íí
Cancer as a Metabolic Disease and Dichloroacetate as a Drug
G1 phase. Metabolic changes prepare the cell for division. At a certain point - the restriction point - the cell is committed to division and moves into the S phase.
S phase. DNA synthesis replicates the genetic material. Each chromosome now consists of two sister chromatids.
G2 phase. Metabolic changes assemble the cytoplasmic materials necessary for mitosis and cytokinesis.
M phase. A nuclear division (mitosis) followed by a cell division (cytokinesis).
The period between mitotic divisions - that is, G1, S and G2 - is known as interphase.https://www2.le.ac.uk/projects/vgec/diagrams/22-Cell-cycle.gif
íí
Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer
Cha Univerisity Korea
Dichloroacetate (DCA), an orphan drug that promotes a shift from glycolysis to oxidative phosphorylation, has been repurposed for cancer therapy. The present study investigated whether DCA may overcome cisplatin resistance in head and neck cancer (HNC). Two cisplatin-resistant HNC cell lines (AMC-HN4R and -HN9R), their parental lines, and other human HNC lines were used. The effect of DCA, alone and in combination with cisplatin, was assessed by measuring cell cycle, viability, death, reactive oxygen species (ROS) production, mitochondrial membrane potential (ªñªÀm), and protein expression in preclinical mouse tumor xenograft models. Increased glycolysis correlated with decreased sensitivity to cisplatin and was reduced by DCA. Cisplatin-resistant cells overexpressed pyruvate dehydrogenase kinase 2 (PDK2). DCA induced HNC cell death by decreasing ªñªÀm and promoting mitochondrial ROS production. This effect was decreased by the antioxidant N-acetyl-L-cysteine or by inhibition of caspase-mediated apoptosis. Activation of mitochondrial glucose oxidation by DCA eventually activated downstream mitochondrial apoptotic signaling, leading to the death of chemoresistant cancer cells. Therefore, DCA significantly sensitized resistant HNC cells to cisplatin in vitro and in vivo. High glycolysis and PDK2 overexpression are closely linked to cisplatin resistance in HNC cells; the latter can be overcome by DCA.
Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer | Request PDF
https://www.researchgate.net/publication/284434401_Activation_of_mitochondrial_oxidation_by_PDK2_inhibition_reverses_cisplatin_resistance_in_head_and_neck_canceríí
Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells .. This in turn increases the flux of pyruvate into the mitochondria, thereby stimulating oxidative phosphorylation over glycolysis. DCA has shown in preclinical work to reverse glycolysis-related suppression of mitochondrial apoptosis in cancer cells (20,216) and boosted the effectiveness of hypoxia-specific che- motherapies in vitro (65,164) and in animal models (19). Resveratrol was reported recently to enhance PDH activity, likely via activation of AMPK, which in turn led to a shift from glycolysis to oxidative phosphorylation in HTC116 colon cancer cells (171). ...
ííSerum pyruvate dehydrogenase kinase as a prognostic marker for cholangiocarcinoma
...Similarly, activation of mitochondrial oxidation following PDK2 inhibition could activate mitochondrial apoptotic signaling, causing the death of chemoresistant cancer cells. 46 Mitochondrial OXPHOS is essential for efficient apoptosis. 47,48 The oncogenic role of mutant KRAS prompts intensive efforts to explore pharmacological approaches. ...íí
íí
Environ Health Perspect. 1998 Aug; 106(Suppl 4): 989¿C994.
Clinical pharmacology and toxicology of dichloroacetate.
Abstract
Dichloroacetate (DCA) is a xenobiotic of interest to both environmental toxicologists and clinicians. The chemical is a product of water chlorination and of the metabolism of various drugs and industrial chemicals. Its accumulation in groundwater and at certain Superfund sites is considered a potential health hazard. However, concern about DCA toxicity is predicated mainly on data obtained in inbred rodent strains administered DCA at doses thousands of times higher than those to which humans are usually exposed. In these animals, chronic administration of DCA induces hepatotoxicity and neoplasia. Ironically, the DCA doses used in animal toxicology experiments are very similar to those used clinically for the chronic or acute treatment of several acquired or hereditary metabolic or cardiovascular diseases. As a medicinal, DCA is generally well tolerated and stimulates the activity of the mitochondrial pyruvate dehydrogenase enzyme complex, resulting in increased oxidation of glucose and lactate and an amelioration of lactic acidosis. By this mechanism, the drug may also enhance cellular energy metabolism. DCA is dehalogenated in vivo to monochloroacetate and glyoxylate, from which it can be further catabolized to glycolate, glycine, oxalate, and carbon dioxide. It remains to be determined whether important differences in its metabolism and toxicology exist in humans between environmentally and clinically relevant doses.
Clinical pharmacology and toxicology of dichloroacetate.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1533324/íí
Â■┬╚ÊÊ╦ßÐ╬(Dichloroacetate)Á─┴┘┤▓Ê®└ÝÂ¥└Ýíú
Â■┬╚ÊÊ╦ßÐ╬ú¿DCAú®╩Ã╗À¥│Â¥└Ýк╝Ê║═┴┘┤▓Ê¢╔·Â╝©ðð╦╚ñÁ─ÊýÍÍ╔·╬´íú©├╗»ÐºãÀ╩Ã╦«┬╚╗»║═©¸ÍÍÊ®╬´║═╣ñÊÁ╗»ÐºãÀ┤·ð╗Á─▓·╬´íú╦³È┌ÁϤ┬╦«║═─│ð®Superfundı¥ÁÒÍðÁ─╗²└█▒╗╚¤╬¬╩ÃÃ▒È┌Á─¢í┐Á╬ú║ªíúÁ½╩Ãú¼ÂÈDCAÂ¥ðÈÁ─ÁúËÃ͸ʬ╩Ã╗¨Ë┌ÊÈDCA╝┴┴┐╩®Ë├Á─¢³¢╗─÷│¦Â»╬´ãÀ¤Á╦¨╗±Á├Á─╩²¥¦ú¼©├╝┴┴┐▒╚╚╦└Ó═¿│ú╦¨▒®┬ÂÁ─╝┴┴┐©▀╩²Ãº▒ÂíúÈ┌ıÔð®Â»╬´Íðú¼│ñã┌À■Ë├DCA╗ßËıÀó©╬Â¥ðÈ║═ÍÎ┴÷ð╬│╔íú¥▀ËðÀÝ┤╠ÊÔ╬ÂÁ─╩Ãú¼Â»╬´Â¥└Ýк╩ÁÐÚÍð╩╣Ë├Á─DCA╝┴┴┐ËÙ┴┘┤▓╔¤Ë├Ë┌╝©ÍÍ╗±Á├ðÈ╗‗Ê┼┤½ðÈ┤·ð╗╗‗ð─Ь╣▄╝▓▓íÁ─┬²ðÈ╗‗╝▒ðÈÍ╬┴ãÁ─╝┴┴┐ÀÃ│ú¤Ó╦ãíúθ╬¬Ê®╬´ú¼DCA═¿│ú¥▀Ëð┴╝║├Á──═╩▄ðÈú¼▓ó┤╠╝ñ¤▀┴ú╠Õ▒¹═¬╦ß═ÐÃÔ├©©┤║¤╬´(PDC)Á─╗¯ðÈú¼┤Ë°Á╝Í┬ã¤╠Ð╠Ã║═╚Ú╦ßÁ─Ч╗»È÷╝Ë▓ó©─╔ã┴╦╚Ú╦ßðÈ╦ßÍðÂ¥íú═¿╣²ıÔÍÍ╗·Íãú¼©├Ê®╬´╗╣┐╔ÊÈÈ÷Ã┐¤©░¹─▄┴┐┤·ð╗íú DCAÈ┌╠Õ─┌═Ð┬▒╬¬Ê╗┬╚ÊÊ╦ߧÑ║═ÊÊ╚®╦ߧÑú¼┤ËÍð┐╔¢°Ê╗▓¢ÀÍ¢Ô┤·ð╗╬¬ÊÊ┤╝╦ߧÑú¼©╩░▒╦ßú¼▓¦╦ߧÑ║═Â■Ч╗»╠╝íúÈ┌╗À¥│║═┴┘┤▓¤Ó╣Ï╝┴┴┐Í«╝õú¼╚╦Á─ð┬│┬┤·ð╗║═Â¥└Ýк╩ÃÀ±┤µÈ┌ÍÏʬ▓¯Êý╔ð┤²╚À¿íúClinical pharmacology and toxicology of dichloroacetate.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1533324/íí
íí
Â■┬╚ÊÊ╦ßÂÈ▒¹═¬╦ß═ÐÃÔ├©╗¯ðÈÁ─╝ñ╗¯©─╔ã┴╦┤¾╩¾ð─ÈÓÍÞ═ú║¾Á─┤µ╗¯║═▒ú╗ñ╔±¥¡¤Á═│
Activation of Pyruvate Dehydrogenase Activity by Dichloroacetate Improves Survival and Neurologic Outcomes After Cardiac Arrest in Rats.
1
Íð╔¢┤¾ÐºÍð╔¢╝═─¯Ê¢È║╝▒ı´┐ãú¼╣Òͦ
2
Íð╔¢┤¾Ðºð─À╬─È©┤╦ıÐð¥┐╦¨ú¼╣Òͦ
3
ÃÓ║ú╩í╚╦├±Ê¢È║╝▒ı´┐ãú¼╬¸─■
4
©Ñ╝¬─ßÐÃ┴¬░¯┤¾Ðºú¼═■¹╦╣ʢкÈ║ú¼═■¹╝▒ı´║═ÍÏÍó╝Ó╗ñÐð¥┐╦¨ú¼©Ñ╝¬─ßÐÃͦ└´╩┐┬·íú
5
©Ñ╝¬─ßÐÃ┴¬░¯┤¾Ðº╝▒ı´Ê¢Ðº¤Áú¼©Ñ╝¬─ßÐÃͦ└´╩┐┬·íú
│Ú¤¾
─┐Ã░╔ð╬ÌÊ®╬´©╔Èñ┤Ù╩®┐╔╬¬╗╝Ëðð─ÈÓÍÞ═úÁ─╗╝ı▀╠ß╣®╔±¥¡▒ú╗ñíúÂ■┬╚ÊÊ╦ßÐ╬ú¿DCAú®╩ÃÊ╗ÍÍ▒¹═¬╦ß═ÐÃÔ├©╝ñ├©(PDK)ÊÍÍã╝┴ú¼┐╔╝ñ╗¯▒¹═¬╦ß═ÐÃÔ├©ú¿PDHú®ú¼▓ó═¿╣²┤┘¢°▒¹═¬╦ß┴¸╚ÙKrebsС╗À└┤È÷╝ˤ©░¹╚²┴Î╦ߤ┘▄ıú¿ATPú®Á─▓·╔·íúÈ┌ıÔ¤¯Ðð¥┐Íðú¼╬Ê├ÃÁ¸▓Ú┴╦ͤ¤óðÈð─ÈÓÍÞ═ú┤¾╩¾─úð═ÍðDCAÂÈ©┤╦ı║¾╔±¥¡¤Á═│╦╔╦Á─Ë░¤ýíúͤ¤óðÈð─ÈÓÍÞ═ú╩Ã═¿╣²ã°╣▄▓Õ╣▄¢¿┴óÁ─íú¢½111Í╗┤¾╩¾╦µ╗·ÀÍ╬¬╚²ÎÚú║╝┘╩Í╩§ÎÚú¼ÂÈııÎÚ║═DCA©╔ÈñÎÚíú DCA©╔ÈñÎÚÁ─»╬´È┌ÎÈÀóС╗Àú¿ROSCú®╗Í©┤║¾15ÀÍÍË©╣─ñ─┌©°ËÞDCAú¼©║║╔╝┴┴┐╬¬80ª╠mg/ kgú¼Â°ÂÈııÎÚÁ─┤¾╩¾¢Ë╩▄Á╚┴┐Á─╔·└ÝÐ╬╦«íú DCAÍ╬┴ãÈ÷╝Ë┴╦3╠ýÁ─╔·┤µ╩▒╝õú¼▓ó¢ÁÁ═┴╦ROSC║¾24íó48║═72 hÁ─╔±¥¡╣ª─▄╚▒╦ã└ÀÍíú═¿╣²╦ı─¥¥½-Ê┴║ý╚¥╔½║═TdT¢ÚÁ╝Á─dUTP╚▒┐┌─®Â╦▒Û╝Ã▓Ô¿ú¼╦³╗╣╝§╚§┴╦║ú┬Ý¢Ã─ññ─ñÊ╗ðË‗Á─¤©░¹Á‗═÷║═╔±¥¡È¬╦╔╦íú┤╦═Ôú¼DCA┐╔¢ÁÁ═ROSC║¾─È║ú┬Ý║═ãñ▓ÒÍÎ┴÷╗Á╦└Ê‗Î˪┴║═░΢Ú╦Ï1ª┬Á─ð┼╩╣RNA▒Ý┤´íú┤╦═Ôú¼DCAÍ╬┴ã¤ÈÎ┼È÷╝Ë┴╦ROSC║¾Á─ATP▓·╔·ú¼PDH╗¯ðÈú¼▓ó¢ÁÁ═┴╦Ь╠Ãú¼╚Ú╦ß║═─È▒¹═¬╦ß╦«ã¢íú╬Ê├ÃÁ─¢ß╣¹▒Ý├¸ú¼DCAÂÈð─ÈÓÍÞ═ú║¾Á──È╦╔╦¥▀Ëð╔±¥¡▒ú╗ñθË├ú¼ãõËðʵθË├ËÙ═¿╣²╝ñ╗¯PDH╗¯ðÈÈ÷╝Ë─Ȥ▀┴ú╠Õ─▄┴┐┤·ð╗Ëð╣ÏíúShock. 2018 Jun;
Author information
1
Department of Emergency Medicine, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou, China.
2
Institute of Cardiopulmonary Cerebral Resuscitation, Sun Yat-sen University, Guangzhou, China.
3
Department of Emergency Medicine, Qinghai Provincial People's Hospital, Xining, China.
4
Weil Institute of Emergency and Critical Care Research, School of Medicine, Virginia Commonwealth University, Richmond, Virginia.
5
Department of Emergency Medicine, Virginia Commonwealth University, Richmond, Virginia.
Abstract
No pharmacological interventions are currently available to provide neuroprotection for patients suffering from cardiac arrest. Dichloroacetate (DCA) is a pyruvate dehydrogenase kinase inhibitor, which activates pyruvate dehydrogenase (PDH), and increases cell adenosine triphosphate (ATP) production by promoting influx of pyruvate into the Krebs cycle. In this study, we investigated the effects of DCA on post-resuscitation neurological injury in an asphyxial cardiac arrest rat model. Asphyxial cardiac arrest was established by endotracheal tube clamping. A total of 111 rats were randomized into three groups: Sham group, Control group, and DCA intervention group. Animals in DCA intervention group were intraperitoneally administered DCA with a loading dose of 80 mg/kg at 15 min after return of spontaneous circulation (ROSC), whereas rats in the Control group received equivalent volume of saline. DCA treatment increased 3-day survival time, and reduced neurologic deficit scores at 24, 48, and 72 h after ROSC. It also attenuated cellular apoptosis and neuronal damage in the hippocampal cornuammonis one region by hematoxylin-eosin staining and TdT-mediated dUTP nick-end labeling assay. In addition, DCA reduced the messenger RNA expression of tumor necrosis factor ª┴ and interleukin 1ª┬ in brain hippocampus and cortex after ROSC. Furthermore, DCA treatment significantly increased ATP production, PDH activity, and decreased blood glucose, lactate, and brain pyruvate levels after ROSC. Our results suggested that DCA has neuroprotective effects on brain injury after cardiac arrest, and its salutary effects were associated with an increase of mitochondrial energy metabolism in the brain through activation of PDH activity.Activation of Pyruvate Dehydrogenase Activity by Dichloroacetate Improves Survival and Neurologic Outcomes After Cardiac Arrest in Rats. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28846566íí
Age-Dependent Kinetics and Metabolism of Dichloroacetate: Possible Relevance to Toxicity
Abstract
Dichloroacetate (DCA) is an investigational drug for certain metabolic diseases. It is biotransformed principally by the ªã-1 family isoform of glutathione transferase (GSTz1), also known as maleylacetoacetate isomerase (MAAI), which catalyzes the penultimate step in tyrosine catabolism. DCA causes a reversible peripheral neuropathy in several species, including humans. However, recent clinical trials indicate that adults are considerably more susceptible to this adverse effect than children. We evaluated the kinetics and biotransformation of DCA and its effects on tyrosine metabolism in nine patients treated for 6 months with 25 mg/kg/day and in rats treated for 5 days with 50 mg/kg/day. We also measured the activity and expression of hepatic GSTz1/MAAI. Chronic administration of DCA causes a striking age-dependent decrease in its plasma clearance and an increase in its plasma half-life in patients and rats. Urinary excretion of unchanged DCA in rats increases with age, whereas oxalate, an end product of DCA metabolism, shows the opposite trend. Low concentrations of monochloroacetate (MCA), which is known to be neurotoxic, increase as a function of age in the urine of dosed rats. MCA was detectable in plasma only of older animals. Hepatic GSTz1/MAAI-specific activity was inhibited equally by DCA treatment among all age groups, whereas plasma and urinary levels of maleylacetone, a natural substrate for this enzyme, increased with age. We conclude that age is an important variable in the in vivo metabolism and elimination of DCA and that it may account, in part, for the neurotoxicity of this compound in humans and other species.Â■┬╚ÊÊ╦ßÁ──Û┴õÊ└└ÁðÈ»┴ªÐº║═┤·ð╗ú║┐╔─▄ËÙÂ¥ðÈËð╣Ï
Â■┬╚ÊÊ╦ßÐ╬ú¿DCAú®╩ÃË├Ë┌─│ð®┤·ð╗╝▓▓íÁ─Ðð¥┐Ê®╬´íú╦³Í¸Ê¬Ë╔╣╚ÙΩ╩Ù─άÊã├©ú¿GSTz1ú®Á─ªã-1╝ÊÎÕ═¼╣ñð═╔·╬´Î¬╗»ú¼Ê▓│ã╬¬┬Ý└┤§úÊʧúÊÊ╦ßÊý╣╣├©ú¿MAAIú®ú¼╦³┤▀╗»└Ê░▒╦ßÀÍ¢Ô┤·ð╗ÍðÁ─Á╣╩²Á┌Â■▓¢íú DCAÈ┌░³└¿╚╦└ÓÈ┌─┌Á─ÂÓÍÍ╬´ÍÍÍðʲã┐╔─µÁ─Í▄╬º╔±¥¡▓í▒õíúÁ½╩Ãú¼Î¯¢³Á─┴┘┤▓╩ÈÐÚ▒Ý├¸ú¼│╔─Û╚╦▒╚¨═»©³╚¦ÊÎ╩▄Á¢ıÔÍÍ▓╗┴╝Ë░¤ýíú╬Ê├Ãã└╣└┴╦DCAÁ─»┴ªÐº║═╔·╬´Î¬╗»╝░ãõÂÈ└Ê░▒╦ß┤·ð╗Á─Ë░¤ýú¼ıÔ9├¹╗╝ı▀¢Ë╩▄25 mg / kg /╠ýÍ╬┴ã6©÷È┬ú¼▓ó¢Ë╩▄50 mg / kg /╠ýÍ╬┴ã5╠ýÁ─┤¾╩¾íú╬Ê├Ã╗╣▓Ô┴┐┴╦©╬GSTz1 / MAAIÁ─╗¯ðÈ║═▒Ý┤´íú│ñã┌À■Ë├DCA╗ßÁ╝Í┬╗╝ı▀║═┤¾╩¾Á─Ь¢¼ÃÕ│²┬╩├¸¤È¤┬¢Áú¼▓ó╦µ─Û┴õÈ÷│ñú¼Ð¬¢¼░Ù╦Ñã┌È÷╝Ëíú DCA▓╗▒õÁ─┤¾╩¾─‗┼┼ð╣┴┐╦µ─Û┴õÈ÷╝Ë°È÷╝Ëú¼Â°▓¦╦ßÐ╬ú¿DCA┤·ð╗Á─Íı▓·╬´ú®È‗¤È╩¥¤ÓÀ┤Á─ø╩ãíúÁ═╝┴┴┐Á─Ê╗┬╚ÊÊ╦ߧÑú¿MCAú®¥▀Ëð╔±¥¡Â¥ðÈú¼╦³╗ß╦µÎ┼╝┴┴┐Á─┤¾╩¾─‗Ê║ÍðÁ──Û┴õ°È÷╝Ëíú¢÷È┌¢¤┤¾Â»╬´Á─Ь¢¼Íð┐╔╝ý▓ÔÁ¢MCAíú DCAÍ╬┴ãÈ┌╦¨Ëð─Û┴õÎÚÍð¥¨═¼Á╚ÁÏÊÍÍã©╬GSTz1 / MAAI╠ÏÊýðÈ╗¯ðÈú¼Â°Ð¬¢¼║═─‗Ê║Íð┬Ý└┤§ú▒¹═¬ú¿©├├©Á─╠ý╚╗ÁÎ╬´ú®╦«ã¢╦µ─Û┴õÈ÷╝Ëíú╬Ê├ÃÁ├│÷¢ß┬█ú¼─Û┴õ╩Ã╠Õ─┌┤·ð╗║═DCA¤¹│²Á─ÍÏʬ▒õ┴┐ú¼▓óÃÊ╦³┐╔─▄▓┐ÀÍ¢Ô╩═┴╦©├╗»║¤╬´ÂÈ╚╦└Ó║═ãõ╦¹╬´ÍÍÁ─╔±¥¡Â¥ðÈíú
Â■┬╚ÊÊ╦ßÁ──Û┴õÊ└└ÁðÈ»┴ªÐº║═┤·ð╗ú║┐╔─▄ËÙÂ¥ðÈËð╣ÏÊ®└ÝкËÙ╩ÁÐÚÍ╬┴ãкÈËÍ¥
Age-Dependent Kinetics and Metabolism of Dichloroacetate: Possible Relevance to Toxicity | Journal of Pharmacology and Experimental Therapeutics
http://jpet.aspetjournals.org/content/324/3/1163íí
Dichloroacetate (DCA) Causes Cancer Cells to Self-Destruct
https://articles.mercola.com/sites/articles/archive/2012/08/04/dca-and-turmeric-on-cancer.aspxSTORY AT-A-GLANCE
1. An experimental cancer drug called DCA (dichloroacetate) shows promise in the fight against cancer by altering cancer cell metabolism and inducing apoptosis (cellular suicide); DCA appears to exert anti-tumor effects against several forms of cancer, including brain, endometrial, cervical, prostate, breast, and colorectal cancers.
2. DCA forces cancer cells to shift from their preferred method of generating energy (glycolysis) to the method normal cells prefer (glucose oxidation) and í░reawakensí▒ cancer cellsí» mitochondria.
3. There are serious side effects reported by some adults self-administering DCA, including peripheral neuropathy and encephalopathy, so more research is needed before DCA can be considered safe.
4. Optimizing your vitamin D level is one of the most important steps you can take to protect yourself from cancer
Certain foods mimic the actions of DCA without ANY side effects, such as broccoli and the spice turmeric
ííDCA Instigates Mass Suicide among Cancer Cells
DCA suppresses PDK (the mitochondrial gatekeeper), and this fires up the cell's mitochondria. Not only does this force the cancer cell to abandon its preferred metabolic process, but it flips the cell's "suicide switch" as well. This happens because mitochondria are the primary regulators of apoptosis, or cellular suicideí¬they are loaded with sensors that react to abnormalities by pushing the cell's self-destruct button.
When a cancer cell's mitochondria realize it's a cancer cell, it spontaneously kills itself. This is the reason chemotherapy and radiation result in such terrible side effectsí¬your healthy cells actually die much more easily because of this self-destruct button.
