Jason B. Zitzer, Nimish B. Vakil, Harry L. T. Mobley, Marilyn Levy, Suhas H. Phadnis, Bruce E. Dunn
Department of Pathology, Medical College of Wisconsin,1
ABSTRACT
Helicobacter pylori, an important etiologic agent in a variety of gastroduodenal diseases, produces large amounts of urease as an essential colonization factor. We have demonstrated previously that urease is located within the cytoplasm and on the surface of H. pylori both in vivo and in stationary-phase culture. The purpose of the present study was to assess the relative contributions of cytoplasmic and surface-localized urease to the ability of H. pylori to survive exposure to acid in the presence of urea. Toward this end, we compared the acid resistance in vitro of H. pylori cells which possessed only cytoplasmic urease to that of bacteria which possessed both cytoplasmic and surface-localized or extracellular urease. Bacteria with only cytoplasmic urease activity were generated by using freshly subcultured bacteria or by treating repeatedly subcultured H. pylori with flurofamide (1 ��M), a potent, but poorly diffusible urease inhibitor. H. pyloriwith cytoplasmic and surface-located urease activity survived in an acid environment when 5 mM urea was present. In contrast, H. pylori with only cytoplasmic urease shows significantly reduced survival when exposed to acid in the presence of 5 mM urea. Similarly,Escherichia coli SE5000 expressing H. pyloriurease and the Ni2+ transport protein NixA, which expresses cytoplasmic urease activity at levels similar to those in wild-typeH. pylori, survived minimally when exposed to acid in the presence of 5 to 50 mM urea. We conclude that cytoplasmic urease activity alone is not sufficient (although cytoplasmic urease activity is likely to be necessary) to allow survival of H. pyloriin acid; the activity of surface-localized urease is essential for resistance of H. pylori to acid under the assay conditions used. Therefore, the mechanism whereby urease becomes associated with the surface of H. pylori, which involves release of the enzyme from bacteria due to autolysis followed by adsorption of the enzyme to the surface of intact bacteria (��altruistic autolysis��), is essential for survival of H. pylori in an acid environment. The ability of H. pylori to survive exposure to low pH is likely to depend on a combination of both cytoplasmic and surface-associated urease activities.
����������ø�������ݸ˾���������������Helicobacter pylori Containing Only Cytoplasmic Urease Is Susceptible to Acid
Helicobacter pylori is a spiral, gram-negative bacterium which is the etiologic agent of chronic superficial gastritis. Infection with H. pylori is strongly associated with peptic ulcer disease, gastric carcinoma, and gastric lymphoma (4, 18, 34-36).Helicobacter pylori Containing Only Cytoplasmic Urease Is Susceptible to Acid | Infection and Immunity
https://iai.asm.org/content/66/11/5060.full��
��
South African Journal of Botany
Volume 119, November 2018, Pages 390-399
South African Journal of Botany
Anti-ulcerogenic and proton pump (H+, K+ ATPase) inhibitory activity of Clematis flammula L. extract
Author links open overlay panelF.YousD.Atmani-KilaniN.Debbache-B
Laboratoire de Biochimie Appliqu��e, Facult�� des Sciences de la Nature et de la Vie, Universit�� de Bejaia, 06000, Algeria
https://doi.org/10.1016/j.sajb.2018.09.036Get rights and content
•
The ethanolic extract of Clematis flammula L. exhibited
•
A good protection of the gastric mucosa
•
A strong antioxidant and anti-inflammatory potential
•
A powerful inhibition of the activity of proton pump
Abstract
Clematis flammula L. (CF) (Ranunculaceae) is widely used traditionally by native Berber populations in Algeria to treat gastrointestinal disorders. The present study was conducted to assess the gastroprotective properties and the underlying mechanisms of CF leaves ethanolic extract (CFEE) in four ulcer models in mice. After total phenol quantification, the gastro-protective effect of the CFEE was evaluated by macroscopic and microscopic analyses, investigation of antioxidant (superoxide dismutase (SOD), catalase (CAT), reduced glutathione (GSH) and pro-oxidant (malondialdehyde (MDA)) parameters. Furthermore, the impact of CFEE pre-treatment on mucus level, myeloperoxidase (MPO) and gastric proton pump (H+/K+-ATPase) activities was determined. Pro-kinetic properties were also assessed.Pharmacological investigation of CFEE showed a dose-dependent gastro-protective potential in different ulceration models, in close correlation with its high antioxidant activity (SOD, CAT and GSH). The most interesting underlying mechanisms were a remarkable reduction of MPO and proton pump activities (90 and 99% inhibition, respectively). Histological examination confirmed the preventive action of CFEE against offensive agents associated with increased mucus production and reduced gastric emptying and intestinal motility.
Our findings provide a scientific basis for the ethno-pharmacological purpose of the studied plant.�Ϸ�ֲ��ѧ��־
��119����2018��11�£�390-399ҳ
�Ϸ�ֲ��ѧ��־
��������ȡ��Ŀ��������ú����ӱ�(H+�� K+ ATPase)���ƻ���
����Ƕ��Ӧ��ʵ���ң������Ǵ�ѧ��Ȼ��������ѧѧԺ�����������ǣ�06000
•�Իƻ�������Ϊ�Բģ��о������Ҵ���ȡ�����ȡ����
•��θճĤ�����õı�������
•���к�ǿ�Ŀ������Ϳ�������
•ǿЧ�������ӱõĻ���
ժҪ
��������(atis flammula L.�� CF) (Ranunculaceae)�ǰ��������ǵ��ذذض��˹㷺ʹ�õĴ�ͳ����θ��������ҩ����о�ּ������CFҶ�Ҵ���ȡ��(CFEE)����������ģ��С���θ�������ü�����ơ��ܷӶ�����ͨ����ۺ��۷������о���������(���������绯ø(SOD)����������ø(CAT)����ԭ������(GSH)�ʹ�������(����ȩ(MDA))����������CFEE��θ�������á����⣬���ǻ��ⶨ��CFEEԤ�������Һˮƽ�����������ø(MPO)��θ���ӱ�(H+/K+-ATPase)���Ե�Ӱ�졣�ٶ�������Ҳ������������CFEE��ҩ��ѧ�о��������ڲ�ͬ������ģ���У�CFEE���м��������Ե�θ������λ���������ĸ߿���������(SOD��CAT��GSH)������ء�����Ȥ��DZ�ڻ�����MPO�����ӱû��Ե���������(�ֱ�Ϊ90%��99%������)����֯ѧ���֤ʵCFEE��������ճҺ����������θ�ſպͳ��䶯��صĽ�����ҩ�����Ԥ�����á�
���ǵķ���Ϊ�о�ֲ�������ҩ�������ṩ�˿�ѧ���ݡ�Anti-ulcerogenic and proton pump (H+, K+ ATPase) inhibitory activity of Clematis flammula L. extract - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0254629918312195��
J Bacteriol. 2018 Sep 1; 200(17): e00124-18.
Noncatalytic Antioxidant Role for Helicobacter pylori Urease
Alan A. Schmalstig,a St��phane L. Benoit,a Sandeep K. Misra,b Joshua S. Sharp,b and Robert J. Maiercorresponding authora
Conrad W. Mullineaux, Editor
Conrad W. Mullineaux, Queen Mary University of London;
Author information Article notes Copyright and License information Disclaimer
aDepartment of Microbiology, University of Georgia, Athens, Georgia, USA
bDepartment of BioMolecular Sciences, School of Pharmacy, The University of Mississippi, Oxford, Mississippi, USA
corresponding authorCorresponding author.
Address correspondence to Robert J. Maier, ude.agu@reiamr.
Citation Schmalstig AA, Benoit SL, Misra SK, Sharp JS, Maier RJ. 2018.
ABSTRACT
The well-studied catalytic role of urease, the Ni-dependent conversion of urea into carbon dioxide and ammonia, has been shown to protect Helicobacter pylori against the low pH environment of the stomach lumen. We hypothesized that the abundantly expressed urease protein can play another noncatalytic role in combating oxidative stress via Met residue-mediated quenching of harmful oxidants. Three catalytically inactive urease mutant strains were constructed by single substitutions of Ni binding residues. The mutant versions synthesize normal levels of urease, and the altered versions retained all methionine residues. The three site-directed urease mutants were able to better withstand a hypochlorous acid (HOCl) challenge than a ��ureAB deletion strain. The capacity of purified urease to protect whole cells via oxidant quenching was assessed by adding urease enzyme to nongrowing HOCl-exposed cells. No wild-type cells were recovered with oxidant alone, whereas urease addition significantly aided viability. These results suggest that urease can protect H. pylori against oxidative damage and that the protective ability is distinct from the well-characterized catalytic role. To determine the capability of methionine sulfoxide reductase (Msr) to reduce oxidized Met residues in urease, purified H. pylori urease was exposed to HOCl and a previously described Msr peptide repair mixture was added. Of the 25 methionine residues in urease, 11 were subject to both oxidation and to Msr-mediated repair, as identified by mass spectrometry (MS) analysis; therefore, the oxidant-quenchable Met pool comprising urease can be recycled by the Msr repair system. Noncatalytic urease appears to play an important role in oxidant protection.
IMPORTANCE Chronic Helicobacter pylori infection can lead to gastric ulcers and gastric cancers. The enzyme urease contributes to the survival of the bacterium in the harsh environment of the stomach by increasing the local pH. In addition to combating acid, H. pylori must survive host-produced reactive oxygen species to persist in the gastric mucosa. We describe a cyclic amino acid-based antioxidant role of urease, whereby oxidized methionine residues can be recycled by methionine sulfoxide reductase to again quench oxidants. This work expands our understanding of the role of an already acknowledged pathogen virulence factor and specifically expands our knowledge of H. pylori survival mechanisms.
KEYWORDS: gastric pathogen, gastritis, methionine, oxidation, oxidative stress, ulcerNoncatalytic Antioxidant Role for Helicobacter pylori Urease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6088170/��
Helicobacter pylori: Physiology and Genetics.
Chapter 17 Ion Metabolism and Transport
Arnoud H. M. van Vliet, Stefan Bereswill, and Johannes G. Kusters.
Author Information
Authors
Arnoud H. M. van Vliet,1 Stefan Bereswill,2 and Johannes G. Kusters1.Nickel
H. pylori expresses large amounts of urease, and levels can reach up to 10% of total cellular protein (3). Urease contains 12 nickel atoms per molecule, and thus H. pylori has a relatively high demand for nickel. Native H. pylori urease is a multimeric protein that consists of six UreA and six UreB subunits (35). The active site of each UreB subunit contains two nickel ions (54, 78), and without these ions the urease apoenzyme is not active (57). Nickel is inserted into the urease apoenzyme by the UreE, UreF, UreG, and UreH proteins (29). Urease plays a central role in the pathogenesis of H. pylori infection (75) and catalyzes the conversion of urea into carbon dioxide and ammonia. The latter is able to neutralize gastric acid and offer protection to H. pylori against the low pH in the stomach (64, 99, 118). In addition, ammonia may be used as a nitrogen source supporting growth of H. pylori (45). Another important H. pylori enzyme, likely to contain nickel, is a hydrogen uptake NiFe hydrogenase (67). Like NiFe hydrogenases from other bacteria, it is involved in electron transfer and respiration and subject to anaerobic activation (67). A detailed discussion of urease structure and function is given in the chapter devoted to this enzyme.Iron
Iron is an essential nutrient for all living organisms, with the exception of some lactobacilli. Iron is a cation that exists in the ferrous (Fe2+) and ferric (Fe3+) states. The redox potential of Fe2+/Fe3+ in biomolecules spans a range from +300 to −500 mV, which makes iron well suited for participating in electron transfer reactions. In addition, iron interacts chemically with oxygen, sulfur, and nitrogen ligands, allowing coordination of the iron atom in the active sites of enzymes with different redox potentials depending on the protein environment surrounding the complexed iron (21, 36). Iron-containing proteins are mostly involved in basic cell metabolism. Ferroprotoporphyrin (heme) groups are essential moieties of many enzymes involved in bacterial respiration, electron transport, and peroxide reduction. Iron-sulfur proteins participate in electron transport reactions, anaerobic respiration, amino acid metabolism, and energy metabolism. Finally, iron-containing non-heme, non-iron-sulfur proteins are required for DNA synthesis, protection from superoxide, and amino acid biosynthesis (36).Nickel Transport
H. pylori has a high demand for nickel as cofactor for its urease enzyme and thus requires high-affinity transport systems to scavenge nickel from environments usually low in nickel. The concentration of this ion in human serum is very low (2 to 11 nM), and the nickel concentration in ingested food varies significantly depending on the diet and on food sources (25, 106). Nickel might be transported by the magnesium transporter CorA when the magnesium concentration is low, but this is thought to be of little relevance under physiological conditions (102). Specific nickel transporters have been described in other bacteria: the one-component transporters like the Ralstonia eutropha HoxN protein, and the multicomponent ABC transporters like the E. coli NikABCDE system (37).
The first nickel transporter identified in H. pylori was the NixA protein (HP1077), a member of the HoxN-like one-component class of nickel transporters (76). NixA is a 37-kDa protein located in the CM and contains eight transmembrane domains (42, 43). Transport through NixA does not seem to be specific for nickel, and it has been suggested that it can transport also cadmium, cobalt, and zinc (42). Nonetheless, the high affinity of NixA for nickel makes it very suitable to supply the high nickel demand of H. pylori (76). A nixA mutant constructed by insertional inactivation displayed significantly decreased nickel transport and urease activity (4), although they were not completely abolished in this mutant, indicating that H. pylori has additional nickel transport systems. Homology searches of the H. pylori genome sequence allowed the identification of the abcC gene (HP1576) (55), which encodes a protein ortholog of the NikD ATPase of E. coli Nik, a nickel ABC transporter system (83). The adjacent abcD gene (HP1577) encodes a protein with low homology to CM permeases. An H. pylori abcD mutant was severely affected in its urease activity, but an abcC mutant showed the same phenotype as the nixA mutant. An abcC nixA double mutant has only residual urease activity, indicating that nickel uptake was almost completely abolished (55). The H. pylori AbcCD system lacks a homolog of the E. coli NikA periplasmic binding protein, which binds nickel ions and carries them to the CM transporter. This suggests that the AbcCD system functions differently from the Nik system or that the homology between the putative H. pylori nickel-periplasmic binding protein and E. coli NikA is too low to identify an H. pylori NikA homolog.
The nixA and abcCD mutational studies demonstrate that H. pylori has two separate nickel acquisition systems, and this manifests the importance of nickel in H. pylori metabolism. A detailed discussion of NixA and nickel transport is also found in chapter 16.
Conclusions
During coevolution with the host, H. pylori has developed unique systems necessary for survival in the human stomach. Concerning competition for nutrients as well as the acidic pH, this niche differs considerably from the environments most other bacteria colonize. Elements of its systems for ion metabolism and transport seem to reflect specific adaptations of H. pylori to its ecological niche, and the functions and mechanisms involved might be different from the paradigms developed for enteric bacteria like E. coli.
H. pylori contains relatively low numbers of ion transport systems, except for nickel and iron. These two ions can be transported by two or more systems, indicating their importance in the physiology of H. pylori. The number of regulatory proteins involved in ion metabolism and uptake is also low, and those that have been identified seem to be involved in the regulation of nickel and iron homeostasis. This is surprising, as the gastric mucosa where H. pylori resides is a challenging environment, in which conditions can vary considerably. Thus, it is possible to infer that systems contributing to ion homeostasis in H. pylori have an extra level of functionality. Further insights into the mechanisms governing ion homeostasis might allow the development of new or improved strategies to prevent or cure infection of this versatile pathogen.Ion Metabolism and Transport - Helicobacter pylori - NCBI Bookshelf
https://www.ncbi.nlm.nih.gov/books/NBK2418/��
Interplay of metal ions and urease
Eric L. Carter a, Nicholas Flugga a, Jodi L. Boer b, Scott B. Mulrooney a and Robert P. Hausinger *abc
aDepartment of Microbiology and Molecular Genetics, Michigan State University, East Lansing, Michigan 48824-4320, USA. E-mail: hausinge@msu.edu
bDepartment of Biochemistry and Molecular Biology, Michigan State University, East Lansing, Michigan 48824-1319, USA
��1.0 Introduction to urease
Urease holds a prominent place in the history of Science. It was the first enzyme to be crystallized1 and the first shown to contain nickel.2 The enzyme catalyzes the deceptively simple hydrolysis of urea into ammonia and carbamic acid. The latter compound spontaneously decomposes into carbonic acid and a second ammonia molecule, as shown in the reactions below:
H2N�CC(O)�CNH2 + H2O �� NH3 + H2N�CCOOH
H2N�CCOOH + H2O �� NH3 + H2CO3
The increase in ammonia levels and consequent elevation of pH associated with this reaction have major medical and agricultural implications.3,4Urease acts as a virulence factor in many pathogenic microorganisms, including those associated with urinary stone formation, pyelonephritis, ammonia encephalopathy, and hepatic coma;5,6 but perhaps the most infamous pathogenic organism that exploits urease is Helicobacter pylori, which takes advantage of the increase in pH associated with the hydrolysis of urea to colonize the acidic stomach.7�C9The active site of urease contains two nickel atoms bridged by a carboxylated lysine and a solvent molecule, as shown for the K. aerogenesenzyme (Fig. 2).19 In addition, Ni1 is coordinated by two His residues plus a terminal water molecule while Ni2 is coordinated by two His, an Asp, and a terminal solvent. Also present at the active site are two His residues thought to participate in substrate binding and/or catalysis. This active site structure is generally consistent with results derived from earlier biophysical investigations of the metallocenter. X-Ray absorption spectroscopy (XAS) of jack bean,20,21K. aerogenes,22 and B. pasteurii23ureases provides evidence for 5�C6 coordinate nickel ions with exclusively O/N ligands, including about two imidazoles per metal, and the K. aerogenes data include a scattering component indicating a Ni�CNi distance of ∼3.26 Å.22 The presence of ��-mercaptoethanol leads to thiolate-to-nickel charge-transfer transitions,24,25 a Ni�CS scattering interaction seen by XAS,21,22 and a unique variable-temperature magnetic circular dichroism feature26 that are all consistent with the inhibitor sulfur atom replacing the bridging solvent molecule in the dinuclear metallocenter. Structural characterization of the ��-mercaptoethanol-bound enzyme directly demonstrates this binding mode.27 Preliminary studies to characterize the magnetic properties of the metals in jack bean urease suggest weak antiferromagnetic exchange coupling between the nickel atoms,28 but saturation magnetization data collected with both jack bean and K. aerogenesureases fail to confirm this result.
Fig. 2 The urease active site. The active site of urease contains two nickel atoms (green) bridged by a carboxylated lysine and ahydroxyl group. Ni1 is also coordinated by two histidine residues and a solvent molecule, while Ni2 is coordinated by twohistidines, an aspartic acid residue, and a water molecule. Waters are red, metal-binding side chains are shown with white carbon atoms, and two nearby histidine residues that function in catalysis are shown with orange carbon atoms.
Fig. 3 Proposals for the urease catalytic mechanism. (A) The hydroxyl group bound to Ni2 attacks urea, whose carbonyl groupis polarized by coordination to Ni1, forming a tetrahedral intermediate that releases ammonia with His 320 (K. aerogenesnumbering) acting as a general acid. (B) The bridging hydroxyl group attacks urea, bound with its carbonyl group coordinated to Ni1 and an amine interacting with Ni2, and the hydroxyl proton transfers to the released ammonia. (C) A merged mechanism in which the bridging water attacks the substrate, but with His 320 acting as a general acid. (D) Elimination mechanism to form a cyanic acid (O
C
Urease, the first enzyme to be crystallized, contains a dinuclear nickel metallocenter that catalyzes the decomposition of urea to produce ammonia, a reaction of great agricultural and medical importance. Several mechanisms of urease catalysis have been proposed on the basis of enzyme crystal structures, model complexes, and computational efforts, but the precise steps in catalysis and the requirement of nickel versus other metals remain unclear. Purified bacterial urease is partially activated via incubation with carbon dioxide plus nickel ions; however, in vitro activation also has been achieved with manganese and cobalt. In vivo activation of most ureases requires accessory proteins that function as nickel metallochaperones and GTP-dependent molecular chaperones or play other roles in the maturation process. In addition, some microorganisms control their levels of urease by metal ion-dependent regulatory mechanisms.Interplay of metal ions and urease - Metallomics (RSC Publishing) DOI:10.1039/B903311D
https://pubs.rsc.org/en/Content/ArticleHtml/2009/MT/b903311d��
Nickel-based Enzyme Systems*
Stephen W. Ragsdale1
From the Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, Michigan 48109-0606
↵1To whom correspondence should be addressed. E-mail: sragsdal@umich.edu.
Abstract
Of the eight known nickel enzymes, all but glyoxylase I catalyze the use and/or production of gases central to the global carbon, nitrogen, and oxygen cycles. Nickel appears to have been selected for its plasticity in coordination and redox chemistry and is able to cycle through three redox states (1+, 2+, 3+) and to catalyze reactions spanning ∼1.5 V. This minireview focuses on the catalytic mechanisms of nickel enzymes, with an emphasis on the role(s) of the metal center. The metal centers vary from mononuclear to complex metal clusters and catalyze simple hydrolytic to multistep redox reactions.
Seven of the eight known nickel enzymes (Table 1) involve the use and/or production of gases (CO, CO2, methane, H2, ammonia, and O2) that play important roles in the global biological carbon, nitrogen, and oxygen cycles (1). CODH2 interconverts CO and CO2; ACS utilizes CO; the nickel ARD produces CO; hydrogenase generates/utilizes hydrogen gas; MCR generates methane; urease produces ammonia; and SOD generates O2.
View this table:
In this window In a new window
TABLE 1
Nickel-containing enzymes
The nickel sites in enzymes exhibit extreme plasticity in nickel coordination and redox chemistry. The metal center in SOD must be able to redox processes with potentials that span from +890 to −160 mV (2), whereas in MCR and CODH, it must be able to reach potentials as low as −600 mV (3); thus, nickel centers in proteins perform redox chemistry over a potential range of ∼1.5 V!Nickel-based Enzyme Systems
https://www.jbc.org/content/284/28/18571.full��
Journal of Advanced Research
Recent advances in design of new urease inhibitors: A review
Author links open overlay panelPawełKafarskiMichałTalma
Department of Bioorganic Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspia��skiego 27, 50-370 Wrocław, Poland
Urease is a nickel-dependent metalloenzyme found in plants, some bacteria, and fungi. Bacterial enzyme is of special importance since it has been demonstrated as a potent virulence factor for some species. Especially it is central to Helicobacter pylori metabolism and virulence being necessary for its colonization of the gastric mucosa, and is a potent immunogen that elicits a vigorous immune response. Therefore, it is not surprising that efforts to design, synthesize and evaluate of new inhibitors of urease are and active field of medicinal chemistry. In this paper recent advances on this field are reviewed.
Recent advances in design of new urease inhibitors: A review - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S2090123218300171��
Home / Archives / Vol 1 (2018): Special issue: Regional Chemical Engineering Undergraduate Congress (RCEUC) / Special issue: Regional Chemical Engineering Undergraduate Congress (RCEUC)
The Efficacy of Active Compound in Garlic as Urease Bio-Inhibitor
Siti Syazwani Samiri
Universiti Teknologi Petronas
Keywords: Urea, Allicin, DADS, Urease, Bio-inhibitor
Abstract
The application of urea fertilizer in agriculture industry has contributed to the advancement of crop production every year. Nevertheless, its low efficiency has led to high nitrogen losses in the form of Ammonia (NH3) and Nitrous Oxide (N2O) gases.Allicin, an organosulfur compound from garlic (Allium savatium L.) is identified as the most potential candidate for urease bio-inhibitor to delay the urease enzyme activity in hydrolysis process. However since it is highly unstable, it will decompose into other stable compounds mostly consist of diallyl disulfide (DADS).
This research mainly aims to evaluate the efficacy of Allicin and DADS in soil urease inhibition. The inhibition study of Allicin and DADS have been conducted by applying both compounds on urea at the soil and the loss of applied urea was quantified by using diacetylmonoxime (DAM) colorimetric method. Next, the stability study is done via High Performance Liquid Chromatography (HPLC) at different soil conditions; pH (3-11), temperature (30-80��C) and moisture content (10-50%).
In overall, the result justified that DADS managed to achieve 20% higher inhibitory action of urease enzyme compare to Allicin. In addition, DADS also exhibited higher stability despite of the variation of soil condition compare to Allicin and the optimum soil condition for higher inhibitory action is identified at 30% moisture, 30��C and pH 7.
Since the condition satisfied the typical soil condition in Malaysia, hence the application of urease bio-inhibitor is highly applicable to improve the utilization of nitrogen in soil.
The Efficacy of Active Compound in Garlic as Urease Bio-Inhibitor | Research Communication in Engineering Science & Technology
https://www.asianscientificresearch.com/journals/index.php/RCEST/article/view/30��
J Physiol Paris. 2001 Jan-Dec;95(1-6):429-36.
Gastric mucosal changes induced by long term infection with Helicobacter pylori in Mongolian gerbils: effects of bacteria eradication.
Keto Y1, Ebata M, Okabe S.
Author information
1 Department of Applied Pharmacology, Kyoto Pharmaceutical University, Misasagi, Yamashina, Kyoto 607-8414, Japan.
Abstract
Helicobacter pylori infection has been reported to induce various mucosal changes, including gastric adenocarcinoma, in Mongolian gerbils 62 weeks after inoculation. Using Mongolian gerbils, this study examined whether or not eradication of the bacteria with drugs at specified times after infection prevents the development of mucosal changes.After orally inoculating with H. pylori (TN2GF4, vacA- and cagA-positive), the animals were killed 18 months later. Four or 8 months after H. pylori inoculation, eradication was performed by concurrent treatment with omeprazole+clarithromycin. Immediately after treatment ended, in both the 5 and 9 month groups, it was verified that H. pylori was completely eradicated. Autopsy performed 18 months after H. pylori inoculation revealed gastric hyperplastic polyps with erosive lesions and ulcers that were grossly visible in the non-treated control group.
In addition, atrophic gastritis, intestinal metaplasia, carcinoids, and adenocarcinomas were histologically observed in the animals. In animals eradicated after 4 months and autopsied after 18 months, however, such mucosal changes were not observed. In contrast, intestinal metaplasia and mucosal atrophy was observed in animals eradicated after 8 months and autopsied after 18 months.
It was concluded that early eradication of H. pylori infection with drug therapy can prevent severe gastric mucosal changes, to include adenocarcinomas, in Mongolian gerbils.
PMID: 11595471 DOI: 10.1016/s0928-4257(01)00059-6Gastric mucosal changes induced by long term infection with Helicobacter pylori in Mongolian gerbils: effects of bacteria eradication. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/11595471��
BISMUTH SUBSALICYLATE
14882-18-9
Bismuth oxysalicylate
Bismuth Subsalicylate is a bismuth salt of salicylic acid. Little absorbed from the gastrointestinal tract, bismuth subsalicylate exerts a local effect on the gastric mucosa, coating it and protecting it from the corrosive effects of acid and pepsin. This agent also has local antimicrobial properties. (NCI04)
NCI Thesaurus (NCIt)
Bismuth subsalicylate appears as white crystalline powder or fluffy white solid. (NTP, 1992)
CAMEO Chemicals
Bismuth subsalicylate is a bismuth salt of salicylic acid. It is a member of salicylates and a bismuth coordination entity.
Bismuth subsalicylate | C7H6BiO4 - PubChem
https://pubchem.ncbi.nlm.nih.gov/compound/Bismuth-subsalicylate��
Drugs Acting on the Gastrointestinal Tract
David H. Shaw, in Pharmacology and Therapeutics for Dentistry (Seventh Edition), 2017
Bismuth Subsalicylate
Bismuth subsalicylate (Pepto-Bismol) is a commonly used OTC agent in the treatment of various gastrointestinal symptoms and diseases, including dyspepsia and acute diarrhea, and in the prevention of traveler��s diarrhea. It is the only OTC bismuth compound available in the United States and is estimated to be used by most American households. It is a crystal complex of bismuth and salicylate suspended in a mixture of magnesium aluminum silicate clay. In the stomach, the bismuth subsalicylate reacts with the hydrochloric acid to form bismuth oxychloride and salicylic acid. The salicylate is readily absorbed into the body, whereas the bismuth passes unaltered and unabsorbed into the feces. Caution is advised if patients are taking aspirin or other salicylate-containing drugs concurrently because toxic levels of salicylate may be reached. Bismuth subsalicylate products are not recommended for patients younger than 12 years because of a lack of studies to prove efficacy in young children.
Bismuth is thought to produce its therapeutic actions in part by stimulating prostaglandin, mucus formation, and bicarbonate secretion. It also has direct antimicrobial effects and may bind to enterotoxins, which accounts for its use in the prevention of traveler��s diarrhea. In addition, bismuth has been used in the home treatment of gastric ulcers because of its ability to coat the ulcer and other gastric erosions, shielding them from the stomach acid and pepsin. In the treatment of acute diarrhea, the salicylate moiety is thought to inhibit intestinal prostaglandin and Cl− secretion, leading to a reduction in stool frequency and liquidity.Bismuth subsalicylate has an excellent safety record, and side effects are minor. Bismuth may cause blackening of the stool or harmless black staining of the tongue, which is thought to be caused by the formation of bismuth sulfide from the reaction between the drug and the bacterial sulfides in the gastrointestinal tract. As noted previously, salicylate-induced adverse reactions may occur after the administration of bismuth subsalicylate (see Chapter 17).
��
Therapy of Viral Gastroenteritis
G. Kang, in Viral Gastroenteritis, 2016
4.2.4 Nonspecific Agents
Bismuth subsalicylate and its degradation products are believed to bind enterotoxin. In trials in Peru and Chile (Figueroa-Quintanilla et al., 1993, Soriano-Brucher et al., 1991), benefits included a reduction in stool frequency and weight and shorter disease duration, but a trial in Bangladesh in patients with acute diarrhoea reported nonsignificant decrease in severity and duration (Chowdhury et al., 2001). There are concerns regarding the toxicity from salicylate absorption, and routine use in children is not recommended (Lewis et al., 2006).
Kaolin-pectin, fibre, and activated charcoal should not be used in the treatment of diarrhoea and dehydration in infants and children. There is no conclusive evidence that they reduce stool losses, duration of diarrhoea or stool frequency. Although nontoxic, disadvantages of their use may include adsorption of nutrients and antibiotics in the intestine and masking the severity of fluid loss. As with other unproven therapies, use should be restricted.Bismuth Subsalicylate - an overview | ScienceDirect Topics
https://www.sciencedirect.com/topics/neuroscience/bismuth-subsalicylate��
SigmaAldrich
Ascorbate, a Serum-Free Medium Supplement, Useful In Biomanufacturing, Tissue Engineering and Specialty Media:
Ascorbic acid is an important water soluble anti-oxidant found in numerous media. These may include media used for biomanufacturing, tissue engineering, vaccine production and other commercially useful applications. Ascorbate is found in BGJb medium; CMRL-1066 medium; Nutrient Medium F-12 (Coon's modificaton); Hbyri-Max (R); McCoy's 5A; Medium 199; Alpha-EMEM; NCTC medium; Waymouth medium MB, William's medium E and various proprietary media. Supplementation of media with ascorbate for serum-free culture of eukaryotic cells is complicated by the fact that ascorbate is very unstable and rapidly oxidizes in aqueous systems. Hence the effective use of ascorbate in cell culture requires an understanding of its chemistry. For a more complete discussion of ascorbate as a cell culture additive go to our Media Expert.
Primary Functions of Ascorbate in Cell Culture Systems:
Ascorbic acid (vitamin C) is an essential vitamin for the growth and maintenance of healthy cells in vivo and in vitro.
Ascorbic acid is a water-soluble antioxidant that protects esterified and non-esterified unsaturated fatty acids from peroxidation. It converts lipid peroxyl radicals and alkoxyl radical into lipid hydroperoxides and lipid hydroxides, respectively. Hydroperoxides are relatively stable in the absence of iron and copper.
Ascorbic acid regenerates membrane bound alpha-tocopherol (vitamin E) that has been oxidized by lipid peroxyl radicals and indirectly limits lipid peroxidation in cell membranes.
Ascorbic acid regenerates urate from the urate radical. Urate is an important aqueous anti-oxidant.
Ascorbic acid facilitates the mobilization of copper from serum ceruloplasmin into cells without producing hydrogen peroxide.
Ascorbic acid is a cofactor for the enzyme prolyl hydroxylase (EC 1.14.11.2) that catalyzes the post translation hydroxylation of proline residues in nascent collagen and elastin molecules. Post-translation hydroxylation of these residues increases the degree of intra-molecular cross-linking of collagen and elastin and facilitates the development of extracellular matrices.
Ascorbic acid is a hydroxylphenylpyruvic acid oxidase (EC 1.2.3.13) cofactor, an enzyme that facilitates the catabolism of amino acids phenylalanine and tyrosine to acetyl-CoA.
Chemical Attributes of Ascorbate that make it a Useful Serum-Free Medium Supplement:
Ascorbic acid is a hexenoic sugar acid, gamma-lactone with an enediol structure at carbons 2 and 3. It can be reversibly oxidized and reduced by losing or gaining reducing equivalents from the oxygen atoms associated with carbons 2 and 3. Ascorbic acid, exists primarily as an ascorbate anion, at physiological pH.
Univalent Chemistry: Ascorbic acid is extremely unstable in vitro. It reduces other radicals including the hydroxyl radical, organic alkoxyl and peroxyl radicals, urate radical, tocopherol radical, and the ferric and cupric ions. It readily oxidizes to dehydro-ascorbate in a two step reaction. When the ascorbate anion loses one electron, it becomes an ascorbate radical, A��.
This ascorbate radical has several possible fates in vitro. It can
loose another electron and become dehydro-ascorbate,
gain an electron and proton to regenerate the ascorbate anion,
react with another radical and form a stable compound, or
two ascorbate radicals can dismutate to form one ascorbate anion and one dehydro-ascorbate molecule.
Univalent Chemistry:
Ascorbic acid is extremely unstable in vitro. It reduces other radicals including the hydroxyl radical, organic alkoxyl and peroxyl radicals, urate radical, tocopherol radical, and the ferric and cupric ions. It readily oxidizes to dehydro-ascorbate in a two step reaction. When the ascorbate anion loses one electron, it becomes an ascorbate radical, A��.
This ascorbate radical has several possible fates in vitro. It can
loose another electron and become dehydro-ascorbate,
gain an electron and proton to regenerate the ascorbate anion,
react with another radical and form a stable compound, or
two ascorbate radicals can dismutate to form one ascorbate anion and one dehydro-ascorbate molecule.
Loss of Ascorbate:
At pH 7.0, 37��C, dehydro-ascorbate breaks down to 2,3-diketo-L-gulonic acid with a half-life of about 6.7 minutes. Superoxides react with dehydro-ascorbate to form oxalic and threonic acids. Hence, when reducing agents that are able to regenerate ascorbate from dehydro-ascorbate or ascorbate radical are missing from the system, ascorbate is lost.
Oxidative Stress:
In the presence of oxygen, under physiological conditions, the ascorbate anion does not have the redox potential to reduce oxygen to the superoxide radical. However, it is capable of reducing the superoxide radical formed by other means to hydrogen peroxide. Ascorbic acid also reduces the transition metals iron and copper. When superoxide radicals and iron or copper are present in a form that can undergo redox cycling, ascorbic acid facilitates the formation of hydroxyl free radicals. These free radicals cause extensive damage to a wide range of biomolecules and initiate lipid peroxidation.
Ascorbate Products that Enhance the Growth of Hybridoma, Chinese Hamster Ovary (CHO) and other Mammalian Eukaryotic Cells in Serum-free Cultures:
Product # Description Add to Cart
A4403 L -����Ѫ�� powder, suitable for cell culture, ��-irradiated
�۸�
A4544 L -����Ѫ�� suitable for cell culture, suitable for plant cell culture, ��98%
�۸�
A4034 (+)-����Ѫ���� L powder, BioReagent, suitable for cell culture
�۸�
Our Cell Culture Media Expert provides in depth discussion of this and other serum-free and protein-free media supplements. The Media Expert contains additional sections on raw materials, component use recommendations, formulation strategies and references. Whenever you have a questions about or problems with your eukaryotic mammalian cell culturing system visit the Media Expert for helpful guidance.
��Ascorbate in Cell Culture | Sigma-Aldrich
https://www.sigmaaldrich.com/life-science/cell-culture/learning-center/media-expert/ascorbate.html��
Published: 18 December 2012
Helicobacter pylori colonization critically depends on postprandial gastric conditions
Roland B��cker, Marina Azevedo-Vethacke, Claudia Groll, D��sir��e Garten, Christine Josenhans, Sebastian Suerbaum & Sören Schreiber
Abstract
The risk of Helicobacter pylori infection is highest in childhood, but the colonization process of the stomach mucosa is poorly understood.We used anesthetized Mongolian gerbils to study the initial stages of H. pylori colonization. Prandial and postprandial gastric conditions characteristic of humans of different ages were simulated.
The fraction of bacteria that reached the deep mucus layer varied strongly with the modelled postprandial conditions. Colonization success was weak with fast gastric reacidification typical of adults. The efficiency of deep mucus entry was also low with a slow pH decrease as seen in pH profiles simulating the situation in babies. Initial colonization was most efficient under conditions simulating the postprandial reacidification and pepsin activation profiles in young children. In conclusion, initial H. pylori colonization depends on age-related gastric physiology, providing evidence from an in vivo infection model that suggests an explanation why the bacterium is predominantly acquired in early childhood.
Helicobacter pylori colonization critically depends on postprandial gastric conditions | Scientific Reports
https://www.nature.com/articles/srep00994��
Curr Opin Microbiol. 2018 Feb; 41: 51�C57.
Colonization, localization, and inflammation: The roles of H. pylori chemotaxis in vivo
Kevin S. Johnson and Karen M. Ottemann*
Author information Copyright and License information Disclaimer
Department of Microbiology and Environmental Toxicology, University of California Santa Cruz, Santa Cruz, CA 95064 USA
Abstract
Helicobacter pylori is a gram-negative bacterium that infects half of the world��s population, causing gastritis, peptic ulcers, and gastric cancer.To establish chronic stomach infection, H. pylori utilizes chemotaxis, driven by a conserved signal transduction system. Chemotaxis allows H. pylori to sense an array of environmental and bacterial signals within the stomach, guiding its motility towards its preferred niche within the gastric mucosa and glands. Fine-tuned localization, regulated by the chemotaxis system, enables robust colonization during the acute stage of infection. During chronic infection, chemotaxis helps maintain bacterial populations and modulates the host immune response.
Given its importance in host colonization and disease, chemotaxis is an attractive target for future treatments against H. pylori infections.
H. pylori colonizes the primate stomach, a harsh environment. The stomach lumen ranges between pH 1�C5 [8], conditions at which H. pylori is viable for only ~30min [9]. Additionally, stomach contents are cleared regularly, and the gastric mucosa undergoes constant turnover [10]. Accordingly, H. pylori must rapidly initiate colonization and localize where the environment is more hospitable: within 15��m from the gastric epithelial cells [11], and deep within gastric glands [12].
Figure 2
Chemotaxis promotes localization and modulates inflammation in vivo. Wild-type (WT) H. pylori robustly colonizes gastric glands early during infection, and can replicate and spread between glands. Conversely, non-chemotactic (Che−) H. pylori fail to colonize gastric glands to the same degree. Concurrently, WT H. pylori initiates a proinflammatory immune response, with a substantial T-helper 17 cell (Th17) response, while non-chemotactic H. pylori promotes a dampened immune response, skewed toward T-regulatory cells (Tregs)....
Chemotaxis modulates host inflammation
Host inflammation is a major disease outcome of H. pylori colonization. Inflammation occurs upon recognition of microbial-associated molecular patterns (MAMPs) or damage associated molecular patterns (DAMPs) by local monocytes, macrophages, and epithelial cells. These cells release pro-inflammatory cytokines and chemokines, the latter of which recruits neutrophils and antigen presenting cells, including macrophages and dendritic cells, to the site of infection [66,67]. Dendritic cells release cytokines and, process and present H. pylori antigens, priming T-cell differentiation [7,66�C71]. H. pylori colonization induces the differentiation of pro-inflammatory T-helper 1 (Th1) and T-helper 17 (Th17) cells and anti-inflammatory T-helper 2 (Th2) and T-regulatory cells (Tregs) [7,66,70,71]. The degree of host inflammation is modulated by the balance of effector T-cell populations. H. pylori induces inflammation via its intimate interaction with the gastric epithelium and the production of virulence factors such as the type 4 secretion system, CagA, NapA, and VacA [7,66,70]. The inflammatory response to H. pylori colonization, however, is also modulated by chemotaxis [14�C16].Highlights
The H. pylori chemotaxis system includes classical and auxiliary chemotaxis proteins
H. pylori senses several host and bacterial ligands through four chemoreceptors
Chemotaxis is critical for colonization during acute infection
Chemotaxis modulates host inflammation during chronic infectionColonization, localization, and inflammation: The roles of H. pylori chemotaxis in vivo
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5862749/��
Gastroenterology. 2018 Apr;154(5):1391-1404.e9. doi: 10.1053/j.gastro.2017.12.008. Epub 2017 Dec 19.
Helicobacter pylori Depletes Cholesterol in Gastric Glands to Prevent Interferon Gamma Signaling and Escape the Inflammatory Response.
Morey P1, Pfannkuch L1, Pang E1, Boccellato F1, Sigal M2, Imai-Matsushima A1, Dyer V1, Koch M1, Mollenkopf HJ1, Schlaermann P1, Meyer TF3.
Author information
1 Department of Molecular Biology, Max Planck Institute for Infection Biology, Berlin, Germany.
2 Department of Molecular Biology, Max Planck Institute for Infection Biology, Berlin, Germany; Department of Hepatology and Gastroenterology, Charit�� University Medicine, Berlin, Germany.
3 Department of Molecular Biology, Max Planck Institute for Infection Biology, Berlin, Germany. Electronic address: meyer@mpiib-berlin.mpg.de.
Abstract
BACKGROUND & AIMS:
Despite inducing an inflammatory response, Helicobacter pylori can persist in the gastric mucosa for decades. H pylori expression of cholesterol-��-glucosyltransferase (encoded by cgt) is required for gastric colonization and T-cell activation. We investigated how cgt affects gastric epithelial cells and the host immune response.
METHODS:
MKN45 gastric epithelial cells, AGS cells, and human primary gastric epithelial cells (obtained from patients undergoing gastrectomy or sleeve resection or gastric antral organoids) were incubated with interferon gamma (IFNG) or interferon beta (IFNB) and exposed to H pylori, including cagPAI and cgt mutant strains. Some cells were incubated with methyl-��-cyclodextrin (to deplete cholesterol from membranes) or myriocin and zaragozic acid to prevent biosynthesis of sphingolipids and cholesterol and analyzed by immunoblot, immunofluorescence, and reverse transcription quantitative polymerase chain reaction analyses. We compared gene expression patterns among primary human gastric cells, uninfected or infected with H pylori P12 wt or P12��cgt, using microarray analysis. Mice with disruption of the IFNG receptor 1 (Ifngr1-/- mice) and C57BL6 (control) mice were infected with PMSS1 (wild-type) or PMSS1��cgt H pylori; gastric tissues were collected and analyzed by reverse transcription quantitative polymerase chain reaction or confocal microscopy.
RESULTS:
In primary gastric cells and cell lines, infection with H pylori, but not cgt mutants, blocked IFNG-induced signaling via JAK and STAT. Cells infected with H pylori were depleted of cholesterol, which reduced IFNG signaling by disrupting lipid rafts, leading to reduced phosphorylation (activation) of JAK and STAT1. H pylori infection of cells also blocked signaling by IFNB, interleukin 6 (IL6), and IL22 and reduced activation of genes regulated by these signaling pathways, including cytokines that regulate T-cell function (MIG and IP10) and anti-microbial peptides such as human ��-defensin 3 (hBD3). We found that this mechanism allows H pylori to persist in proximity to infected cells while inducing inflammation only in the neighboring, non-infected epithelium. Stomach tissues from mice infected with PMSS1 had increased levels of IFNG, but did not express higher levels of interferon-response genes. Expression of the IFNG-response gene IRF1 was substantially higher in PMSS1��cgt-infected mice than PMSS1-infected mice. Ifngr1-/- mice were colonized by PMSS1 to a greater extent than control mice.
CONCLUSIONS:
H pylori expression of cgt reduces cholesterol levels in infected gastric epithelial cells and thereby blocks IFNG signaling, allowing the bacteria to escape the host inflammatory response. These findings provide insight into the mechanisms by which H pylori might promote gastric carcinogenesis (persisting despite constant inflammation) and ineffectiveness of T-cell-based vaccines against H pylori.
