ˇˇ
Br J Pharmacol. 2009 Oct; 158(3): 638¨C651.
Arginase: an emerging key player in the mammalian immune system
Markus Munder
Department of Hematology, Oncology and Rheumatology, University Hospital Heidelberg, and Institute of Immunology, University of Heidelberg, Heidelberg, Germany
Correspondence: Markus Munder, Department of Hematology, Oncology and Rheumatology, University Hospital Heidelberg, Im Neuenheimer Feld 410, 69120 Heidelberg,
Abstract
The enzyme arginase metabolizes L-arginine to L-ornithine and urea. Besides its fundamental role in the hepatic urea cycle, arginase is also expressed the immune system of mice and man. While significant interspecies differences exist regarding expression, subcellular localization and regulation of immune cell arginase, associated pathways of immunopathology are comparable between species. Arginase is induced in murine myeloid cells mainly by Th2 cytokines and inflammatory agents and participates in a variety of inflammatory diseases by down-regulation of nitric oxide synthesis, induction of fibrosis and tissue regeneration. In humans, arginase I is constitutively expressed in polymorphonuclear neutrophils and is liberated during inflammation. Myeloid cell arginase-mediated L-arginine depletion profoundly suppresses T cell immune responses and this has emerged as a fundamental mechanism of inflammation-associated immunosuppression. Pharmacological interference with L-arginine metabolism is a novel promising strategy in the treatment of cancer, autoimmunity or unwanted immune deviation.
Keywords: arginase, L-arginine, inflammation, tumour immunology, myeloid-derived suppressor cells
In the last couple of years literature on arginase in the immune system has increased enormously. This is due to the fact that the enzyme is crucially involved in various aspects of inflammation. Arginase has been shown to be either responsible for or to participate in, for example, inflammation-triggered immune dysfunction, tumour immune escape, fibrosis, immunosuppression and immunopathology of infectious diseases (Bronte and Zanovello 2005b). This review therefore aims to summarize key aspects of arginase in the immune system. After looking back briefly on important historical findings regarding immune cell arginase, the expression and regulation of arginase in the murine and human immune system and its involvement in a variety of immunologically mediated diseases will be discussed. Novel findings on the role of myeloid cell-associated arginase in immunosuppression in general and specifically in the context of tumour immune escape will be analysed in more detail. Finally, pharmacological ways to manipulate arginase and arginine metabolism will be summarized as they hold great promise for the treatment of cancer, autoimmunity and unwanted immunosuppression.
Arginase: from urea cycle enzyme to immune cell component
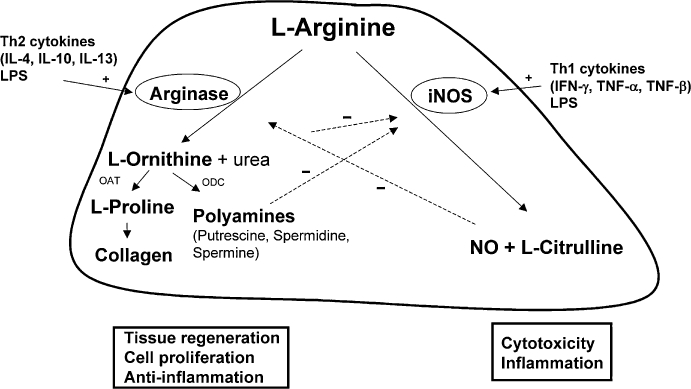
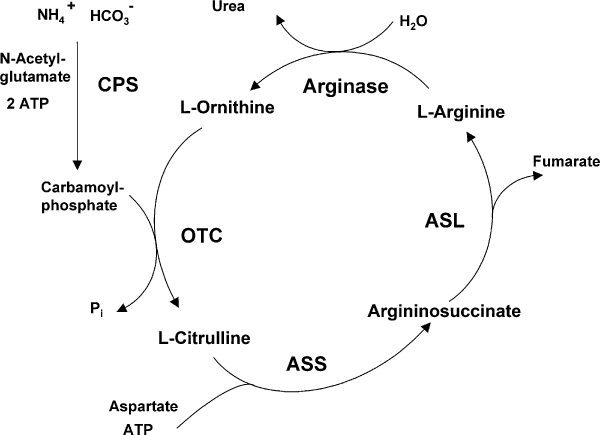
The enzyme arginase hydrolyzes L-arginine to the products L-ornithine and urea. In mammals, two arginase isoenzymes (designated arginase I and II) exist. They catalyse the same biochemical reaction but differ in cellular expression, regulation and subcellular localization (Jenkinson et al., 1996). The isoform arginase I (L-arginine ureahydrolase, AI, EC 3.5.3.1) is expressed in the liver as one of the enzymes of the urea cycle which detoxifies ammonia in mammals (Figure 1). The cycle is distributed over two cellular compartments (mitochondrion/cytosol) with arginase acting as a cytosolic protein (Jenkinson et al., 1996). Rat liver arginase was cristallized as a trimeric enzyme with a binuclear manganese cluster as part of the catalytic site cleft (Kanyo et al., 1996). Human arginase I is a 322-amino acid protein and demonstrates 58% sequence identity to human arginase II. It was cloned more than 20 years ago (Dizikes et al., 1986; Haraguchi et al., 1987) and the gene for human arginase I was localized on chromosome 6q23 (Sparkes et al., 1986). Human arginase II was cloned in 1996 (Gotoh et al., 1996; Vockley et al., 1996) and the gene was mapped to chromosome 14q24.1¨C24.3 (Gotoh et al., 1997a). Arginase II protein is expressed as a mitochondrial protein in a variety of peripheral mammalian tissues, most prominently in kidney, prostate, small intestine and the lactating mammary gland. Due to its generation of L-ornithine, arginase is involved in several important downstream metabolic pathways (Figure 2). L-ornithine can be further metabolized to polyamines (putrescine, spermidine, spermine) via ornithine decarboxylase (ODC). Polyamines are small cationic molecules which participate in a variety of fundamental cellular functions (e.g. proliferation, cell membrane transport). The metabolism of L-ornithine via ornithine aminotransferase (OAT) generates L-proline, which is an essential component of collagen. Alternatively, L-arginine serves as a substrate for nitric oxide synthase (NOS) leading to NO and other reactive nitrogen intermediates (e.g. peroxynitrite). L-arginine can be recycled from L-citrulline via argininosuccinate synthase (ASS) and argininosuccinate lyase (ASL) (Morris, 2002).
Figure 2
Reciprocal regulation of arginase and inducible nitric oxide synthase (iNOS) in murine myeloid cells. Downstream metabolic products of arginase and their association with components of inflammatory responses. OAT, ornithine aminotransferase; ODC, ornithine decarboxylase.
