武汉新冠状病毒研究
欧洲科学家在实验室重组出新冠病毒并激活
新京报
2020-02-23 08:10
进入疫情地图>> 去微公益捐款>>
线上肺炎患者求助专区>>
原标题:欧洲科学家在实验室重组出新冠病毒并激活
新京报快讯(记者 李玉坤)一批欧洲科学家利用反向遗传学技术在实验室重组出新冠病毒。
生物学论文预发布平台BioRxiv1月21日发表一篇名为Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform(利用合成基因组学平台快速重建新冠病毒)的论文手稿,论文的作者来自瑞士伯尔尼的病毒学和免疫学研究所、伯尔尼大学,以及德国、俄罗斯的科研机构。
论文首先证明了基于酵母的合成基因组学平台的完整功能,可用于多种RNA病毒的基因重建,比如,可以重组冠状病毒科,黄病毒科和副粘病毒科的病毒。论文作者表示,反向遗传学已经成为必不可少的工具,它彻底改变了人们对病毒发病机理和疫苗开发的理解。大型RNA病毒基因组(例如冠状病毒),由于大小和偶尔的不稳定的原因,很难在大肠杆菌宿主中进行克隆和操纵,尽管大肠杆菌确实证明了对克隆许多病毒基因组非常有用,但就组装和稳定地维持包括冠状病毒在内的许多RNA病毒均有缺陷。
为了验证合成基因组学平台是否可以应用于其他冠状病毒,论文作者先用了MERS病毒进行了实验,证明合成基因组学平台适合基因修饰冠状病毒基因组,但合成的MERS病毒与细胞培养的相比,复制能力有所降低。
他们将新冠病毒的基因组分割成12个亚基因组片段,大小在0.5-3.4kbp(千碱基对)之间,同时,他们还希望能制造一种表达GFP(绿色荧光蛋白)的新冠病毒,能够在细胞培养中检测,并促进血清学诊断的建立。结果证明,在克隆新冠病毒方面,他们的克隆系统比大肠杆菌系统更有效,因为大肠杆菌系统在复制其中2个片段的时候有问题。基于该平台,他们在收到合成DNA片段后仅一周的时间内,就对最近流行的新冠病毒进行克隆和复活。
作者表示,如果有了新冠病毒毒株,可以建立血清学诊断,开发和评估抗病毒剂和疫苗以及建立适当的体内模型,这是疫情当下迫切需要的。化学合成方法DNA产生的毒株可以绕开病毒毒株的供应限制,还可以对单个基因进行遗传修饰和功能表征。在新冠病毒第一个基因组序列发布时(2020年1月10/11日),尚未有病毒分离株,直到1月底,澳大利亚的科学家分离出了新冠病毒毒株。他们的这种方法可以成为向卫生部门和实验室提供传染性病毒毒株的替代方法,无需获取临床样本。
新京报记者了解到,1月27日,广东省疾病预防控制中心就成功分离出广东省第一株新型冠状病毒毒株,近日,安徽省疾病预防控制中心应用宏转录组基因测序新冠肺炎病例样本,顺利分离到2株新冠病毒毒株,这是继广东、上海、浙江、北京、湖北之后,第六家分离出新冠病毒毒株的省级疾控中心。
重症患者有明显特征!北京地坛医院研究称,年龄≥50岁且淋巴细胞明显降低的新冠肺炎,应尽快收入重症监护室
中国循环杂志
02-13 23:17
2月12日,首都医科大学附属北京地坛医院研究中性粒细胞/淋巴细胞比值人员在医学生物类论文预印本平台medRvix发表的一项研究提示,中性粒细胞/淋巴细胞比值(NLR)有助于早期发现重症新型冠状病毒肺炎(新冠肺炎)患者。
研究者发现,年龄≥50岁且NLR≥3.13的患者进展为重症的可能性大,应尽快转入重症监护病房治疗。
在年龄≥50岁的患者中,NLR≥3.13者一半会进展为重症,而NLR<3.13的患者中仅9.1%进展为重症。
研究者认为,对于新冠肺炎患者,可以根据NLR和年龄进行危险分层和管理。
年龄<50岁且NLR<3.13的患者进展为重症的风险为0,可以在社区医院或家中隔离。
年龄<50岁、NLR≥3.13的患者进展为重症的风险较低,需要住普通隔离病房治疗。
年龄≥50岁、NLR<3.13的患者进展为重症的风险是中等的,应入院隔离治疗,并进行呼吸监测和支持治疗。
年龄≥50岁、NLR≥3.13的患者进展为重症的风险较高,应该积极转入重症监护病房,给予有创呼吸系统支持。
研究者认为,在有大量病例的情况下,这种危险分层和管理方法有助于缓解医疗资源短缺,降低重症患者的死亡率。
该研究前瞻性纳入北京地坛医院于2020年1月13日至1月31日收治的61例新冠肺炎患者,采用LASSO COX回归分析重症患者的预测因素。
分析结果显示,NLR是新冠肺炎患者进展为重症的独立危险因素,且其预测准确性较高(c指数为0.807)。
来源:搜狐Neutrophil-to-Lymphocyte Ratio Predicts Severe Illness Patients with 2019 Novel Coronavirus in the Early Stage. medRvix, Posted February 12, 2020.
Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag
Biorxiv ^ | 01/31/20 | Staff
Posted on 2020/2/1 上午2:32:38 by winoneforthegipper
Abstract We are currently witnessing a major epidemic caused by the 2019 novel coronavirus (2019- nCoV). The evolution of 2019-nCoV remains elusive. We found 4 insertions in the spike glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses. Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV-1 gp120 or HIV-1 Gag. Interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site. The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature. This work provides yet unknown insights on 2019-nCoV and sheds light on the evolution and pathogenicity of this virus with important implications for diagnosis of this virus.Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag
https://www.freerepublic.com/focus/f-news/3812556/posts
2019-nCoV刺突蛋白中独特插入片段与HIV-1 gp120和Gag的离奇相似性
Biorxiv
发表于2020/2/1上午2:32:38通过winoneforthegipper
摘要我们目前正在目睹由2019年新型冠状病毒(2019-nCoV)引起的主要流行病。 2019-nCoV的发展仍然难以捉摸。我们在刺突糖蛋白(S)中发现了4个插入片段,这是2019-nCoV所独有的,其他冠状病毒中没有这些插入片段。重要的是,所有4个插入片段中的氨基酸残基均与HIV-1 gp120或HIV-1 Gag中的氨基酸残基具有相同性或相似性。有趣的是,尽管插入片段在一级氨基酸序列上是不连续的,但2019-nCoV的3D建模表明它们会聚在一起构成受体结合位点。在2019-nCoV中发现4个独特的插入片段,这些插入片段都与HIV-1关键结构蛋白中的氨基酸残基具有同一性/相似性,这在自然界不太可能是偶然的。这项工作提供了关于2019-nCoV的未知见解,并阐明了该病毒的进化和致病性,对诊断该病毒具有重要意义。
2019-nCoV穗蛋白中独特插入片段与HIV-1 gp120和Gag的异常相似性
https://www.freerepublic.com/focus/f-news/3812556/posts
Uncanny similarity of unique inserts in the 2019-nCoV ...
https://www.biorxiv.org/content/10.1101/2020.01.30.927871v1.full.pdf
glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses. Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV- 1 gp120 or HIV-1 Gag.Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag | bioRxiv
https://www.biorxiv.org/content/10.1101/2020.01.30.927871v2
Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag
Prashant Pradhan, Ashutosh Kumar Pandey, Akhilesh Mishra, Parul Gupta, Praveen Kumar Tripathi, Manoj Balakrishna Menon, James Gomes, Perumal Vivekanandan, Bishwajit Kundu
doi: https://doi.org/10.1101/2020.01.30.927871
Abstract
We are currently witnessing a major epidemic caused by the 2019 novel coronavirus (2019- nCoV). The evolution of 2019-nCoV remains elusive. We found 4 insertions in the spike glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses. Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV-1 gp120 or HIV-1 Gag. Interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site. The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature. This work provides yet unknown insights on 2019-nCoV and sheds light on the evolution and pathogenicity of this virus with important implications for diagnosis of this virus.
Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag - New World Order
https://www.tapatalk.com/groups/peakoilpetroleumandpreciousmetals/uncanny-similarity-of-unique-inserts-in-the-2019-n-t41143.htmloronavirus May Have Links to HIV FacebookTwitterPrintMore By Jim Hayek February 1, 2020 Epidemiologist and public health scientist Dr. Eric Feigl-Ding took to Twitter to explain a new study that claims to have found a link between the novel Chinese coronavirus and HIV/AIDS. The study, which comes from Bioxriv, is titled “Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag.” “We found 4 insertions in the spike glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses,” the report’s Abstract section states. “Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV-1 gp120 or HIV-1 Gag.” The report also mentions that “interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site.” Dr. Eric Feigl-Ding, a Chinese-American epidemiologist and public health scientist, referenced the paper in a lengthy Twitter thread explaining the pathology of the virus and the possible misinformation about the epidemic provided by the Chinese government. “BOTTOMLINE: 1) Seafood market not the source,” Feigl-Ding wrote on Twitter. “2) This RNA #coronavirus mutates really fast. 3) has unusual middle segment never seen before in any coronavirus. 4) Not from recent mixing. 5) That mystery middle segment encodes protein responsible for entry into host cells.” The doctor added, “TO BE CLEAR: I am absolutely not saying it’s bioengineering, nor am I supporting any conspiracy theories with no evidence. I’m simply saying scientists need to do more research + get more data. And finding the origin of the virus is an important research priority. Goodnight.” The doctor went on to discuss the possible – but unconfirmed – link between the new coronavirus and human immunodeficiency virus. 16. UPDATE ON 🦠 GENOME 🧬: a very intriguing new paper investigating the aforementioned mystery middle segment w/ “S” spike protein: likely origin from HIV. “Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag” from https://t.co/QAX3usr7vw pic.twitter.com/WeVA948xin — Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020 17. ...WHOA- the authors said the finding was “Unexpectedly” related to genes from HIV virus. Notably there were 4 gene insertions (see figure in above post #16). And so, which HIV gene proteins were found in the new #coronarvirus? Gag protein and Gp120- key HIV proteins... pic.twitter.com/epN66WcObj — Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020 The Bioxrv study concludes by asserting “The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature.” 22. The authors dunked this final conclusion: “This uncanny similarity of novel inserts in the 2019- nCoV spike protein to HIV-1 gp120 and Gag is unlikely to be fortuitous”. Wow, they sure just went straight there! 😱 What a bold paper... I don’t know what to say 🤷🏻♂️ pic.twitter.com/KWcDdknMO4 — Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020 The paper has also been shared online by other public figures. Oh my god. Indian scientists have just found HIV (AIDS) virus-like insertions in the 2019-nCov virus that are not found in any other coronavirus. They hint at the possibility that this Chinese virus was designed [“not fortuitous’]. Scary if true. https://t.co/h6xPX1gYvj pic.twitter.com/kCpd1I00uE — Anand Ranganathan (@ARanganathan72) January 31, 2020 The study has yet to be peer-reviewed, and as of Friday is the only such major paper to link the coronavirus to HIV. Recommended for you
Scientist Explains New Study Revealing Coronavirus May Have Links to HIV · American Truth Today
https://americantruthtoday.com/politics/2020/02/01/scientist-explains-new-study-revealing-coronavirus-may-have-links-to-hiv/
CORONAVIRUS CONTAINS “HIV INSERTIONS”, STOKING FEARS OVER ARTIFICIALLY CREATED BIOWEAPON
‘The virus even responds to treatment by HIV medications’
Zero Hedge - JANUARY 31, 2020 97 评论
Coronavirus Contains "HIV Insertions", Stoking Fears Over Artificially Created Bioweapon
IMAGE CREDITS: NOEL CELIS/AFP VIA GETTY IMAGES.
Over the past few days, the mainstream press has vigorously pushed back against a theory about the origins of the coronavirus that has now infected as many as 70,000+ people in Wuhan alone (depending on whom you believe). The theory is that China obtained the coronavirus via a Canadian research program, and started molding it into a bioweapon at the Institute of Virology in Wuhan. Politifact pointed the finger at Zero Hedge, in particular, though the story was widely shared across independent-leaning media.
The theory is that the virus, which was developed by infectious disease experts to function as a bio-weapon, originated in the Wuhan-based lab of Dr. Peng Zhou, China’s preeminent researcher of bat immune systems, specifically in how their immune systems adapt to the presence of viruses like coronavirus and other destructive viruses. Somehow, the virus escaped from the lab, and the Hunan fish market where the virus supposedly originated is merely a ruse.
Now, a respected epidemiologist who recently caught flack for claiming in a twitter threat that the virus appeared to be much more contagious than initially believed is pointing out irregularities in the virus’s genome that suggests it might have been genetically engineered for the purposes of a weapon, and not just any weapon but the deadliest one of all.
In “Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag“, Indian researchers are baffled by segments of the virus’s RNA that have no relation to other coronaviruses like SARS, and instead appear to be closer to HIV. The virus even responds to treatment by HIV medications.
For those pressed for time, here are the key findings from the paper, which first focuses on the unique nature of 2019-nCoV, and then observe four amino acid sequences in the Wuhan Coronavirus which are homologous to amino acid sequences in HIV1:
Our phylogentic tree of full-length coronaviruses suggests that 2019-nCoV is closely related to SARS CoV [Fig1].
In addition, other recent studies have linked the 2019-nCoV to SARS CoV. We therefore compared the spike glycoprotein sequences of the 2019-nCoV to that of the SARS CoV (NCBI Accession number: AY390556.1). On careful examination of the sequence alignment we found that the 2019- nCoV spike glycoprotein contains 4 insertions [Fig.2]. To further investigate if these inserts are present in any other corona virus, we performed a multiple sequence alignment of the spike glycoprotein amino acid sequences of all available coronaviruses (n=55) [refer Table S.File1] in NCBI refseq (ncbi.nlm.nih.gov) this includes one sequence of 2019-nCoV[Fig.S1]. We found that these 4 insertions [inserts 1, 2, 3 and 4] are unique to 2019-nCoV and are not present in other coronaviruses analyzed. Another group from China had documented three insertions comparing fewer spike glycoprotein sequences of coronaviruses . Another group from China had documented three insertions comparing fewer spike glycoprotein sequences of coronaviruses (Zhou et al., 2020).