Copyright © 2018 AGA Institute. Published by Elsevier Inc. All rights reserved.Helicobacter pylori Depletes Cholesterol in Gastric Glands to Prevent Interferon Gamma Signaling and Escape the Inflammatory Response. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29273450
World J Gastroenterol. 2014 Sep 28;20(36):12767-80. doi: 10.3748/wjg.v20.i36.12767.
Effect of Helicobacter pylori on gastric epithelial cells.
Alzahrani S1, Lina TT1, Gonzalez J1, Pinchuk IV1, Beswick EJ1, Reyes VE1.
Author information
1 Shatha Alzahrani, Taslima T Lina, Irina V Pinchuk, Victor E Reyes, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, United States.
Abstract
The gastrointestinal epithelium has cells with features that make them a powerful line of defense in innate mucosal immunity. Features that allow gastrointestinal epithelial cells to contribute in innate defense include cell barrier integrity, cell turnover, autophagy, and innate immune responses.Helicobacter pylori (H. pylori) is a spiral shape gram negative bacterium that selectively colonizes the gastric epithelium of more than half of the world's population. The infection invariably becomes persistent due to highly specialized mechanisms that facilitate H. pylori's avoidance of this initial line of host defense as well as adaptive immune mechanisms. The host response is thus unsuccessful in clearing the infection and as a result becomes established as a persistent infection promoting chronic inflammation.
In some individuals the associated inflammation contributes to ulcerogenesis or neoplasia. H. pylori has an array of different strategies to interact intimately with epithelial cells and manipulate their cellular processes and functions. Among the multiple aspects that H. pylori affects in gastric epithelial cells are their distribution of epithelial junctions, DNA damage, apoptosis, proliferation, stimulation of cytokine production, and cell transformation.
Some of these processes are initiated as a result of the activation of signaling mechanisms activated on binding of H. pylori to cell surface receptors or via soluble virulence factors that gain access to the epithelium. The multiple responses by the epithelium to the infection contribute to pathogenesis associated with H. pylori.
KEYWORDS:
Apoptosis; Chronic inflammation; Gastric cancer; Gastric diseases; Gastric epithelial cells; Helicobacter pylori; Proinflammatory cytokinesEffect of Helicobacter pylori on gastric epithelial cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25278677��
GastroenterologyVolume 133, Issue 4, October 2007, Pages 1210-1218
Muc1 Mucin Limits Both Helicobacter pylori Colonization of the Murine Gastric Mucosa and Associated Gastritis
Author links open overlay panelMichael A.McGuckin⁎1Alison L.Every‡1Caroline D.Skene‡Sara K.Linden⁎Yok TengChionh‡AgnieszkaSwierczak‡JulieMcAuley⁎StaceyHarbour‡MariaKaparakis��RichardFerrero��PhilipSutton‡
⁎
Mucosal Diseases Program, Mater Medical Research Institute and University of Queensland, Mater Misericordiae Hospitals, Brisbane, Australia
‡
Centre for Animal Biotechnology, School of Veterinary Science, University of Melbourne, Melbourne, Australia
��
Department of Microbiology, Monash University, Clayton, Australia
Background & Aims: The MUC1 mucin is expressed on the cell surface of epithelial cells lining the gastric mucosa. Epidemiologic studies suggest that functional allelic variations in the MUC1 gene may play a role in human susceptibility to Helicobacter pylori-associated pathologies, including gastric adenocarcinoma. We have evaluated the impact of Muc1 expression on the colonization and pathogenesis of gastric Helicobacter infections.Methods: Wild-type and Muc1-deficient mice were infected with H pylori and colonization and gastritis levels determined. Primary gastric cells were used to examine the impact of Muc1 expression on bacterial adherence.
Results: Mice lacking Muc1 were colonized by 5-fold more H pylori within 1 day of infection, and this difference was maintained for at least 2 months postinfection. Mice heterozygous for the null Muc1 allele developed intermediate bacterial colonization. Although wild-type mice developed only a mild gastritis when infected for 2 months with H pylori, Muc1−/− mice developed an atrophic gastritis marked by loss of parietal cells. We demonstrate H pylori adhesion to purified MUC1 and significantly increased adhesion to cultured murine Muc1 null gastric epithelial cells, suggesting that Muc1 acts as a decoy limiting binding to the cell surface.
Conclusions: Muc1 provides a protective barrier, which limits both acute and chronic colonization by H pylori, as well as playing a major role in limiting the inflammation induced by Helicobacter infection. We propose that Muc1 restricts access of H pylori to the epithelial surface, hence reducing exposure of the host to proinflammatory bacterial products.
Muc1 Mucin Limits Both Helicobacter pylori Colonization of the Murine Gastric Mucosa and Associated Gastritis - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0016508507013029��
Yonsei Med J. 2014 Nov 1; 55(6): 1453�C1466.
Helicobacter pylori: Bacterial Strategy for Incipient Stage and Persistent Colonization in Human Gastric Niches
Kwang-Ho Rhee, Jin-Sik Park, and Myung-Je Chocorresponding author
Department of Microbiology, Gyeongsang National University College of Medicine, Research Institute of Life Science, Gyeongsang National University, Jinju, Korea.
Abstract
Helicobacter pylori (H. pylori) undergoes decades long colonization of the gastric mucosa of half the population in the world to produce acute and chronic gastritis at the beginning of infection, progressing to more severe disorders, including peptic ulcer disease and gastric cancer. Prolonged carriage of H. pylori is the most crucial factor for the pathogenesis of gastric maladies. Bacterial persistence in the gastric mucosa depends on bacterial factors as well as host factors. Herein, the host and bacterial components responsible for the incipient stages of H. pylori infection are reviewed and discussed. Bacterial adhesion and adaptation is presented to explain the persistence of H. pylori colonization in the gastric mucosa, in which bacterial evasion of host defense systems and genomic diversity are included.
Keywords: Helicobacter pylori, gastric infection, persistent colonization, pathogenesisHelicobacter pylori: Bacterial Strategy for Incipient Stage and Persistent Colonization in Human Gastric Niches
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4205683/��
Microbiological Research
Volume 218, January 2019, Pages 49-57
Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases
Abstract
Helicobacter pylori (H. pylori) is a bacterial pathogen that resides in more than half of the human population and has co-evolved with humans for more than 58,000 years.This bacterium is orally transmitted during childhood and is a key cause of chronic gastritis, peptic ulcers and two malignant cancers including MALT (mucosa-associated lymphoid tissue) lymphoma and adenocarcinoma.
Despite the strong innate and adaptive immune responses, H. pylori has a long-term survival in the gastric mucosa. In addition to the virulence factors, survival of H. pylori is strongly influenced by the ability of bacteria to escape, disrupt and manipulate the host immune system. This bacterium can escape from recognition by innate immune receptors via altering its surface molecules. Moreover, H. pylori subverts adaptive immune response by modulation of effector T cell.
In this review, we discuss the immune-pathogenicity of H. pylori by focusing on its ability to manipulate the innate and acquired immune responses to increase its survival in the gastric mucosa, leading up to gastrointestinal disorders. We also highlight the mechanisms that resulted to the persistence of H. pylori in gastric mucosa.
Abbreviation
H. pyloriHelicobacter PyloriPAIPathogenicislandsCagACytotoxin-associated gene AVacAVacuolating cytotoxin gene AMALTMucosa-associatedlymphoid tissueKeywords
Adaptive immunityEvasion strategiesInnate immunityH. pyloriHelicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0944501318308358��
Infect Immun. 2000 Jul; 68(7): 4303�C4311.
Helicobacter pylori Modulates Lymphoepithelial Cell Interactions Leading to Epithelial Cell Damage through Fas/Fas Ligand Interactions
Jide Wang,1 Xuejun Fan,1 Catharina Lindholm,2 Michael Bennett,3 Joe O'Connoll,3 Fergus Shanahan,3 Edward G. Brooks,1 Victor E. Reyes,1,4 and Peter B. Ernst1,4,5,*
Departments of Pediatrics1 and Microbiology & Immunology4 and Sealy Center for Molecular Sciences,5 University of Texas Medical Branch, Galveston, Texas, 77555-0366; Department of Medical Microbiology and Immunology, University of Göteborg, Göteborg, Sweden2; and Department of Medicine, National University of Ireland, Cork, Ireland3
ABSTRACT
Helicobacter pylori causes a common chronic infection of humans that leads to epithelial cell damage. Studies have shown that apoptosis of the gastric epithelium is increased during infection and this response is associated with an expansion of gastric T-helper type 1 (Th1) cells.We report that gastric T cells contribute to apoptosis of the epithelium by a Fas/Fas ligand (FasL) interaction. Fas receptor expression was detected on freshly isolated gastric epithelial cells by flow cytometry and immunohistochemistry, and this level of expression was increased during infection with H. pylori. The expression of Fas receptor on three gastric epithelial cell lines was increased by H. pylori, either alone or in combination with gamma interferon or tumor necrosis factor alpha. The role of Fas in apoptosis of gastric epithelial cell lines was evidenced by DNA fragmentation after cross-linking of Fas with specific antibodies.
FasL expression was detected by immunohistochemistry on mononuclear cells in gastric biopsy specimens of infected but not uninfected subjects. Gastric T-cell lines were also shown to express FasL, as evidenced by reverse transcription-PCR and killing of target cells expressing Fas receptor. Moreover, these T-cell lines were capable of killing cultured gastric epithelial target cells and antibodies that block the interaction between Fas receptor and FasL inhibited this cytotoxic activity.
These observations demonstrate that local Th1 cells may contribute to the pathogenesis of gastric disease during H. pylori infection by increasing the expression of Fas on gastric epithelial cells and inducing apoptosis through Fas/FasL interactions.
��
Infect Immun. 1999 Aug; 67(8): 4237�C4242.
Helicobacter pylori Induces Gastric Epithelial Cell Apoptosis in Association with Increased Fas Receptor Expression
Departments of Molecular Microbiology and Medical Genetics1 and Pediatrics,2 University of Toronto, and Research Institute, The Hospital for Sick Children,3 Toronto, Canada
ABSTRACT
The mechanisms involved in mediating the enhanced gastric epithelial cell apoptosis observed during infection with Helicobacter pylori in vivo are unknown.To determine whether H. pylori directly induces apoptosis of gastric epithelial cells in vitro and to define the role of the Fas-Fas ligand signal transduction cascade, human gastric epithelial cells were infected with H. pylori for up to 72 h under microaerophilic conditions.
As assessed by both transmission electron microscopy and fluorescence microscopy, incubation with a cagA-positive, cagE-positive, VacA-positive clinical H. pylori isolate stimulated an increase in apoptosis compared to the apoptosis of untreated AGS cells (16.0% �� 2.8% versus 5.9% �� 1.4%, P < 0.05) after 72 h. In contrast, apoptosis was not detected following infection with cagA-negative, cagE-negative, VacA-negative clinical isolates or a Campylobacter jejuni strain. In addition to stimulating apoptosis, infection with H. pylori enhanced Fas receptor expression in AGS cells to a degree comparable to that of treatment with a positive control, gamma interferon (12.5 ng/ml) (148% �� 24% and 167% �� 24% of control, respectively). The enhanced Fas receptor expression was associated with increased sensitivity to Fas-mediated cell death. Ligation of the Fas receptor with an agonistic monoclonal antibody resulted in an increase in apoptosis compared to the apoptosis of cells infected with the bacterium alone (38.5% �� 7.1% versus 16.0% �� 2.8%, P < 0.05). Incubation with neutralizing anti-Fas antibody did not prevent apoptosis of H. pylori-infected cells.
Taken together, these findings demonstrate that the gastric pathogen H. pylori stimulates apoptosis of gastric epithelial cells in vitro in association with the enhanced expression of the Fas receptor. These data indicate a role for Fas-mediated signaling in the programmed cell death that occurs in response to H. pylori infection.
Helicobacter pylori Induces Gastric Epithelial Cell Apoptosis in Association with Increased Fas Receptor Expression
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC96730/��
World J Gastroenterol. 2004 Aug 15; 10(16): 2334�C2339.
Helicobacter pylori enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in human gastric epithelial cells
Yi-Ying Wu, Hwei-Fang Tsai, We-Cheng Lin, Ai-Hsiang Chou, Hui-Ting Chen, Ping-Ning Hsu, Graduate Institute of Immunology, College of Medicine, National Taiwan University, Taiwan, China
Jyh-Chin Yang, Ping-I Hsu, Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan, China
Abstract
AIM: To investigate the relations between tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Helicobacter pylori (H pylori) infection in apoptosis of gastric epithelial cells and to assess the expression of TRAIL on the surface of infiltrating T-cells in H pylori-infected gastric mucosa.
METHODS: Human gastric epithelial cell lines and primary gastric epithelial cells were co-cultured with H pylori in vitro, then recombinant TRAIL proteins were added to the culture. Apoptosis of gastric epithelial cells was determined by a specific ELISA for cell death. Infiltrating lymphocytes were isolated from H pylori-infected gastric mucosa, and expression of TRAIL in T cells was analyzed by flow cytometry.
RESULTS: The apoptosis of gastric epithelial cell lines and primary human gastric epithelial cells was mildly increased by interaction with either TRAIL or H pylori alone. Interestingly, the apoptotic indices were markedly elevated when gastric epithelial cells were incubated with both TRAIL and H pylori (Control vs TRAIL and H pylori: 0.51 �� 0.06 vs 2.29 �� 0.27, P = 0.018). A soluble TRAIL receptor (DR4-Fc) could specifically block the TRAIL-mediated apoptosis. Further studies demonstrated that infiltrating T-cells in gastric mucosa expressed TRAIL on their surfaces, and the induction of TRAIL sensitivity by H pylori was dependent upon direct cell contact of viable bacteria, but not CagA and VacA of H pylori.
CONCLUSION: H pylori can sensitize human gastric epithelial cells and enhance susceptibility to TRAIL-mediated apoptosis. Modulation of host cell sensitivity to apoptosis by bacterial interaction adds a new dimension to the immunopathogenesis of H pylori infection.
PLoS One. 2012; 7(4): e35072.
Docosahexaenoic Acid Inhibits Helicobacter pylori Growth In Vitro and Mice Gastric Mucosa Colonization
1 Institute of Molecular Pathology and Immunology, University of Porto, Porto, Portugal,
2 Faculty of Medicine, University of Porto, Porto, Portugal,
3 Unit�� de Pathogen��se de Helicobacter, Institut Pasteur, Paris, France,
4 Serviço de Microscopia Electr��nica, Hospital Curry Cabral, Lisboa, Portugal,
5 Plate-forme Transcriptome et Epig��nome, G��nopole Institut Pasteur, Paris, France,
6 Unit�� de Recherche et d'Expertise en Histotechnologie et Pathologie, Institut Pasteur, Paris, France,
7 Centre for Environmental and Marine Studies (CESAM), Aveiro University, Aveiro, Portugal,
Monash University, Australia
Abstract
H. pylori drug-resistant strains and non-compliance to therapy are the major causes of H. pylori eradication failure. For some bacterial species it has been demonstrated that fatty acids have a growth inhibitory effect.Our main aim was to assess the ability of docosahexaenoic acid (DHA) to inhibit H. pylori growth both in vitro and in a mouse model. The effectiveness of standard therapy (ST) in combination with DHA on H. pylori eradication and recurrence prevention success was also investigated. The effects of DHA on H. pylori growth were analyzed in an in vitro dose-response study and n in vivo model.
We analized the ability of H. pylori to colonize mice gastric mucosa following DHA, ST or a combination of both treatments. Our data demonstrate that DHA decreases H. pylori growth in vitro in a dose-dependent manner. Furthermore, DHA inhibits H. pylori gastric colonization in vivo as well as decreases mouse gastric mucosa inflammation.
Addition of DHA to ST was also associated with lower H. pylori infection recurrence in the mouse model.
In conclusion, DHA is an inhibitor of H. pylori growth and its ability to colonize mouse stomach. DHA treatment is also associated with a lower recurrence of H. pylori infection in combination with ST. These observations pave the way to consider DHA as an adjunct agent in H. pylori eradication treatment.
��
H. pylori growth is inhibited by DHA in vitro
...
At higher concentrations of DHA (250 µM or more) the effect on bacterial cultures was drastic with no growth observed for all strains, compared to control (P = 9.41��10−12; P = 1.36��10−08; P = 2.30��10−12 at DHA 250 µM, for strains 26695, SS1 and B128 respectively) (Figure 1).��
DHA inhibits H. pylori gastric colonization in mice
In order to assess the effectiveness of DHA as a therapeutic agent, we investigated the effect of exposure to DHA on the H. pylori colonization of mouse gastric mucosa. The ability of H. pylori strain SS1 to colonize the gastric mucosa was significantly decreased in 50 µM DHA-treated mice as compared to mice from the infected non-treated group at each time-point of infection from 1 to 9 months (P values: 0.028; 0.023; 0.024 and 0.020 at 1, 3, 6 and 9 months respectively) (Figure 3A). In the non-treated group, all animals were successfully colonized by H. pylori. In contrast, amongst DHA supplemented-mice, only 50% of animals are infected, with a median value for the gastric colonization lower than 3 to 4 Log compared to the infected and non DHA-treated group at the same time-point. No toxic effects were observed due to DHA treatment: no weight variations (Figure S2) and no signs of liver toxicity as indicated by histopathology analysis (data not shown). These data showed that also in vivo, DHA inhibits efficiently H. pylori gastric colonization.Mouse gastric mucosa inflammation status
Mouse gastric mucosa at 1 and 3 months of H. pylori infection, did not present significant signs of inflammation. However, at 6 and 9 months post-infection, the gastric mucosa of infected-mice present important inflammatory lesions with infiltrates of polymorphonuclear cells (PMN) and plasmocytes as well as the presence of important lymphocytes aggregates (Figure 4A). Gastric inflammation and histology analysis revealed a significant lower inflammation in the gastric mucosa of H. pylori infected mice treated with DHA, compared to H. pylori infected mice non-treated after both 6 (P = 1.83��10−6) and 9 months (P = 0.0004) of infection (Figure 4B). In addition, according to the semi-quantitative evaluation of inflammatory lesions [24], infiltrates of PMN, plasmocytes and lymphoid aggregates were less abundant in DHA-treated mice than in non-DHA-treated ones (P<0.05) (Figure 4B). These differences were significant at 6 and 9 months in both parts of the stomach, the antrum (P = 1.47��10−4 and P = 1.4��10−3) and the corpus (P = 1.34��10−3 and P = 0.0485), respectively.PGE2 is produced in several pathological conditions by a wide variety of tissues. Increased amount of PGE2 is associated with inflammation and tissue injury [28]. At both time-points, infected mice treated with DHA showed lower serum PGE2 levels compared with infected but non DHA-treated mice (P = 0.0284) (Figure 4C). Altogether, these data showed that in the mouse model of chronic H. pylori infection, DHA addition is accompanied by a reduction in the inflammatory response.
Docosahexaenoic Acid Inhibits Helicobacter pylori Growth In Vitro and Mice Gastric Mucosa Colonization
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3328494/��
Gastric Cancer Risk and Erythrocyte Composition of Docosahexaenoic Acid with Anti-inflammatory Effects
Kiyonori Kuriki, Kenji Wakai, Keitaro Matsuo, Akio Hiraki, Takeshi Suzuki, Yoshitaka Yamamura, Kenji Yamao, Tsuneya Nakamura, Masae Tatematsu and Kazuo Tajima
Divisions of 1Epidemiology and Prevention and 2Oncological Pathology, Aichi Cancer Center Research Institute, Chikusa-ku, Departments of 3Preventive Medicine/Biostatistics and Medical Decision Making, and 4Epidemiology, 5Division of Cancer Genetics, Nagoya University Graduate School of Medicine, Showa-ku, and Departments of 6Gastroenterological Surgery, 7Gastroenterology, and 8Endoscopy, Aichi Cancer Center Hospital, Chikusa-ku, Nagoya, Japan
Abstract
Infection with Helicobacter pylori is linked to inflammation and is the main cause of peptic ulcer, gastritis, and gastric malignancies.To examine associations between gastric cancer risk and the erythrocyte composition of docosahexaenoic acid (DHA), a fatty acid with anti-inflammatory and apoptosis-inducing effects, here we conducted a case-control study of 179 incident gastric cancer cases and 357 noncancer controls (matched by age, sex, and season of sample collection).
Dietary information and blood samples were collected from all subjects, and erythrocyte fatty acid levels were measured using accelerated solvent extraction and gas-liquid chromatography. Gastric cancer risk did not seem to be directly associated with dietary intake of fish and n-3 highly unsaturated fatty acids (HUFAs), such as DHA, derived from fish. However, risk was inversely associated with erythrocyte compositions of n-3 HUFAs [the highest to the lowest tertile, odds ratio (OR), 0.39; 95% confidence interval (95% CI), 0.23-0.68; Ptrend < 0.005] and DHA (OR, 0.47; 95% CI, 0.28-0.79; Ptrend < 0.01). Particularly strong associations were noted for well-differentiated type lesions and n-3 HUFAs (OR, 0.10; 95% CI, 0.03-0.35; Ptrend = 0.0005) as well as DHA (OR, 0.20; 95% CI, 0.07-0.58; Ptrend < 0.01) values.
In conclusion, the erythrocyte composition of DHA was found to be negatively linked to risk of gastric cancer, especially of well-differentiated adenocarcinoma. Further studies are needed to investigate mechanisms of action of DHA relevant to antitumor effects in the stomach. (Cancer Epidemiol Biomarkers Prev 2007;16(11):2406�C15)
��
Figure 1.
Hypothetical mechanism for suppressing gastric cancer related to chronic inflammation according to DHA. Infection with H. pylori to normal gastric mucosa is suggested to induce chronic inflammation in the early life of carriers and cause diseases such as peptic ulceration, gastritis, and gastric cancer after years or decades (1). Infection, therefore, is regarded as a trigger for the sequence of carcinoma development in the stomach. In gastric cancer tissues and cell lines, overexpression of cyclooxygenase-2 linked to the inflammatory responses, angiogenesis, and apoptosis has been observed (2-4, 42, 43). DHA, which is rich in blue skin fish and is n-3 HUFAs, is related to inhibition for the tumorigenesis, like NSAIDs (i.e., here, cyclooxygenase-2 inhibitors). Against the promotion and progression stages of tumor development, DHA is thought to play roles in (a) competitive inhibition of incorporation of arachidonic acid into membrane phospholipids, (b) competitive inhibition of metabolism of arachidonic acid to prostaglandin E2, and (c) apoptosis induction according to the up-regulation of Bax expression (18-20).In the present study, no association was found between the risk of gastric cancer and dietary intakes of fish and other seafood. However, inverse links with erythrocyte compositions of n-3 HUFAs, especially DHA, and a positive association with the arachidonic acid/DHA erythrocyte ratio were evident. We found that the decreased risk with high erythrocyte values for DHA and n-3 HUFAs was particularly strong for well-differentiated type adenocarcinomas.
https://cebp.aacrjournals.org/content/16/11/2406
��
Gut. 1994 Nov;35(11):1557-61.
Inhibitory effect of polyunsaturated fatty acids on the growth of Helicobacter pylori: a possible explanation of the effect of diet on peptic ulceration.
Thompson L1, Cockayne A, Spiller RC.
Author information
1 Department of Therapeutics, University Hospital, Nottingham.
Abstract
Diets high in polyunsaturated fatty acids may protect against duodenal ulcer, possibly through inhibiting the growth of Helicobacter pylori. This hypothesis was tested in vitro by incubating H pylori microaerophilically with a range of polyunsaturated fatty acids. omega-3 Linolenic acid significantly, but reversibly, inhibited growth at 1.8, 2.5, and 5 x 10(-4) M (p < 0.01), while concentrations of 10(-3) M killed virtually all organisms, with cell lysis observed by electron microscopy. Similar inhibitory effects were seen with other polyunsaturated fatty acids, at concentrations of 2.5 x 10(-4) M the relative inhibitory potencies were oleic (C18:1) < linoleic (C18:2) < arachidonic (C20:4) < omega-3 linolenic (C18:3) = omega-6 linolenic (C18:3) = eicosapentanoic (C20:5) acid. Cell fractionation studies with 14C labelled linolenic acid showed that the linolenic acid was associated with the membrane fraction. Commonly ingested dietary polyunsaturated fatty acids inhibit the growth of H pylori in vitro, an effect which deserves further in vivo study.
PMID: 7828972 PMCID: PMC1375611 DOI: 10.1136/gut.35.11.1557Inhibitory effect of polyunsaturated fatty acids on the growth of Helicobacter pylori: a possible explanation of the effect of diet on peptic ulcer... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/7828972/��
J Med Microbiol. 1995 Apr;42(4):276-82.
The effects of unsaturated fatty acids on Helicobacter pylori in vitro.
Khulusi S1, Ahmed HA, Patel P, Mendall MA, Northfield TC.
Author information
1 Department of Cellular and Molecular Sciences, St George's Hospital Medical School, London.
Abstract
The effects of three unsaturated free fatty acids on Helicobacter pylori growth in vitro was determined. Growth of H. pylori in Brucella broth was inhibited in a dose-dependent manner by arachidonic, linoleic and oleic acids. The degree of inhibition at any one concentration was related to the degree of fatty acid unsaturation. Triolein, a triacylglycerol ester of oleic acid did not inhibit growth. Inhibition of H. pylori growth was associated with disruption of cell membranes. Incubation with 14C linoleic acid and 14C oleic acid showed incorporation of these fatty acids into H. pylori cell mass and phospholipids leading to alteration of the phospholipid composition of the organism. Incorporation was greater with linoleic than oleic acid and this was associated with a greater inhibition of growth. These findings indicate that H. pylori is sensitive to unsaturated free fatty acids through their incorporation into phospholipids and membrane destruction. This may have therapeutic implications.
��The effects of unsaturated fatty acids on Helicobacter pylori in vitro. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/7707336��
Open Journal of Medical Microbiology
Vol.3 No.1(2013), Article ID:28667,10 pages DOI:10.4236/ojmm.2013.31011Interaction of Helicobacter pylori Cell Membrane with Non-Esterified Cholesterol and Other Steroids
Hirofumi Shimomura, Kouichi Hosoda, Yoshikazu Hirai
Division of Bacteriology, Department of Infection and Immunity, Jichi Medical University, Tochigi, JapanEmail: shimo@jichi.ac.jp
Email: shimo@jichi.ac.jpReceived December 10, 2012; revised January 19, 2013; accepted February 2, 2013
Keywords: Helicobacter pylori; Cholesterol-��-glucosyltransferase;hosphatidylethanolamineABSTRACT
Helicobacter pylori performs the unique action of assimilating exogenous non-esterified cholesterol into its cell membrane. This bacterium aggressively incorporates non-esterified cholesterol into the membrane, induces its glucosylation, and uses both non-esterified cholesterol and glucosylated cholesterols as membrane lipid compositions.
The reason for this assimilation of non-esterified cholesterol into the cell membrane of H. pylori has eluded investigators for many years. Recent hypotheses posit that the sterol-uptake and sterol-glucosylation contribute to the survival of H. pylori cells in different ways. The incorporation of the non-esterified cholesterol into the cell membrane fortifies the resistance of H. pylori against the antibacterial actions of phosphatidylcholines, antibiotics, and bile salts.
In parallel, the glucosylation of the non-esterified cholesterol incorporated into the cell membrane serves H. pylori in two ways. First, it helps the bacterium evade host immune responses, such as phagocytosis by macrophages and activation of antigen-specific T cells. Second, it detoxifies sterols fatal to the bacterium via a novel action of sterol glucosylation recently described in another report from our group.
The reluctance of H. pylori to absorb esterified cholesterol remains unexplained. A recent study by our group has demonstrated that the phosphatidylethanolamine (PE) in the outer membrane of H. pylori serves as a steroid-binding lipid the incorporation of non-esterified cholesterol into the membrane. We have also discovered that the myristic acid (C14:0) molecule attached to the PE of this bacterium plays an important role in the selective binding of non-esterified cholesterol but not esterified cholesterol.
4.Biological Significance of Cholesterol Uptake by H. pylori
Our study in 2009 revealed that H. pylori cells without non-esterified cholesterol or other cholesterol analogues succumb to the bacteriolytic action of phosphatidylcholines (PCs), while the H. pylori cells with absorbed nonesterified cholesterol or the cholesterol analogue estrone acquire resistance against the bactericidal action of PCs (Figure 2) [27]. H. pylori was incapable of glucosylating estrone absorbed into the cell membrane. Hence, its resistance against the bacteriolytic action of the PCs was acquired independently of the glucosylation of the steroid compounds. PC is the most prevalent glycerophospholipid in mammals and exists in sufficient abundance to kill H. pylori in gastric mucosa and gastric juice of humans [29,30]. In sum, our study demonstrated that H. pylori aggressively incorporates exogenous non-esterified cholesterol into the cell membrane, in order to survive in the presence of PCs.
In separate experiments, McGee et al. (2011) and Trainor et al. (2011) inactivated the CGT of an H. pylori strain by inserting a chloramphenicol-resistant (cat) gene cassette into the hp0421 gene. With this strain, they found that non-glucosylated non-esterified cholesterol absorbed into the cell membrane fortified this bacterium��s resistance to antibacterial agents such as ciprofloxacin, tetracycline, and clarithromycin, and to bile salts and ceragenins [31,32]. The studies by our group and others indicate that non-esterified cholesterol strengthens the cell membrane lipid barrier of H. pylori. Incidentally, McGee et al. have also proposed that the glucosylation of the non-esterified cholesterol absorbed by H. pylori confers further resistance against the bactericidal action of antibacterial peptides such as polymixin B and colistin, at least in part [31].
Interaction of <i>Helicobacter pylori</i> Cell Membrane with Non-Esterified Cholesterol and Other Steroids
https://file.scirp.org/Html/11-2260054_28667.htm��
Antimicrob Agents Chemother. 2003 Dec; 47(12): 3982�C3984.
Efficacy of Sulforaphane in Eradicating Helicobacter pylori in Human Gastric Xenografts Implanted in Nude Mice
Xavier Haristoy,1 Karine Angioi-Duprez,2 Adrien Duprez,3 and Alain Lozniewski1,*
Laboratoire de Bact��riologie-Virologie, UMR CNRS 75-65,1 Laboratoire de Microchirurgie Exp��rimentale,2 Laboratoire d'Anatomie Pathologique EMI INSERM 0014,Facult�� de M��decine, Universit�� Henri-Poincar��, Vandæuvre-les-Nancy, France3
ABSTRACT
Sulforaphane, an isothiocyanate abundant in the form of its glucosinolate precursor in broccoli sprouts, has shown in vitro activity against Helicobacter pylori. We evaluated the effect of sulforaphane in vivo against this bacterium by using human gastric xenografts in nude mice. H. pylori was completely eradicated in 8 of the 11 sulforaphane-treated grafts(0.5 ml of sterile water with 0.5% acetonitrile containing 7.5 ��mol of sulforaphane (approximately 1.33 mg) was administered via catheter once a day for 5 days.). This result suggests that sulforaphane might be beneficial in the treatment of H. pylori-infected individuals.�ܲ�������һ�����������Σ��ڻ�Ҭ��ѿ����������������ε���ʽ���ڡ�����ͨ��������������ֲ��θ����ֲ���������ܲ����ضԸ�ϸ�����������á���11���ܲ����ش�������ֲ���У���8����ȫ���������������˾�����һ�����ʾ�ܲ����ؿ��ܶԸ�Ⱦ���������˾��������档
Efficacy of Sulforaphane in Eradicating Helicobacter pylori in Human Gastric Xenografts Implanted in Nude Mice
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC296232/��
��
Interaction of Helicobacter pylori Cell Membrane with Non-Esterified Cholesterol and Other Steroids
Hirofumi Shimomura, Kouichi Hosoda, Yoshikazu Hirai
Division of Bacteriology, Department of Infection and Immunity, Jichi Medical University, Tochigi, Japan
��ASTRCT
Helicobacter pylori performs the unique action of assimilating exogenous non-esterified cholesterol into its cell membrane. This bacterium aggressively incorporates non-esterified cholesterol into the membrane, induces its glucosylation, and uses both non-esterified cholesterol and glucosylated cholesterols as membrane lipid compositions. The reason for this assimilation of non-esterified cholesterol into the cell membrane of H. pylori has eluded investigators for many years. Recent hypotheses posit that the sterol-uptake and sterol-glucosylation contribute to the survival of H. pylori cells in different ways. The incorporation of the non-esterified cholesterol into the cell membrane fortifies the resistance of H. pylori against the antibacterial actions of phosphatidylcholines, antibiotics, and bile salts. In parallel, the glucosylation of the non-esterified cholesterol incorporated into the cell membrane serves H. pylori in two ways. First, it helps the bacterium evade host immune responses, such as phagocytosis by macrophages and activation of antigen-specific T cells. Second, it detoxifies sterols fatal to the bacterium via a novel action of sterol glucosylation recently described in another report from our group. The reluctance of H. pylori to absorb esterified cholesterol remains unexplained. A recent study by our group has demonstrated that the phosphatidylethanolamine (PE) in the outer membrane of H. pylori serves as a steroid-binding lipid the incorporation of non-esterified cholesterol into the membrane. We have also discovered that the myristic acid (C14:0) molecule attached to the PE of this bacterium plays an important role in the selective binding of non-esterified cholesterol but not esterified cholesterol.��
5. Response of H. pylori Cell Membrane to Other Steroid Compounds
Our group also found that some steroid compounds impair the viability of H. pylori. In particular, progesterone with an oxo group at the carbon 3-position of the steroid framework destabilized the cell membrane structure of H. pylori and ultimately destroyed the cells [41]. The cell membrane of H. pylori apparently interacted with a number of steroid compounds, some of which were even toxic to this bacterium (Figure 3). Intriguingly, a synthetic progesterone derivative acylated by a caproic acid (C6:0) at the carbon 17-position in a progesterone molecule had conspicuously stronger anti-H. pylori activity than the progesterone, whereas a natural progesterone derivative hydroxylated at the same carbon position of a progesterone molecule lacked this activity. This may open the way to the development of a new class of steroidal medicines that lack the hormonal activity but maintain the anti-H. pylori activity, using the progesterone molecule as a basic structure (Figure 4).Progesterone non-reversibly bound to the cell membrane of H. pylori and inhibited the interaction of non-esterified cholesterol to the bacterial cell membrane. Conversely, non-esterified cholesterol protected the H. pylori cells from the bacteriolytic action of progesterone to some degree. This suggested that non-esterified cholesterol and progesterone bind to the identical component on the cell membrane of H. pylori and thereby obstruct each other��s effects on the bacterial cell. We were unable, however, to identify which of the cell membrane components of H. pylori took part in the binding of the steroid compounds in that series of experiments.��
7. A Novel Role of Cholesterol Glucosylation in the Survival of H. pyloriSeveral studies have clarified the respective roles of cholesterol-uptake and cholesterol-glucosylation in H. pylori cells, as mentioned earlier. The uptake of non-esterified cholesterol into the cell membrane is important to H. pylori��s resistance to the antibacterial activity of phosphatidylcholines (PCs), antibiotics, and bile salts. Likewise, the glucosylation of the non-esterified cholesterol absorbed into the cell membrane is important to the regulation of host immune responses against H. pylori.Figure 5. Selective binding of non-esterified cholesterol to H. pylori phosphatidylethanolamine. FC, non-esterified cholesterol; EC, esterified cholesterol; PE, phosphatidylethanolamine.
The most recent study by our group revealed a novel role of cholesterol glucosylation in the survival of H. pylori cells, a role distinct from host immune system evasion [48].The sterol 7-dehydrocholesterol is the direct precursor of non-esterified cholesterol in the cholesterol biosynthetic pathway in mammalian tissues. The non-esterified cholesterol and 7-dehydrocholesterol differ in the number of hydrogen atoms in the steroid framework. The precursor lacks the two hydrogen atoms at carbon positions 7 and 8 of the non-esterified cholesterol molecule. Yet no studies have shown whether 7-dehydrocholesterol is useful or harmful for the growth and/or survival of the H. pylori cell.When H. pylori at a low cell density (106.5 CFU/ml) was incubated in the presence of 7-dehydrocholesterol at a 100 µM concentration, we were intrigued to find that the non-esterified cholesterol precursor induced the bacteriolysis of this bacterium. Put simply, 7-dehydrocholesterol proved to be fatally toxic to H. pylori. Incidentally, non-esterified cholesterol at the same concentration had no influence on the viability or growth of this bacterium. In contrast, when H. pylori at a high cell density (109 CFU/ml) was incubated in the presence of 7-dehydrocholesterol (100 µM), we were surprised to find that the H. pylori cells lasted somehow to the bacteriolytic action of 7-dehydrocholesterol, glucosylated its toxic sterol, and admitted the glucosylated 7-dehydrocholesterols as membrane lipid components. Observing this, we assumed that the H. pylori detoxified the 7-dehydrocholesterol via the glucosylation.Interaction of <i>Helicobacter pylori</i> Cell Membrane with Non-Esterified Cholesterol and Other Steroids
https://file.scirp.org/Html/11-2260054_28667.htm��
.. Cholesterol is essential for maintenance of the epidermal barrier function, and also has a role in the regulation of epidermal differentiation and desquamation [16, 17]. The epidermis is therefore an active site of de novo cholesterol synthesis, which provides a ready source of 7-dehydrocholesterol [18]. 7-Dehydrocholesterol is present in the plasma membrane of cells in both the dermis and epidermis [19], with the highest concentrations in the cells of the stratum basale and stratum spinosum layers of the epidermis [20]. ...
��
.. Although the previtamin D 7-dehydrocholesterol was thought to be produced in the gut wall cells and transported to skin cells, actually skin cells can synthesize their own 7-dehydrocholesterol, which in turn is converted to a provitamin D, cholecalciferol, or vitamin D3, by isomerization upon ultraviolet B (UVB) radiation of sunlight in epidermis [12�C14]. Further photoreaction of vitamin D3 by UVB absorption may generate inactive metabolites. ...
��
Origin of 7-Dehydrocholesterol (Provitamin D) in the Skin
https://www.researchgate.net/publication/44576112_Origin_of_7-Dehydrocholesterol_Provitamin_D_in_the_Skin��
Naturally Treating H. pylori
The earlier the better. As H. pylori is aggressive, this bacteria is best killed early on, but that is hard to do! Not many people infected even know they are infected. Someone can have this bacteria growth underlying years of symptoms and not even know it is H. pylori.
Today we see numerous reports of people with high levels of this bacteria now experiencing great pain and discomfort.
A very recent study published in the journal Cancer Prevention Research, a Johns Hopkins researcher, Jed W. Fahey, M.S., Sc.D., and an international team of scientists concluded that eating broccoli sprouts daily reduced the level of HpSA (a highly specific measure of the presence of H. pylori) by a whopping 40 percent.
The natural compound sulforaphane is found in high levels in broccoli sprouts. In 2002 Dr. Fahey, a nutritional biochemist in the Lewis B. and Dorothy Cullman Cancer Chemoprotection Center at Johns Hopkins University School of Medicine, and his colleagues at Johns Hopkins reported that sulforaphane was a potent weapon against H. pylori. "We know that a dose of a couple ounces a day of broccoli sprouts is enough to elevate the body's protective enzymes," Dr. Fahey explained. "That is the mechanism by which we think a lot of the chemoprotective effects are occurring." Dr. Fahey also stated, "The highlight of the study is that we identified a food that, if eaten regularly, might potentially have an effect on the cause of a lot of gastric problems and perhaps even ultimately help prevent stomach cancer."
Sulforaphane is also found in cabbage and kimchi. Eating cooked cabbage may be soothing. As raw sauerkraut and kimchi are fermented foods, they also contain a great number of useful bacteria that replace H.pylori in the gastric mucosa upon entering the stomach. These healthy microorganisms also help in keeping an ideal pH in the stomach.
Cabbage is naturally high in Vitamin C (thought to inhibit H. Pylori) and also contains glutamine (an amino acid), that can boost stomach circulation. As cabbage is high in sulfur, it is also good for pain and inflammation.
You can cook cabbage, but better yet is the fresh juice of the cabbage. Raw cabbage juice is great, but you can also add a tablespoon of extra virgin olive oil fresh pressed (EXTRA Virgin only), celery, parsley or aloe juice. Newer studies have even found cranberries can inhibit H. pylori from adhering to stomach lining.��
This microorganism spreads quickly through saliva, so is easily transmitted. The family dinner table, the kitchen, child care centers, hospitals, and food establishments are the most common areas of transmittal and infection. Even just having a roommate that shares food or does not do dishes well can leave you with the most unpleasant infection.
��
H. pylori reduces protective mucus in the stomach leading to stomach ulcers
H. pylori neutralizes stomach acids causing a higher pH leading to acid reflux
H. pylori disables the body��s ability to properly break down proteins into amino acids
H. pylori produces the enzyme urease which breaks down the stomachs urea.
H. pylori shuts down production of intrinsic factor, causing a B12 deficiency AKA pernicious anemia.
H. pylori affects the entire digestive process
H. pylori causes depression��
H. Pylori: Tips to Eliminate One Pesky Bacteria �� Karen's Holistic Health
https://www.karensholistichealth.com/blog/2017/10/h-pylori-tips-to-irradiate-the-great-infector-pesky-darn-stubborn-bacteria��
Front Cell Infect Microbiol. 2016; 6: 159.
Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter pylori
Ying Huang,1 Qi-long Wang,2 Dan-dan Cheng,1 Wen-ting Xu,1 and Nong-hua Lu1,*
Author information Article notes Copyright and License information Disclaimer
1Department of Gastroenterology, The First Affiliated Hospital of Nanchang University, Nanchang, China
2Department of General Surgery, Tianjin Haihe Hospital, Tianjin, China
Edited by: D. Scott Merrell, Uniformed Services University, USA
Reviewed by: Robert Maier, University of Georgia, USA; Traci Testerman, University of South Carolina, USA
Abstract
Helicobacter pylori is the main pathogenic bacterium involved in chronic gastritis and peptic ulcer and a class 1 carcinogen in gastric cancer. Current research focuses on the pathogenicity of H. pylori and the mechanism by which it colonizes the gastric mucosa. An increasing number of in vivo and in vitro studies demonstrate that H. pylori can invade and proliferate in epithelial cells, suggesting that this process might play an important role in disease induction, immune escape and chronic infection. Therefore, to explore the process and mechanism of adhesion and invasion of gastric mucosa epithelial cells by H. pylori is particularly important. This review examines the relevant studies and describes evidence regarding the adhesion to and invasion of gastric mucosa epithelial cells by H. pylori.
Keywords: Helicobacter pylori, adhesion, invasion, chronic infection, mechanism
Figure 1
Generally, H. pylori adheres to gastric mucosa epithelial cells via the outer membrane protein and injects CagA into host cells via a type IV secretory system, resulting in changes in cytokine signaling and cell cycle control. When the external environment changes and becomes unfavorable, H. pylori invades the epithelial cells and multiplies in double-layer membrane vesicles to seek refuge. After internalization, autophagosomes form to degrade some of the ingested bacteria. Once the external environment becomes favorable, undegraded H. pylori is released from host cells into the external environment for recolonization.��
Cellular invasion of H. pylori
Observation in Vivo and in Vitro
Adhesion is an important step in H. pylori internalization, and invasion has been confirmed in a variety of samples (Table (Table1).1). Some in vivo studies demonstrate that H. pylori can invade gastric mucosa lesions in different stages. For instance, invasion was observed in patients with gastritis, ulcers, precancerous lesions and gastric cancer, and the bacteria were able to invade epithelial cells of the stomach and duodenum, parietal cells and immune cells, and even the lamina propria (Noach et al., 1994; el-Shoura, 1995; Papadogiannakis et al., 2000; Semino-Mora et al., 2003; Dubois and Bor��n, 2007; Necchi et al., 2007; Ozbek et al., 2010). Animals, such as mice, are also susceptible to H. pylori invasion (Oh et al., 2005), and in vitro studies have shown that H. pylori can enter cells. Although H. pylori is a common pathogenic bacterium of the human digestive system, invasion is not limited to cell lines that are closely related to gastric epithelial cells, such as AGS, MKN45, and SGC-7901. Indeed, invasion has been confirmed in the Huh7, HEp-2, and HeLa cell lines, among others (Björkholm et al., 2000; Amieva et al., 2002; Kwok et al., 2002; Terebiznik et al., 2006; Zhang et al., 2007; Ito K. et al., 2008).
Invasive ability also varies with the host cell type and H. pylori strain, with a rate of approximately 0.01�C0.1% of inoculated bacteria (Rautelin et al., 1995; Burridge and Chrzanowska-Wodnicka, 1996; Amieva et al., 2002; Dubois and Bor��n, 2007; Ito K. et al., 2008; Chu et al., 2010). Zhang et al. (2007) indicated that the strain 88-3887 can invade cells better than strains SS1 and X47, and each strain had the highest invasive ability in SCG-7901 cells. As H. pylori typically does not possess the ability to enter cells freely, most of the bacterial cells are internalized into the cytoplasm via endocytosis (Ozbek et al., 2010). A study from Stanford University (Amieva et al., 2002) showed that due to the innate immune response, cells can form phagocytic vesicles that wrap around H. pylori after cellular entry, even though the vesicles were immature. The encapsulated H. pylori cells were not swallowed or digested; instead, the vesicle acted as a protective barrier, allowing the microbes to evade the immune response.
