ĪĪ
From inflammation to gastric cancer: Role of Helicobacter pylori (Review)
Authors: Xiao-Ying Zhang Pei-Ying Zhang Mourad A.M. Aboul-Soud
View Affiliations
Affiliations: Nanjing University of Chinese Medicine, Information Institute, Nanjing, Jiangsu 221009, P.R. China, Department of Cardiology, Xuzhou Central Hospital, The Affiliated Xuzhou Hospital of Medical College of Southeast University, Xuzhou, Jiangsu 221009, P.R. China,
Chair of Medical and Molecular Genetics Research, Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, King Saud University, Riyadh 11433, Kingdom of Saudi Arabia
Published online on: December 15, 2016
Abstract
Gastric cancer is a multifactorial disease and a leading cause of mortality and the risk factors for this include environmental factors and factors that influence host-pathogen interaction and complex interplay between these factors. Gastric adenocarcinomas are of two types, namely intestinal and diffuse type, and Helicobacter pylori (H. pylori) infection has been suspected of being causally linked to the initiation of chronic active gastritis, which leads to adenocarcinoma of the intestinal type. Even though most individuals with H. pylori infection do not show any clinical symptoms, long‑term infection leads to inflammation of gastric epithelium and approximately 10% of infected patients develop peptic ulcers and 1-3% of patients develop gastric adenocarcinoma. Among the several mechanisms involved in tumorigenesis, CagA and peptidoglycan of H. pylori, which enter the infected gastric epithelial cells play an important role by triggering oncogenic pathways. Inflammation induced by H. pylori in gastric epithelium, which involves the cyclooxygenase-2/prostaglandin E2 pathway and IL-1”┬, is also an important factor that triggers chronic active gastritis and adenocarcinoma. H. pylori infection induced oxidative stress and dysregulated E-cadherin/”┬-catenin/p120 interactions and function also play a critical role in tumorigenesis. Environmental and dietary factors, in particular salt intake, are known to modify the pathogenesis induced by H. pylori. Gastric cancer induced by H. pylori appears to involve several mechanisms, making this mode of tumorigenesis a highly complicated process. Nevertheless, there are many events in this tumorigenesis that remain to be clarified and investigated.
Introduction
Gastric cancer is a leading cause of cancer-related mortality worldwide, with nearly 1 million new cases and approximately 750,000 mortalities annually (1). Gastric cancer is a multifactorial disease and the risk factors for this include environmental factors and factors that influence host-pathogen interaction and complex interplay between these factors. Gastric cancer occurrence is more predominant in developing countries in Eastern Europe, South America, and Asia, accounting for approximately two thirds of all cases globally, with China representing approximately 42% of all new cases (2). Development of gastric cancer likely originates with the onset of chronic active gastritis and follows with atrophic gastritis, intestinal metaplasia, and dysplasia, eventually leading to gastric cancer (3). Besides environmental, diet and genetic factors, gastric cancer is closely associated with Helicobacter pylori (H. pylori) infection (4) and related host gene polymorphisms (5). Gastric adenocarcinomas constitute 90©C95% of gastric cancers and are of two types, intestinal and diffuse type. Although there is no known precursor lesion for the development of diffuse type of gastric cancers, H. pylori infection has been suspected of being causally linked to the initiation of chronic active gastritis, which leads to adenocarcinoma (6). Infection of H. pylori is one of the thoroughly studied risk factors of gastric cancer.
After its identification in 1984, H. pylori was classified as a type I carcinogen and epidemiological studies indicated that H. pylori is the most common etiological agent for cancers that are related to infection (7,8). H. pylori is a gram-negative bacterial pathogen and is colonized in gastric epithelium despite the harsh acidic environment, because of its ability to conduct urease-mediated breakdown of urea to ammonia to release ammonia and neutralize its surrounding environment (9). Even though most individuals with H. pylori infection do not exhibit any clinical symptoms, long-term infection potentially leads to inflammation of gastric epithelium and approximately 10% of infected patients develop peptic ulcers and 1©C3% subjects develop gastric adenocarcinoma (10,11).
In this review, we address the molecular basis by which H. pylori acts as a carcinogen, the potential factors that enhance the risk from H. pylori and the accumulating epidemiological evidence for H. pylori infection and its effect on gastric cancer incidence.
