íí
Mechanisms of Hypoxia-mediated Immune Escape in Cancer
íí
Ivraym B. Barsoum, Madhuri Koti, D. Robert Siemens and Charles H. Graham
DOI: 10.1158/0008-5472.CAN-14-2598 Published December 2014
Department of Biomedical and Molecular Sciences, Queen's University Kingston,
Ontario, Canada.
Abstract
An important aspect of malignant progression is the acquired ability of tumor
cells to avoid recognition and destruction by the immune system (immune escape).
Clinical cancer progression is also associated with the development of tumor
hypoxia, which is mechanistically linked to the acquisition of malignant
phenotypes in cancer cells. Despite the well-established role of hypoxia in
tumor cell invasion and metastasis, and resistance to therapy, relatively few
studies have examined the contribution of hypoxia to cancer immune escape.
Accumulating evidence reveals that hypoxia can impair anticancer immunity by
altering the function of innate and adaptive immune cells and/or by increasing
the intrinsic resistance of tumor cells to the cytolytic activity of immune
effectors. Here, we discuss certain aspects of the contribution of hypoxia to
tumor immune escape and provide evidence for a novel role of cyclic guanosine
monophosphate (cGMP) signaling in the regulation of hypoxia-induced immune
escape. Thus, we propose that activation of cGMP signaling in cancer cells may
have important immunotherapeutic applications. Cancer Res; 74(24); 7185ĘC90.
©2014 AACR.
Mechanisms of Hypoxia-Mediated Immune Escape in Cancer | Cancer Research
https://cancerres.aacrjournals.org/content/74/24/7185
íí
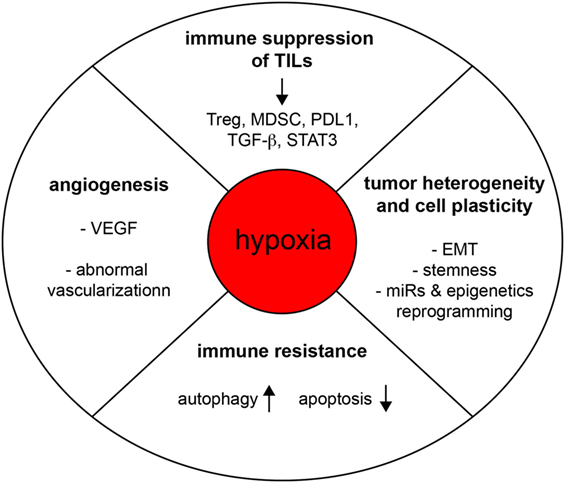
Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity
https://www.frontiersin.org/articles/10.3389/fimmu.2017.01625
017.01625
imageStĘŽphane Terry, imageStĘŽphanie Buart and imageSalem Chouaib*
INSERM UMR 1186, Integrative Tumor Immunology and Genetic Oncology, Gustave
Roussy, EPHE, Fac. de mĘŽdecine ĘC Univ. Paris-Sud, University Paris-Saclay,
Villejuif, France
The microenvironment of a developing tumor is composed of proliferating cancer
cells, blood vessels, stromal cells, infiltrating inflammatory cells, and a
variety of associated tissue cells. The crosstalk between stromal cells and
malignant cells within this environment crucially determines the fate of tumor
progression, its hostility, and heterogeneity. It is widely accepted that
hypoxic stresses occur in most solid tumors. Moreover, cancer cells found within
hypoxic regions are presumed to represent the most aggressive and
therapy-resistant fractions of the tumor. Here, we review evidence that hypoxia
regulates cell plasticity, resistance to cell-mediated cytotoxicity, and immune
suppression. Exposure to hypoxia occurs as a consequence of insufficient blood
supply. Hypoxic cells activate a number of adaptive responses coordinated by
various cellular pathways. Accumulating data also suggest that hypoxic stress in
the tumor microenvironment promotes tumor escape mechanisms through the
emergence of immune-resistant tumor variants and immune suppression. Thus, solid
tumors seem to build up a hostile hypoxic microenvironment that hampers
cell-mediated immunity and dampen the efficacy of the immune response.
íí
The impact of hypoxia on tumor-associated macrophages
íí
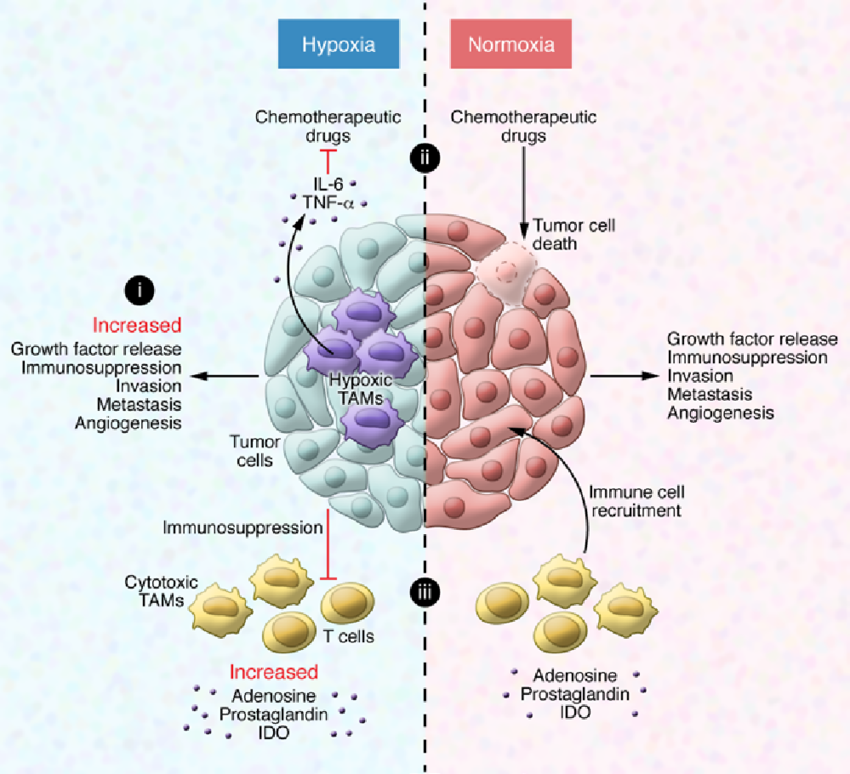
the secretion of immunosuppressive molecules (like lactic acid,TGF-beta, Il-10, VEGF, galectins etc) by tumor cells under conditions of hypoxia provides a survival advantage,
Hypoxic regulation of TAMs in cancer progression and therapy. (i)
Hypoxia-induced release of chemoattractants results in enhanced TAM recruitment,
which further amplifies the protumoral response. (ii) TAMs release survival
factors for cancer cells, which protect them from chemotherapeutics. (iii) The
hypoxic tumor environment is immunosuppressive and prevents an antitumor
response.
