��
Cancer as Metabolic Disease and Metformin is Anticancer Drug
��
metformin enhance mitochondria biosynthesis via FGF21 (in brown adipose tissue)
��
��
��
Diabetes, Cancer and the Drug that Fights them Both - Science in the News
http://sitn.hms.harvard.edu/flash/2017/diabetes-cancer-drug-fights/
HARVARD UNIVERSITY
��
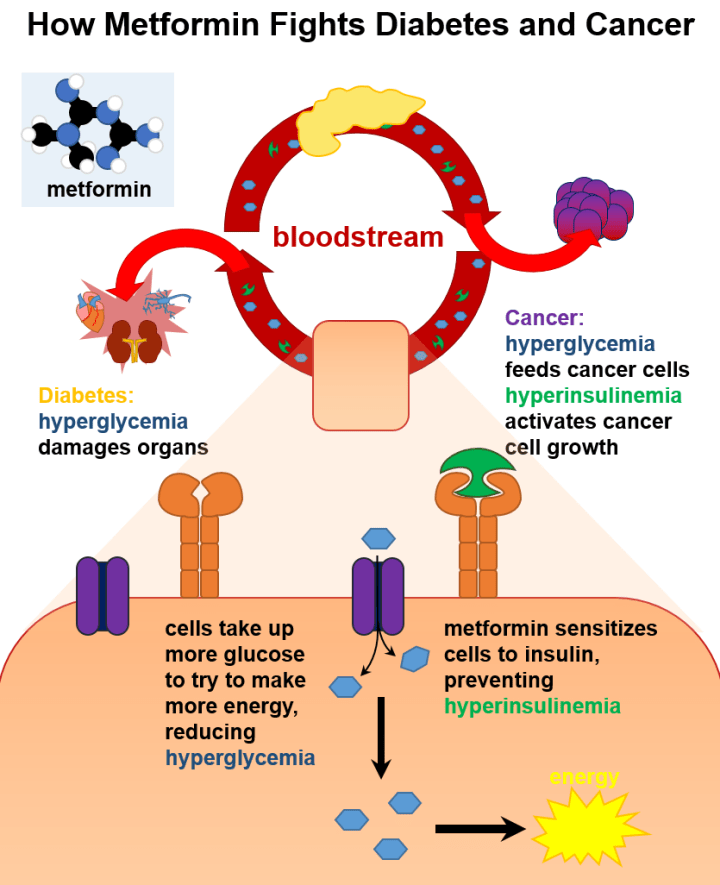
How does Metformin Work?
Metformin accumulates inside the mitochondria, the little energy producing
organelles in our cells. Once inside , metformin inhibits complex I of the
mitochondrial electron transport chain. This in turn activates AMP-Kinase(AMPK),
which then inhibits the mTOR signal pathway which reduces cancer cell
proliferation.(4) Left Image Electron Microscopic Image of Mitochondria.
Metformin Repurposed Anti-Cancer Drug - Jeffrey Dach MD
https://jeffreydachmd.com/2017/07/metformin-repurposed-anti-cancer-drug/
��
Metabolic traits of cancer stem cells
Disease Models & Mechanisms 2018 11: dmm033464 doi:
ABSTRACT
Cancer stem cells are a subpopulation of cells within a tumour believed to
confer resistance to standard cancer therapies. Although many studies have
addressed the specific mechanisms of tumour recurrence driven by cancer stem
cells, cellular metabolism is an often-neglected attribute. The metabolic
features of cancer stem cells are still poorly understood, and they thus
constitute a promising field in cancer research. The findings published so far
point to a distinct metabolic phenotype in cancer stem cells, which might depend
on the cancer type, the model system used or even the experimental design, and
several controversies still need to be tackled. This Review describes the
metabolic phenotype of cancer stem cells by addressing the main metabolic traits
in different tumours, including glycolysis and oxidative, glutamine, fatty acid
and amino acid metabolism. In the context of these pathways, we also mention the
specific alterations in metabolic enzymes and metabolite levels that have a role
in the regulation of cancer stemness. Determining the role of metabolism in
supporting resistance to therapy driven by cancer stem cells can raise the
opportunity for novel therapeutic targets, which might not only eliminate this
resistant population, but, more importantly, eradicate the whole tumour in a
relapse-free scenario.
Metabolic traits of cancer stem cells | Disease Models & Mechanisms
https://dmm.biologists.org/content/11/8/dmm033464
��
eLife. 2014; 3: e02242.
Published online 2014 May 13. doi: 10.7554/eLife.02242
Metformin inhibits mitochondrial complex I of cancer cells to
reduce tumorigenesis
Northwerstern University
There are two postulated mechanisms by which metformin reduces tumor growth. Metformin may act at the organismal level, reducing levels of circulating insulin, a known mitogen for cancer cells. Alternatively, metformin may act in a cancer cell autonomous manner. Metformin is known to inhibit mitochondrial complex I in vitro (Ota et al., 2009; El-Mir et al., 2000; Owen et al., 2000) and it is thus possible that this targeting of the electron transport chain could inhibit tumor cell growth (Birsoy et al., 2012). This latter hypothesis has been questioned as cancer cells have the ability to survive on ATP produced exclusively by glycolysis. Furthermore, cancer cells have been shown to conduct glutamine-dependent reductive carboxylation to generate the TCA cycle intermediates required for cell proliferation when the electron transport chain is inhibited (Mullen et al., 2012; Fendt et al., 2013).
Recent epidemiological and laboratory-based studies suggest that the anti-diabetic drug metformin prevents cancer progression. How metformin diminishes tumor growth is not fully understood. In this study, we report that in human cancer cells, metformin inhibits mitochondrial complex I (NADH dehydrogenase) activity and cellular respiration. Metformin inhibited cellular proliferation in the presence of glucose, but induced cell death upon glucose deprivation, indicating that cancer cells rely exclusively on glycolysis for survival in the presence of metformin. Metformin also reduced hypoxic activation of hypoxia-inducible factor 1 (HIF-1). All of these effects of metformin were reversed when the metformin-resistant Saccharomyces cerevisiae NADH dehydrogenase NDI1 was overexpressed. In vivo, the administration of metformin to mice inhibited the growth of control human cancer cells but not those expressing NDI1. Thus, we have demonstrated that metformin's inhibitory effects on cancer progression are cancer cell autonomous and depend on its ability to inhibit mitochondrial complex I.

Metformin exists as a cation at physiological pH and thus its accumulation within mitochondria is predicted to increase as a function of the mitochondrial membrane potential. The inhibition of electron transport at complex I by metformin should reduce the mitochondrial membrane potential as proton pumping is linked to electron transport.
In summary, our results indicate that metformin reversibly inhibits mitochondrial complex I within cancer cells to reduce tumorigenesis. Metformin inhibits tumorigenesis through multiple mechanisms including the induction of cancer cell death in conditions, when glucose is limited and through inhibition of mitochondrial ROS-dependent signaling pathways that promote tumorigenesis (i.e., HIF). These results indicate that metformin would be most effective in low glucose and oxygen conditions. It will be of interest to determine whether metformin treatment might provide a useful adjunct to therapies that limit glucose uptake (e.g., PI3K inhibitors) or drive tumors to low glucose and oxygen levels (e.g., anti-angiogenic inhibitors).
Metformin inhibits mitochondrial complex I of cancer cells ...
www.ncbi.nlm.nih.gov/pmc/articles/PMC4017650/
Metformin directly acts on mitochondria to alter cellular bioenergetics
��
Goodman Cancer Research Centre, McGill University, 1160
Pine Ave. West, Montr��al, QC, H3A 1A3, Canada
Sylvia Andrzejewski, Simon-Pierre Gravel, Michael Pollak & Julie St-Pierre
Cancer & Metabolism volume 2, Article number: 12 (2014) Cite this article
Abstract
Background
Metformin is widely used in the treatment of diabetes, and there is interest in
��repurposing�� the drug for cancer prevention or treatment. However, the
mechanism underlying the metabolic effects of metformin remains poorly
understood.
Methods
We performed respirometry and stable isotope tracer analyses on cells and
isolated mitochondria to investigate the impact of metformin on mitochondrial
functions.
Results
We show that metformin decreases mitochondrial respiration, causing an increase
in the fraction of mitochondrial respiration devoted to uncoupling reactions.
Thus, cells treated with metformin become energetically inefficient, and display
increased aerobic glycolysis and reduced glucose metabolism through the citric
acid cycle. Conflicting prior studies proposed mitochondrial complex I or
various cytosolic targets for metformin action, but we show that the compound
limits respiration and citric acid cycle activity in isolated mitochondria,
indicating that at least for these effects, the mitochondrion is the primary
target. Finally, we demonstrate that cancer cells exposed to metformin display a
greater compensatory increase in aerobic glycolysis than nontransformed cells,
highlighting their metabolic vulnerability. Prevention of this compensatory
metabolic event in cancer cells significantly impairs survival.
Conclusions
Together, these results demonstrate that metformin directly acts on mitochondria
to limit respiration and that the sensitivity of cells to metformin is dependent
on their ability to cope with energetic stress.
��

Metformin directly acts on mitochondria to alter cellular bioenergetics |
Cancer & Metabolism | Full Text
https://cancerandmetabolism.biomedcentral.com/articles/10.1186/2049-3002-2-12
��
��
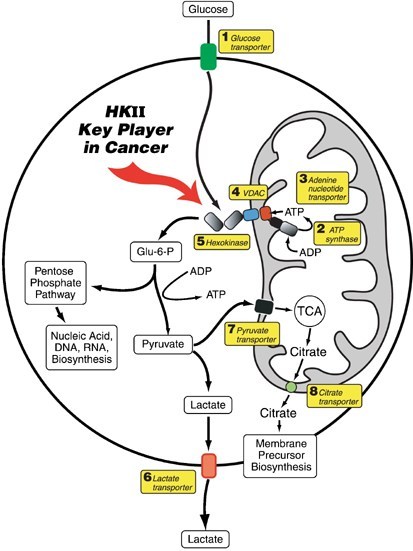
Hexokinase II �C Major Player in the Cancer Cell
Below image schematic showing Hexokinase II attached to VDAC on mitochondrial
membrane, utilizing ATP to convert glucose to G6-P. Courtesy of Mathupala, S.
P., YH and Ko, and P. L. Pedersen. ��Hexokinase II: cancer��s double-edged sword
acting as both facilitator and gatekeeper of malignancy when bound to
mitochondria.�� Oncogene 25.34 (2006): 4777.(58)
hexokinase-HK-II-VDACHexokinase II, the Achilles Heel of the Cancer Cell
As mentioned in my previous article, Cancer as a Metabolic Disease, the cancer
cells are rapidly proliferating in uncontrolled manner. Their metabolic pathways
are massively upregulated to support the rapid proliferation. These metabolic
differences can be exploited to selectively kill cancer cells, leaving normal
cells unharmed. The cancer cell has a voracious appetite for glucose
consumption, and accomplishes this massive glucose utilization by switching to
an embryonic form of the Hexokinase enzyme called Hexokinase II, not normally
present in normal cells. The enzyme Hexokinase II is the first step in
conversion of glucose to glucose-6-Phosphate. (58) Production of Hexokinase II
in the cancer cell is over 100 times upregulated by genetic amplification.(58)
Attachment of HKII to the VDAC in the cancer cell also serves to prevent
mitochondrial apoptosis. Detachment of HKII from the VDAC restores mitochondrial
apoptosis pathways.(58) Below Image showing apoptosis induced by release of
Hexokinase II from VDAC at outer mitochondrial membrane. Fig 3 courtesy of
Mathupala, S. P., YH and Ko, and P. L. Pedersen. et al. (58)
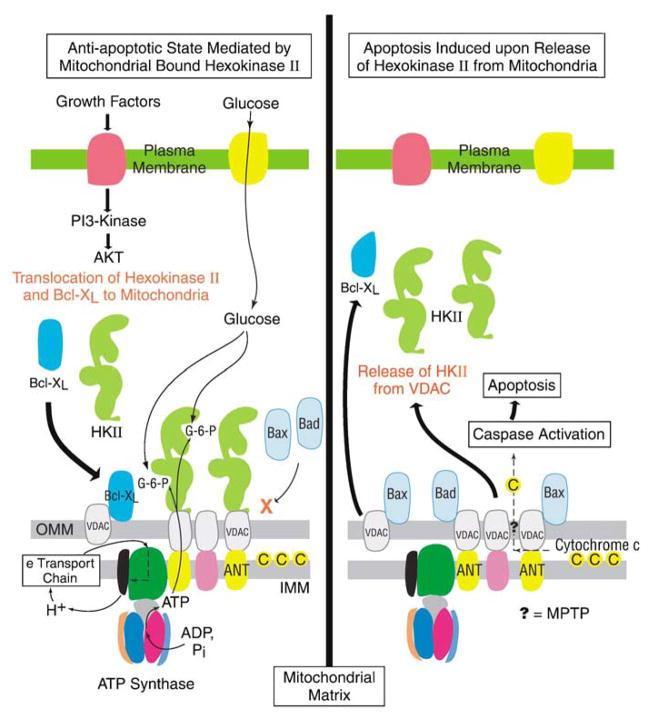
Metformin Docks in Hexokinase Two
In 2013, Barbara Salani��s group from Italy published their in vitro lung cancer
cell study, showing Metformin docks in the Hexokinase II binding site,
effectively blocking its function, resulting in separation Hexokinase II from
the VDAC (voltage dependent anion channel) located on the outer mitochondrial
membrane.(9) Dr Salani says:
This inhibition (of Hexokinase) virtually abolishes cell glucose uptake and
phosphorylation as documented by the reduced entrapment of
18F-fluorodeoxyglucose.(9)
��When HK is released from VDAC ��tumor cells rapidly undergo apoptosis under a
variety of stimuli which were previously ineffective in inducing apoptosis.��
quote from(58)
��
Understanding Cancer Stems Cells
Dr Patricia Sancho in her 2015 article on Pancreatic Cancer Stem Cells explains
Metformin targets pancreatic cancer stem cells (CSCs), but not their
differentiated non-Cancer Stem Cells.(41) Dr. Sancho��s study demonstrates that
non-CSCs are highly glycolytic, while the Cancer Stem Cells (CSCs) are dependent
on oxidative metabolism (OXPHOS) with ��very limited metabolic plasticity��. Thus,
mitochondrial inhibition by metformin creates an energy crisis and induces
cancer stem cell apoptosis.(41) Dr Sanchez states that during treatment with
Metformin, ��resistant Cancer Stem Cell (CSC ) clones eventually emerge with
intermediate glycolytic/respiratory phenotype.��(41) This is very similar to the
findings of the Lisanti group who found emergence of Doxycycline resistant
cancer stem cells which had acquired a purely glycolytic phenotype..(also called
the Warburg Phenotype) .(59) Below image shows effect of metformin on cancer
stem cells mitochondria. Ovoid Pink structures are the mitochondria. ..courtesy
of Patricia Sancho, et al. MYC_PGC1a Determines Metabolic Phenotype Pancreatic
Cancer Stem Cells Patricia Sancho Cell Metabolism 2015 (41) Note Warburg
Phenotype = Glycolytic Phenotype present in non-cancer stem cells. Note cancer
stem cells are OX-Phos dependent unless they develop resistance.

