1. Immune Evasion by Helicobacter pylori is Mediated by Induction of Macrophage Arginase II,upexpression of Arginase2
2. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice.
3.Human Gastric Epithelium Produces IL-4 and IL-4δ2 Isoform Only upon Helicobacter Pylori Infection
4.arginase inhibitors: all alpha-amino acids(non-competitive, di-aminoacids, competitive), especially orthinine(strongly, competitive and non-competiitive), lysine(competitive inhibitor,0.6 times as effective as orthinine), Nitric oxide, CB-1158, borate, l-hydroxyarginine, the most potent physiologic inhibitor of arginase
5.Arginase Inhibitors: A Rational Approach Over One Century
6. L-正缬氨酸(L-norvaline) 增强活化巨噬细胞产生 NO
7.Phenolic Compounds as Arginase Inhibitors: New Insights Regarding Endothelial Dysfunction Treatment.
8.CB-1158 increased tumor-infiltrating CD8+ T cells and NK cells, inflammatory cytokines, and expression of interferon-inducible genes. Patient tumor samples from multiple histologies expressed an abundance of tumor-infiltrating Arg1+ myeloid cells. Plasma samples from cancer patients exhibited elevated Arg1 and reduced L-arginine compared to healthy volunteers.
9. lysine is a potent inhibitor of arginase and hence blocks arginase-mediated urea synthesis from arginine results in increased serum arginine
http://www.jimmunol.org/content/167/11/6533
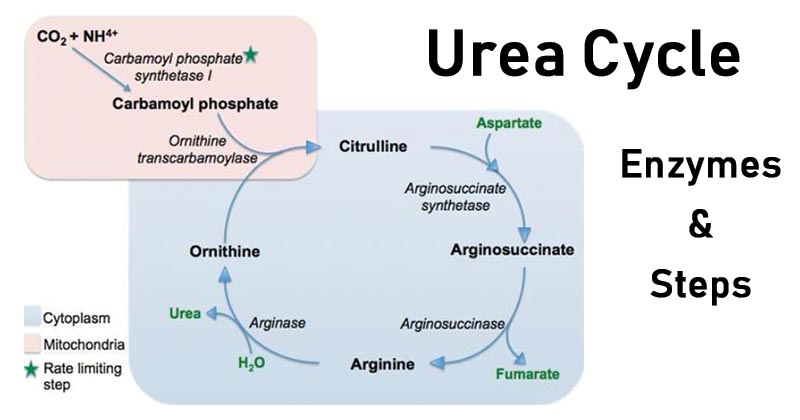
Biosynthesis of nitric oxide
NO biosynthesis is carried out from L-arginine and is catalyzed by NO-synthases, NOS, an heminic enzyme whose structure resembles that of P-450 cytochrome. In the presence of cofactors ( NADPH, oxygen, iron, tetrahydrobiopterine, FAD and FMN), enzyme NO-synthases, convert arginine into hydroxyarginine and finally into citrulline and NO according to the following reactions:
Biosynthesis of NO starting from L-arginine by NO-Synthase
Biosynthesis of NO starting from L-arginine
Citrulline, in presence of arginosuccinate synthethase and aspartate, is then converted into arginosuccinate, fumarate and arginine. Thus, arginine comes from an endogenous renewal and an exogenous source, food.
Nitric oxide - Pharmacorama
https://www.pharmacorama.com/en/Sections/NoNitric_2.php
arginase inhibitors,:orthinine, lysine are competitive inhibitor, norvaline has been shown to improve available resources of arginine and to increase nitric oxide (NO) production.
Georg Thieme Verlag KG Stuttgart · New York.
PMID: 29342480 DOI: 10.1055/s-0044-100398Phenolic Compounds as Arginase Inhibitors: New Insights Regarding Endothelial Dysfunction Treatment. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29342480
Frontiers | Lysine and arginine reduce the effects of ...
https://www.frontiersin.org/articles/10.3389/fnint.2010.00018
In addition, lysine is a potent inhibitor of arginase and hence blocks arginase-mediated urea synthesis from arginine (Egan et al., 1995). Intravenous infusion of lysine resulted in a significant increase in plasma levels of arginine (Kato et al., 1987), suggesting increased availability of arginine for NO synthesis. In general, lysine ...
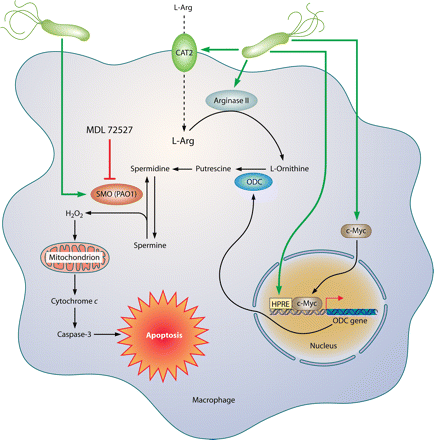
One primordial mechanism for antimicrobial host defense is the generation of high levels of NO derived from the enzyme inducible NO synthase (iNOS) (13). Our laboratory and others have demonstrated that H. pylori induces the expression and activity of iNOS in macrophages both in vivo and in vitro
Arginase enzymes are the endogenous antagonists to iNOS because they compete for the same l-arginine substrate by metabolizing it to urea and l-ornithine (21).
the downstream effects of arginase metabolism can be detrimental to iNOS-mediated host defense against H. pylori. The polyamine spermine inhibits l-arginine uptake in macrophages, thereby blocking NO production and bacterial killing
increased gastritis correlated with decreased bacterial colonization in individual Arg2−/− mice, but not in WT mice. Moreover, we show that in Arg2−/− mice infected with H. pylori there are more iNOS+ gastric macrophages that express increased levels of iNOS, enhanced pro-inflammatory cytokine responses, and decreased levels of macrophage apoptosis.
https://cmr.asm.org/content/23/4/713
J Immunol. Author manuscript; available in PMC 2011 Apr 1.
Immune Evasion by Helicobacter pylori is Mediated by Induction of Macrophage Arginase II1
Nuruddeen D. Lewis,*† Mohammad Asim,*‡ Daniel P. Barry,*‡ Thibaut de Sablet,*‡ Kshipra Singh,*‡ Maria B. Piazuelo,* Alain P. Gobert,*‡§ Rupesh Chaturvedi,*‡ and Keith T. Wilson*†‡,2
Division of Gastroenterology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, 37240
†Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, 37240
‡Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN 37212
§Institut National de la Recherche Agronomique, Unité de Microbiologie UR454, Saint-Genès-Champanelle, France
2Address correspondence and reprint requests to Dr. Keith T. Wilson, Department of Medicine, Division of Gastroenterology, Vanderbilt University School of Medicine, 1030C MRB IV, 2215B Garland Ave., Nashville, TN
Abstract
Helicobacter pylori infection persists for the life of the host due to the failure of the immune response to eradicate the bacterium. Determining how H. pylori escapes the immune response in its gastric niche is clinically important. We have demonstrated in vitro that macrophage NO production can kill H. pylori, but induction of macrophage arginase II (Arg2) inhibits inducible NO synthase (iNOS) translation, causes apoptosis, and restricts bacterial killing. We now determined if Arg2 impairs host defense in vivo, using a chronic H. pylori infection model. In C57BL/6 mice, expression of Arg2, but not arginase I (Arg1), was abundant and localized to gastric macrophages. Arg2−/− mice had increased histologic gastritis and decreased bacterial colonization compared to wild-type (WT) mice. Increased gastritis scores correlated with decreased colonization in individual Arg2−/− mice, but not WT mice. When mice infected with H. pylori were compared, Arg2−/− mice had more gastric macrophages, more of these cells were iNOS+, and these cells expressed higher levels of iNOS protein, as determined by flow cytometry and immunofluorescence microscopy. There was enhanced nitrotyrosine staining in infected Arg2−/− versus WT mice, indicating increased NO generation. Infected Arg2−/− mice exhibited decreased macrophage apoptosis, as well as enhanced IFN-γ, IL-17a, and IL-12p40 expression, and reduced IL-10 levels consistent with a more vigorous Th1/Th17 response. These studies demonstrate that Arg2 contributes to the immune evasion of H. pylori by limiting macrophage iNOS protein expression and NO production, mediating macrophage apoptosis, and restraining pro-inflammatory cytokine responses.
Introduction
Helicobacter pylori is a Gram-negative, microaerophilic bacterium that selectively colonizes the human stomach. All infected individuals exhibit chronic active gastritis and a substantial proportion of subjects develop peptic ulcer disease or gastric adenocarcinoma (1). H. pylori infects approximately 50% of the world’s population, and, more importantly, the associated gastric cancer is the second leading cause of cancer-related death worldwide (2). The infection is usually acquired in childhood and persists for the life of the host despite eliciting a seemingly vigorous immune response (3). Understanding the mechanisms by which H. pylori avoids being eliminated by the immune system is clinically relevant because antibiotic-based eradication regimens are expensive and not always effective, with success rates that can be less than 50% in some regions of the world (4).
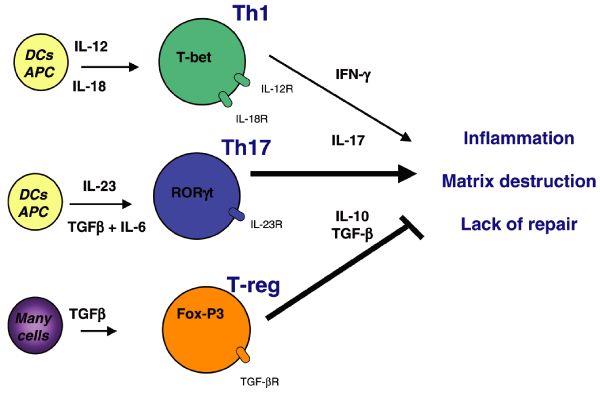
While H. pylori is typically considered to be noninvasive because most of the bacteria reside in the mucus layer of the stomach in contact with the epithelium, studies have demonstrated that the bacterium and its products can be in direct contact with lamina propria immune cells (5–7). Consequently, infection with H. pylori results in a large influx of immune cells that include neutrophils, macrophages, dendritic cells, and lymphocytes, and an associated innate and adaptive immune response (8). While this has been shown to include both Th1 and Th17 components, one hallmark of the response is that there is also a downregulation of effective immunity that appears to involve recruitment of regulatory T cells (Tregs) and B cells (3). Vaccination studies, adoptive transfer of Th1-selected lymphocytes, and efforts to suppress Treg responses have been successful at reducing or clearing the infection in mice (9–12). These studies have provided evidence that the cellular immune response is not vigorous enough to lead to eradication of the infection. One important aspect that remains to be fully elucidated is the role of the innate immune response in the impairment of the host response. We propose that there may be an inability of effector cells to eliminate the infection when given the opportunity to do so.