Events leading to gastric carcinogenesis following H. pylori infection
H. pylori infection of gastric epithelium leads to the development of intestinal-type adenocarcinoma with the primary event being the transition from normal mucosa to chronic superficial gastritis. Subsequently, atrophic gastritis ensues followed by intestinal metaplasia, leading to dysplasia and adenocarcinoma (Fig. 1) (12). Men are twice as susceptible as women to the intestinal type of gastric adenocarcinoma (13). Notably, the location of infection and formation of gastritis influences the outcomes. Thus, corpus-predominant gastritis leads to gastric cancer, probably because of lower acid secretion, whereas, infection of the gastric antrum, which increases acid production predisposes individuals to duodenal ulcer, actually decreases the risk of gastric cancer (14).
Figure 1.
Interaction between host responses, changes in gastric mucosa and environment during gastric carcinogenesis induced by Helicobacter pylori (H. pylori). A combination of several host responses, bacterial pathogen-mediated events, and environmental factors contribute to the precancerous cascade that culminates in gastric adenocarcinoma.
cag pathogenicity island and CagA
Several virulence factors present in H. pylori that are influenced by its genetic heterogeneity, are critical in the pathogenesis of gastric cancer. CagA, which is present in the DNA insertion element, cag pathogenicity island (cagPAI), was found to be important in carcinogenesis and thus, only H. pylori strains that contain cagPAI element enhance the risk of atrophic gastritis and gastric cancer, even though all strains of this bacterium can cause gastritis (15,16). H. pylori CagA is a 120 to 140-kDa protein, which translocates into host cells following attachment of the bacteria to the cell. Inside the host cell, CagA is phosphorylated by Abl and Src kinases, on tyrosine residue at four distinct glutamate-proline-isoleucine-tyrosine-alanine (EPIYA) motifs present at the C-terminal region of the protein, leading to morphological changes in the cell, including increased cell migration (17,18). The number and phosphorylation status of these EPIYA motifs is a determinant and indicator of risk for gastric cancer (19). Tyr-phospho-CagA activates tyrosine phosphatase (SHP-2) in the host cell, leading to sustained activation of ERK1/2, Crk adaptor, and C-terminal Src kinase (20). Interaction between phosphor-CagA and SHP leads to cell elongation by different mechanisms (21). Even non-phosphorylated CagA has pathogenic effects by causing aberrant activation of ”┬-catenin, disruption of apical-junctional complexes, and a loss of cellular polarity (22). Additionally, non-phosphorylated CagA targets E-cadherin, the hepatocyte growth factor receptor c-Met, phospholipase C-”├, the adaptor protein Grb2, and other components that lead to proinflammatory and mitogenic responses, disruption of cell-cell junctions, and loss of cell polarity (Fig. 1) (23). Preclinical studies confirmed a role for CagA in the pathogenesis of gastric cancer, by demonstrating that transgenic mice expressing CagA show gastric epithelial cell proliferation and carcinoma, in a CagA phosphorylation-dependent manner (24).
Peptidoglycan
Along with CagA, H. pylori peptidoglycan can also be delivered into host cells and peptidoglycan binds with NodI (25), which triggers the NF-”╩B dependent pro-inflammatory pathway and interleukin (IL)-8, an inflammatory cytokine, secretion. Peptidoglycan is also shown to activate the PI3K-Akt pathway leading to cell proliferation, migration and prevention of apoptosis (26).
Other virulence factors present in H. pylori include VacA and outer membrane proteins, which are associated with ulceration as well as gastric cancer (27,28).
Inflammatory response to H. pylori infection
COX-2⁄PGE2 pathway
Inflammation of gastric epithelium is known to be associated with the development of gastric cancer (29). There are several mechanisms by which inflammation may promote cancer development and the induction of the cyclooxygenase-2/prostaglandin E2 (COX-2⁄PGE2) pathway and activation of NF-”╩B and Stat3 appear to be major pathways (Fig. 2) (30). Besides these, innate immune responses through the TLR/MyD88 adapter signaling also play a role in tumorigenesis (31,32). In fact, it has been shown that almost all the gastric tumors show an induction of COX-2 expression (33) and H. pylori infection is known to lead to COX-2 induction (34). Inflammation in combination with oncogenic activation, promotes tumorigenesis and also Wnt signaling activation (Fig. 2) with the accumulation of ”┬-catenin, which facilitate tumor growth and this altered signaling has been observed in over 50% of gastric cancers (35). PGE2 signaling, through the EP4 receptor, is known to induce the expansion of CD133+ CD44+ cancer stem cells in intestinal tumors through the activation of PI3K and MAPK signaling (36), which potentially aggravates tumor growth.