We then translated the aligned genome and found that these inserts are present in all Wuhan 2019-nCoV viruses except the 2019-nCoV virus of Bat as a host [Fig.S4]. Intrigued by the 4 highly conserved inserts unique to 2019-nCoV we wanted to understand their origin. For this purpose, we used the 2019-nCoV local alignment with each insert as query against all virus genomes and considered hits with 100% sequence coverage. Surprisingly, each of the four inserts aligned with short segments of the Human immunodeficiency Virus-1 (HIV-1) proteins. The amino acid positions of the inserts in 2019-nCoV and the corresponding residues in HIV-1 gp120 and HIV-1 Gag are shown in Table 1.
The first 3 inserts (insert 1,2 and 3) aligned to short segments of amino acid residues in HIV-1 gp120. The insert 4 aligned to HIV-1 Gag. The insert 1 (6 amino acid residues) and insert 2 (6 amino acid residues) in the spike glycoprotein of 2019-nCoV are 100% identical to the residues mapped to HIV-1 gp120. The insert 3 (12 amino acid residues) in 2019- nCoV maps to HIV-1 gp120 with gaps [see Table 1]. The insert 4 (8 amino acid residues) maps to HIV-1 Gag with gaps.
Why do the authors think the virus may be man-made? Because when looking at the above insertions which are not present in any of the closest coronavirus families, “it is quite unlikely for a virus to have acquired such unique insertions naturally in a short duration of time.” Instead, they can be found in cell identification and membrane binding proteins located in the HIV genome.
Since the S protein of 2019-nCoV shares closest ancestry with SARS GZ02, the sequence coding for spike proteins of these two viruses were compared using MultiAlin software. We found four new insertions in the protein of 2019-nCoV- “GTNGTKR” (IS1), “HKNNKS” (IS2), “GDSSSG” (IS3) and “QTNSPRRA” (IS4) (Figure 2). To our surprise, these sequence insertions were not only absent in S protein of SARS but were also not observed in any other member of the Coronaviridae family (Supplementary figure). This is startling as it is quite unlikely for a virus to have acquired such unique insertions naturally in a short duration of time.
The insertions were observed to be present in all the genomic sequences of 2019-nCoV virus available from the recent clinical isolates. To know the source of these insertions in 2019-nCoV a local alignment was done with BLASTp using these insertions as query with all virus genome. Unexpectedly, all the insertions got aligned with Human immunodeficiency Virus-1 (HIV-1). Further analysis revealed that aligned sequences of HIV-1 with 2019-nCoV were derived from surface glycoprotein gp120 (amino acid sequence positions: 404-409, 462-467, 136-150) and from Gag protein (366-384 amino acid) (Table 1). Gag protein of HIV is involved in host membrane binding, packaging of the virus and for the formation of virus-like particles. Gp120 plays crucial role in recognizing the host cell by binding to the primary receptor CD4.This binding induces structural rearrangements in GP120, creating a high affinity binding site for a chemokine co-receptor like CXCR4 and/or CCR5.
A good recap of the findings was provided by Dr. Feigl-Ding, who started his explanatory thread by pointing out that the transmission rate outside China has surpassed the rate inside China.
A graph is worth a thousand letters. #coronavirus. Source: NYTimes https://t.co/M1K9e6Kgz6 pic.twitter.com/evgM2UHf3U
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
2) Whoa- the rate of increase ***outside of China*** is steeper than inside of China or Wuhan! Figure 1A. From: @TheLancet “Nowcasting and forecasting the potential domestic and international spread of 2019-nCoV https://t.co/SwhxWGeoTj”) pic.twitter.com/u1s4SLEzMv
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
3) “An estimated 75815 individuals have been infected in Wuhan” —> this is substantially higher than current reports or ~10k reports by China 🇨🇳 media. (75k estimate from above Lancet article)
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
4) …”On the present trajectory, 2019-nCoV could be about to become a global epidemic in the absence of mitigation…substantial, even draconian measures that limit population mobility should be seriously and immediately considered in affected areas…” 🤢
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
But the ‘smoking gun’ in this case are pieces of the virus’s genetic code that Indian researchers, led by Prashant Pradhan at the Indian Institute of Technology, found may have been ’embedded’ from HIV, which belongs to an entirely different family of viruses.
16. UPDATE ON 🦠 GENOME 🧬: a very intriguing new paper investigating the aforementioned mystery middle segment w/ “S” spike protein: likely origin from HIV. “Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag” from https://t.co/QAX3usr7vw pic.twitter.com/WeVA948xin
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
17. …WHOA- the authors said the finding was “Unexpectedly” related to genes from HIV virus. Notably there were 4 gene insertions (see figure in above post #16). And so, which HIV gene proteins were found in the new #coronarvirus? Gag protein and Gp120- key HIV proteins… pic.twitter.com/epN66WcObj
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
18. Notably, in 🦠S 🧬, authors say for HIV🧬insertions: “Gag protein of HIV is involved in host membrane binding, packaging of the virus and for the formation of virus-like particles. Gp120 plays crucial role in recognizing the host cell by binding to the primary receptor CD4”
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
19. Again, these are new express published findings and not peer reviewed yet. Let’s not draw conclusions yet. But evidence suggest that 2 different HIV genes 🧬 are present in the #coronarvirus S gene region (that didn’t map to any other coronavirus, according to other studies).
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
20. Further the authors add that “This indicates that these insertions have been preferably acquired by the 2019-nCoV, providing it with additional survival and infectivity advantage. Delving deeper we found that these insertions were similar to HIV-1.” 🤔
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
21. Paper piles on: “these 🧬insertions are present at binding site of 2019-nCoV. Due to presence of gp120 motifs in 2019-nCoV spike glycoprotein at its binding domain, we propose that these motif insertions could have provided an enhanced affinity towards host cell receptors.”🤒
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
22. The authors dunked this final conclusion: “This uncanny similarity of novel inserts in the 2019- nCoV spike protein to HIV-1 gp120 and Gag is unlikely to be fortuitous”. Wow, they sure just went straight there! 😱 What a bold paper… I don’t know what to say 🤷🏻♂️ pic.twitter.com/KWcDdknMO4
— Dr. Eric Feigl-Ding (@DrEricDing) January 31, 2020
The punchline:
9. BOTTOMLINE: 1) Seafood market not the source. 2) This RNA #coronavirus mutates really fast. 3) 🧬 has unusual middle segment never seen before in any coronavirus. 4) Not from recent mixing. 5) That mystery middle segment encodes protein responsible for entry into host cells.
— Dr. Eric Feigl-Ding (@DrEricDing) January 28, 2020
To be sure, Dr. Feigl-Ding insists that he’s not trying to promote any ‘conspiracies’ about the virus being a bioweapon developed by the Chinese, although it is difficult to find a proper name for what appears to be an artificial, weaponized virus.
10. TO BE CLEAR: I am absolutely not saying it’s bioengineering, nor am I supporting any conspiracy theories with no evidence. I’m simply saying scientists need to do more research + get more data. And finding the origin of the virus is an important research priority. Goodnight😴 pic.twitter.com/N4Yp2H8Tst
— Dr. Eric Feigl-Ding (@DrEricDing) January 28, 2020
Another doctor chimed in with what he thought was a solid explanation for the virus’s irregularities…
Dr. @ARanganathan72 might explain. https://t.co/zfOynizRJ6
— Shankara (@fondoflinux) January 31, 2020
Sure. 2019-nCoV is a +ve strand RNA virus that enters human cell and first encodes its RNA-replicase to make -ve stranded RNA that serves a template to make +ve strand RNA that is then translated for daughter nCoV. Drugs Lopinavir and Remdesivir target its protease and replicase.
— Anand Ranganathan (@ARanganathan72) January 31, 2020
…Until he realized something disturbing.
Oh my god. Indian scientists have just found HIV (AIDS) virus-like insertions in the 2019-nCov virus that are not found in any other coronavirus. They hint at the possibility that this Chinese virus was designed [“not fortuitous’]. Scary if true. https://t.co/h6xPX1gYvj pic.twitter.com/kCpd1I00uE
— Anand Ranganathan (@ARanganathan72) January 31, 2020
Coronavirus Contains “HIV Insertions”, Stoking Fears Over Artificially Created Bioweapon
https://www.infowars.com/coronavirus-contains-hiv-insertions-stoking-fears-over-artificially-created-bioweapon/
Normally "inserts" used in the manuscript are "indels" in protein alignments, short for insertions and deletions.
What I think has happened is a group investigating indels in HIV env noticed indels in 2019-nCov. Essentially I think the correlation is spurious - but I haven't test it, but the area of research in understanding indels is certainly valid and important.
What is certain is that indels induce a large structural change to a protein structure and any Gibbs free-energy style calculation will identify this.
Vaccine The spike protein will be the primary candidate to make a 2019-nCov vaccine and that is a very important reason why the sequence was rapidly released. So it is an important protein and the structural changes indels induce mean that a SARS vaccine will probably not provide much protection against 2019-Cov, even apart from the amino acid divergence (below).
Differences 2019-nCov vs HIV In summary, alot. HIV env and particularly HIV gag are very different from coronaviruses, both in the mechanism of genome replication, coronavirus never leaves the cytoplasm, clinical outcomes, tissue tropism and duration of infection.
Similarities HIV env and the glycoprotein spike of coronaviruses are the receptor binding protein to gain entry into a cell. They are called structural proteins. Entry to a cell can be blocked by antibodies and these antibodies are called "neutralizing antibodies". Neutralizing antibodies are catastrophic for a virus. Other antibody responses can be effective, such as IgM, but to clear an infection just using antibodies, you need neutralising antibodies. Both HIV env and the coronavirus spike protein are subject to neutralising antibodies. HIV gag has nothing to do with HIV env, in terms of function or antibody exposure. This is why the spike protein will be the primary vaccine candidate for a subunit vaccine.
Coincidence, law of chance There is large variation of indels in HIV env within HIV and what the authors are inferring is there is a resemblence to that between SARS and 2019-nCov. In my opinion this is a coincidence, because they are comparing a large repertoire of HIV varients against a single indel pattern in the coronaviruses.
Why coronavirus indels?
That is a very good question. Generically indels in viral surface antigen genes are common, much more common in other proteins - such as those involved in virus replication (non-structural proteins). The amino acid identity between SARS and 2019-nCov is 80%, and in any virus, such as flaviviruses 80% identity means indels will be present in surface antigens between the viruses. The answer is it is not unusual in any RNA virus to see indels at a comparatively large amino acid divergence.
What function could they serve
I've briefly looked at indel bioinformatics between flaviviruses (Zika virus, yellow fever virus etc..) notably using envelope (E) protein sequences, and they also occur between African Zika viruses in the E-protein. E-protein being the equivalent of coronavirus spike protein, the receptor-binding protein. No-one has ascribed a function to them and that is the problem with this manuscript.
Hypthoses
One theory is that a structural change in the protein will occur to stop antibody binding.
Another theory is they have functional differences, such as cell tropism
Bioinformatically separating the two theories is extremely hard without wetlab experimentation.
Int J Pept. 2012; 2012: 256294.
Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease
Chris Tikellis * and M. C. Thomas
Division of Diabetic Complications, Baker IDI Heart and Diabetes Institute, P.O. Box 6492 Melbourne, VIC 8008, Australia
Abstract
Angiotensin-converting enzyme 2 (ACE2) shares some homology with angiotensin-converting enzyme (ACE) but is not inhibited by ACE inhibitors. The main role of ACE2 is the degradation of Ang II resulting in the formation of angiotensin 1–7 (Ang 1–7) which opposes the actions of Ang II. Increased Ang II levels are thought to upregulate ACE2 activity, and in ACE2 deficient mice Ang II levels are approximately double that of wild-type mice, whilst Ang 1–7 levels are almost undetectable. Thus, ACE2 plays a crucial role in the RAS because it opposes the actions of Ang II. Consequently, it has a beneficial role in many diseases such as hypertension, diabetes, and cardiovascular disease where its expression is decreased. Not surprisingly, current therapeutic strategies for ACE2 involve augmenting its expression using ACE2 adenoviruses, recombinant ACE2 or compounds in these diseases thereby affording some organ protection.
Figure 1
Schematic representation of the renin-angiotensin system (RAS) and the key balancing role of ACE2. Abbreviations, ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; NEP: neprilysin; AT1: Ang II type 1 receptor; AT2: Ang II type 2 receptor; PEP: prolyl endopeptidase; CAGE: chymostatin-sensitive angiotensin II-generating enzyme.1. Introduction
The renin-angiotensin system (RAS) is a signalling pathway that acts as a homeostatic regulator of vascular function [1]. Its systemic actions include the regulation of blood pressure, natriuresis, and blood volume control. However, the RAS also plays an important local role, regulating regional blood flow and controlling trophic responses to a range of stimuli. The RAS is composed of a number of different regulatory components and effector peptides that facilitate the dynamic control of vascular function, in both health and disease (Figure 1). Many of these components have opposing functions to accommodate a rapid but coordinated response to specific triggers. For example, angiotensin I (Ang I) is metabolised by the dipeptide carboxypeptidase, angiotensin-converting enzyme (ACE) to form angiotensin II (Ang II) and Ang II is metabolised by the carboxypeptidase, ACE2, producing the vasodilator, angiotensin(1–7) (Ang 1–7) [2–4]. Historically, ACE and Ang II have been the key focus for clinical interventions targeting the RAS and its pathogenic actions. However, recent studies have also demonstrated the importance of ACE2 in maintaining the balance of the RAS. Indeed, in some settings, and the cardiovascular system in particular, ACE2 may be more important than ACE in regulating local levels of Ang II and Ang 1–7, and therein the balance of RAS activation. For example, we have shown that acquired or genetic deficiency of ACE2 results in increased tissue and circulating levels of Ang II [5, 6] and reduced levels of Ang 1–7 [6]. By contrast, Ace KO mice have modestly reduced circulating Ang II, while tissue levels are not significantly modified, possibly as substantial amounts of Ang II are generated by non-ACE pathways, while degradation pathways for Ang II are more limited [7]. This paper will specifically examine the actions of ACE2 in the body and discuss their potential role in health and various disease states.