The reason cancer is so fast growing is that the mitochondria have been deactivated, so the cells evade apoptosis, as well as being able to grow in the absence of oxygen (glycolysis)6. DCA reverses this.In effect, DCA directly causes cancer cell apoptosis and works synergistically other cancer therapies, such as radiation, gene therapy, and viral therapy. A number of scientific studies have been performed to date, and most are encouraging.íí
Dichloroacetate (DCA) Causes Cancer Cells to Self-Destruct
Â■┬╚ÊÊ╦ßÐ╬ú¿DCAú®Á╝Í┬░®¤©░¹ÎÈ╗┘
ʬÁÒ
1. ╩ÁÐÚðÈ┐╣░®Ê®╬´DCAú¿Â■┬╚ÊÊ╦ßÐ╬ú®═¿╣²©─▒õ░®¤©░¹Á─ð┬│┬┤·ð╗║═ËıÁ╝¤©░¹Á‗═÷ú¿¤©░¹ÎÈ╔▒ú®¤È╩¥│÷ÂÈ┐╣░®ÍóÁ─¤ú═¹íú DCA╦ã║§ÂÈÂÓÍÍð╬╩¢Á─░®Íó¥▀Ëð┐╣ÍÎ┴÷θË├ú¼░³└¿─È░®ú¼ÎË╣¼─┌─ñ░®ú¼╣¼¥▒░®ú¼Ã░┴ð¤┘░®ú¼╚Ú¤┘░®║═¢ßÍ▒│ª░®íú
2. DCAã╚╩╣░®¤©░¹┤Ëãõ╩ÎÐíÁ──▄┴┐╔·│╔À¢À¿ú¿╠â═¢Ôú®Î¬▒õ╬¬ı²│ú¤©░¹Á─╩ÎÐíÀ¢À¿ú¿ã¤╠Ð╠ÃЧ╗»ú®ú¼▓óí░╗¢ðÐí▒░®¤©░¹Á─¤▀┴ú╠Õíú
3. ─│ð®│╔╚╦ÎÈ╬Ê©°Ê®Á─DCAËðФÍÏÁ─©▒θË├ú¼░³└¿Í▄╬º╔±¥¡▓í▒õ║═─È▓íú¼Ê‗┤╦È┌DCA▒╗╚¤╬¬░▓╚½Í«Ã░╗╣ðÞʬ¢°ðð©³ÂÓÁ─Ðð¥┐íú
4. Ë┼╗»╬¼╔·╦ÏD╦«ã¢╩Ã─·┐╔ÊÈÈñÀ└░®ÍóÁ─ίÍÏʬ▓¢ÍÞÍ«Ê╗
─│ð®╩│╬´─úÀ┬DCAÁ─θË├°├╗Ëð╚╬║╬©▒θË├ú¼└²╚þ╬¸└╝╗¿║═¢¬╗ã¤Ò┴¤íú
DCAÍ·│ñ░®¤©░¹ÎÈ╔▒
DCAÊÍÍãPDKú¿¤▀┴ú╠Õ═°╩Ïú®ú¼▓ó╝ñÀó¤©░¹Á─¤▀┴ú╠ÕíúıÔ▓╗¢÷ã╚╩╣░®¤©░¹À┼ã·ãõã½░«Á─┤·ð╗╣²│╠ú¼Â°ÃÊ╗╣╩╣░®¤©░¹Á─í░ÎÈ╔▒┐¬╣Ïí▒Àó╔·À¡Î¬íúÍ«╦¨ÊÈÀó╔·ıÔÍÍÃÚ┐÷ú¼╩ÃÊ‗╬¬¤▀┴ú╠Õ╩ä©░¹Á‗═÷╗‗¤©░¹ÎÈ╔▒Á─͸ʬÁ¸¢┌╝┴-¤▀┴ú╠ÕÍðÎ░Ëð┤½©ðã¸ú¼ıÔð®┤½©ðã¸┐╔═¿╣²░┤¤┬¤©░¹Á─ÎÈ╗┘░┤┼ÑÂÈÊý│úÎ÷│÷À┤˪íú
Á▒░®¤©░¹Á─¤▀┴ú╠ÕÊÔ╩ÂÁ¢╦³╩Ã░®¤©░¹╩▒ú¼╦³╗ßÎÈÀó╔▒╦└ÎÈ╝║íúıÔ¥═╩Ã╗»Ðº┴ãÀ¿║═À┼╔õ┴ãÀ¿Á╝Í┬╚þ┤╦┐╔┼┬Á─©▒θË├Á─È¡Ê‗-─·Á─¢í┐Á¤©░¹╩Á╝╩╔¤Ë╔Ë┌┤╦ÎÈ╗┘░┤┼Ñ°©³╚¦ÊÎ╦└═÷íú
░®Íó╚þ┤╦Щ╦┘╔·│ñÁ─È¡Ê‗╩ä▀┴ú╠ÕÊÐ¥¡╩º╗¯ú¼Ê‗┤╦¤©░¹┐╔ÊÈ╠Ë▒▄Á‗═÷ú¼▓óÃÊ─▄╣╗È┌├╗ËðЧã°Á─ÃÚ┐÷¤┬╔·│ñú¿╠â═¢Ôú®6íú DCA┼ñά┴╦ıÔÊ╗¥Í├µíú╩Á╝╩╔¤ú¼DCAÍ▒¢ËÁ╝Í┬░®¤©░¹Á‗═÷▓óËÙãõ╦¹░®Íó┴ãÀ¿ú¿└²╚þÀ┼╔õ¤▀ú¼╗¨Ê‗┴ãÀ¿║═▓íÂ¥┴ãÀ¿ú®ð¡═¼Î¸Ë├íú㨢±╬¬Í╣ú¼ÊÐ¥¡¢°ðð┴╦ðÝÂÓ┐ãкÐð¥┐ú¼▓óÃÊ┤¾ÂÓ╩²┴¯╚╦╣─╬ÞíúDichloroacetate (DCA) Causes Cancer Cells to Self-Destruct
https://articles.mercola.com/sites/articles/archive/2012/08/04/dca-and-turmeric-on-cancer.aspxíí
Role of Pyruvate Dehydrogenase Kinase 4 in Regulation of Blood Glucose Levels
In the well-fed state a relatively high activity of the pyruvate dehydrogenase complex (PDC) reduces blood glucose levels by directing the carbon of pyruvate into the citric acid cycle. In the fasted state a relatively low activity of the PDC helps maintain blood glucose levels by conserving pyruvate and other three carbon compounds for gluconeogenesis. The relative activities of the pyruvate dehydrogenase kinases (PDKs) and the opposing pyruvate dehydrogenase phosphatases determine the activity of PDC in the fed and fasted states. Up regulation of PDK4 is largely responsible for inactivation of PDC in the fasted state. PDK4 knockout mice have lower fasting blood glucose levels than wild type mice, proving that up regulation of PDK4 is important for normal glucose homeostasis. In type 2 diabetes, up regulation of PDK4 also inactivates PDC, which promotes gluconeogenesis and thereby contributes to the hyperglycemia characteristic of this disease. When fed a high fat diet, wild type mice develop fasting hyperglycemia but PDK4 knockout mice remain euglycemic, proving that up regulation of PDK4 contributes to hyperglycemia in diabetes. These finding suggest PDK4 inhibitors might prove useful in the treatment of type 2 diabetes.Role of Pyruvate Dehydrogenase Kinase 4 in Regulation of Blood Glucose Levels
In the well-fed state a relatively high activity of the pyruvate dehydrogenase complex (PDC) reduces blood glucose levels by directing the carbon of pyruvate into the citric acid cycle. In the fasted state a relatively low activity of the PDC helps maintain blood glucose levels by conserving pyruvate and other three carbon compounds for gluconeogenesis. The relative activities of the pyruvate dehydrogenase kinases (PDKs) and the opposing pyruvate dehydrogenase phosphatases determine the activity of PDC in the fed and fasted states. Up regulation of PDK4 is largely responsible for inactivation of PDC in the fasted state. PDK4 knockout mice have lower fasting blood glucose levels than wild type mice, proving that up regulation of PDK4 is important for normal glucose homeostasis. In type 2 diabetes, up regulation of PDK4 also inactivates PDC, which promotes gluconeogenesis and thereby contributes to the hyperglycemia characteristic of this disease. When fed a high fat diet, wild type mice develop fasting hyperglycemia but PDK4 knockout mice remain euglycemic, proving that up regulation of PDK4 contributes to hyperglycemia in diabetes. These finding suggest PDK4 inhibitors might prove useful in the treatment of type 2 diabetes.Role of Pyruvate Dehydrogenase Kinase 4 in Regulation of Blood Glucose Levels - ScienceCentral
https://www.e-sciencecentral.org/articles/SC000002734íí
DCA Safety and Side Effects | DCA Watch
https://dcawatch.com/dca-safety-and-side-effects
DCA and the liver. In case of liver failure and severe jaundice doní»t use high doses of DCA because Dichloroacetate is metabolised in the liver. In situations like these, DCA should be administered intravenously and not through the mouth. DCA and the kidneys. Dichloroacetate is safe for patients who have kidney failure.This is the current accurate information on how DCA affects the major organs in the body. We can come to a conclusion that if Sodium Dichloroacetate is administered with care and adequate basic knowledge, its health risks are low and can be almost entirely prevented.
Dichloroacetate toxicokinetics and disruption of tyrosine ...
https://www.osti.gov/biblio/15001591-dichloroacetate-toxicokinetics-disruption...
Jan 01, 2002 íñ Dichloroacetate (DCA) is a by-product of drinking water chlorination. Administration of DCA in drinking water results in accumulation of glycogen in the liver of B6C3F1 mice. To investigate the processes affecting liver glycogen accumulation, male B6C3F1 mice were administered DCA in drinking water at levels varying from 0.1 to 3 g/l for up to ...
DCA, Dichloroacetate for Cancer - Dr. Weil
https://www.drweil.com/health-wellness/body-mind-spirit/cancer/dca-hope-or-hype
Jan 17, 2008 íñ DCA is an organic compound, and a byproduct of TCE (trichloroethylene), a chemical that has been a concern in the development of cancer. In January 2007, researchers at the University of Alberta published a study in the journal Cancer Cell suggesting that DCA showed promise in shrinking tumors in lab rats as well as inhibiting growth of cultured human cancer cells.íí
DCA safety and side effects
íí
Sodium dichloroacetate (commonly referred to as DCA) is a salt of sodium and dichloroacetic acid. The formula of this compound is Cl2 CH COONa. The structure of the molecule is similar to a combination of table salt and vinegar.
Under normal conditions, the material appears in powder form, white color and is soluble in water.
Sodium dichloroacetate is not patentable and has no restrictions on being transported, it can be freely sold without serious regulations.
The compound can be synthesized in laboratories or found as a side product in chlorinated water. DCA can be naturally acquired in a variety of red algae such as Asparagopsis taxiformis.Sodium dichloroacetate has received a lot of its attention by having a beneficial biological effect on living beings.
• Since 1973 Dichloroacetate has been used for treating children with congenital mitochondrial disorders (Ref.) which lead to metabolic lactic acidosis. DCA is able to treat the condition of congenital lactic acidosis and improve the life quality of such patients. The medication works by stimulating the Pyruvate Dehydrogenase complex. This resumes the normal metabolism of carbohydrates through aerobic glucose and lactic acid oxidation. Children treated with DCA achieve lower concentrations of blood serum lactates as well as lower overall metabolic acidosis.
The clinical trials of DCA treatment for congenital lactic acidosis hold a significant role in determining the optimal and safe concentrations for therapeutic purposes. Furthermore, side effects and the overall safety of the drug were carefully observed during the research which permitted the use of Sodium Dichloroacetate in the future. (Ref.)
• Since 1987 till now there are ongoing scientific studies on treating the harmful effects of brain ischemia with the use of DCA. In such conditions, Dichloroacetate can be useful in controlling the post-hypoxic lactic acidosis and the disturbed metabolism in the brain ¿C thus lowering the chances of a poor outcome. (Ref.)
• Since 2007 the anti-cancer effect of Sodium dichloroacetate has been noted. In the early scientific studies DCA had a positive response to implanted tumours of brain, breast and lung cancer. While on the influence of Dichloroacetate, the experimental rodents did not suffer any toxic effects, however, the tumours stopped growing and later on had a drastic size reduction. (link to all DCA studies)
The future in vitro and in vivo experiments had the same positive conclusions which finally lead to a clinical research in 2010. The verdict of the study ¿C DCA has a positive effect on malignant disease outcome, the substance demonstrates potential in the treatment of cancer and it is recommended to continue further clinical observations. (Ref.)
• Since 2009 DCA has been used as an alternative therapy for treating cancer in the first DCA clinics while under professional medical supervision.What is DCA? | DCA Watch
https://dcawatch.com/what-is-dca/íí
Sodium dichloroacetate is considered to be a fairly safe alternative cancer treatment. There have been no cases recorded for DCA to be a cause of death.
Before we begin, we should bear in mind that Sodium dichloroacetate has already demonstrated success in dealing with í«í«Lactic acidosis in children with congenital mitochondrial defectsí░ for some time. The first scientific studies and the usage of the drug began over 40 years ago. (Ref.)
In this time period, DCA has been constantly used as a medication for congenital mitochondrial diseases. The research done by Peter Stacpoole and his colleagues proved that when used for therapy, Sodium dichloroacetate can cause none, mild or moderate side effects. (Ref.)
The probability of adverse reactions is dependent on the dosing and the age of the patient. Larger DCA doses and older patient age (above 40 years) are related to a higher side effect occurrence. (Ref.)
On exceptionally rare occasions, a small portion of the population can metabolize DCA more slowly than the average. For this reason, even the standard DCA doses can cause adverse reactions to appear faster and more prominent in this group of people. In this case, lowering the DCA dose should fix the issues.
If you stop taking DCA, almost all of the side effects disappear in less than a week. The reversible peripheral neuropathy can sometimes take up to 7 or 14 days (rarely) to resolve completely. (Ref.)
According to one of the most famous DCA clinics and their observational data, 44 % of the patients who have taken DCA did not experience side effects.
The most common side effects caused by Dichloroacetate:
▪ Peripheral neuropathy.
(experienced by up to 20% of people who use DCA).
This group of symptoms begins in the fingers, hands and feet. Depending on the intensity of the neuropathy, it can manifest as tingling, numbness, tremor, painful sensations and slightly increased difficulty of coordinated movement.
On less common occasions, neuropathy can emerge in other places and appear as the tingling of eyes, lips and tongue.
Typically, at least a couple of weeks or months are needed for peripheral neuropathy to develop.
This side effect is reversible ¿C its intensity can decrease or it can disappear completely upon lowering the DCA dose or stopping DCA usage. (Ref.)
▪ Sleepiness, mental fogginess, confusion
(experienced by up to 20% of people who use DCA).
This group of symptoms is reversible ¿C you can decrease their intensity or completely make them disappear by lowering the DCA dose or stopping DCA usage.
The rare side effects caused by Dichloroacetate:
▪ Heartburn, nausea, digestive disorders.
Administering Dichloroacetate through the mouth can sometimes cause GI irritability.
▪ Pain at the tumor site (temporary and then resolves).
A very rare adverse reaction. It indicates that due increased apoptosis a lot of cancer cells are dying and indicates that DCA therapy is effective. However, only a couple of Tumor lysis syndrome cases were documented in the most popular DCA administering clinics. This situation is more likely to happen to people who have leukemia, lymphoma or big volume tumors. (Ref.1, Ref.2)
▪ Mild liver enzyme (AST, ALT, GGT) elevation, without symptoms.
A majority of medications can cause mild liver enzyme changes in the blood. DCA can cause minimal liver transaminase and transpeptidase elevations (about 50 ¿C 60 U/l) for 1 % of the patients. These little alterations should not cause any worries.
A similar or bigger liver enzyme increase can be influenced by antibiotic, paracetamol (acetaminophen), some types of medicinal herbs and birth control pills. (Ref.)
▪ Increased anxiety, mood changes, hallucinations.
These effects are temporary and should disappear with the discontinued use of DCA. They are more likely to appear in patients that are using drugs which strongly influence the Central nervous system.
Dichloroacetate influence on different organ systems:
▪ DCA and the brain.
If you are currently using cannabinoids, benzodiazepines, opioids or other drugs which affect the Central nervous system, keep in mind that DCA can amplify the adverse reactions caused by these medications (eg. Delirium, memory problems).
This scenario is more likely to happen if the prescriptions have already caused side effects. If the patient is not experiencing any issues with the CNS affecting drugs ¿C the risk for such interactions with DCA is low.
To minimize the probability of these drugs interacting, we recommend starting with low Sodium dichloroacetate doses and to gradually increase them. (Ref.)
▪ DCA and the heart.
Dichloroacetate seems to have a positive effect for the heart function without increasing the additional demand for oxygen. It also improves the efficiency of energy generation in the heart muscle. The drug is safe to use for people with heart failure and increased risk of cardiac ischemia. (Ref.)
▪ DCA and the liver.
In case of liver failure and severe jaundice doní»t use high doses of DCA because Dichloroacetate is metabolised in the liver. In situations like these, DCA should be administered intravenously and not through the mouth. (Ref.)
▪ DCA and the kidneys.
Dichloroacetate is safe for patients who have kidney failure. The drug has no toxicity for the kidneys.
▪ DCA and diabetes.
Patients who have diabetes can achieve better blood glycemic control with the help of Sodium Dichloroacetate. DCA seems to lower the blood sugar in between meals. (Ref.)
This is the current accurate information on how DCA affects the major organs in the body. We can come to a conclusion that if Sodium Dichloroacetate is administered with care and adequate basic knowledge, its health risks are low and can be almost entirely prevented.
We hope this article answers the most important questions.DCA Safety and Side Effects | DCA Watch
https://dcawatch.com/dca-safety-and-side-effects/íí
DCA is an odourless, colourless, inexpensive, relatively non-toxic, small molecule. And researchers at the University of Alberta believe it may soon be used as an effective treatment for many forms of cancer.
Dr. Evangelos Michelakis, a professor at the U of a Department of Medicine, has shown that dichloroacetate (DCA) causes regression in several cancers, including lung, breast, and brain tumors.
Michelakis and his colleagues, including post-doctoral fellow Dr. Sebastien Bonnet, have published the results of their research in the journal Cancer Cell.
Scientists and doctors have used DCA for decades to treat children with inborn errors of metabolism due to mitochondrial diseases. Mitochondria, the energy producing units in cells, have related to cancer since the 1930s, when researchers first noticed that these organelles dysfunction when cancer is present.
Until recently, researchers believed that cancer-affected mitochondria are permanently damaged and that this damage is the result, not the cause, of the cancer. But Michelakis, a cardiologist, questioned this belief and began testing DCA, which activates a critical mitochondrial enzyme, to í░reviveí▒ cancer-affected mitochondria.
The results astounded him.
Michelakis and his colleagues found that DCA normalized the mitochondrial function in many cancers, showing that their function was actively suppressed by the cancer but was not permanently damaged by it.
More importantly, they found that the normalization of mitochondrial function resulted in a significant decrease in tumor growth both in test tubes and in animal models. Also, they noted that DCA, unlike most currently used chemo therapies, did not have any effects on normal, non-cancerous tissues.
í░I think DCA can be selective for cancer because it attacks a fundamental process in cancer development that is unique to cancer cells,í▒ Michelakis said. í░One of the really exciting things about this compound is that it might be able to treat many different forms of cancerí▒.
Another encouraging thing about DCA is that, being so small, it is easily absorbed in the body, and, after oral intake, it can reach areas in the body that other drugs cannot, making it possible to treat brain cancers, for example.
Also, because DCA has been used in both healthy people and sick patients with mitochondrial diseases, researchers already know that it is a relatively non-toxic molecule that can be immediately tested on patients with cancer.
í░The results are intriguing because they point to the critical role that mitochondria play, they impart a unique trait to cancer cells that can be exploited for cancer therapyí▒
Dario Alteri
Director University of Massachusetts Cancer Center...
This is part of a 2010 press release by the University
DCA is an inexpensive drug that contains dichloroacetic acid, a very small, simple molecule that resembles vinegar. It is mostly used to treat children with a rare metabolic disorder. In 2007, Michelakis and his team published evidence that DCA reverses cancer growth in non-human models by altering the metabolism of the cancer. The drug tricks cancer cells into normal energy production by changing the way they handle nutrient fuels. This causes the cancer cells to í░commit suicide,í▒ without harming healthy cells.
Many researchers around the world have confirmed the University of Alberta teamí»s 2007 findings. Often research that was promising in non-human models does not work outside the lab. However, the U of A team is now reporting success in the next phase of its DCA research, testing the DCA compound in humans. After extracting glioblastomas from 49 patients over a two-year period and studying them within minutes of removal in the operating room, the team verified that the tumors responded to DCA by changing their metabolism.
Comment: So, the University describes Sodium dichloroacetate DCA (NaDCA) as a small little molecule that resembles vinegar and we are not allowed to buy it? The full 2010 press release is coming up below but is heavily censored. The point here is that it worked in humans the same as in the lab....
We all have been led to believe that cancer is a complicated disease, and therefore a death sentence! IT IS NOT! The primary cause of ALL cancer was known in the early 1960í»s.The University of Alberta team proved that Sodium dichloroacetate DCA (NaDCA) could reverse the Warburg effect. Unfortunately for us what they also proved was that all the billions of dollars being spent at the time on research and for most of the last 85 years was useless based on this discovery. Funny as it may seem the useless research has continued to be funded by billions of dollars per year, yet the U of A could not get funding.
...Dr. Michelakis said í░NaDCA did work in Humans exactly the way it was supposed to.í▒ The study only had one patient that had not had chemo, radiation and surgery all of which do irreparable damage. That one person had complete remission including the stem cells. Dr. Michelakis did test NaDCA on the 49 biopsies which proved conclusively that NaDCA works and it worked on all 49 biopsies. The more time you spend on this site the more you will understand why Dr. Michelakis, had to be careful in what he said (PDF of clinical trial can be found here).
...
Fortunately for us, what neither the Canadian Cancer society nor the U of Guelph researcher were aware of at the time was that the University of Leeds in the UK was also doing a study on Nadcapí»s effectiveness on colorectal cancer. The following is the conclusion from that study (read more). Results: Dichloroacetate (20 mM) did not reduce growth of non-cancerous cells but caused significant decrease in
cancer cell proliferation (P=0.009), which was associated with apoptosis and G2 phase cell-cycle arrest.
Conclusions:
Pyruvate dehydrogenase kinase inhibition attenuates glycolysis and facilitates mitochondrial oxidative phosphorylation, leading to reduced growth of colorectal cancer cells but not of non-cancerous cells. Studies will continue to be funded and published discrediting NaDCA, when you come upon a negative study look to see where the funding for that study came from and read the comment stream!
í░That the NCI, with enthusiastic support from the ACS _ the tail that wags the NCI dog _ has effectively blocked funding for research and clinical trials on promising non-toxic alternative cancer drugs for decades, in favor of highly toxic and largely ineffective patented drugs developed by the multibillion dollar global cancer drug industry. Additionally, the cancer establishment has systematically harassed the proponents of non-toxic alternative cancer drugsí▒ Samuel S. Epstein, M.D
Samuel S. Epstein, M.D. is professor emeritus of Environmental and Occupational Medicine at the University of Illinois School of Public Health, and Chairman of the Cancer Prevention Coalition. See Dr. Epsteiní»s biography here: http://www.preventcancer.com/about/epstein.htm#bio...
PET scan as it clearly proves the Warburg effect and the principles behind why if DCA cures one type of cancer it cures ALL cancers.
The Warburg effect states that the difference between a normal cell and a cancer cell is simply that a cancer cell gets its energy from fermentationn of glucoses and a normal cell gets energy from respiration of oxygen, therefore fermentation is common to all cancer cells, although this was proven beyond a doubt in the early 1960í»s the cancer industry in an effort to complicate cancer has been researching all the so called various types of cancer, as if lung cancer is different from breast cancer and also researching treatments for each cancer separately.
However, as a business the cancer industry knew that if they could get a patient earlier in the cancer cycle, they could treat the patient longer and make more money. The Warburg effect was used as the bases behind the PET scan, but not as a direction for research!
With a PET scan the patient is injected with a Glucose-based Radiopharmaceutical, the patient is then put into a PET/CT scanner which identifies the areas were the cancer cells are as they are feeding off the glucose, accurate 3D tumor images can then be seen on a computer screen. Interestingly no research was focused on this simple concept of cancer cell metabolism until the University of Alberta discovered that NaDCA could switch the mitochondria of a cancer cell back on allowing it to commit suicide. The PET scan was developed in the early 70í»s.
If the PET scan works, then the Cancer Industry aggress with Warburgí»s discovery; that all cancer is the same in the way it gets energy is true. Therefore, theoretically NaDCA is a cure for all cancers, it is that simple and is why the researchers made such a bold statement. Why was the U of A discovery made by a cardiologist? In 2001 and 2004 Dr. Michelakis and his team published two papers regarding the virtues of NaDCA in the treatment of heart patients and found that NaDCA would help re open previously clogged arteries, and NaDCA was shown to be helpful in head injury and stroke recovery.
This is the closing paragraph from one of these studies published in the American Heart Journal
DCA is a very attractive drug to be studied in human PHT( Chronic hypoxic pulmonary hypertension (CH-PHT)), particularly because it has already been used in small, short-term human studies without major toxicity.13¿C15 To the best of our knowledge, no other drugs in current clinical use have Kv channel opening properties. DCA may be capable of restoring Kv channel function and expression and thus have benefit in the treatment of pulmonary vascular diseases...
The primary cause of cancer
The biggest breakthrough in early detection is the PET scan machine; it is based on the Warburg effect which is also the bases for the DCA discovery and why such a simple molecule was the missing link in 1966 when Otto Warburg gave the following speech....
