íí
1, Nitric Oxide Inhibits the Replication Cycle of Severe Acute Respiratory Syndrome Coronavirus
2. Endogenous nitric oxide (induced by l-arginine and LPS) inhibits leukotriene B4 release from rat alveolar macrophages
3úČAn Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease-cysteine protease. also malaria parasite cysteine protease. both exogenous and endogenous nitric oxide inhibit viral protease-in Hela cells and macrophage.Since cysteine proteases are critical for virulence or replication of many viruses, bacteria, and parasites, S-nitrosylation of pathogen cysteine proteases may be a general mechanism of antimicrobial host defenses.
íí
J Virol. 2005 Feb; 79(3): 1966ĘC1969.
Nitric Oxide Inhibits the Replication Cycle of Severe Acute Respiratory Syndrome Coronavirus
Sara Åkerström,1 Mehrdad Mousavi-Jazi,2 Jonas Klingström,1,3 Mikael Leijon,2 Åke Lundkvist,1,3 and Ali Mirazimi1,*
Author information Article notes Copyright and License information Disclaimer
Center for Microbiological Preparedness, Swedish Institute for Infectious Disease Control, Solna,1 LightUp Technologies, Huddinge,2 MTC/Karolinska Institutet, Stockholm, Sweden3
ABSTRACT
Nitric oxide (NO) is an important signaling molecule between cells which has been shown to have an inhibitory effect on some virus infections. The purpose of this study was to examine whether NO inhibits the replication cycle of the severe acute respiratory syndrome coronavirus (SARS CoV) in vitro. We found that an organic NO donor, S-nitroso-N-acetylpenicillamine, significantly inhibited the replication cycle of SARS CoV in a concentration-dependent manner. We also show here that NO inhibits viral protein and RNA synthesis. Furthermore, we demonstrate that NO generated by inducible nitric oxide synthase, an enzyme that produces NO, inhibits the SARS CoV replication cycle.Nitric Oxide Inhibits the Replication Cycle of Severe Acute Respiratory Syndrome Coronavirus
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC544093/íí
Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication.
1.Endogenous nitric oxide (induced by l-arginine and LPS) inhibits leukotriene B4 release from rat alveolar macrophages
2úČAn Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease-cysteine protease. also malaria parasite cysteine protease. both exogenous and endogenous nitric oxide inhibit viral protease-in Hela cells and macrophage.Since cysteine proteases are critical for virulence or replication of many viruses, bacteria, and parasites, S-nitrosylation of pathogen cysteine proteases may be a general mechanism of antimicrobial host defenses.
íí
The genomes of many viruses encode a polyprotein that after translation is cleaved into smaller polypeptides by a viral protease. These proteases fall into several categories based on their active site residues, including cysteine proteases, serine proteases, and aspartic proteases (35, 24, 4). NO can inhibit the replication of other viruses that encode cysteine proteases, such as members of the Picornavirus family and the Coronavirus family (45, 56). In contrast, NO does not inhibit the replication of some viruses that encode serine proteases, such as alphaviruses. However, NO also inhibits the replication of some viruses that do not encode cysteine proteases, implying that there exist other viral targets of NO. For example, the EBV transcription factor Zta and ribonucleotide reductase may be targets of NO (46, 49). Nonetheless, to our knowledge there are no viruses that have cysteine proteases that are resistant to NO.
Cysteine proteases are also critical to the replication and virulence of a variety of other micro-organisms.3.viral infection induces expression of NOS2 in Hela cells and macrophages (and monocytes)
4.The Middle East respiratory syndrome coronavirus (MERS-CoV) utilizes host proteases (serine protease) for virus entry into lung cells.a single treatment with camostat(an inhibitor of serine protease ) is sufficient to block MERS-CoV entry into a well-differentiated lung-derived cell line
5.A 23187 was used as stimulus, as rising intracellular Ca2+ activates directly the phospholipase A2 and lipoxygenase pathway. in rat alveolar macrophages, endogenous NO appears to inhibit the release of mediators of the cyclo-oxygenase and lipoxygenase pathway through multiple sites of action.
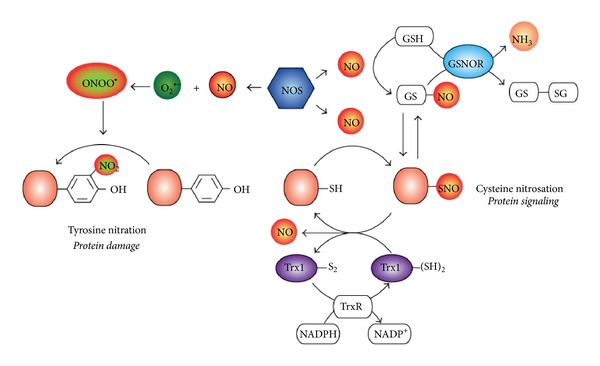
6.Sustained production of NO endows macrophages with cytostatic or cytotoxic activity against viruses, bacteria, fungi, protozoa, helminths, and tumor cells. The antimicrobial and cytotoxic actions of NO are enhanced by other macrophage products such as acid, glutathione, cysteine, hydrogen peroxide, or superoxide.
íí
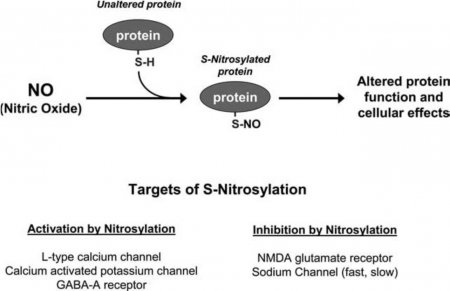
http://aibolita.com/nervous-diseases/113-mechanism-of-action-of-nitric-oxide-s-nitrosylation.html
íí
íí
http://watcut.uwaterloo.ca/webnotes/Pharmacology/noReleasingDrugs.html
íí
http://microbiology.washington.edu/
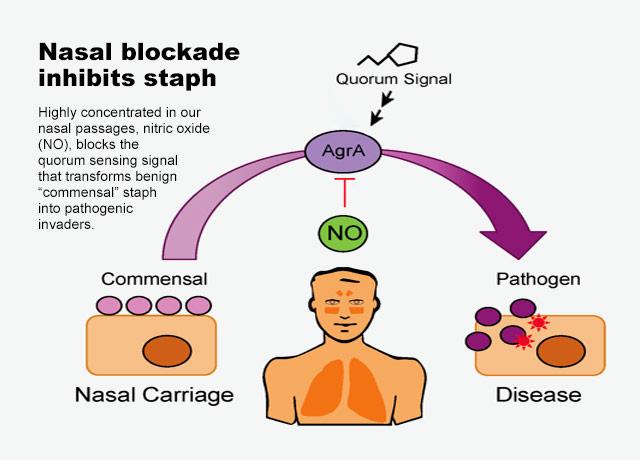
The most common signals detected by Gram-negative and Gram-positive bacteria are acylated homoserine lactones and autoinducing peptides (AIPs), respectively. However, increasing evidence has supported a role for the small molecule nitric oxide (NO) in influencing QS-mediated group behaviors like bioluminescence, biofilm production, and virulence. In this review, we discuss three bacteria that have an established role for NO in influencing bacterial physiology through QS circuits. In two Vibrio species, NO has been shown to affect QS pathways upon coordination of hemoprotein sensors. Further, NO has been demonstrated to serve a protective role against staphylococcal pneumonia through S-nitrosylation of a QS regulator of virulence.
Frontiers | Insights Into Nitric Oxide Modulated Quorum Sensing Pathways | Microbiology
https://www.frontiersin.org/articles/10.3389/fmicb.2019.02174/fullíí
https://www.nature.com/articles/s41598-017-01669-5
íí
íí
íí
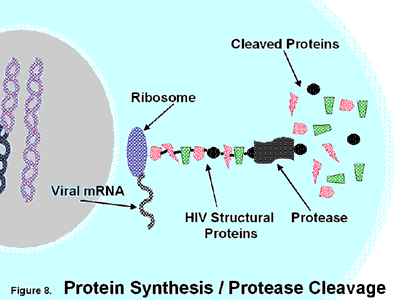
Coxsackievirus, a member of the Picornaviridae, is a nonenveloped virus with a single-stranded RNA genome, whose life cycle includes virion attachment to the host cell, penetration, translation of the viral RNA into a polyprotein, autocleavage of the polyprotein by viral proteases into viral polypeptides, replication of the viral genome, assembly of the virion, and exit from the cell.
íí
Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication.
íí
Abstract
Nitric oxide (NO) has antimicrobial activity against a wide spectrum of infectious pathogens, but an antiviral effect has not been reported.
The impact of NO, from endogenous and exogenous sources, on herpes simplex virus type 1 (HSV 1) replication was studied in vitro.
HSV 1 replication in RAW 264.7 macrophages was reduced 1,806-fold in monolayers induced to make NO by activation with gamma IFN and LPS. A competitive and a noncompetitive inhibitor of nitric oxide synthetase substantially reduced the antiviral effect of activated RAW macrophages.