One primordial mechanism for antimicrobial host defense is the generation of high levels of NO derived from the enzyme inducible NO synthase (iNOS) (13). Our laboratory and others have demonstrated that H. pylori induces the expression and activity of iNOS in macrophages both in vivo and in vitro (14–17). Further, we have reported that macrophages cocultured with H. pylori can kill the bacterium by an NO-dependent mechanism (15, 18). However, this killing is incomplete in vitro, and, moreover, there is clearly a failure of this mechanism in vivo despite the expression of iNOS in the infected mucosa (14, 17). This reasoning has led our laboratory to consider the possibility that iNOS-mediated host defense to H. pylori is suboptimal. We have reported that the generation of NO by macrophages in response to H. pylori is entirely dependent on the availability of its substrate, l-arginine, which enhances expression of iNOS protein, without altering the induction of mRNA expression (15, 18). Specifically, we found that addition of increasing levels of extracellular l-arginine results in a proportionate increase in NO production even at concentrations well above the circulating levels in humans and mice of 0.1 mM and above the Km of the iNOS enzyme for l-arginine, which is in the range of 10 µM (19). In order for macrophages to produce bactericidal amounts of NO when cocultured with H. pylori in our model system, concentrations of l-arginine in the medium that exceeded 0.1 mM were needed. Importantly, infection of iNOS−/− mice with H. pylori results in similar levels of bacterial colonization as wild-type (WT) mice (20), further suggesting a defect in iNOS-dependent host defense.
Arginase enzymes are the endogenous antagonists to iNOS because they compete for the same l-arginine substrate by metabolizing it to urea and l-ornithine (21). The latter is utilized by ornithine decarboxylase to produce the polyamines putrescine, spermidine, and spermine. There are two isoforms of arginase: arginase I (Arg1) is abundant in liver and is important for the urea cycle, and arginase II (Arg2) is abundant in kidney and localizes to mitochondria (22). We have reported that Arg2, but not Arg1, is upregulated in H. pylori-stimulated macrophages (15). Induction of arginase activity by pathogens has been reported to modulate macrophage NO production in the case of both Arg1 and Arg2, which can restrict effective immunity (23–25). These effects have been attributed to substrate competition. However, we have recently reported that induction of macrophage Arg2 by H. pylori inhibits iNOS translation, NO production, and bacterial killing in vitro (15). Furthermore, in mice infected with H. pylori for 48 h, treatment with an arginase inhibitor resulted in increased iNOS protein levels and NO production in gastric macrophages (15). We have also reported that the downstream effects of arginase metabolism can be detrimental to iNOS-mediated host defense against H. pylori. The polyamine spermine inhibits l-arginine uptake in macrophages, thereby blocking NO production and bacterial killing (26). Moreover, back-conversion of spermine into spermidine by the enzyme spermine oxidase, which is also induced by H. pylori, releases hydrogen peroxide that causes apoptosis (27, 28). Consequently, we have found that inhibition of arginase blocks H. pylori-induced macrophage apoptosis in our in vitro studies (29).
We hypothesized that Arg2 expression is detrimental to host defense against H. pylori in vivo by restricting macrophage NO production and inducing macrophage apoptosis. We now report that chronic infection of Arg2−/− mice with H. pylori results in decreased bacterial colonization and increased gastritis as compared to infected WT mice. Importantly, we found that increased gastritis correlated with decreased bacterial colonization in individual Arg2−/− mice, but not in WT mice. Moreover, we show that in Arg2−/− mice infected with H. pylori there are more iNOS+ gastric macrophages that express increased levels of iNOS, enhanced pro-inflammatory cytokine responses, and decreased levels of macrophage apoptosis. Our data suggest that upregulation of macrophage Arg2 is detrimental to the host response against H. pylori and demonstrate a mechanism by which H. pylori evades host immunity.
Materials and Methods
Reagents
All reagents used for RNA extraction were from Invitrogen (Carlsbad, CA). Real-time PCR reagents were from Bio-Rad (Hercules, CA). Isolation of DNA was performed using the DNeasy® Blood & Tissue kit (QIAGEN, Valencia, CA). All other chemicals were from Sigma-Aldrich (St. Louis, MO).
Bacteria
H. pylori SS1, a mouse-adapted human strain, was grown on Brucella blood agar plates under microaerobic conditions as described (16, 28). Prior to infection, bacteria were grown in a Brucella broth culture for 16 – 20 h. Concentrations of bacteria were determined by optical density at 600 nm (OD of 1 = 109 CFU/ml) (16).
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee at Vanderbilt University (Nashville, TN). C57BL/6 Arg2−/− and WT male mice were used at 6–8 wks of age as described (15). Mice were gavaged with 5 × 108 H. pylori SS1 in 100 µl of Brucella broth, or broth vehicle control alone. Some mice were sacrificed at 2 d postinoculation, and stomachs were excised and used for isolation of gastric macrophages, which was performed as described (14, 18). Other mice were sacrificed at 4 mo postinoculation and colonization and gastritis were assessed. H. pylori levels were measured by quantitative DNA PCR for H. pylori ureB, standardized to mouse 18S rRNA copy number (14). Histologic gastritis was quantified by a pathologist (M.B.P.) using a scale of 0 – 3 for acute inflammation and for chronic inflammation in the regions of the gastric antrum and body with the scores added together for a total score of 0 – 12 (14, 30).
Apoptosis detection by annexin V staining
Gastric macrophages were isolated and stained with annexin V conjugated to FITC and 7-aminoactinomycin D (7-AAD; BD Biosciences, San Jose, CA) as described (31, 32).
Real-time PCR
RNA was extracted using the RNeasy Mini Kit from Qiagen (Valencia, CA). PCR methods were performed as described (15, 18, 29, 33). Primer sequences used were as follows: IL-10, 5′-CCAAGCCTTATCGGAAATGA-3′ (forward) and 5′-TCACTCTTCACCTGCTCCAC-3′ (reverse); IL-12p40, 5′-GATTCAGACTCCAGGGGACA-3′ (forward) and 5′-CATCTTCTTCAGGCGTGTCA-3′ (reverse); IL-17a, 5′-GCTCCAGAAGGCCCTCAGA-3′ (forward) and 5′-CTTTCCCTCCGCATTGACA-3′ (reverse); Foxp3, 5′-AGAGCCCTCACAACCAGCTA-3′ (forward) and 5′-CCAGATGTTGTGGGTGAGTG-3′ (reverse); IL-6, 5′-AGTTGCCTTCTTGGGACTGA-3′ (forward) and 5′-TCCACGATTTCCCAGAGAAC-3′ (reverse); and IL-23p19, 5′-CATGGGGCTATCAGGGAGTA-3′ (forward) and 5′-AATAATGTGCCCCGTATCCA-3′ (reverse). Primer sequences for IFN-γ (33), Arg1 (29), Arg2 (15), β-actin (18), and iNOS (18) were as described.
Immunofluorescence staining for F4/80, Arg2, and iNOS, and immunoperoxidase staining for nitrotyrosine and cleaved caspase-3
Immunofluorescence staining for F4/80, and DAPI was performed as described (14). iNOS was detected with a rabbit polyclonal antibody (1:!00 dilution, BD Biosciences, San Diego, CA). Arg2 was detected with a goat polyclonal antibody (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Immunoperoxidase staining was performed as described (14). Nitrotyrosine was detected with a mouse monoclonal anti-vertebrate nitrotyrosine antibody (1:100 dilution; Millipore, Billerica, MA). Cleaved caspase-3 was detected with a rabbit polyclonal antibody (1:200 dilution; Cell Signaling Technology, Danvers, MA). Quantification of cleaved caspase-3 staining was performed by counting all positively-stained cells amongst the inflammatory infiltrate and dividing this number by the total number of inflammatory cells to calculate the percentage of positive cells. Cell counting was performed at 600× magnification under oil immersion.
Gastric Macrophages
Immune cells were isolated from the glandular stomach by enzymatic digestion as described (14, 18, 31). Briefly, mice were sacrificed and the stomach was removed. The forestomach (nonglandular portion) was excised and discarded. The glandular portion of the stomach was washed, cut into 2 mm pieces, and digested for 20 min with 1 mg/ml of dispase, 0.25 mg/ml of collagenase A, and 25 U/ml DNase (Roche Diagnostics, Indianapolis, IN) at 37°C while shaking. The suspension was passed through a 70 µm cell strainer (BD Biosciences, San Diego, CA) and cells harvested by centrifugation. Cells were stained for F4/80 and iNOS and analyzed by flow cytometry. F4/80+ and F4/80+ iNOS+ cell counts were standardized to the weight in grams of the glandular stomach.
Statistical Analysis
Quantitative data are shown as the mean ± SE. Statistical analysis was performed with Prism version 5.0c (GraphPad Software, Inc., San Diego, CA). Where two groups were compared, Student’s t test was used. Data with more than two groups were analyzed by ANOVA and the Student-Newman Keuls post-hoc multiple comparisons test. p < 0.05 was considered statistically significant. The relationship between gastritis and colonization was determined using the Spearman’s correlation test; the correlation coefficient, r, is shown along with the p value.
