Omega-3脂肪酸优先竞争P450酶并取代细胞膜上的OMEGA-6脂肪酸
要点
1.加州大学和哈佛大学的联合研究:OMEGA-3(EPA和DHA)的CY P450代谢产物(EEQs,EDPs)具有抗炎、扩张血管、抑制肿瘤血管生成(阻断VEGFR和FGF血管生成信号通路)、抑制肿瘤生长侵袭和转移的作用,并对心率不齐 、心原性猝死有保护作用。
2.相反,OMEGA-6脂肪酸(AA)的代谢产物有促进炎症、促进肿瘤血管生成和肿瘤生长和转移的作用。
3.EPA和DHA有抗炎和抗过敏作用,可用于哮喘和过敏 和肺部炎症。
4.临床前研究显示,OMEGA-3脂肪酸有保护心血管、扩张血管、抗炎和抗过敏作用。
5.高EPA/DHA饮食可以替换细胞膜上的EETs/AA:52%(左心室)、42%(肾脏)、21%(肝脏)、59%(肺)、47%(胰腺)40%(红细胞)78%(血浆)~与高OMEGA-6饮食相比;伴随EPA/DHA衍生的EEQs和EDPs的增加;
6.脂肪酸氧化的酶包括:COX,LOX 和CY P450酶,磷脂酶A2。其中COX和LOX优先氧化OMEGA-6脂肪酸,而CY P450优先氧化OMEGA-3脂肪酸。
7.目前已经发现,多种癌症过表达CY P450酶。
8. 最近发现,OMEGA-3脂肪酸在体内也可以形成大麻酚(Carnnabinoids),通过神经系统和免疫细胞上的大麻酚受体(CB1和CB2),产生保护神经、抗炎和免疫调节的作用。
9. 研究显示,EPA+DHA的抗炎和抗血管生成作用明显优于阿司匹林,EPA+DHA能够显著降低血浆中的炎症因子和血管生成因子的水平。
10.炎症形成和NSAID的抗炎机制
11.磷脂酶释放细胞膜磷脂双层中的ARA是炎症的开始,OMEGA-3脂肪酸抑制磷脂酶PLA2。 维生素E、虎杖甙(polydatin)和磷酸氯喹也是磷脂酶A2抑制剂
12.动物实验:内毒素LPS增加磷脂酶A2活性。
Omega 3 fatty acids and cardiovascular disease—fishing for a natural treatment | The BMJ
https://www.bmj.com/content/328/7430/30
炎症形成和NSAID的抗炎机制
NSAID是一类阻止炎症过程中前列腺素(PGs),白三烯(LT)或环氧化物(3)增加生成的抗炎药。因此,NSAID通常用于预防多种慢性炎症,包括CVD。参与生物合成前列腺素PG的主要酶是环氧合酶(COX),它被细分为组成型COX-1和诱导型COX-2的形式。这些COX亚型在发炎的组织中显示出不同的活性。 COX-2表达增加10到80倍,而COX-1表达增加2到4倍(4)。两种COX亚型均负责将花生四烯酸(ARA)转化为中间体PG,PGG2和PGH2。 ARA是类花生酸的前体,可被磷脂酶A2(PLA2)从膜磷脂上切割下来。
然后激活血栓烷合酶和各种异构酶,产生血栓烷A2(TxA2)和PG(PGE2,PGF2α,PGD2,PGI2)(5)。这四种PG具有共同的血管舒张功能以及增加膜的通透性(因此由于血流量增加而促进“发红”)。
PGE2和PGF2α主要由单核细胞和巨噬细胞产生,肥大细胞产生的PGD 2和内皮细胞产生PGI2(6)。长期低剂量的NSAIDs治疗也会降低PG的益处(7)。 PGE2和PGF2α控制水和电解质的吸收并维持胃粘膜的分泌。因此,NSAIDs可以减少胃腔和上皮细胞之间粘液-碳酸氢盐屏障的分泌。随后,与胃的低pH值接触,上皮细胞被杀死,粘膜的完整性丧失,引起溃疡(7)。
Omega-3 PUFA vs. NSAIDs for Preventing Cardiac Inflammation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6205954/
Frontiers | Phospholipase A2 – nexus of aging, oxidative stress, neuronal excitability, and functional decline of the aging nervous system? Insights from a snail model system of neuronal aging and age-associated memory impairment | Genetics
https://www.frontiersin.org/articles/10.3389/fgene.2014.00419/full
OMEGA-3多不饱和脂肪酸(ALA,EPA,DHA)对癌症的分子机制
最近的研究表明,脂质信号传导在肿瘤组织中被失调,导致在肿瘤微环境中来自ARA的促炎性和促癌性类花生酸的产量增加。例如,在肿瘤组织中,delta-6-去饱和酶(一种将亚油酸转化为ARA的限速酶)的表达以及从膜磷脂中释放ARA以启动类花生酸生物合成的磷脂酶的表达显着上调38 ,39.据报道,促肿瘤的脂质代谢酶(如COX-2、5-LOX和CYP环氧酶)的表达在肿瘤中被上调[34、36]。而抗致瘤性酶(例如15-LOX-1)被下调34。这些变化共同导致了支持性微环境以支持肿瘤进展。
ω-3PUFA促进健康的重要机制是它们抑制ARA的代谢以生成类花生酸40。在饮食上食用时,ω-3PUFA(包括EPA和DHA)以ω-6ARA为代价掺入膜磷脂中。在细胞刺激下,掺入的ω-3和ω-6PUFA被酶释放以产生细胞内游离脂肪酸(FFA),这些脂肪酸被COX,LOX和CYP酶快速代谢,从而产生ω-3-系列和ω-6-系列LM(脂质调节介质)。 EPA和DHA通过多种机制抑制ARA衍生的ω-6系列LM的形成,包括减少ARA从膜磷脂的释放,抑制代谢酶的酶活性以及与ARA直接竞争酶促转化。除了抑制ARA的酶促代谢外,EPA和DHA还可以作为脂质代谢酶的替代底物,从而导致ω-3系列LM的形成增加。其中一些介体具有抑制炎症,血管生成和癌症的有效作用.ω-3 polyunsaturated fatty acids-derived lipid metabolites on angiogenesis, inflammation and cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4306447/
Arachidonic Acid-metabolizing Cytochrome P450 Enzymes Are Targets of ω-3 Fatty Acids*
+ Author Affiliations
From the ‡Max Delbrueck Center for Molecular Medicine, 13125 Berlin, Germany,
§Lipidomix GmbH, 13125 Berlin, Germany,
the ¶Experimental and Clinical Research Center, Charité Medical Faculty,
Abstract
Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) protect against cardiovascular disease by largely unknown mechanisms. We tested the hypothesis that EPA and DHA may compete with arachidonic acid (AA) for the conversion by cytochrome P450 (CYP) enzymes, resulting in the formation of alternative, physiologically active, metabolites. Renal and hepatic microsomes, as well as various CYP isoforms, displayed equal or elevated activities when metabolizing EPA or DHA instead of AA. CYP2C/2J isoforms converting AA to epoxyeicosatrienoic acids (EETs) preferentially epoxidized the ω-3 double bond and thereby produced 17,18-epoxyeicosatetraenoic (17,18-EEQ) and 19,20-epoxydocosapentaenoic acid (19,20-EDP) from EPA and DHA. We found that these ω-3 epoxides are highly active as antiarrhythmic agents, suppressing the Ca2+-induced increased rate of spontaneous beating of neonatal rat cardiomyocytes, at low nanomolar concentrations. CYP4A/4F isoforms ω-hydroxylating AA were less regioselective toward EPA and DHA, catalyzing predominantly ω- and ω minus 1 hydroxylation. Rats given dietary EPA/DHA supplementation exhibited substantial replacement of AA by EPA and DHA in membrane phospholipids in plasma, heart, kidney, liver, lung, and pancreas, with less pronounced changes in the brain. The changes in fatty acids were accompanied by concomitant changes in endogenous CYP metabolite profiles (e.g. altering the EET/EEQ/EDP ratio from 87:0:13 to 27:18:55 in the heart). These results demonstrate that CYP enzymes efficiently convert EPA and DHA to novel epoxy and hydroxy metabolites that could mediate some of the beneficial cardiovascular effects of dietary ω-3 fatty acids.
Arachidonic Acid-metabolizing Cytochrome P450 Enzymes Are Targets of ω-3 Fatty Acids
http://www.jbc.org/content/285/43/32720.long
Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids
Cosima Arnold, Anne Konkel, Robert Fischer, Wolf-Hagen Schunck
Pharmacological Reports: PR 2010, 62 (3): 536-47
Dietary fish oil omega-3 fatty acids (n-3 PUFAs), such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), protect against arrhythmia and sudden cardiac death using largely unknown mechanisms. EPA and DHA may serve as efficient alternative substrates of arachidonic acid (AA) metabolizing cytochrome P450 (CYP) enzymes. For many of the CYP isoforms, the n-3 PUFAs are the preferred substrates. Moreover, the CYP enzymes oxygenate EPA and DHA with largely different regioselectivities compared to AA.In particular, the omega-3 double bond that distinguishes EPA and DHA from AA is a preferred site of CYP-catalyzed epoxidation reactions. Given the pivotal role of CYP-dependent AA metabolites in the regulation of vascular, renal and cardiac functions, their replacement by unique sets of epoxy- and hydroxy-metabolites derived from EPA and DHA may have far-reaching physiological implications. The currently available data suggest that some of the vasculo- and cardioprotective effects attributed to dietary n-3 PUFAs may be mediated by CYP-dependent metabolites of EPA and DHA.
Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids | Read by QxMD
https://read.qxmd.com/read/20631419/cytochrome-p450-dependent-metabolism-of-omega-6-and-omega-3-long-chain-polyunsaturated-fatty-acids
Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases
a Max Delbrueck Center for Molecular Medicine, Berlin, Germany
b OMEICOS Therapeutics GmbH, Berlin, Germany
c Department of Gastroenterology, Diabetes, Oncology and Rheumatology, Ruppiner Kliniken, Brandenburg Medical School, Neuruppin, Germany
d Lipid Clinic, Experimental and Clinical Research Centre (ECRC), Charité University Medicine and Max Delbrueck Center for Molecular Medicine, Berlin, Germany
Abstract
Numerous benefits have been attributed to dietary long-chain omega-3 polyunsaturated fatty acids (n-3 LC-PUFAs), including protection against cardiac arrhythmia, triglyceride-lowering, amelioration of inflammatory, and neurodegenerative disorders. This review covers recent findings indicating that a variety of these beneficial effects are mediated by “omega-3 epoxyeicosanoids”, a class of novel n-3 LC-PUFA-derived lipid mediators, which are generated via the cytochrome P450 (CYP) epoxygenase pathway. CYP enzymes, previously identified as arachidonic acid (20:4n-6; AA) epoxygenases, accept eicosapentaenoic acid (20:5n-3; EPA) and docosahexaenoic acid (22:6n-3; DHA), the major fish oil n-3 LC-PUFAs, as efficient alternative substrates. In humans and rodents, dietary EPA/DHA supplementation causes a profound shift of the endogenous CYP-eicosanoid profile from AA- to EPA- and DHA-derived metabolites, increasing, in particular, the plasma and tissue levels of 17,18-epoxyeicosatetraenoic acid (17,18-EEQ) and 19,20-epoxydocosapentaenoic acid (19,20-EDP). Based on preclinical studies, these omega-3 epoxyeicosanoids display cardioprotective, vasodilatory, anti-inflammatory, and anti-allergic properties that contribute to the beneficial effects of n-3 LC-PUFAs in diverse disease conditions ranging from cardiac disease, bronchial disorders, and intraocular neovascularization, to allergic intestinal inflammation and inflammatory pain. Increasing evidence also suggests that background nutrition as well as genetic and disease state-related factors could limit the response to EPA/DHA-supplementation by reducing the formation and/or enhancing the degradation of omega-3 epoxyeicosanoids. Recently, metabolically robust synthetic analogs mimicking the biological activities of 17,18-EEQ have been developed. These drug candidates may overcome limitations of dietary EPA/DHA supplementation and provide novel options for the treatment of cardiovascular and inflammatory diseases.
Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0163725817302668
Arachidonic Acid-metabolizing Cytochrome P450 Enzymes Are Targets of ω-3 Fatty Acids*
Abstract
Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) protect against cardiovascular disease by largely unknown mechanisms. We tested the hypothesis that EPA and DHA may compete with arachidonic acid (AA) for the conversion by cytochrome P450 (CYP) enzymes, resulting in the formation of alternative, physiologically active, metabolites. Renal and hepatic microsomes, as well as various CYP isoforms, displayed equal or elevated activities when metabolizing EPA or DHA instead of AA. CYP2C/2J isoforms converting AA to epoxyeicosatrienoic acids (EETs) preferentially epoxidized the ω-3 double bond and thereby produced 17,18-epoxyeicosatetraenoic (17,18-EEQ) and 19,20-epoxydocosapentaenoic acid (19,20-EDP) from EPA and DHA.We found that these ω-3 epoxides are highly active as antiarrhythmic agents, suppressing the Ca2+-induced increased rate of spontaneous beating of neonatal rat cardiomyocytes, at low nanomolar concentrations. CYP4A/4F isoforms ω-hydroxylating AA were less regioselective toward EPA and DHA, catalyzing predominantly ω- and ω minus 1 hydroxylation. Rats given dietary EPA/DHA supplementation exhibited substantial replacement of AA by EPA and DHA in membrane phospholipids in plasma, heart, kidney, liver, lung, and pancreas, with less pronounced changes in the brain. The changes in fatty acids were accompanied by concomitant changes in endogenous CYP metabolite profiles (e.g. altering the EET/EEQ/EDP ratio from 87:0:13 to 27:18:55 in the heart). These results demonstrate that CYP enzymes efficiently convert EPA and DHA to novel epoxy and hydroxy metabolites that could mediate some of the beneficial cardiovascular effects of dietary ω-3 fatty acids.
JBC : Journal of Biological Chemistry
http://m.jbc.org/content/285/43/32720.long
Omega-3脂肪酸及其代谢产物在哮喘和过敏性疾病中的作用
a 日本神奈川县理研综合医学中心代谢组学实验室
b 日本神奈川县横滨市立大学医学生命科学研究科
摘要
Omega-3脂肪酸,二十二碳六烯酸(DHA)和二十碳五烯酸(EPA)天然存在于鱼油中,通常被认为是抗炎营养素,对包括哮喘和过敏在内的炎性疾病具有保护作用。这些作用的机制仍然是未知的,但对其潜在的治疗应用非常感兴趣。进行了大量的流行病学和观察性研究,调查了在怀孕,哺乳期,婴儿期,儿童期和成年期摄入鱼或补充omega-3脂肪酸对哮喘和过敏结果的影响。它们主要表明了保护作用,并暗示了现代化饮食中鱼油摄入减少与哮喘或其他过敏性疾病患者人数增加之间存在因果关系。特殊的促分解介体(SPM:保护素protectins,resolvins和maresins)是通过几种酶促反应从omega-3脂肪酸(例如EPA和DHA)生成的。这些介质反调节呼吸道嗜酸性炎症并促进体内炎症的消退。几篇报道表明,SPM的生物合成受到损害,特别是在严重的哮喘中,这表明肺部的慢性炎症可能是由分解素缺陷引起的。本文着重介绍Omega-3脂肪酸的有益方面,并提供有关其生物活性代谢物(包括分辨素和保护素)的最新见解。
Role of omega-3 fatty acids and their metabolites in asthma and allergic diseases
a
Laboratory for Metabolomics, RIKEN Center for Integrative Medical Sciences, Kanagawa, Japan
b
Graduate School of Medical Life Science, Yokohama City University, Kanagawa, Japan
Received 1 August 2014, Revised 20 August 2014, Accepted 21 August 2014, Available online 27 October 2014.
Abstract
Omega-3 fatty acids, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), are found naturally in fish oil and are commonly thought to be anti-inflammatory nutrients, with protective effects in inflammatory diseases including asthma and allergies. The mechanisms of these effects remain mostly unknown but are of great interest for their potential therapeutic applications. Large numbers of epidemiological and observational studies investigating the effect of fish intake or omega-3 fatty acid supplementation during pregnancy, lactation, infancy, childhood, and adulthood on asthmatic and allergic outcomes have been conducted. They mostly indicate protective effects and suggest a causal relationship between decreased intake of fish oil in modernized diets and an increasing number of individuals with asthma or other allergic diseases. Specialized pro-resolving mediators (SPM: protectins, resolvins, and maresins) are generated from omega-3 fatty acids such as EPA and DHA via several enzymatic reactions. These mediators counter-regulate airway eosinophilic inflammation and promote the resolution of inflammation in vivo. Several reports have indicated that the biosynthesis of SPM is impaired, especially in severe asthma, which suggests that chronic inflammation in the lung might result from a resolution defect. This article focuses on the beneficial aspects of omega-3 fatty acids and offers recent insights into their bioactive metabolites including resolvins and protectins.Role of omega-3 fatty acids and their metabolites in asthma and allergic diseases - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1323893014000100
Prostaglandins Other Lipid Mediat. Author manuscript; available in PMC 2015 Oct 1.
ω-3 polyunsaturated fatty acids-derived lipid metabolites on angiogenesis, inflammation and cancer
1Department of Food Science, University of Massachusetts-Amherst, Amherst, MA 01003
2Center for Vascular Biology Research and Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115
3Department of Biomedical Engineering, University of California, Davis, CA 95616
4Department of Entomology and Comprehensive Cancer Center, University of California, Davis, CA 95616
Abstract
Epidemiological and pre-clinical studies support the anti-tumor effects of ω-3 PUFAs; however, the results from human trials are mixed, making it difficult to provide dietary guidelines or recommendations of ω-3 PUFAs for disease prevention or treatment. Understanding the molecular mechanisms by which ω-3 PUFAs inhibit cancer could lead to better nutritional paradigms and human trials to clarify their health effects. The ω-3 PUFAs exert their biological activities mainly through the formation of bioactive lipid metabolites. Here we discuss the biology of cyclooxygenase, lipoxygenase and cytochrome P450 enzymes-derived ω-3-series lipid metabolites on angiogenesis, inflammation and cancer.
Keywords: ω-3 polyunsaturated fatty acids, cyclooxygenase, lipoxygenase, cytochrome P450
Introduction
In the U.S., there are ~1,665,540 new cases and ~585,720 deaths from cancers expected in 2014. It is estimated that 30% of cancer in developed countries are diet-related1. Human studies support that dietary ω-3 polyunsaturated fatty acids (PUFAs), in particular eicosapentaenoic acid (EPA, 20:5ω-3) and docosahexaenoic acid (DHA, 22:6ω-3), may reduce cancer risks2–10; while ω-6 PUFAs, such as linoleic acid (18:2ω-6) and arachidonic acid (ARA, 20:4ω-6), could promote tumor progression11–16. For example, in the VITamins And Lifestyle (VITAL) Cohort, current use of fish oil, but not other dietary supplements, was associated with reduced risks of breast cancer10. The breast cancer patients with high tissue levels of DHA also respond better to chemotherapeutic drugs17. In contrary to the effects of ω-3 PUFAs, studies carried out in Mexico, Sweden, Singapore and China showed that dietary intake of ω-6 PUFAs was associated with increased breast cancer risks11, 14, 16, 18. This is important because current western diet contains 20–30 times more ω-6 than ω-3 PUFAs19. Validation of the anti-tumor effects of ω-3 PUFAs will have significant impact on public health. However, there are inconsistent results from human studies, which showed that ω-3 PUFAs had no effects20, 21 or detrimental effects on cancers22, making it difficult provide dietary guidelines or recommendations of ω-3 PUFAs for cancer prevention or treatment.