Figure 1
Arginase as part of the hepatic urea cycle. ASL, argininosuccinate lyase; ASS, argininosuccinate synthetase; CPS, carbamoyl phosphate synthetase; OTC, ornithine transcarbamoylase.
Myeloid cell-associated arginase was described as an immunosuppressive principle already 30 years ago. The addition of peritoneal cells to murine mixed lymphocyte reaction (MLR) assays suppressed splenocyte cytotoxicity which correlated with (i) inducible expression of arginase within the peritoneal cells/macrophages and (ii) complete depletion of arginine in cell culture medium of suppressed cultures (Kung et al., 1977). In fact, a complete urea cycle was demonstrated in murine bone marrow and rat peritoneal macrophages (Hofmann et al., 1978). The proliferation of mouse lymphoma cells and immune functions of murine splenocytes were inhibited by the release of arginase from activated murine macrophages into the cell culture medium with subsequent degradation of L-arginine (Chen and Broome, 1980). Murine macrophage arginase was characterized as a high molecular weight protein (110 kDa) with alkaline pH optimum and a preference for manganese as cofactor (Chen and Broome, 1980). A little later, a lymphokine with arginase-inducing potential during murine secondary MLR reactions was partially characterized (Dy et al., 1983). Several reports also demonstrated that the replication of malignant cells (Currie, 1978), parasites (Olds et al., 1980) and viruses (Wildy et al., 1982) is inhibited by the depletion of L-arginine (Schneider and Dy, 1985). Summarizing all these data, a first review on arginase in the immune response was published in 1985, concluding that macrophages can be activated to express arginase leading to L-arginine depletion in the microenvironment (Schneider and Dy, 1985). In this review it was already argued that L-arginine deficiency might lead to suppression of T lymphocyte proliferation (immunosuppression) on the one hand and to enhanced killing of malignant cells, parasites and viruses on the other hand.
Expression and regulation of arginase in the immune system
The view on the role of L-arginine in the immune response shifted completely when murine macrophages were found to express the inducible isoform of nitric oxide synthase (iNOS) upon stimulation by cytokines or microbial products (Nathan and Xie, 1994; MacMicking et al., 1997). Numerous reports demonstrated that activated macrophages consume L-arginine, convert it to L-citrulline, NO and reactive nitrogen species and that this is the primary mechanism of cytostatic or cytotoxic activity of macrophages against viruses, bacteria, fungi, protozoa, helminths and tumour cells (MacMicking et al., 1997). Interest in arginase slowly increased again when it was reported that the expression of both L-arginine-degrading enzymes in macrophages is regulated by T cell cytokines. While macrophage iNOS is induced by the prototypical T helper 1 (Th1) cytokine Interferon (IFN)-¦Ă (Nathan and Xie, 1994), arginase expression in murine macrophages is inducible by activation of the cells with the Th2 cytokines Interleukin (IL)-4, IL-10 and IL-13 (Corraliza et al., 1995; Modolell et al., 1995; Munder et al., 1998; Louis et al., 1999; Munder et al., 1999; Chang et al., 2000). Synergism among Th1 (Nathan and Xie, 1994) and Th2 cytokines (Munder et al., 1998) regarding their induction of iNOS and arginase, respectively, was demonstrated and reciprocal suppression of the alternative L-arginine-metabolizing enzyme was shown. Specifically, the NOS intermediate NG-hydroxy-L-arginine (NOHA) was shown to inhibit co-expressed macrophage arginase activity, thereby increasing L-arginine availability for NO production (Hecker et al., 1995). Th2-mediated induction of arginase I is a general feature of murine myeloid cell types and was also found in dendritic cells (Munder et al., 1999; Mayer et al., 2008) and granulocytes (Munder et al., 2005). Also, the cytokine IL-21 acts as an amplifier of Th2-mediated arginase induction by increasing the expression of the IL-4R¦Á and IL-13R¦Á1 chains in murine macrophages (Pesce et al., 2006). The dichotomous regulation of iNOS versus arginase is likely an old evolutionary program as it can already be detected in leukocytes of fish (Joerink et al., 2006). The role of macrophage arginase in inflammation is also clearly demonstrated by the fact that the murine enzyme is inducible by pathogen-derived macromolecules like lipopolysaccharide (LPS) or lipoproteins (Corraliza et al., 1995), by a broad range of inflammatory stimuli (Hrabak et al., 2006) and in rat alveolar macrophages by the reactive oxygen product hydrogen peroxide (H2O2) (Matthiesen et al., 2008). Recently, the induction of arginase I in murine macrophages by oxidized and acetylated lipoproteins was demonstrated (Gallardo-Soler et al., 2008) connecting macrophage alternative activation and an inflammatory phenotype with lipid metabolism and vascular atherosclerosis. Arginase expression is regulated by glucocorticoids in a cell type and species-specific way. While rat liver arginase is induced by glucocorticoids via the binding of the transcription factor CCAAT/enhancer binding protein ¦Â (C/EBP¦Â) to the arginase I enhancer (Gotoh et al., 1997b), both the LPS-induced up-regulation of rat alveolar macrophage arginase (Klasen et al., 2001) as well as the IL-4/IL-13-mediated up-regulation of rat airway fibroblast arginase (Lindemann and Racke, 2003) are inhibited by dexamethasone. The constitutive expression of arginase I in human polymorphonuclear granulocytes (PMN) is not modulated in vitro by a variety of pro- and anti-inflammatory stimuli including glucocorticoids (Munder et al., 2005).
Several groups have addressed the intracellular signalling mechanisms responsible for arginase induction in murine cells. Th2-mediated induction of arginase I is regulated by the coordinated action of the transcription factors PU.1, signal transducer and activator of transcription (STAT)-6 and C/EBP¦Â (Gray et al., 2005) at an enhancer 3 kilobases (kb) upstream of the basal promotor (Pauleau et al., 2004). The cAMP-mediated induction of murine arginase I (Corraliza et al., 1997; Morris et al., 1998) is mediated by Protein Kinase A Type I (PKAI) (and not PKA-II or EPAC) involving histone deacetylation (Haffner et al., 2008), while arginase I induction by modified lipoproteins is mediated via Peroxisome Proliferator-Activated Receptor (PPAR¦Ă and PPAR¦Ä) (Gallardo-Soler et al., 2008). The Src-homology 2 (SH2)-containing inositol-5ˇä-phosphatase SHIP1 restrains the inducibility of murine macrophage arginase I as macrophages are skewed towards an M2 phenotype with high levels of arginase I in SHIP1¨C/¨C mice (Rauh et al., 2004).
