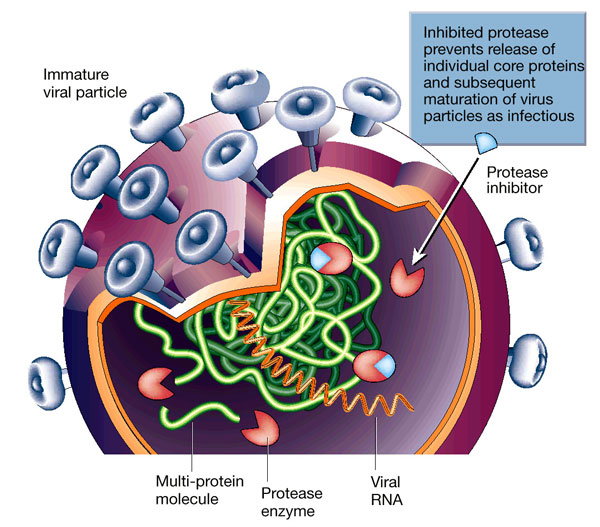
ĄĄ
ĄĄ
Natural inhibitors
ĄĄ
1.curcumin is a weak inhibitor of the viral protease.
ĄĄ
http://www.medscape.org/viewarticle/416524
ĄĄ
Human Immunodeficiency Virus and Heparan Sulfate: From Attachment to Entry Inhibition
Front Immunol. 2013; 4: 385.
Published online 2013 Nov 20. Prepublished online 2013 Oct 15. doi: 10.3389/fimmu.2013.00385
PMCID: PMC3834540
PMID: 24312095
Bridgette J. Connell1,2,3 and Hugues Lortat-Jacob1,2,3,*
Author information Article notes Copyright and License information Disclaimer
This article has been cited by other articles in PMC.
Go to:
Abstract
By targeting cells that provide protection against infection, HIV-1 causes acquired immunodeficiency syndrome. Infection starts when gp120, the viral envelope glycoprotein, binds to CD4 and to a chemokine receptor usually CCR5 or CXCR4. As many microorganisms, HIV-1 also interacts with heparan sulfate (HS), a complex group of cell surface associated anionic polysaccharides. It has been thought that this binding, occurring at a step prior to CD4 recognition, increases infectivity by pre-concentrating the virion particles at the cell surface. Early work, dating from before the identification of CCR5 and CXCR4, showed that a variety of HS mimetics bind to the gp120 V3 loop through electrostatic interactions, compete with cell surface associated HS to bind the virus and consequently, neutralize the infectivity of a number of T-cell line-adapted HIV-1 strains. However, progress made to better understand HIV-1 attachment and entry, coupled with the recent identification of additional gp120 regions mediating HS recognition, have considerably modified this view. Firstly, the V3 loop from CXCR4-using viruses is much more positively charged compared to those using CCR5. HS inhibition of cell attachment is thusHuman Immunodeficiency Virus and Heparan Sulfate: From Attachment to Entry Inhibition
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3834540/ĄĄ
http://lookfordiagnosis.com/mesh_info.php?term=Receptors%2C+Ccr5&lang=1
ĄĄ
http://microbewiki.kenyon.edu/index.php/CCR5,_Delta-32_Mutation_and_the_HIV_Infection_Pathway
ĄĄ
Soraphen A A broad-spectrum antiviral natural product with potent anti-hepatitis C virus activity
Author links open overlay panelGeorgeKoutsoudakis1#InĻĶsRomero-Brey2†CarolaBerger2†GemmaPĻĶrez-VilarĻŪ1PaulaMonteiro Perin34Florian Wolfgang RudolfVondran45MarkusKalesse6KirstenHarmrolfs7RolfMĻđller7Javier P.Martinez8ThomasPietschmann34RalfBartenschlager29MarkBrönstrup6AndreasMeyerhans810JuanaDĻŠez1 Show more https://doi.org/10.1016/j.jhep.2015.06.002Get rights and content Background & Aims
1Molecular Virology Group, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona, Spain
2Department of Infectious Diseases, Molecular Virology, Heidelberg University, Heidelberg, Germany
3TWINCORE ĻC Institute of Experimental Virology, Centre for Experimental and Clinical Infection Research, Hannover, Germany
4German Centre for Infection Research (DZIF), partner site Hannover-Braunschweig, Germany
ĄĄ
5ReMediES, Department of General, Visceral and Transplantation Surgery, German Centre for Infection Research Hannover Medical School, Hannover, Germany
6Department of Chemical Biology, Helmholtz Centre for Infection Research, Braunschweig, Germany
7Helmholtz Institut fĻđr Pharmazeutische Forschung Saarland, SaarbrĻđcken, Germany
8Infection Biology Group, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona, Spain
9German Centre for Infection Research (DZIF), partner site Heidelberg, Germany
10InstituciĻŪ Catalana de Recerca i Estudis Avançats (ICREA), Barcelona, Spain
Soraphen A (SorA) is a myxobacterial metabolite that inhibits the acetyl-CoAcarboxylase, a key enzyme in lipid biosynthesis. We have previously identified SorA to efficiently inhibit the human immunodeficiency virus (HIV).
The aim of the present study was to evaluate the capacity of SorA and analogues to inhibit hepatitis C virus (HCV) infection.
Methods
SorA inhibition capacity was evaluated in vitro using cell culture derived HCV, HCV pseudoparticles and subgenomic replicons. Infection studies were performed in the hepatoma cell line HuH7/Scr and in primary human hepatocytes. The effects of SorA on membranous web formation were analysed by electron microscopy.
Results
SorA potently inhibits HCV infection at nanomolar concentrations. Obtained EC50 values were 0.70 nM with a HCV reporter genome, 2.30 nM with wild-type HCV and 2.52 nM with subgenomic HCV replicons. SorA neither inhibited HCV RNA translation nor HCV entry, as demonstrated with subgenomic HCV replicons and HCV pseudoparticles, suggesting an effect on HCV replication. Consistent with this, evidence was obtained that SorA interferes with formation of the membranous web, the site of HCV replication. Finally, a series of natural and synthetic SorA analogues helped to establish a first structureĻCactivity relationship.
Conclusions SorA has a very potent anti-HCV activity. Since it also interferes with the membranous web formation, SorA is an excellent tool to unravel the mechanism of HCV replication.
Soraphen A: A broad-spectrum antiviral natural product with potent anti-hepatitis C virus activity - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0168827815003918ĄĄ
The Myxobacterial Metabolite Soraphen A Inhibits HIV-1 by Reducing Virus Production and Altering Virion Composition
Eric Fleta-Soriano,a KatarĻŠna SmutnĻĒ,a Javier P. MartĻŠnez,corresponding authora Cristina Lorca OrĻŪ,b S. Kashif Sadiq,a Gilles Mirambeau,b Carmen Lopez-Iglesias,c Marta Bosch,d Albert Pol,d,g Mark Brönstrup,e Juana Diez,f and Andreas Meyerhanscorresponding authora,g
ABSTRACT
Soraphen A is a myxobacterial metabolite that blocks the acetyl-coenzyme A carboxylase of the host and was previously identified as a novel HIV inhibitor. Here, we report that soraphen A acts by reducing virus production and altering the gp120 virion content, impacting entry capacity and infectivity. These effects are partially reversed by addition of palmitic acid, suggesting that inhibition of HIV envelope palmitoylation is one of the mechanisms of antiviral action.
KEYWORDS: soraphen A, HIV, fatty acid synthesis, host factor, broad-spectrum antiviral, human immunodeficiency virus
TEXT
Cellular lipids play an important role in the propagation of diverse viruses (1). A key pathway in lipid metabolism is de novo fatty acid synthesis mediated by acetyl-coenzyme A carboxylase (ACC) and multifunctional fatty acid synthase. Blockage of these enzymes by small molecules leads to broad-spectrum inhibition of several viruses, including hepatitis C virus (HCV), West Nile virus, dengue virus, yellow fever virus, rotavirus, human cytomegalovirus, vesicular stomatitis virus, and influenza virus (2,ĻC7). A highly potent inhibitor of ACC is soraphen A (SorA), a myxobacterial secondary metabolite that we previously identified as an HIV inhibitor in an antiviral screening assay (8) and recently showed to efficiently inhibit HCV with a large therapeutic window (9). Here, we sought to further determine the anti-HIV properties of SorA.
To analyze the antiviral potency of SorA, TZM-bl cells or primary peripheral blood mononuclear cells (PBMCs) were infected with HIVLAI and HIVBaL wild-type strains and with a primary HIV isolate from clade A under increasing SorA concentrations (Fig. 1A to toD).D). The production of infectious virus was then tested by titrating the culture supernatants on TZM-bl cells with a luciferase readout (10, 11). The effect of SorA on cell viability was assessed in parallel by a commercial ATP assay. SorA reduced infectious virus production in a dose-dependent manner. The calculated 50% effective concentration (EC50) ranged from ∼0.2 to 2 ĶĖM, depending on the cells and virus used. No SorA-mediated toxicity was detected up to the 50 ĶĖM concentration tested.The Myxobacterial Metabolite Soraphen A Inhibits HIV-1 by Reducing Virus Production and Altering Virion Composition. - Abstract - Europe PMC
http://europepmc.org/article/PMC/5527598ĄĄ
Sci Rep. 2016; 6: 34190.
Functional organization of the HIV lipid envelope
Nerea Huarte,a,1,* Pablo Carravilla,1,* Antonio Cruz,2 Maier Lorizate,1,3 Jon A. Nieto-Garai,1 Hans-Georg Kräusslich,3 JesĻēs PĻĶrez-Gil,2 Jose Requejo-Isidro,b,1 and JosĻĶ L. Nievac,1
1Biophysics Unit (CSIC, UPV/EHU) and Department of Biochemistry and Molecular Biology, University of the Basque Country (UPV/EHU), P.O. Box 644, 48080 Bilbao, Spain
2Department of Biochemistry, Faculty of Biology, and Research Institute Hospital 12 de Octubre, Universidad Complutense, Madrid, Spain
3Department of Infectious Diseases, Virology, University Hospital Heidelberg, 69120 Heidelberg, Germany
Abstract
The chemical composition of the human immunodeficiency virus type 1 (HIV-1) membrane is critical for fusion and entry into target cells, suggesting that preservation of a functional lipid bilayer organization may be required for efficient infection. HIV-1 acquires its envelope from the host cell plasma membrane at sites enriched in raft-type lipids. Furthermore, infectious particles display aminophospholipids on their surface, indicative of dissipation of the inter-leaflet lipid asymmetry metabolically generated at cellular membranes. By combining two-photon excited Laurdan fluorescence imaging and atomic force microscopy, we have obtained unprecedented insights into the phase state of membranes reconstituted from viral lipids (i.e., extracted from infectious HIV-1 particles), established the role played by the different specimens in the mixtures, and characterized the effects of membrane-active virucidal agents on membrane organization. In determining the molecular basis underlying lipid packing and lateral heterogeneity of the HIV-1 membrane, our results may help develop compounds with antiviral activity acting by perturbing the functional organization of the lipid envelope.Functional organization of the HIV lipid envelope
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5039752/ĄĄ
Lipids in HIVĄŊs Envelope Help the Virus to Spread
Freelance Science Writer, San Diego, California, United States of America
Dendritic cells (DCs) are a type of immune cell that patrol tissues, on the lookout for microbial invaders. When DCs encounter a pathogen, they chop it up into tiny pieces and then carry samples of it to local lymph nodes. There, they display their finds to another kind of immune cell, the T cell, which then mounts a full-fledged immune response against the invader.ĄĄ
Left: Gangliosides in HIV's lipid envelope (red) promote uptake of the virus (or experimental substitutes) by dendritic cells.