The inconsistent results could be due to many reasons. In previous ω-3 human trials, different types, doses and treatment durations of ω-3 PUFAs were used, making it difficult to compare and analyze the results of different ω-3 studies. Besides the heterogeneity of experimental designs, the mixed results could be, in part, due to inter-individual variations to metabolize ω-3 PUFAs to generate bioactive ω-3 lipid mediators (LMs)23–26. The ω-3 PUFAs act in part via formation of certain LMs such as cyclooxygenase (COX)-derived prostaglandin E3 (PGE3)27, lipoxygenase (LOX)-derived 4-hydroxy-docosahexaenoic acid (4-HDHA)28, cytochrome P450 (CYP)-derived epoxydocosapentaenoic acids (EDPs)29, as well as unique ω-3 lipid autacoids such as resolvins and protectins30, 31. These ω-3 LMs serve as autocrine and/or paracrine mediators to regulate inflammation and homeostasis32; many of these mediators are short-lived, locally produced and locally acting in response to cellular stimuli, followed with degradation or metabolism to maintain homeostasis33. The polymorphisms in the genes encoding ω-3 metabolism enzymes could affect the ω-3 metabolism, leading to different levels of ω-3/ω-6 LMs in tissues and varied biological responses to ω-3 supplementation. Indeed, recent research showed that there is a high degree of inter-individual variability in metabolizing ω-3 PUFAs to generate LMs upon dietary intake of ω-3 PUFAs23. Clearly, it is important to elucidate the specific lipid metabolizing enzymes and metabolites required for the anti-tumor effects of ω-3 PUFAs. The identified enzymes and metabolites could serve as biomarkers to screen the sub-populations which are most likely to respond to ω-3 PUFAs, or develop personalized doses for ω-3 supplementation. In addition, the bioactive ω-3 LMs could serve as biotemplates to design more potent and safer therapeutic drugs32.
The enzymatic metabolism of ω-6 ARA leads to formation of predominately though not exclusively pro-inflammatory and pro-tumorigenic LMs, which have been shown to play a central role in tumor progression34–36. Compared with the ω-6-series LMs, the roles of ω-3-series LMs in angiogenesis, inflammation and cancer are less known. The ω-3 LMs were thought to be less-active mediators, while emerging evidences support that certain classes of ω-3 LMs have potent effects to modulate inflammation, angiogenesis and tumorigenesis. In this review we will discuss the biology of COX, LOX and CYP-derived ω-3 LMs on angiogenesis, inflammation and cancer. The ω-3 PUFAs also act as precursors for biosynthesis of unique lipid autacoids such as resolvins and protectins, which also regulate multiple cellular processes including inflammation, angiogenesis and cancer31. These ω-3 autacoids have been discussed in several recent reviews30–32 and will not be discussed here.
ω-3 and ω-6 PUFAs
The ω-3 and ω-6 PUFAs are polyunsaturated fatty acids which have a double bond at the third and the sixth carbon atom from the end of the carbon chain respectively. Linoleic acid (LA, 18:2ω-6), which is an essential fatty acid and is highly abundant in common vegetable oils, is the major source of dietary ω-6 PUFA in the western diet. The average adult intake of LA in the U.S. ranges from 12–17 g/day for men and 9–11 g/day for women. LA can be further converted to arachidonic acid (ARA, 20:4ω-6), which is an important PUFA involved in cell signaling by generation of ω-6-series LMs (termed eicosanoids). Most research of ω-3 PUFAs have focused on EPA and DHA. Food sources of EPA and DHA include fish and fish oil supplements. Fish oil is among the most popular dietary supplements in United States. It is the most popular nonvitamin/nonmineral supplements in adults and the second most popular in children. In addition, major food companies are increasingly adding ω-3 PUFAs to various foods as value-added ingredients. FDA has approved Lovaza®, a mixture of EPA and DHA ethyl ester, as a prescription drug to treat hypertriglyceridemia (high levels of triglycerides). Another drug Vascepa® that is a pure EPA ethyl ester is currently seeking approval from FDA targeting hypertriglyceridemia. Recent technology development, using transgenic yeast, algae or supercritical carbon dioxide separation, allows the industry to prepare large-scale of highly purified EPA or DHA. Diets with a ω-6-to-ω-3 PUFA ratio of 1 are recommended by nutritionists, however, current western diets have a ratio of 20–30 due to too much consumption of LA and too low consumption of ω-3 PUFAs19. Due to the high dietary intake of ω-6 PUFAs, ARA is among the most abundant PUFAs in most tissues. The ω-3 PUFAs are highly enriched in retina and brain tissues, mainly in the form of DHA, the tissue levels of EPA are usually low37.
Molecular mechanisms of ω-3 PUFAs on cancer
Recent research shows that the lipid signaling is deregulated in tumor tissues, leading to increased production of pro-inflammatory and pro-tumorigenic eicosanoids from ARA in the tumor microenvironment. For example, the expressions of delta-6-desaturase which is a rate-limiting enzyme to convert linoleic acid to ARA, as well as phospholipases which release ARA from membrane phospholipids to initiate the biosynthesis of eicosanoids, are significantly up-regulated in tumor tissues38, 39. The expressions of pro-tumorigenic lipid metabolizing enzymes, such as COX-2, 5-LOX and CYP epoxygenases, have been reported to be up-regulated in tumors34, 36; while the anti-tumorigenic enzymes such as 15-LOX-1 are down-regulated34. Together, these changes lead to a supportive microenvironment to support tumor progression.
An important mechanism for the health-promoting effects of ω-3 PUFAs is that they suppress the metabolism of ARA to generate eicosanoids40. Upon dietary consumption, ω-3 PUFAs, including EPA and DHA, are incorporated into the membrane phospholipids at the expense of ω-6 ARA. Upon cellular stimulation, the incorporated ω-3 and ω-6 PUFAs are enzymatically released to generate intracellular free fatty acids (FFAs), which are rapidly metabolized by COX, LOX and CYP enzymes to generate ω-3-series and ω-6-series LMs. EPA and DHA inhibit the formation of ARA-derived ω-6-series LMs via multiple mechanisms, including reduced release of ARA from membrane phospholipids, inhibition of the enzymatic activities of the metabolizing enzymes, and direct competition with ARA for the enzymatic conversions. Besides inhibition of enzymatic metabolism of ARA, EPA and DHA also serve as alternative substrates of the lipid metabolism enzymes, leading to increased formation of ω-3-series LMs. Some of these mediators have potent effects to inhibit inflammation, angiogenesis and cancer28, 29, and will be discussed below.
COX-derived ω-3 LMs in angiogenesis, inflammation and cancer
The COX-2 pathway plays a critical role in angiogenesis, inflammation and cancer (Figure 1)34. The COX-2 metabolite of ARA, prostaglandin E2 (PGE2), is widely known to promote inflammation, neovascularization, primary tumor growth and metastasis. Increased expression of COX-2 has been observed in many tumor tissues34. COX-2 inhibitors, including non-steroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase (COX)-2 selective inhibitors (coxibs), have been shown to reduce cancer risks41, 42. However, the life-threatening cardiovascular risks as well as other adverse effects induced by long-term and high-dose use of these drugs have jeopardized their therapeutic applications43, 44.
Figure 1
The ω-3 PUFAs reduce cancer risks via COX-2-dependent mechanisms. Human studies support that polymorphisms in the genes encoding COX-2 modulate the anti-tumor effects of ω-3 PUFAs45, 46. EPA and DHA reduced the formation of COX-2-derived PGE2, which contributes to the beneficial effects of ω-3 PUFAs40. DHA is widely believed not to be a substrate of COX enzymes, although it has been reported that DHA is converted by COX-2 to form hydroxyl DHA, which is further metabolized to generate electrophilic LMs with anti-inflammatory actions47. EPA has been shown to be an alternative substrate of COX-2, which converts it to the ω-3-series of prostaglandin termed prostaglandin E3 (PGE3) and other LMs. Compared with ARA, EPA is a poor substrate for COX enzymes48, 49. Previous studies have shown that PGE3 has less detrimental or even beneficial effects on cancer, the biology of PGE3 was discussed in a recent review27. PGE3 has been shown to have less pro-inflammatory and pro-angiogenic effects than PGE2. In NIH 3T3 fibroblasts, PGE2 induced cell proliferation while PGE3 had no effect in the same dose range. Both PGE2 and PGE3 induced COX-2 transcription in NIH 3T3 cells and IL-6 production in RAW 264.7 cells, but PGE3 had a significantly reduced pro-inflammatory effect50. In human endothelial cells, PGE2 further increased Ang2 expression induced by a combination of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF), while PGE3 had no such effect51. PGE3 has also been shown to inhibit cancer cell proliferation and invasion. At a dose of 1 µM, PGE2 had no effect, while PGE3 inhibited proliferation of lung cancer A549 cells52. PGE3 also inhibited proliferation and induced apoptosis in B16F10 melanoma cells via mechanisms involving increased expression of PTEN53. In a Matrigel-based Boyden chamber assay, PGE3 inhibited cell invasion in the highly aggressive brain-metastatic melanoma 70W cell line54. The cellular receptors of PGE3 have not been confirmed; some studies have shown that PGE3 binds to the same receptors as PGE2 with reduced affinity and potency27, 55. Further, a recent study showed that Δ12-prostaglandin J3 (Δ12-PGJ3), which is a novel COX-derived metabolite of EPA, potently inhibited progression of leukemia in animal models56.
LOX-derived ω-3 LMs in angiogenesis, inflammation and cancer
The metabolism of PUFAs by LOX enzymes leads to the formation of leukotrienes and hydroxyl fatty acids (Figure 2)33. The LOX pathway in cancer is more complicated as there are multiple isoforms of LOX enzymes. It is generally believed that 5-LOX and 12-LOX and their metabolites promote cancer, while 15-LOX-1 and 15-LOX-2 have anti-tumor effects34, 57, 58. 5-HETE, a 5-LOX metabolite of ARA, has been shown to induce angiogenesis, inflammation, and tumor progression. Pharmacological inhibitors of the 5-LOX enzyme have been shown to suppress tumor progression in animal models34. Since both COX-2 and 5-LOX are up-regulated in tumor tissues, dual inhibition of COX-2 and 5-LOX has been shown to cause enhanced anti-tumor effect59, 60.
Figure 2
The 5-LOX metabolites of ARA are generally believed to promote inflammation and angiogenesis34. Surprisingly, 5-LOX was recently shown to play a central role in the anti-angiogenic effect of DHA via formation of an anti-angiogenic metabolite 4-hydroxydocosahexaenoic acid (4-HDHA)28. Dietary supplementation of DHA has been shown to suppress retinal angiogenesis in an oxygen-induced retinopathy model61. Transgenic deletion of 5-LOX greatly reduced the anti-angiogenic effect of DHA, while deletion of COX-1/2 or 12/15-LOX had little effect, suggesting a central role of 5-LOX in the anti-angiogenic effect of DHA28. The 5-LOX enzyme mediates the anti-angiogenic effect of DHA via formation of 4-HDHA, which inhibited angiogenesis through a PPAR-γ-dependent mechanism28. Considering the importance of angiogenesis in tumor progression, it would be important to study the effect of the 4-HDHA pathway in tumor angiogenesis and associated tumor progression and metastasis. EPA and DHA are also substrates of 15-LOX, which convert them to 15-hydroxyeicosapentaenoic acid (15-HEPE) and 17-hydroxy-docosahexaenoic acid (17-HDHA) respectively62, 63. Both 15-HEPE and 17-HDHA have been shown to inhibit the enzymatic activity of 5-LOX (a major enzyme to generate pro-inflammatory LMs), suggesting their potential anti-inflammatory effects62, 64. Further animal studies demonstrate the potent anti-inflammatory effects of 17-HDHA in colitis models. In a dextran sulfate sodium (DSS)-or 2,4,6-trinitrobenzene sulfonic acid-induced colitis model, treatment with 0.1–1 µg/animal/day 17-HDHA significantly reduced the disease activity index, body weight loss, colonic damage and polymorphonuclear infiltration in both colitis models. 17-HDHA also reduced levels of pro-inflammatory cytokines such as TNF-α, IL-1β, MIP-2, and CXCL1/KC and mRNA expression of NF-κB and adhesion molecules in colon tissue65. In another study, 17-HDHA also suppressed DSS-induced colitis in mice. In murine macrophage RAW264.7 cells, 17-HDHA increased phagocytosis in macrophages and promoted polarization towards an anti-inflammatory M2 phenotype66. These studies demonstrate the potent anti-inflammatory effect of 17-HDHA, indicating a critical role of the 15-LOX enzyme in the biological activities of DHA. 17-HDHA is a precursor for the biosynthesis of resolvins, also shown to have potent anti-inflammatory effects in recent reviews25, 26. The 15-LOX metabolites of EPA and DHA have also been shown to directly inhibit cancer cell proliferation. EPA-derived 15-HEPE inhibited the formation of PGE2 and 5-HETE, as well as cancer cell proliferation in PC-3 and LNCaP cells67. DHA-derived 17-HDHA inhibited the proliferation of prostate cancer cells (PC-3, LNCaP and DU145) at doses much lower than the corresponding metabolite of ARA (15-HETE) and DHA68. The other 15-LOX metabolites of DHA, including 17-hydroperoxy-, 10,17-dihydroxy- and 7,17-dihydroxy-DHA, also inhibited cell proliferation via mechanisms involving activation of PPAR-γ and syndecan-1 signaling in prostate cancer cells68. The 15-LOX-mediated metabolism is required for the effect of DHA to induce syndecan-1 signaling and apoptosis in prostate cancer cells69. In another study, 17-hydroperoxy-DHA inhibited cell proliferation in neuroblastoma cells with an IC50 of 3–6 µM, compared with 12–15 µM for DHA70. Human studies support a critical role of LOX in the effects of ω-3 PUFAs. Carriers of variant 5-LOX genotypes have been shown to have increased risks for inflammation and atherosclerosis compared with the carriers of common allele. Dietary intake of ω-6 PUFAs increased, while intake of ω-3 PUFAs decreased, the risk of atherosclerosis only in the carriers of variant alleles but not in the common alleles71. This study suggests that 5-LOX may be a biomarker to distinguish people who respond to the anti-atherosclerotic benefits of ω-3 PUFAs from non-responders71. In terms of cancer, Wang et al. showed that there is a significant interaction of the polymorphism in the genes encoding 5-LOX-activating protein (ALOX5AP) and dietary intake of ω-6 PUFA LA in terms of breast cancer risks in a population-based case-control study in San Francisco bay area12. Among the women with high dietary intake of LA, carrying the ALOX5AP −4900 AA genotype was associated with higher risk of breast cancer compared with other genotypes. No such correlations were observed in women consuming low levels of LA12. It would be interesting to test whether dietary intervention with ω-3 PUFAs would inhibit breast cancer progression in this high-risk sub-population. These two human studies demonstrate a strong diet-gene interaction, supporting the critical importance of PUFA metabolism enzyme in the biological activity of ω-3 and ω-6 PUFAs.
CYP-derived ω-3 LMs in angiogenesis, inflammation and cancer
The CYP pathway has two branches, converting ARA to epoxyeicosatrienoic oacids (EETs) by CYP epoxygenases (largely CYP2C and CYP2J) and 20-hydroxyeicosatetraenoic acid (20-HETE) by CYP ω-hydroxylase (largely CYP4A and CYP4F)72. EETs have been shown to have an array of beneficial actions, including anti-inflammatory, vasodilatory, anti-hypertensive, renal-protective, cardio-protective, and tissue regenerative actions. Pharmacological inhibitors of soluble epoxide hydrolase (sEH, the major enzyme to degrade EETs) which stabilize and increase EETs, are being developed to treat many human disorders72. 20-HETE has been shown to have predominately detrimental effects, inducing inflammation, vasoconstriction, hypertension, and cardiovascular problems. Pharmacological inhibitors of 20-HETE biosynthesis have been shown to alleviate many disease states in animal models73.
The roles of EETs and 20-HETE in cancer have not been well studied. EETs are mildly pro-angiogenic, stimulating endothelial cell proliferation, migration, invasion and tube formation72, as well as tissue regeneration74. Due to the pro-angiogenic actions, increased levels of EETs stimulate primary tumor growth and metastasis in implantable and spontaneous transgenic murine tumor models75. On the other hand, EETs are anti-inflammatory, where increased level of EETs inhibited colon cancer progression and tumor-associated inflammation in inflammation-driven colon cancer models76, 77. 20-HETE has been shown to increase primary tumor growth in murine tumor models, in part via induction of tumor angiogenesis and inflammation. Pharmacological inhibitors targeting 20-HETE biosynthesis have been shown to inhibit tumor progression in animal models35.
EPA and DHA have been shown to be highly efficient alternative substrates of CYP epoxygenases, leading to the formation of epoxygenated ω-3 PUFAs termed epoxyeicosatetraenoic acids (EEQs) and epoxydocosapentaenoic acids (EDPs) respectively72, 78. CYP epoxygenases selectively catalyze the epoxidation of the terminal double bond of ω-3 PUFAs, leading to the predominate formation of 17,18-EEQ from EPA and 19,20-EDP from DHA78–81. Compared with other DHA epoxide regioisomers, 19,20-EDP is the poorest substrate of sEH, which increases the relative proportion of 19,20-EDP in tissues82. Many studies have shown that ω-3 supplementation significantly increased levels of EEQs and EDPs in animal and human plasma and tissues78, 83–87.
EEQs and EDPs have similar or more potent effects for vasodilation, anti-inflammation and analgesia than EETs. EPA-derived 17,18-EEQ, as well as ARA-derived 14–15-EET, inhibited TNF-α-induced inflammation in human bronchi via NF-κB- and PPAR-γ-related mechanisms88, 89. In a carrageenan-induced inflammatory pain model in rats, all epoxygenated PUFAs (EETs, EEQs and EDPs) inhibited inflammatory pain, while the effects of EEQs were less potent than those of EETs and EDPs82. Also, a 12-LOX-derived metabolite of 17,18-EEQ, 12- hydroxy-17,18-epoxyeicosatetraenoic acid (12-OH-17,18-EEQ), inhibited LTB4-induced neutrophil chemotaxis and polarization in vitro at a low nM range90. In terms of vasodilation, EDPs are among the most potent vasodilators ever discovered (dilation EC50 = 0.5–24 pM)91. Direct treatment of EDPs suppressed Angiotensin II-induced hypertension in mice92. The CYP-mediated formation of EDPs has been hypothesized to contribute to the anti-hypertensive effects of DHA, as shown by transgenic deletion of CYP1A1 (a CYP epoxygenase enzyme) which attenuated the anti-hypertensive effects of DHA93.
Opposite to the pro-angiogenic effects of EETs, the ω-3-series fatty acid epoxides (EEQs and EDPs) have been shown to inhibit angiogenesis. 17,18-EEQ, but not other EEQ regioisomers, inhibited cell proliferation in the immortalized endothelial cell line bEND.3 at a dose of 10 µM, while EETs at the same dose range showed opposite effects to increase cell proliferation in bEND.3 cells94. This study suggests a potential anti-angiogenic effect of EEQs, however, more studies are needed to characterize their effects on angiogenesis, in particular in animal models of neovasculization. Our recent study showed that EDPs potently inhibited angiogenesis, primary tumor growth and metastasis29. In a Matrigel plug assay in mice, all EDP regioisomers (except 4,5-EDP which is chemically unstable) inhibited VGEF-induced angiogenesis. 19,20-EDP, which is a major EDP isomer in tissues, inhibited VEGF-induced angiogenesis with an EC50 value of 0.3 µg/animal, suggesting its potent anti-angiogenic effect. 19,20-EDP also suppressed basic fibroblast growth factor (bFGF)-induced angiogenesis in mice, suggesting a potential broad-spectrum anti-angiogenic effect. In human endothelial cells, 19,20-EDP inhibited endothelial tube formation, migration, and production of matrix metalloproteinases, via a mechanism involving VEGF receptor 2 (VEGFR2)-dependent signaling. Given that tumor metastasis causes 90% of human cancer deaths, anti-metastatic agents are very important therapeutic agents95. We demonstrated that two EDP regioisomers (i.e.,16,17-EDP and 19,20-EDP, dose = 0.05 mg/kg/day), when stabilized in circulation by co-administration of a selective sEH inhibitor, suppressed ~70% of tumor metastasis in mice29. In fact, EDPs are the first fatty acid metabolites to be shown to have anti-metastatic activities. Moreover, the stabilized EDP also inhibited Met-1 breast tumor growth (a highly aggressive triple-negative breast cancer model) in mice by ~70%29. Our findings demonstrate potent effects of EDPs on tumor angiogenesis, however, two recent studies showed that EDPs did not impact angiogenesis in retinal angiogenesis models96, 97. More studies are needed to characterize the effects and mechanisms of ω-3-series epoxides and diols on angiogenesis in different disease models as it is likely that the effects of these LMs may be disease- and tissue-specific.