Source publication
The role of tumor-associated macrophages (TAMs) in cancer is often correlated
with poor prognosis, even though this statement should be interpreted with care,
as the effects of macrophages primarily depend on their localization within the
tumor. This versatile cell type orchestrates a broad spectrum of biological
functions and exerts very complex and even opposing functions on cell death,
immune stimulation or suppression, and angiogenesis, resulting in an overall
pro- or antitumoral effect. We are only beginning to understand the
environmental cues that contribute to transient retention of macrophages in a
specific phenotype. It has become clear that hypoxia shapes and induces specific
macrophage phenotypes that serve tumor malignancy, as hypoxia promotes immune
evasion, angiogenesis, tumor cell survival, and metastatic dissemination.
Additionally, TAMs in the hypoxic niches within the tumor are known to mediate
resistance to several anticancer treatments and to promote cancer relapse. Thus,
a careful characterization and understanding of this macrophage differentiation
state is needed in order to efficiently tailor cancer therapy.
(PDF) The impact of hypoxia on tumor-associated macrophages
https://www.researchgate.net/publication/305786494_The_impact_of_hypoxia_on_tumor-associated_macrophages
íí
Targeting the renin-angiotensin system to improve cancer
treatment: Implications for immunotherapy
Matthias Pinter1,2 and Rakesh K. Jain1,*
1Edwin L. Steele Laboratories for Tumor Biology, Department of Radiation
Oncology, Harvard Medical School and Massachusetts General Hospital, Boston, MA
02114, USA.
2Division of Gastroenterology and Hepatology, Department of Internal Medicine
III, Medical University of Vienna, Vienna, A-1090, Austria.
Abstract
Renin-angiotensin system (RAS) inhibitors (RASi)í¬widely prescribed for the
treatment of cardiovascular diseasesí¬have considerable potential in oncology.
The RAS plays a crucial role in cancer biology and affects tumor growth and
dissemination directly and indirectly by remodeling the tumor microenvironment.
We review clinical data on the benefit of RASi in primary and metastatic tumors
and propose that, by activating immunostimulatory pathways, these inhibitors can
enhance immunotherapy of cancer.

INTRODUCTION
The circulating renin-angiotensin system (RAS) is mainly known for its pivotal
role in maintaining cardiovascular homeostasis and fluid and electrolyte
balance. In addition, a local RAS is expressed in many tissues and mainly acts
at the cellular level, where it mediates cell proliferation, growth, and
metabolism. The local RAS works synergistically and independently of the
systemic RAS. Angiotensin II (AngII) is the main effector and maintains tissue
homeostasis by exerting regulatory and counterregulatory effects through its
different receptors. Alternative peptide-receptor axes also assist in
maintaining this balance (1ĘC7). Figure 1 provides an overview of the main
components of the RAS. Dysregulation of the RAS, for example, by overexpression
of certain RAS components [such as renin, Ang-converting enzyme (ACE), or AngII
type 1 receptor (AT1R)], can be involved in the pathophysiology and progression
of a broad range of diseases, such as arterial hypertension, kidney disease, and
other cardiovascular conditions (5, 8, 9).
íí
Targeting the renin-angiotensin system to improve cancer treatment:
Implications for immunotherapy | Science Translational Medicine
https://stm.sciencemag.org/content/9/410/eaan5616.full
íí
Intratumoral Immune Landscape: Immunogenicity to
Tolerogenicity
Abir K Panda, Sayantan Bose, Sreeparna Chakraborty, Kirti Kajal and Gaurisankar
Sa*
Division of Molecular Medicine, Bose Institute, India
*Corresponding author: Gaurisankar Sa, Division of Molecular Medicine, Bose
Institute, P-1/12, CIT Scheme VII M, Kolkata-700054, India
Received: April 13, 2015; Accepted: September 05, 2015; Published: September 10,
2015
Abstract
Immune system possesses distinct innate (less specific) and adaptive (more
specific) branches which act in a collaborative way to eliminate cancer from the
host. In spite of the presence of immune response, tumors develop in the body
spontaneously through different immune escape strategies. During the progression
of cancer, immune cells become paralyzed and altered. In tumor microenvironment
both innate (macrophage and NK cells) and adaptive (CTLs and effector T cells)
immune cells are unable to recognize and induce specific effector response
against cancer to eradicate it. Tumor cells release different types of
chemokines, cytokines, growth factors that can modulate immune cells to become
tolerogenic and allow tumor cells to grow rapidly without any restriction.
Immune cells also cannot discriminate the tumor antigens as they are concealed
in stroma and are also less immunogenic. The immune cells thus become dormant
and effective immune responses against tumors could not be elicited. Tumor cells
exploit the plethora of immunosuppressive mechanisms which include abnormalities
of antigen processing and presentation, induction of negative co-stimulatory
signals that helps to establish tumor immune evasion. In addition, infiltration
of T-regulatory cells, immature and tolerogenic Dendritic Cells (DCs),
tumor-associated macrophages, and myeloid-derived stromal cells foster
suppressive, tolerogenic condition. The understanding of different immune
evasion mechanisms will help to design effective immunotherapies to overcome
tolerogenic condition and elicit tumor regression.
Keywords: Immune cell dysfunction; Immunogenicity; Tolerogenicity; Tumor immune
evasion; Tumor micro-environment
íí
íí
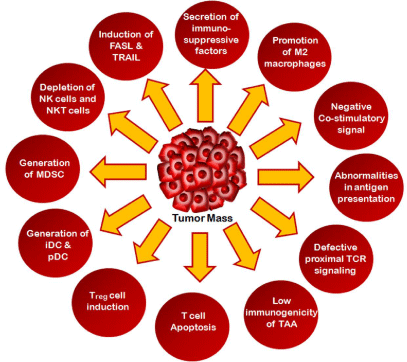
Figure 1: Major strategies adopted by tumor cells for immune evasion.