��
Dr Sancho found that cancer stem cells developed resistance to Metformin (see
above diagram), and she states that combining Metformin with c-MYC inhibitor
overcomes this resistant phenotype.
��Alternatively, combining metformin with c-MYC inhibition, prevented or
reversed, respectively, resistance to metformin by enforcing their dependence on
OXPHOS, suggesting a new multimodal approach for targeting the distinct
metabolic features of pancreatic CSCs.��
��
��
�����ؽ���c-MYC Artesunate Degrades c-MYC
The anti-malaria drug Artesunate is now first line treatment for severe malaria
in third word countries, and is commonly infused intravenously for millions of
patients with virtually no adverse effects. (See my article on Artemisinin)
Artesunate is also an effective anti-cancer agent which degrades the c-MYC
protein. (65-66) According to Dr Lu in his 2010 article ��Dihydroartemisinin
accelerates c-MYC oncoprotein degradation and induces apoptosis in
c-MYC-overexpressing tumor cells.�� Dr Lu found Artesunate and Dihydroartemisinin
(DHA) induce significant apoptosis in cancer cell lines over-expressing c-MYC
protein. Dr Lu found that DHA (and Artesunate) irreversibly down-regulated the
protein level of c-MYC and accelerated degradation of c-MYC protein in the
cancer cells. Dr Lu concluded that Artesunate would be useful in the treatment
of c-MYC-overexpressing cancer cell types, as c-Myc could serve as biomarker
candidate for prediction of antitumor efficacy of Artesunate.(65-66)
Over-Expression of c-MYC Associated with Aggressive Biology and Poor Prognosis
The c-Myc gene is a transcription factor regulating proliferation, growth, and
apoptosis. Overexpression or amplification of the c-Myc protein is associated
with aggressive cancer cell biology with poor prognosis. (67-70) Indeed, Dr Yi
studied a series of Mantle B-Cell Lymphoma patients with c-MYC overexpression in
Oncotarget 2015, stating:
��Intensive chemotherapy such as HyperCVAD/MA �� R did not improve the survival of
(lymphoma patients) with a c-MYC abnormality, and a new treatment strategy
should be developed.��
Dr Yi found that the highly aggressive biology of the c-MYC abnormality rendered
intensive chemotherapy futile, providing brief remission with no survival
benefit. The combination of an OX-Phos inhibitors (such as Metformin or
Doxyxyxline) targeting cancer stem cells along with the c-Myc inhibitor,
Artesunate, might represent such a new treatment strategy. We await NIH funded
confirmatory studies.
Metformin Targets Cancer Stem Cells
As mentioned above. cancer stems cells utilize mitochondrial OX-PHOS (oxidative
phosphorylation) for their energetic migratory and metastatic capacity.(11)
Indeed, Dr Diana Whitaker-Menezes in Cell Cycle 2011, reported hyperactive
oxidative mitochondrial metabolism in cancer cells was blocked by Metformin.(11)
Metformin treatment serves to induce purely glycolytic phenotype in surviving
cancer stem cells, now rendered sensitive to glucose starvation with a second
agent such as 2DG or high dose intravenous vitamin C, creating synthetic
lethality. (59)(5)(9-10)
��

��
Dr Sancho found that cancer stem cells developed resistance to Metformin (see
above diagram), and she states that combining Metformin with c-MYC inhibitor
overcomes this resistant phenotype.
��Alternatively, combining metformin with c-MYC inhibition, prevented or
reversed, respectively, resistance to metformin by enforcing their dependence on
OXPHOS, suggesting a new multimodal approach for targeting the distinct
metabolic features of pancreatic CSCs.��
��
Metformin Repurposed Anti-Cancer Drug - Jeffrey Dach MD
https://jeffreydachmd.com/2017/07/metformin-repurposed-anti-cancer-drug/
��
voltage-dependent anion channel (VDAC)-bound HK-II contributes to the inhibition of apoptosis by suppressing the formation of mitochondrial permeability transition pores (mPTPs) [38]. On the other hand, numerous studies have indicated that inhibitors [e.g.
Department of Otolaryngology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
Oncotarget | Warburg effect, hexokinase-II, and radioresistance of laryngeal
carcinoma
http://www.oncotarget.com/index.php?journal=oncotarget&page=article&op=view&path%5B%5D=13044&path%5B%5D=41337
��
Metformin Repurposed Anti-Cancer Drug
Posted on July 27 2017
Metformin_500mg_Tablets_Jeffrey Dach MDMetformin Repurposed Anti-Cancer Drug
Metformin, FDA approved in 1994, is known as ��the Good Anti-Diabetic Drug�� ,
taken by 150 million people worldwide for control of blood sugar in Type Two
Diabetes.(57) Remarkably, metformin is also an anti-cancer drug. In 2005, Dr
Evans made the observation that Diabetic patients on Metformin have a 23%
reduction in cancer.(4) Others have found a 30-50 per cent reduction in risk for
cancer in metformin users.(1-2) (6) Since 2005, there has been considerable
effort to elucidate the anti-cancer mechanism of metformin in both the
laboratory and clinical setting.(3)(23) Left Image Metformin Courtesy of
Wikimedia Commons.
How does Metformin Work?
Metformin accumulates inside the mitochondria, the little energy producing
organelles in our cells. Once inside , metformin inhibits complex I of the
mitochondrial electron transport chain. This in turn activates AMP-Kinase(AMPK),
which then inhibits the mTOR signal pathway which reduces cancer cell
proliferation.(4) Left Image Electron Microscopic Image of Mitochondria.
Hexokinase II �C Major Player in the Cancer Cell
Below image schematic showing Hexokinase II attached to VDAC on mitochondrial
membrane, utilizing ATP to convert glucose to G6-P. Courtesy of Mathupala, S.
P., YH and Ko, and P. L. Pedersen. ��Hexokinase II: cancer��s double-edged sword
acting as both facilitator and gatekeeper of malignancy when bound to
mitochondria.�� Oncogene 25.34 (2006): 4777.(58)
hexokinase-HK-II-VDACHexokinase II, the Achilles Heel of the Cancer Cell
As mentioned in my previous article, Cancer as a Metabolic Disease, the cancer
cells are rapidly proliferating in uncontrolled manner. Their metabolic pathways
are massively upregulated to support the rapid proliferation. These metabolic
differences can be exploited to selectively kill cancer cells, leaving normal
cells unharmed. The cancer cell has a voracious appetite for glucose
consumption, and accomplishes this massive glucose utilization by switching to
an embryonic form of the Hexokinase enzyme called Hexokinase II, not normally
present in normal cells. The enzyme Hexokinase II is the first step in
conversion of glucose to glucose-6-Phosphate. (58) Production of Hexokinase II
in the cancer cell is over 100 times upregulated by genetic amplification.(58)
Attachment of HKII to the VDAC in the cancer cell also serves to prevent
mitochondrial apoptosis. Detachment of HKII from the VDAC restores mitochondrial
apoptosis pathways.(58) Below Image showing apoptosis induced by release of
Hexokinase II from VDAC at outer mitochondrial membrane. Fig 3 courtesy of
Mathupala, S. P., YH and Ko, and P. L. Pedersen. et al. (58)
Metformin Docks in Hexokinase Two
In 2013, Barbara Salani��s group from Italy published their in vitro lung cancer
cell study, showing Metformin docks in the Hexokinase II binding site,
effectively blocking its function, resulting in separation Hexokinase II from
the VDAC (voltage dependent anion channel) located on the outer mitochondrial
membrane.(9) Dr Salani says:
This inhibition (of Hexokinase) virtually abolishes cell glucose uptake and
phosphorylation as documented by the reduced entrapment of
18F-fluorodeoxyglucose.(9)
��When HK is released from VDAC ��tumor cells rapidly undergo apoptosis under a
variety of stimuli which were previously ineffective in inducing apoptosis.��
quote from(58)
Metformin, the Monkey Wrench
The Metformin molecule is a ��Monkey Wrench�� sabotaging the machinery of the
cancer cell.
See this video of 3-D computer rendering of Metformin docking in Hexokinase II
by the Salani Group (9). Click Here to view Video.
Left Image monkey wrench courtesy of wikimedia commons.
Understanding Cancer Stems Cells
Dr Patricia Sancho in her 2015 article on Pancreatic Cancer Stem Cells explains
Metformin targets pancreatic cancer stem cells (CSCs), but not their
differentiated non-Cancer Stem Cells.(41) Dr. Sancho��s study demonstrates that
non-CSCs are highly glycolytic, while the Cancer Stem Cells (CSCs) are dependent
on oxidative metabolism (OXPHOS) with ��very limited metabolic plasticity��. Thus,
mitochondrial inhibition by metformin creates an energy crisis and induces
cancer stem cell apoptosis.(41) Dr Sanchez states that during treatment with
Metformin, ��resistant Cancer Stem Cell (CSC ) clones eventually emerge with
intermediate glycolytic/respiratory phenotype.��(41) This is very similar to the
findings of the Lisanti group who found emergence of Doxycycline resistant
cancer stem cells which had acquired a purely glycolytic phenotype..(also called
the Warburg Phenotype) .(59) Below image shows effect of metformin on cancer
stem cells mitochondria. Ovoid Pink structures are the mitochondria. ..courtesy
of Patricia Sancho, et al. MYC_PGC1a Determines Metabolic Phenotype Pancreatic
Cancer Stem Cells Patricia Sancho Cell Metabolism 2015 (41) Note Warburg
Phenotype = Glycolytic Phenotype present in non-cancer stem cells. Note cancer
stem cells are OX-Phos dependent unless they develop resistance.
Metformin Metabolic-Phenotype-and-Plasticity-of-Pancreatic-Cancer-Stem-CellsDr
Sancho found that cancer stem cells developed resistance to Metformin (see above
diagram), and she states that combining Metformin with c-MYC inhibitor overcomes
this resistant phenotype.
��Alternatively, combining metformin with c-MYC inhibition, prevented or
reversed, respectively, resistance to metformin by enforcing their dependence on
OXPHOS, suggesting a new multimodal approach for targeting the distinct
metabolic features of pancreatic CSCs.��
Artesunate Degrades c-MYC
The anti-malaria drug Artesunate is now first line treatment for severe malaria
in third word countries, and is commonly infused intravenously for millions of
patients with virtually no adverse effects. (See my article on Artemisinin)
Artesunate is also an effective anti-cancer agent which degrades the c-MYC
protein. (65-66) According to Dr Lu in his 2010 article ��Dihydroartemisinin
accelerates c-MYC oncoprotein degradation and induces apoptosis in
c-MYC-overexpressing tumor cells.�� Dr Lu found Artesunate and Dihydroartemisinin
(DHA) induce significant apoptosis in cancer cell lines over-expressing c-MYC
protein. Dr Lu found that DHA (and Artesunate) irreversibly down-regulated the
protein level of c-MYC and accelerated degradation of c-MYC protein in the
cancer cells. Dr Lu concluded that Artesunate would be useful in the treatment
of c-MYC-overexpressing cancer cell types, as c-Myc could serve as biomarker
candidate for prediction of antitumor efficacy of Artesunate.(65-66)
Over-Expression of c-MYC Associated with Aggressive Biology and Poor Prognosis
The c-Myc gene is a transcription factor regulating proliferation, growth, and
apoptosis. Overexpression or amplification of the c-Myc protein is associated
with aggressive cancer cell biology with poor prognosis. (67-70) Indeed, Dr Yi
studied a series of Mantle B-Cell Lymphoma patients with c-MYC overexpression in
Oncotarget 2015, stating:
��Intensive chemotherapy such as HyperCVAD/MA �� R did not improve the survival of
(lymphoma patients) with a c-MYC abnormality, and a new treatment strategy
should be developed.��
Dr Yi found that the highly aggressive biology of the c-MYC abnormality rendered
intensive chemotherapy futile, providing brief remission with no survival
benefit. The combination of an OX-Phos inhibitors (such as Metformin or
Doxyxyxline) targeting cancer stem cells along with the c-Myc inhibitor,
Artesunate, might represent such a new treatment strategy. We await NIH funded
confirmatory studies.
Metformin Targets Cancer Stem Cells
As mentioned above. cancer stems cells utilize mitochondrial OX-PHOS (oxidative
phosphorylation) for their energetic migratory and metastatic capacity.(11)
Indeed, Dr Diana Whitaker-Menezes in Cell Cycle 2011, reported hyperactive
oxidative mitochondrial metabolism in cancer cells was blocked by Metformin.(11)
Metformin treatment serves to induce purely glycolytic phenotype in surviving
cancer stem cells, now rendered sensitive to glucose starvation with a second
agent such as 2DG or high dose intravenous vitamin C, creating synthetic
lethality. (59)(5)(9-10)
Synthetic Lethality with Glucose Starvation
My previous article discussed the combination of Doxycycline with High Dose
Vitamin C as reported by the Lisanti Group��s work from Italy.(59) Dr Lisanti��s
group showed that converting cancer stem cells to a purely glycolytic phenotype
using repeated passages through higher doses of Doxycycline renders the cancer
stem cells sensitive to synthetic lethality with a second metabolic inhibitor.