The role of myeloid cell arginase in disease models
Based on the regulatory principles (see above) and the different metabolic pathways associated with up-regulated immune cell arginase, it was hypothesized that the enzyme might causally be involved in disease pathogenesis because of (i) suppression of NO-mediated cytotoxicity via L-arginine consumption, (ii) enhanced collagen synthesis and fibrosis via proline generation and (iii) enhancement of cellular proliferation via polyamine generation. Consecutively, arginase expression was analysed in various disease models. In vivo, the enzyme was demonstrated in murine inflammatory cell infiltrates in experimental glomerulonephritis (Waddington et al., 1998), schistosomiasis (Hesse et al., 2001), trypanosomiasis (Gobert et al., 2000; Duleu et al., 2004), leishmaniasis (Iniesta et al., 2001; Kropf et al., 2005), autoimmune encephalomyelitis (Xu et al., 2003), asthma (Meurs et al., 2002; Zimmermann et al., 2003; Maarsingh et al., 2006), several viral (Mistry et al., 2001; Wang et al., 2005) and bacterial (Gobert et al., 2002; Bussiere et al., 2005) infections, lung fibrosis (Liu et al., 2005), sepsis (Carraway et al., 1998), trauma (Makarenkova et al., 2006) and tumours (Chang et al., 2001; Bronte et al., 2003; Liu et al., 2003; Kusmartsev et al., 2004; Rodriguez et al., 2004; Sinha et al., 2005). The reciprocal regulation of iNOS and arginase that was found in vitro is replicated in various inflammatory pathologies in vivo. In murine disease models that follow the Th1/Th2 paradigm with regard to disease susceptibility or resistance iNOS is induced in the context of a Th1-dominated resistant phenotype while macrophage arginase is up-regulated during Th2-mediated disease progression (e.g. Leishmaniasis; Iniesta et al., 2001; Kropf et al. 2005). In chronic murine schistosomiasis, arginase is similarly induced in Th2 granuloma-associated macrophages and is critical for enhanced granuloma size and fibrosis (Hesse et al., 2001). On the other hand, arginase I expressing alternatively activated macrophages protect the host during acute schistosomiasis by reducing massive Th1-mediated immunopathology and iNOS activity, as demonstrated by 100% mortality of mice with a macrophage/neutrophil ¨C targeted deletion of the IL4R¦Á chain (i.e. absence of alternatively activated macrophages) during acute infection with Schistosoma mansoni (Herbert et al., 2004). Tumour growth or rejection can also be dictated by the reciprocal expression of iNOS or arginase (Sinha et al., 2005). Genes of arginine metabolism like cationic amino acid transporter (CAT)-1, arginase I and arginase II are highly up-regulated in the lung in different murine asthma models (Zimmermann et al., 2003) and arginase has been linked pathogenetically with bronchial hyperreactivity by the depletion of L-arginine and consecutively reduced synthesis of the bronchodilatory agent NO (Meurs et al., 2002; Maarsingh et al., 2006). A pathogenetically oriented summary of arginase in different disease models is presented in Table 1.
Table 1
Examples of pathogenetic association of arginase and downstream metabolic consequences with different diseases
Disease model Mechanism Species Reference
Depletion of arginine/reduced synthesis of nitric oxide
Allergen-induced hyperresponsiveness Deficiency of cNOS/nNOS-derived NO gp Meurs et al., 2002; Maarsingh et al., 2006
Trypanosoma brucei brucei infection Reduced synthesis of NO due to high arginase activity in macrophages m Gobert et al., 2000; Duleu et al., 2004
Leishmania mexicana infection Parasite-encoded arginase: Suppression of macrophage microbicidal activity by reducing NO production via arginine depletion) m Gaur et al., 2007
Psoriasis Arginase overexpression in psoriatic epidermis ¨C reduction of anti-proliferative NO hu Bruch-Gerharz et al., 2003
Apoptosis Induction of arginase II in macrophages upon phagocytosis of apoptotic cells m Johann et al., 2007
Cystic fibrosis Sputum arginase activity inversely correlated with exhaled NO hu Grasemann et al., 2005
Toxoplasma gondii infection Stat6-dependent macrophage arginase induction: depletion of L-arginine m Rutschman et al., 2001; El Kasmi et al., 2008
Mycobacterium tuberculosis infection TLR-mediated induction of macrophage arginase I: L-arginine substrate competition m El Kasmi et al., 2008
Synthesis of collagen via proline
Schistosomiasis Th2-mediated induction of arginase I m Hesse et al., 2001
Lung allograft fibrosis TGF-¦Â-mediated induction of arginase; inhibition of arginase = reduction of fibrosis r Liu et al., 2005
Synthesis of polyamines
Helicobacter pylori (H.p.) infection Inhibition of iNOS translation = NO production via spermine (generated via arginase-ODC pathway) ¨C persistence of H.p. m Bussiere et al., 2005
Helicobacter pylori infection Induction of macrophage apoptosis via arginase-ODC-pathway: spermidine/spermine synthesis m Gobert et al., 2002
Leishmania infantum/major infection N¦Ř-hydroxy-L-arginine: inhibition of arginase = infection control m Iniesta et al., 2001; Kropf et al., 2005; Muller et al., 2008
Colitis Protective role of arginase within the intestinal inflammatory milieu m Gobert et al., 2004
Breast adenocarcinoma Macrophage arginase: providing polyamines for growth of tumour cells hu Chang et al., 2001
Inhibition of protein synthesis due to arginine depletion
Pseudorabies infection Reduced synthesis of viral structural proteins and folding m Wang et al., 2005
Inhibition of T cell activation via arginine depletion
Tumours Tumour progress: correlation with TCR¦Ć down-regulation m/hu Zea et al., 2005; Rodriguez et al., 2004; Sinha et al., 2005
Colon carcinoma/fibrosarcoma L-arginine limitation ˇú ROS production via NOS reductase domain ˇú Suppression of T cell activation m Bronte et al., 2003; Kusmartsev et al., 2004
Pregnancy correlation with TCR¦Ć down-regulation hu Kropf et al., 2007
Helicobacter pylori infection H. pylori arginase: down-regulation of T cell TCR¦Ć hu Zabaleta et al., 2004
Hepatitis B Inhibition of CD8 T cell proliferation and IL-2 production hu Das et al., 2008
Trauma correlation with TCR¦Ć down-regulation m Makarenkova et al., 2006
Host protection in infectious disease
Acute Schistosoma mansoni infection Down-regulation of detrimental Th1 inflammatory response m Herbert et al., 2004
Heligmosomoides polygyrus infection Intestinal Worm expulsion m Anthony et al., 2006
Nippostrongylus brasiliensis infection Intestinal smooth muscle hypercontractility: worm expulsion m Zhao et al., 2008
Open in a separate window
CD, cluster of differentiation; gp, guinea pig; hu, human; IL, interleukin; iNOS, inducible nitric oxide synthase; m, murine; NO, nitric oxide; ODC, ornithine decarboxylase; r, rat; ROS, reactive oxygen species; TCR, T cell receptor; TGF, transforming growth factor; Th, T helper; TLR, toll like receptor.