These investigators also used differential immune fluorescence staining and live video microscopy to further investigate the physiological activities of internalized H. pylori and found that internalization occurred within 45 min of bacterial attachment to the cell surface. The bacterial cells then entered vacuoles and remained viable for at least 48 h. However, replication of the internalized H. pylori cells was not observed in the study, perhaps because of the short monitoring time and because the intracellular milieu was not suitable for bacterial replication (Amieva et al., 2002). Nonetheless, a most recent study found that H. pylori could proliferate in cells: two cells lines (AGS and MKN45) were evaluated in gentamicin protection experiments, revealing that H. pylori could proliferate after entering the cells, with the maximum number of bacterial cells observed after 6�C12 h (van der Wouden et al., 1999; Rokkas et al., 2009; Chu et al., 2010). Zhang et al. (2015) also observed this phenomenon in their study, whereby H. pylori attached to the cell membrane and combined with cellular microvilli after 2.5 h of infection. After invasion, double-layer membrane vesicles surrounded the H. pylori cells (4 h). At 6 h after infection, invasion reached a peak, with many bacteria observed in the cytoplasm. Bacterial adhesion, invasion, division and lysis were observed after 12 h, and most of the intracellular bacterial cells had lysed at 24 h. Another study confirmed that autophagosomes form to degrade ingested H. pylori through the lysosomal killing mechanism at 24 h after invasion (Chu et al., 2010). H. pylori possesses sophisticated mechanisms. For example, the bacterium seeks refuge inside host cells when the external environment changes and becomes unsuitable for survival; once the external conditions become suitable for survival, undegraded H. pylori is released from host cells into the external environment at the appropriate time. In vitro experiments have shown that when gentamicin was removed, viable H. pylori repopulates the extracellular environment, in parallel with a decrease in the number of intravacuolar bacteria (Amieva et al., 2002). Regardless, how H. pylori exits the intracellular space remains unclear. It is possible that viable bacteria are released as infected cells die. Vacuoles containing live H. pylori resemble multivesicular bodies, which, in some cell types, are capable of exocytosis and thus release their contents into the extracellular medium (Amieva et al., 2002). In brief, the number of bacteria in the cell is maintained through a dynamic process involving invasion, proliferation, apoptosis and release (Figure (Figure11).
To some extent, these findings provide a clear explanation for repeated infection by H. pylori: bacterial cells could escape the immune response of epithelial cells after invasion, and the amount and pathogenicity of the bacteria do not decrease during this process. Such a process might be a strategy for H. pylori to escape immune surveillance and remain alive in vivo (Amieva et al., 2002; Dubois and Bor��n, 2007), and it contributes to the complication of eradication. To determine whether H. pylori internalization plays a role in treatment failure, one study (Wang et al., 2016) carried out in our laboratory applied an invasion assay to evaluate the levels of H. pylori invasion of GES-1 cells. The results showed that the internalization levels of the failing strains were higher than those of the successful strains. However, there is no consensus regarding whether the level of internalization is related to antibiotic resistance, with some studies considering that resistant strains are associated with significantly higher internalization activity than susceptible strains (Lai et al., 2006) but Wang et al. (2016) reporting no evidence of this. Regardless of the correlation, once therapy fails, more consideration should be given to H. pylori internalization activity.Association with pathogenesis
The pathogenicity of bacteria that colonize the human mucosa is influenced by their capacity to invade and survive within epithelial cells. However, whether H. pylori internalization affects pathogenicity remains controversial. An in vivo study showed a positive correlation between the invasive ability of H. pylori and disease severity, and the average invasion rates of H. pylori strains found in gastric cancer and ulcers were higher than the rate of strains found in gastritis (Zhang et al., 2015). Moreover, many studies have confirmed that internalized H. pylori is pathogenic and able to express the virulence proteins Cag A and Vac A (Semino-Mora et al., 2003; Dubois and Bor��n, 2007; Necchi et al., 2007; Ito T. et al., 2008; Chu et al., 2010). Internalized H. pylori can also activate the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-��B) signaling pathway and induce interleukin-8 (IL-8) secretion, which suggests that invasion by this bacterium might be an important strategy in the development of H. pylori-associated diseases (Zhang et al., 2015). In addition, in vivo and in vitro studies show an association between internalized H. pylori and cell damage as well as cell disintegration (el-Shoura, 1995; Wilkinson et al., 1998). Ito T. et al. (2008) even found that H. pylori-induced gastric epithelial damage allows the bacterial cells to invade the lamina propria and translocate to the gastric lymph nodes, which may chronically stimulate the immune system. Additionally, the bacterial cells, alive or not, captured by macrophages may contribute to the induction and development of H. pylori-induced chronic gastritis.Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter pylori
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5118847/��
Invasion and Multiplication of Helicobacter pylori in Gastric Epithelial Cells and Implications for Antibiotic Resistance
Yen-Ting Chu, Ya-Hui Wang, Jiunn-Jong Wu, Huan-Yao Lei
Departments of Microbiology and Immunology
2Institute of Basic Medical Sciences
3Medical Laboratory Science and Biotechnology, College of Medicine, National Cheng Kung University, Tainan, Taiwan, Republic of China
ABSTRACT
Helicobacter pylori is a Gram-negative, spiral-shaped bacterium that infects more than 50% of the human population and can cause gastritis, peptic ulcer, or gastric malignancies. It is generally viewed as an extracellular microorganism. In a gentamicin protection assay on AGS or MKN45 cells, H. pylori could invade the epithelial cells and multiply within double-layer vesicles either on the plasma membrane or in the cytoplasm. A 5-fold increase in the number of bacteria was recultured from the infected cells at 12 h, compared with the number of invading cells at 2.5 h postinfection. The autophagic vesicles induced by H. pylori are the sites of replication and also of the degradation of the replicating bacteria after fusion with lysosomes. Many H. pylori bacteria in coccoid form associated with the plasma membrane can be released into culture. Only cell-penetrating antibiotics can enhance the intracellular killing of the replicating bacteria. The multiplication of H. pylori within cells provides a niche for its resistance to antibacterial therapy and has a significant impact on its biological life cycle.
Helicobacter pylori is a Gram-negative, flagellated, microaerophilic bacterium that selectively colonizes the gastric mucosa. It infects people worldwide and is correlated with socioeconomic conditions (24). The prevalence among middle-aged adults is over 80% in many developing countries. Overt disease, however, occurs in only 10 to 20% of infected individuals. The most common pathology associated with H. pylori infection is chronic active gastritis and peptic ulceration. A long-term chronic infection will increase the risk of gastric adenocarcinoma and mucosa-associated lymphoid-tissue lymphoma (19). Gastric mucosa is well protected against bacterial infections. However, H. pylori adapts and resides in the mucus and achieves attachment to epithelial cells, evasion of the immune responses, and persistent colonization in the stomach. It is not well understood why the immune system fails to clear H. pylori infection. Furthermore, the mechanisms controlling the induction and maintenance of the H. pylori-induced chronic inflammation are only partly understood.
Although H. pylori is generally viewed as a noninvasive pathogen, a number of in vivo and in vitro studies have shown that H. pylori is invasive, and it can reside in the vacuole in the cytoplasm or even replicate on the cell membrane to form a microcolony (2, 11, 25). This suggests that H. pylori can be considered a facultative intracellular organism (6, 20). We have reported that H. pylori can multiply in macrophages and bone marrow-derived dendritic cells with autophagy induction (27, 28). In this study, we further extended this line of research to epithelial cells and found that H. pylori could invade and replicate in epithelial cells. Thus, H. pylori can be considered an intracellular microorganism, and this has an impact on its own biological life cycle and its resistance to antibiotics....
some coccoid forms of H. pylori were present on the plasma membrane and were abundant at 18 h p.i. The coccoid form of H. pylori represents a transformation from the spiral one under the stress of the host cells. The ratio of the coccoid form of membrane-associated H. pylori increased as infection proceeded. H. pylori bacteria were attached to the plasma membrane, suggesting that they can freely exit the cell membrane or detach from the membrane and release into the culture supernatant, as shown in Fig. 2e.
Antibiotic resistance of H. pylori-infected epithelial cells.The effect of antibiotics on the intracellular multiplication of H. pylori was evaluated. For the gentamicin protection assay, gentamicin at 25 ��g/ml was included in the culture. Various concentrations of clarithromycin, metronidazole, or amoxicillin were then used to replace the gentamicin at this stage. For HP238, the MIC of clarithromycin is 0.06 ��g/ml. Higher doses (10 to 20 ��g/ml, or 166 to 333 times the MIC) of clarithromycin are needed to inhibit the multiplication of H. pylori in AGS cells. Lower doses (1.2 to 2 ��g/ml, or 20 to 33 times the MIC) are not effective (Fig. 4). This also applied to another clarithromycin-sensitive strain, ATCC 43504 (MIC of 0.03 ��g/ml), where a higher dose of clarithromycin (at least 333 times the MIC) enhanced the intracellular killing of the bacteria. For metronidazole-resistant HP238 (MIC of 32 ��g/ml) or metronidazole-sensitive HP917 (MIC of 4 ��g/ml), a higher dose of metronidazole (10 to 20 times the MIC) enhanced the clearance of the invading H. pylori. On the other hand, amoxicillin, which like gentamicin is unable to penetrate into cells, even at 20 to 40 times the MIC (HP238; MIC of 0.06 ��g/ml) did not have any effect. This suggests that the intracellular growth of H. pylori provides a niche to escape being killed by extracellular antibiotics. However, if the antibiotic can diffuse into the cells, such as with clarithromycin or metronidazole, there is still an inhibitory effect on the intracellular growth of H. pylori, but a higher dose of antibiotic is required.
DISCUSSION
Using the gentamicin protection assay on AGS cells, we demonstrated that H. pylori can invade the epithelial cells and multiply either on the plasma membrane or in double-layer vesicles. The invasion rate is around 0.01 to 0.1% of the bacteria inoculated. The 5-fold CFU increment between 2.5 h and 12 h p.i. indicates that the bacteria can replicate in the cells. The doubling time of the cell-associated H. pylori is calculated to be 5 to 6 h. The autophagic vesicles induced by H. pylori provide the site for its replication. The multiple vacuoles that fused from the H. pylori-containing autophagic vesicles with lysosomes will degrade the bacteria. Many H. pylori bacteria in coccoid form associated with the plasma membrane. This study is consistent with the report of Tan et al. that H. pylori can form microcolonies on the plasma membrane and use it as the replicative niche, with a doubling time of 4 to 6 h (25). However, we further showed that these autophagic vesicles are the site not only of replication but also of degradation when they are fused into autolysosomes.
Although H. pylori is generally viewed as a noninvasive pathogen present only in the lumen of the stomach and attached to gastric epithelial cells, many studies have shown that H. pylori is invasive and can be considered a facultative intracellular organism (6, 20). However, no direct demonstration of its replication in cells has been provided by the conventional gentamicin protection assay. We have found that H. pylori can multiply not only in macrophages and bone marrow-derived dendritic cells (27, 28) but also in epithelial cells as shown in this study. The key to this finding is that the increase in the CFU count is transient, observed only from 6 to 12 h p.i., while most of the other studies have determined the bacterial number at 1 to 3 days p.i. Another difference is the strain of H. pylori used. This new finding has several implications for the biological life cycle of H. pylori in the host. H. pylori can be considered a kind of intracellular microorganism because it can invade cells, undergo replication within cells, and leave the infected cells for further spread. Kwok et al. reported the entry of H. pylori into the AGS cells via a zipper-like mechanism, involving intimate contact with AGS cell microvilli and surface membrane pseudopod structures (11). The internalized H. pylori bacteria are found in LAMP1-containing vacuoles. The CagA protein can be translocated to the plasma membrane and form rafts with the invading H. pylori (12).
Autophagy is a component of the innate cellular immune responses against not only intracellular but also extracellular microorganisms (4, 5, 8, 14, 18). After internalization of the microorganism, the autophagosomes are formed to degrade the ingested bacterium by the lysosomal killing mechanism. Several intracellular bacteria have developed different mechanisms to evade the autophagic surveillance in macrophages. The autophagic vesicles are induced, but their maturation into autophagolysosomes is arrested or delayed (1, 22). The autophagy is induced by H. pylori in either phagocytic THP-1 cells or dendritic cells (27, 28) or nonphagocytic AGS epithelial cells. All H. pylori bacteria reside within the autophagosome, with no escape from the autophagic vesicle. On invasion of epithelial cells, some of the H. pylori bacteria were internalized by EEA1+ early endosome followed by the LAMP1+ late endosome/lysosomes. Multiple-vesicle vacuoles formed at 18 or 24 h p.i. that resulted in the destruction of the bacteria by fusion with lysosomes. Amieva et al. have reported viable H. pylori bacteria in large cytoplasmic vacuoles (2). They reasoned that bacteria can repopulate the extracellular environment in parallel with the disappearance of intravacuolar bacteria in the absence of gentamicin, and the intravacuolar niche is a place for the release of the bacteria and the place for evasion of the hostile microenvironment of the host. Recently, they have also reported that the plasma membranes are the site for H. pylori to replicate (25). We found many degrading bacteria in the multiple vacuoles, especially at 24 h p.i. The autophagy inducer rapamycin enhanced the clearance of the H. pylori (data not shown). We reasoned that the autophagic flux will clear the H. pylori in the autophagosome after fusion with the lysosomes. The multiple vacuoles are the place for the destruction of the invading and replicating bacteria. With regard to the release of H. pylori into the culture supernatant from the infected AGS cells, as shown in Fig. 2e, it is the plasma membrane-associated H. pylori bacteria that are released and which can be recovered as viable bacteria in the absence of gentamicin. H. pylori is spiral in nature, but it transforms into a coccoid form under stress conditions (3). It is generally accepted that coccoid forms of H. pylori are viable but are not culturable. Many coccoid forms of H. pylori were found on the membrane of the infected AGS cells, and the ratio of coccoid to bacillary forms increased as infection proceeded (Fig. 3a).
Intracellular H. pylori collections have been demonstrated in a small subset of gastric epithelial progenitors after infection of acid-producing parietal cell-deficient mice (16, 17) or in preneoplastic or cancer tissue (15, 23). H. pylori bacteria were detectable in endosome, cytoplasm vacuole, or plasma membrane or in membrane-associated coccoid form, which is consistent with our observation that H. pylori is replicating in the autophagosome or plasma membrane.
A triple-therapy regimen for 7 to 14 days is used as the first-line treatment for H. pylori-infected patients, but the failure rate is around 20 to 30% (7, 9). The failure of antibiotic treatment has been assumed to be due to the selection of an antibiotic-resistant mutant. The second-line treatment with quadruple therapy or even a third-line levofloxacin-based empirical regimen can be used to retreat the failed patients, and high cumulative eradication rates can be achieved (9, 10, 13, 21, 26). Our finding that H. pylori is capable of multiplication within infected epithelial cells provides an explanation of why the treatment failure may not be exclusively caused by the recurrence of antibiotic-resistant mutants. More likely, it may be due to the existence of an ecological niche for the organism to survive both the hostile gastric environment and the antibacterial therapy. The dormant coccoid form on the plasma membrane is resistant to antibiotic and can spread to infect other cells in the absence of an effective concentration of antibiotic. Thus, the antibiotic concentration needs to be high and sustained longer as it not only is needed to kill the extracellular H. pylori but also must be capable of penetrating the epithelial cells to kill the intracellular H. pylori. Cell membrane-penetrating antibiotics such as clarithromycin or metronidazole are effective, whereas non-cell membrane-penetrating amoxicillin or gentamicin is not effective. Interestingly, when the H. pylori bacteria derived from the clarithromycin-treated AGS cells were recultured on the plate, the colony size was much smaller than colonies from metronidazole- or gentamicin-treated cells (data now shown). In summary, H. pylori usurps the autophagic vesicles as the site for replication, and the autolysosomes after fusion will also degrade the replicating bacteria. This raises a distinct potential for new anti-H. pylori drugs that target the autophagy to enhance the clearance of intracellular bacteria in addition to the conventional use of antibiotics.��
Invasion and Multiplication of Helicobacter pylori in Gastric Epithelial Cells and Implications for Antibiotic Resistance | Infection and Immunity
https://iai.asm.org/content/78/10/4157/article-info��
World J Gastroenterol. Jul 21, 2017; 23(27): 4867-4878
Significance of dormant forms of Helicobacter pylori in ulcerogenesis
Vasiliy Ivanovich Reshetnyak, Tatiana Magomedalievna Reshetnyak
Vasiliy Ivanovich Reshetnyak, VA Negovsky Research Institute of General Reanimatology of Federal Research and Clinical Center of Intensive Care Medicine and Rehabilitology, 107031 Moscow, Russia
Tatiana Magomedalievna Reshetnyak, Department of Systemic Connective Tissue Diseases, VA Nasonova Research Institute of Rheumatology, 115522 Moscow, Russia
Nearly half of the global population are carriers of Helicobacter pylori (H. pylori), a Gram-negative bacterium that persists in the healthy human stomach. H. pylori can be a pathogen and causes development of peptic ulcer disease in a certain state of the macroorganism. It is well established that H. pylori infection is the main cause of chronic gastritis and peptic ulcer disease (PUD). Decontamination of the gastric mucosa with various antibiotics leads to H. pylori elimination and longer remission in this disease. However, the reasons for repeated detection of H. pylori in recurrent PUD after its successful eradication remain unclear. The reason for the redetection of H. pylori in recurrent PUD can be either reinfection or ineffective anti-Helicobacter therapy.The administration of antibacterial drugs can lead not only to the emergence of resistant strains of microorganisms, but also contribute to the conversion of H. pylori into the resting (dormant) state.
The dormant forms of H. pylori have been shown to play a potential role in the development of relapses of PUD. The paper discusses morphological H. pylori forms, such as S-shaped, C-shaped, U-shaped, and coccoid ones. The authors proposes the classification of H. pylori according to its morphological forms and viability.
��
The Coccoid and dormant forms of H.pylori
Morphological Form of H.pylori
All living organisms are equipped with mechanisms that allow extended survival in adverse environments. For a number of them, this response involves, besides metabolic adaptations, changes in cell morphology[62]. H. pylori is mainly present as a spiral-shaped form in human gastric biopsy specimens. On aging, the bacterial cells lose their typical spiral-shaped form and convert to coccoid ones (Figure 2)[54]. When influenced by adverse factors (temperature or pH changes, prolonged fasting when cultivated, or use of antibacterial drugs), non-spore-forming microorganisms can be transformed into a latent coccoid form[63].
��
Figure 2 Microscopic images of coccoid forms of Helicobacter pylori after 6-d exposure to antibiotics. From: Faghri et al[65].
The ability of H. pylori to transform from the spiral-shaped form to the coccoid form is one of its most important, but not exclusive properties. Through the course of evolution, H. pylori has evolved special adaptive mechanisms and acquired vital physiological properties allowing it to survive extreme situations in the human organism, when cultivated, and to survive in the external environment[64].
The bacterium transforms from spiral to coccoid under mild circumstances, whereas under extreme ones it is unable to undergo shape modification. This strongly supports the view that transformation into the coccoid form is an active, biologically led process, switched on by the bacterium as a protection mechanism[62,65]. This study demonstrates that the coccoid shape is in fact a manifestation of cell adaptation to less than optimum environments.The C-shaped and/or U-shaped forms of H. pylori are an intermediate bacterial type transforming into an inactive phase (dormancy). Dormancy is understood to be a reversible state of bacterial cells with a low metabolic activity, in which they can be for a long time without replication[63,70,71]. In microbiology, this condition has, until recently, been associated with the presence of forms, such as spores and cysts. In the late 20th century, the literature began to discuss the possibility of formation of dormant (resting) forms by non-spore-forming bacteria[63,72] that encompass most Gram-negative pathogenic bacteria, including H. pylori. Dormancy is characterized by the increased resistance of bacterial cells to extreme stresses (deficiency of nutrients, effects of antisecretory drugs, antibiotics, etc.) and favors their survival[73]. The ability of H. pylori to go into a dormant state may be an important factor in the epidemiology and spread of helicobacteriosis. The C-shaped and U-shaped forms of H. pylori can be considered truly a dormant form capable of reinfection[53]. The role of these forms in the pathogenesis and transmission of infection needs to be clarified.
The C-shaped and/or U-shaped forms of H. pylori keep the polar membrane associated with the flagellar basal complex[56,65]. Only a small number of intermediate forms of H. pylori possesses a complete set of flagella and retains metabolic activity to ensure mobility comparable to that of spiral-shaped forms[68].
The fully-formed coccoid forms maintain the basic pattern of a bacterial cell structure (Figure 4). They have a cell wall, cytoplasmic membrane and cytoplasm[56,62,67,69,74]. The coccoid cells differ in details of the cell wall structure, which leads to impairment of recognition of the bacteria by the host immune system (bacterial mimicry)[68].
The accumulated scientific data suggest that there are three morphological forms of H. pylori: (1) S-shaped forms; (2) U-shaped and C-shaped (intermediate or transitional) forms; and (3) Coccoid forms.
The intermediate and coccoid forms can coexist in the mucosa or in the culture in various ratios[75]. Their ratio depends on the time of H. pylori being present under adverse conditions and on the level of exposure to adverse factors. Spiral-shaped forms were predominant in a 3-d culture of H. pylori; about half of the bacteria are coccoid forms at 6 d[76-78]. Mouery et al[67] show the graphical distribution of the ratio of morphological forms of H. pylori after 12, 24 and 48 h of cultivation. The distributions of morphologies of more than 100 cells of each genotype for each time point are shown[67]. The number of coccoid forms increases with the longer time of cultivation. This happens due to the transition of C-shaped and U-shaped forms of H. pylori to coccoid ones.The C-shaped, U-shaped and coccoid forms of H. pylori lose enzyme activity and show a lower metabolism[79]. The urease activities of resting (dormant) and coccoid cells were found to be lower than those of the spiral-shaped form of H. pylori[80,81]. A significant transformation of H. pylori into coccoid forms may result in loss of urease activity. However, urease-encoding genes continue to be identified in H. pylori by polymerase chain reaction (PCR)[69,81,82]. The minimization of enzyme activity and energy metabolism in the transformable H. pylori forms is adaptive and aimed at preserving the viability of microorganisms[65,83]. Bacterial viability has been confirmed by the fact that the transformed forms of H. pylori can be detected by acridine orange staining[69,84]. The C-shaped, U-shaped and coccoid forms of H. pylori tolerate a wider pH range to a greater extent than the spiral-shaped forms, are resistant to unfavorable factors and antibiotics, and cannot lose virulence. All this creates favorable conditions for the preservation of bacteria in the human body or in the external environment.
The triggers of H. pylori transformation from spiral-shaped to coccoid forms in the environment may be physical factors: higher insolation, low humidity, and lack of food substrates[77,85]. During bacteriological cultivation, transformation into the coccoid forms occurs due to the depletion of adequate components of the nutrient medium[85,86]. H. pylori transforms in the human body due to changes in the habitat conditions upon exposure to antisecretory and antibacterial drugs. Khomeriki et al[69] have studied the time course of changes in the transformation of H. pylori in the GM. They have indicated that the spiral-shaped forms transform into the coccoid ones a few hours after adhesion to the cell surface of GM cells[69].
The conversion of the spiral forms of H. pylori to its C-shaped, U-shaped and coccoid forms in the GM and in the nutrient medium is due to the accumulation of toxic metabolic products of H. pylori vital functions. Reactive oxygen species generated by phagocytes or by H. pylori itself in the presence of specific pyridine nucleotides may trigger the formation of transitional and coccoid forms in the GM of untreated patients[69]. When an unfavorable situation occurs, there is accumulation of factors that induce the conversion of cells in the bacterial populations to a dormant state.
Loginov et al[8] carried out a comparative analysis in which H. pylori in GM biopsy specimens from patients with duodenal ulcer was detected by a quantitative urease test and PCR before and a month after anti-Helicobacter therapy. Prior to anti-Helicobacter therapy, the detection rate of H. pylori in the GM biopsy specimens from patients with PUD was 93.4% and 98.7%, as shown by the quantitative urease test and PCR, respectively (Table 1). The difference of 5.3% in the detection rate of H. pylori may be due to the different sensitivities of these methods.One month after eradication therapy, these patients had H. pylori detected by the quantitative urease test in 11.1% of cases and by PCR in 24.1%. The difference between the methods was 13%, i.e., almost twice that as before the treatment. These findings suggest that H. pylori were not completely eliminated in some patients at 1 mo after the anti-Helicobacter therapy, and the H. pylori diagnosed by PCR were at least partially in a dormant (resting) state and partly in coccoid forms. The low urease activity (or lack thereof) of coccoid H. pylori forms precludes identifying them by the quantitative urease test. The data presented allow us to indirectly suggest that after anti-Helicobacter therapy, the dormant forms of H. pylori are present in patients as a result of their incomplete elimination[7,87].
��
Figure 5 Major physiological states and forms of Helicobacter pylori[101]. H. pylori: Helicobacter pylori.The C-shaped and U-shaped forms of H. pylori are most likely in a dormant (resting) state and can be a viable and culturable (Figure 5)[101].
And if so, once under favorable conditions, the dormant (resting) forms of H. pylori can revert to a vegetative spiral-shaped form. By using electron microscopy, Konstantinova et al[102] have shown that there are various defects in the cell wall of the transformed forms of H. pylori. The authors point out that before the reversion of the dormant forms of H. pylori to vegetative forms, there is a need for certain conditions for the repair of cellular damages.
��Reanimated�� H. pylori can play an important role in the development of recurrent PUD after anti-Helicobacter treatment[81,96]. The ��revived�� forms of H. pylori are able to colonize the GM to subsequently develop peptic ulcer relapse[78,95].��
Significance of dormant forms of Helicobacter pylori in ulcerogenesis
https://www.wjgnet.com/1007-9327/full/v23/i27/4867.htm��
World J Gastroenterol. 2014 Sep 28; 20(36): 12767�C12780.
Effect of Helicobacter pylori on gastric epithelial cells
Shatha Alzahrani, Taslima T Lina, Jazmin Gonzalez, Irina V Pinchuk, Ellen J Beswick, and Victor E Reyes
Shatha Alzahrani, Taslima T Lina, Irina V Pinchuk, Victor E Reyes, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, United States
Jazmin Gonzalez, MSIII, School of Medicine, University of Texas Medical Branch, Galveston, TX 77555, United States
Irina V Pinchuk, Department of Internal Medicine, University of Texas Medical Branch, Galveston, TX 77555, United States
Ellen J Beswick, Departments of Molecular Genetics and Microbiology, University of New Mexico Health Sciences Center, Albuquerque, NM 87131, United States
Victor E Reyes, Departments of Pediatricsand and Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, United States
Author contributions: All authors contributed to the writing of the different sections of this review; Alzahrani S took the leading role in organizing the topic and Reyes VE was responsible for the outline and editing.
Correspondence to: Victor E Reyes, PhD, Departments of Pediatricsand and Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX 77555, United States. ude.bmtu@seyerv
Abstract
The gastrointestinal epithelium has cells with features that make them a powerful line of defense in innate mucosal immunity. Features that allow gastrointestinal epithelial cells to contribute in innate defense include cell barrier integrity, cell turnover, autophagy, and innate immune responses. Helicobacter pylori (H. pylori) is a spiral shape gram negative bacterium that selectively colonizes the gastric epithelium of more than half of the world��s population. The infection invariably becomes persistent due to highly specialized mechanisms that facilitate H. pylori��s avoidance of this initial line of host defense as well as adaptive immune mechanisms. The host response is thus unsuccessful in clearing the infection and as a result becomes established as a persistent infection promoting chronic inflammation. In some individuals the associated inflammation contributes to ulcerogenesis or neoplasia. H. pylori has an array of different strategies to interact intimately with epithelial cells and manipulate their cellular processes and functions. Among the multiple aspects that H. pylori affects in gastric epithelial cells are their distribution of epithelial junctions, DNA damage, apoptosis, proliferation, stimulation of cytokine production, and cell transformation. Some of these processes are initiated as a result of the activation of signaling mechanisms activated on binding of H. pylori to cell surface receptors or via soluble virulence factors that gain access to the epithelium. The multiple responses by the epithelium to the infection contribute to pathogenesis associated with H. pylori.IMPACT OF H. PYLORI AND GASTRIC EPITHELIUM INTERACTIONS ON PATHOGENESIS
H. pylori and GECs interactions represent a complex set of multiple events, many of which induce pathogenesis and sustain the infection. Although only about 20% of H. pylori are attached to the epithelial cells at any point in time via different adhesions[110], adhesion is essential for the stimulation of the inflammatory cascade. Specifically, adhesion is a prerequisite for interleukin 8 (IL8) productions by gastric epithelial cells[111]. Also, adherence induces the development of severe diseases. For example, BabA adhesion, which binds the Lewis b blood group antigen on the gastric epithelium, is associated with development of duodenal ulcer, distal gastric cancer and more severe gastritis[112]. Attachment to epithelial cells via virulence factors can also induce tissue injury. Binding of vacA to the epithelial cells leads to eventual pore formation in the lysosomal membrane, generation of vacuoles, and increases in anion permeability[113]. Patients who are infected with H. pylori that have a functional cag-PAI have increased IL-8 levels in the gastric mucosa, resulting in significant neutrophil infiltration, and an increased risk of peptic ulcer and gastric cancer[114].
Figure 3
Cytokines production by gastric epithelial cell during Helicobacter pylori Infection. Helicobacter pylori (H. pylori) leads to production of proinflammatory cytokines by gastric epithelial cells (GECs). Interactions between the translocated cagA and gastric epithelial cells lead to activation of nuclear factor (NF)-��B, alteration in gene transcription in the GECs, and secretion of IL-8 by GECs, which leads to recruitment of neutrophils. H. pylori urease induces the production of IL-6 and tumor necrosis factor alpha (TNF-��) by GECs. Other cytokines secreted by GECs during H. pylori infection such as, TNF-��, IL-1��, IL-1��, granulocyte-macrophage colony-stimulating factor, monocyte chemoattractant protein-1 (MCP-1), migration inhibitory factor and tumor growth factor (TGF)-��.
Keywords: Helicobacter pylori, Apoptosis, Gastric epithelial cells, Proinflammatory cytokines, Chronic inflammation, Gastric diseases, Gastric cancerEffect of Helicobacter pylori on gastric epithelial cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4177462/��
Cancers (Basel). 2019 Apr; 11(4): 547.
Novel Insights of Lymphomagenesis of Helicobacter pylori-Dependent Gastric Mucosa-Associated Lymphoid Tissue Lymphoma
Sung-Hsin Kuo,1,2,3,4 Ming-Shiang Wu,5 Kun-Huei Yeh,1,2,3,4 Chung-Wu Lin,6 Ping-Ning Hsu,5 Li-Tzong Chen,7,8,9 and Ann-Lii Cheng2,3,4,5,*
1Department of Oncology, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei 100, Taiwan; wt.ude.utn@101oukhs (S.-H.K.); wt.ude.utn@heyhk (K.-H.Y.)
2Cancer Research Center, National Taiwan University College of Medicine, Taipei 100, Taiwan
3Graduate Institute of Oncology, National Taiwan University College of Medicine, Taipei 100, Taiwan
4National Taiwan University Cancer Center, National Taiwan University College of Medicine, Taipei 106, Taiwan
5Department of Internal Medicine, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei 100, Taiwan; wt.ude.utn@gnaihsgnim (M.-S.W.); wt.ude.utn@5368ushp (P.-N.H.)
6Department of Pathology, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei 100, Taiwan; moc.oohay@niluwgnuhc
7National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan; wt.gro.irhn@nehcoel
8Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 807, Taiwan
9Department of Internal Medicine, National Cheng-Kung University Hospital, Tainan 704, Taiwan
Abstract
Gastric mucosa-associated lymphoid tissue (MALT) lymphoma is the most common subtype of gastric lymphoma. Most gastric MALT lymphomas are characterized by their association with the Helicobacter pylori (HP) infection and are cured by first-line HP eradication therapy (HPE).Several studies have been conducted to investigate why most gastric MALT lymphomas remain localized, are dependent on HP infection, and show HP-specific intratumoral T-cells (e.g., CD40-mediated signaling, T-helper-2 (Th2)-type cytokines, chemokines, costimulatory molecules, and FOXP3+ regulatory T-cells) and their communication with B-cells. Furthermore, the reason why the antigen stimuli of these intratumoral T-cells with tonic B-cell receptor signaling promote lymphomagenesis of gastric MALT lymphoma has also been investigated. In addition to the aforementioned mechanisms, it has been demonstrated that the translocated HP cytotoxin-associated gene A (CagA) can promote B-cell proliferation through the activation of Src homology-2 domain-containing phosphatase (SHP-2) phosphorylation-dependent signaling, extracellular-signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), B-cell lymphoma (Bcl)-2, and Bcl-xL. Furthermore, the expression of CagA and these CagA-signaling molecules is closely associated with the HP-dependence of gastric MALT lymphomas (completely respond to first-line HPE).
In this article, we summarize evidence of the classical theory of HP-reactive T-cells and the new paradigm of direct interaction between HP and B-cells that contributes to the HP-dependent lymphomagenesis of gastric MALT lymphomas. Although the role of first-line HPE in the treatment of HP-negative gastric MALT lymphoma remains uncertain, several case series suggest that a proportion of HP-negative gastric MALT lymphomas remains antibiotic-responsive and is cured by HPE.
Considering the complicated interaction between microbiomes and the genome/epigenome, further studies on the precise mechanisms of HP- and other bacteria-directed lymphomagenesis in antibiotic-responsive gastric MALT lymphomas are warranted.
Keywords: MALT lymphoma, Helicobater pylori, T-cell, Tregs, CagANovel Insights of Lymphomagenesis of Helicobacter pylori-Dependent Gastric Mucosa-Associated Lymphoid Tissue Lymphoma
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6520890/��
Helicobacter pylori Induces Apoptosis of Macrophages in Association with Alterations in the Mitochondrial Pathway
Rena J. Menaker, Peter J. M. Ceponis, Nicola L. Jones
1Research Institute, Hospital for Sick Children
2Department of Physiology
3Department of Laboratory Medicine and Pathobiology
4Department of Paediatrics, University of Toronto, Toronto, Canada
ABSTRACT
Helicobacter pylori is a gastric bacterial pathogen that evades host immune responses in vivo and is associated with the development of gastritis, peptic ulcer disease, and gastric cancers. Induction of macrophage apoptosis is a method employed by multiple pathogens to escape host immune responses.Therefore, we hypothesized that H. pylori induces apoptosis of infected macrophages. RAW 264.7 cells were infected with H. pylori strain 60190, and apoptosis was assessed. Transmission electron microscopy and fluorescence microscopy showed that infected macrophages displayed morphological features characteristic of apoptosis. Quantification by acridine orange-ethidium bromide fluorescent-dye staining showed that apoptosis was dose and time dependent, and apoptosis was further confirmed by increased binding of annexin V-fluorescein isothiocyanate (FITC) to externalized phosphatidylserine of infected but not of control macrophages.
Macrophages infected with isogenic mutants of H. pylori strain 60190 deficient in either cagA or vacA induced significantly less apoptosis than the parental strain, as assessed by increased binding of annexin V-FITC. Western blot analysis of whole-cell protein lysates revealed that infection with strain 60190 induced a time-dependent increase in cleavage of procaspase 8 and disappearance of full-length Bid compared with uninfected cells. Furthermore, pharmacological inhibition of caspase 8 caused a decrease in levels of apoptosis.
Finally, infection caused a time-dependent increase in mitochondrial-membrane permeability and release of cytochrome c into the cytosol.
These results suggest that H. pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Elimination of this key immunomodulatory cell may represent a mechanism employed by the bacterium to evade host immune responses.
In summary, this study showed that H. pylori infection induces apoptosis in macrophages through a caspase 8-, Bid-, and mitochondrion-dependent mechanism. This apoptotic signaling cascade is mediated in part by VacA and the cag PAI gene cagA. Thus, in addition to other strategies to evade host immune responses, such as disruption of phagosome maturation (72) and disruption of cytokine signaling (10), induction of macrophage apoptosis may represent a mechanism by which H. pylori usurps the host immune response to establish chronic infection in humans.
Helicobacter pylori Induces Apoptosis of Macrophages in Association with Alterations in the Mitochondrial Pathway | Infection and Immunity
https://iai.asm.org/content/72/5/2889
Proposed mechanism of H. pylori-induced macrophage apoptosis in macrophages by polyamines
Fig. 7. H. pylori induces mitochondrial membrane depolarization, translocation of cytochrome c from mitochondria to cytosol, and activation of caspase-3 that is inhibited by MDL 72527. RAW 264.7 cells were treated with HPL in the absence and presence of MDL 72527 (250 ^M). (A) Mitochondrial membrane potential was measured by flow cytometry using MitoCapture dye at the times indicated. Summary data of mean relative fluorescence, expressed as negative values to indicate depolarization is shown. **p < 0.01 vs control, ���p < 0.01 vs HPL + MDL 72527. (B) Western blot analysis for cytochrome c (15 kDA protein). Upper panel: mito-chondrial fraction; middle panel: cytosolic fraction. Equal amounts of protein were loaded per lane, and equal loading was verified by staining of membranes with Ponceau S and immunoblot-ting for ß-actin (lower panel). (C) Cells were treated with HPL and caspase-3 activity measured by colorimetric assay at the times indicated. (D) In cells assessed at 24 h after stimulation, MDL 72527 (250 ^M) blocked the caspase-3 activation. For (C) and (D), **p < 0.01 vs control, ���p < 0.01 vs HPL. (Data shown are from ref. 41.)
Macrophage Apoptosis
Fig. 8. Proposed mechanism of H. pylori-induced macrophage apoptosis in macrophages by polyamines. H. pylori stimulates expression of ODC, generating polyamines, and the increased spermine is acted upon by SMO(PAO1) that is also upregulated by H. pylori. The H2O2 so generated causes mitochondrial membrane depolarization, release of cytochrome c from mitochondria to cytosol, and activation of caspase-3, leading to apoptosis.
and additional studies are in progress in our laboratory to determine the response to other pathogenic stimuli.
5.2. Further Insights Into the Induction of Macrophage Apoptosis by Polyamines
Previous work from our laboratory has implicated specific H. pylori-derived factors in the activation of macrophages, including urease (3). This enzyme serves a biochemically essential function for the bacterium of converting urea to ammonia and carbon dioxide, creating an alkaline microenvironment required for the organism to survive in the acid milieu of the stomach. However, urease is also an immunologically active protein, and we have found that urease appears to mediate the induction of ODC in macrophages because isogenic mutant strains of H. pylori lacking urease exhibited significant attenuation of induction of ODC expression and activity, and recombinant urease stimulated ODC activation (43).
ODC expression has been shown to be enhanced by binding of the transcription factor c-Myc to the ODC promoter (6). We have substantial evidence that H. pylori and its urease induces ODC by a c-Myc-dependent process, because H. pylori induces c-Myc expression and inhibition of c-Myc binding prevents activation of the ODC promoter, ODC mRNA expression, ODC activity, and apoptosis (43). Additionally, we have recently found that the activation of c-Myc and induction of ODC in response to H. pylori is dependent on phosphorylation of extracellular signalregulated kinase (ERK) 1/2 (M. Asim, R. Chaturvedi, and K.T. Wilson, manuscript in preparation).
6. Involvement of SMO(PAO1) in the Pathogenic Aspects of Other Model Systems
We have conducted additional studies that indicate that SMO(PAO1) activity is important in other disease states. Specifically, we have recently reported that as in macrophages, SMO(PAO1) expression is upregulated in gastric epithelial cells exposed to H. pylori and that inhibition with MDL72527 or SMO(PAO1) siRNA significantly attenuated the induction of apoptosis (44). Importantly, we also demonstrated that DNA damage induced by H. pylori in these cells was mediated by H2O2 derived from induction of SMO(PAO1), which may have direct relevance to H. pylori-associated gastric cancer (44). Additionally, we reported that polyamines are beneficial in mouse colitis (45), and we now have evidence that SMO(PAO1) correlates with the severity of disease in several colitis models, suggesting that oxidative stress derived from spermine oxidation may play an important role either from apoptosis or other effects that we are now investigating.
7. The Balance of Spermine and its Oxidation Products, Determined by ODC and SMO(PAO1) is Important in the Regulation of Innate Immune Response
We have previously reported that in a coculture model in which H. pylori is separated from macrophages by a filter support the bacteria are killed by a NO-dependent mechanism (46). However, in the stomach, the bacteria persists for the life of the host, despite the expression of iNOS in the gastric mucosa (39). Because spermine has been shown to downregulate cytokine production by lipopolysaccharide-stimulated macrophages (47), we have assessed the effect of spermine on iNOS (48). Spermine inhibited NO production from H. pylori-stimulated macrophages in a concentration-dependent manner. Intriguingly, there was no effect on the induction of iNOS mRNA, but rather there was potent inhibition of iNOS protein translation. Consistent with this, knockdown of ODC by siRNA increased iNOS protein expression and NO production in response to H. pylori. This enhanced killing of the bacterium, whereas spermine inhibited killing (48). Recently, we have found that knockdown of SMO(PAO1) in H. pylori-stimulated macrophages prevents depletion of spermine, and thus inhibits iNOS protein levels, NO production, and bacterial killing (R. Chaturvedi and K.T. Wilson, manuscript in preparation).
8. Summary
Evidence has been presented in this review indicating that polyamines can have either a pro- or antiapoptotic effect in immune cells. This most likely depends greatly on the inducing stimuli, the cell type, and other factors in the intracellular environment. We have presented our work in the H. pylori model that has identified a pathway of macrophage apoptosis in which the activation of arginase, ODC, and SMO(PAO1) is required, with the latter resulting in generation of H2O2 that activates the mitochondr-ial pathway of apoptosis. We suggest that the balance of spermine and its oxidation is a critical aspect of innate immune response to pathogens, in that spermine accumulation inhibits responses such as iNOS-derived NO and cytokine production, whereas spermine catabolism leads to oxidative stress, apoptosis, and DNA damage.
9. Acknowledgments
Supported by grants to K. Wilson from the National Institutes of Health (R01
DK53620 and R21 DK63626), the Office of Medical Research, Department of Veterans
Affairs, and the Crohn's and Colitis Foundation of America.��
Fig. 7. H. pylori induces mitochondrial membrane depolarization, translocation of cytochrome c from mitochondria to cytosol, and activation of caspase-3 that is inhibited by MDL 72527. RAW 264.7 cells were treated with HPL in the absence and presence of MDL 72527 (250 ^M).
��
Fig. 8. Proposed mechanism of H. pylori-induced macrophage apoptosis in macrophages by polyamines. H. pylori stimulates expression of ODC, generating polyamines, and the increased spermine is acted upon by SMO(PAO1) that is also upregulated by H. pylori. The H2O2 so generated causes mitochondrial membrane depolarization, release of cytochrome c from mitochondria to cytosol, and activation of caspase-3, leading to apoptosis.
A - Cancer Research - European Medical Alliance
https://www.europeanmedical.info/cancer-research/a-1.html��
��
��
MDL 72527 ��98% (HPLC) | CPC-200 | Sigma-Aldrich
MDL 72527is an inhibitor of polyamine oxidase (spermine oxidase SMO). It blocks the production of H2O2 and increases the cells survival. Polyamine catabolism is one of the major causes of the ROS production of prostate cells in general and androgen-induced enhancement of ROS in androgen-dependent CaP cells in particular.
��
Ornithine Decarboxylase in Macrophages Exacerbates Colitis ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6072585
Aug 01, 2018 �� In colitis tissues, immunofluorescence demonstrated strong double-staining for ODC and macrophages, and positive selection studies showed that macrophages represent the myeloid subpopulation with an increase in Odc expression. These data indicate that macrophages are the main source of ODC during colitis, although basal ODC in other immune cells could have a role.
Cited by: 9
Publish Year: 2018
Author: Kshipra Singh, Lori A. Coburn, Mohammad Asim, Daniel P. Barry, Margaret M. Allaman, Chanjuan Shi, M....
Ornithine Decarboxylase in Macrophages Exacerbates Colitis ...
https://www.ncbi.nlm.nih.gov/pubmed/29853605
Aug 01, 2018 �� Ornithine decarboxylase (ODC) is the rate-limiting enzyme for polyamine biosynthesis and restricts M1 macrophage activation in gastrointestinal (GI) infections. However, the role of macrophage ODC in colonic epithelial-driven inflammation is unknown.
Cited by: 9
Publish Year: 2018
Author: Kshipra Singh, Lori A. Coburn, Mohammad Asim, D��
Ornithine decarboxylase regulates M1 macrophage activation ...
Jan 31, 2017 �� Here, we demonstrate that ornithine decarboxylase (ODC), the rate-limiting enzyme in polyamine synthesis, regulates M1 activation during Helicobacter pylori and Citrobacter rodentium infection. Deletion of Odc in macrophages resulted in increased inflammation and decreased bacterial persistence in mouse models.