íí
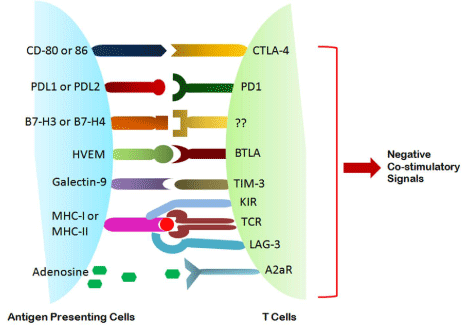
Figure 2: Negative Co-stimulatory signals between APC and Tcells in tumor microenvironment.
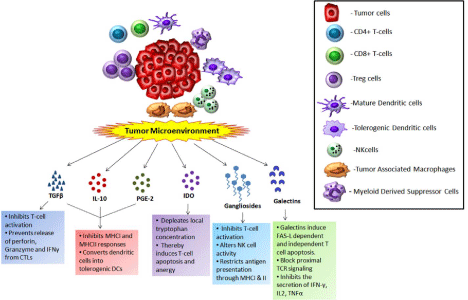
Figure 3: Different immunosuppressive factors and their interaction with immune cells in the tumor microenvironment.

Figure 4: Modulation
of different immune cells in the tumor microenvironment.
Intratumoral Immune Landscape: Immunogenicity to Tolerogenicity
https://austinpublishinggroup.com/clinical-immunology/fulltext/ajci-v2-id1025.php
íí
Tumor escape mechanisms.
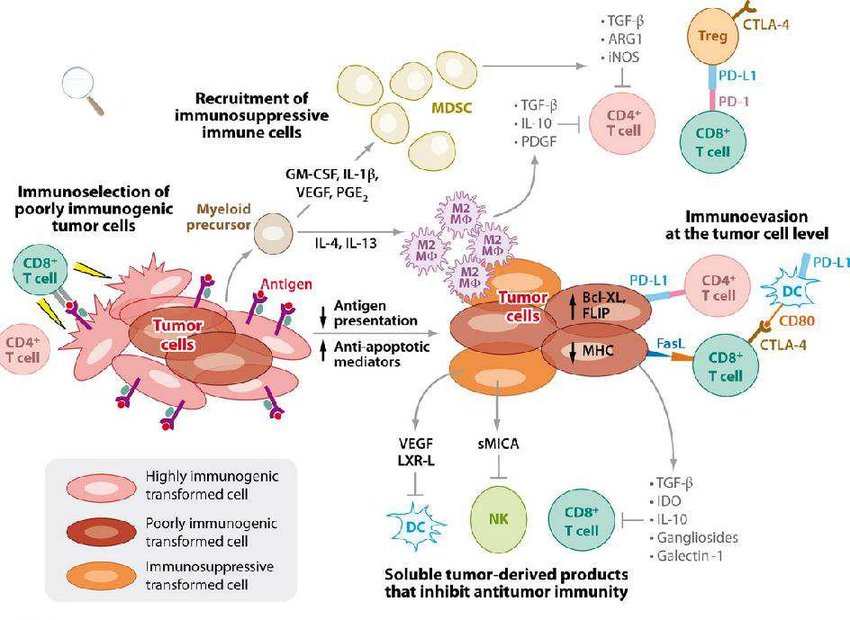
The immune system exerts selective pressure on tumors through a variety of processes, including the destruction of antigen-positive tumor cells by CD8 + T cells. As a result, immunogenic tumor cells are eliminated, leaving behind tumor cell variants more adept at evading immune-mediated destruction (i.e., immunoselection). Over time, tumors evolve mechanisms to elude or inhibit immunity by both intrinsic and extrinsic means. Intrinsic alterations within tumor cells evade immunity by downregulating antigen presentation (MHC), upregulating inhibitors of apoptosis (Bcl- XL, FLIP), or expressing inhibitory cell surface molecules that directly kill cytotoxic T cells (PD-L1, FasL). In addition, tumor cells secrete factors that inhibit effector immune cell functions (TGF-Ž┬, IL-10, VEGF, LXR-L, IDO, gangliosides, or soluble MICA) or recruit regulatory cells to generate an immunosuppressive microenvironment (IL-4, IL-13, GM-CSF, IL-1Ž┬, VEGF, or PGE2). Once recruited, regulatory cells attenuate antitumor immunity through the liberation of immunosuppressive cytokines and alterations in the nutrient content of the microenvironment. Specifically, secretion of IL-4 and IL-13 leads to recruitment and polarization of M2 macrophages (M2 MŽÁ) from myeloid precursors, which express TGF-Ž┬, IL-10, and PDGF that inhibit T cells. The release of colony-stimulating factors, IL-1Ž┬, VEGF, or PGE2 by tumor cells results in the accumulation of MDSCs that can block T cell function by expressing TGF-Ž┬, ARG1, and iNOS. Regulatory T cells (Tregs) can also inhibit effector T cells through multiple mechanisms, including expression of CTLA-4. (Abbreviations: ARG1, arginase 1; Bcl-XL, B cell lymphoma extra-long; CTLA-4, cytotoxic T lymphocyte associated protein-4; DC, dendritic cell; FasL, Fas ligand; FLIP, apoptosis-stimulating fragment-associated protein with death domain-like interleukin-1 converting enzyme-like inhibitory protein; GM-CSF, granulocyte macrophage colonyĘCstimulating factor; IDO, indoleamine 2,3-deoxygenase; IL, interleukin; iNOS, inducible nitric oxide synthase; LXR-L, liver X receptor ligand; MDSC, myeloid-derived suppressor cells; MHC, major histocompatibility complex; MICA, MHC class I polypeptide-related sequence A; PDGF, platelet-derived growth factor; PD-L1, programmed cell death 1 ligand 1; PGE2, prostaglandin-E2; TGF-Ž┬, transforming growth factor-Ž┬; Treg, regulatory T cell; VEGF, vascular endothelial growth factor) (Vesely et al. 2011).
íí
Mechanisms of Hypoxia-mediated Immune Escape in Cancer
Ivraym B. Barsoum, Madhuri Koti, D. Robert Siemens and Charles H. Graham
DOI: 10.1158/0008-5472.CAN-14-2598 Published December 2014
Department of Biomedical and Molecular Sciences, Queen's University Kingston,
Ontario, Canada.