In contrast to its involvement in host-detrimental immunopathology, myeloid cell arginase can also serve a crucial host-protective function by down-regulating excessive Th1-induced inflammation (Table 1). Finally, data have emerged recently which demonstrate a host-protective effect of macrophage arginase I by itself during infections. Infection with different nematodes leads to induction of Th2 immunity, intestinal smooth muscle hypercontractility, increased luminal fluid secretion and consecutive worm expulsion. Infection of mice with the nematodes Heligmosomoides polygyrus (Anthony et al., 2006) or Nippostrongylus brasiliensis (Reese et al., 2007; Zhao et al., 2008) leads to the IL-4/IL-13/STAT6-mediated infiltration of alternatively activated (CD206+, arginase 1+, FIZZ1+, Ym1+) macrophages into the intestinal wall. Depletion of these alternatively activated macrophages with clodronate-liposomes or pharmacological inhibition of macrophage arginase with S-(2-boronoethyl)-l-cysteine (BEC) both inhibit helminth expulsion (Anthony et al., 2006; Zhao et al., 2008). The impaired nematode clearance from the intestinal lumen due to arginase inhibition is associated with abrogated smooth muscle hypercontractility during N. brasiliensis infection (Zhao et al., 2008). Although the exact mechanism of control of intestinal smooth muscle contractility by macrophage-associated arginase is still unclear, it is reminiscent of the arginase-mediated airway hyperresponsiveness during asthmatic inflammation (Maarsingh et al., 2006). Additionally, metabolic products downstream of arginase-induced ornithine might participate in the anti-helminth immune response.
A mouse model for arginase I deficiency was published (Iyer et al., 2002), but as the animals die between postnatal days 10¨C14 it cannot be used to verify hypotheses on the role of arginase I in murine immunology. It is therefore of great interest that a model of macrophage/neutrophil-specific deletion of arginase I was published recently (El Kasmi et al., 2008). Infection of this mouse with Toxoplasma gondii or Mycobacterium tuberculosis demonstrated decreased bacterial load, correlating with increased NO production, upon ablation of myeloid cell-specific arginase I (El Kasmi et al., 2008). These data are compatible with the notion of an anti-inflammatory pathway utilized by invading pathogens to suppress anti-microbial, NO-based effector pathways. In contrast, murine viability in the context of sepsis models (by LPS challenge or Streptococcus pneumoniae inoculation) was unaltered in the context of macrophage arginase-deficiency (El Kasmi et al., 2008).
While myeloid cell arginase I is induced in a variety of infectious diseases via a Th2 driven adaptive immune response (see above) it became clear that arginase I can also be directly up-regulated in macrophages via Pathogen-Associated Molecular Patterns (PAMP) so that the nature of the pathogen dictates the type of the evolving innate immune response. Chitin, a widespread polymer of N-acetyl-¦Â-D-glucosamine and part of, for example, fungal cell walls and helminths, leads to arginase I expression and the acquisition of an alternative activation phenotype of resident murine macrophages during infection with Nippostrongylus brasiliensis (Reese et al., 2007). The ensuing, leukotriene B4-mediated tissue infiltration of IL-4 expressing innate immune cells (eosinophils/basophils) then leads to the Th2-mediated, STAT6-dependent induction of chitinase-like proteins acidic mammalian chitinase (AMCase), Ym1 and Ym2, which are characteristic of alternatively activated macrophages. Interestingly, chitin degradation by AMCase is able to abrogate Th2 inflammatory cell infiltration so that a possible feedback inhibition of chitin-induced allergic or helminth-induced innate immune response by AMCase is likely (Reese et al., 2007). Another pathogen with direct arginase-inducing potential is Mycobacterium tuberculosis, which induces arginase I in murine macrophages through the toll like receptor (TLR) ¨C MyD88 pathway. This induction is independent of STAT6 but involves the up-regulation of C/EBP¦Â and binding of this transcription factor to the C/EBP¦Â site within the upstream enhancer of the murine arginase I gene (Pauleau et al., 2004).
The analysis of the role of myeloid cell-associated arginase during infection is further complicated by the fact that various pathogens express an enzymatically active arginase themselves (McGee et al., 2004; Viator et al., 2008). In parallel to the myeloid cell-expressed arginase of the host, the pathogen-encoded arginase might contribute, for example, to the down-regulation of T lymphocyte functions in the context of Helicobacter pylori infection (Zabaleta et al., 2004) or to the down-regulation of macrophage microbicidal activity by reducing NO production via L-arginine depletion in Leishmania mexicana infection (Gaur et al., 2007). Another critical component in the system of arginase-mediated L-arginine metabolism clearly is the capacity and regulation of L-arginine transport via the cell membrane. The transport system y+ is selective for the transport of cationic amino acids like L-arginine (Closs et al., 2004). Murine macrophages express CAT-1 constitutively and up-regulate CAT-2 upon classical (IFN-¦Ă) or alternative (IL-4, IL-10) activation (Louis et al., 1999; Rodriguez et al., 2003; Yeramian et al., 2006) so that enhanced catabolism via induced iNOS or arginase, respectively, is met by coordinated increased cellular uptake capacity. On the other hand, murine infections with the helminth parasite Schistosoma mansoni or the protozoan pathogen Toxoplasma gondii are exacerbated in the absence of CAT-2, demonstrating a crucial regulatory role of L-arginine membrane transport for immune responses (Thompson et al., 2008). The expression and regulation of L-arginine transport systems in cells of the human immune system is largely unresolved so far.