S-nitroso-L-acetyl penicillamine (SNAP) is a donor of NO and was added to the media of infected monolayers to assess the antiviral properties of NO in the absence of gamma IFN and LPS. A single dose of S-nitroso-L-acetyl penicillamine 3 h after infection inhibited HSV 1 replication in Vero, HEp2, and RAW 264.7 cells in a dose-dependent manner.
Neither virucidal nor cytocidal effects of NO were observed under conditions that inhibited HSV 1 replication. Nitric oxide had inhibitory effects, comparable to that of gamma IFN/LPS, on protein and DNA synthesis as well as on cell replication. This report demonstrates that, among its diverse properties, NO has an antiviral effect.
J Clin Invest. 1993 Jun; 91(6): 2446ĘC2452.
Department of Internal Medicine, University of Cincinnati College of Medicine, OH 45267-0560.
K D Croen
Author information Copyright and License information Disclaimer
This article has been cited by other articles in PMC.
íí
doi: [10.1172/JCI116479]
PMCID: PMC443304
PMID: 8390481
Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC443304/
íí
An Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease
Author links open overlay panelMartaSaura1CarlosZaragoza1AudreyMcMillan1Richard AQuick1ChristineHohenadl2John MLowenstein3Charles JLowenstein1íý
1
Division of Cardiology, Department of Medicine, The Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA
2
GSF-Forschungszentrum fĘ╣r Umwelt und Gesundheit, Institut fĘ╣r Molekulare Virologie, OberschleiĘóheim D85758, Germany
3
Department of Biochemistry, Brandeis University, Waltham, Massachusetts 02254, USA
Received 8 July 1998, Revised 16 November 1998, Available online 11 April 2001.
Abstract
Although nitric oxide (NO) kills or inhibits the replication of a variety of intracellular pathogens, the antimicrobial mechanisms of NO are unknown.Here, we identify a viral protease as a target of NO. The life cycle of many viruses depends upon viral proteases that cleave viral polyproteins into individual polypeptides. NO inactivates the Coxsackievirus protease 3C, an enzyme necessary for the replication of Coxsackievirus. NO S-nitrosylates the cysteine residue in the active site of protease 3C, inhibiting protease activity and interrupting the viral life cycle.
Substituting a serine residue for the active site cysteine renders protease 3C resistant to NO inhibition. Since cysteine proteases are critical for virulence or replication of many viruses, bacteria, and parasites, S-nitrosylation of pathogen cysteine proteases may be a general mechanism of antimicrobial host defenses.
Introduction
Nitric oxide (NO) is an antiviral effector of the immune system (45, 14, 44, 56). Viral infection can induce expression of the inducible nitric oxide synthase (iNOS) in cells and in animals. NO inhibits the replication of a variety of viruses (9, 30, 7, 8, 26, 1, 33, 65). We and others have previously shown that NO inhibits the replication of Coxsackievirus and other Picornaviruses (33, 42, 50, 15, 21, 68, 69). Coxsackievirus B3 (CVB3) infection induces iNOS expression in macrophages in mice, and CVB3 replicates more rapidly to higher titers and causes more tissue damage in mice lacking iNOS, compared to infections of wild-type mice. Exogenous NO inhibits replication of Coxsackievirus and poliovirus in vitro, reducing viral RNA and protein synthesis. Thus, iNOS and NO are important components of the immune response to CVB3 infection. However, the viral targets of NO are unknown.
Results
To explore the mechanism by which NO inhibits CVB3 replication, we first determined that NO inhibits CVB3 replication by blocking a step early in the viral life cycle. We infected HeLa cells with CVB3 and then at various times treated the infected cells with NO donors or control compounds; after 10 hr, we measured the amount of CVB3 produced during one viral life cycle. The NO donor S-nitroso-acetyl-penicillamine (SNAP) reduces the amount of CVB3 produced by infected HeLa cells, and the maximum effect of SNAP occurs when the NO donor is added 1ĘC2 hr after infection (Figure 1). In contrast, the control compound acetyl-penicillamine (AP) has no effect. When SNAP is added to HeLa cells, most of the NO is released within 2 hr (data not shown). Since the life cycle of CVB3 lasts approximately 8 hr, these data suggest that NO inhibits an early step in the life cycle of CVB3.
Download : Download high-res image (105KB)Download : Download full-size image
Figure 1. The NO Donor SNAP Inhibits CVB3 Replication
HeLa cells were infected with CVB3 at a multiplicity of infection of 10. SNAP (200 Ž╠M) was added at various times after infection as indicated. The amount of CVB3 after 10 hr of infection was measured by the plaque assay. To one set of infected cells (C), 200 Ž╠M AP was added 1 hr after infection. To another set of cells (-1), SNAP was added 1 hr before infection. n = 3 í└ SD.
Coxsackievirus, a member of the Picornaviridae, is a nonenveloped virus with a single-stranded RNA genome, whose life cycle includes virion attachment to the host cell, penetration, translation of the viral RNA into a polyprotein, autocleavage of the polyprotein by viral proteases into viral polypeptides, replication of the viral genome, assembly of the virion, and exit from the cell. Since protease activity is a critical step early in the life cycle of Coxsackievirus, we hypothesized that NO inhibits CVB3 proteases.
Exogenous NO Inhibits Protease Processing of an Endogenous Viral Polyprotein
In order to examine the effect of NO upon viral protease processing of viral proteins, we added the NO donor SNAP or its control AP to infected HeLa cells; cell lysates were harvested at various times after infection, fractionated by SDS-PAGE, and analyzed by immunoblotting for viral polypeptides. NO inhibits proteolysis of the viral protein precursor 2ABC, delaying and reducing the amount of the cleavage products 2BC and 2C detected by immunoblotting (Figure 2A). Since 3Cpro is responsible for cleavage of 2ABC, one interpretation of these data is that exogenous NO inhibits the proteolysis of the viral polypeptide 2BC.
Download : Download high-res image (259KB)Download : Download full-size image
Figure 2. Figure 2. NO Inhibits Proteolysis of CVB3 Polypeptide 2BC in Infected Cells
(A) Exogenous NO donor SNAP inhibits proteolysis of CVB3 polypeptide. HeLa cells were infected with CVB3 at a multiplicity of infection of 10. After 1 hr, 200 Ž╠M of SNAP (+) or AP (-) was added, and after 3.5 hr guanidine was added to inhibit new viral RNA synthesis. HeLa cells were harvested at various times indicated after guanidine treatment. Cell lysates were fractionated by SDS-PAGE and immunoblotted with an antibody to CVB3 polypeptide 2C. These experiments were repeated twice with similar results.
(B) Endogenous NO from activated macrophages inhibits proteolysis of CVB3 polypeptide. HeLa cells were infected with CVB3 at a multiplicity of infection of 10. After 1 hr, plastic inserts containing resting (R) or LPS/IFN-stimulated (S) macrophages were added to the wells containing infected HeLa cells. Some cultures were also treated with the NOS inhibitor NAME (0ĘC10 mM). Cells were harvested 5 hr after infection and cell lysates analyzed for CVB3 polypeptide 2C as above. This experiment was repeated five times with similar results.
(C) LPS or IFN do not directly affect CVB3 replication. HeLa cells were infected with CVB3 at a multiplicity of infection of 10. After 1 hr, LPS or IFN or media alone was added to the wells containing infected HeLa cells. Some cultures were also treated with the NOS inhibitor NAME (0ĘC10 mM). Cells were harvested 5 hr after infection and cell lysates analyzed for CVB3 polypeptide 2C as above.
Endogenous NO Inhibits Protease Processing of an Endogenous Viral Polyprotein
To answer the question of whether or not endogenous NO produced by iNOS would also interfere with viral proteolytic processing, we added macrophages that had been induced to express iNOS to infected cells. First, HeLa cells were infected with CVB3, and then cell culture inserts were added that contained RAW 264.7 macrophages, activated or not with lipopolysaccharide and interferon-Ž├. The macrophages were separated from the infected HeLa cells by a 0.4 Ž╠M pore size membrane, which would allow the passage of NO from macrophages to infected cells. The NOS inhibitor nitro-arginine-methyl-ester (NAME) was added to some cocultures of infected HeLa cells and activated macrophages, in order to confirm that any antiviral effect of activated macrophages was a result of NO, and not some other mechanism. Cells were harvested 5 hr after infection, and the cleavage of the viral polyprotein 2ABC was measured by immunoblotting as above.
Activated macrophages block proteolytic processing of the CVB3 polyprotein 2ABC, resulting in a decrease in the amount of 2BC and 2C (Figure 2B). Infected HeLa cells alone (lane 2) contain three prominent polypeptides recognized by the antibody to 2C (as well as another band also found in noninfected cells [lane 1]). These three polypeptides are 2ABC and its two proteolytic products 2BC and 2C. Resting macrophages (lane 3) do not affect the intensity of the three viral polypeptides. However, macrophages activated by lipopolysaccharide (LPS) and interferon-Ž├ (IFNŽ├) (lane 4) release a soluble mediator that diffuses across the 4 Ž╠M pores and inhibits the processing of 2ABC into 2BC and 2C. This soluble mediator may be NO, since increasing concentrations of the NOS inhibitor NAME are associated with decreasing amounts of NO and also with increasing proteolytic processing of the viral polypeptide (lanes 5ĘC7). (LPS or IFN alone have no effect upon viral replication in HeLa cells [Figure 2C].) Thus, NO synthesized by activated macrophages can diffuse into infected cells and inhibit the viral proteases that process the viral polyproteins.