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.001.jpg
Figure 1
Schematic representation of the renin-angiotensin system (RAS) and the key balancing role of ACE2. Abbreviations, ACE: angiotensin-converting enzyme; ACE2: angiotensin-converting enzyme 2; NEP: neprilysin; AT1: Ang II type 1 receptor; AT2: Ang II type 2 receptor; PEP: prolyl endopeptidase; CAGE: chymostatin-sensitive angiotensin II-generating enzyme.
2. Angiotensin-Converting Enzyme (ACE2)
ACE2 is a type 1 integral membrane glycoprotein [8] that is expressed and active in most tissues. The highest expression of ACE2 is observed in the kidney, the endothelium, the lungs, and in the heart [2, 8]. The extracellular domain of ACE2 enzyme contains a single catalytic metallopeptidase unit that shares 42% sequence identity and 61% sequence similarity with the catalytic domain of ACE [2]. However, unlike ACE, it functions as a carboxypeptidase, rather than a dipeptidase, and ACE2 activity is not antagonized by conventional ACE inhibitors [4]. The major substrate for ACE2 appears to be (Ang II) [2–4], although other peptides may also be degraded by ACE2, albeit at lower affinity. For example, ACE2 is able to cleave the C-terminal amino acid from angiotensin I, vasoactive bradykinin (1–8), des-Arg-kallidin (also known as des-Arg10 Lys-bradykinin) [2], Apelin-13 and Apelin-36 [9] as well as other possible targets [10]. The noncatalytic C-terminal domain of ACE2 shows 48% sequence identity with collectrin [11], a protein recently shown to have an important role in neutral amino acid reabsorption from the intestine and the kidney [12]. This is highly consistent with ACE2's actions as a carboxypeptidase, as the removed amino acid then becomes available for reabsorption. The cytoplasmic tail of ACE2 also contains calmodulin-binding sites [13] which may influence shedding of its catalytic ectodomain. In addition, ACE2 has also been associated with integrin function, independent of its angiotensinase activity.
3. ACE2 and Atherosclerosis
Abnormal activation of the RAS contributes to the development and progression of atherosclerotic vascular disease [14–16]. Independent and additional to the induction of systemic hypertension and vasoconstriction, Ang II has a number of direct proatherosclerotic effects on the vascular wall [17–19], including promoting inflammation [20], endothelial dysfunction [21], oxidative stress, endothelial cell, and vascular smooth muscle cell migration, growth, proliferation [22], and thrombosis. By contrast, the major product of ACE2, Ang 1–7, has a range of anti-inflammatory and antioxidant effects [23, 24] that oppose those of Ang II in the vasculature. Indeed, an infusion of Ang 1–7 is able to attenuate vascular dysfunction and atherosclerosis in genetically susceptible apolipoprotein E knockout (apoE KO) mice [25], possibly by increased activation of the Mas receptor and the type 2 angiotensin receptor (AT2). It is thought that the balance of Ang II and Ang 1–7 represents an important driving factor for vascular disease progression. Consequently, ACE2 is also likely to play an important role in atherosclerotic plaque development. Certainly, ACE2 expression is reduced in established atherosclerotic plaques [26] and in proatherosclerotic states, such as diabetes [27]. However, direct evidence for ACE2 in the development and progression of atherosclerotic plaques has only recently become available [5].
We have shown that in apoE KO mice, deficiency of ACE2 is associated with increased plaque accumulation (Figure 2), comparable to that observed following angiotensin II infusion [19]. This possibly relates to an increased proinflammatory responsiveness [5], as leukocyte recruitment and adhesion to the nascent atherosclerotic lesion is generally regarded as one of the first steps toward plaque formation. While a healthy endothelium does not in general support binding of white blood cells, we show that the aortic endothelium of apoE/Ace2 double KO mice shows increased adhesion of labeled leukocytes [5]. In addition, genetic ACE2 deficiency is associated with upregulation of putative mediators of atherogenesis, such as cytokines and adhesion molecules. The role of the RAS in these actions is further emphasized by the finding that RAS blockade is able to prevent atherogenesis in apoE/Ace2 double KO mice. Such data emphasize the potential utility of ACE2 repletion as a strategy to reduce atherosclerosis, particularly in combination with ACE inhibition and other interventions to reduce activation of the RAS (see below).
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.002.jpg
Figure 2
Increased plaque area accumulation in the aorta of Apoe/Ace2 double KO mice when compared to control Apoe KO mice [5]. *vs control Apoe KO mice P < 0.05.
4. ACE2 and Hypertension
Activation of the RAS is known to be a key mediator of hypertension, and interventions to block RAS activation are the most widely used of all blood pressure lowering agents. The antihypertensive efficacy of these agents is partly mediated by their ability to reduce Ang II or its signalling. However, the antihypertensive effects of conventional RAS blockade are also partly determined by the ability of both ACE inhibitors and angiotensin receptor blockers (ARBs) to increase circulating levels of Ang 1–7 [28]. Moreover, inhibiting the vascular actions of Ang 1–7 in spontaneously hypertensive rats (SHRs) receiving RAS blockade, attenuates the antihypertensive response to these agents [28, 29]. Given that the major source of Ang 1–7 is ACE2, this data suggests that ACE2, consequently influences not only the development of hypertension, but also potentially the response to its treatment. Certainly, ACE2 expression is abnormal in SHRs, in which one genetic component of this phenotype tracks to the Ace2 locus. In addition, ACE2 deficiency is associated with modest systolic hypertension [30], although the mouse genetic background significantly alters the cardiovascular phenotype [30–33]. Ace2 KO mice also have a heightened hypertensive response to Ang II infusion associated with exaggerated accumulation of Ang II in the kidney [30].
The RAS and ACE2 are also implicated in the pathogenesis of central hypertension. In particular, the rostral ventrolateral medulla (RVLM) is a relay point that provides supraspinal excitatory input to sympathetic preganglionic neurons in the regulation of blood pressure. In the SHRs, ACE2 expression is reduced in the RVLM [34], and persistent overexpression of ACE2 in the RVLM results in a significant attenuation of high blood pressure in this model [35, 36]. In addition, injections of the ACE2 inhibitor MLN4760 into the nucleus tractus solitarii reduce reflex bradycardia in response to the baroreceptor stimulation in rats [37], suggesting an additional role for central ACE2 in controlling baroreceptor responsiveness.
5. ACE2 in Heart Failure
In addition to effects on blood pressure, natriuresis and atherogenesis, the RAS plays a critical pathophysiological role in the maintaining and subsequently subverting cardiac function in the setting of progressive heart failure [38]. The cardiac RAS is upregulated in almost all models of cardiac injury, including volume overload [39], myocardial infarction [40], and heart failure [41]. As in the kidney, RAS upregulation appears to be a homeostatic response to restore cardiac function. For example, Ang II is an inotropic and growth factor for cardiac myocytes, stimulating compensatory hypertrophy [42]. Ang II is also important in left ventricular remodeling following myocardial infarction or with after-load-induced cardiac hypertrophy [43]. However, in the long term such actions lead to progressive functional loss and cardiac fibrosis [42], as the synthesis of extracellular matrix is increased by Ang II [44]. The key role of RAS activation in the development and progression of cardiac failure is supported by findings in a number of different models in which blockade of the RAS was able to attenuate or prevent cardiac damage, independent of blood pressure lowering [45].
In the heart, ACE2 represents the primary pathway for the metabolism of Ang II [46, 47]. ACE2 deficiency in mice results in early cardiac hypertrophy (Figure 3) [32] and accelerates adverse postmyocardial infarction ventricular remodeling [48]. Furthermore, this appears to be through the activation of the NAPDH oxidase system with the p47(phox) subunit playing a critical role [49]. In some, but not all models, ACE2 deficiency also results in progressive cardiac fibrosis with aging and/or cardiac pressure overload [33, 50, 51]. Again, these changes are reversed following treatment with ACE inhibitors or AT1 receptor blockers [33, 50, 51] suggesting that the balance of ACE and ACE2 in the heart is an important driving factor for progressive cardiac disease.
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.003.jpg
Figure 3
Increased LV mass in Ace2 KO mice versus C57bl6 mice (unpublished data). *vs control C57Bl6 mice, P < 0.05.
6. ACE2 and Chronic Kidney Disease (CKD)
The RAS also plays an important role in renal physiology and pathophysiology. In the adult kidney [2], ACE2 is predominantly expressed in the proximal tubule at the luminal brush border. Despite the presence of unopposed ACE activity and elevated Ang II levels, both kidney function and renal development are normal in the Ace2 knockout mouse [33]. By comparison, ACE, angiotensinogen, and AT1 receptor deficiency results in a number of alterations in kidney morphology [52]. This suggests that, at least in the healthy state, ACE2 may have a limited role in regulating renal development. However, the actions of ACE2 appear to come into its own in states of RAS activation. This is much like Ang 1–7, its major product, which shows very limited renal effects in the healthy state but profound benefits in the diabetic kidney and other states associated with renal damage and activation [10, 53]. For example, ACE2 deficient mice have been reported to show increased age-related glomerulosclerosis in susceptible mouse models [54] and enhanced renal Ang II-induced renal oxidative stress, resulting in greater renal injury [55]. Similarly, in the diabetic kidney, downregulation of tubular ACE2 (Figure 4) [27] is associated with albuminuria or tubular injury, while further inhibition of ACE2 results in augmented renal damage [56, 57]. Indeed, in most forms of CKD, including diabetes, expression of ACE2 has been reported to be reduced in tubules. However, some studies have reported that glomerular ACE2 expression may be increased in human kidney disease [58]. It is possible that this differential expression pattern of glomerular and tubular ACE2 is an important determinant for progressive renal disease.
An external file that holds a picture, illustration, etc.
Object name is IJPEP2012-256294.004.jpg
Open in a separate window
Figure 4
Reduced ACE2 expression (arrows) in renal cortical tubules of diabetic mice (b) when compared to control mice (a) [27].
7. ACE2 and the Lung
RAS activity is intrinsically high in the lung, which is a major source of ACE and therefore a major site of systemic Ang II synthesis. ACE2 is also highly expressed in the lung. Pulmonary ACE2 appears to have a role in regulating the balance of circulating Ang II/Ang 1–7 levels. Ang II induces pulmonary vasoconstriction in response to hypoxia, which is important in preventing shunting in patients with pneumonia or lung injury [59]. Locally increased Ang II production also triggers increasing vascular permeability facilitating pulmonary edema [60]. In Acute respiratory distress syndrome (ARDS), the RAS appears crucial in maintaining oxygenation, possibly as widespread lung injury would otherwise result in complete pulmonary shutdown. Certainly in ARDS models, ACE2 knockout mice displayed more severe symptoms of this disease compared with wild-type mice [60] while overexpression appears protective (see below). Interestingly, ACE2 protein also appears to be the entry-point receptor for the severe acute respiratory syndrome (SARS) coronavirus [61, 62].
8. Replenishing ACE2 as a Potential Therapeutic
Given the key role of ACE2, degrading Ang II and generating Ang 1–7, a number of studies have explored its potential as a treatment strategy using human recombinant ACE2 (rhACE2) or adenoviral (Ad)-ACE2 in animal disease models. For example, overexpression of ACE2 in human endothelial cells attenuates Ang II-induced oxidative stress and subsequent increase in monocyte adhesion [63]. Similarly, in rabbits, a recombinant ACE2 expressing vector stabilized atherosclerotic plaques induced by balloon injury to the abdominal aorta [64]. Treatment with a lentiviral vector containing ACE2 resulted in lower blood pressure in hypertensive mice [65, 66] or following an Ang II infusion [67]. Strategies to upregulate or replenish ACE2 are thought to be beneficial in diabetic nephropathy. For example, in diabetes the replenishment of ACE2 with rhACE2 in a mouse model of type 1 diabetes attenuated diabetic kidney injury as well as reducing in blood pressure [68]. The use of (Ad)-ACE2 has had similar beneficial effects in streptozotocin-induced diabetes, where it was shown to attenuate glomerular mesangial cell proliferation, blood pressure, oxidative stress, and fibrosis [69].
In contrast to these studies, the potential utility of ACE2 supplementation in cardiac disease remains controversial. The expression of ACE2 in the failing human heart is generally increased [70–72], consistent with the finding of elevated levels of Ang 1–7 in the same setting [73]. More importantly, overexpression of ACE2 in cardiac myocytes resulted in conduction disturbances by 2 weeks of age, ultimately leading to lethal ventricular arrhythmias and severe fibrosis [74, 75]. This may be because ACE2 is not normally expressed in high levels in myocytes, although it is present in the endocardium and other cardiac cells. However, other studies using transgenic overexpression of cardiac ACE2 have demonstrated partial protection in the heart from ischemia-induced heart failure [76]. Indeed, more recent studies using rhACE2 have shown beneficial cardiac effects [77]. However, the indication for ACE2 that appears most likely to be first tested in the clinic is the treatment of ARDS. In murine models, treatment with catalytically active recombinant ACE2 protein improved the symptoms of acute lung injury in wild-type mice as well as in ACE2 knockout mice [60]. Clinical trials in this often fatal condition are now underway.