Results
H. pylori infection induces Arg2, not Arg1, in gastric lamina propria F4/80+ macrophages
Mice were infected for 4 mo, a time point at which we have demonstrated consistent development of chronic active gastritis with strain SS1 (14). Arg2 mRNA expression was increased by >9–fold in H. pylori-infected WT mice when assessed by real-time PCR, while Arg1 was not induced (Fig. 1A, 1B). Furthermore, Arg1 was not upregulated in Arg2−/− mice (Fig. 1B). Additionally, iNOS mRNA expression increased in infected WT mice, and this was further enhanced in Arg2−/− mice (Fig. 1C).
An external file that holds a picture, illustration, etc.
Object name is nihms277366f1.jpg
Open in a separate window
FIGURE 1
H. pylori infection induces iNOS (in Arg2−/− mice )and Arg2, but not Arg1, in gastric tissues. C57BL/6 WT and Arg2−/− mice were infected with H. pylori strain SS1. At 4 mo postinfection, mice were sacrificed, gastric tissues were removed, and RNA was extracted. iNOS (A), Arg2 (B), and Arg1 (C) mRNA levels were measured by real-time PCR from the antral portion of the stomach. All real-time PCR data were standardized to β-actin and presented as fold increase versus uninfected WT mice. Data are the mean ± SEM, n = 6 in the uninfected mice and n = 10 in each of the infected mice groups. ***p < 0.001 compared with WT uninfected mice. §p < 0.05; §§§p < 0.001 compared with WT infected mice. D, Photomicrographs, displayed at 200× and 600×, demonstrating Arg2 immunofluorescence staining in F4/80+ macrophages. F4/80 was detected with tetramethyl rhodamine isothiocyanate (red), and Arg2 was detected with FITC. Data are representative of 3–4 mice per group.
To determine which cell type expressed Arg2 in the stomach, we immunostained formalin-fixed, paraffin-embedded gastric tissue sections from WT and Arg2−/− mice (Fig. 1D). This staining demonstrated increased Arg2 levels in H. pylori-infected tissues from WT mice that localized to F4/80+ macrophages. Arg2 staining was not present in macrophages in the Arg2−/− tissues.
Chronically-infected Arg2−/− mice have increased gastritis and decreased H. pylori colonization
To determine the role of Arg2 during H. pylori infection, we infected WT and Arg2−/− mice for 4 mo and assessed gastritis levels and bacterial colonization. With H. pylori infection, there was a significant increase in overall gastritis in WT mice that was further increased in the Arg2−/− mice (Fig. 2A). In addition to enhanced gastritis levels, we found a significant decrease in H. pylori colonization levels in Arg2−/− mice (Fig. 2B). Notably, increased gastritis correlated significantly with decreased bacterial colonization in Arg2−/− mice (p = 0.006, r = −0.491), but there was no such effect in WT mice (p = 0.636, r = −0.088).
An external file that holds a picture, illustration, etc.
Object name is nihms277366f2.jpg
Open in a separate window
FIGURE 2
Increased gastritis and decreased bacterial colonization in Arg2−/− mice. C57BL/6 WT and Arg2−/− mice were infected with H. pylori for 4 mo, sacrificed, and their stomachs were removed. A, Hematoxylin and eosin-stained slides were prepared from a strip of the glandular stomach containing both the antrum and body. Acute and chronic inflammation were each scored 0–3 in the antrum and body, and the scores added for a 0–12 scale. **p < 0.01. B, DNA was extracted from the body of the stomach and bacterial colonization was quantified by real-time PCR for the H. pylori gene ureB normalized to 18S rRNA. **p < 0.01. C, Gastritis scores and bacterial colonization were plotted for individual WT and Arg2−/− mice to determine if a correlation exists between gastritis and bacterial colonization. Linear regression lines are shown. For WT mice, Spearman’s correlation coefficient r = −0.088 and p = 0.636. For Arg2−/− mice, Spearman’s correlation coefficient r = −0.491 and p = 0.006. D, Representative hematoxylin and eosin-stained sections are shown for both uninfected and infected WT and Arg2−/− mice. Photomicrographs are depicted at 200× magnification and 600× for the inset. E, Photographs were taken to demonstrate the gross anatomy of the glandular portion of the stomach. The red rectangle highlights the transition zone (TZ) of the stomach.
Representative photomicrographs of hematoxylin and eosin-stained gastric sections are shown for uninfected and infected WT and Arg2−/− mice (Fig. 2D). These demonstrate that in the transition zone between the body and the antrum of the stomach where H. pylori-induced inflammation is typically most severe, there was more extensive acute and chronic inflammatory cell infiltration in Arg2−/− mice. There was no spontaneous inflammation in the uninfected Arg2−/− mice. Additionally, we observed gross thickening of the gastric mucosa in the transition zone of Arg2−/− mice that was indicative of increased gastric inflammation (Fig. 2E).
Chronic infection of H. pylori induces pro-inflammatory cytokine production that is further enhanced in Arg2−/− mice
To assess alterations in cytokine production during H. pylori infection and determine differences in the immune response, we analyzed mRNA expression of various cytokines in uninfected and infected WT and Arg2−/− mice. IFN-γ, IL-12p40, and IL-17a were increased in infected WT mice and were further upregulated in Arg2−/− mice (Fig. 3A, 3B, 3C, respectively). IL-10 expression, a hallmark of the counter-regulatory response to Th1 and Th17 responses in H. pylori infection (11, 34), was increased in infected WT, but not Arg2−/− mice (Fig. 3D). IL-23p19, IL-6, and Foxp3 were each modestly increased upon infection with H. pylori, but there was no difference between the WT and Arg2−/− mice (Supplemental Fig. 1).
An external file that holds a picture, illustration, etc.
Object name is nihms277366f3.jpg
Open in a separate window
FIGURE 3
Chronic infection with H. pylori induces proinflammatory cytokine production that is further enhanced in Arg2−/− mice. mRNA was extracted from the gastric antrum of uninfected and infected WT and Arg2−/− mice at 4 mo postinoculation, converted to cDNA, and real-time PCR was performed for IFN-γ (A), IL-12p40 (B), IL-17a (C), and IL-10 (D). Data were standardized to β-actin and presented as fold increase versus uninfected WT mice. *p < 0.05; **p < 0.01; ***p < 0.001 compared to uninfected WT mice. §p < 0.05; §§p < 0.01 compared to infected WT mice. For uninfected mice, n = 3–6 per group, and for infected mice, n = 5–10 per group.
Arg2−/− macrophages are more abundant, express more iNOS, and have increased nitrotyrosine staining as compared to WT macrophages during H. pylori infection
We have previously demonstrated that H. pylori stimulation induces Arg2 expression in macrophages and this expression attenuates iNOS translation in vitro (15). Furthermore, we have shown that Arg2−/− gastric macrophages isolated 48 h postinoculation with H. pylori express more iNOS protein and produce more NO as compared to WT macrophages (15). Therefore, to determine if such an effect occurred during chronic infection with H. pylori, we analyzed levels of the macrophage surface marker F4/80, and iNOS protein expression by immunofluorescence in WT and Arg2−/− gastric tissues 4 mo postinoculation (Fig. 4), along with the appropriate isotype controls (Supplemental Fig. 2). Consistent with our recent report (14), there was increased macrophage staining in H. pylori-infected versus uninfected tissues in WT mice. The abundance of this F4/80 staining was significantly increased in the infected Arg2−/− mice. Similarly, with H. pylori infection, iNOS staining was increased in WT mice, but substantially potentiated in the Arg2−/− mice (Fig. 4). When the merged images were assessed, the iNOS staining was found to localize predominantly to the F4/80+ cells, and there were more iNOS+ macrophages in the Arg2−/− mice (Fig. 4). This staining was present in the lamina propria, with trails of iNOS+ macrophages migrating towards the lumen, as well as in the submucosal region.
An external file that holds a picture, illustration, etc.
Object name is nihms277366f4.jpg
Open in a separate window
FIGURE 4
Arg2−/− mice have increased iNOS+ macrophages during chronic infection with H. pylori. Representative immunofluorescence staining from uninfected and infected WT and Arg2−/− mice are shown. The macrophage marker F4/80 was detected with tetramethyl rhodamine isothiocyanate (red), iNOS was detected with FITC (green), and nuclei were stained with DAPI (blue); co-localization is shown in merged images by the yellow color. Photomicrographs are shown at 200× magnification and 600× for the inset.
To confirm our observation that there are more iNOS+ macrophages in infected Arg2−/− mice as compared to infected WT mice, we isolated gastric immune cells and analyzed F4/80 and iNOS expression by flow cytometry. In accordance with our immunofluorescence data, we found a significant increase in both the quantity of macrophages (Fig. 5A) and iNOS+ macrophages (Fig. 5B) in infected Arg2−/− mice compared to WT mice. Representative flow cytometric dot plots are also shown demonstrating an increased percentage of F4/80+ iNOS+ cells in infected Arg2−/− mice versus WT mice (Fig. 5C). Importantly, the Arg2−/− gastric macrophages expressed more iNOS protein than the cells from the WT mice (Fig. 5D, 5E).
An external file that holds a picture, illustration, etc.
Object name is nihms277366f5.jpg
Open in a separate window
FIGURE 5
Arg2−/− macrophages are more abundant, express more iNOS, and have increased nitrotyrosine staining as compared to WT macrophages during H. pylori infection. Gastric cells were isolated from infected WT and Arg2−/− mice and analyzed by flow cytometry for the expression of the macrophage marker F4/80 and iNOS. The total number of F4/80+ cells (A, **p < 0.01 and ***p < 0.001 vs uninfected WT macrophages; §p < 0.05 vs infected WT macrophages, n = 3 for uninfected groups, and n = 9–10 for infected groups) and F4/80+ iNOS+ cells (B, *p < 0.05) are shown. C, Representative flow cytometric dot plots are shown demonstrating iNOS and F4/80 staining in cells isolated from infected WT and Arg2−/− mice. D, In the F4/80+ cells, iNOS mean fluorescence units (MFU) are shown to demonstrate the level of iNOS expression in macrophages. *p < 0.05. E, Representative histogram demonstrating iNOS fluorescence in macrophages from uninfected and infected WT and Arg2−/− mice. F, Immunoperoxidase staining for nitrotyrosine is shown for uninfected control mice and infected WT and Arg2−/− mice at 200× and 600× magnification. Data are representative of 2 mice in the control group and 4–6 in the infected groups.