Arginase in the human immune system
In humans, arginase was detected in the peripheral blood mononuclear cell (PBMC) fraction after injury (Ochoa et al., 2001), inflammatory synovial fluid macrophages (due to arginase II) of patients with arthritis (Corraliza and Moncada, 2002), inflammatory cells of bronchoalveolar lavage fluid of asthmatic patients (Zimmermann et al., 2003), psoriatic lesions (Bruch-Gerharz et al., 2003), in activated monocytes of patients with autoimmune diseases (Rouzaut et al., 1999) and in the PBMC fraction of patients with active pulmonary tuberculosis (Zea et al., 2006). We have shown that among peripheral circulating human leukocytes of normal blood donors only PMN express arginase (Munder et al., 2005). By biochemical fractionation and immunoelectron microscopy we demonstrated that the enzyme is constitutively present in azurophil granules of human PMN, where it constitutes a novel oxygen-independent anti-microbial defense mechanism (Munder et al., 2005). After fusion of azurophil granules with a phagosome, arginase is present in the phagosome and likely depletes the intraphagosomal microenvironment of L-arginine during phagocytosis of pathogenic microorganisms which enhances the fungicidal activity of human PMN. Interestingly, Saccharomyces cerevisiae and Candida albicans up-regulate genes of their endogenous L-arginine biosynthetic pathways upon phagocytosis by human neutrophils (Rubin-Bejerano et al., 2003). This transcriptional response likely reflects the L-arginine-deprived intraphagosomal micromilieu of PMN and is not detectable upon phagocytosis by human monocytes (Rubin-Bejerano et al., 2003) which do not express arginase (Munder et al., 2005). Another study confirmed the expression of arginase I in human PMN but localized the enzyme to the gelatinase granules (Jacobsen et al., 2007). The discrepancy in results is still unclear at the moment. In vitro, constitutive human PMN arginase activity was not modulated by a variety of pro- and anti-inflammatory stimuli, including cytokines that typically lead to arginase induction in murine myeloid cells (Munder et al., 2005). In contrast, arginase is inducible in a variety of other human cell types like, for example, endothelium, epithelial cells and smooth muscle. Arginase I shares this feature with other important constitutive PMN proteins or peptides involved in inflammation and microbial defense like human cationic anti-microbial protein of 18 kDa (hCAP18), neutrophil gelatinase-associated lipocalin (NGAL), bactericidal/permeability-increasing protein (BPI) and the defensins (Borregaard et al., 2007). The fundamental disrepancies of arginase expression and regulation between murine and human immune cells fit into a growing list of differences in the immune systems of both species (Mestas and Hughes, 2004). This must be kept in mind when data from animal models are extrapolated to the human situation. Also, data on arginase expression in the human PBMC fraction without further purification need to be interpreted with caution. It remains to be analysed if arginase protein and activity is really induced in monocytes within the PBMC fraction. Alternatively, activated PMN are known to aberrantly co-purify within the PBMC fraction of patients with tumours or inflammation (Schmielau and Finn, 2001), so that de novo arginase activity in the PBMC population under conditions of inflammation might actually be confined to the neutrophil subset (M. Munder, unpublished).
Arginase: an endogenous immunosuppressive pathway
Inflammation is often associated with immunosupression locally within the inflammatory micromilieu as well as systemically (Nathan, 2002). While inflammation-induced immunosuppression has likely evolved as a homeostatic mechanism to prevent excessive tissue destruction during inflammation (Baniyash, 2004) it might be detrimental in situations of infection- and tumour-associated inflammation by impeding the clearance of the relevant microorganism or inhibiting tumour cytotoxicity. During the last couple of years, arginase expression and L-arginine depletion have emerged as a powerful immunosuppressive pathway of the mammalian immune system (Bronte and Zanovello 2005b). Several years ago, Ochoa et al. analysed the influence of L-arginine deficiency on the function of human T lymphocytes and found a down-regulation of the T cell receptor (TCR) ¦Ć chain, a critical signaling element of the TCR, as a possible mechanism for the impaired T cell function under conditions of L-arginine depletion (Rodriguez et al., 2002). Also, an arrest of T cells in the G0-G1 phase of the cell cycle, associated with the absence of up-regulated cyclin D3 and cyclin-dependent kinase 4 (cdk4) was seen upon L-arginine depletion (Rodriguez et al., 2007). Murine macrophages express arginase after Th2 stimulation (Munder et al., 1998) and this also leads to depletion of extracellular L-arginine and consecutive down-regulation of the TCR¦Ć chain in activated T cells (Rodriguez et al., 2003). T cell hyporesponsiveness associated with down-regulated TCR¦Ć chain can be the consequence of various circumstances (Baniyash, 2004) and transcriptional (Tsokos et al., 2003), posttranscriptional (Rodriguez et al., 2002) or posttranslational (Bronstein-Sitton et al., 2003) mechanisms of TCR¦Ć down-regulation have been described. In humans, immunosuppression in association with T cells that have partially down-regulated their TCR¦Ć chain is a recurrent finding in patients with cancer, autoimmunity or chronic infections (Baniyash, 2004). The mechanism(s) and cells that induce the observed T cell phenotype and/or the associated immunosuppression in vivo are largely unknown.
As intracellular constituents are liberated from dying PMN and accumulate in the micromilieu we hypothesized that human T cell activation should be blunted in the case of arginase liberation from human PMN. In fact, of all naturally occuring amino acids only L-arginine is depleted (and L-ornithine and urea synthesized) within the extracellular micromilieu of dying PMN (Munder et al., 2006). Within such an L-arginine-depleted milieu human T lymphocytes remained viable, but stopped proliferation and secretion of cytokines, while TCR activation-induced transcription of cytokine genes remained intact. Interestingly, human purulent exudate contains extraordinarily high arginase activities and liberated PMN arginase I fully accounts for the profound T cell suppressive properties of human pus by L-arginine depletion (Munder et al., 2006). Arginase-mediated T cell hyporesponsiveness is also involved in the suppression of the maternal anti-fetal immune reponse. A successful pregnancy depends on largely unknown mechanisms by which the immune system of the mother is made tolerant to the semi-allogeneic fetus. Myometrial arginase activity in the vicinity of the placental implantation site is >25 higher than in myometrium from non-pregnant guinea pigs (Weiner et al., 1996). In humans, arginase activity is also highly up-regulated in term placenta and increased in the peripheral blood of pregnant women (Kropf et al., 2007). While placental arginase might be important in the supply of polyamines, the local depletion of L-arginine via PMN-expressed arginase clearly dampens invading T lymphocytes (Kropf et al., 2007), possibly by down-regulating their TCR¦Ć chain. Arginase I is also expressed constitutively in human erythrocytes (Kim et al., 2002) and is liberated into the extracellular milieu upon hemolysis. It was recently shown that the immune suppression associated with transfusion of packed red blood cells might be due to the liberation of arginase from erythrocytes during storage and consecutive systemic L-arginine hydrolysis upon transfusion (Bernard et al., 2008).