Since exogenous and endogenous NO inhibit proteolytic processing of Coxsackievirus polyproteins, we therefore focused our attention on the effect of NO upon Coxsackievirus proteases.
NO Inhibits CVB3 Protease Activity
Picornaviruses encode two cysteine proteases, 2Apro and 3Cpro (35, 24, 55, 11). The protease 2Apro cleaves the viral polyprotein at the boundary between the structural proteins and the nonstructural proteins, VP1 and 2A, producing two polyproteins. The protease 3Cpro is thought to cleave the resulting polyproteins into ten viral polypeptides, including the cleavage of 2BC into 2B and 2C (27, 22, 51). The protease 3Cpro is a critical component of the Picornavirus life cycle: mutation of any of the three amino acids in the active site of 3Cpro inactivates the protease and abolishes viral replication (27, 22, 51). One of the residues of the catalytic site of 3Cpro is cysteine at amino acid position 147, and since NO can nitrosylate cysteine residues (61, 62, 60), we hypothesized that NO inhibits the CVB3 3Cpro.
We therefore prepared pure 3Cpro and various protease substrates. We subcloned the cDNA encoding 3Cpro into a bacterial expression vector, expressed a fusion protein consisting of 3Cpro tagged with His6 in bacteria, and then purified 3Cpro (Figure 3A) (Ghosh and Lowenstein 1996). To prepare an authentic protease substrate, we subcloned, expressed, and purified a fusion polypeptide, consisting of glutathione-S-transferase, the entire CVB3 3B polypeptide, and the amino acid residues 1ĘC140 of 3C; we refer to this fusion protein as GST-3B-3CT. This fusion protein contains an authentic 3Cpro cleavage site between 3B and 3C, consisting of the 3Cpro recognition motif AXXQG (where Q is the P1 residue) (35, 24, 38, 55). (The fragment of 3C included in the fusion protein consists of the first 140 amino acids of 3C. It lacks the Cys147 of the active site and is thus incapable of protease activity.)
Download : Download high-res image (200KB)Download : Download full-size image
Figure 3. NO Inhibits Cleavage of a Viral Target by Purified 3Cpro
(A) 3Cpro was expressed in bacteria and purified. Bacterial lysates were subjected to SDS-PAGE: lane 1, noninduced bacteria; lane 2, bacteria induced with IPTG; lane 3, high-speed supernatant of induced bacteria; lane 4, flow-through from a Ni+ column; lanes 5ĘC9, sequential column washes with 10 mM imidazole; and lanes 10ĘC13, fractions from a Ni+ column eluted with 500 mM imidazole.
(B) The purified fusion protein GST-3B-3CT, which contains an authentic 3Cpro cleavage site, was incubated with buffer, purified 3Cpro, or 3Cpro with the control compound AP, or the NO donor SNAP, or SNAP and DTT. The reaction mixture was analyzed by immunoblot with an antibody to GST and quantitated by densitometry. n = 3 í└ SD, and * p < 0.05.
(C) The cleavage experiment was then repeated using spermine NONOate (SP-NO) as an NO donor or its control compound spermine (SP). n = 3 í└ SD, and * p < 0.05.
To test the activity of recombinant protease, purified 3Cpro was incubated with the protease substrate GST-3B-3CT and the reaction mixture analyzed by immunoblotting with an antibody to GST. Purified 3Cpro is capable of cleaving the polyprotein GST-3B-3CT, as detected by immunoblotting (Figure 3B, immunoblot lanes 1 and 2).
In order to measure the susceptibility of 3Cpro to NO, we added a variety of NO donors or their controls to 3Cpro and a protease substrate. The NO donor SNAP inhibits the proteolytic activity of 3Cpro in a dose-dependent manner, while its control acetyl-penicillamine (AP), which has no nitroso group, has no effect (Figure 3B). DTT, an agent capable of reducing the nitrosylation of cysteine residues, reverses the NO inhibition of 3Cpro. Another NO donor, spermine NONOate, also inhibits 3Cpro cleavage of GST-3B-3CT. DTT also reverses this effect (Figure 3C).
To confirm these results, we used luciferase as another protease substrate (25, 19, 64). Firefly luciferase serves as a 3Cpro substrate because it contains the 3Cpro motif AXXQG. Cleavage of luciferase was measured by monitoring luciferase activity. 3Cpro cleaves luciferase in a dose- and time-dependent manner (Figure 4A and Figure 4B). The NO donors SNAP or spermine NONOate but not their corresponding control drugs inhibit 3Cpro activity, and these inhibitions are reversed by DTT (Figure 4C). It was previously shown that the nonphysiological reagents iodoacetamide and N-ethylmaleimide can inhibit 3Cpro (19, 54). Thus, NO inhibits the activity of 3Cpro.
Download : Download high-res image (123KB)Download : Download full-size image
Figure 4. NO Inhibits 3Cpro Cleavage of Luciferase
(A) Purified 3Cpro was added to luciferase, which contains a 3Cpro consensus cleavage motif, and the activity of luciferase remaining at various times was assayed using a luminometer. n = 3 í└ SD.
(B) Increasing amounts of 3Cpro were added to luciferase for 3 hr, and the amount of luciferase assayed as above. n = 3 í└ SD.
(C) Luciferase was incubated alone, or in the presence of 3Cpro with the following additions: the control compound AP, the NO donor SNAP, SNAP and DTT, the control compound spermine (SP), the NO donor spermine NONOate (SP-NO), or spermine NONOate and DTT. n = 3 í└ SD.
NO Nitrosylates the CVB3 Protease Active Site Cysteine
3Cpro has only one cysteine, namely Cys147 in the active site. To determine whether or not NO nitrosylates this cysteine, we first measured the effect of NO donors upon its thiol group with the Ellman assay (13, 70). Purified 3Cpro was incubated with NO donors for 1 hr and then dialyzed. 5,5íń- Dithiobis (2-nitrobenzoic acid) was added, the increase of absorbance at 412 nm was measured, and the concentration of thiols was calculated from the molar extinction coefficient of the nitrothiobenzoate ion. NO donors but not their corresponding control compounds eliminate the sulfhydryl group from purified 3Cpro over time (Figure 5A).
Download : Download high-res image (158KB)Download : Download full-size image
Figure 5. NO Eliminates a Sulfhydryl Group from 3Cpro and Nitrosylates 3Cpro
Purified 3Cpro was incubated with AP, SNAP, spermine, or spermine NONOate. (A) Analysis of the 3Cpro sulfhydryl group. Treated 3Cpro was incubated with an excess of 5,5 dithiobis(2-nitrobenzoate) (DNTB) and its absorption measured at 412 nm. n = 4 í└ SD. (B) Nitrosylation of the 3Cpro active site Cys147. 3Cpro was exposed to NO donors or their controls, then incubated with HgCl2, and the amount of nitrite released was measured by the Griess reaction. n = 4 í└ SD.
We next measured the effect of NO donors upon the formation of a nitroso-thiol group by the Saville assay (58, 70). Purified 3Cpro was incubated with various NO donors or their controls, and excess NO donors and nitrite were removed from the sample with Sephadex G-25 column chromatography. 3Cpro was then incubated with HgCl2 and the amount of nitrite was measured in the Griess reaction (Stamler et al. 1992a). For every 1 mole of 3Cpro added to the reaction, approximately 1 mole of nitrite is detected (Figure 5B). These data suggest that NO nitrosylates 3Cpro on a cysteine residue. Since the active site Cys147 is the only cysteine residue in 3Cpro, the data imply that NO nitrosylates the active site cysteine residue.
Mutating the CVB3 Protease Active Site Cysteine to Serine Renders the Protease Resistant to NO
To confirm that nitrosylation of the active site Cys147 is responsible for the inhibition of 3Cpro, we created a mutant protease by PCR-directed mutagenesis, substituting a serine for the cysteine at position 147. The resultant mutant protease, C147S3Cpro, still has catalytic activity, although at a level greatly reduced from wild-type protease, as others have shown (27, 22, 51). The effect of NO upon protease activity was determined by adding NO donors and luciferase to either wild-type or mutant protease. NO reduces wild-type 3Cpro activity, and DTT restores wild-type protease activity (Figure 6). However, NO has no effect upon mutant C147S3Cpro activity, nor does DTT change mutant protease activity. Thus, the Cys147 of the catalytic triad is critical in determining the susceptibility of 3Cpro to NO.
Download : Download high-res image (151KB)Download : Download full-size image
Figure 6. Mutation of the 3Cpro Active Site Cys147 Renders 3Cpro Resistant to NO
A mutant 3Cpro with serine substituted for cysteine at amino acid residue 147 was purified from bacteria. Wild-type 3Cpro (1 Ž╠g/ml) and mutated 3Cpro (5 Ž╠g/ml) were incubated for 3 hr with luciferase and AP, SNAP, or SNAP and DTT, and the activity of luciferase assayed in a luminometer. n = 3 í└ SD, repeated twice with similar results.