Future Directions
The ω-3 PUFAs are among the most intensively studied nutritional compounds, as demonstrated by epidemiological and pre-clinical studies. However, after decades of ω-3 PUFA research, many of the health claims of ω-3 PUFAs remain controversial and have therefore had limited impact in disease prevention and treatment. The mixed results obtained with the use of ω-3 PUFAs in human trials may result in part from failing to recognize the importance of ω-3 PUFA metabolism. As we have discussed in this review, the enzymatic metabolism of ω-3 PUAFs generates ω-3-series LMs, which have potent actions to regulate inflammation, angiogenesis and tumor progression. The ω-3 LMs, rather than the parent ω-3 PUFAs (EPA or DHA), are more likely to be the ultimate bioacitve species interacting with cellular targets to exert the biological effects of ω-3 PUFA supplementation. However, the vast majority of previous ω-3 PUFA research has focused on tissue levels of ω-3 PUFAs, instead of ω-3 LMs, as biomarkers to establish the nutritional or therapeutic effects of ω-3 PUFAs.
As discussed above, it is critical to elucidate the lipid metabolizing enzymes and metabolites which are required for the biological effects of ω-3 PUFAs. We expect that increased dietary intake of ω-3 PUFAs is associated with reduced cancer risks among those with genetic variant that result in increased activity of the required ω-3 metabolizing enzymes. Recently, the development of transgenic animal models, LC-MS/MS-based lipidomics and standards for LMs has greatly facilitated the study of LMs. In the past decade, transgenic animal models with deletion or over-expression of lipid metabolism enzymes (COX-2, COX-1, 5-LOX, 12/15-LOX, CYP epoxygenase, CYP ω-hydroxylase, sEH, etc) have been developed and many of them are commercially available. Multiple laboratories in the U.S. have developed LC-MS/MS-based lipidomics methods, which can systematically analyze >100 LMs derived from ω-3/ω-6 PUFAs using minimal plasma or tissues98, 99. Many ω-3/ω-6 LM have been chemically synthesized and some of these LMs are commercially available, allowing cell culture and animal experiments to directly study their effects and mechanisms. These resources will greatly help to elucidate the roles of specific lipid metabolism pathway(s) and metabolite(s) in the effects of ω-3 and ω-6 PUFAs in different disease states. The knowledge obtained in pre-clinical models must be further verified in human trials. The identified lipid metabolism enzymes or metabolites could be used as biomarkers to distinguish "ω-3 responders" from "non-responders", leading to targeted human trials. Such knowledge could also help to provide personalized dietary recommendations. For example, sub-populations carrying certain genotypes of 5-LOX or 5-LOXAP could be educated to optimize their diet12, 71. An in-depth understanding of the molecular mechanisms of PUFAs, together with utilization of nutrigenomic and metabolomic approaches, could lead to targeted nutritional paradigms to better understand the metabolic individuality and nutrition effects of ω-3 PUFAs on human health100.
Figure 3ω-3 polyunsaturated fatty acids-derived lipid metabolites on angiogenesis, inflammation and cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4306447/
UC Davis HealthNewsroom Fatty acid metabolite shows promise against cancer in ...
NEWS | April 2, 2013
Fatty acid metabolite shows promise against cancer in mice
UC Davis discovery demonstrates mechanism in dietary omega-3 fatty acids (fish oils)
(SACRAMENTO) —
A team of UC Davis scientists has found that a product resulting from a metabolized omega-3 fatty acid helps combat cancer by cutting off the supply of oxygen and nutrients that fuel tumor growth and spread of the disease.
The scientists report their discovery in the Proceedings of the National Academy of Sciences (PNAS). The groundbreaking study was a collaboration among multiple UC Davis laboratories and Harvard University.
The metabolite is epoxy docosapentaenoic acid (EDP), an endogenous compound produced by the human body from the omega-3 fatty acid named docosahexaenoic acid (DHA), which is found in fish oil and breast milk. In animal studies, the UC Davis scientists found that EDP inhibits angiogenesis, the formation of new blood vessels in the body.
Tumors grow and spread by hijacking the normal biological process of angiogenesis, which plays a role in wound repair as well in growth and development. The UC Davis researchers determined that by inhibiting angiogenesis, EDP reduces the growth and spread (metastasis) of tumors in mice. The research provides the first scientific evidence about EDP’s potent anti-cancer, anti-metastatic effects.
EDP works by a different mechanism than many current anti-cancer drugs that block angiogenesis.
“Our investigation opens up a new understanding of the pathways by which omega-3 fatty acids exert their biologic effects,” said Guodong Zhang, the lead author of the article and a postdoctoral researcher in the laboratory of Bruce Hammock in the Department of Entomology and the UC Davis Comprehensive Cancer Center.
The researchers said that future studies hopefully will determine that stabilized EDP can be safely and effectively combined with other current anti-angiogenesis drugs in the treatment of cancer.
“As far as we know, EDPs are the first signaling lipids that have been discovered to have such potent anti-cancer effects. Researchers may be able to use EDPs as structural targets to develop stable analogs as anti-cancer agents,” Zhang said.
“The study by Zhang and colleagues has uncovered a previously unrecognized anti-cancer effect of omega-3 fatty acids, which are an important lipid component of diets that have been developed to prevent heart disease and cancer,” said Jonathan R. Lindner, professor of medicine at Oregon Health & Sciences University.
“The authors have demonstrated that metabolites of these lipids can act to suppress the growth of new blood vessels that are necessary to feed tumor growth,” added Lindner, who was not involved in the study. “By shutting off a tumor’s blood supply, these compounds can act to dramatically slow tumor growth and prevent spread. The results from this study suggest that new drug strategies for fighting cancer could emerge from knowledge of how the body uses nutrition to promote health.”
The EDPs are broken down in the body by the enzyme soluble epoxide hydrolase (sEH). In previous research, Hammock’s lab showed that inhibitors of the sEHI enzyme (sEHIs) help to normalize physiological activity. In the current study, UC Davis researchers determined that the addition of sEHI stabilized EDP in circulating blood thereby producing EDPs’ anti-tumor effects. The FDA-approved anti-cancer drugs sorafenib and regorafenib are also potent sEHIs.
“It may be possible to improve the efficacy of these anti-cancer drugs by combining them with a diet high in omega-3 and low in omega-6 fatty acids,” Hammock said.
The researchers also found that a metabolite of arachidonic acid (ARA), an omega-6 fatty acid, has the opposite effect of EDP. The ARA metabolite, epoxyeicosatrienoic acids (EETs), slightly increases angiogenesis and tumor progression in mice.
“There is no free lunch,” said Katherine W. Ferrara, professor in the UC Davis Department of Biomedical Engineering. “The EETs encourage wound healing, while the EDPs block the growth and metastasis of solid tumors.
“Our results designate EDPs and EETs as unique mediators of an angiogenic switch to regulate tumorigenesis,” Ferrara said. "They also implicate a novel mechanistic linkage between omega-3 and omega-6 fatty acids and cancers."
UC Davis scientists determined that EDP starves tumors by inhibiting vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 (FGF-2)-induced angiogenesis in mice. In laboratory cultures, EDP also suppresses the endothelial cell migration needed for new blood vessels.
Thus, EDP-based angiogenesis inhibitors offer an advantage over angiogenesis inhibitors that target the VEGF-VEGFR2 pathway. The drugs that target the VEGF-VEFGFR2 pathway increase patients’ risk for high blood pressure.
Because EDPs widen the blood vessels, a medication based on the UC Davis researchers’ discovery should not increase the patient’s risk for high blood pressure.
Harvard researchers Mark Kieran and Dipak Panigrahy conducted the metastasis studies. The in vivoimaging work that allowed the scientists to monitor tumors in living mice was done in Ferrara’s UC Davis laboratory.Fatty acid metabolite shows promise against cancer in mice
https://health.ucdavis.edu/publish/news/newsroom/7671Study: Fatty Acid Metabolite Shows Promise Against Cancer
https://www.nutritioninsight.com/news/Study-Fatty-Acid-Metabolite-Shows-Promise-Against-Cancer.html
Omega-3 Fatty Acids Reduce Inflammation through Cannabinoids
March 13, 2019 by Kim Stewart
The chemical compounds called cannabinoids, found in cannabis and hemp, are also produced naturally in the body from omega-3 fatty acids. A new study in animal tissue reveals the cascade of chemical reactions that convert omega-3 fatty acids into cannabinoids that have anti-inflammatory benefits. The findings are published in the Proceedings of the National Academy of Sciences.
Foods such as meat, eggs, fish and nuts contain omega-3 and omega-6 fatty acids, which the body converts into endocannabinoids – cannabinoids that the body produces naturally, said Aditi Das, a University of Illinois professor of comparative biosciences and biochemistry, who led the study.
“Cannabinoids in marijuana and endocannabinoids produced in the body can support the body’s immune system and therefore are attractive targets for the development of anti-inflammatory therapeutics,” said Das.
Cannabinoids bind to two types of cannabinoid receptors in the body – one that is found predominantly in the nervous system and one in the immune system, Das said. “Some cannabinoids, such as THC in marijuana or endocannabinoids can bind to these receptors and elicit anti-inflammatory and anti-pain action,” she said.
Clinical studies suggest that diets rich in ω-3 polyunsaturated fatty acids (PUFAs) provide beneficial anti-inflammatory effects, in part through their conversion to bioactive metabolites. This study reports on the endogenous production of a previously unknown class of ω-3 PUFA–derived lipid metabolites that originate from the crosstalk between endocannabinoid and cytochrome P450 (CYP) epoxygenase metabolic pathways. The ω-3 endocannabinoid epoxides are derived from docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) to form epoxyeicosatetraenoic acid-ethanolamide (EEQ-EA) and epoxydocosapentaenoic acid-ethanolamide (EDP-EA), respectively.
“Our team discovered an enzymatic pathway that converts omega-3-derived endocannabinoids into more potent anti-inflammatory molecules that predominantly bind to the receptors found in the immune system,” Das said. “This finding demonstrates how omega-3 fatty acids can produce some of the same medicinal qualities as marijuana, but without a psychotropic effect.”
Epidemiological evidence suggests that a diet rich in the ω-3 fatty acids (ω-3 FAs) docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) promotes beneficial cardiovascular, neurological, and anti-inflammatory health effects. The biochemical mechanisms facilitating these beneficial effects are yet to be fully elucidated. Mounting evidence, such as what is presented in this study, suggests that these actions are mediated through both oxidative and nonoxidative routes of metabolism that convert ω-3 FAs into bioactive lipid metabolites. See Figure 1
Figure 1
Overview of the CYP epoxygenase-mediated metabolism of endocannabinoids. Both ω-6 and ω-3 dietary fatty acids are stored in plasma membrane and can be converted to the ω-6 and ω-3 endocannabinoids AEA, DHEA, and EPEA. AEA, EPEA, and DHEA are substrates for CYP epoxygenases. The metabolism of AEA produces EET-EA metabolites (the 14,15-EET-EA regioisomer is shown). The metabolism of EPEA and DHEA by CYP epoxygenases leads to the formation of EEQ-EA and EDP-EA, respectively. The terminal endocannabinoid epoxide regioisomer is shown, and other possible epoxides at each double bond are denoted by the numbering system.
Conclusion /The health benefits of ω-3 fatty acids are mediated, in part, through metabolic conversion to bioactive epoxides. Here researchers detail the discovery and initial characterization of naturally occurring ω-3–derived endocannabinoid epoxides that are formed via enzymatic oxidation of ω-3 endocannabinoids by cytochrome P450s. These dual functional ω-3 endocannabinoid epoxides are anti-inflammatory and vasodilatory and reciprocally modulate platelet aggregation. By virtue of their physiological properties, they are expected to play important roles in neuroinflammation and in cerebrovascular diseases such as stroke.
Omega-3 Fatty Acids Reduce Inflammation through Cannabinoids
https://todayspractitioner.com/cannabis/omega-3-fatty-acids-reduce-inflammation-through-cannabinoids/#.Xb-KDtM6ujg
World J Cardiovasc Dis. Author manuscript; available in PMC 2013 Jan 1.
Published in final edited form as:
World J Cardiovasc Dis. 2012 Jan 1; 2(1): 14–19.
The Effects of EPA+DHA and Aspirin on Inflammatory Cytokines and Angiogenesis Factors
1Department of Community and Preventive Medicine, the University of Rochester School of Medicine and Dentistry, Rochester, New York
2Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Albany, New York
3Cardiovascular Health Research Center, Department of Internal Medicine and Basic Biomedical Sciences, Sanford School of Medicine of the University of South Dakota, Sioux Falls, SD
4Department of Epidemiology, Biostatistics and Occupational Health, McGill University, Montréal, Québec
5Augustana College, Sioux Falls, SD
6Pulmonary and Critical Care Division, Department of Medicine, the University of Rochester School of Medicine and Dentistry, Rochester, New York
7College of Medicine, King Saud University, Riyadh, Saudi Arabia
Corresponding Author: Robert C. Block, MD, MPH, Division of Epidemiology, Department of Community and Preventive Medicine, The University of Rochester School of Medicine and Dentistry, 601 Elmwood Avenue, Box 644, Rochester, New York 14642, (office) 585.275.3356, (fax) 585.461.4532, ude.retsehcor.cmru@kcolb_trebor
Abstract
Objective
In a recent study, we showed that the combination of aspirin plus the omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) synergistically inhibited platelet function. As aspirin, EPA, and DHA have demonstrated anti-inflammatory properties, we hypothesized that the ingestion of EPA and DHA, with and without aspirin, would reduce plasma levels of inflammatory cytokines and angiogenesis factors more than aspirin alone and before aspirin was ingested.
Methods
Using multiplex technology, we investigated the effects of aspirin (single-dose 650 mg on day 1), EPA+DHA (3.4 g/d for days 2-29), and aspirin with EPA+DHA (day 30) on plasma levels of inflammatory cytokines and angiogenesis factors in healthy adults.
Results
Aspirin alone had no effect on any factor versus baseline, but EPA+DHA, with and without aspirin, significantly reduced concentrations of 8 of 9 factors. Although EPA+DHA plus aspirin reduced concentrations of a subset of the factors compared to baseline, neither aspirin alone nor the combination significantly reduced the level of any analyte more robustly than EPA+DHA alone.
Conclusions
These data suggest that EPA+DHA has more pronounced down-regulatory effects on inflammation and angiogenesis than aspirin. The implications of these findings for the use of combined therapy for cardiovascular disease remain to be clarified.
Figure 1
The effects of aspirin and EPA+DHA on plasma cytokine levels. Plasma concentrations of the indicated cytokines were evaluated at baseline, 1 day after an oral dose of 650 mg of regular aspirin (aspirin), after 28 days of treatment with 3.4 mg/day of prescription EPA+DHA (EPA+DHA), and after 1 day of combined treatment with EPA+DHA and aspirin (aspirin & EPA+DHA). Units are in picogram/mL and error bars represent 1 standard deviation. Single asterisks indicate a p-value of <0.1 and double asterisks indicate a p-value of <.05 as compared to baseline. Single crosses indicate a p-value <0.1 and double crosses indicate a p-value <0.05 as compared to the effects of aspirin alone. Aspirin did not significantly alter concentrations of any cytokine when ingested alone, compared to baseline, or in combination with EPA+DHA when compared to EPA+DHA alone.
Figure 2
The effects of aspirin and EPA+DHA on plasma angiogenesis factor levels. Plasma concentrations of b-FGF and VEGF were evaluated as described for Figure 1. Single asterisks indicate a p-value of <0.1 and double asterisks indicate a p-value of <.05 as compared to baseline. Single crosses indicate a p-value <0.1 and double crosses indicate a p-value <0.05 as compared to the effects of aspirin alone. Aspirin did not significantly alter the concentrations of any angiogenesis factor when ingested alone, compared to baseline, or in combination with EPA+DHA, when compared to EPA+DHA alone.DISCUSSION
The key finding of this study is that a brief 28-day supplementation with EPA+DHA led to a significant reduction in plasma levels of cytokines and angiogenesis factors implicated in atherosclerosis. To the best of our knowledge, no prior studies have compared the effects of aspirin and EPA+DHA, alone, and in combination, on all of these analytes simultaneously. EPA and DHA have been shown to exert potent anti-angiogenic effects by inhibiting the production of important inflammatory and angiogenic mediators, namely VEGF, PDGF, platelet-derived endothelial cell growth factor (PD-ECGF), COX-2, prostaglandin-E2 (PGE2), nitric oxide, NF-κB, matrix metalloproteinases and beta-catenin[8]. PDGF concentrations in plasma in our study were reduced by consumption of 3.4g/d of EPA+DHA. This is in contrast to the results of an earlier study of 63 healthy participants in which the consumption of 0.3, 0.6 or 0.9 g/day of EPA+DHA for 8 weeks had no effect on serum PDGF levels[9]. A dose of 0.6 mg/day is consistent with slightly more than 2 portions of fatty fish per week, and 1 g/day is consistent with about 4 fatty fish meals per week. These differences in the effects of EPA+DHA on PDGF suggest that doses of EPA+DHA higher than those typically achieved through diet may be necessary to achieve downregulation of PDGF. In support of this, a dose of 7 g/day for 4 weeks has been shown to suppress adherence-activated and non-activated mononuclear cell-mediated production of PDGF and MCP in humans[10]. To date, the modulation of blood concentrations of angiogenesis activator b-FGF in humans by EPA and DHA has not been reported.
It is known from in vitro studies that EPA and DHA downregulate the production of TNF-α, IL-6, IL-8, and IL-1ß via modulation of nuclear factor (NF)-B[11], while also downregulating monocyte[12] and MCP-1[13] activity. However, the doses used and duration of administration vary considerably from study-to-study in humans. While some of the studies provided <2 g EPA+DHA per day[11], others have examined the effects of higher doses[14]. Perhaps because of these differences, the amount of EPA+DHA required to exert beneficial effects is not clear. Most of these previous studies used ex vivo techniques to examine immune cell function, unlike the current study in which we directly measured plasma levels in human subjects taking approved doses of EPA+DHA.
The formation of vasa vasorum through angiogenesis has been associated with plaque instability and rupture, as micro-vessel formation has a predilection for the shoulder regions of atherosclerotic plaques[15]. Stimulators of angiogenesis that would induce the growth of new blood vessels and thus potentially reduce ischemic burden in the heart and limbs have been considered promising[16], however, despite promising results in preclinical models, data from clinical trials have been inconclusive, and evidence suggests that angiogenic factors actually promote atherosclerosis and potentially destabilize coronary plaques[2]. EPA and DHA have been shown to inhibit pathologic angiogenesis[17].
In contrast to our expectations, IL-10 concentrations decreased with EPA+DHA treatment. In an observational study involving 1123 subjects[18], lower DHA plasma levels were strongly associated with lower IL-10 concentrations. To our knowledge, there are no published data on the effects of pharmaceutical-grade EPA+DHA given at FDA-approved doses on IL-10. Interestingly, the inhibitory effect of EPA+DHA on plasma IL-10 levels was more modest in our study than for most of the other cytokines. Although we cannot know if oral doses of DHA alone would alter IL-10 levels, nor whether, in our study, it was the high dose of omega-3 fatty acids or the provision of EPA that lowered this marker, it seems possible that EPA and/or DHA on IL-10 may affect this cytokine differently depending on the dose and preparation used.