Because we found that gastric macrophages isolated from H. pylori-infected Arg2−/− mice express higher iNOS protein levels than infected WT mice, we sought to determine if this resulted in increased NO production. To assess this, we used nitrotyrosine staining as a marker of NO synthesis in the mucosa. Nitrotyrosine formation occurs when tyrosine residues react with peroxynitrite, which is formed by the reaction of NO with superoxide (O2−), and this has been used as a marker of the production of reactive nitrogen species (23, 35–37). We stained tissues from chronically-infected WT and Arg2−/− mice and found that inflammatory cells in Arg2−/− mice had increased nitrotyrosine staining as compared to WT mice (Fig. 5F). This staining was most intense in mononuclear immune cells of the lamina propria near the luminal surface, similar in location to the macrophages that we identified in Fig. 1D and Fig. 4.
H. pylori infection increases macrophage apoptosis that is abolished in Arg2−/− mice
To determine whether increased survival of macrophages could be a mechanism responsible for increased iNOS+ macrophages in Arg2−/− mice, we isolated gastric macrophages and measured apoptosis by annexin V staining. We have previously shown that H. pylori induces macrophage apoptosis in vitro that is Arg2-dependent (29), therefore, we sought to confirm this in vivo. To pursue this, we used a 48 h model of infection, as we have previously demonstrated that there is both maximal macrophage infiltration into the stomach, and maximal apoptosis at this timepoint postinoculation with H. pylori (31). Additionally, we have demonstrated that Arg2 restricts NO production in gastric macrophages at this timepoint (15). Consistent with our in vitro data, we found that H. pylori infection induced macrophage apoptosis, and that this was abolished in Arg2−/− macrophages (Fig. 6A). Representative flow cytometric dot plots demonstrating annexin V and 7-AAD staining in infected WT and Arg2−/− macrophages are shown in Fig. 6B.
An external file that holds a picture, illustration, etc.
Object name is nihms277366f6.jpg
Open in a separate window
FIGURE 6
Arg2−/− macrophages undergo less apoptosis than WT macrophages during H. pylori infection. A, Gastric macrophages from WT and Arg2−/− mice were isolated 48 h postinoculation with H. pylori SS1 and stained for flow cytometry with annexin V-FITC and 7-AAD. The percentage of annexin-V+ 7-AAD− cells are shown. **p < 0.01 compared to uninfected WT mice. §§p < 0.01 compared to infected WT mice, n = 4 mice per group. B, Representative flow cytometric dot plots are shown demonstrating staining for annexin V and 7-AAD in gastric macrophages isolated from infected WT and Arg2−/− mice. C, Photomicrographs of slides from chronically-infected WT and Arg2−/− mice stained for cleaved caspase-3. The arrows are used to highlight apoptotic bodies. Data are representative of 7–8 mice per group. D, Quantification of cleaved caspase-3 staining. The percentage of positively-stained inflammatory cells is shown.
We also sought to corroborate this finding in our chronic infection model by immunostaining tissues for cleaved caspase-3, a marker for apoptosis in H. pylori gastritis tissues (38). In infected WT mice there was abundant staining in the mononuclear inflammatory cells with marked staining of cells with apoptotic bodies (Fig. 6C). In contrast, cleaved caspase-3 staining in Arg2−/− mice was less intense and less frequent, which correlates with our findings with the annexin V staining of isolated gastric macrophages in Fig. 6A. Additionally, quantification of this staining revealed a decrease in cleaved caspase-3 staining among the inflammatory cells in the Arg2−/− mice compared to WT mice (Fig. 6D).
Discussion
H. pylori infection induces a vigorous immune response, and in the murine model there is a rapid influx of macrophages (31, 39) and neutrophils (39) 48 h postinfection, followed by infiltration of lymphocytes 10 d postinfection (39). This produces a smoldering gastritis that persists as long as the bacteria reside in the gastric niche. Despite this robust immune response, the bacterium typically persists for the life of the host. It is generally assumed that the immune response is not vigorous enough to eliminate the infection, due to demonstration of clearance of the bacterium in adoptive transfer experiments and IL-10−/− mice, both of which exhibit enhanced gastric inflammation (9, 34). We sought to determine if the macrophage response is inhibited by H. pylori. In the current report, we demonstrate that H. pylori upregulates macrophage Arg2, thereby restricting iNOS protein levels and NO production, and enhancing macrophage apoptosis. Consequently, this restricts host defense against H. pylori. This is the first report to demonstrate that macrophage Arg2 expression has a deleterious impact on the effectiveness of host immunity by impairing the inflammatory response in vivo.
Herein, we have demonstrated that induction of Arg2 during chronic H. pylori infection restricts macrophage iNOS protein levels, limits the pro-inflammatory immune response, and increases bacterial colonization. These data confirm our recent studies showing that inhibition of macrophage arginase in vitro enhances iNOS translation and NO production and, consequently, causes more bacterial killing (15). We have now shown that infection with H. pylori causes upregulation of Arg2 that localizes to lamina propria and submucosal macrophages. We have previously reported that Arg2 expression is induced in RAW 264.7 cells and peritoneal macrophages stimulated ex vivo with H. pylori (15, 29). Furthermore, we have shown that Arg2 gene expression is upregulated in human gastric tissues infected with H. pylori (29). Simultaneous induction of both iNOS and arginase in macrophages is uncommon, as studies have demonstrated that induction of one usually leads to the inhibition of the other (40, 41). Nevertheless, several pathogens have devised strategies to upregulate arginase to suppress iNOS-dependent host defense. For example, downregulation of NO production by macrophages has been attributed to induction of Arg1 by the parasites Leishmania major (24) and Toxoplasma gondii (23) and the bacterium Mycobacterium tuberculosis (23); and to induction of Arg2 by the parasite Trypanosoma brucei brucei (42) and the bacteria Chlamydia psittaci and Chlamydia pneumoniae (25). We now demonstrate that H. pylori upregulates Arg2 in vivo leading to an impaired macrophage immune response.
Our data suggest that the enhanced inflammation induced by H. pylori has no benefit for reducing bacterial colonization under normal circumstances. When we analyzed the gastritis scores and colonization levels in WT mice, we found that there was no correlation between these two parameters and the linear regression line was almost flat. This was surprising because it is generally assumed that in H. pylori infection enhanced inflammation will decrease bacterial colonization (9, 10, 34). In fact, persistence of the bacterial infection is primarily thought to be due to an immune response that is not vigorous enough (8). However, our data demonstrate that even in WT mice with very high levels of gastritis, there was no noticeable ability of this response to reduce bacterial colonization. These data suggest that there is a defect in the immune response against H. pylori and that effector mechanisms responsible for clearance of the bacterial infection are inhibited. In contrast, mice deficient in Arg2 showed a beneficial inverse correlation between gastritis and bacterial colonization, producing a negative linear regression line, thus demonstrating that mice with high levels of inflammation had less bacterial colonization. These data suggest that Arg2 induction contributes to the defective immune response in WT mice. However, in Arg2-deficient mice, the bacterial infection was decreased but not eliminated. Linear regression analysis of our Arg2−/− mice suggest that after removal of the Arg2 ‘block’, the effectiveness of the immune response is restored, but enhancement of other responses may be needed to completely eliminate the infection, which correlates with what others have demonstrated with lymphocyte adoptive transfer experiments (9, 43).
We previously demonstrated that Arg2 localizes to mitochondria while iNOS resides in the cytoplasm (15). Although these two enzymes are physically separated, Arg2 is still able to restrict NO production by its inhibitory effect on iNOS translation; as such, NO production in response to H. pylori can be enhanced in vitro in macrophages by inhibition of arginase activity, knockdown of Arg2, or use of Arg2−/− cells (15). These previous findings are substantiated by our current in vivo data that gastric macrophages isolated from H. pylori-infected Arg2−/− mice express more iNOS protein than WT macrophages. We also demonstrated enhanced nitrotyrosine staining in these cells which is a signature of increased NO production. Our data indicate that the competition for intracellular l-arginine is important for modulating host immunity against H. pylori.
It has been reported that Arg2−/− mice have higher levels of serum l-arginine than WT mice under basal conditions (44). Such an effect could enhance T cell responses because it has been reported that T cell responses in vitro are inhibited under conditions of limited l-arginine availability (45). Furthermore, it has been shown that Arg1-expressing macrophages can inhibit both the re-expression of T cell receptors and T cell proliferation in vitro (46) and T cell proliferation in vivo (47). We have considered the possibility that the effects we have described with our Arg2−/− mice may have been due to enhanced T cell responses that could ensue from increased l-arginine availability. However, when we supplemented WT mice with l-arginine in their drinking water, we found no reduction in h. pylori colonization when assessed at 4 mo postinoculation (data not shown). This lack of effect of l-arginine treatment may be due to the presence of Arg2, which impairs NO-dependent antimicrobial host defense. Our data suggest impairment of classical (M1) activation (48) of macrophages in H. pylori infection. However, the competing alternative (M2) activation nomenclature has been used to refer to cells with induction of Arg1 rather than Arg2 (48). We have shown that upregulation of Arg1 does not occur in H. pylori infection, and intriguingly, Arg2 induction has recently been associated with M1 responses in a murine model of atherosclerosis (49). Another factor in the utilization of l-arginine in host defense is that its uptake into macrophages is required to allow generation of NO (18, 50). We have reported that while the l-arginine transporter, cationic amino acid transporter 2, is upregulated in gastric macrophages upon infection with H. pylori, l-arginine uptake is actually inhibited by the polyamine spermine, which is generated downstream of Arg2 (14). We are currently investigating the role of macrophage cationic amino acid transporter 2 during chronic H. pylori infection.