One such second metabolic inhibitor is high dose IV vitamin C (Ascorbate), which
serves as a potent glycolysis inhibitor, 10 times more potent than 2-DG
(2-deoxy-glucose).(38-39)(59)
Similarly, by blocking mitochondrial oxidative phophorylation, Metformin
converts cancer stem cells to a purely glycolytic phenotype. Since mechanisms
differ, one might speculate a more robust result with combined use of both
Doxycycline and Metformin. Doxycycline impairs ribosomal protein production in
the mitochondria while, as mentioned above, metformin blocks the Hexokinase II
enzyme, and impairs Complex One in the electron transport chain (E.T.C.) in the
mitochondria.(58) Indeed, a clinical trial of the Doxycycline/ Metformin
combination is underway.(22)
Metformin Inhibits Progression of B Cell Lymphocytic Leukemia
In her 2015 article in Oncotarget, Dr Silvia Bruno, Silvia reports that
Metformin inhibits cell cycle progression of B-cell chronic lymphocytic leukemia
cells.�� (6) She reports that Metformin slowed the proliferation rate of the
cancer cells, as measured by the Ki-67 iindex:
��the fraction of Ki-67 positive cells was significantly lower in
metformin-treated CLL (cancer) cells than in untreated controls, in a
dose-dependent way.��(6)
In addition, the stimulated cancer cells had a 10 fold increase in glucose
uptake compared to quiescent cancer cells. This rise in glucose uptake was
remarkably inhibited by metformin.(6)
Metformin Inhibits B Cell Lymphoma
AMPK (AMP Kinase) activity is completely lost in lymphoma cells. (8) Dr W.Y. Shi
reported in Cell Death 2012 that metformin restores AMPK activity and blocks
lymphoma cell growth via inhibition of the mTOR pathway.(8) Metformin remarkably
blocked tumor growth in murine lymphoma xenografts at a concentration of 10
mM.(8)
Metformin Down-Regulates Inflammatory Cytokines, Enhances Immune System,
Inhibits Angiogenesis
Metformin down regulates inflammatory cytokines used for cancer growth and
signalling.(16) In addition, Metformin inhibits cancer cell induced
angiogenesis. (44-45) Moreover, Metformin has a beneficial effect on the immune
system by enhancing Killer T Cell anti-cancer activity. Dr. Kim reports in 2014
(16):
Metformin has been shown to decrease the production of inflammatory cytokines,
including TNF-a, interleukin-6, and vascular endothelial growth factor (VEGF),
through the inactivation of NF-KB and HIF-1a ��. metformin treatment inhibits
neoplastic angiogenesis, resulting in the reduction of tumor growth.(16)
Metformin Degrades Cyclin D1
Over-expression of the cell cycle regulator Cyclin D1 is a frequent feature in
cancer, and predicts early metastatic spread with poor prognosis.(19) Dr Gwak
reports in 2017 that Metformin degrades Cyclin D1 in ovarian cancer cell model
irrespective of p53 status.(19) This is done via metformin��s ability to
upregulate the AMPK/GSK3ß signaling axis . (19)
Metformin inhibits WNT pathway
In 2016 Dr Kamal reported that Metformin inhibits the WNT pathway in cancer
cells at commonly used doses.(20) This is indirect inhibition via the AMPK �C
MTOR signalling pathway. Downstream mediators of the WNT pathway are Cyclin D1
and C-Myc.(20)
Metformin Activates Immune Response to Cancer
Dr Chae reports in 2016 , ��metformin activates the T cell mediated immune
response against cancer cells.�� (23) In a 2015 report, Dr Eikawa��s group studied
the Immune-mediated antitumor effect of metformin using a mouse xenograft
model.(24) The authors state:
Metformin increased the number of CD8(+) tumor-infiltrating lymphocytes (TILs)
and protected them from apoptosis and exhaustion characterized by decreased
production of IL-2, TNFa, and IFN. CD8(+) TILs (tumor infiltrating lymphocytes)
capable of producing multiple cytokines were mainly PD-1(-)Tim-3(+), an Effector
Memory T Cell subset responsible for tumor rejection. �� (24)
Metformin for BRCA Gene Carriers
Metformin has been suggested for prevention and treatment of BRCA gene
carriers.(27)
Combination of Metformin with Propranolol (Beta Blocker)
The Beta-Blocker, Propranolol has been re purposed as an anti-cancer drug. Mode
of action is both directly on cancer cell metabolism as well as cancer
micro-environment, disrupting catecholammine cancer signalling. (36)(60-64) The
combination of metformin and propranolol has been found synergistic in Triple
Negative breast cancer cell lines studied in vitro.(36)(48)
Combined with Chemotherapy or Hyperthermia
Metformin was found synergistic with conventional chemotherapy providing better
results than chemo alone. This was thought to be due to metformin��s ability to
target cancer stem cells. (13) Hyperthermia, or use of a sauna, increased
Metformin cytotoxicity against cancer stem cells.(14)(29)
Conclusion: The evidence for Metformin as anti-cancer drug is overwhelming. It
is best used in combination with other agents such as Artesunate, Doxycycline,
and IV vitamin C to create Synthetic Lethality and overcome resistant cell
types. There is an urgent need for NIH funding for studies confirming this
combination approach to eradicating cancer stem cells.
Jeffrey Dach MD
7450 Griffin Road
Suite 190
Davie, Fl 33314
954 792-4663
Links to Articles with Related Interest:
Artemisinin Anti-Cancer Gift from China
IV Vitamin C as Cancer Chemotherapy
Doxycycline IV Vitamin C Anticancer Combination
Metformin the Anti-Aging Miracle Drug
Cancer as a Metabolic Disease
This article is part two. For part one, click here.
Links and References
METFORMIN as Anticancer Drug
1) Evans, Josie MM, et al. ��Metformin and reduced risk of cancer in diabetic
patients.�� Bmj 330.7503 (2005): 1304-1305.
2) Kasznicki, Jacek, Agnieszka Sliwinska, and J��zef Drzewoski. ��Metformin in
cancer prevention and therapy.�� Annals of translational medicine 2.6 (2014).
numerous meta-analyses that confirmed that metformin reduces cancer incidence by
30-50%.
3) Sacco, Francesca, et al. ��The cell-autonomous mechanisms underlying the
activity of metformin as an anticancer drug.�� British journal of cancer 115.12
(2016): 1451.
4) Chen, Chuan-Mu, et al. ��Repurposing Metformin for Lung Cancer Management.�� A
Global Scientific Vision-Prevention, Diagnosis, and Treatment of Lung Cancer.
InTech, 2017.
In this article, we introduced the background knowledge of lung cancer
management and considered repurposing old drugs to overcome therapy bottleneck.
We chose metformin to prove both its antihyperglycemia and antitumor formation
effects. Based on the metformin-related AMPK-dependent pathway, we tried to
explore the AMPK-independent pathway in inhibition of lung tumorigenesis by
metformin.
Initially, Evans et al. [2] observed that patients with type 2 diabetes mellitus
(DM) under metformin treatment had a reduction of cancer incidence. It caused a
23% reduction of risk of any cancer for the metformin group.
Metformin can accumulate within the matrix of mitochondria, and it could exert
the inhibition of the complex I of the mitochondrial electron transport chain.
Metformin can activate AMPK to initiate the downstream signal transduction to
affect the transcription of tumor suppressor liver kinase B1 (LKB1) [14]. When
metformin-related AMPK dependent pathway is affected, the inhibition of mTOR
signal transduction and reduction of cancer cell proliferation are achieved [
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
5) Menendez, Javier A., et al. ��Metformin is synthetically lethal with glucose
withdrawal in cancer cells.�� Cell cycle 11.15 (2012): 2782-2792.
we recently hypothesized that stress-energy mimickers such as the AMPK agonist
metformin should produce metabolic
synthetic lethality in a glucose-starved cell culture milieu imitating the
adverse tumor growth conditions in vivo.
representative cell models of breast cancer heterogeneity underwent massive
apoptosis (by > 90% in some cases) when glucose-starved cell cultures were
supplemented with metformin.
the preferential killing of cancer stem cells (CSC) by metformin may simply
expose the best-case scenario for its synthetically lethal activity because an
increased dependency on Warburg-like aerobic glycolysis (hyperglycolytic
phenotype) is critical to sustain CSC stemness and immortality;
6) Bruno, Silvia, et al. ��Metformin inhibits cell cycle progression of B-cell
chronic lymphocytic leukemia cells.�� Oncotarget 6.26 (2015): 22624.
Recent studies have provided evidence that diabetic patients receiving metformin
have a reduced risk of developing cancer and decreased cancer mortality [13,
14].
metformin reduces tumor growth not only indirectly (systemic effect: glucose and
insulin lowering) but also by direct inhibition of energetic metabolism [18] and
inhibition of pathways involved in cell proliferation [18�C20], through both
AMPK-dependent [21, 22] and -independent mechanisms [23�C27].
the fraction of Ki-67 positive cells was significantly lower in
metformin-treated CLL cells than in untreated controls, in a dose-dependent way
(Figure2B).
Flow cytometric single-cell data of 2-NBDG fluorescence indicated that the
average uptake of 2-NBDG after 48 hours CD40L-stimulation was almost ten fold
the uptake of 2-NBDG in quiescent CLL cells (Figure ?(Figure5D).5D). The
presence of metformin during CLL cell activation remarkably inhibited this rise
(Figure ?(Figure5D5D).
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
7) Gu, Juan J., et al. ��Up-regulation of hexokinase II (HK) alters the glucose
metabolism and disrupts the mitochondrial potential in aggressive b-cell
lymphoma contributing to rituximab-chemotherapy resistance and is a clinically
relevant target for future therapeutic development.�� (2014): 1767-1767.
8) Shi, W. Y., et al. ��Therapeutic metformin/AMPK activation blocked lymphoma
cell growth via inhibition of mTOR pathway and induction of autophagy.�� Cell
death & disease 3.3 (2012): e275.
In vivo, metformin induced AMPK activation, mTOR inhibition and remarkably
blocked tumor growth in murine lymphoma xenografts. Of note, metformin was
equally effective when given orally.
As shown in Figure 1, the AMPK activity was completely lost in lymphoma cells.
Consistent with the downregulation of AMPK expression, increased phosphorylation
of mTOR, p70S6K and 4EBP1 were present in 77.3%, 66.7% and 69.7% of B-lymphoma
cases
��
In primary lymphoma cells, metformin resulted in significant growth
inhibition from the concentration of 10mM (Figure 2d). However, proliferation of
CD34+ cells isolated from human cord blood, a population relatively enriched in
hematopoietic progenitor cells, was not affected even at the concentrations up
to 120?mM, suggesting that metformin exerted no major cytotoxic effect on normal
hematopoietic precursors (Figure 2e).
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
HEXOKINASE II
very important!! Use figure 2��shows molecular binding of Metformin into
hexokinase 2
LUNG CANCER CELL MODEL
9) Salani, Barbara, et al. ��Metformin impairs glucose consumption and survival
in Calu-1 cells by direct inhibition of hexokinase-II.�� Scientific reports 3
(2013).
The anti-hyperglycaemic drug metformin has important anticancer properties as
shown by the direct inhibition of cancer cells proliferation. Tumor cells avidly
use glucose as a source for energy production and cell building blocks. Critical
to this phenotype is the production of glucose-6-phosphate (G6P), catalysed by
hexokinases (HK) I and II, whose role in glucose retention and metabolism is
highly advantageous for cell survival and proliferation. Here we show that
metformin impairs the enzymatic function of HKI and II in Calu-1 cells. This
inhibition virtually abolishes cell glucose uptake and phosphorylation as
documented by the reduced entrapment of 18F-fluorodeoxyglucose.
In-silico models indicate that this action is due to metformin capability to
mimic G6P features by steadily binding its pocket in HKII. The impairment of
this energy source results in mitochondrial depolarization and subsequent cell
death. These results could represent a starting point to open effective
strategies in cancer prevention and treatment.
One of the primary metabolic changes observed in malignant transformation is an
increased catabolic glucose metabolism characterized by high rates of anaerobic
glycolysis regardless of oxygen concentration1. Critical to this phenotype is
glucose cellular entrapment by its conversion to glucose-6-phosphate (G6P). In
normal tissues, this basic process is regulated by four different hexokinase
(HK) isoforms indicating that regulation of glucose phosphorylation can vary in
different tissues under different condition2. In cancer cells, this reaction is
mainly catalysed by HK II whose glucose affinity and mitochondrial localization
are highly advantageous for cancer survival and growth3. Inhibition of HKII
enzymatic activity and its mitochondrial localization, are associated with
cancer cells death4,5.
Metformin effect on glucose metabolism in cancer cells
Metformin effect on cancer metabolism was evaluated by estimating Calu-1 cells
capability to retain FDG. Metformin treatment decreased tracer uptake in a dose
and time dependent manner up to its virtual abolition after 24 hours exposure to
10 mM drug concentration (32.7 �� 1.0% in controls vs 3.1 �� 0.4% in treated
cells, p < 0.0001)
Figure 2
Molecular mechanism of HK II inhibition by metformin.
metformin is thus prefigured as an uncompetitive (Figure S1F) and allosteric
inhibitor of HK II as only the enzyme-substrate complex can be bound.
reduced FDG uptake reflects a selective metformin induced impairment of glucose
phosphorylation.
Figure 3. Metformin displaces HK II from Mitochondria.
In conclusion the key finding of the present study is that metformin inhibits HK
II in Calu-1 cells through an allosteric modification of its molecular structure
blocking the synthesis of G6P. Moreover, our results demonstrate that HK II
inhibition by metformin causes release of this enzyme from the outer membrane of
mitochondria, thus leading to the activation of apoptotic signals.
BREAST CANCER CELL MODEL (SAME GROUP)
10) Marini, Cecilia, et al. ��Direct inhibition of hexokinase activity by
metformin at least partially impairs glucose metabolism and tumor growth in
experimental breast cancer.�� Cell cycle 12.22 (2013): 3490-3499.
Recently, we demonstrated that metformin impairs cancer energy asset in vitro
via a direct and selective enzymatic inhibition of HK isoforms I and II.19
Metformin strikingly impaired glucose consumption of MDA-MB-231 in a dose- and
time-dependent manner. Maximal effect occurred with exposure to 10 mM drug
concentration that progressively reduced FDG uptake down to its minimum values
after 48 h (Fig. 1A).
11) Whitaker-Menezes, Diana, et al. ��Hyperactivation of oxidative mitochondrial
metabolism in epithelial cancer cells in situ: visualizing the therapeutic
effects of metformin in tumor tissue.�� Cell cycle 10.23 (2011): 4047-4064.
Similar results were obtained with NADH activity staining, which measures
Complex I activity, and succinate dehydrogenase (SDH) activity staining, which
measures Complex II activity. COX (Cytochrome C Oxidase) and NADH activities
were blocked by electron transport inhibitors, such as Metformin. This has
mechanistic and clinical implications for using Metformin as an anti-cancer
drug, both for cancer therapy and chemo-prevention.