Middle: a space-filling model of a ganglioside. Right: a schematic of a simple ganglioside's head group. Image credit: F.-Xabier Contreras, Maier Lorizate, and Nuria Izquierdo-Useros.ĄĄ
Unfortunately, HIV, the virus that causes AIDS, can exploit the DC surveillance network to its own advantage: HIV is picked up by DCs that patrol mucosal tissues, but avoids being killed by them, and instead hitches a ride to lymph nodes. There, the virus transfers into its favorite host cell type, the T cell. Until now, it wasnĄŊt clear how DCs recognize HIV for uptakeĄŠa mystery thatĄŊs now been solved thanks to a joint effort by Spanish and German research groups. The groups, headed by Nuria Izquierdo-Useros and Javier Martinez-Picado in Spain, and Maier Lorizate and Hans-Georg Kräusslich in Germany.
ItĄŊs no mystery how DCs detect many pathogens: the microbes betray themselves by displaying certain pathogen-specific compounds on their surfaces, which DCs recognize using dedicated receptors. HIV also displays a few viral proteins on its surface (it hides the rest away under a lipid bilayer envelope that it acquires from the plasma membrane of infected cells). However, previous studies indicated that viral proteins displayed on the surface may not be essential for DC capture. Instead, prior work from Martinez-PicadoĄŊs group led the authors to theorize that host cell glycosphingolipids incorporated into the virus envelope could be critical in triggering DC recognition.
There are many types of glycosphingolipids, and one kind had previously been observed in the membranes of several retroviruses, including HIV. This lipid, GM3, is from a subclass of glycosphingolipids called gangliosides. To follow up on these findings, Izquierdo-Useros, Lorizate, and colleagues first examined whether other gangliosides might also be present in HIVĄŊs envelope. Indeed, they found several other gangliosides thereĄŠan observation that prompted the researchers to examine what role gangliosides might play in HIV recognition and capture by DCs.
To determine whether gangliosides are needed for HIV uptake by DCs, the authors examined DC uptake of artificial virus-like particles (VLPs), which carry a lipid envelope and the inner structural proteins, but lack HIV surface glycoproteins and viral genetic material. They found that VLPs containing gangliosides were able to get into DCs, but those lacking gangliosides could not do so. Next, the researchers tested which gangliosides can promote DC uptake by using artificial lipid globules called liposomes, each containing a different ganglioside. Similar to VLPs, DCs easily took up liposomes containing gangliosides, but were unable to take up liposomes devoid of all gangliosides. Both liposomes and VLPs, the authors showed, use the same method to enter DCsĄŠa method that depends not on viral proteins but on the presence of gangliosides in the viral envelope.
These data suggest that there is some special feature of gangliosides that promotes their specific recognition by DCs. While much of a ganglioside is buried in the lipid bilayer, a portion of it, called the head group, is exposed at the membrane surface. Ganglioside head groups contain one or more copies of sialic acid, a sugar, with different ganglioside types each sporting distinct arrangements of their sialic acids. The authorsĄŊ investigation revealed that itĄŊs these head groupsĄŠparticularly sialic acids attached to a lactose groupĄŠthat promote DC recognition. But, while DC uptake requires sialic acid molecules, gangliosides whose head groups are too complex canĄŊt be recognized by DCs.
These findings all point to the idea that gangliosides are essential for HIV uptake by DCs. In fact, the authors showed that addition of free GM3 head groups to cell cultures prevented DCs from picking up intact HIV. They also demonstrated that DCs exposed to ganglioside-depleted HIV canĄŊt pick up the virus and so canĄŊt efficiently transfer it to T cells. Therefore, gangliosides may also affect HIVĄŊs ability to establish infection in the body. Of course, further studies are needed to understand how DCs recognize gangliosides in the viral envelope, how HIV avoids destruction after ganglioside-mediated DC uptake, and how the virus later escapes to infect T cells. Nonetheless, gangliosides may represent a useful therapeutic target for preventing or limiting infection by HIV and other ganglioside-containing viruses.ĄĄ
Lipids in HIV's Envelope Help the Virus to Spread - ScienceBuzz
https://www.sciencebuzz.com/lipids-in-hivs-envelope-help-the-virus-to-spread/Lipids in HIV's Envelope Help the Virus to Spread
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3339841/ĄĄ
Which microorganisms are killed by Monolaurin?:
While Monolaurin is most widely used as an anti-viral agent, it
also has beneficial effects against pathogenic bacteria, yeasts
and fungi; other fatty acids such as caprylic and sorbic acids are
more effective against yeasts, but ineffective against viruses.
In a study performed at the CDC, which focused on Monolaurin tested
strains of viruses, Monolaurin was able to solubulize the
enveloped membrane of 14 human RNA and DNA viruses (3).
These include influenza, RSV, Rubeola, NewcastleĄŊs, Coronavirus,
Herpes Simplex types 1 & 2, Epstein-Barr Virus (EBV)
and cytomegalovirus. (Monolaurin has no effect on
naked viruses, such as polio, encephalitis virus, coxsachie, or pox
viruses.) Monolaurin works by disintegrating the lipid
envelope coat of viruses. Data from these studies suggest that the
loss of virus infectivity is associated with the solubilization
of Monolaurin into the envelope. The virus absorbs the fatty acid
for its own replication, but winds up destroying its own protective
coat.☯ * Subject: Coronavirus and Monolaurin: Destroys Viral Lipid Coatings:
https://www.beyond-the-fringe.com/showthread.php?tid=3334ĄĄ
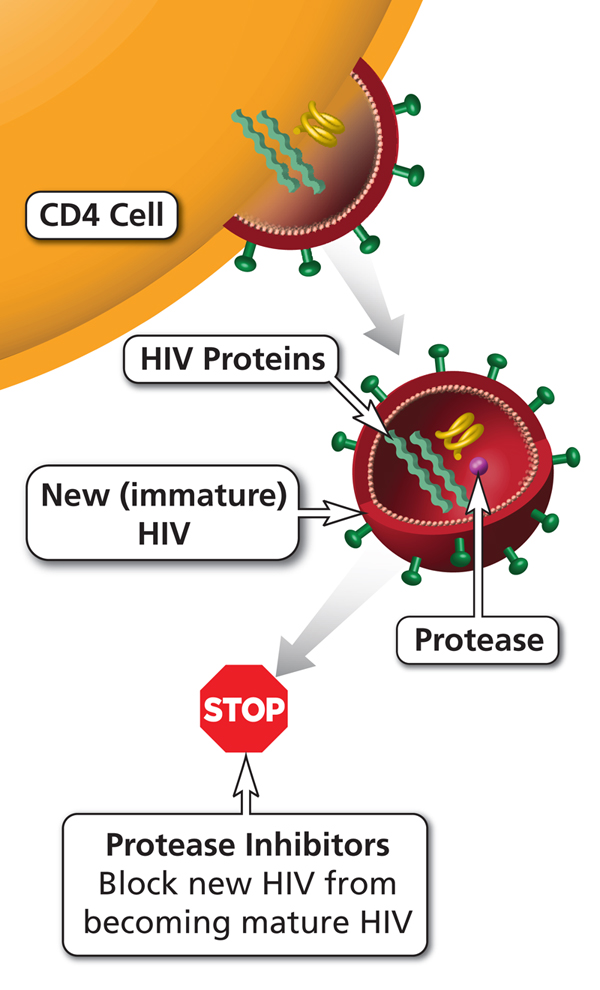
ĄĄ
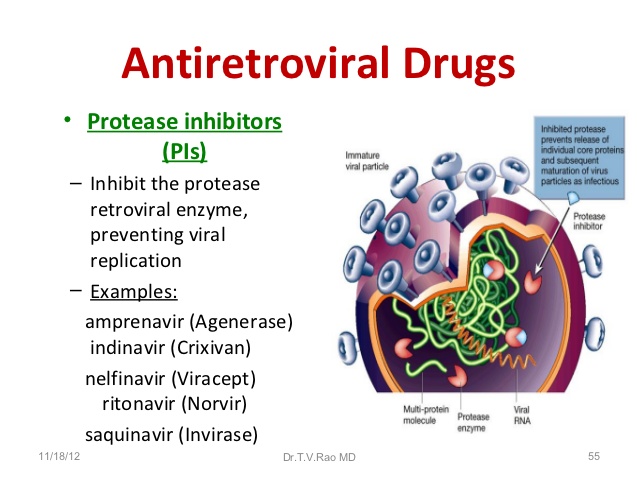
Antiretroviral (ARV) HIV drug class. Protease inhibitors (PIs) block protease (an HIV enzyme). By blocking protease, PIs prevent new (immature) HIV from becoming a mature virus that can infect other CD4 cells.
ĄĄhttps://aidsinfo.nih.gov/education-materials/glossary/603/protease-inhibitor
ĄĄ
Viruses 2012, 4(2), 309-324; https://doi.org/10.3390/v4020309
Review
Novel Approaches to Inhibit HIV Entry
by Chukwuka A. Didigu and Robert W. Doms *
Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, 3610 Hamilton Walk, Philadelphia, PA 19104, USA
Abstract: Human Immunodeficiency Virus (HIV) entry into target cells is a multi-step process involving binding of the viral glycoprotein, Env, to its receptor CD4 and a coreceptorĄŠeither CCR5 or CXCR4. Understanding the means by which HIV enters cells has led to the identification of genetic polymorphisms, such as the 32 base-pair deletion in the ccr5 gene (ccr5∆32) that confers resistance to infection in homozygous individuals, and has also resulted in the development of entry inhibitorsĄŠsmall molecule antagonists that block infection at the entry step. The recent demonstration of long-term control of HIV infection in a leukemic patient following a hematopoietic stem cell transplant using cells from a ccr5∆32 homozygous donor highlights the important role of the HIV entry in maintaining an established infection and has led to a number of attempts to treat HIV infection by genetically modifying the ccr5 gene. In this review, we describe the HIV entry process and provide an overview of the different classes of approved HIV entry inhibitors while highlighting novel genetic strategies aimed at blocking HIV infection at the level of entry.