Additionally, we have found that induction of Arg2 enhances H. pylori-induced apoptosis in gastric macrophages, consistent with our previous in vitro findings (29). This apoptosis was associated with decreased abundance of macrophages in the gastric mucosa in WT mice when compared to Arg2−/− mice. Another contributing factor to the increase in macrophages in the Arg2−/− mice may be increased stimulation of mononuclear cell infiltration, since enhanced Th1 and Th17 responses, as we have detected, have been correlated with increased inflammatory cells in the gastric mucosa (9, 10, 34). Taken together, our data suggest that the limited numbers of macrophages associated with Arg2 induction results in diminished gastric inflammation and pro-inflammatory cytokine production. Consequently, the inhibition of H. pylori-induced macrophage apoptosis that we achieved by using mice deficient in Arg2 caused an increase in the number of surviving infiltrating macrophages, and an associated increase in gross and histologic gastritis (increased neutrophils and lymphocytes), and proinflammatory cytokine production. Other reports have demonstrated that macrophages mediate gastric inflammation during H. pylori infection, since depletion of macrophages from mice with clodronate-loaded liposomes resulted in reduced levels of histologic gastritis (51). It should be noted that the reduction in macrophages had no effect on H. pylori colonization levels in that study (51), which is consistent with our current findings that WT mice with higher gastritis scores exhibited no reduction in colonization, and further supports the concept that altered macrophage immune function is a hallmark of H. pylori infection, since loss of cells would not be expected to have an effect on host defense if the cells are already defective.
In summary, our data indicate that induction of Arg2 by H. pylori is a mechanism by which the pathogen escapes the host innate immune response and contributes to the immunopathogenesis of the infection. However, we also recognize that another possibility is that the Arg2 component of the innate immune response in macrophages may serve to protect the host from unrestrained inflammation, and as such Arg2 could prevent overabundant nitrosative stress and its associated mutagenic potential that would derive from unrestricted NO production (52). However, it should be noted that the increased nitrotyrosine staining in the Arg2−/− mice that we observed did not appear to involve epithelial cells in our model, indicating that Arg2 may be dispensable in protecting epithelial cells from nitrosative stress. Because we have reported that Arg2 is upregulated in H. pylori gastritis tissues from human subjects, insights into the importance of Arg2 could be gained from molecular epidemiology studies of Arg2 levels in human subject groups, such as in persons from Latin America where regions of low versus high risk of gastric cancer have been described, despite similarly high prevalence rates of H. pylori (53–55). Studies related to this issue may be a promising area for future investigation.Immune Evasion by Helicobacter pylori is Mediated by Induction of Macrophage Arginase II
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3069806/
PLoS One. 2013; 8(9): e75192.
Gastric Epithelial Expression of IL-12 Cytokine Family in Helicobacter pylori Infection in Human: Is it Head or Tail of the Coin?
Fadi Al-Sammak, 1 Thomas Kalinski, 2 Sönke Weinert, 3 Alexander Link, 1 Thomas Wex, 1 , 4 , * and Peter Malfertheiner 1
Josep Bassaganya-Riera, Editor
Author information Article notes Copyright and License information Disclaimer
1 Department of Gastroenterology, Hepatology and Infectious Diseases, Otto-von-Guericke University, Magdeburg, Germany,
2 Department of Pathology, Otto-von-Guericke University, Magdeburg, Germany,
3 Department of Cardiology, Angiology and Pneumology, Otto-von-Guericke University, Magdeburg, Germany,
4 Medical Laboratory for Clinical Chemistry, Microbiology and Infectious Diseases, Department of Molecular Genetics, Magdeburg, Germany,
ASTRACT
Recently, there has been a growing interest in an expanding group of cytokines known as “IL-12 family”. The so far gained knowledge about these cytokines, as crucial playmakers in mucosal immunity, has not yet been sufficiently investigated in the context of Helicobacter pylori infection.All genes encoding the monomeric components of these cytokines and their corresponding receptors were examined in gastric epithelial cell lines (AGS and MKN-28) after being infected with 4 H. pylori strains: BCM-300, P1 wild-type, and P1-derived isogenic mutants lacking cytotoxin-associated gene A (cagA) or virulence gene virB7 (multiplicity of infection=50).
Both infected and uninfected samples were analyzed after 24h and 48h using real-time quantitative polymerase chain reaction (RT-qPCR). Gene expression analysis demonstrated a strong upregulation of IL23A (encodes p19) by infection, whereas IL23R, Epstein–Barr virus-induced gene 3 (EBI3), IL6ST, IL12A, and IL27RA were found to be expressed, but not regulated, or to a lesser extent. Transcripts of IL12RB2, IL12B, IL12RB1, and IL27A were not detected.
Interestingly, P1 resulted in stronger alterations of expression than CagA mutant and BCM-300, particularly for IL23A (59.7-fold versus 32.4- and 6.7-fold, respectively in AGS after 48h, P<.05), whereas no changes were seen with VirB7 mutant.
In a proof-of-principle experiment, we demonstrated epithelial-derived expression of IL-12, p19, and Ebi3 in gastric mucosa of gastritis patients using immunohistochemistry (IHC). Unlike IL-12 and Ebi3, increased immunostaining of p19 was observed in H. pylori gastritis. Herein, we highlight the potential role of gastric epithelial cells in mucosal immunity, not only because they are predominant cell type in mucosa and initial site of host-bacterial interaction, but also as a major contributor to molecules that are thought to be primarily expressed by immune cells so far. Of these molecules, p19 was the most relevant one to H. pylori infection in terms of expression and localization.
In a nutshell, the most obvious finding to emerge from this study is the contribution of gastric epithelial cells to molecules that belong to the IL-12 family of cytokines, which is against the dogma that these molecules are primarily produced by immune cells. Since epithelial cells are the predominant cell type in gastric mucosa, hence, these findings provide support for a conceptual premise that epithelial cells could have a major impact on the type of immune response at any given point via epithelial-derived immune mediators whose exact function remains elusive. Furthermore, our gene expression and IHC/IF studies were in agreement to identify the importance of p19 as the most relevant molecule in IL-12 family to H. pylori infection in terms of expression and localization.Gastric Epithelial Expression of IL-12 Cytokine Family in Helicobacter pylori Infection in Human: Is it Head or Tail of the Coin?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3775749/
Serum Helicobacter pylori CagA antibody as a biomarker for ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3044821
At this point, anti-CagA antibody alone is not a superior biomarker to the anti-H. pylori antibody alone. It is necessary to evaluate the availability of anti-H. pylori antibody plus anti-CagA antibody for screening the risk of gastric cancer. As described above, the relationship between anti-CagA antibody and gastric cancer varied in each study.
Arginase Structure and Inhibition: Catalytic Site Plasticity Reveals New Modulation Possibilities
Jérémie Mortier, Julien R. C. Prévost, Dominique Sydow, Sabine Teuchert, Christian Omieczynski, Marcel Bermudez, Raphaël Frédérick & Gerhard Wolber
Scientific Reports volume 7, Article number: 13616 (2017) Cite this article
Abstract
Metalloenzyme arginase is a therapeutically relevant target associated with tumor growth. To fight cancer immunosuppression, arginase activity can be modulated by small chemical inhibitors binding to its catalytic center. To better understand molecular mechanisms of arginase inhibition, a careful computer-aided mechanistic structural investigation of this enzyme was conducted. Using molecular dynamics (MD) simulations in the microsecond range, key regions of the protein active site were identified and their flexibility was evaluated and compared. A cavity opening phenomenon was observed, involving three loops directly interacting with all known ligands, while metal coordinating regions remained motionless. A novel dynamic 3D pharmacophore analysis method termed dynophores has been developed that allows for the construction of a single 3D-model comprising all ligand-enzyme interactions occurring throughout a complete MD trajectory. This new technique for the in silico study of intermolecular interactions allows for loop flexibility analysis coupled with movements and conformational changes of bound ligands. Presented MD studies highlight the plasticity of the size of the arginase active site, leading to the hypothesis that larger ligands can enter the cavity of arginase. Experimental testing of a targeted fragment library substituted by different aliphatic groups validates this hypothesis, paving the way for the design of arginase inhibitors with novel binding patterns.
Arginase Structure and Inhibition: Catalytic Site Plasticity Reveals New Modulation Possibilities | Scientific Reports
https://www.nature.com/articles/s41598-017-13366-4
Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment
Susanne M. Steggerda, Mark K. Bennett, Jason Chen, Ethan Emberley, Tony Huang, Julie R. Janes, Weiqun Li, Andrew L. MacKinnon, Amani Makkouk, Gisele Marguier, Peter J. Murray, Silinda Neou, Alison Pan, Francesco Parlati, Mirna L. M. Rodriguez, Lee-Ann Van de Velde, Tracy Wang, Melissa Works, Jing Zhang, Winter Zhang & Matthew I. Gross
Journal for ImmunoTherapy of Cancer volume 5, Article number: 101 (2017) Cite this article
Abstract
Background
Myeloid cells are an abundant leukocyte in many types of tumors and contribute to immune evasion. Expression of the enzyme arginase 1 (Arg1) is a defining feature of immunosuppressive myeloid cells and leads to depletion of L-arginine, a nutrient required for T cell and natural killer (NK) cell proliferation. Here we use CB-1158, a potent and orally-bioavailable small-molecule inhibitor of arginase, to investigate the role of Arg1 in regulating anti-tumor immunity.
Methods
CB-1158 was tested for the ability to block myeloid cell-mediated inhibition of T cell proliferation in vitro, and for tumor growth inhibition in syngeneic mouse models of cancer as a single agent and in combination with other therapies. Tumors from animals treated with CB-1158 were profiled for changes in immune cell subsets, expression of immune-related genes, and cytokines. Human tumor tissue microarrays were probed for Arg1 expression by immunohistochemistry and immunofluorescence. Cancer patient plasma samples were assessed for Arg1 protein and L-arginine by ELISA and mass spectrometry, respectively.