Perhaps, the most clinically interesting, however, is the potential for rACE2 to augment the vasculoprotective effects of ACE inhibition or ARBs, in the millions of patients that take these agents, worldwide. In theory, this would be achieved by preventing feedback escape for RAS blockade or enhancing the generation of Ang 1–7, and subsequent signaling through the Mas receptor and or AT2 receptor. Certainly, ACE2 inhibition attenuates the effects of RAS blockade, both in vitro [78] and in vivo [6]. But could rACE2 make the response to conventional RAS blockade more effective or durable? The problem is that conventional RAS blockade is highly effective in animal models of vascular and renal disease, meaning that it is difficult to explore the potential for further improvements. However, chronic intravenous infusion of ANG-(1–7), or the nonpeptide mas receptor agonist, AVE-0991, are able to improve salt-induced suppression of endothelium-dependent vasodilatation in the mesenteric arteries of male Sprague-Dawley rats, and these actions are not modified by the angiotensin receptor blocker, losartan [79], suggesting that the effects of enhancing the Ang 1–7 mas axis may be beneficial, even in the setting of conventional RAS blockade. Although it enhances the generation of Ang 1–7, whether rACE2 can also provide synergistic benefits, remains to be established.9. ACE2 Augmenters: A New Kind of Intervention
Rather than providing exogenous ACE2, an alternative approach for augmenting ACE2 has been to increase its endogenous expression. For example, in hypertensive SHRs, all-trans retinoic acid, which increases ACE2 expression, lowers blood pressure levels, and prevents vascular damage [80]. Unfortunately retinoic acid has broader actions that make its potential utility as a therapeutic limited. However, compounds that increase activity of ACE2 could also be beneficial as a treatment in conditions where ACE2 activity is decreased. One exemplar is xanthenone (XNT). This molecule was selected following structure-based screening on compounds that would stabilize the activated form of ACE2, thereby enhancing its catalytic efficacy [81]. In experimental studies, this compound has been shown to enhance ACE2 activity in a dose-dependent manner and significantly decreased blood pressure in both SHRs rats and wild-type WKY rats [81]. Furthermore, improvements in cardiac function and reversal of myocardial, perivascular, and renal fibrosis in the SHRs were also observed [81, 82]. XNT has also shown promise in treating pulmonary hypertension (PH). For example, in a rat model of PH, treatment with XNT was shown to reduce elevated right ventricular systolic pressure, right ventricular hypertrophy, increased pulmonary vessel wall thickness, and interstitial fibrosis [83]. In a model of thrombus formation using SHRs and WKY rats, XNT has also shown antithrombotic action, reducing platelet attachment, and reducing thrombus formation [84]. This compound will not come to clinical trials because of issues of solubility that restrict its formulation. However, other drugs of the same class may prove more suitable.
10. Conclusion
ACE2 is an integral component of the RAS. It is highly expressed in the vasculature, the kidney, lungs, and heart where its actions on peptide signals balance and offset those of ACE. Its actions appear critical in a variety of disease states, including hypertension, diabetes, ageing, renal impairment, and cardiovascular disease. ACE2 deficiency leads to modest physiological changes. However, in states of RAS activation, the loss of ACE2 appears far more important in the development and progression of disease. By contrast, augmentation of ACE2 expression, either directly with recombinant ACE2 or indirectly via agonists like XNT, may have important benefits relevant in the treatment of a range of conditions.Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3321295/
The Secret Life of ACE2 as a Receptor for the SARS Virus
Dimiter S Dimitrov Human Immunovirology and Computational Biology Group, LECB, CCR, NCI-Frederick, NIH, Frederick, MD 21702 USA
Open ArchiveDOI:https://doi.org/10.1016/S0092-8674(03)00976-0 AbstractAbstract
The membrane-associated carboxypeptidase angiotensin-converting enzyme 2 (ACE2) is an essential regulator of heart function. Now, Li at al. identify and characterize an unexpected second function of ACE2 as a partner of the SARS-CoV spike glycoprotein in mediating virus entry and cell fusion.
Main Text
Many cell surface-associated molecules with diverse sequences, structures, and cellular functions are usurped by viruses for use as their receptors. Receptor identification is important for understanding virus tropism, pathogenicity, and mechanisms of entry, and may help in the development of therapeutics and vaccines, but remains a challenging task. Although the number of identified receptors for human viruses has increased rapidly over the past two decades, the receptors for most of the several hundred known human viruses remain elusive. The receptor for one of the three known human coronaviruses, HCoV-229E, was identified as the human aminopeptidase N (hAPN, CD13) more than a decade ago (Yeager et al., 1992), but the functional receptor for another human coronavirus, HCoV-OC43, remains unknown. However, the overall pace of research on the third human coronavirus, the SARS-CoV, has been amazingly rapid, and, in keeping with this, just months after the virus itself was discovered, the angiotensin-converting enzyme 2 (ACE2) was identified as its receptor (Li et al., 2003).
Li et al. used a straightforward approach—coimmunoprecipitation of the virus attachment glycoprotein (S1) with lysates from cells that are susceptible to virus infection (Vero E6) followed by mass spectrometry analysis of the coimmunoprecipitated proteins. To express the SARS-CoV full-length glycoprotein (S) and S1 in sufficient amounts required for coimmunoprecipitation and functional characterization, they synthesized a codon-optimized gene based on the published sequence of the Urbani isolate (Rota et al., 2003). The observations that ACE2 specifically binds to S1, supports formation of syncytia due to cell fusion mediated by the interaction with S, and mediates infection of cells otherwise inefficient for virus replication that can be inhibited by an anti-ACE2 antibody provide convincing evidence for its receptor function.
In a remarkable series of experiments, Li et al. not only identified the virus receptor, but also demonstrated key characteristics of the membrane fusion process mediated by the ACE2 interaction with S. First, they showed that expression of recombinant ACE2 and S resulted in cell fusion at neutral pH. This finding suggests that low pH and other viral proteins are not required for fusion. The S glycoprotein from another SARS-CoV isolate (Tor2) can also mediate fusion at neutral pH (Xiao et al., 2003), suggesting that the absence of a low pH requirement to trigger fusion is not strain-specific, although more isolates should be tested. It has also been demonstrated that expression of recombinant S from some coronaviruses can lead to syncytia formation at neutral pH (Lai and Cavanagh, 1997). However, it remains possible that low pH is important for uptake of cell-free virus. Second, the S glycoprotein was not cleaved to any measurable degree, but effects of cleavage at the cell surface by proteases on fusion cannot be excluded. Recent biochemical and functional data showed that coronavirus S glycoprotein is a class I fusion protein (Bosch et al., 2003); the lack of cleavage sets apart the SARS-CoV S glycoprotein and spike proteins from other coronaviruses from a prototype class I fusion protein, which is cleaved. Third, the receptor binding domain (RBD) is within the N-terminal fragment containing amino acid (aa) residues 12–672, which Li et al. define as S1. The RBD was recently localized between residues 303 and 537 (Xiao et al., 2003) and is therefore similar to the RBD of the HCoV-229E, which is within a fragment containing residues 407 to 547 (Breslin et al., 2003); whether this reflects any similarity in structure and mechanism of binding of these human coronaviruses is unknown. Finally, Li et al. developed a fusion assay based on syncytia formation that can be used to study mechanisms and to test inhibitors without the need to work with a lethal virus. A pseudovirus-based assay would be a useful complement to control for differences between cell fusion and virus entry.
Preliminary experiments reported by Li et al. also give some initial clues to the molecular mechanism of the ACE2 interaction with S. Two mutations of the ACE2 catalytic site did not affect syncytia formation, indicating that the S binding site on ACE2 is located in a different region and that the enzymatic function of ACE2 is not required for fusion. Although normal cellular function is not usually required for a virus receptor function, further experiments are needed to validate this finding; one possible reason for the lack of effect is related to the long time (48 hr) of syncytia formation (see the supplementary information to Li et al.), which could lead to saturation. The fact that the ACE2-S1 association endured the perils of the coimmunoprecipitation procedure also suggests it may be a high-affinity interaction. The precise affinities of other coronavirus spike-receptor interactions have not been determined (Gallagher and Buchmeier, 2001). However, for most known virus-receptor interactions (but not all), high-affinity binding suggests the possibility of receptor-induced conformational changes in the viral proteins. Whether the SARS-CoV S glycoprotein will follow this rule remains to be seen.
In trying to predict the implications of the receptor identification and future research directions, it may be useful to consider parallels with the history of HIV research. However, the speed could well be an order of magnitude faster if the research continues at the pace set by Li et al. An immediate question is whether there are other receptors or coreceptors—for HIV it took more than a decade to identify the elusive coreceptors. ACE2 is expressed at significant levels in heart and other tissues (Donoghue et al., 2000), where SARS-CoV replication has not been reported. Does it need a coreceptor(s) that is absent in cells from these tissues but not in 293T cells, or there are other factors that prevent replication in these tissues?
Perhaps the most urgent question is whether soluble ACE2 (sACE2), and various fusion constructs or fragments, can serve as potent inhibitors of the virus infection in vivo. The analogy to HIV could help to avoid costly clinical trials and save time. After the failure of recombinant soluble CD4 (sCD4) to affect HIV replication in humans, it took more than a decade to develop a potent multivalent CD4-IgG fusion protein, which is now showing promising results in recent clinical trials. Like sCD4, sACE2 is likely to have a short half-life in vivo, and may not be a very potent inhibitor in a monovalent form. Multivalent sACE2-immunoglobulin proteins might be much better inhibitors of SARS-CoV infection in vivo than sACE2. Antibodies, other proteins, and perhaps peptides and small molecules disrupting the ACE2 interaction with the S glycoprotein could also be viable tools in the treatment of SARS-CoV infections (although existing ACE inhibitors are unlikely to be useful). The solution of the crystal structure of the receptor and its complex with receptor binding fragments of S1 will provide a detailed understanding of its interactions with the viral protein and could help in the development of such inhibitors. Finally, soluble forms of the S glycoprotein ectodomain, the RBD, and even receptor-bound conformations of the S glycoprotein may have potential as vaccine immunogens that elicit neutralizing antibodies; such receptor-bound conformations of the HIV-1 gp120 have been recently proposed as vaccine immunogens that could elicit potent broadly neutralizing antibodies. The rapid pace of research and the acute self-limiting nature of the SARS-CoV infection (unlike HIV infection) could lead to significantly faster development of therapeutics and vaccines than for HIV, and this could be another unexpected but welcome surprise.The Secret Life of ACE2 as a Receptor for the SARS Virus: Cell
https://www.cell.com/cell/fulltext/S0092-8674(03)00976-0
PLoS One. 2011; 6(8): e23710.
Published online 2011 Aug 22. doi: 10.1371/journal.pone.0023710
Inhibition of SARS Pseudovirus Cell Entry by Lactoferrin Binding to Heparan Sulfate Proteoglycans
Jianshe Lang, Ning Yang, Jiejie Deng, Kangtai Liu, Peng Yang, Guigen Zhang, and Chengyu Jiang *
Robert J. Geraghty, Editor
Author information Article notes Copyright and License information Disclaimer
State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Peking Union Medical College, Tsinghua University, Beijing, People's Republic of China,
University of Minnesota, United States of America
* E-mail: nc.ude.cmup@gnaij
乳铁蛋白结合硫酸乙酰肝素蛋白聚糖对SARS伪病毒细胞进入的抑制。
郎建社,杨宁,邓杰杰,刘康泰,杨鹏,张贵根和江成玉*
Robert J. Geraghty,编辑
作者信息文章注释版权和许可信息免责声明
清华大学北京协和医学院,中国医学科学院基础医学研究所,医学分子生物学国家重点实验室,北京,
美国明尼苏达大学
图9
SARS-CoV(冠状病毒)细胞进入的模型和乳铁蛋白在SARS-CoV感染中的保护作用。
(A)HSPG在SARS-CoV细胞进入过程中起重要作用。 HSPG提供的锚定位点允许SARS-CoV与宿主细胞之间的初始接触以及细胞表面病毒颗粒的浓度。 SARS-CoV通过与HSPG结合而滚动到细胞膜上,并扫描特定的进入受体,从而导致随后的细胞进入。 (B)LF通过与HSPG结合来阻断SARS-CoV的感染。当SARS-CoV感染人体时,LF表达可能会上调。 LF定位于细胞表面HSPG,可防止病毒与宿主细胞之间的初步相互作用以及随后的内在化过程。
Abstract
It has been reported that lactoferrin (LF) participates in the host immune response against Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) invasion by enhancing NK cell activity and stimulating neutrophil aggregation and adhesion. We further investigated the role of LF in the entry of SARS pseudovirus into HEK293E/ACE2-Myc cells.Our results reveal that LF inhibits SARS pseudovirus infection in a dose-dependent manner. Further analysis suggested that LF was able to block the binding of spike protein to host cells at 4°C, indicating that LF exerted its inhibitory function at the viral attachment stage. However, LF did not disrupt the interaction of spike protein with angiotensin-converting enzyme 2 (ACE2), the functional receptor of SARS-CoV.
Previous studies have shown that LF colocalizes with the widely distributed cell-surface heparan sulfate proteoglycans (HSPGs). Our experiments have also confirmed this conclusion. Treatment of the cells with heparinase or exogenous heparin prevented binding of spike protein to host cells and inhibited SARS pseudovirus infection, demonstrating that HSPGs provide the binding sites for SARS-CoV invasion at the early attachment phase.
Taken together, our results suggest that, in addition to ACE2, HSPGs are essential cell-surface molecules involved in SARS-CoV cell entry. LF may play a protective role in host defense against SARS-CoV infection through binding to HSPGs and blocking the preliminary interaction between SARS-CoV and host cells.
Our findings may provide further understanding of SARS-CoV pathogenesis and aid in treatment of this deadly disease.
Introduction
Severe acute respiratory syndrome (SARS) is an infectious disease that emerged in Guangdong Province, China in November 2002 [1]. This deadly disease rapidly spread to many countries around the world, with a mortality rate of approximately 10%. SARS presents as an atypical pneumonia that often leads to acute respiratory distress syndrome and respiratory failure, the main cause of death [2]. This unusual infectious disease has become a major threat to public health and social stability since its outbreak. To date, there is no effective therapeutic strategy or preventive vaccine available for the treatment of SARS.
In April 2003, a novel coronavirus, SARS coronavirus (SARS-CoV), was identified as the etiological agent of SARS [3]. SARS-CoV is an enveloped, positive-strand RNA virus. Its large RNA genome is approximately 30000 nucleotides in length and encodes a nonstructural replicase complex and structural proteins, including spike (S), envelope (E), membrane (M) and nucleocapsid (N) proteins [4]. Spike protein is the envelope protein responsible for invasion of host cells. Further three-dimensional structure analyses suggest that spike protein is composed of two subunits: S1, which mediates SARS-CoV binding to receptors on host cells, and S2, which triggers virus and host cell membrane fusion [5]. Angiotensin-converting enzyme 2 (ACE2), a metallopeptidase, has been identified as one of the functional receptors of SARS-CoV and is responsible for binding to spike protein and mediating SARS-CoV entry into host cells [6]. Crystallographic studies have shown that a segment containing amino acids 318–510 of S1 is the critical receptor-binding domain for the ACE2 receptor [6]. ACE2 is highly expressed on human lung alveolar epithelial cells, enterocytes of the small intestine and the brush border of the proximal tubular cells of the kidney. These locations of ACE2 expression are consistent with the tissue tropism and pathogenesis of SARS-CoV infection [7]. Other coreceptors or cellular molecules may be required to facilitate SARS-CoV invasion.