��
��
Helicobacter pylori Induces ERK-dependent Formation of a Phospho-c-Fos.c-Jun Activator Protein-1 Complex That Causes Apoptosis in Macrophages
Journal of Biological Chemistry 285(26):20343-57 �� June 2010
Macrophages are essential components of innate immunity, and apoptosis of these cells impairs mucosal defense to microbes. Helicobacter pylori is a gastric pathogen that infects half of the world population and causes peptic ulcer disease and gastric cancer. The host inflammatory response fails to eradicate the organism.
We have reported that H. pylori induces apoptosis of macrophages by generation of polyamines from ornithine decarboxylase (ODC), which is dependent on c-Myc as a transcriptional enhancer.
We have now demonstrated that expression of c-Myc requires phosphorylation and nuclear translocation of ERK, which results in phosphorylation of c-Fos and formation of a specific activator protein (AP)-1 complex. Electromobility shift assay and immunoprecipitation revealed a previously unrecognized complex of phospho-c-Fos (pc-Fos) and c-Jun in the nucleus. Fluorescence resonance energy transfer demonstrated the interaction of pc-Fos and c-Jun. The capacity of this AP-1 complex to bind to putative AP-1 sequences was demonstrated by oligonucleotide pulldown and fluorescence polarization. Binding of the pc-Fos.c-Jun complex to the c-Myc promoter was demonstrated by chromatin immunoprecipitation. A dominant-negative c-Fos inhibited H. pylori-induced expression of c-Myc and ODC and apoptosis. H. pylori infection of mice induced a rapid infiltration of macrophages into the stomach. Concomitant apoptosis depleted these cells, and this was associated with formation of a pc-Fos.c-Jun complex. Treatment of mice with an inhibitor of ERK phosphorylation attenuated phosphorylation of c-Fos, expression of ODC, and apoptosis in gastric macrophages. A unique AP-1 complex in gastric macrophages contributes to the immune escape of H. pylori.
��
Pathway for induction of apoptosis in H. pylori-stimulated macrophages by an AP-1-depen- dent mechanism leading to polyamine-derived oxidative stress. As demonstrated in the current report, H. pylori causes activation of ERK1/2 phosphorylation by MEK1/2 that leads to nuclear translocation of pERK. This results in the induction of both c-Fos and c-Jun and the phosphorylation of c-Fos. pc-Fos and c-Jun form an AP-1 complex that binds to the c-Myc gene leading to expression of c-Myc mRNA and protein. This c-Myc protein in the cytoplasm translocates to the nucleus and enhances ODC transcription (9) in concert with activation of a purported H. pylori response element (HPRE). This induction of ODC leads to increased generation of the polyamines putrescine, spermidine, and spermine. In parallel, H. pylori up-regulates the expression and activity of the enzyme spermine oxidase (SMO), which back-converts spermine to spermidine, a process that generates H 2 O 2 , which causes apoptosis through mitochondrial membrane depolarization (8).
��
Omega-3 fatty acids and the treatment of depression: a ...
It is proposed that omega-3 fatty acids have a role in transcriptional regulation by phosphorylation inhibition of JNK, ERK, and MAPK proteins, which downregulates protein-1 expression. 26 Furthermore, omega-3 fatty acids are proposed to have a role in restraining nuclear factor-��B nuclear translocation secondary to I��B phosphorylation 27 .
��
��
Cancer Res. 2018 Aug 1;78(15):4303-4315. doi: 10.1158/0008-5472.CAN-18-0116. Epub 2018 May 31.
Ornithine Decarboxylase in Macrophages Exacerbates Colitis and Promotes Colitis-Associated Colon Carcinogenesis by Impairing M1 Immune Responses.
Singh K1,2, Coburn LA1,2,3, Asim M1, Barry DP1, Allaman MM1, Shi C4,5, Washington MK4,5, Luis PB6, Schneider C2,6, Delgado AG1, Piazuelo MB1,2, Cleveland JL7, Gobert AP1,2, Wilson KT8,2,3,4,5.
Author information
1 Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee.
2 Center for Mucosal Inflammation and Cancer, Vanderbilt University Medical Center, Nashville, Tennessee.
3 Veterans Affairs Tennessee Valley Healthcare System, Nashville, Tennessee.
4 Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee.
5 Vanderbilt Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, Tennessee.
6 Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee.
7 Department of Tumor Biology, Moffitt Cancer Center and Research Institute, Tampa, Florida.
8 Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee. keith.wilson@vanderbilt.edu.
Abstract
Ornithine decarboxylase (ODC) is the rate-limiting enzyme for polyamine biosynthesis and restricts M1 macrophage activation in gastrointestinal (GI) infections. However, the role of macrophage ODC in colonic epithelial-driven inflammation is unknown.Here, we investigate cell-specific effects of ODC in colitis and colitis-associated carcinogenesis (CAC). Human colonic macrophages expressed increased ODC levels in active ulcerative colitis and Crohn's disease, colitis-associated dysplasia, and CAC.
Mice lacking Odc in myeloid cells (Odc��mye mice) that were treated with dextran sulfate sodium (DSS) exhibited improved survival, body weight, and colon length and reduced histologic injury versus control mice. In contrast, GI epithelial-specific Odc knockout had no effect on clinical parameters. Despite reduced histologic damage, colitis tissues of Odc��mye mice had increased levels of multiple proinflammatory cytokines and chemokines and enhanced expression of M1, but not M2 markers.
In the azoxymethane-DSS model of CAC, Odc��mye mice had reduced tumor number, burden, and high-grade dysplasia. Tumors from Odc��mye mice had increased M1, but not M2 macrophages. Increased levels of histone 3, lysine 9 acetylation, a marker of open chromatin, were manifest in tumor macrophages of Odc��mye mice, consistent with our findings that macrophage ODC affects histone modifications that upregulate M1 gene transcription during GI infections. These findings support the concept that macrophage ODC augments epithelial injury-associated colitis and CAC by impairing the M1 responses that stimulate epithelial repair, antimicrobial defense, and antitumoral immunity. They also suggest that macrophage ODC is an important target for colon cancer chemoprevention.
Significance: Ornithine decarboxylase contributes to the pathogenesis of colitis and associated carcinogenesis by impairing M1 macrophage responses needed for antitumoral immunity; targeting ODC in macrophages may represent a new strategy for chemoprevention. Cancer Res; 78(15); 4303-15. ©2018 AACR.
©2018 American Association for Cancer Research.��
... Arginase is present in the cell in two isoforms; arginase 1 (ARG1) is abundantly found in liver and is involved in the urea cycle, and arginase 2 (ARG2) is found in kidney and localizes to the mitochondria. Ornithine is then converted into putrescine by ODC in the cytosol (Pegg 2006;Asim et al. 2010). Putrescine can then be converted into spermidine by spermidine synthase (SRM), and spermidine converted into spermine by spermine synthase (SMS) (Fig. 1). ...
... Polyamines are pleiotropic polycations that have many cellular functions, including the regulation of gene transcription, protein translation, cell growth, proliferation, and differentiation ( Hardbower et al. 2017;Pegg 2006Pegg , 2009. The production of the three major polyamines-putrescine, spermidine, and spermine-is tightly regulated and centers on the rate-limiting enzyme, ornithine decarboxylase (ODC1, hereafter referred to as ODC) (Asim et al. 2010). ODC uses the substrate Lornithine to produce putrescine via a decarboxylation reaction ( Asim et al. 2010). ...
... The production of the three major polyamines-putrescine, spermidine, and spermine-is tightly regulated and centers on the rate-limiting enzyme, ornithine decarboxylase (ODC1, hereafter referred to as ODC) (Asim et al. 2010). ODC uses the substrate Lornithine to produce putrescine via a decarboxylation reaction ( Asim et al. 2010). ODC has been implicated in several malignancies, including breast, colorectal, and gastric cancers. ...
... Most of the studies related to ODC have been focused on its role in epithelial cell function. However, ODC expression is upregulated in macrophages by H. pylori in vitro ( Asim et al. 2010) and in infected gastritis tissues of mice and humans (Hardbower et al. 2017). Importantly, macrophage ODC is immunosuppressive, impairing M1-dependent host defense against H. pylori; mice with myeloidspecific deletion of Odc exhibited marked upregulation of M1 responses, including NOS2 expression/NO production; M1 gene signatures (Nos2, Il1b, Tnf, Il6, Il12a, Il12b) and M1 protein responses (IL-1�� and TNF-��) as well as pro-inflammatory chemokines and IL-17A ( Hardbower et al. 2017). ...Ornithine Decarboxylase in Macrophages Exacerbates Colitis and Promotes Colitis-Associated Colon Carcinogenesis by Impairing M1 Immune Responses. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29853605��
Evolutionary Roots of Arginase Expression and Regulation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4224125Arginase expression is the default mode of tissue macrophages, but can also be amplified by signals, such as IL-4/13 or transforming growth factor-�� (TGF-��) that accelerates wound healing and tissue repair. In worms, the induction of collagen gene is coupled with induction of immune response genes, both depending on the same TGF-��-like pathway.
��
��
Pivotal Advance: Arginase�\1�\independent polyamine production stimulates the expression of IL�\4�\induced alternatively activated macrophage markers while inhibiting LPS�\induced expression of inflammatory genes
Jan Van den Bossche Wouter H. Lamers Eleonore S. Koehler Jan M. C. Geuns Leena Alhonen Anne Uimari Sini Pirnes�\Karhu Eva Van Overmeire Yannick Morias Lea Brys �� See all authors
In macrophages, basal polyamine (putrescine, spermidine, and spermine) levels are relatively low but are increased upon IL�\4 stimulation. This Th2 cytokine induces Arg1 activity, which converts arginine into ornithine, and ornithine can be decarboxylated by ODC to produce putrescine, which is further converted into spermidine and spermine.Recently, we proposed polyamines as novel agents in IL�\4�\dependent E�\cadherin regulation in AAMs. Here, we demonstrate for the first time that several, but not all, AAM markers depend on polyamines for their IL�\4�\induced gene and protein expression and that polyamine dependency of genes relies on the macrophage type. Remarkably, Arg1�\deficient macrophages display rather enhanced IL�\4�\induced polyamine production, suggesting that an Arg1�\independent polyamine synthesis pathway may operate in macrophages.
On the other side of the macrophage activation spectrum, LPS�\induced expression of several proinflammatory genes was increased significantly in polyamine�\depleted CAMs. Overall, we propose Arg1 independently produced polyamines as novel regulators of the inflammatory status of the macrophage.
Indeed, whereas polyamines are needed for IL�\4�\induced expression of several AAM mediators, they inhibit the LPS�\mediated expression of proinflammatory genes in CAMs.
Pivotal Advance: Arginase�\1�\independent polyamine production stimulates the expression of IL�\4�\induced alternatively activated macrophage markers while inhibiting LPS�\induced expression of inflammatory genes - Van den Bossche - 2012 - Journal of Leukocyte Biology - Wiley Online Library
https://jlb.onlinelibrary.wiley.com/doi/full/10.1189/jlb.0911453��
Front Immunol. 2014; 5: 544.
Evolutionary Roots of Arginase Expression and Regulation
Jolanta Maria Dzik1,*
1Department of Biochemistry, Faculty of Agriculture and Biology, Warsaw University of Life Sciences �C SGGW, Warszawa, Poland
Edited by: Charles Dudley Mills, BioMedical Consultants, USA
Reviewed by: Andrew Tasman Hutchinson, University of Technology Sydney, Australia; Dalil Hannani, APCure SAS, France
Abstract
Two main types of macrophage functions are known: classical (M1), producing nitric oxide, NO, and M2, in which arginase activity is primarily expressed. Ornithine, the product of arginase, is a substrate for synthesis of polyamines and collagen, important for growth and ontogeny of animals. M2 macrophages, expressing high level of mitochondrial arginase, have been implicated in promoting cell division and deposition of collagen during ontogeny and wound repair. Arginase expression is the default mode of tissue macrophages, but can also be amplified by signals, such as IL-4/13 or transforming growth factor-�� (TGF-��) that accelerates wound healing and tissue repair. In worms, the induction of collagen gene is coupled with induction of immune response genes, both depending on the same TGF-��-like pathway. This suggests that the main function of M2 ��heal�� type macrophages is originally connected with the TGF-�� superfamily of proteins, which are involved in regulation of tissue and organ differentiation in embryogenesis. Excretory�Csecretory products of metazoan parasites are able to induce M2-type of macrophage responses promoting wound healing without participation of Th2 cytokines IL-4/IL-13. The expression of arginase in lower animals can be induced by the presence of parasite antigens and TGF-�� signals leading to collagen synthesis. This also means that the main proteins, which, in primitive metazoans, are involved in regulation of tissue and organ differentiation in embryogenesis are produced by innate immunity. The signaling function of NO is known already from the sponge stage of animal evolution. The cytotoxic role of NO molecule appeared later, as documented in immunity of marine mollusks and some insects. This implies that the M2-wound healing promoting function predates the defensive role of NO, a characteristic of M1 macrophages. Understanding when and how the M1 and M2 activities came to be in animals is useful for understanding how macrophage immunity, and immune responses operate.
Keywords: arginase, evolution, hemocytes, M1/M2 macrophages, nitric oxide synthase, parasites, TGF-��, wound healingEvolutionary Roots of Arginase Expression and Regulation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4224125/
Human Gastric Epithelium Produces IL-4 and IL-4��2 Isoform Only upon Helicobacter Pylori Infection
B. Orsini, J.R. Vivas, B. Ottanelli, ...
Abstract
Recent evidence suggests that interleukin-4 (IL-4) is related to mucosal tolerance by which an injurious immune response is prevented, suppressed or shifted to a non-injurious response. We investigated the expression of IL-4 and its splice variant isoform IL-4��2 in gastric epithelial cells of healthy subjects and gastritis patients infected with Helicobacter pylori (H. pylori) with or without the cag pathogenicity island (cag-PAI). IL-4 and IL-4��2 mRNAs were evaluated in microdissected gastric epithelium and in AGS cell lines co-cultured with H. pylori B128 or SSI strains. IL-4 mRNA was consistently detected in microdissected gastric epithelial cells from healthy subjects. The IL-4 mRNA expression was low in H. pylori-infected patients, and markedly reduced in cag-PAI-positive ones. IL-4��2 mRNA was expressed on gastric epithelium of H. pylori-infected patients, but not in healthy subjects. The IL-452 expression was lower in cag-PAI-positive than in cag-PAI-negative H. pylori infected patients. AGS cells also produced IL-4 mRNA upon SSI strain stimulation, whereas IL-4��2 mRNA expression was detected in AGS co-cultured with either SSI or B128 strains. An inverse correlation was documented between IL-4 and IL-482 mRNA expression by microdissected gastric epithelial cells and the score of gastritis. IL-4, but not IL-452, is expressed by gastric epithelium of healthy subjects, whereas IL-452 and lesser IL-4 mRNA are detectable in the gastric epithelium of H. pylori-infected patients. Data suggest that gastric epithelial cells might regulate the balance between tolerance and immune response by the fine tuning of IL-4 and IL-4��2 expression.Human Gastric Epithelium Produces IL-4 and IL-4��2 Isoform Only upon Helicobacter Pylori Infection - B. Orsini, J.R. Vivas, B. Ottanelli, A. Amedei, E. Surrenti, A. Galli, S. Milani, P. Pinzani, G. Del Prete, C. Surrenti, C.T. Baldari, E. Touati, M.M. D'Elios, 2007
https://journals.sagepub.com/doi/abs/10.1177/039463200702000417��
Infect Immun. 2010 Jan; 78(1): 108�C114.
Helicobacter pylori Promotes the Production of Thymic Stromal Lymphopoietin by Gastric Epithelial Cells and Induces Dendritic Cell-Mediated Inflammatory Th2 Responses▿
Masahiro Kido,1,2 Junya Tanaka,1,2 Nobuhiro Aoki,1,2 Satoru Iwamoto,1,2 Hisayo Nishiura,1,2 Tsutomu Chiba,1 and Norihiko Watanabe1,2,*
Department of Gastroenterology and Hepatology,1 Center for Innovation in Immunoregulative Technology and Therapeutics, Graduate School of Medicine, Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan2
*Corresponding author. Mailing address: Department of Gastroenterology and Hepatology, Kyoto University,
ABSTRACT
Helicobacter pylori colonizes the stomach and induces strong, specific local and systemic humoral and cell-mediated immunity, resulting in the development of chronic gastritis in humans. Although H. pylori-induced chronic atrophic gastritis is characterized by marked infiltration of T helper type 1 (Th1) cytokine-producing CD4+ T cells, almost all of the inflamed gastric mucosae also contain focal lymphoid aggregates with germinal centers. In addition, typical H. pylori-induced chronic gastritis in children, called follicular gastritis, is characterized by B-cell follicle formation in the gastric mucosa. The aim of this study was to examine whether thymic stromal lymphopoietin (TSLP), an epithelial-cell-derived cytokine inducing a dendritic cell (DC)-mediated inflammatory Th2 response, is involved in Th2 responses triggering B-cell activation in H. pylori-induced gastritis. Here, we show that H. pylori triggered human gastric epithelial cells to produce TSLP, together with the DC-attracting chemokine MIP-3�� and the B-cell-activating factor BAFF. After DCs were incubated with supernatants from H. pylori-infected epithelial cells, the conditioned cells expressed high levels of costimulatory molecules, such as CD80, and triggered naïve CD4+ T cells to produce high levels of the Th2 cytokines interleukin-4 and interleukin-13 and of the inflammatory cytokines tumor necrosis factor alpha and gamma interferon.In contrast, after incubation of the supernatants with the neutralizing antibodies to TSLP, the conditioned DCs did not prime T cells to produce high levels of Th2 cytokines. These results, together with the finding that TSLP was expressed by the epithelial cells of human follicular gastritis, suggest that H. pylori can directly trigger epithelial cells to produce TSLP. It also suggests that TSLP-mediated DC activation may be involved in Th2 responses triggering B-cell activation in H. pylori-induced gastritis.
Helicobacter pylori Promotes the Production of Thymic Stromal Lymphopoietin by Gastric Epithelial Cells and Induces Dendritic Cell-Mediated Inflammatory Th2 Responses
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2798197/��
Pathog Dis. 2013 Feb;67(1):46-53. doi: 10.1111/2049-632X.12014. Epub 2013 Jan 22.
Dendritic cell function in the host response to Helicobacter pylori infection of the gastric mucosa.
Shiu J1, Blanchard TG.
Author information
1
Department of Pediatrics, University of Maryland School of Medicine, Baltimore, MD, USA.
Abstract
Dendritic cells (DCs) play an important role as antigen-presenting cells that direct the nature of the adaptive immune response. Subtypes are differentiated by lineage, tissue, marker expression and function. Their function in promoting regulatory T cells in the gut to maintain immunologic homeostasis is well documented, but their role in the Helicobacter pylori-infected stomach is less clear.Some analyses of bone marrow-derived DCs stimulated with H. pylori have demonstrated proinflammatory potential based on secretion of IL-12 or IL-23 or activation of Th1 and Th17 cells. Other analyses indicate that H. pylori-activated DCs are less responsive compared with other gastrointestinal bacteria and activate DCs to promote Treg development.
DC depletion in mice supports a role for DCs in down-regulating H. pylori-induced gastritis. These data indicate that gastric DCs recognize H. pylori much like DCs in the gut that recognize commensal organisms and promote a regulatory T-cell response. This is consistent with a growing body of literature documenting the prevalence and function of Treg cells in the host response to H. pylori. Research is now focused on characterizing how H. pylori induces such activity in DCs and identifying the mechanisms by which H. pylori-activated DCs activate Treg cells.
© 2012 Federation of European Microbiological Societies. Published by Blackwell Publishing Ltd. All rights reservedDendritic cell function in the host response to Helicobacter pylori infection of the gastric mucosa. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/23620119��
TOLEROGENIC ACTIVITIES OF H. PYLORI
Studies indicate that H. pylori-derived factors are capable to inhibit T-cell proliferation. Using normal T cells and Jurkat cells, a human T-cell line, it was demonstrated that VacA interfered with calcium-signaling events inside the cell and prevented activation of the calcium-dependent phosphatase calcineurin[75,76]. The subsequent dephosphorylation of nuclear factor of activated T cells (NFAT), a transcription factor that regulates immune responses, was suppressed resulting in inhibition of IL-2 expression and proliferation of T cells. Similar anti-proliferative effect on T cells was reported for the ��-glutamyl transpeptidase (��-GGT), this immunosuppressive factor inhibits T-cell proliferation by induction of a cell cycle arrest in the G(1) phase[77]. In addition to VacA and ��-GGT, H. pylori arginase can impair T-cell function during infection. Using Jurkat T cells and human normal lymphocytes, it was found that a wild type H. pylori strain, but not an arginase mutant strain, inhibited T-cell proliferation, depleted L-arginine, and reduced the expression of the CD3 chain of the T-cell receptor[78]. Most (80%-90%) H. pylori strains display Lewis blood-group antigens on their LPS, and these are similar to the Lewis blood-group antigens that are expressed on the mucosal surface of the human stomach[79]. Lewis positive H. pylori variants are able to bind to the C-type lectin DC-SIGN and present on gastric DCs, and demonstrate that this interaction blocks Th1 development[80].
In addition to suppress T cell activation, H. pylori has been demonstrated to decrease the functioning of the innate immune system. For instance, efficient phagocytosis and killing of H. pylori is prevented by the presence of the cag pathogenicity island[81,82] and H. pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity[83]. Importantly, at the opposite to the LPS and flagellins of others gram-negative bacteria, the LPS and flagellins of H. pylori do not adequately activate the antigen presenting cells via the Toll-like receptors[84,85].
Collectively, H. pylori counteract innate and T cell responses and clearly exhibited tolerogenic activities on the immune system. It can be suggested that these tolerogenic activities participate to the H. pylori persistence within the stomach mucosa.Immune responses to Helicobacter pylori infection
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4024767/��
World J Gastroenterol. 2014 May 21; 20(19): 5583�C5593.
Immune responses to Helicobacter pylori infection
Mati Moyat and Dominique Velin
Mati Moyat, Dominique Velin, Service of Gastroenterology and Hepatology, Department of Medicine, Lausanne University Hospital, CH-1011 Lausanne, Switzerland
Abstract
Helicobacter pylori (H. pylori) infection is one of the most common infections in human beings worldwide. H. pylori express lipopolysaccharides and flagellin that do not activate efficiently Toll-like receptors and express dedicated effectors, such as ��-glutamyl transpeptidase, vacuolating cytotoxin (vacA), arginase, that actively induce tolerogenic signals. In this perspective, H. pylori can be considered as a commensal bacteria belonging to the stomach microbiota. However, when present in the stomach, H. pylori reduce the overall diversity of the gastric microbiota and promote gastric inflammation by inducing Nod1-dependent pro-inflammatory program and by activating neutrophils through the production of a neutrophil activating protein. The maintenance of a chronic inflammation in the gastric mucosa and the direct action of virulence factors (vacA and cytotoxin-associated gene A) confer pro-carcinogenic activities to H. pylori. Hence, H. pylori cannot be considered as symbiotic bacteria but rather as part of the pathobiont. The development of a H. pylori vaccine will bring health benefits for individuals infected with antibiotic resistant H. pylori strains and population of underdeveloped countries.
Keywords: Helicobacter pylori, Vaccine, Immune response, Peptic ulcer, Gastric cancer
Core tip: Helicobacter pylori (H. pylori) infection is one of the most common infections in human beings worldwide. H. pylori actively induce tolerogenic signals and can be considered as a commensal bacteria belonging to the stomach microbiota. However, H. pylori also promote a chronic inflammation in the gastric mucosa and the direct action of virulence factors confers pro-carcinogenic activities to H. pylori. Hence, H. pylori cannot be considered as symbiotic bacteria but rather as part of the pathobiont. The development of a H. pylori vaccine will bring health benefits for individuals infected with antibiotic resistant H. pylori strains and population of underdeveloped countries.
INTRODUCTION
Helicobacter pylori (H. pylori) infection is one of the most common infections in human beings worldwide[1]. After entering the stomach, this spiral, Gram-negative, micro-aerophilic bacterium penetrates the mucus gastric layer[2] but does not traverse the epithelial barrier[3], and therefore it is considered as a non-invasive bacteria. Most of H. pylori organisms are free living in the mucus layer, but some organisms attach to the apical surface of gastric epithelial cells[3] and small numbers have been shown to invade epithelial cells[4]. Humans carry an estimated of 104 to 107 H. pylori CFU per gram of gastric mucus[5].
Upon infection, H. pylori uses urease and ��-carbonic anhydrase to generate ammonia and HCO32- which mitigate the effects of low pH[6,7]. Moreover, thanks to its flagella and shape, H. pylori penetrate the mucus layer. H. pylori null mutant defective in production of flagella are unable to colonize gnotobiotic piglets[8]. Once established in the inner mucus layer, several outer membrane proteins, including BabA, SabA, AlpA, AlpB and HopZ can mediate bacterial adherence to gastric epithelial cells. Once attached, bacterial effector molecules, both secreted [vacuolating cytotoxin (VacA) and cytotoxin- associated gene A (CagA)] or attached [components of the type IV secretion system (CagL)], modulate gastric epithelial cell behaviour leading to loss of cell polarity, release of nutrients and chemokines [e.g., interleukin (IL)-8], and regulation of acid secretion via control of gastrin and H+/ K+ ATPase[9,10].
The infections are acquired during childhood; frequent clonal transmission of H. pylori between first degree relatives demonstrates intra-familial transmission of H. pylori in developed countries. In developing world, members of the same family can be infected with widely diverse strains, and multiple infections were common arguing for horizontal transmission of H. pylori infection[11]. After ingestion, there is a period of intense bacterial proliferation and gastric inflammation. Concomitant with the intense gastritis is hypochlorhydria. Fecal shedding of H. pylori is maximal during this period, facilitating transmission to new hosts. Ultimately, the inflammatory response is reduced to a low-level stable state, normal gastric pH is restored, and most of the infected person becomes asymptomatic[12]. This outcome persists for years or decades and appears to predominate in the population. Depending on H. pylori virulence factors, environmental factors and the host response to bacterial infection, H. pylori infection can be associated with several clinical complications such as gastritis, peptic ulcer disease, gastric cancer and mucosa-associated lymphoid tissue (MALT) lymphoma[13-15]. H. pylori eradication therapies have revolutionised the natural course of peptic ulcer disease[13]. Antibiotic treatment of H. pylori infection is relatively successful, with the organism being eradicated from around 80% of patients[16].Immune responses to Helicobacter pylori infection
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4024767/��
Gastroenterology. 2010 Mar;138(3):1046-54. doi: 10.1053/j.gastro.2009.11.043. Epub 2009 Nov 18.
Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice.
Kao JY1, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J.
Author information
1
Department of Internal Medicine, Division of Gastroenterology, University of Michigan Health System, 6520A MSRB I, SPC 5682, 1150 West Medical Center Drive, Ann Arbor, MI 48109-5682, USA. jykao@umich.edu
Abstract
BACKGROUND & AIMS:
Helicobacter pylori infection increases gastric regulatory T cell (Treg) response, which may contribute to H pylori immune escape. We hypothesize that H pylori directs Treg skewing by way of dendritic cells (DCs) and thus inhibits interleukin-17(+) helper T cells (Th17) immunity.
METHODS:
Two-photon microscopy was used to locate DCs in gastric lamina propria of mice. The induction of Th17 and Treg responses by bacteria-pulsed murine bone marrow-derived DCs was analyzed by cytokine production and stimulation of T-cell proliferation. The effect of VacA, CagA, transforming growth factor-beta (TGF-beta), and IL-10 on Th17/Treg balance was assessed. The in vivo significance of Tregs on the H pylori-specific Th17 response and H pylori density was determined by using anti-CD25 neutralizing antibodies to deplete Tregs in mice.
RESULTS:
We showed that mucosal CD11c(+) DCs are located near the surface of normal gastric epithelium, and their number increased after H pylori infection. Study of the direct interaction of DCs with H pylori showed a Treg-skewed response. The Treg skewing was independent of H pylori VacA and CagA and dependent on TGF-beta and IL-10. In vivo Treg skewing by adoptive transfer of H pylori-pulsed DCs reduces the ratio of gastric IL-17/Foxp3 mRNA expressions. The depletion of CD25(+) Tregs results in early reduction of H pylori density, which is correlated with enhanced peripheral H pylori-specific Th17, but not Th1, response.
CONCLUSIONS:
Overall, our study indicates that H pylori alters the DC-polarized Th17/Treg balance toward a Treg-biased response, which suppresses the effective induction of H pylori-specific Th17 immunity.
Copyright 2010 AGA Institute. Published by Elsevier Inc. All rights reserved.
PMID: 19931266 PMCID: PMC2831148 DOI: 10.1053/j.gastro.2009.11.043Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/19931266��
Th17 Cells Require You to Chew before You Swallow: Immunity
Jan 17, 2017 �� Th17 Cells Require You to Chew before You Swallow How immunity is regulated at distinct epithelial tissues that vary in microbial occupancy and environmental and tissue specific cues isn��t clear. Dutzan et al. (2017)
��
��
��
Th17 Cells in Immunity and Autoimmunity
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3886602Dec 26, 2013 �� Th17 cells have earned a well-deserved reputation as key modulators in autoimmunity; however, the Th17 lineage has pleiotropic effects on a variety of cell types including epithelial, endothelial, and fibroblastic cells . The Th17 lineage provides a unique mechanism for protection against bacterial and fungal pathogens through production and induction of inflammatory cytokines and other ��
��
��
The Immunomodulator VacA Promotes Immune Tolerance and Persistent Helicobacter pylori Infection through Its Activities on T-Cells and Antigen-Presenting Cells
by Aleksandra Djekic and Anne M��ller *
Institute of Molecular Cancer Research, University of Z��rich, Winterthurerstr. 190, 8057 Z��rich, Switzerland
Abstract
VacA is a pore-forming toxin that has long been known to induce vacuolization in gastric epithelial cells and to be linked to gastric disorders caused by H. pylori infection. Its role as a major colonization and persistence determinant of H. pylori is less well-understood. The purpose of this review is to discuss the various target cell types of VacA and its mechanism of action; specifically, we focus on the evidence showing that VacA targets myeloid cells and T-cells to directly and indirectly prevent H. pylori-specific T-cell responses and immune control of the infection. In particular, the ability of VacA-proficient H. pylori to skew T-cell responses towards regulatory T-cells and the effects of Tregs on H. pylori chronicity are highlighted.The by-stander effects of VacA-driven immunomodulation on extragastric diseases are discussed as well.
Keywords: persistent infection; myeloid cells; professional antigen-presenting cells; T-cell priming; effector T-cells; regulatory T-cells; immune tolerance
Figure 1. VacA prevents phagosome maturation in professional antigen-presenting cells. Most strains of H. pylori express the secreted virulence factors VacA to affect phagosome maturation and lysosome fusion. The receptor for VacA on myeloid cells is not known. It has been proposed that H. pylori-containing phagosomes fail to mature and instead resemble early endosomes that have resisted lysosome fusion; this process appears to be specific to type I strains and is dependent on VacA expression [12]. Treatment with recombinant IFN-�� allows phagocytes to overcome their block in phagosome maturation and to kill intracellular type I strains of H. pylori.
Figure 2. H. pylori exerts immunomodulatory effects at extragastric sites. H. pylori exclusively inhabits the gastric mucosa but has systemic immunomodulatory effects that manifest in the airways, lower gastrointestinal tract, and possibly the skin. Gastric tissue-resident DCs presumably sample H. pylori antigens locally at the site of infection and subsequently migrate to the stomach-draining and mesenteric lymph nodes (MLNs), where they prime T-effector and regulatory T-cell responses. VacA is believed to skew this process towards Treg differentiation [2]. MLN-derived, H. pylori-induced Tregs enter the circulation and, due to cross-talk between the GI tract and lung immune systems, may accumulate in both the gastric mucosa and the lung. According to current models, allergen-specific Th2 cells in the lung are suppressed by H. pylori-induced Tregs via soluble mediators (such as IL-10) and contact-dependent mechanisms (reviewed in [43]). Tregs further suppress Th1 and Th17 responses directed against H. pylori in the gastric mucosa [30,44]. DC: dendritic cell; Th1/2/17: T-helper cell subsets; Treg: regulatory T-cell. Please note that, due to space restrictions, the MLN shown here is not in its correct anatomical location, which would be in the mesentery, linking the intestines to the abdominal wall.
5. Conclusions
Despite being better known for its vacuolating and cytotoxic activity, VacA has also recently received considerable attention for its immunomodulatory properties. A range of effects on several types of immune cells has been attributed to VacA in vitro and in vivo; the protein��s ability to insert into lipid bilayers to form anion-selective channels appears to be a prerequisite of both its cytotoxic and immunomodulatory activity. Whether and how VacA targets immune cells in the gastric mucosa remains to be determined; an in vivo tracking system using GFP- or otherwise tagged VacA would be a highly useful tool in this respect. It is conceivable that VacA secreted from bacteria residing in the gastric mucus, or in close proximity to gastric epithelial cells, is readily sampled by intraepithelial DCs and macrophages that extend protrusions into the gastric lumen. It is less straightforward to envision VacA targeting T-cells, which reside exclusively in the lamina propria. A direct interaction of VacA with T-cells would require disruption of epithelial tight junctions and the subsequent loss of epithelial barrier function, processes that are known to occur during H. pylori infection of polarized monolayers [45,46]. More evidence is needed from both naturally infected humans hosts as well as experimental animals to better understand the immunological consequences of VacA-positive H. pylori infection. In particular, more work is needed to clarify the differential cytotoxic and immunomodulatory activity of s1i1m1 VacA relative to its less toxigenic counterparts encoded by other alleles. Recent work on H. pylori strains in which the endogenous allele was exchanged for one of two alternative, naturally occuring alleles [3] to generate isogenic strains, differing only in the type of VacA they produce, represents a step in the right direction and model for future work. The identification of a (putative) VacA receptor on immune cells other than T-cells is also of great importance, as dendritic cells and macrophages likely represent the first line of defence (and first target cell of VacA) encountered by H. pylori. A better understanding of VacA��s target cells and mechanism of action will facilitate its targeting and exploitation for new treatment and prevention strategies.Toxins | Free Full-Text | The Immunomodulator VacA Promotes Immune Tolerance and Persistent Helicobacter pylori Infection through Its Activities on T-Cells and Antigen-Presenting Cells | HTML
https://www.mdpi.com/2072-6651/8/6/187/html��
��
Cell Microbiol. 2013 Jan; 15(1): 145�C156.
Published online 2012 Nov 7. doi: 10.1111/cmi.12039
Heme oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells
Alain P. Gobert,1,4,5 Thomas Verriere,1 Thibaut de Sablet,1,4 Richard M. Peek, Jr.,1,2,4 Rupesh Chaturvedi,1,4 and Keith T. Wilson1,2,3,4,*1Division of Gastroenterology, Department of Medicine, Vanderbilt University, Nashville, TN, USA
2Department of Cancer Biology, Vanderbilt University, Nashville, TN, USA
3Department of Pathology, Microbiology and Immunology, Vanderbilt University, Nashville, TN, USA
4Veterans Affairs TN Valley Healthcare System, Nashville, TN, USA
5 Institut National de la Recherche Agronomique, UR454 Unit�� de Microbiologie, Saint-Gen��s-Champanelle, France
Summary
The cytotoxin-associated gene A protein (CagA) plays a pivotal role in the etiology of Helicobacter (H.) pylori-associated gastric diseases. CagA is injected into the cytoplasm of host cells by a type IV secretion system, and is phosphorylated on tyrosine residues by the host enzyme c-Src.We previously reported that the enzyme heme oxygenase-1 (HO-1) inhibits IL-8 secretion by H. pylori-infected cells. However, the cellular mechanism by which HO-1 regulates the innate immune function of infected cells remains unknown. We now show that nitric oxide and hemin, two inducers of HO-1, decrease the level of phosphorylated CagA (p-CagA) in H. pylori-infected gastric epithelial cells and this is blocked by either pharmacologic inhibition of HO-1 or siRNA knockdown of hmox-1. Moreover, forced expression of HO-1 by transfection of a plasmid expressing hmox-1 also results in a strong attenuation of CagA phosphorylation.
This occurs through the inhibition of H. pylori-induced c-Src phosphorylation/activation by HO-1. Consequently, H. pylori-induced cytoskeletal rearrangements and activation of the pro-inflammatory response mediated by p-CagA are inhibited in HO-1-expressing cells.
These data highlight a mechanism by which the innate immune response of the host can restrict the pathogenicity of H. pylori by attenuating CagA phosphorylation in gastric epithelial cells.
��
HO-1 suppresses c-Src activation
HO-1 downregulates H. pylori-induced NF-��B, p38, and ERK1/2 signaling in gastric epithelial cells
Discussion
The subversion of gastric epithelial cell signaling by the H. pylori virulence factor CagA, which is phosphorylated by the host enzyme c-Src, is a striking example of a co-evolution of a pathogenic bacterium with its host (Atherton and Blaser, 2009). Although the complexity of this interaction in the context of a long-term infection remains not well understood, human epidemiological studies and animal models have demonstrated a correlation between the presence of CagA and the level of gastritis and the development of gastric cancer (Blaser et al., 1995; Parsonnet et al., 1997; Ogura et al., 2000). Therefore, strategies to counteract the phosphorylation of CagA might be useful to prevent H. pylori-associated diseases and malignancies. We demonstrated in this work that the inducible enzyme HO-1 inhibits the activation of c-Src in gastric epithelial cells and therefore decreases CagA phosphorylation. Consequently, p-CagA-dependent signal transduction, involving p38, ERK1/2 and NF-��B, was decreased in cells expressing HO-1. Therefore, our work highlights an unrecognized role for HO-1: the inhibition of the post-translational activation of a virulence factor from a pathogenic bacterium. Interestingly, we have found that NO directly blocks the production of the virulence factor Shiga-toxin by enterohemorrhagic Escherichia coli (Vareille et al., 2007). Herein, we report that NO also causes the attenuation of the virulence of another pathogen, H. pylori, but indirectly through the induction of HO-1.Heme oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3524338/��
Helicobacter. 2003 Jun;8(3):235-43.
CagA tyrosine phosphorylation in gastric epithelial cells caused by Helicobacter pylori in patients with gastric adenocarcinoma.
Lai YP1, Yang JC, Lin TZ, Wang JT, Lin JT.
Author information
Abstract
BACKGROUND:
Tyrosine phosphorylation of Helicobacter pylori cytotoxin-associated protein of in gastric epithelial cells is reported. The goals of this study are first to examine the occurrence of CagA tyrosine phosphorylation in H. pylori strains isolated from patients with gastric adenocarcinoma and gastritis, and second to clarify the relationship between the diversity of tyrosine phosphorylation motifs and the presence of CagA tyrosine phosphorylation.
METHODS:
Fifty-eight clinical isolates of H. pylori from patients with gastric adenocarcinoma (29 cases) and gastritis (29 cases) were studied for CagA tyrosine phosphorylation by Western blotting. Sequence diversity of tyrosine phosphorylation motifs was analysed among positive- or negative-CagA tyrosine phosphorylation isolates.
RESULTS:
Positive CagA tyrosine phosphorylation was found in 93.1% (27 of 29) of strains from gastric adenocarcinoma patients and 51.7% (15 of 29) of strains from gastritis patients (p < 0.001). Intact motifs were found in H. pylori isolates with CagA tyrosine phosphorylation. Of the 16 negative CagA tyrosine phosphorylation isolates, intact tyrosine phosphorylation motifs were found in 15 isolates.
CONCLUSIONS:
CagA tyrosine phosphorylation, which is significantly greater in strains from gastric adenocarcinoma patients, may play a role in gastric carcinogenesis, and could be a better marker of more virulent strains than the cag pathogenicity island in Asia, where the cag pathogenicity island is present in nearly all H. pylori strains. Sequence diversity of tyrosine phosphorylation motifs on CagA was not related to the presence of tyrosine phosphorylation. The absence of tyrosine phosphorylation motif might result in negative tyrosine phosphorylation phenotypes, but such motifs are not the sole factors associated with CagA tyrosine phosphorylation.CagA tyrosine phosphorylation in gastric epithelial cells caused by Helicobacter pylori in patients with gastric adenocarcinoma. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/12752736
Nitric Oxide Induces Heme Oxygenase-1 Gene Expression and Carbon Monoxide Production in Vascular Smooth Muscle Cells
William Durante , Michael H. Kroll , Nick Christodoulides , Kelly J. Peyton , and Andrew I. Schafer
Houston VA Medical Center (W.D., M.H.K., N.C., K.J.P., A.I.S.) and the Departments of Medicine (W.D., M.H.K., N.C., A.I.S.) and Pharmacology (W.D.), Baylor College of Medicine, Houston, Tex.
Abstract
Since recent studies demonstrate that vascular smooth muscle cells synthesize two distinct guanylate cyclase�Cstimulatory gases, NO and CO, we examined possible regulatory interactions between these two signaling molecules. Treatment of rat aortic smooth muscle cells with the NO donors, sodium nitroprusside, S-nitroso-N-acetyl-penicillamine, or 3-morpholinosydnonimine, increased heme oxygenase-1 (HO-1) mRNA and protein levels in a concentration- and time-dependent manner.Both actinomycin D and cycloheximide blocked NO-stimulated HO-1 mRNA and protein expression. Nuclear run-on experiments demonstrated that NO donors increased HO-1 gene transcription between 3- and 6-fold. In contrast, NO donors had no effect on the stability of HO-1 mRNA.
Incubation of vascular smooth muscle cells with the membrane-permeable cGMP analogues, dibutyryl cGMP and 8-bromo-cGMP, failed to induce HO-1 gene expression. Treatment of vascular smooth muscle cells with NO donors also stimulated the production and release of CO, as demonstrated by the CO-dependent increase in intracellular cGMP levels in coincubated platelets.
Finally, incubating vascular smooth muscle cells with interleukin-1�� and tumor necrosis factor-�� induced NO synthesis and also significantly increased the level of HO-1 protein.
The cytokine-stimulated production of both NO and HO-1 protein in smooth muscle cells was blocked by the NO synthase inhibitor methyl-l-arginine.
These results demonstrate that exogenously administered or endogenously released NO stimulates HO-1 gene expression and CO production in vascular smooth muscle cells. The ability of NO to induce HO-catalyzed CO release from vascular smooth muscle cells provides a novel mechanism by which NO might modulate soluble guanylate cyclase and, thereby, vascular smooth muscle cell and platelet function.
Nitric Oxide Induces Heme Oxygenase-1 Gene Expression and Carbon Monoxide Production in Vascular Smooth Muscle Cells | Circulation Research
https://www.ahajournals.org/doi/full/10.1161/01.RES.80.4.557��
PLoS One. 2014; 9(6): e99134.
Pharmacological Preconditioning with Vitamin C Attenuates Intestinal Injury via the Induction of Heme Oxygenase-1 after Hemorrhagic Shock in Rats
Bing Zhao,# 1 Jian Fei,# 2 Ying Chen, 1 Yi-Lin Ying, 1 Li Ma, 3 Xiao-Qin Song, 1 Lu Wang, 1 Er-Zhen Chen, 1 , * and En-Qiang Mao 1 , *
Prasun K. Datta, Editor
1 Department of Emergency Intensive Care Unit, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China,
2 Department of Surgery, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China,
3 Department of Emergency Intensive Care Unit, the Third People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China,
Temple University, United States of America,
Abstract
Pre-induction of heme oxygenase (HO)-1, which is regarded as an effective method of ��organ preconditioning��, exerts beneficial effects during hemorrhagic shock (HS). However, the available HO-1 inducers exhibit disadvantages such as toxicity or complex technical requirements. Therefore, a safe and convenient HO-1 inducer would be promising and could be exploited in the treatment of foreseeable hemorrhaging, such as prior to major surgery.Here we investigated the effect of vitamin C (VitC), a common antioxidant, on intestinal HO-1 expression and examined whether VitC pretreatment prevented HS related intestinal tissue injuries after HO-1 induction.
First, we conducted an in vitro study and found that HO-1 expression in rat intestinal epithelial cells (IEC-6) was induced by non-toxic VitC in a time and concentration dependent manner, and the mechanism was related to the activation of extracellular signal-regulated kinase 1/2 (ERK1/2).
Next, we conducted an in vivo study and found that VitC induced intestinal HO-1 protein expression (mainly observed in the intestinal epithelial cells) and HO-1 activity in normal SD rats, and that these HO-1 levels were further enhanced by VitC in a rat model of HS.
The HS related intestinal injuries, including histological damage, pro-inflammatory cytokine levels (tumor necrosis factor and interleukin-6), neutrophil infiltration and apoptosis decreased after VitC pretreatment, and this alleviating of organ injuries was abrogated after the inhibition of HO-1 activity by zinc protoporphyrin-IX. It was of note that VitC did little histological damage to the intestine of the sham rats. These data suggested that VitC might be applied as a safe inducer of intestinal HO-1 and that VitC pretreatment attenuated HS related intestinal injuries via the induction of HO-1.