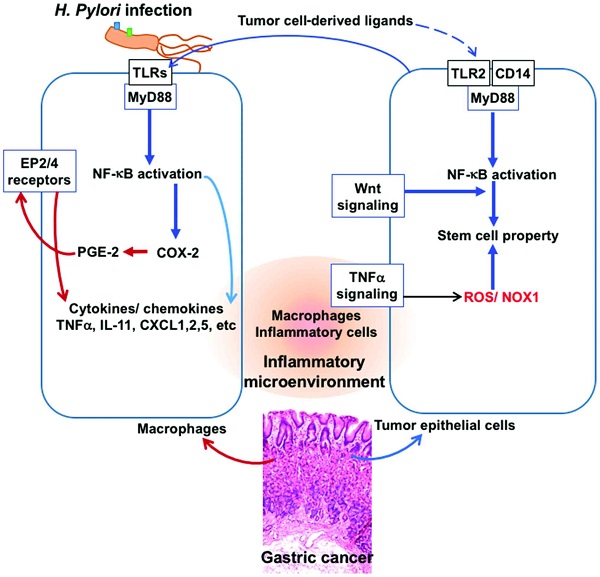
Figure 2.
Tumor inflammatory microenvironment: Interplay of factors derived from H. pylori and tumor cells. A crosstalk between tumor cell-derived inflammatory factors and macrophage-derived factors during infection of Helicobacter pylori (H. pylori) results in aggravation of the inflammatory microenvironment and the tumor cells acquiring stem cell property and progression of tumor in gastric epithelium. Signaling through the TLR/MyD88/NF-”╩B pathways to activate cyclooxygenase-2 (COX-2) and production of prostaglandin E2 (PGE-2), release of cytokines such as TNF-”┴ and production of ROS, in macrophages and in tumor cells facilitate this tumor inflammatory environment.
Infection of H. pylori induces inflammation through CagA injection into host cells followed by the activation of SHP and TLRs leading to chronic active gastritis and eventually gastric cancer. However, the expression pattern of inflammation markers is not always comparable between gastritis and gastric cancer. Thus, IL-8 and IL-11 expression is predominantly induced in gastric cancer, whereas in gastritis mostly TNF-”┴ expression is increased. It has been suggested that once tumor growth starts, tumor cells also contribute to the inflammation of local microenvironment through different pathways, known as Ī«tumor-elicited inflammationĪ», which is different from infection-induced inflammation, thereby resulting in different cytokine profiles from H. pylori infection-induced gastritis (29). Nevertheless, gastritis and gastric cancer demonstrate common increases in inflammatory cytokines CXCL1, CXCL2, CXCL5, CCL3, CCL4, and TLR2 (Fig. 2) (29). Inasmuch as these cytokines are effective in causing immune suppression, the Ī«infection-associatedĪ» and Ī«tumor- elicitedĪ» inflammation appears to promote and accelerate gastric tumorigenesis by activating the COX-2/PGE2 pathway and subsequent induction of tumor-promoting cytokines.
IL-1”┬
Another important cytokine, IL-1”┬ is known to play a role in a variety of cellular activities such as inflammatory response and acid secretion by gastric epithelium (37). Disturbances in the regulation of IL-1”┬ are observed in several cancer types and in particular, in IL-1”┬ gene polymorphisms including IL-1”┬ −31 (T>C) and IL-1”┬ −511 (C>T) which are closely related to gastric cancer (Fig. 1) (38,39). Of note, it has been shown that IL-1”┬-511T polymorphism is present in all the Mozambican subjects with intestinal metaplasia (40). This polymorphism is also associated with the prevalence of dysplasia (41), indicating that the IL-1”┬ T alleles are related to premalignant gastric lesions. Apparently, the same polymorphism of IL-1”┬ is involved in the intestinal type of gastric cancers, which are triggered by H. pylori infection and not diffusive type (42). IL-1”┬ gene polymorphisms also increase the production of IL-1”┬, which suppresses gastric acid secretion, and is related to the grade of gastric atrophy in patients with H. pylori infection (43). Additionally, H. pylori infection leads to elevated secretion of IL-1”┬ and reduction in acid secretion (44). It has been suggested that a combination of IL-1”┬-511T/T polymorphism and H. pylori infection aggravates the development of gastric tumor more than either of these agents alone (45). Thus, infection of H. pylori promotes the expression of IL-1”┬, which leads to gastric carcinogenesis through its actions on both inflammatory and epithelial cells (46). Even though the precise molecular basis of these actions is not clear, it seems that hypochlorhydria and atrophic gastritis induced by IL-1”┬ polymorphisms, which depends on H. pylori infection are critical in gastric cancer development (47).