Abstract
An important aspect of malignant progression is the acquired ability of tumor
cells to avoid recognition and destruction by the immune system (immune escape).
Clinical cancer progression is also associated with the development of tumor
hypoxia, which is mechanistically linked to the acquisition of malignant
phenotypes in cancer cells. Despite the well-established role of hypoxia in
tumor cell invasion and metastasis, and resistance to therapy, relatively few
studies have examined the contribution of hypoxia to cancer immune escape.
Accumulating evidence reveals that hypoxia can impair anticancer immunity by
altering the function of innate and adaptive immune cells and/or by increasing
the intrinsic resistance of tumor cells to the cytolytic activity of immune
effectors. Here, we discuss certain aspects of the contribution of hypoxia to
tumor immune escape and provide evidence for a novel role of cyclic guanosine
monophosphate (cGMP) signaling in the regulation of hypoxia-induced immune
escape. Thus, we propose that activation of cGMP signaling in cancer cells may
have important immunotherapeutic applications. Cancer Res; 74(24); 7185ĘC90.
©2014 AACR.
Introduction
Hypoxia, a characteristic of many solid cancers, develops from an imbalance
between oxygen consumption and oxygen supply. Although hypoxia is an important
driver of tumor invasion and metastasis, as well as resistance to therapy (1),
there is limited knowledge on the contribution of hypoxia to tumor cell escape
from destruction by innate and adaptive immune effector mechanisms.
Immune escape in cancer is a multifaceted process resulting from the suppression
of immune effector mechanisms and/or the acquisition of intrinsic tumor cell
resistance to the cytotoxic activity of immune effectors. Hypoxia can influence
these aspects of immune escape by modifying the intrinsic properties of tumor
cells and of the stromal compartment. Here, we review some of the mechanisms by
which hypoxia contributes to immune escape in cancer. Furthermore, we propose
that activation of cyclic guanosine monophosphate (cGMP) signaling in cancer
cells, via administration of low doses of nitric oxide (NO) mimetic drugs, may
be a novel therapeutic approach to interfere with hypoxia-induced immune escape.
Hypoxia-Induced Release of Immunosuppressive Molecules by Tumor Cells
Upon exposure to hypoxia, tumor cells release a variety of immunosuppressive
molecules. For example, in the severely hypoxic tumor microenvironment, dying
cells release ATP that is metabolized to adenosine by the ectonucleotidases CD73
and CD39 (2). Soluble adenosine in the extracellular matrix binds specific
receptors on T cells to increase their intracellular levels of cAMP, which, in
turn, suppresses T-cell functions (3). Tumor-derived cytokines released under
hypoxic conditions, such as IL10 and TGFŽ┬, induce the differentiation of
tumor-associated macrophages (TAM) into M2 macrophages with immune-suppressive
activities (4). TGFŽ┬ released by tumor cells also inhibits T-cell proliferation
and effector function, promotes the generation of regulatory T cells (Treg), and
blocks the expression of receptors required for the cytotoxic function of
natural killer (NK) cells (5). In addition, TGFŽ┬ negatively regulates the
antigen presentation function of dendritic cells (DC), resulting in the
inhibition of T-cell function and differentiation (5).
Interestingly, emerging evidence links hypoxia-induced angiogenesis with immune
tolerance (6, 7). Hypoxia drives angiogenesis within the tumor microenvironment
by inducing the secretion of vascular endothelial growth factor (VEGF) and other
proangiogenic molecules by tumor cells. Tumor-derived VEGF suppresses the
maturation of DCs and blocks the presentation of tumor-associated antigens to
helper T cells, thereby promoting immune escape (6). Moreover, in response to
tumor-derived VEGF, DCs increase their expression of the programmed death ligand
1 (PD-L1 or B7-H1), a negative regulator of T-cell function (7). VEGF promotes
the accumulation of myeloid-derived suppressor cells (MDSC) in tumor tissues and
secondary lymphoid organs (6). MDSCs are potent suppressors of anticancer T-cell
responses and also contribute to tumor progression by releasing factors that
promote angiogenesis and metastasis (for a review on MDSCs see refer. 8).
Consequently, VEGF is a potential target for immune therapy. In support of this,
anti-VEGF therapy was shown to be associated with increased numbers of activated
DCs and heightened T-cell function in patients with cancer (9). However,
targeting VEGF as an immunotherapeutic approach may lead to tumor hypoxia via
inhibition of angiogenesis, thereby resulting in the activation of other
hypoxia-induced immune escape pathways.
Tumor cells can secrete proteins such as CC-chemokine ligand 22 (CCL22) and
various chemokines that inhibit effector T-cell responses and promote the
generation and recruitment of immunosuppressive Tregs (10). In an ovarian cancer
model, hypoxia was shown to promote the recruitment of Tregs via increased tumor
cell expression of CCL28 (11). Tregs in turn can also secrete VEGF, thereby
contributing to the VEGF pool in the tumor microenvironment that contributes to
immune tolerance (11).
Tumor cells can also produce galectin-1 and galectin-3 to induce apoptosis of
activated lymphocytes (12, 13). In patients with melanoma, there was a strong
correlation between expression of galectin-3 and apoptosis of tumor-infiltrating
lymphocytes (TIL; ref. 12). In Wilms tumors and Schwannomas, galectin-3 was
shown to colocalize with the transcription factor hypoxia-inducible factor-1Ž┴
(HIF1Ž┴; ref. 14). In addition, it was reported that galectin-1 expression is
transcriptionally regulated by HIF1 in colorectal cancers (15) and head and neck
squamous cell carcinomas (16).
Hypoxia was also shown to induce immunosuppression by upregulating COX-2
expression in tumor cells; and HIF1-mediated upregulation of COX-2 increased
colorectal tumor cell survival and VEGF production (17). COX-2 is a
proinflammatory enzyme that converts arachidonic acid into prostaglandin E2
(PGE2). The latter causes immunosuppression by increasing adenosine/cAMP
signaling in effector T cells (18). PGE2 secreted by tumor cells can also
inhibit antitumor immunity by inhibiting the maturation of DCs (19). Also, PGE2
enhances the suppressive activity of Tregs and supports the differentiation of
Tregs (20). Finally, PGE2 can stimulate the immunosuppressive functions of MDSC
by binding to EP-4 receptors on these cells (21). Hypoxia-induced immune
suppression via COX-2 can explain why chronic administration of indomethacin, a
COX-2 inhibitor, in the drinking water of mice led to significant reduction in
the growth rate and metastasis of mammary tumors as well as restoration of
splenic NK cell activity (22). A recent study revealed that use of NSAIDs
reduced recurrence of breast cancer in overweight and obese women (23).
