ACEs AND ACE INHIBITORS
ĪĪ
Classification
Distribution
Function
Dycfunction
ĪĪ
ACE2 is a type 1 integral membrane glycoprotein [8] that is expressed and active in most tissues. The highest expression of ACE2 is observed in the kidney, the endothelium, the lungs, and in the heart [2, 8].
Angiotensin-Converting Enzyme (ACE) | Lab Tests Online
https://labtestsonline.org/tests/angiotensin-converting-enzyme-ace
Angiotensin-converting enzyme (ACE) is an enzyme that helps regulate blood pressure. An increased blood level of ACE is sometimes found in sarcoidosis, a systemic disorder of unknown cause that often affects the lungs but may also affect many other body organs, including the eyes, skin, nerves, liver, and heart., This test measures the amount of ACE in the blood.ĪĪ
ACE2: from vasopeptidase to SARS virus receptor
July 2004 Īż Trends in Pharmacological Sciences
Anthony J. TurnerJulian A HiscoxNigel M. Hooper
The zinc metallopeptidase angiotensin-converting enzyme 2 (ACE2) is the only known human homologue of the key regulator of blood pressure angiotensin-converting enzyme (ACE). Since its discovery in 2000, ACE2 has been implicated in heart function, hypertension and diabetes, with its effects being mediated, in part, through its ability to convert angiotensin II to angiotensin-(1-7). Unexpectedly, ACE2 also serves as the cellular entry point for the severe acute respiratory syndrome (SARS) virus and the enzyme is therefore a prime target for pharmacological intervention on several disease fronts.ACE2: from vasopeptidase to SARS virus receptor
https://www.researchgate.net/publication/8540237_ACE2_from_vasopeptidase_to_SARS_virus_receptorĪĪ
Angiotensin-Converting Enzyme (ACE) is a key enzyme in cardiovascular pathophysiology (Yang et al., 1970, Ehlers et al., 1989, Bernstein et al., 2005). ACE is critically involved in endothelial homeostasis by controlling the circulating levels of bradykinin and angiotensin II and thereby affects vascular development, tone, and permeability, particularly in the lung, the organ with the highest level of endothelial cell ACE expression. Lung and blood activity of ACE is also a sensitive marker of endothelial dysfunction, including PAH and lung vascular injury (Kay et al., 1982, Keane et al., 1982, Oparil et al., 1988, Muzykantov and Danilov, 1991, Morrell et al., 1995a, Atochina et al, 1997, Orfanos et al., 2000).
REDUCED EXPRESSION OF ANGIOTENSIN I-CONVERTING ENZYME IN CAVEOLIN-1 KNOCKOUT MOUSE LUNGS
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2919634/ĪĪ
Effect of flow and surface area on angiotensin-converting ...
https://www.physiology.org/doi/abs/10.1152/jappl.1987.62.5.2042
Pulmonary angtiotensin-converting enzyme (ACE) is located on the luminal surface of pulmonary microvasculature. Multiple indicator-dilution techniques have been used to measure pulmonary ACE activity in vivo and in isolated lungs. These studies suggest that ACE activity is depressed in several forms of acute lung injury.
Cited by: 9
Publish Year: 1987
Author: R. Moalli, B. R. Pitt, C. N. GillisĪĪ
Angiotensin Peptides and Lung Cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3314267
Angiotensin converting enzyme (ACE), produced primarily in the epithelial cells of the lung, catalyzes the conversion of Ang I to Ang II and attenuation of the enzyme actions by selective inhibitors, such as enalopril, captopril, perindopril, is among the therapeutic modalities commonly used to treat patients with high blood pressure.
Cited by: 29
Publish Year: 2011
AutĪĪ
Curr Cancer Drug Targets. Author manuscript; available in PMC 2012 Mar 28.
Published in final edited form as:
Curr Cancer Drug Targets. 2011 May; 11(4): 394©C404.
Angiotensin Peptides and Lung Cancer
P.E. Gallagher,*,1 K. Cook,1,2 D. Soto-Pantoja,3 J. Menon,4 and E.A. Tallant1,2
1Hypertension and Vascular Research Center
2Molecular Medicine and Translational Science Program, Wake Forest University School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157, USA
3Laboratory of Pathology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
4Ben May Department of Cancer Research, University of Chicago, Chicago, IL 60637, USA
*Address correspondence to this author at the Hypertension and Vascular Research Center, Wake Forest University School of Medicine, Medical Center Boulevard, Winston-Salem, NC 27157, USA; Tel: (336) 716-4455; Fax: (336) 716-2456; ude.cmbufw@hgallagp
Abstract
Lung cancer is a leading cause of death in both men and women, with over 1,000,000 new cases diagnosed worldwide annually and a 5-year survival rate of only 14%, a figure that has improved little in the past thirty years. This poor prognosis suggests a need for novel approaches for the treatment and prevention of lung cancer. The renin-angiotensin system is an established, primary regulator of blood pressure, homeostasis, and natriuresis; however, compelling evidence indicates that the angiotensin peptides also play a role in cell proliferation and inflammation.Angiotensin II is a vasoconstrictor, a mitogen, and an angiogenic factor, while angiotensin-(1-7) has vasodilator, anti-proliferative, and anti-angiogenic properties. This review focuses on studies examining the renin-angiotensin system in pulmonary cancers and whether clinical intervention of this pathway may serve as an effective chemotherapeutic and/or chemopreventive modality for lung cancer.
Keywords: Renin-angiotensin system, angiotensin, lung cancer, pulmonary cancer, angiotensin-(1-7), angiotensin converting enzyme, angiogenesis, angiotensin receptor blockerEFFECT OF ACE INHIBITORS AND ARBS ON LUNG CARCINOGENESIS
In a retrospective study of 5207 patients in Scotland, the relative risks of incident and fatal cancer among the 1559 patients treated with ACE inhibitors were significantly reduced, to 0.72 and 0.65, respectively [77]. The relative risk was lowest in patients with lung, colon, or sex-specific cancer, as compared with other sites. Inhibition of ACE activity decreases the vasoconstrictor and growth stimulator Ang II and increases Ang-(1-7), a heptapeptide with vasodilator and anti-proliferative properties [78, 79] (Fig. 2). This suggests that the reduced risk of cancer could result not only from decreased Ang II but also from the elevation in Ang-(1-7). Since this clinical study demonstrated a reduced incidence of lung cancer, the alteration in the angiotensin peptide levels by ACE inhibitors may not only inhibit the growth of lung cancer cells, but also prevent lung tumor formation. In a similar retrospective study, patients with advanced non-small-cell lung cancer that received a therapy regimen of an ACE inhibitor or ARB with a first-line platinum-based chemotherapeutics had a 3.1 month longer median survival than patients not receiving the anti-hypertensive medication [80]. These results suggest that ACE inhibitors or ARBs may provide enhanced efficacy for lung cancer in combination with chemotherapeutics.
Pre-clinical studies with ACE inhibitors or ARBs also demonstrate a role for the RAS in lung carcinogenesis as well as prevention of lung tumor formation. Prontera et al. showed that the combination of the matrix metalloproteinase inhibitor batimastat and the ACE inhibitor captopril markedly reduced the mean volume, mean metastasis and survival time of Lewis lung tumors in syngeneic C57BL/6 mice as compared to control animals [59]. In addition, cyclosporin-enhanced pulmonary metastases were reduced to control levels in mice following treatment with the AT1 receptor antagonist losartan [62]. Attoub et al. reported that the ACE inhibitor captopril reduced the growth of human LNM35 lung tumors as well as lymph node metastases in mice by more than 50% as compared to tumors in control animals with no appreciable side-effects [72]. Immunohistochemical analysis of tumor tissue sections showed that captopril administration markedly reduced the cell proliferation marker Ki67. Captopril induced apoptotic morphological changes in LNM35 lung cancer cells, suggesting that the anti-proliferative effect of this ACE inhibitor was due to induction of programmed cell death. Taken together, these studies suggest that administration of ACE inhibitors or ARBs may effectively reduce lung cancer proliferation as well as lung tumor metastases to prevent further tumor formation.
EFFECT OF ACE INHIBITORS AND ARBS ON LUNG TUMOR ANGIOGENESIS
Ang II increased the growth of vascular smooth muscle cells in vitro and stimulated blood vessel formation in several in vivo models of angiogenesis [81-84]; conversely, ACE inhibitors that block endogenous Ang II production or ARBs that attenuate Ang II activity reduce angiogenesis. The AT1 receptor antagonist candesartan (TCV-116, CV11974) significantly inhibited the growth of pulmonary metastases from renal cell carcinoma with an associated decrease in vascular endothelial growth factor (VEGF) and inhibition of angiogenesis as compared to control animals [63]. In similar studies, the lung metastasis of intravenously injected Lewis lung carcinoma cells was markedly inhibited following candesartan or ACE inhibitor lisinopril treatment with a significant decrease in tumor-associated blood vessel formation [61]. A significant reduction in VEGF-A mRNA and protein in association with attenuated tumor growth was also observed in Lewis lung tumors following candesartan administration [52], Incubation of Lewis lung carcinoma cells with Ang II caused a significant increase in VEGF-A mRNA and protein which was prevented by co-administration of an AT1 receptor antagonist. While these studies demonstrate that Ang II stimulates angiogenesis in lung tumors through activation of AT1 receptors, the molecular signaling pathways involved in the process are not known.ĪĪ
AT2 RECEPTOR AND LUNG CARCINOGENESIS
As illustrated in Fig. (1), Ang II activates both AT1 and AT2 receptors, two distinct pharmacological classes of seven transmembrane, G protein-coupled receptors [16, 17]. While the physiological role of the AT1 receptor in blood pressure regulation, diuresis, and mitogenesis is established, the precise function of the AT2 receptor is not clear for many tissues. Several studies demonstrate that Ang II binding to the AT2 receptor may be involved in lung carcinogenesis. Kanehira et al. showed that AT2 receptor-null mice had reduced tumor number and multiplicity following treatment with NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone] as compared to control [64]. Co-incubation of A549 human lung cancer cells with lung fibroblasts from the AT2 receptor-null mice decreased colony count with an associated increase in transforming growth factor-”┬ (TGF-”┬) production, suggesting that the AT2 receptor on lung fibroblasts may be involved in chemical carcinogen-induced lung tumorigenesis. The immunostain for both the AT1 and AT2 receptors was enhanced in NNK-induced tumor sections, [65] indicating a potential role not only for the angiotensins receptors on lung cancer cells but also on cells of the tumor microenvironment in lung carcinogenesis. In support, the volume of Lewis lung tumor xenografts was significantly reduced in mice following administration of the AT2 receptor antagonist PD123,319 (20 mg/kg/day) with an associated reduction in VEGF [60]. Inhibition of the parent cell growth was also observed following blockade of the AT2 receptor with an associated reduction in VEGF. These studies suggest that activation of the AT2 receptor may promote lung tumorigenesis by increasing cell proliferation and tumor angiogenesis. Conversely, Pickel et al. [85] found that over-expression of the AT2 receptor using nanoparticle vectors reduced the growth, increased the number of apoptotic cells and activated caspase 3 in the human adenocarcinoma cell line A549 and bronchioalveolar carcinoma line H358. These results suggest that activation of the AT2 receptor may inhibit lung cancer proliferation and are in conflict with the studies described above in carcinogen-induced lung tumorigenesis and in the Lewis lung tumor xenografts. The differences may be reflective of the AT2 receptor number as an 80-fold increase in the AT2 receptor mRNA was detected in the A549 transfected cell line [85]. While the protein concentration of the receptor was not determined, the mRNA data suggest that the level of the AT2 receptor achieved by transfection using the nanoparticle vectors may be considerably higher than occurs by normal, physiological regulation. Further studies are certainly needed to rectify this conflicting data before the AT2 receptor could be considered as a target for lung cancer intervention.
ANG-(1-7) AND LUNG CANCER
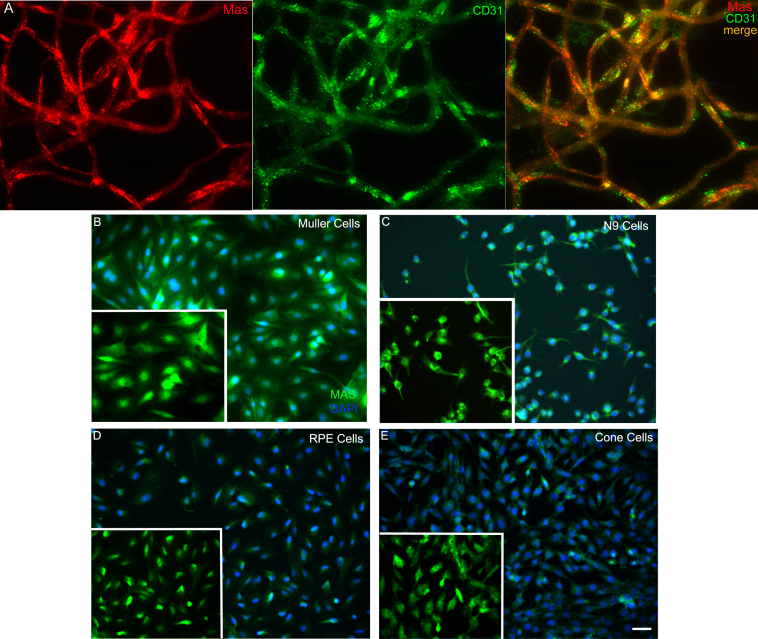
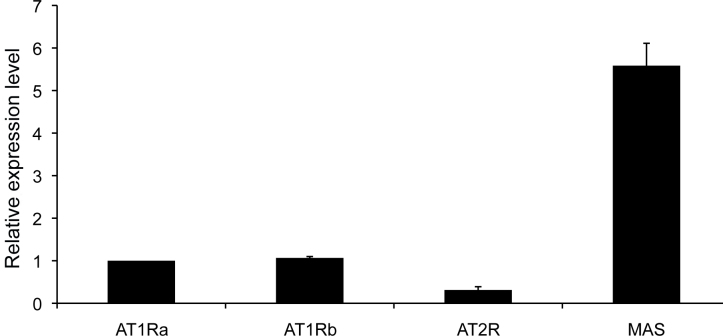
As discussed above, Ang-(1-7) is an endogenous, seven amino acid peptide hormone of the RAS with vasodilator, anti-proliferative, and anti-thrombotic properties. Ang-(1-7) mediates biological properties through activation of a unique, G-protein-coupled AT(1-7) receptor, mas. We showed that this heptapeptide inhibits the growth of human lung cancer cells through a reduction in MAP kinase [53]. Ang-(l-7) caused a significant decrease in serum-stimulated growth of human SK-LU-1, A549, and SK-MES-1 lung cancer cells with a dose- and time-dependent reduction in DNA synthesis and IC50Ī»s in the sub-nanomolar range. Other angiotensin peptides, Ang I, Ang II, Ang-(2-8), Ang-(3-8) and Ang-(3-7), did not attenuate mitogen-stimulated DNA synthesis of SK-LU-1 cells, demonstrating that Ang-(l-7) selectively inhibits the growth of these human cancer cells. The Ang-(l-7) receptor antagonist [D-Ala7]-Ang-(l-7) blocked the attenuation of serum-stimulated DNA synthesis in SK-LU-1 cells by Ang-(l-7), while neither AT1 nor AT2 angiotensin receptor subtype antagonists prevented the response to the heptapeptide. Mas mRNA and protein were detected in the three lung cancer cell lines, suggesting that mas mediated the anti-proliferative response. Pretreatment of SK-LU-1 cells with 10 nM Ang-(l-7) reduced serum-stimulated phosphorylation of ERK1 and ERK2 (by 61% and 68%, respectively), indicating that the anti-proliferative effects may occur, at least in part, through inhibition of the ERK signal transduction pathway. These results suggest that Ang-(l-7) inhibits lung cancer cell growth through activation of an angiotensin peptide receptor and may represent a novel therapeutic and/or preventive treatment for lung cancer.