================
12) Metformin��an Adjunct Antineoplastic Therapy��Divergently Modulates Tumor
Metabolism and Proliferation, Interfering with Early Response Prediction by
18F-FDG PET Imaging
Peiman Habibollahi*,1, Nynke S. van den Berg*,1, Darshini Kuruppu1, Massimo
Loda2 and Umar Mahmood1
1Division of Nuclear Medicine and Molecular Imaging, Department of Radiology,
Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts;
and 2Department of Pathology, Dana-Farber Cancer Institute, Harvard Medical
School, Boston, Massachusetts
MET, through activation of the AMPK pathway, produces a dose-dependent increase
in tumor glucose uptake while decreasing cell proliferation in human and murine
colon cancer cells.
Cancer Stem Cells
13) Bradford, Sherry A., and A. Khan. ��Individualizing chemotherapy using the
anti-diabetic drug, metformin, as ��adjuvant��: an exploratory study.�� J Cancer
Sci Ther 5.6 (2013). Individualizing chemotherapy using metformin Bradford
Sherry J Cancer Sci Ther 2013
when metformin was combined(with chemo) , a synergistic effect was observed
resulting in high sensitivity (high cell kill);
metformin suppressed the generation of the breast cancer stem cell phenotype by
regulating stem cell properties including the epithelial-mesenchymal transition
status.
14) Lee, Hyemi, et al. ��Response of breast cancer cells and cancer stem cells to
metformin and hyperthermia alone or combined.�� PloS one 9.2 (2014): e87979.
In the present study, we show that metformin is preferentially cytotoxic to
Cancer Stem Cells (CSCs) relative to non-CSCs and that hyperthermia markedly
increases the metformin cytotoxicity against CSCs. For the first time, we
observed that hyperthermia activates AMPK, thereby suppressing mTOR. Such an
activation of AMPK by hyperthermia appeared to play an important role in the
hyperthermia-induced potentiation of metformin cytotoxicity against cancer
cells, particularly against CSCs.
15) Song, Chang W., et al. ��Metformin kills and radiosensitizes cancer cells and
preferentially kills cancer stem cells.�� Scientific reports 2 (2012): 362.
16) Kim, Tae Hun, et al. ��Metformin against cancer stem cells through the
modulation of energy metabolism: special considerations on ovarian cancer.��
BioMed research international 2014 (2014).
Activation of AMPK provides a metabolic barrier to reprogramming somatic cells
into stem cells [70]. The AMPK activators established a metabolic barrier to
reprogramming that could not be bypassed, even through p53 deficiency, a
fundamental mechanism to greatly improve the efficiency of stem cell production.
Metformin interferes with oxidative phosphorylation via interactions with
respiratory complex I, resulting in reduced ATP production and metabolic stress.
Metformin lowers plasma glucose levels by decreasing gluconeogenesis and glucose
uptake, resulting in lower circulating insulin and IGF-1 levels.
Furthermore, LKB1-deficient cells were more sensitive to metformin-induced
energy stress when cultured at low glucose concentrations and were unable to
compensate for the decreased cellular ATP concentration, causing cell death
[86]. These cytotoxic effects of metformin arise only in the context of a
genetic defect, such as loss of p53 and/or LKB1, that is present in the cancer
but not in the normal host tissue, providing opportunities for ��synthetic
lethality��
Metformin has been shown to decrease the production of inflammatory cytokines,
including TNF-a, interleukin-6, and vascular endothelial growth factor, through
the inactivation of NF-KB and HIF-1a [92�C94]. Emerging results demonstrating the
capacity of AMPK to inhibit the inflammatory responses suggest that metformin
may also target the inflammatory component present in the tumor microenvironment
[95]. In addition, several reports demonstrated that metformin treatment
inhibits neoplastic angiogenesis, resulting in the reduction of tumor growth
Complex I inhibition is partially involved in metformin��s growth inhibition of
EOC, possibly by increasing ROS and sensitizing cancer to additional oxidative
stress.
Metformin has been demonstrated to augment the effects of various
chemotherapeutic regimens by improving their efficacy as well as overcoming the
chemoresistance in EOC (Table 1) [63�C65, 67]. In fact, most in vitro studies
used doses of metformin between 1 and 40?mM, which is well above the feasible
therapeutic plasma levels (2.8�C15?µM) in humans [98]. Whereas the cytotoxic
effect of metformin alone was achieved at millimolar concentrations in most
studies, Erices et al. observed cytotoxicity with micromolar metformin in
combination with chemotherapy at concentrations where the chemotherapy alone
produced no loss in viability.
��
Metformin Inhibits Inflammation Needed by Cancer Stem Cells 17) Hirsch,
Heather A., Dimitrios Iliopoulos, and Kevin Struhl. ��Metformin inhibits the
inflammatory response associated with cellular transformation and cancer stem
cell growth.�� Proceedings of the National Academy of Sciences 110.3 (2013):
972-977. Metformin, the first-line drug for treating diabetes, inhibits cellular
transformation and selectively kills cancer stem cells in breast cancer cell
lines. In a Src-inducible model of cellular transformation, metformin inhibits
the earliest known step in the process, activation of the inflammatory
transcription factor NF-KappaBeta. Metformin strongly delays cellular
transformation in a manner similar to that occurring upon a weaker inflammatory
stimulus. Conversely, inhibition of transformation does not occur if metformin
is added after the initial inflammatory stimulus. The antitransformation effect
of metformin can be bypassed by overexpression of Lin28B or IL1ß, downstream
targets of NF-KB. Metformin preferentially inhibits nuclear translocation of
NF-KB and phosphorylation of STAT3 in cancer stem cells compared with non-stem
cancer cells in the same population. The ability of metformin to block tumor
growth and prolong remission in xenografts in combination with doxorubicin is
associated with decreased function of the inflammatory feedback loop. Lastly,
metformin-based combinatorial therapy is effective in xenografts involving
inflammatory prostate and melanoma cell lines, whereas it is ineffective in
noninflammatory cell lines from these lineages. Taken together, our observations
suggest that metformin inhibits a signal transduction pathway that results in an
inflammatory response. As metformin alters energy metabolism in diabetics, we
speculate that metformin may block a metabolic stress response that stimulates
the inflammatory pathway associated with a wide variety of cancers. IL-6 The
transformed cells contain a minority population of CSCs that have an enhanced
inflammatory loop that results in overproduction of IL6 (22, 24). The CSCs and
non-stem cancer cells (NSCCs) within the transformed population are in a dynamic
equilibrium that involves IL6 secretion Taken together, our observations suggest
that metformin inhibits the inflammatory pathway necessary for transformation
and CSC formation. 18) Hirsch, Heather A., et al. ��Metformin selectively targets
cancer stem cells, and acts together with chemotherapy to block tumor growth and
prolong remission.�� Cancer research 69.19 (2009): 7507-7511. Here, we show that
metformin selectively kills cancer stem cells in four genetically different
types of breast cancer. The combination of metformin and doxorubicin, a
well-defined chemotherapeutic drug, kills both cancer stem cells and non�Cstem
cancer cells in culture, and reduces tumor mass and prolongs remission much more
effectively than either drug alone in a xenograft mouse model. These
observations constitute independent support for the cancer stem cell hypothesis,
and they provide a rationale for why the combination of metformin and
chemotherapeutic drugs might improve treatment of patients with breast (and
possibly other) cancers.�� Metformin Degrades Reduces Cyclin D1 19) Gwak, HyeRan,
et al. �� Metformin induces degradation of cyclin D1 via AMPK/GSK3ß axis in
ovarian cancer. �� Molecular carcinogenesis 56.2 (2017): 349-358. Metformin,
which is widely used as an anti-diabetic drug, reduces cancer related morbidity
and mortality. However, the role of metformin in cancer is not fully understood.
Here, we first describe that the anti-cancer effect of metformin is mediated by
cyclin D1 deregulation via AMPK/GSK3ß axis in ovarian cancer cells. Metformin
promoted cytotoxic effects only in the cancer cells irrespective of the p53
status and not in the normal primary-cultured cells. Metformin induced the G1
cell cycle arrest, in parallel with a decrease in the protein expressions of
cyclin D1 without affecting its transcriptional levels. Using a proteasomal
inhibitor, we could address that metformin-induced decrease in cyclin D1 through
the ubiquitin/proteasome process. Cyclin D1 degradation by metformin requires
the activation of GSK3ß, as determined based on the treatment with GSK3ß
inhibitors. The activation of GSK3ß correlated with the inhibitory
phosphorylation by Akt as well as p70S6K through AMPK activation in response to
metformin. These findings suggested that the anticancer effects of metformin was
induced due to cyclin D1 degradation via AMPK/GSK3ß signaling axis that involved
the ubiquitin/proteasome pathway specifically in ovarian cancer cells. Metformin
Inhibits WNT pathway 20) Ahmed, Kamal, et al. ��A second WNT for old drugs: Drug
repositioning against WNT-dependent cancers.�� Cancers 8.7 (2016): 66. A recent
study revealed that anti-proliferative actions of metformin are also associated
with the indirect inhibition of the WNT pathway. Surprisingly, its effects are
mediated through its original target��AMPK, which then employs the MTOR signaling
pathway to promote the ubiquitination and proteasomal degradation of DVL3, one
of the principal WNT transducers [186]. This is very encouraging as it means
that the drug can be used at its normal dose to exert its anti-WNT effects, and
indeed the doses of metformin reported in the study corresponded to those found
for AMPK activation in human tissues [187]. Metformin Glioblastoma Stem Cells
21) Gritti, Marta, et al. ��Metformin repositioning as antitumoral agent:
selective antiproliferative effects in human glioblastoma stem cells, via
inhibition of CLIC1-mediated ion current.�� Oncotarget 5.22 (2014): 11252.
Clinical Trial 22) Metformin Hydrochloride and Doxycycline in Treating Patients
With Localized Breast or Uterine Cancer . Verified May 2017 by Sidney Kimmel
Cancer Center at Thomas Jefferson University 23) Chae, Young Kwang, et al.
��Repurposing metformin for cancer treatment: current clinical studies.��
Oncotarget 7.26 (2016): 40767. Preclinical studies have demonstrated several
anticancer molecular mechanisms of metformin including mTOR inhibition,
cytotoxic effects, and immunomodulation. Clinical trials in pre-surgical
endometrial cancer patients exhibited a significant decrease in Ki67 with
metformin monotherapy. Another interesting observation was made in patients with
breast cancer, wherein a trend towards improvement in cancer proliferation
markers was noted in patients without insulin resistance. metformin activates
the T cell mediated immune response against cancer cells. Animal models of
pancreatic cancer fed with metformin showed inhibition of insulin like growth
factor-1 (IGF-1) and mTOR, along with an increase in phosphorylated AMPK In
tobacco carcinogen induced lung cancer mice, the inhibition of insulin like
growth factor 1 receptor/insulin receptor (IGF- 1R/IR) by metformin decreased
the downstream signaling through Akt pathway. This reduced the activation of
mTOR in lung tissue which corresponded to a 72% reduction in tumor burden [13].
24) Proc Natl Acad Sci U S A. 2015 Feb 10;112(6):1809-14. Immune-mediated
antitumor effect by type 2 diabetes drug, metformin. Eikawa S1, Nishida M1,
Mizukami S1, Yamazaki C1, Nakayama E2, Udono H3. Metformin, a prescribed drug
for type 2 diabetes, has been reported to have anti-cancer effects; however, the
underlying mechanism is poorly understood. Here we show that this mechanism may
be immune-mediated. Metformin enabled normal but not T-cell-deficient SCID mice
to reject solid tumors. In addition, it increased the number of CD8(+)
tumor-infiltrating lymphocytes (TILs) and protected them from apoptosis and
exhaustion characterized by decreased production of IL-2, TNFa, and IFN?. CD8(+)
TILs capable of producing multiple cytokines were mainly PD-1(-)Tim-3(+), an
effector memory subset responsible for tumor rejection. Combined use of
metformin and cancer vaccine improved CD8(+) TIL multifunctionality. The
adoptive transfer of antigen-specific CD8(+) T cells treated with metformin
concentrations as low as 10 µM showed efficient migration into tumors while
maintaining multifunctionality in a manner sensitive to the AMP-activated
protein kinase (AMPK) inhibitor compound C. Therefore, a direct effect of
metformin on CD8(+) T cells is critical for protection against the inevitable
functional exhaustion in the tumor microenvironment. ���������������������������������C from
Targeting Cancer Stem Cells with NonToxic Therapies
��
25) Metformin Supplementation and Cancer Treatment
Feb 19, 2013 Brian D. Lawenda, M.D.
26) Bednar, Filip, and Diane M. Simeone. ��Metformin and cancer stem cells: old
drug, new targets.�� Cancer Prevention Research 5.3 (2012): 351-354.
Metformin for BRCA GENE Carriers
27) Cell Cycle. 2017 Jun 3;16(11):1022-1028. Metformin inhibits RANKL and
sensitizes cancer stem cells to denosumab. Cuy��s E1,2, Martin-Castillo B3,
Bosch-Barrera J4,5, Menendez JA1,2.
The increased propensity of BRCA1 mutation carriers to develop aggressive breast
tumors with stem-like properties begins to be understood in terms of
osteoprotegerin (OPG)-unrestricted cross-talk between RANKL-overproducing
progesterone-sensor cells and cancer-initiating RANK+ responder cells that
reside within pre-malignant BRCA1mut/+ breast epithelial tissue. We recently
proposed that, in the absence of hormone influence, cancer-initiating cells
might remain responsive to RANKL stimulation, and hence to the therapeutic
effects of the anti-RANKL antibody denosumab because genomic instability induced
by BRCA1 haploinsufficiency might suffice to cell-autonomously hyperactivate
RANKL gene expression. Here we report that the biguanide metformin prevents
BRCA1 haploinsufficiency-driven RANKL gene overexpression, thereby disrupting an
auto-regulatory feedback control of RANKL-addicted cancer stem cell-like states
within BRCA1mut/- cell populations. Moreover, metformin treatment elicits a
synergistic decline in the breast cancer-initiating cell population and its
self-renewal capacity in BRCA1-mutated basal-like breast cancer cells with bone
metastasis-initiation capacity that exhibit primary resistance to denosumab in
mammosphere assays. The specific targeting of RANKL/RANK signaling with
denosumab is expected to revolutionize prevention and treatment strategies
currently available for BRCA1 mutation carriers. Our findings provide a
rationale for new denosumab/metformin combinatorial strategies to clinically
manage RANKL-related breast oncogenesis and metastatic progression.