The Prime Cause and Prevention of Cancer (Revised Lindau Lecture) By OTTO WARBURG Director, Max Planck Institute for Cell Physiology, Berlin-Dahlem, Germany) English Edition by DEAN BURK, National Cancer Institute, Bethesda, Maryland. Note by DEAN BURK: Adapted from a lecture originally delivered by O. Warburg at the 1966 annual meeting of Nobelists in Lindau, Germany. O. Warburg won the Nobel Prize in Medicine in 1931 for his discovery of the oxygen-transferring enzyme of cell respiration and was voted a second Nobel Prize in 1944 for his discovery of the active groups of the hydrogen transferring enzymes. Many universities, like Harvard, Oxford, Heidelberg has offered him honorary degrees. He is a Foreign member of the Royal Society of London, a Knight of the Order of Merit founded by Frederick the Great and was awarded the Great Cross with Star and Shoulder ribbon of the Bundesrepublik. His main interests are Chemistry and Physics of Life. In both fields no scientists have been more successful. There are prime and secondary causes of diseases. For example, the prime cause of the plague is the plague bacillus, but secondary causes of the plague are filth, rats, and the fleas that transfer the plague bacillus from rats to man. By a prime cause of a disease I mean one that is found in every case of the disease. Cancer, above all other diseases, has countless secondary causes. But, even for cancer, there is only one prime cause. Summarized in a few words, the prime cause of cancer is the replacement of the respiration of oxygen in normal body cells by a fermentation of sugar. All normal body cells meet their energy needs by respiration of oxygen, whereas cancer cells meet their energy needs in great part by fermentation. All normal body cells are thus obligate aerobes, whereas all cancer cells are partial anaerobes. From the standpoint of the physics and chemistry of life this difference between normal and cancer cells is so great that one can scarcely picture a greater difference. Oxygen gas, the donor of energy in plants and animals is dethroned in the cancer cells and replaced by an energy yielding reaction of the lowest living forms, namely, a fermentation of glucose. The key to the cancer problem is accordingly the energetics of life, which has been the field of work of the Dahlem institute since its initiation by the Rockefeller Foundation about 1930. In Dahlem the oxygen transferring and hydrogen transferring enzymes were discovered and chemically isolated. In Dahlem the fermentation of cancer cells was discovered decades ago; but only in recent years has is been demonstrated that cancer cells can grow in the body almost with only the energy of fermentation. Only today can one submit, with respect to cancer, all the experiments demanded by PASTEUR and KOCH as proof of the prime causes of a disease. If it is true that the replacement of oxygen-respiration by fermentation is the prime cause of cancer, then all cancer cells without exception must ferment, and no normal growing cell ought to exist that ferments in the body. Comment: Two things to note here, one is that all cancer cells are the same in the way they get energy from fermentation of sugar, therefore a rational approach to curing cancer would be to obstruct the cells access to sugar, if it caní»t feed it caní»t grow and caní»t multiply and by cutting off the glucose and oxygenating the cell we now know thanks to the U of A, it will revert back to oxygen respiration and because the cell is bad it creates apoptosis (commits suicide). In simple terms that is exactly what NaDCA can do! The second thing to note here is that this work was carried on at the Rockefeller foundations Dahlem Institute. This would be like an oil company discovering that saltwater could replace gasoline and your car would run fine. If one puts embryonic mouse cells into a suitable culture medium saturated with physiological oxygen pressures, they will grow outside the mouse body, in vitro, and indeed as pure aerobes, with a pure oxygen respiration, without a trace of fermentation. However, if during the growth one provides and oxygen pressure so reduced that the oxygen respiration is partially inhibited, the purely aerobic metabolism of the mouse embryonic cells is quantitatively altered within 48 hours, in the course of two cell divisions, into the metabolism characteristic of fermenting cancer cells. Fig. 2 illustrates the very simple experimental procedure involved.
ííThe key to the cancer problem is accordingly the energetics of life, which has been the field of work of the Dahlem institute since its initiation by the Rockefeller Foundation about 1930. In Dahlem the oxygen transferring and hydrogen transferring enzymes were discovered and chemically isolated. In Dahlem the fermentation of cancer cells was discovered decades ago; but only in recent years has is been demonstrated that cancer cells can grow in the body almost with only the energy of fermentation. Only today can one submit, with respect to cancer, all the experiments demanded by PASTEUR and KOCH as proof of the prime causes of a disease. If it is true that the replacement of oxygen-respiration by fermentation is the prime cause of cancer, then all cancer cells without exception must ferment, and no normal growing cell ought to exist that ferments in the body.
Comment: Two things to note here, one is that all cancer cells are the same in the way they get energy from fermentation of sugar, therefore a rational approach to curing cancer would be to obstruct the cells access to sugar, if it caní»t feed it caní»t grow and caní»t multiply and by cutting off the glucose and oxygenating the cell we now know thanks to the U of A, it will revert back to oxygen respiration and because the cell is bad it creates apoptosis (commits suicide). In simple terms that is exactly what NaDCA can do! The second thing to note here is that this work was carried on at the Rockefeller foundations Dahlem Institute. This would be like an oil company discovering that saltwater could replace gasoline and your car would run fine. If one puts embryonic mouse cells into a suitable culture medium saturated with physiological oxygen pressures, they will grow outside the mouse body, in vitro, and indeed as pure aerobes, with a pure oxygen respiration, without a trace of fermentation. However, if during the growth one provides and oxygen pressure so reduced that the oxygen respiration is partially inhibited, the purely aerobic metabolism of the mouse embryonic cells is quantitatively altered within 48 hours, in the course of two cell divisions, into the metabolism characteristic of fermenting cancer cells. Fig. 2 illustrates the very simple experimental procedure involved.
If one then brings such cells, in which during their growth under reduced oxygen pressure a cancer cell metabolism has been produced, back under the original high oxygen pressure, and allows the cell to grow further, the cancer metabolism remains. The transformation of embryonic cell metabolism into cancer cell metabolism can thus be irreversible, and important result, since the origin of cancer cells from normal body cells is an irreversible process. It is equally important that these body cells whose metabolism has thus been transformed into cancer metabolism now continue to grow in vitro as facultative anaerobes. The duration of our experiments is still too limited to have yielded results of tests of inoculation of such cells back into mice, but according to all previous indications such cells will later grow as anaerobes upon transplantation into animals.
In any case, these experiments belong to the most important experiments in the field of cancer investigation since the discovery of the fermentation of tumors. For cancer metabolism, heretofore, measured so many thousand of times, has now been induced artificially in body cells by the simplest conceivable experimental procedure, and with this artificially induced cancer metabolism the body cells divide and grow as anaerobes in vitro.
Comment: This was a simple experiment proving the primary cause of every cancer is a lack of oxygen in cell tissue. THEY ACTUALLY CAUSED CANCER CELLS by reducing the oxygen supply to normal cells. Keep in mind this is prior to 1966í¡ Although they knew how to cause cancer now it was irreversible. It is my belief as well as others that the Rockefellerí»s Dahlem Institute continued on to discover the cure for cancer in the late 60í»s which was non patentable and therefore buried it.
Also realize that this transcript of Otto Warburgí»s lecture was translated and published by Dean Burk who co-founded the US National Cancer Institute in 1937 and headed its Cytochemistry department for over three decades. Burk left NCI in 1974 claiming they had falsified testing on a natural cancer treatment Laetrile and the FDA was blocking what many considered the foremost treatment for cancer at the time.íí
Comment: If you are still reading then you are as convinced as we are that NaDCA treatment for all cancers, if taken as a supplement it must then prevent cancer also. It is good for your heart and in theory could prevent or reverse the effects of coronary artery disease. Of course, if tests are never done to prove any of this it will never be made available.
Time to re read this excerpt from the May 2010 press release belowí¡
In 2007 the U of A team led by Dr Michelakis, published evidence that DCA reverses cancer growth in non-human models and test tubes. The team showed then that DCA achieves these antitumor effects by altering the metabolism of cancer. By altering the way cancer handles its nutrient fuels, specifically the sugars, DCA was able to take away cancerí»s most important strength, the resistance to death. Since then, several independent groups across the world have confirmed the Alberta teamí»s findings. In December 2009, the editors of í░Scienceí▒ predicted that cancer metabolism is one of only 5 areas across all scientific disciplines, to í░watch for major breakthroughsí▒ in 2010.The U of A team set out to show that the way that DCA works in actual patients is the same with the way it works in the lab. In addition, researchers wanted to show whether DCA is safe and possibly effective in very sick patients with brain cancer.
By extracting glioblastomas from 49 patients over a period of 2 years and studying them within minutes of removal in the operating room, the team showed that tumors respond to DCA by changing their metabolism. Then, the team treated 5 patients with advanced glioblastoma and secured tumor tissues before and after the DCA therapy. By comparing the two, the team showed that DCA works in these tumors exactly as was predicted by test tube experiments. This is very important because often the results in non-human models tested in the lab do not agree with the results in patients. In addition, the team showed that DCA has anti-cancer effects by altering the metabolism of glioblastoma cancer stem cells, the cells thought responsible for the recurrences of cancer.
In the 5 patients tested, the drug took 3 months to reach blood levels high enough to alter the tumor metabolism. At those levels, there were no significant adverse effects. However, at some of the higher doses tested, DCA caused nerve malfunction, i.e. numbing of toes and fingers. Importantly, in some patients there was also evidence for clinical benefit, with the tumors either regressing in size or not growing further during the 18-month study.
No conclusions can be made on whether the drug is safe or effective in patients with this form of brain cancer, due to the limited number of patients tested by the studyí»s leads Drs Michelakis and Petruk. Researchers emphasize that use of DCA by patients or physicians, supplied from for-profit sources or without close clinical observation by experienced medical teams in the setting of research trials, is not only inappropriate but may also be dangerous. The U of A results are encouraging and support the need for larger clinical trials with DCA. This work is also one of the first in humans to support the emerging idea that altering the metabolism of tumors is a new direction in the treatment of cancer, Michelakis and Petruk said.
Comment: This comment í░ no conclusioní▒ and í░the warning not to use on your owní▒ was required most likely by Health Canada, as NaDCA is so easy to use and is so safe that it is a crazy statement. What could happen? We could all get healthy, that would be bad, I guess they are counting on none of us doing the research and finding out how harmless NaDCA really is.
The research team hopes to secure additional funding to continue the ongoing trials with DCA at the University of Alberta. Further studies would include more patients with brain cancer, and test the combination of DCA and standard chemotherapies, eventually including patients from other academic health sciences centres.
Comment: So, the only thing they didní»t know in 1966 was that the mitochondria could be turned back on in a cancerous cell allowing the cell to cause apoptosis (cell suicide). This is exactly what NaDCA does, NaDCA does not kill the cancer! It turns the mitochondria back on so that the cancer cell kills itself. I caní»t find any mention of the Phase 1 study on dosages and I assume the dosage was set by Health Canada in the protocol for the clinical trial, which was probably a low dosage of around 10mg per kg of body weight and the reason why it took 3 month to build up in the system. The most amazing item of note is that NaDCA altered the metabolism of the Glioblastoma cancer Stem Cellsí¡í¡.No current cancer treatment is known to do this!
What can you do?
If you are recently diagnosed with cancer, please re-read the information again on the lack of proof as to the effectiveness of the í░standard therapiesí▒ and consider the damage to your body that these treatments are documented to cause. I woní»t tell you not to take Chemo, Radiation or surgery as that is your choice, I will say that you will be pressured by the medical community, but always ask questions, it is your health and the statistics you are usually quoted are biased and usually the information is supplied to your doctor by the Drug Companies or Cancer Societies. When quoted a statistic always ask if the result is a relative or absolute statistic as the difference is huge! Please provide your doctor with the NaDCA info page, you do have freedom of choice in your treatment and should be able to ask to have your progress monitored, if your Doctor refuses, find another Doctor!
If you have had cancer and are concerned about it coming back you have a very valid concern as the chance of that happening is greater than 90%, and as you have seen if you have read this far the drugs that most cancer survivors are currently taking offer virtually no benefit at all and are carcinogenic
themselves. NaDCA may be your solution to clearing any remaining cancer cells from your body and enjoying the rest of your life without the worry of a cancer recurrence!
There is enough evidence out there now to support the claims that NaDCA is effective in treating cancer. It is quite possibly the powers that be will simply continue to try and discredit the effectiveness and let the few of us that do the research continue to get the product. The unfortunate truth is that most people will continue to follow whatever their doctor suggests, not realizing how tied his/her hands are in what they can tell you without risking their license.
Take NaDCA as a supplement, my family and I take NaDCA daily to avoid ever getting cancer and for the benefits to the heart. After about a week we noticed clearer thinking and increased energy and better circulation. See the study on NaDCA for athletic enhancement here.
As a result of NaDCAí»s low molecular weight it could travel to all parts of the bodiesí» tissue keeping arteries and small veins clear and oxygenated? In the process allowing your own immune system to operate at peak levels leaving our organs to heal themselves.
My 74-year-old father who has smoked for 55 years suffers from bad circulation and had a black spot beginning on one of his toes; his Doctor was saying if it was to get any worse it would need to be amputated. The black spot was gone in about 10 days. He also suffers from COPD and claims to have much more energy and greater endurance.
Send an Email to your local government representative, state or provincial, and federal. Tell them you are upset about the lack of support that this nontoxic, unpatented discovery is receiving and ask for their help, include a link to this site. Most politicians are completely unaware of this discovery and they have most likely been touched in their own families by cancer Tell your friends, it is hard to explain, the best way to help someone is to send them to this website, my intent is to share it on Facebook once a month because people tend not to pay attention until it hits close to home and the unfortunate truth is that every month in North America another 130,000 people are diagnosed with cancer and over 1 million people world wide.
The World Health Organization (WHO) stated that in 2000 over 10 million people were diagnosed with a malignant tumor and 6.2 million people died of a malignant tumor, these numbers are expected to increase by 50% by 2020. Not sure why blood cancers were not included but they were not.
If you start taking NaDCA, please report your progress on this site you have no idea who you may help by doing so!
What if DCA is a molecule that adds oxygen to our blood and balances out our sugar and PH levels? There has long been a theory that when we are í░run downí▒ we are more susceptible to illness and that illness and disease can not survive when our PH is more alkaline than acidic. Disease will not take hold in a PH balanced body! A swimming pool is a perfect analogy. I have had two homes with swimming pools, If I kept my PH perfect the water would be crystal clear, if it slipped below 7 on the PH scale it would start to get murky and if left that way for a few days it turned green from algae. It didní»t matter if all the algae were the same, as I am sure many different algae could be identified if analyzed. Much like cancer, however once I started adding chlorine the water would start to clear up and when it was back to proper PH all the algae were gone. I didní»t have to kill the specific type of algae just create an
environment in which it couldní»t survive. Our bodies are 60% water and our blood are 90% water, how important do you think our PH level is?
Is it possible that DCA is the chlorine puck for our bodies? The scientists will have a hay day with that statement; however, they were the same people spreading the propaganda that DCA was a poisonous chemotherapy drug that none of us should use.
What is interesting is sodium dichloroacetate is just a simple molecule that could easily be taken as a dietary supplement by perfectly healthy people and is considered by the EPA to have no mutagenic effect. It has been studied as a supplement for athletes, and to greatly reduce insulin requirements for diabetics. It has been proven effective in the treatment of heart and stroke patients and I would suggest could be useful in the prevention of heart attacks and strokes. All studies either in vitro or in vivo turned out great results; however, the non patentable nature of the compound stopped the studies from progressing to clinical trials as there would be no money in it. SOUND FAMILIAR? There is enough credible evidence that DCA cures cancer that I believe at this point the adverse publicity of forcing these sites to close could be more costly to big pharma then letting them continue to sell DCA. At some point this may change, as in 2007 when the FDA forced all sites to close.
What if a simple molecule could cure all cancers? A good friend of mine said to me that there is no way! If a cure for cancer was discovered the medical community would tell us!
As I said to her í░They Didí▒ the University of Alberta told us in 2007 and again in 2010, and I am telling you now!
The discovery was accidental and not expected by the industry, after all no one was working on a cancer cure; research by big Pharma has always been focused on expensive ways to treat the symptom not the cause. There is no money in curing someone especially with a simple molecule that cost a few dollars per day and no hospital stay.
They caní»t afford to let you know how simple DCA is and how beneficial it is to be balancing our body, my hope is the information we have put together saves your life.
DCA watch does not provide medical advice, diagnosis or treatment. The information provided on this site is for the purposes of information only. You should not use this information to diagnose, cure or treat any health problem without consulting with a qualified and licensed healthcare professional.Home | DCA Watch
http://dcawatch.com/home/íí
ÂȤ╚╠ýðȤ▀┴ú╠Õ├©╚▒ÀªÊ²ãÁ─╚Ú╦ß╦ßÍðÂ¥ú¼│²┴╦Â■┬╚ÊÊ╦ßÐ╬Í«═Ôú¼─┐Ã░╔ð╬Ìãõ╦³╠µ┤·Ðíȱíú¥í╣▄Â■┬╚ÊÊ╦ßÐ╬Ê▓▓╗─▄©¨Í╬©├▓íú¼Á½¥═©─╔ã╦ßÍðÂ¥ÍóÎ┤║═┐ÏÍã▓óÀóÍó°ÐÈú¼┤╦Ê®▒╚╠╝╦ßÃÔ─ã║═©┤║¤╬¼╔·╦Ï¥▀Ëð©³╗²╝½Á─Í╬┴ãÊÔÊÕíúËÙ╠╝╦ßÃÔ─ã¤Ó▒╚ú¼Â■┬╚ÊÊ╦ß─ã▓╗Á½─▄╣╗╗║¢Ô╦ßÍðÂ¥ú¼Â°ÃÊ─▄╣╗©─╔ãЬÊ║»┴ªÐºÍ©▒Û[8]íúÊ╗ð®│ñã┌À■Ë├Â■┬╚ÊÊ╦ßÐ╬Á─¨═»╗╝ı▀ú¼▓╗¢÷╦ßÍðÂ¥ÍóÎ┤Á├Á¢╗║¢Ôú¼ÃÊãõ╔·│ñÀó˲Î┤┐÷ÊÓÁ├Á¢©─╔ãú¼▓óÃÊ╬┤Àó¤ÍÂ■┬╚ÊÊ╦ßÐ╬ʲãÁ─Â¥©▒θË├[1,13]íúÂÈË┌Àó╔·È┌©╬ÈÓÊãÍ▓╣²│╠ÍðÁ─╚Ú╦ß╦ßÍðÂ¥ú¼Â■┬╚ÊÊ╦ßÐ╬ËÙ╠╝╦ßÃÔ─ã▓óË├─▄╣╗░▓╚½ËððºÁÏ╝§╔┘╚Ú╦ßð¯╗²ú¼╗║¢Ô╦ßÍðÂ¥ú¼═¼╩▒┐╔╝§╔┘╠╝╦ßÃÔ─ãÁ─Ë├┴┐ú¼¢ÁÁ═©▀Ь─ãÍóÁ─Àó╔·┬╩[14]íúÂ■┬╚ÊÊ╦ßÐ╬╔ð┐╔ÊÈÍ╬┴ãФÍÏ┼▒╝▓ʲãÁ─╚Ú╦ß╦ßÍðÂ¥ú¼Â°ÃÊ▓╗Ë░¤ý┐╣┼▒Ê®┐³─■Á─Ê®┤·Â»┴ªÐº[15,16]íú
íí
The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury
Abstract
Our previous studies demonstrated that some degree of neuronal death is caused by hypoglycemia, but a subsequent and more severe wave of neuronal cell death occurs due to glucose reperfusion, which results from the rapid restoration of low blood glucose levels. Mitochondrial dysfunction caused by hypoglycemia leads to increased levels of pyruvate dehydrogenase kinase (PDK) and suppresses the formation of ATP by inhibiting pyruvate dehydrogenase (PDH) activation, which can convert pyruvate into acetyl-coenzyme A (acetyl-CoA). Sodium dichloroacetate (DCA) is a PDK inhibitor and activates PDH, the gatekeeper of glucose oxidation. However, no studies about the effect of DCA on hypoglycemia have been published. In the present study, we hypothesized that DCA treatment could reduce neuronal death through improvement of glycolysis and prevention of reactive oxygen species production after hypoglycemia. To test this, we used an animal model of insulin-induced hypoglycemia and injected DCA (100 mg/kg, i.v., two days) following hypoglycemic insult. Histological evaluation was performed one week after hypoglycemia. DCA treatment reduced hypoglycemia-induced oxidative stress, microglial activation, blood¿Cbrain barrier disruption, and neuronal death compared to the vehicle-treated hypoglycemia group. Therefore, our findings suggest that DCA may have the therapeutic potential to reduce hippocampal neuronal death after hypoglycemia.Â■┬╚ÊÊ╦ß─ãÂÈÁ═Ь╠ÃʲãÁ─╦╔╦║¾¤▀┴ú╠Õ╣ª─▄ı¤░¡║═╔±¥¡È¬╦└═÷Á─Ë░¤ý
╬Ê├ÃÊÈÃ░Á─Ðð¥┐▒Ý├¸ú¼Á═Ь╠ÃÍó╗ßÁ╝Í┬Ê╗¿│╠Â╚Á─╔±¥¡È¬╦└═÷ú¼Á½╩ÃË╔Ë┌Á═Ь╠Ã╦«ã¢Á─┐ý╦┘╗Í©┤ú¼ã¤╠Ð╠ÃÈ┘╣ÓÎó╗ßÁ╝Í┬╦µ║¾║═©³Ð¤ÍÏÁ─╔±¥¡È¬¤©░¹╦└═÷└╦│▒íúÁ═Ь╠ÃʲãÁ─¤▀┴ú╠Õ╣ª─▄ı¤░¡Á╝Í┬▒¹═¬╦ß═ÐÃÔ├©╝ñ├©ú¿PDKú®╦«ã¢╔²©▀ú¼▓ó═¿╣²ÊÍÍã▒¹═¬╦ß═ÐÃÔ├©ú¿PDHú®╝ñ╗¯ÊÍÍãATPÁ─ð╬│╔ú¼▒¹═¬╦ß═ÐÃÔ├©┐╔ÊÈ¢½▒¹═¬╦ßά╗»╬¬Êʧú©¿├©Aú¿Êʧú©¿├©Aú®íúÂ■┬╚ÊÊ╦ß─ãú¿DCAú®╩ÃPDKÊÍÍã╝┴ú¼┐╔╝ñ╗¯PDHú¿ã¤╠Ð╠ÃЧ╗»Á─╩Ï├┼È▒ú®íúÁ½╩Ãú¼╔ð╬┤Àó▒ÝËð╣ÏDCAÂÈÁ═Ь╠ÃÁ─Ë░¤ýÁ─Ðð¥┐íúÈ┌▒¥Ðð¥┐Íðú¼╬Ê├Ã╝┘╔ÞDCAÍ╬┴ã┐╔═¿╣²©─╔ã╠â═¢Ô║═ÈñÀ└Á═Ь╠Ã║¾▓·╔·╗¯ðÈЧ└┤╝§╔┘╔±¥¡È¬╦└═÷íú╬¬┴╦▓Ô╩ÈıÔÊ╗ÁÒú¼╬Ê├Ã╩╣Ë├┴╦Ê╚Á║╦ÏËıÀóÁ─Á═Ь╠ÃÁ─»╬´─úð═ú¼▓óÈ┌¢ÁЬ╠Ã║¾Îó╔õ┴╦DCAú¿100 mg / kgú¼¥▓┬÷Îó╔õú¼┴¢╠ýú®íúÁ═Ь╠Ã║¾Ê╗Í▄¢°ððÎÚͻкã└╣└íúËÙ╚▄├¢Í╬┴ãÁ─Á═Ь╠ÃÎÚ¤Ó▒╚ú¼DCAÍ╬┴ã╝§╔┘┴╦Á═Ь╠ÃʲãÁ─Ч╗»Ëª╝ñú¼ðí¢║Í╩¤©░¹╗¯╗»ú¼Ð¬─Èã┴ı¤ãã╗Á║═╔±¥¡È¬╦└═÷íúÊ‗┤╦ú¼╬Ê├ÃÁ─Àó¤Í▒Ý├¸DCA┐╔─▄¥▀Ëð¢ÁÁ═Á═Ь╠Ã║¾║ú┬Ý╔±¥¡È¬╦└═÷Á─Í╬┴ãÃ▒┴ªíúView Full-Text
Keywords: hypoglycemia; sodium dichloroacetate; pyruvate dehydrogenase kinase; pyruvate dehydrogenase; oxidative stress; neuron death
Cells | Free Full-Text | The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury
https://www.mdpi.com/2073-4409/8/5/405íí
Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock
Abstract
Hemorrhagic shock is a leading cause of death in people under the age of 45 and accounts for almost half of trauma-related deaths. In order to develop a treatment strategy based on potentiating mitochondrial function, we investigated the effect of the orphan drug dichloroacetate (DCA) on survival in an animal model of hemorrhagic shock in the absence of fluid resuscitation. Hemorrhagic shock was induced in rats by withdrawing 60% of the blood volume and maintaining a hypotensive state. The studies demonstrated prolonged survival of rats subjected to hemorrhagic injury (HI) when treated with DCA. In separate experiments, using a fluid resuscitation model we studied mitochondrial functional alterations and changes in metabolic networks connected to mitochondria following HI and treatment with DCA. DCA treatment restored cardiac mitochondrial membrane potential and tissue ATP in the rats following HI. Treatment with DCA resulted in normalization of several metabolic and molecular parameters including plasma lactate and p-AMPK/AMPK, as well as Ach-mediated vascular relaxation. In conclusion we demonstrate that DCA can be successfully used in the treatment of hemorrhagic shock in the absence of fluid resuscitation; therefore DCA may be a good candidate in prolonged field care following severe blood loss.│÷ЬðÈð¦┐╦║¾Ë├Â■┬╚ÊÊ╦ß░ð¤‗¤▀┴ú╠Õ┐╔©─╔ãÈñ║¾
ííı¬Ê¬
╩ºÐ¬ðÈð¦┐╦╩Ã45╦ÛÊȤ┬╚╦╚║╦└═÷Á─͸ʬȡÊ‗ú¼╝©║§ı╝ËÙ┤┤╔╦Ëð╣ÏÁ─╦└═÷Á─Ê╗░Ùíú╬¬┴╦Íã¿╗¨Ë┌È÷Ã┐¤▀┴ú╠Õ╣ª─▄Á─Í╬┴ã▓▀┬Èú¼╬Ê├ÃÐð¥┐┴╦È┌╬ÌÊ║╠Õ©┤╦ıÁ─╩ºÐ¬ðÈð¦┐╦»╬´─úð═Íðú¼╣┬¨ʮÂ■┬╚ÊÊ╦ßÐ╬ú¿DCAú®ÂÈ┤µ╗¯┬╩Á─Ë░¤ýíú═¿╣²│Ú│÷60úÑÁ─Ь╚¦┴┐▓ó╬¼│ÍÁ═ЬÐ╣Î┤╠¼ú¼È┌┤¾╩¾Íðʲã│÷ЬðÈð¦┐╦íúıÔð®Ðð¥┐▒Ý├¸ú¼¢Ë╩▄DCAÍ╬┴ãÁ─┤¾╩¾Ê‗│÷ЬðÈ╦╔╦ú¿HIú®┐╔ÊÈÐË│ñ╔·┤µã┌íúÈ┌ÁÑÂ└Á─╩ÁÐÚÍðú¼╬Ê├Ã╩╣Ë├┴¸╠Õ©┤╦ı─úð═Ðð¥┐┴╦HI║═DCAÍ╬┴ã║¾¤▀┴ú╠Õ╣ª─▄©─▒õ║═ËÙ¤▀┴ú╠Õ¤Ó╣ÏÁ─┤·ð╗═°┬þÁ─▒õ╗»íú DCA┤ª└Ý┐╔©─╔ãHI║¾┤¾╩¾Á─ð─ÈÓ¤▀┴ú╠Õ─ñÁþ╬╗║═ÎÚÍ»ATPíúË├DCA¢°ððÍ╬┴ã┐╔╩╣░³└¿Ð¬¢¼╚Ú╦ß║═p-AMPK / AMPKÈ┌─┌Á─Ê╗ð®┤·ð╗║═ÀÍÎË▓╬╩²ı²│ú╗»ú¼ÊÈ╝░Ach¢ÚÁ╝Á─Ь╣▄╩µı┼íúÎ▄Í«ú¼╬Ê├ÃÍñ├¸┴╦DCA┐╔ÊÈÈ┌├╗ËðÊ║╠Õ©┤╦ıÁ─ÃÚ┐÷¤┬│╔╣ªË├Ë┌│÷ЬðÈð¦┐╦Á─Í╬┴ãíúÊ‗┤╦ú¼DCA┐╔─▄╩ÃФÍÏ╩ºÐ¬║¾│ñã┌¢°ðð¤Í│í╗ñ└ÝÁ─┴╝║├Ðíȱíú
│÷ЬðÈð¦┐╦║¾Ë├Â■┬╚ÊÊ╦ß░ð¤‗¤▀┴ú╠Õ┐╔©─╔ãÈñ║¾┐ãк▒¿©µ
https://www.nature.com/articles/s41598-017-02495-5
Mitochondrial targeting by dichloroacetate improves outcome following hemorrhagic shock | Scientific Reports
https://www.nature.com/articles/s41598-017-02495-5íí
Dichloroacetate Stabilizes Mitochondrial Fusion Dynamics in Models of Neurodegeneration
Darren Oí»Hara, Gavin M. Davis, Natalie A. Adlesic, Jerrard M. Hayes and Gavin P. Davey*
School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland
Mitochondrial dysfunction is a recognized hallmark of neurodegenerative diseases and abnormal mitochondrial fusion-fission dynamics have been implicated in the pathogenesis of neurodegenerative disorders. This study characterizes the effects of metabolic flux inhibitors and activators on mitochondrial fusion dynamics in the neuronal cell culture model of differentiated PC12 cells. Using a real time confocal microscopy assay, it was found that the carnitine palmitoyltransferase I (CPTI) inhibitor, etomoxir, reduced mitochondrial fusion dynamics in a time-dependent manner. Etomoxir also decreased JO2, ªñªÀm and reactive oxygen species (ROS) production rates. The mitochondrial pyruvate carrier (MPC) inhibitor, UK5099, reduced fusion dynamics and in combination with etomoxir these inhibitory effects were amplified. Use of the pyruvate dehydrogenase (PDH) kinase inhibitor dichloroacetate, which is known to increase metabolic flux through PDH, reversed the etomoxir-induced effects on fusion dynamics, JO2, ªñªÀm but not ROS production rates. Dichloroacetate also partially reversed inhibition of mitochondrial fusion dynamics caused by the parkinsonian-inducing neurotoxin, MPP+. These results suggest that dichloroacetate-induced activation of metabolic flux in the mitochondrion may be a mechanism to restore normal mitochondrial fusion-fission dynamics in metabolically challenged cells.íí
Frontiers | Dichloroacetate Stabilizes Mitochondrial Fusion Dynamics in Models of Neurodegeneration | Frontiers in Molecular Neuroscience
https://www.frontiersin.org/articles/10.3389/fnmol.2019.00219/fullíí
A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth.