Keywords: HIV entry; gene therapy; CCR5
Viruses | Free Full-Text | Novel Approaches to Inhibit HIV Entry | HTML
https://www.mdpi.com/1999-4915/4/2/309/htmĄĄ
ĄĄ
ĄĄ
https://www.mdpi.com/1999-4915/4/12/3859
ĄĄ
EVIEW ARTICLE
Year : 2012 |
Pathogenesis and treatment of human immunodeficiency virus lipodystrophy
Suyog Subhash Jain1, Karuna Balwant Ramteke1, Girish Tulsidas Raparti1, Sanjay Kalra2
1 Department of Pharmacology, Government Medical College, Miraj, Maharashtra, India
2 Department of Endocrinology, Bharti Hospital and B.R.I.D.E., Karnal, India
Abstract
Enhanced understanding about the way human immunodeficiency virus (HIV) infects and causes infection in humans has led to invention and use of newer more effective antiretroviral drugs. As treatment for HIV is long term, side effects of the antiretrovirals become an important area of research focus. Antiretrovirals can cause severe metabolic abnormalities, collectively known as HIV lipodystrophy syndrome. If untreated, these metabolic abnormalities have the potential to increase stroke and cardiac ischemia. Management includes choice of nonoffending drugs, switch over to less toxic drugs, hypolipidemics, oral antidiabetics including thiazolidinediones, metformin and growth hormone analogs and finally facial surgeries. Updated knowledge about HIV lipodystrophy, and the hormone-related drugs used to treat it, is essential for physicians and endocrinologists to be able to diagnose the patients and effectively treat them.
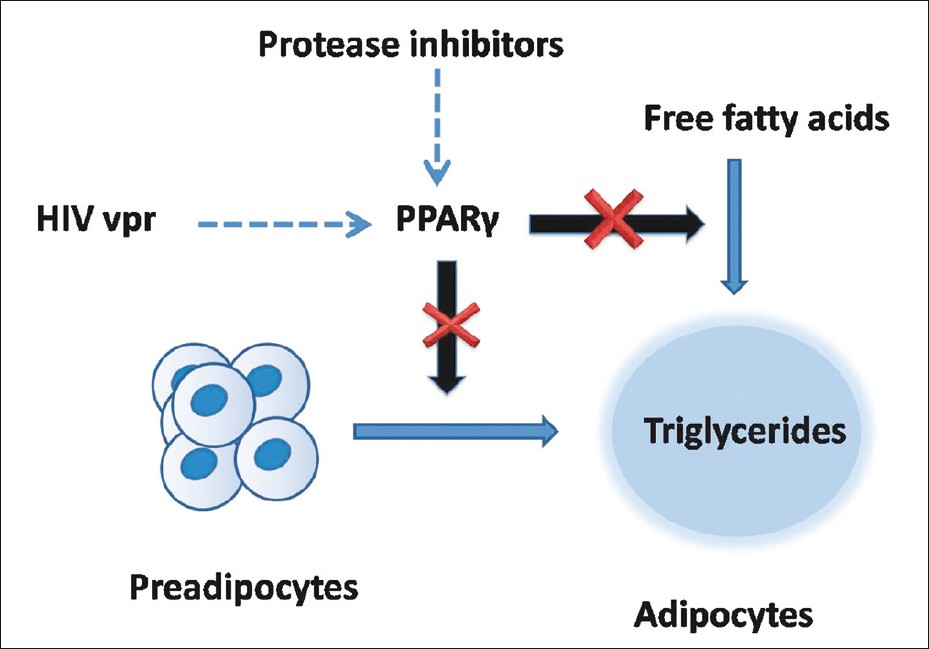
ĄĄHIV lipodystrophy is a disorder of fat metabolism which has diverse clinical and biochemical manifestations including lipoatrophy, lipohypertrophy and redistribution of fat. [5] Hypertrophic changes consist of excess fat deposition, most commonly around the abdominal viscera, but also around the neck, in the supraclavicular region, around the scapulae. Fat deposition may also take the form of bilateral symmetrical lipomatosis or breast enlargement. Atrophic changes consist of subcutaneous fat atrophy in the legs, buttocks, arms, and nasolabial and malar fat pads. Metabolic abnormalities include insulin resistance, raised triglycerides, and increased total cholesterol. Lipodystrophy-induced cosmetically disfiguring changes may have significant impact on quality of life. [6] The endocrinologist plays an important role in the management of HIV lipodystrophy in association with the primary care physician or HIV specialist.
ĄĄ
Keywords: Antiretroviral, human immunodeficiency virus lipodystrophy, protease inhibitors, tesamorelin
Figure 2: Human immunodeficiency virus protease inhibitors and HIV viral proteins accessory protein inhibit intracellular peroxisome proliferator-activated receptor-ĶÃ, which is necessary for differentiation of preadipocytes to mature adipocytes
ĄĄ
ĄĄ
PLoS Pathog. 2009 Jul; 5(7): e1000511.
Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel
Konstantin Pervushin, 1 , 2 , * Edward Tan, 1 Krupakar Parthasarathy, 1 Xin Lin, 1 Feng Li Jiang, 3 Dejie Yu, 3 Ardcharaporn Vararattanavech, 1 Tuck Wah Soong, 3 Ding Xiang Liu, 4 and Jaume Torres 1 , *
Ralph S. Baric, Editor
Author information Article notes Copyright and License information Disclaimer
1 School of Biological Sciences, Nanyang Technological University, Singapore,
2 Biozentrum of University Basel, Basel, Switzerland,
3 Center for Life Sciences, Department of Physiology, National University of Singapore, Singapore,
4 Institute of Molecular Cell Biology, Proteos, Singapore,
University of North Carolina, United States of America
* E-mail: gs.ude.utn@nihsuvrepk or hc.sabinu@nihsuvrep.nitnatsnok (KP); gs.ude.utn@serrotj (JT)
Conceived and designed the experiments: K. Pervushin, T. Soong, D. Liu, J. Torres. Performed the experiments: K. Pervushin, E. Tan, K. Parthasarathy, F. Jiang, D. Yu, A. Vararattanavech. Analyzed the data: K. Pervushin, E. Tan, X. Lin, F. Jiang, J. Torres. Contributed reagents/materials/analysis tools: K. Pervushin, K. Parthasarathy, A. Vararattanavech, T. Soong, J. Torres. Wrote the paper: K. Pervushin, J. Torres.
Abstract
The envelope (E) protein from coronaviruses is a small polypeptide that contains at least one ĶÁ-helical transmembrane domain. Absence, or inactivation, of E protein results in attenuated viruses, due to alterations in either virion morphology or tropism. Apart from its morphogenetic properties, protein E has been reported to have membrane permeabilizing activity. Further, the drug hexamethylene amiloride (HMA), but not amiloride, inhibited in vitro ion channel activity of some synthetic coronavirus E proteins, and also viral replication. We have previously shown for the coronavirus species responsible for severe acute respiratory syndrome (SARS-CoV) that the transmembrane domain of E protein (ETM) forms pentameric ĶÁ-helical bundles that are likely responsible for the observed channel activity. Herein, using solution NMR in dodecylphosphatidylcholine micelles and energy minimization, we have obtained a model of this channel which features regular ĶÁ-helices that form a pentameric left-handed parallel bundle. The drug HMA was found to bind inside the lumen of the channel, at both the C-terminal and the N-terminal openings, and, in contrast to amiloride, induced additional chemical shifts in ETM. Full length SARS-CoV E displayed channel activity when transiently expressed in human embryonic kidney 293 (HEK-293) cells in a whole-cell patch clamp set-up. This activity was significantly reduced by hexamethylene amiloride (HMA), but not by amiloride. The channel structure presented herein provides a possible rationale for inhibition, and a platform for future structure-based drug design of this potential pharmacological target....
All coronaviruses express the envelope (E) protein, a typically short polypeptide that in SARS-CoV is 76 amino acids long, and which contains at least one ĶÁ-helical transmembrane domain (ETM). In SARS-CoV E the transmembrane domain spans ∼25 residues [21], approximately from residue 10 to 35. Coronavirus E proteins are incorporated into the virion lipidic envelope, along with the spike protein (S) and the membrane protein (M). While the S protein is involved in fusion with host membranes during entry into cells, and the M protein is important in envelope formation and budding, E protein is not essential for in vitro and in vivo coronavirus replication. However, its absence results in an attenuated virus, as shown for SARS-CoV [22]. Recently, using a transgenic mouse model expressing the SARS-CoV receptor human angiotensin converting enzyme-2 (hACE-2), SARS coronavirus lacking gene E was shown to be attenuated and, in contrast to the wild type virus, did not grow in the central nervous system [23]. In other coronaviruses, E protein affects viral morphogenesis, i.e., virus-like particle (VLP) formation and release [24]ĻC[29]. Indeed, mutations in the extramembrane domain of E protein impaired viral assembly and maturation in MHV [30]. In TGEV, the absence of E protein resulted in a blockade of virus trafficking in the secretory pathway and prevention of virus maturation [31],[32].
ĄĄ
Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2702000/ĄĄ
Coronavirus
Structure of Coronavirus
Coronaviruses fall in the virus family Coronaviridae, order Nidovirales.
Coronaviruses are enveloped, 120 to 160 nm particles that contain an unsegmented genome of single-stranded positive-sense RNA (27ĻC32 kb).
The large, plus-stranded RNA genome associates with the N protein to form a helical nucleocapsid.
The helical nucleocapsid is 9ĻC11 nm in diameter.
There are 20 nm long club or petal-shaped projections that are widely spaced on the outer surface of the envelope, suggestive of a solar corona.
Structure of Coronavirus
Source: Nature Reviews \ Microbiology
The viral structural proteins include a 50ĻC60 kDa phosphorylated nucleocapsid (N) protein, a 20ĻC35 kDa membrane (M) glycoprotein that serves as a matrix protein embedded in the envelope lipid bilayer and interacting with the nucleocapsid, and the spike (S) a 180ĻC220 kDa glycoprotein that makes up the petal-shaped peplomers.
Some viruses, including human coronavirus OC43 (HCoV-OC43), contain a third glycoprotein (HE; 65 kDa) that causes hemagglutination and has acetylesterase activity.
Coronavirus genomes are monopartite, single-stranded, positive-sense, polyadenylated, and capped RNAs ranging from 27 to 32 kb in length.
The 5Ąä approximately 20 to 22 kb carries the replicase gene, which encodes multiple enzymatic activities.
The replicase gene products are encoded within two very large open reading frames, ORFs 1a and 1b.
The order of the genes encoding the viral RNA-dependent RNA polymerase and the four common structural proteins, the spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins are indicated as Pol-S-E-M-N.
Coronavirus genomes also include a variety of additional open reading frames (ORFs) that encode two to four nonstructural proteins of unknown functions.
In the genome, a common intergenic sequence (IS) of about 7 bases is found at the 5Ąä end of each gene.
This IS sequence is essential for the formation of subgenomic RNAs.
Coronavirus-infected cells contain multiple overlapping subgenomic, capped, and polyadenylated mRNAs with a common 3Ąä end.
Each subgenomic mRNA and the viral genomic RNA, which also serves as an mRNA, is translated to yield only the protein encoded by the 5Ąä gene on the mRNA.