Our study found no effect of a 650mg dose of aspirin alone on plasma cytokines and pro-angiogenesis factors. This dose of aspirin was chosen because we originally wanted to study the potential effects of aspirin and EPA+DHA on platelet function[19]. It remains possible that other doses of aspirin, or ingestion of aspirin for longer time points, might affect some of the inflammatory cytokines or proangiogenesis factors measured. Alternatively, while the clearance of many cytokines and angiogenesis factors occurs quickly with half-lives of < 3 hours, it is possible that the effects of aspirin on molecules with longer half-lives could be detected in studies with longer periods of aspirin use. In addition, the data in this study should be cautiously interpreted as the number of participants was relatively small. Their age was also quite low and they were quite healthy, limiting the ability to predict the process of atherosclerosis and clinical events. Thus, different results could be found in a much larger cohort with older, or diseased, participants. In addition, we were not able to determine if the angiogenesis stimulating factors of cigarette smoking[20] and endothelial progenitor cells[21] influence the effects that EPA+DHA or aspirin had on the pro-angiogenesis molecules that we measured. Future studies will be needed to investigate these possibilities.
In conclusion, our findings support the idea that the omega-3 fatty acids EPA+DHA have anti-inflammatory and anti-angiogenesis effects in vivo, which may contribute to the beneficial effects of fish oil supplementation in susceptible human subjects who take or do not take aspirin.
Keywords: eicosapentaenoic acid, docosahexaenoic acid, lipid mediators, fatty acids, angiogenesis, hemostasis, platelet function, cytokines, aspirinThe Effects of EPA+DHA and Aspirin on Inflammatory Cytokines and Angiogenesis Factors
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3331709/
Carcinogenesis. 2010 Sep; 31(9): 1584–1591.
Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines
Cancer Medicine Research Programme, Translational Medical Sciences, Division of Applied Medicine, School of Medicine and Dentistry, University of Aberdeen, Polwarth Building, Aberdeen, AB25 2ZD, UK
1Neurobiology Research Programme, School of Medical Sciences, University of Aberdeen, Polwarth Building, Aberdeen, AB25 2ZD, UK
2Faculty of Medicine, Hebrew University, Ein Kerem Campus, 91120 Jerusalem, Israel
Abstract
The omega-3 fatty acid ethanolamides, docosahexaenoyl ethanolamide (DHEA) and eicosapentaenoyl ethanolamide (EPEA), displayed greater anti-proliferative potency than their parent omega-3 fatty acids, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), in LNCaP and PC3 prostate cancer cells.DHEA and EPEA activated cannabinoid CB1 and CB2 receptors in vitro with significant potency, suggesting that they are endocannabinoids.
Both LNCaP and PC3 cells expressed CB1 and CB2 receptors, and the CB1- and CB2-selective antagonists, AM281 and AM630, administered separately or together, reduced the anti-proliferative potencies of EPEA and EPA but not of DHEA or DHA in PC3 cells and of EPA but not of EPEA, DHEA or DHA in LNCaP cells. Even so, EPEA and EPA may not have inhibited PC3 or LNCaP cell proliferation via cannabinoid receptors since the anti-proliferative potency of EPEA was well below the potency it displayed as a CB1 or CB2 receptor agonist. Indeed, these receptors may mediate a protective effect because the anti-proliferative potency of DHEA in LNCaP and PC3 cells was increased by separate or combined administration of AM281 and AM630. The anandamide-metabolizing enzyme, fatty acid amide hydrolase (FAAH), was highly expressed in LNCaP but not PC3 cells. Evidence was obtained that FAAH metabolizes EPEA and DHEA and that the anti-proliferative potencies of these ethanolamides in LNCaP cells can be enhanced by inhibiting this enzyme. Our findings suggest that the expression of cannabinoid receptors and of FAAH in some tumour cells could well influence the effectiveness of DHA and EPA or their ethanolamide derivatives as anticancer agents.
In this investigation, we explored the possibility that the anticancer effects of DHA and EPA depend, at least in part, on their conversion to their ethanolamides: DHA to docosahexaenoyl ethanolamide (DHEA) and EPA to eicosapentaenoyl ethanolamide (EPEA). The difference in the structures of EPEA, DHEA and anandamide can be seen in Figure 1A. Because there is evidence that anandamide acts through CB1 receptors to inhibit the proliferation of human breast and prostate cancer cells, we tested the hypothesis that DHA, EPA, DHEA and/or EPEA produce their anticancer effects by activating cannabinoid receptors in these cells (5,6). There is evidence that anandamide can be catabolized by fatty acid amide hydrolase (FAAH) and that inhibition of this enzyme can potentiate some effects of anandamide (7,8). Accordingly, we explored the possibility that the anticancer effects of DHA and EPA and/or of their ethanolamides can be potentiated by inhibiting this enzyme.
Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2930808/
Omega-3 PUFA vs. NSAIDs for Preventing Cardiac Inflammation
炎症形成和NSAID的抗炎机制
NSAID是一类阻止炎症过程中前列腺素(PGs),白三烯(LT)或环氧化物(3)增加生成的抗炎药。因此,NSAID通常用于预防多种慢性炎症,包括CVD。
参与生物合成前列腺素PG的主要酶是环氧合酶(COX),它被细分为组成型COX-1和诱导型COX-2的形式。这些COX亚型在发炎的组织中显示出不同的活性。 COX-2表达增加10到80倍,而COX-1表达增加2到4倍(4)。两种COX亚型均负责将花生四烯酸(ARA)转化为中间体PG,PGG2和PGH2。 ARA是类花生酸的前体,可被磷脂酶A2(PLA2)从膜磷脂上切割下来。
然后激活血栓烷合酶和各种异构酶,产生血栓烷A2(TxA2)和PG(PGE2,PGF2α,PGD2,PGI2)(5)。这四种PG具有共同的血管舒张功能以及增加膜的通透性(因此由于血流量增加而促进“发红”)。
PGE2和PGF2α主要由单核细胞和巨噬细胞产生,肥大细胞产生的PGD 2和内皮细胞产生PGI2(6)。长期低剂量的NSAIDs治疗也会降低PG的益处(7)。 PGE2和PGF2α控制水和电解质的吸收并维持胃粘膜的分泌。因此,NSAIDs可以减少胃腔和上皮细胞之间粘液-碳酸氢盐屏障的分泌。随后,与胃的低pH值接触,上皮细胞被杀死,粘膜的完整性丧失,引起溃疡(7)。
阿司匹林
阿司匹林是一种广泛使用的消炎药。阿司匹林通过乙酰化COX上的羟基(具体作用于丝氨酸残基)来抑制COX活性。这导致对COX的不可逆抑制,并导致ARA结合受限(8)。
阿司匹林可轻松快速地吸收到胃肠道中,并在胃和肠中水解为水杨酸(SA)。但是,SA和阿司匹林可以与白蛋白强烈结合。这避免了阿司匹林的水解过快(9),因为在急性炎症下白蛋白浓度通常会由于复合白蛋白-透明质酸的形成或白蛋白合成的减少而急剧降低(9、10)。因此,SA和阿司匹林在急性炎症下水解更快。在这种情况下,调节SA(或阿司匹林剂量)浓度范围至关重要,以避免进一步的不良影响。阿司匹林的半衰期相对较短,在成年人中为15–20分钟。阿司匹林抑制PGs的产生主要是由于COX-2的阻滞。但是,阿司匹林还抑制胃粘膜中的细胞保护性PG,从而损害上皮细胞的完整性并破坏溶酶体膜的稳定性(11)。Omega-3 PUFA vs. NSAIDs for Preventing Cardiac Inflammation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6205954/
在心脏疾病中使用NSAID的最新问题
像阿司匹林这样的非甾体抗炎药已经使用了数十年,以防止心血管疾病中的轻度炎症。最初被认为对影响心血管系统的全身性炎症是安全有效的(16、17)。但是,获益的证据并不一致(18),并受到胃肠道出血等主要副作用的困扰,这可能使这种治疗方法在弱势人群中值得怀疑(19)。本质上,NSAID的核心作用是抑制COX活性。但是,COX活性,尤其是COX-2,也负责维持主动脉功能。 COX2破坏可导致主动脉硬化,导致主动脉纤维化(20)和动脉粥样硬化(21)。此外,抑制COX2会降低他汀类药物对心脏保护的作用(22)。这可能是为什么近年来除阿司匹林以外的所有非甾体抗炎药(如布洛芬和萘普生)均已显示出包括心脏病发作在内的心血管效应(23、24)。更重要的是,根据最近的一项研究,长期服用阿司匹林的人在停药后发生心血管事件的机会增加了30%以上(25)。因此,在包括CVD在内的慢性炎性疾病中,有必要避免长期使用NSAID的预防性治疗。
欧米茄3 PUFA
Omega-3 PUFA是多不饱和脂肪酸,在从末端甲基开始的第三个碳上具有第一个双键。鱼和亚麻籽油富含omega-3 PUFA,对心脏(26),肝脏(27)和大脑(28)具有保护功能。鱼油补充剂中包含的主要脂肪酸是二十碳五烯酸(EPA)和二十二碳六烯酸(DHA),它们是omega-3家族的长链成员。相反,亚麻籽油主要由母体omega-3 PUFAα-亚麻酸(ALA)组成。
抗炎反应的机制
与NSAID相似,omega-3 PUFA,尤其是EPA和DHA抑制促炎性类花生酸的产生。但是,他们使用相同的COX而不是阻止COX活性,而是通过提供不同的底物来增加抗炎类花生酸的产生。二十碳omega-3 PUFA和ARA相互竞争使用COX酶。这增加了来自EPA的抗炎介质如LTB5和PGE3的产生,同时限制了来自ARA的炎性LTB4和PGE2的产生(29)。在这种情况下,尽管减少了粘膜保护性PG(PGE2),但仍然可用。结果,由抗炎药引起的副作用被大大降低。鉴于NSAID和omega-3 PUFA在阻断COX的机理与为COX酶提供替代底物的机理上存在根本差异,因此行动的时间表截然不同。由于直接的酶促阻断作用,NSAID的作用更为急性,其中omega-3 PUFA逐渐取代膜磷脂ARA的作用较慢,这可能需要数周甚至数月的时间才具有生物学上的合理性。显然,对于因身体伤害或外伤引起的急性炎症挑战,首选NSAIDs,而omega-3 PUFA最好是长期,温和的抗炎溶液。因此,在解决长期慢性炎症阶段时,食用omega-3补充剂可被视为预防性治疗,可替代NSAIDs,其主要区别如表11所示(表11)。除了调节PG外,EPA和DHA还产生负责抗发炎和消退的脂质介体:resolvins和保护素(66)。由于RESOLVINS生产的底物不同,它们分为EPA的E系列和DHA的D系列(66)。尽管它们来自不同的来源,但它们在预防炎症方面显示出非常相似的效果。随着阿司匹林乙酰化COX-2的刺激,阿司匹林的存在或EPA / DHA的消耗量均增加。如前所述,取决于不同的底物,COX-2是促进促炎性和抗炎性类花生酸合成的限速酶。通过与LTB4竞争白三烯B4受体(BLT1),依赖COX-2的RESOLVIN减轻炎症反应并阻止人中性粒细胞跨内皮迁移(67)。保护素是另一组新的促分解和抗炎脂质介体,仅源自DHA(66)。它们阻止T细胞的迁移,促进T细胞凋亡,并降低有效的炎症因子TNFα(68)。
除了直接影响类花生酸途径外,ALA,EPA和DHA还有助于减少促炎性细胞因子,包括TNFα,IL-1和IL-6(30)。这些有效的细胞因子在损伤后引发级联的促炎性介质,包括细胞因子,趋化因子和粘附分子。这导致免疫细胞,例如嗜中性粒细胞,单核细胞,B细胞和T细胞的高募集。在大多数急性和慢性炎性疾病中,已证明omega-3 PUFA通过降低炎性标志而减弱了炎性反应(30)。在心脏疾病中,omega-3 PUFA可以抑制脂多糖(LPS)的分泌。这限制了LPS诱导的炎症途径的启动,包括NF-κB和toll样受体4(TLR4)(69)。随着对促炎性信号传导的这种抑制,一氧化氮(NO)的产生增加(45)。这导致内皮功能的改善(70)。
考虑到炎症和氧化应激之间的密切关系,omega-3 PUFA还可以通过增加细胞的抗氧化能力来降低氧化应激。但是,仅每天消耗3.4 g以上的EPA / DHA才能达到这一结果(71)。另一方面,由于omega-3 PUFA具有多个易于氧化的双键,因此高剂量的omega-3 PUFA会引起过多的脂肪酸蓄积,这也可能会增加氧化应激。
Front Cardiovasc Med. 2018; 5: 146.
Omega-3 PUFA vs. NSAIDs for Preventing Cardiac Inflammation
Introduction
Inflammatory cell accumulation occurs in the cardiac muscle during cardiac injury and repair (1). From an evolutionary perspective, inflammation is required for immunosurveillance and host defense. However, such cardinal signs of acute inflammation, such as redness, pain, swelling etc., due to injury or infection. Might be typically absent in chronic low-grade inflammation (LGI). Current literature suggests that chronic low-grade inflammation (LGI) is a primary causative factor behind chronic diseases like cardiovascular diseases (CVD), non-alcoholic fatty liver disease (NAFLD) and obesity (2).
Common mechanisms for NSAIDs
NSAIDs are anti-inflammatory drugs which as a class block the generation of prostaglandins (PGs), leukotrienes (LT) or epoxides (3), which are upregulated during inflammation Therefore, NSAIDs are commonly used for prevention of multiple chronic inflammatory conditions including CVD. The major enzyme participating in PG biosynthesis is cyclooxygenase (COX), which is subdivided as constitutive COX-1 and inducible COX-2 forms. These isoforms of COX show differential activity in inflamed tissues. COX-2 is expressed 10- to 80-fold, whereas COX-1 expressed 2- to 4-fold (4). Both COX isoforms are responsible for converting arachidonic acid (ARA) to intermediate PGs, the PGG2, and the PGH2. ARA is the precursor of eicosanoids which is cleaved by phospholipase A2 (PLA2) from membrane phospholipids. Then thromboxane synthase and various isomerases are activated which generates thromboxane A2 (TxA2) and PGs (PGE2, PGF2α, PGD2, PGI2) (5). These four PGs have the common function of vasodilation as well as increasing permeability of membranes (thus promoting “redness” due to increased blood flow). PGE2 and PGF2α are mainly produced by monocytes and macrophages, mast cells produce PGD2 and endothelial cells produce PGI2 (6). Long-term treatment with NSAIDs lower beneficial PGs as well (7). PGE2 and PGF2α control water and electrolyte absorption and maintain secretion in gastric mucosa. Thus, NSAIDs can decrease the secretion of mucous-bicarbonate barrier between the gastric lumen and epithelial cells. Subsequently, in contact with low pH of the stomach, epithelial cells are killed and the integrity of the mucosa is lost, causing ulceration (7).
Omega-3 PUFA
Omega-3 PUFA are polyunsaturated fatty acids with the first double bond on the third carbon from the terminal methyl end. Fish and flaxseed oils are rich in omega-3 PUFA with protective functions for the heart (26), liver (27), and brain (28). The major fatty acids contained in the fish oil supplement are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), the long chain members of the omega-3 family. In contrast, flaxseed oil is mainly composed of alpha-linolenic acid (ALA), the parent omega-3 PUFA.
Mechanism of anti-inflammatory response
Similar to NSAIDs, omega-3 PUFA, especially EPA and DHA inhibit the production of pro-inflammatory eicosanoids. However, instead of blocking COX activity, they use the same COX to increase the production of anti-inflammatory eicosanoids by providing a different substrate. Twenty-carbon omega-3 PUFA and ARA compete with each other for the use of the COX enzyme. This increases the production of the anti-inflammatory mediators like LTB5 and PGE3 from EPA and at the same time limits the inflammatory LTB4 and PGE2 production from ARA (29). In this case, the mucosa protective PGs (PGE2) are still available, albeit reduced. As a result, the side effects caused by anti-inflammatory drugs are drastically reduced. Given the basic differences in the mechanism of NSAIDs and omega-3 PUFAs in blocking COX vs. providing an alternate substrate to COX enzyme, the timeline for actions are vastly different. NSAIDs are more acute in action due to direct enzymatic blockade, whereeas omega-3 PUFAs act slower due to its gradual replacement of membrane phospholipid ARA, which might take weeks if not months to have a biologically plausible effect. Clearly, NSAIDs are preferred for acute inflammatory challenges resulting from physical injury or trauma whereas omega-3 PUFAs are at best a long term, mild anti-inflammatory solution. Thus, consuming omega-3 supplement can be considered as a preventative therapy, alternate to NSAIDs on resolving long-term chronic inflammatory stage, with some major differences as listed in Table Table11 (65).In addition to modulating PGs, EPA and DHA produce lipid mediators responsible for anti-inflammation and resolution: resolvins and protectins (66). Due to the different substrates of resolvins' production, they are divided into E-series from EPA and D-series from DHA (66). Although they are from different sources, they show very similar effects on preventing inflammation. Both of them increase with the presence of aspirin or with higher EPA/DHA consumption, which are stimulated by the aspirin acetylated COX-2. As mentioned before, COX-2 is the rate-limiting enzyme promoting the synthesis of pro and anti-inflammatory eicosanoids, depending on the different substrates. COX-2 dependent resolvins attenuate inflammation and block the human neutrophil transendothelial migration by competing for the leukotriene B4 receptors (BLT1) with LTB4 (67). Protectins are the other group of new pro-resolving and anti-inflammatory lipid mediators, which are derived from DHA only (66). They block the immigration of T-cells, promote the T cell apoptosis, and reduce the potent inflammatory factor TNFα (68).
Other than directly affecting the eicosanoid pathway, ALA, EPA, and DHA also helps to reduce pro-inflammatory cytokines, including TNFα, IL-1, and IL-6 (30). These potent cytokines initiate the cascade of pro-inflammatory mediators, including cytokines, chemokines and adhesion molecules following injury. This leads to the high recruitment of immune cells, such as neutrophils, monocytes, B cells and T cells. In most acute and chronic inflammatory diseases, it has been shown that omega-3 PUFA attenuates the inflammatory response by reducing the inflammatory hallmarks (30). In cardiac diseases, omega-3 PUFA can inhibit the secretion of lipopolysacchrides (LPS). This limits the initiation of LPS-induced inflammatory pathway, including NF-κB and toll-like receptor 4 (TLR4) (69). Along with such inhibition of pro-inflammatory signaling, nitric oxide (NO) production increases (45). This leads to improved endothelial function (70).
Considering of the close relation between inflammation and oxidative stress, omega-3 PUFA can also lower oxidative stress through increased cellular antioxidant capacity. However, this result can only be reached with over 3.4 g/day EPA/DHA consumption (71). Having a high dose of omega-3 PUFA on the other hand can cause excess fatty acid accumulation, which potentially can also increase oxidative stress, given that omega-3 PUFAs have multiple double bonds amenable to oxidation.NSAIDs and omega-3 PUFA in combination?
An intriguing idea would be to use both low dose NSAID and long chain omega-3s like DHA/EPA in combination for prevention of cardiac and other LGI states. In theory, as both these classes of drugs act on the same COX/LOX pathway, the requirements/dosing of each might be lower due to their synergistic effects. The problem with such an approach is that long term safety of omega-3 supplementation in the pill form still remains unestablished in patients with various LGI states including CVD. With recent reports of long term ill effects of NSAIDs at the current dosing levels in cardiac patients, there is evidence that it might be risky to carry on such a trial for potential negative effects on coagulation (78).
