Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk
Lydia E. Wroblewski1*, Richard M. Peek Jr.1,2,3 and Keith T. Wilson1,2,3
1Division of Gastroenterology, Department of Medicine
2Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, Tennessee 37232
3Department of Veterans Affairs Medical Center, Nashville, Tennessee 37212
SUMMARY
Summary: Helicobacter pylori is a gastric pathogen that colonizes approximately 50% of the world's population. Infection with H. pylori causes chronic inflammation and significantly increases the risk of developing duodenal and gastric ulcer disease and gastric cancer. Infection with H. pylori is the strongest known risk factor for gastric cancer, which is the second leading cause of cancer-related deaths worldwide. Once H. pylori colonizes the gastric environment, it persists for the lifetime of the host, suggesting that the host immune response is ineffective in clearing this bacterium. In this review, we discuss the host immune response and examine other host factors that increase the pathogenic potential of this bacterium, including host polymorphisms, alterations to the apical-junctional complex, and the effects of environmental factors. In addition to host effects and responses, H. pylori strains are genetically diverse. We discuss the main virulence determinants in H. pylori strains and the correlation between these and the diverse clinical outcomes following H. pylori infection. Since H. pylori inhibits the gastric epithelium of half of the world, it is crucial that we continue to gain understanding of host and microbial factors that increase the risk of developing more severe clinical outcomes....
Macrophages as effector cells.Macrophages constitute a potentially powerful line of defense against H. pylori through their own effector function, yet, intriguingly, these capabilities fail the host. One such pathway is the generation of nitric oxide (NO) derived from the enzyme inducible NO synthase (iNOS or NOS2), which has been shown to be upregulated by H. pylori in macrophages in vitro (42, 117, 118, 339) and in vivo (103, 192) (Fig. 3). Coculture studies demonstrate that H. pylori organisms can be eliminated by macrophages even when the bacteria are physically separated from these cells and that this antimicrobial defense is NO dependent (42, 117). The arginase enzyme possessed by H. pylori, encoded by the rocF gene, can compete sufficiently with macrophages for the iNOS substrate l-arginine (l-Arg) that host NO production is impaired, leading to enhanced survival of the bacterium through this mechanism (117). Moreover, this competition can deplete l-Arg sufficiently to impair the synthesis of iNOS protein, since its translation is highly dependent on l-Arg availability inside the macrophage (46). Bacterial arginase serves to generate urea from l-Arg, which is then utilized by urease to synthesize ammonia that is required to neutralize gastric acid.
Pathways involved in regulation of macrophage iNOS synthesis and NO production in response to H. pylori. The translation of iNOS protein depends on the availability of l-arginine (l-Arg). Pathogenic mechanisms that inhibit l-Arg availability for iNOS include (i) the consumption of extracellular l-Arg by H. pylori itself, through its bacterial arginase activity; (ii) the upregulation of macrophage arginase II, which depletes intracellular l-Arg; and (iii) induction of ODC that generates the polyamine spermine, which blocks uptake of l-Arg into macrophages by CAT2. The resulting effect is limitation of iNOS protein synthesis and NO production, despite high levels of iNOS mRNA. Arginase and ODC are novel targets for therapeutic intervention to enhance antimicrobial NO production and hence reduce persistent colonization that leads to chronic inflammation and cancer risk.
FIG. 3. | Clinical Microbiology Reviews
https://cmr.asm.org/content/23/4/713/F3
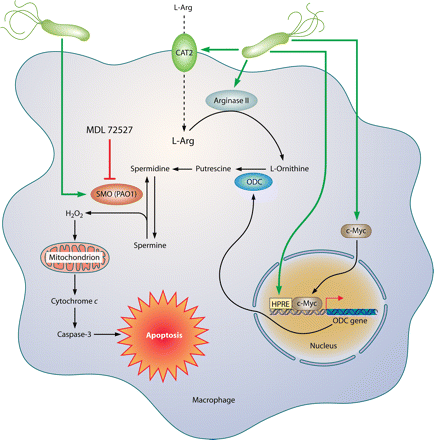
Mechanism of macrophage apoptosis caused by H. pylori. This pathway is dependent on the activities of the enzymes arginase II, ODC, and SMO. Induction of arginase II enhances synthesis of l-ornithine, which is converted into polyamines by ODC via a process that requires both H. pylori activation of the ODC promoter and c-Myc as a transcriptional enhancer. Production of the polyamine spermine provides a substrate for SMO, which is also upregulated by H. pylori. SMO generates H2O2, which causes mitochondrial membrane depolarization, cytochrome c release from mitochondria to the cytosol, and caspase-3 activation, followed by apoptosis. Induction of macrophage apoptosis leads to impairment of mucosal immunity to H. pylori, chronic inflammation, and cancer risk (48, 50, 116).
Relationships between H. pylori, inflammation, and acid secretion. H. pylori infection can reduce acid secretion and increase inflammation via multiple intermediates. Increased production of IL-1β and TNF-α from inflammatory cells inhibits acid secretion from parietal cells. Acid secretion is also inhibited by repression of H+K+ ATPase α-subunit promoter activity, in addition to VacA-induced proteolysis of ezrin.
Chronic H. pylori infection may result in hypochlorhydria or hyperchlorhydria, depending on the severity and distribution of gastritis. Most patients infected long-term develop pangastritis associated with hypochlorydria, which may progress to gastric ulceration and/or adenocarcinoma. Conversely, antral predominant gastritis occurs in approximately 12% of chronically infected patients and is characterized by hyperchlorhydria, which may lead to duodenal ulcer disease (23).Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk | Clinical Microbiology Reviews
https://cmr.asm.org/content/23/4/713
Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk | Clinical Microbiology Reviews
https://cmr.asm.org/content/23/4/713