Natural outbreaks of colds caused by coronaviruses occur predominantly during the winter months, although in children, two peaks in late autumn to early winter and early summer were detected.
It is estimated that coronaviruses cause 15ĻC30% of all colds.
Studies using virus detection or serology have shown that HCoV 229E, OC43, and NL63 occur worldwide.
The contribution of each HCoV may vary widely from year to year, for example, 229E contributing as little as 1% to acute respiratory infections in the community in one year and up to 35% in the next.
The incidence of coronavirus infections varies markedly from year to year, ranging in one 3-year study from 1% to 35%.
Replication of Coronavirus
Natural infection of humans with human respiratory coronaviruses occurs through exposure to respiratory secretions.
Coronaviruses attach to their glycoprotein receptors on host cells via their S proteins.
Viral entry is mediated by fusion of the viral envelope with the host cell membrane or by receptor-mediated endocytosis.
Group 1 coronaviruses 229E and NL63 bind to the metalloproteases, human aminopeptidase N and angiotensin-converting enzyme 2 (ACE-2) respectively.
The receptors for OC43 and HKU-1 have not been yet identified.
The fusion of the viral and cell membranes (either at the cell surface or within the endocytic vesicle) is mediated by the S2 portion of the virus spike protein which functions as a class 1 fusion protein.
Once the viral RNA is released into the cytoplasm, translation of the positive-strand genomic RNA gives rise to a large polyprotein that undergoes proteolytic processing to generate an RNA-dependent RNA polymerase.
An RNA-dependent RNA polymerase translated from the plus-stranded viral genomic RNA makes a negative-strand that serves as the template for a nested set of five to seven subgenomic mRNAs.
Translation of subgenomic mRNAs gives rise to structural viral proteins.
The N protein and newly synthesized genomic RNA assemble to form helical nucleocapsids.
Membrane glycoprotein M is inserted in the endoplasmic reticulum (ER) and anchored in the Golgi apparatus.
The nucleocapsid (N plus genomic RNA) binds to M protein at the budding compartment (ERGIC).
E and M proteins interact to trigger the budding of virions, enclosing the nucleocapsid.
These newly formed virions are transported via the Golgi apparatus to the plasma membrane where they are released by exocytosis.
Pathogenesis of Coronavirus
The primary route of transmission of human coronaviruses is via the respiratory tract, most likely spread by aerosols and in large droplets (e.g., sneezes).
Infection with the common-cold coronaviruses leads to loss of ciliary action (ciliostasis) and degenerative changes affecting the cilia of epithelial cells of the respiratory tract.
Infection remains localized to the upper respiratory tract because the optimum temperature for viral growth is 33Ąã C to 35Ąã C and may lead to the lower respiratory tract.
HCoV-OC43 is generally associated with mild upper respiratory tract infections, although it has been shown to have neuroinvasive properties.
Clinical manifestations of Coronavirus
HCoVs in both the 229E- and OC43-related serogroups cause upper respiratory signs and symptoms in adults and children that vary in frequency and severity.
HCoVs cause respiratory infections(bronchiolitis and pneumonia), but gastroenteritis and neurological disorders can also occur.
The human coronaviruses produce Ą°common colds,Ąą usually afebrile, in adults.
The symptoms include nasal discharge and malaise.
Other symptoms include rhinorrhea, headache, malaise, chills, sore throat, and cough.
The incubation period is from 2 to 5 days.
Symptoms last for a mean of 7 days, with a range of 3 to 18 days.
Patients with symptomatic coronavirus infection show a rise in neutralizing and complement fixation antibody titers in the serum after inoculation that waned after months.
The lower respiratory tract is seldom involved, although pneumonia may occur.
Asthmatic children may suffer wheezing attacks, and respiratory symptoms may be exacerbated in adults with chronic pulmonary disease.
HCoV-OC43 can infect neurons and cause encephalitis.
Lab Diagnosis of Coronavirus
Specimen: respiratory secretions, stool (HKU1)
Virus isolation- The human hepatoma cell-line HUH7 has been recently used for primary isolation of OC43, 229E and HKU-1 viruses from clinical specimens and NL63 has been isolated in LLC-MK2 and Vero B4 cells.
Detection of viral RNA by RT-PCR.
Electron microscopy of negatively stained stool specimens is useful for the detection of enteric coronaviruses.
Complement fixation, ELISA assays, immunofluorescence or virus neutralization tests have been used for serological diagnosis.
The serologic diagnosis of infections with strain 229E is possible using a passive hemagglutination test in which red cells coated with coronavirus antigen are agglutinated by antibody-containing sera.
HCoV-OC43ĻCrelated virions that express a HE glycoprotein on the viral envelope can also be detected by hemagglutination and acetyl esterase assays.
Treatment of Coronavirus
There is no proven treatment for human coronavirus infections and no vaccine.
Prevention and control of Coronavirus
Washing hands often with soap and water.
Avoid touching eyes, nose, or mouth with unwashed hands.
Avoiding close contact with people who are sick.
Covering mouth and nose with a tissue while coughing or sneezing, then throw the tissue in the trash and washing hands.Coronavirus | Online Microbiology Notes
https://microbenotes.com/coronavirus/ĄĄ
MERS, SARS, and emerging Coronaviruses: theoretical considerations and a proposal for critical care parenteral oxygen/ozone therapy
© May 2013, revised 2014, by GĻĶrard V. Sunnen, M.D.
Abstract
SARS (Severe Acute Respiratory Syndrome) is a global disease of significant lethality with a variable incidence and prevalence base. Of massive public health importance because of the unpredictability of its cycles and its lethality, SARS presents supremely challenging problems in light of its pathogenic capacity and mutational potential.
MERS (Middle East Respiratory Syndrome) belongs to the same Family as SARS and is more recent on the world stage. Much uncertainty remains as to its modes of transmission and the nature of its animal reservoirs. Its lethality, however, is well established.
There are neither vaccines nor antiviral agents available for SARS nor MERS, only supportive and often intensive measures such as cardiopulmonary assistance and the maintenance of physiological homeostasis.
Ozone, via a number of possible mechanisms enumerated in this paper, possesses recognized anti-viral properties. The technology of interfacing oxygen/ozone mixtures with biological fluids has long been mastered, as has been the technology of administering this gaseous mixture to the systemic circulation.
Ozone, because of its special biological properties, has theoretical and practical attributes to make it a viable candidate as a MERS and SARS inactivator, through a variety of physicochemical and immunological mechanisms. Bereft of all known therapeutic strategies, a proposal is herewith made for the parenteral administration of calibrated oxygen/ozone gaseous mixtures in the critical care of MERS, SARS, and related Coronavirus infections.
The Family of Coronaviruses
The SARS and MERS viruses belong to the viral family Coronaviridae. Which includes two genera, coronavirus and togovirus, each showing similar replication mechanisms and genomic organization but distinct genomic lengths and viral architecture. First identified in the 60ĄŊs, this family identifies itself by large, enveloped, positive-stranded RNA virions. Their appearance is characteristically distinct, with envelopes endowed with host cell membrane-tropic petal shaped spikes (peplomers). The large, amply spaced peplomers on the virion surface suggests a coronal (crown-like) appearance.
Prior to SARS and MERS, Coronaviridae were responsible for relatively mild cold-like syndromes in humans corresponding to their predilection for the ciliary epithelium of the trachea, nasal mucosa, and alveolar cells of the lungs. At times they were only rarely implicated in serious respiratory illnesses in frail older adults (Falsey 2002). SARS and MERS represent a quantum leap in Coronaviridae infectivity by way of their significant lethality. Widely seen in nature, coronaviruses infect a spectrum of animal hosts and are responsible for avian infectious bronchitis, murine hepatitis, and porcine gastroenteritis, among others. Of probable significance to humans is that animal coronaviruses are able to penetrate into the central nervous system.
SARS and MERS: Virion architecture and molecular biology
The SARS virion differs from other members of the Coronaviridae family in its genomic composition. The other viral structures, however, are similar, including virion architecture, and the fundamental composition of structural and non-structural proteins.
The software for viral replication is the nucleic acid core, a single strand long chain RNA nucleotide. The core is surrounded by the nucleic acid coat or capsid. The capsid is rigid and determines the shape of the virus; it is made of repeating units called capsomeres. The SARS viral nucleocapsid is tubular with a helical symmetry.
An envelope that forms the outer layer of the virion and maintains intimate contact with host bodily fluids surrounds the nucleocapsid. As such, it is sensitive to the composition and alterations in its milieu, such as temperature, pH, and ionic balance. The viral envelope is formed at the time of budding, an intricate process in which the nucleocapsid exits the host cell. In order to do this, it fuses with the host cell membrane, appropriating its components to form its own envelope. It is known that the lipid composition of viral membranes reflects the lipid composition through which the particles exit. Viral envelopes are composed of lipid bilayers associated with a union of carbohydrates and proteins, glycoproteins, and lipids and phosphates, phospholipids. Up to 60% of the lipid component of the envelope is composed of phospholipid and the remainder is mostly cholesterol. This lipid-carbohydrate envelope is closely articulated with the peplomers, which determine attachment and penetration into host cells.
The genome composition and sequence of the SARS virus has been identified (Marra 2003; Rota 2003). Marra et al. described a viral genome configuration of 29,727 nucleotides in length, within which exists a gene order similar to other coronaviruses. However, because the genetic composition of SARS does not closely resemble any of the three known classes of coronaviruses, they propose a new and fourth class of coronaviruses, the SARS-CoV. Postulated, is a hypothesis that an animal virus recently mutated to successfully infect humans, or that the SARS virus mutated from a common human coronavirus.
Rota et al. reported a nucleotide sequence of 29,727 in SARS-CoV, with 11 open reading frames. Phylogenetic analyses and sequence comparisons showed that the SARS virus is not closely related to any of the previously characterized coronaviruses.
Virion structural proteins are essential elements in determining the morphological and functional dimensions of the SARS virus. Coronavirus structural proteins include the N nucleocapsid phosphoprotein which binds to viral RNA; the membrane glycoprotein M which forms the shell of the internal viral core and is responsible for triggering viral assembly; the protein E associated with the virion envelope; the spike glycoprotein S which binds to specific cellular receptors and elicits cell-mediated immunity; and the hemagglutinin-esterase glycoprotein HE forming small spikes on the coronavirus envelope (Knipe 2001).
SARS: Viral replication
The viral replication cycle follows the pattern seen in mammalian viruses and may be divided into several stages (Cann 1997; Evans 1997; Knipe 2001). The coronavirus attaches to the membrane of the host cells by binding the S and HE proteins of its peplomers to receptor glycoproteins or glycans.
Once cell entry is achieved, the virion sheds its envelope to commence its replication in the host cell cytoplasm. It binds to cellular ribosomes and released viral polymerase begins the RNA replication cycle. Newly formed nucleocapsids continue their assembly with the acquisition of new envelopes by means of budding through membranes of the cell's endoplamic reticulum.
Virions are then released into the general blood and lymphatic circulation, ready to infect new cells, other organ systems, and new hosts.