http://complementaryoncology.com/reports/others/a-mitochondria-k-channel-axis-is-suppressed-in-cancer-and-its-normalization-promotes-apoptosis-and-inhibits-cancer-growth/íí
íí
2019-5-29 íñ Insulin, insulin receptors, and cancer. ... Since many years, it is known that many cancer cells require insulin for optimal in vitro growth. Recent data indicate that: (1) insulin stimulates growth mainly via its own receptor and not the IGF-1 receptor; (2) in many cancer cells, the IR is overexpressed and the A isoform, which has a ...
https://www.ncbi.nlm.nih.gov/pubmed/27368923
Cancer Cells LOVE Sugar 44 Times More Than Healthy í¡À¡ÊÙ┤╦Ê│
2013-4-8 íñ Since cancer has 46 insulin receptor sites compared to 2 on a healthy cell, the cancer cells gobble up the radioactive sugar solution first. The end result is an image with little light bulbs glowing in areas of the body that are afflicted with cancer cells. ... í░Ití»s been known since 1923 that tumor cells use a lot more glucose than normal ...
https://breastcancerconqueror.com/cancer-cells...
Insulin enhances metabolic capacities of cancer cells í¡À¡ÊÙ┤╦Ê│
Cited by: 69
Publish Year: 2013
Author: Mohd Askandar Iqbal, Farid Ahmad Siddiqui, Vibhor Gupta, Shilpi Chattopadhyay, Prakasam Gopinath, Bh...
θı▀: Mohd Askandar Iqbal
2013-7-9 íñ Insulin is tightly associated with cancer progression; however, mechanistic insights into such observations are poorly understood. Recent studies show that metabolic transformation is critical to cancer cell proliferation. Here, we attempt to understand the role of insulin in promotion of cancer metabolism. To this end, the role of insulin in regulating glycolytic enzyme pyruvate kinase M2 ...
https://molecular-cancer.biomedcentral.com/articles/10.1186/1476-4598-12-72
The Insulin Receptor: A New Target for Cancer TherapyÀ¡ÊÙ┤╦Ê│
Cited by: 57
Publish Year: 2011
Author: Roberta Malaguarnera, Antonino Belfiore
╬╗Í├: 8600 Rockville Pike, Bethesda, MD
2011-12-6 íñ A large body of evidences have shown that both the IGF-I receptor (IGF-IR) and the insulin receptor (IR) play a role in cancer development and progression. In particular, IR overactivation by IGF-II is common in cancer cells, especially in dedifferentiated/stem-like cells. In spite of these findings ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3356071
Insulin Receptor: Trends in Endocrinology & MetabolismÀ¡ÊÙ┤╦Ê│
Cited by: 52
Publish Year: 1997
Author: Vincenzo Papa, Angela Costantino, Antonino Belfiore
This article reviews evidence indicating that (a) insulin receptors are overexpressed in human breast cancer, (b) insulin stimulates growth in breast cancer cells, (c) cells transfected with human insulin receptor may acquire a ligand-dependent transformed phenotype, and (d) breast cancer is associated with insulin resistance and hyperinsulinemia.
https://www.cell.com/trends/endocrinology-metabolism/references/S1043-2760(97)00114-8
Insulin Receptor: What Role in Breast Cancer? - í¡À¡ÊÙ┤╦Ê│
Cited by: 52
Publish Year: 1997
Author: Vincenzo Papa, Angela Costantino, Antonino Belfiore
This article reviews evidence indicating that (a) insulin receptors are overexpressed in human breast cancer, (b) insulin stimulates growth in breast cancer cells, (c) cells transfected with human insulin receptor may acquire a ligand-dependent transformed phenotype, and (d) breast cancer is associated with insulin resistance and hyperinsulinemia.
https://www.sciencedirect.com/science/article/pii/S1043276097001148
Elevated insulin levels trigger cancer cell growth ...À¡ÊÙ┤╦Ê│
Elevated insulin levels caused a 45 times greater risk, leading researchers to conclude that hyperinsulinemia could even be a í░key factorí▒ in the initiation and promotion of cancer cell growth. High levels of insulin also raise risk of prostate cancer malignancies by 2.55-fold, with a 5.62-fold risk of locally advanced tumors.
https://www.naturalhealth365.com/insulin-cancer-2502.htmlíí
A schematic of central carbon metabolism is presented to show how glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway, and glutamine metabolism are interconnected in cells. The major points of enzymatic regulation, along with the enzymes discussed in the text that have been demonstrated to be important in cancer, are shown for orientation within the pathways, Glut, glucose transporter; HK, hexokinase; PFK, phosphofructokinase; PK, pyruvate kinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; IDH, isocitrate dehydrogenase; SDH, succinate dehydrogenase; FH, fumarate hydratase. The site of regulation within these pathways by some of the major oncogenes and tumor suppressor genes is also shown.
Cancer Metabolism | Oncohema Key
https://oncohemakey.com/cancer-metabolism-2/íí
WHY IS LACTATE DEHYDROGENASE (LDH) RELEASE A GOOD MEASURE FOR CYTOTOXICITY?
When treated with a cytotoxic compound, living cells may face one of two fates. They could either stop growing and dividing, or die through either of two distinct processes - necrosis or apoptosis. Basically, cells undergoing necrosis (accidental cell death) swell and lose membrane integrity before shutting down and releasing their intracellular contents into the surrounding environment. This type of cell death is usually triggered by external factors such as toxic chemical or traumatic physical events.
On the other hand, cells undergoing apoptosis (normal or programmed cell death) go through a series of well-defined events such as the shrinking of the cytoplasm, cleavage of DNA into smaller fragments, etc. before being engulfed by white blood cells.
When the cell membranes are compromised or damaged in any way, lactate dehydrogenase (LDH), a soluble yet stable enzyme found inside every living cell, is released into the surrounding extracellular space. Since this only happens when cell membrane integrity is compromised, the presence of this enzyme in the culture medium can be used as a cell death marker. The relative amounts of live and dead cells within the medium can then be quantitated by measuring the amount of released LDH using a colorimetric or fluorometric LDH cytotoxicity assay.
Other enzymes such as adenylate kinase and glucose-6-phosphate may also be used to measure cytotoxicity but these enzymes are not stable and lose their activity during cell death assays.
Why Is Lactate Dehydrogenase (LDH) Release A Good Measure For Cytotoxicity?
https://info.gbiosciences.com/blog/why-is-lactate-dehydrogenase-ldh-release-a-good-measure-for-cytotoxicityíí
Pyruvate dehydrogenase function depends on thiamine (B1 ...
https://forums.phoenixrising.me/threads/pyruvate-dehydrogenase-function-depends-on...
Jan 15, 2017 íñ The pyruvate dehydrogenase (PDH) and alpha ketoglutarate dehydrogenase(a-KGDH) enzyme complexes are important thiamine dependent enzyme complexes that help liberate energy from glucose in the citric acid cycle of mitochondria. During glycolysis in the cytosol, glucose is converted in to 2 pyruvate molecules that enter the mitochondria.íí
The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents
Cancer cells exhibit an altered metabolism which is characterized by a preference for aerobic glycolysis more than mitochondrial oxidation of pyruvate. This provides anabolic support and selective growth advantage for cancer cells. Recently, a new concept has arisen suggesting that these metabolic changes may be due, in part, to an attenuated mitochondrial function which results from the inhibition of the pyruvate dehydrogenase complex (PDC). This mitochondrial complex links glycolysis to the Krebs cycle and the current understanding of its regulation involves the cyclic phosphorylation and dephosphorylation by specific pyruvate dehydrogenase kinases (PDKs) and pyruvate dehydrogenase phosphatases (PDPs).
The mitochondrial pyruvate dehydrogenase complex (PDC) acts as a gatekeeper enzyme for energy metabolism by catalyzing irreversible decarboxylation of pyruvate into acetyl®\CoA. The activity of PDC is highly regulated, at least in part, by reversible phosphorylation through pyruvate dehydrogenase kinases (PDKs) and pyruvate dehydrogenase phosphatases (PDPs) the functions of which are regulated by cellular nutrient cues.
Cancer cells exhibit an altered metabolism which is characterized by a preference for aerobic glycolysis more than mitochondrial oxidation of pyruvate. This provides anabolic support and selective growth advantage for cancer cells. Recently, a new concept has arisen suggesting that these metabolic changes may be due, in part, to an attenuated mitochondrial function which results from the inhibition of the pyruvate dehydrogenase complex (PDC). This mitochondrial complex links glycolysis to the Krebs cycle and the current understanding of its regulation involves the cyclic phosphorylation and dephosphorylation by specific pyruvate dehydrogenase kinases (PDKs) and pyruvate dehydrogenase phosphatases (PDPs).Four PDK isoenzymes (PDK1, PDK2, PDK3 and PDK4) have been identified in mammalian tissues. They exhibit 70% identity amongst themselves and possess apparent molecular masses ranging from 45 kDa (PDK1) to 48 kDa (PDK2, PDK3 and PDK4). High resolution crystallographic structures are available only for PDK2 and PDK3.
The activity of PDK is stimulated rapidly by ATP, NADH and acetyl®\CoA and it is inhibited by ADP, NAD+, CoA®\SH and pyruvate (Fig. 1).
Several studies have shown that PDKs play major roles in the metabolic adaptations that occur during the acquisition of the tumor metabolic phenotype. Furthermore, the expression and/or the activities of the PDKs are strongly regulated by oncogenes. In cancer cells, the expression of the PDK1 gene, as well as that of several glycolytic enzymes, is upregulated by c®\Myc and hypoxia®\inducible factor®\1ª┴ (HIF®\1ª┴).13, 14 The transcription of PDK3 also has been reported to be induced by HIF®\1ª┴, which leads to the inhibition of mitochondrial respiration in cancer cells.15 The tumor suppressor protein p53 negatively regulates PDK2. his is a new mechanism in which p53 suppresses tumorigenesis by acting at the level of cancer cell metabolism.16 Taken together, these data suggest that the phosphorylation of the PDC is an important factor for the formation and/or progression of tumors.17 Consistent with this hypothesis, PDK1 appears to be upregulated in many cancer cells. The expression of PDK2 is correlated significantly with metastasis and it is a strong, independent prognostic factor in cervical cancers.18 PDK3 expression is markedly increased in colon cancer and its level is positively associated with the severity of cancer and negatively associated with disease®\free survival.19 The expression of PDK4 has been found to be quite variable depending upon the type of the cancer cell. Global analyses of gene expression in cancer cell types have demonstrated that PDK4 is highly expressed in renal, ovarian, prostate, lung and skin cancer cells.20 In contrast, in 35 studies available through Oncomine21, 22 the amount of PDK4 mRNA was found to be significantly lower, as compared to the corresponding normal tissues, in human breast, ovarian, lung, and colon tumors. In these tumors, PDC might be activated and acetyl®\CoA, the end product of the reaction, could be utilized by the truncated TCA cycle of the cells which leads to the accumulation of citrate and, consequently, to increased fatty acid synthesis, a hallmark of cancer cells Thus, the variability in the level of PDK, depending upon the tumor type, could result from an adaptation of the PDC flux rates during different stages of tumor development. This adaptation could depend upon the extent to which distinct metabolic pathways in the tumor are used for energy production and anabolic biosynthesis.The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents - Saunier - 2016 - International Journal of Cancer - Wiley Online Library
https://onlinelibrary.wiley.com/doi/full/10.1002/ijc.29564íí
New Connections between Old Pathways: PDK1 Signaling Promotes Cellular Transformation through PLK1-Dependent MYC Stabilization
Summary: Limited understanding of the functional link between multiple oncogenic pathways is a major barrier in the ongoing effort of cancer biologists to design an effective therapeutic approach to treat malignancies characterized by driver oncogenic network signals. In this issue of Cancer Discovery, Tan and colleagues elucidate a novel PDK1¿CPLK1¿CMYC signaling pathway connecting two fundamental oncogenic programs, phosphoinositide 3-kinase and MYC. They define the functional role for PDK1¿CPLK1¿CMYC signaling in cancer cell survival and tumor formation and show the therapeutic benefit of inhibiting PDK1 and PLK1 pharmacologically in cancer, tackling the most undruggable tumors defined by elevated levels of the MYC oncoprotein.Cancer Discov; 3(10); 1099¿C1102. ©2013 AACR.
New Connections between Old Pathways: PDK1 Signaling Promotes Cellular Transformation through PLK1-Dependent MYC Stabilization | Cancer Discovery
https://cancerdiscovery.aacrjournals.org/content/3/10/1099íí
MYC Activation Is a Hallmark of Cancer Initiation and Maintenance
Division of Oncology, Departments of Medicine and Pathology, Stanford University School of Medicine, Stanford, California 94305
ííAbstract
The MYC proto-oncogene has been implicated in the pathogenesis of most types of human tumors. MYC activation alone in many normal cells is restrained from causing tumorigenesis through multiple genetic and epigenetically controlled checkpoint mechanisms, including proliferative arrest, apoptosis, and cellular senescence. When pathologically activated in a permissive epigenetic and/or genetic context, MYC bypasses these mechanisms, enforcing many of the í░hallmarkí▒ features of cancer, including relentless tumor growth associated with DNA replication and transcription, cellular proliferation and growth, protein synthesis, and altered cellular metabolism. MYC mandates tumor cell fate, by inducing stemness and blocking cellular senescence and differentiation. Additionally, MYC orchestrates changes in the tumor microenvironment, including the activation of angiogenesis and suppression of the host immune response. Provocatively, brief or even partial suppression of MYC back to its physiological levels of activation can result in the restoration of intrinsic checkpoint mechanisms, resulting in acute and sustained tumor regression, associated with tumor cells undergoing proliferative arrest, differentiation, senescence, and apoptosis, as well as remodeling of the tumor microenvironment, recruitment of an immune response, and shutdown of angiogenesis. Hence, tumors appear to be í░addictedí▒ to MYC because of both tumor cell¿Cintrinsic, cell-autonomous and host-dependent, immune cell¿Cdependent mechanisms. Both the trajectory and persistence of many human cancers require sustained MYC activation. Multiscale mathematical modeling may be useful to predict when tumors will be addicted to MYC. MYC is a hallmark molecular feature of both the initiation and maintenance of tumorigenesis.
The MYC proto-oncogene was first discovered as an etiologic agent of retrovirally mediated tumorigenesis. Later, MYC was illustrated to be activated through chromosomal translocation in Burkitt lymphoma (see Conacci-Sorrell et al. 2014). MYC is also commonly activated in tumorigenesis as a consequence of both oncogenic and epigenetic events (Boxer and Dang 2001; Eilers and Eisenman 2008; Dang 2012). Indeed, MYC is overexpressed and/or activated in more than half of human cancers (Escot et al. 1986; Ladanyi et al. 1993; Gamberi et al. 1998; Kawate et al. 1999; Stock et al. 2000; Boxer and Dang 2001).
MYC largely functions as a transcription factor that coordinates many biological processes (Dang et al. 1999). MYC activation can usurp these programs, resulting in the cardinal hallmark features of cancer. Thus, MYC activation contributes to autonomous proliferation and growth, relentless DNA replication, increased protein biogenesis, global changes in cellular metabolism, activation of the angiogenic switch, suppression of the response to autocrine and paracrine regulatory programs, and a restraint of host immune responses (Felsher 2003; Shachaf and Felsher 2005; van Riggelen et al. 2010b; Bachireddy et al. 2012; Dang 2012). Hence, MYC activation appears to be a molecular hallmark of cancer.BRIEF OR PARTIAL SUPPRESSION OF MYC CAN REVERSE TUMORIGENESIS
A brief 2-day or partial (twofold decrease) suppression of MYC can result in sustained tumor regression. Brief suppression of MYC is associated with an irreversible change in the cellular program; in some contexts the tumors cannot be restored on MYC reactivation (Jain et al. 2002). Similarly, a twofold decrease in oncogenic levels of MYC was sufficient to induce tumor regression (Shachaf and Felsher 2005). This is tumor type specific, because lymphoma and osteosarcoma exhibit this phenotype (Jain et al. 2002; Giuriato et al. 2006), whereas epithelial tumors such as hepatocellular or breast carcinoma do not (Boxer et al. 2004; Shachaf et al. 2004).
In osteogenic sarcoma MYC suppression results in terminal cellular differentiation from osteoblasts into differentiated osteocytes (which are associated with bone formation in vivo) (Jain et al. 2002). The reactivation of MYC not only fails to restore the cancer but either has no consequence or is associated with apoptosis. Microarray analysis revealed that MYC suppression is associated with irreversible changes in gene expression as a result of the inability of MYC to bind to the promoters of many of these genes. In particular, MYC suppression results in the permanent shutdown of genes related to ribosome biosynthesis and protein synthesis (Wu et al. 2008; van Riggelen et al. 2010b).
Partial suppression of MYC can also result in sustained tumor regression. Notably, in this case the levels of MYC were below that of human tumor-derived cell lines and above that of proliferating normal human cells or Epstein¿CBarr virus¿Ctransformed lymphocytes (Shachaf et al. 2008). Thus, there appears to be a threshold level of MYC that is required to sustain a malignant phenotype (Shachaf et al. 2008). Protein and gene expression analysis identified many specific changes, but notably, ribosomal gene products were suppressed. Collectively, these results suggest that a global shift in protein biogenesis is an important feature of how MYC suppression results in tumor regression (Ruggero and Pandolfi 2003).
MYC activation is also associated with global changes in the energy metabolism of cancer cells. These changes may make tumors particularly susceptible to the inhibition of enzymes that are essential for energy metabolism (Dang 1999, 2013; Dang et al. 2009; Oí»Shea and Ayer 2013). Hence, the addiction to MYC observed in many cancer cells could at least in part relate to acute changes in metabolism. The suppression of MYC may induce tumor regression by acutely disrupting the ability of tumor cells to maintain sufficient metabolism to sustain survival and/or by directly regulating death signaling (see Dang 2013; Oí»Shea and Ayer 2013; Morrish and Hockenbery 2014).
An important implication of these results is that it may be sufficient to partially and/or briefly suppress MYC expression in at least some tumor types to induce a sustained clinical effect on human disease. That transient inactivation of MYC is effective may be due to the dependence of MYC-associated oncogene addiction on molecular features that are dictated shortly after oncogene inactivation (Tran et al. 2011). Whether tumor inhibition is entirely cell autonomous or results from a delayed host-dependent mechanism remains to be determined. For example, the host immune system seems to be critical for tumor regression on withdrawal of MYC.Table 1.
Models illustrating MYC-associated oncogene addiction
Oncogene Conditional mouse model Reference MYC T-cell acute lymphoblastic leukemia Felsher and Bishop 1999a íí Hepatocellular carcinoma Shachaf et al. 2004 íí Osteosarcoma Jain et al. 2002 íí T- and B-cell acute lymphoblastic leukemia Marinkovic et al. 2004 íí Mammary adenocarcinoma Dí»Cruz et al. 2001 íí Islet tumors Lawlor et al. 2006 RAS Melanoma (HRAS) Chin et al. 1999 íí Lung adenocarcinoma (KRAS) Fisher et al. 2001 BRAF Thyroid cancer Chakravarty et al. 2011 íí Lung adenocarcinoma Ji et al. 2007 EGFR Lung adenocarcinoma Li et al. 2007 BCR-ABL B-cell acute lymphoblastic leukemia Huettner et al. 2000 íí
íí
íí
Figure 2.
Consequences of MYC inactivation in multiple types of cancer. MYC inactivation elicits oncogene addiction by multiple mechanisms that differ depending on tumor type. MYC inactivation in lymphoma induces proliferative arrest, differentiation/senescence, and widespread apoptosis. MYC inactivation in osteosarcoma induces proliferative arrest and differentiation/senescence but not apoptosis. MYC reactivation does not restore tumorigenesis. MYC inactivation in liver adenocarcinoma induces proliferative arrest, differentiation/senescence, and apoptosis. MYC reactivation can result in restoration of the tumor.
íí
Figure 3.
MYC inactivation elicits tumor regression through both cell-autonomous and non-cell-autonomous mechanisms of tumor regression. MYC activation leads to tumorigenesis through suppression of critical safeguards such as apoptosis, proliferative arrest, differentiation, and senescence. Activation of MYC also facilitates engagement of hallmarks of tumor growth, as well as cell-extrinsic phenomena such as host immunity. TGF, transforming growth factor.