How does L-arginine depletion translate into T cell suppression? The regulation of gene expression by amino acid availability is a fundamental regulatory mechanism in lower eukaryotes. In yeast, amino acid deprivation induces the accumulation of uncharged tRNA which leads to the phosphorylation of the ¦Á-subunit of eukaryotic Initiation Factor 2 (eIF2¦Á). As a consequence, the synthesis of the 43S pre-initiation complex (Met-tRNA, GTP, eIF2) is impaired and protein translation inhibited (Hinnebusch, 1994). In contrast, translation of the transcription factor GCN4 is augmented which results in the induction of more than 30 genes in multiple biosynthetic pathways. Amino acid deprivation therefore does not globally repress protein translation but specifically interferes with selected cellular activation programs via a general control response (Fafournoux et al., 2000). Mammalian cells express multiple eIF2¦Á kinases (e.g. GCN2 kinase) which are activated upon different types of cellular stress. Furthermore, various transcription factors (ATF-2, ATF-4, CHOP) are up-regulated and the mTOR-p70S6Kinase pathway is blunted in different mammalian cell types upon amino acid withdrawal (Rohde et al., 2001; Averous et al., 2004). This amino acid depletion finally inhibits the progression of various mammalian cell types through the cell cycle. The specificity of the response to amino acid withdrawal is at least partially mediated via amino acid response elements (AARE) in the promotor region of certain transcription factors (Averous et al., 2004). In murine macrophages the induction of arginase I via IL-13 leads to intra- and extracellular depletion of L-arginine and this inhibits the translation of inducible nitric oxide synthase (iNOS). This inhibitory effect seems to be specific as total protein synthesis of the cells is unaltered and, for example, the synthesis of the proinflammatory cytokine TNF-¦Á is unimpaired (El-Gayar et al., 2003). Translation of iNOS is also inhibited in astrocytes by L-arginine depletion via activation of GCN2K and increased eIF2¦Á-phosphorylation (Lee et al., 2003). The induction of amino acid transport proteins (e.g. CAT-1, -2) upon intracellular amino acid depletion is a likely compensatory mechanism (Aulak et al., 1999). In murine T lymphocytes it was demonstrated that the suppression of T cell proliferation upon depletion of L-tryptophan via indoleamine dioxygenase (IDO) is mediated through the activation of GCN2 Kinase with consecutive induction of the transcription factor CHOP (Munn et al., 2005). It is unclear so far, if human T lymphocytes also use this ancient nutrient sensing system to regulate their activation program. No published data are available on L-arginine depletion and the GCN2K-eIF2¦Á-system in human T cells in particular and on the protein expression profile of human T cells upon L-arginine depletion in general. Alternatively, mammalian cellular responses to amino acid deficiency might be regulated by micro-RNA binding. Human hepatocarcinoma cells up-regulate CAT-1 upon extracellular L-arginine depletion. This is mediated by replacement of CAT-1 mRNA-associated microRNA miR-122 and consecutive relief from translational inhibition (Bhattacharyya et al., 2006).
Arginase and cancer
Research over the last couple of years has convincingly demonstrated a crucial role for arginase in tumour immunobiology (Sica and Bronte, 2007; Rodriguez and Ochoa, 2008). Earlier reports focused on the expression of arginase in murine or human primary cancer tissue as well as malignant cell lines (Wu et al., 1996; Mumenthaler et al., 2008) and emphasized its potential role in the promotion of tumour growth via polyamine synthesis or down-regulation of NO-mediated tumour cytotoxicity. It also became clear that malignant tumours have evolved strategies to evade an effective tumour-cytotoxic immune response by inducing pathways of inflammation-associated immunosuppression (Smyth et al., 2001; Rabinovich et al. 2007; Sica and Bronte, 2007). The fate of a developing tumour is dictated not only by the properties of the malignant cells but also by the phenotype of tumour-infiltrating and tumour-interacting myeloid cells (Lewis and Pollard, 2006). Leukocyte-tumour interaction can result in tumour destruction as well as promotion of tumour growth, tissue invasion or metastasis (Lewis and Pollard, 2006). A key mechanism of tumour evasion from immune-mediated destruction is the induced impairment of T cell functions (Kershaw et al., 2005). Alternatively, tumour progression can be enhanced by infiltrating CD4+ T cells and a reduced carcinogenesis interestingly correlated with decreased infiltration of neutrophils (Daniel et al., 2003) recapitulating an earlier report on the tumour growth-promoting potential of PMN (Pekarek et al., 1995).
In tumour-bearing mice a heterogenous mixture of myeloid cells expands at various stages of development. This population is characterized by expression of CD11b and Gr-1 and efficiently suppresses T cell immune functions (Serafini et al., 2006a; Rodriguez and Ochoa, 2008). Murine MDSC clearly comprise monocyte-like cells as well as PMN-like cells at different stages of maturation (Movahedi et al., 2008). MDSC can suppress T cell immune functions by constitutive expression of arginase with consecutive L-arginine depletion (Bronte et al., 2003; Liu et al., 2003; Kusmartsev et al., 2004; Rodriguez et al., 2004; Sinha et al., 2005; Gallina et al., 2006; Serafini et al., 2006b). The immunosuppressive function of murine MDSC is enhanced further by IL-4-mediated increased expression of arginase (Bronte et al., 2003). When arginase of tumour-associated MDSC (Rodriguez et al., 2004; De Santo et al., 2005; Sinha et al., 2005) or tumour-infiltrating Gr-1¨C mature myeloid cells (Rodriguez et al., 2004) is inhibited in various murine tumour models, T cell functions are restored and tumour growth is inhibited. Additionally, murine MDSC can suppress T cells also via production of reactive nitrogen (Bronte et al., 2003) or oxygen (Bronte et al., 2003; Kusmartsev et al., 2004) intermediates or by cooperation of the different pathways, for example, by producing peroxynitrite from O2¨C and NO under conditions of L-arginine limitation (Bronte et al., 2003; Gallina et al. 2006; Sica and Bronte, 2007).
Human arginase-expressing myeloid suppressor cells are CD13+, CD33+, HLA-DRlow/¨C cells, that variably express monocyte (CD14) or granulocyte markers (CD15). Cells with this phenotype were described in patients with renal cell carcinoma (Zea et al., 2005) and the suppressive cells resemble PMN morphologically and by surface marker profile (CD11b+, CD14-, CD15+) although they appear in the PBMC fraction within the monocyte gate. They express arginase and down-regulate the TCR¦Ć chain in tumour-infiltrating T cells via L-arginine depletion. Depletion of the myeloid suppressor cells re-establishes T cell receptor- and costimulation-induced T cell activation (proliferation, IFN-¦Ă secretion and TCR¦Ć chain expression) in cell culture experiments (Zea et al., 2005). Arginase was also shown to participate in the suppression of tumour-infiltrating lymphocytes in patients with prostate carcinoma (Bronte et al., 2005a), non small cell lung carcinoma (Rodriguez et al., 2004) and multiple myeloma (Serafini et al., 2006b).