In conclusion, the effectiveness of NSAIDs for acute inflammation has not translated to a safe strategy for long term prevention of CVD. Controversy also surrounds the long term impact of omega-3 PUFA as a preventative measure against chronic low grade inflammation (77). However, in patients unable to take NSAIDs in the long term due to GI or bleeding problems, due to the similarity in their mechanism of action, low dose omega-3 PUFA could be a substitute to prevent LGI associated with cardiovascular diseases.Omega-3 PUFA vs. NSAIDs for Preventing Cardiac Inflammation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6205954/
Arachidonic Acid Released by Phospholipase A2 Activation Triggers Ca2+-dependent Apoptosis through the Mitochondrial Pathway
Article (PDF Available) in Journal of Biological Chemistry
We studied the effects of the divalent cation ionophore A23187 on apoptotic signaling in MH1C1 cells. Addition of A23187 caused a fast rise of cytosolic Ca(2+) ([Ca(2+)](c)), which returned close to the resting level within about 40 s. The [Ca(2+)](c) rise was immediately followed by phospholipid hydrolysis, which could be inhibited by aristolochic acid or by pretreatment with thapsigargin in Ca(2+)-free medium, indicating that the Ca(2+)-dependent cytosolic phospholipase A(2) (cPLA(2)) was involved. These early events were followed by opening of the mitochondrial permeability transition pore (PTP) and by apoptosis in about 30% of the cell population. In keeping with a cause-effect relationship between addition of A23187, activation of cPLA(2), PTP opening, and cell death, all events but the [Ca(2+)](c) rise were prevented by aristolochic acid. The number of cells killed by A23187 was doubled by treatment with 0.5 microm MK886 and 5 microm indomethacin, which inhibit arachidonic acid metabolism through the 5-lipoxygenase and cyclooxygenase pathway, respectively. Consistent with the key role of free arachidonic acid, its levels increased within minutes of treatment with A23187; the increase being more pronounced in the presence of MK886 plus indomethacin. Cell death was preceded by cytochrome c release and cleavage of caspase 9 and 3, but not of caspase 8. All these events were prevented by aristolochic acid and by the PTP inhibitor cyclosporin A. Thus, A23187 triggers the apoptotic cascade through the release of arachidonic acid by cPLA(2) in a process that is amplified when transformation of arachidonic acid into prostaglandins and leukotrienes is inhibited. These findings identify arachidonic acid as the causal link between A23187-dependent perturbation of Ca(2+) homeostasis and the effector mechanisms of cell death.
Figure 1. Mitochondrial Ca2+ transport pathways in energized mitochondria. The figure shows the principle players of mitochondrial Ca2+ homeostasis. Ca2+ enters mitochondria through an electrogenic pathway, the "Ca2+ uniporter," recently shown to be a selective channel (Kirichok et al., 2004). The Ca2+ efflux pathways are also schematically shown: the H+-Ca2+ and Na+-Ca2+ exchangers, as well as the permeability transition pore (PTP).
Regulation of cell death by Ca2+ is a problem of great complexity. First proposed in the early 1970s as a mediator of cell demise in heart ischemia (1, 2), Ca2+ is today considered a key to both necrosis and apoptosis through its modulation of transcriptional factors, kinase and phosphatase signaling cascades, activation of enzymes involved in protein degradation and phospholipid metabolism, and modulation of mitochondrial function (3).
Mitochondria are central to Ca2+ signaling. This is true of both the response to physiological stimuli that pertains to normal cell function and the response to apoptotic signals that leads to cell demise (4). A key switch between cell survival and cell death is the mitochondrial PTP,1 a high conductance channel that may open in response to mitochondrial Ca2+ uptake (5). The PTP participates in the release of proapoptotic proteins through at least two mechanisms: swelling-dependent rupture of the outer mitochondrial membrane and remodeling of mitochondrial cristae, which increases the availability of cytochrome c for release through specific pathways activated by outer membrane tBID insertion (6, 7). PTP opening is not always followed by cell death, and the relationship between the two events appears to correlate with the pore open time. Only openings that last long enough to cause measurable depolarization are followed by cytochrome c release and detrimental effects on cell survival (8, 9).
Mitochondrial Ca2+ homeostasis in health and disease
https://scielo.conicyt.cl/scielo.php?script=sci_arttext&pid=S0716-97602004000400022
Arachidonic Acid Released by Phospholipase A2 Activation Triggers Ca2+-dependent Apoptosis through the Mitochondrial Pathway
http://www.jbc.org/content/279/24/25219.full
(PDF) Arachidonic Acid Released by Phospholipase A2 Activation Triggers Ca2+-dependent Apoptosis through the Mitochondrial Pathway
https://www.researchgate.net/publication/8630669_Arachidonic_Acid_Released_by_Phospholipase_A2_Activation_Triggers_Ca2-dependent_Apoptosis_through_the_Mitochondrial_Pathway
Differential decoding of Ca2+-linked stimuli evoking the activation of cell metabolism or apoptosis.
Calcium and apoptosis: ER-mitochondria Ca 2+ transfer in the control of apoptosis | Oncogene
https://www.nature.com/articles/onc2008308
ER and mitochondria have a close structural relationship. Mitochondria are thus capable of sensing microdomains of high [Ca2+] generated in close proximity to ER Ca2+ release channels (Rizzuto et al., 1998), that are sufficient to cause accumulation through the low-affinity transporters of the inner membrane, the "uniporter," recently shown to be a selective Ca2+ channel (Fig.1).
Figure 1. Mitochondrial Ca2+ transport pathways in energized mitochondria. The figure shows the principle players of mitochondrial Ca2+ homeostasis. Ca2+ enters mitochondria through an electrogenic pathway, the "Ca2+ uniporter," recently shown to be a selective channel (Kirichok et al., 2004). The Ca2+ efflux pathways are also schematically shown: the H+-Ca2+ and Na+-Ca2+ exchangers, as well as the permeability transition pore (PTP).
Mitochondrial Ca2+ homeostasis in health and disease
https://scielo.conicyt.cl/scielo.php?script=sci_arttext&pid=S0716-97602004000400022
Mitochondrial calcium: a crucial hub for cancer cell metabolism?
Wolfgang F. Graier, Roland Malli
Institute of Molecular Biology and Biochemistry, Medical University of Graz, Graz, Austria
Correspondence to: Wolfgang F. Graier, PhD. Chair and Full Professor of Molecular Biology, Institute of Molecular Biology and Biochemistry, Medical University of Graz, Neue Stiftingtalstrasse 6/6, 8010 Graz, Austria. Email: wolfgang.graier@medunigraz.at.
Provenance: This is an invited Editorial commissioned by Section Editor Zheng Li (Department of Gynecologic Oncology, The Third Affiliated Hospital of Kunming Medical University, Kunming, China).
Comment on: Chakraborty PK, Mustafi SB, Xiong X, et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun 2017;8:14634.
INTRODUCTION
According to the current concept, a typical tumor contains “driver gene” mutations that establish a growth advantage to the tumor cell (1). It was proposed that one of the first mutations must have either a mutation that introduces a selective growth advantage to a normal cell (1) or one of the first mutations results in genomic instability, thus, increasing the chance for the accumulation of further mutations (2). Recent data indicate that metabolic reprogramming by (somatic) mutations of key metabolic genes promotes cancer growth (3). In this context metabolic adaptations upon aging need to be considered as hallmark of cancer (4). Since the landmark discovery of cancer-specific enhancement of glycolysis with concurrent small respiration (5), abnormalities in mitochondrial functions as a key phenomenon of the metabolic peculiarities of cancer cells has been postulated. Otto Warburg assumed that the accumulation of lactate in cancer tissue is the result of enhanced anaerobic glycolysis and dysfunctional mitochondrial respiration. This assumption was lately challenged by still controversial findings of a reduced conversion of phosphoenolpyruvate to pyruvate by the less active, cancer-specific isoform pyruvate kinase 2 (PKM2) (6), which might limit the production of ATP from glycolysis, but boosts the accumulation of intermediate products like nucleic acids and phospholipids for cancer cell growth (7). Nevertheless, Otto Warburg was prophetic in his view on the role of metabolism for cancer. Recent work highlights alterations of mitochondria-associated genes to play an essentially role in the metabolic reprogramming in cancer cells that play a supportive, if not causative role in tumorigenesis (8). The latter hypothesis was stressed by a recent work where mitochondria/cytosolic transfer from a cancer cell into a somatic cell turned the latter one to a cancer cell as well (9). While the interpretation of this work is complex, mitochondrial metabolic and signal functions that undergo a cancer-specific transformation (4) involving mitochondria-endoplasmic reticulum (ER) settings (10,11) are known to be essential to meet the enhanced energy demand of a cancer cell. The study “MICU1 drives glycolysis and chemoresistance in ovarian cancer” by Chakraborty et al. recently published in Nature Communications (12) describes important new findings supporting the current hypothesis on mechanistic/functional mitochondrial adaptations to be crucial for cancer cell development. Importantly this work points to a crucial role of mitochondrial calcium uptake 1 (MICU1), the gatekeeper of mitochondrial Ca2+ uniport (MCU) (13), for glycolysis and chemoresistance in ovarian cancer. Notably, this work considerably fuels current hypothesis on the importance of mitochondrial Ca2+ uptake for cancer cell survival and growth (10,11,14) and points to adaptations in mitochondrial Ca2+ as a hallmark in the adjustment of mitochondrial activity in cancer cells.
All about mitochondrial calcium?Other Section
Chakraborty and coworkers also described an obvious role of MICU1 in the regulation of pyruvate dehydrogenase (PDH) by affecting the phosphoPDH: PHD ratio. They further describe a strong impact of MICU1 depletion on the anti-tumor effects of cisplatin. Notably, these particular findings were acquired under conditions of MICU1 silencing, conditions where mitochondrial Ca2+ uptake is strongly enhanced (12), due to the protein’s gatekeeper function in most cells (15). Hence, because the activity of the mitochondrial oxidative phosphorylation (OXPHOS) crucially depends on Ca2+-dependent dehydrogenases in the citrate cycle in the mitochondrial matrix, Ca2+ acts a main regulator for mitochondrial ATP synthesis (16), thus, a regulatory role of mitochondrial Ca2+ appears likely even in case of PDH activity (17). Such central role of mitochondrial Ca2+ in the regulation of mitochondrial processes and function appears a consequence of the tight physical and functional coupling of ER-released Ca2+ and mitochondrial Ca2+ sequestration. Recently, an optimized physical coupling between the ER and mitochondria was described in cancer cells (10,11). Notably, while the constitutive Ca2+ flux from the ER to mitochondria was demonstrated to be essential to maintain viability of cancer cells with high proliferation activity (10), mitochondrial Ca2+ overload (Figure 1) due to e.g., uncontrolled mitochondrial Ca2+ uptake, like under conditions of MICU1 depletion, yields activation of apoptotic pathways and, ultimately, cell death (11). The importance of the ER-mitochondrial Ca2+ transfer in cancer cells was further highlighted by the finding that certain oncogenes manipulate contact sites between these two organelles thereby affecting mitochondria-associated-ER membranes (MAMs) (18). In contrast the cancer testis antigen, FATE1 antagonizes cancer apoptosis by physically uncoupling the ER from mitochondria (19).
Mitochondrial calcium: a crucial hub for cancer cell metabolism? - Graier - Translational Cancer Research
http://tcr.amegroups.com/article/view/15469/htmlFigure 1 ATP and ROS production is under the control of mitochondrial Ca2+ uptake. MICU1 prevents mitochondrial Ca2+ loading under resting conditions (left blue panels). Physiological Ca2+ signals activate MCU via Ca2+ binding to MICU1, which allows controlled mitochondrial Ca2+ sequestration and consequently stimulates mitochondrial ATP production (middle green panels). Diminished MICU1 expression levels can lead to an uncontrolled mitochondrial Ca2+ overload, which has the potency to trigger enhanced ROS production and cell death pathways (right red panels). MICU1, mitochondrial calcium uptake 1; MCU; mitochondrial Ca2+ uniport.
Accordingly, there is a large body of evidence that mitochondrial Ca2+ is of utmost importance for (cancer) cells homeostasis, functions, growth, migration, and, ultimately, survival. The contribution of Chakraborty et al. convincingly demonstrates that besides cancer cell survival, their glycolytic activity and chemoresistance are also critically associated with mitochondrial Ca2+ uptake (12).
Mitochondrial Ca2+ uptake in cancer: the balance between energy and deathOther Section
Chakraborty et al. described a poor prognosis of cancer patient suffering from ovarian cancer with elevated expression of MICU1 (12). These data point to a protective effect of elevated MICU1 on cancer cell survival by preventing mitochondrial Ca2+ overload and, subsequently the initiation of the machinery for apoptotic cell death (Figure 1). These data further indicate that cancer cells benefit from reduced mitochondrial Ca2+ uptake, thus, illustrating the obvious risk of tumor cells for mitochondrial Ca2+ overload. Recently, cancer cells were found to exhibit a stronger tethering between mitochondria and ER in comparison to non-cancerous cells (11). While this enforced intra-organelle communication fosters e.g., mitochondrial ATP supply for the ER’s protein folding machinery and, thus, meets the high energy demand of cancer cells, such settings makes them more vulnerable for mitochondrial Ca2+ overload.
Notably, cancer cells developed multiple unique strategies to protect themselves against lethal mitochondrial Ca2+ overload:
Via the protein arginine methyl transferase 1 (PRMT1) MICU1 gets methylated yielding a strong sensitivity loss of the mitochondrial Ca2+ uptake machinery. In cancer cells methylated MICU1 protects from mitochondrial Ca2+ overload (14).
Tumor cell physiology and plasticity is regulated by cancer-specific mitochondrial dynamics (20).
Most cancer cells develop a unique redox handling by enhancing NADPH-dependent scavenging enzymes (21).
Cancer cells can actively untether their mitochondria from the ER (22).
Cancer cells modulate ER Ca2+ release by regulating inositol-1,4,5 trisphosphate receptors (IP3R), the constitutive receptors for ER Ca2+ release (23).
The permeability of the outer mitochondrial membrane (VDAC) is regulated (24).
Cancer cell suppress mitochondrial K+-channels and its normalization promotes apoptosis and hampers cancer growth (25).
This incomplete list illustrates the flexibility of cancer cells to address the issue of keeping the balance between enough mitochondrial Ca2+ uptake to meet the cell’s ATP demand while to avoid mitochondrial Ca2+ overload in order to stay clear from the initiation of apoptotic processes that ultimately would cause cancer cell death.
Mitochondrial Ca2+ uptake as therapeutic targetOther Section
Many authors including Chakraborty et al. (12,26) have indicated the potential of targeting mitochondrial Ca2+ uptake to fight against cancer growth. In this regard, three approaches appear most promising to be effective to introduce mitochondrial Ca2+ overload leading to apoptotic cancer cell death:
Directly affecting constituents of the MCU complex yielding uncontrolled Ca2+ uptake;
Manipulating the vicinity between the mitochondria and the ER resulting in an harmful enhancement of mitochondrial Ca2+ sequestration;
Disturbing the main Ca2+ regulators of the ER (i.e., SERCA and IP3R) to trigger exhaustive ER Ca2+ depletion and subsequently mitochondrial Ca2+ overload.CONCLUSION
While controlled mitochondrial Ca2+ uptake promotes cancer cell growth, the adequate transfer of Ca2+ into mitochondria is also of utmost importance for the proper functioning of somatic cells and tissues. Therefore, future work essentially needs to be aimed on the identification of cancer specificities in mitochondrial Ca2+ handling that can be therapeutically targeted to selectively fight cancer cells without affecting non-cancerous cells.Mitochondrial calcium: a crucial hub for cancer cell metabolism? - Graier - Translational Cancer Research
http://tcr.amegroups.com/article/view/15469/html
磷脂酶A2活性测试 - 大平原实验室公司
https://www.greatplainslaboratory.com/phospholipase-a2
磷脂酶A2活性测试
一般
The Great Plains Laboratory, Inc. is excited about our recently developed phospholipase A2 (PLA2) test that measures the activity of PLA2 in urine. We are the only commercial lab currently offering PLA2 level measurement as a urine test. PLA2 is elevated in a wide range of inflammation-related disorders and is considered a good marker for increased risk of developing or worsening of inflammatory conditions. PLA2 levels can easily be sampled along with the organic acid test (full or microbial), offering a powerful new combination of clinical insights.
Bee stings and venomous snake bites cause immediate inflammation, resulting in pain and swelling. The enzyme in venom that triggers this immune response is phospholipase A2. This enzyme is also present in human tissue. During infection, PLA2 activates a cascade that results in the destruction of the cell membranes of invading microbes. The products of the PLA2 reaction responsible for eliminating these organisms, lysolecithins and free fatty acids, are powerful detergents that damage cell membranes, denature proteins, and disrupt their function. However, this killing mechanism comes at a cost to the human host. Lysolecithin products are involved in the pain response and cause hemolysis of erythrocytes. The most common free fatty acid produced by PLA2is arachidonic acid, which can increase the production of powerful mediators of inflammation: prostaglandins, leukotrienes, and thromboxanes, collectively called eicosanoids. Release of these mediators initiates the pain, swelling, and other unpleasant symptoms we experience as part of an inflammatory response.
Excess PLA2 not only causes local damage, but can be transported by the blood vessels to other parts of the body, causing widespread tissue damage. Normal amounts of PLA2 are involved in remodeling cell membranes and changing cell architecture, but sustained release of PLA2 and the resulting inflammation is implicated in the development of chronic conditions including multiple sclerosis, cardiovascular disease, rheumatoid arthritis, gastrointestinal disorders, allergies, and neuropsychiatric disorders.
什么原因引起PLA2升高?
A large dose of PLA2 is delivered in venom from snakes, spiders, and bees (experienced as pain and stinging) and is responsible for much of the toxicity of these venoms. Microorganisms such as Candida albicans and certain Clostridia species produce PLA2, which increases the ability of the microorganism to infect the host. This same enzyme is produced in the body as a response to invading microorganisms and foreign proteins such as allergens, particularly house dust and cats. Physical trauma may also cause significant increases in PLA2, contributing to tissue damage and brain injury.
Phospholipase A2 is in fact a family of enzymes that are categorized by location and function. They have strong antibacterial and antiviral properties as part of the innate immune system, but also participate in many other biochemical processes that include cell signaling, cell proliferation and differentiation, cell migration and apoptosis, and modulation of inflammatory response. PLA2 is not only released in tissues and cells of the immune system, it is also produced by the pancreas and released into the small intestine following a fatty meal, to assist in the digestion and absorption of phospholipids. Additionally, PLA2 is expressed in neuronal tissue and is involved in the degranulation process that releases neurotransmitters from neurons. Research efforts have focused on the role that derangement of normal PLA2 activity plays in the etiology of many chronic illnesses. The specific roles, interactions, and interdependencies of PLA2 have been a major area of interest as it relates to chronic inflammatory conditions, cardiovascular disease, and cancer.
PLA2及炎症性疾病
研究已牵涉于神经变性疾病的病理生理学PLA2如多发性硬化症(MS)和阿尔茨海默氏病(AD)。多发性硬化症同时涉及抗原特异性机制和导致炎症反应的先天免疫系统的组分。升高的PLA2活性被发现是正在进行的MS患者中,在患者的疾病进展测量的最高水平。在阿尔茨海默氏病的发展,异常PLA2水平似乎对氧化信令涉及NADPH氧化酶和产生活性氧物种的途径导致损伤和神经元和神经胶质细胞的炎症的破坏有关。
炎症是类风湿性关节炎(RA)的标志,一个联合破坏性自身免疫性疾病。 PLA 2是在RA-受影响的个体的关节液及在RA患者的软骨中发现相比于从骨关节炎和正常个体的软骨。
PLA 2的测量正在成为用于评价心血管疾病(CVD),包括未来中风,心肌梗塞,心脏衰竭,以及其它血管事件的可能性的重要工具。 PLA 2似乎比hsCRP的更具体的为心血管疾病的风险,并且还可以具有如心血管病理的介质发挥关键作用。在动脉粥样硬化,PLA2不仅激活巨噬细胞和形成泡沫细胞,但它也水解LDL和HDL,产卵亲致动脉粥样小LDL颗粒的数量增加,并且损害抗动脉粥样硬化的HDL。 PLA2活性甚至可能诱发的粥样硬化斑块出血。
PLA 2是表示通常在胰,胆囊和GI上皮细胞,但在炎症性胃肠疾病的显著增加。在溃疡性结肠炎和克罗恩病,所有肠细胞类型增加PLA 2的表达,从而增加肠渗透性和实际上可能有助于感染性。
PLA2和癌症
PLA2的海拔已在胃肠道癌症,包括结肠腺瘤和癌和胰腺癌ductogenic癌等等被发现。肺癌患者的肿瘤呈阳性PLA2了大大提高肿瘤的生长速度和显着降低成活率。肺癌患者也有PLA2的血浆水平较高比例良性结节。类似的模式已经在前列腺癌中观察到,虽然转移性肿瘤表达比原发肿瘤低PLA2。作为PLA 2释放花生四烯酸和从细胞膜的其它脂肪酸,它们启动下游生产肿瘤促进花生酸类。在癌症,肿瘤细胞从原发肿瘤的体内辅助站点的蔓延是涉及细胞增殖和迁移,基底膜,侵袭,粘附和血管生成的降解一个复杂的过程。在癌症PLA2表达不断研究肯定会揭示有价值的新见解。
什么降落时PLA2?