Ang-(1-7) administration markedly attenuated the growth of human A549 lung cancer xenografts [57]. Tumor volume was reduced by 30% following medication with the heptapeptide as compared with the size before treatment; in contrast, tumor size in the saline-treated animals increased 2.5-fold. No adverse side effects, including changes in heart and body weight, heart rate or blood pressure, were observed following Ang-(1-7) administration. The tumor inhibition correlated with a reduction in the proliferation marker Ki67 as well as cyclooxygenase 2 (COX-2) mRNA and protein in tumors from the Ang-(1-7)-medicated animals as compared with the saline control tumor tissue. In contrast, the heptapeptide had no effect on COX-1 mRNA in xenograft tumors. Similar results for COX regulation were observed with the parent A549 human lung cancer cells in tissue culture. These results suggest that Ang-(1-7) may decrease COX-2 activity and pro-inflammatory prostaglandins to inhibit lung tumor growth. Because Ang-(1-7) reduces growth through activation of a selective AT(1-7) receptor, the heptapeptide may serve as a first-in-class, targeted therapy for lung cancer by reducing COX-2.
COXs, the key enzymes in the conversion of arachidonic acid to prostaglandins (PGs) and other bioactive lipids, are up-regulated by a wide variety of mitogens and tumor promoting agents involved in the regulation of normal growth responses and in aberrant cellular growth. Selective COX-2 inhibitors provided a promising treatment for lung cancer. COX-2 is over-expressed in lung tumors [86-89] COX-2 inhibitors prevent lung cancer in experimental animals, [86, 87, 90, 91] and epidemiological studies suggest that regular use of NSAIDs can reduce incidence of lung cancer [92]. Unfortunately, a number of these drugs were withdrawn from the market due to an increased risk of cardiovascular events with long-term drug treatment for colon cancer [93]. The APPROVe (Adenomatous Polyp Prevention on VIOXX) study was stopped after 18 months since patients taking rofecoxib had twice the risk of a myocardial infarction compared with those receiving placebo. Increased incidence of thrombotic events (myocardial infarction, angina, stroke, transient ischemic events, etc.) was reported in previous clinical trials with Vioxx (VIGOR, with 8076 patients) and Celecoxib (CLASS, with 8059 patients); additionally, in 23,407 patients in primary prevention trials, the increased incidence of cardiovascular events was 0.74% and 0.80% with rofecoxib and celecoxib, respectively, compared to the normal population (0.52%) [93]. Since Ang-(1-7), an endogenous peptide hormone, causes a significant but not complete reduction in COX-2, treatment with the heptapeptide or drugs that elevate endogenous Ang-(1-7) may represent a novel mechanism to reduce COX-2 activity and inhibit lung cancer cell growth and tumor formation. Ang-(1-7) has anti-thrombotic properties, which will oppose any increase in thrombosis by reduced COX-2 activity. Ang-(1-7) caused a decrease in thrombus weight following vena cava occlusion as well as reduced collagen adhesion to platelets, in 2-kidney, 1-clip hypertensive rats [94]. Yoshida et al. [95] reported an increase in plasminogen activated inhibitor-1 (PAI-1) and tissue plasminogen activator in cultured human umbilical vein endothelial cells treated with Ang-(1-7). These results suggest that Ang-(1-7) may reduce COX-2 to inhibit lung cancer cell growth as well as provide anti-thrombotic protection against cardiovascular events due to decreased COX-2 in blood vessels.
ANG-(1-7) AND ANGIOGENESIS
Ang-(1-7) inhibited angiogenesis in a murine sponge model, a technique representative of the formation of new blood vessels from pre-existing blood vessels during wound healing [82]. In this model, a cannulated sponge disc was implanted subcutaneously in the dorsa of mice to induce a wound repair response. Infusion of Ang-(1-7) reduced hemoglobin content, blood flow, and proliferative activity in the disc, as compared to a disc-containing vehicle. The anti-angiogenic effect of the heptapeptide in this model was regulated by a [D-Ala7]-Ang-(1-7)-specific receptor and was blocked by pre-incubation with either aminoguanidine or NG-nitro-Z-arginine methyl ester (L-NAME), indicating that the response was mediated by nitric oxide [96]. While this is a model of angiogenesis during wound healing, it suggests that Ang-(1-7) may inhibit the angiogenesis that occurs during tumor formation, to supply necessary nutrients for tumor growth.
We showed that subcutaneous injection of Ang-(1-7) not only caused a significant reduction in human A549 lung tumor xenograft growth in mice but also markedly decreased vessel density [58], suggesting that the heptapeptide inhibits angiogenesis to reduce tumor size. VEGF-A, a primary angiogenic factor, was reduced approximately 85% in lung tumor xenografts from mice medicated with Ang-(1-7) as compared to tumors from saline-treated animals. An associated decrease in VEGF-A mRNA suggested that a transcriptional regulatory mechanism was involved in the reduction of angiogenic factor by the heptapeptide. Similar results were obtained with the parent A549 human lung cancer cells in culture with a maximal decrease in VEGF-A after 12 h treatment with Ang-(1-7). Antagonists specific for the Ang-(1-7) receptor completely blocked the reduction in VEGF-A by the heptapeptide. We previously showed that intravenous infusion of 24 ”╠g/kg/h of Ang-(1-7) for 28 days markedly attenuated human A549 lung cancer xenografts [57]. As shown in Fig. (3), VEGF-A was also reduced in the tumors from animals administered Ang-(1-7), suggesting that the decrease in the angiogenic factor by the heptapeptide was not affected by different drug scheduling modalities. Taken together, these results suggest that Ang-(1-7) attenuates tumor angiogenesis by reducing VEGF-A.
Additional in vitro and in vivo models of angiogenesis were used to assess the anti-angiogenic properties of Ang-(1-7).[58] A decrease in human endothelial cell tubule formation in Matrigel was observed following a 16 h incubation with Ang-(1-7), with a maximal reduction at a 10 nmol/L concentration. The Ang-(1-7)-mediated effects were blocked by the specific Ang-(1-7) receptor antagonist [D-proline(7)]-Ang-(1-7). Similar results were obtained using human EA.hy.926 cells, a transformed human endothelial cell line derived from umbilical vein endothelial cells.[97] EA.hy.926 cells retain their endothelial phenotype and were previously used to study angiogenesis.[98,99] The cells were seeded onto Matrigel with or without 10 nM Ang-(1-7) and after 16 h, the EA.hy.926 cells were photographed to visualize and quantify tubule formation. As shown in Panel A of Fig. (4), EA.hy.926 cells formed multiple tube-like structures on Matrigel. In contrast, EA.hy.926 cells seeded in the presence of Ang-(1-7) formed fewer tube-like structures and the length of the tubes decreased compared to those in cells cultured in the absence of the heptapeptide. Tube formation was reduced significantly in the presence of increasing concentrations of Ang-(1-7) (Fig. 4, Panel B), suggesting that the heptapeptide inhibits angiogenesis in a dose-dependent manner. The chorioallantoic membrane (CAM) assay also was used to assess the effect of Ang-(1-7) on vascularization in vivo.[58] Control embryos displayed extensive neovascularization, while a marked reduction in vessel formation and branching was observed following 2-day incubation with 100 nM Ang-(1-7). These effects were blocked completely by the specific Ang-(1-7) receptor antagonist [D-proline(7)]-Ang-(1-7), suggesting that the anti-angiogenic actions of the heptapeptide were mediated by an AT(1-7) receptor. Taken together, these preclinical studies suggest that Ang-(1-7) may be a first-in-class compound for the treatment of lung cancer, providing combination therapy as a selective COX-2 and angiogenic inhibitor, targeting a specific AT(1-7) receptor mas.PERSPECTIVE
The studies discussed above suggest that the RAS not only may be an effective target for chemotherapeutic/chemopreventive intervention but also may be involved in lung carcinogenesis. Undoubtedly, controversies prevail and additional research is required. Since ACE inhibitors and ARBs are currently prescribed for hypertension control and the safety profile of these drugs is known, properly designed clinical trials with lung cancer patients assessing these two medications is certainly feasible. Of note, there are multiple formulations of ACE inhibitors and ARBs and their efficacy may differ depending on the drug structure chosen. It is unlikely that ACE inhibitors or ARBs will be a single drug treatment for lung cancer; however, these medications may provide synergistic effects to existing chemotherapies by reducing Ang II-mediated mitogenesis and angiogenesis as well as increasing the anti-proliferative and anti-angiogenic effects of Ang-(1-7). In addition, the tortuous structure of tumor blood vessels leads to vasoconstriction, thereby limiting drug delivery. Administration of either ACE inhibitors or ARBs could cause dilation of the tumor vessels, leading to improved overall drug delivery. The use of these two anti-hypertensive agents in combination with other chemotherapeutic agents is worthy of investigation.
Both arms of the RAS must be considered in the development of new therapeutic medications. Drugs that limit the synthesis or activity of Ang II will potentially decrease tumor growth by attenuating the mitogenic and angiogenic properties of the peptide. On the other hand, increased production of Ang-(1-7), administration of the heptapeptide, or activation of the mas receptor with synthetic agonists should inhibit tumor growth by initiating signaling pathways that reduce cell growth and new blood vessel formation. Since Ang-(1-7) counteracts the anti-proliferative effects of Ang II as well as other mitogens [31], drugs that activate the Ang-(1-7) arm of the RAS may be more effective. Based on our preclinical data discussed above, we initiated a Phase I clinical trial to examine the toxicity of Ang-(1-7) in patients with solid tumors. The Phase I trial was completed in 18 months and the results were published in Clinical Cancer Research [100]. The drug was well-tolerated with no hypertension, bleeding disorders, or drug-related deaths. Of the 15 evaluable patients treated with the heptapeptide, 4 patients displayed clinical benefit with an associated reduction in circulating placental growth factor, while the remainder of the patients did not. This is in agreement with our preclinical data demonstrating the anti-angiogenic effects of Ang-(1-7) [58]. These results suggest that Ang-(1-7), a drug that would increase endogenous synthesis of Ang-(1-7), or a synthetic agonist of the mas receptor may represent a novel cancer treatment. However, it is essential that the cardiovascular effects are considered when designing a chemotherapeutic drug targeting components of the RAS, as this pathway plays a critical role in blood pressure regulation, homeostasis and natriuresis.ĪĪ
Fig. (1)
The renin-angiotensin system.ĪĪ
Fig. (2)
Effect of ACE inhibition on angiotensin peptides.ĪĪ
Fig. (3)
Reduction of VEGF-A in human A549 lung tumor xenografts infused with saline or Ang-(1-7). n = 3 for each group.
Fig. (4)
Ang-(1-7) inhibition of endothelial cell tubule formation. Panel A ©C A representative photograph of endothelial cells incubated with or without Ang-(1-7) on Matrigel. Panel B ©C Quantification of tubule number following 16 h treatment with various doses of Ang-(1-7). *p<0.05; n = 3-5.ĪĪ
Table 1
Summary of Pre-Clinical Studies Supporting a Role for the RAS in Lung Cancer
Source Key Finding Reference Human NCI-H345, H209, H146,
H82, H69, H187, H2122 and A549Neprilysin is reduced in small cell and non-small cell lung cancer lines; transfection of the
neprilysin gene resulted in reduced proliferation.[49] Murine Lewis lung carcinoma cells Ang II increased Lewis lung cell VEGF-A which was prevented by addition of an AT1 receptor
antagonist CV11974.[52] Human SK-LU-1, A549, SK-MES-1
lung cancer cellsAng-(l-7) caused a significant decrease in serum-stimulated growth of human lung cancer cells
with an associated reduction in MAP kinase.[53] Human A549 lung tumor cells Ang II activation of the AT1 receptor increased cytosolic free calcium in human A549 lung cancer cells. [54] ĪĪ Ang II promoted apoptosis in lung cancer cells; treatment with ACE inhibitors or ARBs
inhibited the response.[55,56] Human A549 lung tumor xenografts Ang-(1-7) reduced COX-2 mRNA and protein in human A549 lung cancer cells. [57] ĪĪ Subcutaneous injection of Ang-(1-7) markedly decreased vessel density with a concomitant
reduction in VEGF.[58] Murine Lewis lung carcinoma
xenograftBatimastat and AT1 receptor blocker captopril reduced the mean volume and mean metastasis
of Lewis lung tumors.[59] ĪĪ Attenuated lung tumor growth and a decrease in VEGF-A was observed following ACE
inhibitor candesartan administration.[52] ĪĪ Tumor xenografts were reduced following AT2 receptor antagonist treatment with an
associated reduction in VEGF.[60 ] ĪĪ Lewis lung carcinoma metastasis was inhibited following candesartan or lisinopril treatment
with a decrease in blood vessel formation.[61] Murine lung metastasis from renal
cell carcinomaCyclosporin-enhanced pulmonary metastases were reduced in mice following treatment with
losartan.[62] ĪĪ Candesartan inhibited the growth of pulmonary metastases with a decrease in VEGF and
inhibition of angiogenesis.[63] Chemically-induced mouse lung
tumorAT2 receptor-null mice had reduced tumor number and multiplicity following treatment with NNK. [64] ĪĪ AT2 receptor on lung fibroblasts may be involved in chemical carcinogen-induced lung
tumorigenesis.[64] ĪĪ Immunostaining for the AT1 and AT2 receptors was enhanced in NNK-induced tumor sections. [65] ĪĪ
Angiotensin Peptides and Lung Cancer
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3314267/ĪĪ
Angiotensin-(1-7) Induces Mas Receptor Internalization
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3141282
Angiotensin (Ang) (1-7) is the endogenous ligand for the G protein-coupled receptor Mas, a receptor (R) associated with cardiac, renal and cerebral protective responses. Physiological evidence suggests that Mas R undergoes agonist-dependent desensitization, but the underlying molecular mechanism regulating R activity is unknown.
Cited by: 91
Publish Year: 2011
Author: Mariela M. Gironacci, Hugo P. Adamo, Gerardo CĪĪ
Int J Pept. 2012; 2012: 256294.
Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease
Chris Tikellis * and M. C. Thomas
Division of Diabetic Complications, Baker IDI Heart and Diabetes Institute, P.O. Box 6492 Melbourne, VIC 8008, Australia
Abstract
Angiotensin-converting enzyme 2 (ACE2) shares some homology with angiotensin-converting enzyme (ACE) but is not inhibited by ACE inhibitors. The main role of ACE2 is the degradation of Ang II resulting in the formation of angiotensin 1©C7 (Ang 1©C7) which opposes the actions of Ang II. Increased Ang II levels are thought to upregulate ACE2 activity, and in ACE2 deficient mice Ang II levels are approximately double that of wild-type mice, whilst Ang 1©C7 levels are almost undetectable. Thus, ACE2 plays a crucial role in the RAS because it opposes the actions of Ang II. Consequently, it has a beneficial role in many diseases such as hypertension, diabetes, and cardiovascular disease where its expression is decreased. Not surprisingly, current therapeutic strategies for ACE2 involve augmenting its expression using ACE2 adenoviruses, recombinant ACE2 or compounds in these diseases thereby affording some organ protection.
Figure 1
Schematic representation of the renin-angiotensin system (RAS) and the key balancing role of ACE2. Abbreviations, ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; NEP: neprilysin; AT1: Ang II type 1 receptor; AT2: Ang II type 2 receptor; PEP: prolyl endopeptidase; CAGE: chymostatin-sensitive angiotensin II-generating enzyme.1. Introduction
The renin-angiotensin system (RAS) is a signalling pathway that acts as a homeostatic regulator of vascular function [1]. Its systemic actions include the regulation of blood pressure, natriuresis, and blood volume control. However, the RAS also plays an important local role, regulating regional blood flow and controlling trophic responses to a range of stimuli. The RAS is composed of a number of different regulatory components and effector peptides that facilitate the dynamic control of vascular function, in both health and disease (Figure 1). Many of these components have opposing functions to accommodate a rapid but coordinated response to specific triggers. For example, angiotensin I (Ang I) is metabolised by the dipeptide carboxypeptidase, angiotensin-converting enzyme (ACE) to form angiotensin II (Ang II) and Ang II is metabolised by the carboxypeptidase, ACE2, producing the vasodilator, angiotensin(1©C7) (Ang 1©C7) [2©C4]. Historically, ACE and Ang II have been the key focus for clinical interventions targeting the RAS and its pathogenic actions. However, recent studies have also demonstrated the importance of ACE2 in maintaining the balance of the RAS. Indeed, in some settings, and the cardiovascular system in particular, ACE2 may be more important than ACE in regulating local levels of Ang II and Ang 1©C7, and therein the balance of RAS activation. For example, we have shown that acquired or genetic deficiency of ACE2 results in increased tissue and circulating levels of Ang II [5, 6] and reduced levels of Ang 1©C7 [6]. By contrast, Ace KO mice have modestly reduced circulating Ang II, while tissue levels are not significantly modified, possibly as substantial amounts of Ang II are generated by non-ACE pathways, while degradation pathways for Ang II are more limited [7]. This paper will specifically examine the actions of ACE2 in the body and discuss their potential role in health and various disease states.