Discussion
The major finding of this study is that NO inhibits Coxsackievirus replication at least in part by nitrosylating the cysteine residue in the active site of a viral protease, thereby drastically reducing the activity of this enzyme. This protease is critical to the life cycle of Picornaviruses. Nitrosylation of cysteine residues is one mechanism by which NO can directly modify a polypeptide, and NO in theory can reduce the activity of an enzyme if its catalytic site includes a cysteine residue (62, 63, 60). For example, NO inhibits activity of the cysteine protease caspase-3 (31, 40, 47, 52). Although proteins capable of being nitrosylated at a cysteine residue contain a consensus nitrosylation motif, the 3Cpro lacks this sequence (Stamler et al. 1997). Coxsackievirus may have a selective advantage if its viral protease lacks a nitrosylation consensus sequence, decreasing cysteine reactivity to NO. The other protease of Coxsackievirus, 2Apro, is also a cysteine protease, and may be a target of NO as well.
Previously, we showed that NO inhibits Coxsackievirus replication in vitro and that NO reduces both viral RNA synthesis and viral protein synthesis (Zaragoza et al. 1997). NO inactivation of 3Cpro could explain this reduction in both viral RNA and viral protein levels, given the characteristics of the Coxsackievirus life cycle. After Coxsackievirus enters the cell, viral RNA is translated into a large polyprotein. The 3Cpro excises itself from the polyprotein and cleaves the polyprotein into its components, including 3Dpol. This RNA-dependent RNA polymerase 3Dpol then replicates the viral genome. Thus, 3Cpro processing acts upstream of viral RNA synthesis and is necessary to generate 3Dpol. Therefore, nitrosylation and inactivation of 3Cpro would be expected to block not only viral protein processing but also viral RNA synthesis.
Although our data suggest that NO inhibits activity of Coxsackievirus protease 3Cpro, there are other potential viral targets of NO as well. For example, NO could affect the RNA polymerase activity of 3Dpol. Although 3Dpol lacks the typical molecular targets of NO, cysteine residues or iron in its active site, NO could affect its activity by other mechanisms. It is also possible that NO could inactivate host proteins necessary for viral replication. The lack of effect of NO upon viral replication when NO is added to cells prior to infection argues against this possibility (Figure 1). Nonetheless, NO can affect a variety of host proteins; however, the net effect of NO upon host processes either harmful or beneficial to viral replication is unknown.
The genomes of many viruses encode a polyprotein that after translation is cleaved into smaller polypeptides by a viral protease. These proteases fall into several categories based on their active site residues, including cysteine proteases, serine proteases, and aspartic proteases (35, 24, 4). NO can inhibit the replication of other viruses that encode cysteine proteases, such as members of the Picornavirus family and the Coronavirus family (45, 56). In contrast, NO does not inhibit the replication of some viruses that encode serine proteases, such as alphaviruses. However, NO also inhibits the replication of some viruses that do not encode cysteine proteases, implying that there exist other viral targets of NO. For example, the EBV transcription factor Zta and ribonucleotide reductase may be targets of NO (46, 49). Nonetheless, to our knowledge there are no viruses that have cysteine proteases that are resistant to NO.
Cysteine proteases are also critical to the replication and virulence of a variety of other micro-organisms. Inhibitors of cysteine proteases block the replication of Plasmodium falciparum, Plasmodium berghei, Leishmania major, Schistosoma mansoni, and Trypanosoma cruzi. (5, 12, 23, 36, 39, 48, 67, 59). Furthermore, inhibitors of cysteine proteases also reduce the virulence of Streptococcus pyogenes, Porphyromonas gingivalis, Schistosoma mansoni, Naegleria fowler, and Plasmodium flaciparum. (28, 29, 3, 6, 18, 53, 67, 37, 43, 20). (For example, the malaria parasite uses its cysteine protease falcipain to degrade hemoglobin for use as a source of amino acids, and cysteine protease inhibitors block the ability of the parasite to infect erythrocytes and to replicate [18, 10].) It is striking that NO inhibits the replication of all of these parasites (45, 14, 44, 56). NO production may thus be a general mechanism by which the host defends itself against infection by viruses and other pathogens whose life cycle depends upon cysteine proteases.An Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1074761300800035íí
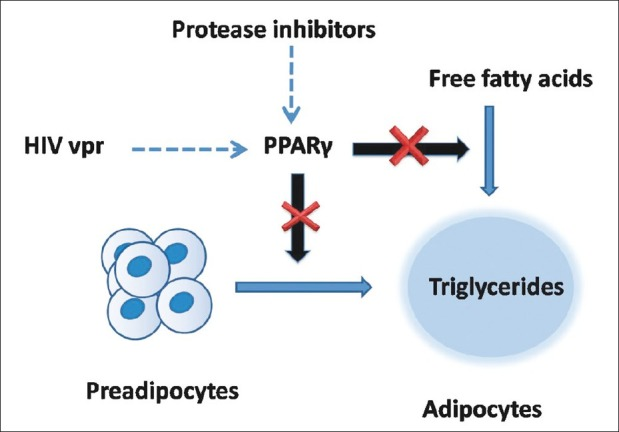
Pathogenesis and treatment of human immunodeficiency virus lipodystrophy
Enhanced understanding about the way human immunodeficiency virus (HIV) infects and causes infection in humans has led to invention and use of newer more effective antiretroviral drugs. As treatment for HIV is long term, side effects of the antiretrovirals become an important area of research focus. Antiretrovirals can cause severe metabolic abnormalities, collectively known as HIV lipodystrophy syndrome. If untreated, these metabolic abnormalities have the potential to increase stroke and cardiac ischemia. Management includes choice of nonoffending drugs, switch over to less toxic drugs, hypolipidemics, oral antidiabetics including thiazolidinediones, metformin and growth hormone analogs and finally facial surgeries. Updated knowledge about HIV lipodystrophy, and the hormone-related drugs used to treat it, is essential for physicians and endocrinologists to be able to diagnose the patients and effectively treat them.
Human immunodeficiency virus protease inhibitors and HIV viral proteins accessory protein inhibit intracellular peroxisome proliferator-activated receptor-Ž├, which is necessary for differentiation of preadipocytes to mature adipocytes
íí
(PDF) Pathogenesis and treatment of human immunodeficiency virus lipodystrophy
https://www.researchgate.net/publication/225375999_Pathogenesis_and_treatment_of_human_immunodeficiency_virus_lipodystrophyíí
European Journal of Pharmacology
Endogenous nitric oxide inhibits leukotriene B4 release from rat alveolar macrophages
Author links open overlay panelGernotBrunnaClaudiaHeybIgnazWesslercKurtRackĘŽb
a
Department of Pharmacology, University of Frankfurt, Theodor-Stern-Kai 7, D-60590 Frankfurt, Germany
b
Institute of Pharmacology and Toxicology, University of Bonn, Reuterstraße 2b, D-53113 Bonn, Germany
c
Department of Pharmacology, University of Mainz, Obere Zahlbacher Straße 67, D-55101 Mainz, Germany
Received 7 November 1996, Revised 18 February 1997, Accepted 19 February 1997, Available online 3 September 1997.
Abstract
Effects of endogenous nitric oxide (NO) on the release of mediators of the lipoxygenase and cyclo-oxygenase pathway from rat alveolar macrophages were studied.Alveolar macrophages, freshly isolated or after 18-h culture, were incubated in (amino acid-free) Krebs medium and labelled with [3H]arachidonic acid. The release of [3H]leukotriene B4 and [3H]prostanoids (separated by high performance liquid chromatography) was determined.
A 23187 was used as stimulus, as rising intracellular Ca2+ activates directly the phospholipase A2 and lipoxygenase pathway. A 23187 (10 Ž╠M) enhanced [3H]leukotriene B4 release from freshly prepared alveolar macrophages about 65-fold, but only 5- to 6-fold from cultured alveolar macrophages. Evoked [3H]leukotriene B4 release and spontaneous [3H]prostanoid release were inhibited when l-arginine (300 Ž╠M) was added to the Krebs incubation medium of alveolar macrophages, in which marked NO synthase had been induced by culture with lipopolysaccharides (10 Ž╠g/ml). Inhibitory effects of l-arginine were prevented by NG-monomethyl-l-arginine (l-NMMA, 100 Ž╠M). Inhibition of NO synthase during the culture period by l-NMMA (culture medium, in contrast to Krebs medium, already contains the substrate ofNO synthase, l-arginine), resulted in attenuation of the `culture-dependent' decline of the evoked release of [3H]leukotriene B4 and allowed lipopolysaccharides to cause an increase in spontaneous [3H]prostanoid release (i.e., to induce cyclo-oxygenase activity).
In conclusion, in rat alveolar macrophages, endogenous NO appears to inhibit the release of mediators of the cyclo-oxygenase and lipoxygenase pathway through multiple sites of action.
Endogenous nitric oxide inhibits leukotriene B4 release from rat alveolar macrophages - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0014299997001362íí
Annu Rev Immunol. 1997;15:323-50.
Nitric oxide and macrophage function.
MacMicking J1, Xie QW, Nathan C.
Department of Medicine, Cornell University Medical College, New York, NY 10021, USA.