Pharmacological modulation of L-arginine metabolism
Pharmacological strategies that interfere with L-arginine metabolism hold great promise for the treatment of inflammatory diseases and cancer. Two opposing treatment options seem reasonable depending on the type of inflammation and tumour:
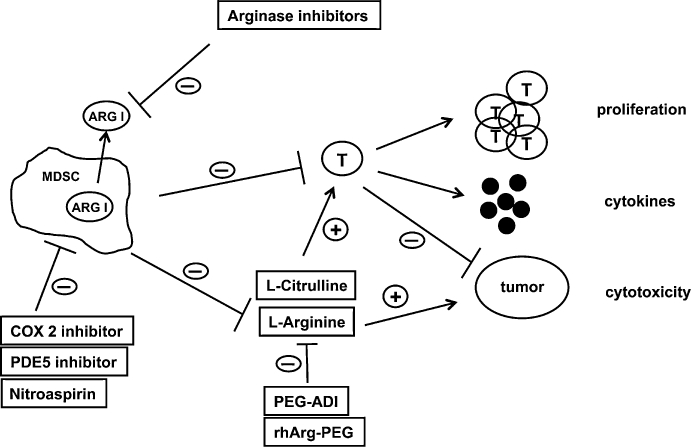
Arginase-mediated L-arginine depletion can lead to direct tumour cell death (Cavanaugh and Nicolson, 2000; Philip et al., 2003) so that recombinant L-arginine degrading enzymes (arginase, arginine deiminase) have been suggested as anti-tumour agents (Philip et al., 2003; Cheng et al., 2007), analogous to the clinically employed anti-leukemia agent L-asparaginase (Figure 3). Pegylated arginine deiminase (PEG-ADI) depleted L-arginine (by producing L-citrulline) and demonstrated clinical activity in patients with malignant melanoma (Ascierto et al., 2005). PEG-ADI-mediated L-arginine depletion induces autophagy and caspase-independent apoptosis in human prostate cancer cells (Kim et al., 2009) and inhibits growth of human pancreatic cancer xenografts in mice (Bowles et al., 2008). Interestingly, tumour susceptibility to L-arginine deprivation was associated with lack of tumour-expressed ASS as a possible L-citrulline-L-arginine recycling pathway for the tumour cells (Bowles et al., 2008). Pegylated recombinant human arginase (rhArg-PEG) demonstrated anti-tumour activity in ASS-expressing hepatocellular carcinoma (Cheng et al., 2007) as, in contrast to the L-citrulline generating enzyme ADI, arginase hydrolyses L-arginine to L-ornithine. Autoimmunity and unwanted inflammatory reactions might also be treated with approaches to lower local or systemic L-arginine concentrations. Clearly, problems of L-citrulline-to-L-arginine recycling and tolerability of low L-arginine concentrations over extended periods of time need to be addressed before such treatment options can be successfully implemented into clinical practice.
Figure 3
Opposing roles of L-arginine in tumour biology. L-arginine can directly support tumour growth and myeloidcell arginase-mediated or pharmacological depletion (PEG-ADI/rhArg-PEG) of the amino acid inhibits tumour growth. L-arginine deficiency, in contrast, can also suppress anti-tumour immune responses by down-regulating key functions of T lymphocytes. Pharmacological interference with arginase-mediated L-arginine depletion is demonstrated here in the context of myeloid-derived suppressor cell (MDSC)-associated immune suppression. T cell functions are restored by direct inhibition of cell-bound or liberated arginase I by arginase inhibitors, by supplementation of L-arginine or L-citrulline (for regeneration of L-arginine via the L-citrulline-L-arginine cycle) and by inhibition of various arginase-inducing mechanisms in MDSC. ARG I, arginase I; COX 2, cyclooxygenase 2; PDE5, phosphodiesterase 5; PEG-ADI, pegylated arginine deiminase; rhArg-PEG, recombinant human pegylated arginase; T, T lymphocyte.
While L-arginine depletion is a potential direct anti-tumour mechanism against certain malignant entities, the reverse scenario holds true for other tumour types due to the tumour-induced arginase-mediated immune dysfunction (as outlined above in section ˇ®Arginase and CancerˇŻ). It clearly depends on the tumour type, biochemical signature of the tumour cells (e.g. ASS expression) and the inflammatory context within the tumour micromilieu if L-arginine inhibits or suppresses tumour growth within the complex situation of cancer in vivo. Arginase-mediated L-arginine depletion might be adressed pharmacologically by either L-arginine supplementation or by inhibition of the enzyme. While pronounced local depletion of L-arginine in the inflammatory microenvironment of different immunopathologies is evident, L-arginine concentration in the systemic circulation can also be reduced due to liberated arginase. The normal concentrations of L-arginine in human plasma are 60¨C140 µM (Schwedhelm et al., 2008). Reduced plasma levels of L-arginine and elevated plasma arginase were reproducibly described in patients after surgery, trauma or during sepsis (Luiking et al., 2004). Various animal studies or small clinical trials and several randomized, placebo-controlled patient trials have analysed the influence of oral or parenteral L-arginine supplementation on different parameters of the immune system. In summary, no clear-cut reproducible advantage regarding immune functions or clinical outcome emerged so far (Nieves and Langkamp-Henken, 2002). More promising results were seen in trials to improve vascular physiology that was impaired in various diseases associated with L-arginine deficiency and impaired NO bioavailability, like, for example, in patients with pulmonary hypertension due to hemolysis and liberation of erythrocyte arginase in sickle cell disease (Morris et al., 2003) or thalassemia (Morris et al., 2005). In a recently published clinical trial, L-arginine supplementation could increase L-arginine plasma concentrations and improve endothelial NO bioavailability in patients with falciparum malaria and endothelial dysfunction due to hypoargininemia secondary to hemolysis-associated erythrocyte arginase release (Yeo et al., 2007). While L-arginine is extensively metabolized by arginases in gut and liver (Nieves and Langkamp-Henken, 2002) and also possibly by liberated inflammation-associated arginase (Munder et al., 2005), no significant intestinal degradation of L-citrulline takes place and its supplementation was already tested as alternative to L-arginine (Curis et al., 2005). Physiologically, L-citrulline is derived in the intestine from L-glutamine through the L-glutamate-L-ornithine pathway. Normally, the kidneys take up ca. 80% of intestinally released L-citrulline, which contributes about 10¨C15% of whole body L-arginine synthesis. L-arginine can be recycled from L-citrulline via the enzymes ASS and ASL (Morris, 2002), both enzymes colocalize in various tissues and are inducible by a variety of inflammatory stimuli. L-Citrulline can be taken up and partially recycled to L-arginine in J774 murine macrophages upon activation with IFN-¦Ă and LPS, but this mechanism is unable to sustain maximal nitric oxide production (Baydoun et al., 1994). In LPS-stimulated rat alveolar macrophages L-arginine is metabolized to L-citrulline (presumably via nitric oxide synthase) and L-ornithine (presumably via arginase), whereas no metabolism of exogenously applied L-citrulline to L-arginine takes place despite induction of ASS (Hammermann et al., 1998). ASS is also expressed constitutively in murine bone marrow-derived macrophages (Hofmann et al., 1978) and inducibly in the RAW264.7 macrophage cell line (Nussler et al., 1994). The induction of ASS was also demonstrated in the immortalized human Jurkat T lymphocyte line (Bansal et al., 2004) suggesting a potential metabolic bypass for T cells under conditions of L-arginine limitation. The oral administration of L-citrulline significantly increased L-arginine plasma concentrations without relevant side effects in a double-blind, placebo-controlled study (Schwedhelm et al., 2008).