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.001.jpg
Figure 1
Schematic representation of the renin-angiotensin system (RAS) and the key balancing role of ACE2. Abbreviations, ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; NEP: neprilysin; AT1: Ang II type 1 receptor; AT2: Ang II type 2 receptor; PEP: prolyl endopeptidase; CAGE: chymostatin-sensitive angiotensin II-generating enzyme.
2. Angiotensin-Converting Enzyme (ACE2)
ACE2 is a type 1 integral membrane glycoprotein [8] that is expressed and active in most tissues. The highest expression of ACE2 is observed in the kidney, the endothelium, the lungs, and in the heart [2, 8]. The extracellular domain of ACE2 enzyme contains a single catalytic metallopeptidase unit that shares 42% sequence identity and 61% sequence similarity with the catalytic domain of ACE [2]. However, unlike ACE, it functions as a carboxypeptidase, rather than a dipeptidase, and ACE2 activity is not antagonized by conventional ACE inhibitors [4]. The major substrate for ACE2 appears to be (Ang II) [2©C4], although other peptides may also be degraded by ACE2, albeit at lower affinity. For example, ACE2 is able to cleave the C-terminal amino acid from angiotensin I, vasoactive bradykinin (1©C8), des-Arg-kallidin (also known as des-Arg10 Lys-bradykinin) [2], Apelin-13 and Apelin-36 [9] as well as other possible targets [10]. The noncatalytic C-terminal domain of ACE2 shows 48% sequence identity with collectrin [11], a protein recently shown to have an important role in neutral amino acid reabsorption from the intestine and the kidney [12]. This is highly consistent with ACE2's actions as a carboxypeptidase, as the removed amino acid then becomes available for reabsorption. The cytoplasmic tail of ACE2 also contains calmodulin-binding sites [13] which may influence shedding of its catalytic ectodomain. In addition, ACE2 has also been associated with integrin function, independent of its angiotensinase activity.
3. ACE2 and Atherosclerosis
Abnormal activation of the RAS contributes to the development and progression of atherosclerotic vascular disease [14©C16]. Independent and additional to the induction of systemic hypertension and vasoconstriction, Ang II has a number of direct proatherosclerotic effects on the vascular wall [17©C19], including promoting inflammation [20], endothelial dysfunction [21], oxidative stress, endothelial cell, and vascular smooth muscle cell migration, growth, proliferation [22], and thrombosis. By contrast, the major product of ACE2, Ang 1©C7, has a range of anti-inflammatory and antioxidant effects [23, 24] that oppose those of Ang II in the vasculature. Indeed, an infusion of Ang 1©C7 is able to attenuate vascular dysfunction and atherosclerosis in genetically susceptible apolipoprotein E knockout (apoE KO) mice [25], possibly by increased activation of the Mas receptor and the type 2 angiotensin receptor (AT2). It is thought that the balance of Ang II and Ang 1©C7 represents an important driving factor for vascular disease progression. Consequently, ACE2 is also likely to play an important role in atherosclerotic plaque development. Certainly, ACE2 expression is reduced in established atherosclerotic plaques [26] and in proatherosclerotic states, such as diabetes [27]. However, direct evidence for ACE2 in the development and progression of atherosclerotic plaques has only recently become available [5].
We have shown that in apoE KO mice, deficiency of ACE2 is associated with increased plaque accumulation (Figure 2), comparable to that observed following angiotensin II infusion [19]. This possibly relates to an increased proinflammatory responsiveness [5], as leukocyte recruitment and adhesion to the nascent atherosclerotic lesion is generally regarded as one of the first steps toward plaque formation. While a healthy endothelium does not in general support binding of white blood cells, we show that the aortic endothelium of apoE/Ace2 double KO mice shows increased adhesion of labeled leukocytes [5]. In addition, genetic ACE2 deficiency is associated with upregulation of putative mediators of atherogenesis, such as cytokines and adhesion molecules. The role of the RAS in these actions is further emphasized by the finding that RAS blockade is able to prevent atherogenesis in apoE/Ace2 double KO mice. Such data emphasize the potential utility of ACE2 repletion as a strategy to reduce atherosclerosis, particularly in combination with ACE inhibition and other interventions to reduce activation of the RAS (see below).
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.002.jpg
Figure 2
Increased plaque area accumulation in the aorta of Apoe/Ace2 double KO mice when compared to control Apoe KO mice [5]. *vs control Apoe KO mice P < 0.05.
4. ACE2 and Hypertension
Activation of the RAS is known to be a key mediator of hypertension, and interventions to block RAS activation are the most widely used of all blood pressure lowering agents. The antihypertensive efficacy of these agents is partly mediated by their ability to reduce Ang II or its signalling. However, the antihypertensive effects of conventional RAS blockade are also partly determined by the ability of both ACE inhibitors and angiotensin receptor blockers (ARBs) to increase circulating levels of Ang 1©C7 [28]. Moreover, inhibiting the vascular actions of Ang 1©C7 in spontaneously hypertensive rats (SHRs) receiving RAS blockade, attenuates the antihypertensive response to these agents [28, 29]. Given that the major source of Ang 1©C7 is ACE2, this data suggests that ACE2, consequently influences not only the development of hypertension, but also potentially the response to its treatment. Certainly, ACE2 expression is abnormal in SHRs, in which one genetic component of this phenotype tracks to the Ace2 locus. In addition, ACE2 deficiency is associated with modest systolic hypertension [30], although the mouse genetic background significantly alters the cardiovascular phenotype [30©C33]. Ace2 KO mice also have a heightened hypertensive response to Ang II infusion associated with exaggerated accumulation of Ang II in the kidney [30].
The RAS and ACE2 are also implicated in the pathogenesis of central hypertension. In particular, the rostral ventrolateral medulla (RVLM) is a relay point that provides supraspinal excitatory input to sympathetic preganglionic neurons in the regulation of blood pressure. In the SHRs, ACE2 expression is reduced in the RVLM [34], and persistent overexpression of ACE2 in the RVLM results in a significant attenuation of high blood pressure in this model [35, 36]. In addition, injections of the ACE2 inhibitor MLN4760 into the nucleus tractus solitarii reduce reflex bradycardia in response to the baroreceptor stimulation in rats [37], suggesting an additional role for central ACE2 in controlling baroreceptor responsiveness.
ĪĪ5. ACE2 in Heart Failure
In addition to effects on blood pressure, natriuresis and atherogenesis, the RAS plays a critical pathophysiological role in the maintaining and subsequently subverting cardiac function in the setting of progressive heart failure [38]. The cardiac RAS is upregulated in almost all models of cardiac injury, including volume overload [39], myocardial infarction [40], and heart failure [41]. As in the kidney, RAS upregulation appears to be a homeostatic response to restore cardiac function. For example, Ang II is an inotropic and growth factor for cardiac myocytes, stimulating compensatory hypertrophy [42]. Ang II is also important in left ventricular remodeling following myocardial infarction or with after-load-induced cardiac hypertrophy [43]. However, in the long term such actions lead to progressive functional loss and cardiac fibrosis [42], as the synthesis of extracellular matrix is increased by Ang II [44]. The key role of RAS activation in the development and progression of cardiac failure is supported by findings in a number of different models in which blockade of the RAS was able to attenuate or prevent cardiac damage, independent of blood pressure lowering [45].
In the heart, ACE2 represents the primary pathway for the metabolism of Ang II [46, 47]. ACE2 deficiency in mice results in early cardiac hypertrophy (Figure 3) [32] and accelerates adverse postmyocardial infarction ventricular remodeling [48]. Furthermore, this appears to be through the activation of the NAPDH oxidase system with the p47(phox) subunit playing a critical role [49]. In some, but not all models, ACE2 deficiency also results in progressive cardiac fibrosis with aging and/or cardiac pressure overload [33, 50, 51]. Again, these changes are reversed following treatment with ACE inhibitors or AT1 receptor blockers [33, 50, 51] suggesting that the balance of ACE and ACE2 in the heart is an important driving factor for progressive cardiac disease.
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.003.jpg
Figure 3
Increased LV mass in Ace2 KO mice versus C57bl6 mice (unpublished data). *vs control C57Bl6 mice, P < 0.05.
6. ACE2 and Chronic Kidney Disease (CKD)
The RAS also plays an important role in renal physiology and pathophysiology. In the adult kidney [2], ACE2 is predominantly expressed in the proximal tubule at the luminal brush border. Despite the presence of unopposed ACE activity and elevated Ang II levels, both kidney function and renal development are normal in the Ace2 knockout mouse [33]. By comparison, ACE, angiotensinogen, and AT1 receptor deficiency results in a number of alterations in kidney morphology [52]. This suggests that, at least in the healthy state, ACE2 may have a limited role in regulating renal development. However, the actions of ACE2 appear to come into its own in states of RAS activation. This is much like Ang 1©C7, its major product, which shows very limited renal effects in the healthy state but profound benefits in the diabetic kidney and other states associated with renal damage and activation [10, 53]. For example, ACE2 deficient mice have been reported to show increased age-related glomerulosclerosis in susceptible mouse models [54] and enhanced renal Ang II-induced renal oxidative stress, resulting in greater renal injury [55]. Similarly, in the diabetic kidney, downregulation of tubular ACE2 (Figure 4) [27] is associated with albuminuria or tubular injury, while further inhibition of ACE2 results in augmented renal damage [56, 57]. Indeed, in most forms of CKD, including diabetes, expression of ACE2 has been reported to be reduced in tubules. However, some studies have reported that glomerular ACE2 expression may be increased in human kidney disease [58]. It is possible that this differential expression pattern of glomerular and tubular ACE2 is an important determinant for progressive renal disease.
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.004.jpg
Open in a separate window
Figure 4
Reduced ACE2 expression (arrows) in renal cortical tubules of diabetic mice (b) when compared to control mice (a) [27].
7. ACE2 and the Lung
RAS activity is intrinsically high in the lung, which is a major source of ACE and therefore a major site of systemic Ang II synthesis. ACE2 is also highly expressed in the lung. Pulmonary ACE2 appears to have a role in regulating the balance of circulating Ang II/Ang 1©C7 levels. Ang II induces pulmonary vasoconstriction in response to hypoxia, which is important in preventing shunting in patients with pneumonia or lung injury [59]. Locally increased Ang II production also triggers increasing vascular permeability facilitating pulmonary edema [60]. In Acute respiratory distress syndrome (ARDS), the RAS appears crucial in maintaining oxygenation, possibly as widespread lung injury would otherwise result in complete pulmonary shutdown. Certainly in ARDS models, ACE2 knockout mice displayed more severe symptoms of this disease compared with wild-type mice [60] while overexpression appears protective (see below). Interestingly, ACE2 protein also appears to be the entry-point receptor for the severe acute respiratory syndrome (SARS) coronavirus [61, 62].ACE2║═Ę╬
RAS╗Ņąįį┌Ę╬ųą▒Šų╩╔ŽĮŽĖ▀Ż¼šŌ╩ŪACEĄ─ų„ę¬└┤į┤Ż¼ę“┤╦ę▓╩ŪŽĄ═│ąįAng II║Ž│╔Ą─ų„ę¬▓┐╬╗ĪŻ ACE2į┌Ę╬ųąę▓Ė▀Č╚▒Ē┤’ĪŻĘ╬ACE2╦Ų║§į┌Ą„Į┌čŁ╗ĘĄ─Ang II / Ang 1-7╦«ŲĮĄ─ŲĮ║ŌųąŲū„ė├ĪŻ Ang IIę²ŲĄ─╚▒č§Ę┤ė”┐╔ę²ŲĘ╬č¬╣▄╩š╦§Ż¼šŌČįė┌įżĘ└Ę╬čū╗“Ę╬╦╔╦╗╝š▀Ą─Ęų┴„║▄ųžę¬[59]ĪŻ Ang II▓·╔·Ą─Šų▓┐į÷╝ėę▓┤źĘóč¬╣▄═©═Ėąįį÷╝ėŻ¼┤┘Į°Ę╬╦«ųū[60]ĪŻį┌╝▒ąį║¶╬³ŠĮŲ╚ū█║Žš„Ż©ARDSŻ®ųąŻ¼RAS╦Ų║§Čį╬¼│ųč§║Žū„ė├ų┴╣žųžę¬Ż¼┐╔─▄╩Ūė╔ė┌╣ŃĘ║Ą─Ę╬╦╔╦╗ߥ╝ų┬═Ļ╚½Ą─Ę╬╣ž▒šĪŻĄ▒╚╗Ż¼į┌ARDS─Żą═ųąŻ¼ėļę░╔·ą═ąĪ╩¾ŽÓ▒╚Ż¼ACE2╗∙ę“Ū├│²ąĪ╩¾▒ĒŽų│÷Ė³čŽųžĄ─šŌųų╝▓▓Īųóū┤[60]Ż¼Č°╣²▒Ē┤’į“Š▀ėą▒Ż╗żąįŻ©╝¹Ž┬╬─Ż®ĪŻėą╚żĄ─╩ŪŻ¼ACE2Ą░░ū╦Ų║§ę▓╩ŪčŽųž╝▒ąį║¶╬³ŽĄ═│ū█║ŽųóŻ©SARSŻ®╣┌ū┤▓ĪČŠĄ─Ūą╚ļĄŃ╩▄╠Õ[61Ż¼62]ĪŻ
8. Replenishing ACE2 as a Potential Therapeutic
Given the key role of ACE2, degrading Ang II and generating Ang 1©C7, a number of studies have explored its potential as a treatment strategy using human recombinant ACE2 (rhACE2) or adenoviral (Ad)-ACE2 in animal disease models. For example, overexpression of ACE2 in human endothelial cells attenuates Ang II-induced oxidative stress and subsequent increase in monocyte adhesion [63]. Similarly, in rabbits, a recombinant ACE2 expressing vector stabilized atherosclerotic plaques induced by balloon injury to the abdominal aorta [64]. Treatment with a lentiviral vector containing ACE2 resulted in lower blood pressure in hypertensive mice [65, 66] or following an Ang II infusion [67]. Strategies to upregulate or replenish ACE2 are thought to be beneficial in diabetic nephropathy. For example, in diabetes the replenishment of ACE2 with rhACE2 in a mouse model of type 1 diabetes attenuated diabetic kidney injury as well as reducing in blood pressure [68]. The use of (Ad)-ACE2 has had similar beneficial effects in streptozotocin-induced diabetes, where it was shown to attenuate glomerular mesangial cell proliferation, blood pressure, oxidative stress, and fibrosis [69].
In contrast to these studies, the potential utility of ACE2 supplementation in cardiac disease remains controversial. The expression of ACE2 in the failing human heart is generally increased [70©C72], consistent with the finding of elevated levels of Ang 1©C7 in the same setting [73]. More importantly, overexpression of ACE2 in cardiac myocytes resulted in conduction disturbances by 2 weeks of age, ultimately leading to lethal ventricular arrhythmias and severe fibrosis [74, 75]. This may be because ACE2 is not normally expressed in high levels in myocytes, although it is present in the endocardium and other cardiac cells. However, other studies using transgenic overexpression of cardiac ACE2 have demonstrated partial protection in the heart from ischemia-induced heart failure [76]. Indeed, more recent studies using rhACE2 have shown beneficial cardiac effects [77]. However, the indication for ACE2 that appears most likely to be first tested in the clinic is the treatment of ARDS. In murine models, treatment with catalytically active recombinant ACE2 protein improved the symptoms of acute lung injury in wild-type mice as well as in ACE2 knockout mice [60]. Clinical trials in this often fatal condition are now underway.