Abstract
At the interface between the innate and adaptive immune systems lies the high-output isoform of nitric oxide synthase (NOS2 or iNOS).This remarkable molecular machine requires at least 17 binding reactions to assemble a functional dimer. Sustained catalysis results from the ability of NOS2 to attach calmodulin without dependence on elevated Ca2+.
Expression of NOS2 in macrophages is controlled by cytokines and microbial products, primarily by transcriptional induction.
NOS2 has been documented in macrophages from human, horse, cow, goat, sheep, rat, mouse, and chicken. Human NOS2 is most readily observed in monocytes or macrophages from patients with infectious or inflammatory diseases.
Sustained production of NO endows macrophages with cytostatic or cytotoxic activity against viruses, bacteria, fungi, protozoa, helminths, and tumor cells. The antimicrobial and cytotoxic actions of NO are enhanced by other macrophage products such as acid, glutathione, cysteine, hydrogen peroxide, or superoxide.
Although the high-output NO pathway probably evolved to protect the host from infection, suppressive effects on lymphocyte proliferation and damage to other normal host cells confer upon NOS2 the same protective/destructive duality inherent in every other major component of the immune response.
PMID: 9143691 DOI: 10.1146/annurev.immunol.15.1.323Nitric oxide and macrophage function. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/9143691
Antiviral Res. Author manuscript; available in PMC 2016 Apr 1.
Protease Inhibitors Targeting Coronavirus and Filovirus Entry
Yanchen Zhou,1,2 Punitha Vedantham,3 Kai Lu,1 Juliet Agudelo,1 Ricardo Carrion,4 Jerritt W. Nunneley,4 Dale Barnard,5 Stefan Pöhlmann,6 James H. McKerrow,7,8 Adam R. Renslo,3 and Graham Simmons1,2,*
Author information Copyright and License information Disclaimer
1Blood Systems Research Institute, San Francisco, CA 94118, USA
2Department of Laboratory Medicine, University of California, San Francisco, San Francisco, CA 94118, USA
3Small Molecule Discovery Center and Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, CA 94158, USA
4Texas Biomedical Research Institute, San Antonio, TX, 78227
5Institute for Antiviral Research, Department of Animal, Dairy and Veterinary Science, Utah State University, Logan, Utah 84322, USA
6Infection Biology Unit, German Primate Center, 37077 Göttingen, Germany
7Department of Pathology and Center for Discovery and Innovation in Parasitic Diseases, University of California, San Francisco, San Francisco, CA 94158, USA
*Corresponding author: Graham Simmons. Blood Systems Research Institute, 270 Masonic Avenue, San Francisco, CA 94118. 415-901-0748. FAX 415-567-5899. gro.smetsysdoolb@snommisg
8Present address: Skaggs School of Pharmacy and Pharmaceutical Sciences UCSD, San Diego CA
Abstract
In order to gain entry into cells, diverse viruses, including Ebola virus, SARS-coronavirus and the emerging MERS-coronavirus, depend on activation of their envelope glycoproteins by host cell proteases. The respective enzymes are thus excellent targets for antiviral intervention. In cell culture, activation of Ebola virus, as well as SARS- and MERS-coronavirus can be accomplished by the endosomal cysteine proteases, cathepsin L (CTSL) and cathepsin B (CTSB). In addition, SARS- and MERS-coronavirus can use serine proteases localized at the cell surface, for their activation. However, it is currently unclear which protease(s) facilitate viral spread in the infected host. We report here that the cysteine protease inhibitor K11777, ((2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-methylpiperazine-1-carbonyl]amino}-3-phenylpropanamide) and closely-related vinylsulfones act as broad-spectrum antivirals by targeting cathepsin-mediated cell entry. K11777 is already in advanced stages of development for a number of parasitic diseases, such as Chagas disease, and has proven to be safe and effective in a range of animal models. K11777 inhibition of SARS-CoV and Ebola virus entry was observed in the sub-nanomolar range. In order to assess, whether cysteine or serine proteases promote viral spread in the host, we compared the antiviral activity of an optimized K11777-derivative with that of camostat, an inhibitor of TMPRSS2 and related serine proteases. Employing a pathogenic animal model of SARS-CoV infection, we demonstrated that viral spread and pathogenesis of SARS-CoV is driven by serine rather than cysteine proteases and can be effectively prevented by camostat. Camostat has been clinically used to treat chronic pancreatitis, and thus represents an exciting potential therapeutic for respiratory coronavirus infections. Our results indicate that camostat, or similar serine protease inhibitors, might be an effective option for treatment of SARS and potentially MERS, while vinyl sulfone-based inhibitors are excellent lead candidates for Ebola virus therapeutics.
Keywords: Vinylsulfones, Coronavirus, Filovirus, Cathepsin
1. Introduction
Emerging viral diseases pose a unique risk to public health. Ebola virus, severe acute respiratory syndrome coronavirus (SARS-CoV) and members of the Henipavirus genus of paramyxoviruses are all highly pathogenic viruses that have arisen in the past 40 years and caused, or threaten to cause, major outbreaks. New viral threats continue to emerge, most recently demonstrated by a novel beta-coronavirus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), which was identified in 2012 [1ĘC3]. There are currently no approved vaccines or therapeutics for many of the highly pathogenic viruses potentially dependent on cathepsins, including Ebola virus, Nipah virus (NiV), MERS-CoV and SARS-CoV. Broad-spectrum antiviral drugs, with overlapping therapeutic indications, would facilitate rapid responses to new or changing pandemic threats, potentially even without precise identification of the agent. Targeting host factors involved in viral entry provides an excellent avenue for such drug development, due to the limited number of pathways involved [4].
The glycoproteins of corona-, filo- and paramyxoviruses facilitate viral entry into target cells by binding to receptors and by driving fusion of viral and host cell membranes. However, the glycoproteins are synthesized as inactive precursors and depend on activation by host cell proteases to acquire a fusion active form. As a consequence, the respective enzymes are potential targets for broad-spectrum antiviral intervention. Cell culture studies demonstrated that endosomal cysteine proteases, in particular cathepsin B (CTSB) and/or L (CTSL), can activate the glycoproteins of filoviruses, SARS-CoV, other coronaviruses, and NiV and Hendra (HeV) viruses to facilitate entry into certain cell lines. In addition, activation of coronaviruses can also be accomplished by TMPRSS2, or other serine proteases located at the cell surface, or secreted into the extracellular space [5]. However, the respective roles of endosomal and cell surface proteases in viral spread in the infected host is unknown.
The development of protease inhibitors able to inhibit CTSL, CTSB and related proteases would be an excellent starting point for development of broad-spectrum antiviral therapies [4]. We describe here the discovery of K11777 and its related compounds, as broad-spectrum antivirals targeting endosomal proteases involved in viral entry. K11777, a cysteine protease inhibitor, blocked infection when viral entry did not require activating serine proteases, as is the case with ebolavirus (EBOV). K11777 also fully inhibited coronavirus infection, but only when target cell lines lacking activating serine proteases were used. If cells expressed cell-surface serine proteases known to activate coronaviruses, both K11777 and a serine protease inhibitor, such as camostat were required for full inhibition. Thus, both compounds were deployed to examine which activation pathway is predominant in vivo. Camostat displayed antiviral activity in a pathogenic animal model for SARS-CoV infection, indicating that serine protease inhibitors are suitable for treatment of SARS and potentially MERS. The predicted effect of K11777 and related cysteine protease inhibitors versus Ebola virus in vivo must await studies in approved biocontainment facilities.Protease Inhibitors Targeting Coronavirus and Filovirus Entry
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4774534/íí
J Virol. 2013 Dec;87(23):12552-61. doi: 10.1128/JVI.01890-13. Epub 2013 Sep 11.
Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2.
Shirato K1, Kawase M, Matsuyama S.Department of Virology III, National Institute of Infectious Diseases, Murayama Branch, Musashi-Murayama, Tokyo, Japan.
Abstract
The Middle East respiratory syndrome coronavirus (MERS-CoV) utilizes host proteases for virus entry into lung cells.In the current study, Vero cells constitutively expressing type II transmembrane serine protease (Vero-TMPRSS2 cells) showed larger syncytia at 18 h after infection with MERS-CoV than after infection with other coronaviruses. Furthermore, the susceptibility of Vero-TMPRSS2 cells to MERS-CoV was 100-fold higher than that of non-TMPRSS2-expressing parental Vero cells.
The serine protease inhibitor camostat, which inhibits TMPRSS2 activity, completely blocked syncytium formation but only partially blocked virus entry into Vero-TMPRSS2 cells. Importantly, the coronavirus is thought to enter cells via two distinct pathways, one mediated by TMPRSS2 at the cell surface and the other mediated by cathepsin L in the endosome.
Simultaneous treatment with inhibitors of cathepsin L and TMPRSS2 completely blocked virus entry into Vero-TMPRSS2 cells, indicating that MERS-CoV employs both the cell surface and the endosomal pathway to infect Vero-TMPRSS2 cells.