During SARS-CoV infection, a host immune response against the virus is triggered. The innate immune response plays an essential role in the inhibition of viral infection. It has been reported that many genes involved in the innate immune response, such as those encoding lactoferrin (LF), S100A9 and Lipocalin 2, participate in SARS-CoV clearance [8]. Among these up-regulated genes, LF expression was elevated by approximately 150 fold in SARS patients compared with healthy controls. That study demonstrated that LF exerted its function in the process of SARS-CoV infection by enhancing NK cell activity and stimulating neutrophil aggregation and adhesion [8]. However, the bioactivity of lactoferrin is not completely understood.
LF is a multifunctional protein present in external secretions, including saliva, tears, milk, nasal and bronchial secretions, gastrointestinal fluids and urine mucosal secretions, and is an important constituent of the neutrophilic granules of leukocytes [9]. LF possesses strong antiviral activity against a broad spectrum of RNA and DNA viruses, such as Sindbis virus [10], cytomegalovirus [11], [12], herpes simplex virus [13], Semliki forest virus [14], human polyomavirus [15], human papillomavirus [16], echovirus [17], human immunodeficiency virus [18] and rotavirus [19]. These viruses typically utilize common molecules on the cell membrane to facilitate their invasion into cells. These molecules, including heparan sulfate proteoglycans (HSPGs), provide the first anchoring sites on the cell surface and help the virus make primary contact with host cells [20]. It has been shown that LF is able to prevent the internalization of some viruses by binding to HSPGs, which is present on most cells [21]. This property of LF confers protection to the host against viral infections. Based on these findings, we hypothesize that another underlying mechanism for the anti-SARS-CoV effect of LF involves its capability to bind to the extensively distributed HSPGs molecule on host cells. Our results indicate that HSPGs provide the preliminary docking sites on the cell surface and play an important role in the process of SARS pseudovirus cell entry. LF can block the infection of SARS pseudovirus by binding to HSPGs, suggesting that it may exert a protective role in host immune defense against SARS-CoV invasion.
Materials and Methods
Plasmids and cell lines
The plasmid pQCXIX, the SARS-CoV spike protein-encoding vector sh-2, and the gag/pol expression plasmid were kindly provided by Dr. Wenhui Li [22]. VSV-G plasmid encoding Vesicular stomatitis virus (VSV) G glycoprotein was kindly provided by Dr. Xiaozhong Peng (Peking Union Medical College). The HEK293E/ACE2-Myc, HEK293E/S1190-Fc and HEK293E/Fc cell lines, stably expressing ACE2-Myc, S1190-Fc and human IgG Fc fragment, respectively, were generated in our laboratory as described previously [22], [23]. The human colon carcinoma-derived Caco-2 cells and the African green monkey kidney-derived Vero E6 cells were provided by American Type Culture Collection (ATCC). Cells were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Hyclone) supplemented with 10% fetal bovine serum (FBS) at 37°C in humidified incubators with 5% CO2.
Preparation of pseudotyped viruses
SARS pseudotyped viruses were produced as reported previously [22]. Briefly, HEK293T cells at 70% confluence in 10-cm dishes were co-transfected with 4 µg of pQCXIX, 2 µg of sh-2, and 4 µg of the gag/pol expression plasmid using Lipofectamine 2000 (Invitrogen). After a 48-hour transfection, viral supernatants were harvested and filtered through screens with 0.45-µm pore size. Viral particles without glycoprotein as negative control were prepared with pQCXIX and gag/pol plasmids by the same method as above. pQCXIX, gag/pol and VSV-G plasmids were used for VSV-G pseudotyped virus packaging in the same way.Viral stocks were aliquoted and frozen at −80°C for long-term storage. Short-term storage at 4°C did not dramatically affect viral titers. Viral titers were determined as previously described [24].
Pseudovirus infection assay
A total of 2×105 HEK293E/ACE2-Myc cells were seeded into 12-well plates. After 24 hours, the cells were washed with phosphate-buffered saline (PBS) two times and the culture was replaced with fresh DMEM without FBS. Bovine LF (Wako) or heparin (Sigma-Aldrich) at the indicated concentration was added to the cells at 37°C for 1 h or 10 min, respectively. Subsequently, 1×105 transducing units (TUs) of SARS pseudoviruses were added to the cells and incubated at 37°C for 4 h. The concentration of LF or heparin was maintained throughout the process of viral infection. Simultaneously, 15 µM bovine serum albumin (BSA) (Sigma-Aldrich) or PBS was used as control by the same method as described above, respectively. Unbound pseudovirions were removed by three washes with PBS. Then, the cells were cultured with fresh DMEM with 10% FBS at 37°C for 48 h. The SARS pseudovirus-infected GFP-expressing HEK293E/ACE2-Myc cells were observed by fluorescence microscopy (Nikon Eclipse TE 2000-U) and counted by flow cytometry (Beckman Coulter Epics Elite EST). The tests of SARS pseudovirus on Vero E6 and Caco-2 cells were performed by the same method as described above. The infection of HEK293E/ACE2-Myc cells by VSV-G pseudotyped virus in the presence of LF or heparin was carried out in the same way. Viral particles without glycoprotein were incubated with the cells to test whether they had the capability of infection or not.
Western blotting
HEK293E/ACE2-Myc cells (2×105) were seeded into 12-well plates. The next day, after incubation at 37°C for 1 h with LF at concentrations of 0.34 µM, 1.3 µM, 4 µM and 12 µM, HEK293E/ACE2-Myc cells were treated with 1×105 SARS pseudovirus particles for 4 h at 37°C. 15 µM BSA was used as control. Then, the pseudovirus-containing supernatant was removed, and the cells were washed three times with PBS. After growth in fresh DMEM with 10% FBS at 37°C for 48 h, the cells were harvested to analyze GFP expression level by western blotting. Band density was calculated from western blots using Quantity One software.
S1190-Fc binding assay
S1190-Fc and human IgG Fc was prepared as described by Hongliang Wang et al. in our laboratory [22]. Briefly, S1190-Fc and Fc were expressed by HEK293E/S1190-Fc and HEK293E/Fc cells, respectively. S1190-Fc is a recombinant protein composed of the soluble extracellular region (amino acids 1–1190) of SARS spike protein with human IgG Fc fused to its C terminus. The proteins were purified using a protein A column (GE Healthcare), and the protein concentration was measured with a BCA assay kit from Bio-Rad. HEK293E/ACE2-Myc cells at 80% confluence were harvested from 10-cm dishes. HEK293E/ACE2-Myc cells (2×105) were exposed to 5 nM S1190-Fc at 4°C for 1 h after incubation with LF or heparin at 37°C for 1 h or 10 min, respectively. The concentration of LF or heparin was maintained throughout the process of incubation. Fc protein was used as a control. Unbound S1190-Fc was removed by three washes with PBS. Cell surface bound S1190-Fc was detected with a FITC-labeled mouse anti-human IgG Fc secondary antibody (Santa Cruz). After three washes with PBS, the mean fluorescence intensity (MFI) of each group was measured by flow cytometry. The results were analyzed by FlowJo software.
ELISAs
96-well immunosorbent plates were coated with 100 µl of 20 µg/ml S1190-Fc or Fc in sodium carbonate buffer (0.1 M NaHCO3, pH 8.6) at 4°C overnight, followed by blocking for 2 h at 37°C with 300 µl of 3% bovine serum albumin (BSA) in PBS. Wells coated with Fc and PBS were used as negative controls and blank controls, respectively. HEK293E/ACE2-Myc cells were lysed to harvest the ACE2-Myc containing supernatant. To determine whether LF interferes with the interaction between S1190-Fc and ACE2-Myc, both S1190-Fc and ACE2-Myc were treated with LF. 20 µM LF was added to the S1190-Fc coated wells and to the ACE2-Myc containing supernatant, respectively, at the same time and incubated for 1 h at 37°C. LF untreated ACE2-Myc and S1190-Fc were used as control. Then, the LF pretreated or untreated ACE2-Myc was added to S1190-Fc coated wells with or without LF at 37°C for 1 h to allow them to interact with each other. The wells were washed six times with PBST (phosphate-buffered saline with 0.1% Tween 20) and then incubated with 100 µl of mouse anti-Myc antibody (Santa Cruz) diluted 1∶500 in PBS for 1 h. After the plates were washed, horseradish peroxidase-conjugated goat anti-mouse antibody (Santa Cruz) was added into each well, incubated at 37°C for 1 h and washed six times with PBST. The plates were developed with 100 µl of tetramethylbenzidine substrate. The reaction was terminated with 50 µl of 2.0 M H2SO4, and the absorbance was read at 450 nm.
Confocal microscopy
Oregon Green 488-labeled LF was prepared according to the manual provided by Invitrogen and used at a concentration of 0.5 µM in PBS (pH 7.4). HEK293E/ACE2-Myc cells were grown on coverslips in 24-well plates. After incubation with Oregon Green-labeled LF at 4°C for 1 h, unbound LF in the culture supernatant was removed by three washing steps. Then, the cells were fixed with 4% paraformaldehyde for 15 min. Subsequently, cell membrane and nuclei were stained with 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (Sigma-Aldrich) and Hoechst33342 (Sigma-Aldrich), respectively. The subcellular localization of LF was observed using confocal laser-scanning microscopy (Leica TCS SP2), and the images were analyzed using Leica confocal software.
Enzymatic degradation of cell-surface heparan sulfate and chondroitin sulfate
Heparan sulfate was removed from the cell surface of HEK293E/ACE2-Myc cells by treatment with heparinase I (Sigma-Aldrich). Cells were grown to 80% confluence in 12-well plates, washed three times with DMEM without FBS, and incubated with 10 units of heparinase I per ml for 1 h at 37°C in heparinase I buffer (20 mM Tris–HCl, 50 mM NaCl, 4 mM CaCl2, 0.01% BSA, pH 7.5). After three washes with PBS, the cells were resuspended in fresh DMEM without FBS and subjected to the subsequent SARS pseudovirus, VSV-G pseudovirus or S1190-Fc treatment as described above. The same method was also applied in the treatment of Vero E6 or Caco-2 cells. Chondroitin sulfate on the cell surface were digested by Chondroitinase ABC (Sigma-Aldrich) in the same way.
Statistical analysis
All data are presented as the mean±SD from at least three independent experiments. Statistical analyses were performed using Student's t-test. Differences with P<0.05 were considered significant.
Results
Lactoferrin inhibits entry of SARS pseudovirus into HEK293E/ACE2-Myc cells
In addition to replication-competent viruses, pseudoviruses have become an ideal tool to investigate cell entry of SARS-CoV without safety concerns [22], [25], [26]. SARS pseudovirus possesses the morphological characteristics of replication-competent SARS-CoV, with SARS-CoV spike protein on the envelope membrane, and can mimic SARS-CoV in the process of cell entry. We use three plasmids to produce the SARS pseudovirus particles: gag/pol, pQCXIX and sh-2. The gag/pol plasmid carries the lentiviral gene gag-pol for the expression of the capsid proteins and the enzymes for replication. pQCXIX encodes the lentiviral packaging signal and the gene for GFP. sh-2 is responsible for SARS spike protein expression. Coexpression of the three plasmids in HEK293T cells results in the incorporation of SARS-CoV spike protein into the budding lentiviral particle envelope along with the reporter GFP, enabling the analysis of spike-mediated entry into host cells.
It has been reported that LF is able to inhibit a broad range of viruses at the early attachment stage [27]. To establish the antiviral effects of LF on SARS-CoV, we utilized SARS pseudovirus and HEK293E/ACE2-Myc cells to perform a series of a transduction assays in the presence or absence of bovine LF. To facilitate the detection of SARS pseudovirus internalization, our laboratory has generated the cell line HEK293E/ACE2-Myc, which stably expresses surface-localized ACE2 with a Myc tag fused to its C terminus. As demonstrated in previous studies, the HEK293E/ACE2-Myc cell line is a perfect tool to study the interaction of SARS-CoV and host cells, particularly at the stage of SARS-CoV cell entry [22], [28], [29].
We incubated HEK293E/ACE2-Myc cells with different concentrations of LF at 37°C for 1 h. Then SARS pseudoviruses were added to the LF-treated cells to test the effect of LF on SARS pseudovirus infection. As shown in Figure 1A–1D, the number of GFP expressing cells decreased sharply with increasing concentration of LF. Because cell entry of SARS pseudoviruses leads to GFP expression, this result suggests that the infection of HEK293E/ACE2-Myc cells by SARS pseudovirus can be dramatically inhibited in the presence of LF. We further utilized western blotting to examine GFP expression in the LF-treated cells after incubation with SARS pseudovirus. We found that the amount of GFP protein was reduced in the presence of LF (Fig. 1E and 1F). The degree of inhibition was correlated with the concentration of LF. At the same time, the GFP expressing cells were tested by flow cytometry after incubation with LF and SARS pseudovirus. Figures 1G and 1H demonstrate that LF inhibits SARS pseudovirus infection in a dose-dependent manner. The 50% inhibitory concentration (IC50) is approximately 0.7 µM. However, there was no GFP expression in the cells incubated with viral particles without spike protein (Fig.1I), indicating that SARS pseudovirus cell entry is mediated by spike protein. The data described above suggest that LF can prevent SARS pseudovirus from infecting host cells and that the inhibitory effect is dose dependent.
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g001.jpg
Open in a separate window
Figure 1
Lactoferrin inhibits SARS pseudovirus infection of HEK293E/ACE2-Myc cells.
(A–D) Fluorescence microscopy illustrates that the number of SARS pseudovirus-infected GFP-expressing HEK293E/ACE2-Myc cells decreases in the presence of LF. HEK293E/ACE2-Myc cells were treated with LF for 1 h at 37°C at the concentration of 1 µM (B), 3 µM (C) or 10 µM (D). BSA (10 µM) was used as control (A). The LF-pretreated cells were treated with SARS pseudovirus as described in Methods. (E and F) Western blotting reveals that LF markedly reduces GFP expression in HEK293E/ACE2-Myc cells incubated with SARS pseudovirus. Statistical analysis of the relative band density ratio of GFP to actin was performed using a t-test. Error bars represent the SD of three independent experiments. **P<0.01 and *P<0.05. (G and H) Flow cytometry demonstrates that LF is able to inhibit the infection of HEK293E/ACE2-Myc cells by SARS pseudovirus. The concentration of LF was 0.625 µM, 1.25 µM, 2.5 µM, 5 µM or 10 µM. BSA (10 µM) served as control. The percentage of GFP expressing cells in the total population was calculated by flow cytometry. The relative viral infection ratio was measured by comparing the percentage of GFP expressing cells in each group with that of the BSA control. Error bars represent the SD of three independent experiments. (I) No GFP expression can be detected in the cells treated with viral particles without spike protein. The percentage of GFP expressing cells in the total population was calculated by flow cytometry as described above. Error bars represent the SD of three independent experiments.