Perhaps, the most clinically interesting, however, is the potential for rACE2 to augment the vasculoprotective effects of ACE inhibition or ARBs, in the millions of patients that take these agents, worldwide. In theory, this would be achieved by preventing feedback escape for RAS blockade or enhancing the generation of Ang 1©C7, and subsequent signaling through the Mas receptor and or AT2 receptor. Certainly, ACE2 inhibition attenuates the effects of RAS blockade, both in vitro [78] and in vivo [6]. But could rACE2 make the response to conventional RAS blockade more effective or durable? The problem is that conventional RAS blockade is highly effective in animal models of vascular and renal disease, meaning that it is difficult to explore the potential for further improvements. However, chronic intravenous infusion of ANG-(1©C7), or the nonpeptide mas receptor agonist, AVE-0991, are able to improve salt-induced suppression of endothelium-dependent vasodilatation in the mesenteric arteries of male Sprague-Dawley rats, and these actions are not modified by the angiotensin receptor blocker, losartan [79], suggesting that the effects of enhancing the Ang 1©C7 mas axis may be beneficial, even in the setting of conventional RAS blockade. Although it enhances the generation of Ang 1©C7, whether rACE2 can also provide synergistic benefits, remains to be established.9. ACE2 Augmenters: A New Kind of Intervention
Rather than providing exogenous ACE2, an alternative approach for augmenting ACE2 has been to increase its endogenous expression. For example, in hypertensive SHRs, all-trans retinoic acid, which increases ACE2 expression, lowers blood pressure levels, and prevents vascular damage [80]. Unfortunately retinoic acid has broader actions that make its potential utility as a therapeutic limited. However, compounds that increase activity of ACE2 could also be beneficial as a treatment in conditions where ACE2 activity is decreased. One exemplar is xanthenone (XNT). This molecule was selected following structure-based screening on compounds that would stabilize the activated form of ACE2, thereby enhancing its catalytic efficacy [81]. In experimental studies, this compound has been shown to enhance ACE2 activity in a dose-dependent manner and significantly decreased blood pressure in both SHRs rats and wild-type WKY rats [81]. Furthermore, improvements in cardiac function and reversal of myocardial, perivascular, and renal fibrosis in the SHRs were also observed [81, 82]. XNT has also shown promise in treating pulmonary hypertension (PH). For example, in a rat model of PH, treatment with XNT was shown to reduce elevated right ventricular systolic pressure, right ventricular hypertrophy, increased pulmonary vessel wall thickness, and interstitial fibrosis [83]. In a model of thrombus formation using SHRs and WKY rats, XNT has also shown antithrombotic action, reducing platelet attachment, and reducing thrombus formation [84]. This compound will not come to clinical trials because of issues of solubility that restrict its formulation. However, other drugs of the same class may prove more suitable.
10. Conclusion
ACE2 is an integral component of the RAS. It is highly expressed in the vasculature, the kidney, lungs, and heart where its actions on peptide signals balance and offset those of ACE. Its actions appear critical in a variety of disease states, including hypertension, diabetes, ageing, renal impairment, and cardiovascular disease. ACE2 deficiency leads to modest physiological changes. However, in states of RAS activation, the loss of ACE2 appears far more important in the development and progression of disease. By contrast, augmentation of ACE2 expression, either directly with recombinant ACE2 or indirectly via agonists like XNT, may have important benefits relevant in the treatment of a range of conditions.Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3321295/ĪĪ
Cardiovasc Res. 2007 Feb 1;73(3):463-9. Epub 2006 Sep 19.
ACE2 of the heart: From angiotensin I to angiotensin (1-7).
Keidar S1, Kaplan M, Gamliel-Lazarovich A.
Author information
The Lipid Research Laboratory, The Bruce Rappaport Faculty of Medicine, Technion, Israel. skeidar@rambam.health.gov.il
Abstract
Angiotensin II (Ang II), a bioactive peptide of the renin-angiotensin system (RAAS), plays an important role in the development of cardiovascular diseases (CVD). Pharmacological inhibition of angiotensin-converting enzyme (ACE), the Ang II forming enzyme, or specific blockade of Ang II binding to angiotensin type 1 receptor (AT1R) through which it exerts its deleterious effects, were shown to provide some protection against progression of CVD.The ACE-Ang II-AT1R axis has been challenged over the last few years with RAAS components able to counterbalance the effects of the main axis. The ACE homologue ACE2 efficiently hydrolyses Ang II to form Ang (1-7), a peptide that exerts actions opposite to those of Ang II. In contrast to the Ang II axis, the role of the ACE2-Ang (1-7) axis in cardiac function is largely obscure. Ang (1-7) is present in the viable myocardium, and its formation depends on Ang II as a substrate. The expression of this peptide is associated with cardiac remodeling: it is lost in the infarcted area and significantly increased in the border area. Low doses of Ang (1-7) improve cardiac output and antagonize Ang II-induced vasoconstriction. The type of Ang (1-7) biological activity is tissue specific and dose dependent.
These findings point to a possible protective role for Ang (1-7) in abating the Ang II-induced actions. The elevated expression of Ang (1-7) in failing heart tissue paralleled the expression of its forming enzyme, ACE2. Several observations and experimental evidence suggest a beneficial role for ACE2 in cardiovascular function. Elevated ACE2 expression at the initial stage of several pathologies which decline with progression of disease might indicate a protective role for ACE2. Genetic manipulation of ACE2 expression, either targeted disruption or overexpression, point to the possible significance of this enzyme in cardiac function.
Based on the above, a therapeutic approach that will amplify the ACE2-Ang (1-7) axis could provide further protection against the development of CVD. It turns out that the merits of currently used drugs--ACE inhibitors, AT1R blockers and mineralocorticoid receptor blockers (MRB) - lay beyond their direct effects on suppression of the ACE-Ang II-AT1R axis as they also increase cardiac ACE2 and Ang (1-7) significantly.
PMID: 17049503 DOI: 10.1016/j.cardiores.2006.09.006ACE2 of the heart: From angiotensin I to angiotensin (1-7). - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/17049503ĪĪ
ĪĪ
Sci Rep. 2014; 4: 7027.
Published online 2014 Nov 13. doi: 10.1038/srep07027
Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury
Penghui Yang,1,2,* Hongjing Gu,1,* Zhongpeng Zhao,1 Wei Wang,3 Bin Cao,4 Chengcai Lai,1 Xiaolan Yang,1 LiangYan Zhang,1 Yueqiang Duan,1 Shaogeng Zhang,2 Weiwen Chen,5 Wenbo Zhen,5 Maosheng Cai,6 Josef M. Penninger,7 Chengyu Jiang,a,3 and Xiliang Wangb,1
Author information Article notes Copyright and License information Disclaimer
1State Key Laboratory of Pathogens and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Beijing 100071, China
2Beijing 302 Hospital, Beijing, 100039, China
3State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Department of Biochemistry and Molecular Biology, Peking Union Medical College, Tsinghua University, Beijing 100005, China
4Beijing Chao-Yang Hospital, Beijing Institute of Respiratory Medicine, Capital Medical University, Beijing 100020, China
5Quanzhou First Hospital, Fujian 362321, China
6Shishi Hospital, Fujian 362700, China
7Institute of Molecular Biotechnology in the Austrian Academy of Sciences, Vienna, A-1030, Austria
aEmail: nc.ude.cmup@gnaij
bEmail: moc.621@wgnailix
*These authors contributed equally to this work.Abstract
Since March 2013, the emergence of an avian-origin influenza A (H7N9) virus has raised concern in China. Although most infections resulted in respiratory illness, some severe cases resulted in acute respiratory distress syndrome (ARDS), which is a severe form of acute lung injury (ALI) that further contributes to morbidity. To date, no effective drugs that improve the clinical outcome of influenza A (H7N9) virus-infected patients have been identified. Angiotensin-converting enzyme (ACE) and ACE2 are involved in several pathologies such as cardiovascular functions, renal disease, and acute lung injury. In the current study, we report that ACE2 could mediate the severe acute lung injury induced by influenza A (H7N9) virus infection in an experimental mouse model. Moreover, ACE2 deficiency worsened the disease pathogenesis markedly, mainly by targeting the angiotensin II type 1 receptor (AT1). The current findings demonstrate that ACE2 plays a critical role in influenza A (H7N9) virus-induced acute lung injury, and suggest that might be a useful potential therapeutic target for future influenza A (H7N9) outbreaks.
ĪĪ
Avian influenza A (H7N9) virus is a viral subtype that was detected in birds previously. However, it had not been reported in either animals or humans prior to its identification in China in March 2013Ż¼1,2,3. The first wave of cases occurred between February and May 2013Ż¼4,5,6. Reports of human infections then decreased during the summertime, but increased subsequently from October, demonstrating the occurrence of a second wave of infections. The disease caused by the H7N9 virus is characterized by rapidly progressing severe pneumonia. Complications include acute respiratory distress syndrome (ARDS), septic shock, and multi-organ failure that require intensive care and mechanical ventilation. To date, most influenza A (H7N9) virus-infected patient deaths were due to acute lung injury (ALI) and ARDS7,8,9,10.
The rennin-angiotensin system (RAS) is a complex network that plays a major role in maintaining blood pressure, electrolyte and fluid homeostasis, and fluid and salt balance11,12,13. Angiotensin-converting enzyme-2 (ACE2) was discovered as a homolog of ACE that regulated RAS negatively by converting angiotensin (Ang)-II to Ang-1©C714. Previous reports identified ACE2 as the receptor for the severe acute respiratory syndrome (SARS) coronavirus15. Recently, it was reported that ACE2 modulated innate immunity and influenced the composition of the gut microbiota16. Interestingly, ACE2 is also involved in the severe ALI and failure that is induced by sepsis, acid aspiration, SARS, and lethal avian influenza A H5N1 virus17. As such recombinant soluble ACE2 is currently being tested in phase 2 clinical trials as a potential therapy for the treatment of acute lung injury in humans18,19. Of note, we demonstrated recently that serum Ang II levels were elevated in H5N1- and H7N9-infected patients20. More importantly, plasma Ang II levels were linked to disease severity and predicted a fatal outcome in H7N9-infected patients21. Therefore, the aim of the current study was to further determine whether interfering with RAS could influence the severity of avian influenza A (H7N9) virus-induced lung injury in an experimental mouse model.Methods
Animals
Four-week-old wild-type (WT) C57BL/6 (abbreviated B6) mice (Experimental Animal Center of Academy Military Medical Sciences, Beijing, China), and 4-week-old ACE2 knockout (abbreviated KO) mice (B6 background, a gift from Professor Josef M. Penninger) were housed in the animal facility at the Beijing Institute of Microbiology and Epidemiology in accordance with institutional guidelines.
All experimental protocols were approved by the Institutional Animal Care and Use Committee of Academy Military Medical Sciences (ID: SYXK2010-005). Live-virus experiments were performed in Bio-safety Level 3 facilities in accordance with governmental and institutional guidelines.
Experimental mouse models of acute lung injury
The influenza A H7N9 virus (A/Hebei/01/2013, abbreviated Hb01/H7N9) used in this study was isolated from a confirmed H7N9-infected patient. The genomic sequences of Hb01/H7N9 are available in the Global Initiative on Sharing All Influenza Data (GISAID) database under the accession numbers EPI509120©CEPI509127. Live virus experiments were performed in Biosafety Level 3 facilities in accordance with governmental and institutional guidelines. For influenza A H7N9 virus-induced acute lung injury, 4-week-old WT B6 mice were anesthetized with 50-”╠l 1% (w/v) pentobarbital sodium, and then inoculated intranasally (i.n.) with 2 Ī┴ 103.5 of the 50% tissue culture infectious dose (TCID50) of Hb01/H7N9 virus or mock-infected control allantoic fluid (AF). The survival, weight loss, acute pulmonary edema (wet-to-dry ratio), and histological measurements were performed as described previously22.
AT1/AT2 receptor inhibitors
For inhibitor experiments, mice were injected intraperitoneally with the AT1 inhibitor losartan (15 mg/kg), the AT2 inhibitor PD123.319 (15 mg/kg), or PBS 30 min before Hb01/H7N9 virus infection.
Angiotensin II levels and western blotting
Ang II levels were detected as described previously21. Rat polyclonal anti-ACE2 antibodies (R&D Systems) were used for western blotting.
Histological examination
After being anesthetized with pentobarbital sodium, 4-week-old B6 mice were treated i.n. with 2 Ī┴ 103.5 TCID50 virus, and were then sacrificed at various days post-infection (DPI). The lungs of each group of mice (five mice per group) were fixed in formalin, and then embedded in paraffin. The number of inflammatory cells was counted, and the data are presented as the number of cell per 200Ī┴ field.
Lung wet-to-dry ratio
To assess the extent of acute pulmonary edema, the lung wet-to-dry weight ratios were calculated. WT 4-week-old B6 mice were anesthetized using sodium pentobarbital, and were then inoculated intranasally with 2 Ī┴ 103.5 TCID50 virus. At 5 DPI, the wet weights of the lungs were then measured from eight mice per group. The lungs were then heated to 68ĪŃC overnight, and the dry weights were measured.
Quantitative real-time PCR
RNA was extracted from the lungs of 4-week-old B6 mice infected with 2 Ī┴ 103.5 TCID50 virus using an RNeasy Mini Kit (Qiagen). Real-time RT-PCR was performed using an ABI 7500 Real-Time PCR system (Applied Biosystems) with primers and a TaqMan one-step RT-PCR master mix (Applied Biosystems). Primers that could detect the influenza NP gene specifically were used. The expression of target genes was normalized to that of the control gene GAPDH. Relative amounts of mRNA were calculated using the comparative CT method.
Viral titration
Virus titers were measured in the supernatants of mouse lung homogenates from ACE2 KO or WT mice on day 5 post-infection. Briefly, samples were added to 96-well plates containing MDCK cells, and then diluted 10-fold. The infected cells were then cultured for 96 h. The viral titers were calculated using the Reed and Muench method, and data were expressed as log10 TCID50/g of lung tissue.
Statistical analyses
All data are presented as means Ī└ SEM. Measurements at single time-points were analyzed using ANOVA, and survival data were analyzed using Kaplan-Meier survival analysis. All statistical analyses were performed using the GraphPad Prism 5 software. A value of p < 0.05 was considered to indicate statistical significance.Results
RAS is dysregulated during mouse H7N9 infection
Two research groups cloned the ACE homolog ACE2 independently in 200023. ACE2 negatively regulates RAS by inactivating Ang II24. To assess the effect of influenza virus infection on RAS directly, mice were infected with live influenza Hb01/H7N9 virus. Influenza A (H7N9) virus infection in WT mice caused the development of pulmonary edema and an increase in lung viral titers. Importantly, the WT mice infected with influenza H7N9 virus downregulated ACE2 protein markedly on day 3 after infection. In contrast, the expression of ACE in the lungs was comparable between H7N9-infected and control mice (Figure 1A).
ĪĪ
Figure 1
ACE2 plays a critical role in 2013 influenza H7N9 virus-induced ALI.
(A) Downregulated ACE2 expression in the lungs of Hb01/H7N9 virus-infected mice. Lung tissue homogenates prepared from control and Hb01/H7N9 virus-infected WT mice on day 3 were analyzed by western blotting. (B) Plasma levels of Ang II in control and Hb01/H7N9 virus-infected ACE2 KO or WT mice on day 3 (n = 8). (C) Ang II levels in the lungs of control and Hb01/H7N9 virus-infected WT mice on day 3 were measured using enzyme immunoassays (n = 8). (D) Plasma levels of Ang II in healthy and H7N9 virus-infected patients were measured using enzyme immunoassays. *, p < 0.05; ** p < 0.01 between groups.
Next, Ang II levels were measured in Hb01/H7N9 virus-infected mice on day 3 post-infection. Hb01/H7N9 virus infection increased Ang II levels in plasma (Figure 1B) of WT mice (p < 0.01) markedly. Similarly, there was also a significant increase in Ang II levels in the lungs of Hb01/H7N9 virus-infected mice (Figure 1C). It was noticeable that ACE2 protein expression was downregulated in WT mice following influenza A (H7N9) virus infection. Therefore, this experimental mouse model suggests that the ALI that was induced by influenza A (H7N9) virus infection resulted in decreased ACE2 expression and elevated Ang II levels.