Pharmacological Preconditioning with Vitamin C Attenuates Intestinal Injury via the Induction of Heme Oxygenase-1 after Hemorrhagic Shock in Rats
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4057195/��
L-ascorbate attenuates methamphetamine neurotoxicity ...
https://www.ncbi.nlm.nih.gov/pubmed/23022510Dec 01, 2012 �� Pharmacological inhibition of HO activity abolished suppressive effects of Vit. C on METH-induced ROS production and attenuated neurotoxicity. We conclude that induction of HO-1expression contributes to the attenuation of METH-induced ROS production and neurotoxicity by Vit. C. We suggest that HO-1 induction by Vit.
��
��
J Clin Biochem Nutr. 2009 Jul; 45(1): 9�C13.
The Expression of Heme Oxygenase-1 Induced by Lansoprazole
Tomohisa Takagi, Yuji Naito,* and Toshikazu Yoshikawa
Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan
Abstract
Our previous studies have demonstrated that lansoprazole inhibits acute inflammatory reactions as well as intestinal mucosal injuries induced by ischemia-reperfusion or indomethacin administration in rats. Thus, proton pump inhibitors such as lansoprazole have been demonstrated to prevent gastrointestinal mucosal injury by mechanisms independent of acid inhibition.In our in vitro study, lansoprazole induced the expression of heme oxygenase-1 (HO-1) on rat gastric epithelial cells (RGM-1 cells), and exerted anti-inflammatory effect on the dependent of HO-1 expression. Furthermore, NF-E2-related factor-2 (Nrf2) played an important role in HO-1 expression induced by lansoprazole.
In this review, we focused on lansoprazole-induced HO-1 expression, its anti-inflammatory action, and the role of Nrf2 in its expression.
Keywords: Lansoprazole, Heme Oxygenase-1 (HO-1), NF-E2-related factor-2 (Nrf2)
The Expression of Heme Oxygenase-1 Induced by Lansoprazole
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2704331/��
J Biol Chem. 2011 Nov 11;286(45):39387-402. doi: 10.1074/jbc.M111.279893. Epub 2011 Sep 9.
Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury.
Bindu S1, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, Bandyopadhyay U.
1 Department of Infectious Diseases and Immunology, Indian Institute of Chemical Biology, 4 Raja S. C. Mullick Road, Jadavpur, Kolkata 700032, West Bengal, India.
Erratum in
Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. [J Biol Chem. 2015]
Abstract
The mechanism of action of heme oxygenase-1 (HO-1) in mitochondrial oxidative stress (MOS)-mediated apoptotic tissue injury was investigated. MOS-mediated gastric mucosal apoptosis and injury were introduced in rat by indomethacin, a non-steroidal anti-inflammatory drug. Here, we report that HO-1 was not only induced but also translocated to mitochondria during gastric mucosal injury to favor repair mechanisms. Furthermore, mitochondrial translocation of HO-1 resulted in the prevention of MOS and mitochondrial pathology as evident from the restoration of the complex I-driven mitochondrial respiratory control ratio and transmembrane potential. Mitochondrial translocation of HO-1 also resulted in time-dependent inhibition of apoptosis. We searched for the plausible mechanisms responsible for HO-1 induction and mitochondrial localization. Free heme, the substrate for HO-1, was increased inside mitochondria during gastric injury, and mitochondrial entry of HO-1 decreased intramitochondrial free heme content, suggesting that a purpose of mitochondrial translocation of HO-1 is to detoxify accumulated heme. Heme may activate nuclear translocation of NF-E2-related factor 2 to induce HO-1 through reactive oxygen species generation. Electrophoretic mobility shift assay and chromatin immunoprecipitation studies indicated nuclear translocation of NF-E2-related factor 2 and its binding to HO-1 promoter to induce HO-1 expression during gastric injury. Inhibition of HO-1 by zinc protoporphyrin aggravated the mucosal injury and delayed healing. Zinc protoporphyrin further reduced the respiratory control ratio and transmembrane potential and enhanced MOS and apoptosis. In contrast, induction of HO-1 by cobalt protoporphyrin reduced MOS, corrected mitochondrial dysfunctions, and prevented apoptosis and gastric injury. Thus, induction and mitochondrial localization of HO-1 are a novel cytoprotective mechanism against MOS-mediated apoptotic tissue injury.Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochon... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/21908612��
Immune Cells May Induce Gastritis During H. Pylori Infection
Date: May 27, 2008
Source: American Society for Microbiology
Summary:
Researchers have examined the inflammatory response induced by macrophages that may contribute to the development of gastritis during Helicobacter pylori infection in mice. H. pylori is the causative agent of human chronic gastritis, a condition that often leads to gastrointestinal ulcers and cancer.
FULL STORY
For the first time researchers from Australia and The Netherlands examine the inflammatory response induced by macrophages that may contribute to the development of gastritis during Helicobacter pylori infection in mice.
H. pylori is the causative agent of human chronic gastritis, a condition that often leads to gastrointestinal ulcers and cancer. The gastric mucus found in the human stomach of a H. pylori infection-free adult or child is populated by very few macrophages, however previous research shows that macrophages increase in number in response to H. pylori infection. The mouse model of Helicobacter infection mimics human H. pylori disease in many ways and offers researchers an opportunity to analyze the adaptive immune regulation of Helicobacter-induced chronic gastritis.
Macrophages are defined as white blood cells that are key players in the immune response to foreign invaders. In the study mice were intravenously injected with drug-loaded liposomes to deliberately deplete their spleens and stomachs of macrophages and then infected with H. pylori. Results showed that elimination of macrophages reduced the gastric pathology induced by H. pylori, however it did not affect bacterial presence in the stomach.
"This study demonstrates that macrophages have a central role in Helicobacter infection-induced gastritis but do not affect H. pylori-specific antibody responses," say the researchers. "In identifying a role for macrophages in the initiation of gastritis during H. pylori infection, this study may assist in future studies targeting the inhibition of gastritis in the host and provide a stimulus to study the capacity of macrophage-modifying drugs to reduce the gastritis associated with Helicobacter disease.Immune Cells May Induce Gastritis During H. Pylori Infection -- ScienceDaily
https://www.sciencedaily.com/releases/2008/05/080523071035.htm��
cagA + strains of H. pylori activate the transcription factor NF-��B in gastric epithelial cells (15), which results in the upregulated expression of several target genes, including interleukin-8 (IL-8), cyclooxygenase 2, and nitric oxide synthase 2, whose products (e.g., IL-8, prostaglandins, and nitric oxide, respectively) are important in host innate antimicrobial defense (9, 13, 16). However, those mediators are not effective in clearing H. pylori. Thus, infection persists despite an inflammatory host response characterized by an influx of polymorphonuclear leukocytes that are chemoattracted by IL-8 and other chemokines and an influx of T cells that produce gamma interferon (IFN-��) and other Th1 cytokines (reviewed in reference3).
Regulation of Human ��-Defensins by Gastric Epithelial Cells in Response to Infection with Helicobacter pylori or Stimulation with Interleukin-1 | Infection and Immunity https://iai.asm.org/content/68/9/5412
��
J Clin Invest. 2016 Sep 1;126(9):3296-312. doi: 10.1172/JCI83585. Epub 2016 Aug 2.
EGFR regulates macrophage activation and function in bacterial infection.
Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villag��mez D, Barry DP, Allaman MM, Washington MK, Peek RM Jr, Piazuelo MB, Wilson KT.
Abstract
EGFR signaling regulates macrophage function, but its role in bacterial infection has not been investigated. Here, we assessed the role of macrophage EGFR signaling during infection with Helicobacter pylori, a bacterial pathogen that causes persistent inflammation and gastric cancer. EGFR was phosphorylated in murine and human macrophages during H. pylori infection. In human gastric tissues, elevated levels of phosphorylated EGFR were observed throughout the histologic cascade from gastritis to carcinoma. Deleting Egfr in myeloid cells attenuated gastritis and increased H. pylori burden in infected mice. EGFR deficiency also led to a global defect in macrophage activation that was associated with decreased cytokine, chemokine, and NO production. We observed similar alterations in macrophage activation and disease phenotype in the Citrobacter rodentium model of murine infectious colitis. Mechanistically, EGFR signaling activated NF-��B and MAPK1/3 pathways to induce cytokine production and macrophage activation. Although deletion of Egfr had no effect on DC function, EGFR-deficient macrophages displayed impaired Th1 and Th17 adaptive immune responses to H. pylori, which contributed to decreased chronic inflammation in infected mice. Together, these results indicate that EGFR signaling is central to macrophage function in response to enteric bacterial pathogens and is a potential therapeutic target for infection-induced inflammation and associated carcinogenesis.
EGFR regulates macrophage activation and function in bacterial infection. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/27482886��
Gastroenterology. 2014 Jun;146(7):1739-51.e14. doi: 10.1053/j.gastro.2014.02.005. Epub 2014 Feb 13.
Activation of EGFR and ERBB2 by Helicobacter pylori results in survival of gastric epithelial cells with DNA damage.
Chaturvedi R1, Asim M2, Piazuelo MB1, Yan F3, Barry DP2, Sierra JC1, Delgado AG1, Hill S4, Casero RA Jr5, Bravo LE6, Dominguez RL7, Correa P1, Polk DB8, Washington MK9, Rose KL4, Schey KL10, Morgan DR1, Peek RM Jr11, Wilson KT12.
Author information
1 Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee.
2 Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Veterans Affairs Tennessee Valley Healthcare System, Nashville, Tennessee.
3 Division of Gastroenterology, Department of Pediatrics, Vanderbilt University Medical Center, Nashville, Tennessee.
4 Mass Spectrometry Research Center, Vanderbilt University Medical Center, Nashville, Tennessee.
5 Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, Maryland.
6 Department of Pathology, Universidad del Valle School of Medicine, Cali, Colombia.
7 Hospital de Occidente, Santa Rosa de Copan, Copan, Honduras.
8 Division of Gastroenterology, Department of Pediatrics, Vanderbilt University Medical Center, Nashville, Tennessee; Department of Pediatrics, University of Southern California Keck School of Medicine, Los Angeles, California.
9 Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee.
10 Mass Spectrometry Research Center, Vanderbilt University Medical Center, Nashville, Tennessee; Department of Biochemistry, Vanderbilt University Medical Center, Nashville, Tennessee.
11
Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Veterans Affairs Tennessee Valley Healthcare System, Nashville, Tennessee; Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, Tennessee.
12 Division of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Veterans Affairs Tennessee Valley Healthcare System, Nashville, Tennessee; Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee; Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, Tennessee. Electronic address: keith.wilson@vanderbilt.edu.
Abstract
BACKGROUND & AIMS:
The gastric cancer-causing pathogen Helicobacter pylori up-regulates spermine oxidase (SMOX) in gastric epithelial cells, causing oxidative stress-induced apoptosis and DNA damage. A subpopulation of SMOX(high) cells are resistant to apoptosis, despite their high levels of DNA damage. Because epidermal growth factor receptor (EGFR) activation can regulate apoptosis, we determined its role in SMOX-mediated effects.
METHODS:
SMOX, apoptosis, and DNA damage were measured in gastric epithelial cells from H. pylori-infected Egfr(wa5) mice (which have attenuated EGFR activity), Egfr wild-type mice, or in infected cells incubated with EGFR inhibitors or deficient in EGFR. A phosphoproteomic analysis was performed. Two independent tissue microarrays containing each stage of disease, from gastritis to carcinoma, and gastric biopsy specimens from Colombian and Honduran cohorts were analyzed by immunohistochemistry.
RESULTS:
SMOX expression and DNA damage were decreased, and apoptosis increased in H. pylori-infected Egfr(wa5) mice. H. pylori-infected cells with deletion or inhibition of EGFR had reduced levels of SMOX, DNA damage, and DNA damage(high) apoptosis(low) cells. Phosphoproteomic analysis showed increased EGFR and erythroblastic leukemia-associated viral oncogene B (ERBB)2 signaling. Immunoblot analysis showed the presence of a phosphorylated (p)EGFR-ERBB2 heterodimer and pERBB2; knockdown of ErbB2 facilitated apoptosis of DNA damage(high) apoptosis(low) cells. SMOX was increased in all stages of gastric disease, peaking in tissues with intestinal metaplasia, whereas pEGFR, pEGFR-ERBB2, and pERBB2 were increased predominantly in tissues showing gastritis or atrophic gastritis. Principal component analysis separated gastritis tissues from patients with cancer vs those without cancer. pEGFR, pEGFR-ERBB2, pERBB2, and SMOX were increased in gastric samples from patients whose disease progressed to intestinal metaplasia or dysplasia, compared with patients whose disease did not progress.
CONCLUSIONS:
In an analysis of gastric tissues from mice and patients, we identified a molecular signature (based on levels of pEGFR, pERBB2, and SMOX) for the initiation of gastric carcinogenesis.
Copyright © 2014 AGA Institute. Published by Elsevier Inc. All rights reserved.
KEYWORDS:
Marker; Prognostic Factor; Risk; Signal Transduction Pathways��
Helicobacter pylori CagA Phosphorylation-Independent Function in Epithelial Proliferation and Inflammation
CagA, a major virulence factor of Helicobacter pylori (Hp), is delivered into gastric epithelial cells and exists in phosphorylated and nonphosphorylated forms. The biological activity of the phosphorylated form is well established; however, function(s) of the nonphosphorylated form remain elusive. Here, we report that a conserved motif in the C-terminal region of CagA, which is distinct from the EPIYA motifs used for phosphorylation and which we designate CRPIA (conserved repeat responsible for phosphorylation-independent activity), plays pivotal roles in Hp pathogenesis. The CRPIA motif in nonphosphorylated CagA was involved in interacting with activated Met, the hepatocyte growth factor receptor, leading to the sustained activation of phosphatidylinositol 3-kinase/Akt signaling in response to Hp infection. This in turn led to the activation of ��-catenin and NF-��B signaling, which promote proliferation and inflammation, respectively. Thus, nonphosphorylated CagA activity contributes to the epithelial proliferative and proinflammatory responses associated with development of chronic gastritis and gastric cancer.Helicobacter pylori CagA Phosphorylation-Independent Function in Epithelial Proliferation and Inflammation: Cell Host & Microbe
https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(08)00402-2��
Free heme toxicity and its detoxification systems in human ...
https://www.sciencedirect.com/science/article/pii/S0378427405000883Hx, by transporting heme inside the cells reduces the level of free heme in serum and at the same time provides heme for the maturation of hemoprotein. Heme bound in Hx is taken up by human leukemia HL60 cells, as mediated by Hx-receptors and that the internalized heme used for maturation of myeloperoxidase (Taketani et al., 1987). Toxic effects of hemin are found to be inhibited by Hx.
A Central Role for Free Heme in the Pathogenesis of Severe ...
Sep 29, 2010 �� Once the concentration of HPT and/or HPX in serum decreases below a certain threshold level, free heme accumulates in plasma and can sensitize cells in parenchymal tissues to undergo programmed cell death in response to a variety of proinflammatory agonists.
Hemolysis and free hemoglobin revisited: exploring ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3578950Feb 21, 2013 �� Hemin release allows for transfer of the reactive porphyrin to cell membranes or soluble plasma proteins and lipids and provides free hemin as a ligand for molecular signaling interactions. As a hydrophobic molecule, it is unlikely that significant quantities of free, monomeric hemin can be ��
Dysfunction of the heme recycling system in heme oxygenase ...
Dec 23, 2010 �� Hemopexin (Hpx) is a heme-binding serum protein that scavenges free heme in the circulation, 6,7 and haptoglobin (Hp) binds free Hb, 8,9 whereupon the Hb-Hp complex is endocytosed through the CD163 receptor 10 and is metabolized by macrophages.
Evaluation of Unbound Free Heme in Plant Cells by ...
See more on academic.oup.com
high sensitivity for heme was observed under conditions in which the defined medium was free of serumand where a chelator of iron was added to the medium to diminish the synthesis of endogenous heme. Heme endogenously generated from exogenous &aminolevulinic acid also inhibited the induction;
Heme-based catalytic properties of human serum albumin
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4991842Under physiological and pathological conditions, HSA has a pivotal role in heme scavenging. 3,4 In fact, although heme regulates gene expression and is the prosthetic group of heme proteins under physiological conditions, high levels of free heme: (i) catalyze the synthesis of toxic free hydroxyl radicals, (ii) affect the integrity of erythrocyte membranes, (iii) induce the enrollment to the vascular endothelium of red blood cells��
��
��
Sci Rep. 2015; 5: 10336.
Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways
Wenjin Hao,1,* Xuan Yuan,2,* Lina Yu,1 Caixia Gao,1 Xiling Sun,1 Dong Wang,b,3 and Qiusheng Zhenga,1,4
1Binzhou medical University, Yantai, 264003, Shandong, China
2The Affiliated Hospital of Qingdao University, Qingdao, 266003, Shandong, China
3Qianfoshan Hospital of Shandong University, Jinan, 250014, China
4Key Laboratory of Xinjiang Endemic Phytomedicine Resources, Ministry of Education, School of Pharmacy, Shihezi University, Shihezi, 832002, Xinjiang, China
Abstract
Both phosphatidylinositol 3-kinase (PI3K)/AKT and mitogen activated protein kinase (MAPK) signaling cascades play an important role in cell proliferation, survival, angiogenesis, and metastasis of tumor cells. In the present report, we investigated the effects of licochalcone A (LA), a flavonoid extracted from licorice root, on the PI3K/AKT/mTOR and MAPK activation pathways in human gastric cancer BGC-823 cells. LA increased reactive oxygen species (ROS) levels, which is associated with the induction of apoptosis as characterized by positive Annexin V binding and activation of caspase-3, and cleavage of poly-ADP-ribose polymerase (PARP). Inhibition of ROS generation by N-acetylcysteine (NAC) significantly prevented LA-induced apoptosis. Interestingly, we also observed that LA caused the activation of ERK, JNK, and p38 MAPK in BGC-823 cells. The antitumour activity of LA-treated BGC-823 cells was significantly distinct in KM mice in vivo. All the findings from our study suggest that LA can interfere with MAPK signaling cascades, initiate ROS generation, induce oxidative stress and consequently cause BGC cell apoptosis.Licochalcone A (����ͪA)ͨ������ROS�鵼��MAPKs��PI3K/AKT�ź�ͨ·�յ���θ��BGC-823ϸ������
��֬������3-��ø(PI3K)/AKT��˿��ԭ�����ø(MAPK)�źż�����ϸ����ֳ����Ѫ�����ɺ�����ϸ��ת���з�����Ҫ���á��ڱ������У������о��˴Ӹʲ�����ȡ�Ļ�ͪ�����licochalcone A (LA)����θ��BGC-823ϸ����PI3K/AKT/mTOR��MAPK����ͨ·��Ӱ�졣LA�����˻�����(ROS)��ˮƽ������������յ��йأ���������Annexin V�����Խ�Ϻ�caspase-3�ļ���Լ���adp -���Ǿۺ�ø(PARP)���ѽ⡣n -�������װ���(NAC)��ROS���ɵ����ƿ�������ֹla�յ���ϸ����������Ȥ���ǣ����ǻ��۲쵽LA��BGC-823ϸ���м�����ERK��JNK��p38 MAPK��la������BGC-823ϸ����KMС�����ڵĿ������������Բ�ͬ�����ǵ��о����������LA���Ը���MAPK�źż���������ROS���ɣ��յ�����Ӧ�����Ӷ�����BGCϸ��������
Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4434846/
Int J Mol Sci. 2014 Sep; 15(9): 15754�C15765.
Curcumin Induced Human Gastric Cancer BGC-823 Cells Apoptosis by ROS-Mediated ASK1-MKK4-JNK Stress Signaling Pathway
Tao Liang, Xiaojian Zhang,* Wenhua Xue, Songfeng Zhao, Xiang Zhang, and Jianying Pei
Abstract
The signaling mediated by stress-activated MAP kinases (MAPK), c-Jun N-terminal kinase (JNK) has well-established importance in cancer. In the present report, we investigated the effects of curcumin on the signaling pathway in human gastric cancer BGC-823 cells. Curcumin induced reactive oxygen species (ROS) production and BGC-823 cells apoptosis. Inhibition of ROS generation by antioxidant (NAC or Trion) significantly prevented curcumin-mediated apoptosis. Notably, we observed that curcumin activated ASK1, a MAPKKK that is oxidative stress sensitive and responsible to phosphorylation of JNK via triggering cascades, up-regulated an upstream effector of the JNK, MKK4, and phosphorylated JNK protein expression in BGC-823 cells. However, curcumin induced ASK1-MKK4-JNK signaling was attenuated by NAC. All the findings confirm the possibility that oxidative stress-activated ASK1-MKK4-JNK signaling cascade promotes the apoptotic response in curcumin-treated BGC-823 cells.
��������ͨ��ros�鵼��ASK1-MKK4-JNKӦ���ź�ͨ·�յ���θ��BGC-823ϸ������
��������С����Ѧ�Ļ������ɷ壬���裬�ὨӢ
ժҪ
Ӧ�������MAP��ø(MAPK)��c-Jun n -ĩ�˼�ø(JNK)�鵼���źŴ����ڰ�֢�о�����Ҫ���塣�ڱ������У������о��˽����ض���θ��BGC-823ϸ���ź�ͨ·��Ӱ�졣�������յ�������(ROS)���ɺ�BGC-823ϸ����������������(NAC��Trion)��ROS���ɵ����ƿ�������ֹ�����ؽ鵼��ϸ��������ֵ��ע����ǣ����ǹ۲쵽�����ؼ�����ASK1������һ������Ӧ�����е�MAPKKK��ͨ������������Ӧ����JNK���ữ���ϵ���BGC-823ϸ����JNK��MKK4������ЧӦ�������ữ��JNK���ı��Ȼ�����������յ���ASK1-MKK4-JNK�źű�NAC���������еĽ����֤ʵ������Ӧ�������ASK1-MKK4-JNK�źż����ٽ������ش�����BGC-823ϸ��������Ӧ�Ŀ����ԡ�
Keywords: curcumin, gastric cancer, ROS, ASK1-MKK4-JNK, apoptosis
Astragalus saponins affect proliferation, invasion and apoptosis of gastric cancer BGC-823 cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3818446/
��Journal of Molecular and Cellular Cardiology
Volume 135, October 2019, Pages 40-51
Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis
ShuaiYanga1Yuan-pengXiaa1Xue-yingLuoaShao-liChenaBo-weiLiaZi-mingYeabSheng-caiChenaLingMaoaHui-juanJinaYa-nanLiaBoHua
a Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
b Department of Neurology, The First Affiliated Hospital, Guangxi Medical University, Nanning, Guangxi, China
Highlights
• CagA-positive H. pylori infection does not initiate the development of atherosclerosis but can accelerate its progression by promoting macrophage-derived foam cell formation.
• CagA-positive H. pylori infection promotes atherogenesis by releasing CagA-containing EVs and exosomal CagA mediates this effect.
• CagA suppresses the transcription of cholesterol efflux transporters by downregulating the expression of transcriptional factors PPAR�� and LXR�� and thus enhances foam cell formation.
Abstract
Background
Seroepidemiological studies have highlighted a positive relation between CagA-positive Helicobacter pylori (H. pylori), atherosclerosis and related clinic events. However, this link has not been well validated. The present study was designed to explore the role of H. pylori PMSS1 (a CagA-positive strain that can translocate CagA into host cells) and exosomal CagA in the progression of atherosclerosis.
Methods
To evaluate whether H. pylori accelerates or even induces atherosclerosis, H. pylori-infected C57/BL6 mice and ApoE−/− mice were maintained under different dietary conditions. To identify the role of H. pylori-infected gastric epithelial cells-derived exosomes (Hp-GES-EVs) and exosomal CagA in atherosclerosis, ApoE−/− mice were given intravenous or intraperitoneal injections of saline, GES-EVs, Hp-GES-EVs, and recombinant CagA protein (rCagA).
Findings
CagA-positive H. pylori PMSS1 infection does not induce but promotes macrophage-derived foam cell formation and augments atherosclerotic plaque growth and instability in two animal models. Meanwhile, circulating Hp-GES-EVs are taken up in aortic plaque, and CagA is secreted in Hp-GES-EVs. Furthermore, the CagA-containing EVs and rCagA exacerbates macrophage-derived foam cell formation and lesion development in vitro and in vivo, recapitulating the pro-atherogenic effects of CagA-positive H. pylori. Mechanistically, CagA suppresses the transcription of cholesterol efflux transporters by downregulating the expression of transcriptional factors PPAR�� and LXR�� and thus enhances foam cell formation.
Interpretation
These results may provide new insights into the role of exosomal CagA in the pathogenesis of CagA-positive H. pylori infection-related atherosclerosis. It is suggested that preventing and eradicating CagA-positive H. pylori infection could reduce the incidence of atherosclerosis and related events.Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S002228281930152X��
J Immunol. Author manuscript; available in PMC 2011 Apr 1.
Immune Evasion by Helicobacter pylori is Mediated by Induction of Macrophage Arginase II1
Nuruddeen D. Lewis,*† Mohammad Asim,*‡ Daniel P. Barry,*‡ Thibaut de Sablet,*‡ Kshipra Singh,*‡ Maria B. Piazuelo,* Alain P. Gobert,*‡�� Rupesh Chaturvedi,*‡ and Keith T. Wilson*†‡,2
*Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, 37240
†Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, 37240
‡Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN 37212
��Institut National de la Recherche Agronomique, Unit�� de Microbiologie UR454, Saint-Gen��s-Champanelle, France
2Address correspondence and reprint requests to Dr. Keith T. Wilson, Department of Medicine, Division of Gastroenterology, Vanderbilt University School of Medicine, 1030C MRB IV, 2215B Garland Ave., Nashville, TN 37232.
Abstract
Helicobacter pylori infection persists for the life of the host due to the failure of the immune response to eradicate the bacterium.Determining how H. pylori escapes the immune response in its gastric niche is clinically important. We have demonstrated in vitro that macrophage NO production can kill H. pylori, but induction of macrophage arginase II (Arg2) inhibits inducible NO synthase (iNOS) translation, causes apoptosis, and restricts bacterial killing.
We now determined if Arg2 impairs host defense in vivo, using a chronic H. pylori infection model. In C57BL/6 mice, expression of Arg2, but not arginase I (Arg1), was abundant and localized to gastric macrophages. Arg2−/− mice had increased histologic gastritis and decreased bacterial colonization compared to wild-type (WT) mice. Increased gastritis scores correlated with decreased colonization in individual Arg2−/− mice, but not WT mice. When mice infected with H. pylori were compared, Arg2−/− mice had more gastric macrophages, more of these cells were iNOS+, and these cells expressed higher levels of iNOS protein, as determined by flow cytometry and immunofluorescence microscopy. There was enhanced nitrotyrosine staining in infected Arg2−/− versus WT mice, indicating increased NO generation. Infected Arg2−/− mice exhibited decreased macrophage apoptosis, as well as enhanced IFN-��, IL-17a, and IL-12p40 expression, and reduced IL-10 levels consistent with a more vigorous Th1/Th17 response.
These studies demonstrate that Arg2 contributes to the immune evasion of H. pylori by limiting macrophage iNOS protein expression and NO production, mediating macrophage apoptosis, and restraining pro-inflammatory cytokine responses.
Immune Evasion by Helicobacter pylori is Mediated by Induction of Macrophage Arginase II
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3069806/��
The problem - infection with Helicobacter pylori
H.p doest not activate TLR4/5 on macrophages
...Alongside the capability of breaching the cellular and physical barriers, H. pylori has additionally developed ways of avoiding both the innate and the adaptive immune response of the host organism.
Helicobacter pylori are helix-shaped, microaerophilic bacteria with polar flagella that live near the surface of the human gastric mucosa. Approximately half of the world`s population harbors H. pylori in their upper gastrointestinal tract, with 3rd world countries being most affected. There is evidence of co-evolution of human and H. pylori, tracing the migration of human race in the last tens of thousands of years (Covacci et al., 1999).
Helicobacter pylori
Global prevalence of H. pylori infection
Infection with H. pylori is usually acquired early in childhood and lasts for a lifetime, and the majority of those infected (80%�C90%) remain asymptomatic. However, infection with H. pylori is known to be the strongest risk factor for the development of peptic ulcers. Furthermore, H. pylori was the first bacterium to be classified as a definite carcinogen by the WHO. Thus, from the medical point of view, H. pylori represents a challenging pathogen responsible for much mortality worldwide. The generally accepted route of transmission for H. pylori is from person to person, e.g. by oral-oral transmission, since H. pylori can only survive for short periods of time because it is rapidly killed by higher oxygen tension and light. Therefore, infection with H. pylori is considered as a "kissing disease". Infection with H. pylori can lead to several clinical outcomes. By far the most common gastric phenotype is the »simple gastritis« which is characterized by mild inflammation of the stomach lining and little disruption of gastric acid secretion. The second main phenotype is the duodenal ulcer phenotype (duodenum is the beginning portion of the small intestine) which accounts for up to 15 % in infected subjects. Subjects with this phenotype have defective inhibitory control of gastric acid secretion, which leads to very high acid output and consequently the development of peptic ulcers.
If untreated, peptic ulcers can commonly result in haemorrhage into the
stomach or the abdominal cavity which often leads to severe blood loss
and even death.
Image from http://medicalimages.allrefer.com/large/ulcer-emergencies. jpg
The third and most serious phenotype is the »gastric cancer phenotype«, which is, in contrast to the previous two phenotypes, characterized with hypo- or achlorhydria (low acid secretion) and affects up to 5% of infected subjects. Gastric cancer is a formidable disorder, primarily because most patients are diagnosed at an advanced stage of disease. Despite the curative surgical resection vast majority of affected individuals often die of recurrent disease.
Interestingly, H. pylori is the only known organism capable of colonizing the severe environment of the human stomach, but even this bacterium can withstand low pH for only short periods of time. H. pylori has developed several mechanisms to avoid bactericidal activity of HCl. It uses its polar flagella for motility in chemotactic response to pH gradient, by which it stays in close proximity to the surface of the epithelium, where pH is near neutral. Besides that, H. pylori was shown to be capable of adhering to the surface and even invading the epithelial cells so as to protect itself from gastric contents and mechanical clearance to promote perseverance. H. pylori also possesses artificial means of increasing pH in its direct proximity by generating large quantities of cytosolic and cell surface-associated urease, an enzyme capable of the breakdown of urea into ammonia and carbon dioxide.
Alongside the capability of breaching the cellular and physical barriers, H. pylori has additionally developed ways of avoiding both the innate and the adaptive immune response of the host organism. To date, several bacterial strategies have been described (some of them are illustrated in table and figure below). For example, H. pylori flagellar proteins have developed in a way that masks them from the Toll-like receptor 5 (an innate immune receptor, that recognizes various other bacterial flagellins). LPS from H. pylori is 1000 times less pyrogenic and 500-fold less toxic than that of gram-negative enteric bacteria due to modifications that mimic human glycans and therefore induces less inflammation. H. pylori is also capable of obtaining cholesterol directly from epithelial cell membranes and incorporating it into its own lipid layer. To escape macrophage phagocytosis it modifies obtained cholesterol by adding a glucosyl group (a reaction catalysed by cholesterol-�� glucosyltransferase). Additionally, H. pylori reduces generation of bactericidal nitric oxide by the macrophage. Its enzyme arginase competes with macrophages inducible nitric oxide synthase for substrate L-arginine that is used by the bacteria for the synthesis of urea (a substrate for urease). Moreover, exposure to H. pylori products upregulates transcription of arginase II in macrophages, which catalyses breakdown of L-arginine into urea and L-ornithine that is subsequently metabolized into polyamines that trigger apoptosis in macrophages.
��
Helicobacter pylori modifies its LPS and flagellin so that they do not activate Toll-like receptors 4 and 5��opposite to LPS and flagellin of Escherichia coli, which results in no activation of the immune system cells.
Summary of strategies that H. pylori employs to avoid detection of the immune system
MECHANISM EXAMPLE
Bypassing the TLR system - Flagellin fails to activate TLR5 (Gewirtz et al., 2004)
- Tetraacylated LPS poorly activates TLR4 and may actually work as an antagonist (Moran and Aspinall, 1998)
- DNA is rich in A/T nucleotides and frequently methylated, making TLR9 response more unprobable (Blaser and Atherton, 2004)
Minimization of innate immunity - Modification of LPS (long 3-hydroxy fatty acids of lipid A, unusual phosphorylation pattern��) (Moran et al., 1997, 1998)
Mimicry of host antigens - Lewis expression (O antigen region of H. pylori LPS) (Wirth et al., 1997)
Antigenic variation CagY (Aras et al., 2003)
Host gene expression modulation - Upregulation of specific inflammatory and immune mediators including ��-defensin, protease inhibitor, chemokine receptor, interleukin-1��, tumor necrosis factor-��-inducible protein�� (Israel and Peek, 2006)
Downregulation of immune effectors - Blocking the proliferation of T cell through VacA and B cell proliferation through CagA-induced supression- Interference with phagocytosis (Baldari et al., 2005)
Avoidance of attack by ROIs and RNIs - Enzymes involved in ROI scavenging, such as catalase and superoxide dismutase
- Arginase regulates NO synthesis (Baldari et al., 2005)
Ability to colonize gastric environment - Neutralizing pH around the organism with urease enzyme
- Interaction between adhesins and local cell receptors
- Expressing mucolytic molecules
- Relative absence of immune cells in gastric mucosa and rare competition with commensal bacteria
High mutational and recombinational frequency, high diversity - Rapid development in the bacterial population of high-level resistance to commonly used antibiotics
- High competence for uptake of DNA from other H. pylori strains (Sansonetti and Di Santo, 2007)
There is also experimental evidence linking bacterial adhesion and disease progression as an outcome of delivery of toxins, for instance VacA (pore-forming cytotoxin) that is characterized as a potent inhibitor of T-cell function and believed to be capable of inserting into the membranes of late endosomal vesicles to perturb antigen presentation. VacA can also disrupt the barrier function of tight junctions and thereby causes leakage of ions and small molecules from host cells, so that H. pylori can acquire nutrients across an intact epithelial barrier.
Current medical treatment of peptic ulcers and H. pylori infection consists mainly of three- and four-drug regimes, which combine two antibiotics with either proton pump inhibitors to reduce the production of gastric acid and/or cytoprotective agents that protect cells from toxic chemicals or other stimuli. Although these regimes are very effective at eradicating H. pylori with cure rates higher than 90%, there remains a serious problem of increasing antibiotic resistance. In addition, re-infection with H. pylori after antimicrobal therapy occurs frequently in countries where infection rates are high. Therefore, the only way to effectively prevent H. pylori (re)infection seems to be through long-term vaccination programs.
In the past 25 years since H. pylori has been first described and cultured, more than 20.000 publications have appeared on the subject. Understanding how this organism interacts with its host is of fundamental importance for preparation an intelligent strategy to prevent and cure H. pylori infections. To date, some promising approaches in vaccine development have been tested but still an effective vaccine against H. pylori remains to be demonstrated. Current knowledge of immunology, especially TLR activation and subsequent signaling, has enabled our team to design and test out some innovative ideas in vaccine development.��
�쳣�����ƻ��������ݸ˾�
����ͻ��ϸ�����������ϵ������⣬���������˾��������˱������������������߷�Ӧ����Ӧ�����߷�Ӧ�ķ�����
��ĿǰΪֹ���Ѿ������˼���ϸ������(����һЩ������ı���ͼ�н�����˵��)�����磬
���������˾���ë���ķ�չʹ���Dz���toll������(һ��ʶ����������ϸ����ë����������������)��Ӱ�졣
���������˾���LPS�²��Աȸ��������Գ���ϸ���ĵ�1000�������Աȸ��������Գ���ϸ���ĵ�500����
���������˾�����ֱ�Ӵ���Ƥϸ��Ĥ��õ��̴������������ϵ�������֬�ʲ��С�
���Ѿ���ϸ����������ͨ������glucosyl�Ļ�õ��̴���(cholesterol-����Ӧ�������ǻ�ת��ø)��
���⣬�����ݸ˾����ܼ��پ���ϸ��������ɱ����һ��������NO)��
���ľ�����ø�����ϸ���յ���һ�������ϳ�ø��������L-�����ᣬ���߱�ϸ�������ϳ�����(����ø�ĵ���)��
���⣬��¶�����������˾��IJ�����ϵ�����ϸ���о�����øII��ת¼���Ӷ���L -������ֽ�Ϊ���غ�L -���ᣬ�������л�ɶష���Ӷ���������ϸ���ĵ�����
��
Cooperation between Innate and Adaptive Immune Response
Schematic representation of immune response against pathogens based on the cooperation between innate and adaptive immunity components.
Immunity is a state of sufficient biological defenses to avoid infection, disease or other unwanted biological invasion. It is divided in a non-specific (innate) and specific (adaptive) components. The term innate immunity is due to its constant presency in healthy individuals and generally refers to responses that do not require previous exposures to the pathogen, whereas adaptive immunity develops more slowly and is specific to a particular pathogen and involves immunological memory. The adaptive immunity consists of humoral immunity in which antibodies neutralize and eradicate extracellular microbes and toxins and cell-mediated immunity in which T-lymphocytes play a key role. The most important is the cooperation between innate and adaptive immunity.
Until recently innate immunity was largely ignored as the adaptive immunity was thought to play a key role in the immune response. Now it became clear that innate immunity has an important role as the first line of defense against pathogens as well as in development of an adaptive immune response. The polarization of immune response largely depends on the initial signals that are brought by innate immunity. There are some essential signals, required to induce T- and B-cell responses. Innate immune signals modulate the quantity and the quality of adaptive immune response and are required to initiate an effective immune response, that mainly depends on the nature of the pathogen. There are two separated ways of antigen processing, which enable the immune system to appropriately respond against extracellular and intracellular pathogens, respectively.
Briefly, we will focus on the immune response against extracellular pathogens. As the pathogen invades the host, its pathogen associated molecular pattern (PAMP) is being recognised by the pathogen recognition receptor (PRR) on APC, for instance dendritic cell (signal 1 in immune activation). Pathogen recognition and APC activation is the first signal needed for activation of immune response. After internalisation (phagocytosis) antigens are processed into peptides. These antigen-derived peptides are presented by major histocompatibility complex MHC class 2 (MHC II) molecules on APC surface for recognition by CD4+ T lymphocytes that have T-cell receptor specific for the same peptides. TCR recognition of specific peptide represents signal 2 in immune activation. The last step - signal 3 in immune activation is co-stimulation, which depends on the interactions between costimulatory molecules, such as B7 and CD40 on APC and CD-28 and CD40L on CD4+ T. All this is happening at the first stages of immune response and leads to the expression of appropriate immune mediators, such as cytokines, that enable the polarization of immune response in a direction, that is potentially optimal for pathogen eradication.
Schematic representation of immune response against pathogens based on the cooperation between innate and adaptive immunity components.
For example, when IL-12 is absent or set of interleukins like IL-4 and IL-10 are present, T-helper 2 (Th2) cell polarization occurs. Th2 cells produce interleukin IL-4, IL-5, IL-6, IL-10, IL-13 and participate in phagocytosis-independent responses. They provide B-cell help and the induction of IgG1, IgE and IgA, which all provide the defense against extracellular pathogens. For instance, IL-4, that is produced by Th2 cells, promotes IgE heavy chain class switching. These antibodies are effective against helminths, since IgE has Fc, which is recognized by Fc-��RI on eosinophills, that finally eradicate the parasitic infection.
So it is obvious, that interactions between Th and B-cells is of a great importance. B-cells are a special type of APC's that are a part of adaptive immune response. In contrast to macrophages and dendritic cells, which have extracellulary exposed PAMP's, B-cells retain specific B cell receptors (BCR) that are basically membrane-bound immunoglobulins, which bind a particular antigen. After B cell encounters and recognizes a fitting (cognate) native antigen with its B-cell receptor, internalization, antigen processing and presentation of antigen derived peptides in MHC II molecules occurs (for B cell is APC). This leads to the interaction with previously activated Th cell that carries T-cell receptor, specific for the same antigenic component, that is presented in MHC II molecules of B cell. Only after this specific interaction B-cell receives an additional signals from a T-helper cell which leads to a clonal expansion and differentiation of a B-cell that carries an appropriate BCR. The net result is a production of antibodies of the same specifity and later on, also affinity maturation and heavy chain class switching, which all is necessary for effective humoral response. The final stage is an establishment of a memory B-cell repertoire, that quickly responds to a subsequent exposition to the cognate antigen.
In contrary, defense against intracellular pathogens is carried out by different strategy since humoral response is not effective against intracellular patogens, such as viruses and intracellular bacteria. In that case, cytosolic antigens are presented within MHC class 1 (MHC I) molecules, which can be found on all nucleated cells, for all these cells could be infected by viruses. Antigenic peptides, presented in MHC I molecules, are recognised by CD8+ T cells, which then, with help of Th cells differentiate into CTL. These kill infected cells and eradicate infection.
Very simplified, we can say, that the result (polarization) of immune response depends on which player of innate branch of immune response (e.g., dendritic cell) recognized invading pathogen, how the peptides derived from antigenic components were presented (MHC I or MHC II molecules) and which immunomodulatory molecules were produced (e.g. IL-12). Therefore, cooperation between initial innate and subsequent adaptive response is needed for effective functioning of the immune system and immune response.Team:Slovenia/Background/The problem - 2008.igem.org
http://2008.igem.org/Team:Slovenia/Background/The_problemTeam:Slovenia/Background/Immune response - 2008.igem.org
http://2008.igem.org/Team:Slovenia/Background/Immune_response��
Am J Transl Res. 2016; 8(10): 4184�C4194.
Comparative analysis of the interaction of HSPs in dendritic cells, macrophages, RGM-1 cells infected by Helicobacter pylori
Yongliang Yao,1 Jianhong Wu,1 Tao Gu,1 Yang Cheng,1 and Guangxin Li2
Author information Article notes Copyright and License information Disclaimer
1Department of Clinical Laboratory, Kunshan First People��s Hospital, Affiliated to Jiangsu University, Kunshan 215300, People��s Republic of China
2Department of Pathology, Chongqing Cancer Institute, Chongqing 400030, People��s Republic of China
Address correspondence to: Guangxin Li, Department of Pathology, Chongqing Cancer Institute, Chongqing 400030, People��s Republic of China.
Abstract
Helicobacter pylori may cause chronic gastritis, even gastric cancer, however, antigen-presenting cells (APCs) are most important immune cells involved in the induction and expression of the underlying inflammatory responses to resist H. pylori. To study the interaction of HSPs in dendritic cells (DCs), macrophages and RGM-1 cells infected with H. pylori, HSP-27, HSP-60, HSP-70 and HSP-90 proteins were analyzed in the mucosa tissue or serum of gastritis patients caused by H. pylori, and in cell supernatant of DCs, macrophages, RGM-1 infected by H. pylori, or in above host cells. We found that HSP-27, HSP-60, HSP-70 and HSP-90 decreased in gastric epithelial cells, but increased significantly in DCs, macrophages. Meanwhile, inflammation associated proteins iNOS-2 and COX-2 were participated in the expression of HSPs in the process of host cells defensing against H. pylori infection. These findings contribute to understand the functions of HSP-27, HSP-60, HSP-70 and HSP-90 in H. pylori infection APCs and gastric epithelial cells indicating that HSPs would be diagnostic markers for H. pylori infection.
Keywords: HSPs, Helicobacter pylori, dendritic cells, macrophages, RGM-1
Introduction
Helicobacter pylori infection is the major cause of chronic gastritis, peptic ulcers and gastric cancer [1]. At present approximately half of the world��s population is infected with this bacterium, however, only a minority of infection individuals develop gastric cancer [2,3]. Gastric mucosa is protected by a complex defense system, which includes the production of surface mucus and bicarbonate, the regulation of gastric mucosal blood flow, the acceleration of epithelial regeneration, and the preservation of epithelial homeostasis to defense H. pylori infection [4-6].
HSPs are groups of stress-response proteins which are either constitutively expressed or induced through the transcriptional action of heat shock factor (HSF), which have been proved to play a cytoprotective role in gastrointestinal tract [7,8]. HSPs are classified into four major families according to their biological activities and apparent molecular weights; HSP90, HSP70, HSP60 expressed constitutively, and small HSPs including HSP27 and HSP10 induced by various conditions, including heat, oxidative stress, or bacterium infection [9,10]. A number of studies have consistently demonstrated that H. pylori infection delays gastric mucosal healing by disrupting the balance in cell apoptosis and proliferation, decreasing the migration of epithelial cells, decreasing blood flow or angiogenesis within the gastric mucosa, and HSPs could reverse these limitation and inferiorities in mucosal healing.