In contrast, a single camostat treatment suppressed MERS-CoV entry into human bronchial submucosal gland-derived Calu-3 cells by 10-fold and virus growth by 270-fold, although treatment with both camostat and (23,25)-trans-epoxysuccinyl-L-leucylamindo-3-methylbutane ethyl ester, a cathepsin inhibitor, or treatment with leupeptin, an inhibitor of cysteine, serine, and threonine peptidases, was no more efficacious than treatment with camostat alone. Further, these inhibitors were not efficacious against MERS-CoV infection of MRC-5 and WI-38 cells, which were derived from lung, but these characters differed from those of mature pneumocytes.
These results suggest that a single treatment with camostat is sufficient to block MERS-CoV entry into a well-differentiated lung-derived cell line.
PMID: 24027332 PMCID: PMC3838146 DOI: 10.1128/JVI.01890-13Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/24027332íí
Antiviral Res. 2015 Mar;115:21-38. doi: 10.1016/j.antiviral.2014.12.015. Epub 2014 Dec 29.
The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds.
BĘóez-Santos YM1, St John SE1, Mesecar AD2.
Author information
1
Department of Biological Sciences, Purdue University, West Lafayette, IN, USA; Department of Chemistry, Purdue University, West Lafayette, IN, USA; Center for Drug Discovery, Purdue University, West Lafayette, IN, USA; Center for Cancer Research, Purdue University, West Lafayette, IN, USA.
2
Department of Biological Sciences, Purdue University, West Lafayette, IN, USA; Department of Chemistry, Purdue University, West Lafayette, IN, USA; Center for Drug Discovery, Purdue University, West Lafayette, IN, USA; Center for Cancer Research, Purdue University, West Lafayette, IN, USA. Electronic address: amesecar@purdue.edu.
Abstract
Over 10 years have passed since the deadly human coronavirus that causes severe acute respiratory syndrome (SARS-CoV) emerged from the Guangdong Province of China. Despite the fact that the SARS-CoV pandemic infected over 8500 individuals, claimed over 800 lives and cost billions of dollars in economic loss worldwide, there still are no clinically approved antiviral drugs, vaccines or monoclonal antibody therapies to treat SARS-CoV infections. The recent emergence of the deadly human coronavirus that causes Middle East respiratory syndrome (MERS-CoV) is a sobering reminder that new and deadly coronaviruses can emerge at any time with the potential to become pandemics. Therefore, the continued development of therapeutic and prophylactic countermeasures to potentially deadly coronaviruses is warranted. The coronaviral proteases, papain-like protease (PLpro) and 3C-like protease (3CLpro), are attractive antiviral drug targets because they are essential for coronaviral replication. Although the primary function of PLpro and 3CLpro are to process the viral polyprotein in a coordinated manner, PLpro has the additional function of stripping ubiquitin and ISG15 from host-cell proteins to aid coronaviruses in their evasion of the host innate immune responses. Therefore, targeting PLpro with antiviral drugs may have an advantage in not only inhibiting viral replication but also inhibiting the dysregulation of signaling cascades in infected cells that may lead to cell death in surrounding, uninfected cells. This review provides an up-to-date discussion on the SARS-CoV papain-like protease including a brief overview of the SARS-CoV genome and replication followed by a more in-depth discussion on the structure and catalytic mechanism of SARS-CoV PLpro, the multiple cellular functions of SARS-CoV PLpro, the inhibition of SARS-CoV PLpro by small molecule inhibitors, and the prospect of inhibiting papain-like protease from other coronaviruses. This paper forms part of a series of invited articles in Antiviral Research on "From SARS to MERS: 10years of research on highly pathogenic human coronaviruses."
Copyright © 2014 Elsevier B.V. All rights reserved.
KEYWORDS:
3C-like protease; MERS-CoV; Nsp3; Papain-like protease; SARS-CoV; UbiquitinThe SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25554382íí
Front Immunol. 2019 May 7;10:944. doi: 10.3389/fimmu.2019.00944. eCollection 2019.
Pyruvate Dehydrogenase Kinase Is a Metabolic Checkpoint for Polarization of Macrophages to the M1 Phenotype.
Min BK1,2, Park S3, Kang HJ2, Kim DW3, Ham HJ3, Ha CM1, Choi BJ1, Lee JY3, Oh CJ2, Yoo EK3, Kim HE3, Kim BG3, Jeon JH4, Hyeon DY5, Hwang D5,6, Kim YH7, Lee CH7, Lee T8, Kim JW9, Choi YK4, Park KG4, Chawla A10, Lee J11, Harris RA12, Lee IK1,2,3,4.
Author information
1
BK21 Plus KNU Biomedical Convergence Programs, Department of Biomedical Science, Kyungpook National University, Daegu, South Korea.
2
Research Institute of Aging and Metabolism, Kyungpook National University, Daegu, South Korea.
3
Leading-Edge Research Center for Drug Discovery and Development for Diabetes and Metabolic Disease, Kyungpook National University Hospital, Daegu, South Korea.
4
Department of Internal Medicine, School of Medicine, Kyungpook National University, Kyungpook National University Hospital, Daegu, South Korea.
5
Department of Biological Sciences, Seoul National University, Seoul, South Korea.
6
Center for Plant Aging Research, Institute for Basic Science, Daegu Gyeongbuk Institute of Science and Technology, Daegu, South Korea.
7
Laboratory Animal Resource Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea.
8
College of Pharmacy, Kyungpook National University, Daegu, South Korea.
9
Department of Biological Sciences, The University of Texas at Dallas, Richardson, TX, United States.
10
Department of Medicine, University of California, San Francisco, San Francisco, CA, United States.
11
Soonchunhyang Institute of Medi-Bio Science, Soon Chun Hyang University, Cheonan, South Korea.
12
Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, United States.
Abstract
Metabolic reprogramming during macrophage polarization supports the effector functions of these cells in health and disease. Here, we demonstrate that pyruvate dehydrogenase kinase (PDK), which inhibits the pyruvate dehydrogenase-mediated conversion of cytosolic pyruvate to mitochondrial acetyl-CoA, functions as a metabolic checkpoint in M1 macrophages. Polarization was not prevented by PDK2 or PDK4 deletion but was fully prevented by the combined deletion of PDK2 and PDK4; this lack of polarization was correlated with improved mitochondrial respiration and rewiring of metabolic breaks that are characterized by increased glycolytic intermediates and reduced metabolites in the TCA cycle. Genetic deletion or pharmacological inhibition of PDK2/4 prevents polarization of macrophages to the M1 phenotype in response to inflammatory stimuli (lipopolysaccharide plus IFN-Ž├). Transplantation of PDK2/4-deficient bone marrow into irradiated wild-type mice to produce mice with PDK2/4-deficient myeloid cells prevented M1 polarization, reduced obesity-associated insulin resistance, and ameliorated adipose tissue inflammation. A novel, pharmacological PDK inhibitor, KPLH1130, improved high-fat diet-induced insulin resistance; this was correlated with a reduction in the levels of pro-inflammatory markers and improved mitochondrial function. These studies identify PDK2/4 as a metabolic checkpoint for M1 phenotype polarization of macrophages, which could potentially be exploited as a novel therapeutic target for obesity-associated metabolic disorders and other inflammatory conditions.
KEYWORDS:
dichloroacetate; high-fat diet; inflammation; insulin resistance; macrophage polarization; metabolic reprogramming; pyruvate dehydrogenase kinase
PMID: 31134063 PMCID: PMC6514528 DOI: 10.3389/fimmu.2019.00944The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25554382íí
Int J Mol Sci. 2019 Mar; 20(6): 1345.
Plant Serine Protease Inhibitors: Biotechnology Application in Agriculture and Molecular Farming
Marina Clemente,* Mariana G. Corigliano, SebastiĘón A. Pariani, Edwin F. SĘónchez-LĘ«pez, Valeria A. Sander, and Vʬctor A. Ramos-Duarte
Abstract
The serine protease inhibitors (SPIs) are widely distributed in living organisms like bacteria, fungi, plants, and humans. The main function of SPIs as protease enzymes is to regulate the proteolytic activity.In plants, most of the studies of SPIs have been focused on their physiological role. The initial studies carried out in plants showed that SPIs participate in the regulation of endogenous proteolytic processes, as the regulation of proteases in seeds. Besides, it was observed that SPIs also participate in the regulation of cell death during plant development and senescence. On the other hand, plant SPIs have an important role in plant defense against pests and phytopathogenic microorganisms. In the last 20 years, several transgenic plants over-expressing SPIs have been produced and tested in order to achieve the increase of the resistance against pathogenic insects. Finally, in molecular farming, SPIs have been employed to minimize the proteolysis of recombinant proteins expressed in plants. The present review discusses the potential biotechnological applications of plant SPIs in the agriculture field.
Keywords: serine protease inhibitors, plants, pathogen resistance, molecular farmingPlant Serine Protease Inhibitors: Biotechnology Application in Agriculture and Molecular Farming
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6471620/íí
J Innate Immun. 2020;12(1):4-20. doi: 10.1159/000503030. Epub 2019 Oct 14.
Innate Immune Evasion by Human Respiratory RNA Viruses.
Kikkert M1.
Author information
1
Department of Medical Microbiology, Leiden University Medical Center, Molecular Virology Laboratory, Leiden, The Netherlands, m.kikkert@lumc.nl.