SARS: Clinical findings
Recently, the clinical manifestations of SARS have been comprehensively described (Peiris 2003). In this study of 50 hospitalized patients, fever, chills, myalgia, and dry cough were the most frequent presenting complaints. Also reported, were rhinorrhea, sore throat, and gastrointestinal symptoms.
Radiological examination showed evidence of pulmonary consolidation approximately 5 days after the onset of symptoms. Laboratory examination showed leucopenia and lymphopenia, despite the presence of fever; also anemia, thrombocytopenia, liver enzyme elevations (alanine aminotransferase), and skeletal and heart muscle enzyme elevation (creatinine phosphokinase). All these features point to severe systemic inflammatory insults.
The incubation of SARS is 2 to 10 days, and in some patients perhaps longer. The respiratory route achieves viral transmission where it may infect the new host through aerosol and droplet contact with mucosal surfaces of the mouth, nose, throat, and probably the conjunctiva. SARS virions have been found in feces and the importance of this route of transmission is being evaluated, as it is known that several animal coronaviruses use this propagation venue. Moreover, since it is appreciated that SARS particles remain viable on fomites for 48 hours or longer, any eradication effort must address the infectivity of objects in the environment.
The syndrome progresses to severe disease with respiratory distress and oxygen desaturation requiring ventilatory support in over a third of patients, approximately 8 days after symptom onset. Mortality has been noted to vary according to transmission clusters, ranging from 3 to 20%. This suggests that the etiology of SARS depends upon a heterogenous population of viral quasispecies with variable degrees of virulence.
SARS and MERS: Genetic creativity
As is the case in the majority of RNA viruses, coronaviruses mutate at a high rate (Steinhauer 1986). Within any one afflicted individual, coronaviruses particles do not show a homogeneous population. Instead, they function as a pool of genetically variant strains known as quasispecies. This is due to the high error frequency of RNA polymerases, the presence of deletion mutants, the high frequency of RNA recombination and point mutations, and the occurrence of defective-interfering RNA (DI RNA). The net result of these diverse and complex mechanisms is the continuous spawning of novel virions and divergent quasispecies. Some of the genetic creations will find themselves at an advantage in negotiating new host antibody responses and pharmacological antiviral countermeasures; and they will propagate accordingly, thus expanding their ecological terrain. Other genetic creations will be too lethal to their hosts, work against their own survival, and will prove to be non-adaptive. If we can speak of a viral psychology, an efficient survival balance aims somewhere between defeat by host defenses on one hand, and viral suicide through aggressive lethality on the other.MERS, SARS, and emerging Coronaviruses:
http://triroc.com/sunnen/topics/sars.htmlĄĄ
Inhibition of dengue virus by curcuminoids - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0166354218303875
The results revealed that curcumin is a weak inhibitor of the viral protease. However, the analogues exhibited more potent inhibition of DENV infectivity in plaque assays suggesting that the cellular pathway (s) required for viral replication and/or assembly are targeted by these compounds.
Cited by: 3
Publish Year: 2019
Author: Anuradha Balasubramanian, Rajendra Pilankatta, Tadahisa Teramoto, Ayyiliath M. Sajith, Evaristus Nwu...
A Biotin Analog Inhibits Acetyl-CoA Carboxylase Activity ...
www.jbc.org/content/277/19/16347.full
May 10, 2002 ĄĪ Acetyl-CoA carboxylase is a biotin-dependent enzyme that catalyzes the following two-step mechanism. The first half-reaction is carried out by the biotin carboxylase component of acetyl-CoA carboxylase and involves the ATP-dependent carboxylation of biotin with bicarbonate serving as the source of CO 2.
Published in:
Journal of Biological Chemistry ĄĪ 2002
Authors:
Keith L Levert ĄĪ Grover L Waldrop ĄĪ Jacqueline M Stephens
Affiliation:
Louisiana State UniversityĄĄ
ĄĄ
ĄĄ
Viruses 2012, 4(2), 309-324; https://doi.org/10.3390/v4020309
Novel Approaches to Inhibit HIV Entry
by Chukwuka A. Didigu and Robert W. Doms *
Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, 3610 Hamilton Walk, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
Received: 16 December 2011; in revised form: 17 January 2012 / Accepted: 7 February 2012 / Published: 21 February 2012
Abstract:Human Immunodeficiency Virus (HIV) entry into target cells is a multi-step process involving binding of the viral glycoprotein, Env, to its receptor CD4 and a coreceptorĄŠeither CCR5 or CXCR4. Understanding the means by which HIV enters cells has led to the identification of genetic polymorphisms, such as the 32 base-pair deletion in the ccr5 gene (ccr5∆32) that confers resistance to infection in homozygous individuals, and has also resulted in the development of entry inhibitorsĄŠsmall molecule antagonists that block infection at the entry step. The recent demonstration of long-term control of HIV infection in a leukemic patient following a hematopoietic stem cell transplant using cells from a ccr5∆32 homozygous donor highlights the important role of the HIV entry in maintaining an established infection and has led to a number of attempts to treat HIV infection by genetically modifying the ccr5 gene. In this review, we describe the HIV entry process and provide an overview of the different classes of approved HIV entry inhibitors while highlighting novel genetic strategies aimed at blocking HIV infection at the level of entry.
Keywords: HIV entry; gene therapy; CCR5
1. Introduction
Since its identification as the etiologic agent of acquired immune deficiency syndrome (AIDS), human immunodeficiency virus (HIV) has claimed the lives of millions around the world. Although the introduction of antiretroviral therapy (ART) has dramatically altered the disease course of treated HIV-infected individuals by delaying the progression to AIDS [1,2,3], often times for many years, the considerable morbidities associated with ART (reviewed in [4]) still drive efforts to develop a cure for HIV. As one of the most intensely studied pathogens of the last decade, much is known about the life cycle of HIV, which begins with entry of the virus into susceptible cells. To enter a cell, HIV must first bind to its primary receptor CD4, and then to one of two coreceptorsĄŠCCR5 or CXCR4 (Figure 1) [5,6,7,8,9,10,11,12,13]. The choice of coreceptor used by the virus is intimately linked to disease acquisition and pathogenesis as the majority of transmitted viruses use CCR5 to enter cells [14,15,16,17], while the appearance of viruses capable of using CXCR4 during infection is associated with a more rapid progression to AIDS [18,19,20,21]. Additionally, there is a naturally occurring mutation in ccr5 (ccr5∆32) that prevents expression of CCR5 on the cell surface, and individuals homozygous for this mutation are highly resistant to infection with viruses capable of using CCR5, although their cells remain susceptible to infection with CXCR4-using viruses [22,23,24]. Interest in a cure for HIV was recently reignited following the report of an HIV-infected maleĄŠthe berlin patientĄŠwho developed acute myelogenous leukemia and received a hematopoietic stem cell (HSC) transplant using cells from a ccr5∆32 homozygous donor [25,26]. Following his transplant, he was taken off ART and his virus has remained undetectable for greater than four years, suggesting that long-term control and a possible functional cure of his disease have been achieved. This remarkable report highlights how our understanding of a very basic questionĄŠhow a virus enters its host cellĄŠhas led to the development of new antiviral drugs and therapeutic approaches that have brought us a step closer to controlling the global HIV pandemic. In this review, we provide an overview of HIV entry and its impact on disease pathogenesis, and report on novel treatment approaches targeting the entry process.
2. HIV Entry: The Basics
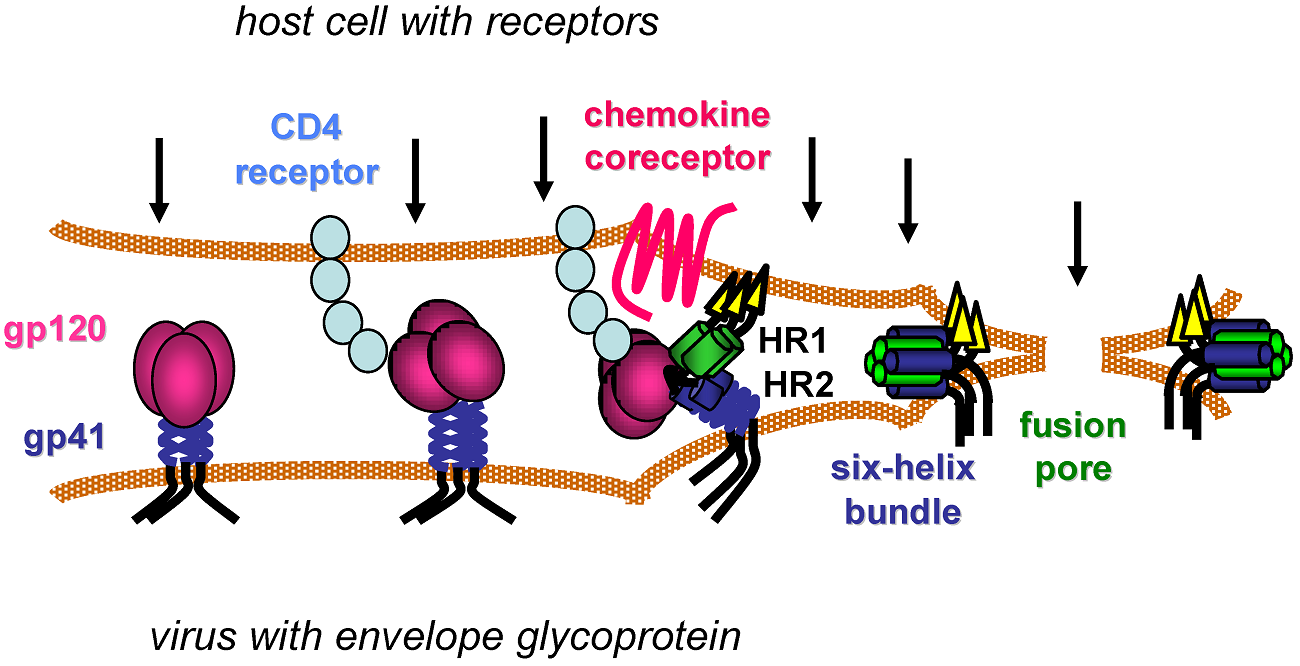
Entry of HIV into target cells is mediated by the type I integral membrane viral glycoprotein Env. Env is synthesized as a polypeptide precursor termed gp160 which undergoes several of modifications within the cell as it is transported to the cell surface, including extensive N-linked glycosylation, and cleavage by cellular proteases into the extracellular gp120 and the membrane-spanning gp41 subunits [27,28,29]. The extensive glycosylation of Env contributes to its ability to evade humoral immune responses, as the carbohydrate moieties covering its surface are poorly immunogenic and may shield potentially antigenic epitopes on the glycoprotein from recognition by the immune system [30,31,32]. Following gp160 cleavageĄŠan event required for subsequent membrane fusionĄŠgp120 and gp41 maintain their association via non-covalent interactions and are transported to the cell surface where they exist as a trimer of heterodimers that is ultimately incorporated into the viral membrane. Prior to CD4 bindingĄŠthe first essential step in HIV entryĄŠa number of cell-surface molecules are capable of mediating Env-dependent attachment of the virion to target cells. One such attachment factor is the C-type lectin CD209 or dendritic cell-specific intercellular adhesion molecule (ICAM) grabbing non-integrin (DC-SIGN). Expressed on dendritic cells, DC-SIGN and several other lectins are capable of binding Env and boosting infection in vitro by facilitating trans-infection of surrounding CD4 T cells by dendritic cell-bound virions (reviewed in [33]). More recently, monomeric gp120 from some HIV strains has been shown to bind the gut homing integrin ĶÁ4ß7 [34,35], which is expressed on activated CD4 T cells. This finding is of particular interest as the depletion of CD4 T cells in the gut-associated lymphoid tissue (GALT) early in infection is a hallmark of HIV disease [36,37,38] and ß7 integrins mediate trafficking of lymphocytes to the gut mucosa [39]. However, it is not clear whether ĶÁ4ß7 supports binding of trimeric Env on the surface of virions, and thus its relevance to the gut pathology associated with HIV infection remains unknown. Further, while interactions with these and other attachment factors influence HIV infection in vitro, little is known regarding their significance in vivo.