The above studies indicate that the secretion of immunosuppressive molecules by
tumor cells under conditions of hypoxia provides a survival advantage, and
therefore support the concept that hypoxia represents a selection pressure
driving immune escape.
Direct Effects of Hypoxia on Immune Effectors
Hypoxia can also directly impair antitumor immune responses. For example,
hypoxia in the tumor microenvironment can induce the release of VEGF by M2
macrophages (24). Furthermore, TAMs suppress T-cell function in a manner
dependent on HIF1 (25), and TAMs in hypoxic regions of tumors exhibit increased
expression of M2-promoting molecules, such as TGFŽ┬ (26). Hypoxia inhibits the in
vitro cytolytic activity of other immune effectors such as the NK cellĘCmediated
killing of hepatocellular carcinoma cells and multiple myeloma cells (27, 28).
Hypoxia was shown to decrease T-cell survival (29), and incubation of naïve T
cells under hypoxia decreases their secretion of the trophic cytokine IL2 in a
HIF1-dependent manner (30). CD4+ and CD8+ T cells derived from HIF1Ž┴-deficient
mice exhibit increased proliferation, produce higher levels of interferon-Ž├, and
display increased antitumor responses (31). HIF1 was also shown to mediate Treg
differentiation via increased expression of FoxP3 (32). Increased numbers of
Tregs in the tumor stroma have been associated with poor survival of patients
with various cancers (33, 34).
Another mechanism of tumor cell immune escape involves binding of the cytotoxic
T lymphocyte antigen-4 (CTLA-4; an immune checkpoint regulator) to its natural
receptors, CD80 (B7.1) and CD86 (B7.2). Interestingly, hypoxia was shown to
increase the expression of CD86 by bone marrowĘCderived mouse DCs in a
HIF1-independent manner (35). Studies revealed that CTLA-4 blockade attenuates
the growth of several mouse tumors (36), reduces tumor-infiltrating Tregs, and
promotes effector T-cell function in humans (37).
It is important to note that not all of the reported effects of hypoxia on T
cells are detrimental to their function. Hypoxia was reported to upregulate
CD137, a member of the TNF receptor family that is known for its costimulatory
activity on T cells (38). Expression of CD137 on activated mouse T cells was
shown to be stimulated by hypoxia (39), and tumors from HIF1Ž┴-deficient mice
exhibited undetectable numbers of CD137+ TILs (39). In tumor growth assays,
hypoxia induced the activation of T cells via the upregulation of surface CD137
in a HIF1-dependent manner, which, in turn, resulted in improved immune response
and slower tumor growth (39).
Hypoxia Induces Immune Tolerance via Regulation of Tumor Cell-Associated Immune
Checkpoint Molecules
In addition to decreasing the cytolytic potential of immune effectors, hypoxia
increases the intrinsic resistance of tumor cells to immune-mediated killing.
One strategy that tumor cells use to avoid immune detection and destruction is
to alter their expression of cell-surface immune checkpoint regulators. For
example, tumor cells may shed stress-induced MHC class I chain-related proteins
A and B (MICA/B) from their surface to avoid interaction with NKG2D receptors on
NK cells, Ž├Ž─ T cells, and CD8+ Ž┴Ž┬ T cells (40), thereby escaping cytolysis (41).
We have shown that exposure of tumor cells to hypoxia leads to the shedding of
surface MICA, which, in turn, results in increased resistance to lysis by innate
immune effectors (42). We also showed that the hypoxia-induced release of MICA
and resistance of tumor cells to lysis required HIF1-mediated expression of the
metalloproteinase ADAM 10 in the tumor cells (Fig. 1; ref. 43).

Figure 1.
Proposed mechanisms of hypoxia-induced tumor cell escape from innate and
adaptive immunity. Hypoxia increases the accumulation of HIF1Ž┴ in tumor cells,
which, in turn, leads to higher levels of ADAM 10 and PD-L1 on the surface of
tumor cells. ADAM 10 cleaves MICA from the cell surface to limit binding to
NKG2D receptors on NK cells, leading to escape from innate immunity, whereas
interaction of PD-L1 with PD-1 or CD80 on activated CTLs causes apoptosis in the
CTLs and escape from adaptive immunity. NO/cGMP signaling is proposed to block
the effect of hypoxia on ADAM 10 and PD-L1 upregulation by inhibiting HIF1Ž┴
accumulation.
As discussed earlier, there is evidence that tumor cells can suppress cytotoxic
T lymphocyte (CTL) function through the interaction of inhibitory costimulatory
molecules with their ligands. Certain members of the B7 family of costimulatory
molecules expressed on the surface of tumor cells provide signals that suppress
CTL responses. For example, binding of PD-L1 with PD-1 or with CD80 (B7.1) on
activated CTLs leads to suppression of immune responses via mechanisms that
include induction of apoptosis and anergy (nonresponsiveness to antigen) in the
T cells (44). Recent clinical studies revealed that therapy with blocking
anti-PD-1 antibody (nivolumab) produced objective responses in patients with
nonĘCsmall-cell lung cancer, melanoma, or renal-cell cancer (45). Also,
reinduction therapy with anti-PD-1 antibody for late tumor recurrence showed
durable remissions in patients with colorectal cancer, renal cell cancer, and
melanoma (46). In another study, concurrent therapy with anti-CTLA-4 antibody
(ipilimumab) and nivolumab resulted in tumor regression in a substantial
proportion of patients with unresectable, stage III or IV melanoma (47). We
recently provided evidence that, when exposed to hypoxia, human and mouse cancer
cells increased their expression of PD-L1 and acquired resistance to
CTL-mediated lysis in a manner dependent on HIF1Ž┴ (Fig. 1; ref. 48).
Furthermore, the hypoxia-induced expression of PD-L1 in tumor cells led to
increased apoptosis of cocultured CTLs as well as Jurkat T cells (48).