MYC Activation Is a Hallmark of Cancer Initiation and Maintenance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4031954/íí
Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis
Highlights
•
Loss of nutrient-sensing bHLHZ MondoA is synthetic lethal with deregulated Myc
•
Myc and MondoA transcriptionally coregulate cancer cell metabolic reprogramming
•
MondoA suppresses Myc-dependent metabolic stress and promotes proliferation
•
High expression of Myc/MondoA targets correlates with poor outcome in human cancers
Summary
Deregulated Myc transcriptionally reprograms cell metabolism to promote neoplasia. Here we show that oncogenic Myc requires the Myc superfamily member MondoA, a nutrient-sensing transcription factor, for tumorigenesis. Knockdown of MondoA, or its dimerization partner Mlx, blocks Myc-induced reprogramming of multiple metabolic pathways, resulting in apoptosis. Identification and knockdown of genes coregulated by Myc and MondoA have allowed us to define metabolic functions required by deregulated Myc and demonstrate a critical role for lipid biosynthesis in survival of Myc-driven cancer. Furthermore, overexpression of a subset of Myc and MondoA coregulated genes correlates with poor outcome of patients with diverse cancers. Coregulation of cancer metabolism by Myc and MondoA provides the potential for therapeutics aimed at inhibiting MondoA and its target genes.
Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis: Cancer Cell
https://www.cell.com/cancer-cell/fulltext/S1535-6108(14)00475-9íí
Blocking Lactate Export by Inhibiting the Myc Target MCT1 ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3946415
Feb 01, 2014 íñ Myc oncoproteins induce genes driving aerobic glycolysis, including lactate dehydrogenase-A that generates lactate.
Cited by: 165
Publish Year: 2014íí
Myc-induced glutaminolysis bypasses HIF-driven glycolysis in hypoxic small cell lung carcinoma cells
We previously demonstrated that small cell lung carcinoma (SCLC) cells lack HIF-2ª┴ protein expression, whereas HIF-1ª┴ in these cells is expressed at both acute and prolonged hypoxia. Here we show that low HIF2A expression correlates with high expression of MYC genes. Knockdown of HIF1A expression had no or limited effect on cell survival and growth in vitro. Unexpectedly, hypoxic ATP levels were not affected by HIF-1ª┴ knockdown and SCLC cell viability did not decrease upon glucose deprivation. In line with these in vitro data, xenograft tumor-take and growth were not significantly affected by repressed HIF1A expression. Glutamine withdrawal drastically decreased SCLC cell proliferation and increased cell death at normoxia and hypoxia in a HIF-independent fashion and the dependence on glutaminolysis was linked to amplification of either MYC or MYCL. Downregulation of GLS expression, regulating the first step of the glutaminolysis pathway, in MYC/MYCL overexpressing SCLC cells resulted in both impaired growth and increased cell death. Our results suggest that MYC/MYCL overexpression in SCLC cells overrides the need of HIF-1 activity in response to hypoxia by inducing glutaminolysis and lipogenesis. Targeting the glutaminolysis pathway might hence be a novel approach to selectively kill MYC amplified SCLC cells in vivo.
Oncotarget | Myc-induced glutaminolysis bypasses HIF-driven glycolysis in hypoxic small cell lung carcinoma cells
http://www.oncotarget.com/index.php?journal=oncotarget&page=article&op=view&path[]=16904&path[]=54085íí
Gamabufotalin triggers c-Myc degradation via induction of WWP2 in multiple myeloma cells
Zhenlong Yu1,*, Tao Li2,*, Chao Wang1, Sa Deng1, Baojing Zhang1, Xiaokui Huo1, Bo Zhang3, Xiaobo Wang1, Yuping Zhong4, Xiaochi Ma1
1College of Pharmacy, Academy of Integrative Medicine, Dalian Medical University, Dalian, China
2Department of Biology, College of Chemistry and Life Sciences, Zhejiang Normal University, Zhejiang, China
3Department of Neurosurgery, The Second Affiliated Hospital of Dalian Medical University, Dalian, China
4Department of Hematology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, China
ABSTRACT
Deciding appropriate therapy for multiple myeloma (MM) is challenging because of the occurrence of multiple chromosomal changes and the fatal nature of the disease. In the current study, gamabufotalin (GBT) was isolated from toad venom, and its tumor-specific cytotoxicity was investigated in human MM cells. We found GBT inhibited cell growth and induced apoptosis with the IC50 values <50 nM. Mechanistic studies using functional approaches identified GBT as an inhibitor of c-Myc. Further analysis showed that GBT especially evoked the ubiquitination and degradation of c-Myc protein, thereby globally repressing the expression of c-Myc target genes. GBT treatment inhibited ERK and AKT signals, while stimulating the activation of JNK cascade. An E3 ubiquitin-protein ligase, WWP2, was upregulated following JNK activation and played an important role in c-Myc ubiquitination and degradation through direct protein-protein interaction. The antitumor effect of GBT was validated in a xenograft mouse model and the suppression of MM-induced osteolysis was verified in a SCID-hu model in vivo. Taken together, our study identified the potential of GBT as a promising therapeutic agent in the treatment of MM.
Oncotarget | Gamabufotalin triggers c-Myc degradation via induction of WWP2 in multiple myeloma cells
http://www.oncotarget.com/index.php?journal=oncotarget&page=article&op=view&path[]=7398&path[]=26694íí
MYC on the Path to Cancer
Cell. Author manuscript; available in PMC 2013 Mar 30.
Published in final edited form as:
Cell. 2012 Mar 30; 149(1): 22¿C35.
Abstract
The MYC oncogene contributes to the genesis of many human cancers. Recent insights into its expression and function have led to new cancer therapeutic opportunities. MYCí»s activation by bromodomain proteins could be inhibited by drug-like molecules, resulting in tumor inhibition in vivo. Tumor growth can also be curbed by pharmacologically uncoupling bioenergetic pathways involving glucose or glutamine metabolism from Myc-induced cellular biomass accumulation. Other approaches to halt Myc on the path to cancer involve targeting Myc-Max dimerization or Myc-induced microRNA expression. Here the richness of our understanding of MYC is reviewed, highlighting new biological insights and opportunities for cancer therapies.
Figure 1 A. The MYC protooncogene is depicted downstream of receptor signal transduction pathways, which elicit positive or negative regulation of the MYC gene. MYC produces the transcription factor Myc, which dimerizes with Max and bind target DNA sequences or E-boxes (with the sequence 5íõ-CANNTG-3íõ) to regulate transcription of genes involved in cell growth and proliferation. The WNT pathway is depicted with APC negatively regulating ª┬-catenin, which upon nuclear translocation participates in the transactivation of MYC, such that loss of APC results in constitutive oncogenic MYC expression. B. When MYC is deregulated, by gene amplication, chromosomal translocation or loss of upstream regulators, such as APC, acute sustained oncogenic MYC expression results in checkpoint activation p53 or Arf. Loss of checkpoint regulation through mutations of p53 or Arf, for example, uncloaks MYCí»s full tumorigenic potential.
MYC on the Path to Cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3345192/íí
TGF-ª┬ ¿C an excellent servant but a bad master | Journal of Translational Medicine | Full Text
https://translational-medicine.biomedcentral.com/articles/10.1186/1479-5876-10-183íí
TGF-ª┬ Signaling in Myeloid Cells Is Required for Tumor Metastasis
Abstract
TGF-ª┬ is overexpressed in advanced human cancers. It correlates with metastasis and poor prognosis. However, TGF-ª┬ functions as both a tumor suppressor and a tumor promoter. Here, we report for the first time that genetic deletion of Tgfbr2 specifically in myeloid cells (Tgfbr2MyeKO) significantly inhibited tumor metastasis. Reconstitution of tumor-bearing mice with Tgfbr2MyeKO bone marrow recapitulated the inhibited metastasis phenotype. This effect is mediated through decreased production of type II cytokines, TGF-ª┬1, arginase 1, and inducible nitric oxide synthase, which promoted IFN-ª├ production and improved systemic immunity. Depletion of CD8 T cells diminished the metastasis defect in the Tgfbr2MyeKO mice. Consistent with animal studies, myeloid cells from patients with advanced-stage cancer showed increased TGF-ª┬ receptor II expression. Our studies show that myeloid-specific TGF-ª┬ signaling is an essential component of the metastasis-promoting puzzle of TGF-ª┬. This is in contrast to the previously reported tumor-suppressing phenotypes in fibroblasts, epithelial cells, and T cells.
Significance: Our study identifies myeloid-specific TGF-ª┬ signaling as a critical mediator in tumor metastasis, distinct from the tumor-suppressive effect of TGF-ª┬ signaling in epithelial cells, fibroblasts, and T cells. We further provide mechanistic insight into host antitumor immunity and suggest a cell type¿Cspecific cancer-targeting strategy. Cancer Discov; 3(8); 936¿C51. ©2013 AACR.íí
The MAPK Signaling Pathway
The RAS-RAF-MEK-ERK pathway is far more complex than once thought. Figure 1 presents a simplified description of its basic components. The pathwayí»s general structure includes a small G protein (RAS) and three protein kinases (RAF, MEK, ERK). (A kinase is an enzyme that catalyzes transfer of a phosphate group from a donor molecule to an acceptor.) The starting point for this pathway is the binding of ligand to a transmembrane protein, a receptor tyrosine kinase (RTK). The resulting signaling cascade culminates with translocation of ERK (MAPK) to the nucleus, where ERK activates transcription factors that result in gene expression.The MAPK (ERK) Pathway
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3628180/íí
Dichloroacetate, a Metabolic Modulator, Prevents and Reverses Chronic Hypoxic Pulmonary Hypertension in Rats
Role of Increased Expression and Activity of Voltage-Gated Potassium Channels
Evangelos D. Michelakis , M. Sean McMurtry , Xi-Chen Wu , Jason R.B. Dyck , Rohit Moudgil , Teresa A. Hopkins , Gary D. Lopaschuk , Lakshmi Puttagunta , Ross Waite , and Stephen L. Archer
Originally published15 Jan 2002 https://doi.org/10.1161/hc0202.101974Circulation. 2002;105:244¿C250
Abstract
Backgroundí¬ Chronic hypoxic pulmonary hypertension (CH-PHT) is associated with suppressed expression and function of voltage-gated K+ channels (Kv) in pulmonary artery (PA) smooth muscle cells (SMCs) and a shift in cellular redox balance toward a reduced state. We hypothesized that dichloroacetate (DCA), a metabolic modulator that can shift redox balance toward an oxidized state and increase Kv current in myocardial cells, would reverse CH-PHT.
Methods and Resultsí¬ We studied 4 groups of rats: normoxic, normoxic+DCA (DCA 70 mg íñ kg−1 íñ d−1 PO), chronically hypoxic (CH), and CH+DCA. CH and CH+DCA rats were kept in a hypoxic chamber (10% Fio2) for 2 to 3 weeks. DCA was given either at day 1 to prevent or at day 10 to reverse CH-PHT. We used micromanometer-tipped catheters and measured hemodynamics in closed-chest rats on days 14 to 18. CH+DCA rats had significantly reduced pulmonary vascular resistance, right ventricular hypertrophy, and PA remodeling compared with the CH rats. CH inhibited IK, eliminated the acute hypoxia¿Csensitive IK, and decreased Kv2.1 channel expression. In the short term, low-dose DCA (1 ª╠mol/L) increased IK in CH-PASMCs. In a mammalian expression system, DCA activated Kv2.1 by a tyrosine kinase¿Cdependent mechanism. When given long-term, DCA partially restored IK and Kv2.1 expression in PASMCs without altering right ventricular pyruvate dehydrogenase activity, suggesting that the beneficial effects of DCA occur by nonmetabolic mechanisms.
Conclusionsí¬ DCA both prevents and reverses CH-PHT by a mechanism involving restoration of expression and function of Kv channels. DCA has previously been used in humans and may potentially be a therapeutic agent for pulmonary hypertension.
Exposure of animals or humans to chronic hypoxia (CH) leads to the development of chronic hypoxic pulmonary hypertension (CH-PHT) by an unknown mechanism. CH-PHT is characterized by pulmonary arterial (PA) vasoconstriction and remodeling.1 Although the role of endothelium in the pathogenesis of CH-PHT is important, the role of vascular smooth muscle cells (SMCs) is increasingly recognized.2 Both the contractile status and proliferative status of SMCs are regulated by the levels of the intracellular Ca2+ ([Ca2+]i). [Ca2+]i levels are determined in part by influx of Ca2+ through the voltage-gated, L-type Ca2+ channels, the gating of which is controlled by the SMC membrane potential. In the PASMCs, the membrane potential is regulated by voltage-gated K+ channels (Kv), including Kv1.5 and Kv2.1.3 These channels, as well as Kv1.2, Kv3.1b, and Kv9.1, are O2-sensitive and can be inhibited by hypoxia in expression systems (for review see Reference 3). Inhibition of one or more of these channels by acute hypoxia contributes to the initiation of hypoxic pulmonary vasoconstriction.3
CH reduces K+ current density in PASMCs, resulting in a state of depolarization,4,5 which elevates [Ca2+]i and thus promotes contraction and proliferation.6 CH is associated with impaired expression of certain Kv channels (eg, Kv1.5 and Kv2.1), although the expression of many other channels is unaltered.4,7 The mechanism for Kv channel downregulation is unclear, but recent work suggests that this channel remodeling may relate to the altered redox state induced by CH.4 Lungs of rats with CH-PHT are in a more reduced redox state than those of normoxic controls, as indicated by a sustained reduction in activated oxygen species production and increased levels of reduced glutathione.4 A reduced redox state has potential for both short-term hemodynamic effects (through modulation of K+ channel function8) and long-term effects (because several oxygen-responsive genes, including hypoxia-inducible factor, are redox regulated9).
We hypothesized that downregulation of Kv channel function and expression is causally related to the development and maintenance of CH-PHT. We sought to enhance expression and function of Kv channels using dichloroacetate (DCA), a metabolic modulator, which has been shown to increase whole-cell K+ current (IK) in cardiac myocytes from a rat myocardial infarction model.10 DCA inhibits mitochondrial pyruvate dehydrogenase kinase (PDK)11 and, by increasing the pyruvate/lactate ratio, might promote an oxidized state in PASMCs.12 Thus, we speculated that DCA would reverse the reduced redox state in the PASMCs of CH-PHT rats and thereby enhance the activity and expression of Kv channels and reverse CH-PHT, mimicking the benefits of a return to normoxia.Dichloroacetate, a Metabolic Modulator, Prevents and Reverses Chronic Hypoxic Pulmonary Hypertension in Rats | Circulation
https://www.ahajournals.org/doi/10.1161/hc0202.101974íí
¤▀┴ú╠Õ-K +═¿Á└ÍßÈ┌░®ÍóÍð▒╗ÊÍÍãú¼ãõı²│ú╗»┤┘¢°¤©░¹Á‗═÷▓óÊÍÍã░®ÍóÁ─╔·│ñíú
A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth
CANCER CELL, JANUARY 2007
╝Ë─├┤¾░¼▓«╦■╩í░¼▓«╦■┤¾ÐºÀ╬»┬÷©▀Ð╣╝ã╗«║═Ь╣▄╔·╬´ÐºðíÎÚú¼AB T6G 2B7ú¼╝Ë─├┤¾
ı¬Ê¬
░®ÍóÁ─Â└╠Ï┤·ð╗╠Ïı¸ú¿ËðЧ╠â═¢Ôú®┐╔─▄©│ËÞ¤©░¹Á‗═÷┐╣ðÈú¼▓ó¥▀ËðÍ╬┴ãıÙÂÈðÈíúËÙı²│ú¤©░¹¤Ó▒╚ú¼╝©ÍÍ╚╦└Ó░®Íó¥▀Ë𢤩▀Á─¤▀┴ú╠Õ─ñÁþ╬╗ú¿ªñªÀmú®║═K +═¿Á└Kv1.5Á─Á═▒Ý┤´ú¼┴¢ı▀¥¨ËðÍ·Ë┌¤©░¹Á‗═÷íúÂ■┬╚ÊÊ╦ßú¿DCAú®ÊÍÍã¤▀┴ú╠Õ▒¹═¬╦ß═ÐÃÔ├©╝ñ├©ú¿PDKú®ú¼¢½┤·ð╗┤Ë╠â═¢Ôά▒õ╬¬ã¤╠Ð╠ÃЧ╗»ú¼¢ÁÁ═ªñªÀmú¼È÷╝ˤ▀┴ú╠ÕH2O2ú¼▓ó╝ñ╗¯╦¨ËðÁ½ÀÃı²│ú¤©░¹ÍðÁ─Kv═¿Á└ú╗ DCA═¿╣²NFAT1Ê└└ÁðÈ╗·Íã╔¤Á¸Kv1.5íú DCAËıÁ╝¤©░¹Á‗═÷ú¼╝§╔┘È÷Í│ú¼▓óÊÍÍãÍÎ┴÷╔·│ñú¼Â°├╗Ëð├¸¤ÈÁ─Â¥ðÈíú siRNAÂÈPDK2Á─ÀÍÎËÊÍÍãθË├└Ó╦ãË┌DCAíú¤▀┴ú╠Õ-NFAT-KvÍß║═PDK╩Ã░®ÍóÁ─ÍÏʬÍ╬┴ã░ðÁÒíú┐┌À■DCA╩ÃÊ╗ÍÍËðÃ░═¥Á─ÐíȱðÈ┐╣░®Ê®íú
Pulmonary Hypertension Program and Vascular Biology Group, University of Alberta, Edmonton, AB T6G 2B7, CanadaSummary
The unique metabolic profile of cancer (aerobic glycolysis) might confer apoptosis resistance and be therapeutically targeted. Compared to normal cells, several human cancers have high mitochondrial membrane potential (ªñªÀm) and low expression of the K+ channel Kv1.5, both contributing to apoptosis resistance. Dichloroacetate (DCA) inhibits mitochondrial pyruvate dehydrogenase kinase (PDK), shifts metabolism from glycolysis to glucose oxidation, decreases ªñªÀm, increases mitochondrial H2O2, and activates Kv channels in all cancer, but not normal, cells; DCA upregulates Kv1.5 by an NFAT1-dependent mechanism. DCA induces apoptosis, decreases proliferation, and inhibits tumor growth, without apparent toxicity. Molecular inhibition of PDK2 by siRNA mimics DCA. The mitochondria-NFAT-Kv axis and PDK are important therapeutic targets in cancer; the orally available DCA is a promising selective anticancer agent.íí
Figure 1A Reversible Metabolic-Electrical Remodeling in Cancer Contributes to Resistance to Apoptosis and Reveals Several Potential Therapeutic Targets
In cancer, mitochondrial glucose oxidation is inhibited and energy production relies on the cytoplasmic glycolysis. This í░inactivityí▒ of the mitochondria likely induces a state of apoptosis resistance. Activation of PDH by DCA increases glucose oxidation by promoting the influx of acetyl-CoA into the mitochondria and the Krebs cycle, thus increasing NADH delivery to complex I of the electron transport chain, increasing the production of superoxide, which in the presence of MnSOD is dismutated to the more stable H2O2. Sustained increase in ROS generation can damage the redox-sensitive complex I, inhibiting H+ efflux and decreasing ªñªÀm. Opening of the ªñªÀm-sensitive mitochondrial transition pore (MTP) allows the efflux of cytochrome c and apoptosis inducing factor (AIF). Both cytochrome c and H2O2 open the redox-sensitive K+ channel Kv1.5 in the plasma membrane and hyperpolarize the cell (increased Em), inhibiting a voltage-dependent Ca2+ entry. The decreased [Ca2+]i suppresses a tonic activation of NFAT, resulting in its removal from the nucleus, thus increasing Kv1.5 expression. The increased efflux of K+ from the cell decreases the tonic inhibition of [K+]i on caspases, further enhancing apoptosis. DCA's selectivity is based on its ability to target the unique metabolic profile that characterizes most cancers, and its effectiveness is explained by its dual mechanism of apoptosis induction, both by depolarizing mitochondria (proximal pathway) and activating/upregulating Kv1.5 (distal pathway).
SIGNIFICANCE
The small molecule DCA is a metabolic modulator that has been used in humans for decades in the treatment of lactic acidosis and inherited mitochondrial diseases. Without affecting normal cells, DCA reverses the metabolic-electrical remodeling that we describe in several cancer lines (hyperpolarized mitochondria, activated NFAT1, downregulated Kv1.5), inducing apoptosis and decreasing tumor growth. DCA in the drinking water at clinically relevant doses for up to 3 months prevents and reverses tumor growth in vivo, without apparent toxicity and without affecting hemoglobin, transaminases, or creatinine levels. The ease of delivery, selectivity, and effectiveness make DCA an attractive candidate for proapoptotic cancer therapy which can be rapidly translated into phase II¿CIII clinical trials.
Introduction
Cancer progression and its resistance to treatment depend, at least in part, on suppression of apoptosis. Although mitochondria are recognized as regulators of apoptosis, their importance as targets for cancer therapy has not been adequately explored or clinically exploited. In 1930, Warburg suggested that mitochondrial dysfunction in cancer results in a characteristic metabolic phenotype, that is, aerobic glycolysis (Warburg, 1930). Positron emission tomography (PET) imaging has now confirmed that most malignant tumors have increased glucose uptake and metabolism. This bioenergetic feature is a good marker of cancer but has not been therapeutically pursued, as it is thought to be a result and not a cause of cancer; that is, the cells rely mostly on glycolysis for energy production because of permanent mitochondrial damage, preventing oxidative phosphorylation. However, whether the mitochondria in cancer are indeed damaged and whether this is reversible remain unknown.
The metabolic hypothesis of cancer has recently been rekindled. Gatenby and Gillies recently proposed that because early carcinogenesis occurs in a hypoxic microenvironment, the transformed cells initially have to rely on glycolysis for energy production (Gatenby and Gillies, 2004). However, this early metabolic adaptation appears to also offer a proliferative advantage, suppressing apoptosis. Furthermore, the í░byproductsí▒ of glycolysis (i.e., lactate and acidosis) contribute to the breakdown of the extracellular matrix, facilitate cell mobility, and increase the metastatic potential. Therefore, even though the tumors eventually become vascularized and O2 levels increase, the glycolytic phenotype persists, resulting in the í░paradoxí▒ of glycolysis during aerobic conditions (the Warburg effect). Metabolic and apoptotic pathways that converge in the mitochondria are not independent from each other, and it appears that glycolytic phenotype is indeed associated with a state of apoptosis resistance (Plas and Thompson, 2002). Many glycolytic enzymes have been recognized to also regulate apoptosis, and several oncoproteins induce the expression of glycolytic enzymes (Kim and Dang, 2005). For example, Akt, which stimulates glycolysis and induces resistance to apoptosis (Elstrom et al., 2004), activates hexokinase, an enzyme catalyzing the first and irreversible step in glycolysis. Via its downstream mediator glycogen synthase kinase 3 (GSK3), Akt induces the translocation of hexokinase to the mitochondrial membrane where it binds to the voltage-dependent anion channel (VDAC), suppressing apoptosis (Kim and Dang, 2005, Pastorino et al., 2005). Inhibition of GSK3 in cancer cells causes unbinding of hexokinase from VDAC, induces apoptosis, and increases sensitivity to chemotherapy (Pastorino et al., 2005). This suggests that perhaps the metabolic phenotype in cancer is due to a potentially plastic mitochondrial remodeling that results in suppressed oxidative phosphorylation, enhanced glycolysis, and suppressed apoptosis.
Whether the metabolism of glucose will end with glycolysis in the cytoplasm (converting pyruvate to lactate) or continue with glucose oxidation in the mitochondria is controlled by a gate-keeping mitochondrial enzyme, pyruvate dehydrogenase (PDH) (Figure 1). PDH converts pyruvate to acetyl-CoA which, along with the acetyl-CoA from the fatty acid ª┬-oxidation, is fed to the Krebs cycle, producing the electron donors NADH and FADH2. NADH donates electrons to complex I of the electron transport chain (ETC) (and FADH2 to complex III). The flux of electrons down the ETC is associated with production of reactive oxygen species (ROS) and with the efflux of H+, which causes a negative mitochondrial membrane potential (ªñªÀm). The F1F0-ATP synthase uses the stored energy of the ªñªÀm to synthesize ATP; thus the ªñªÀm reflects ETC activity and mitochondrial function. PDH is inhibited by phosphorylation by PDH kinase (PDK). The role of PDH and PDK in cancer is unknown.Mitochondrial remodeling has multiple downstream effects, beyond energy production, because mitochondria regulate several critical functions including [Ca2+]i and ROS-redox control. Through the release of ROS, mitochondria regulate the opening of plasma-membrane ion channels and through the control of [Ca2+]i, regulate Ca2+-sensitive transcription factors. Some of these downstream pathways are also important in apoptosis and might contribute to the apoptosis resistance in cancer. For example, inhibition or downregulation of K+ channels results in increased [K+]i, by decreasing the tonic efflux of K+ down its intracellular/extracellular gradient (145/5 mEq). Because [K+]i exerts a tonic inhibitory effect on caspases, K+ channel inhibition or downregulation suppresses apoptosis in several cell types, including cancer (Andersson et al., 2006, Remillard and Yuan, 2004, Wang et al., 2002, Yu et al., 1997). The voltage-gated family of K+ channels (Kv) is redox sensitive and therefore can be regulated by mitochondria. For example, mitochondria-derived H2O2 (a relatively stable ROS) can activate Kv1.5 (Caouette et al., 2003). Furthermore, the mitochondria-derived proapoptotic mediator cytochrome c activates, whereas the antiapoptotic bcl-2 inhibits, Kv channels (Remillard and Yuan, 2004). This mitochondria-ROS-Kv channel axis is now recognized as a basis of an important O2-sensing mechanism in many tissues (Michelakis et al., 2004).
