��
��
��
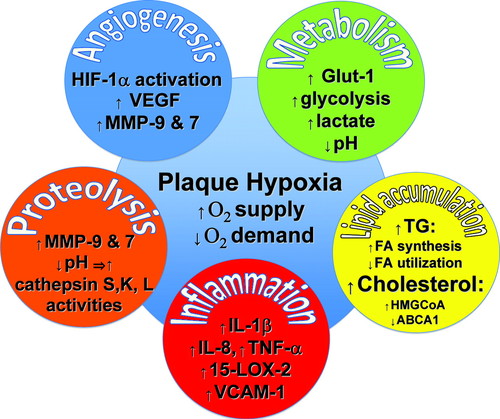
Figure. Hypoxia in atherogenesis. This figure illustrates selected examples
of the manifold effects of hypoxia in atheromata; see text for explanation.
15-LOX-2 indicates 15-lipoxygenase-2; ABCA1, ATP-binding cassette A 1; FA, fatty
acid; GLUT-1, glucose transporter 1; HIF-1��, hypoxia-inducible factor 1��;
HMG-CoA, hydroxymethylglutaryl coenzyme A; IL, interleukin; MMP, matrix
metalloproteinase; VCAM-1, vascular cell adhesion molecule 1; VEGF, vascular
endothelial growth factor.
��
Hypoxia Stimulates Plaque Angiogenesis
Pathologists have long appreciated the microvasculature of plaques. The rise of
Judah Folkman's concept of tumor angiogenesis as a growth-promoting mechanism in
malignancy stimulated parallel thinking in atherosclerosis research.5 Plaque
neovessels may stimulate lesion growth and provide a portal with a large surface
area for penetration of inflammatory cells. Fragile neovessels in atheromata, as
in the diabetic retina, may prove prone to hemorrhage. Extravasated erythrocytes
furnish a local depot of cholesterol-rich red cell membranes and of heme, a
source of iron��which is a catalyst for oxidative stress. Thrombosis in situ may
elicit cycles of thrombin-mediated smooth-muscle cell (SMC) migration and
proliferation and hence lesion growth. Thus the neovessels stimulated through
the hypoxia-inducible factor (HIF)/vascular endothelial growth factor (VEGF)
axis in response to hypoxia may promote intraplaque hemorrhage, lesion growth,
recruitment of inflammatory cells, and oxidative stress.
Hypoxia Alters Glucose Metabolism in the Atherosclerotic Plaque
Through the regulation of glucose transporters (eg, GLUT-1) and enzymes that
capture glucose within the cell (hexokinases), hypoxia augments glucose
utilization by plaque cells�� notably, the mononuclear phagocytes that abound in
many lesions.6 A shift to anaerobic glycolysis leads to lactate overproduction
and lowers the pH prevailing in plaques. Hypoxia-driven increases in glucose
uptake, incidentally, provide an opportunity to image plaque metabolism.
Fluorodeoxyglucose (FdG), a tracer commonly used in tracking tumors using
positron emission tomography, accumulates in some atherosclerotic plaques.7 The
avidity of atheromata for FdG uptake may provide a clinical window on some of
the metabolic shifts associated with hypoxia.6
Could accelerated glucose utilization, and energy substrate depletion due to
reduced delivery, alter plaque biology? Anerobic glycolysis yields much less
adenosine triphosphate (ATP) per glucose molecule than does oxidative
metabolism. Reduced ATP availability may promote mitochondrial and
extramitochondrial pathways of apoptosis in the plaque. Furthermore, hypoxia
causes an imbalance between electron transport and the intracellular O2
concentration that leads to the production of reactive oxygen species and
oxidative stress, further predisposing to cell death.8
Apoptosis of macrophages in atheromatous lesions favors formation of the
��necrotic core�� of the plaque, a structure associated with disruption of human
atheromata and thrombosis. SMCs in the plaque manufacture most of the
interstitial collagen that lends tensile strength to the plaque's protective
fibrous cap; hence, SMC apoptosis can impair the cap's integrity.9 Fracture of
the fibrous cap causes most fatal acute myocardial infarctions in humans. Thus,
sensitization of macrophages and SMCs to apoptosis by hypoxic conditions could
contribute to the thrombotic complications of this disease.
Does Hypoxia Promote Plaque Proteolysis?
Proteolysis drives dissolution of the plaque extracellular matrix. Accelerated
catabolism of extracellular matrix constituents likely contributes decisively to
plaque evolution and complication. Outward remodeling (also called compensatory
enlargement)��characteristic of arteries that harbor growing atheromata��requires
reshaping of the extracellular matrix, a process that probably involves both
elastolysis and collagenolysis. The penetration of microvessels from the
adventitia into the plaque likewise requires digestion of extracellular
matrix.10 Excessive degradation of collagen may predispose toward plaque rupture
by decreasing the collagen content of the plaque's protective fibrous cap. The
catabolism of nonfibrillar collagen in the basement membrane of the arterial
intima may set the stage for superficial erosion of the endothelial
monolayer��another common mechanism of thrombosis complicating human
atherosclerotic plaques��by altering the subendothelial matrix, thereby
sensitizing these cells to death by anoikis.