28) Metformin suppresses triple-negative breast cancer stem cells by targeting
KLF5 for degradation Cell Discovery 3, Article number: 17010 (2017) Peiguo Shi,
Wenjing Liu, Tala, Haixia Wang, Fubing Li, Hailin Zhang, Yingying Wu, Yanjie
Kong, Zhongmei Zhou, Chunyan Wang, Wenlin Chen, Rong Liu & Ceshi Chen
metformin significantly decreased the percentage of TNBC stem cells in two cell
lines. Metformin inhibits mitochondrial complex I, which results in a decrease
of ATP and the accumulation of AMP [32]. Accumulated AMP inhibits the generation
of cAMP [32]. It has been established that cAMP activates PKA [32] and that
activated PKA promotes mammary tumorigenesis [43]. Activated PKA also induces
tamoxifen resistance in breast cancer [44]. We found that PKA has an important
role in metformin-induced breast cancer stem cell suppression and that PKA is
highly activated in triple-negative breast tumors. In agreement with our
findings, metformin was reported to suppress breast cancer stem cells through
the disruption of ATP production [45].
Synergy with Hyperthermia
29) Lee, Hyemi, et al. ��Response of breast cancer cells and cancer stem cells to
metformin and hyperthermia alone or combined.�� PloS one 9.2 (2014): e87979.
Metformin, the most widely prescribed drug for treatment of type 2 diabetes, has
been shown to exert significant anticancer effects. Hyperthermia has been known
to kill cancer cells and enhance the efficacy of various anti-cancer drugs and
radiotherapy. We investigated the combined effects of metformin and hyperthermia
against MCF-7 and MDA-MB-231 human breast cancer cell, and MIA PaCa-2 human
pancreatic cancer cells. Incubation of breast cancer cells with 0.5�C10 mM
metformin for 48 h caused significant clonogenic cell death. Culturing breast
cancer cells with 30 µM metformin, clinically relevant plasma concentration of
metformin, significantly reduced the survival of cancer cells. Importantly,
metformin was preferentially cytotoxic to CD44high/CD24low cells of MCF-7 cells
and, CD44high/CD24high cells of MIA PaCa-2 cells, which are known to be cancer
stem cells (CSCs) of MCF-7 cells and MIA PaCa-2 cells, respectively. Heating at
42��C for 1 h was slightly toxic to both cancer cells and CSCs, and it markedly
enhanced the efficacy of metformin to kill cancer cells and CSCs. Metformin has
been reported to activate AMPK, thereby suppressing mTOR, which plays an
important role for protein synthesis, cell cycle progression, and cell survival.
For the first time, we show that hyperthermia activates AMPK and inactivates
mTOR and its downstream effector S6K. Furthermore, hyperthermia potentiated the
effect of metformin to activate AMPK and inactivate mTOR and S6K. Cell
proliferation was markedly suppressed by metformin or combination of metformin
and hyperthermia, which could be attributed to activation of AMPK leading to
inactivation of mTOR. It is conclude that the effects of metformin against
cancer cells including CSCs can be markedly enhanced by hyperthermia.
30) Metformin targets multiple signaling pathways in cancer
Yong Lei†, Yanhua Yi†, Yang Liu, Xia Liu, Evan T. Keller, Chao-Nan Qian, Jian
Zhang Chinese Journal of Cancer 2017 36:17
31) A phase II clinical trial of metformin as a cancer stem cell targeting agent
in stage IIc/III/IV ovarian, fallopian tube, and primary peritoneal cancer.
Meeting:2017 ASCO Annual Meeting Abstract No:5556
Poster Board Number:Poster Session (Board #378)
Citation:J Clin Oncol 35, 2017 (suppl; abstr 5556)
Author(s): Ronald J. Buckanovich, Jason Brown, Jessica Shank, Kent A. Griffith,
R. Kevin Reynolds, Carolyn Johnston, Karen McLean, Shitanshu Uppal, J. Rebecca
Liu, Lourdes Cabrera, Geeta Mehta; Department of Internal Medicine, University
of Michigan, Ann Arbor, MI; Department of Obstetrics and Gynecology, Naval
Medical Center San Diego, San Diego, CA; Department of Biostatistics, University
of Michigan, Ann Arbor, MI; Department of Obstetrics and Gynecology, University
of Michigan, Ann Arbor, MI; Department of Bioengineering, University of
Michigan, Ann Arbor, MI
Background: Epidemiologic and preclinical studies suggest that Metformin has
antitumor effects which may be due to an impact on cancer stem-like cells (CSC).
We present a phase II trial of metformin administered in combination with
chemotherapy for patients with advanced stage epithelial ovarian cancer (EOC).
Primary endpoints were 18 month progression free survival (PFS) and CSC number
in Metformin treated tumors. Methods: Thirty-eight patients with confirmed stage
IIC(n=1)/III(n=25)/IV(n=12) EOC were treated with either neoadjuvant metformin
followed primary debulking surgery and adjuvant Metformin+chemotherapy, or
neo-adjuvant metformin+chemotherapy, followed by interval debulking and adjuvant
chemotherapy+Metformin. Patients were evaluated for side effects, PFS and
overall survival (OS). Metformin treated tumors were evaluated for the presence
of CSC via FACS and sphere assays. Results: Thirty-two patients (84%) completed
at least six cycles of metformin+chemotherapy. Metformin was well tolerated with
only one grade III/IV treatment-related adverse event (3%) noted. Common adverse
effects were diarrhea (18%) and nausea (16%). Eighteen month PFS was 65.4% (95%
confidence interval 47.9-78.3), Median PFS was 21.7 months (CI-17-26.7).
Estimated three year OS was 73.5% (CI-54.7-84.3) with median OS not reached
after a media follow-up of 33 months. Finally, tumors treated with metformin
were noted to have a 3-fold decrease in ALDH+ CSC at baseline, increased
sensitivity to Cisplatin in vitro, and a reduced ability to amplify ALDH+ CSC
with passage in vitro. Conclusions: This is the first prospective study of
Metformin in EOC patients. Translational studies confirm an impact of metformin
on CSC. Metformin was well tolerated and outcome results were favorable,
supporting the use of Metformin in phase-III studies. Clinical trial
information: NCT01579812
32) Leão, Ricardo, et al. ��Cancer Stem Cells in Prostate Cancer: Implications
for Targeted Therapy.�� Urologia Internationalis (2017).
33) DORAN, Elena, and Andrew P. HALESTRAP. ��Evidence that metformin exerts its
anti-diabetic effects through inhibition of complex 1 of the mitochondrial
respiratory chain.�� Biochemical Journal 348.3 (2000): 607-614. Metformin exerts
effects through inhibition of complex 1 of the mitochondrial respiratory chain
DORAN Elena Biochemical Journal 2000
34) Ward, N. P., et al. ��Complex I inhibition augments dichloroacetate
cytotoxicity through enhancing oxidative stress in VM-M3 glioblastoma cells.��
PloS one 12.6 (2017): e0180061.
The robust glycolytic metabolism of glioblastoma multiforme (GBM) has proven
them susceptible to increases in oxidative metabolism induced by the pyruvate
mimetic dichloroacetate (DCA). Recent reports demonstrate that the anti-diabetic
drug metformin enhances the damaging oxidative stress associated with DCA
treatment in cancer cells. We sought to elucidate the role of metformin��s
reported activity as a mitochondrial complex I inhibitor in the enhancement of
DCA cytotoxicity in VM-M3 GBM cells. Metformin potentiated DCA-induced
superoxide production, which was required for enhanced cytotoxicity towards
VM-M3 cells observed with the combination. Similarly, rotenone enhanced
oxidative stress resultant from DCA treatment and this too was required for the
noted augmentation of cytotoxicity. Adenosine monophosphate kinase (AMPK)
activation was not observed with the concentration of metformin required to
enhance DCA activity. Moreover, addition of an activator of AMPK did not enhance
DCA cytotoxicity, whereas an inhibitor of AMPK heightened the cytotoxicity of
the combination. Our data indicate that metformin enhancement of DCA
cytotoxicity is dependent on complex I inhibition. Particularly, that complex I
inhibition cooperates with DCA-induction of glucose oxidation to enhance
cytotoxic oxidative stress in VM-M3 GBM cells.
These data suggest that complex I inhibition cooperates with DCA activation of
oxidative glucose metabolism to promote catastrophic oxidative stress in VM-M3
glioblastoma cells.
136) Wheaton, William W., et al. ��Metformin inhibits mitochondrial complex I of
cancer cells to reduce tumorigenesis.�� Elife 3 (2014): e02242.
35) Griss, Takla, et al. ��Metformin antagonizes cancer cell proliferation by
suppressing mitochondrial-dependent biosynthesis.�� PLoS biology 13.12 (2015):
e1002309.
Metformin Propranolol Combination
36) also see (48) Rico, Mar��a, et al. ��Metformin and propranolol combination
prevents cancer progression and metastasis in different breast cancer models.��
Oncotarget 8.2 (2017): 2874. Metformin and propranolol combination prevents
cancer progression and metastasis in different breast cancer models.
37) Saengboonmee, Charupong, et al. ��Metformin Exerts Antiproliferative and
Anti-metastatic Effects Against Cholangiocarcinoma Cells by Targeting STAT3 and
NF-KB.�� Anticancer research 37.1 (2017): 115-123.
38) Zhu, Jie, et al. ��Targeting cancer cell metabolism: The combination of
metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38
MAPK/JNK signaling pathway.�� American journal of translational research 8.11
(2016): 4812.
Targeting cancer cell metabolism is a new promising strategy to fight cancer.
Metformin, a first-line treatment for type 2 diabetes mellitus, exerts
anti-cancer and anti-proliferative action. 2-deoxyglucose (2-DG), a glucose
analog, works as a competitive inhibitor of glycolysis. In this study, we show
for the first time that metformin in combination with 2-DG inhibited growth,
migration, invasion and induced cell cycle arrest of ovarian cancer cells in
vitro. Moreover, metformin and 2-DG could efficiently induce apoptosis in
ovarian cancer cells, which was achieved by activating p38 MAPK and JNK
pathways. Our study reinforces the growing interest of metabolic interference in
cancer therapy and highlights the potential use of the combination of metformin
and 2-DG as an anti-tumor treatment in ovarian cancer.
39) Ben, Sahra I., et al. ��Targeting cancer cell metabolism: the combination of
metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer
cells.�� Cancer research 70.6 (2010): 2465.
Metformin targets cancer stem cells
40) Bost, F., et al. ��Energy disruptors: rising stars in anticancer therapy?.��
Oncogenesis 5.1 (2016): e188. Energy disruptors: rising stars in anticancer
therapy?
Biguanides Metformin target cancer stem cells
Cancer stem cells (CSCs) are localized in tumors, resistant to chemotherapy, and
capable of self-renewal and differentiation. Importantly, CSCs are the cause of
disease relapse. Biguanides appear to target this cancer cell population. The
combination of metformin with chemotherapy has been shown to be more efficient
than either drug alone in xenograft models using several cancer cell lines, and
this treatment specifically targets CSCs. Furthermore, treatment with both drugs
significantly prolongs the remission following xenograft implantation.33, 34
This specific effect was confirmed in several other cancer models, including
pancreas, breast and ovary.35, 36, 37 Interestingly, Sancho et al. have shown
that CSCs rely mainly on OXPHOS and are unable to effectively induce glycolysis
to compensate for reduced ATP production upon mitochondrial inhibition. The
level of MYC expression controls this metabolic characteristic of CSCs; low MYC
expression allows high PGC1-a expression, which results in enhanced
mitochondrial biogenesis. Consequently, the observation that metformin
specifically affects the viability of CSCs to a greater extent than non-CSCs is
not surprising.38
��
Metformin and Cancer Stem cells
41) Patricia Sancho, et al. ��MYC/PGC-1a Balance Determines the Metabolic
Phenotype and Plasticity of Pancreatic Cancer Stem Cells.�� Cell Metabolism 22
(2015): 1-16. MYC_PGC1a Determines Metabolic Phenotype Pancreatic Cancer Stem
Cells Patricia Sancho Cell Metabolism 2015
The anti-diabetic drug metformin targets pancreatic cancer stem cells (CSCs),
but not their differentiated progenies (non-CSCs), which may be related to
distinct metabolic phenotypes. Here we conclusively demonstrate that while
non-CSCs were highly glycolytic, CSCs were dependent on oxidative metabolism
(OXPHOS) with very limited metabolic plasticity. Thus, mitochondrial inhibition,
e.g., by metformin, translated into energy crisis and apoptosis. However,
resistant CSC clones eventually emerged during treatment with metformin due to
their intermediate glycolytic/respiratory phenotype. Mechanistically,
suppression of MYC and subsequent increase of PGC-1a were identified as key
determinants for the OXPHOS dependency of CSCs, which was abolished in resistant
CSC clones. Intriguingly, no resistance was observed for the mitochondrial ROS
inducer menadione and resistance could also be prevented/reversed for metformin
by genetic/pharmacological inhibition of MYC. Thus, the specific metabolic
features of pancreatic CSCs are amendable to therapeutic intervention and could
provide the basis for developing more effective therapies to combat this lethal
cancer.
Metformin Enhances Rituxan in B-cell Lymphoma
42) http://ascopubs.org/doi/abs/10.1200/jco.2015.33.15_suppl.e19513
Metformin enhances the activity of rituximab in B-cell lymphoma pre-clinical
models. Priyank P. Patel, Juan J Gu, Cory Mavis, Myron Stefan Czuczman,
Francisco J. Hernandez-Ilizaliturri
Background: The Warburg effect is primarily observed in rapidly growing tumors
including aggressive B-cell lymphomas and is thought to be a consequence of the
progression to cancer rather than the cause of it and altering the glucose
metabolism in cancer cells appears to be an attractive strategy in cancer
medicine. Retrospective studies have shown survival benefit in solid tumors and
diffuse large B-cell lymphoma cohorts who were on metformin for type-2 diabetes.
In an attempt to characterize the mechanism by which metformin affects the
biology of B-cell lymphoma, we studied its effect on rituximab activity.
Methods: A panel of B-cell lymphoma cells was exposed to metformin +/- rituximab
or isotype control, changes in cell cycle distribution or induction of apoptosis
was determined by flow cytometry. Antibody-dependent cellular cytotoxicity
(ADCC) and complement mediated cytotoxicity (CMC) assays were performed to
demonstrate changes in sensitivity to rituximab following metformin exposure.