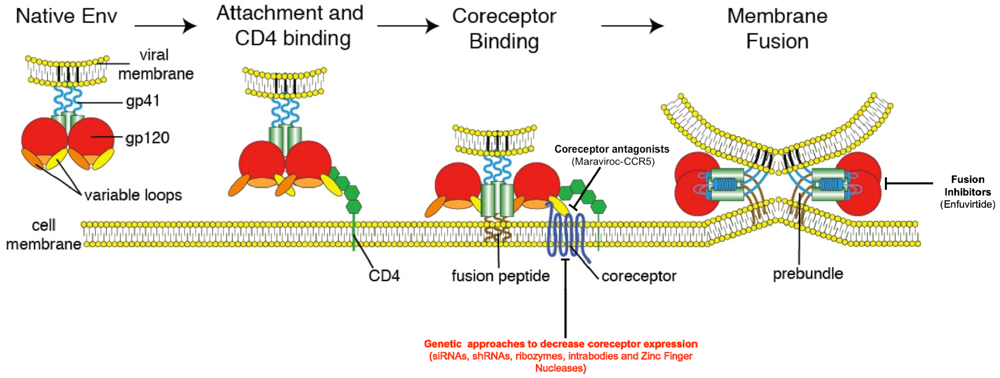
Viruses 04 00309 g001 1024 Figure 1. The HIV Entry Process. The figure below outlines a model for HIV Entry. The entry process begins with binding of gp120 (red) to its primary cellular receptor CD4 (green). CD4 binding results in conformational changes that allow binding of gp120 to the coreceptor-either CCR5 or CXCR4. Coreceptor binding results in triggering of the fusion machinery and formation of the six-helix bundle required to drive fusion of the viral and host cell membranes. Also pictured are the two main steps that have been successfully targeted (coreceptor binding and viral fusionĄŠapproved therapeutics appear in parentheses) along with the primary target of most genetic therapies aimed at preventing HIV entryĄŠthe HIV coreceptors (in red). Adapted from Antiviral Research Vol 85, Tilton J.C. and Doms R.W, Ą°Entry inhibitors in the treatment of HIV-1 infection,Ąą 91-100, Copyright 2009, with permission from Elsevier.
The gp120 subunit of Env is composed of five relatively conserved (C1ĻCC5) and five more variable (V1ĻCV5) domains [40]. The conserved regions form the proximal core of gp120 while intrachain disulfide bonds in the variable regions of gp120 result in the formation of five variable ĄŪloopĄŊ structures that make up the most exterior portion of the gp120 ectodomain [41]. The CD4 binding site on gp120 is a well conserved cavity formed at the interface of the inner and outer domains of the glycoprotein [42]. Following binding of gp120 to CD4, a series of conformational changes occur including the rearrangement of two pairs of ß-sheets from the gp120 inner and outer domains that come together to form a four-stranded ß-sheet structure termed the bridging sheet. The bridging sheet links the inner and outer domains of gp120 and interacts with the viral coreceptor, be it CCR5 or CXCR4 [42,43]. gp120 binding to CD4 also results in enhanced exposure and reorientation of the V1/V2 and V3 loops of gp120, outward rotation of each gp120 monomer to reveal the gp41 stalk, and hinge-like movements in CD4 that are thought to bring the viral membrane in close proximity to the target cell [42,44,45]. Together, all of these events culminate in the creation and exposure of the coreceptor-binding site.
The HIV coreceptors belong to the family of chemokine receptorsĄŠseven-transmembrane G-protein coupled receptors with prominent roles in immune cell trafficking. These receptors have three extracellular and intracellular loops, extracellular N-termini, and intracellular C-termini. While several different chemokine receptors are capable of mediating HIV entry in vitro, current evidence suggests that the CCR5 and CXCR4 chemokine receptors are the most frequently utilized in vivo [46,47,48]. Viruses capable of utilizing CCR5 alone, CXCR4 alone, or both coreceptors, are labeled R5-tropic, X4-tropic, and RX54 or dual-tropic viruses respectively. CCR5 is the primary coreceptor for the majority of HIV-1 isolates and is expressed on CD4+ T-cell subsets, macrophages and dendritic cells, while CXCR4 is less commonly used, but is expressed on a wide variety of cells both within and outside the immune system [49]. For reasons that remain unclear, the majority of transmitted viruses utilize CCR5 irrespective of the route of transmission and despite the availability of target cells expressing CXCR4 [17,50,51]. Multiple lines of evidence including mutational analyses, studies of small molecule inhibitors, and inhibition by coreceptor-specific blocking antibodies suggest that both the second extracellular loop (ECL2) and sulfated tyrosines within the N-terminus of the coreceptor interact with the V3 loop of gp120 and mediate coreceptor binding [52,53,54,55,56,57]. The V3 loop is also known to be a key determinant of coreceptor preference as the presence of positively charged amino acids at positions 11 and or 24/25 of V3 is correlated with CXCR4 usage [58,59].
The HIV fusion machinery is contained within the gp41 subunit of Env, which is comprised of a large cytoplasmic domain, a membrane-spanning segment, and an ectodomain that maintains contact with gp120. The ectodomain contains a typical fusion peptideĄŠa stretch of hydrophobic amino acids at the N-terminus [60,61]ĄŠalong with two ĶÁ-helical heptad repeats (HR), the N-terminal HR1 and the C-terminal HR2 repeats [62,63]. The current model of gp41-mediated fusion is based on studies performed using HIV fusion inhibitors, crystal structures, and structural similarities between gp41 and other well-characterized type I membrane fusion proteins including the influenza virus glycoprotein, hemagglutinin (HA) [63,64,65,66]. In this model, the sequential interaction of Env with CD4 and a coreceptor results in exposure of the fusion peptide, which then inserts into the plasma membrane of the host cell, causing gp41 to physically link both membranes. Subsequently, the three HR1 domains of the Env trimer interact with one another to form a coiled coil, and the three HR2 segments fold back on the HR1 trimer creating a six-helix bundle that brings the viral and host cell membranes in close contact with one another, allowing for mixing of the two membranes and formation of the fusion pore. Fusion between Env and the host cell was long thought to occur at the plasma membrane as HIV entry occurs in a pH-independent manner [67] and Env is capable of mediating fusion between neighboring cells, provided that they express CD4 and an appropriate coreceptor [68]. However, recent work using trans dominant-negative mutants of proteins involved in clathrin-mediated endocytosis [69] along with elegant studies using single-virion imaging [70] have demonstrated a clear role for components of the endocytic pathway in HIV entry in a number of cell lines. These studies were performed using immortalized cell lines, and as such, the role of the endocytic pathway in HIV entry into relevant cell types in vivo will need to be determined.
3. Inhibition of HIV Entry
The multi-step process by which HIV enters cells provides a series of unique targets for interventions to prevent viral entry including receptor and coreceptor binding, and membrane fusion. Efforts to inhibit these steps have led to the discovery of a new class of anti-HIV drugsĄŠthe HIV entry inhibitors (reviewed in [71]). A number of CCR5 inhibitors have been developed and display anti-HIV activity both in vitro and in vivo. These drugs are believed to work by binding to CCR5 at a site distinct from the gp120-binding site and subsequently alter the conformation of the CCR5 extracellular loops required for entry of R5-tropic HIV variants. One such CCR5 antagonist, maraviroc, is licensed for use in the United States and in Europe [43,72]. A variety of CXCR4 antagonists have also been developed, and while they exhibit potent anti-HIV activity in vitro (against X4 but not R5 virus strains), administration of these drugs in vivo results in mobilization of HSCs from the bone marrow to the peripheral blood, highlighting the important role of CXCR4 in HSC homing [73,74,75]. Although this side effect limits their use in HIV-infected individuals, the CXCR4 antagonist plerixafor, is currently used to mobilize HSCs for subsequent autologous transfer in patients with non-HodgkinĄŊs lymphoma and multiple myeloma [76].
gp41-mediated membrane fusion presents another drug target in the HIV entry process. Synthetic peptides based on the sequence of HR2 display significant antiviral activity against HIV in vitro but for many years, the mechanism of this antiviral activity remained unknown [77]. However, both the observation that these peptides display higher antiviral activity as dimers, and the elucidation of the structure of the gp41 fusion machinery have led to a model for their mechanism of action [63,64,78]. These drugs are now believed to act in a dominant negative fashion by competing with the HR1 and HR2 domains of gp41 and ultimately preventing the formation of the six-helix bundle required for membrane fusion. One such peptide, enfuvirtide, is the only FDA-approved HIV fusion inhibitor, and is indicated for use in combination with standard antiretroviral therapy. However, the twice-daily subcutaneous dosing schedule of the drug makes it an unattractive choice for many patients and care providers.
As is the case for most anti-HIV drugs, viral variants resistant to all of the entry inhibitors have been identified. The appearance of maraviroc resistant viruses in vitro is a well-established phenomenon and these viruses either adapt to recognize the drug-bound conformation of CCR5, or more commonly, acquire the ability to use CXCR4 in addition to CCR5 (reviewed in [79]). In vivo, resistance to maraviroc most commonly results from outgrowth of pre-existing X4 or R5X4 variants that are sometimes present at very low levels prior to the onset of therapy. As such, tropism testing is indicated prior to administration of maraviroc. In the case of enfuvirtide, mutations within HR1 and HR2 rapidly select for viruses with a dramatically decreased sensitivity to the drug [80,81]. HIV drug resistance is not unique to entry inhibitors, but is a widespread problem seen with all classes of HIV chemotherapeutics, and while ART increases survival in HIV-infected individuals, the problems of drug resistance, impaired immune function despite ART, long-term financial cost and drug-associated toxicities of ART continue to fuel the search for curative therapies for HIV infection. Although the search for a cure has long been met with an aura of skepticism, the success of the berlin patient has provided hope that a cure is indeed achievable. After living with HIV for at least 10 years, and experiencing control of viremia on ART for 4 years, the berlin patient developed acute myelogenous leukemia and received a HSC transplant using cells from a ccr5∆32 homozygous donor. His ART was stopped 1 day before the transplant and despite being able to detect proviral HIV DNA for up to 60 days post-transplant, he never experienced a rebound of his viral load [26]. In the 4 years that have followed this transplant, his viral load has remained undetectable as determined by viral RNA and DNA assays of multiple tissues including the gut, bone marrow, peripheral blood and brain. He has also experienced a repopulation of his gut mucosa CD4+ T cells along with a decrease in antibodies directed against HIV.