In addition, hypoxia may induce immune escape in cancer cells via epigenetic
mechanisms. For example, tumor cells can upregulate miR210 in lung cancer and
melanoma (49). In turn, miR210 was shown to block the susceptibility of tumor
cells to lysis by antigen-specific CTLs. This effect was mediated via increased
expression of protein tyrosine phosphatase, nonreceptor type I (PTPN1), homeobox
A1 (HOXA1), and tumor protein p53-inducible protein 11 (TP53I11; ref. 49).
Further studies are required to elucidate the mechanisms used by these molecules
to suppress CTL activity.
Exposure of tumor cells to hypoxia also resulted in resistance to autologous
CTL-mediated lysis in a manner dependent on the signal transducer and activator
of transcription (STAT) 3 (50). STAT3 modulates the cross-talk between tumor and
immune cells (51). A small-molecule inhibitor of STAT3, WP1066, was reported to
reverse immune tolerance in patients with malignant glioma (52). Another STAT3
inhibitor, sunitinib, reduced the immunosuppressive phenotype of renal cell
carcinomas (53) and reversed MDSC-mediated immune suppression via increased
recruitment of CD4+CD8+ cytotoxic T cells (54).
Regulation of Immune Tolerance via Hypoxia-Induced Autophagy
Cancer cells often rely on autophagy as a mechanism of survival under conditions
of stress including hypoxia, nutrient starvation, growth factor withdrawal, and
chemotherapy (55, 56). However, the mechanisms by which autophagy enables
survival of normal or malignant cells are not well known.
Hypoxia-induced autophagy is partly dependent on the HIF1/BNIP3ĘCBNIP3LĘCBeclin1
axis (57), and partly on HIF1/platelet-derived growth factor receptor signaling
(58). Through the activating transcription factor 4 and C/EBP homologous protein
(CHOP), hypoxia increases the expression of microtubule-associated protein 1
light chain 3 (LC3) and autophagy protein 5 (ATG5) involved in formation and
maturation of autophagosomes (59).
Hypoxia-induced autophagy is known to promote tumor cell survival via several
mechanisms, including the removal of damaged mitochondria that produce cytotoxic
reactive oxygen species (57) and the degradation of harmful protein aggregates
(59). Activation of autophagy in cancer cells during hypoxia or exposure to
other microenvironmental stressors may also lead to inhibition of death signals
such as those triggered by CTLs (60). Furthermore, stress-induced release of the
molecular pattern molecule HMGB1 induces cytoprotective autophagy and leads to
recruitment of Tregs (60).
Autophagy can also promote activation of anticancer immunity. For example,
autophagy has been shown to be crucial for proliferation of immune cells as well
as for their effector functions such as antigen presentation and T cellĘCmediated
tumor cell cytotoxicity (61). In T cells, autophagy is activated upon TCR
engagement in both CD4+ and CD8+ T-cell subtypes (62). The knockdown of the
essential autophagy-related genes, ATG5 or ATG7, during TCR stimulation leads to
a significant decrease in cellular proliferation demonstrating the importance of
autophagy during T-cell activation (62, 63). Furthermore, culture of DCs under
low-oxygen results in the stabilization of HIF1Ž┴, which initiates BNIP3
expression and promotes survival of mature DCs, possibly due to induction of
autophagy (64). Hypoxia-induced autophagy in antigen-presenting cells
infiltrating a tumor can occur via Toll-like receptor (TLR) signaling (65).
Together, the above findings indicate a dual role for autophagy in cancer immune
escape. Therefore, immunotherapeutic strategies designed to target autophagy
will need to consider its impact on the immune system.
Nitric Oxide/cGMPĘCMediated Inhibition of Hypoxia-Induced Immune Escape
Our research over the last 15 years has revealed that classical NO signaling
involving cGMP production functions as an O2-sensing mechanism playing a key
role in tumor cell adaptations to hypoxia (42, 43, 48, 66ĘC69). On the basis of
our findings, we postulated that an important aspect of the mechanism by which
cancer cells adapt to hypoxia involves inhibition of endogenous NO/cGMP
signaling. Our research demonstrates that low concentrations of NO mimetics
[e.g., glyceryl trinitrate (GTN), DETA/NO], known to selectively activate
soluble guanylyl cyclase (sGC), inhibit malignant adaptations to hypoxia such as
increased invasiveness, metastatic ability, and drug resistance (66ĘC69).
Moreover, because NO production is dependent on O2 availability, endogenous NO
generation is severely limited in cells exposed to hypoxia (70, 71). This is
despite the fact that hypoxia was shown to increase the expression of inducible
NO synthase (iNOS) in the same cells (RAW 264.7 macrophages; ref. 71). We
previously reported that cGMP levels are decreased in MDA-MB-231 breast tumor
cells incubated for 6 hours in 0.5% O2 (68). This observation is consistent with
the more recent findings of Hickok and colleagues (71), who reported decreased
sGC activation in a murine macrophage line incubated under 5% O2. It is likely
that our observed effects of NO/cGMP signaling on hypoxia-induced malignant
phenotypes are at least partly mediated via inhibition of HIF1 transcriptional
activity. This conclusion is based on evidence that NO mimetics, including the
cGMP analogue 8-bromo-cGMP, inhibit the accumulation of HIF1Ž┴ in cells exposed
to hypoxia (43, 72). Our research has also revealed that NO mimetics interfere
with the HIF1-mediated upregulation of ADAM10 expression involved in the
shedding of MICA from the tumor cell surface and resistance to immune-mediated
lysis (Fig. 1; ref. 43). In that same study, treatment of mice with GTN
attenuated the growth of transplanted prostate tumors via a mechanism dependent
on innate immune effectors. More recently, we demonstrated that low
concentrations of GTN interfere with hypoxia-induced escape from T cellĘCmediated
immunity in tumor cells by preventing the HIF1-dependent expression of PD-L1
(Fig. 1; ref. 48). Together, these studies indicate that activation of NO/cGMP
signaling may have important applications in the prevention and/or treatment of
cancer.