Results
CB-1158 blocked myeloid cell-mediated suppression of T cell proliferation in vitro and reduced tumor growth in multiple mouse models of cancer, as a single agent and in combination with checkpoint blockade, adoptive T cell therapy, adoptive NK cell therapy, and the chemotherapy agent gemcitabine. Profiling of the tumor microenvironment revealed that CB-1158 increased tumor-infiltrating CD8+ T cells and NK cells, inflammatory cytokines, and expression of interferon-inducible genes. Patient tumor samples from multiple histologies expressed an abundance of tumor-infiltrating Arg1+ myeloid cells. Plasma samples from cancer patients exhibited elevated Arg1 and reduced L-arginine compared to healthy volunteers.
Conclusions
These results demonstrate that Arg1 is a key mediator of immune suppression and that inhibiting Arg1 with CB-1158 shifts the immune landscape toward a pro-inflammatory environment, blunting myeloid cell-mediated immune evasion and reducing tumor growth. Furthermore, our results suggest that arginase blockade by CB-1158 may be an effective therapy in multiple types of cancer and combining CB-1158 with standard-of-care chemotherapy or other immunotherapies may yield improved clinical responses.Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment | Journal for ImmunoTherapy of Cancer | Full Text
https://jitc.biomedcentral.com/articles/10.1186/s40425-017-0308-4
Planta Med. 2018 Mar;84(5):277-295. doi: 10.1055/s-0044-100398. Epub 2018 Jan 17.
Phenolic Compounds as Arginase Inhibitors: New Insights Regarding Endothelial Dysfunction Treatment.
Minozzo BR1, Fernandes D2, Beltrame FL1.
Author information
1 Department of Pharmaceutical Sciences, State University of Ponta Grossa, Ponta Grossa, Paraná, Brazil.
2 Department of Pharmacology, Federal University of Santa Catarina, Florianópolis, Santa Catarina, Brazil.
Abstract
Endothelial dysfunction is characterised by the low bioavailability of nitric oxide with a relevant negative impact on the nitric oxide/cGMP pathway. The loss of nitric oxide/cGMP signaling may be caused by an increased arginase activity. Plant-derived substances, especially polyphenols, are compounds that have the potential to inhibit arginase activity and they may represent an attractive therapeutic option to combat clinical outcomes related to endothelial dysfunction. An extensive review was carried out using all available data published in English in the Pubmed database, and without restriction regarding the year of publication. Despite the increased number of new substances that have been tested as arginase inhibitors, it is rare to find a compound that satisfies all the toxicological criteria to be used in the development of a new drug.On the other hand, recent data have shown that substances from plants have great potential to be applied as arginase inhibitors, most of which are polyphenols. Of the relevant mechanisms in this process, the inhibition of arginase by natural products seems to act against endothelial dysfunction by reestablishing the vascular function and elevating nitric oxide levels (by increasing the amounts of substrate (L-arginine, and endothelial nitric oxide synthase activation and stabilisation) as well as decreasing the generation of reactive species (formed by uncoupledendothelial nitric oxide synthase).
This review summarises several topics regarding arginase inhibition by natural substances as well as indicating this pathway as an emergent strategy to elevate nitric oxide levels in disorders involving endothelial dysfunction. In addition, some aspects regarding structural activity and future perspectives are discussed.
As an arginase inhibitor, orthinine, lysine are competitive inhibitor, norvaline has been shown to improve available resources of arginine and to increase nitric oxide (NO) production.
Georg Thieme Verlag KG Stuttgart · New York.
PMID: 29342480 DOI: 10.1055/s-0044-100398Phenolic Compounds as Arginase Inhibitors: New Insights Regarding Endothelial Dysfunction Treatment. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29342480
L-正缬氨酸增强活化巨噬细胞产生 NO
L -正缬氨酸 arginase inhibitor | Sigma-Aldrich
https://www.sigmaaldrich.com/catalog/product/sigma/n7627?lang=zh®ion=CN
L-Norvaline Side Effects - When You Take Too Much L-Norvaline
https://www.esupplements.com/ingredients/l-norvaline/l-norvaline-side-effects
L-Norvaline is a unique amino acid. It’s actually a form of the BCAA valine, commonly taken to improve muscle growth. Norvaline has an extra hydrogen molecule distinguishing it from valine. It plays a special role in pre-workouts and nitric oxide.
L-Norvaline - Evolutionary.org
https://www.evolutionary.org/l-norvaline
L-Norvaline is a form of the Branched Chain Amino Acid L-Valine, but aside from L-Valine's normal benefits, L-Norvaline has been shown to positively effect circulation and nitric oxide production. The difference, and reason for the the “nor” prefix only makes note that Norvaline is one hydrogen molecule longer than the original protein Valine.
Author: Rick Vallejo
What is L-Norvaline? – ATP Nutrition
https://www.atpnutrition.com/what-is-l-norvaline
What Is L-Norvaline?L-Norvaline and CytotoxicityThe Takeaway on L-Norvaline and Nitric OxideReferences
L-Norvaline is a derivative of the branched-chain amino acid (BCAA) valine that has been noted in research to be a mixed arginase inhibitor. Arginase is the enzyme in the body that degrades arginine — the primary substrate used to generate nitric oxide, increase blood flow, boost muscle pumps.The theory goes that by limiting the actions of arginase, you’re removing the “governor” plate in your body allowing for unrestricted nitri…
L-Norvaline Reverses Cognitive Decline and Synaptic Loss ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6277292
Oct 04, 2018 · L-Norvaline Ameliorated Memory Deficits in the 3×Tg Mice To investigate whether L-norvaline treatment affected learning and memory in AD pathogenesis, a set of behavioral tests was administered following L-norvaline …
Cited by: 11
Publish Year: 2018
Author: Baruh Polis, Kolluru D. Srikanth, Evan Elliott, Ha
Neurotherapeutics. 2018 Oct; 15(4): 1036–1054.
L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease
Baruh Polis,corresponding author1,2 Kolluru D. Srikanth,2,3 Evan Elliott,3 Hava Gil-Henn,2 and Abraham O. Samson1
1Drug Discovery Laboratory, The Azrieli Faculty of Medicine, Bar-Ilan University, 1311502 Safed, Israel
2Laboratory of Cell Migration and Invasion, The Azrieli Faculty of Medicine, Bar-Ilan University, 1311502 Safed, Israel
3Laboratory of Molecular and Behavioral Neuroscience, The Azrieli Faculty of Medicine, Bar-Ilan University, 8th Henrietta Szold Street, P.O. Box 1589, 1311502 Safed, Israel
Abstract
The urea cycle is strongly implicated in the pathogenesis of Alzheimer’s disease (AD). Arginase-I (ARGI) accumulation at sites of amyloid-beta (Aβ) deposition is associated with L-arginine deprivation and neurodegeneration.An interaction between the arginase II (ARGII) and mTOR-ribosomal protein S6 kinase β-1 (S6K1) pathways promotes inflammation and oxidative stress. In this study, we treated triple-transgenic (3×Tg) mice exhibiting increased S6K1 activity and wild-type (WT) mice with L-norvaline, which inhibits both arginase and S6K1.
The acquisition of spatial memory was significantly improved in the treated 3×Tg mice, and the improvement was associated with a substantial reduction in microgliosis.
In these mice, increases in the density of dendritic spines and expression levels of neuroplasticity-related proteins were followed by a decline in the levels of Aβ toxic oligomeric and fibrillar species in the hippocampus. The findings point to an association of local Aβ-driven and immune-mediated responses with altered L-arginine metabolism, and they suggest that arginase and S6K1 inhibition by L-norvaline may delay the progression of AD.
L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6277292/
Georg Thieme Verlag KG Stuttgart · New York.
PMID: 29342480 DOI: 10.1055/s-0044-100398Phenolic Compounds as Arginase Inhibitors: New Insights Regarding Endothelial Dysfunction Treatment. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/29342480
ACS Applied Materials & Interfaces 10(6) · January 2018
Gold Nanoparticle-Based Photoluminescent Nanoswitch Controlled by Host-Guest Recognition and Enzymatic Hydrolysis for Arginase Activity Assay福建医科大学
The development of simple yet powerful methods for monitoring enzyme activity is of great significance. Herein, a facile, convenient, cost-effective, and continuous fluorescent method for the detection of arginase and its inhibitor has been reported based on a host-guest interaction- and enzymatic hydrolysis-controlled luminescent nanoswitch.The fluorescence intensity of 6-aza-2-thiothymine-stabilized gold nanoparticle (ATT-AuNP) is enhanced by L-arginine, owing to the formation of supramolecular host-guest assembly between the guanidine group of L-arginine and ATT molecules capped on AuNP surface.
However, hydrolysis of L-arginine, catalyzed by arginase, leads to a decrease of fluorescence intensity of L-arginine/ATT-AuNPs hybrids. Upon incorporation of the arginase inhibitor L-norvaline, the fluorescence of the ATT-AuNP-based detecting system is restored. The linear range of arginase activity determination is from 0.0625 to 1.15 U/mL and the limit of detection is 0.056 U/mL. The half-maximal inhibition value IC50 of L-norvaline is determined to be 5.6 mM. The practicability of this luminescent nanoswitch is validated by assaying arginase activity in rat liver and monitoring the response of rat liver arginase to pharmacological agent. Compared to the existing fluorescent method of arginase activity assay, the approach demonstrated here does not involve any complicated technical manipulation, thereby greatly simplifying the detection steps. We propose that this AuNP-based luminescent nanoswitch would find wide applications in the field of life sciences and medicine.