Glucocorticoids such as the natural hormone cortisol and pharmaceutical agents such as dexamethasone inhibit the production of phospholipase, decreasing harm caused by the enzyme but also decreasing the benefits of the enzyme in killing harmful microorganisms. Thus, excess glucocorticoids can reduce inflammation in a patient with tuberculosis while reducing the effects of PLA2 against the bacteria resulting in spread of the illness. Lithium at pharmacological doses, carbamazepine, and the antimalarial drug chloroquine are all PLA2 inhibitors. Vitamin E is also an inhibitor of PLA2. In addition, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) (belonging to the omega-3 class of fatty acids) inhibit PLA2.
Cytidine 5-diphospho-choline (CDP-choline), a precursor in the formation of phospholipids, is a potent inhibitor of PLA2 that has been used as a nutritional supplement at doses ranging from 500-4000 mg per day in the treatment of patients with a variety of disorders including Parkinson's disease, memory disorders, vascular cognitive impairment, vascular dementia, senile dementia, schizophrenia, Alzheimer's disease (especially effective in those with the epsilon-4 apolipoprotein E genotype), head trauma, and ischemic stroke. A trial in patients with Alzheimer's disease indicated that citicoline (1,000 mg/day) is well tolerated and improves cognitive performance, cerebral blood perfusion, and the brain bioelectrical activity pattern. No side effects were noticed at the lower doses of CDP-choline and only some mild gastrointestinal symptoms were found using higher doses. No abnormal blood chemistry or hematology values were found after the use of CDP-choline.
测试PLA2
Because PLA2 is a relatively small enzyme (about 14 KD), it is able to be excreted in urine. 10 mL of the first morning urine before food or drink is suggested for testing. There are no dietary restrictions. This test is convenient to include with the organic acids test. Since chelating agents might interfere with the test, they should not be used for at least 48 hours prior to testing.
PLA2测试被推荐用于以下病症:
多发性硬化症
类风湿关节炎
克罗恩病
胰腺炎
溃疡性结肠炎
过敏
心血管疾病包括atherosclerosi
神经退行性疾病
精神分裂症
双相抑郁症,亚型精神病
念珠菌感染
脓血症
长期抑郁症
哮喘
慢性阻塞性肺疾病(COPD)磷脂酶A2活性测试 - 大平原实验室公司
https://www.greatplainslaboratory.com/phospholipase-a2
Natural Inibitors of phospholipase
Translocation of the G proteins and PKCs that are required for PLD activation; therefore, its major effect could be indirect. Natural products were also found to inhibit PLD activity, such as honokiol, which blocks Ras-dependent PLD activity in human cancer cells through an indirect method [69].
Targeting phospholipase D with small-molecule inhibitors as a potential therapeutic approach for cancer metastasis
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2814819/
injury. 1999 Jan;30(1):9-14.
Subcellular membrane impairment and application of phospholipase A2 inhibitors in endotoxic shock.
Song SM1, Lu SM, Wang ZG, Liu JC, Guo SQ, Li Z.
Author information
1
Research Institute of Surgery, Chongqing, People's Republic of China.
Abstract
The study aims at elucidating the mechanism involved in the cell dysfunction or impairment and the protective effects of phospholipase A2 (PLA2) inhibitors in endotoxin shock.Thirty-four rabbits were divided randomly into four groups: (1) normal control group (NC, n = 6), receiving saline intravenously; (2) endotoxin shock group (ES, n = 12), receiving 3 mg/kg of E. coli endotoxin; (3) chloroquine pretreated group (CQ, n = 8), receiving 3 mg/kg of chloroquine 3 min before endotoxin injection and (4) chlorpromazine pretreated group (CPZ, n = 8), receiving 0.3 mg/kg of chlorpromazine 30 min before endotoxin injection.
Hepatic mitochondria were extracted either 8 h after commencement of the experiment or when the animals died for detecting PLA2 activity, membrane fluidity, membrane bound succinate dehydrogenate (SDH) and malondialdehyde (MDA). Mitochondria of the lung, heart and kidney were also used for detection of the membrane fluidity. It was revealed that the survival rate of 8 h was 100% (NC), 58% (ES), 87.5% (CQ) and 75% (CPZ), respectively.
Mean arterial pressure (MAP) dropped soon after endotoxin injection and descended continuously afterwards in the ES group (P < 0.01). Fluorescence polarization, microviscosity and anisotrophy with a DPH probe were elevated above control levels (P < 0.01). SDH was decreased obviously following endotoxin infusion (P < 0.01). Chloroquine and chlorpromazine, serving as PLA2 inhibitors, could abate cellular dysfunction and increase survival rate. It is proposed that PLA2 plays a pivotal role in cellular injury in endotoxin shock. PLA2 inhibitor might serve as a useful adjunct in combating sepsis and shock.
PMID: 10396448 DOI: 10.1016/s0020-1383(98)00178-8Subcellular membrane impairment and application of phospholipase A2 inhibitors in endotoxic shock. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/10396448
Circ Shock. 1990 Jan;30(1):43-9.
Mechanisms of myocardial membrane alterations in endotoxin shock: roles of phospholipase and phosphorylation.
Liu MS1.
Author information
1
Department of Physiology, St. Louis University School of Medicine, MO 63104.
Abstract
Current progress in the studies of myocardial membrane alterations during endotoxin shock indicates that endotoxin administration impairs (Na(+) + K(+)-ATPase enzyme system by disrupting the coordination of the ouabain receptor subunit and the catalytic subunit of the enzyme system, and that the disruption is due to an alteration in the lipid microenvironment and a decrease in the phosphorylated intermediate of the enzyme cycle. Studies of the membrane lipid profile provide evidence that endotoxin administration modifies the molecular structure of cardiac membrane lipids in association with the activation of phospholipases A1 and A2 and with the inhibition of phospholipid methylating enzymes. Using liposomes as a membrane model for investigation, endotoxin was found to be capable of modifying the physical property of membrane phospholipids by altering the molecular packing and the phase transition temperature of lipid bilayers. Further studies with Na(+)-Ca2+ exchange system in cardiac sarcolemma have established the roles of phospholipase A2 and protein phosphorylation on the endotoxin-induced derangement in myocardial Na(+)-Ca2+ exchange. Based on these studies, it is concluded that endotoxin administration exerts multiple injuries in different membrane-associated enzyme/receptor systems and that the mechanisms responsible for the endotoxin-induced membrane damage can be categorized into two conceptual frameworks: namely, changes in membrane lipid microenvironment in response to phospholipase A activation and alterations in the phosphorylation of the enzyme/receptor proteins.Mechanisms of myocardial membrane alterations in endotoxin shock: roles of phospholipase and phosphorylation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/2154341
J Surg Res. 1988 Nov;45(5):472-80.
Activation of phospholipases A1 and A2 in heart, liver, and blood during endotoxin shock.
Liu MS1, Kang GF, Ghosh S.
Author information
Department of Physiology, St. Louis University School of Medicine, Missouri 63104.
Abstract
The effects of endotoxin shock on the activities of phospholipases A1 and A2 were studied in canine heart sarcolemma, liver plasma membrane, and blood serum.Results obtained from control dogs indicate that phospholipases A1 and A2 were equally active in cardiac sarcolemma. In liver plasma membrane the activity of phospholipase A1 was twice that of phospholipase A2, while in blood serum phospholipase A1 activity was only half that of phospholipase A2.
Results obtained 4 hr after endotoxin administration (0.5 mg/kg, iv) demonstrate that phospholipase A1 activities were activated by 117% in cardiac sarcolemma, 188% in liver plasma membrane, and 150% in blood serum, while phospholipases A2 activities were stimulated by 71% in cardiac sarcolemma, 175% in liver plasma membrane, and 31% in blood serum.
The in vitro incubation of all three enzyme preparations from control dogs with endotoxin (5-15 micrograms/0.5 ml) stimulated both phospholipase A1 and A2 activities and the stimulation was concentration dependent. These data suggest that endotoxin may have a direct stimulatory effect on phospholipases A1 and A2 activities in the cell membranes of heart and liver, and also in blood serum.
Since phospholipases A play an important role in the regulation of membrane structure and function, the observed activation of phospholipases A1 and A2 may have a physiological significance in the pathogenesis of cellular dysfunction in endotoxin shock.
PMID: 3054324 DOI: 10.1016/0022-4804(88)90198-9
Home / Medicinal Chemistry Reviews - Online, Volume 2, Number 5
Inhibition of Brain Phospholipase A2 by Antimalarial Drugs: Implications for Neuroprotection in Neurological Disorders
Abstract
Phospholipases A2 belong to a large family of enzymes involved in the generation of several second messengers that play an important role in signal transduction processes associated with normal brain function. The phospholipase A2 family includes secretory phospholipase A2, cytosolic phospholipase A2, calcium-independent phospholipase A2, and plasmalogen-selective phospholipase A2.
The systemic administration of kainic acid to rats results in seizures and subsequent degeneration of specific neurons in the hippocampus and striatum. The kainic acid-induced neurodegeneration is accompanied by upregulation of PLA2 activity and immunoreactivity. Stimulation of PLA2 activity results in the degradation of phospholipids in neuronal membranes with the generation of arachidonic acid and lysophospholipids. These products are further metabolized to potent inflammatory mediators such as eicosanoids and platelet activating factor. Although an inflammatory response can be induced by many different means, phospholipase A2-generated inflammatory mediators are closely associated with the pathogenesis of inflammation and oxidative stress in neurodegenerative diseases.
Peroxidation of arachidonic acid, a PLA2 reaction product, also results in generation of 4-hydroxy-2,3-nonenal (4- HNE), an α, β-aldehyde with neurotoxic properties. In kainic acid-mediated neurotoxicity, the treatment of brain slices with antimalarial drugs, quinacrine, chloroquine, hydroxychloroquine, and quinine, inhibits neurodegeneration and reduces PLA2 and 4-HNE immunoreactivities. This suggests that antimalarial drugs can be used as neuroprotectants and anti-inflammatory agents in neurodegenerative diseases. In vitro and in vivo studies indicate that antimalarial drugs can also be used for the treatment of prion diseases, ischemic injury, and experimental Parkinson disease (PD). These drugs maintain blood pressure, decrease infarct size, reduce inflammation, and inhibit neurodegeneration in focal and global models of cerebral ischemia and protect dopaminergic neurons from neurodegeneration in experimental PD.
Initial attempts to treat Creutzfeldt-Jakob disease (CJD) with quinacrine in humans indicate that this antimalarial drug may have some transient beneficial effects in advanced CJD patients. Antimalarial drugs have no beneficial effects in Alzheimer disease.
Keywords: and parkinson disease; antimalarial drugs; arachidonic acid; eicosanoids; ischemia; neurodegeneration; phospholipase a; prion disease
Document Type: Review Article
Affiliations: Department of Molecular and Cellular Biochemistry, The Ohio State University, 1645 Neil Ave, Room 409, Columbus, OH 43210-1218, U.S.A.
Publication date: 2005年10月1日Inhibition of Brain Phospholipase A2 by Antimalarial Drugs: Impli...: Ingenta Connect
https://www.ingentaconnect.com/content/ben/mcro/2005/00000002/00000005/art00002
中国创伤学
Effect of polydatin(虎杖甙) on phospholipase A2 in lung tissues in rat with endotoxic shock
重庆医科大学
Abstract
To study the effect of polydatin on phospholipase A(2) in lung tissues in rats with endotoxic shock. Thirty-two healthy male Wistar rats were employed in this study. A total of 8 rats received normal saline intravenously (control group), 8 rats received 10 mg/kg of endotoxin (endotoxic shock group), 8 rats received 1 mg/kg of polydatin after endotoxin injection (polydatin treatment group), and 8 rats received 1 mg/kg of polydatin (polydatin prevention group) 30 minutes before endotoxin injection. Mean arterial pressure was measured once half an hour. Lung tissues were collected 6 hours later. Phospholipase A(2) activity was measured with acid titration. The gene expression of secretory phospholipase A(2) type IIA was detected with reverse transcription polymerase chain reaction. Meanwhile, the histological changes of the lungs among four groups were compared through microscopic examination. Phospholipase A(2) activity and the gene expression of secretory phospholipase A(2) type IIA increased after endotoxin injection, but polydatin could inhibit these effects of endotoxin. Obvious morphological evidence could be found in the lung pathological sections and the protective effect of polydatin was most significant in the polydatin prevention group. Polydatin has prophylactic and therapeutic effects (the former is more distinct than the latter) on acutely injured lungs in rats with endotoxic shock and which suggests that polydatin may be a phospholipase A(2) inhibitor.select more than one item to compare
Effect of polydatin on phospholipase A2 in lung tissues in ...
https://www.researchgate.net/publication/8416792_Effect_of_polydatin_on_phospholipase...Chloroquine and chlorpromazine, serving as PLA2 inhibitors, could abate cellular dysfunction and increase survival rate. It is proposed that PLA2 plays a pivotal role in cellular injury in ...
Repurposing quinacrine for treatment-refractory cancer
Author links open overlay panelDerek B.Oiena1Christopher L.Pathoulasa1UpasanaRaya1PrabhuThirusanguaEleftheriaKalogerabVijiShridhara
a
Division of Experimental Pathology and Laboratory Medicine, Mayo Clinic, Rochester, MN, United States
b
Division of Gynecologic Surgery, Mayo Clinic, Rochester, MN, United States
Abstract
Quinacrine, also known as mepacrine, has originally been used as an antimalarial drug for close to a century, but was recently rediscovered as an anticancer agent. The mechanisms of anticancer effects of quinacrine are not well understood. The anticancer potential of quinacrine was discovered in a screen for small molecule activators of p53, and was specifically shown to inhibit NFκB suppression of p53. However, quinacrine can cause cell death in cells that lack p53 or have p53 mutations, which is a common occurrence in many malignant tumors including high grade serous ovarian cancer. Recent reports suggest quinacrine may inhibit cancer cell growth through multiple mechanisms including regulating autophagy, FACT (facilitates chromatin transcription) chromatin trapping, and the DNA repair process. Additional reports also suggest quinacrine is effective against chemoresistant gynecologic cancer. In this review, we discuss anticancer effects of quinacrine and potential mechanisms of action with a specific focus on gynecologic and breast cancer where treatment-refractory tumors are associated with increased mortality rates. Repurposing quinacrine as an anticancer agent appears to be a promising strategy based on its ability to target multiple pathways, its selectivity against cancer cells, and the synergistic cytotoxicity when combined with other anticancer agents with limited side effects and good tolerability profile.
Keywords
QuinacrineDrug repurposingOvarian cancerEndometrial cancerBreast cancerRepurposing quinacrine for treatment-refractory cancer - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S1044579X19302263
Front. Genet., 04 December 2014 | https://doi.org/10.3389/fgene.2014.00419
Phospholipase A2 – nexus of aging, oxidative stress, neuronal excitability, and functional decline of the aging nervous system? Insights from a snail model system of neuronal aging and age-associated memory impairment
Petra M. Hermann1,2, Shawn N. Watson1 and Willem C. Wildering1,2,3*
1Department of Biological Sciences, University of Calgary, Calgary, AB, Canada
2Department of Physiology and Pharmacology, University of Calgary, Calgary, AB, Canada
3Hotchkiss Brain Institute, University of Calgary, Calgary, AB, Canada
The aging brain undergoes a range of changes varying from subtle structural and physiological changes causing only minor functional decline under healthy normal aging conditions, to severe cognitive or neurological impairment associated with extensive loss of neurons and circuits due to age-associated neurodegenerative disease conditions. Understanding how biological aging processes affect the brain and how they contribute to the onset and progress of age-associated neurodegenerative diseases is a core research goal in contemporary neuroscience. This review focuses on the idea that changes in intrinsic neuronal electrical excitability associated with (per)oxidation of membrane lipids and activation of phospholipase A2 (PLA2) enzymes are an important mechanism of learning and memory failure under normal aging conditions. Specifically, in the context of this special issue on the biology of cognitive aging we portray the opportunities offered by the identifiable neurons and behaviorally characterized neural circuits of the freshwater snail Lymnaea stagnalis in neuronal aging research and recapitulate recent insights indicating a key role of lipid peroxidation-induced PLA2 as instruments of aging, oxidative stress and inflammation in age-associated neuronal and memory impairment in this model system. The findings are discussed in view of accumulating evidence suggesting involvement of analogous mechanisms in the etiology of age-associated dysfunction and disease of the human and mammalian brain.
Lipid Membranes as a Key Theater of Aging and Age-Associated Decline of the Nervous System
A large body of evidence indicates that oxidative stress is a prominent feature of both pathological and non-pathological forms of nervous system aging (Choi, 1995; Mattson and Magnus, 2006; Torres et al., 2011; Lopez et al., 2013; Orr et al., 2013; Haddadi et al., 2014). How oxidative stress contributes to age-associated disease processes of the brain and translates to functional decline of normal healthy aging neurons is not well understood. Here we explore the theory that processes associated with the high level of unsaturation of the lipid portion of neuronal membrane constitute a key link between aging, oxidative stress, and the age-associated physiological and behavioral decline frequently observed in normal healthy aging animals and the etiology of age-associated disorders of the brain. Central to this idea are the inordinate sensitivity of methylene-interrupted PUFAs to electrophilic attacks and subsequent activation of PLA2’s, a family of enzymes involved in maintenance of membrane phospholipid integrity and membrane-associated lipid signaling processes (Figure 2).
Eukaryote cell and organelle membranes are functionally complex, dynamically regulated assemblies of mostly glycerophospholipids, fatty acids (FAs), sterols, proteins, glycoproteins, and glycolipids (Alberts et al., 2002). While most of life’s structures are sensitive to oxidation, few are as susceptible to oxidative damage as phospholipid bilayer membranes (Zimniak, 2011). This elevated susceptibility to oxidation is due to a number of factors that conspire, as a manner of speaking, to create “perfect storm” conditions. First, under normal conditions solubility of molecular oxygen and many reactive oxygen and nitrogen species (RONS) in the lipid microenvironment of membranes is quite high (Subczynski and Wisniewska, 2000). Second, as mentioned, PUFAs are inordinately sensitive to attack by RONS and other electrophiles (Hulbert et al., 2007). Third, PUFAs that have undergone free radical attack are themselves reactive free radicals and are, because of the tightly packed molecular arrangement of phospholipids in bilayer membranes, capable of propagating potentially catastrophic lipid peroxidation cascades (Forlenza et al., 2007; Ballatori et al., 2009; Currais and Maher, 2013; Sultana et al., 2013). Neuronal membranes contain high levels of PUFAs such as arachidonic acids (AA), docosahexaenoic acids (DHA), and eicosapentaenoic acids (EPA), and are therefore particularly sensitive to (per)oxidation making lipid peroxidation potentially one of the major instruments of free radical-mediated injury and cell death in the nervous system.
Parenthetically, the idea that phospholipid membranes are one of the primary molecular theaters of aging and longevity has gained substantial traction in recent decades (for reviews see Pamplona, 2008; Zimniak, 2011). In fact, the observation that the level of lipid membrane unsaturation correlates inversely with longevity in mammals, birds, and even invertebrates led to formulation of the “Membrane hypothesis of aging” which essentially sees lipid membranes as one of the weakest and therefore highest maintenance links in the biochemical armature of living organisms (Hulbert et al., 2007; Ungvari et al., 2011, 2013; Munro and Blier, 2012; Munro et al., 2013).Lipid Peroxidation Induced Phospholipase A2 Activity and its Consequences
One characteristic cellular response triggered by lipid peroxidation is activation of one or more PLA2 family members that catalyze cleavage of FAs from the sn-2 position of glycero-phospholipids in plasma- and organelle membranes (Brown et al., 2003; Sun et al., 2004; Farooqui and Horrocks, 2006; Chen et al., 2008; Dennis et al., 2011). Although the precise underlying molecular mechanisms of lipid-peroxidation induced PLA2 activation are still a matter of debate (see for instance Greenberg et al., 2008), it is generally thought to involve peroxidation induced changes in fatty acyl chain geometry (Niki, 1990; Salgo et al., 1992; Rashba-Step et al., 1997; Greenberg et al., 2008).