For in vivo studies, SCID mice were inoculated via tail vein injection (iv) with
Raji cells (day 0) and assigned to observation, rituximab (at 10mg/kg/dose on
days +3,7,10 and 14), metformin (at 2mg/ml in drinking water) or metformin and
rituximab. Differences in survival (measured at the time for limb paralysis
development) were evaluated by log-rank test between treatment arms. Results: In
vitro exposure to metformin resulted in S/G1 cell cycle arrest and induction of
apoptosis in a dose-dependent manner. Metformin enhanced the anti-proliferative
effects of mAbs targeting CD20. Moreover, pre-incubation of lymphoma cells
enhanced rituximab-mediated CMC. In vivo, significant improvement in survival
was observed in metformin + rituximab arm (mean survival not reached at 69+/-
5.3 days) compared to rituximab (mean survival 57.1 +/- 4.2 days) (p = 0.05).
Conclusions: Our data suggests that metformin inhibits the proliferation of
B-cell lymphoma cell lines and enhances the anti-tumor activity of rituximab.
Our finding highlights a potential role for metformin in the treatment of B-cell
malignancies.
43) Rodr��guez-Lirio, A., et al. ��Metformin induces cell cycle arrest and
apoptosis in drug-resistant leukemia cells.�� Leukemia research and treatment
2015 (2015).
=======================================
Anti Angiogenesis
44) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4792577/
Wang, Jichang, et al. ��Suppression of tumor angiogenesis by metformin treatment
via a mechanism linked to targeting of HER2/HIF-1a/VEGF secretion axis.��
Oncotarget 6.42 (2015): 44579.
45) https://www.ncbi.nlm.nih.gov/pubmed/25196138/
Int J Cancer. 2015 Mar 15;136(6):E534-44. doi: 10.1002/ijc.29193. Epub 2014 Sep
18. The biguanides metformin and phenformin inhibit angiogenesis, local and
metastatic growth of breast cancer by targeting both neoplastic and
microenvironment cells. Orecchioni S1, Reggiani F, Talarico G, Mancuso P,
Calleri A, Gregato G, Labanca V, Noonan DM, Dallaglio K, Albini A, Bertolini F.
The human white adipose tissue (WAT) contains progenitors with cooperative roles
in breast cancer (BC) angiogenesis, local and metastatic progression. The
biguanide Metformin (Met), commonly used for Type 2 diabetes, might have
activity against BC and was found to inhibit angiogenesis in vivo. We studied
Met and another biguanide, phenformin (Phe), in vitro and in vivo in BC models.
In vitro, biguanides activated AMPK, inhibited Complex 1 of the respiratory
chain and induced apoptosis of BC and WAT endothelial cells. In coculture,
biguanides inhibited the production of several angiogenic proteins. In vivo,
biguanides inhibited local and metastatic growth of triple negative and HER2+ BC
in immune-competent and immune-deficient mice orthotopically injected with BC.
Biguanides inhibited local and metastatic BC growth in a genetically engineered
murine model model of HER2+ BC. In vivo, biguanides increased pimonidazole
binding (but not HIF-1 expression) of WAT progenitors, reduced tumor microvessel
density and altered the vascular pericyte/endothelial cell ratio, so that cancer
vessels displayed a dysplastic phenotype. Phe was significantly more active than
Met both in vitro and in vivo. Considering their safety profile, biguanides
deserve to be further investigated for BC prevention in high-risk subjects, in
combination with chemo and/or targeted therapy and/or as post-therapy
consolidation or maintenance therapy for the prevention of BC recurrence.
VEGF Overexpressed in Mantle Cell Lymphoma
46) Anticancer Res. 2002 Sep-Oct;22(5):2899-901.
Immunohistochemical detection of C-kit (CD117) and vascular endothelial growth
factor (VEGF) overexpression in mantle cell lymphoma. Potti A1, Ganti AK, Kargas
S, Koch M.
Mantle cell lymphoma (MCL) is a low-grade lymphoproliferative malignancy that is
extremely refractory to chemotherapy. Commonly used treatments have yielded
unfavorable response rates (30% complete remission). We evaluated the incidence
of c-kit (CD117) and vascular endothelial growth factor (VEGF) overexpression in
patients with MCL in an effort to identify possible targets for therapeutic
Patients with a diagnosis of MCL based on CD5 positivity associated with cyclin
D1 positivity and CD23 negativity on the lymph node/bone marrow specimen were
included in our retrospective study. CD117 overexpression was performed using
immunohistochemistry on archival specimens. VEGF expression was detected by the
avidin-biotin-complex method.
RESULTS:Between 1997 and 2001, we identified 17 patients with MCL (9 males, 8
females) with a mean age of 57 years (age range: 42-66 years). The mean overall
survival was 34 months (range: 11-60 months). VEGF expression was identified in
7 out of 17 (41.18%) patients with MCL. Among the VEGF-positive patients (n = 7,
41.1%), the mean survival was 24 months (range: 11-42 months), while patients
without VEGF expression (n = 10, 58.9%) had a mean survival of 44 months (range:
21-60 months). CD117 expression was identified in only 2 out of 17 (1.17%)
patients in our study.
CONCLUSION:Our study evaluated the role of c-kit and VEGF overexpression in MCL.
Although CD117 may not be of therapeutic significance, target-directed signal
transduction inhibition therapy using VEGF-inhibitors may be a distinct
possibility in a select group of patients with MCL. Future larger studies are
urgently needed to elaborate the role of VEGF in MCL.
metformin with 2DG
47) http://www.fasebj.org/content/31/1_Supplement/824.5
Anti-angiogenic Effects of Metformin in 2-Deoxyglucose Treated Microvascular
Endothelial Cells: Role of Thrombospondin-1
Samson Mathews Samuel, Suparna Ghosh, Yasser Majeed, Hong Ding and Chris R
Triggle. Pharmacology, Weill Cornell Medicine-Qatar, Doha, Qatar
Background & objective The biguanide metformin, which is widely used in the
management of type 2 diabetes, has received considerable interest as a potential
anti-cancer agent in many forms of cancer. However, the effect of metformin in
tumor endothelial cells (TECs) has not been studied. TECs play a key role in
tumor angiogenesis thereby supporting tumor growth, cancer cell survival and
metastasis and hence targeting TECs in order to inhibit angiogenesis could prove
to be a potential anti-angiogenic cancer therapy in a wide range of cancers.
Reports show that metformin increases the levels of anti-angiogenic
thrombospondin-1 (TSP1) in the serum of women with polycystic ovarian syndrome
(1). Data from our preliminary studies in Mouse Microvascular Endothelial Cells
(MMECs) that overexpress angiogenic VEGF (CRL-2460; ATCC; sarcoma cell line;
cell type: endothelial) has revealed that metformin (2mM) significantly
increased TSP1 in glucose-starved cells when compared to metformin treated
glucose exposed cells. We therefore investigated the effects of metformin on
angiogenesis, in MMECs in combination with the glycolytic inhibitor,
2-deoxyglucose (2DG).
Materials & methods MMECs were treated with 2DG (5mM) for 48h in the presence or
absence of metformin (2mM) & western blot analysis was performed to assess the
status of angiogenic and anti-angiogenic marker proteins. Alternatively, the
putative AMPK activator, A769662 (150mM), was also used instead of metformin.
Cell proliferation assay, cell migration assays and wound healing assays were
also performed.
Results We observed a significant up-regulation of TSP1, while the levels of
pVEGFR2 (Y1175) were markedly decreased; in 2DG-exposed cells treated with
metformin (2mM) when compared to cells maintained in normal glucose exposed
cells that were treated with metformin. A769662, did not show any effect on
TSP-1 levels in normal glucose or 2DG-exposed cells. Furthermore, treatment with
metformin (2mM) in 2DG exposed cells significantly increased the levels of pRap
(S792), which in turn should have caused the inhibition of the mTOR pathway as
evidenced by the significant decrease in the levels of pmTOR (S2448), p4E-BP1
(T36/47), pS6 (S235/236) and pS6 (240/244) when compared to metformin treated
normal glucose exposed cells. Levels of cell cycle related proteins such as
pCycB1 (S147), CycD1 and CycD2 significantly decreased in cells treated with a
combination of 2DG and metformin when compared to cells that were treated with
either 2DG or metformin alone. The rate of cell proliferation and endothelial
cell migration also significantly decreased in cells treated with a combination
of 2DG and metformin when compared to cells that were treated with either 2DG or
metformin alone.
Conclusion Our findings show that using metformin in combination with 2DG has an
anti-angiogenic activity associated with a significant up-regulation of
thrombospondin-1 and could prove to be therapeutic strategy in a wide range of
cancers.
Metformin and Propranolol
48) Rico, Mar��a, et al. ��Metformin and propranolol combination prevents
cancer progression and metastasis in different breast cancer models.�� Oncotarget
8.2 (2017): 2874.
Taken together our results suggest that metformin plus propranolol combined
treatment might be beneficial for triple negative breast cancer control, with no
symptoms of toxicity.
The indirect anticancer effect of Met involves insulin dependent actions,
associated with a reduction of insulin circulating levels that lead to a
decrease in the mitogenic and antiapoptotic potential of insulin. In this way,
Met may diminish the pro-stimulatory effect of insulin on cancer cells. Met
direct effects are linked to inhibition of mitochondrial complex I [16]. This
inhibition interrupts mitochondrial respiration, decreasing proton-driven
synthesis of ATP, causing cellular energetic stress and elevation of the AMP:ATP
ratio which, in turn, activates AMP-activated protein kinase (AMPK), a key
cellular energy sensor kinase [10]. AMPK activation leads to a reduction in
mammalian target of rapamycin (mTOR) signaling, protein synthesis and
proliferation [16�C18].
Propranolol (Prop) is a noncardioselective ß-adrenergic receptor blocker with
reported antioxidant and anti-inflammatory properties, used traditionally for
hypertension, angina pectoris, myocardial infarction, migraines, anxiety
disorders, and tremor [19]. It was previously shown that Prop reduces
intracellular calcium levels, Bax-mediated cytochrome C release and inhibits
protein kinase C (PKC) activity in a ß-adrenoreceptor independent manner
[19�C21], and it induces cell cycle arrest and apoptosis via Akt/MAPK pathway in
melanoma cells [22]. Many studies in humans have demonstrated its efficacy for
the treatment of infantile haemangioma. In this regard, it seems that Prop
exerts its suppressive effects acting through the HIF-1a-VEGF-A angiogenesis
axis, with effects mediated through the PI3K/Akt and p38/MAPK pathways [23]. In
relation to breast cancer, retrospective studies reported an improved survival
with reduction in the risk of recurrence in woman receiving this ß-blocker
therapy [24].����
49) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3248132/
Oncotarget. 2010 Nov; 1(7): 466�C469. Beta-adrenergic signaling, a novel target
for cancer therapy? Hildegard M. Schuller
itraconazole
50) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4703001/
Head, Sarah A., et al. ��Antifungal drug itraconazole targets VDAC1 to modulate
the AMPK/mTOR signaling axis in endothelial cells.�� Proceedings of the National
Academy of Sciences 112.52 (2015): E7276-E7285.
free pdf
51) Tsubamoto, Hiroshi, et al. ��Repurposing itraconazole as an anticancer
agent.�� Oncology Letters 14.2 (2017): 1240-1246.
Itraconazole may be a promising agent for targeting CSCs in relapsed disease of
multiple types of cancer; therefore, further preclinical studies on CSCs and the
surrounding stroma cells are warranted.
52) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4406527/
Pantziarka, Pan, et al. ��Repurposing Drugs in Oncology (ReDO)��itraconazole as an
anti-cancer agent.�� ecancermedicalscience 9 (2015).
========================================
53) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4096030/
Pantziarka, Pan, et al. ��The repurposing drugs in oncology (ReDO) project.��
ecancermedicalscience 8 (2014).
Mebendazole Anthelminthic Threadworm infections Generic
Nitroglycerin Vasodilator Angina Generic
Cimetidine H2-receptor antagonist Peptic ulcer Generic
Clarithromycin Antibiotic Respiratory tract infection Generic
Diclofenac NSAID Pain relief Generic
Itraconazole Antifungal Broad spectrum antifungal Generic
pdf
55) Pantziarka, Pan, et al. ��Repurposing drugs in your medicine cabinet:
untapped opportunities for cancer therapy?.�� Future oncology 11.2 (2015):
181-184.
56) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4488459/
McCabe, Bronagh, Fabio Liberante, and Ken I. Mills. ��Repurposing medicinal
compounds for blood cancer treatment.�� Annals of hematology 94.8 (2015):
1267-1276.
57) Cao, Jia, et al. ��Low concentrations of metformin suppress glucose
production in hepatocytes through AMP-activated protein kinase (AMPK).�� Journal
of Biological Chemistry 289.30 (2014): 20435-20446.
58) Mathupala, S. P., YH and Ko, and P. L. Pedersen. ��Hexokinase II: cancer��s
double-edged sword acting as both facilitator and gatekeeper of malignancy when
bound to mitochondria.�� Oncogene 25.34 (2006): 4777.
59) De Francesco, E. M., Michael Lisanti et al. ��Vitamin C and Doxycycline: a
synthetic lethal combination therapy targeting metabolic flexibility in cancer
stem cells (CSCs).�� Oncotarget (2017). Vitamin C and Doxycycline: A synthetic
lethal combination therapy targeting metabolic flexibility in cancer stem cells
(CSCs).
60) Sci Rep. 2016; 6: 18673. Aspirin and atenolol enhance metformin activity
against breast cancer by targeting both neoplastic and microenvironment cells
Giovanna Talarico,1,* Stefania Orecchioni,1,* Katiuscia Dallaglio,2 Francesca
Reggiani,1 Patrizia Mancuso,1 Angelica Calleri,1 Giuliana Gregato,1 Valentina
Labanca,1 Teresa Rossi,2 Douglas M. Noonan,3,4 Adriana Albini,3,* and Francesco
Bertolinia,1,*
61) http://jnm.snmjournals.org/content/55/3/439.full
Propranolol Inhibits Glucose Metabolism and 18F-FDG Uptake of Breast Cancer
Through Post-transcriptional Downregulation of Hexokinase-2. Fei Kang1, Wenhui
Ma1, Xiaowei Ma1, Yahui Shao1, Weidong Yang1, Xiaoyuan Chen2, Liwen Li1,3 and
Jing Wang1 Author Affiliations. 1Department of Nuclear Medicine, Xijing
Hospital, Fourth Military Medical University, Xi��an, China
62) ��-adrenoceptor can influence the 18F-FDG PET imaging of breast cancer
through its regulation to the posttranscriptional level of hexokinase-2. Fei
Kang1, Xiaowei Ma1, Wenhui Ma1, Yahui Shao1, Liwen Li1, Weidong Yang1 and Jing
Wang1
63) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5102691/
Ecancer medical science. 2016; 10: 680. Repurposing Drugs in Oncology
(ReDO)��Propranolol as an anti-cancer agent. Pan Pantziarka,1,2 Gauthier Bouche,1
Vidula Sukhatme,3 Lydie Meheus,1 Ilse Rooman,1,4 and Vikas P Sukhatme3,5
Dosage
The PRO dose varies by indication. The anti-hypertensive dose is in the range
160 �C 320 mg/day, starting at 80 mg and increasing as required to a maintenance
dose that is generally 160 mg �C 240 mg, in divided doses or as once a day use of
extended release tablets. For angina the dose is 120 �C 240 mg/day. Migraine
prophylaxis is in the range 80 �C 240 mg/day [4].