Conclusions
Although there is evidence that hypoxia can activate certain components of
pathways involved in antitumor immunity, most studies indicate that hypoxia is a
major contributor to cancer immune escape. Hypoxia-induced tumor cell escape
from innate and adaptive immunity is likely a consequence of multiple mechanisms
operating in a complementary, and sometimes redundant, manner. Thus, targeting
individual mechanisms of hypoxia-induced immune escape will likely prove to be
ineffective as a therapeutic strategy. However, it is clear that several
mechanisms of such immune escape rely on the transcriptional activity of HIF1.
This raises the possibility that interference with hypoxia response pathways
involving HIF1 activity may be a fruitful immunotherapeutic approach.
Interference with such pathways could be achieved through the use of molecules
that directly inhibit HIF1 activity or block HIF1Ž┴ accumulation in hypoxia. Our
studies on the inhibitory effect of NO/cGMP signaling on HIF1Ž┴ accumulation and
malignant adaptations to hypoxia, including tumor cell escape from innate and
adaptive immunity, support the therapeutic potential of NO mimetic agents. In
this review, we highlighted some key mechanisms of hypoxia-mediated immune
escape. However, because tumor cell avoidance of immune destruction is
multifaceted, it is likely that hypoxia influences escape mechanisms not
described herein. The role of the hypoxic tumor microenvironment on other key
aspects of cancer immune surveillance, such as antigen presentation, additional
immune checkpoints and effector mechanisms of tumor cell destruction, warrants
investigation.
Mechanisms of Hypoxia-Mediated Immune Escape in Cancer | Cancer Research
https://cancerres.aacrjournals.org/content/74/24/7185
íí
Little bang for the Big Cancer? Nitroglycerin in the
anti-cancer arsenal
James Bond on a lilo couldní»t match this relaxation agent
By Pan Pantziarka 27 Oct 2015 at 09:28 37 Reg comments SHARE Ęő
Big Bang
It's often the case that when people talk of wonder drugs in cancer they most
often think of the latest exquisitely engineered molecules that closely target
very specific biochemical pathways. Think high cost, think high science, and
think high hopes.
And yet there's evidence mounting that one of our oldest and most widely used
medical treatments is something of a wonder drug too.
Another paper published in the British Journal of Cancer (Aspirin as a
neoadjuvant agent during preoperative chemoradiation for rectal cancer) is
reporting on a clinical trial that showed that the humble aspirin had a positive
effect on overall survival when used during the chemoradiation treatment of
rectal cancer patients.
It is one of a number of studies which show that aspirin has positive effects as
a cancer treatment, not just as a cancer prevention agent.
All this from a drug that has existed in its modern form for more than 115 years
ĘC Ií»d say that qualifies aspirin as something of a wonder drug.
But while the story of aspirin as an anticancer drug is gaining increasing
public attention, less well known is the story of another venerable old drug ĘC
glyceryl trinitrate; better known as nitroglycerin.
Like aspirin, this is a drug with a long history ĘC the original report of its
medical application dates back to a paper in the Lancet by William Murrell in
1879, so it predates aspirin by about 20 years. A medical application for a
compound better known as a potent explosive ĘC most of us know of nitroglycerin
as the active ingredient in dynamite ĘC was something of a surprise.
But then there are a lot of surprising things about nitroglycerin, not the least
of which is that it might enable us to tackle one of the most difficult problems
in cancer: treatment resistance.
It is this possibility that attracted our attention at the Repurposing Drugs in
Oncology project and explored in a recent paper (Repurposing Drugs in Oncology
(ReDO) ĘC nitroglycerin as an anti-cancer agent). Before looking at what we found
we need to back up a little bit and take a look at what nitroglycerin does when
ití»s not blowing things up, and at what treatment failure means in cancer.
Nitroglycerin is used as a vasodilator ĘC that is, it eases the muscle cells that
surround blood vessels so they relax and open up. Ití»s this property which makes
it useful as treatment for hypertension, angina and congestive heart failure. As
an aside ití»s this property that makes the related drug amyl nitrite (the
recreational drug known as poppers) of interest in certain sexual subcultures.
In the case of nitroglycerin it can be used in an acute situation ĘC usually
delivered via a sublingual tablet or spray ĘC or as a chronic treatment, where it
is delivered via transdermal patches, which are worn during the day. Ití»s a
safe, cheap and well-known drug with decades of use behind it.
You would think, therefore, that we would know all there is to know about how it
works its magic. But thatí»s not case. The actual mechanism by which it works at
the molecular level has still not been fully worked out. The question is how do
we go from the drug to the action of vasodilation?
The drug was developed at a time when we had very little knowledge of
fundamental biochemistry ĘC for a long time drug development was a rather
empirical process in comparison with the targeted molecular approach we now use.
In fact, even now there are competing theories to explain how it works ĘC but
that it works is not in doubt.
What has all this got to do with cancer? Well, potentially rather a lot. Two of
the mainstays of cancer treatment are high dose chemotherapy and radiotherapy.
Even though the anti-cancer armoury has expanded and continues to expand, most
cancer treatments will involve one or both of these.
Both of these treatments work by massively killing tumour cells ĘC and we need to
do this quickly before resistance evolves and before we kill the patient. If
thereí»s one thing we know about cytotoxic chemotherapy in particular is that
thereí»s a lot of collateral damage ĘC to cells in the gastrointestinal tract
(vomiting), immune system (neutropenia), hair follicles (hair loss) and so on.
Radiotherapy carries its own set of side effects, and there too we have the
problem that some tumours can become radio-resistant. The treatment resistance
is a major problem ĘC because once the first-line treatments fail the second
often fail too.
There are numerous factors involved in these forms of resistance, and some of
them are architectural. Tumours need a supply of nutrients, they need oxygen,
they need all the support systems that normal tissues need ĘC and they need lots
of them.
However, these tumours are very good are kicking off a process called
angiogenesis ĘC the sprouting of new blood vessels to keep themselves going.
Angiogenesis is a normal physiological function in wound healing, foetal growth,
and general development; but what happens in cancer is that this process is
hijacked. The result though is a highly disordered and chaotic vasculature
around a tumour, with immature blood vessels, some areas starved of oxygen and
nutrients and some areas well-supplied with both.
The problem is we need that blood supply to get the chemo drugs deep inside
tumours. And we need oxygen to react with the radiotherapy to cause the cell
damage to kill cancer cells. Therefore, if we can tackle this then we can get
more bang for our buck ĘC the chemotherapy drugs make it deeper into the tumours,
the radiotherapy causes more reactive oxygen to kill resistant cells. And this
is where nitroglycerin comes in.