Activation of PLA2 leads to the production of (peroxidized) free FAs (FFAs) and lysophospholipids (LPLs), both classes of molecules with potentially substantial biological activity (Figure 6; see below; Brown et al., 2003; Farooqui and Horrocks, 2006). For example, the molecular geometry of LPLs and FFAs is quite different from their mutual parent molecule (i.e., a phospholipid). They may therefore also cause changes in the micro-architecture and biophysical properties of phospholipid membranes and potentially affect membrane-associated signaling functions (Figure 2; Yu et al., 1992; Choi and Yu, 1995; Choe et al., 1995; Patel et al., 2001; reviewed in Piomelli et al., 2007). In addition, PLA2 mediated FFA liberation is the starting point for the generation of an extensive array of lipid-based neuromodulators. That includes FFAs themselves (e.g., AA and DHA) and LPLs (e.g., lysolecithin) but also encompasses an extensive array of biologically active molecules such as eicosanoids, isoprostanes, and neuroprostanes generated by a variety of lipid metabolizing pathways. Many of these pathways act as direct effectors of signal transduction in the nervous system through either direct interaction with ion channels or binding to G-protein-coupled or nuclear receptors (reviewed in Piomelli et al., 2007; Sultana et al., 2013).
FIGURE 6
www.frontiersin.org
FIGURE 6. Mechanisms of PUFAs peroxidation and hydrolysis. Neuronal glycerophospholipids contain high levels of methylene-separated PUFAs such as the 20:4 omega-6 fatty acid arachidonic acid (AA) shown attached at the second carbon of the phospholipid glycerol backbone (the sn-2 position) in the diagram. These PUFAs are very sensitive to free radical attack at their bis-allylic C-H bonds (indicated by *) and subsequent (per)oxidation. This process starts with (I) the formation of a C-centered radical (indicated by •) followed by (II) recombination with molecular oxygen that is normally present in abundance in lipid bilayers. The lipid peroxide radicals formed in step II can start a peroxidation chain reaction unless (IV) defused by reduction through for instance donation of a proton by vitamin E or neutralization by other chemical mechanisms. Peroxidation of these PUFAs (V) recruits members of the PLA2 family of phospholipases. PLA2 mediated hydrolysis of peroxidized PUFAs (VI) generates peroxidized FFAs and LPLs.
Poly-unsaturated fatty acid peroxidation generally also leads to the formation of electrophilic aldehydes such as acrolein, malondialdehyde (MDA), 4-hydroxy-2-nonenal (4-HNE; Pamplona, 2008, 2011). These highly reactive aldehydes can seriously compromise molecular integrity of cells and tissues through the formation of molecular adducts with phospholipids, proteins, and DNA. However, recent evidence suggests that these lipid-derived aldehydes may also induce adaptive responses driven to decrease oxidative damage and improve antioxidant defenses (see for instance Pizzimenti et al., 2013).
Lipid Peroxidation and (Brain) Aging
Considering that neuronal membranes tend to have a high PUFA content it is not surprising that lipid peroxidation and its byproducts are increasingly implicated in brain aging. For instance, accumulation of MDA, 4-HNE, acrolein is commonly reported in aged brains of vertebrate and invertebrate model systems (Terman and Brunk, 2006; Zhu et al., 2006; Watson et al., 2012b). Moreover, PLA2s are also increasingly implicated in normal brain aging and the etiology of age-associated neurodegenerative disorders (Farooqui and Horrocks, 2006; Ong et al., 2010). How the products of PLA2 activity exert their detrimental effects in the aging brain is, however, not always clear but may involve one or more of the scenarios outlined in the preceding sections.
PLA2 – A Lipase at the Nexus of Aging, Oxidative Stress, Neuronal Electrical Excitability Control and Functional Decline of the Aging Nervous System?
In the preceding paragraphs we explored the prospect for interplay between oxidative stress, lipid membrane factors, PLA2, and changes in intrinsic neuronal excitability as a factor in functional decline of normal aging neurons. In this section we review the evidence that lipid peroxidation-induced PLA2 activity lays at the root of inflammation-, age-, and oxidative stress-associated functional impairment of the nervous system in Lymnaea and put our findings in the context of a growing literature suggesting that we are dealing with an evolutionary widely conserved mechanism.
As described above, aging Lymnaea display associative LTM impairment in two functionally and anatomically distinct (i.e., appetitive and respiratory) learning paradigms (Hermann et al., 2007, 2013; Watson et al., 2012a, 2013, 2014; Beaulieu et al., 2014). In both instances, LTM failure was associated with reduced spontaneous action potential activity and enhanced spike frequency adaptation of the two key interneurons underlying both execution and plasticity of the respective behaviors (i.e., CGCs and RPeD1). Both behavioral and electrophysiological facets of age-associated respiratory and appetitive LTM impairment could be reproduced in young animals through treatment with 2,2′- azobis-2-methyl-propanimidamide (AAPH), a water-soluble free radical generator commonly used to induce PUFA peroxidation, furthering the notion of plasmamembrane lipid peroxidation as a significant agent of neuronal and behavioral aging in Lymnaea (Figure 8). Evidence of increased FFA mobilization from brains and other tissues was found in both old and AAPH-treated young animals (Watson et al., 2013; Beaulieu et al., 2014). Moreover, treatment with the lipid-domain anti-oxidant α-tocopherol reversed decline in neuronal excitability in both experimental oxidative stressed and naturally aged neurons (Watson et al., 2012a,b). Remarkably, all these behavioral, electrophysiological, and biochemical symptoms of aging and experimental oxidative stress could be reversed by treatment with aristolochic acid, a broad spectrum PLA2 inhibitor that inhibits both Ca2+-dependent cPLA2 and Ca2+-independent iPLA2 (Figure 8; Vishwanath et al., 1988; Lopes et al., 1998; Carnevale and Cathcart, 2001).
No evidence was found of significant age-associated impairment or experimental oxidative stress-induced repression of transcription-independent short/intermediate term memory (S/ITM) in either of the two behavioral conditioning paradigms, suggesting that transcription-independent forms of memory appear impervious to aging or oxidative stress. As yet, we have no definitive answer to the question why LTM is selectively susceptible. However, preliminary evidence from our laboratory suggests that both aging and AAPH-induced oxidative stress in participation with PLA2 alters aspects of mitochondrial functions that may affect their ability to contribute to intracellular Ca2+-buffering. The molecular pathways underlying LTM formation in Lymnaea involve “traditional” conserved cAMP- and Ca2+-sensitive CREB- and MAPK-dependent pathways (Ribeiro et al., 2003; Wagatsuma et al., 2006; Guo et al., 2010). It is therefore conceivable that dysregulation of intracellular Ca2+ homeostasis disrupts transcription-regulation pathways involved in LTM expression. Furthermore, the observation that PLA2 inhibition rescues LTM is particularly intriguing in view of evidence implicating FFAs and PLA2 in mitochondrial uncoupling in pancreatic β-cells (Ježek et al., 2010, 2014). Although speculative at this point in time, it is not unreasonable to postulate that a similar mechanism may underlie the PLA2-associated LTM impairment in Lymnaea.
Intriguingly, selective appetitive LTM impairment could also be induced in young Lymnaea through systemic activation of their cellular immune system (Hermann et al., 2013). As before in naturally aged and experimentally oxidation-stressed young animals, inflammation-associated LTM impairment could be rescued by means of PLA2 inhibition (Hermann et al., 2013). Together, our data indicate that the processes underlying Lymnaea’s transcription-dependent forms of memory are particularly vulnerable to aging, oxidative stress, and inflammation and that PLA2 plays a pivotal role in each of these models of associative LTM failure.
PLA2’s as Therapeutic Targets?
PLA2’s and deregulation of lipid metabolism are increasingly implicated in human age-, oxidative stress-, and inflammation-associated conditions including cardiovascular diseases and age-associated neurodegenerative diseases such as Alzheimer’s disease (Sun et al., 2004; Farooqui and Horrocks, 2006; Adibhatla and Hatcher, 2008; Sanchez-Mejia et al., 2008; Chalimoniuk et al., 2009; Sanchez-Mejia and Mucke, 2010; Desbene et al., 2012; Gentile et al., 2012; Hui, 2012; reviewed in Sun et al., 2014). They are therefore, not surprisingly, increasingly targeted or considered as targets for therapeutic intervention (see for instance, Garcia-Garcia and Serruys, 2009; Quach et al., 2014). Particularly, it is intriguing that lipoprotein-associated phospholipase A2 (Lp-PLA2), also known as platelet-activating factor acetylhydrolase (PAF-AH) was identified as a potential factor in cerebrovascular oxidative stress and inflammation and risk factor for human dementia and subsequently drew considerable interest as a therapeutic target in Alzheimer’s disease (Davidson et al., 2012; Acharya et al., 2013; Fitzpatrick et al., 2014). However, there are still a lot of unknowns surrounding the pathways, processes and conditions leading to PLA2 activation in different cell types that should be resolved before effective and safe therapeutic targets and agents for the treatment and prevention of these increasingly germane age-associated public health concerns (Sun et al., 2014). For instance, whereas many studies implicate PLA2 hyperactivity as a factor in the etiology of Alzheimer’s disease (Sanchez-Mejia and Mucke, 2010; Gentile et al., 2012; Hui, 2012), other studies suggest that hypoactivity of intracellular PLA2s (cPLA2 and iPLA2) are a contributing factor to memory impairment and Alzheimer’s neuropathology (reviewed by Schaeffer et al., 2009).
Relevance and Future Directions
The evidence reviewed here infers close ties between inflammation, oxidative stress, lipid-peroxidation, PLA2-activation, neuronal excitability, and AMI in Lymnaea and echo a rapidly growing evidence from basic, clinical, and epidemiological research that PLA2s, inflammation and oxidative stress are important factors in age-related cognitive decline and impairment in humans and other mammals (Sun et al., 2004, 2014; Phillis et al., 2006; Adibhatla and Hatcher, 2008; Lee et al., 2011). Considering this remarkable conservation we believe that Lymnaea’s extraordinary suitability for detailed, direct, and tightly integrated molecule-to-behavior dissection of the neurobiological substrates of AMI will in addition to furthering understanding of the biological foundations and parameters of aging potentially also generate valuable insights into mechanisms of and possible solutions for human AMI and other age-associated afflictions of our own nervous system. Before that goal can be achieved many questions need to be answered.
Among the unanswered questions requiring further attention is the identity of the ionic mechanisms through which the products of PLA2-mediated fatty acyl hydrolysis exert their inhibitory influence on neuronal excitability. As outlined in the preceding pages, there are many possibilities to test ranging from FA dependent modulation of ion channels to alterations in autacoid modulation of synaptic functions and hypotheses encompassing elements of membrane architecture dependent regulation of neuronal electrical properties.
Another important puzzle to solve is what inhibition of PLA2, one of the main tools of membrane homeostasis and repair, does to long-term cell/tissues homeostatic integrity. Considering the impact lipid peroxidation is expected to have on membrane structure and function, our finding that PLA2 inhibition restored rather than aggravated neuro-physiological and behavioral correlates of aging, experimental oxidative stress, and inflammation is puzzling and urges further study.
Yet another intriguing perspective we are currently exploring is mitochondrial involvement in the lipid peroxidation- and PLA2-dependent declining excitability and plasticity of aging Lymnaea neurons. Neurons critically depend on mitochondria to provide the energy to establish and maintain membrane excitability and engage in their quintessential and metabolically costly electrochemical signaling activities (Kann and Kovács, 2007; Mattson et al., 2008). Mitochondria are also a critical partner in intracellular free Ca2+ homeostasis and are one of the main sources of RONS and other pro-oxidants. There are many tight reciprocal links between neuronal electrical and mitochondrial activities and many neuronal excitability disorders appear to be associated with mitochondrial aberrancies (Kann and Kovács, 2007; Mattson et al., 2008; Surmeier et al., 2012). Moreover, there is also a substantial body of evidence indicating involvement of mitochondria-associated oxidative stress, lipid signaling, Ca2+ dysregulation in normal aging, and the pathogenesis of common age-associated neurological diseases including AD and PD in which lipid peroxidation and PLA2 may be key partners (reviewed in Thibault et al., 2007; Mattson et al., 2008; Toescu and Vreugdenhil, 2010; Waldbaum and Patel, 2010). The Lymnaea model system we have developed provides an excellent platform to investigate these interactions between neuronal energy metabolism, electrical excitability, and plasticity in great detail.
The Lymnaea research platform we portrayed here will also be a great tool to investigate fundamental mechanisms of neuronal and behavioral aging and test other existing and emerging aging theories. For example, there is growing evidence that insulin and other insulin-like peptides (ILPs) modulate aspects of plasticity in the Lymnaea CNS and enhance learning abilities in older learning-impaired snails (Murakami et al., 2013; Pirger et al., 2014). These findings resonate very well with a rapidly expanding literature that ILPs and insulin resistance are a major endocrine factor in aging in general and aging of the nervous system in particular (Kim and Feldman, 2012; Alcedo et al., 2013). Moreover, because of its identified neurons the model system will allow us to explore many important fundamental scientific questions about aging neurons and brains like: do neurons age according to “plan” or do they fail at random? Are some neuronal physiological phenotypes more vulnerable to aging than others? Can we reverse the effects of aging without repercussion? What are the principles and mechanisms underlying robust and reliable complex brains? Are all changes observed in aging neurons mal-adaptive or are some perhaps compensatory and adaptive? Are neurons and brains life cycles subject to the same resource constraints and trade-offs as other types of cells and tissues in the animal body?Frontiers | Phospholipase A2 – nexus of aging, oxidative stress, neuronal excitability, and functional decline of the aging nervous system? Insights from a snail model system of neuronal aging and age-associated memory impairment | Genetics
https://www.frontiersin.org/articles/10.3389/fgene.2014.00419/full
http://spot.colorado.edu/~falke/figures/fig13/figure13.html
Revue des Maladies Respiratoires
Volume 25, Issue 9, November 2008, Page 1190
Role of cytosolic phospholipase A2 in Pseudomonas aeruginosa-induced inflammation
Author links open overlay panelY.WuD.LeducI.Garcia-VerdugoV.BalloyM.ChignardL.Touqui
Défense innée et Inflammation – Inserm U874, Institut Pasteur, Paris, France.
Introduction
Phospholipase A2 (PLA2) belongs to a superfamily of enzymes that hydrolyse phospholipids. This family can be classified into secretory PLA2 (sPLA2), cytosolic PLA2 (cPLA2) and Ca2+-independent PLA2 (iPLA2). cPLA2 selectively release arachidonic acid (AA) from membrane phospholipids. AA metabolites play a role in inflammation and have cytotoxic effects. Pseudomonas aeruginosa, a Gram-negative bacteria, is an opportunistic pathogen that infects immuno-compromised and cystic fibroblast patients. This results in high mortality due to the notorious resistance to antibiotics of this bacterium. But, the down-stream enzymes of the host involved in the pathogenesis of lung inflammation induced by P aeruginosa have not been identified. We postulated that cPLA2 plays an important role in P aeruginosa-induced inflammation.
Methodology
Alveolar epithelial cells A459 were pre-incubated with the indicated drugs 1 h before infection with laboratory P aeruginosa strains PAK or PAO1 or stimulation with bacterial toxins, PMA or TNFα A release was measured using [3H]-AA labeled cells. IL-8 and PGE2 levels were determined using ELISA methods. Activation of MAPKs and expression of COX enzymes were analyzed by western blot.
Results
results showed that incubation of A549 epithelial cells with a wild-type strain of P aeruginosa, PAK, led to an increased cPLA2 phosphorylation which reflects its activation. This suggests that cPLA2 may be involved in P aeruginosa-induced inflammation. We found that either PAK or another P aeruginosa strain, PAO1, induced a time-dependent AA release from A549 cells. The production of prostaglandin E2 (PGE2), a downstream metabolite of AA, was increased significantly after stimulation with PAK PAK also induced IL-8 synthesis. In addition, our results showed that PAK induced p38MAPK phosphorylation in a time- and concentration- dependent manner. SB2035803, a specific inhibitor of p38 MAPK, abolished the effect of PAK on both PGE2 and IL-8 synthesis. Neither Erk nor Junk MAPK was involved in this process. The expression of cyclooxygenase (COX), which converts AA into PGE2, was increased after PAK stimulation. Finally, the effects of different virulence factors isolated from P aeruginosa, including LPS, flagellin and pili were investigated. LPS had no effect on PGE2 release and IL-8 synthesis. Flagellin stimulation increased IL- 8 expression but had no effect on PGE2 release. The δflic strain, in which the flagellin gene was deleted, had less effect on IL-8 synthesis but had similar effect on PGE2, as compared to the PAK strain.