64) http://www.tandfonline.com/doi/full/10.3109/13880209.2014.892513
İşeri, Özlem Darcansoy, et al. ��beta-Adrenoreceptor antagonists reduce cancer
cell proliferation, invasion, and migration.�� Pharmaceutical biology 52.11
(2014): 1374-1381.
Artesunate Degrades c-MYC and Inhibits WNT
65) Lu, Jin-Jian, et al. ��Dihydroartemisinin accelerates c-MYC oncoprotein
degradation and induces apoptosis in c-MYC-overexpressing tumor cells.��
Biochemical pharmacology 80.1 (2010): 22-30.
Artemisinin and its derivatives (ARTs) are effective antimalarial drugs and also
possess profound anticancer activity. However, the mechanism accounted for its
distinctive activity in tumor cells remains unelucidated. We computed Pair wise
Pearson correlation coefficients to identify genes that show significant
correlation with ARTs activity in NCI-55 cell lines using data obtained from
studies with HG-U133A Affymetrix chip. We found c-myc is one of the genes that
showed the highest positive correlation coefficients among the probe sets
analyzed (r=0.585, P<0.001). Dihydroartemisinin (DHA), the main active
metabolite of ARTs, induced significant apoptosis in HL-60 and HCT116 cells that
express high levels of c-MYC. Stable knockdown of c-myc abrogated DHA-induced
apoptosis in HCT116 cells. Conversely, forced expression of c-myc in NIH3T3
cells sensitized these cells to DHA-induced apoptosis. Interestingly, DHA
irreversibly down-regulated the protein level of c-MYC in DHA-sensitive HCT116
cells, which is consistent to persistent G1 phase arrest induced by DHA. Further
studies demonstrated that DHA accelerated the degradation of c-MYC protein and
this process was blocked by pretreatment with the proteasome inhibitor MG-132 or
GSK 3beta inhibitor LiCl in HCT116 cells. Taken together, ARTs might be useful
in the treatment of c-MYC-overexpressing tumors. We also suggest that c-MYC may
potentially be a biomarker candidate for prediction of the antitumor efficacies
of ARTs.
66) Oncotarget. 2015 Jun 30;6(18):15857-70.
Structurally diverse c-Myc inhibitors share a common mechanism of action
involving ATP depletion. Wang H1, Sharma L1, Lu J1, Finch P1, Fletcher S2,3,
Prochownik EV1,4,5.
The c-Myc (Myc) oncoprotein is deregulated in a large proportion of diverse
human cancers. Considerable effort has therefore been directed at identifying
pharmacologic inhibitors as potential anti-neoplastic agents. Three such groups
of small molecule inhibitors have been described. The first is comprised of
so-called ��direct�� inhibitors, which perturb Myc��s ability to form productive
DNA-binding heterodimers in association with its partner, Max. The second group
is comprised of indirect inhibitors, which largely function by targeting the
BET-domain protein BRD4 to prevent the proper formation of transcriptional
complexes that assemble in response to Myc-Max DNA binding. Thirdly, synthetic
lethal inhibitors cause the selective apoptosis of Myc over-expressing either by
promoting mitotic catastrophe or altering Myc protein stability. We report here
a common mechanism by which all Myc inhibitors, irrespective of class, lead to
eventual cellular demise. This involves the depletion of ATP stores due to
mitochondrial dysfunction and the eventual down-regulation of Myc protein. The
accompanying metabolic de-regulation causes neutral lipid accumulation, cell
cycle arrest, and an attempt to rectify the ATP deficit by up-regulating
AMP-activated protein kinase (AMPK). These responses are ultimately futile due
to the lack of functional Myc to support the requisite anabolic response.
Finally, the effects of Myc depletion on ATP levels, cell cycle arrest,
differentiation and AMPK activation can be mimicked by pharmacologic inhibition
of the mitochondrial electron transport chain without affecting Myc levels.
Thus, all Myc inhibitors promote a global energy collapse that appears to
underlie many of their phenotypic consequences.
67) Patricia Sancho, et al. MYC_PGC1a Determines Metabolic Phenotype Pancreatic
Cancer Stem Cells Patricia Sancho Cell Metabolism 2015
However, eventually tumors relapsed under metformin due to emergence of
resistant CSCs with an intermediate metabolic phenotype with increased c-MYC
expression.
Mechanistically, we found that low MYC expression in human CSCs allowed high
PGC1A expression levels, which resulted in enhanced mitochondrial biogenesis,
strong mitochondrial activity and antioxidant properties, and subsequently low
mitochondrial ROS levels, as a prerequisite for their stemness functions.
Intriguingly, sustained suppression of MYC was required for maintaining stemness
but rendered CSCs unable to substantially activate glycolysis and thus highly
susceptible to mitochondrial targeting, e.g., by metformin or menadione.
MYC promotes a Warburg-like glycolytic phenotype, probably via dual mechanism:
(1) upregulation of key glycolytic enzymes and (2) suppression of PGC1A . The
latter is a transcriptional co-activator for nuclear receptor PPARs , which
allows the protein to interact with multiple transcription factors, e.g., cAMP
response element-binding protein (CREB) and nuclear respiratory factors (NRFs)
(Fernandez-Marcos and Auwerx, 2011 ). While it is known that proteasomal
degradation inhibits PGC-1 a(Ahuja et al.,2010), we here show for the first time
a direct inhibitory effect of MYC on PGC-1 a at the transcriptional level, upon
MYC binding to the PGC1A promoter.
Interestingly, while MYC has been linked to stemness properties in other tumors,
e.g., hepatocellular carcinoma, breast, and lung cancer (
Akita et al., 2014; Zhao et al., 2015 ), we found in PDAC that MYC was
associated with a more differentiated phenotype and MYC overexpression actually
reduced stemness in CSCs (Figure 7). Notably, while suppression of MYC was
essential for maintaining CSC phenotypes, the mere inhibition of MYC in non-CSCs
did not equip them with stemness features.
68) Yi, Shuhua, et al. ��High incidence of MYC and BCL2 abnormalities in mantle
cell lymphoma, although only MYC abnormality predicts poor survival.�� Oncotarget
6.39 (2015): 42362. In multivariate analysis, the MYC abnormality was the
independent adverse factor for both PFS and OS, and intensive chemotherapy did
not improve the outcome of these patients.
In the literatures, patients with a MYC abnormity always had extensive bone
marrow involvement or leukemic presentation. So, cases of MCL with a MYC
abnormality represent a relatively unique group with highly aggressive clinical
and biological behavior.
Then, we determined if the HyperCVAD/MA �� R regimen could improve the survival
of patients with a MYC abnormality. Unfortunately, this regimen had no impact on
the survival of these patients.
��Intensive chemotherapy such as HyperCVAD/MA �� R did not improve the survival of
patients with a MYC abnormality, and a new treatment strategy should be
developed.��
69) Histopathology. 2016 Feb;68(3):442-9. MYC overexpression correlates with MYC
amplification or translocation, and is associated with poor prognosis in mantle
cell lymphoma.
Choe JY1,2, Yun JY1,2, Na HY2,3, Huh J4, Shin SJ4, Kim HJ5, Paik JH1, Kim YA6,
Nam SJ3, Jeon YK3, Park G7, Kim JE2,6.
We aimed to investigate MYC expression and chromosomal aberration in mantle cell
lymphoma (MCL), and the clinical significance of these factors.
METHODS AND RESULTS: Sixty-five patients with MCL, including 54 classic, nine
blastoid and two pleomorphic variants, were enrolled. Expression of MYC, Ki67
and p53 was assessed by immunohistochemistry. MYC amplification or translocation
was examined by fluorescence in-situ hybridization. MYC expression was higher in
blastoid/pleomorphic MCL variants (mean, 19.0%) than in classic MCL (mean, 1.9%;
P < 0.001). Expression of p53 and Ki67 was also significantly higher in these
variants. MYC amplification was found in two of 53 cases tested, both of which
were blastoid variants with high MYC expression (29.7% and 20.4%). MYC
translocation was found in two of 52 cases tested, both of which were
pleomorphic variants with remarkably high MYC expression (68.5% and 71.0%). High
MYC or p53 expression was significantly associated with shortened overall
survival and progression-free survival in univariable and multivariable analyses
(all P < 0.05).
CONCLUSIONS: MYC overexpression is a negative predictor of MCL patient outcomes.
MYC gene amplification or translocation might be related to the pathogenesis of
MCL, particularly in blastoid/pleomorphic variants.
70) Nguyen, Lynh, Peter Papenhausen, and Haipeng Shao. ��The Role of c-MYC in
B-Cell Lymphomas: Diagnostic and Molecular Aspects.�� Genes 8.4 (2017): 116.
c-MYC is one of the most essential transcriptional factors, regulating a diverse
array of cellular functions, including proliferation, growth, and apoptosis.
Dysregulation of c-MYC is essential in the pathogenesis of a number of B-cell
lymphomas, but is rarely reported in T-cell lymphomas. c-MYC dysregulation
induces lymphomagenesis by loss of the tight control of c-MYC expression,
leading to overexpression of intact c-MYC protein, in contrast to the somatic
mutations or fusion proteins seen in many other oncogenes. Dysregulation of
c-MYC in B-cell lymphomas occurs either as a primary event in Burkitt lymphoma,
or secondarily in aggressive lymphomas such as diffuse large B-cell lymphoma,
plasmablastic lymphoma, mantle cell lymphoma, or double-hit lymphoma. Secondary
c-MYC changes include gene translocation and gene amplification, occurring
against a background of complex karyotype, and most often confer aggressive
clinical behavior, as evidenced in the double-hit lymphomas. In low-grade B-cell
lymphomas, acquisition of c-MYC rearrangement usually results in transformation
into highly aggressive lymphomas, with some exceptions. In this review, we
discuss the role that c-MYC plays in the pathogenesis of B-cell lymphomas, the
molecular alterations that lead to c-MYC dysregulation, and their effect on
prognosis and diagnosis in specific types of B-cell lymphoma.
Double Hit Lymphomas Venetoclax
71) Friedberg, Jonathan W. ��How I treat�� Double Hit�� lymphoma.�� Blood (2017):
blood-2017. How I treat Double Hit Lymphoma c-MYC Friedberg Jonathan W Blood
2017
The 2016 revision of the WHO classification for lymphoma classification has
included a new category of lymphoma, separate from diffuse large B-cell lymphoma
termed High grade B-cell lymphoma with translocations involving myc and bcl-2 or
bcl-6. These lymphomas, which occur
in less than 10% of cases of diffuse large B-cell lymphoma, have been referred
to as ��double hit�� lymphomas (or triple hit lymphomas if all three
rearrangements are present). It is important to differentiate these lymphomas
from the larger group of ��double expressor�� lymphomas which have increased
expression of MYC and BCL-2 and/or BCL-6 by immunohistochemistry, using variable
cut-off percentages to define positivity. Double hit lymphomas have a poor
prognosis when treated with standard chemoimmunotherapy, and have increased risk
of central nervous system involvement and progression.
Novel agents with particular promise in patients with double hit DLBCL may
include small molecule inhibitors of BCL-2 such as venetoclax, which has
demonstrated in vivo efficacy against aggressive myc-driven mouse lymphomas 64
and has been studied in patients with
relapsed lymphoma with limited activity in aggressive histologies.65
72) Rattan, Ramandeep, et al. ��Metformin suppresses ovarian cancer growth and
metastasis with enhancement of cisplatin cytotoxicity in vivo.�� Neoplasia 13.5
(2011): 483IN26-491IN28.
Ovarian cancer is the most lethal gynecologic cancer in women. Its high
mortality rate (68%) reflects the fact that 75% of patients have extensive
(>stage III) disease at diagnosis and also the limited efficacy of currently
available therapies. Consequently, there is clearly a great need to develop
improved upfront and salvage therapies for ovarian cancer. Here, we investigated
the efficacy of metformin alone and in combination with cisplatin in vivo. A2780
ovarian cancer cells were injected intraperitoneally in nude mice; A2780-induced
tumors in nude mice, when treated with metformin in drinking water, resulted in
a significant reduction of tumor growth, accompanied by inhibition of tumor cell
proliferation (as assessed by immunohistochemical staining of Ki-67, Cyclin D1)
as well as decreased live tumor size and mitotic cell count. Metformin-induced
activation of AMPK/mTOR pathway was accompanied by decreased microvessel density
and vascular endothelial growth factor expression. More importantly, metformin
treatment inhibited the growth of metastatic nodules in the lung and
significantly potentiated cisplatin-induced cytotoxicity resulting in
approximately 90% reduction in tumor growth compared with treatment by either of
the drugs alone. Collectively, our data show for the first time that, in
addition to inhibiting tumor cell proliferation, metformin treatment inhibits
both angiogenesis and metastatic spread of ovarian cancer. Overall, our study
provides a strong rationale for use of metformin in ovarian cancer treatment.
link to this article: http://wp.me/p3gFbV-59p
Jeffrey Dach MD
7450 Griffin Road, Suite 190
Davie, Fl 33314
954-792-4663
https://jeffreydachmd.com
http://www.drdach.com
http://www.naturalmedicine101.com
http://www.truemedmd.com
��
Diabetes, Cancer and the Drug that Fights them Both
by Megan L Norris
figures by Bradley Wierbowski
The emerging link between cancer and diabetes
In the early 2000s, observations that diabetics are more likely to get cancer
than non-diabetics began piling up. Was this because diabetes and cancer share
general risk factors such as diet, aging and obesity? Or was there a direct link
between them, with cancer benefiting from the sugar-rich and inflamed
environment brought on by diabetes? Making bad news worse, it became apparent
that cancer thrives in the presence of excess insulin, like that injected by
many diabetics as therapy. Thus, one of the ways to treat diabetes could be
making the cancer risk even worse.