Lactoferrin blocks spike protein binding to HEK293E/ACE2-Myc cells by an ACE2-independent pathway
Previous studies have revealed that the infection of host cells by SARS-CoV is driven by spike protein, which is the only envelop protein responsible for attachment and fusion of the viral and the cellular membranes [30]. Thus, the preventive effect of LF on SARS pseudovirus infection may occur through targeting the attachment or fusion step. To determine whether the inhibitory effect was due to LF blocking the interaction between spike protein and host cells, we incubated S1190-Fc with the LF pre-treated HEK293E/ACE2-Myc cells at 4°C for 1 h. S1190-Fc is a soluble, truncated form of SARS CoV spike protein that retains the extracellular region (amino acids 1–1190) with human IgG Fc fused to its C terminus. After the treatment described above, the MFI of each group was analyzed by flow cytometry. As shown in Figure 2A, LF can effectively block S1190-Fc binding to HEK293E/ACE2-Myc cells at 4°C, suggesting that LF exerts its inhibitory effect on SARS pseudovirus internalization at the initial attachment stage.
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g002.jpg
Figure 2
Lactoferrin blocks the interaction between spike protein and HEK293E/ACE2-Myc cells in an ACE2-independent fashion.
(A) LF inhibits the binding of S1190-Fc to HEK293E/ACE2-Myc cells. Before incubation of S1190-Fc with HEK293E/ACE2-Myc cells at 4°C for 1 h, the cells were treated with LF at 37°C for 1 h at concentrations of 1 µM, 3 µM and 10 µM. Fc protein was used as a control. S1190-Fc binding to the cells was detected by flow cytometry as described in Methods. Error bars represent the SD of three independent experiments. ***P<0.001, **P<0.01 and *P<0.05. (B) LF does not disrupt the binding of S1190-Fc to ACE2-Myc. Error bars represent the SD of three independent experiments. ** P<0.01.
The cell entry of SARS CoV is a rather complex process triggered by the interaction of spike protein with some receptors and/or cofactors on the cell surface. It has been established that ACE2 serves as a functional receptor for SARS-CoV infection [30]. The lectin DC-SIGN (dendritic cell-specific ICAM-grabbing non-integrin), or other related molecules, may also play an important role in SARS-CoV invasion [31]. To date, the underlying mechanism of viral entry is not fully understood, and further investigations are needed to elucidate this subtle process. To test whether LF prevents spike protein binding to the main receptor ACE2, we added LF to S1190-Fc and ACE2-Myc containing supernatants. After incubation at 37°C for 1 h, we mixed these solutions together in immunosorbent plates to examine the interaction between S1190-Fc and ACE2-Myc by ELISA. The results suggest that LF does not disrupt the binding of S1190-Fc to ACE2-Myc (Fig. 2B). Thus, LF may employ other mechanisms to inhibit the attachment of SARS pseudovirus to host cells.
Subcellular localization of lactoferrin
Spike protein is the protein on the SARS-CoV envelope responsible for entry into host cells. The binding of spike protein to the ACE2 receptor can initiate fusion between the viral and cellular membranes [6]. Based on these facts, we deduced that if LF did not block the binding of spike protein to ACE2, it could interact with another molecule on the cell surface, thereby playing an inhibitory role against SARS pseudovirus cell entry. To determine the subcellular localization of LF, we labeled LF with Oregon Green fluorescent dye and incubated the labeled LF with HEK293E/ACE2-Myc cells. At the same time, cell membrane was stained with the red membrane dye DiI. According to the confocal microscopy results, LF was present on the cell membrane (Fig. 3A).
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g003.jpg
Open in a separate window
Figure 3
Lactoferrin localizes to the cell membrane by targeting HSPGs.
(A) LF is present on the cell surface. Oregon Green-labeled LF localization was observed under confocal microscopy. LF untreated cells were used as control. Cell membrane and nuclei were stained with DiI and Hoechst33342, respectively. Scale bar, 8 µm. (B) Heparin inhibits LF binding to HEK293E/ACE2-Myc cells. HEK293E/ACE2-Myc cells were incubated with 0.5 µM Oregon Green-labeled LF at 4°C for 1 h after pretreatment with heparin at the concentration of 3 µM, 10 µM or 30 µM. The MFI was measured for each group by flow cytometry as described above. Error bars represent the SD of three independent experiments. **P<0.01.
Previous studies have established that LF mediates inhibition of some viral infections by interfering with virus-cell interactions after binding to the widespread family of cell-surface molecules, the HSPGs [32]–[34]. HSPGs are complex macromolecules consisting of unbranched heparan sulfate (HS) polysaccharide chains, composed of repeating disaccharide subunits of hexuronic acid and hexosamine, covalently linked to the core protein through O-glycosidic linkages [20]. The binding of LF to HSPGs prevents the first contact between virus and host cells and thus prevents subsequent infection. To ascertain whether LF interacts with the cell-surface HSPG molecules, we used heparin to interfere with the binding of LF to target cells. Due to their similar structural compositions, exogenous heparin has been widely utilized as a soluble HS analog to competitively inhibit the interaction between HSPGs and their ligands [35]. Prior to incubation with Oregon Green-labeled LF, HEK293E/ACE2-Myc cells were treated with heparin at different concentrations. The MFI was analyzed to detect the amount of LF binding for each group. As shown in Figure 3B, exogenous heparin efficiently prevented LF binding to HEK293E/ACE2-Myc cells, indicating that LF localizes to the cell surface mainly through interactions with HSPGs.
Lactoferrin inhibits spike protein binding to HEK293E/ACE2-Myc cells and SARS pseudovirus infection by binding to cell-surface HSPGs
To further investigate whether LF inhibits S1190-Fc binding to HEK293E/ACE2-Myc cells due to its binding to cell-surface HSPGs, we added S1190-Fc after incubating heparin pre-treated HEK293E/ACE2-Myc cells with LF. Figure 4A shows that heparin neutralized the inhibitory effect of LF on the binding of S1190-Fc to host cells in a dose-dependent manner, suggesting that the anchoring sites for LF on the cell surface are provided by the HS moieties of HSPGs. The HS analog heparin competed for LF binding to HSPGs, preventing cell binding.
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g004.jpg
Figure 4
Lactoferrin exerts its inhibitory effect on spike protein and SARS pseudovirus by binding to cell-surface HSPGs.
(A) Heparin neutralizes the LF-mediated inhibition of S1190-Fc binding to HEK293E/ACE2-Myc cells. Before treatment with 10 µM LF for 1 h at 37°C, HEK293E/ACE2-Myc cells were incubated with heparin for 10 min at the concentration of 1 µM, 3 µM or 10 µM. S1190-Fc was added to each group to detect S1190-Fc binding to the cells as described above. Error bars represent the SD of three independent experiments. ***P<0.001, **P<0.01 and *P<0.05. (B) Inhibition of SARS pseudovirus infection of HEK293E/ACE2-Myc cells by LF can be partially neutralized by heparin. HEK293E/ACE2-Myc cells were treated with heparin for 10 min at the concentration of 1 µM, 3 µM or 10 µM. Then, 10 µM LF was added to each group and incubated at 37°C for 1 h. The GFP-expressing HEK293E/ACE2-Myc cells in the total population were analyzed as described above. The relative viral infection ratio was measured by comparing the percentage of GFP expressing cells of each group with that of the BSA control. Error bars represent the SD of three independent experiments. *** P<0.001, ** P<0.01 and *P<0.05.
In the next experiment, we examined whether heparin could neutralize the LF-mediated inhibition of SARS pseudovirus infection of HEK293E/ACE2-Myc cells in the same manner. Prior to SARS pseudovirus infection, we added heparin to the cells and incubated the heparin-treated cells with LF for 1 h at 37°C. As shown in Figure 4B, adding heparin increased the infectivity of SARS pseudovirus in the presence of LF compared with controls. This evidence further supports the conclusion that LF blocks adsorption and internalization of SARS pseudovirus through binding to HSPGs on the cell surface. Interestingly, another phenomenon also captured our attention. Adding heparin alone also prevented the cell entry of SARS pseudovirus into host cells (Fig. 4B). This result is consistent with the previous finding that heparin can reduce the infection of Vero E6 cells by replication-competent SARS-CoV [36]. This evidence implies that HSPGs play an important role in SARS-CoV cell entry. Furthermore, we found that, instead of increasing cell entry, incubating HEK293E/ACE2-Myc cells with 10 µM heparin and 10 µM LF curtailed the infectivity of SARS pseudovirus, compared with the cells treated with 3 µM heparin and 10 µM LF. One reasonable explanation for this result is that excessive heparin competes with the HS chain on the cell surface and prevents adsorption of SARS pseudovirus to host cells. These data indicate that HSPGs are important molecules involved in SARS pseudovirus cell entry.
HSPGs provide docking sites for spike protein on the cell surface and play an important role in SARS pseudovirus infection
To further prove the involvement of HSPGs in the SARS-CoV entry process, we first utilized S1190-Fc to test whether HSPGs participated in the viral adsorption step. Prior to incubation with S1190-Fc at 4°C for 1 h, HEK293E/ACE2-Myc cells were treated with heparin for 10 min. The mean fluorescence intensity analysis demonstrates that the binding of S1190-Fc to the cells was efficiently blocked by the addition of heparin (Fig. 5A). Then, we used heparinase I to degrade the HS chain on the cell surface. We found that enzymatic removal of HS polysaccharides resulted in a reduced ability of S1190-Fc to bind to host cells at 4°C (Fig. 5B), suggesting that in addition to ACE2, HSPGs act as another docking site for SARS-CoV on the cell surface. The presence of ACE2 and heparinase-resistant oligosaccharides may give rise to the incomplete inhibition [37].
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g005.jpg
Figure 5
Addition of exogenous heparin and enzymatic removal of HS chains by heparinase reduce spike protein binding to HEK293E/ACE2-Myc cells.
(A) Heparin blocks the binding of S1190-Fc to HEK293E/ACE2-Myc cells. HEK293E/ACE2-Myc cells were incubated with heparin for 10 min at the concentration of 10 µM, 30 µM or 100 µM. S1190-Fc was added to each group and incubated at 4°C for 1 h. The MFI of each group was measured as described above. Error bars represent the SD of three independent experiments. *** P<0.001, **P<0.01 and *P<0.05. (B) Enzymatic degradation of cell-surface heparan sulfate (HS) chains reduces S1190-Fc binding. After treatment with 10 U of heparinase I, the cells were incubated with S1190-Fc at 4°C for 1 h. The MFI test was performed using the same method as above.
To provide further supporting evidence of the requirement for cell-surface HSPGs during SARS-CoV infection, we added heparin to HEK293E/ACE2-Myc cells before incubating the cells with SARS pseudovirus. The treatment of host cells with a cellular HS analog dramatically decreased the infectivity of SARS pseudovirus (Fig. 6A), indicating that SARS pseudovirus cell entry is blocked by competitive inhibition on the part of heparin and that the binding sites provided by HSPGs are important for effective infection. The enzymatic degradation of HS chains from the cell surface also led to resistance of HS-deficient cells to SARS pseudovirus infection (Fig. 6B), demonstrating that the HS chains of HSPGs are involved in SARS pseudovirus cell entry. Because HSPGs are widely distributed on the cell membrane, it is very likely that they serve as the initial anchoring site, facilitating the preliminary contact between the virus and host cells and the subsequent concentration of viruses on the cell surface. According to previous studies, viral particles are then transferred from the low affinity HSPGs to specific high affinity entry receptors, which results in internalization into host cells [37]. HSPGs themselves are not sufficient for SARS-CoV entry. HSPGs are widely expressed on most mammalian cells. However, SARS-CoV can be found only in limited host cells, such as lung alveolar epithelial cells, enterocytes of the small intestine and the brush border of the proximal tubular cells of the kidney [3]. Therefore, HSPGs may play an important role in promoting SARS-CoV infection by facilitating access to a specific entry receptor. However, when chondroitin sulfate was removed from cell surface by enzymatic digestion, SARS pseudovirus could still effectively infect the host cells, suggesting that compared with HSPG, chondroitin sulfate may have much less impact on SARS pseudovirus cell entry.
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g006.jpg
Figure 6
Incubation with heparin and degradation of HS polysaccharides on the cell surface by heparinase inhibits SARS pseudovirus infection of HEK293E/ACE2-Myc cells.
(A) Heparin inhibits SARS pseudovirus entry into HEK293E/ACE2-Myc cells. Before incubation with SARS pseudovirus at 37°C for 4 h, HEK293E/ACE2-Myc cells were treated with heparin for 10 min at the concentration of 0.625 µM, 1.25 µM, 2.5 µM, 5 µM or 10 µM. GFP-expressing HEK293E/ACE2-Myc cells in the total population were analyzed by flow cytometry. The relative viral infection ratio was measured by comparing the percentage of GFP expressing cells of each group to that of the BSA control. Error bars represent the SD of three independent experiments. (B) Lysis of cell-surface HS by heparinase blocks the infection of HEK293E/ACE2-Myc cells by SARS pseudovirus. After incubation with 10 U of heparinase I or chondroitinase ABC for 1 h at 37°C, the cells were treated with SARS pseudovirus as described above. The relative viral infection ratio was calculated by the same method. *P<0.05.
We further investigated the role of HSPGs in SARS pseudovirus cell entry into Caco-2 and Vero E6 cells. As shown in Figure 7, SARS pseudovirus infection was efficiently blocked by LF, heparin or cell surface HSPGs degradation, respectively. Again, these results provide further supporting evidence of the involvement of HSPGs in the SARS-CoV cell entry.
An external file that holds a picture, illustration, etc.