Conclusions
Collectively, our results show that cPLA2 is involved in P aeruginosa-induced inflammation through a process probably involving p38MAPK. However, the virulence factors involved in cPLA2 activation remains to be identified.Role of cytosolic phospholipase A2 in Pseudomonas aeruginosa-induced inflammation - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0761842508750599
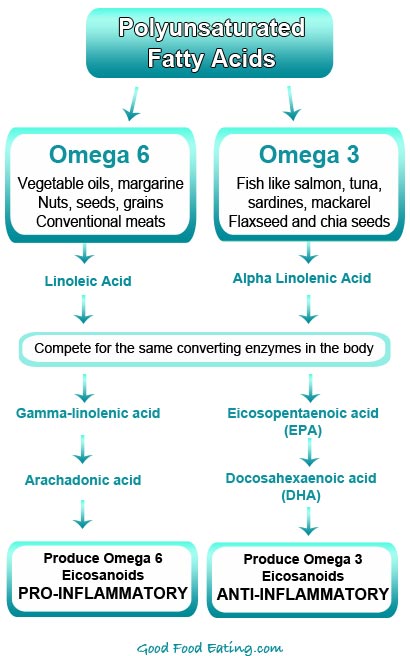

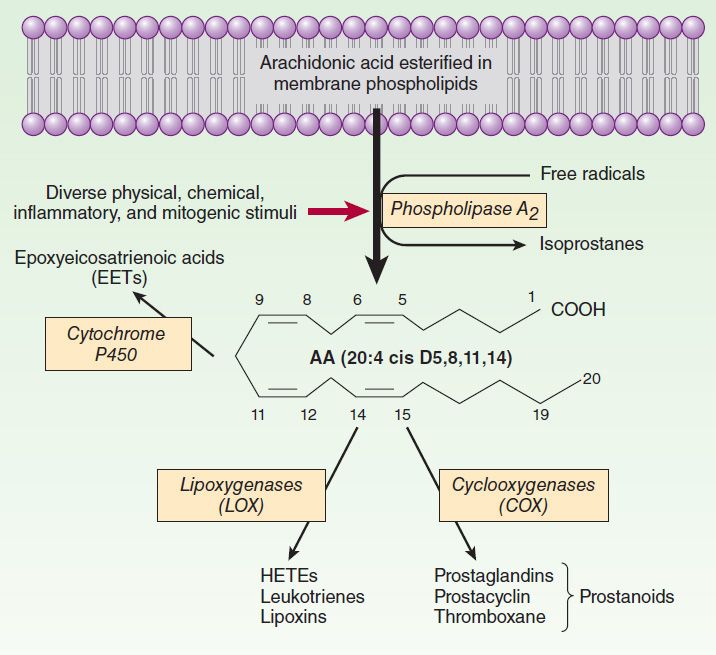
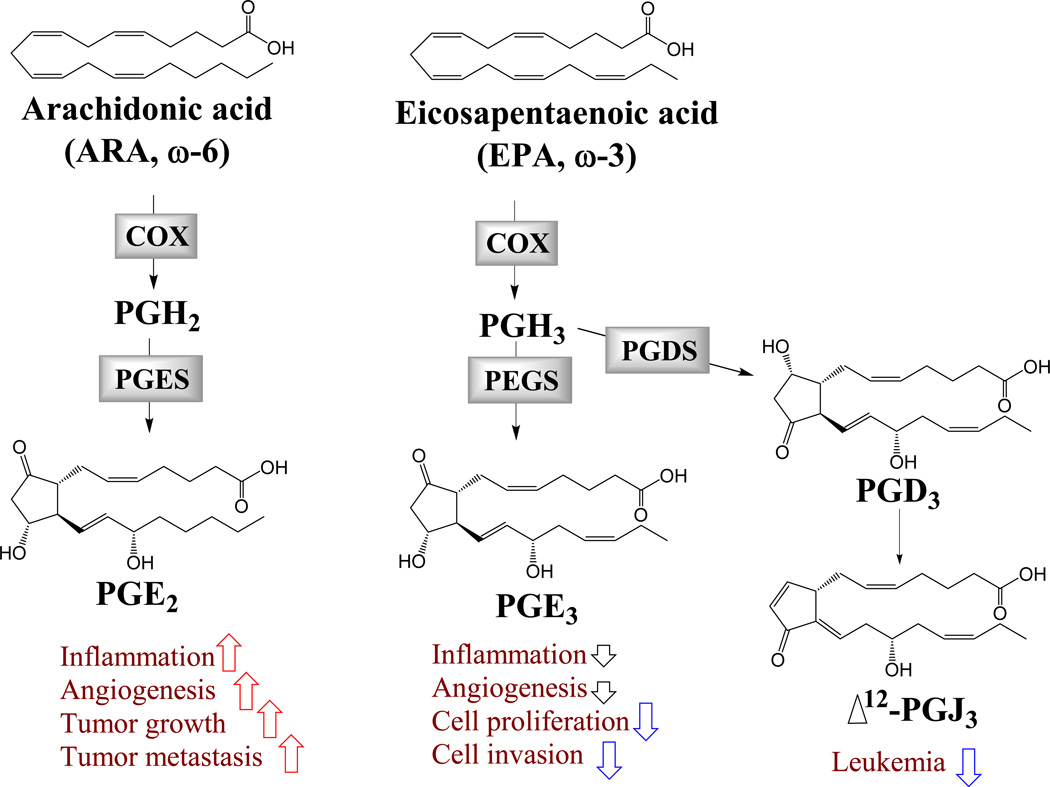
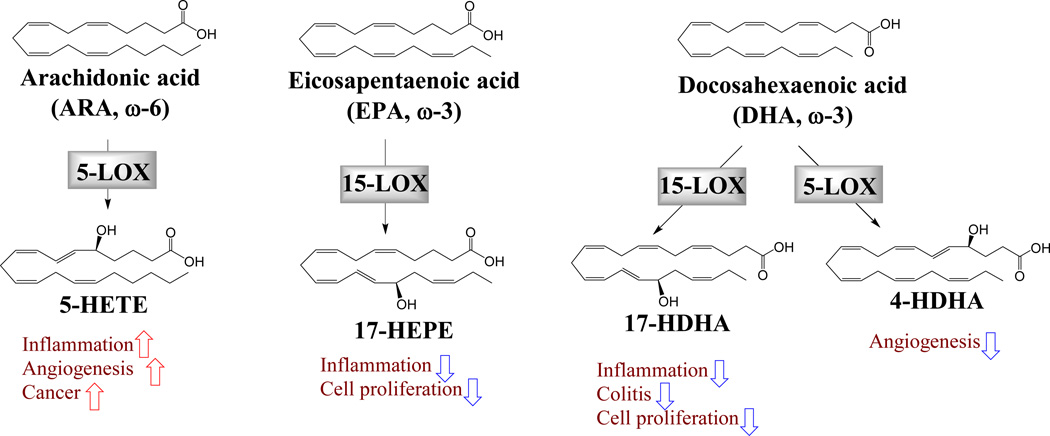
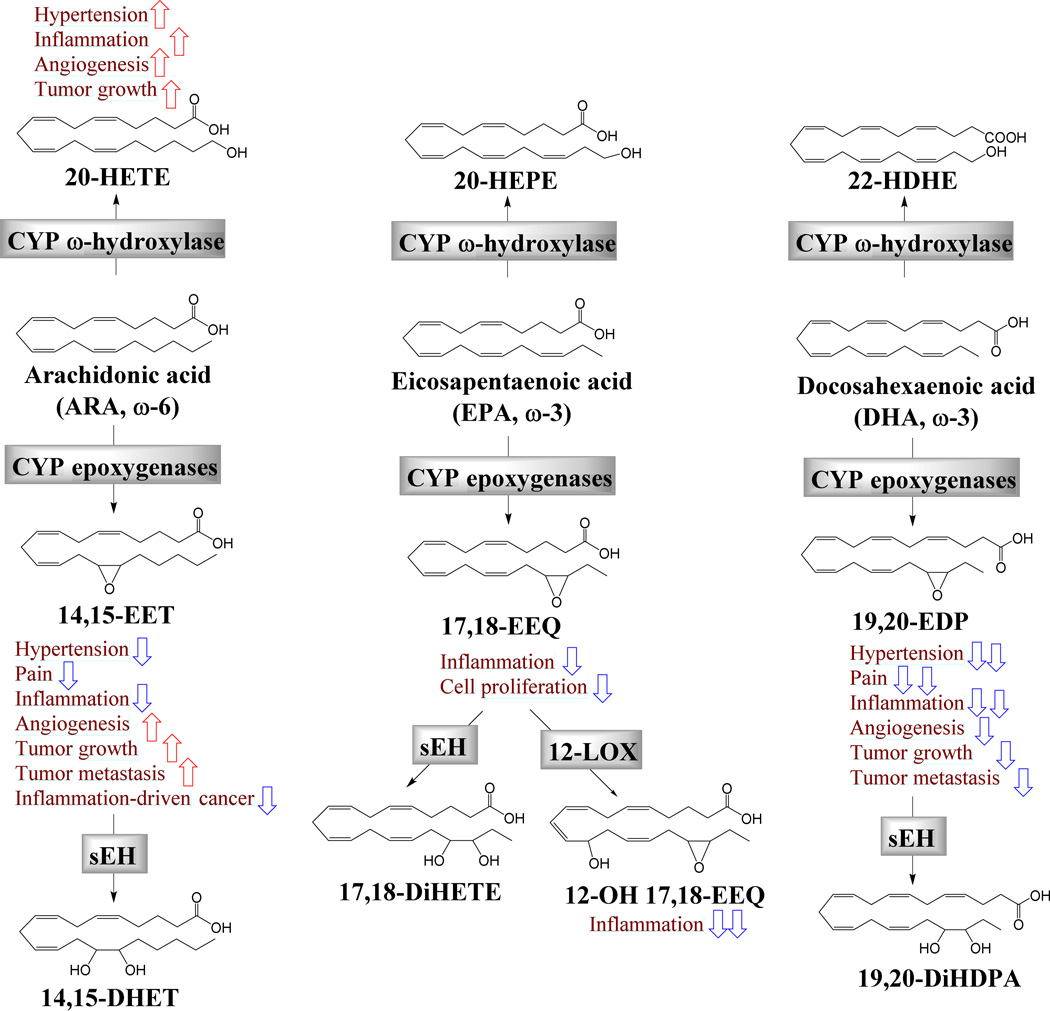
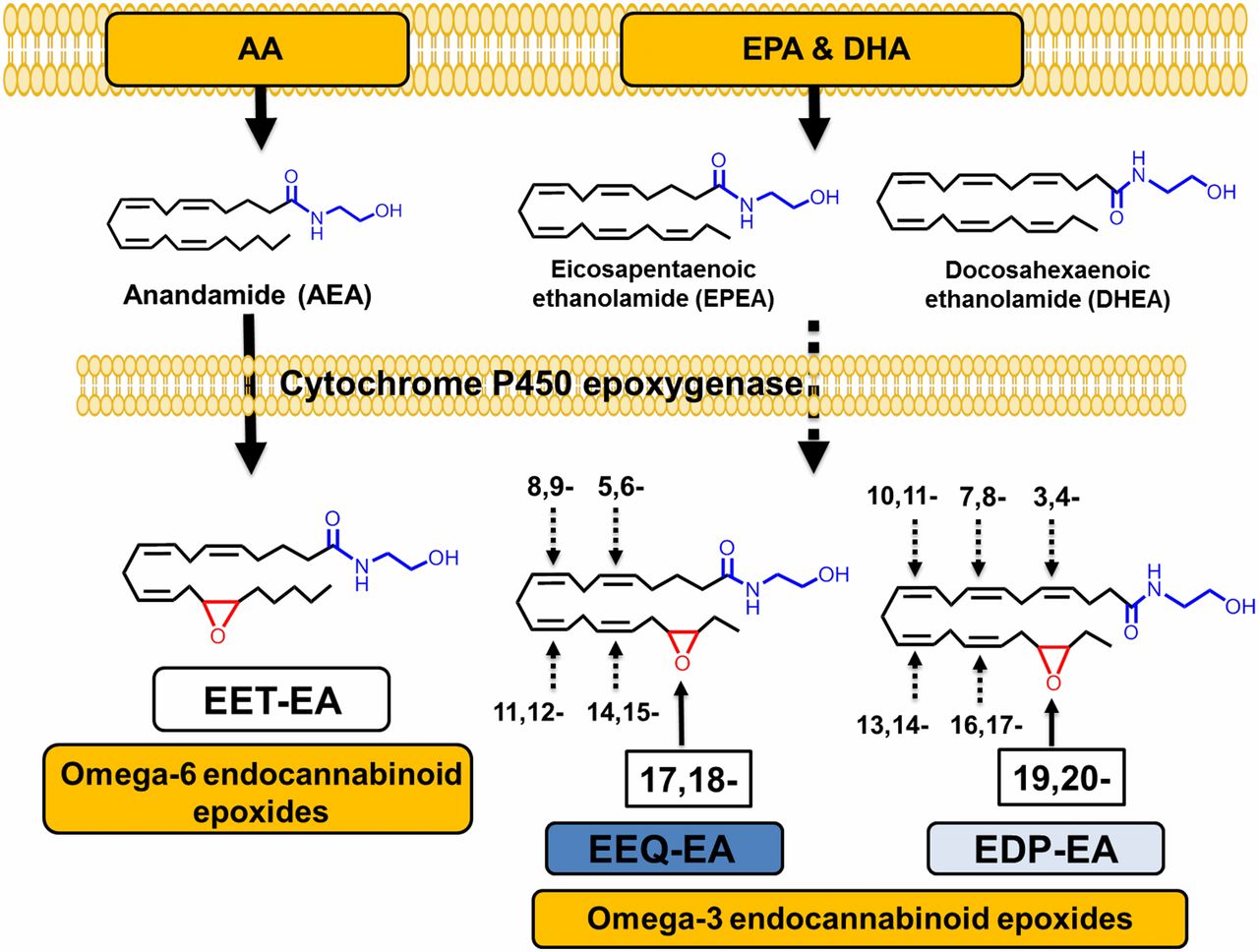
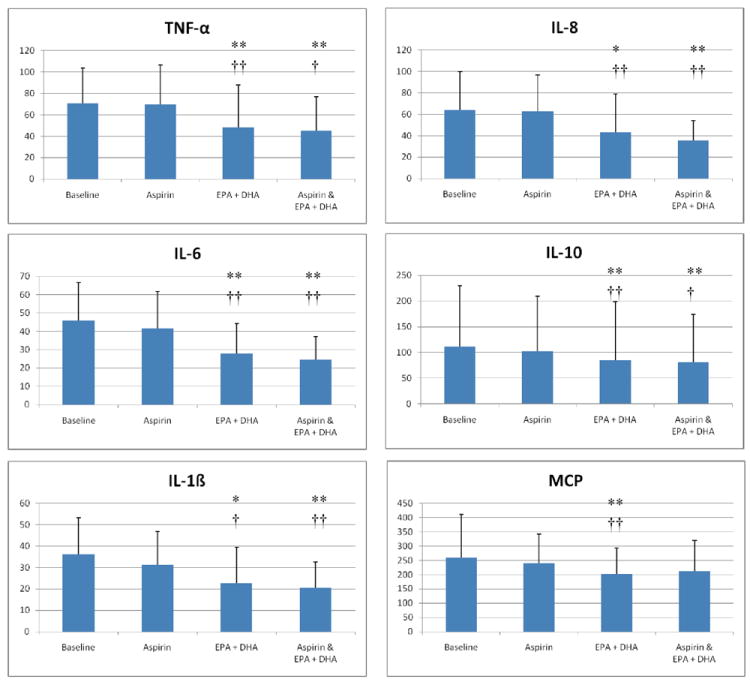

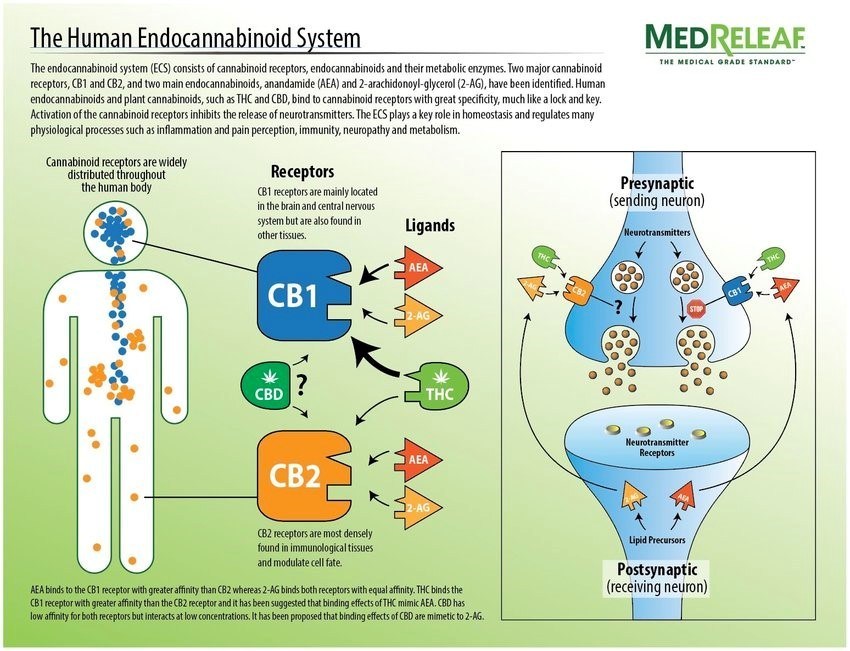
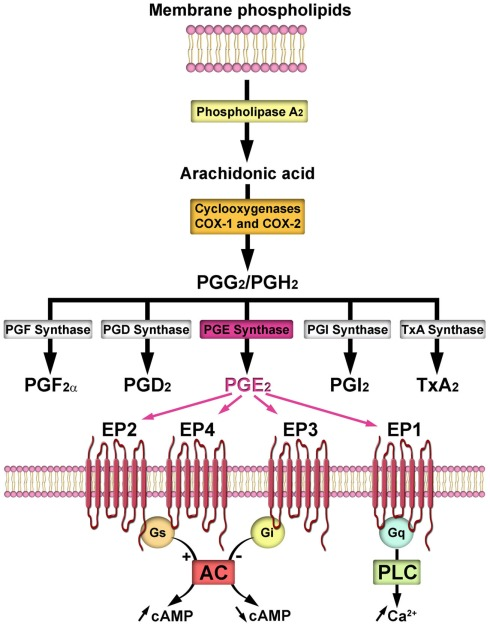
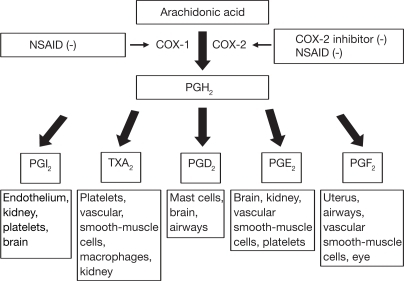
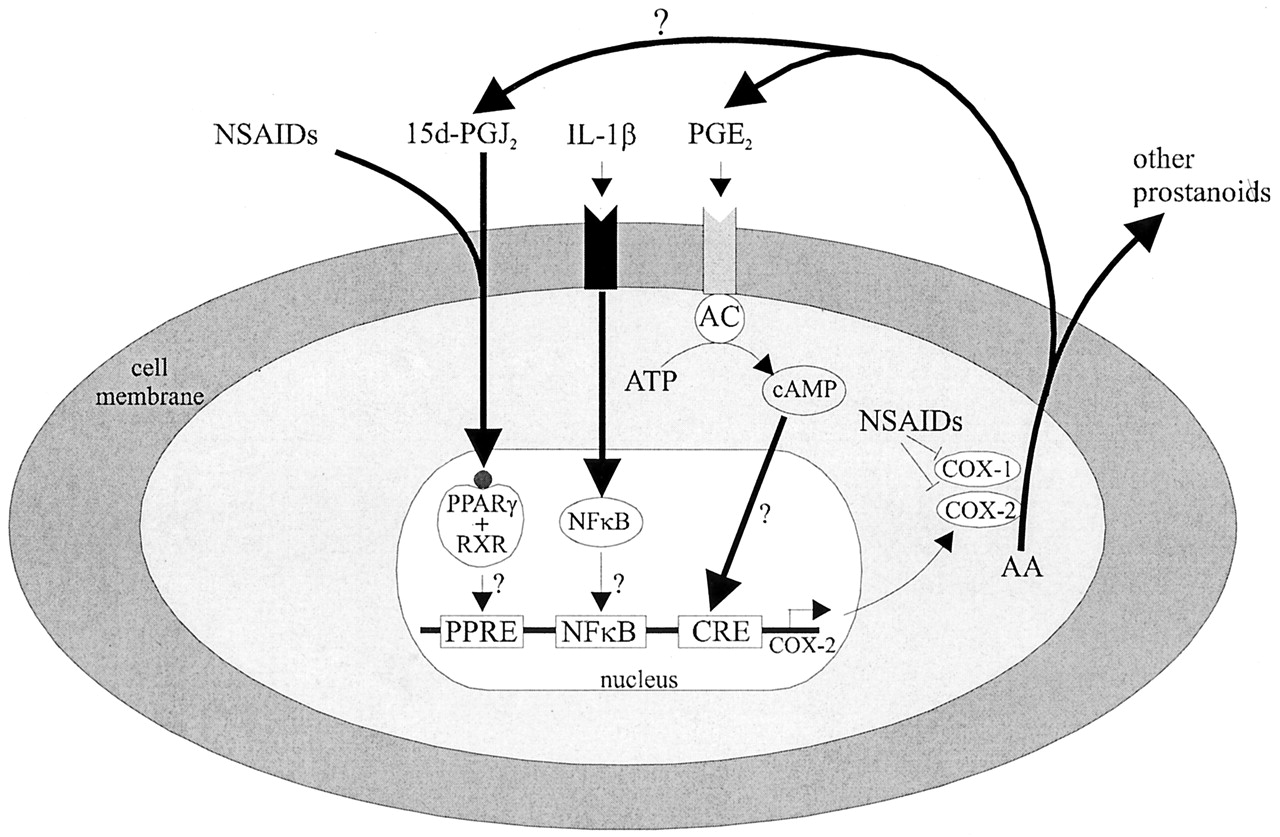

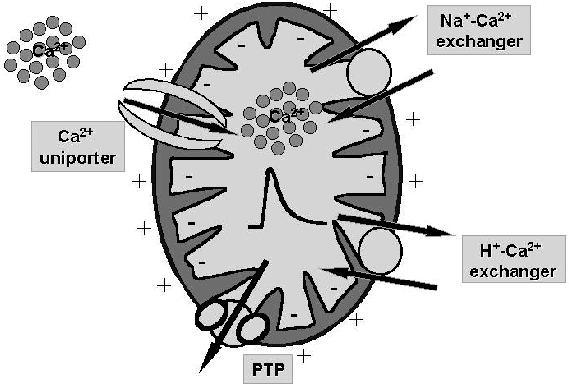

.png)
.jpg)