Abstract
The impact of respiratory virus infections on the health of children and adults can be very significant. Yet, in contrast to most other childhood infections as well as other viral and bacterial diseases, prophylactic vaccines or effective antiviral treatments against viral respiratory infections are either still not available, or provide only limited protection. Given the widespread prevalence, a general lack of natural sterilizing immunity, and/or high morbidity and lethality rates of diseases caused by influenza, respiratory syncytial virus, coronaviruses, and rhinoviruses, this difficult situation is a genuine societal challenge. A thorough understanding of the virus-host interactions during these respiratory infections will most probably be pivotal to ultimately meet these challenges. This review attempts to provide a comparative overview of the knowledge about an important part of the interaction between respiratory viruses and their host: the arms race between host innate immunity and viral innate immune evasion. Many, if not all, viruses, including the respiratory viruses listed above, suppress innate immune responses to gain a window of opportunity for efficient virus replication and setting-up of the infection. The consequences for the host's immune response are that it is often incomplete, delayed or diminished, or displays overly strong induction (after the delay) that may cause tissue damage. The affected innate immune response also impacts subsequent adaptive responses, and therefore viral innate immune evasion often undermines fully protective immunity. In this review, innate immune responses relevant for respiratory viruses with an RNA genome will briefly be summarized, and viral innate immune evasion based on shielding viral RNA species away from cellular innate immune sensors will be discussed from different angles. Subsequently, viral enzymatic activities that suppress innate immune responses will be discussed, including activities causing host shut-off and manipulation of stress granule formation. Furthermore, viral protease-mediated immune evasion and viral manipulation of the ubiquitin system will be addressed. Finally, perspectives for use of the reviewed knowledge for the development of novel antiviral strategies will be sketched.
© 2019 The Author(s) Published by S. Karger AG, Basel.
KEYWORDS:
2í»O-methylation; Coronavirus; Endoribonuclease; Guanylate-binding proteins; HRV; IAV; Interferon; Replication organelles; Respiratory syncytial virus; Vaccine
PMID: 31610541 PMCID: PMC6959104 DOI: 10.1159/000503030Plant Serine Protease Inhibitors: Biotechnology Application in Agriculture and Molecular Farming
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6471620/íí
Coronavirus | In the Pipeline
https://blogs.sciencemag.org/pipeline/archives/2020/01/27/coronavirus
Jan 27, 2020 íĄ Interesting one person trial with desired effect 😉 Earlier in this blog I hinted at the possibilty of non-specific covalent inhibition of Coronavirus cysteine protease with the drug dimethyl-fumarate. Garlic delivers other covalent inhibitors (activity in low-micromolar range) of cys proteases such as Allicin and diallyl-disulfide.
Therapeutic options for the 2019 novel coronavirus (2019-nCoV)
https://www.nature.com/articles/d41573-020-00016-0
Feb 10, 2020 íĄ HIV protease belongs to the aspartic protease family, whereas the two coronavirus proteases are from the cysteine protease family.
Author: Guangdi Li, Erik De Clercq
Author: Nature Editorial
Publish Year: 2020íí
Disulfiram can inhibit MERS and SARS coronavirus papain ...
https://www.sciencedirect.com/science/article/pii/S0166354217306101
PL pro s are cysteine proteases that use the thiol group of cysteine as a nucleophile to attack the carbonyl group of the scissile peptide bond (Chou et al., 2014, Han et al., 2005, Verma et al., 2016). Inhibition can be expected if the catalytic cysteine of a PL pro is interfered with or modified (Cheng et al., 2015, Chou et al., 2008).
Cited by: 4
Publish Year: 2018
Auíí
Structural Basis of SARS-CoV-2 3CLpro and Anti-COVID-19 ...
https://www.preprints.org/manuscript/202002.0193/v1
The recent outbreak of coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 in December 2019 raised global health concerns. The viral 3-chymotrypsin-like cysteine protease (3CLpro) enzyme, which controls coronavirus replication and is essential for its life cycle, is a proven drug discovery target in the case of severe acute respiratory syndrome coronavirus (SARS-CoV) and middle east ...
How can AI accelerate the COVID-19 research? - Causaly
https://www.causaly.com/blog/how-can-ai-accelerate...
Vincent, Martin J., et al. "Chloroquine is a potent inhibitor of SARS coronavirus infection and spread." Virology journal 2.1 (2005): 69. Kawase, Miyuki, et al. "Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry."
Protease Inhibitors Targeting Coronavirus and Filovirus Entry
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4774534
The availability of a novel, highly potent and largely non-toxic cysteine protease inhibitor, SMDC256160, afforded the opportunity to assess whether the activity of cysteine or serine proteases is required for viral spread in vivo. For this, a mouse model for lethal SARS-CoV infection was employed.
Cited by: 75
Publish Year: 2015
Author: Yanchen Zhou, Yanchen Zhou, Punitha Vedantham, Kai Lu, Juliet Aíí
Induction and Regulation of Nitric Oxide Synthase in Airway Epithelial Cells by Respiratory Syncytial Virus
YACHU J. KAO , PEDRO A. PIEDRA , GARY L. LARSEN , and GIUSEPPE N.
In this study, we evaluated the effects of respiratory syncytial virus (RSV) infection on nitric oxide (NO) production in human airway epithelial cells. In addition, we evaluated whether T-helper type 1 (Th1)- and Th2-type cytokines modulate the release of NO in response to RSV infection. To do this, we infected monolayers of A549 cells with RSV and determined nitrite levels in the supernatant fluids. We also measured nitrite levels in human small-airway epithelial cells (SAEC) in primary culture and in the bronchoalveolar lavage fluid (BALF) obtained from Balb / c mice after RSV infection. To further support our observations in these analyses, we performed immunocytochemistry and Western blot analysis for inducible nitric oxide synthase (iNOS) in A549 cells. To evaluate the regulation of NO production in response to RSV, we performed experiments in the absence and presence of the Th1 and Th2 type cytokines: interferon (IFN)- Ž├ , interleukin (IL)-4, and IL-13. In addition, we assessed the inhibitory effect of dexamethasone on iNOS in RSV infected A549 cells. Results were expressed in terms of nmol/mg protein and shown as percents of control values (mean í└ SE). RSV increased the release of nitrites in A549 cells, SAEC, and BALF. The increase in nitrite levels was supported by immunocytochemistry and Western blot analysis for iNOS protein in A549 cells, indicating activation of iNOS in response to RSV infection. IFN- Ž├ and IL-13 did not affect the RSV-induced increase in NO production. By contrast, IL-4 and dexamethasone suppressed the release of NO in response to RSV infection. These observations show that RSV infection leads to activation of iNOS within the airway epithelium and that IL-4 and dexamethasone inhibit the production of NO in response to RSV infection.║˘╬ŘÁ└║¤░ű╠ň▓íÂżÂďĂ°Á└╔¤ĂĄ¤Ş░űĎ╗Ч╗»Á¬║¤├ŞÁ─ËŇÁ╝║═Á¸┐ě
ď┌▒żĐđż┐ÍđúČ╬Ď├ăĂ└╣└┴╦║˘╬ŘÁ└║¤░ű╠ň▓íÂż(RSV)Şđ╚żÂď╚╦Ă°Á└╔¤ĂĄ¤Ş░űĎ╗Ч╗»Á¬(NO)▓˙╔˙Á─Ë░¤ý┤╦═ÔúČ╬Ď├ă╗╣Ă└╣└┴╦T -ŞĘÍ˙đď1đ═(Th1)║═Th2đ═¤Ş░űϲÎË╩ăĚ˝Á¸Ż┌NOÁ─╩═Ě┼ĎďËŽÂďRSVŞđ╚żíú
╬¬┤╦úČ╬Ď├ăË├RSVŞđ╚żA549¤Ş░űÁą▓ŃúČ▓ó▓ÔÂĘ╔¤ăňĎ║ÍđÁ─Đ㤧╦ßĐ╬╦«ĂŻíú╬Ď├ă╗╣▓Ô┴┐┴╦ďş┤˙┼ÓĐ°Á─╚╦đíĂ°Á└╔¤ĂĄ¤Ş░ű(SAEC)║═RSVŞđ╚ż║ˇBalb / cđí╩ˇÍžĂ°╣▄Ě╬┼Ţ╣Ó¤┤Ď║(BALF)ÍđÁ─Ď╗Ч╗»Á¬╦«ĂŻíú
╬¬┴╦Ż°Ď╗▓ŻÍž│Í╬Ď├ăď┌ŇÔđęĚÍ╬÷ÍđÁ─╣█▓ýúČ╬Ď├ăÂďA549¤Ş░űÍđËŇÁ╝đ═Ď╗Ч╗»Á¬║¤├Ş(iNOS)Ż°đđ┴╦├ÔĎ▀¤Ş░ű╗»Đž║═Western blotĚÍ╬÷íú
Ă└╣└Á─╣ŠÂĘď┌ËŽÂďRSV├╗Ëđ╔˙▓˙,╬Ď├ăď┌├╗ËđŻ°đđ╩ÁĐÚ,Th1íóTh2đ═¤Ş░űϲÎËÁ─┤Šď┌:Ş╔╚┼╦ě(IFN) -Ž├,░ÎŻÚ╦ě(IL) 4, IL-13íú
┤╦═ÔúČ╬Ď├ă╗╣Ă└╣└┴╦Áě╚ű├Î╦╔ÂďRSVŞđ╚żA549¤Ş░űÍđiNOSÁ─ĎÍÍĂθË├íú
Żß╣űĎďnmol/mgÁ░░Î▒Ý┤´úČĎď┐ěÍĂÍÁ░┘ĚÍ▒╚(ż¨╩ří└▒ŕÎ╝▓ţ)▒Ý╩żíúRSVď÷╝Ë┴╦A549¤Ş░űíóSAEC║═BALFÍđĎ╗Ч╗»Á¬Á─╩═Ě┼íúA549¤Ş░űÍđiNOSÁ░░ÎÁ─├ÔĎ▀¤Ş░ű╗»Đž║═Western blotĚÍ╬÷Íž│Í┴╦Ď╗Ч╗»Á¬╦«ĂŻÁ─╔řŞ▀úČ╠ß╩ż┴╦Ď╗Ч╗»Á¬║¤├ŞÂďRSVŞđ╚żÁ─╝Ą╗ţíúŞ╔╚┼╦ě-Ž├║═IL-13▓ó▓╗Ë░¤ýRSV-inducedď÷╝Ë├╗Ëđ╔˙▓˙íú¤Ó▒╚Í«¤┬úČIL-4║═Áě╚ű├Î╦╔ď┌RSVŞđ╚ż╩▒ĎÍÍĂNOÁ─╩═Ě┼íú