In preliminary experiments, we compared several cancer with normal cell lines and found that cancer cells had more hyperpolarized mitochondria and were relatively deficient in Kv channels. If this metabolic-electrical remodeling is an adaptive response, then its reversal might increase apoptosis and inhibit cancer growth. We used dichloroacetate (DCA), a small, orally available small molecule and a well-characterized inhibitor of PDK (Bowker-Kinley et al., 1998, Knoechel et al., 2006, Stacpoole, 1989). As seen in Figure 1, inhibition of PDK shifts pyruvate metabolism from glycolysis and lactate production to glucose oxidation in the mitochondria. The ability of DCA to decrease lactate production has been used for more than 30 years in the treatment of lactic acidosis that complicates inherited mitochondrial diseases in humans (Stacpoole et al., 1988, Stacpoole et al., 2006).
We hypothesized that the downstream effects of the DCA-induced shift in metabolism will have beneficial effects in cancer therapy (Figure 1). We show that, as predicted, DCA changes the metabolism of cancer cells from the cytoplasm-based glycolysis to the mitochondria-based glucose oxidation. This is associated with increased production of ROS and decreased ªñªÀm in all cancer, but not normal, cells, efflux of proapoptotic mediators from the mitochondria, and induction of mitochondria-dependent apoptosis. DCA also reverses the inhibition and downregulation of Kv1.5 in all cancer, but not normal, cells. The resultant efflux of K+, and decrease in intracellular K+, further increases the proapoptotic effects of DCA. DCA effectively decreases tumor growth in vitro and in vivo. We show that a metabolic-electrical remodeling regulates apoptosis resistance in cancer. Moreover, this abnormality is easily reversible by a simple drug that is already used in humans....
DCA in the Drinking Water Induces Apoptosis and Decreases Tumor Growth In Vivo
We studied nude athymic rats implanted subcutaneously with 3 í┴ 106 A549 cells. The rats had free access to water with or without DCA (75 mg/l). In the first set of experiments (protocol a), 21 animals were divided into three groups: untreated controls (n = 5), DCA-prevention rats (n = 8), which received DCA just after cell injection for 5 weeks, and DCA-reversal rats (n = 8), which received DCA 2 weeks post-cell injection for 3 more weeks. The untreated rats rapidly developed tumors with a constant exponential tumor growth (Figure 8A). Both DCA-treated groups had a significant decrease in tumor size, measured by tumor weight at sacrifice and maximal diameter using calipers; in some rats, in vivo magnetic resonance imaging allowed us to visualize the tumors in vivo and calculate their volume. The decrease in tumor growth by DCA was associated with an increase in apoptosis (TUNEL) and a decrease in proliferation (PCNA) (Figure 8B). There was an inverse correlation between apoptosis and tumor size in the treated rats (Figure 8B). Kv1.5 was upregulated and survivin was downregulated in the DCA-treated rats (Figure 8C), confirming our in vitro data (Figure 4, Figure 6).
ííFigure 8Decreased Tumor Size in DCA-Treated Nude Rats Is Due to an Increase in Apoptosis and Decrease in Cell Proliferation
(A) Injection of A549 cells into the flank of nude rats results in the development of measurable tumors within 1 week. DCA-treated rats in both the prevention and reversal groups of protocols a and b (see Results) have smaller tumors. The size of the tumors was assessed by weight, calipers, or magnetic resonance imaging in vivo and at the time of euthanasia, as shown.
(B) DCA-treated rats had smaller tumors due to a significant increase in apoptosis (TUNEL) and a decrease in proliferation (PCNA), as shown by triple-staining. A significant correlation was observed between % TUNEL and both tumor diameter and weight (the higher the % of TUNEL-positive cells, the smaller the tumor).
(C) In agreement with our in vitro data, DCA increases Kv1.5 and decreases survivin expression, as shown by both immunohistochemistry and immunoblotting.
(D) DCA-treated rats did not have any sign of liver (AST), kidney (creatinine), or blood (hemoglobin) toxicity.
∗p < 0.05 versus untreated controls.íí
In a second set of experiments (protocol b), we studied whether the effects of DCA were sustained for longer periods of time and whether DCA would have a similar effect in more advanced tumors. We followed three groups of rats (n = 6/group) for 12 weeks: an untreated control group, a prevention group (rats given DCA at the time of tumor cell injection), and a reversal group where rats were given DCA at week 10 for 2 weeks. As in protocol a, at all times rats in the prevention group had significantly smaller tumors compared to the untreated controls; DCA at week 10 inhibited tumor growth immediately, with a significant decrease even after 1 week of treatment. DCA therapy did not have any toxic effects, as measured by several blood tests (Figure 8D; also see McMurtry et al., 2004).
íí
DCA Causes Efflux of Proapoptotic Factors from Mitochondria and Increases ROS Production
Whereas the untreated cancer cells (A549) showed cytochrome c and apoptosis-inducing factor (AIF) restricted to the mitochondria (colocalized with mitotracker red), in the DCA-treated cells cytochrome c was diffusely present in the cytoplasm and AIF was translocated to the nucleus (Figure 3A), both indicating induction of apoptosis.
DCA increased H2O2 production in a dose-dependent manner; this increase was inhibited by rotenone, suggesting that it was based on complex I of the ETC (Figure 3B, n = 5 plates/group). We also measured NADH levels in isolated mitochondria and showed that DCA increased the intramitochondrial NADH (Figure 3C, 5 plates/experiment, n = 5). The DCA-induced decrease in ªñªÀm was limited by the VDAC (an important component of the mitochondrial transition pore; MTP) inhibitor 4íõ-diisothiocyano-2,2íõ-disulfonic acid stilbene (DIDS; 0.5 mM) (Granville and Gottlieb, 2003) (Figure 3D, n = 5, ∼60 cells/group).
To determine whether cancer cells are less dependent on the ETC and oxidative phosphorylation, we studied the effects of low-dose cyanide (a complex-IV inhibitor and a well-known poison for normal cells). Cyanide's effects on mitochondria (as measured by ªñªÀm) were much less pronounced in cancer compared to the DCA-treated cells (Figure 3E, n = 5, ∼60 cells/group).DCA Activates Kv Channels in Cancer Cells by an H2O2-Dependent Mechanism
Using whole-cell patch clamping, we showed that in all untreated cancer cell lines the outward K+ current (Ik) was small and essentially voltage independent. DCA increased the Ik significantly in all cancer cell lines, but did not alter the Ik in the noncancerous SAEC (Figure 4A), PASMC, or fibroblasts (not shown) (Figure 4A, n = 7¿C8/group). The increase in Ik occurred as early as 5 min and persisted after 48 hr of DCA exposure. Most of the increased Ik was voltage dependent and blocked by 4-aminopyridine, a specific Kv channel inhibitor. The increased Ik caused hyperpolarization of the plasma-membrane Em. DCA also decreased cell capacitance, an electrophysiologic surrogate of cell size/volume, consistent with the cell shrinkage that characterizes early apoptosis (Figure 4A).
The DCA-induced increase in Ik was blocked by intracellular catalase, delivered through the patch pipette (i.e., due to H2O2), and by rotenone (i.e., due to complex I-produced H2O2), but not by thenoyltrifluoroacetone (TTFA; an inhibitor of complex II of the ETC) (Figure 4B, n = 5). It was also not blocked by the human ether-a-go-go-related gene (HERG) inhibitor E4031 (50 nM) (Wang et al., 2002) (not shown). The activation of Kv channels by DCA resulted in a decrease in intracellular K+, due to efflux of K+ down its concentration gradient. When this gradient was diminished by the addition of KCl, the intracellular K+-lowering effects of DCA were inhibited (Figure 4C, n = 20).DCA Induces Mitochondria-Dependent Apoptosis and Decreases Proliferation In Vitro
DCA increases annexin expression, causes a ∼6-fold increase in the percentage of TUNEL-positive nuclei, and activates both caspase 3 and 9 in A549 cells (Figures 6A and 6D). Eliminating highly proliferative cells by the induction of apoptosis, and by decreasing [Ca2+]i levels, DCA decreases indices of proliferation (Figures 6B and 6D) including BrdU incorporation and expression of proliferating cell nuclear antigen (PCNA). In addition, DCA decreases the expression of survivin (Figure 6B).
DCA-induced apoptosis proceeds by two pathways, one in the mitochondria, where depolarization activates mitochondria-dependent apoptosis, and the other at the plasmalemmal level, where activation/upregulation of Kv1.5 channels decreases [K+]i, activating caspases. To determine the relative importance of the two mechanisms, we compared the apoptosis induced by DCA to the apoptosis induced by a primary increase in Kv1.5 expression, using adenoviral gene transfer (Figures 6C and 6D). Compared to the adenovirus carrying green fluorescent protein (GFP) only, the adenovirus carrying GFP and cloned human Kv1.5 (Pozeg et al., 2003) significantly increased the percentage of TUNEL-positive cells. However, although the increase in Ik achieved with the gene transfer was higher than the increase achieved by DCA (Figure 6C; compare with Figure 4A), the increase in apoptosis achieved by the gene transfer was significantly less than that achieved by DCA (Figure 6D). We then measured the DCA-induced apoptosis in the presence of 4-aminopyridine (5 mM), a blocker of the whole Kv family. In addition to the A549 cells we also studied glioblastoma, an excitable cell type in which Kv channels might be more important in apoptosis regulation compared to the epithelial A549 cells. In the presence of 4-aminopyridine, DCA induced 68% of the apoptosis (% TUNEL-positive cells) induced by DCA alone (Figure 6D). Similarly, in glioblastoma cells, DCA + 4-aminopyridine induced 62% of the apoptosis induced by DCA alone (Figure S4). Moreover, 4-aminopyridine did not limit DCA's ability to cause efflux of cytochrome c from mitochondria, initiating mitochondria-based apoptosis (Figure S4). These data underlie the preponderant importance of the mitochondrial component of DCA's proapoptotic actions.
That NFAT1 is a distal mediator in DCA's anticancer effects was supported by the fact that VIVIT increased apoptosis and decreased proliferation in a manner similar to DCA (Figure S3B). For imaging studies, we studied four random fields per slide for ∼30 slides/group, and for patch clamping, 6¿C8 cells/group.Molecular Inhibition of PDK2 Mimics DCA
To confirm that inhibition of PDK is the major mechanism for the effects of DCA, we determined whether molecular inhibition of PDK2 by siRNA mimics DCA. We chose PDK2 because it is the only ubiquitously expressed isoenzyme; PDK1 and 3 are restricted in the heart and testis, respectively, and PDK4 is mostly expressed in skeletal muscle and heart. PDK2 is the most active of all and also has the lowest Ki for DCA (0.2 mM) (Bowker-Kinley et al., 1998). siRNA for PDK2 inhibited the expression of PDK2 in a dose-dependent manner, inhibiting mRNA up to 80% and protein expression (measured by both immunoblots and immunohistochemistry) by ∼70% (Figure S5). We tested three commercially available PDK2 siRNAs, which all inhibited the gene in a similar manner. Scrambled siRNA for PDK2 as well as siRNA for PDK1 did not decrease PDK2 expression (Figure S5). Whereas the scrambled siRNA had no effect on A549 cells, PDK2 siRNA decreased ªñªÀm and increased mitochondrial ROS in a manner identical to DCA (Figures 7A and 7B, n ∼ 20 plates/group). DCA added to siRNA-treated cells had no additional effects (data not shown). Inhibition of PDK2 by siRNA also increased apoptosis and decreased proliferation in cancer cells (Figure 7C, n ∼ 30 plates/group). To further prove that DCA activates PDH by inhibiting PDK, we immunoprecipitated PDH and showed that DCA increased the nonphosphorylated fraction (i.e., active) of the catalytic subunit (E1ª┴) (Figure 7D).íí
Discussion
Here we show that a metabolic-electrical remodeling (hyperpolarized mitochondria, downregulated Kv channels) regulates the apoptosis resistance that characterizes multiple human cancers. DCA, a small molecule that targets mitochondria, reverses this remodeling, inducing apoptosis and decreasing cancer growth in vitro and in vivo. These beneficial effects occur without affecting noncancerous cells or eliciting systemic toxicity. DCA treatment significantly increases glucose oxidation (which only occurs in functional mitochondria), indicating that the well-recognized, metabolic cancer signature (aerobic glycolysis) is reversible, rather than a consequence of permanent mitochondrial damage. DCA exerts its beneficial effects by two pathways, both of which induce apoptosis: first, by mitochondrial depolarization and efflux of proapoptotic mediators, and second, by an increase in Kv channel expression/function. DCA increases Kv channel expression by inhibiting NFAT1, a calcium-sensitive transcription factor that regulates cell-differentiation programs in many cell types but which has previously been unexplored in cancer. The mitochondria-NFAT-Kv pathway in cancer offers several new candidate targets for proapoptotic therapy that would be predicted to have high therapeutic selectivity.
Glycolysis and Cancer: Not Just an Epiphenomenon
It is now well accepted that most cancers have a glycolytic phenotype. Warburg suggested, but did not prove, that this was due to í░abnormal mitochondriaí▒ (Warburg, 1930); that is, cancer cells are forced to use inefficient, nonmitochondrial means of generating ATP. Our data suggest that this apparent mitochondrial í░dysfunctioní▒ is in fact reversible. Oxidative metabolism in cancer could be actively suppressed; the resultant shift to glycolysis may lead to apoptosis resistance (Plas and Thompson, 2002), offering a survival advantage in the transformed cells (Gatenby and Gillies, 2004). This suggests that a novel way to reverse apoptosis resistance might be to undo this metabolic/mitochondrial remodeling. We show that the glycolytic phenotype in cancer is easily altered by promoting oxidative phosphorylation (Figure 2C). This is associated with mitochondrial depolarization, which facilitates apoptosis and inhibits tumor growth. All the human cancer cell lines studied had more negative ªñªÀm compared to several noncancerous cell lines (Figure 2A), suggesting that this might be a hallmark of malignancy. Although apoptosis is not always associated with mitochondrial depolarization, our data are in agreement with the observation that cationic lipophilic drugs preferentially accumulate to tumor mitochondria (Don and Hogg, 2004). In addition, the positively charged rhodamine-based dyes (like TMRM) have been tried as í░carriersí▒ for selective delivery of drugs in cancer. More than 200 carcinomas were screened and were shown to accumulate rhodamine much more than noncarcinoma cells; these findings were first reviewed in 1988 (Chen, 1988), and although the mechanism was not clear then, it likely reflects the more negative ªñªÀm of cancer compared to noncancerous cells. Our work directly shows that this relative increase in ªñªÀm is associated with increased resistance to apoptosis, and its í░normalizationí▒ increases apoptosis and decreases cancer growth. Furthermore, it has just been shown that ªñªÀm of colon cancer cells predicts the aggressiveness of the tumor cells, that is, the more hyperpolarized the ªñªÀm, the more aggressive and metastatic the tumor (Heerdt et al., 2005), in agreement with our proposal. Studying ªñªÀm in fresh tumor specimens might be a convenient means to predict resistance to proapoptotic chemotherapies, with important implications in clinical decision making.
How Does DCA Alter Metabolism, Depolarize Mitochondria, and Initiate Apoptosis?
The shift in the metabolism of pyruvate away from lactate and toward acetyl-CoA and the Krebs cycle (Figures 2C and 2D), caused by DCA or molecular inhibition of PDK2 (Figure 7), increases the intramitochondrial production of the electron-donor NADH (Figure 3C), a substrate of the ETC complex I, leading to increased complex I-based ROS production (Figures 3B and 7B) (Kushnareva et al., 2002). A sustained increase in the ROS production can cause oxidative damage in the ETC, particularly complex I. This megacomplex is the most sensitive of all ETC complexes to ROS damage because it is by far the largest (46 subunits), and has at least nine ROS-sensitive iron-sulfur centers and seven mitochondrial DNA-encoded subunits (Brandt, 2006), which are very susceptible to oxidative damage. The ROS-induced complex-I dysfunction can limit the efflux of H+, decreasing ªñªÀm. Upon sustained and significant decrease in ªñªÀm, the voltage-sensitive MTP opens (Zamzami and Kroemer, 2001), allowing the efflux of many proapoptotic factors and the initiation of apoptosis (Figures 3A, 6, and 8B). This further increases the production of mitochondrial ROS, likely reinforcing a positive feedback loop enhancing apoptosis (Zamzami and Kroemer, 2001).
This í░complex I-centeredí▒ proposed mechanism has a precedent in congenital mitochondrial syndromes and neurodegenerative diseases. Patients with congenital complex-I deficiency have decreased ªñªÀm and increased ROS production (Pitkanen and Robinson, 1996). Inhibition of complex I in cell lines is associated with decreased ªñªÀm and increased ROS production in a dose-dependent manner, that is, the higher the percent complex-I inhibition, the higher the ROS and the lower the ªñªÀm (Barrientos and Moraes, 1999). A similar mechanism where dose-dependent inhibition of complex I leads to dose-dependent efflux of cytochrome c and apoptosis (Clayton et al., 2005) is proposed in the pathogenesis of neurodegenerative diseases such as Parkinson's, where complex-I dysfunction and ROS-mediated oxidative damage are well described (Bao et al., 2005, Schon and Manfredi, 2003).
Inhibition of VDAC limited the DCA-induced decrease in ªñªÀm (Figure 3D). VDAC (along with the adenine nucleotide translocase) is involved in the translocation of ADP (a substrate for the F1F0-ATPase) from the cytoplasm into the mitochondria. Inhibition of the VDAC would thus inhibit the function of the F1F0-ATPase, which would lead to accumulation of H+ in the intermembrane mitochondrial space, promoting hyperpolarization of the ªñªÀm, thus limiting the depolarizing effects of DCA. This is supported by findings from Thompson's group (Vander Heiden et al., 1999), although the role of VDAC in the regulation of ªñªÀm and initiation of apoptosis remains controversial (Shimizu et al., 1999) (reviewed in Granville and Gottlieb, 2003), and some of these mechanisms might only be relevant to specific experimental conditions, such as growth-factor withdrawal (Vander Heiden et al., 1999). An additional intriguing possibility is that, because DCA is itself an anion (see structure in Figure 1), it likely enters the mitochondria via the VDAC, explaining, at least in part, why its inhibition limits the effects of DCA on ªñªÀm.
Unexpectedly, but consistent with its therapeutic benefit, DCA decreased the expression of survivin, an inhibitor of apoptosis, both in vitro and in vivo (Figure 6, Figure 8). Survivin has recently emerged as a major antiapoptotic oncoprotein. The mechanism by which survivin is downregulated is unclear. Recent observations describing the direct involvement of a mitochondrial survivin pool in the suppression of apoptosis suggest that survivin might participate in the mitochondrial remodeling of cancer (Dohi et al., 2004, McMurtry et al., 2005).
A Mitochondria-NFAT-Kv Channel Axis in Cancer Is Normalized by DCA, Contributing to the Proapoptotic and Antiproliferative Effects of DCA
The apoptosis resistance in cancer likely involves multiple mechanisms. The current findings highlight the contribution of Kv channel inhibition/downregulation, due to impaired mitochondrial signaling, to this resistance. Closing of K+ channels or decreasing their expression results in an increase in [K+]i which increases the tonic inhibition that cytosolic K+ exerts on caspases. Kv1.5 gene transfer directly activated apoptosis in A549 cells (Figures 6C and 6D). Functional inhibition of all Kv channels by 4-aminopyridine limited the DCA-induced apoptosis by ∼32% in A549 cells and by ∼38% in glioblastoma cells (Figure 6D; Figure S4), suggesting that although the majority of apoptosis in DCA-treated cells is a direct result of efflux of proapoptotic mediators from cancer cells, the secondary effects on Kv channels also play an important role.
The precise role of K+ channels in cancer remains unclear, and although K+ channel opening promotes apoptosis in several tumors, the opposite result has also been noted (reviewed in Wang, 2004). Perhaps this relates to the type of tumor or the well-known diversity of K+ channel families. Specific K+ channels are now emerging as important regulators of apoptosis in different cell types. For example, HERG, a Kv channel, mediates H2O2-dependent apoptosis in various cancer cell lines (i.e., low HERG expression reduces apoptosis and enhances proliferation) (Wang et al., 2002). Kv1.5 regulates apoptosis in PASMC (Remillard and Yuan, 2004) and is downregulated in the proliferative and apoptosis-resistant vascular media in pulmonary hypertension (McMurtry et al., 2004, McMurtry et al., 2005, Pozeg et al., 2003). A teleological advantage of Kv1.5 as a regulator of apoptosis in cancer, and a factor which focused our attention on this channel, is its very short turnover time, less than 8 hr from transcription to functional expression (Levitan et al., 1995).
We show that cancer cells are deficient in ETC complex I-based production of H2O2, a Kv1.5 channel opener (Figure 3B). Perhaps more importantly, Kv1.5 is downregulated in cancer cell lines (Figure 4D), and Kv1.5 expression correlates inversely with histologic grade in a cohort of patients with non-small-cell lung cancer (more aggressive tumors have less Kv1.5) (Figure S2). We have identified NFAT1 as an important transcription factor responsible for this Kv1.5 downregulation (more aggressive cancers have more activated NFAT1) (Figure 5C).
The cellular environment in cancer is favorable for NFAT activation. A549 cells have increased [Ca2+]i (Figure 5A), a direct activator of calcineurin and thus NFAT (Macian, 2005). This increase in [Ca2+]i is, at least in part, due to the increased Ca2+ influx that results from the Kv channel deficiency (Figure 4). In addition, calcineurin is inhibited by increased ROS levels (Namgaladze et al., 2005); thus, the low mitochondrial ROS in cancer (Figures 3B and 7B) promote NFAT activation. Furthermore, the acidotic environment in cancer (due to aerobic glycolysis) (Figure 2E) would further promote NFAT activation (Komarova et al., 2005). It is remarkable that all of these mechanisms are reversed by DCA, which increases ROS, increases pH, and decreases [Ca2+]i, explaining its impressive effects on NFAT (Figure 5B). The upregulation of Kv1.5 by a drug that directly affects mitochondrial function suggests the presence of the mitochondria-NFAT-Kv1.5 axis, which is suppressed in cancer. Our work suggests that potential effects on Kv channels should be considered in cancer therapies targeting the mitochondria.Metabolic Modulation in Cancer by DCA: Possibility of Prompt Translation to Clinical Oncology
Our work suggests that metabolic modulators could be beneficial in human cancer, either alone or in combination with traditional chemotherapies, as apoptosis sensitizers. By targeting a fundamental and unique property of cancer cells, this approach may combine efficacy and selectivity. DCA (in clinically relevant doses; Stacpoole et al., 2006) was effective in preventing and inhibiting tumor growth in established tumors both early (week 2) and late (week 10) in their development (Figure 8A). DCA's effects in the reversal protocols were immediate, with significant effects even after 1 week of treatment. The relative specificity of DCA to target a metabolic (mitochondria) and electric (K+ channels) remodeling was confirmed by microarray experiments, where pathway analysis revealed a short list of altered mitochondrial apoptosis cell cycle and ion channel genes (Supplemental Results; Figure S6).
A very attractive property of DCA is its selectivity, evident by the lack of any systemic toxicity in this (Figure 8D) and other recent animal (McMurtry et al., 2004) and human studies (Stacpoole et al., 2006). DCA's ability to í░restoreí▒ ªñªÀm might explain why it is effective preferentially in cells that have very high ªñªÀm, such as cancer cells, but has no effects in normal cells (epithelial, fibroblasts, or PASMC). Preferential expression of PDK might also contribute to its selectivity. In a recent study of non-small-cell lung cancer specimens, cancer cells had increased PDK2 and decreased PDH expression (compatible with a glycolytic phenotype) compared to neighboring nonmalignant cells (Koukourakis et al., 2005).
The small size of DCA results in excellent tissue penetration after oral intake, including the central nervous system (Stacpoole et al., 2003), relevant to the difficult-to-treat glioblastoma, one of the tumors that we studied in vitro. In addition, DCA decreases tumor lactic-acid production and increases intracellular pH (Figure 2E); future studies need to address the hypothesis that this will decrease tumor invasiveness and metastatic potential (Gatenby and Gillies, 2004).
Our work identifies the mitochondria-NFAT-Kv channel axis and PDK as critical components of the metabolic-electrical remodeling that characterizes many human cancers and offers a tantalizing suggestion that DCA may have selective anticancer efficacy in patients. The very recent report of the first randomized long-term clinical trial of oral DCA in children with congenital lactic acidosis (at doses similar to those used in our in vivo experiments) showing that DCA was well tolerated and safe (Stacpoole et al., 2006) suggests a potentially easy translation of our work to clinical oncology.A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth: Cancer Cell
https://www.cell.com/cancer-cell/fulltext/S1535-6108(06)00372-2íí
The repopulating cancer cells in melanoma are characterized by increased mitochondrial membrane potential
Cancer Letters 2016
a
T Cell Therapy Unit, Eutilex Research Institute of Biomedicine, 222 Banpo-daero, Seocho-gu, Seoul, Republic of Korea
b
Cancer Immunology Branch, Division of Cancer Biology, National Cancer Center, Goyang, Republic of Korea
c
Department of Biological Sciences, Sungkyunkwan University, Suwon, Republic of Korea
d
Section of Clinical Immunology, Allergy, and Rheumatology, Department of Medicine, Tulane University Health Sciences Center, New Orleans, LA, USA
Highlights
•
B16-F10 melanoma cells have mitochondrial heterogeneity under stress conditions.