To further elucidate the role of ACE2 in influenza A (H7N9) virus-induced ACI, we next examined the levels of Ang II in the plasma of H7N9-infected patients. Six patients who were PCR-positive for the 2013 influenza A (H7N9) virus were recruited from Beijing Chaoyang hospital and Quanzhou hospital, China. The characteristics underlying the conditions and the outcomes of the six patients are described in Supplementary Table S1. A significant increase in plasma Ang II levels was observed in influenza A (H7N9) virus-infected patients (Figure 1D). In addition, plasma Ang II levels were measured in influenza A (H7N9)-infected patients at different time-points post-infection, and data revealed that Ang II concentrations increased continuously increased from ~350 to 6100 pg/mL; the peak levels were detected when the patient was on the verge of death (Figure S1A). The kinetics of Ang II expression was also quantified in patients who recovered from H7N9 infection (Figure S1B; Table S2 and S3). Data revealed that plasma Ang II levels decreased sharply from the early to the late phase of influenza A (H7N9) virus infection in the recovered population. Therefore, we hypothesized that ACE2 might play a critical role in the host response against 2013 influenza A (H7N9) infection via Ang II.
ACE2 deficiency increased the severity of H7N9-induced lung injury
We next investigated whether ACE2 could mediate or protect mice from 2013 H7N9 influenza infection-induced ALI. ACE2 KO mice that were infected with influenza Hb01/H7N9 virus exhibited survival rates that were reduced significantly compared with WT mice (Figure 2A). Specifically, the survival rate of WT mice was 20%, whereas no KO mice survived for at least 9 days after influenza Hb01/H7N9 virus infection. Similarly, the lung histopathology and lung injury scores, as defined by leukocyte infiltration cell counts, were worsened significantly in ACE2 KO mice compared with WT (Figure 2B). Moreover, lung edema, defined by the wet-to-dry weight ratio of lung tissue, was much more severe in ACE2 KO mice (Figure 2C). In addition, the influenza Hb01/H7N9 virus nucleoprotein (NP) copy number was also increased greatly in the KO mice (Figure 2D). Consistent with this, the lung viral titers of the ACE KO mice were also increased significantly (Figure 2E). Therefore, these results suggest that ACE2 plays an important role in mediating H7N9 influenza-induced ALI, and that interfering with ACE2 expression might attenuate the disease severity and increase survival following respiratory H7N9 infection.
Figure 2
Loss of ACE2 expression worsens H7N9-induced ALI.
(A) The survival rates of WT and ACE2 KO mice (n = 10). (B) H&E staining and infiltrating cell counts (n = 200 fields) in the lung tissues of WT B6 and ACE2 KO mice (n = 5) at day 5 post-infection. (C) The wet-to-dry ratio of lungs from WT B6 and ACE2 KO mice (n = 8) at day 5 post-infection. (D) Detection of Hb01/H7N9 virus NP RNA from WT B6 and ACE2 KO mice (n = 8) at day 5 post-infection. (E) Lung viral titers in Hb01/H7N9 virus-infected WT B6 and ACE2 KO mice (n = 8) at day 5 post-infection.
The Ang II receptor AT1 affects the severity of H7N9-induced lung injury
We next attempted to rescue the ALI in WT mice infected with influenza Hb01/H7N9 virus using specific AT1 and AT2 receptor blockers. Inhibiting AT1 alleviated the severity of influenza Hb01/H7N9 virus-induced lung injury significantly in WT mice, as determined by the lung wet-to-dry ratio and histopathology (Figures 3A and 3B). Similarly, the NP copy number and viral titers were decreased significantly in the lungs of H7N9 virus-infected WT mice treated with AT1 receptor blockers (Figures 3C and D). In contrast, inhibiting AT2 had no significant effect on the ALI phenotypes (Figure S2), suggesting that actions of Ang II via the AT1 receptor play a critical role in influenza A (H7N9) virus-induced ALI.
Figure 3
The Ang II receptor AT1 regulates H7N9-induced lung injury.
(A) The wet-to-dry ratio of the lungs of WT mice treated with control or AT1 inhibitor (losartan, 15 mg/kg) 30 min before Hb01/H7N9 virus infection (n = 8). (B) H&E staining and infiltrating cell counts (n = 200 fields) in the lung tissue of Hb01/H7N9 influenza virus-infected B6 mice treated with PBS control or AT1 inhibitor (losartan, 15 mg/kg) at day 5 post-infection. (C) Detection of Hb01/H7N9 virus NP RNA in WT mice treated with PBS control or AT1 inhibitor (losartan, 15 mg/kg) 30 min before Hb01/H7N9 virus infection (n = 8) at day 5 post-infection. NP mRNA expression was quantified using real-time PCR, and was normalized to GAPDH (n = 10). (D) Lung viral titers in WT mice treated with PBS control or AT1 inhibitor (losartan, 15 mg/kg) before Hb01/H7N9 virus infection (n = 8) at day 5 post-infection. **p < 0.01(two-tailed t-test).
Inhibiting AT1 attenuates H7N9-induced lung injury in ACE2 KO mice
Similarly, the lung wet-to-dry ratio of H7N9 virus-infected ACE2 KO mice treated with At1 receptor blockers was decreased and the pathological lung alterations were improved (Figures 4A and 4B). Similarly, the NP copy number and viral titers were decreased significantly in the lungs of H7N9 virus-infected ACE2 KO mice treated with AT1 receptor blockers (Figure 4C and D). Consistent with our observations in WT mice, there was no apparent effect on lung edema and histopathology when ACE2 KO mice were treated with AT2 receptor blockers (data not shown). These results suggest that ACE2 might play a crucial role in influenza A (H7N9) virus infection-induced ALI via the AT1 receptor. Based on these data, schematic diagrams of the RAS in influenza A (H7N9) virus-induced ALI have been proposed (Figure 4E). Taken together, these data suggest that the novel influenza H7N9 virus causes severe ALI in a experimental mouse model at least in part by altering the RAS via ACE2 expression that targets AT1.
Figure 4
Inhibiting AT1 attenuates H7N9-induced lung injury in ACE2 KO mice.
(A) The wet-to-dry ratio of the lungs of ACE2 KO mice treated with PBS control or AT1 inhibitor (losartan, 15 mg/kg) 30 min before Hb01/H7N9 virus infection (n = 8). (B) H&E staining and infiltrating cell counts (n = 200 fields) in the lung tissue of Hb01/H7N9 virus-infected ACE2 KO mice treated with PBS control or AT1 inhibitor (losartan, 15 mg/kg) at day 5 post-infection. **p < 0.01 (two-tailed t-test). (C) Detection of Hb01/H7N9 virus NP RNA in ACE2 KO mice treated with PBS control or AT1 inhibitors at day 5 post-infection. NP mRNA expression was assessed using real-time PCR, and was normalized to GAPDH (n = 10). (D) Lung viral titers in ACE2 KO mice treated with PBS control or AT1 inhibitor at day 5 post-infection. **p < 0.01 (two-tailed t-test). (E) Schematic diagram of the role of the renin-angiotensin system in ALI and influenza A H7N9 virus infection.
Discussion
ARDS is the most severe stage of ALI during pathogenic infections25. ARDS is characterized by lung edema, increased viral titers, and the accumulation of inflammatory cells. Consistent with previous studies21, the current results revealed elevated levels of Ang II in plasma samples from H7N9-infected patients in China. Nevertheless, a considerably larger cohort of H7N9-infected individuals is required to confirm these observations. Influenza A (H7N9) virus-induced ALI results in the significant downregulation of ACE2, which regulates the RAS. Moreover, the current findings provide a molecular explanation for these causes of death and the mechanism of ALI in patients infected with H7N9 influenza. Ang II is upregulated after the downregulation of ACE2, and it then causes severe lung injury via the AT1 receptor during influenza A (H7N9) virus infections.
The data presented here also demonstrated that ACE2 deficiency aggravated influenza A (H7N9) virus-induced ALI in mice. Nevertheless, further studies assessing the potential therapeutic effects of recombinant human ACE2 protein are required using different animal models to verify the protective effects of ACE2 against H7N9-induced lung pathologies. The combination of clinical findings and mice experiments has revealed a critical role for the RAS in the pathogenesis of influenza A (H7N9) virus-induced ALI, and demonstrated that ACE2 plays a key role in the development and progression of H7N9 influenza. In addition, the molecular basis by which influenza H7N9 virus infection decreases ACE2 expression should be investigated in future studies. Taken together, these data raise the intriguing possibility that treatments targeting ACE2 and RAS might be viable strategies for treating influenza A (H7N9) virus infections.Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4229671/ĪĪ
Science China Life Science. 2015 Feb;58(2):208-11. doi: 10.1007/s11427-015-4814-7. Epub 2015 Feb 7.
Angiotensin II receptor blocker as a novel therapy in acute lung injury induced by avian influenza A H5N1 virus infection in mouse.
Yan Y1, Liu Q, Li N, Du J, Li X, Li C, Jin N, Jiang C.
Author information
1
State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences; Department of Biochemistry and Molecular Biology, Peking Union Medical College, Tsinghua University, Beijing, 100005, China.
Dear Editor,
Since the first recognized case of human infection with avian influenza H5N1 virus in 1997, the worldwide spread and re-emergence of this highly pathogenic influenza virus have led to 694 human infection cases. Of these cases, 402 died from 2003 to 2015 (http://www.who.int/influenza/human_animal_interface/EN_GIP_20150106CumulativeNumberH5N1cases.pdf?ua=1),
with a morbidity rate of 57.9%, which is much higher than that of seasonal influenza H1N1 virus. The outbreak of the H7N9 virus epidemic in 2013 has been a warning sign that the occurrence of new influenza pandemics and the sustained circulation of these viruses in poultry and other animals pose a persistent threat to public health. In addition, recent studies on influenza virus transmission proved that only a few amino acid mutations are required to make H5N1 virus transmissible among ferrets, an animal model that resembles human response to influenza virus infection [1,2]. Considering the potential of mutated avian influenza virus being both easily transmitted and highly pathogenic to human, it is time to take measures before the next coming influenza pandemic. After entering its host cells, influenza virus first mas-sively replicates, and then it induces acute and severe immune
responses, which lead to serious respiratory injuries and consequently acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) [3]. At present, due to limited therapeutic strategies for influenza virus infection or ARDS, the mortality rate for humans infected with high pathogenic H5N1 remains high. Therefore, to find effective prophylactic or therapeutic agents that potentially alleviate H5N1 virus-induced lung injury should be of great importance.
Re-investigating existing drugs is an economical and time-saving approach to satisfy unmet medical needs because the safety, pharmacology, formulation and potential toxicity of shelved drugs have been largely established. Our previous study has shown that angiotensin II receptor blocker (ARB) losartan could effectively reduce ALI induced by SARS-CoV [4]. We recently reported that avian influenza A H5N1 virus infection disrupted host renin-angiotensin system (RAS) in mice and human plasma level of angiotensin II was markedly elevated in both H5N1 and H7N9 infected patients [5,6]. Consisting with that, we found administration of recombinant human ACE2 protein effectively ameliorated the murine ALI induced by H5N1 virus infection. Dysregulated angiotensin II level is highly associated with diseases such as hypertension and cardiac disease [7] and two classes of drugs have already been developed and clinically applied to maintain the normal function of RAS. The first class is ACE inhibitors (ACEis), which inhibit ACE from cleaving angiotensin I to generate angiotensin II. The second class is angiotensin II receptor
blockers (ARBs), which block the interaction between angiotensin II and its type I receptor (AT1R) to suppress the downstream immune responses [8]. We thus hypothesized that these drugs, which demonstrated the same therapeutic consequence as increased ACE2 protein expression, may also be effective for the treatment of H5N1 infections. Previous studies have shown that ARBs perform better than ACEi in respiratory disorders such as ARDS induced by bacterial infection, as angiotensin II can be synthesized by some alternative enzymes such as chymase, cathepsin and trypsin [9,10]. Thus in our study, we focused on losartan,
the first orally active non-peptide selective ARB, to further investigate its therapeutic role in ALI induced by
H5N1 virus. To test the potential therapeutic effect of losartan on ALI induced by live H5N1 virus, we used Losartan developed by DuPont Merck Pharmaceutical Co. under the code DuP753 or MK954, which is a potassium salt of 2-n-butyl-4-chloro5-hydroxymethyl-1-{(2Īõ-[1H-tetrazol-5-yl]biphenyl-4-yl)me thyl}imidazole [11], with a molecular mass of 461 Da. We found that administration of losartan at a dose clinically equivalent to human significantly improved the survival rate
in H5N1 virus-infected mice (Figure 1A). Then, the severity of lung injury was determined by lung histopathology andthe wet/dry lung weight ratio, which is an indicator of lung edema. Mice treated with losartan showed alleviated lung edema and improved lung histopathology, demonstrated as lowered lung injury scores and a reduced number of infiltrating leukocytes (Figure 1B and C). Taken together, these results suggest that losartan, a conventional anti-hypertension medication, is a promising strategy to ameliorate ALI and improve survival of mice infected with live avian influenza A H5N1 virus. Though effective when treated with Losartan, the viral load in the lung tissue of infected mice was not reduced after losartan treatment, indicating that losartan may not influence H5N1 viral entry or replication (Figure 1D). Since the binding of angiotensin II with AT1R promotes inflammation and vascular permeability of lung tissue [12], which at least partially accounts for ALI induced by H5N1 virus,
we supposed that the effect of losartan may result from a prevention of excessive downstream inflammatory responses.In agreement with that, IL-6, one of the primary contributor cytokines downstream of AT1R, was also significantly down-regulated in losartan-treated group (Figure 1E). Furthermore,a remarkable recovery of ACE2 expression and down-regulation of ACE in lung tissues of losartan-treated mice were also observed 48 h after H5N1 virus infection (Figure 1F). These data support the above notion that the mechanism of action of losartan in the context of H5N1 influenza virus infection is mediated via RAS pathway.
Dr. David Fedson has recently called for the use of statin, ARB, and ACEi in next influenza pandemic [13], and previous
population-based study has clearly demonstrated that ARB inpatient application for severe pneumonia could significantly
reduce 30-day mortality rate in hospital [14].Here, we further provided experimental evidence that losartan
and perhaps other ARBs should be considered as candidate therapies for clinical treatment of H5N1-infected patients.
Herein randomized clinical trials on ARB treatment for ARDS are indeed in need.Angiotensin II receptor blocker as a novel therapy in acute lung injury induced by avian influenza A H5N1 virus infection in mouse.
https://link.springer.com/content/pdf/10.1007/s11427-015-4814-7.pdfĪĪ
ĪĪ
Angiotensin II receptor blocker as a novel therapy in acute lung injury induced by avian influenza A H5N1 virus infection in mouse. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/25655897ĪĪ
ĪĪ
ĪĪ
ĪĪ
Angiotensin-(1©C7) is an endogenous ligand for the G ...
https://www.pnas.org/content/100/14/8258
Jul 08, 2003 Īż Ang-(1©C7) has become an angiotensin of interest in the past few years, because its cardiovascular and baroreflex actions counteract those of Ang II. Unique angiotensin-binding sites specific for this heptapeptide and studies with a selective Ang-(1©C7) antagonist indicated the existence of a distinct Ang-(1©C7) receptor.
Cited by: 1276
Publish Year: 2003
Author: Robson A. S. Santos, Ana C. Simoes e Silva, Christine Maric, Denise M. R. Silva, Raquel Pillar Macha...
Angiotensin (1-7) and other angiotensin peptides.
https://www.ncbi.nlm.nih.gov/pubmed/23176220
Ang (1-7), produced by angiotensin-converting enzyme 2 (ACE2), has received attention because ACE2-deficient mice have heart failure. In addition, the proto-oncogene mas and insulin regulatory aminopeptidase (IRAP) have been identified as receptors for Ang (1-7) and Ang IV, respectively, accelerating investigations into both peptides.