(PDF) Gold Nanoparticle-Based Photoluminescent Nanoswitch Controlled by Host-Guest Recognition and Enzymatic Hydrolysis for Arginase Activity Assay
https://www.researchgate.net/publication/322728337_Gold_Nanoparticle-Based_Photoluminescent_Nanoswitch_Controlled_by_Host-Guest_Recognition_and_Enzymatic_Hydrolysis_for_Arginase_Activity_Assay
Arginase Inhibitors: A Rational Approach Over One Century
Marc Pudlo Céline Demougeot Corine Girard‐Thernier
First published: 15 November 2016 https://doi.org/10.1002/med.21419Citations: 25
Abstract
Arginase (EC 3.5.3.1) is the bimanganese enzyme that converts L‐arginine into ornithine and urea. This enzyme was discovered more than a century ago and early α‐amino acids were identified as weak inhibitors. It was only during the 90s, after nitric oxide (NO) was reported as one of the most important biological mediators and when tight interrelation of arginase and NO synthase was found, that the development of arginase inhibitors was accelerated.The regulation of arginase activity by the N‐hydroxy‐L‐arginine (3, NOHA) intermediate of the NO synthesis was the starting point of the N‐hydroxy‐nor‐arginine (21, nor‐NOHA) that proved to be the first micromolar inhibitor. The previously known manganese and arginase binding by borate inspired the 2(S)‐amino‐6‐boronohexanoic acid (39, ABH) and S‐(2‐boronoethyl)‐L‐cysteine (40, BEC) now both considered as reference compounds in arginase inhibition. The high‐resolution crystal structure of arginase and molecular modeling has rendered possible the recent design of (53) the strongest α,α‐disubstituted derivatives of ABH. Simultaneously, traditional medicinal plants have contributed as a source of molecular diversity to the discovery of arginase inhibitors. This rational, step‐by‐step approach serves as guide in the present review where emphasis is placed on structure activity relationships.
Highlights
exhaustive review on arginase inhibitors
highlight is made on rational approach to conception and structure activity relationships
evaluation model is systematically mentioned with results.Arginase Inhibitors: A Rational Approach Over One Century - Pudlo - 2017 - Medicinal Research Reviews - Wiley Online Library
https://onlinelibrary.wiley.com/doi/abs/10.1002/med.21419
Frontiers | Therapeutic Potential of the Nitrite-Generated NO Pathway in Vascular Dysfunction | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2013.00174/full
Gastroenterology. 2010 Mar;138(3):1046-54. doi: 10.1053/j.gastro.2009.11.043. Epub 2009 Nov 18.
Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice.
Kao JY1, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J.
Author information
1
Department of Internal Medicine, Division of Gastroenterology, University of Michigan Health System, 6520A MSRB I, SPC 5682, 1150 West Medical Center Drive, Ann Arbor, MI 48109-5682, USA. jykao@umich.edu
Abstract
BACKGROUND & AIMS:
Helicobacter pylori infection increases gastric regulatory T cell (Treg) response, which may contribute to H pylori immune escape. We hypothesize that H pylori directs Treg skewing by way of dendritic cells (DCs) and thus inhibits interleukin-17(+) helper T cells (Th17) immunity.
METHODS:
Two-photon microscopy was used to locate DCs in gastric lamina propria of mice. The induction of Th17 and Treg responses by bacteria-pulsed murine bone marrow-derived DCs was analyzed by cytokine production and stimulation of T-cell proliferation. The effect of VacA, CagA, transforming growth factor-beta (TGF-beta), and IL-10 on Th17/Treg balance was assessed. The in vivo significance of Tregs on the H pylori-specific Th17 response and H pylori density was determined by using anti-CD25 neutralizing antibodies to deplete Tregs in mice.
RESULTS:
We showed that mucosal CD11c(+) DCs are located near the surface of normal gastric epithelium, and their number increased after H pylori infection. Study of the direct interaction of DCs with H pylori showed a Treg-skewed response. The Treg skewing was independent of H pylori VacA and CagA and dependent on TGF-beta and IL-10. In vivo Treg skewing by adoptive transfer of H pylori-pulsed DCs reduces the ratio of gastric IL-17/Foxp3 mRNA expressions. The depletion of CD25(+) Tregs results in early reduction of H pylori density, which is correlated with enhanced peripheral H pylori-specific Th17, but not Th1, response.
CONCLUSIONS:
Overall, our study indicates that H pylori alters the DC-polarized Th17/Treg balance toward a Treg-biased response, which suppresses the effective induction of H pylori-specific Th17 immunity.
Copyright 2010 AGA Institute. Published by Elsevier Inc. All rights reserved.
PMID: 19931266 PMCID: PMC2831148 DOI: 10.1053/j.gastro.2009.11.043Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/19931266
Human Gastric Epithelium Produces IL-4 and IL-4δ2 Isoform Only upon Helicobacter Pylori Infection
B. Orsini, J.R. Vivas, B. Ottanelli, ...Department of Clinical Pathophysiology, Gastroenterology Unit, University of Florence, Italy
Abstract
Recent evidence suggests that interleukin-4 (IL-4) is related to mucosal tolerance by which an injurious immune response is prevented, suppressed or shifted to a non-injurious response. We investigated the expression of IL-4 and its splice variant isoform IL-4δ2 in gastric epithelial cells of healthy subjects and gastritis patients infected with Helicobacter pylori (H. pylori) with or without the cag pathogenicity island (cag-PAI). IL-4 and IL-4δ2 mRNAs were evaluated in microdissected gastric epithelium and in AGS cell lines co-cultured with H. pylori B128 or SSI strains. IL-4 mRNA was consistently detected in microdissected gastric epithelial cells from healthy subjects. The IL-4 mRNA expression was low in H. pylori-infected patients, and markedly reduced in cag-PAI-positive ones. IL-4δ2 mRNA was expressed on gastric epithelium of H. pylori-infected patients, but not in healthy subjects. The IL-452 expression was lower in cag-PAI-positive than in cag-PAI-negative H. pylori infected patients. AGS cells also produced IL-4 mRNA upon SSI strain stimulation, whereas IL-4δ2 mRNA expression was detected in AGS co-cultured with either SSI or B128 strains. An inverse correlation was documented between IL-4 and IL-482 mRNA expression by microdissected gastric epithelial cells and the score of gastritis. IL-4, but not IL-452, is expressed by gastric epithelium of healthy subjects, whereas IL-452 and lesser IL-4 mRNA are detectable in the gastric epithelium of H. pylori-infected patients. Data suggest that gastric epithelial cells might regulate the balance between tolerance and immune response by the fine tuning of IL-4 and IL-4δ2 expression.Human Gastric Epithelium Produces IL-4 and IL-4δ2 Isoform Only upon Helicobacter Pylori Infection - B. Orsini, J.R. Vivas, B. Ottanelli, A. Amedei, E. Surrenti, A. Galli, S. Milani, P. Pinzani, G. Del Prete, C. Surrenti, C.T. Baldari, E. Touati, M.M. D'Elios, 2007
https://journals.sagepub.com/doi/abs/10.1177/039463200702000417
Increased expression of mRNA encoding interleukin (IL)-4 ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1782524
IL-4 functions at very low concentrations, and has a low mRNA copy number, a very short half-life and a splice variant (IL-4δ2) that acts as a competitive antagonist. 1, 2 Appropriate methods have shown increased expression of both IL-4 and IL-4δ2 in fresh unstimulated peripheral blood mononuclear cells (PBMC) from tuberculosis (TB) patients, and the level of IL-4 mRNA correlates with disease severity. 3 …
Cited by: 83
Publish Year: 2004
Author: Helen A. Fletcher, Patrick Owiafe, David Jeffrie
Dendritic cells and NK cells
Author links open overlay panelAmy K.WesaRobbie B.Mailliard
Show more
https://doi.org/10.1016/B978-0-12-370454-2.00018-1Get rights and content
Publisher Summary
Dendritic cells (DCs) are antigen-presenting cells (APCs) controlling two reciprocal functions: maintenance of self-tolerance and initiation of immune responses to foreign antigens. DCs play a central role in the initiation and regulation of the immune response. Acting as the bridge between the innate and adaptive branches of immunity, DCs convert environmental cues they gather at sites of inflammation into the development of polarized adaptive immune responses once they reach draining lymph nodes. An important aspect in this system lies with the ability of DCs and natural killer (NK) cells to exchange bi-directional signals capable of influencing the functional status of each cell type as well as the overall nature of the immune response.DC activation of NK cells through their production of such factors as IL-12, IL-18, IL-15, and type-1 interferons (IFNs), and the NK cell modulation of DC function through their production of TNF-α and IFN-γ can result from direct cell-to-cell contact involving NK cell receptors such as NKp30, NKp46, and NKG2D or through indirect means of bystander activation. Moreover, while NK cells can act as “helper” cells to promote DC function, they can also serve to police DC activity through a selective elimination process resulting in DC death. Importantly, the net immunological effects of the interactions of these cells are largely dependant on the activation status of both cell types as well as the context in which they meet. It discusses the functional properties of the DC, with particular attention focusing on the immunoregulatory impact of their interactions with NK cells. A suitable paradigm for the role of DCs in the induction of cellular immunity is represented by the function of skin Langerhans cells (LC).
Dendritic cells and NK cells - ScienceDirect
https://www.sciencedirect.com/science/article/pii/B9780123704542000181
Dynamic interactions between T cells and dendritic cells and their derived cytokines/chemokines in the rheumatoid synovium
Pierre Miossec
Arthritis Research & Therapy volume 10, Article number: S2 (2008) Cite
Abstract
This review focuses on the contributions made by interactions between dendritic cells (DCs) and T cells, and by local production of cytokines and chemokines to the pathogenesis of rheumatoid arthritis (RA) synovitis. DCs are efficient professional antigen-presenting cells, which are critical for the development of innate and adaptative immune responses through interactions with T cells. Cytokines from DCs play a key role in the switch inside effector T-cell pathways. Chemokines are important mediators of the immune response because they regulate leucocyte recruitment to tissue, and they play a key role in inflammatory diseases by acting on T-cell and DC migration. Furthermore, the recently discovered T-helper-17 proinflammatory cytokines, present in syno-vium samples, are associated with the migration, differentiation and maturation of inflammatory cells, and they facilitate a network of interactions between all components of the immune response. An understanding of such interactions is essential because it is the key to therapeutic application.
