Arthritis Res Ther. 2007; 9(Suppl 2): S2.
How does endothelial cell injury start? The role of endothelin in systemic sclerosis
David Abrahamcorresponding author1 and Oliver Distler2
Author information Article notes Copyright and License information Disclaimer
1Department of Medicine, Centre for Rheumatology and Connective Tissue Diseases, Royal Free Hospital and University College, Rowland Hill Street, London, NW3 2PF, UK
2Center of Experimental Rheumatology and Department of Rheumatology, University Hospital Zürich, Gloriastrasse, CH-8032 Zürich, Switzerland
Abstract
A considerable amount of research time has been invested in studies aimed at elucidating pathogenic processes in systemic sclerosis (SSc). Despite this, major challenges for biomedical science remain, such as identification of the key factors that determine susceptibility to SSc, and elucidation of the precise nature of the initiating event that causes endothelial cell injury and ultimately brings about the biological cascade(s) that lead to the pathologic vascular changes. Involved factors are likely to include genetic perturbations, environmental cues, tissue injury, infection and hypoxia/oxidative stress. As important as determining the initiating events are the identification and characterization of key factors that are functionally important in driving vascular disease progression, because these factors are potential targets for therapeutic intervention. This article reviews the role of endothelin as an example of a pleiotropic mediator with effects on various aspects of SSc pathogenesis, such as inflammation, vasculopathy and tissue remodelling.
Introduction
In order to understand the initiators of endothelial injury and how the ensuing vascular dysfunction contributes to the development of systemic sclerosis (SSc), it is necessary to consider the normal biology of the endothelium and of the myriad of biological molecules and biological functions under its control, and to assess which specific processes are dysregulated in disease. In SSc, the vasculopathy is one of the earliest pathological events, characterized by endothelial cell activation and altered vascular tone. These pathological changes are accompanied by the presence of pro-inflammatory cytokines and angiogenic regulatory factors, and the loss of redox control, leading to oxidative stress and hypoxia. This complex array of molecular interactions involves a number of cell types including the endothelial cells and their attendant perivascular supporting cells (pericytes and vascular smooth muscle cells [vSMCs]), and inflammatory cells, and they are profoundly influenced by the presence of growth factors, cytokines, chemokines and potent vasoactive factors. Together, this diverse range of factors is believed to initiate and drive the vascular pathogenesis that leads to severe vessel disease and occlusion. Here we focus on one particularly attractive candidate, endothelin, and critically examine the cellular and molecular activities mediated by endothelin and its receptors that are intimately associated with endothelial cell injury and vascular dysfunction in SSc.
The endothelium
The vascular endothelium is a complex, highly specialized and metabolically active organ, which performs a number of essential biological functions. The endothelium provides a compatible interface to facilitate blood circulation, it inherently inhibits excessive platelet aggregation and leucocyte adhesion, and it produces a balance of vasoconstrictive and vasodilatory molecules that coordinate vascular tone and serve to inhibit extracellular matrix (ECM) deposition and prevent smooth muscle cell proliferation. The endothelium also serves as a multifunctional interface between blood and all internal organs, selectively determining the movement of macromolecules and governing the recruitment of circulating cells from the blood into the extravascular tissues. The prominent endocrine functions of the endothelium work to regulate vascular tone, through the production of vasoconstrictive (for instance, endothelin [ET]-1) and vasodilatory (for example, nitric oxide and prostaglandins) molecules; to maintain blood fluidity; to regulate platelet function; and to control inflammation. Healthy functioning of the endothelium is critical for remodelling of blood vessels, through angiogenesis and vasculogenesis, during times of tissue growth and repair. Thus, the endothelial cell phenotype must be regulated, and this is achieved by environmental cues and mechanical forces such as shear stress, as well as inflammatory and angiogenic stimuli [1].
Triggers of endothelial cell injury and dysfunction
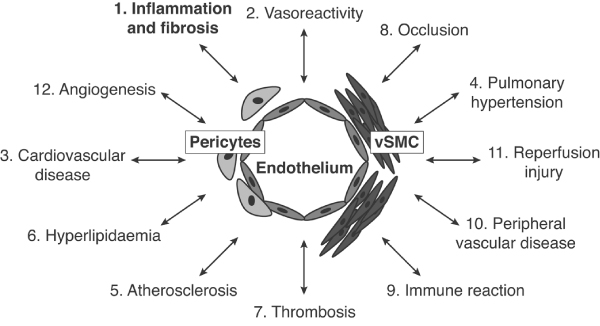
Endothelial dysfunction is an important component of a number of common human diseases, including those characterized by inflammation and fibrosis (Figure (Figure1).1). Our particular interest is in the contribution of endothelial dysfunction to the pathophysiology of inflammatory and fibrotic connective tissue diseases and, in particular, SSc. Endothelial dysfunction is likely to result from endothelial cell injury triggered via a number of different mechanisms, including the following [2]: bacterial or viral infection; oxidative stress through abnormal regulation of reactive oxygen species, hypoxia, turbulent blood flow and shear stress; environmental irritants such as tobacco; and hyperlipidaemia. These factors all lead to the generation of an inflammatory process and endothelial cell activation.
Figure 1
The myriad of potential consequences of endothelial dysfunction. Many human diseases, and in particular some of the most common diseases, are characterized by abnormal changes that take place within the blood vessel and alter vascular responses and function. vSMC, vascular smooth muscle cells.