The development of arginase inhibitors for clinical use is of prime importance in light of all the accumulated data on the role of arginase in tumour-associated MDSC and its pathogenetic role in inflammation-induced immunosuppression. Several specific arginase inhibitors have already been developed and tested in vitro. The intermediate of NO-synthesis, NG-hydroxy-L-arginine (NOHA) is a well-known arginase inhibitor (Hecker et al., 1995). It was successfully employed, for example, in human prostate carcinoma organ cultures, inhibited arginase activity and restored reactivity of tumour-infiltrating lymphocytes in cooperation with a NOS inhibitor (Bronte et al., 2005a). The L-arginine derivative N¦Ř-hydroxy-nor-L-arginine (nor-NOHA) was, for example, able to completely reverse PMN-mediated T cell suppression in purulent inflammation (Munder et al., 2006) and to restore airway responsiveness in an arginase-mediated asthma animal model (Maarsingh et al., 2006). The boronic acids 2(S)-amino-6-boronohexanoic acid (ABH) and BEC are potent inhibitors of both arginase isoforms at physiologic pH, binding with much higher affinity than the natural substrate (KM L-arginine 80.000 nM; Kd ABH 5 nM; KdBEC 270 nM) (Ash, 2004; Christianson, 2005). Human arginase I was cristallized in association with both inhibitors and new insights into the catalytic mechanism have been gained (Di Costanzo et al., 2005). Arginase inhibitors can potentially interfere with the urea cycle in the liver causing hyperammonemia and associated clinical problems. Another approach is therefore to inhibit the induction of iNOS and arginase in tumour-promoting MDSC. The NO-liberating compound nitroaspirin (NCX 4016) inhibits iNOS via NO feedback and arginase most likely indirectly by interfering with the arginase inducing pathways (De Santo et al., 2005). Nitroaspirin corrected the MDSC-mediated T cell immune dysfunction and thereby restrained tumour growth of a murine colon carcinoma (De Santo et al., 2005). Alternatively, inhibition of cyclooxygenase-2 (expressed by murine 3LL lung carcinoma) and prevention of Prostaglandin E2-mediated arginase induction in MDSC led to effective tumour control (Rodriguez et al., 2005). In a similar approach, phosphodiesterase-5 inhibitors were shown to down-regulate arginase I in murine MDSC and this led to an increased spontaneous anti-tumour response as well as to a more efficient adoptive T cell therapy (Serafini et al., 2006b). Finally, molecular interference by inhibition of nuclear factor kappaB (NF-¦ĘB) signaling in arginase I-expressing tumour-associated macrophages transformed them into tumour-cytotoxic effector cells (Hagemann et al., 2008).
In summary, arginase has emerged as a key player in the mammalian immune system and the enzyme is involved in various aspects of inflammation. Pharmacological interference with arginase specifically and L-arginine metabolism in general (Figure 3) therefore holds great promise for the treatment of cancer, autoimmunity and unwanted immunosuppression in clinical medicine.Arginase: an emerging key player in the mammalian immune system
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2765586/ˇˇ
Trends Pharmacol Sci. Author manuscript; available in PMC 2016 Jun 1.
Arginase: an old enzyme with new tricks
Ruth B. Caldwell,a,b,c Haroldo A. Toque,d S. Priya Narayanan,b,c,e and R. William Caldwellb,d
Author information Copyright and License information Disclaimer
aVA Medical Center, One Freedom Way, Augusta, GA, 30904, USA
bVision Discovery Institute, School of Allied Health Sciences, Medical College of Georgia, Georgia Regents University, 1459 Laney Walker Boulevard, Augusta, 30912, USA
cVascular Biology Center, School of Allied Health Sciences, Medical College of Georgia, Georgia Regents University, 1459 Laney Walker Boulevard, Augusta, 30912, USA
dDepartment of Pharmacology & Toxicology, School of Allied Health Sciences, Medical College of Georgia, Georgia Regents University, 1459 Laney Walker Boulevard, Augusta, 30912, USA
eDepartment of Occupational Therapy, School of Allied Health Sciences, Medical College of Georgia, Georgia Regents University, 1459 Laney Walker Boulevard, Augusta, 30912, USA
Ruth B. Caldwell: ude.urg@lewdlacr; Haroldo A. Toque: ude.urg@euqotserolfh; S. Priya Narayanan: ude.urg@nanayaranp; R. William Caldwell: ude.urg@lewdlacw
Abstract
Arginase has roots in early life forms. It converts L-arginine to urea and ornithine. The former provides protection against NH3; the later serves to stimulate cell growth and other physiological functions. Excessive arginase activity in mammals has been associated with cardiovascular and nervous system dysfunction and diseases. Two relevant aspects of this elevated activity may be involved in these disease states. First, excessive arginase activity reduces the supply of L-arginine needed by nitric oxide (NO) synthase to produce NO. Second, excessive production of ornithine leads to vascular structural problems and neural toxicity. Recent research has identified inflammatory agents and reactive oxygen species (ROS) as drivers of this pathologic elevation of arginase activity and expression. Here we review involvement of arginase in cardiovascular and nervous system dysfunction and discuss potential therapeutic interventions targeting excess arginase.
Keywords: arginase, vascular dysfunction, neurodegeneration, polyamine, oxidative stress, nitric oxide, peroxynitrite, superoxideˇˇ
Figure 1
Scheme for arginase catabolism of L-arginine to L-ornithine/urea or L-citrulline/NO, production of polyamines and anabolism and catabolism of proline. Also shown are the pathway for synthesis of L-arginine from L-glutamine, the reversible pathway between L-ornithine and L-glutamine, and the recycling of L-citrulline into L-arginine. Abbreviations: ASL, aminosuccinate lyase; ASS, aminosuccinate synthase; NOS, nitric oxide synthase; OAT; ornithine aminotransferase; ODC, ornithine decarboxylase; OTC, ornithine transcarbamylase. Arginase (bottom, left) is the final enzyme in the urea cycle within the liver, which restarts the cycle through the synthesis of L-citrulline from carbamoyl-phosphate (1) and L-ornithine (2) by OTC (center). It should be noted that that these reactions do not all occur within any given cell. In particular, the urea cycle is independent of the other reactions; i.e., L-arginine produced within the urea cycle is not a substrate for NOS and L-ornithine produced within the urea cycle is not a substrate for OAT or ODCArginase: an old enzyme with new tricks
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4461463/ˇˇ