Object name is pone.0023710.g007.jpg
Figure 7
LF, heparin or enzymatic removal of cell surface HSPGs can prevent SARS pseudovirus entry into Vero E6 or Caco-2 cells.
(A) Interference of the interaction between Vero E6 and SARS pseudovirus by LF, heparin and heparinase leads to reduction of viral infection. Vero E6 cells were treated by the same methods above with 10 µM LF, 10 µM heparin or 10 U of heparinase I, respectively. Then, the cells were incubated with SARS pseudovirus as described in Methods. GFP-expressing cells in the total population were analyzed by flow cytometry. The relative viral infection ratio was measured by comparing the percentage of GFP expressing cells of each group to that of the control. Error bars represent the SD of three independent experiments. *** P<0.001 and **P<0.01. (B) Incubation with LF or heparin, or degradation of HSPGs by heparinase inhibits SARS pseudovirus infection of Caco-2 cells. The Caco-2 cells were treated by same methods as described above. *** P<0.001, **P<0.01 and *P<0.05.
VSV-G pseudotyped virus was usually used as control in other virus studies. Thus, the role of HSPGs in VSV-G pseudotyped virus cell entry was investigated in our experiments. Surprisingly, we found that soluble heparin and enzymatic removal of cell surface HSPGs effectively inhibited VSV-G virus particle infection (Fig. 8), which is consistent with previous reports using replication-competent VSV and VSV-G pseudotyped virus on other cell lines, including human Hela, avian CER, human bronchial 16HBE-S1 and tracheal CFT1-C2 epithelial cells, etc. [38]–[41]. Furthermore, previous reports have shown that LF could exert an inhibitory effect on VSV infection [42], which is also confirmed by our experiment result (Fig. 8). All these data suggest that HSPGs also play an important role in the process of VSV cell entry. The binding of LF to cell surface HSPGs may account for the reduction of VSV infection. To date, many investigations have been performed to elucidate this rather complicated process of viral cell entry. To sum up the findings of the underlying mechanism of viral infection, we speculated that it might be an universal phenomenon that enveloped viruses tend to utilize cell-surface HSPGs as primary anchoring sites to initiate the subsequent cell entry.
Figure 8
HSPGs are also involved in VSV-G virus cell entry.
HEK293E/ACE2-Myc cells were treated with 10 µM LF, 10 µM heparin or 10 U of heparinase I, respectively, in the same way. And the same methods were used for the subsequent assay as described above. GFP-expressing cells in the total population were analyzed by flow cytometry. The relative viral infection ratio was measured by comparing the percentage of GFP expressing cells of each group to that of the control. Error bars represent the SD of three independent experiments. *** P<0.001 and **P<0.01.
Discussion
Recent studies have revealed that viral entry is a highly complex process that usually involves various molecules on the cell surface [43]. The primary step is often initiated by low-affinity binding to attachment sites, which promotes the concentration of virions on the cell surface. The subsequent binding to a high-affinity receptor triggers cell entry [44], [45]. The ubiquitous HSPGs, which are widely distributed on mammalian cells, have been identified as the initial docking site for a number of viruses, such as herpes virus [46], hepatitis C virus [47], dengue virus [48], human immunodeficiency virus type 1 [20], foot and mouth disease virus [20], human papillomavirus [49] and hepatitis B virus [50]. It has been revealed that HSPGs acts as primary binding sites, promoting viral docking and facilitating subsequent interaction with the specific receptors. Furthermore, HSPGs can function as storage sites on non-permissive cells and mediate “in trans” infection by presenting the viruses to their target cells [51].
Previous studies have indicated that in addition to ACE2, other coreceptors or cellular molecules are required for SARS-CoV infection [7]. Our results suggest that the efficient entry of SARS-CoV into host cells requires the involvement of HSPGs in concert with ACE2. Increased infectivity of SARS pseudovirus was associated with binding to HS. Elimination of cell-surface HS by heparinase or the addition of exogenous heparin reduced the ability of the virus to bind to the cell surface and increased cellular resistance to infection. Furthermore, it has been demonstrated that murine coronavirus utilizes HSPGs as a receptor for cell entry [52], suggesting that HSPGs may also play an important role in the process of SARS-CoV infection due to the similarity of the spike protein structures of these two viruses. Thus, it is very likely that HSPGs serve as SARS-CoV attachment sites and facilitate the concentration of the virus on the cell surface as well as access to specific entry receptors. At the preliminary stage, the docking of the virus to host cells is mediated by the interaction between the spike protein and HS chains of HSPGs. This association facilitates the further binding of SARS-CoV to its cell-surface receptor, ACE2. The current widely accepted model for the role of HSPGs suggests that the transport of extracellular virus particles from the low affinity anchoring sites to the high affinity specific entry receptors is a ubiquitous phenomenon in viral invasion, which is termed ‘viral surfing’ [48]. Through the weak and reversible interaction between the virus and the docking sites, the virus binds to host cells and scans the cell surface for the specific receptors. SARS-CoV may also adopt a similar strategy for cell entry. Figure 9 illustrates the hypothetical process of SARS-CoV infection. However, SARS-CoV attachment to HSPGs alone does not enable viral entry. HSPGs are widely distributed on the surface of mammalian cells. However, the target cells of SARS-CoV are limited to lung alveolar epithelial cells, enterocytes of the small intestine and the brush border of the proximal tubular cells of the kidney, among others [3]. Therefore, HSPGs play an important and complex role in promoting SARS-CoV infection. The entry process also requires the interaction of viral spike protein with one or more co-receptor molecules, such as ACE2, on the cell surface.
Figure 9
A model of SARS-CoV cell entry and the protective role of Lactoferrin in SARS-CoV infection.
(A) HSPGs play an important role in the process of SARS-CoV cell entry. The anchoring sites provided by HSPGs permit initial contact between SARS-CoV and host cells and the concentration of virus particles on cell surface. SARS-CoV rolls onto the cell membrane by binding to HSPGs and scans for specific entry receptors, which leads to subsequent cell entry. (B) LF blocks the infection of SARS-CoV by binding to HSPGs. LF expression may be up-regulated when SARS-CoV infects the human body. LF locates to cell-surface HSPGs and prevents the preliminary interaction between the virus and host cells and the subsequent internalization process.
It has been reported that SARS-CoV can utilize DC-SIGN to enhance infectivity [31]. Studies have confirmed that LF blocks the interaction between the DC-SIGN receptor on dendritic cells and HIV glycoprotein 120, resulting in inhibition of subsequent virus transmission [53]. These findings suggest that LF may prevent SARS-CoV spread in the human body through the same mechanism.
It has been revealed that LF is highly up-regulated (by 150 fold) in the peripheral blood mononuclear cells of SARS patients [8]. LF is usually present in plasma and external secretions and exerts a wide variety of biological functions, such as inhibiting infection by microbes, including bacteria, viruses, protozoa and fungi. LF is a highly conserved protein, with approximately 70% sequence homology between the bovine and human forms [54]. Therefore, bovine LF is often utilized as a substitute for human LF because of their similar functions [55], [56]. Our results suggest that LF can inhibit the entry of SARS pseudovirus into HEK293E/ACE2-Myc cells in a dose-dependent manner. LF does not disrupt the interaction between the spike protein and the ACE2 receptor in vitro. However, LF inhibits spike protein binding to HEK293E/ACE2-Myc cells. The target molecules on the cell membrane that LF interacts with are HSPGs. Previous three-dimensional structure analyses have revealed that LF carries highly cationic charge on its molecular surface, particularly in the N-terminal domain [57]. The highly positive surface charge is a characteristic feature of this multifunctional protein. Many studies have demonstrated that LF binds to HSPGs on the cell membrane with a high affinity through its N-terminal glycosaminoglycan-binding domain [58], [59]. HSPGs are composed of a core protein that is covalently connected with linear, polysulfated heparan sulfate polysaccharides. Due to their abundant carboxyl and sulfate groups, HSPGs are a major source of the highly negatively charged macromolecules that surround almost all mammalian cells [51]. This strong net negative charge permits HSPGs to bind to the N-terminal glycosaminoglycan-binding domain of LF through electrostatic attraction [58]. Our experiments revealed that LF-mediated inhibition of SARS-CoV infection occurs through LF competitively localizing to the SARS-CoV anchoring sites provided by HSPGs. The binding of LF to HSPGs prevented preliminary contact between SARS-CoV and host cells and, thus, prevented subsequent cell entry. In summary, LF may exert its protective functions against SARS-CoV invasion in two ways at the same time. On the one hand, as reported in previous research, LF enhances NK cell activity and stimulates neutrophil aggregation and adhesion in immune defense [8]; on the other hand, LF curtails the entry of SARS-CoV into host cells during SARS-CoV infection. LF may have potential therapeutic applications as a drug candidate for the treatment of SARS disease.
Since the SARS outbreak in November 2002, there is still no effective preventive vaccine or antiviral therapeutic strategy available to combat this deadly virus [59]. Although great progress has been made in understanding the molecular mechanism of the SARS-CoV life cycle, further investigations should be performed to elucidate the subtle process of SARS-CoV cell entry, which will provide deeper insight into rational drug design for the treatment of SARS disease.Inhibition of SARS Pseudovirus Cell Entry by Lactoferrin Binding to Heparan Sulfate Proteoglycans
Inhibition of SARS Pseudovirus Cell Entry by Lactoferrin Binding to Heparan Sulfate Proteoglycans
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3161750/
Heparan sulfate proteoglycans (HSPGs) are cell-surface and extracellular matrix macromolecules that are composed of a core protein decorated with covalently linked glycosaminoglycan (GAG) chains. In vitro studies have demonstrated the roles of these molecules in many cellular functions, and recent in vivo studies have begun to clarify their essential functions in development. In particular, HSPGs play crucial roles in regulating key developmental signaling pathways, such as the Wnt, Hedgehog, transforming growth factor-β, and fibroblast growth factor pathways. This review highlights recent findings regarding the functions of HSPGs in these signaling pathways during development.
硫酸肝素蛋白聚糖(HSPG)是细胞表面和细胞外基质大分子,由装饰有共价连接的糖胺聚糖(GAG)链的核心蛋白组成。体外研究表明这些分子在许多细胞功能中的作用,最近的体内研究已开始阐明它们在发育中的基本功能。特别是,HSPG在调节关键的发育信号传导途径(例如Wnt,Hedgehog,转化生长因子-β和成纤维细胞生长因子途径)中起关键作用。这篇综述突出了有关HSPG在发育过程中在这些信号通路中功能的最新发现。
Functions of heparan sulfate proteoglycans in cell signaling during development | Development
https://dev.biologists.org/content/131/24/6009
A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence
Published: 09 November 2015
A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence
Vineet D Menachery, Boyd L Yount Jr, Kari Debbink, Sudhakar Agnihothram, Lisa E Gralinski, Jessica A Plante, Rachel L Graham, Trevor Scobey, Xing-Yi Ge, Eric F Donaldson, Scott H Randell, Antonio Lanzavecchia, Wayne A Marasco, Zhengli-Li Shi & Ralph S Baric
Nature Medicine volume 21, pages1508–1513(2015)Cite this article
Abstract
The emergence of severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome (MERS)-CoV underscores the threat of cross-species transmission events leading to outbreaks in humans. Here we examine the disease potential of a SARS-like virus, SHC014-CoV, which is currently circulating in Chinese horseshoe bat populations1. Using the SARS-CoV reverse genetics system2, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone. The results indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Additionally, in vivo experiments demonstrate replication of the chimeric virus in mouse lung with notable pathogenesis. Evaluation of available SARS-based immune-therapeutic and prophylactic modalities revealed poor efficacy; both monoclonal antibody and vaccine approaches failed to neutralize and protect from infection with CoVs using the novel spike protein. On the basis of these findings, we synthetically re-derived an infectious full-length SHC014 recombinant virus and demonstrate robust viral replication both in vitro and in vivo. Our work suggests a potential risk of SARS-CoV re-emergence from viruses currently circulating in bat populations.
Main
The emergence of SARS-CoV heralded a new era in the cross-species transmission of severe respiratory illness with globalization leading to rapid spread around the world and massive economic impact3,4. Since then, several strains—including influenza A strains H5N1, H1N1 and H7N9 and MERS-CoV—have emerged from animal populations, causing considerable disease, mortality and economic hardship for the afflicted regions5. Although public health measures were able to stop the SARS-CoV outbreak4, recent metagenomics studies have identified sequences of closely related SARS-like viruses circulating in Chinese bat populations that may pose a future threat1,6. However, sequence data alone provides minimal insights to identify and prepare for future prepandemic viruses. Therefore, to examine the emergence potential (that is, the potential to infect humans) of circulating bat CoVs, we built a chimeric virus encoding a novel, zoonotic CoV spike protein—from the RsSHC014-CoV sequence that was isolated from Chinese horseshoe bats1—in the context of the SARS-CoV mouse-adapted backbone. The hybrid virus allowed us to evaluate the ability of the novel spike protein to cause disease independently of other necessary adaptive mutations in its natural backbone. Using this approach, we characterized CoV infection mediated by the SHC014 spike protein in primary human airway cells and in vivo, and tested the efficacy of available immune therapeutics against SHC014-CoV. Together, the strategy translates metagenomics data to help predict and prepare for future emergent viruses.