Cited by: 16
Publish Year: 2013
Author: Mitsuru Ohishi, Koichi Yamamoto, Hiromi Rakugi
Angiotensin-(1-7) Decreases Cell Growth and Angiogenesis ...
https://mct.aacrjournals.org/content/15/1/37
Jan 01, 2016 Īż Angiotensin-(1-7) [Ang-(1-7)] is an endogenous, heptapeptide hormone acting through the Mas receptor (MasR), with antiproliferative and antiangiogenic properties. Recent studies have shown that Ang-(1-7) has an antiproliferative action on lung adenocarcinoma cells and prostate cancer cells.
Cited by: 12
Publish Year: 2016
Author: Pei N, Pei N, Wan R, Chen X, Li A, Zhang Y, Li J, Du H, ChenĪĪ
Natural ACE Inhibitors - peaktestosterone.com
https://www.peaktestosterone.com/Natural_ACE_Inhibitors.aspx
Pomegranate juice is so powerful, for example, that it can alter absorption rates of certain pharmaceuticals. 1. Pomegranate Juice . Pomegrantate Juice has a natural ACE Inhibitor (among other powerful phytochemicals) that can lower blood pressure. I call pomegranate juice ĪŁ
The Algae that Outperform 14 Kinds of Ace Inhibitors
https://www.institutefornaturalhealing.com/2015/07/the-algae-that-outperform-14-kinds...
Jul 27, 2015 Īż ThatĪ»s a two point greater reduction in systolic blood pressure and four point reduction in diastolic blood pressure than 14 different types of ACE inhibitors.2. And the research only gets better from thereĪŁ. Remember, when it comes to heart disease, high blood pressure is only part of the story.ĪĪ
Natural Alternative Remedies Instead of ACE Inhibitors
There has been a lot in the news recently about pharmaceutical drug recalls. Most affected have been the class of drugs known as ACE inhibitors and Angiotensin Receptor Blockers (ARBĪ»s).
These drugs are most often used for hypertension, but also for people after a heart attack and those who suffer from congestive heart failure.
The most common side effect of this drug class is a cough. But recently, reports and FDA recalls have alerted the public to significant cancer risk with these drugs AND cancer-causing additives in the drug.
A recent study found a 14% higher risk of lung cancer in people who used ACE inhibitors for 5 years and 31% for those who used for 10 years!
Drugs are not just the drug. They are the drug PLUS the Ī░inactiveĪ▒ ingredients.
Why Not Do It Naturally?
The following is a list of phytonutrients found in plants that perform the same function as the ACE inhibitor, but they do so naturally.
Remember, you are not deficient in pharmaceuticals. Your blood pressure can be from nutrient deficiencies and not enough of the following:
Delphinidin (pomegranate, berries)
Cyanidin (pomegranate, berries)
Apigenin (parsley, celery, chamomile)
Luteolin (thyme, oregano, and mint)
Luteolin-7-O-glucopyranoside (dandelion)
Kaempferol (veggies, ginkgo, moringa)
Black cumin seed or oil
Cacao- the seed that makes chocolate, when sugar is added.
Green tea
Capers/onions (quercetin)- love adding capers to my homemade organic salad dressing
Nattokinase-eat organic Natto, a fermented soybean popular in the Japanese culture.
Many studies show that whey protein leads to ACE inhibition naturally. This is one of the many reasons we created our Daily Defense Shake Mix made with whey protein from New Zealand grass-fed cowĪ»s milk.
Daily Defense contains the whey AND dozens of vitamins/minerals/phytonutrients to stay healthy in a 21st century world. Delicious chocolate and vanilla. Buy the shake mix TODAY are reap the benefits immediately.Natural Alternative Remedies Instead of ACE Inhibitors
https://thedrswolfson.com/natural-alternative-remedies-instead-of-ace-inhibitors/ĪĪ
Hypertension. Author manuscript; available in PMC 2012 Aug 1.
Published in final edited form as:
Hypertension. 2011 Aug; 58(2): 176©C181.
Published online 2011 Jun 13. doi: 10.1161/HYPERTENSIONAHA.111.173344
PMCID: PMC3141282
NIHMSID: NIHMS305555
PMID: 21670420
Angiotensin-(1-7) Induces Mas Receptor Internalization
Mariela M. Gironacci, Hugo P. Adamo, Gerardo Corradi, Robson A. Santos, Pablo Ortiz, and Oscar A. Carretero
Author information Copyright and License information Disclaimer
The publisher's final edited version of this article is available at Hypertension
See other articles in PMC that cite the published article.Dpto. Qu©¬mica Biol©«gica, IQUIFIB-CONICET, Facultad de Farmacia y Bioqu©¬mica, Universidad de Buenos Aires, Buenos Aires, Argentina (M.M.G., H.P.A and G.C.); Hypertension and Vascular Research Division, Henry Ford Hospital, Detroit, USA (P.O.C and O.A.C.), and Department of Physiology, Federal University of Minas Gerais, Belo Horizonte, Brazil (R.A.S.)
Corresponding autor: Mariela M. Gironacci, IQUIFIB-CONICET, Facultad de Farmacia y Bioqu©¬mica, Universidad de Buenos Aires, Jun©¬n 956, 1113 Buenos Aires, Argentina. TE: 5411-49648290; FAX: 5411- 49625457; ra.abu.byff.bq@aleiram
Go to:
Abstract
Angiotensin (Ang) (1-7) is the endogenous ligand for the G protein-coupled receptor Mas, a receptor (R) associated with cardiac, renal and cerebral protective responses. Physiological evidence suggests that Mas R undergoes agonist-dependent desensitization, but the underlying molecular mechanism regulating R activity is unknown. We investigated the hypothesis that Mas R desensitizes and internalizes upon stimulation with Ang-(1-7). For this purpose, we generated a chimera between the Mas R and the fluorescent protein YFP (MasR-YFP). MasR-YFP transfected HEK 293T cells were incubated with Ang-(1-7) and the relative cellular distribution of MasR-YFP was observed by confocal microscopy. In resting cells, MasR-YFP was mostly localized to the cell membrane. Ang-(1-7) induced a redistribution of MasR-YFP to intracellular vesicles of various sizes after 5 min. Following the time course of [125I]Ang-(1-7) endocytosis we observed that half of MasR-YFP underwent endocytosis after 10 min and this was blocked by a Mas R antagonist. MasR-YFP colocalized with Rab5, the early endosome antigen 1 and the adaptor protein complex 2, indicating that the R is internalized through a clathrin-mediated pathway and targeted to early endosomes after Ang-(1-7) stimulation. A fraction of MasR-YFP also colocalized with caveolin-1 suggesting that at some point MasR-YFP traverses caveolin-1 positive compartments. In conclusion, Mas R undergoes endocytosis upon stimulation with Ang-(1-7) and this event may explain the desensitization of Mas R responsiveness. In this way, Mas R activity and density may be tightly controlled by the cell.
Keywords: receptor internalization, desensitization, angiotensin-(1-7), Mas receptor, trafficking
Go to:
Introduction
The renin angiotensin system (RAS) consists of 2 distinct and counterregulatory axes. The classic angiotensin converting enzyme (ACE)/angiotensin (Ang) II/AT1 receptor (R) axis is responsible for the vasoconstrictive, proliferative, hypertensive, and fibrotic actions of the RAS. Its hyperactivity is associated with hypertension and cardiovascular diseases such as cardiac hypertrophy, heart failure, stroke, coronary artery disease, and end-stage renal disease. This axis is the primary target for the antihypertensive therapy.1 The ACE2/Ang-(1-7)/Mas R axis constitutes an alternative axis that represents an intrinsic mechanism to induce vasoprotective actions by counterregulating the ACE/AngII/AT1R axis, thus inducing many beneficial effects in cardiovascular diseases. This vasoprotective axis of the RAS could be targeted for novel therapeutics strategies.1,2
Ang-(1-7) is the endogenous ligand for the G protein-coupled receptor (GPCR) Mas.3 Prolonged stimulation of Mas R with Ang-(1-7) or with high concentrations of the ligand caused an attenuation of R responsiveness,4-6 suggesting receptor desensitization. Receptor desensitization represents an important physiological Ī░feedbackĪ▒ mechanism that protects against both acute and chronic R overstimulation and is the consequence of a combination of different mechanisms.7 These mechanisms include the uncoupling of the R from heterotrimeric G proteins in response to R phosphorylation followed by the internalization of cell surface receptors to intracellular membranous compartments. Once internalized, the R may be recycled back to the cell surface in a resensitized state competent for signaling, or may be sorted to lysosomes or proteasome for degradation, a process important for signal termination.7-9 Thus, R trafficking has critical function in signal termination and propagation as well as receptor resensitization. The rates of GPCR internalization, recycling and lysosomal sorting differ widely among receptors, suggesting that different mechanisms control trafficking of distinct R.8 Thus, the spatial and temporal control of GPCRs determines the specificity of receptor-mediated signal transduction among the distinct downstream effectors and the ultimate cellular response.
The underlying molecular mechanism of Mas R desensitization is unknown. In this study we investigated the early fate of Mas R following agonist exposure. To better image receptor trafficking we generated a chimera between the Mas R and the fluorescent protein YFP (MasR-YFP).
Go to:
Methods
Materials
Fetal bovine serum, penicilin-streptomycin, Lipofectamine 2000, goat anti-mouse antibody coupled to Alexa 594 and Dulbecco's modified Eagle's medium (DMEM) were purchased from Invitrogen (Carlsbad, CA, USA). Bovine seroalbumin (BSA), paraformaldehyde, phosphate buffered saline (PBS) and the protease inhibitors cocktail were from Sigma Chemical Co. (St. Louis, MO, USA). Mouse anti-GFP monoclonal antibody was from Clontech. Mouse anti-AP50, anti Rab5, anti-caveolin-1 (Cav-1) or anti-early endosome antigen 1 (EEA1) antibodies were purchased from BD Biosciences. [3H]arachidonic acid was from Perkin Elmer, Boston, MA. Ang-(1-7) and [D-Ala7]-Ang-(1-7) were synthesized in our laboratory by the Merrifield solid-phase procedure, as previously described.10 Peptide purity (> 97%) was confirmed by matrix assisted laser desorption mass spectrometry. All other chemicals were analytical grade reagents of the highest purity available.DNA construction
The yellow fluorescent protein (YFP) cDNA was attached to the carboxy-terminus end of the cDNA encoding the Mas receptor by the megaprimer method11 into XhoI-ApaI sites of pEYFP-N1 plasmid (Clontech). The construct was verified by DNA sequencing.
Cell culture and transfection
Human embryonic kidney (HEK) 293T cells were grown in DMEM high glucose supplemented with 10% heat inactivated fetal bovine serum and penicilin-streptomycin at 37 ĪŃC in a humidified atmosphere at 95% air and 5% CO2. Cells were transiently transfected using Lipofectamine 2000 according to instructions of the manufacturer and were used 36 h post-transfection.
MasR-YFP expression
The expression of Mas R fused to YFP in transfected HEK 293T cells was evaluated by Western-blot as previously described10. MasR-YFP expression was evaluated with a mouse anti-GFP monoclonal antibody (dilution 1/1000).
MasR-YFP expression was also evaluated by laser scanning confocal microscopy (Olympus Fluoview FV1000, Japan).
[125I]Ang-(1-7) binding studies
Thirty six hours post-transfection, cells on 12-well plates were rinsed two times with DMEM and equilibrated on ice with incubation buffer (DMEM containing 0.2% BSA and a protease inhibitors cocktail, pH: 7.4) for 30 min. Subsequently, the plates were incubated at 4ĪŃC for 60 min with incubation buffer containing 0.5 nmol/L [125I]Ang-(1-7) (labeled in our laboratory as previously described12). Incubation was stopped by rinsing the cells three times with ice-cold PBS. Cells were solubilized by incubation with 0.1 mol/L NaOH for 60 min and the radioactivity was measured. Nonspecific binding was determined in the presence of 10 ”╠mol/L unlabeled Ang-(1-7), which was no higher than 15 %. Specific binding was calculated by the subtraction of nonspecific binding from total binding. Competition binding experiments were performed and Ki was calculated using Graphpad Prism (Graphpad Software Inc., San Diego, CA).
Arachidonic acid (AA) release
Twenty four hours posttransfection cells were labeled with [3H]AA (0.2 ”╠Ci/well) for 16 h as described previously3. Then, cells were washed with DMEM containing 2% BSA and incubated with Ang-(1-7) during different times at 37 ĪŃC. Radioactivity in the supernatant was measured. For total cellular radioactivity, cells in each well were solubilized with 1 mol/L NaOH and counted. [3H]AA released into the medium was expressed as percent of the total cellular radioactivity and referred to as fractional release.
Receptor internalization assay
It was measured according to the method described by Thomas et al.13 Briefly, thirty six hours posttansfection, cells on 12-well plates were rinsed two times with DMEM, preincubated with incubation buffer (DMEM containing 0.2% BSA and a protease inhibitors cocktail, pH: 7.4) for 30 min and then incubated with 0.5 nmol/L [125I]Ang-(1-7) (labeled in our laboratory as previously described12 during different times. After two washes with ice-cold PBS, surface-bound [125I]Ang-(1-7) associated with non-internalized receptors was separated by treating the cells with an ice-cold acid solution (0.2 mol/L acetic acid, 0.5 mol/L NaCl, pH:5.5) for 5 min on ice and radioactivity was determined (acid-sensitive fraction). Cells, which contain the internalized [125I]Ang-(1-7), were lysed by incubation with 0.1 mol/L NaOH for 60 min and the radioactivity was measured (acid-insensitive fraction). The index of receptor internalization was determined as acid-insensitive cpm as a percentage of the total binding (acid-sensitive + acid-insensitive). Non-receptor-mediated [125I]Ang-(1-7) and surface binding was measured in the presence of 10 ”╠mol/L unlabeled Ang-(1-7) and subtracted from total binding to calculate the specific values. Curve fitting was performed with Graphpad Prism (Graphpad Software Inc., San Diego, CA).
In another set of experiments, transfected cells were incubated with the Mas R antagonist [D-Ala7]-Ang-(1-7) (100 nmol/L) during 15 min and then internalized [125I]Ang-(1-7) was determined as described.
MasR-YFP trafficking
Trafficking pathway was evaluated by colocalization between MasR-YFP and endocytosis markers signals by immunocytochemistry. Briefly, thirty six hours posttansfection, cells were incubated with 10 ”╠mol/L Ang-(1-7) (synthetized in our lab by the Merrifield method as previously described10) for 15 min at 37 ĪŃC. After two washes with PBS, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS and incubated in blocking solution (PBS/0.2% Triton X-100/3% BSA) for 30 min at room temperature. Cells were then incubated with different primary antibodies (anti-AP50, anti Rab5, anti-Cav-1 or anti-EEA1 mouse monoclonal antibodies, diluted 1:150 in blocking solution) overnight at 4 ĪŃC. The samples were rinsed twice in PBS/0.2% Triton X-100, and exposed to the secondary antibody (goat anti-mouse antibody coupled to Alexa 594, dilution 1:600 in blocking solution) for 2h at room temperature.
Samples were mounted and imaged using an Olympus Fluoview FV1000 spectral laser scanning confocal microscope with a 60Ī┴ oil immersion lens using dual excitation (473 nm for YFP and 559 nm for Alexa 594). Due to the spectral properties of the scan head, fluorescence emission was collected between 520 and 550 nm for YFP and 600-660 nm for Alexa 594. Images were obtained using sequential scanning for each channel to eliminate the cross-talk of chromophores. Quantitative colocalization was estimated by Pearson's correlation coefficient and overlap coefficient according to Manders14, which were calculated using Image-Pro Plus software (MediaCybernetics Inc.). Negative controls consisted of mocked transfected cells treated with blocking solution in the absence of the primary antibody. Some images (Figure 3) were acquired using a laser scanning confocal system (Visitech International, England) mounted on a Nikon TE2000-eclipse microscope, 100Ī┴ 1.3 NA lens, and visualized at 514 nm excitation, 550 LP emission. Identical laser, slit, and acquisition settings were used to obtain all images.
An external file that holds a picture, illustration, etc.
Object name is nihms305555f3.jpg
Open in a separate window
Figure 3
Agonist-induced subcellular distribution of MasR-YFP
Subcellular localization of the MasR-YFP fusion protein in HEK 293T transfected cells and stimulated with 1 ”╠mol/L Ang-(1-7) for the defined time periods. Scale bar = 10 ”╠m.