ŇÔđę╣█▓ýŻß╣ű▒Ý├¸úČRSVŞđ╚ż┐╔Á╝Í┬║˘╬ŘÁ└╔¤ĂĄ─┌Á─iNOS╝Ą╗ţúČIL-4║═Áě╚ű├Î╦╔┐╔ĎÍÍĂRSVŞđ╚ż╩▒NOÁ─▓˙╔˙íúInduction and Regulation of Nitric Oxide Synthase in Airway Epithelial Cells by Respiratory Syncytial Virus | American Journal of Respiratory and Critical Care Medicine
https://www.atsjournals.org/doi/full/10.1164/ajrccm.163.2.9912068íí
Human alveolar epithelial cells induce nitric oxide synthaseę\2 expression in alveolar macrophages
D.V. Pechkovsky, G. Zissel, C. Stamme, T. Goldmann, H. Ari Jaffe, M. Einhaus, C. Taube, H. Magnussen, M. Schlaak, J. MĘ╣llerQuernheim
European Respiratory Journal 2002 19: 672-683; DOI: 10.1183/09031936.02.00682001a
ArticleFigures & DataInfo & Metrics PDF
Abstract
It was hypothesized that celltocell interaction between human alveolar macrophages (AM) and alveolar epithelium, might be an important factor leading to nitric oxide synthaseę\2 (NOS2) messenger ribonucleic acid (mRNA) and protein expression by constituent cells of the alveolar wall and/or AM.
NOS2 mRNA and the protein expression patterns of human AM and alveolar epithelial cells type II (AECII) isolated from normal parts of lung resections of patients with pulmonary malignancies were determined. In addition, NOS2 mRNA expression in human AM cocultured with autologous AECII in the presence of proinflammatory cytokines interleukin (IL)ę\1Ž┬, tumour necrosis factor (TNF)ę\Ž┴, interferon (IFN)ę\Ž├ or lipopolysaccharide (LPS) was investigated. The effect of human surfactant proteinę\A (SPę\A) on IFNę\Ž├ę\mediated NOS2 mRNA expression in human AM was also studied.
Neither NOS2 mRNA nor NOS2 protein could be detected in freshly isolated, unstimulated or cytokinestimulated AECII. In contrast, freshly isolated AM from bronchoalveolar lavage or lung tissue samples expressed immunoreactivity for NOS2 protein, but no NOS2 mRNA could be detected by reverse transcriptase polymerase chain reaction. All stimuli tested failed to induce NOS2 mRNA expression in human AM in vitro. Only AMAECII coculture in the presence of IFNę\Ž├ led to NOS2 mRNA and NOS2 protein expression. In situ hybridization of NOS2 mRNA on lung tissue explants and immunohistochemical staining of cytospin preparations of AMAECII cocultures demonstrated that NOS2 is expressed in AM but not in AECII. This coculture effect could not be reproduced by substitution of AECII with SPę\A.
These data give evidence of a regulatory network controlling human nitric oxide synthaseę\2 expression in the lower respiratory tract.Human alveolar epithelial cells induce nitric oxide synthaseę\2 expression in alveolar macrophages | European Respiratory Society
https://erj.ersjournals.com/content/19/4/672íí
Crit Rev Immunol. 2001;21(5):399-425.
Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue.
Mills CD1.
Author information
1
Department of Surgery and Diabetes Institute for Immunology and Transplantation, University of Minnesota Hospitals and Clinics, Minneapolis 55455, USA. mills002@tc.umn.edu
Abstract
Macrophages can metabolize arginine to nitric oxide in quantities that inhibit pathogens or nearby host cells. They can instead metabolize arginine to ornithine (a precursor of polyamines and collagen) in quantities that stimulate pathogens or nearby host cells.Macrophages are essentially the only circulating cells that can make these life or death decisions with arginine. Macrophages expressing these destructive or constructive phenotypes have been termed M-1 or M-2 because they also stimulate TH1 or TH2 responses, respectively.
Factors that influence whether a macrophage expresses the M-1 or M-2 phenotype and the real or potential impact on immune responses and other host processes are discussed.
PMID: 11942557
[Indexed for MEDLINE]https://www.ncbi.nlm.nih.gov/pubmed/11942557
íí
Front Immunol. 2019; 10: 1569.
Bone Marrow NK Cells: Origin, Distinctive Features, and Requirements for Tissue Localization
Valentina Bonanni,1 Giuseppe SciumĘĘ,1 Angela Santoni,1,2 and Giovanni Bernardini1,*
1Department of Molecular Medicine, Sapienza University of Rome, Laboratory Affiliated to Institute Pasteur-Italia, Rome, Italy
Abstract
NK cell maturation is a continuous process, which initiates in the bone marrow and proceeds in peripheral tissues, where NK cells follow distinct differentiation routes. Drastic phenotypic changes are observed during progression from precursors to mature NK cells, including changes of expression and functionalities of several chemoattractant receptors. Upon differentiation, mature NK cells migrate outside the bone marrow; as well, peculiar subsets of NK cells can also home back to or localize in this anatomic compartment to play specific functions. In humans, NK cells with a tissue resident phenotype have been identified in bone marrow, sharing similarities with tissue resident memory CD8+ T cells; while in mouse, long-lived NK cells undergo homeostatic proliferation in this site during viral infections. The mechanisms underlying NK cell subset localization in the bone marrow have only recently started to be investigated, especially in pathological settings such as tumors or infections. In this review, we discuss the phenotype and function of NK cells as well as their requirements for bone marrow maintenance and/or homing.
Introduction
Natural Killer (NK) cells are innate lymphocytes able to recognize and kill cancer or virus-infected cells (1). They account for 5ĘC20% of the mononuclear cells of the peripheral blood and the spleen. They also produce cytokines, among which interferon (IFN)-Ž├ delivers signals to the innate component of the immune system, which activate the inflammatory process in defense of the organism. Activation of NK cell function following interaction with a target cell is the result of the integration of signals generated by inhibitory and activating receptors expressed simultaneously by NK cells and engaged by the ligands present on the target cells (2). By acting early during cell infection or transformation, before and independently of specific immunity, they take part to the first line of the immune response. These characteristics make them fundamental as a defense mechanism.
Recently NK cells have been re-categorized as part of the innate lymphoid cells (ILCs). ILCs have been characterized in three groups. The group 1 comprises cells expressing the transcription factors T-BET and producing the T helper cell type 1 (Th1)-associated cytokine IFN-Ž├, including NK cells (3).
Conventional NK cells appear to be the only cytotoxic cells, while all the other ILCs follow the pattern of helper CD4 T cells and produce cytokines and other soluble factors that help adaptive immune response development.
Keywords: natural killer cells, bone marrow (bm), infectionĘCimmunology, innate lymphoid cell, chemokine receptorsBone Marrow NK Cells: Origin, Distinctive Features, and Requirements for Tissue Localization
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6635729/íí
An Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease
Although nitric oxide (NO) kills or inhibits the replication of a variety of intracellular pathogens, the antimicrobial mechanisms of NO are unknown. Here, we identify a viral protease as a target of NO. The life cycle of many viruses depends upon viral proteases that cleave viral polyproteins into individual polypeptides. NO inactivates the Coxsackievirus protease 3C, an enzyme necessary for the replication of Coxsackievirus. NO S-nitrosylates the cysteine residue in the active site of protease 3C, inhibiting protease activity and interrupting the viral life cycle. Substituting a serine residue for the active site cysteine renders protease 3C resistant to NO inhibition. Since cysteine proteases are critical for virulence or replication of many viruses, bacteria, and parasites, S-nitrosylation of pathogen cysteine proteases may be a general mechanism of antimicrobial host defenses.
An Antiviral Mechanism of Nitric Oxide: Inhibition of a Viral Protease - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1074761300800035
íííí

.jpg)