Almost as soon as this dark cloud began to loom, rays of light broke through
from an unexpected source. Research on a popular type II diabetes treatment
called metformin revealed that metformin actually seemed to lower the risk for
colorectal and other cancers in diabetics. Though it may seem paradoxical that
metformin and insulin injections, two treatments for the same disease, could
have such opposite effects on cancer, years of research in both the clinic and
the laboratory has begun to pull back the curtain on this mystery. Broadly
speaking, metformin makes the body more sensitive to the insulin it already is.
For type II diabetics, not only does this increased insulin sensitivity treat
diabetes, but it drains the fuel on which some cancers may thrive.
Figure 1: An overview of diabetes. In non-diabetics, sugar (glucose) from food
exits the bloodstream in response to insulin and is made into cellular energy.
In type I diabetics, the pancreatic beta-cells in charge of making insulin fail,
causing a toxic build-up of glucose in the bloodstream called hyperglycemia.
Type I diabetes can be treated with injection of insulin, which is significantly
less precise than what a healthy pancreas can do. In type II diabetes, insulin
is created but not sensed properly causing hyperglycemia. In response, the
pancreas creates even more insulin resulting in high blood insulin levels called
hyperinsulinemia.
Figure 1: An overview of diabetes. In non-diabetics, sugar (glucose) from food
exits the bloodstream in response to insulin and is made into cellular energy.
In type I diabetics, the pancreatic beta-cells in charge of making insulin fail,
causing a toxic build-up of glucose in the bloodstream called hyperglycemia.
Type I diabetes can be treated with injection of insulin, which is significantly
less precise than what a healthy pancreas can do. In type II diabetes, insulin
is created but not sensed properly causing hyperglycemia. In response, the
pancreas creates even more insulin resulting in high blood insulin levels called
hyperinsulinemia.
Diabetes is a pervasive disease
Diabetes is the 7th leading cause of death in the US and is on the rise. More
than 10% of Americans over 20 years old have diabetes, with type II diabetes
accounting for 95% of those cases. Untreated, diabetes leads to excessive
amounts of glucose in the blood, a state called hyperglycemia, which can damage
organs such as the heart, kidney and nerves.
In a non-diabetic individual following a meal, sugars and starches break down
into glucose, which circulates in the blood. Beta-cells in the pancreas release
insulin to trigger the movement of glucose out of the bloodstream and into cells
where it can be used or stored. As shown in Figure 1, when no insulin is
produced (type I diabetes) or is produced but not sensed correctly (type II
diabetes), glucose amounts remain elevated in the blood causing dangerous
hyperglycemia.
In addition to high blood glucose, diabetics also have a condition called
hyperinsulinemia, which refers to excessively high amounts of insulin in their
bloodstream. Since type I diabetes is marked by the body��s inability to produce
insulin, a common treatment is insulin injection. Fine-tuning both the
concentration and timing of these injections, and properly mimicking what a
non-diabetic body can do, has proven a great challenge. As a result, these
injections lead to significantly higher insulin levels than a non-diabetic would
ever experience, resulting in hyperinsulinemia. Conversely, type II diabetics
can make their own insulin, but their body isn��t sensitive to it. As a result,
their bodies often pump out more and more insulin, resulting in a nearly
constant state of hyperinsulinemia.
Additionally, this constant demand on the beta-cells can cause them to wear out
and lose their ability to make insulin at all; patients who experience this are
often given the insulin injections more common to type I diabetics, perpetuating
their hyperinsulinemia through a different means. These elevated insulin levels
are much less toxic to organs as compared to excess glucose, so the fact that
treating hyperglycemia can lead to hyperinsulinemia, though not ideal, has been
historically viewed as a considerably lesser evil.

Figure 2: The relationship between cancer and diabetes. Scenario I: Diabetics
may have an increased occurrence of cancer because traits that make them
susceptible to diabetes also make them susceptible to cancer. Scenario II: Side
effects of diabetes, like hyperglycemia and hyperinsulinemia, may directly
increase the ability of cancer to grow in a diabetic��s body.
Cancer runs on insulin
How could diabetes be affecting cancer? The simplest possibility is that what
makes a person predisposed for diabetes is the same as what makes them
predisposed for certain cancers, such as obesity and age. In this scenario,
shown in Figure 2, one disease isn��t causing the other. Rather, both diseases
simply capitalize on the same health situations. Though shared predispositions
and risk factors may certainly explain some of the co-occurrence between
diabetes and cancer, doctors and researchers are collecting mounting evidence
that implicates diabetes as having a more direct role in increasing the risk of
cancer. For one, cancer cells require a lot of energy to grow, divide, and
spread. Importantly, one of cancer��s most vital energy sources is glucose,
meaning that the high blood glucose levels in many diabetics could greatly
benefit the growth and progression of cancer. More striking, however, is that
insulin has been found to promote tumor growth and metastasis. Though this
happens in diabetics and non-diabetics alike, people with diabetes have
especially high levels of insulin in their blood, which could make for extra
fertile grounds for cancer progression (Figure 2). As such, the insulin
injections aimed at treating diabetes may also be increasing cancer risk.
Metformin treats diabetes without giving cancer a leg up
With the excess glucose and insulin present in diabetics and cancer potentially
thriving off of both, must diabetics just live with an increased cancer risk?
Enter metformin: the first-choice drug to treat type II diabetes. The exact way
that metformin works is still being explored, but in the last two decades we
have learned that metformin seems to have two important, and likely overlapping,
methods of action. First, it sensitizes the body to insulin and, second, it
coerces cells to make more energy (see Figure 3).
Figure 3: Metformin affects insulin signaling and energy production. Metformin
sensitize the body to insulin already present in the bloodstream. As a result of
better insulin signaling, cells begin to remove glucose from the blood and
produce cellular energy.
Figure 3: Metformin affects insulin signaling and energy production. Metformin
sensitize the body to insulin already present in the bloodstream. As a result of
better insulin signaling, cells begin to remove glucose from the blood and
produce cellular energy.
By increasing sensitivity to insulin, metformin helps type II diabetics start
using the insulin already present in their bodies. This results in decreased
insulin levels, which many researchers believe is how metformin is affecting
cancer. This also means that metformin requires some insulin to be present in
order to work best, so it alone may not be able to treat type I diabetics.
However, it has recently been proposed to treat type I diabetes with a mix of
insulin therapy and metformin, with the hope that lower concentrations of
insulin would be required. In either case, after cells are treated with
metformin and begin responding better to insulin, the cells can remove glucose
from the blood to make cellular energy, thereby decreasing hyperglycemia and
treating diabetes.
Cancer as a metabolic disease
Insulin and glucose are both metabolites, meaning that they are produced as
byproducts of metabolism �� the processes that all cells perform to obtain
energy. The realization that cancer is tightly linked to metabolism only came to
light in the early 2000s from unexpected, and seemingly unrelated, sources.
While one group of researchers in Scotland was studying the basics of metabolism
in yeast, separate groups across the world were studying cancer in rats, humans
and frogs. Unexpectedly, the groups realized that they were all studying the
same genes. Nearly simultaneously, one of the same genes was predicted to be the
target of metformin. When all the pieces were put together, the idea that
metformin could treat diabetes and cancer began to make sense.
Today, the data demonstrating the efficacy of metformin protecting against
cancer is clear. A 2015 paper in the journal Cancer revealed metformin treatment
decreased the chance of colorectal cancer in diabetics by 12%. Other studies
demonstrated metformin selectively blocks tumor growth, can suppress
cancer-promoting genes and provides a reduced risk of cancer in general.
Intriguingly, some data suggests that, for certain cancers, this protective
effect is only seen in diabetic settings. That is, cancers that are thriving
from the hyperinsulinemic environment were treatable with metformin, while other
cancers were not affected. Research into which cancers are likely to be
susceptible to metformin is ongoing and relies heavily on understanding with
more detail just how metformin is working. Ideally, we will soon know exactly
how metformin is impacting insulin, metabolism and cancer.
Basic science makes big discoveries
The case of metformin is not unique. The more scientists follow the rabbit hole
of basic research, which is research targeted at fundamental biology as opposed
to medicine or disease, the more we equip our researchers in the clinic to make
important, perhaps startling, medical advancements. Though possibly a pervasive
idea, it is inaccurate to think we would be better off if we focused all our
research efforts on disease. We have certainly come a long way, but with each
decade of basic research, we uncover things cells do that we could have never
predicted the decade before. Though aiming research directly at a disease is of
course useful, persevering in our understanding of the foundations of biology is
both crucial and expedient to making meaningful medical discoveries.
Megan L Norris is a 5th year PhD student in the department of Molecules, Cells
and Organisms at Harvard University.
For more information:
Drug discovery: metformin and the control of diabetes
LKB1 and AMPK and the cancer-metabolism link �C ten years after
Cover image modified from Sugar Cubes by David Pacey [CC BY 2.0], via Wikimedia
Commons and Metastatic Melanoma Cells by Julio C. Valencia [CC BY-NC 2.0] via
NIH Image Gallery flickr
Diabetes, Cancer and the Drug that Fights them Both - Science in the News
http://sitn.hms.harvard.edu/flash/2017/diabetes-cancer-drug-fights/
��
��
��
.jpg)
Pleiotropic Benefits of Metformin: Macrophage Targeting Its Anti-inflammatory
Mechanisms | Diabetes
https://diabetes.diabetesjournals.org/content/64/6/1907
��
��
����˫��ͨ����������ؽ���PD-L1�ٽ����������ߡ�
Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated
Degradation of PD-L1
University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
ǿ��
•����˫��ͨ�����PD-L1 / PD-1����ǿ������CTL������
•����˫�Ҽ����AMPK��S195ֱ����PD-L1��ϲ����ữ
•pS195�յ���PD-L1�ǻ����쳣����ERAD����PD-L1
•����˫�ҺͿ�CTLA4�������ƾ���Эͬ����������
ժҪ
�ݱ���������˫�Ҿ��п��������Բ�ά��ϸ������T�ܰ�ϸ����CTL���߶�����⡣���ǣ���δ��ȫ�˽����˫���ڰ�֢�����еĹ��ܺ���ϸ���ơ������������ʾ����˫��ͨ�����ͳ�������������1��PD-L1�����ȶ��Ժ�Ĥ��λ������CTL���ԡ����⣬���Ƿ��ֱ�����˫�Ҽ����AMP����ĵ���ø��AMPK��ֱ�����ữPD-L1��S195��
S195���ữ�յ��쳣��PD-L1�ǻ�����������ER���ۺ���ER��صĵ����ʽ��⣨ERAD����һ�µأ�����˫�����Ƶ����ٰ����ߵ�������֯��AMPK�����±��ֳ����͵�PD-L1ˮƽ��ͨ������˫�����PD-L1�������źſ���ǿ���ϸ����CTL���ԡ����ǵķ���ȷ����ͨ��ERAD;������PD-L1���µ��ڻ��ƣ�����������˫��-CTLA4��ϼ���Ͼ�����������Ʒ���Ч��DZ����
Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated
Degradation of PD-L1 - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1097276518305999#!
��
Biomed Pharmacother. 2019 Mar;111:1156-1165. doi:
10.1016/j.biopha.2019.01.021. Epub 2019 Jan 12.
Metformin enhances mitochondrial biogenesis and thermogenesis
in brown adipocytes of mice.
Karise I1, Bargut TC2, Del Sol M3, Aguila MB4, Mandarim-de-Lacerda CA5.
Author information
1
Laboratory of Morphometry, Metabolism and Cardiovascular Disease, Biomedical
Center, Institute of Biology, The University of the State of Rio de Janeiro, Rio
de Janeiro, Brazil. Electronic address: iarakarise@hotmail.com.
2
Laboratory of Morphometry, Metabolism and Cardiovascular Disease, Biomedical
Center, Institute of Biology, The University of the State of Rio de Janeiro, Rio
de Janeiro, Brazil. Electronic address: therezabargut@gmail.com.
3
Doctoral Program in Morphological Sciences, Universidad de La Frontera, Temuco,
Chile. Electronic address: mariano.delsol@ufrontera.cl.
4
Laboratory of Morphometry, Metabolism and Cardiovascular Disease, Biomedical
Center, Institute of Biology, The University of the State of Rio de Janeiro, Rio
de Janeiro, Brazil. Electronic address: mbaguila@uerj.br.
5
Laboratory of Morphometry, Metabolism and Cardiovascular Disease, Biomedical
Center, Institute of Biology, The University of the State of Rio de Janeiro, Rio
de Janeiro, Brazil. Electronic address: mandarim@uerj.br.
Abstract
AIMS:
We studied the effect of metformin on the brown adipose tissue (BAT) in a
fructose-rich-fed model, focusing on BAT proliferation, differentiation, and
thermogenic markers.
MAIN METHODS:
C57Bl/6 mice received isoenergetic diets for ten weeks: control (C) or
high-fructose (F). For additional eight weeks, animals received metformin
hydrochloride (M, 250 mg/kg/day) or saline. After sacrifice, BAT and white fat
pads were prepared for light microscopy and molecular analyses.
KEY FINDINGS:
Body mass gain, white fat pads, and adiposity index were not different among the
groups. There was a reduction in energy intake in the F group and energy
expenditure in the F and FM groups. Metformin led to a more massive BAT in both
groups CM and FM, associated with a higher adipocyte proliferation
(��1-adrenergic receptor, proliferating cell nuclear antigen, and vascular
endothelial growth factor), and differentiation (PR domain containing 16, bone
morphogenetic protein 7), in part by activating 5' adenosine
monophosphate-activated protein kinase. Metformin also enhanced thermogenic
markers in the BAT (uncoupling protein type 1, peroxisome proliferator-activated
receptor gamma coactivator-1 alpha) through adrenergic stimuli and fibroblast
growth factor 21. Metformin might improve mitochondrial biogenesis in the BAT
(nuclear respiratory factor 1, mitochondrial transcription factor A), lipolysis
(perilipin, adipose triglyceride lipase, hormone-sensitive lipase), and fatty
acid uptake (lipoprotein lipase, cluster of differentiation 36, adipocyte
protein 2).
SIGNIFICANCE:
Metformin effects are not linked to body mass changes, but affect BAT
thermogenesis, mitochondrial biogenesis, and fatty acid uptake. Therefore, BAT
may be a metformin adjuvant target for the treatment of metabolic disorders.
Copyright © 2019 Elsevier Masson SAS. All rights reserved.
KEYWORDS:
Brown adipose tissue; Fructose; Metformin; Mouse; Thermogenesis
Metformin enhances mitochondrial biogenesis and thermogenesis in brown
adipocytes of mice. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/30841429
��
��