Hypoxia may regulate the enzymes involved in catabolism of the plaque's
extracellular matrix in several ways. Hypoxic conditions may augment the
activity of matrix metalloproteinases (MMPs), a family that includes
interstitial collagenases that weaken the fibrous cap and gelatinases capable of
catabolizing nonfibrillar collagen, to which endothelial cells adhere.11�C13
Hypoxia-induced MMP-7 may participate critically in atherothrombosis. In
addition to directly contributing to extracellular matrix remodeling, this
metalloproteinase can elicit proatherogenic molecules such as tumor necrosis
factor �� (TNF-��),14 and promotes thrombogenicity by degrading tissue factor
pathway inhibitor.15 In a recently recognized novel twist, MMP-14 can augment
HIF-1 activity by a non-proteolytic mechanism and increase macrophage ATP
production, simulating hypoxic alterations in glucose metabolism.16
In addition, the drop in pH in hypoxic portions of plaques in lesions favors the
activity of lysosomal hydrolases.17 Notably, cysteinyl elastases��such as
cathepsins S, K, and L��localize in plaques and contribute to lesion evolution.18
These potent elastases may participate in remodeling of arteries during
atherogenesis, among other functions.8 Thus hypoxic regulation of proteolytic
activity may have multiple consequences for plaque evolution and complication.
Hypoxic Conditions May Incite Inflammation in Plaques
Hypoxia can foster the formation of proinflammatory cytokines and leukotrienes,
and activate Akt (Figure).3,12 Ultimately, hypoxia and inflammation conspire to
promote the evolution and clinical complications of atherosclerosis.
Lipid Accumulation
Mononuclear phagocytes subjected to hypoxia accumulate triglyceride, due to
increased production of, and from reduced oxidation of, fatty acids.3,19
Augmented expression of stearoyl-coenzyme A desaturase (SCD-1) may promote fatty
acid synthesis in hypoxic mononuclear phagocytes. In this issue of Circulation
Research, Parathath and colleagues show that hypoxic conditions augment cellular
content of sterols as well as triglycerides. They implicate both increased
production due to augmented hydroxymethylglutaryl coenzyme A (HMG-CoA)
expression and decreased efflux mediated by ATP-binding cassette A (ABCA1)
function.20 Thus, hypoxia modulates the metabolism of both triglycerides and
sterols��lipids that accumulate in macrophage foam cells, a hallmark of
atheromata.
Implications of Plaque Hypoxia
Increased recognition of the low oxygen tension in regions of atheromata and its
metabolic consequences has considerable implications for contemporary
atherosclerosis research. In vitro experiments indubitably have advanced the
understanding of mechanisms relevant to atherogenesis. Yet, most studies
cultivate SMCs and macrophages under normoxic conditions. Our usual laboratory
culture conditions strive to buffer the pH to maintain neutrality. Normoxia and
pH 7.4 represent conditions far afield from those found in regions of the
atheroma. Moreover, much contemporary experimental work in atherosclerosis
relies on the use of mice. Due to their smaller size, mouse lesions may harbor
less hypoxia than their human counterparts. While exceedingly informative,
studies of cultured cells and of mouse atheromata should be considered in light
of these important differences with conditions pertaining to human plaques.
Increased recognition of plaque hypoxia also has some pathophysiological
implications, beyond these technical experimental points. The great German
biochemist Otto Warburg described overutilization of glucose by cancer cells and
constructed a unified theory of cancer related to some of the metabolic
consequences of hypoxia. Warburg's unitary view vastly oversimplified the
complex and multifactorial diseases lumped together as ��cancer.�� Our concepts of
the pathogenesis of atherosclerosis have likewise witnessed similar cycles of
enthusiasm for specific mechanisms: bland lipid storage, mechanical injury,
neoplastic-like SMC proliferation, oxidative stress, and inflammation. Hypoxia
now garners recognition as a modulator of mechanisms that drive atherogenesis
and its clinical consequences. Although Warburg's scientific insight stands, his
monomaniacal view of cancer has fallen. We need to recognize that no one
instigator or pathway explains atherogenesis in its full complexity. We stand to
learn more about the disease, and have a greater chance of mastering it, if we
appreciate its multifactorial mechanisms, including hypoxia.
Footnotes
The opinions expressed in this article are not necessarily those of the editors
or of the American Heart Association.
Correspondence to Peter Libby, MD,
Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women's
Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, NRB741, Boston, MA
02115.
E-mail: plibby@rics.bwh.harvard.edu
Tension in the Plaque: Hypoxia Modulates Metabolism in Atheroma | Circulation
Research
https://www.ahajournals.org/doi/full/10.1161/res.0b013e31823bdb84
��