Dynamic interactions between T cells and dendritic cells and their derived cytokines/chemokines in the rheumatoid synovium | Arthritis Research & Therapy | Full Text
https://arthritis-research.biomedcentral.com/articles/10.1186/ar2413
Nature
Published: 08 August 1964
In vivo Effect of L-Lysine on Rat Liver Arginase
DOMENICO CITTADINI, CONCETTA PIETROPAOLO, DOMENICO DE CRISTOFARO & MARIA D'AYJELLO-CARACCIOLO
Institute of Biological Chemistry, Medical School, University of Naples, Italy
Abstract
PREVIOUS work carried out in this Laboratory was devoted to explaining the effective physiological role of ornithine cycle reactions in urea biosynthesis1,2. On the basis of the results obtained, serious doubts arose concerning the role of argininosuccinate synthetase in this process as well as with regard to the position of carbamylphosphate as an intermediate in nitrogen transfer from protein to urea. So far as the problem of urea biosynthesis is concerned, we are now investigating the role of arginase in this process. This communication reports in vivo inhibition of rat liver arginase activity by a strong competitive arginase inhibitor, L-lysine3,4.In vivo Effect of L -Lysine on Rat Liver Arginase | Nature
https://www.nature.com/articles/203643a0
ORIGINAL RESEARCH ARTICLE
Front. Integr. Neurosci., 14 June 2010 | https://doi.org/10.3389/fnint.2010.00018
Lysine and arginine reduce the effects of cerebral ischemic insults and inhibit glutamate-induced neuronal activity in rats
Takashi Kondoh 1,2, Makiko Kameishi 1, Hruda Nanda Mallick 3, Taketoshi Ono 4 and Kunio Torii 1*
1 Institute of Life Sciences, Ajinomoto Co., Inc., Kawasaki, Japan
2 AJINOMOTO Integrative Research for Advanced Dieting, Graduate School of Agriculture, Kyoto University, Kyoto, Japan
3 Department of Physiology, All India Institute of Medical Sciences, New Delhi, India
4 System Emotional Science, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama, Japan
Intravenous administration of arginine was shown to be protective against cerebral ischemic insults via nitric oxide production and possibly via additional mechanisms. The present study aimed at evaluating the neuroprotective effects of oral administration of lysine (a basic amino acid), arginine, and their combination on ischemic insults (cerebral edema and infarction) and hemispheric brain swelling induced by transient middle cerebral artery occlusion/reperfusion in rats. Magnetic resonance imaging and 2,3,5-triphenyltetrazolium chloride staining were performed 2 days after ischemia induction. In control animals, the major edematous areas were observed in the cerebral cortex and striatum. The volumes associated with cortical edema were significantly reduced by lysine (2.0 g/kg), arginine (0.6 g/kg), or their combined administration (0.6 g/kg each). Protective effects of these amino acids on infarction were comparable to the inhibitory effects on edema formation. Interestingly, these amino acids, even at low dose (0.6 g/kg), were effective to reduce hemispheric brain swelling. Additionally, the effects of in vivo microiontophoretic (juxtaneuronal) applications of these amino acids on glutamate-evoked neuronal activity in the ventromedial hypothalamus were investigated in awake rats. Glutamate-induced neuronal activity was robustly inhibited by microiontophoretic applications of lysine or arginine onto neuronal membranes.Taken together, our results demonstrate the neuroprotective effects of oral ingestion of lysine and arginine against ischemic insults (cerebral edema and infarction), especially in the cerebral cortex, and suggest that suppression of glutamate-induced neuronal activity might be the primary mechanism associated with these neuroprotective effects.
Arginine and Neuroprotection
Protective effects of arginine against ischemic damages have been studied during the last 20 years by many researchers in terms of the “arginine–NO production–vasodilation” hypothesis. For example, intravenous administration of arginine increases cerebral blood flow both in baseline (Morikawa et al., 1992 ; Sadoshima et al., 1997 ; Caramia et al., 1998 ) and ischemic conditions (Dalkara et al., 1994 ; Sadoshima et al. 1997 ). Recovery of cerebral blood flow for more than 30% is associated with signal reappearance in electroencephalograms (Dalkara et al., 1994 ). The d-form of arginine (Morikawa et al., 1992 ; Dalkara et al., 1994 ) and the non-selective smooth muscle relaxant papaverine (Zhang and Iadecola, 1994 ) produce no such effects, suggesting that the observed actions are induced specifically by the l-form of arginine. Administration of NO donors such as sodium nitroprusside and 3-morpholinosydnonimine produce effects similar to those of arginine (Zhang and Iadecola, 1994 ), while administration of NOS inhibitors exacerbates ischemic damages (Kuluz et al., 1993 ). NO acts neuroprotector via S-nitrosylation of NMDA receptor thiol and suppression of NMDA receptor activity in the presence of conditions leading to the synthesis of nitrosonium ion (NO+) (Satoh and Lipton, 2007 ).
In contrast to the protective effects mentioned above, different lines of evidence suggest that arginine exacerbates ischemic infarction (Zhao et al. 2003 ) and NOS inhibitors ameliorate it (Buisson et al., 1992 ; Kozniewska et al., 1995 ; Quast et al., 1995 ). One plausible explanation for such discrepancy may be that the arginine effect depends on the redox state of the body (Lipton et al., 1993 ). For example, under conditions of preferential production of the NO radical (NO·), arginine is cytotoxic via production of toxic free radicals, such as peroxynitrite anion (ONOO−), which lead to indiscriminate damage to biological macromolecules including DNA, proteins, and membrane lipids (Halliwell et al., 1992 ; Lewén et al., 2000 ). The redox versatility of NO may explain its interconversion from neuroprotective to neurotoxic species by a change in the ambient milieu.
Taken together, actions of arginine (and NO) are complex and the observed results may largely depend on a variety of factors, such as tissue regions, concentration and timing of NO production, and redox state of the body.Lysine and Neuroprotection
Although l-lysine shares many of the chemical properties of arginine (Smulders et al., 1997 ), effects of lysine on cerebral ischemic injury have not been investigated previously. We have demonstrated for the first time that oral administration of lysine alone or in combination with arginine is effective against cerebral ischemic insults. The effectiveness of lysine is of interest with respect to the following three issues: (1) lysine is not a precursor for NO synthesis, (2) the mechanism of neuroprotection may be distinct from the one used by arginine, and (3) l-lysine is the limiting amino acid in proteins of feed grains such as wheat and corn (Torii et al., 1987 ) and therefore the risk of lysine deficiency is high in low socioeconomic human populations who depend predominantly on wheat for their protein supply (Young and Pellett, 1990 ). Therefore, deficiency of dietary lysine might increase the risk of stroke and hence fortification of lysine on foods might be beneficial for preventing ischemic insults. Additionally, combined ingestion of lysine with arginine may enhance the neuroprotective effects. Suppression of stress-induced anxiety has been shown to be enhanced by a combination of lysine and arginine in rats and humans (Smriga and Torii, 2003b ; Smriga et al., 2007 ).
Frontiers | Lysine and arginine reduce the effects of cerebral ischemic insults and inhibit glutamate-induced neuronal activity in rats | Frontiers in Integrative Neuroscience
https://www.frontiersin.org/articles/10.3389/fnint.2010.00018/full
2014,
Inhibitory Effect of L-Canavanine and L-Lysine on Arginase
Ataturk University
Abstract
Arginase (L-arginine amidinohydrolase; (E.C.3.5.3.1) is the last enzyme of urea cycle and it catalyzes the hydrolysis of L-arginine to urea and L-ornithine in livers of animals. It helps to the excretion process of urea from the body, which is the most soluble and non-toxic form of nitrogen. Along with its essential role in urea synthesis, arginase is also found to have important roles in ornithine production for polyamine, proline and glutamate synthesis and in immune system activation. Mostly found in liver tissue, arginase enzyme is also found in non-ureolytic tissues like heart, rumen, skeletal muscle, kidney, intestine, brain, spleen, thyroid gland, salivary gland, erythrocyte, fibroblast, macrophage, mammary gland in lactation, testicle. With this study, it was aimed to assess the inhibitory effects of arginase in sheep spleen tissue on L-canavanine and L-lysine. Consequently, it was observed that L-lysine as a L-amino acid and guanidine compound L-canavanine inhibits enzyme activity non-competitively.
Inhibitory Effect of L-Canavanine and L-Lysine on Arginase ...
https://www.researchgate.net/publication/274252577...The Km for L-arginine was 10.5 mM and for L-canavanine 50mM, and L-lysine exhibited a competitive type of inhibition with a Ki of 4.4mM. L-Homoarginine was not a substrate for liver arginase. View
Journal of Inorganic Biochemistry
Volume 77, Issues 3–4, 30 November 1999, Pages 163-167
Journal of Inorganic Biochemistry
Manganese-dependent inhibition of human liver arginase by borate
NelsonCarvajalMónicaSalasVasthiLópezElenaUribePaulaHerreraJuanCerpaMarciaFuentes
Departamento de Biologı́a Molecular, Facultad de Ciencias Biológicas, Universidad de Concepción, Casilla 160-C, Concepción, Chile
Abstract
Full activation of human liver arginase (EC 3.5.3.1), by incubation with 5 mM Mn2+ for 10 min at 60°C, resulted in increased Vmax and a higher sensitivity of the enzyme to borate inhibition, with no change in the Km for arginine. Borate behaved as an S-hyperbolic I-hyperbolic non-competitive inhibitor and had no effect on the interaction of the enzyme with the competitive inhibitors l-ornithine (Ki=2±0.5 mM), l-lysine (Ki=2.5±0.4 mM), and guanidinium chloride (Ki=100±10 mM). The pH dependence of the inhibition was consistent with tetrahedral B(OH)4− being the inhibitor, rather than trigonal B(OH)3. We suggest that arginase activity is associated with a tightly bound Mn2+ whose catalytic action may be stimulated by addition of a more loosely bound Mn2+, to generate a fully activated enzyme form. The Mn2+ dependence and partial character of borate inhibition are explained by assuming that borate binds in close proximity to the loosely bound Mn2+ and interferes with its stimulatory action. Although borate protects against inactivation of the enzyme by diethyl pyrocarbonate (DEPC), the DEPC-sensitive residue is not considered as a ligand for borate binding, since chemically modified species, which retain about 10% of enzymatic activity, were also sensitive to the inhibitor.Manganese-dependent inhibition of human liver arginase by borate - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0162013499001877