The Berlin patient received two interventionsĄŠconditioning chemotherapy and radiation, and an infusion of HIV resistant cellsĄŠall of which of which could have contributed to his dramatic control of the infection. However, as similar chemotherapy regimens and HSC transplants using cells from normal donors have always been followed by a recrudescence of infection following cessation of ART (reviewed in [82]), the control of infection seen in the Berlin patient was clearly dependent upon the genetic absence of CCR5, reinforcing the important role of HIV entry in the maintenance of an established infection. Despite the urgent need for a cure for HIV infection, the risks associated with chemotherapy and radiation, and the relatively low frequency of ccr5∆32 homozygous individuals, makes it unlikely that allogeneic HSC transplants using cells from ccr5∆32 homozygous donors will become a widespread treatment option, and has prompted attempts to mimic the genetic knockout of CCR5 that was achieved in the Berlin patient.
4. Genetic Knockout of CCR5
Several studies have successfully decreased expression of CCR5 in primary human HSCs, CD4 T cells and macrophages in vitro using a variety of unique genetic approaches. Dominant negative forms of CCR5, anti-CCR5 intracellular antibodies (intrabodies), and a number of RNA-based approaches including RNA interference (RNAi), short-hairpin RNAs (shRNA), and ribozymes have been all been shown to decrease CCR5 expression [83,84,85,86,87,88]. In one recent study, the investigators took a combinatorial approach by simultaneously targeting CCR5, the viral genes tat and rev, and the viral trans-activating region (TAR) [83]. Using this approach, the authors successfully introduced an shRNA construct targeting a tat/rev exon, a TAR decoy that localized to the nucleolus, and a CCR5-targeting ribozyme into human HSCs ex vivo using a lentiviral vector, and showed that the cells expressing the constructĄŠdubbed Triple-RĄŠwere protected from infection in vitro and provided protection from infection in a humanized mouse model of HIV infection. Following this promising pre-clinical study, Triple-R was tested in a Phase I trial where four patients with AIDS-related lymphoma who were scheduled to receive autologous HSC transplants had a fraction of their infused stem cells modified by the Triple-R construct [89]. The gene-modified cells engrafted in all four patients, and were readily detectable at a consistently low level for up to 24 months. While these cells did not have any detectable effect on infection, possibly due to the low number of gene-modified cells, the study was successfully completed without any toxicity related to the genetic modification of HSCs suggesting that modification of these long-term progenitor cells is a safe and viable option for future genetic therapies aimed at eliminating HIV infection.
As with most gene therapy approaches, achieving lasting expression of the transgene of interest in progenitor cell populations such as HSCs is a challenge, and there remains a need for the development of vector systems that will allow for stable transgene expression in different cell populations over long periods of time. A limitation of a number of these studies has been the need for repeated administration of many of these agents due to their transient effects. As such, recent work has sought to introduce somatic mutations into the ccr5 gene with the ultimate goal of phenocopying the natural ccr5∆32 mutation by rendering a fraction of cells ccr5 negative. These mutations were introduced using zinc finger nucleases (ZFNs)ĄŠchimeric proteins composed of a DNA-binding zinc-finger protein (ZFP) fused to the catalytic domain of a FokI restriction endonuclease (Figure 2) [90]. Following binding of a ZFN pair to the intended target sequence, the nuclease domains of the ZFN pair dimerize and introduce a double stranded break (DSB) in the DNA molecule. This break is repaired by the error-prone cellular non-homologous end joining (NHEJ) machinery and typically results in the introduction of insertions and deletions that render the gene product non-functional [91,92,93]. As the ZFP domain of a ZFN can be engineered to bind unique DNA sequences, a pair of ZFNs that recognize the portion of the ccr5 gene encoding the first transmembrane segment were designed and shown to specifically modify 20ĻC40% of the CCR5 alleles in human CD4 T cells in vitro [94]. These gene-modified cells appeared indistinguishable from wildtype cells in culture, but following challenge with an R5-tropic virus, the gene-modified cells had a selective growth advantage as they were protected from infection. In vivo experiments using a humanized mouse model of HIV infection also showed that CCR5-modified CD4 T cells engrafted into the animals at levels similar to unmodified CD4 cells, and are capable of controlling viral load and maintaining CD4 counts following infection with R5-tropic viruses. More recently, CCR5-specific ZFNs have been shown to be capable of disrupting the CCR5 gene in human HSCs ex vivo, with CCR5-modified HSCs displaying normal multi-lineage differentiation into a variety of CCR5-negative hematopoietic cell subsets that recapitulate the protective phenotype seen in the in vitro and humanized mouse studies using CCR5-modified CD4 T cells [95]. The ultimate goal of ZFN therapy is the ex vivo modification of CD4 T cells and HSCs from HIV infected individuals, with the hope that after reinfusion, these cells will control HIV infection in the absence of ART. To this end, the safety and tolerability of CCR5-ZFN modified CD4 T cells is currently being tested in two ongoing human clinical trials (NCT00842634, NCT01044654; Clinicaltrials.gov) and preliminary results presented at the 2011 Conference on Retroviruses and Opportunistic Infections (CROI) suggest that infusion of ZFN-modified CD4 T cells into patients currently on ART is safe, and these gene modified cells appear to initially expand and persist at a stable level over time. While these human studies provide evidence for the safety of this approach and its ability to be scaled up, the effect of infusing CCR5-ZFN modified cells on HIV disease can only be assessed in the absence of ART. As such, structured treatment interruptions, where patients are taken off ART following infusions of CCR5-ZFN modified cells, have been built into the human trials. One of the benefits of the ZFN approach is the permanent modification of the ccr5 gene in a subset of cells, thereby getting around the problem of longevity that is intrinsic to approaches targeting CCR5 at the level of RNA. Additionally, the ex vivo modification step bypasses the need for systemic in vivo delivery of the gene therapy vector and thus potentially limits toxicity while ensuring that only the intended cells receive the CCR5 ZFN.
Viruses 04 00309 g002 1024 Figure 2. Zinc Finger Nucleases (ZFNs) bind and cleave DNA. A ZFN consists of a DNA-binding zinc finger protein (ZFP) domain fused to the catalytic domain of a FokI endonuclease. Each ZFP array in the image (ZFP left & right) contacts 12bp of DNA for a total DNA specificity of 24 base pairs. The members of the ZFN pair depicted are separated by a 5 to 6 base pair spacer and concomitant binding of each ZFN results in dimerization of the nuclease domains and the introduction of a double stranded break. Adapted by permission from Macmillian Publishers Ltd: Nature Biotechnology. Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nature biotechnology 2007, 25, 778-785. Copyright 2007.
5. Future Directions
Despite their promise, genetic approaches targeting CCR5 are currently incapable of achieving the complete genetic knockout of CCR5 that exists in ccr5∆32 homozygous individuals and was accomplished in the Berlin patient. Whether the incomplete ablation of CCR5 provided by these genetic approaches will be able to provide long-term, or indeed, any control of established HIV infection remains to be seen. Additionally, it is unclear how the virus will respond to the partial ablation of one of its primary coreceptors. One possible outcome is a switch from R5 to X4-tropism or an outgrowth of a pre-existing X4-tropic virus reservoir following removal of ccr5. As the coreceptor switch correlates with an accelerated disease progression, a possible coreceptor switch following the removal of ccr5 may be minimized by performing tropism testing to exclude patients with detectable levels of X4-tropic virus prior to administering treatments that reduce levels of CCR5 expression, or by the use of approaches that simultaneously target both coreceptors [96]. However, this latter goal may prove challenging because of the important role of CXCR4 in immune cell trafficking. Additionally, HIV is capable of using a number of other chemokine receptors in vitro, and recent reports have identified variants of the closely related simian immunodeficiency virus (SIVsmm) that utilizes CCR5 but not CXCR4 in vitro, yet still infects sooty mangabeys lacking functional CCR5ĄŠsuggesting in vivo usage of coreceptors other than CCR5 and CXCR4 [97]. This raises the possibility that another potential mechanism of escape from therapies involving the genetic ablation of the major coreceptors is viral evolution to use some of the less commonly utilized alternative coreceptors. Although a number of questions remain regarding the feasibility, efficacy, and long-term toxicities of these approaches to genetically modify the HIV coreceptors, as well as their effects on viral evolution, genetic ablation of the entry factor CCR5 remains the only proven means of providing long-term control of infection in the absence of ART and as such, continues to provides hope that a cure for HIV can be achieved.Viruses | Free Full-Text | Novel Approaches to Inhibit HIV Entry | HTML
https://www.mdpi.com/1999-4915/4/2/309/htmĄĄ
Sci Rep. 2016; 6: 27539.
Published online 2016 Jun 10. doi: 10.1038/srep27539
Curcumin inhibits HIV-1 by promoting Tat protein degradation
Amjad Ali1 and Akhil C. Banerjeaa,11Laboratory of Virology, National Institute of Immunology, New Delhi, India
Abstract
HIV-1 Tat is an intrinsically unfolded protein playing a pivotal role in viral replication by associating with TAR region of viral LTR. Unfolded proteins are degraded by 20S proteasome in an ubiquitin independent manner. Curcumin is known to activate 20S proteasome and promotes the degradation of intrinsically unfolded p53 tumor suppressor protein. Since HIV-1 Tat protein is largerly unfolded, we hypothesized that Tat may also be targeted through this pathway. Curcumin treated Tat transfected HEK-293T cells showed a dose and time dependent degradation of Tat protein. Contrary to this HIV-1 Gag which is a properly folded protein, remained unaffected with curcumin. Semi-quantitative RT-PCR analysis showed that curcumin treatment did not affect Tat gene transcription. Curcumin increased the rate of Tat protein degradation as shown by cycloheximide (CHX) chase assay. Degradation of the Tat protein is accomplished through proteasomal pathway as proteasomal inhibitor MG132 blocked Tat degradation. Curcumin also decreased Tat mediated LTR promoter transactivation and inhibited virus production from HIV-1 infected cells. Taken together our study reveals a novel observation that curcumin causes potent degradation of Tat which may be one of the major mechanisms behind its anti HIV activity.