The endothelial response to injury can be divided into two 'levels' of response: an initial rapid response and a slower, phenotypic response. The initial rapid response involves (among other factors) changes in levels of nitric oxide, prostaglandins, ET-1, von Willebrand factor and tissue plasminogen activator. The slower response depends on fundamental changes in cell surface characteristics, and alterations in the underlying basement membrane and the smooth muscle cells that surround the endothelium. These changes are brought about by molecules that are particularly potent growth factors involved in the deposition of ECM, and activation and proliferation of the vSMCs, pericytes and other mesenchymal cell types associated with the blood vessel wall. This ultimately results in vessel remodelling, with profound changes in cellular architecture [3].
Pericytes are associated with microvessels and capillaries (about 30 μm diameter) and smooth muscle cells are associated with intermediate (> 50 μm) and large (1 to 5 mm) vessels and muscular arteries [4]. In terms of endothelial cells and blood vessels, one must consider these as functional units, which also involve both pericytes and vSMCs. In SSc there are a number of important endothelium-derived mediators that have been shown to be important in the early disease process. It is important to determine which of these link vascular damage to the ensuing fibrogenic or pathogenic tissue remodelling process and the characteristic signs and symptoms of SSc.
Pathogenesis of systemic sclerosis
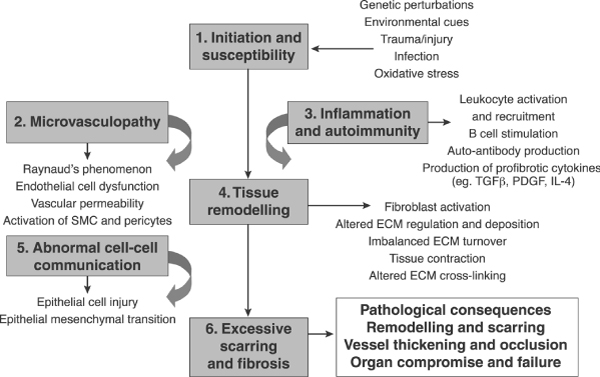
Over the past two decades considerable research effort has been directed toward investigating and elucidating the pathogenic processes of connective tissue diseases such as SSc. Factors that determine susceptibility to these diseases, and the precise nature of the initiating event, remain largely unknown. However, they are likely to involve host genetics, the impact of environmental cues, tissue injury, infection, hypoxia and inability to control oxidative stress adequately [5] (Figure (Figure22).
Figure 2
Sequence of pathological events leading to the development of SSc. ECM, extracellular matrix; IL, interleukin; PDGF, platelet-derived growth factor; SMC, smooth muscle cell; SSc, systemic sclerosis; TGF, transforming growth factor.
Patients with SSc often exhibit early signs of vasculopathy, with many experiencing Raynaud's phenomenon, often for many years before developing overt signs of skin fibrosis. Consistent with this, morphological changes in capillaries are detectable before or at disease onset, which can be used for early diagnosis using nail-fold capillaroscopy. Changes in the blood vessels of the SSc patient are frequently associated with stimulation of both the innate and adaptive immune responses, resulting in B-cell and T-cell activation and, in many cases, autoantibody production. Inflammation is believed to be a primary driver that leads to tissue remodelling and gives rise to the characteristic features of excess matrix production, caused by an imbalance in turnover and increased deposition of various extracellular proteins [6]. The impact of this inflammation probably depends on the organs involved, with evidence for a stronger association of inflammation and fibrosis, for example, in the lungs, whereas significant inflammatory infiltrates in the skin are limited to perivascular areas in early-stage disease [7]. However, the expression of profibrotic mediators is not limited to inflammatory cells in the skin, and resident cells, such as dermal fibroblasts, express increased levels of these mediators in later stage disease [8].
These changes result in a sequence of pathological events that includes impaired cell-cell communication between epithelial cells and fibroblasts, and development of fibrotic lesions that disrupt the normal tissue architecture and lead to impaired expression of the correct molecular cues and cellular mediators that maintain normal tissue structure and function. Ultimately, these abnormalities lead to vascular and interstitial fibrogenic processes. This so-called 'replacement fibrosis' replaces normal tissue architecture and often leads to organ compromise and failure [9].
Because early disease is initiated in the vasculature, and many of the manifestations of SSc are vascular in nature (capillary abnormalities, vessel occlusion and fibrosis, and digital ulceration), identification of the components of the pathway from the initial vascular insult to the downstream fibrotic phenotype is critical. However, characterization of the early vascular events is challenging in SSc, because patients are usually available only in later stages of disease when the disease is already established and because animal models only incompletely reflect the human condition [10,11]. Regardless of the precise mechanisms that underlie the initial vascular injury, it might be even more important to identify and characterize the array of mediators driving the vascular remodelling that leads to the vascular features typical of SSc. Those mediators that are over-produced (beyond normal levels of production) by endothelial cells of SSc patients include growth factors, cytokines, and other mediators of hyperplasia and hypertrophy. These include ET-1, connective tissue growth factor and transforming growth factor (TGF)-β, and matrix-modulating proteins such as matrix metalloproteinases (MMPs) and natural tissue inhibitors of metalloproteinases (TIMPs) [3,12]. They are also important regulators of the activation and function of inflammatory mediators, including tumour necrosis factor (TNF), interleukins and some chemokines. These mediators in turn have a significant impact on the progression of connective tissue diseases.How does endothelial cell injury start? The role of endothelin in systemic sclerosis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2072886/