Dendritic cells (DCs), and macrophages are both myeloid antigen-presenting cells (APCs), and are present in the H. pylori-infected mucosa and are likely involved in both the induction and maintenance of H. pylori specific immune responses and inflammatory response. Macrophages or DCs were originated from a common myeloid progenitor, monocytes and could be differentiated into tissue when crossing the endothelial barrier and have a high capacity to kill H. pylori [11,12]. Dendritic cells, and macrophages are specialized in antigen capture, processing, and presentation to naïve T cells after migration to the draining lymph nodes [13], and are widely distributed in human tissues, including the gastric mucosa, and are capable of penetrating the gut epithelial monolayers to sample, e.g., luminal bacteria [14]. Exosomes derived from are dendritic cells, macrophages and tumor cells are lipid bilayer vesicles of 30 to 100 nm in size, which contain microbial components [15,16] and could altered the expression of cellular inflammatory signaling proteins such as CD63, CD81 and HSP-90 [17].
Here, we intend to investigate the functions of HSPs in dendritic cells, macrophages, RGM-1 infected with H. pylori and the protective mechanisms of HSPs in H. pylori infection.Comparative analysis of the interaction of HSPs in dendritic cells, macrophages, RGM-1 cells infected by Helicobacter pylori
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5095311/��
Comparative Analysis of the Interaction of Helicobacter pylori with Human Dendritic Cells, Macrophages, and Monocytes
Michael Fehlings, Lea Drobbe, Verena Moos, Pablo Renner Viveros, Jana Hagen, Macarena Beigier-Bompadre, Ervinna Pang, Elena Belogolova, Yuri Churin, Thomas Schneider, Thomas F. Meyer, Toni Aebischer, Ralf Ignatius
S. R. Blanke, Editor
aInstitute of Tropical Medicine and International Health, Charit��-Universitätsmedizin Berlin, Berlin, Germany
bDepartment of Molecular Biology, Max Planck Institute for Infection Biology, Berlin, Germany
cMedical Clinic I, Charit��-Universitätsmedizin Berlin, Berlin, Germany
dRobert Koch Institute, Berlin, Germany
ABSTRACT
Helicobacter pylori may cause chronic gastritis, gastric cancer, or lymphoma. Myeloid antigen-presenting cells (APCs) are most likely involved in the induction and expression of the underlying inflammatory responses. To study the interaction of human APC subsets with H. pylori, we infected monocytes, monocyte-derived dendritic cells (DCs), and monocyte-derived (classically activated; M1) macrophages with H. pylori and analyzed phenotypic alterations, cytokine secretion, phagocytosis, and immunostimulation.Since we detected CD163+ (alternatively activated; M2) macrophages in gastric biopsy specimens from H. pylori-positive patients, we also included monocyte-derived M2 macrophages in the study. Upon H. pylori infection, monocytes secreted interleukin-1�� (IL-1��), IL-6, IL-10, and IL-12p40 (partially secreted as IL-23) but not IL-12p70. Infected DCs became activated, as shown by the enhanced expression of CD25, CD80, CD83, PDL-1, and CCR7, and secreted IL-1��, IL-6, IL-10, IL-12p40, IL-12p70, and IL-23.
However, infection led to significantly downregulated CD209 and suppressed the constitutive secretion of macrophage migration inhibitory factor (MIF). H. pylori-infected M1 macrophages upregulated CD14 and CD32, downregulated CD11b and HLA-DR, and secreted mainly IL-1��, IL-6, IL-10, IL-12p40, and IL-23. Activation of DCs and M1 macrophages correlated with increased capacity to induce T-cell proliferation and decreased phagocytosis of dextran. M2 macrophages upregulated CD14 and CD206 and secreted IL-10 but produced less of the proinflammatory cytokines than M1 macrophages.
Thus, H. pylori affects the functions of human APC subsets differently, which may influence the course and the outcome of H. pylori infection. The suppression of MIF in DCs constitutes a novel immune evasion mechanism exploited by H. pylori.
Fig 1
M2 macrophages are present in the gastric mucosal tissue of H. pylori patients. (A) Immunohistochemical analysis of the gastric mucosa of H. pylori-positive patients and healthy controls. Paraffin sections of biopsy specimens were analyzed for CD68, CD163, and stabilin-1 expression (red cells). One representative example of three experiments is shown. (B) Cell counts were determined from three biopsy specimens per sampling date as the mean cell count of 10 high-power fields of 0.237 mm2 each. *, P < 0.05 (nonparametric Mann-Whitney test).In conclusion, a comparative analysis of four different types of APCs in vitro revealed considerable variation in phenotype, cytokine secretion, phagocytosis and T cell stimulating capabilities in these cells in response to H. pylori infection. During the course of infection, monocytes may be the first APCs attracted to the site of infection and may first encounter H. pylori. The secretion of IL-1�� and IL-6 by these cells may recruit further immune cells, including DCs, to the site of inflammation and contribute to the maturation of DCs. DCs likely pick up antigen and migrate to the draining lymph nodes, where they induce CD4+ Th1 (by IL-12p70), Th17 (by IL-23), and Treg (by IL-10) responses. In this context, H. pylori may further hamper the induction of excessive proinflammatory responses by the suppression of MIF secretion in DCs. The peripheral inflammatory environment during chronic infection might be dominated by macrophages, where M2 macrophages may limit excessive inflammation in favor of a chronic inflammatory response.
Comparative Analysis of the Interaction of Helicobacter pylori with Human Dendritic Cells, Macrophages, and Monocytes | Infection and Immunity
https://iai.asm.org/content/80/8/2724��
The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides
Alexandra Grubman Maria Kaparakis J��rôme Viala Cody Allison Luminita Badea Abdulgader Karrar Ivo G. Boneca Lionel Le Bourhis Shane Reeve Ian A. Smith Elizabeth L. Hartland Dana J. Philpott Richard L. Ferrero
Summary
The cytosolic innate immune molecule, NOD1, recognizes peptidoglycan (PG) delivered to epithelial cells via the Helicobacter pylori cag pathogenicity island (cagPAI), and has been implicated in host defence against cagPAI+H. pylori bacteria. To further clarify the role of NOD1 in host defence, we investigated NOD1�\dependent regulation of human �©\defensins (DEFBs) in two epithelial cell lines. Our findings identify that NOD1 activation, via either cagPAI+ bacteria or internalized PG, was required for DEFB4 and DEFB103 expression in HEK293 cells. To investigate cell type�\specific induction of DEFB4 and DEFB103, we generated stable NOD1��knockdown�� (KD) and control AGS cells. Reporter gene assay and RT�\PCR analyses revealed that only DEFB4 was induced in an NOD1�\/cagPAI�\dependent fashion in AGS cells.Moreover, culture supernatants from AGS control, but not AGS NOD1 KD cells, stimulated with cagPAI+H. pylori, significantly reduced H. pylori bacterial numbers. siRNA studies confirmed that human �©\defensin 2 (hBD�\2), but not hBD�\3, contributes to the antimicrobial activity of AGS cell supernatants against H. pylori. This study demonstrates, for the first time, the involvement of NOD1 and hBD�\2 in direct killing of H. pylori bacteria by epithelial cells and confirms the importance of NOD1 in host defence mechanisms against cagPAI+H. pylori infection.
��Ȼ���߷��ӣ�NOD1������ֱ��ɱ�������ݸ˾�������
�ܽ�
ϸ���ڹ������߷���NOD1ʶ��ͨ�������ݸ˾�Cag�²��Ե�(cagPAI)���ݵ���Ƥϸ�����ľ���(PG)��������������cagPAI+�ݸ˾�ϸ���ķ�������һ������NOD1����������������,���ǵ�����NOD1���������defensinsӦ�е����(DEFBs)������Ƥϸ��ϵ�����ǵ��о����֣�������ͨ��cagPAI+ϸ�������ڻ�PG����NOD1������HEK293ϸ����DEFB4��DEFB103����������ġ�Ϊ���о�ϸ�����Ͷ�DEFB4��DEFB103���������յ������Dz������ȶ���NOD1'knockdown ' (KD)��������AGSϸ����������������RT - PCR������ʾ����AGSϸ���У�ֻ��DEFB4��NOD1�\/cagPAI�\�����ķ�ʽ���յ���
���⣬��cagPAI+Hp�̼���������AGS���յ�����Һ��������AGS nod1kdϸ�������Լ����������ݸ˾���ϸ�����������о�֤ʵ,����±ؾ�defensin 2 (hBD���2),������hBD��3������AGSϸ������Һ�����������˾��Ŀ������ԡ����о��״�֤ʵ��NOD1��hBD�\2��������Ƥϸ��ֱ��ɱ�������ݸ˾��Ĺ��̣���֤ʵ��NOD1���������������ж�cagPAI+�ݸ˾���Ⱦ����Ҫ�ԡ�The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides - Grubman - 2010 - Cellular Microbiology - Wiley Online Library https://onlinelibrary.wiley.com/doi/full/10.1111/j.1462-5822.2009.01421.x
��
The Helicobacter pylori Virulence Effector CagA Abrogates Human ��-Defensin 3 Expression via Inactivation of EGFR Signaling
Antimicrobial peptides are constituents of the first-line innate mucosal defense system that acts as a barrier to establishment of infection. The highly successful human gastric pathogen, Helicobacter pylori, is able to persistently colonize its host despite inducing expression of several antimicrobial peptides, including human ��-defensin 3 (hBD3). We find that hBD3 is highly active against H. pylori in vitro and is rapidly induced during early infection via EGFR-dependent activation of MAP kinase and JAK/STAT signaling.
However, during prolonged infection, hBD3 was subsequently downregulated by the H. pylori virulence determinant CagA. Upon translocation into host cells, CagA activated the cellular tyrosine phosphatase, SHP-2, terminating EGFR activation and downstream signaling and increasing bacterial viability. Chemical inhibition and knockdown of SHP-2 expression rescued hBD3 synthesis and bactericidal activity. Thus, we reveal how cagPAI-positive H. pylori strains use CagA to evade a key innate mucosal defense pathway to support the establishment of persistent infection.
Highlights
H. pylori is highly susceptible to the bactericidal activity of hBD3
hBD3 is initially induced by H. pylori via activation of EGFR/MAPK/JAK signaling
H. pylori evades killing by downregulating hBD3 via CagA-mediated SHP-2 activation
CagA virulence factor is a potent inhibitor of an immune effector mechanismThe Helicobacter pylori Virulence Effector CagA Abrogates Human ��-Defensin 3 Expression via Inactivation of EGFR Signaling: Cell Host & Microbe https://www.cell.com/cell-host-microbe/fulltext/S1931-3128(12)00135-7
��
��
��
Regulation of Human ��-Defensins by Gastric Epithelial Cells in Response to Infection with Helicobacter pylori or Stimulation with Interleukin-1 | Infection and Immunity https://iai.asm.org/content/68/9/5412
��
Helicobacter pylori VacA, acting through receptor protein tyrosine phosphatase ��, is crucial for CagA phosphorylation in human duodenum carcinoma cell line AZ-521
Masayuki Nakano, Kinnosuke Yahiro, Eiki Yamasaki, Hisao Kurazono, Junko Akada, Yoshio Yamaoka, Takuro Niidome, Masanori Hatakeyama, Hidekazu Suzuki, Taro Yamamoto, Joel Moss, Hajime Isomoto, Toshiya Hirayama
1Department of Bacteriology, Institute of Tropical Medicine, Nagasaki University, 1-12-4, Sakamoto, Nagasaki 852-8523, Japan
ABSTRACT
Helicobacter pylori, a major cause of gastroduodenal diseases, produces vacuolating cytotoxin (VacA) and cytotoxin-associated gene A (CagA), which seem to be involved in virulence. VacA exhibits pleiotropic actions in gastroduodenal disorders via its specific receptors. Recently, we found that VacA induced the phosphorylation of cellular Src kinase (Src) at Tyr418 in AZ-521 cells.Silencing of receptor protein tyrosine phosphatase (RPTP)��, a VacA receptor, reduced VacA-induced Src phosphorylation. Src is responsible for tyrosine phosphorylation of CagA at its Glu-Pro-Ile-Tyr-Ala (EPIYA) variant C (EPIYA-C) motif in Helicobacter pylori-infected gastric epithelial cells, resulting in binding of CagA to SHP-2 phosphatase. Challenging AZ-521 cells with wild-type H. pylori induced phosphorylation of CagA, but this did not occur when challenged with a vacA gene-disrupted mutant strain. CagA phosphorylation was observed in cells infected with a vacA gene-disrupted mutant strain after addition of purified VacA, suggesting that VacA is required for H. pylori-induced CagA phosphorylation. Following siRNA-mediated RPTP�� knockdown in AZ-521 cells, infection with wild-type H. pylori and treatment with VacA did not induce CagA phosphorylation. Taken together, these results support our conclusion that VacA mediates CagA phosphorylation through RPTP�� in AZ-521 cells. These data indicate the possibility that Src phosphorylation induced by VacA is mediated through RPTP��, resulting in activation of Src, leading to CagA phosphorylation at Tyr972 in AZ-521 cells.
Fig. 7.
Putative model showing VacA-induced CagA phosphorylation during H. pylori infection in AZ-521 cells. H. pylori injects CagA into gastric epithelial cells by a type-IV secretion system and secretes VacA into the extracellular space (step 1). Secreted VacA binds to the extracellular domain of RPTP�� (step 2). VacA induces Src phosphorylation at Tyr418 through RPTP��, resulting in activation of Src (step 3). It has been shown that activated Src promotes tyrosine phosphorylation at Tyr972 of an EPIYA-C motif in translocated CagA and pCagA then inactivates Src (Selbach et al., 2003; Mueller et al., 2012) (step 4). Finally, pCagA interacts with other host molecules, including SHP2 phosphatase (step 5).http://dmm.biologists.org/content/9/12/1473
��
European Journal of Medicinal Chemistry
Volume 181, 1 November 2019, 111512
European Journal of Medicinal Chemistry
Review article
Curcumin as tyrosine kinase inhibitor in cancer treatment
Author links open overlay panelA.GolonkoaH.LewandowskabR.ŚwisłockacU.T.Jasi��skaaW.PriebedW.Lewandowskia
a
Institute of Agricultural and Food Biotechnology, Rakowiecka 36, 02-532, Warsaw, Poland
b
Institute of Nuclear Chemistry and Technology, Centre for Radiobiology and Biological Dosimetry, Dorodna 16, 03-195, Warsaw, Poland
c
Bialystok University of Technology, Faculty of Civil Engineering and Environmental Engineering, Department of Chemistry, Biology and Biotechnology, Wiejska 45E, 15-351, Bialystok, Poland
d
Department of Experimental Therapeutics, University of Texas MD, Anderson Cancer Center, Houston, TX, USA
Highlights
• Our article summarizes knowledge about the mechanisms of curcumin activity directed to signaling pathways of tyrosine kinases.
• We have attempted to explain how the antitumor activity can be enhanced by modifications in the structure of the molecule.
• We have discussed the possibility of using curcumin in anti-cancer therapy and problems with its bioavailability.
Abstract
Curcumin is a natural substance known for ages, exhibiting a multidirectional effect in cancer prevention and adjuvant cancer therapies. The great advantage of using nutraceuticals of vegetable origin in comparison to popular cytostatic drugs is the minimized side effect and reduced toxicity. The targets in oncological therapy are, among others, tyrosine kinases, important mediators of signaling pathways whose impaired expression is observed in many types of cancer. Unfortunately, the hydrophobic nature of the curcumin molecule often limits its bioavailability, which is why many studies focus on the chemical modification of this compound. Current research is aimed at modifying structures that improve the pharmacokinetic parameters of curcumin, e.g. the formation of nanoparticles, complexes with metals or the synthesis of curcumin derivatives with functional substituents that allow tumor targeting. The article is a review and analysis of current literature on the properties of curcumin and its derivatives in the treatment of cancers directed to signaling pathways of tyrosine kinases and confronts the problem of low assimilation of curcumin with potential therapeutic effects.Curcumin as tyrosine kinase inhibitor in cancer treatment - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0223523419306361��
The Immune Battle against Helicobacter pylori Infection: NO Offense
Alain P. Gobert
Keith T. WilsonDivision of Gastroenterology, Hepatology, and Nutrition, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Trends
Macrophages are an important source of NOS2-dependent NO production in gastric tissues of H. pylori-infected humans and in experimental animal models.
Animal models, nitrosative damage in gastric epithelial cells of H. pylori-infected patients, and molecular epidemiology demonstrate that NO is involved in H. pylori-mediated carcinogenesis.
Several unique proteins, such as the NO reductase, NorH, and biochemical pathways, including metal acquisition or urease-dependent synthesis of CO2, confer on H. pylori a partial resistance to reactive nitrogen intermediates (RNI).
The lack of major effectors allowing resistance to high concentrations of RNI in H. pylori is compensated by a strong ability to inhibit host cell NO production. H. pylori dampens macrophage NO synthesis by transcriptional and translational regulation of NOS2 expression and control of l-arginine substrate availability.
Helicobacter pylori is a successful pathogen of the human stomach. Despite a vigorous immune response by the gastric mucosa, the bacterium survives in its ecological niche, thus favoring diseases ranging from chronic gastritis to adenocarcinoma. The current literature demonstrates that high-output of nitric oxide (NO) production by the inducible enzyme NO synthase-2 (NOS2) plays major functions in host defense against bacterial infections. However, pathogens have elaborated several strategies to counteract the deleterious effects of NO; this includes inhibition of host NO synthesis and transcriptional regulation in response to reactive nitrogen species, allowing the bacteria to face the nitrosative stress. Moreover, NO is also a critical mediator of inflammation and carcinogenesis. In this context, we review the recent findings on the expression of NOS2 in H. pylori-infected gastric tissues and epithelial cells, the role of NO in H. pylori-related diseases and H. pylori gene expression, and the mechanisms whereby H. pylori regulates NO synthesis by host cells.��
��
��
��
RocF:Arginase2, Odc: orthinine decarboxylase, Spd: spermindine, Spm:spermine, ADMA:
Helicobacter pylori arginase mutant colonizes arginase II ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3160534The Immune Battle against Helicobacter pylori Infection: NO Offense - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0966842X1600041X��
J Bacteriol. 1999 Dec;181(23):7314-22.
Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity.
McGee DJ1, Radcliff FJ, Mendz GL, Ferrero RL, Mobley HL.
1 Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA. dmcge001@umaryland.edu
Abstract
Arginase of the Helicobacter pylori urea cycle hydrolyzes L-arginine to L-ornithine and urea. H. pylori urease hydrolyzes urea to carbon dioxide and ammonium, which neutralizes acid. Both enzymes are involved in H. pylori nitrogen metabolism. The roles of arginase in the physiology of H. pylori were investigated in vitro and in vivo, since arginase in H. pylori is metabolically upstream of urease and urease is known to be required for colonization of animal models by the bacterium. The H. pylori gene hp1399, which is orthologous to the Bacillus subtilis rocF gene encoding arginase, was cloned, and isogenic allelic exchange mutants of three H. pylori strains were made by using two different constructs: 236-2 and rocF::aphA3. In contrast to wild-type (WT) strains, all rocF mutants were devoid of arginase activity and had diminished serine dehydratase activity, an enzyme activity which generates ammonium. Compared with WT strain 26695 of H. pylori, the rocF::aphA3 mutant was approximately 1, 000-fold more sensitive to acid exposure. The acid sensitivity of the rocF::aphA3 mutant was not reversed by the addition of L-arginine, in contrast to the WT, and yielded a approximately 10, 000-fold difference in viability. Urease activity was similar in both strains and both survived acid exposure equally well when exogenous urea was added, indicating that rocF is not required for urease activity in vitro. Finally, H. pylori mouse-adapted strain SS1 and the 236-2 rocF isogenic mutant colonized mice equally well: 8 of 9 versus 9 of 11 mice, respectively. However, the rocF::aphA3 mutant of strain SS1 had moderately reduced colonization (4 of 10 mice). The geometric mean levels of H. pylori recovered from these mice (in log(10) CFU) were 6.1, 5.5, and 4.1, respectively. Thus, H. pylori rocF is required for arginase activity and is crucial for acid protection in vitro but is not essential for in vivo colonization of mice or for urease activity.Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. McGee DJ(1), Radcliff FJ, Mendz GL, Ferrero RL, Mobley HL. Cited by: 159 Publish Year: 1999 Au
��Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/10572136��
Arginase: an emerging key player in the mammalian immune system
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2765586/��
Helicobacter pylori arginase mutant colonizes arginase II ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3160534
Jul 28, 2011 �� Urease is an enzyme made by H. pylori the converts urea to ammonia and carbon dioxide. Arginase is an enzyme that converts arginine to ornithine and urea and is made by humans, mice and H. pylori. Knockout refers to a gene that is disrupted and no longer functional.
Cited by: 5
Publish Year: 2011
Author: Songhee H Kim, Melanie L Langford, Jean-Luc Bouc��
The Immune Battle against Helicobacter pylori Infection: NO Offense: Trends in Microbiology
https://www.cell.com/trends/microbiology/fulltext/S0966-842X(16)00041-X��
The Immune Battle against Helicobacter pylori Infection: NO Offense - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0966842X1600041X��
Arginase of Helicobacter Gastric Pathogens Uses a Unique Set of Non-catalytic Residues for Catalysis: Biophysical Journal
National Institute of Immunology, New Delhi, India
https://www.cell.com/biophysj/fulltext/S0006-3495(17)30168-6
Helicobacter pylori arginase, a bimetallic enzyme, is crucial for pathogenesis of the bacterium in human stomach. Despite conservation of the signature motifs in all arginases, the H. pylori homolog has a non-conserved motif (153ESEEKAWQKLCSL165), whose role was recently shown to be critical for its stability and function.The sequence analysis also reveals the presence of this motif with critical residues in the homolog of other Helicobacter gastric pathogens. However, the underlying mechanism for its significance in catalytic function is not clearly understood. Using H. pylori arginase, our studies reveal that the interactions of His122 and Tyr125 with Trp159 are indispensable for tertiary structural intactness through optimal positioning of the motif and thus for the catalytic function. The single and double mutants of His122 and Tyr125 not only enhanced the solvent accessibility and conformational flexibility of Trp159 in the holo protein, but also showed complete loss of catalytic activity. An intact bimetallic center and unaltered substrate binding indicate that proper positioning of the motif by aromatic-aromatic contact is vital for the generation of a catalytically active conformation.
Additionally, the metal ions provide higher stability to the holo protein. We also identified the presence of these two residues exclusively in arginase of other Helicobacter gastric pathogens, which may have similar function.
Therefore, to the best of our knowledge, these findings highlight for the first time that
arginase of all Helicobacter gastric pathogens utilizes a unique non-catalytic triad for catalysis, which could be exploited for therapeutics.��
INTRODUCTION
Helicobacter pylori is a human gastric pathogen that infects half of the world-wide populations, causing chronic active gastritis, peptic ulcer, gastric adenocarcinoma, and mucosa-associated lymphoid tissue lymphoma (1, 2, 3, 4, 5). Chronic gastritis induced by H. pylori is the strongest known risk factor for the gastric adenocarcinoma, the second leading cause of cancer-related death (6, 7, 8). The bacterium has a complete urea cycle and contains the rocF gene encoding arginase (9, 10). Alterations in the availability of L-arginine and its metabolism into polyamines contribute significantly to the dysregulation of the host immune response to H. pylori infection (11). The binuclear Mn2+-metalloenzyme arginase, a member of the ureohydrolase family, catalyzes the conversion of L-arginine to L-ornithine and urea (9), where ornithine is further converted into polyamines, which are essential for various critical metabolic processes. The product urea is further utilized by urease to produce ammonia. This provides acid resistance and is thus important for colonization of the bacterium in the gastric epithelial cells (12, 13). Arginase of H. pylori also plays a role in evasion of the pathogen from the host immune system mainly by various proposed mechanisms, as suggested below. RocF competes with host-inducible nitric oxide (NO) synthase for the common substrate L-arginine, and thus reduces the synthesis of NO, an important component of innate immunity and an effective antimicrobial agent that is able to kill the invading pathogens directly (14, 15). The enzyme is involved in the inhibition of human T cell proliferation and T cell CD3�� expression and thus efficiently reduces host cellular immune response (16). Therefore, RocF could inhibit the host innate defense and adaptive immune response (17) and also facilitate the pathogenesis. Thus, this enzyme is considered to be a new virulence factor of H. pylori (18).
Figure 2(A) Surface view of the simulated structure of H. pylori apo-arginase at the 450 ns timescale showing the insertion motif in purple and Trp159 in red. (B) Enlarged view of the boxed region in (A). Trp159 is in close contact with the surrounding aromatic amino acids His122 and Tyr125 near the active site. Trp159 also shows a hydrogen-bonding contact with Asp126 at a distance of 2.65 Å. The two metal ions have been depicted based on the closest homologous structure of H. pylori arginase. To see this figure in color, go online.Using a combinational approach that includes site-directed mutagenesis, steady-state and time-resolved fluorescence measurements, anisotropy decay kinetics, heat-induced denaturation, circular dichroism, metal analysis, and kinetic assays, our data indicate that a network of interactions involving Trp159, His122, and Tyr125 retains proper positioning of the motif, which is crucial for tertiary structural intactness of the protein. The disruption of this set of interactions also leads to loss of catalytic activity, implying that maintaining proper tertiary structure through the above network is vital for its function. Moreover, we found that a bimetallic center is important for providing higher stability to the holo protein. Thus, this study not only highlights the presence of a novel non-catalytic triad of arginase in Helicobacter gastric pathogens, but also shows its evolutionary importance for function.
Arginase of Helicobacter Gastric Pathogens Uses a Unique Set of Non-catalytic Residues for Catalysis: Biophysical Journal
https://www.cell.com/biophysj/fulltext/S0006-3495(17)30168-6��
J Immunol. Author manuscript; available in PMC 2010 Mar 18.
Arginase II Restricts Host Defense to Helicobacter pylori by Attenuating Inducible Nitric Oxide Synthase Translation in Macrophages
Nuruddeen D. Lewis,*† Mohammad Asim,*‡ Daniel P. Barry,*‡ Kshipra Singh,*‡ Thibaut de Sablet,*‡ Jean-Luc Boucher,�� Alain P. Gobert,*¶ Rupesh Chaturvedi,*‡ and Keith T. Wilson*†‡
Author information Copyright and License information Disclaimer
* Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37240
† Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN 37240
‡ Veterans Affairs, Tennessee Valley Healthcare System, Nashville, TN 37212
�� Universit�� Ren�� Descartes, Paris
¶ Institut National de la Recherche Agronomique, Unit�� de Microbiologie UR454, Saint-Gen��s-Champanelle, France
Abstract
Helicobacter pylori infection of the stomach causes peptic ulcer disease and gastric cancer. Despite eliciting a vigorous immune response, the bacterium persists for the life of the host.An important antimicrobial mechanism is the production of NO derived from inducible NO synthase (iNOS). We have reported that macrophages can kill H. pylori in vitro by an NO-dependent mechanism, but supraphysiologic levels of the iNOS substrate L-arginine are required. Because H. pylori induces arginase activity in macrophages, we determined if this restricts NO generation by reducing L-arginine availability.
Inhibition of arginase with S-(2-boronoethyl)-L-cysteine (BEC) significantly enhanced NO generation in H. pylori-stimulated RAW 264.7 macrophages by enhancing iNOS protein translation but not iNOS mRNA levels. This effect resulted in increased killing of H. pylori that was attenuated with an NO scavenger.
In contrast, inhibition of arginase in macrophages activated by the colitis-inducing bacterium Citrobacter rodentium increased NO without affecting iNOS levels. H. pylori upregulated levels of arginase II (Arg2) mRNA and protein, which localized to mitochondria, whereas arginase I was not induced. Increased iNOS protein and NO levels were also demonstrated by small interfering RNA knockdown of Arg2 and in peritoneal macrophages from C57BL/6 Arg2−/− mice.
In H. pylori-infected mice, treatment with BEC or deletion of Arg2 increased iNOS protein levels and NO generation in gastric macrophages, but treatment of Arg2−/− mice with BEC had no additional effect.
These studies implicate Arg2 in the immune evasion of H. pylori by causing intracellular depletion of L-arginine and thus reduction of NO-dependent bactericidal activity.
Arginase II Restricts Host Defense to Helicobacter pylori by Attenuating Inducible Nitric Oxide Synthase Translation in Macrophages
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2841360/��
J Biol Chem. 2005 Jan 28;280(4):2409-12. Epub 2004 Nov 17.
Spermine causes loss of innate immune response to Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation.
Bussi��re FI1, Chaturvedi R, Cheng Y, Gobert AP, Asim M, Blumberg DR, Xu H, Kim PY, Hacker A, Casero RA Jr, Wilson KT.
Author information
1 Department of Medicine, Division of Gastroenterology, and Greenebaum Cancer Center, University of Maryland School of Medicine, Baltimore, USA.
Abstract
Helicobacter pylori infection of the stomach elicits a vigorous but ineffective host immune and inflammatory response, resulting in persistence of the bacterium for the life of the host. We have reported that in macrophages, H. pylori up-regulates inducible NO synthase (iNOS) and antimicrobial NO production, but in parallel there is induction of arginase II, generating ornithine, and of ornithine decarboxylase (ODC), generating polyamines. Spermine, in particular, has been shown to restrain immune response in activated macrophages by inhibiting proinflammatory gene expression. We hypothesized that spermine could prevent the antimicrobial effects of NO by inhibiting iNOS in macrophages activated by H. pylori. Spermine did not affect the up-regulation of iNOS mRNA levels but in a concentration-dependent manner significantly attenuated iNOS protein levels and NO production. Reduction in iNOS protein was due to inhibition of iNOS translation and not due to iNOS degradation. ODC knockdown with small interfering (si) RNA resulted in increased H. pylori-stimulated iNOS protein expression and NO production without altering iNOS mRNA levels. When macrophages were cocultured with H. pylori, killing of bacteria was enhanced by transfection of ODC siRNA and prevented by addition of spermine. These results identify a mechanism of immune dysregulation induced by H. pylori in which stimulated spermine synthesis by the arginase-ODC pathway inhibits iNOS translation and NO production, leading to persistence of the bacterium and risk for peptic ulcer disease and gastric cancer.
��Spermine causes loss of innate immune response to Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/15548540��
Structure of Arginase
A common feature of all arginases studied so far, whether eukaryotic or prokaryotic, is the requirement of cations with a positive two charge for activity. Manganese2+ is, by far, the most common activator of arginase, although the divalent cation requirements for some forms of the protein have been reportedly satisfied by cobalt, nickel, and iron [check the ��Manganese�� box to the left to view the location of these ions]. Experiments have shown that fully manganese-activated arginase contains two manganese (II) ions per subunit and that these ions form electron paramagnetic resonance (EPR) spin-coupled binuclear centers. EPR is a technique that has been proven to be beneficial to examining and understanding chemicals that have one or more unpaired electrons (7). EPR is comparable to the processes of nuclear magnetic resonance (NMR), except it analyzes the spin of the unpaired electrons instead of the spin of atomic nuclei (7). Detailed analyses of the temperature dependence of the manganese (II) EPR properties indicate a separation of 3.36�C3.57 Šbetween manganese ions in the native enzyme.
Figure 4. This figure concisely illustrates the effects of increased arginase activity. (12)
Arginase
https://chemistry.berea.edu/~biochemistry/2011/lb_dw/��
Arginase inhibitors
The first arginase inhibitors to be developed had non-specific actions and many side effects because of the high concentrations required [8]. For example, norvaline is a substrate for amidotransferases [88]. Similarly, while N��-hydroxy-L-arginine (NOHA) is a potent inhibitor for arginase, it is also an intermediate precursor in the production NO from L-arginine by NOS. An analog, Nor-NOHA is also a potent arginase inhibitor, but has a much longer half-life than NOHA. Neither inhibits NOS. ��-Difluoromethylornithine (DFMO) is a non-specific weak inhibitor of arginase, but is a potent inhibitor for ornithine decarboxylase [8]. Thus, the effects of DFMO in increasing NO production in models of elevated arginase activity are likely due to increased accumulation of ornithine which is known to inhibit arginase [89]. Besides producing urea, arginase is also involved in synthesis of polyamines and amino acids such as ornithine, proline and glutamate. In fact, ornithine, leucine, valine, lysine, isoleucine and nor-valine inhibit arginase with ornithine being the most potent [90]. Also, L-citrulline is an allosteric inhibitor of arginase [91], but it also increases NO formation.
Recently, competitive inhibitors of arginase have been developed which have greater specificity for the enzyme. Initial development of these compounds involved determination of the crystal structure of arginase. Christianson and colleagues found that a binuclear manganese cluster was required for catalytic activity [92]. They also identified the full structures of human A1 and A2 [93]. Boronic acid analogs of L-arginine [S-(2-boronoethyl)-L-cysteine (BEC) and 2(S)-amino-6-boronohexanoic acid (ABH)] are highly selective arginase inhibitors. Both contain a boronic acid or N-hydroxyguanidinium head which binds the manganese cluster, the active catalytic site in arginase [49]. Several inhibitors of arginase are commercially available, including nor-NOHA, BEC and ABH.
Although several specific arginase inhibitors have been developed, none of those currently available are isoform selective. This is a significant limitation of the field in light of the growing evidence that arginase activity can be damaging in some contexts and protective in others. For example, activity of A2-positive macrophages has been implicated in the development and progression of atherosclerosis, whereas A1-positive macrophages may promote plaque resolution [33�C36, 94]. A1-positive macrophages are clearly involved in repair functions after tissue injury, whereas excessive A2 activity appears to be damaging in a number of CNS disease/injury conditions. In the absence of isoform selective inhibitors, RNA interference has been used to specifically inhibit expression of A1 or 2. For example, administration of short hairpin RNA (shRNA) against A1 greatly reduced IL-13 induced airway hyper-responsiveness with a concomitant decrease in A1 mRNA and protein levels in the lungs of mice [95]. Direct delivery of anti-sense A1 using adeno-virus vector in corpus cavernosum improved erectile function in aged mice [96]. Anti-sense A1 also increased NO production in endothelial cells exposed to high glucose [12]. The search for more potent and isoform selective inhibitors is ongoing and involves structure-based design and plant extracts [38].
So far studies using arginase inhibitors in humans with cardiovascular disease has been limited to small-scale clinical ��proof-of-concept�� studies involving local administration of arginase inhibitors by cutaneous microdialysis or intra-arterial infusion. However, promising results have been reported in patients with coronary artery disease and type 2 diabetes [30, 97], heart failure [98], hypertension [99] and following resuscitation after cardiac arrest [100]. These observations suggest that inhibiting arginase activity could be beneficial in the treatment of cardiovascular disease. Systemic treatment with arginase inhibitors is used in the treatment of parasitic disease without significant adverse effects. However further study is needed to demonstrate the safety of long-term treatment in patients with cardiovascular disease. Given the important role that A1 plays in detoxification of ammonia in the urea cycle, it will be important to make sure that the treatment does not suppress hepatic arginase activity enough to adversely impact the urea cycle. Another potential limitation is that suppression of A1 may further limit tissue repair function which is already suppressed in many cardiovascular diseases.Arginase: an old enzyme with new tricks
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4461463/��
��
https://www.slideshare.net/SiuKeeLee/redox-reactions-9065170
��
https://www.slideshare.net/nazimah55/redox-quiz-part-1-with-answers
��
��
��
��
Helicobacter pylori: Physiology and Genetics.
Chapter 15 Evasion of the Toxic Effects of Oxygen
Stuart L. Hazell, Andrew G. Harris, and Mark A. Trend.
Stuart L. Hazell, Andrew G. Harris, and Mark A. Trend.
Affiliations
School of Science, Food and Horticulture, College of Science, Technology and Environment, University of Western Sydney, Sydney, Australia
Oxygen as a Toxic Species
Oxygen is an efficient terminal electron acceptor in respiratory pathways. During aerobic respiration the electron transport chain generates free radical oxygen species as a result of electron leakage; this generation of toxic species is proportional to the oxygen tension (51). In addition, toxic oxygen species (TOS) may be formed exogenously, for example, by chemical processes or through radiation. TOS also result from the oxidative burst of polymorphonuclear leukocytes (PMN). Infection with Helicobacter pylori induces an inflammatory response (gastritis), which leads to an increase in the level of TOS in the gastric mucosa and the gastric juice (4, 24�C26, 59). This increase in the level of toxic metabolites is probably the result of the generation of the superoxide anion (O2��− ), a reactive TOS, formed as part of the oxidative burst of PMN and enzymic activities of gastric epithelial cells. There is evidence that H. pylori infection leads to increased production of O2��− via NADPH oxidase in gastric cells, stimulated by lipopolysaccharide as well as xanthine oxidase, another mechanism for the generation of oxygen-derived free radicals (8, 80). In response to increased superoxide anion production in gastric tissue, changes have been detected in the level of expression of human superoxide dismutase (SOD) (12). Human gastric SOD exists as a cytoplasmic copper-zinc-superoxide dismutase (Cu, Zn-SOD) found in gland cells of the gastric body and antral mucosa, and as a manganese-superoxide dismutase (Mn-SOD) within mitochondria (63). An increase in the amount and activity of Mn-SOD has been observed in response to H. pylori infection and gastritis, whereas the amount and activity of the Cu, Zn-SOD remained constant or decreased slightly (39). It has been suggested that the induction of Mn-SOD is in response to increased cytokine production within the inflamed gastric mucosa (39). This situation is reversed following successful treatment of the infection (38). The data suggest that within the gastric environment H. pylori may be exposed to increased levels of TOS. In such an environment it is important for bacterial survival that the impact of such TOS be neutralized.
Conclusion
H. pylori is a microaerophile that colonizes the inflamed gastric mucosa of humans. These two facts suggest the presence of a network of systems needed to manage both the oxygen to which H. pylori is exposed and the oxidative stress induced by endogenous and exogenous processes. That oxygen and TOS are constant companions of H. pylori in vivo is reflected in the enzymes expressed to manage them and the regulatory and repair mechanisms developed by the bacterium to cope with this type of stress. Nonetheless, our understanding of how H. pylori evades and avoids toxic oxygen effects is far from complete; and despite the importance of the topic, the management of oxygen and oxidative stress in H. pylori is a relatively neglected subject area.Evasion of the Toxic Effects of Oxygen - Helicobacter pylori - NCBI Bookshelf
https://www.ncbi.nlm.nih.gov/books/NBK2416/
Helicobacter pylori Biofilm Formation and Its Potential Role in Pathogenesis
Skander Hathroubi, Stephanie L. Servetas, Ian Windham, D. Scott Merrell, Karen M. Ottemann
SUMMARY
Despite decades of effort, Helicobacter pylori infections remain difficult to treat. Over half of the world's population is infected by H. pylori, which is a major cause of duodenal and gastric ulcers as well as gastric cancer. During chronic infection, H. pylori localizes within the gastric mucosal layer, including deep within invaginations called glands; thanks to its impressive ability to survive despite the harsh acidic environment, it can persist for the host's lifetime. This ability to survive and persist in the stomach is associated with urease production, chemotactic motility, and the ability to adapt to the fluctuating environment. Additionally, biofilm formation has recently been suggested to play a role in colonization. Biofilms are surface-associated communities of bacteria that are embedded in a hydrated matrix of extracellular polymeric substances. Biofilms pose a substantial health risk and are key contributors to many chronic and recurrent infections. This link between biofilm-associated bacteria and chronic infections likely results from an increased tolerance to conventional antibiotic treatments as well as immune system action. The role of this biofilm mode in antimicrobial treatment failure and H. pylori survival has yet to be determined. Furthermore, relatively little is known about the H. pylori biofilm structure or the genes associated with this mode of growth. In this review, therefore, we aim to highlight recent findings concerning H. pylori biofilms and the molecular mechanism of their formation. Additionally, we discuss the potential roles of biofilms in the failure of antibiotic treatment and in infection recurrence.
...
IMMUNE EVASION AND TREATMENT FAILURE
Studies of H. pylori Biofilms and Antibiotic Resistance
While there are still many unanswered questions regarding H. pylori biofilm formation, the implications of biofilm-associated infections are well documented for other species (3, 83, 100). The possibility that H. pylori may adopt a biofilm mode of growth during colonization of the gastric mucosa and gastric glands could have a profound impact on antimicrobial treatment. For multiple bacteria, biofilms are known to render infections of multiple bacteria more difficult to eradicate with antimicrobial therapy (3, 8, 83, 100, 101). Bacterial biofilms resist antimicrobial treatments by using a transient and noninheritable mechanism called tolerance, which is related to the physiological state of the biofilm cells and the physical barrier formed by the extracellular matrix (8, 100, 101). In addition to antibiotic tolerance, biofilms often represent a reservoir of genetic diversity by promoting genetic exchange between different subpopulations. Several studies have documented the increased dissemination of antibiotic resistance genes in biofilms through horizontal gene transfer, integrative conjugative elements, and natural transformation (102�C104). Additionally, bacterial cells within biofilms undergo several stressful conditions, including major nutrient starvation, which could increase the mutation frequency and the emergence of antibiotic-resistant mutant strains (8, 69, 105, 106).
Biofilm-associated H. pylori was shown to be more resistant in vitro to clarithromycin, which is one of the common antibiotics used to treat H. pylori infections (107). Specifically, the MIC increased by 16-fold and the minimum bactericidal concentration (MBC) increased by up to 4-fold in biofilm cells compared to planktonic ones (107). H. pylori biofilm cells also showed an increased mutation rate, which could promote clarithromycin-resistant mutations (107).
The contribution of H. pylori biofilms to the spread of antibiotic resistance has not been fully explored; however, the emergence of resistance to clarithromycin, levofloxacin, and metronidazole urges us to develop new strategies that take into account not only bacterial resistance but also the mode of growth.USE OF NAC FOR H.pylori BIOFILM
While the field of H. pylori biofilm research is fairly new, groups have already begun to explore alternative therapeutic approaches that may target and eradicate biofilms. The use of the mucolytic, thiol-containing compound N-acetylcysteine (NAC) has shown promise in both humans and mice (21, 108). In early studies that looked at H. pylori-infected mice, daily treatment with NAC (120 mg for 14 days) successfully reduced the H. pylori load by almost 1 log compared to the nontreated group. Pretreatment with NAC (40 mg/day) significantly reduced the H. pylori load but did not completely prevent colonization (108). Furthermore, NAC was also shown to augment the activity of clarithromycin and lansoprazole, a proton pump inhibitor (PPI) dual therapy. H. pylori-positive patients that received both dual therapy and NAC showed significantly better eradication of H. pylori than did those on dual therapy only (109). The mechanism by which NAC functions in the treatment of H. pylori is not clear; however, subsequent studies showed that NAC was effective at dispersing preexistent in vitro H. pylori biofilms and at preventing their formation (21). NAC is believed to function by disrupting disulfide bonds that cross-link glycoproteins in the mucus. While the exact proteins targeted by NAC are not known, it is possible that NAC targets the proteins of the biofilm matrix and leads to their destruction. In a clinical trial, NAC was demonstrated to similarly disperse H. pylori biofilms during in vivo infections (21). Indeed, subjects with a history of at least 4 H. pylori eradication failures who were treated with NAC before receiving a traditional antibiotic regimen demonstrated better clearance of H. pylori (65%) than did the non-NAC group (20%) (21). The exact molecular mechanisms underlying the reported therapeutic effect of NAC are not clear, since this molecule seems to be able to target both H. pylori biofilms and the gastric mucus. Further studies are required to decipher NAC's activity against H. pylori and its biofilms and to help assess its efficacy for possible future antimicrobial therapies.��
Helicobacter pylori Biofilm Formation and Its Potential Role in Pathogenesis | Microbiology and Molecular Biology Reviews
https://mmbr.asm.org/content/82/2/e00001-18��
Journal of Medical Microbiology
Interaction of Helicobacter pylori Cell Membrane with Non-Esterified Cholesterol and Other Steroids
Hirofumi Shimomura, Kouichi Hosoda, Yoshikazu Hirai
Division of Bacteriology, Department of Infection and Immunity, Jichi Medical University, Tochigi, JapanKeywords: Helicobacter pylori; Cholesterol-��-glucosyltransferase; Phosphatidylethanolamine
ABSTRACT
Helicobacter pylori performs the unique action of assimilating exogenous non-esterified cholesterol into its cell membrane. This bacterium aggressively incorporates non-esterified cholesterol into the membrane, induces its glucosylation, and uses both non-esterified cholesterol and glucosylated cholesterols as membrane lipid compositions. The reason for this assimilation of non-esterified cholesterol into the cell membrane of H. pylori has eluded investigators for many years.
Recent hypotheses posit that the sterol-uptake and sterol-glucosylation contribute to the survival of H. pylori cells in different ways.
The incorporation of the non-esterified cholesterol into the cell membrane fortifies the resistance of H. pylori against the antibacterial actions of phosphatidylcholines, antibiotics, and bile salts. In parallel, the glucosylation of the non-esterified cholesterol incorporated into the cell membrane serves H. pylori in two ways. First, it helps the bacterium evade host immune responses, such as phagocytosis by macrophages and activation of antigen-specific T cells. Second, it detoxifies sterols fatal to the bacterium via a novel action of sterol glucosylation recently described in another report from our group.