Data analysis
All average results are presented as mean Ī└ SEM. One-way ANOVA computation combined with the Bonferroni test was used to analyze data with unequal variance between each group. A probability level of 0.05 was considered significant.
Go to:
Results
Characterization of MasR-YFP
To better image Mas R trafficking we generated the chimera C-terminally YFP tagged Mas R. Since the fusion of YFP to the C-terminal of the R may alter its correct folding and hence its functionality, we first characterized the C-terminally tagged MasR-YFP. HEK293T cells transfected with the DNA coding for the chimera showed that MasR-YFP was found predominantly in the plasma membrane (Figure 1A). However, intracellular localization of the fusion protein was also detectable, especially in cells showing higher levels of R expression. In some cells, the MasR-YFP was present around the nucleus, presumably in the endoplasmic reticulum, and probably represents newly synthesized molecules passing through the secretory pathway, but intranuclear localization of the receptor was not observed.
An external file that holds a picture, illustration, etc.
Object name is nihms305555f1.jpg
Figure 1
Characterization of MasR-YFP
A, Expression and cellular localization of the MasR-YFP fusion protein in transfected HEK 293T cells. B, Immunoblot of extracts of HEK 293T cells transfected with empty vector (lane 1), the DNA encoding MasR-YFP (lanes 2 and 3) or YFP (lane 4). C, Specific binding of [125I]Ang-(1-7) to MasR-YFP transfected cells in the presence of Ang-(1-7) or the Mas R antagonist (D-Ala-Ang-(1-7). Values are meanĪ└SE.
MasR-YFP expression was also investigated by Western-blot. Figure 1B shows that transfected cells expressed a protein with a molecular weight corresponding to the MasR-YFP chimera (68 kDa).
The functional integrity of the YFP tagged receptor was investigated by Ang-(1-7) binding. As shown in Figure 1C, increasing concentrations of unlabeled Ang-(1-7) as well as the Mas receptor antagonist [D-Ala7]-Ang-(1-7) displaced the binding of [125I]Ang-(1-7) to transfected cells (Ki: 4.92Ī└0.12 Ī┴10-8 mol/L for Ang-(1-7) and 4.96 Ī└0.18Ī┴10-9 mol/L for D-Ala7]-Ang-(1-7)). These results demonstrate that MasR-YFP attained a correct folding in the plasma membrane and binds its physiological ligand as well as its antagonist. Cells transfected with the empty vector showed undetectable Ang-(1-7) binding (total binding: 480Ī└68 cpm vs nonspecific binding 395Ī└87 cpm).
Receptor functionality and coupling were evaluated by measuring Mas R signaling. Mas R activation is coupled to AA release.3,5 To determine the functionality of MasR-YFP, HEK 293T cells transfected with the MasR-YFP construct and labeled with [3H]AA were exposed to increasing concentration of Ang-(1-7). As shown in Figure 2, 100 nmol/L Ang-(1-7) caused an increase in [3H]AA release in MasR-YFP transfected cells. A higher concentration (1 ”╠mol/L) produced a smaller response, suggesting receptor desensitization. When cells were incubated with Ang-(1-7) for a prolonged period, a decrease in the Ang-(1-7)-induced [3H]AA release was observed (Figure 2), suggesting receptor desensitization. Cells transfected with the empty vector (mock) showed no changes in [3H]AA release upon Ang-(1-7) stimulation (Figure 2).
An external file that holds a picture, illustration, etc.
Object name is nihms305555f2.jpg
Open in a separate window
Figure 2
A, [3H]AA release from mocked transfected cells or cells transfected with the DNA coding for MasR-YFP in the absence (C) and presence of Ang-(1-7) during 15 min. Results are presented as the percentage of the response detected in control, taking them as 100%. Values are mean Ī└ SEM. * P < 0.05 compared with control. B, [3H]AA release from MasR-YFP transfected cells in the presence of 100 nmol/L or 1 ”╠mol/L Ang-(1-7) (Ang) during 10, 20 and 30 min. Results are presented as the percentage of the response detected in control, taking them as 100%. Values are mean Ī└ SEM. * P < 0.05 compared with control.
Collectively, these data show that MasR-YFP is properly expressed and fully active. Furthermore, our results show that higher concentrations or longer times of stimulation with Ang-(1-7) caused a decrease in its response (AA release) demonstrating receptor desensitization.
Ang-(1-7) induces MasR-YFP internalization
To investigate whether Ang-(1-7) induces Mas receptor internalization, MasR-YFP transfected cells were incubated with 1 ”╠mol/L Ang-(1-7) during different times and the relative cellular distribution of MasR-YFP was observed by laser scanning confocal microscopy. In resting cells, MasR-YFP was mostly localized on the cell membrane and in the endoplasmic reticulum. Treatment of cells with Ang-(1-7) induced redistribution in fluorescence after 5 min stimulation changing the localization of MasR-YFP to intracellular vesicles of various sizes (Figure 3), suggesting internalization of the receptor upon agonist stimulation.
The capacity of MasR-YFP to internalize in response to Ang-(1-7) was determined by following the time course of [125I]Ang-(1-7) endocytosis. Half of MasR-YFP underwent endocytosis after 10 min, and this internalization was partially blocked by the Mas receptor antagonist, [D-Ala7]-Ang-(1-7) (Figure 4).
An external file that holds a picture, illustration, etc.
Object name is nihms305555f4.jpg
Figure 4
Agonist-induced internalization of MasR-YFP
MasR-YFP transfected cells were incubated with [125I]Ang-(1-7) in the absence (circles) or presence of the Mas R antagonist [D-Ala7]-Ang-(1-7) (squares) and internalized [125I]Ang-(1-7) was evaluated as described in Methods. An index of internalization was calculated by expressing the internalized radioactivity (acid-resistant) as a percentage of the total binding (acid-resistant plus acid-susceptible). Data are meansĪ└SE for four independent determinations.
MasR-YFP trafficking
The Rab family of small GTPases is integral in determining the fate of a GPCR.15 Different Rab GTPase family members selectively associate with specific intracellular structures including both recycling and sorting endosomes, where they mediate multiple steps of vesicular membrane trafficking including vesicle budding, docking and fusion.15 Rab 5 plays a central role in endocytosis via clathrin-coated pits and subsequent fusion of vesicles with early endosomes.15 To investigate whether MasR-YFP was targeted to early endosome, MasR-YFP transfected cells were stimulated with Ang-(1-7) during 15 min and then co-localization of MasR-YFP with Rab5 was investigated. A fraction of MasR-YFP colocalized with Rab5 (Figure 5A) showing that the R was internalized and targeted to early endosomes. In addition to Rab5, early endosomes contain Rab5 effectors and regulator proteins, including EEA1.9 To provide further confirmation that MasR-YFP is targeted to early endosome, colocalization studies between MasR-YFP and the EEA1 marker protein after agonist stimulation was performed. Figure 5B shows that MasR-YFP also colocalized with EEA1, confirming that MasR-YFP is targeted to early endosomes after Ang-(1-7) stimulation.
An external file that holds a picture, illustration, etc.
Object name is nihms305555f5.jpg
Open in a separate window
Figure 5
MasR-YFP trafficking
Colocalization of MasR-YFP (green) and early endosomes markers (Rab 5 and EEA1), clathrin-coated pits (AP50) or caveolin-1 (caveolae marker) (red) in MasR-YFP transfected HEK293T cells stimulated with Ang-(1-7) during 15 min. Three independent experiments were analyzed. See text for analysis. Scale bar = 5 ”╠m.
Most GPCR are endocyted through clathrin-mediated endocytosis.8,9,16 However, some GPCRs preferentially localize to and/or internalize via specialized lipid raft/caveolae microdomains of the plasma membrane.9,17,18 Caveolae are Cav-1-enriched smooth invaginations of the plasma membrane that form a subdomain of lipid rafts and represent a non-clathrin internalization pathway. To begin studying the mechanisms by which Mas R is internalized, MasR-YFP transfected cells were stimulated with Ang-(1-7) and then co-localization of MasR-YFP with markers of the clathrin or caveolin pathway were investigated. We evaluated whether MasR-YFP is endocyted through clathrin-coated pits by analyzing its colocalization with the adaptor protein complex 2 (AP-2, ”╠ or AP50), which is an essential component of the clathrin-coated vesicle machinery.9 Upon Ang-(1-7) stimulation, a fraction of MasR-YFP colocalized with AP-50 (Figure 5C), which suggests that MasR-YFP is internalized via a clathrin-mediated pathway.
To further study whether the caveolae-dependent pathway was involved in the endocytosis of MasR-YFP, colocalization with Cav-1 was evaluated. After Ang-(1-7) stimulation, colocalization of MasR-YFP and Cav-1 specific signal was observed (Figure 5D).
Altogether, these results indicate that MasR-YFP is internalized into early endosomes via a clathrin-dependent pathway. However co-localization with Cav-1 suggests that at some point in the receptor trafficking, MasR-YFP traverses Cav-1 positive compartments.
Go to:
Discussion
The endocytic pathway tightly controls the activity of GPCR. Ligand-induced endocytosis can drive R into divergent lysosomal and recycling pathways, producing essentially opposite effects on the strength and duration of cellular signaling, allowing for the fine-tuning of signal magnitude and duration.9,19 Our study shows that Ang-(1-7) induces Mas R internalization, a process that is involved in the feedback desensitization of GPCR responsiveness that protects against R overstimulation.7 This event may explain the decrease in Ang-(1-7) response in releasing AA from tissue lipids and in stimulating 6-keto-prostaglandin F1”┴ production in rabbit aortic smooth muscle cells,5 phosphatydylcholine biosynthesis in the rat renal cortex4 and the attenuation in responsiveness in the Ang-(1-7)-induced proliferation of endothelial progenitor cells from sham or infarcted rodents.6
GPCR desensitization also acts to filter information from multiple receptor inputs into an integrated and meaningful biological signal. Present results show that MasR-YFPs are internalized together with Ang-(1-7). This event may be important for directing Ang-(1-7) to certain cellular locations for full expression of its biological response, as it happens with Ang II at the renal tissue.20,21 Renal AT1 receptors are responsible for internalizing Ang II, and the presence of substantial Ang II in endosomes in both control and Ang II-infused hypertensive rats supports their internalization into a protected compartment that prevents degradation of some of the internalized Ang II.20,21 In this way, the internalized Ang II activates various signaling pathways, contributing to fibrogenic proliferative responses while also migrating to the nucleus to exert transcriptional effects.21
The fact that a fraction of 20% of the binding of radiolabeled Ang-(1-7) was not completely displaced by Ang-(1-7) or its antagonist or that its endocytosis was not completely prevented by the Mas R antagonist suggests that some of the Ang-(1-7) may be bound to some AT1 or AT2 R or an unknown R or be internalized by another mechanism. For instance, Gonzalez-Villalobos et al22 have shown that the scavenger receptor megalin binds and internalizes Ang-(1-7).
GPCR endocytosis, in addition to playing a role in receptor desensitization, has been shown to have other important functions in regulating and even promoting GPCR signaling.19,23 Recent studies indicate that GPCRs can continue signaling after internalization together with their agonists.19,23 GPCR endocytosis appears to be required for efficient mitogen activated protein kinase (MAPK) signaling by certain GPCRs.19 Growing evidence shows that caveolae is not only involved in endocytosis but also functions as cell surface signal transduction domain by affecting both signaling selectivity and coupling efficacy,17,18 as it happens for the AT1 R.24,25 After agonist binding, the AT1R moves to caveolae and this event is involved in the reactive oxygen species-dependent AT1 R signaling regulating vascular smooth muscle cells hypertrophy.24,25 It is increasingly evident that endocytosis and signaling are not only connected but likely inextricably intertwined. Our present results show that Mas R is internalized upon ligand stimulation into early endosomes via a clathrin-dependent pathway; however, co-localization with Cav-1 suggests that at some point in the receptor trafficking MasR-YFP traverses Cav-1 positive compartments (present results). The relative contribution of both clathrin- and caveolae-dependent pathways in Mas R desensitization deserves further future investigation.
Perspectives
Through desensitization and internalization Mas R activity and density may be tightly controlled by the cell. The broad data available reveal a degree of specificity and plasticity in the cellular regulation of GPCRs by endocytic membrane trafficking. We showed that Mas R is internalized upon ligand stimulation into early endosomes via a clathrin-dependent pathway; however, at some point in the receptor trafficking MasR-YFP traverses Cav-1 positive compartments. To our knowledge this is the first report on Mas R regulation and it opens the way for a better understanding of Mas R biochemistry. Within the cardiovascular system, regulation of GPCR endocytosis and trafficking is of fundamental importance both for physiological homeostasis and molecular response to physiological perturbation.16,19 Many studies suggest that the endocytic trafficking of GPCR is highly controlled and has profound functional consequences in vivo.16,19 Elucidating those mechanisms will grow our understanding of GPCR pharmacology and function, and open new opportunities for the development of strategies to therapeutically manipulate GPCR function in diseases associated with altered GPCR signaling, such as hypertension and congestive heart failure.26,27Angiotensin-(1-7) Induces Mas Receptor Internalization
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3141282/ĪĪ
Published: 10 September 2019
Stimulation of the ACE2/Ang-(1©C7)/Mas axis in hypertensive pregnant rats attenuates cardiovascular dysfunction in adult male offspring
Amanda S. M. Bessa, Érika F. Jesus, Allancer D. C. Nunes, Carolina N. R. Pontes, Ismaley S. Lacerda, Jaqueline M. Costa, Elisângela J. Souza, Ruy S. Lino-J©▓nior, Manoel F. Biancardi, Fernanda C. A. dos Santos, Gustavo R. Pedrino, Diego B. Colugnati, Renata Mazaro-Costa, Elizabeth P. Mendes & Carlos H. Castro
Hypertension Research volume 42, pages1883©C1893(2019)Cite this article
Abstract
The aim of this study was to investigate whether treatment with diminazene aceturate (DIZE), a putative ACE2 activator, or with angiotensin-(1©C7) during pregnancy could attenuate the development of cardiovascular dysfunction in the adult offspring of spontaneously hypertensive rats (SHRs). For this, pregnant SHRs received DIZE or Ang-(1©C7) throughout gestation. The systolic blood pressure (SBP) was measured in the male offspring from the 6th to16th weeks of age by tail-cuff plethysmography. Thereafter, the left ventricular contractile function and coronary reactivity were evaluated by the Langendorff technique. Samples of the left ventricles (LVs) and kidneys were collected for histology and western blot assay in another batch of adult rat offspring. Maternal treatment with DIZE or Ang-(1©C7) during pregnancy attenuated the increase in SBP in adult offspring. In addition, both DIZE and Ang-(1©C7) treatments reduced the cardiomyocyte diameter and fibrosis deposition in the LV, and treatment with Ang-(1©C7) also reduced the fibrosis deposition in the kidneys. Maternal treatment with DIZE, as well as Ang-(1©C7), improved the coronary vasodilation induced by bradykinin in isolated hearts from adult offspring. However, no difference was observed in the contractile function of the LVs of these animals. The expression levels of AT1 and Mas receptors, ACE, ACE2, SOD, and catalase in the LV were not modified by maternal treatment with Ang-(1©C7), but this treatment elicited a reduction in AT2 expression. These data show that treatment with DIZE or Ang-(1©C7) during gestation promoted beneficial effects of attenuating hypertension and cardiac remodeling in adult offspring.Stimulation of the ACE2/Ang-(1©C7)/Mas axis in hypertensive pregnant rats attenuates cardiovascular dysfunction in adult male offspring | Hypertension Research
https://www.nature.com/articles/s41440-019-0321-8ĪĪ
Renin©Cangiotensin system blockade: a novel therapeutic approach in chronic obstructive pulmonary disease
Dinesh Shrikrishna ; Ronan Astin ; Paul R. Kemp ; Nicholas S. Hopkinson
Clin Sci (Lond) (2012) 123 (8): 487©C498.