•
B16-F10 melanoma cells with enhanced mitochondrial membrane potential more efficiently drive tumor formation.
•
Enhanced mitochondrial membrane potential may be a marker for cancer-initiating cells and be used to isolate the live cancer-initiating cells.
Abstract
Although considerable effort has been expended in identifying definitive markers for cancer stem cells (CSCs) or cancer-initiating cells (CICs), the phenotypic plasticity of these cells obviates simple characterization using cell surface markers. We hypothesized that these cells could be characterized by their metabolic properties because they are in a quiescent state with low energy needs. We examined whether cancer cells differ in mitochondrial membrane potential (ªñªÎm) when they are under stress. The ªñªÎm of B16-F10 melanoma cells increased when they were exposed in vitro to serum starvation and chemotherapeutic agents, but not when exposed to hypoxia. Such TMREhigh cells were also present in tumor tissue. They primarily used glucose and/or lactate, and were superior to TMRElow B16-F10 cells in their ability to drive tumor growth. These findings suggest that CSCs or CICs could be identified in heterogeneous melanoma populations by measuring ªñªÎm.
Keywords
Mitochondrial membrane potentialRepopulating cancer cellsMetabolismMelanomaChemotherapeutics
Abbreviations
CSCcancer stem cellTMREtetramethylrhodamine ethyl esterªñªÎmmitochondrial membrane potentialESCsembryonic stem cellsMTGmitotracker green FMCCCPcarbonyl cyanide m-chlorophenyl hydrazineBPTESbis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfideETXetomoxir2-DG2-deoxy-D-glucoseCTXcyclophosphamideMTXmethotrexateíí
Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy
Michael D Forrest Ph.D.August 21, 2015.
Abstract
Cancer cells have a more hyperpolarised mitochondrial membrane potential (ªÀIM) than normal cells. ªÀIM = ∼−220 mV in cancer cells as compared to ∼−140 mV in normal cells. Until now it has not been known why. This paper explains this disparity, in a mathematical framework, and identifies molecular targets and operations unique to cancer cells. These are thence prospective cancer drug targets. BMS-199264 is proposed as an anti-cancer drug. It inhibits the reverse, proton-pumping mode of ATP synthase, which this paper identifies as crucial to cancer cells but not to healthy, normal adult cells. In the cancer cell model, the adenine nucleotide exchanger (ANT) is inversely orientated in the mitochondrial inner membrane as compared to normal cells. This predicts it to have a different drug interaction profile, which can be leveraged for cancer therapy. Uncouplers, which dissipate the proton motive force, are proposed as anti-cancer medicines e.g. 2,4-dinitrophenol.
During aerobic respiration, the movement of electrons along the respiratory chain pumps protons across the inner mitochondrial membrane to build a proton motive force (pmf) [1-3]. The pmf is electrochemical, consisting of a hyperpolarised transmembrane voltage (ªÀIM, negative inside) and a proton concentration gradient (ªÊpH, alkali inside). In vivo, with high concentrations of Pi, ª─pH is minor as compared to ªÀIM [4]. Protons move down this electrochemical gradient, through ATP synthase, to generate ATP. Eukaryote cells must maintain a hyperpolarized voltage across their inner mitochondrial membranes. If this hyperpolarisation dissipates, the voltage-sensitive permeability transition pore (PTP) will open and release pro-apoptotic agents (e.g. cytochrome c) into the cytoplasm and drive apoptotic cell death [5].
In a normal cell, ªÀIM flickers between -108 and -159 mV: with a mean value of -139 mV [6, 7]. Thermodynamically, the optimal ªÀIM for maximal ATP production is between 130 to 140 mV, a rule that applies for all living organisms [7]. 10% value alterations in ªÀIM, above or below its optimum, results in a ∼90% decrease in ATP synthesis and a ∼90% increase in harmful reactive oxygen species (ROS) [7].
Cancer cells have a more hyperpolarised ªÀIM than normal cells [8-17]. The more invasive and dangerous the cancer, the more hyperpolarised its ªÀIM is observed to be [14-16]. The hyperpolarisation of ªÀIM can be >50% greater in cancer cells than normal cells [14] e.g. ªÀIM = ∼-210 mV in Neu4145 cancer cells [18]. The ªÀIM hyperpolarisation in cancer cells can even be double that of normal cells [19]. So, generally, the ªÀIM of cancer cells is extremely suboptimal for ATP production. However, these cancer cells arení»t using aerobic respiration and arení»t using ªÀIM in the same way as normal cells.
Aerobic respiration is O2 dependent and uses glycolysis, the Krebs cycle and oxidative phosphorylation (OXPHOS) to produce ATP [1-3]. Aerobic glycolysis is the sole use of glycolysis to produce ATP, even in the presence of O2. Cancer cells can use aerobic glycolysis (Warburg effect) some or all of the time [18-39]. I propose that when in this mode, they have a more hyperpolarised ªÀIM. Indeed, experimentally, when cancer cells are switched out of aerobic glycolysis, into aerobic respiration, their ªÀIM is returned to that of normal cells [18-19].
The fact that there is a disparity in ªÀIM between normal and cancer cells is well established and has already been leveraged in human drug trials [40-41]. Delocalized lipophilic cations (DLCs) can cross membranes and their positive charge means they are drawn to, and accumulate in, the mitochondrial matrix (negative inside, because of hyperpolarised ªÀIM). Cancer cells have a more hyperpolarised ªÀIM and so DLCs are more attracted to, and better retained by, their mitochondria than that of normal cells [12]. Using the Nernst equation [14], if the ªÀIM of a cancer cell is 60 mV more hyperpolarised than that of a normal cell ¿C which is within the range of observation [14, 18-19] ¿C then a single charged DLC will accumulate 10 times more in the mitochondrial matrix of cancer cells than normal cells (T=300 K). DLCs with a double charge will accumulate 100 times more [42]. So, DLC poisons are more targeted to cancer cells and this means there are likely to be doses that can kill cancer cells, but leave normal cells unharmed. Different DLCs have been shown to accumulate in and selectively kill cancer cells, in vitro and in vivo [11, 43-47]. This affirms that cancer cells do have a more hyperpolarised ªÀIM, although no DLC has been successful in clinical trialling to date. For example, MKT-077 caused renal toxicity in Phase 1 trials [40-41].
It is not known why or how cancer cells have a more hyperpolarised ªÀIM. I provide a quantitative explanation, which identifies molecular targets and operations unique to cancer cells. These can be leveraged as cancer drug targets. To understand how ªÀIM generation differs in cancer cells, we must first explain it for normal cells.íí
The biophysics of ªÀIM in normal cells [4, 48-50]
Mitochondrial ATP synthase (F0F1-ATPase) can synthesise or hydrolyse ATP. Protons can flow í░downhillí▒ through the ATPase, to generate ATP, or be pumped í░uphillí▒ by the ATPase, using ATP. The mitochondrial Adenine Nucleotide Transporter (ANT) can export ATP4− for the import of ADP3−, or conduct the inverse. So, both ATPase and ANT catalyse reversible processes. Their directionality is governed by the mitochondrial membrane potential (ªÀIM) in relation to their reversal potential, Erev_ATPase and Erev_ANT respectively (mV). Which are set by the concentrations of the participating reactants, as shown in Equations 1-11 [4, 50].
During OXPHOS, protons are pumped by the complexes of the respiratory chain out of the mitochondrial matrix and into the mitochondrial intermembrane space. This hyperpolarises ªÀIM and makes it more negative than Erev_ATPase and Erev_ANT (the green coloured í░A-spaceí▒ of Figure 1). With ªÀIM hyperpolarised to Erev_ATPase, ATPase works in its í░forwardí▒ mode and synthesises ATP. With ªÀIM hyperpolarised to Erev_ANT, ANT works in its í░forwardí▒ mode and exports mitochondrial matrix ATP for the import of cytoplasmic ADP. So, the mitochondrion produces and exports ATP. The í░forwardí▒ operation of ANT and ATPase is a depolarising force to ªÀIM. ATP4− export for ADP3− import is depolarising and so are protons flowing í░downhillí▒ through ATPase. However, ªÀIM doesní»t depolarise because at the same time protons are being continually pumped í░uphillí▒ by the respiratory chain complexes, which is a hyperpolarising drive to ªÀIM. Actually, ªÀIM doesní»t remain constant during OXPHOS ¿C but í░flickersí▒ (as much as >100 mV) [48-50] as these depolarising and hyperpolarising forces wrestle back and forth for a temporary net dominance.
If ªÀIM is more positive (depolarised) than Erev_ATPase and Erev_ANT, they both work in their í░reverseí▒ mode (the grey coloured í░C-spaceí▒ of Figure 1). ATPase hydrolyses ATP and ANT imports cytoplasmic ATP for the exchange of mitochondrial matrix ADP. So, the mitochondrion imports and consumes ATP. In this state, ATPase pumps protons into the intermembrane space which hyperpolarises ªÀIM. In addition, ANT imports ATP4− and exports ADP3−, so a negative charge is gained on the matrix side which hyperpolarises ªÀIM.
During OXPHOS, ªÀIM is more negative than Erev_ATPase and Erev_ANT. If aerobic respiration is switched off, for example if the cell switches into aerobic glycolysis, then there will no longer be the hyperpolarising offset to the depolarising, í░forwardí▒ action of ANT and ATPase: ªÀIM will depolarise. Erev_ATPase is more negative than Erev_ANT and so ªÀIM will depolarise past this reversal potential first. In this case, ATPase will switch into its í░reverseí▒ mode and ANT will remain in its í░forwardí▒ mode (the orange coloured í░B-spaceí▒ of Figure 1). So, the ANT action will remain depolarising but the ATPase action will switch to being hyperpolarising ¿C pumping protons out rather passing protons in. However, this reverse ATPase action requires ATP and with ANT pumping ATP out, there is little to be had. Furthermore, near the reversal potential of ATPase there is little driving force for an ATPase action. Hence, depolarising forces dominate and ªÀIM depolarises further. When ªÀIM is equal to Erev_ANT then ANT does no í░forwardí▒ or í░reverseí▒ ATP/ADP exchange and its effect on ªÀIM is lost. At this point: With no ATP coming into the matrix, ATPase can no longer hydrolyse ATP to pump protons and its hyperpolarising action is also lost. In the absence of these forces, ensuing proton leak will depolarise the membrane potential. This will then make ªÀIM more depolarised than Erev_ANT and permit ANT to conduct a hyperpolarising exchange of ATP/ADP. The more depolarised it is past Erev_ANT, the more drive there is for this hyperpolarising exchange and the more that will occur. The resultant ATP entry will permit ATPase to conduct a hyperpolarising pumping of protons. So, there are hyperpolarising forces, conducted by ATPase and ANT, that come into play to prevent further depolarisation past Erev_ANT. They cannot hyperpolarise ªÀIM to be more negative than Erev_ANT because these forces are largely lost at this point, but they can prevent further depolarisation past this point. The result is that ªÀIM will oscillate around Erev_ANT. So, at the loss aerobic respiration ªÀIM will converge to Erev_ANT. Hence, at the loss of aerobic respiration there is a í░safety netí▒ of mechanisms to prevent the collapse of ªÀIM and the mass consumption of cytoplasmic ATP through mitochondrial proton pumping by ATPase.
At certain mitochondrial matrix [ATP]in/[ADP]in ratios, it is possible for ATPase to be in í░forwardí▒ operation and ANT to be in í░reverseí▒ operation (the whine coloured í░D-spaceí▒ of Figure 1). The former is depolarising, the latter hyperpolarising. However, it may be unlikely for mitochondria to have such a hyperpolarised ªÀIM and a low matrix [ATP]in/[ADP]in ratio. So, it may be that this part of the graph has no biological representation and can be discounted [4, 50]. The ªÀIM in the í░D-spaceí▒ prompts ATPase to create ATP and ANT to import ATP. The ensuing rise in the [ATP]in/[ADP]in ratio would push the system out of the í░D-spaceí▒ and into another area of the graph (Figure 1).
If proton pumping by the respiratory chain is stopped, but the Krebs cycle still persists, ªÀIM will depolarise past Erev_ATP but not all the way to Erev_ANT. The Krebs cycle can produce ATP (or GTP) in its succinyl CoA to succinate step. Once ªÀIM is less negative than Erev_ATP, the ATP produced by the Krebs cycle may support the í░reverseí▒ ATP hydrolysing, proton pumping, hyperpolarising action of ATPase [4, 48-50]. This action will í░holdí▒ ªÀIM in this range between Erev_ATP and Erev_ANT. However, during aerobic glycolysis: both OXPHOS and the Krebs cycle are shunted. So, this situation will not apply in this case. As aforementioned, ªÀIM should converge upon and oscillate around Erev_ANT.
IF-1 is a physiological protein, expressed by some tissues of some organisms, that inhibits the consumption of ATP by the F0F1-ATPase [4]. So, it prevents the í░reversalí▒ of the ATPase upon depolarisation of ªÀIM past Erev_ATP. Its blockage isní»t complete, but increases with matrix [ATP], decreased matrix pH (acidification) and dissipated ªÀIM. With F0F1-ATPases unable to í░reverseí▒, to confer a hyperpolarising pump of protons, they offer little resistance to an external, imposed depolarisation. So, this imposed depolarisation can converge relatively unopposed to Erev_ANT. Depolarisation past this point switches the ANT into producing a hyperpolarising exchange and this tries to í░holdí▒ ªÀIM at Erev_ANT, as described earlier. The result is that ªÀIM will oscillate around Erev_ANT. Unless the imposed depolarisation is strong enough to overcome this resistance, in which case the continued depolarisation will eventually open the voltage-dependent PTP and apoptosis is then all but assured.
íí...
Why cancer cells have a more hyperpolarised ªÀIM is mysterious
As aforementioned, I suggest that cancer cells have a more hyperpolarised ªÀIM when they are utilising aerobic glycolysis. I suggest that this mode is a function of cancer proliferation and so the more aggressive and dangerous the cancer, the more time they spend in this operating state. During aerobic glycolysis, the Krebs cycle and OXPHOS are shunted and arení»t used. By the reasoning of the previous section, ªÀIM should thus converge upon ¿C and oscillate around ¿C Erev_ANT. However, there is a problem. Refer to Figure 1 and note that Erev_ANT is in a range around ∼−120 mV (−115 mV at matrix [ATP]in/[ADP]in ratio = 1.5). But the ªÀIM of cancer cells is much more hyperpolarised: e.g. the ªÀIM of Neu4145 cancer cells is ∼-210 mV [18].ANT2 is essential to cancer cells; ANT1 and ANT3 kill cancer cells
It might still be that ANT2 transports glycolytic ATP into mitochondria in cancer cells, but it isní»t the only pathway for this. There may be a redundancy in this cancer system. Or ANT2 may have some other role. ANT2 does seem crucial to cancer cells.
I propose that cancer cell metabolism is similar to that of embryonic stem (ES) cells. Indeed, they share genetic expression fingerprints [67-68] and ES cells have a hyperpolarised ªÀIM also [69]. They both employ aerobic glycolysis some or all of the time [18-39, 70], are immortal (divide forever without limit) [71-72] (as a function of using aerobic glycolysis [73]), respond to ROS damage by apoptosis rather than repair [19, 74] and can proliferate rapidly. So, with caution, we can learn more about cancer from ES cells and vice versa. In mice, ANT2 deficiency is embryonically lethal [75]. ANT2 is crucial to ES cells and we extrapolate from this to suggest that it is crucial to cancer cells. Indeed, ANT2 knockdown (RNA interference, shRNA) represses cancer proliferation and induces apoptotic death to cancer cells in vitro and in vivo [76]. Although others have reported ANT2 knockdown to have no such effect [77]; but this earlier, alternative report can be considered an inferior study because it used siRNA rather than shRNA; shRNA produces a more complete, robust, long lasting, long term knockdown [76].
In cancer cells, whereas ANT2 is anti-apoptotic [76], ANT1 is pro-apoptotic [78]. Overexpression of ANT1 induces apoptosis in cultured cancer cells by collapsing ªÀIM and opening the voltage-dependent PTP [78]. Indeed, ANT1 transfection significantly suppresses tumor growth in vivo [78]. ANT3 is pro-apoptotic also [79]. Interestingly, over-expression of ANT1 is lethal to embryo cells [80], like it is to cancer cells.
The biophysics of ªÀIM in cancer cells
In cancer cells ªÀIM is hyperpolarised at ∼-220 mV. I suggest because Erev_ATP is ∼-220 mV in cancer cells. And that ªÀIM oscillates around this point. When more depolarised, ATPase is in it í░reverseí▒ mode and pumping protons at the expense of ATP hydrolysis to ADP and Pi. ANT2 imports ATP to service this hydrolysis and exports the ensuing ADP. The ATP-Mg/Pi carrier (APC) imports further ATP and exports Pi. Glycolysis in the cytoplasm synthesises ATP from this ADP and Pi. APC is electroneutral. The í░reverseí▒ mode of ATPase is hyperpolarising as is the ATP4− import and ADP3− export by ANT2. These hyperpolarising forces drive ªÀIM to Erev_ATP and then past it at which point ATPase switches into its depolarising í░forwardí▒ mode. This then depolarises ªÀIM back towards Erev_ATP. So ªÀIM oscillates around Erev_ATP (∼-220 mV). At Erev_ATP precisely there is no drive for proton conductance or pumping through ATPase, so oscillating around this point ensures little ATP generation but not much ATP hydrolysis either. It is a í░cheapí▒ way to hold ªÀIM at a hyperpolarised potential, safely well away from í░dangerousí▒ depolarised potentials that could open PTP and drive apoptosis. In cancer cells, ANT1 and ANT3 are expressed at low levels and so are irrelevant. However, if by an experimental intervention they are expressed at significant levels, they can kill the cancer cell. In cancer cells, the Erev_ANT of ANT1 and ANT3 are abnormally depolarised. This means that their depolarising í░forwardí▒ mode of operation ¿C ATP4− export, ADP3− import ¿C depolarises ªÀIM towards their í░dangerouslyí▒ depolarised Erev_ANT value. What is more, their export of ATP undermines the import of ATP by ANT2 and denies it to ATPase. Hence the hyperpolarising í░reverseí▒ mode of ATPase, wherein it needs ATP to pump protons, is compromised. Thus, it isní»t able to combat the depolarisation conveyed by ANT1 and/or ANT3. They have such depolarised Erev_ANT values that the driving force for their depolarising exchange is immense at even rather modestly hyperpolarised potentials e.g. -100 mV.Cancer cells reduce ROS at source and sink
I propose that during aerobic glycolysis, ROS are confronted at source and sink. OXPHOS is shunted leading to less ROS generation and the mitigation system is upregulated leading to more ROS mitigation. ROS generation is determined by the redox state of NAD+, while the NADP+ redox state is pivotal to antioxidant defence. As compared to normal cells, cancer cells decrease NADH and increase NADPH levels. The latter may carry over to confer greater protection if the cancer cell periodically switches into aerobic respiration. Investigators have reported cancer cells to have higher NADH levels than normal cells [97]. But their spectroscopy caní»t discriminate between NADH and NADPH and I suggest they are actually observing higher NADPH levels in cancer cells. Indeed, later studies with a spectroscopy that can distinguish between these two species reports higher NADPH, rather than NADH, in cancer cells [98]. Cancer may be combatted by increasing NADH [34] and/or lowering NADPH. This could be achieved by transfecting cancer cells with a mutant lactate dehydrogenase (LDH) that uses NADPH rather than NADH. Such a form has been engineered for a prokaryote LDH [99]. In a prior paper, I propose exogenous NADH as a cancer medicine [34]....
The cytoplasm is, and needs to be, neutral in normal and cancer cells [118-120]. Tumours are acidic, normal tissue is neutral [120-122]. This is likely because cancer cells, unlike normal cells, are using aerobic glycolysis and excreting lactate and protons through the monocarboxylate symporter (a promising cancer drug target). The more aggressive the cancer is, the more acidic its tumour [123]. So, cancer cells, unlike normal cells, must maintain their intracellular pH above their extracellular acidity and protonophores will shuttle protons, undermine this homeostasis and kill cancer cells; a prediction.
Uncouplers can kill cancer cells
2,4-dinitrophenol (DNP) and FCCP are uncouplers. In cancer cells, they cause cell cycle arrest at low doses and apoptosis at higher doses, via depolarising ªÀIM which opens PTP [124-125]. The uncouplers: moronone [126], CCCP [127], clusianone [128] and hyperforin [129] also kill cancer cells. So, uncouplers can kill cancer cells. However, the crux issue is: can they kill cancer cells whilst leaving normal cells unharmed?
In vitro, F16 ¿C a lipophilic cation uncoupler (a DLC) ¿C kills cancer cells but not normal cells [11]. In vitro, nemorosone ¿C a lipophilic anion uncoupler ¿C kills HepG2 cancer cells (∼75% cell death) but not non-cancer human embryonic kidney HEK293T cells to the same degree (∼10% cell death) [130]. So, there is a possible selectivity of action; which might be enhanced if cancer and normal cells were to be tested side-by-side in the same in vitro assay. Because cancer cells, with their greater affinity for a charged lipophilic protonophore (as previously discussed), may accumulate and sequester it from the normal cells. A DLC derivative of gallic acid, TPP+C10, can uncouple and selectively kill cancer cells in vitro and in a singenic mouse model [131].
Valinomycin depolarises ªÀIM; nigericin hyperpolarises ªÀIM [3]. As aforementioned, depolarisation or hyperpolarisation of ªÀIM may kill cancer cells and these drugs both demonstrate an anti-cancer activity [115, 132]. Together, valinomycin and nigericin can uncouple H+, while K+ cycles around the membrane [3]. This combined uncoupling activity should be tested against cancer cells. Coumarins comprise a structurally diverse group of natural compounds found in a variety of plant sources [133-135]. Some coumarin molecules (mammea A/BA, mammea A/BB) can reduce tumour weight by 83% in test animals by halting the cell cycle and inducing apoptosis selectively in cancer cells, by an unknown mechanism [133]. Possibly by uncoupling ªÀIM: mammea A/BB collapses the ªÀIM of the Leishmania amazonensis parasite [134]. A different coumarin molecule (mammea E/BB), with an anti-cancer action, has been shown to be an anionic protonophore with an uncoupling potency equivalent to that of FCCP [135].
Uncoupling chemicals can shuttle protons alone (e.g. DNP) or in interaction with a transmembrane protein in the inner mitochondrial membrane e.g. ANT [136]. There are other, further conceivable mechanisms to uncouple e.g. neutralising a negative molecular species residing in the mitochondrial matrix, shuttling a negative species out of the matrix, stimulating UCP activity or the intrinsic, basal uncoupling activity of other inner mitochondrial membrane proteins e.g. ANT.