The reluctance of H. pylori to absorb esterified cholesterol remains unexplained. A recent study by our group has demonstrated that the phosphatidylethanolamine (PE) in the outer membrane of H. pylori serves as a steroid-binding lipid the incorporation of non-esterified cholesterol into the membrane. We have also discovered that the myristic acid (C14:0) molecule attached to the PE of this bacterium plays an important role in the selective binding of non-esterified cholesterol but not esterified cholesterol.
Figure 2. How H. pylori acquires resistance to the bacteriolytic action of phosphatidylcholine by incorporating steroid compounds into the cell membrane. FC, non-esterified cholesterol; E1, estrone; PC, phosphatidylcholine.
http://file.scirp.org/Html/11-2260054_28667.htm
��
��
https://link.springer.com/article/10.1007/s00535-014-0938-y
��
��
Phytother Res. 2018 Jul;32(7):1289-1296. doi: 10.1002/ptr.6057. Epub 2018 Feb 26.
Astragaloside IV inhibits TGF-��1-induced epithelial-mesenchymal transition through inhibition of the PI3K/Akt/NF-��B pathway in gastric cancer cells.
Zhu J1, Wen K1.
Author information
1 Department of Ultrasound, Huaihe Hospital of Henan University, Kaifeng, 475000, Henan, China.
Abstract
Astragaloside IV (AS-IV) has been reported to possess anti-metastasis activity in cancer cells. However, it is unknown whether AS-IV could inhibit epithelial-mesenchymal transition (EMT), a cellular de-differentiation program that promotes metastasis, in cancer cells.The aim of this study was to study the effect and mechanism of AS-IV on EMT in gastric cancer (GC) cells. The results showed that AS-IV significantly inhibited cell viability, invasion, and migration of GC cells.
The E-cadherin to N-cadherin switch and expression of Vimentin and metastasis-related genes were induced by transforming growth factor ��1 (TGF-��1), whereas AS-IV reversed the induction. In addition, AS-IV inhibited TGF-��1-induced activation of PI3K/Akt/NF-��B. Inhibition of the PI3K/Akt/NF-��B pathway reversed TGF-��1-induced EMT. In conclusion, AS-IV inhibited TGF-��1-induced EMT through inhibition of the PI3K/Akt/NF-��B pathway in GC cells. AS-IV might be an effective candidate for the treatment for GC.
Copyright © 2018 John Wiley & Sons, Ltd.
KEYWORDS:
NF-��B; PI3K/Akt; astragaloside IV; epithelial-mesenchymal transition; gastric cancer
PMID: 29480652 DOI: 10.1002/ptr.6057��
Astragaloside IV inhibits TGF-��1-induced epithelial-mesenchymal transition through inhibition of the PI3K/Akt/NF-��B pathway in gastric cancer cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29480652��
Korean J Intern Med. 2016 Jan; 31(1): 30�C35.
The antibacterial effect of fatty acids on Helicobacter pylori infection
Sung Woo Jung and Sang Woo Lee
Division of Gastroenterology and Hepatology, Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea
Abstract
Eradication of Helicobacter pylori is recommended for the management of various gastric diseases, including peptic ulcers and mucosa-associated lymphoid tissue lymphoma. Because of the increasing prevalence of antibiotic resistance, the eradication rates of antibiotic-based therapies have decreased. Therefore, alternative treatments should be considered. The antibacterial properties of fatty acids (FAs) have been investigated in various organisms, including H. pylori. Some FAs, particularly polyunsaturated FAs, have been shown to have bactericidal activity against H. pylori in vitro; however, their antibacterial effects in vivo remain controversial. Poor solubility and delivery of FAs may be important reasons for this discrepancy. Recently, a series of studies demonstrated the antibacterial effects of a liposomal formulation of linolenic acid against H. pylori, both in vitro and in vivo. Further research is needed to improve the bioavailability of FAs and apply them in clinical use.
Keywords: Helicobacter pylori, Fatty acids, Anti-bacterial agents, Liposomes, Drug delivery systems
��The antibacterial effect of fatty acids on Helicobacter pylori infection
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4712431/��
Mediators Inflamm. 2014;2014:128919. doi: 10.1155/2014/128919. Epub 2014 Jun 2.
Anti-inflammatory mechanism of polyunsaturated fatty acids in Helicobacter pylori-infected gastric epithelial cells.
Lee SE1, Lim JW1, Kim JM2, Kim H1.
Author information
1 Department of Food and Nutrition, Brain Korea 21 PLUS Project, College of Human Ecology, Yonsei University, Seoul 120-749, Republic of Korea.
2 Department of Microbiology, Hanyang University College of Medicine, Seoul 133-791, Republic of Korea.
Abstract
Helicobacter pylori is an important risk factor for gastric inflammation, which is mediated by multiple signaling pathways. The aim of this study was to investigate the effects of polyunsaturated fatty acids (PUFAs), such as linoleic acid (LA), alpha-linolenic acid (ALA), and docosahexaenoic acid (DHA), on the expression of the proinflammatory chemokine interleukin-8 (IL-8) in H. pylori-infected gastric epithelial AGS cells. To investigate whether PUFAs modulate H. pylori-induced inflammatory signaling, we determined the activation of epidermal growth factor receptor (EGFR), protein kinase C-�� (PKC ��), mitogen-activated protein kinases (MAPKs), nuclear factor-kappa B (NF- ��B), and activator protein-1 (AP-1) as well as IL-8 expression in H. pylori-infected gastric epithelial cells that had been treated with or without PUFAs. We found that PUFAs inhibited IL-8 mRNA and protein expression in H. pylori-infected cells. ��-3 fatty acids (ALA, and DHA) suppressed the activation of EGFR, PKC ��, MAPK, NF- ��B, and AP-1 in these infected cells. LA did not prevent EGFR transactivation and exhibited a less potent inhibitory effect on IL-8 expression than did ALA and DHA. In conclusion, PUFAs may be beneficial for prevention of H. pylori-associated gastric inflammation by inhibiting proinflammatory IL-8 expression.
PMID: 24987192 PMCID: PMC4060060 DOI: 10.1155/2014/128919��
��
https://www.ibc.sinica.edu.tw/wp-content/uploads/2020/03/20200313-Lin-CH-paper-photo-1.jpg
Tumor and Stem Cell Biology
IFN-�� Inhibits Gastric Carcinogenesis by Inducing Epithelial Cell Autophagy and T-Cell Apoptosis
Shui Ping Tu, Michael Quante, Govind Bhagat, Shigeo Takaishi, Guanglin Cui, Xiang Dong Yang, Sureshkumar Muthuplani, Wataru Shibata, James G. Fox, D. Mark Pritchard and Timothy C. Wang
Departments of 1Medicine and 2Pathology and Cell Biology, College of Physicians & Surgeons, Columbia University, New York;
3Department of Chemical Biology, Center for Cancer Prevention, Susam L. Cullman Laboratory of Cancer Research, Rutgers University, Piscataway, New Jersey;
4Division of Comparative Medicine, Massachusetts Institute of Technology, Cambridge, Massachusetts; and 5Division of Gastroenterology, School of Clinical Sciences, University of Liverpool, Liverpool, United Kingdom
Abstract
IFN-�� mediates responses to bacterial infection and autoimmune disease, but it is also an important tumor suppressor. It is upregulated in the gastric mucosa by chronic Helicobacter infection; however, whether it plays a positive or negative role in inflammation-associated gastric carcinogenesis is unexplored.To study this question, we generated an H+/K+-ATPase-IFN-�� transgenic mouse that overexpresses murine IFN-�� in the stomach mucosa. In contrast to the expected proinflammatory role during infection, we found that IFN-�� overexpression failed to induce gastritis and instead inhibited gastric carcinogenesis induced by interleukin-1beta (IL-1��) and/or Helicobacter infection. Helper T cell (Th) 1 and Th17 immune responses were inhibited by IFN-�� through Fas induction and apoptosis in CD4 T cells. IFN-�� also induced autophagy in gastric epithelial cells through increased expression of Beclin-1. Finally, in the gastric epithelium, IFN-�� also inhibited IL-1��- and Helicobacter-induced epithelial apoptosis, proliferation, and Dckl1+ cell expansion.
Taken together, our results suggest that IFN-�� coordinately inhibits bacterial infection and carcinogenesis in the gastric mucosa by suppressing putative gastric progenitor cell expansion and reducing epithelial cell apoptosis via induction of an autophagic program. Cancer Res; 71(12); 4247�C59. ©2011 AACR.
��IFN-�� Inhibits Gastric Carcinogenesis by Inducing Epithelial Cell Autophagy and T-Cell Apoptosis | Cancer Research
https://cancerres.aacrjournals.org/content/71/12/4247��
J Immunol. 2010 Mar 1;184(5):2572-82. doi: 10.4049/jimmunol.0902436. Epub 2010 Jan 22.
Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages.
Lewis ND1, Asim M, Barry DP, Singh K, de Sablet T, Boucher JL, Gobert AP, Chaturvedi R, Wilson KT.
Author information
1 Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37240, USA.
Abstract
Helicobacter pylori infection of the stomach causes peptic ulcer disease and gastric cancer. Despite eliciting a vigorous immune response, the bacterium persists for the life of the host. An important antimicrobial mechanism is the production of NO derived from inducible NO synthase (iNOS). We have reported that macrophages can kill H. pylori in vitro by an NO-dependent mechanism, but supraphysiologic levels of the iNOS substrate l-arginine are required. Because H. pylori induces arginase activity in macrophages, we determined if this restricts NO generation by reducing l-arginine availability. Inhibition of arginase with S-(2-boronoethyl)-l-cysteine (BEC) significantly enhanced NO generation in H. pylori-stimulated RAW 264.7 macrophages by enhancing iNOS protein translation but not iNOS mRNA levels. This effect resulted in increased killing of H. pylori that was attenuated with an NO scavenger. In contrast, inhibition of arginase in macrophages activated by the colitis-inducing bacterium Citrobacter rodentium increased NO without affecting iNOS levels. H. pylori upregulated levels of arginase II (Arg2) mRNA and protein, which localized to mitochondria, whereas arginase I was not induced. Increased iNOS protein and NO levels were also demonstrated by small interfering RNA knockdown of Arg2 and in peritoneal macrophages from C57BL/6 Arg2(-/-) mice. In H. pylori-infected mice, treatment with BEC or deletion of Arg2 increased iNOS protein levels and NO generation in gastric macrophages, but treatment of Arg2(-/-) mice with BEC had no additional effect. These studies implicate Arg2 in the immune evasion of H. pylori by causing intracellular depletion of l-arginine and thus reduction of NO-dependent bactericidal activity.
PMID: 20097867 PMCID: PMC2841360 DOI: 10.4049/jimmunol.0902436Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/20097867��
Why Your Stomach Will Love Turmeric and Licorice
Add Spice and Sweetness to Your Life
These ancient medicinal remedies have been
shown to kill H. pylori, the cause of most ulcers
By Hyla Cass, M.D.H. pylori Can Kill People