The majority of cellular protein degradation occurs through the two pathways, the lysosomal and the proteasomal pathway1,2. Proteasomal pathway is a specific and controlled process in which the substrate protein is tagged with ubiquitin and degraded by 26S proteasome complex3. Proteins are also degraded through the 20S proteasomes where the degradation is independent of the ubiquitination process4,5. Partially or completely unstructured proteins as well as oxidized proteins are degraded through this pathway6,7. The unstructured proteins are normally stabilized in the cell by associating with other proteins or protein complexes8. The unstructured proteins are also protected by NADH bound NAD(P)H:quinone oxidoreductase 1 (NQO1) protein present on the 20S proteasome4,8. Competitive inhibitors of NADH, like dicoumarol and curcumin release NADH from NQO1 rendering the 20S proteasome active for the degradation of unstructured proteins9,10,11. Many of the cellular proteins which are either partially or completely unfolded in their native condition, are degraded through this pathway including p53, p73, BimEL, PGC1 alpha etc10,11,12,13,14.
The pathogenic potential of HIV-1 is due to its rapid replication, spread and successful neutralization of host restriction factors which is mediated by its regulatory and accessory proteins15. HIV-1 Tat increases viral replication, by binding with TAR region in viral RNA and enhancing its transcription16. Tat is a small (86ĻC101 amino acids) and intrinsically unfolded protein which helps Tat to interact with multiple cellular proteins influencing multiple cellular pathways17,18.
Curcumin has long been known to possess anti HIV activity due to its effect on the HIV-1 protease, integrase and LTR19,20,21,22. Curcumin is an inhibitor of protease, integrase and it also inhibits NF-ĶĘB pathway which is important for HIV-1 gene expression20,21,22. The p53 tumor suppressor protein that possesses intrinsically unfolded regions is also degraded through ubiquitin independent 20S proteasomal pathway by curcumin4,11. Given the fact that curcumin is an activator of ubiquitin independent protein degradation pathway and Tat is an intrinsically unfolded protein18, in this report we have investigated whether curcumin also degrades HIV-1 Tat. Our results show that curcumin increases the rate of Tat protein degradation and the degradation process is carried out by the proteasomal pathway. The effect of curcumin mediated Tat degradation is also reflected in its functions namely the HIV-1 LTR promoter transactivation and virion production. Furthermore, curcumin treatment of chronically infected HIV-1 cells also results in potent inhibition of virus production.Curcumin inhibits HIV-1 by promoting Tat protein degradation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4901322/ĄĄ
Sci Rep. 2016; 6: 27539.
Published online 2016 Jun 10. doi: 10.1038/srep27539
Curcumin inhibits HIV-1 by promoting Tat protein degradation
Amjad Ali1 and Akhil C. Banerjeaa,1
Author information Article notes Copyright and License information Disclaimer
1Laboratory of Virology, National Institute of Immunology, New Delhi, India
aEmail: ni.ser.iin@lihka
Abstract
HIV-1 Tat is an intrinsically unfolded protein playing a pivotal role in viral replication by associating with TAR region of viral LTR. Unfolded proteins are degraded by 20S proteasome in an ubiquitin independent manner. Curcumin is known to activate 20S proteasome and promotes the degradation of intrinsically unfolded p53 tumor suppressor protein. Since HIV-1 Tat protein is largerly unfolded, we hypothesized that Tat may also be targeted through this pathway. Curcumin treated Tat transfected HEK-293T cells showed a dose and time dependent degradation of Tat protein. Contrary to this HIV-1 Gag which is a properly folded protein, remained unaffected with curcumin. Semi-quantitative RT-PCR analysis showed that curcumin treatment did not affect Tat gene transcription. Curcumin increased the rate of Tat protein degradation as shown by cycloheximide (CHX) chase assay. Degradation of the Tat protein is accomplished through proteasomal pathway as proteasomal inhibitor MG132 blocked Tat degradation. Curcumin also decreased Tat mediated LTR promoter transactivation and inhibited virus production from HIV-1 infected cells. Taken together our study reveals a novel observation that curcumin causes potent degradation of Tat which may be one of the major mechanisms behind its anti HIV activity.
The majority of cellular protein degradation occurs through the two pathways, the lysosomal and the proteasomal pathway1,2. Proteasomal pathway is a specific and controlled process in which the substrate protein is tagged with ubiquitin and degraded by 26S proteasome complex3. Proteins are also degraded through the 20S proteasomes where the degradation is independent of the ubiquitination process4,5. Partially or completely unstructured proteins as well as oxidized proteins are degraded through this pathway6,7. The unstructured proteins are normally stabilized in the cell by associating with other proteins or protein complexes8. The unstructured proteins are also protected by NADH bound NAD(P)H:quinone oxidoreductase 1 (NQO1) protein present on the 20S proteasome4,8. Competitive inhibitors of NADH, like dicoumarol and curcumin release NADH from NQO1 rendering the 20S proteasome active for the degradation of unstructured proteins9,10,11. Many of the cellular proteins which are either partially or completely unfolded in their native condition, are degraded through this pathway including p53, p73, BimEL, PGC1 alpha etc10,11,12,13,14.
The pathogenic potential of HIV-1 is due to its rapid replication, spread and successful neutralization of host restriction factors which is mediated by its regulatory and accessory proteins15. HIV-1 Tat increases viral replication, by binding with TAR region in viral RNA and enhancing its transcription16. Tat is a small (86ĻC101 amino acids) and intrinsically unfolded protein which helps Tat to interact with multiple cellular proteins influencing multiple cellular pathways17,18.
Curcumin has long been known to possess anti HIV activity due to its effect on the HIV-1 protease, integrase and LTR19,20,21,22. Curcumin is an inhibitor of protease, integrase and it also inhibits NF-ĶĘB pathway which is important for HIV-1 gene expression20,21,22. The p53 tumor suppressor protein that possesses intrinsically unfolded regions is also degraded through ubiquitin independent 20S proteasomal pathway by curcumin4,11. Given the fact that curcumin is an activator of ubiquitin independent protein degradation pathway and Tat is an intrinsically unfolded protein18, in this report we have investigated whether curcumin also degrades HIV-1 Tat. Our results show that curcumin increases the rate of Tat protein degradation and the degradation process is carried out by the proteasomal pathway. The effect of curcumin mediated Tat degradation is also reflected in its functions namely the HIV-1 LTR promoter transactivation and virion production. Furthermore, curcumin treatment of chronically infected HIV-1 cells also results in potent inhibition of virus production.Discussion
HIV-1 Tat is a structurally unfolded protein and gains its structure when associates with its partner cellular proteins18,34. In the cellular environment Tat protein must be present both in free form and in association with its partner proteins. The unassociated free Tat protein is unstructured and prone to degradation through ubiquitin independent 20S proteasomal pathway. NQO1 inhibits the degradation of Tat protein whereas dicoumarol which is an inhibitor of NQO1 enzymatic activity promotes its degradation24. NAD(P)H bound NQO1 protects many intrinsically unstructured proteins from degradation through 20S proteasomal pathway9,10,11,12. Dicoumarol and curcumin inhibit the NQO1 enzymatic activity and also displace NAD(P)H from NQO1 promoting the degradation of various proteins including p5311. Curcumin promotes the degradation of various proteins including cyclin D1, p185ErbB2, c-Jun, C/EBPĶÁ,, C/EBPĶÂ and PGC1ĶÁ, however the mechanism of degradation has not been worked out11,35. Besides it is clearly established that p53 is degraded through ubiquitin independent mechanism11. Curcumin also inhibits the proteasomal enzymes as well as deubiquitinating enzymes hence promote the accumulation of ubiquitinated proteins in general36. Thus general inhibition of 20S proteasomal pathway fails to inhibit the degradation of proteins many of which have intrinsically unstructured regions. These reports thus suggest that curcumin might be working separately as an inhibitor of proteasomal enzymes that leads to accumulation of ubiquitinated proteins, as well as inhibitor of the NAD(P)H-NQO1 interaction that leads to the degradation of unstructured proteins like p53, cyclin D1, p185ErbB2, c-Jun, C/EBPĶÁ,, C/EBPĶÂ and HIV-1 Tat.
Our studies have clearly shown that curcumin promotes the degradation of Tat protein but leaving its mRNA level unaffected. The CHX chase assay and inhibition of degradation by MG132 clearly suggests the degradation of Tat protein is independent of the mRNA transcription which was further validated by demonstrating that Tat protein supernatant treated cells also showed degradation of Tat by curcumin. The treatment of Pyr-41 or expression of HA-Ubiquitin KO and subsequent treatment with curcumin clearly suggested that the degradation is independent of ubiquitination process. Tat protein gets ubiquitinated with wild type ubiquitin however the ubiquitinated fraction of Tat protein also gets reduced in response to curcumin treatment as like the total Tat protein. Curcumin also inhibits the functional activity of Tat namely HIV-1 LTR promoter transactivation. Tat independent LTR transactivation is also inhibited by curcumin albeit at a low rate since curcumin is an inhibitor of NF-ĶĘB transcription activity21. Curcumin treatment resulted in a decrease of p24 protein level both by western blotting and ELISA in both pNL4-3 transfected cells and chronically infected J1.1 cells showed the adverse functional effect of Tat degradation. The decrease in Tat level as well as p24 protein level is reversible as removal of curcumin led to reappearance of Tat protein as well as p24 protein. Thus the degradation of Tat is not due to the death of treated cells as there is no PARP cleavage after 8 hrs of treatment and PARP was cleaved when treatment was performed for extended time periods (30 hrs.). Curcumin has already been reported to inhibit the HIV-1 replication through a variety of mechanisms namely by the inhibition of its protease activity or by inhibiting the NF-ĶĘB mediated LTR transactivation19,20,21,22. Using multiple experimental approaches we have shown that curcumin rapidly induces specific degradation of Tat and this may explain why curcumin is a potent inhibitor of HIV-1 replication. In summary, our study establishes that curcumin mediated rapid degradation of HIV-1 Tat protein must be the major mechanism contributing to its potent anti-HIV-1 activity.
ĄĄCurcumin inhibits HIV-1 by promoting Tat protein degradation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4901322/ĄĄ
ĄĄ
ĄĄ
ĄĄ
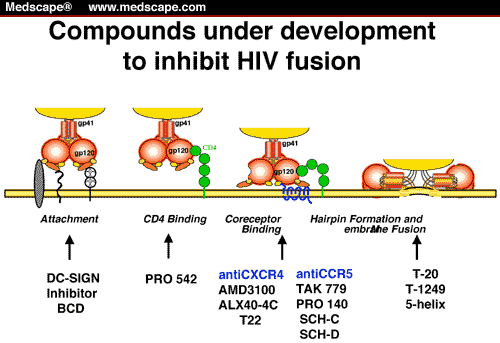



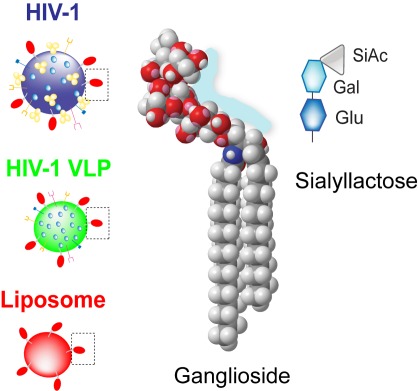
-600.jpg)