Oxidative stress induced by H. pylori
A primary factor that is important in the events that lead to the progression of the inflammation-to-carcinoma is oxidative DNA damage induced by H. pylori infection (48), which is probably due to infiltrating neutrophils, and also direct effects of H. pylori (49). Production of reactive oxygen species in the H. pylori-infected gastric epithelium is linked to the presence of cagPAI and contribute to the oxidative stress response in gastric epithelial cells (50). It is well known that H. pylori infection causes elevated level of polyamines, in particular spermine and this is associated with an induction of spermine oxidase (51). Action of spermine oxidase on spermine leads to the production of elevated levels of hydrogen peroxide, which is a powerful oxidizing agent and also contributes to the production of free radicals such as hydroxyl radical (52). Besides, H. pylori also activates macrophages which show a significant upregulation of spermine oxidase, contributing to oxidative stress and damage to the gastric epithelial cells (53). Besides, altered polyamine metabolism and overexpression of arginase enzyme in the infected gastric epithelium leads to lowered NO production and increased production of spermine and hydrogen peroxide.
H. pylori and E-cadherin
E-cadherin, which is an adhesion molecule in epithelial tissues that is important in maintaining proper cellular architecture, is regulated by the binding of p120 to the cadherin juxtamembrane domain (54). Furthermore, the cytoplasmic domain of E-cadherin interacts with ”┬-catenin and p120, which, in turn, interact with the cytoskeletal component actin. It has been documented that there is a loss of E-cadherin function in gastric cancer, and in fact promoter methylation of E-cadherin gene is induced by H. pylori infection, leading to reduction in E-cadherin expression (55). Following H. pylori infection, the translocated CagA in the gastric epithelial cells binds with E-cadherin, resulting in the dissociation of the E-cadherin-”┬-catenin complex and accumulation of ”┬-catenin in cytoplasm and nucleus, where it transactivates ”┬-catenin-dependent genes involved in carcinogenesis (23,56). Along with the downregulation of E-cadherin, a decreased expression or aberrant subcellular localization of p120, from membrane to the cytosol or nucleus, is commonly seen in gastric cancer (57). In the cytoplasm, p120 interacts with Rho GTPases and promotes motility and metastasis (58). Aberrant localization of p120 to the nucleus in gastric epithelia infected with H. pylori has been reported and p120 in nucleus can relieve transcriptional repression of the mmp-7 gene, which is involved in gastric tumorigenesis, leading to its enhanced expression (59).
Environmental factors and H. pylori-mediated gastric carcinogenesis
Gastric adenocarcinoma is strongly influenced by dietary salt intake, with high salt intake aggravating tumorigenesis (60). Epidemiological studies indicated that high salt intake increases the prevalence of H. pylori infection (61) and the incidence of gastric adenocarcinoma in infected patients (62). Experimental studies indicated that a high-salt diet and H. pylori infection exert synergistic effects on the development of premalignant lesions or gastric cancer (63), probably by elevating the production of inflammatory cytokines IL-1, IL-6 and TNF-”┴ (64). However, the precise molecular events that underlie this synergistic effect on cancer development are not known. It has been suggested that high salt increases the expression of CagA, the potential carcinogen in H. pylori (65), which may be the reason for the observed synergy between H. pylori and salt for gastric cancer induction (Fig. 1).
In addition to salt, other factors that influence H. pylori infection-associated gastric cancer includeOn the other hand, cigarette smoking is a potential risk factor for enhancing the tumorigenesis induced by H. pylori infection (21).
Conclusion
Intestinal type gastric adenocarcinomas are known to be causally linked to H. pylori infection, which leads to the initiation of chronic active gastritis, and adenocarcinoma. Even though most individuals with H. pylori infection do not show any clinical symptoms, 1©C3% people with long-term infection develop gastric adenocarcinoma. Of the several mechanisms of the tumorigenesis induced by H. pylori, CagA and peptidoglycan of H. pylori, inflammation, oxidative stress and dysregulated E-cadherin/”┬-catenin/p120 interactions play an important role. Environmental and dietary factors, in particular salt intake and cigarette smoking are known to aggravate H. pylori-induced carcinogenesis. Thus, H. pylori infection appears to invoke multi-thronged mechanisms, to induce gastric adenocarcinoma. Nevertheless, many events in this tumorigenic process remain to be clarified and investigated.From inflammation to gastric cancer: Role of Helicobacter pylori (Review)
https://www.spandidos-publications.com/10.3892/ol.2016.5506ĪĪ