By causing vasodilation, nitroglycerin causes the immature blood vessels to
become more leaky ĘC so the chemo drugs which are circulating in the blood seep
out more readily into the tumour. While it doesní»t molecularly target cancer
cells specifically, it does mean that you get more chemo where you want it and
less where you doní»t. Nitroglycerin also has the nice property that it increases
the oxygenation of tissues which are relatively hypoxic (lacking oxygen).
Hypoxia is a factor in treatment resistance to both chemo and radiotherapy, and
ití»s also a selective pressure associated with the evolution of more aggressive
and metastatic disease. Ití»s a major target of drug research at the moment,
although from the evidence that weí»ve uncovered in our paper it looks like
nitroglycerin does a good job there too.
The good news is that there is some clinical work already going on to prove that
this works in practice. For example, we are supporting a clinical trial in the
Netherlands in non-small cell lung cancer in which patients being treated with
combined chemo and radiotherapy wear a transdermal nitroglycerin patch.
There are other trials too, including a Phase III trial in prostate cancer in
which transdermal nitroglycerin is used as a standalone therapy in men who show
biochemical signs of recurrence after treatment.
Our hope is that in the future patients undergoing standard cancer treatments
will also be wearing transdermal nitroglycerin patches in those cancers in which
we have proved that doing so increases the overall survival. The potential
impact might be explosive. ®
Little bang for the Big C? Nitro in the anti-cancer arsenal • The Register
https://www.theregister.co.uk/2015/10/27/nitroglycerin_cancer_treatment/
íí
Glyceryl trinitrate‑induced cytotoxicity of
docetaxel‑resistant prostatic cancer cells is associated with differential
regulation of clusterin
Laboratoire d'Immunologie et ImmunothĘŽrapie des Cancers, EPHE, PSL Research
University, F‑75000 Paris, France, Centre Georges‑François Leclerc, F‑21000
Dijon, France
Metastatic castration resistant prostate cancer (mCRPC) relapse due to acquired
resistance to chemotherapy, such as docetaxel, remains a major threat to patient
survival. Resistance of mCRPC to docetaxel can be associated with elevated
levels of soluble clusterin (sCLU) and growth differentiation factor‑15
(GDF‑15). Any strategies aiming to modulate sCLU and/or GDF‑15 in
docetaxel‑resistant prostate cancer cells present a therapeutic interest.
The present study reports the cytotoxic effect of a nitric oxide donor,
glyceryl trinitrate (GTN), on docetaxel‑resistant mCRPC human cell lines and
demonstrates that GTN displays greater inhibition of cell viability toward
docetaxel‑resistant mCRPC cells than on mCRPC cells. It is also demonstrated
that GTN modulates the level of expression of clusterin (CLU) which is dependent
of GDF‑15, two markers associated with docetaxel resistance in prostate cancer.
The results indicate that GTN represses the level of expression of the
cytoprotective isoform of CLU (sCLU) and can increase the level of expression of
the cytotoxic isoform (nuclear CLU) in docetaxel resistant cells. Furthermore,
it was observed that GTN differentially regulates the level of the precursor
form of GDF‑15 between resistant and parental cells, and that recombinant GDF‑15
can modulate the expression of CLU isoforms and counteract GTN‑induced
cytotoxicity in resistant cells. A link was established between GDF‑15 and the
expression of CLU isoforms. The present study thus revealed GTN as a potential
therapeutic strategy to overcome docetaxel‑resistant mCRPC.
Glyceryl trinitrate‑induced cytotoxicity of docetaxel‑resistant prostatic cancer
cells is associated with differential regulation of clusterin
https://www.spandidos-publications.com/10.3892/ijo.2019.4708
íí
Chemosensitization of Cancer In vitro and In vivo by Nitric Oxide Signaling
Department of Pathology and Molecular Medicine, Queen's University
Lisa J. Frederiksen, Richard Sullivan, Lori R. Maxwell, Shannyn K.
Macdonald-Goodfellow, Michael A. Adams, Brian M. Bennett, D. Robert Siemens and
Charles H. Graham
DOI: 10.1158/1078-0432.CCR-06-1807 Published April 2007
ArticleFigures & DataInfo & Metrics PDF
Abstract
Purpose: Hypoxia contributes to drug resistance in solid cancers, and studies
have revealed that low concentrations of nitric oxide (NO) mimetics attenuate
hypoxia-induced drug resistance in tumor cells in vitro. Classic NO signaling
involves activation of soluble guanylyl cyclase, generation of cyclic GMP
(cGMP), and activation of cGMP-dependent protein kinase. Here, we determined
whether chemosensitization by NO mimetics requires cGMP-dependent signaling and
whether low concentrations of NO mimetics can chemosensitize tumors in vivo.
Experimental Design: Survival of human prostate and breast cancer cells was
assessed by clonogenic assays following exposure to chemotherapeutic agents. The
effect of NO mimetics on tumor chemosensitivity in vivo was determined using a
mouse xenograft model of human prostate cancer. Drug efflux in vitro was
assessed by measuring intracellular doxorubicin-associated fluorescence.
Results: Low concentrations of the NO mimetics glyceryl trinitrate (GTN) and
isosorbide dinitrate attenuated hypoxia-induced resistance to doxorubicin and
paclitaxel. Similar to hypoxia-induced drug resistance, inhibition of various
components of the NO signaling pathway increased resistance to doxorubicin,
whereas activation of the pathway with 8-bromo-cGMP attenuated hypoxia-induced
resistance. Drug efflux was unaffected by hypoxia and inhibitors of drug efflux
did not significantly attenuate hypoxia-induced chemoresistance. Compared with
mice treated with doxorubicin alone, tumor growth was decreased in mice treated
with doxorubicin and a transdermal GTN patch. The presence of GTN and GTN
metabolites in plasma samples was confirmed by gas chromatography.
Conclusion: Tumor hypoxia induces resistance to anticancer drugs by interfering
with endogenous NO signaling and reactivation of NO signaling represents a novel
approach to enhance chemotherapy.
Chemosensitization of Cancer In vitro and In vivo by Nitric Oxide Signaling |
Clinical Cancer Research
https://clincancerres.aacrjournals.org/content/13/7/2199
íí