ACE (angiotensin-converting enzyme) inhibitors and ARBs (angiotensin II receptor blockers) are already widely used for the treatment and prevention of cardiovascular disease and their potential role in other disease states has become increasingly recognized. COPD (chronic obstructive pulmonary disease) is characterized by pathological inflammatory processes involving the lung parenchyma, airways and vascular bed. The aim of the present review is to outline the role of the RAS (renin©Cangiotensin system) in the pathogenesis of COPD, including reference to results from fibrotic lung conditions and pulmonary hypertension. The review will, in particular, address the emerging evidence that ACE inhibition could have a beneficial effect on skeletal muscle function and cardiovascular co-morbidity in COPD patients. The evidence to support the effect of RAS blockade as a novel therapeutic approach in COPD will be discussed.Renin©Cangiotensin system blockade: a novel therapeutic approach in chronic obstructive pulmonary disease | Clinical Science | Portland Press
https://portlandpress.com/clinsci/article-abstract/123/8/487/69059/Renin-angiotensin-system-blockade-a-novel?redirectedFrom=fulltextĪĪ
ACE and ACE2: a tale of two enzymes | European Heart ...
https://academic.oup.com/eurheartj/article/26/4/322/439241
Jan 24, 2005 Īż Northern blot analysis demonstrated that, in contrast to the near ubiquitous expression of somatic ACE, ACE2 gene expression was localized predominantly to the heart, kidney, and testes. Subsequent studies using quantitative PCR have shown that ACE2 gene expression also occurs in the gastrointestinal tract and,...
Cited by: 12
Publish Year: 2005
Author: Lawrence S. Zisman
ACE2 gene expression is up-regulated in the human failing ...
https://bmcmedicine.biomedcentral.com/articles/10.1186/1741-7015-2-19
May 19, 2004 Īż ACE2 is a novel homologue of angiotensin converting enzyme (ACE). ACE2 is highly expressed in human heart and animal data suggest that ACE2 is an essential regulator of cardiac function in vivo . Since overactivity of the renin-angiotensin system contributes to the progression of heart failure, this investigation assessed changes in gene expression of ACE2, ACE, AT 1 receptor and ĪŁ
Cited by: 156
Publish Year: 2004
Author: Andrew B Goulter, Martin J Goddard, Jennifer CĪĪ
Mol Vis. 2014; 20: 1443©C1455.
Expression and cellular localization of the Mas receptor in the adult and developing mouse retina
Tuhina Prasad, Amrisha Verma, and Qiuhong Licorresponding authorDepartment of Ophthalmology, College of Medicine, University of Florida, Gainesville, FL
Purpose
Recent studies have provided evidence that a local renin-angiotensin system (RAS) exists in the retina and plays an important role in retinal neurovascular function. We have recently shown that increased expression of ACE2 and angiotensin (1-7) [Ang (1-7)], two components of the protective axis of the RAS, in the retina via adeno-associated virus (AAV)-mediated gene delivery, conferred protection against diabetes-induced retinopathy. We hypothesized that the protective molecular and cellular mechanisms of Ang (1-7) are mediated by its receptor, Mas, and the expression level and cellular localization dictate the response to Ang (1-7) and activation of subsequent protective signaling pathways. We tested this hypothesis by examining the expression and cellular localization of the Mas receptor in adult and developing mouse retinas.
Methods
The cellular localization of the Mas receptor protein was determined with immunofluorescence of the eyes of adult and postnatal day 1 (P1), P5, P7, P15, and P21 mice using the Mas receptor-specific antibody, and mRNA was detected with in situ hybridization of paraffin-embedded sections. Western blotting and real-time reverse-transcription (RT)©CPCR analysis were performed to determine the relative levels of the Mas protein and mRNA in adult and developing retinas, as well as in cultured retinal M©╣ller glial and RPE cells.
Results
In the adult eye, the Mas receptor protein was abundantly present in retinal ganglion cells (RGCs) and photoreceptor cells; a lower level of expression was observed in endothelial cells, M©╣ller glial cells, and other neurons in the inner nuclear layer of the retina. In the developing retina, Mas receptor mRNA and protein expression was detected in the inner retina at P1, and the expression levels increased with age to reach the adult level and pattern by P15. In the adult mouse retina, Mas receptor mRNA was expressed at a much higher level when compared to angiotensin II (Ang II) type I (AT1R) and type II (AT2R) receptor mRNA.
Conclusions
The Mas receptor is expressed in developing and adult mouse retinas, and is more abundant in retinal neurons than in endothelial and M©╣ller glial cells. These observations suggest that Mas receptor-mediated signaling may play important roles that extend beyond mediating the vascular effects of Ang (1-7) in developing and adult retinas. In addition, the relatively high expression of the Mas receptor when compared to AT1R suggests that they may play a more important role in maintaining normal retinal physiology than previously considered.Introduction
The renin-angiotensin system (RAS) plays a vital role in regulating the normal physiologic functions of the cardiovascular and renal systems. The RAS was classically viewed as a circulating endocrine system with angiotensin II (Ang II) as the main peptide effector hormone, which mediates its effects primarily through activation of the angiotensin type I receptor (AT1R). Recent studies have confirmed the presence of an additional local organ-specific RAS in almost all organs including the retina [1-8]. The discovery of the angiotensin-converting enzyme (ACE) homolog ACE2 resulted in the identification of an important pathway responsible for angiotensin (1-7) [Ang (1-7)] synthesis [9-11]. This enzyme can form Ang (1-7) from Ang II or less efficiently through hydrolysis of Ang I to Ang (1-9) with subsequent Ang (1-7) formation by ACE. Ang-(1-7) is now recognized as a biologically active component of the RAS that plays a critical role in counteracting the effects mediated by Ang II. Ang-(1-7) induces vasodilation, improves insulin sensitivity, and has antiproliferative, antioxidative, and anti-inflammatory activities [8,12-15]. In addition, it is now well established that Ang (1-7) is an endogenous ligand for the G protein-coupled receptor Mas [16]. There is growing evidence indicating that this endogenous counter-regulatory axis of the RAS, composed of ACE2, Ang (1-7), and the Mas receptor, has protective effects in many tissues and organs, including the neurovascular system of the retina and the brain [8,15,17-19]. Increasing evidence indicates that a balance between activation of the ACE/Ang II/AT1R axis and the ACE2/Ang (1-7)/Mas receptor axis plays a critical role in maintaining normal function in different organs and that an imbalance in these opposing pathways toward the ACE/Ang II/AT1R axis predisposes the organism to many pathological conditions, including retinal vascular diseases such as retinopathy of prematurity, diabetic retinopathy (DR), a common diabetic neurovascular complication, choroidal neovascularization, glaucoma, and ocular inflammation [8,17,18,20-23]. Our previous studies have also shown that the increased expression of Ang (1-7) and ACE2 in the retina has a protective role against the development of diabetic retinopathy [17,18] and increased Ang (1-7) levels and ACE2 activities protected ocular inflammation in mice [20,23].
The Mas gene codes for a G-protein-coupled cell surface receptor and was initially discovered by Young et al. in 1986 [24]. They used an RNase protection assay to show high levels of Mas expression in the cerebral cortex and hippocampal regions of the rat brain. The cellular localization and distribution of Mas mRNA in the rat brain was further investigated with in situ hybridization that showed strong specific signals in the dentate gyrus, CA3 and CA4 areas of the hippocampus, piriform cortex, and olfactory bulb, and moderate labeling was observed in the frontal lobe [25,26]. In the mouse, the distribution of Mas mRNA in the brain is comparable to that in the rat, highest in the hippocampus and the piriform cortex. In the adult central nervous system (CNS), Mas mRNA is most abundant in hippocampal pyramidal neurons and dentate granule cells. A study of Mas expression during development showed that Mas is first expressed in the developing rat CNS at postnatal day 1 (P1) [26]. Recently, Mas expression was also discovered in the cardiovascular regions of the brain with western blot and immunofluorescence [27]. However, Mas has a role not only in cardiovascular functions but also in neuronal signaling [19,28-30]. It has been shown by using Mas-deficient mice that Mas has important functions in regulating behavior and the cardiovascular system [31]. Ang (1-7) increases the excitability of CA1 neurons in the hippocampus [28] and enhances hippocampal, long-term potentiation (LTP), whereas Mas-deficient mice did not show Ang (1-7)-induced enhancement of hippocampal LTP [28]. Mas immunoreactivity has also been detected in endothelial cells of blood vessels in the CNS and other organs suggesting that they mediate the vasodilating and vasoprotective actions of Ang (1-7). Recent studies using the Mas-deficient mouse and Mas antagonists have shown that the Mas receptor not only mediates the vascular actions of Ang (1-7) but also is a critical element needed for nitric oxide-mediated vasodilation induced by RAS-dependent and RAS-independent agonists [32].
In this study, we aimed to investigate the expression and cellular localization of Mas receptor mRNA and protein in developing and adult mouse retinas to better understand the molecular and cellular mechanism by which Ang (1-7) attenuates the deleterious effects of Ang II in retinal pathophysiology. Mas receptor protein expression and cell type specificity were determined with immunofluorescence using an antibody against the Mas receptor in combination with other retinal cell-specific markers. Mas receptor mRNA expression and cell type specificity were determined with in situ hybridization. Real-time reverse-transcription (RT)©CPCR was performed to quantify the relative levels of Mas receptor expression in the mouse retina compared to other RAS receptors.ĪĪ
Differential levels of Mas expression in retinal blood vessels and cultured retinal cells
Ang (1-7) has a known vasodilatory function that is mediated by the binding to the specific receptor Mas. Previous Mas receptor expression studies in the brain and other organs have shown that Mas receptors are expressed in blood vessels. Here we show for the first time that the Mas receptor is also expressed strongly in retinal blood vessels. Trypsin digestion was used to isolate retinal blood vessels, and coimmunostaining with CD31, an endothelial cell marker, showed that the Mas receptor protein is strongly expressed in the endothelial cells and pericytes of the retinal vessels (Figure 7A). Four cell lines derived from specific retinal cell types were used to examine the presence or absence of Mas receptor expression in different cell types. The immunofluorescence study showed that the Mas receptor protein is strongly expressed in M©╣ller cells and microglial cells, and a lower level of expression was observed in RPE cells and cone cells (Figure 7B©CE).
Figure 7
Mas expression in retinal vessels and cultured cell lines. A: The Mas receptor protein detected with immunofluorescence and colocalization with endothelial cell marker (CD31) in trypsin digested retinal vasculature preparation. B©CE: Mas expression detected with immunofluorescence in (B) human M©╣ller cells, (C) mouse N9 microglial cells, (D) human retinal pigment epithelial (RPE, ARPE19) cells, and (E) cone photoreceptor cells (661W). Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI). B©CE: Inserts show Mas expression alone. Scale bar=20 ”╠m.Discussion
Previous studies that used RT©CPCR have shown that Mas receptor mRNA is expressed in the retina and the ciliary body of rodent eyes [18,35-37]. However, the studies did not establish the type of retinal cell in which the Mas receptor is localized. To understand the mechanism by which the protective axis of the RAS comprised of ACE2, Ang (1-7), and Mas receptor functions in the retina, it is essential to identify the specific cell types that express the Mas receptor. In the present study, immunofluorescence and in situ hybridization techniques were used to identify for the first time, to our knowledge, the type of cells in the retina that express the Mas receptor as well as the developmental pattern of the expression.
Our study confirmed that Mas receptor mRNA and protein are expressed in the mouse retina. Studying the expression pattern across developmental time points showed that Mas receptor mRNA and protein expression begins as early as P1 and continues to adulthood, with the adult level and pattern reached by P15. Mas receptor mRNA and protein are highly expressed in retinal ganglion cells and photoreceptors. A much lower level of expression was observed in M©╣ller glial and endothelial cells of the retinal blood vessels since their expression becomes easily evident only when they are isolated from retinal tissue in the form of the M©╣ller cell line and trypsin digest preparation of retinal vasculature. This suggests that the ACE2/Ang (1-7)/Mas receptor axis may play a vital protective role in the neurovascular system in developing and adult retinas. Immunofluorescence studies have shown that the Mas receptor and AT1R, the main receptor of Ang II, are expressed in the same cells in the retina, consistent with the counteracting effects mediated by the two receptors [38]. Ang (1-7) has been detected in M©╣ller cells in the human retina [39]. The presence of high levels of Mas receptor in retinal neurons thus strongly suggests that the Mas receptor may also be involved in mediating neuron glial interaction. This makes the Mas receptor an important potential therapeutic target for the treatment of retinal diseases characterized by neurovascular dysfunction such as diabetic retinopathy.
The relative expression level and cellular localization of RAS receptors may have an important role in their functionality. Real-time RT©CPCR results showed that the Mas receptor is expressed in the retina at a much higher level than AT1R and AT2R. The functional significance of these differences in unclear, but may contribute to differential responses in physiologic and pathological conditions. The localization of Mas receptors in specific retinal cells that also express other RAS receptors suggests the possibility of direct interaction at the level of the receptor or an indirect interaction occurring between the corresponding downstream signaling pathways. Previous studies suggested that AT1R and AT2R heterodimerize and/or homodimerize. More recent studies show that similar to other G-protein coupled receptors these receptors are more likely to undergo homodimerization and that AT2R counteracts the AT1R effect via negative crosstalk in the cytoplasm [40]. A similar interaction is possible between AT1R and the Mas receptor and must be elucidated in future studies. The relatively high level of Mas mRNA expression when compared to AT1Ra, AT1Rb, and AT2R strongly suggests that the Mas receptor may play a more important role in the pathophysiology of the retina than previously envisaged.
Our previous study showed that intraocular administration of adenoassociated virus (AAV)-mediated gene delivery of ACE2 or Ang (1-7) to diabetic rats and mice attenuated diabetes-induced retinal vascular leakage, infiltrating inflammatory cells and oxidative damage, thus providing protection against diabetic retinopathy [18]. It has also been shown that intravitreal treatment with Ang (1-7) in rabbits decreases intraocular pressure without affecting the aqueous humor outflow [37]. Since these protective effects of ACE2 and Ang (1-7) are mediated through the activation of the Mas receptor, we need to investigate the effect of manipulating the expression or activity level of the Mas receptor on the development of various retinal neurovascular disorders in future studies. In addition, we also need to study the downstream signaling pathway of Mas receptor activation.
Figure 8
Relative levels of angiotensin receptor expression in the mouse retina. The relative mRNA levels of the AT1Ra, AT1Rb, AT2R, and Mas receptors were examined in adult mouse retinal samples with real-time RT©CPCR and normalized to ”┬-actin. Sample size, n=3.Conclusion
Immunocytochemistry, immunofluorescence, and in situ hybridization showed that Mas receptor protein and mRNA are expressed locally in the retina. The Mas receptor protein is specifically expressed in retinal ganglion cells, nerve fibers, M©╣ller cells, and endothelial cells. The Mas receptor protein is expressed at a high level in the mouse inner retina from early development to adulthood suggesting that they may play an important role in neurovascular function, not only in adult retinas but also in developing retinas. Immunofluorescence in the cell lines showed that the Mas receptor and AT1R are expressed in the same cells in the retina, consistent with the counteracting effects mediated by the two receptors. The Mas receptor expression level was about five times higher than AT1Ra and AT1Rb in the adult mouse retina, thus suggesting that it plays a central role in the pathophysiology of the retinal neurovascular system.Expression and cellular localization of the Mas receptor in the adult and developing mouse retina
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4203581/ĪĪ


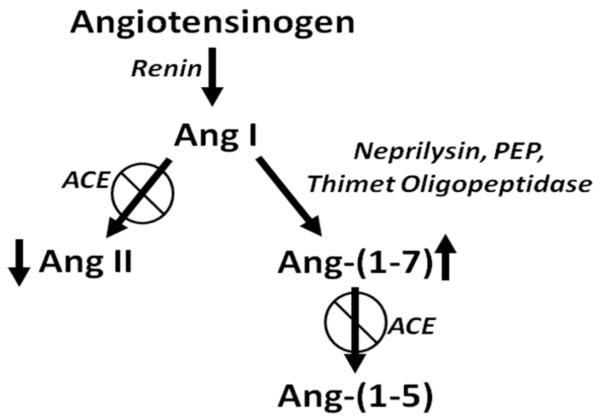
.%20n%20=%203%20for%20each%20group..jpg)
%20inhibition%20of%20endothelial%20cell%20tubule%20formation..jpg)