Now that we know all about licorice and turmeric, it��s time to see what has brought them together in this article. It turns out that both of them��but turmeric especially��are effective killers of one of the most destructive bacteria known to humankind: Helicobacter pylori.2 This malevolent bug infects the gastrointestinal tracts of about half the people on earth. It��s the primary cause of gastric and duodenal ulcers (collectively called peptic ulcers), as well as gastritis and stomach cancer.
The World Health Organization classifies H. pylori as a Class 1 carcinogen (��known human carcinogen��). Although most infected people remain asymptomatic, they are believed to have low-level gastritis, and about 5�C10% of them eventually develop severe gastric or duodenal disease��mainly ulcers, but also cancer.
Turmeric and Licorice Kill H. pylori
An international team of researchers from England, Sri Lanka, Italy, and France recently conducted laboratory studies of the effects of 25 culinary and medicinal plants against H. pylori.2 By boiling the appropriate plant parts in water, they obtained simple aqueous extracts simulating the ones used in primitive cultures, which lack the more sophisticated chemical extraction techniques used in industrialized countries. They used these aqueous extracts against a standard reference strain of H. pylori and against six fresh clinical strains obtained from Italian patients with peptic ulcer disease. The idea was to measure the killing efficiency of the extracts and the time required for maximum action.
Turmeric emerged as the clear champion in these tests, killing 100% of the bacteria within 15 minutes. Licorice too killed 100%, although it took longer: up to 60 minutes. Curiously, the effects in all 25 plants were all-or-nothing within the 60-minute limit: 17 plants killed H. pylori with 100% efficiency or extremely close to it (high 90s), and 8 plants were duds, with 0% killing. The results of these experiments confirmed what had been published previously regarding the bactericidal effects of turmeric3 and licorice4,5against H. pylori.
If You Don��t Kill Them, Flush Them
The researchers also measured the plant extracts�� ability to inhibit the adhesion of H. pylori to the inner lining of the stomach��a biochemical process that��s critical for the bacteria��s ability to take up residence there and cause long-term infection. This experiment represents an interesting and potentially useful therapeutic approach, complementary to the outright killing of the bacteria. If one could prevent them from taking up residence in the GI tract, it would not even be necessary to kill them��they would just be flushed out, so to speak.Why Your Stomach Will Love Turmeric and Licorice - Life Enhancement Products
https://life-enhancement.com/pages/why-your-stomach-will-love-turmeric-and-licorice��
World J Gastroenterology. 2020 Jul 7;
Type I and type II Helicobacter pylori infection status and their impact on gastrin and pepsinogen level in a gastric cancer prevalent area
1Department of Gastroenterology and Hepatology, People's Hospital of Zhengzhou University, Henan Province, China.
Background: Type I Helicobacter pylori (H. pylori) infection causes severe gastric inflammation and is a predisposing factor for gastric carcinogenesis. However, its infection status in stepwise gastric disease progression in this gastric cancer prevalent area has not been evaluated; it is also not known its impact on commonly used epidemiological gastric cancer risk markers such as gastrin-17 (G-17) and pepsinogens (PGs) during clinical practice.
Aim: To explore the prevalence of type I and type II H. pylori infection status and their impact on G-17 and PG levels in clinical practice.
Methods: Thirty-five hundred and seventy-two hospital admitted patients with upper gastrointestinal symptoms were examined, and 523 patients were enrolled in this study. H. pylori infection was confirmed by both 13C-urea breath test and serological assay. Patients were divided into non-atrophic gastritis (NAG), non-atrophic gastritis with erosion (NAGE), chronic atrophic gastritis (CAG), peptic ulcers (PU) and gastric cancer (GC) groups. Their serological G-17, PG I and PG II values and PG I/PG II ratio were also measured.
Results: A total H. pylori infection rate of 3572 examined patients was 75.9%, the infection rate of 523 enrolled patients was 76.9%, among which type I H. pylori infection accounted for 72.4% (291/402) and type II was 27.6%; 88.4% of GC patients were H. pylori positive, and 84.2% of them were type I infection, only 11.6% of GC patients were H. pylori negative. Infection rates of type I H. pylori in NAG, NAGE, CAG, PU and GC groups were 67.9%, 62.7%, 79.7%, 77.6% and 84.2%, respectively. H. pylori infection resulted in significantly higher G-17 and PG II values and decreased PG I/PG II ratio. Both types of H. pylori induced higher G-17 level, but type I strain infection resulted in an increased PG II level and decreased PG I/PG II ratio in NAG, NAGE and CAG groups over uninfected controls. Overall PG I levels showed no difference among all disease groups and in the presence or absence of H. pylori; in stratified analysis, its level was increased in GC and PU patients in H. pylori and type I H. pylori-positive groups.
Conclusion: Type I H. pylori infection is the major form of infection in this geographic region, and a very low percentage (11.6%) of GC patients are not infected by H. pylori. Both types of H. pylori induce an increase in G-17 level, while type I H. pylori is the major strain that affects PG I and PG IIs level and PG I/PG II ratio in stepwise chronic gastric disease. The data provide insights into H. pylori infection status and indicate the necessity and urgency for bacteria eradication and disease prevention in clinical practice.
Keywords: Helicobacter pylori; Chronic gastric diseases; Gastric cancer; Gastrin-17; Pepsinogen.����Jθ����ѧ�� 2020��7��7��;
θ�����е�����I�ͺ�II�������ݸ˾���Ⱦ״�������θ���غ�θ����øԭˮƽ��Ӱ��
1����֣�ݴ�ѧ����ҽԺ�����ڿƣ�����
������I�������ݸ˾���H. pylori����Ⱦ�������ص�θ����֢����θ������������Ȼ������δ������θ�����е���������θ����չ�еĸ�Ⱦ״����������������ٴ�ʵ���жԳ��õ����в�ѧθ��Σ��ָ�꣨��θ����17��G-17����θ����øԭ��PGs������Ӱ�졣
Ŀ�ģ�̽���ٴ�ʵ����I�ͺ�II�������ݸ˾���Ⱦ״�������G-17��PGˮƽ��Ӱ�졣
��������3572��סԺ����������֢״���߽����˼�飬������523�����ߡ������ݸ˾���Ⱦͨ��13C-���غ��������Ѫ��ѧ�ⶨ��ȷ֤�����߷�Ϊ��ή����θ�ף�NAG������ή����������θ�ף�NAGE��������ή����θ�ף�CAG��������������PU����θ����GC���顣�����������ǵ�Ѫ��G-17��PG I��PG IIֵ�Լ�PG I / PG II�ȡ���������ܼ���3572�������ݸ˾��ܸ�Ⱦ��Ϊ75.9����523�����黼�ߵĸ�Ⱦ��Ϊ76.9��������I�������ݸ˾���Ⱦռ72.4����291/402����II�������ݸ˾���ȾΪ27.6�� ��; GC��������88.4��Ϊ�����ݸ˾����ԣ�����I��ȾΪ84.2����GC������ֻ��11.6��Ϊ�����ݸ˾����ԡ� NAG��NAGE��CAG��PU��GC���I�������ݸ˾���Ⱦ�ʷֱ�Ϊ67.9����62.7����79.7����77.6����84.2���������ݸ˾���Ⱦ����G-17��PG IIֵ�������ߣ���PG I / PG II��ֵ���͡��������͵������ݸ˾����յ��ϸߵ�G-17ˮƽ������δ��Ⱦ�Ķ�������ȣ�NAG��NAGE��CAG���е�I�;����Ⱦ����PG IIˮƽ���ߺ�PG I / PG II�Ƚ��͡�����PG Iˮƽ�����м�����֮���Լ��Ƿ���������ݸ˾������졣�ڷֲ�����У������ݸ˾���I�������ݸ˾��������GC��PU��������ˮƽ���ߡ�
���ۣ�I�������ݸ˾���Ⱦ�Ǹõ����������Ҫ��Ⱦ��ʽ������ֻ�м��Ͱٷֱȵ�θ�����ߣ�11.6����δ�ܵ������ݸ˾���Ⱦ���������͵������ݸ˾����յ�G-17ˮƽ���ߣ���I�������ݸ˾���Ӱ�����������θ����PG I��PG IIsˮƽ�Լ�PG I / PG II�ȵ���Ҫ���ꡣ��Щ�����ṩ���й������ݸ˾���Ⱦ״���ļ��⣬��ָ�������ٴ�ʵ���и���ϸ����Ԥ�������ı�Ҫ�Ժͽ����ԡ�Type I and type II Helicobacter pylori infection status and their impact on gastrin and pepsinogen level in a gastric cancer prevalent area - PubMed
https://pubmed.ncbi.nlm.nih.gov/32742135/��
Lactoferrin in a Context of Inflammation-Induced Pathology
imageMarian L. Kruzel1, imageMichal Zimecki2 and imageJeffrey K. Actor1*
1McGovern Medical School, University of Texas, Health Science Center, Houston, TX, United States
2Polish Academy of Sciences, Institute of Immunology and Experimental Therapy, Wrocław, PolandLTF�Ŀ���������ѱ����֤�����������ֻ�����ɣ�һ�����������Եģ�������LTF�����ĸ��������־��ԣ�������һ����������LTF��֬���ǣ�LPS����������ֱ�����á����������Ծ���ɱ������С�仯�����絥�������̬�ԣ����ܻ�Ӱ��ֿ���ԭ��Ľ����23��24���� LTF����롰Σ���źš�ʶ���ϸ���������������[���磬�շ������壨TLR��4��CD14��CD22]��8�������������ϣ�LTF���Կ���������������Ϊ����ϸ������������ϸ������������з�Ӧ�����⣬ͨ�������Ĥ����ż����LTFӰ�쵥��ϸ��/����ϸ���ġ�Σ���źš�������磬��CD14�����ϵ�LTF��ϸ��LPS������ϸ���IJ��������25�������ҿ��Լ���NF-��B�յ��ĸ�����֢���ʵĻ���ת¼��26������ˣ�LTF�ڻ�е�ϲ��ֳ䵱������֢�ķ������ʡ�ͼ1ʾ���Եر�ʾ����LTFΪ�ؼ����ʵļ�����֢��չ�����е�һϵ���¼���
https://www.frontiersin.org/articles/256587
��
Have Chest Pains? It Could Just Be H Pylori. - HubPages
https://discover.hubpages.com/health/My-Experience-With-H-Pylori��
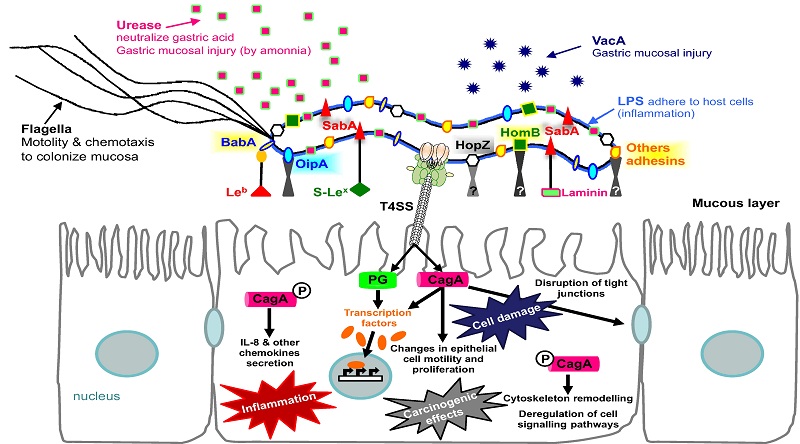

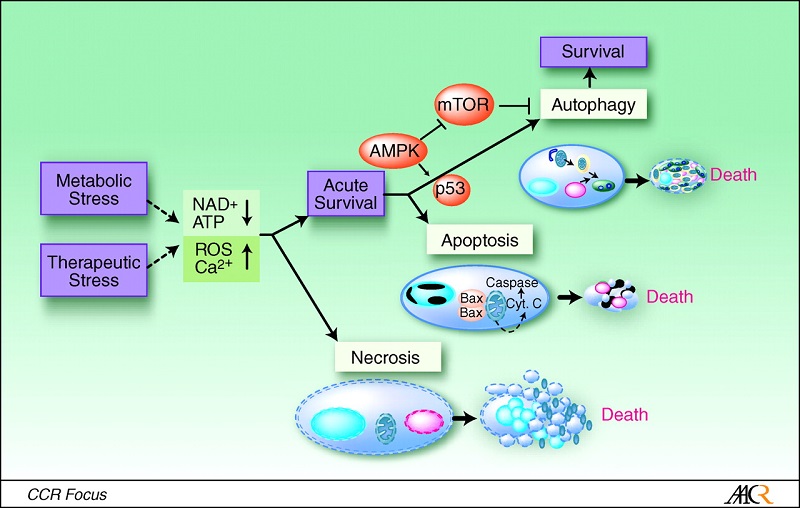
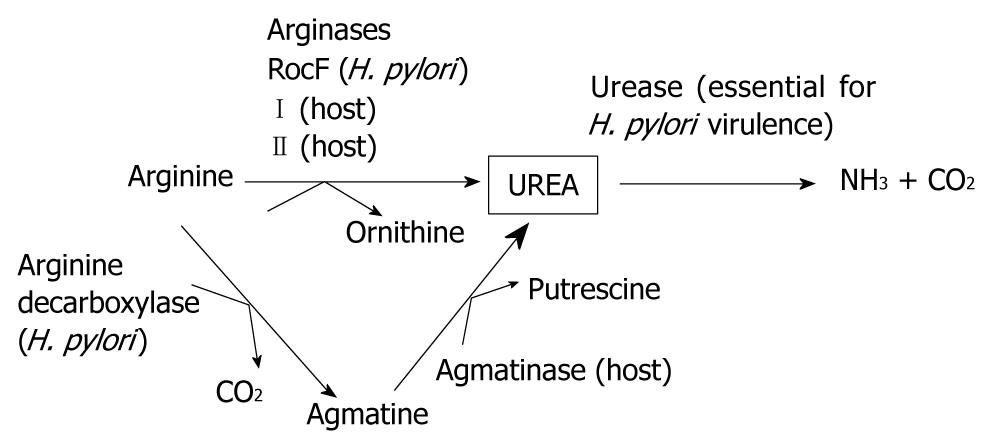


.png)

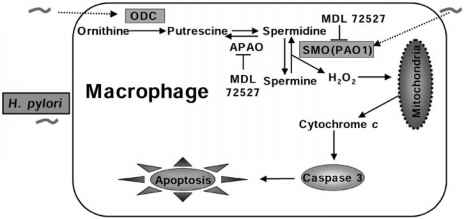
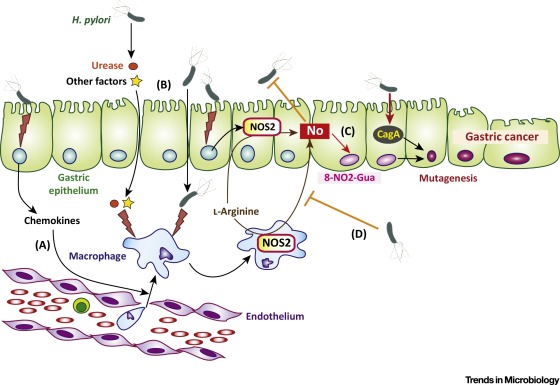
.jpg)


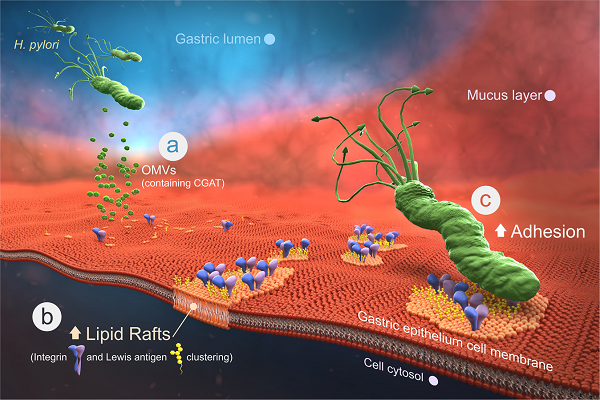
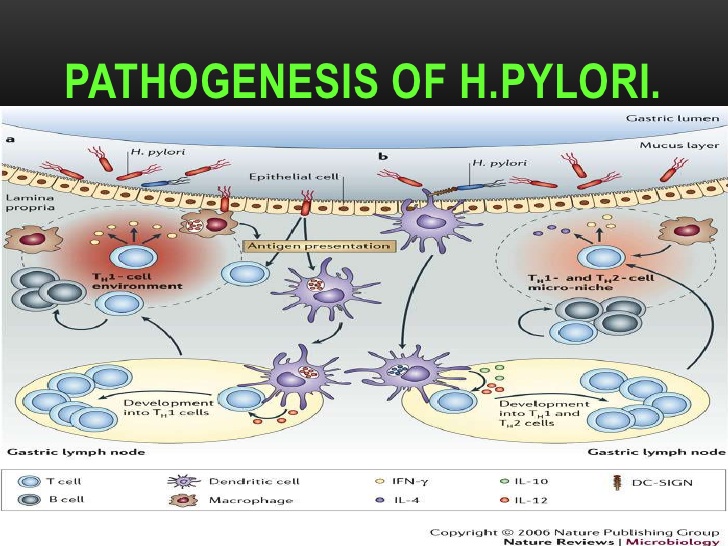
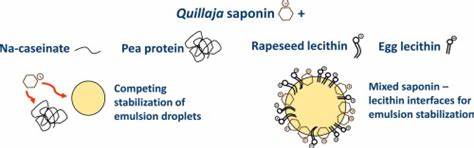
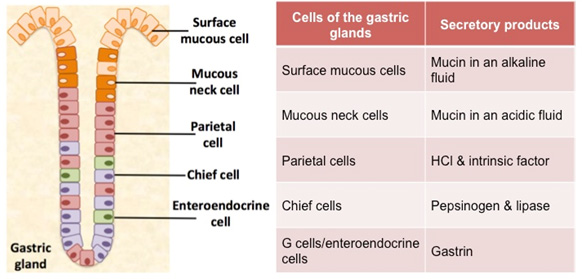
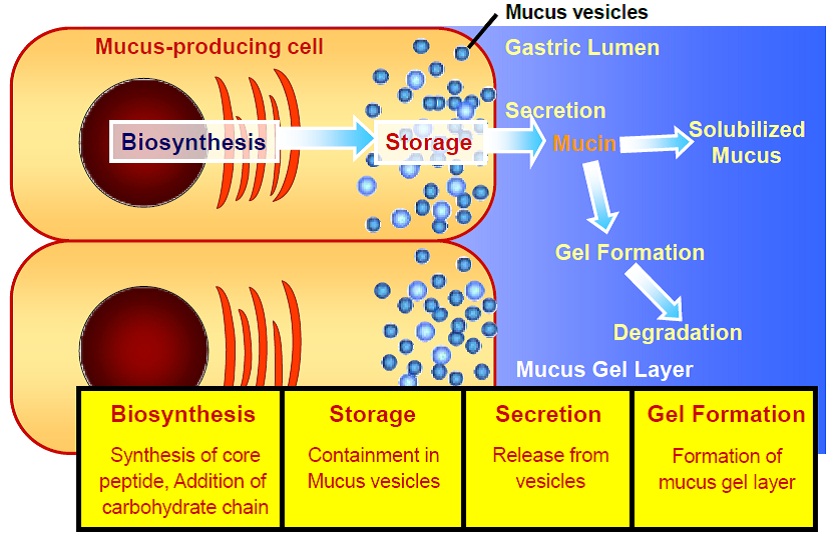
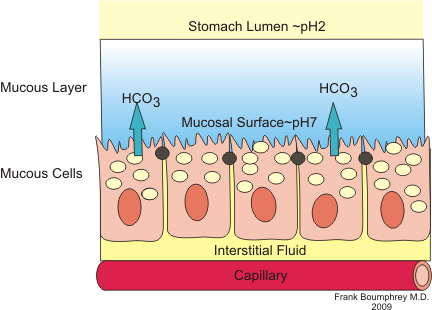
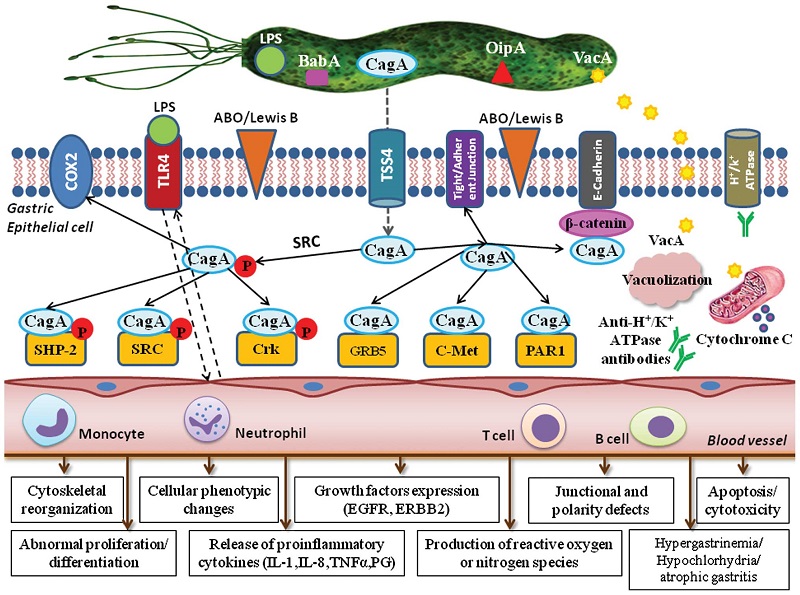
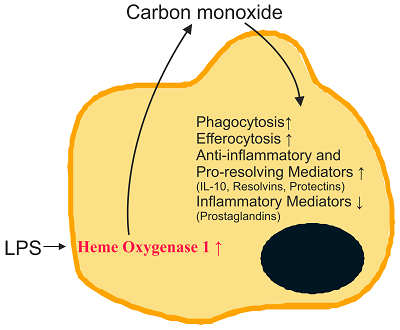
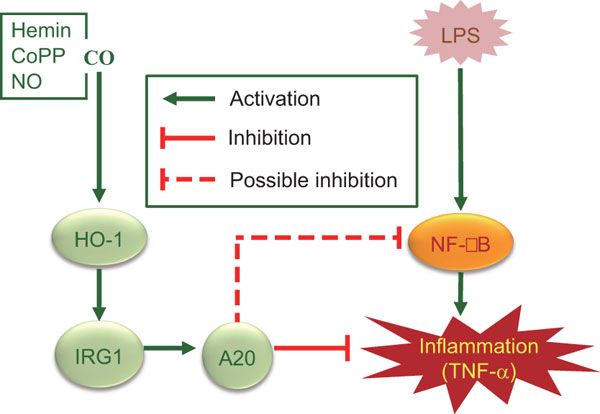
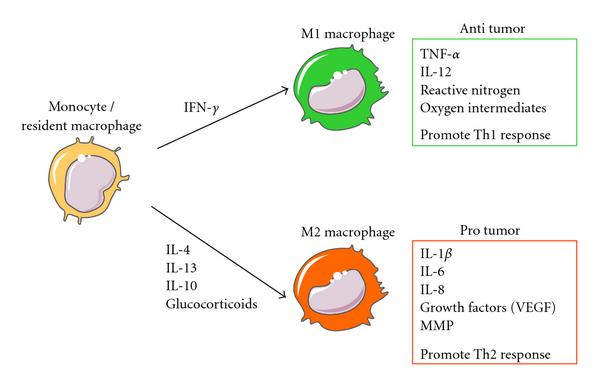
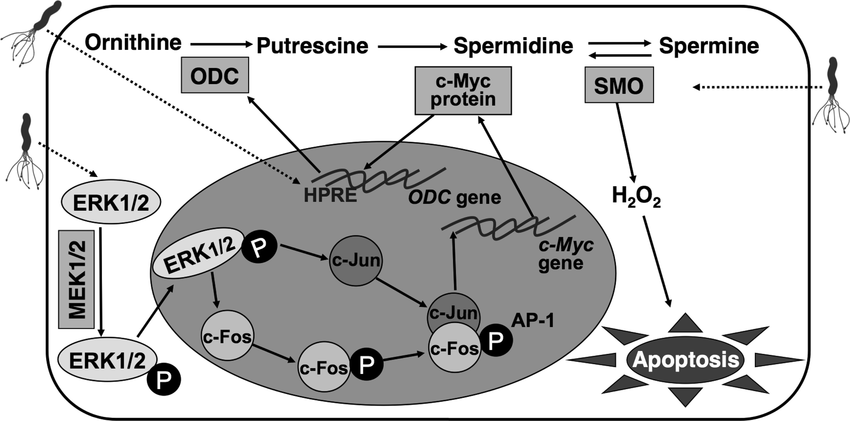
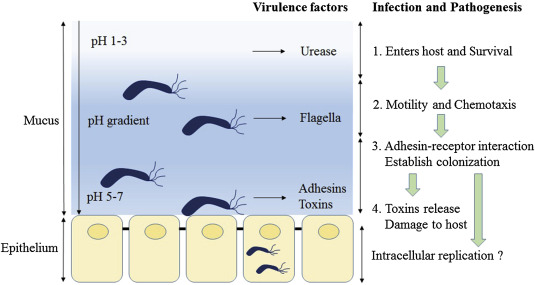
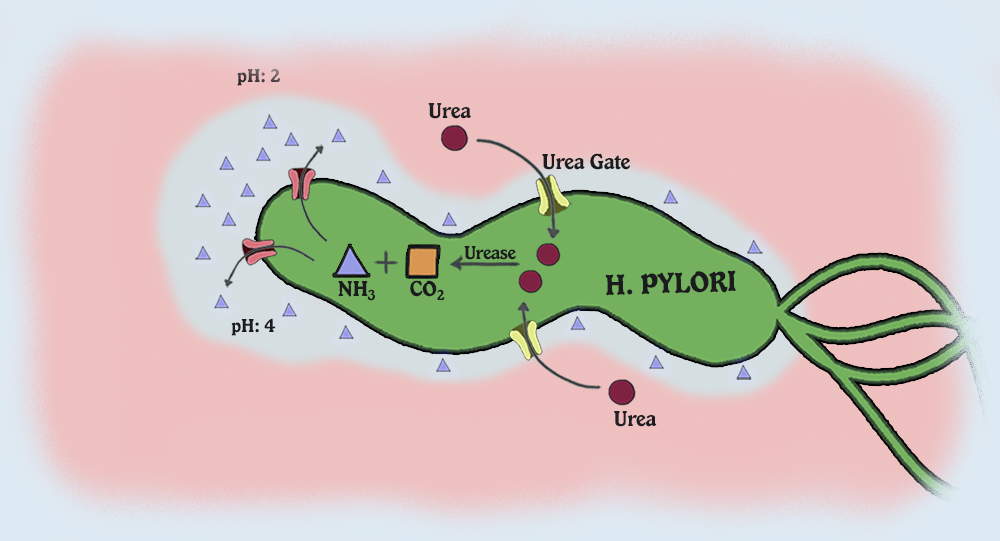
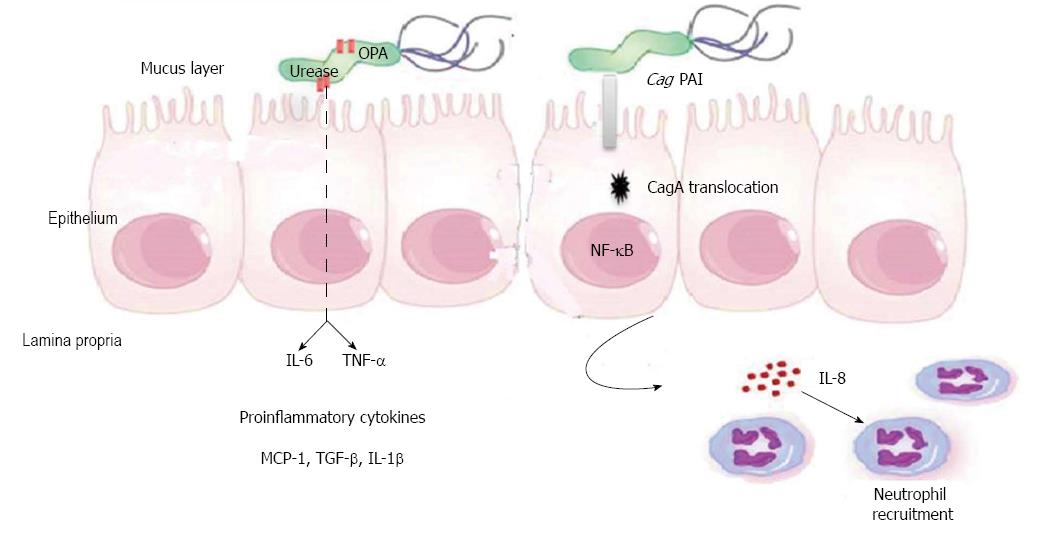

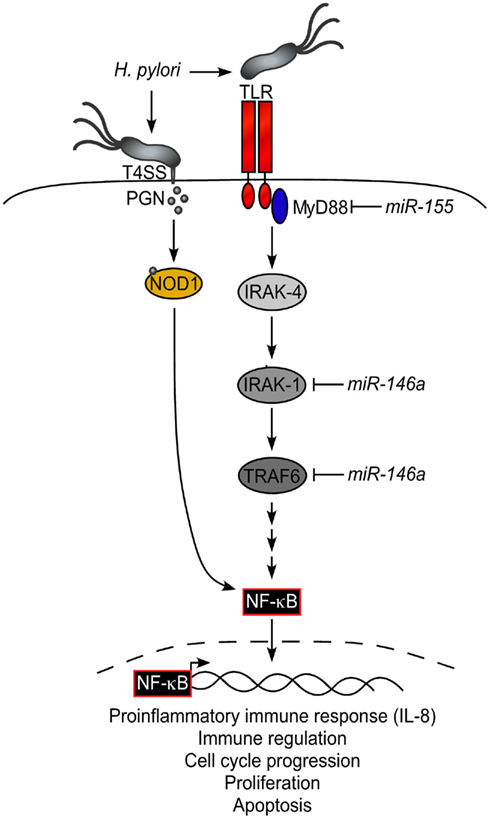
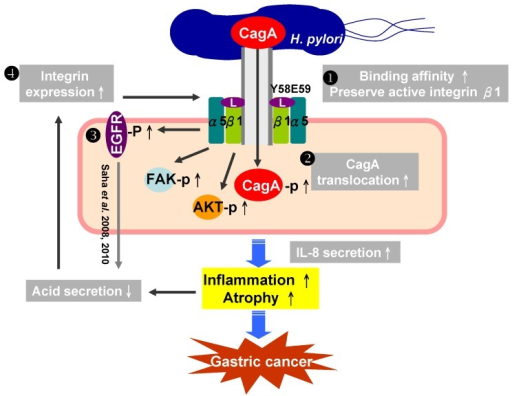
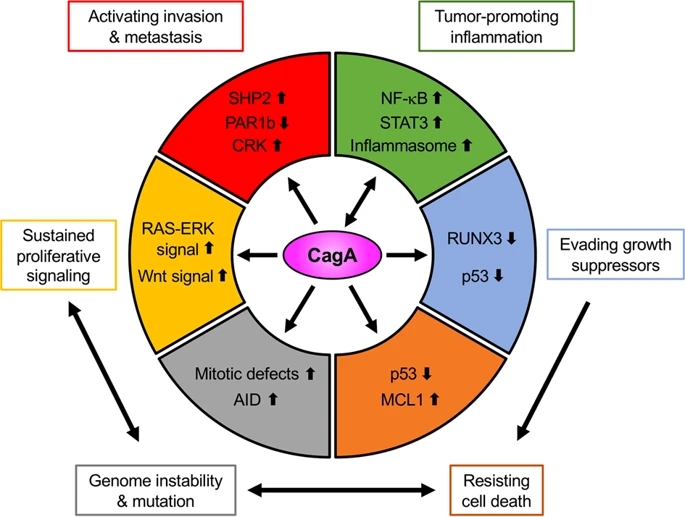
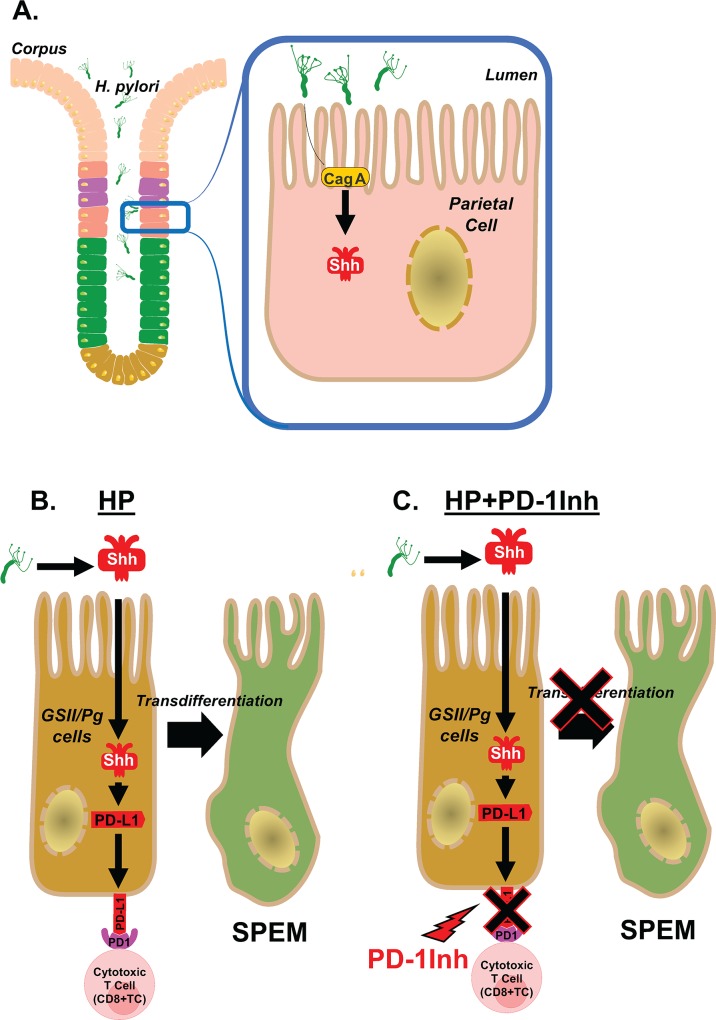

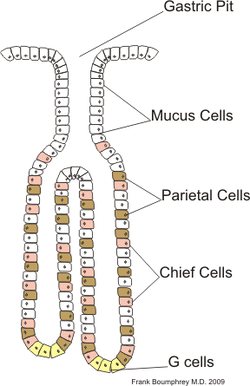
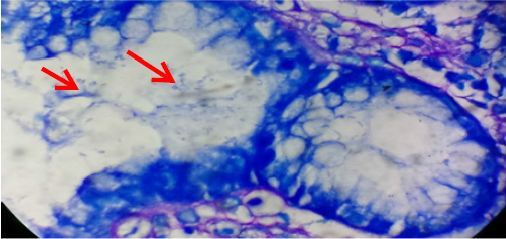
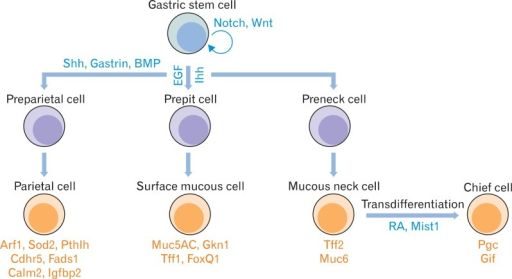
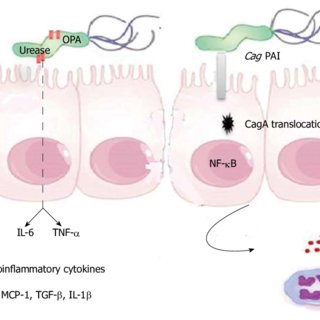
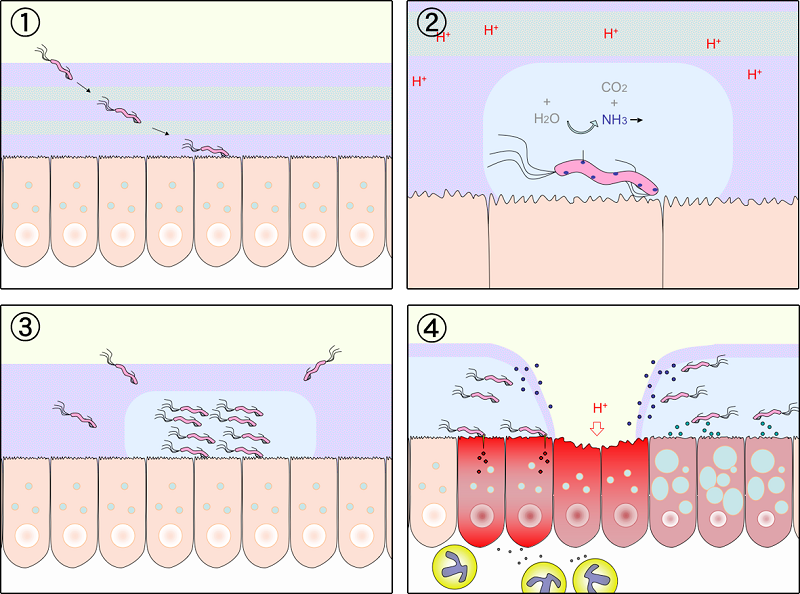
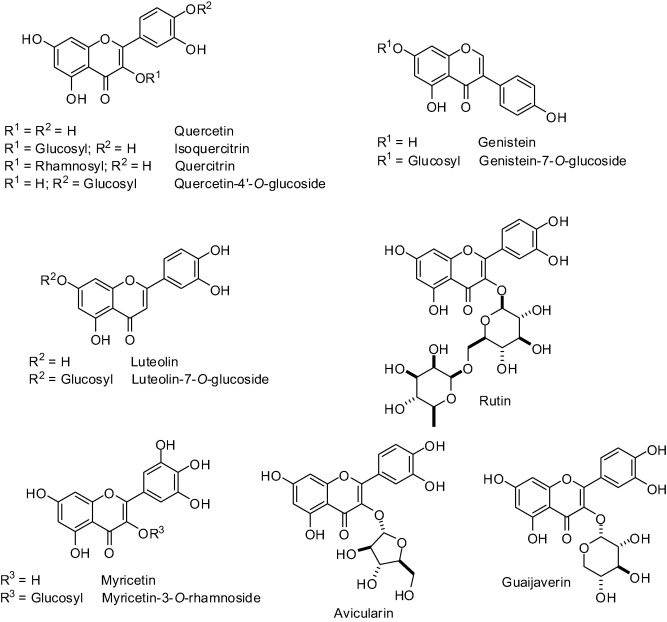
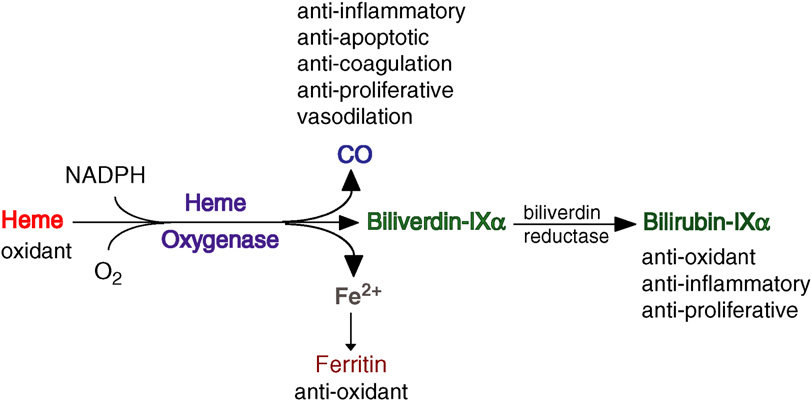
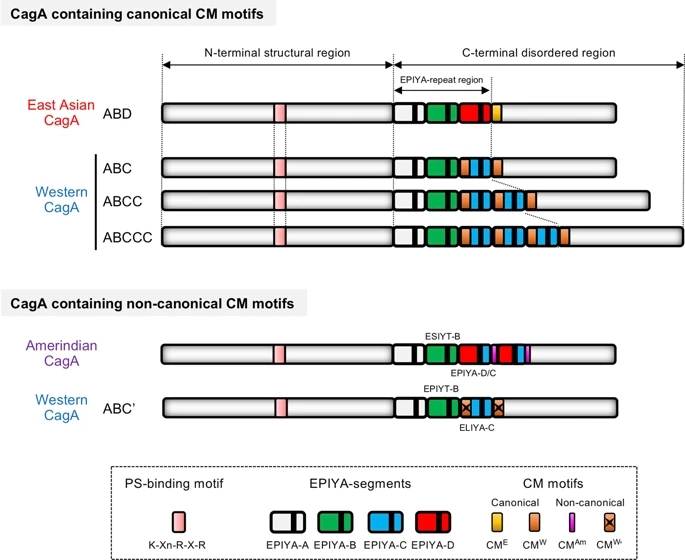
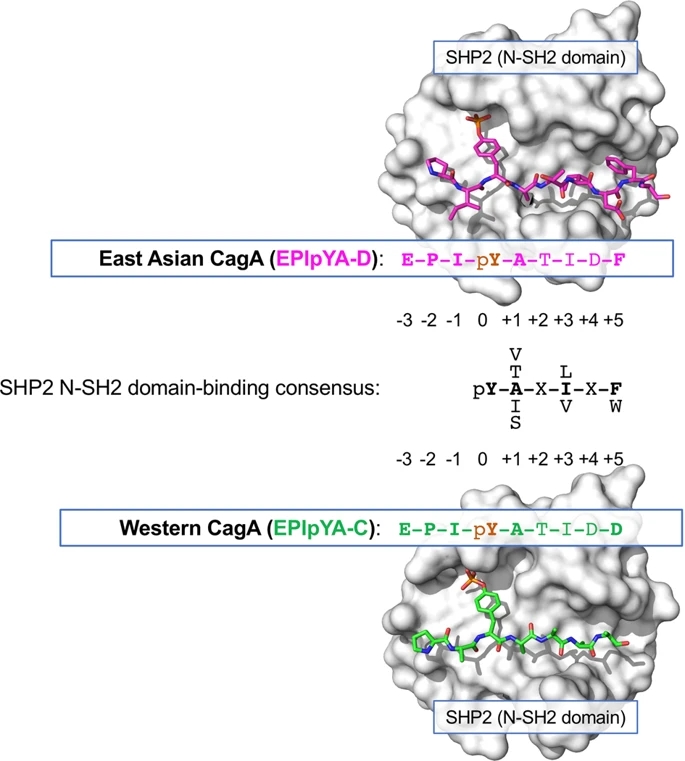
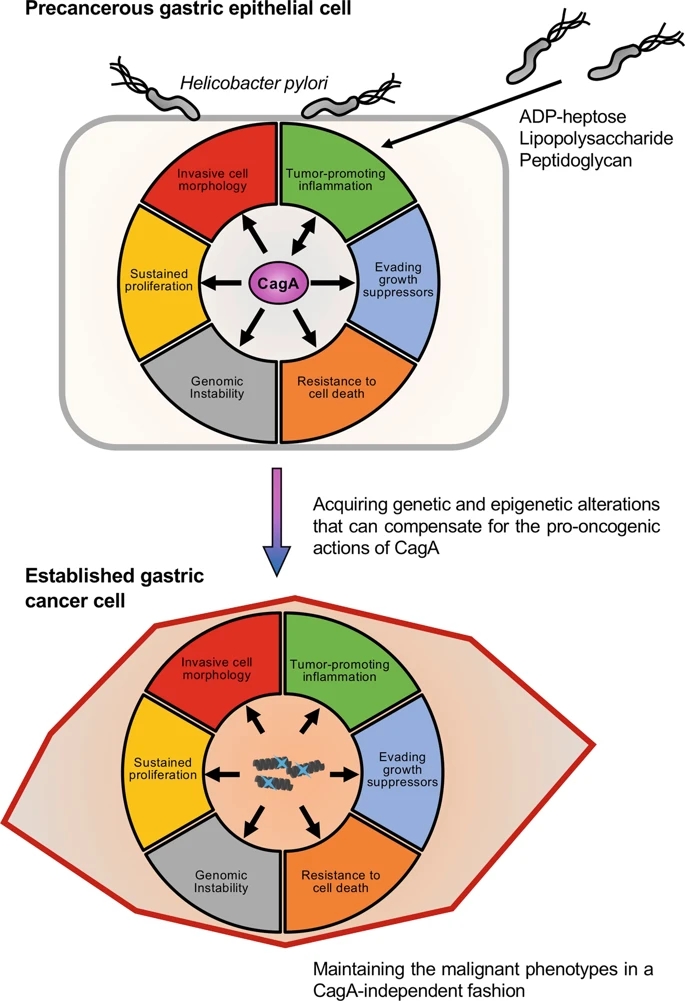
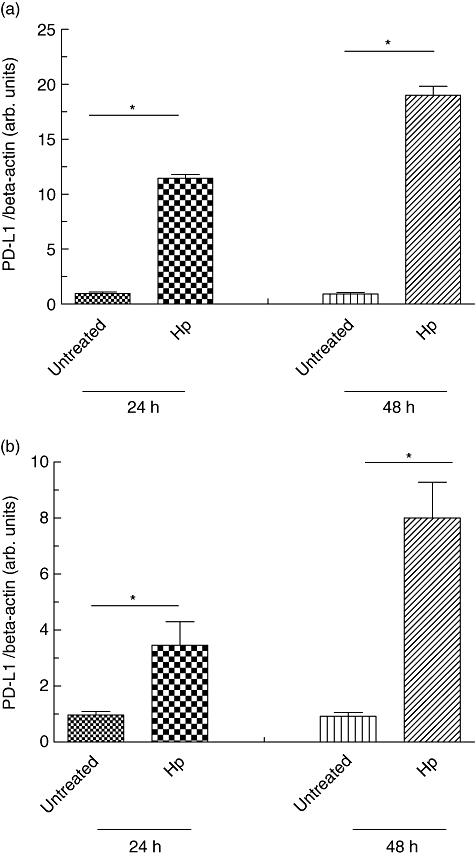
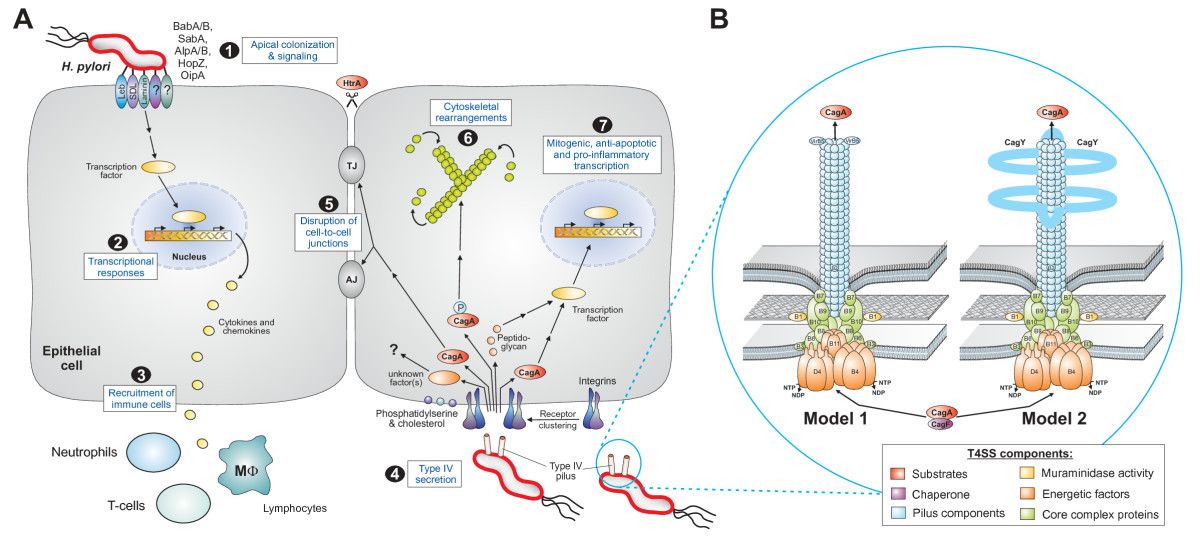

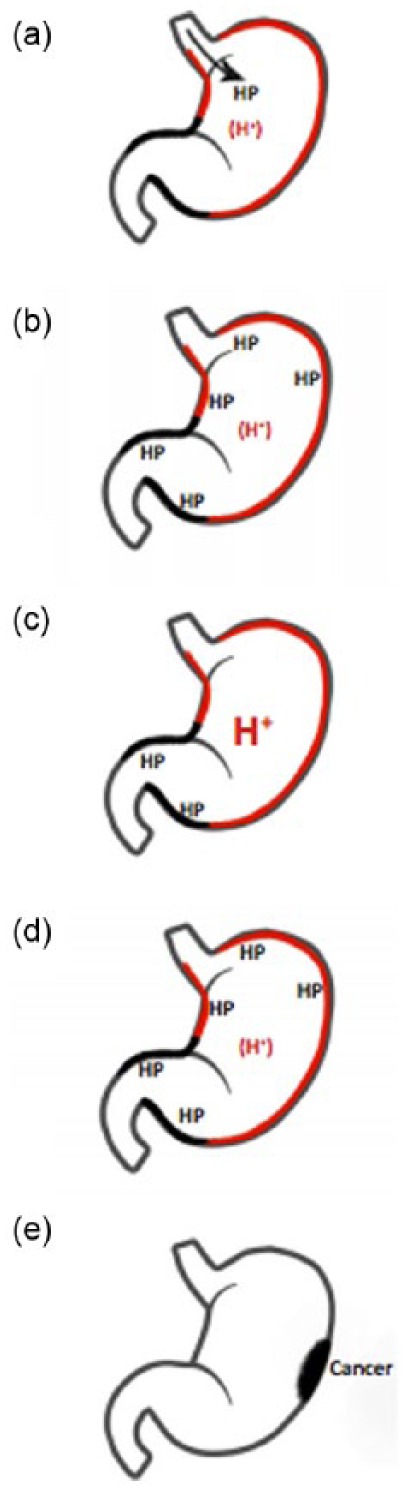
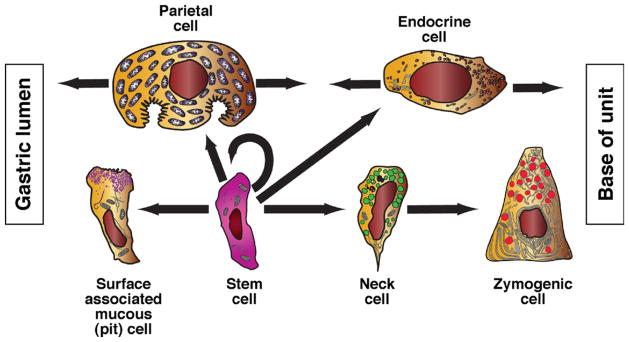
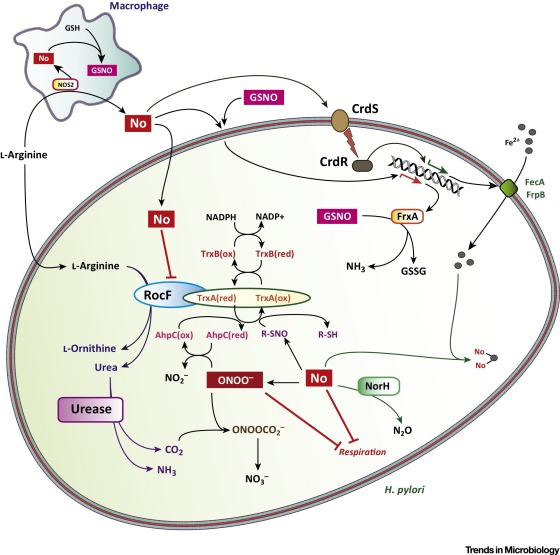
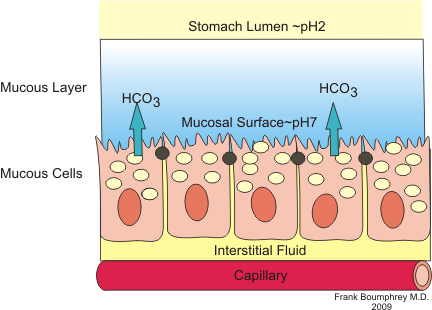
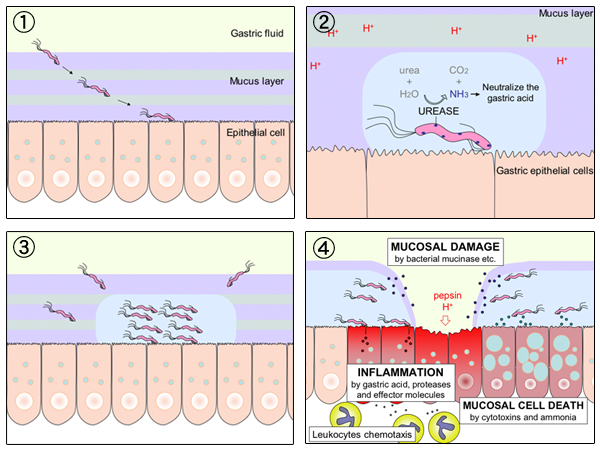
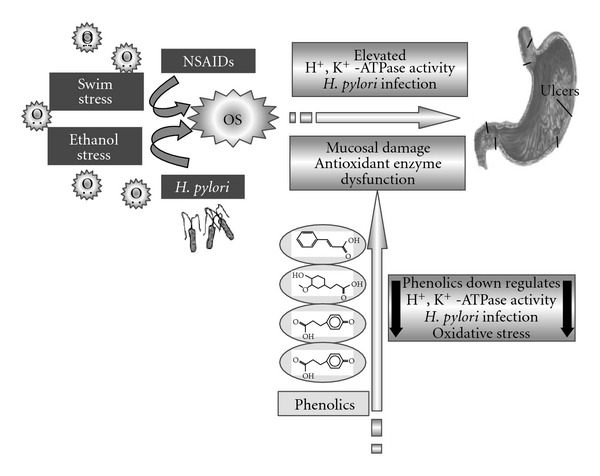
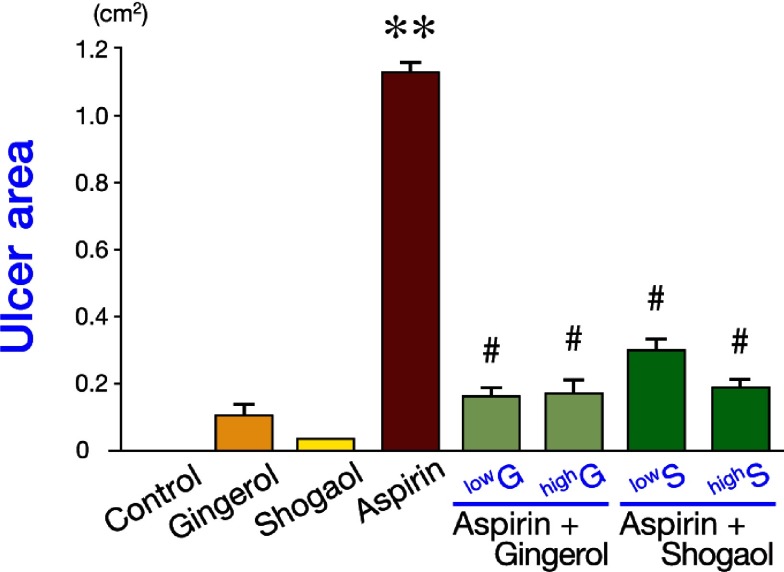
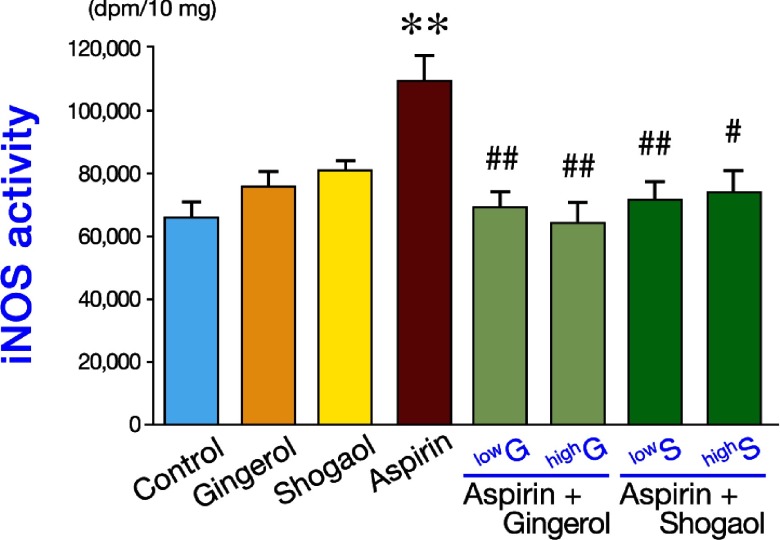
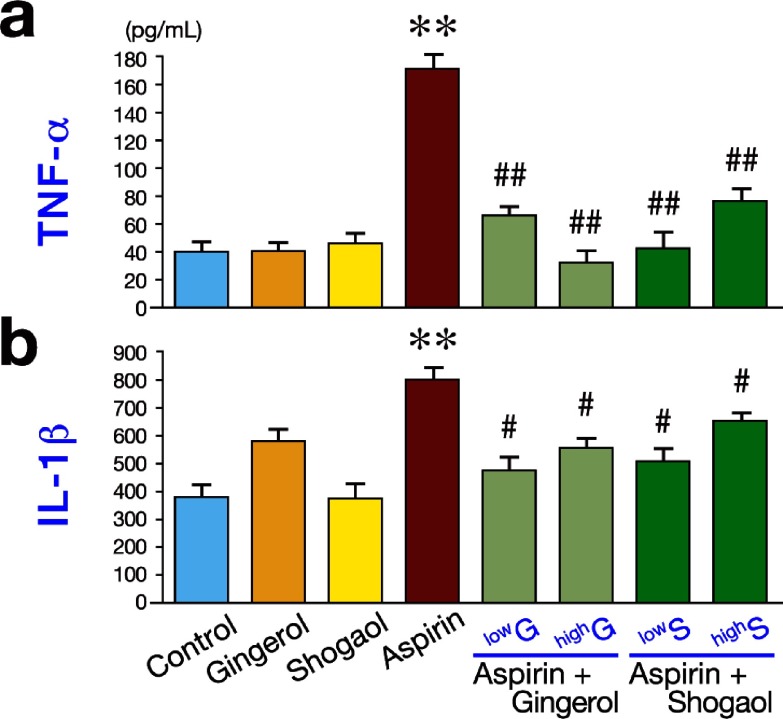
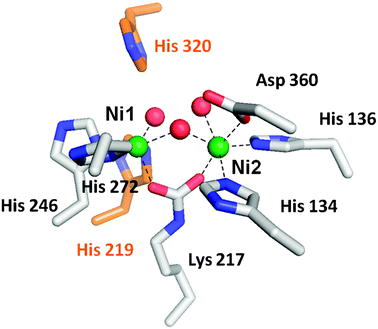
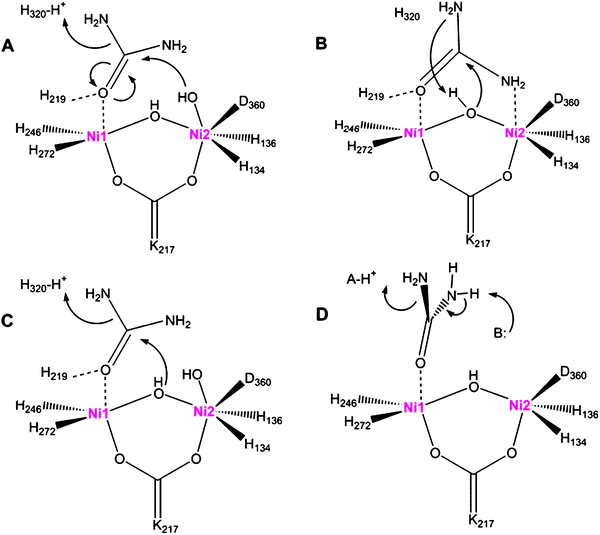
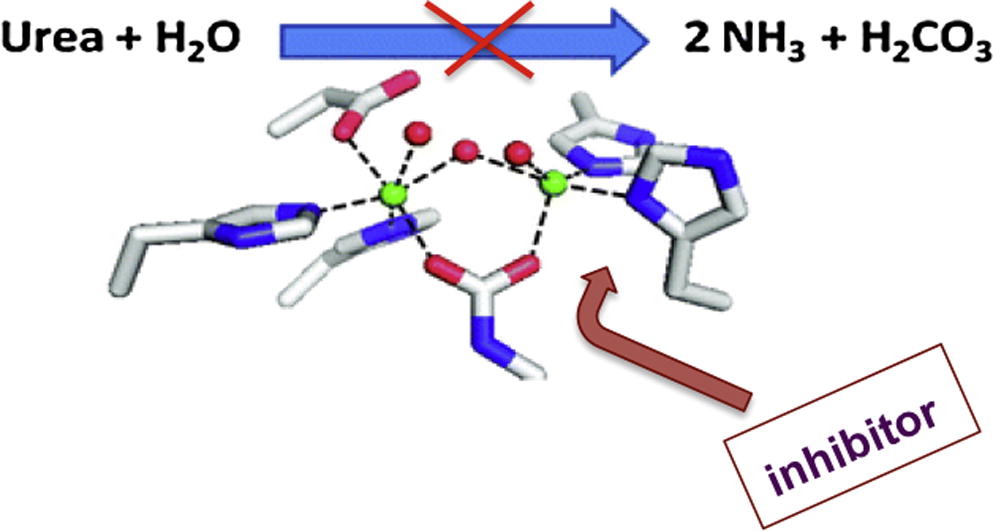
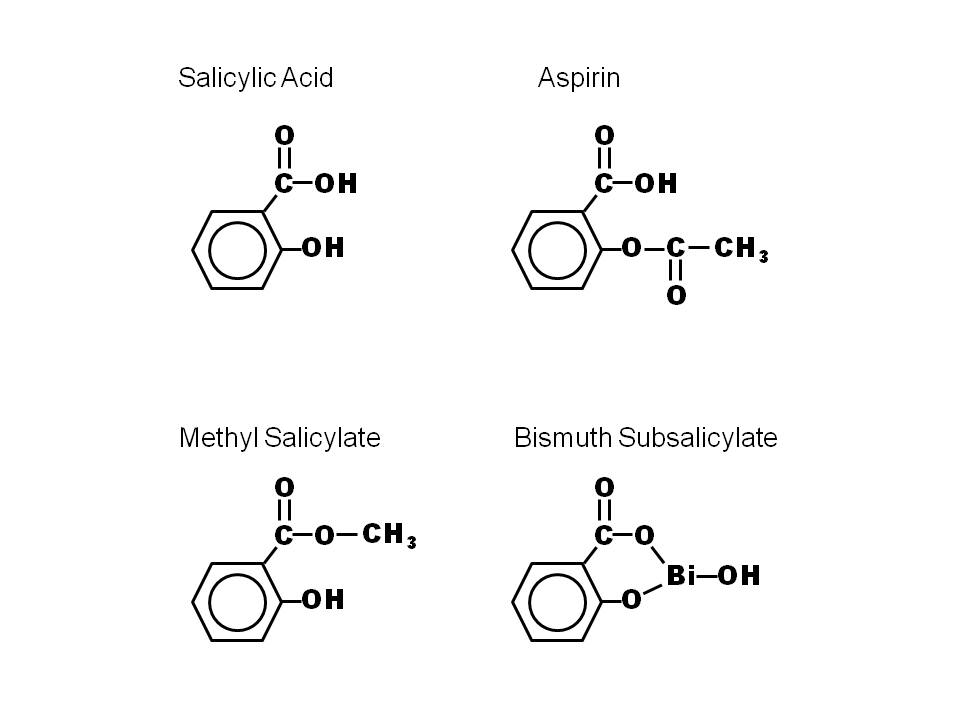
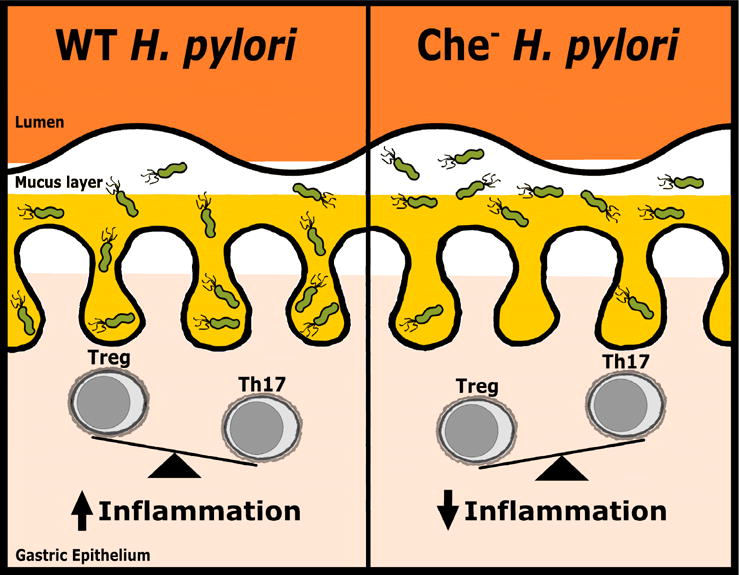
.jpg)
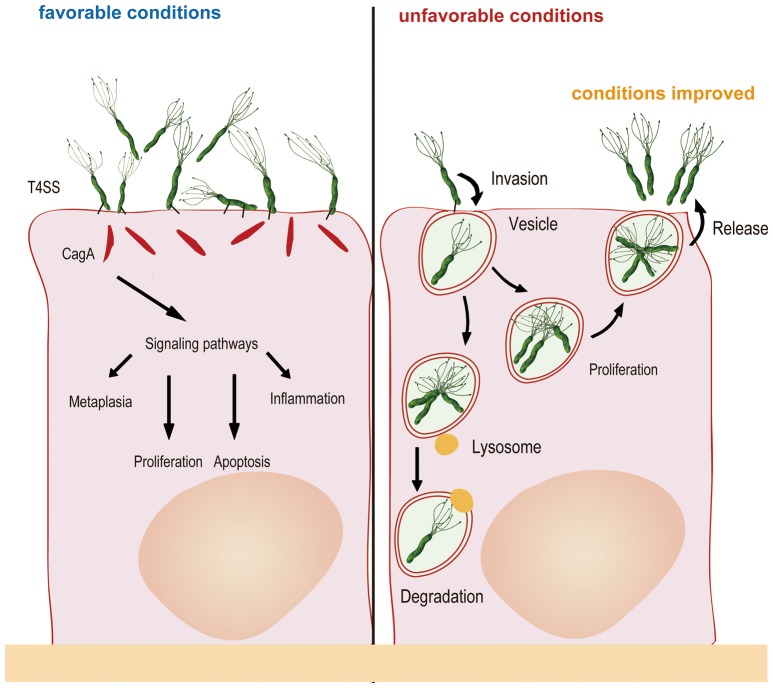
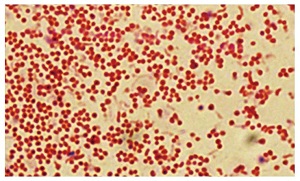
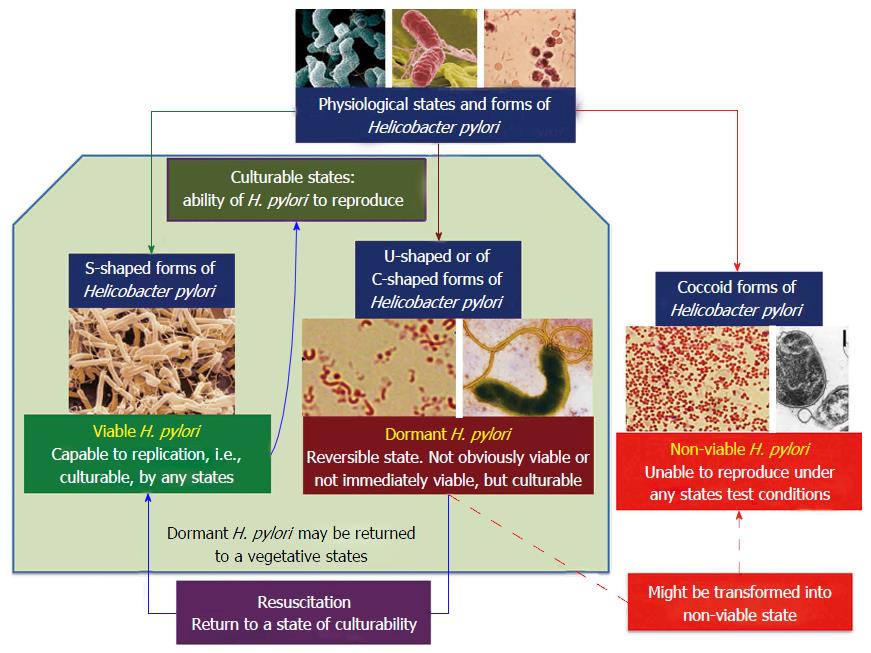
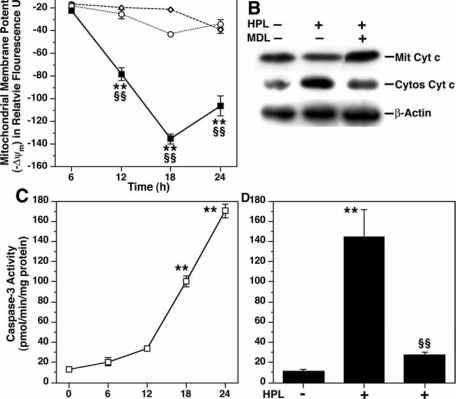
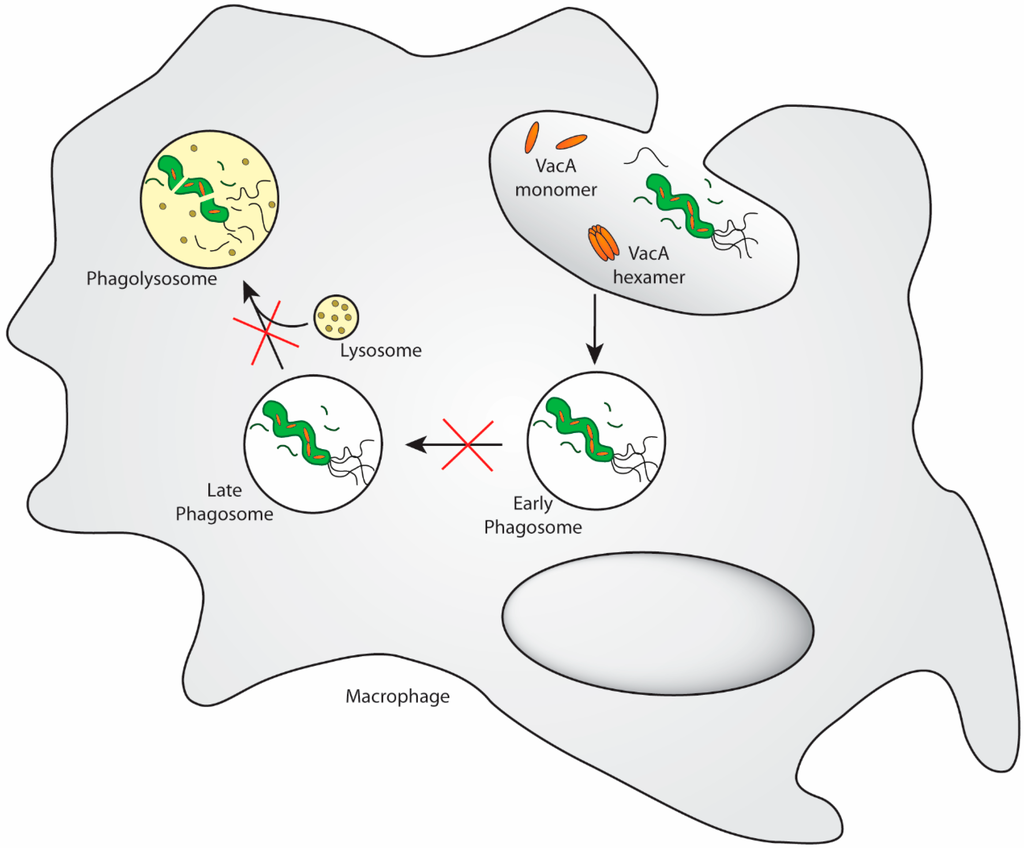
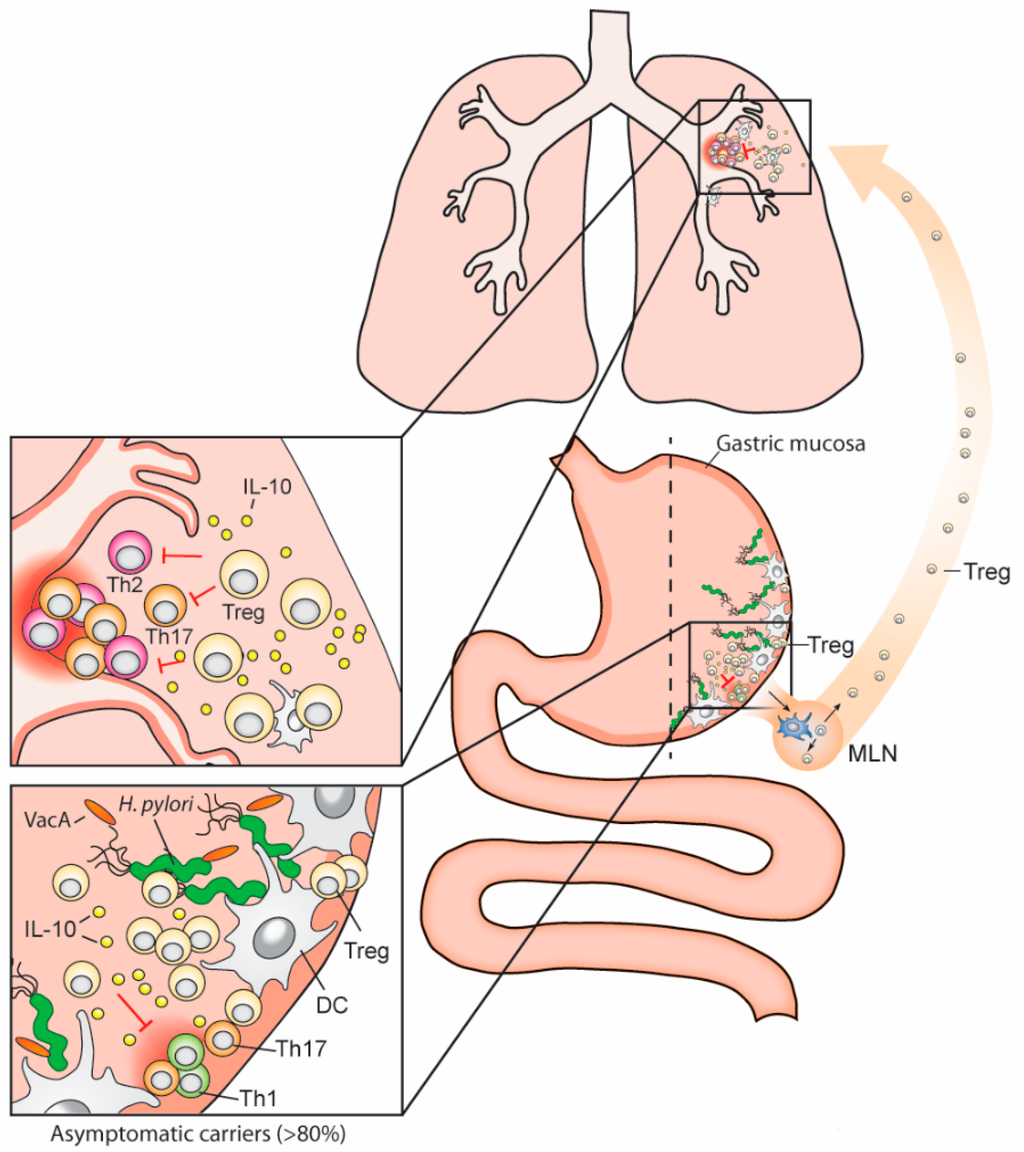
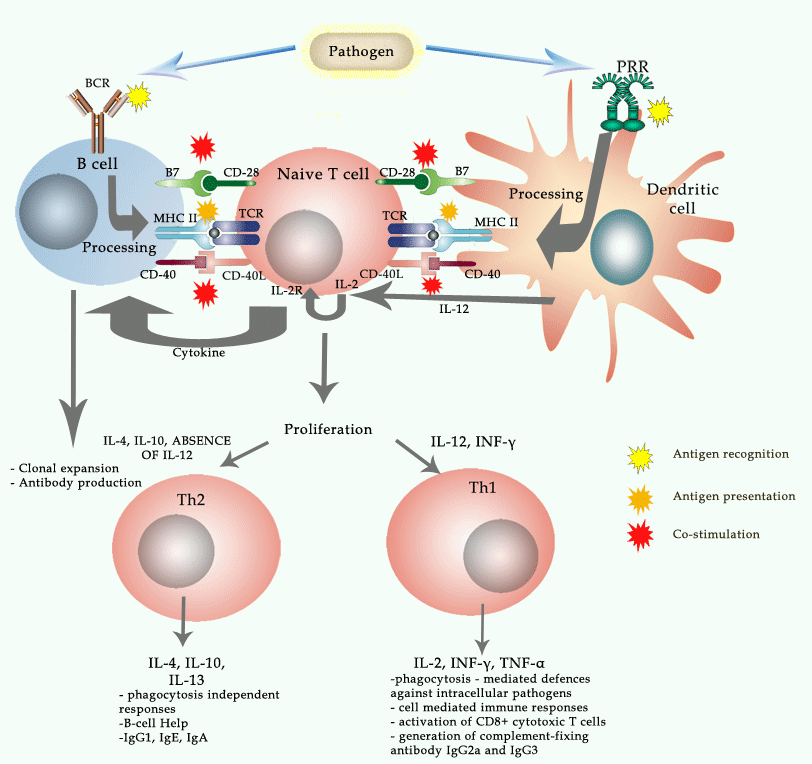




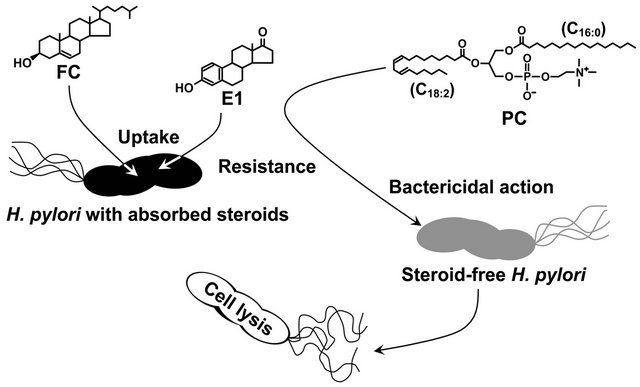

{kind=link}