Phenols, benzimidazoles, N-phenylanthranilates, salicylanilides, phenylhydrazones, salicylic acids, acyldithiocarbazates, cumarines, and aromatic amines can induce uncoupling [137]. I anticipate that these will have an anti-cancer activity and merit investigation.¢Ô┼╝┴¬╝┴┐╔ÊÈ╔▒╦└░®¤©░¹
2,4-Â■¤§╗¨▒¢ÀËú¿DNPú®║═FCCP╩âÔ┼╝┴¬╝┴íúÈ┌░®¤©░¹Íðú¼╦³├Ã═¿╣²┤‗┐¬PTPÁ─╚Ñ╝½╗»ªÀIM╩╣Á═╝┴┴┐╩▒Á─¤©░¹Í▄ã┌═úÍ═║═©▀╝┴┴┐╩▒Á─¤©░¹Á‗═÷[124-125]íú¢Ô┼╝┴¬╝┴ú║─¬┬Ì═¬[126]ú¼CCCP [127]ú¼clusianone [128]║═hyperforin [129]Ê▓┐╔ÊÈ╔▒╦└░®¤©░¹íúÊ‗┤╦ú¼¢Ô┼╝┴¬╝┴┐╔ÊÈ╔▒╦└░®¤©░¹íú╚╗°ú¼╣Ï╝³╬╩╠Ô╩Ãú║╦³├Ã┐╔ÊÈ╔▒╦└░®¤©░¹ú¼Â°▓╗╗ßãã╗Áı²│ú¤©░¹┬ú┐
È┌╠Õ═Ôú¼F16╩ÃÊ╗ÍÍÃÎͼðÈж└ÙÎË¢Ô┼╝┴¬╝┴ú¿DLCú®ú¼┐╔ÊÈ╔▒╦└░®¤©░¹ú¼Á½▓╗─▄╔▒╦└ı²│ú¤©░¹[11]íúÈ┌╠Õ═Ôú¼nemorosoneú¿Ê╗ÍÍÃÎͼðÈʧ└ÙÎË¢Ô┼╝┴¬╝┴ú®┐╔ÊÈ╔▒╦└HepG2░®¤©░¹ú¿È╝75úÑÁ─¤©░¹╦└═÷ú®ú¼Á½╔▒╦└ÀÃ░®ðÈ╚╦└Ó┼▀╠Ñ╔÷HEK293T¤©░¹Á─│╠Â╚¤Ó═¼ú¿È╝10úÑÁ─¤©░¹╦└═÷ú®[130]íúÊ‗┤╦ú¼┐╔─▄┤µÈ┌ÐíȱðÈÁ─θË├íú╚þ╣¹Ê¬È┌═¼Ê╗╠Õ═Ô╩ÈÐÚÍð═¼╩▒╝ý▓Ô░®¤©░¹║═ı²│ú¤©░¹ú¼È‗┐╔─▄╗ßÈ÷Ã┐╝ý▓Ôðº╣¹íúÊ‗╬¬░®¤©░¹ÂÈ┤°ÁþÁ─ÃÎͼðÈÍ╩ÎË╠Õ¥▀Ëð©³┤¾Á─ÃÎ║═┴ªú¿╚þÃ░╦¨╩÷ú®ú¼┐╔─▄╗ß┤Ëı²│ú¤©░¹Íð╗²¥█║═©¶└Ù╦³íú├╗╩│ÎË╦ßÁ─DLCÐ▄╔·╬´TPP + C10┐╔ÊÈÈ┌╠Õ═Ô║═│╔─Ûðí╩¾─úð═Íð¢Ô┼╝┴¬▓óÐíȱðÈ╔▒╦└░®¤©░¹[131]íúþË░▒╦ß├╣╦Ï╩╣ªÀIM╚Ñ╝½╗»ú╗─ß╚ı└¹ÐÃ├╣╦Ï╩╣polarIM [3]│¼╝½╗»íú╚þÃ░╦¨╩÷ú¼ªÀIMÁ─╚Ñ╝½╗»╗‗│¼╝½╗»┐╔─▄╗ß╔▒╦└░®¤©░¹ú¼▓óÃÊıÔð®Ê®╬´Â╝¤È╩¥│÷┐╣░®╗¯ðÈ[115ú¼132]íú═▀└¹├╣╦Ï║═─ß╚ı└¹ÐÃ├╣╦Ï┐╔ÊÈ╩╣H +¢Ô┼╝┴¬ú¼Â°K +È┌─ñÍ▄╬ºÐ¡╗À[3]íúıÔÍÍ¢ß║¤Á─¢Ô┼╝┴¬╗¯ðÈ˪ıÙÂÈ░®¤©░¹¢°ðð▓Ô╩Èíú¤ÒÂ╣╦Ï░³└¿È┌©¸ÍÍÍ▓╬´└┤È┤ÍðÀó¤ÍÁ─¢ß╣╣▓╗═¼Á─Ê╗ÎÚ╠ý╚╗╗»║¤╬´[133-135]íú═¿╣²╬┤ͬÁ─╗·Íãú¼─│ð®¤ÒÂ╣╦ÏÀÍÎËú¿▓©╚Ú»╬´A / BAú¼▓©╚Ú»╬´A / BBú®┐╔ÊÈ═¿╣²═úÍ╣¤©░¹Í▄ã┌▓óÐíȱðÈËıÁ╝░®¤©░¹Á‗═÷└┤╩╣╩ÈÐÚ»╬´Á─ÍÎ┴÷ÍÏ┴┐╝§Ãß83úÑ[133]íú┐╔─▄═¿╣²¢Ô┼╝┴¬ªÀIMú║╚Ú¤┘A / BB╩╣└¹╩▓┬³È¡│µÐÃ┬ÝÐÀ╝─╔·│µÁ─ªÀIM▒└└ú[134]íú¥▀Ëð┐╣░®Î¸Ë├Á─┴ÝÊ╗ÍͤÒÂ╣╦ÏÀÍÎËú¿╚Ú¤┘E / BBú®ÊÐ▒╗Íñ├¸╩ÃÊ╗ÍÍʧ└ÙÎËÍ╩ÎË╠Õú¼ãõ╚Ñ┼╝┴¬ðº┴ªËÙFCCP¤ÓÁ▒[135]íú
¢Ô┼╝┴¬Á─╗»Ðº╬´Í╩┐╔ÁÑÂ└┤®╦¾Í╩ÎËú¿└²╚þDNPú®╗‗ËÙ¤▀┴ú╠Õ─┌─ñÍðÁ─┐þ─ñÁ░░ΤÓ╗ÑθË├ú¼└²╚þ┤®╦¾Í╩ÎËíú┬ýʤ[136]íú╗╣Ëðãõ╦¹Ê╗ð®┐╔ÊȤÙÁ¢Á─╗·Íã└┤¢Ô±¯└²╚þÍð║═┤µÈ┌Ë┌¤▀┴ú╠Õ╗¨Í╩ÍðÁ─ʧðÈÀÍÎË╬´Í╩ú¼¢½Ê§ðÈ╬´Í╩┤®╦¾Ë┌╗¨Í╩Í«═Ôú¼┤╠╝ñUCP╗¯ðÈ╗‗ãõ╦¹─┌▓┐¤▀┴ú╠Õ─ñÁ░░Îú¿└²╚þH.O.ú®Á──┌È┌ú¼╗¨┤í¢Ô┼╝┴¬╗¯ðÈíú┬ýʤíú
▒¢ÀËú¼▒¢▓ó▀õ▀‗ú¼N-▒¢╗¨┴┌░▒╗¨▒¢╝Î╦ߧÑú¼╦«Ð¯§ú▒¢░Àú¼▒¢hydrú¼╦«Ð¯╦ßú¼§ú╗¨Â■┴‗┤·░▒╗¨╝Î╦ߧÑú¼¤ÒÂ╣╦Ï║═À╝¤Ò░À╗ßʲã¢Ô┼╝┴¬[137]íú╬ÊÈñ╝ãıÔ𮢽¥▀Ëð┐╣░®╗¯ðÈ║═Ë┼ÁÒÐð¥┐íúíí
An uncoupler in combination therapy with dichloroacetate (DCA)
DCA inhibits pyruvate dehydrogenase kinase (PDK). This decreases PDK inhibition of pyruvate dehydrogenase (PDH) and permits pyruvate to enter the Krebs cycle and OXPHOS to proceed [19]. DCA selectively kills cancer cells in vitro and in vivo [19], and has caused much excitement [163], but its breakdown products can cause neuropathy [164-166]. DCA acts by constitutively switching on OXPHOS and the ROS produced kills cancer cells [19]. An uncoupler will synergise DCA action by increasing the OXPHOS and ROS production rate. So, it will permit lower DCA concentrations to be used, which will diminish DCA side effects. DCA will reciprocally permit lower uncoupler concentrations to be used (e.g. DNP), which will diminish uncoupler side effects. In a prior paper I proposed exogenous NADH as an anti-cancer drug [34]. I suggest it kills cancer cells as DCA does, by constitutively switching on OXPHOS (by conveying it substrate) [34]. However, unlike DCA, there is likely to be few side-effects as ití»s a natural metabolite. It could be used in combination with DCA and/or an uncoupler.¢Ô┼╝┴¬╝┴ËÙÂ■┬╚┤Î╦ßÐ╬ú¿DCAú®┴¬║¤Í╬┴ã
DCAÊÍÍã▒¹═¬╦ß═ÐÃÔ├©╝ñ├©ú¿PDKú®íúıÔ¢ÁÁ═┴╦▒¹═¬╦ß▒¹═¬╦ß═ÐÃÔ├©ú¿PDHú®Á─PDKÊÍÍãθË├ú¼▓óÈ╩ðÝ▒¹═¬╦ߢ°╚Ù┐╦└Î▓╝╦╣С╗À║═OXPHOS¢°ðð[19]íú DCAÈ┌╠Õ─┌═ÔÐíȱðÈ╔▒╔╦░®¤©░¹[19]ú¼▓óʲãðÝÂÓð╦À▄[163]ú¼Á½ãõÀÍ¢Ô▓·╬´┐╔ʲã╔±¥¡▓í▒õ[164-166]íú DCA═¿╣²ÎÚ│╔ðÈÁÏ¢Ë═¿OXPHOS°ãθË├ú¼▓·╔·Á─ROS╔▒╦└░®¤©░¹[19]íú¢Ô±¯ã¸¢½═¿╣²È÷╝ËOXPHOS║═ROSÁ─▓·╔·╦┘┬╩└┤ð¡═¼DCAÁ─θË├íúÊ‗┤╦ú¼╦³¢½È╩ðÝ╩╣Ë├¢¤Á═Á─DCA┼¿Â╚ú¼ıÔ¢½╝§╔┘DCAÁ─©▒θË├íú DCA¢½¤Ó╗ÑÈ╩ðÝ╩╣Ë├¢¤Á═Á─¢Ô±¯╝┴┼¿Â╚ú¿└²╚þDNPú®ú¼ıÔ¢½╝§╔┘¢Ô±¯╝┴Á─©▒θË├íúÈ┌ÊÈÃ░Á─┬█╬─Íðú¼╬Ê╠ß│÷┴╦═ÔÈ┤ðÈNADHθ╬¬┐╣░®Ê®[34]íú╬Ê¢¿ÊÚ╦³¤±ÎÚ│╔DCAÊ╗Ш╔▒╦└░®¤©░¹ú¼À¢À¿╩ÃÎÚ│╔ðÈÁÏ┐¬ã¶OXPHOSú¿═¿╣²¤‗╗¨Í╩╩õ╦═ú®[34]íúÁ½╩Ãú¼ËÙDCA▓╗═¼ú¼Ë╔Ë┌╦³╩Ã╠ý╚╗┤·ð╗▓·╬´ú¼Ê‗┤╦╝©║§├╗Ëð©▒θË├íú╦³┐╔ÊÈËÙDCA║═/╗‗¢Ô±¯ã¸¢ß║¤╩╣Ë├íúConclusion
Cancer cells have a more hyperpolarised ªÀIM than normal cells (∼-220 mV compared to ∼-140 mV). This discrepancy suggests that different processes generate ªÀIM in cancer cells, which may be compromised to selectively kill them. This paper identifies these processes and prospective anticancer drugs, which I hope will be entered into animal and clinical studies. For example, BMS-199264, which blocks the reverse, ATP hydrolysing, but not the forward, ATP synthesising, operation of ATP synthase.¢ß┬█
░®¤©░¹¥▀Ëð▒╚ı²│ú¤©░¹©³©▀Á─│¼╝½╗»ªÀIMú¿〜-220 mVú¼Â°〜-140 mVú®íúıÔÍÍ▓¯Êý▒Ý├¸ú¼▓╗═¼Á─╣²│╠╗ßÈ┌░®¤©░¹Íð▓·╔·ªÀIMú¼ıÔ┐╔─▄╗ß╩▄Á¢╦║ªÊÈÐíȱðÈ╔▒╦└╦³├Ãíú▒¥╬─╚À¿┴╦ıÔð®╣²│╠║═Ã░ı░ðÈ┐╣░®Ê®╬´ú¼¤ú═¹¢½ãõË├Ë┌»╬´║═┴┘┤▓Ðð¥┐íú└²╚þú¼BMS-199264ú¼╦³ÎÞÍ╣ATP║¤│╔├©Á─À┤¤‗ATP╦«¢Ôú¼Á½ÎÞÍ╣ı²¤‗ATP║¤│╔íúFunding
The author wrote this paper without any financial support.
Footnotes
Email: mikeforrest{at}hotmail.com
Copyright
The copyright holder for this preprint is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission.Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy | bioRxiv
https://www.biorxiv.org/content/10.1101/025197v1.fullíí
Biochimica et Biophysica Acta (BBA) - Bioenergetics
Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism
íía
CNRS, UMR6214, F-49000 Angers, France; INSERM, U771, F-49000 Angers, France
b
CHU d'Angers, D¿ªpartement de Biochimie et G¿ªn¿ªtique, Angers, F-49000, France
c
INRA U1019, Unit of Human Nutrition, St Gene's Champanelle, France F-63000
Abstract
Adenine nucleotide translocase (ANT), a mitochondrial protein that facilitates the exchange of ADP and ATP across the mitochondrial inner membrane, plays an essential role in cellular energy metabolism. Human ANT presents four isoforms (ANT1-4), each with a specific expression depending on the nature of the tissue, cell type, developmental stage and status of cell proliferation. Thus, ANT1 is specific to muscle and brain tissues; ANT2 occurs mainly in proliferative, undifferentiated cells; ANT3 is ubiquitous; and ANT4 is found in germ cells. ANT1 and ANT3 export the ATP produced by oxidative phosphorylation (OxPhos) from the mitochondria into the cytosol while importing ADP. In contrast, the expression of ANT2, which is linked to the rate of glycolytic metabolism, is an important indicator of carcinogenesis. In fact, cancers are characterized by major metabolic changes that switch cells from the normally dual oxidative and glycolytic metabolisms to an almost exclusively glycolytic metabolism. When OxPhos activity is impaired, ANT2 imports glycolytically produced ATP into the mitochondria. In the mitochondrial matrix, the F1F0-ATPase complex hydrolyzes the ATP, pumping out a proton into the intermembrane space. The reverse operations of ANT2 and F1F0-ATPase under glycolytic conditions contribute to maintaining the mitochondrial membrane potential, ensuring cell survival and proliferation. Unlike the ANT1 and ANT3 isoforms, ANT2 is not pro-apoptotic and may therefore contribute to carcinogenesis. Since the expression of ANT2 is closely linked to the mitochondrial bioenergetics of tumors, it should be taken into account for individualizing cancer treatments and for the development of anticancer strategies. This article is part of a Special Issue entitled: Bioenergetics of Cancer.
Research Highlights
► Mitochondrial adenine nucleotide translocase isoforms (ANTs) have a main role in cellular energy metabolism.► In normal cell, ANT1 and ANT3 export OXPHOS mitochondrial ATP to the cytosol.
► In tumoral cell, aerobic glycolysis is associated with defective mitochondrial ATP production.
► Glycolytic ATP is imported into mitochondria by ANT2 to maintain the mitochondrial membrane potential (ªñªÀm) and to prevent apoptosis.
íí
ANT2 is specifically expressed either in undifferentiated cells, such as lymphocytes, or in tissues that are able to proliferate and regenerate, such as those of the kidney and liver [18], [21]. The expression of ANT2, which has been shown to be growth-dependent [24], is considered as a marker of cell proliferation [25]. The ANT2 gene expression is down-regulated in differentiated cell lines and remains unexpressed, or only slightly expressed, in most tissues [18]. Consequently, ANT2 expression has been widely studied in cancer cells and cell lines. Unlike the ANT1 and ANT3 isoforms, ANT2 is strongly overexpressed in various types of human cancer cells and in several hormone-dependent cancers compared with normal human fibroblasts and hepatocytes [23], [26], [27].
Cancer cells are known to have a glycolytic phenotype that favors cellular accumulation of intermediates such as lactate. An increase in the glycolytic metabolism is associated with mitochondrial OxPhos damage or inactivation due to the intrinsic conditions of transformed tumoral cells, their undifferentiated state and their microenvironment [43], [44], [45]. In vitro studies have shown that some tumoral cell lines maintain the ability to use their mitochondrial energetic background. Interestingly, the expression of ANT isoforms is closely related to the energetic metabolic properties of tumoral cells [46]. The induction of ANT2 expression in cancer cells is directly related to the higher glycolytic metabolism whereas ANT3 is not affected [23]. Thus, undifferentiated tumoral cell line derived from osteosarcoma overexpressing ANT2 develops in hypoxic conditions whereas the hepatocarcinoma tumoral cell line, with a more differentiated phenotype and lower ANT2 expression, was arrested at the G1/S checkpoint [47]. Under conditions of hypoxic stress, cell growth depends on the rapid metabolic adaptation to decreased mitochondrial ATP production. Oxygen deprivation leads to the arrest of the mitochondrial respiratory chain activity, causing a collapse of the mitochondrial membrane potential (ªñªÀm), resulting in cell death. Cancer cells, which can survive a complete OxPhos suppression by using the glycolytic metabolism [48], depend mainly on the ATP uptake to generate their mitochondrial ªñªÀm [23]. It has been recently shown that the glycolytic phenotype in cancer can be altered by promoting oxidative phosphorylation with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase [49]. In human non-small-cell lung cancer, glioblastoma and breast cancer cell lines, the use of dichloroacetate increased the production of reactive oxygen species and decreased ªñªÎm leading to cell death. Similarly, a highly glycolytic cell line unable to reactivate the OxPhos metabolism was found to be more sensitive to chloroethylnitrosourea, an anticancer agent, as compared to a tumoral cell line conserving at least partial mitochondrial OxPhos activity [50]. In most transformed tumoral cells, mitochondrial ATP consumption imposes the reversal of the cellular ATP phosphoryl pathway. ANT2 is co-expressed with hexokinase II (HK II) and ATPsynª┬, an essential subunit of mitochondrial F0F1-ATPase, suggesting a mechanism for the regulation of ATP import [23] (Fig. 1B). The HK II isoform was shown to be the predominantly overexpressed form of HK in rapidly growing tumors and under hypoxic conditions [47], [51]. To ensure the maintenance of energetic states in membrane-bound processes, most glycolytic enzymes are associated with the cytoplasmic membrane and the membranes of the intracellular compartments [52]. Glycolytic enzymes thus contribute to intracellular phosphoryl transfer and spatial distribution [53]. Cytoplasmic HK, which initiates glycolysis by phosphorylating glucose to glucose-6P (G6P), is inhibited by the product; in contrast, HK II, bound to the outside of the mitochondrial outer membrane, is not inhibited by G6P. According to our hypothesis, HK II, which is bound to the voltage-dependent anion channel (VDAC) located on the mitochondrial outer membrane [54] uses cytosolic G6P to provide mitochondria with ATP through an inverse reaction.
This involves the transfer of glycolytic ATP through the VDAC and ANT2 toward the matrix side of the mitochondrial inner membrane. Within the inner membrane, ANT and F0F1-ATPase associate with the inorganic phosphate carrier (PiC) to form a complex called the ATP-synthasome [55]. This complex supports a mechanism in which the release and exit of ATP/ADP and P(i) are highly localized and tightly coordinated events. The imported ATP may then be used either for the essential intra-mitochondrial enzymatic pathways or hydrolyzed to ADP by the F1 part of the F0F1-ATPase to maintain the ªñªÀm [23].
This type of ATP hydrolysis by the reverse action of mitochondrial F1F0-ATPase has also been associated with pathological conditions when the respiratory chain is inhibited as in the ischemic phase of some neurodegenerative diseases [56], [57], [58], as well as in macrophages activated by interferon gamma and lipopolysaccharide that consequently inhibit OxPhos with NO [59]. The mitochondrial import of glycolytic ATP is essential for maintaining ªñªÀm and preventing apoptotic cell death. Since ANT2 has been reported to be up-regulated in OxPhos-deficient liver pathology [60], a detailed investigation of the expression of ANT isoforms should prove useful in such disorders.íí
7. Conclusion
The metabolic changes following the switch from the dual oxidative and glycolytic metabolism in normal cells to a mainly glycolytic metabolism plays a significant role in the survival and proliferation of cancer cells. Further investigation of the glycolytic phenotype, an early manifestation of carcinogenesis, should offer novel therapeutic approaches. Thus, the silencing of ANT2 so as to promote cell death, or the sensitization of tumoral cells to apoptotic agents could prove useful in anticancer treatment. The chemotherapeutic efficiency may depend on the capacity of cancer cells to adjust the energy balance between glycolysis and oxidative phosphorylation. The ANT2/ANT3 expression ratio may be used to predict whether the up-regulation of glycolysis or the activation of OxPhos would be more effective in maintaining mitochondrial ªñªÀm. In any case, the mitochondrial bioenergetic background of tumors should be taken into account for the conception of individualized cancer treatments and for the development of anticancer strategies.Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0005272810007164íí
íí
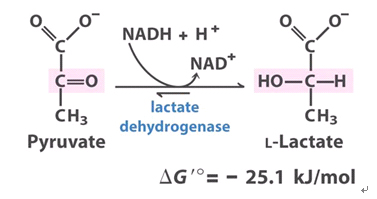
.jpg)
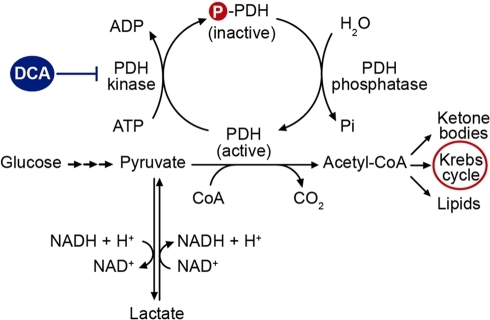



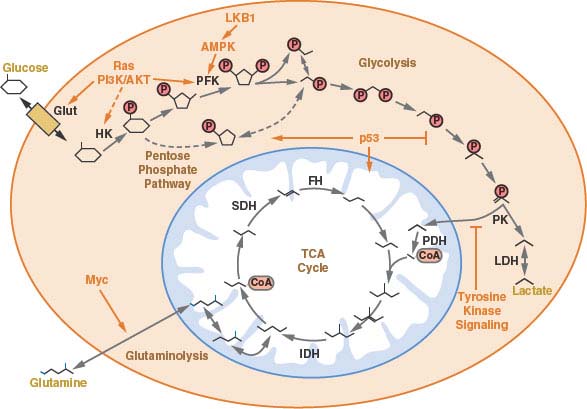
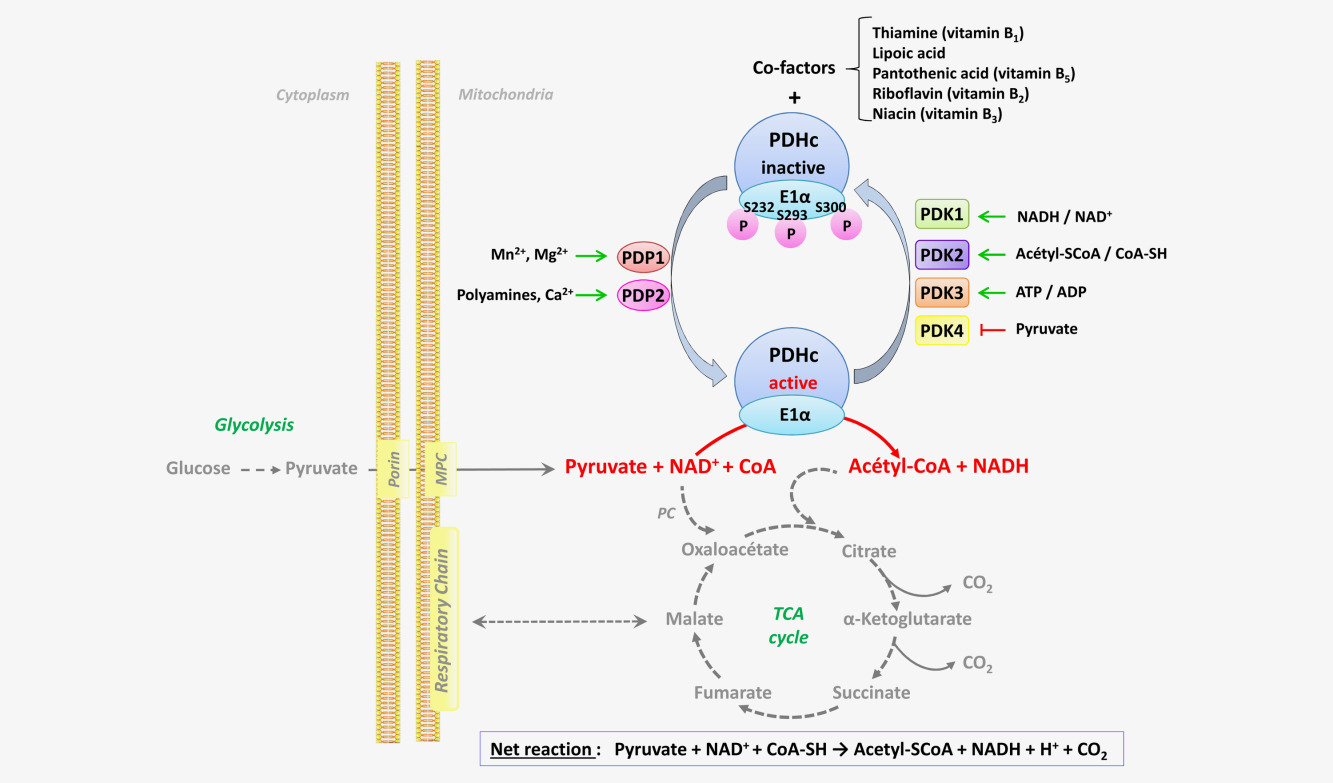

.jpg)
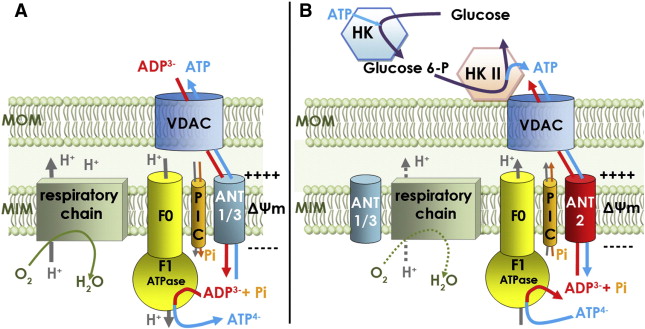
{kind=link}