The sequences of SHC014 and the related RsWIV1-CoV show that these CoVs are the closest relatives to the epidemic SARS-CoV strains (Fig. 1a,b); however, there are important differences in the 14 residues that bind human ACE2, the receptor for SARS-CoV, including the five that are critical for host range: Y442, L472, N479, T487 and Y491 (ref. 7). In WIV1, three of these residues vary from the epidemic SARS-CoV Urbani strain, but they were not expected to alter binding to ACE2 (Supplementary Fig. 1a,b and Supplementary Table 1). This fact is confirmed by both pseudotyping experiments that measured the ability of lentiviruses encoding WIV1 spike proteins to enter cells expressing human ACE2 (Supplementary Fig. 1) and by in vitro replication assays of WIV1-CoV (ref. 1). In contrast, 7 of 14 ACE2-interaction residues in SHC014 are different from those in SARS-CoV, including all five residues critical for host range (Supplementary Fig. 1c and Supplementary Table 1). These changes, coupled with the failure of pseudotyped lentiviruses expressing the SHC014 spike to enter cells (Supplementary Fig. 1d), suggested that the SHC014 spike is unable to bind human ACE2. However, similar changes in related SARS-CoV strains had been reported to allow ACE2 binding7,8, suggesting that additional functional testing was required for verification. Therefore, we synthesized the SHC014 spike in the context of the replication-competent, mouse-adapted SARS-CoV backbone (we hereafter refer to the chimeric CoV as SHC014-MA15) to maximize the opportunity for pathogenesis and vaccine studies in mice (Supplementary Fig. 2a). Despite predictions from both structure-based modeling and pseudotyping experiments, SHC014-MA15 was viable and replicated to high titers in Vero cells (Supplementary Fig. 2b). Similarly to SARS, SHC014-MA15 also required a functional ACE2 molecule for entry and could use human, civet and bat ACE2 orthologs (Supplementary Fig. 2c,d). To test the ability of the SHC014 spike to mediate infection of the human airway, we examined the sensitivity of the human epithelial airway cell line Calu-3 2B4 (ref. 9) to infection and found robust SHC014-MA15 replication, comparable to that of SARS-CoV Urbani (Fig. 1c). To extend these findings, primary human airway epithelial (HAE) cultures were infected and showed robust replication of both viruses (Fig. 1d). Together, the data confirm the ability of viruses with the SHC014 spike to infect human airway cells and underscore the potential threat of cross-species transmission of SHC014-CoV.
Figure 1: SARS-like viruses replicate in human airway cells and produce in vivo pathogenesis.
(a) The full-length genome sequences of representative CoVs were aligned and phylogenetically mapped as described in the Online Methods. The scale bar represents nucleotide substitutions, with only bootstrap support above 70% being labeled. The tree shows CoVs divided into three distinct phylogenetic groups, defined as α-CoVs, β-CoVs and γ-CoVs. Classical subgroup clusters are marked as 2a, 2b, 2c and 2d for the β-CoVs and as 1a and 1b for the α-CoVs. (b) Amino acid sequences of the S1 domains of the spikes of representative β-CoVs of the 2b group, including SARS-CoV, were aligned and phylogenetically mapped. The scale bar represents amino acid substitutions. (c,d) Viral replication of SARS-CoV Urbani (black) and SHC014-MA15 (green) after infection of Calu-3 2B4 cells (c) or well-differentiated, primary air-liquid interface HAE cell cultures (d) at a multiplicity of infection (MOI) of 0.01 for both cell types. Samples were collected at individual time points with biological replicates (n = 3) for both Calu-3 and HAE experiments. (e,f) Weight loss (n = 9 for SARS-CoV MA15; n = 16 for SHC014-MA15) (e) and viral replication in the lungs (n = 3 for SARS-CoV MA15; n = 4 for SHC014-MA15) (f) of 10-week-old BALB/c mice infected with 1 × 104 p.f.u. of mouse-adapted SARS-CoV MA15 (black) or SHC014-MA15 (green) via the intranasal (i.n.) route. (g,h) Representative images of lung sections stained for SARS-CoV N antigen from mice infected with SARS-CoV MA15 (n = 3 mice) (g) or SHC014-MA15 (n = 4 mice) (h) are shown. For each graph, the center value represents the group mean, and the error bars define the s.e.m. Scale bars, 1 mm.
To evaluate the role of the SHC014 spike in mediating infection in vivo, we infected 10-week-old BALB/c mice with 104 plaque-forming units (p.f.u.) of either SARS-MA15 or SHC014-MA15 (Fig. 1e–h). Animals infected with SARS-MA15 experienced rapid weight loss and lethality by 4 d post infection (d.p.i.); in contrast, SHC014-MA15 infection produced substantial weight loss (10%) but no lethality in mice (Fig. 1e). Examination of viral replication revealed nearly equivalent viral titers from the lungs of mice infected with SARS-MA15 or SHC014-MA15 (Fig. 1f). Whereas lungs from the SARS-MA15–infected mice showed robust staining in both the terminal bronchioles and the lung parenchyma 2 d.p.i. (Fig. 1g), those of SHC014-MA15–infected mice showed reduced airway antigen staining (Fig. 1h); in contrast, no deficit in antigen staining was observed in the parenchyma or in the overall histology scoring, suggesting differential infection of lung tissue for SHC014-MA15 (Supplementary Table 2). We next analyzed infection in more susceptible, aged (12-month-old) animals. SARS-MA15–infected animals rapidly lost weight and succumbed to infection (Supplementary Fig. 3a,b). SHC014-MA15 infection induced robust and sustained weight loss, but had minimal lethality. Trends in the histology and antigen staining patterns that we observed in young mice were conserved in the older animals (Supplementary Table 3). We excluded the possibility that SHC014-MA15 was mediating infection through an alternative receptor on the basis of experiments using Ace2−/− mice, which did not show weight loss or antigen staining after SHC014-MA15 infection (Supplementary Fig. 4a,b and Supplementary Table 2). Together, the data indicate that viruses with the SHC014 spike are capable of inducing weight loss in mice in the context of a virulent CoV backbone.
Given the preclinical efficacy of Ebola monoclonal antibody therapies, such as ZMApp10, we next sought to determine the efficacy of SARS-CoV monoclonal antibodies against infection with SHC014-MA15. Four broadly neutralizing human monoclonal antibodies targeting SARS-CoV spike protein had been previously reported and are probable reagents for immunotherapy11,12,13. We examined the effect of these antibodies on viral replication (expressed as percentage inhibition of viral replication) and found that whereas wild-type SARS-CoV Urbani was strongly neutralized by all four antibodies at relatively low antibody concentrations (Fig. 2a–d), neutralization varied for SHC014-MA15. Fm6, an antibody generated by phage display and escape mutants11,12, achieved only background levels of inhibition of SHC014-MA15 replication (Fig. 2a). Similarly, antibodies 230.15 and 227.14, which were derived from memory B cells of SARS-CoV–infected patients13, also failed to block SHC014-MA15 replication (Fig. 2b,c). For all three antibodies, differences between the SARS and SHC014 spike amino acid sequences corresponded to direct or adjacent residue changes found in SARS-CoV escape mutants (fm6 N479R; 230.15 L443V; 227.14 K390Q/E), which probably explains the absence of the antibodies' neutralizing activity against SHC014. Finally, monoclonal antibody 109.8 was able to achieve 50% neutralization of SHC014-MA15, but only at high concentrations (10 μg/ml) (Fig. 2d). Together, the results demonstrate that broadly neutralizing antibodies against SARS-CoV may only have marginal efficacy against emergent SARS-like CoV strains such as SHC014.
Figure 2: SARS-CoV monoclonal antibodies have marginal efficacy against SARS-like CoVs.
figure2
(a–d) Neutralization assays evaluating efficacy (measured as reduction in the number of plaques) of a panel of monoclonal antibodies, which were all originally generated against epidemic SARS-CoV, against infection of Vero cells with SARS-CoV Urbani (black) or SHC014-MA15 (green). The antibodies tested were fm6 (n = 3 for Urbani; n = 5 for SHC014-MA15)11,12 (a), 230.15 (n = 3 for Urbani; n = 2 for SHC014-MA15) (b), 227.15 (n = 3 for Urbani; n = 5 for SHC014-MA15) (c) and 109.8 (n = 3 for Urbani; n = 2 for SHC014-MA15)13 (d). Each data point represents the group mean and error bars define the s.e.m. Note that the error bars in SARS-CoV Urbani–infected Vero cells in b,c are overlapped by the symbols and are not visible.
Full size image
To evaluate the efficacy of existing vaccines against infection with SHC014-MA15, we vaccinated aged mice with double-inactivated whole SARS-CoV (DIV). Previous work showed that DIV could neutralize and protect young mice from challenge with a homologous virus14; however, the vaccine failed to protect aged animals in which augmented immune pathology was also observed, indicating the possibility of the animals being harmed because of the vaccination15. Here we found that DIV did not provide protection from challenge with SHC014-MA15 with regards to weight loss or viral titer (Supplementary Fig. 5a,b). Consistent with a previous report with other heterologous group 2b CoVs15, serum from DIV-vaccinated, aged mice also failed to neutralize SHC014-MA15 (Supplementary Fig. 5c). Notably, DIV vaccination resulted in robust immune pathology (Supplementary Table 4) and eosinophilia (Supplementary Fig. 5d–f). Together, these results confirm that the DIV vaccine would not be protective against infection with SHC014 and could possibly augment disease in the aged vaccinated group.
In contrast to vaccination of mice with DIV, the use of SHC014-MA15 as a live, attenuated vaccine showed potential cross-protection against challenge with SARS-CoV, but the results have important caveats. We infected young mice with 104 p.f.u. of SHC014-MA15 and observed them for 28 d. We then challenged the mice with SARS-MA15 at day 29 (Supplementary Fig. 6a). The prior infection of the mice with the high dose of SHC014-MA15 conferred protection against challenge with a lethal dose of SARS-MA15, although there was only a minimal SARS-CoV neutralization response from the antisera elicited 28 d after SHC014-MA15 infection (Supplementary Fig. 6b, 1:200). In the absence of a secondary antigen boost, 28 d.p.i. represents the expected peak of antibody titers and implies that there will be diminished protection against SARS-CoV over time16,17. Similar results showing protection against challenge with a lethal dose of SARS-CoV were observed in aged BALB/c mice with respect to weight loss and viral replication (Supplementary Fig. 6c,d). However, the SHC014-MA15 infection dose of 104 p.f.u. induced >10% weight loss and lethality in some aged animals (Fig. 1 and Supplementary Fig. 3). We found that vaccination with a lower dose of SHC014-MA15 (100 p.f.u.), did not induce weight loss, but it also failed to protect aged animals from a SARS-MA15 lethal dose challenge (Supplementary Fig. 6e,f). Together, the data suggest that SHC014-MA15 challenge may confer cross-protection against SARS-CoV through conserved epitopes, but the required dose induces pathogenesis and precludes use as an attenuated vaccine.
Having established that the SHC014 spike has the ability to mediate infection of human cells and cause disease in mice, we next synthesized a full-length SHC014-CoV infectious clone based on the approach used for SARS-CoV (Fig. 3a)2. Replication in Vero cells revealed no deficit for SHC014-CoV relative to that for SARS-CoV (Fig. 3b); however, SHC014-CoV was significantly (P < 0.01) attenuated in primary HAE cultures at both 24 and 48 h after infection (Fig. 3c). In vivo infection of mice demonstrated no significant weight loss but showed reduced viral replication in lungs of full-length SHC014-CoV infection, as compared to SARS-CoV Urbani (Fig. 3d,e). Together, the results establish the viability of full-length SHC014-CoV, but suggest that further adaptation is required for its replication to be equivalent to that of epidemic SARS-CoV in human respiratory cells and in mice.
Figure 3: Full-length SHC014-CoV replicates in human airways but lacks the virulence of epidemic SARS-CoV.
(a) Schematic of the SHC014-CoV molecular clone, which was synthesized as six contiguous cDNAs (designated SHC014A, SHC014B, SHC014C, SHC014D, SHC014E and SHC014F) flanked by unique BglI sites that allowed for directed assembly of the full-length cDNA expressing open reading frames (for 1a, 1b, spike, 3, envelope, matrix, 6–8 and nucleocapsid). Underlined nucleotides represent the overhang sequences formed after restriction enzyme cleavage. (b,c) Viral replication of SARS-CoV Urbani (black) or SHC014-CoV (green) after infection of Vero cells (b) or well-differentiated, primary air-liquid interface HAE cell cultures (c) at an MOI of 0.01. Samples were collected at individual time points with biological replicates (n = 3) for each group. Data represent one experiment for both Vero and HAE cells. (d,e) Weight loss (n = 3 for SARS-CoV MA15, n = 7 for SHC014-CoV; n = 6 for SARS-Urbani) (d) and viral replication in the lungs (n = 3 for SARS-Urbani and SHC014-CoV) (e) of 10-week-old BALB/c mice infected with 1 × 105 p.f.u. of SARS-CoV MA15 (gray), SHC014-CoV (green) or SARS-CoV Urbani (black) via the i.n. route. Each data point represents the group mean, and error bars define the s.e.m. **P < 0.01 and ***P < 0.001 using two-tailed Student's t-test of individual time points.
During the SARS-CoV epidemic, links were quickly established between palm civets and the CoV strains that were detected in humans4. Building on this finding, the common emergence paradigm argues that epidemic SARS-CoV originated as a bat virus, jumped to civets and incorporated changes within the receptor-binding domain (RBD) to improve binding to civet Ace2 (ref. 18). Subsequent exposure to people in live-animal markets permitted human infection with the civet strain, which, in turn, adapted to become the epidemic strain (Fig. 4a). However, phylogenetic analysis suggests that early human SARS strains appear more closely related to bat strains than to civet strains18. Therefore, a second paradigm argues that direct bat-human transmission initiated SARS-CoV emergence and that palm civets served as a secondary host and reservoir for continued infection (Fig. 4b)19. For both paradigms, spike adaptation in a secondary host is seen as a necessity, with most mutations expected to occur within the RBD, thereby facilitating improved infection. Both theories imply that pools of bat CoVs are limited and that host-range mutations are both random and rare, reducing the likelihood of future emergence events in humans.
Figure 4: Emergence paradigms for coronaviruses.
Coronavirus strains are maintained in quasi-species pools circulating in bat populations. (a,b) Traditional SARS-CoV emergence theories posit that host-range mutants (red circle) represent random and rare occurrences that permit infection of alternative hosts. The secondary-host paradigm (a) argues that a nonhuman host is infected by a bat progenitor virus and, through adaptation, facilitates transmission to humans; subsequent replication in humans leads to the epidemic viral strain. The direct paradigm (b) suggests that transmission occurs between bats and humans without the requirement of an intermediate host; selection then occurs in the human population with closely related viruses replicating in a secondary host, permitting continued viral persistence and adaptation in both. (c) The data from chimeric SARS-like viruses argue that the quasi-species pools maintain multiple viruses capable of infecting human cells without the need for mutations (red circles). Although adaptations in secondary or human hosts may be required for epidemic emergence, if SHC014 spike–containing viruses recombined with virulent CoV backbones (circles with green outlines), then epidemic disease may be the result in humans. Existing data support elements of all three paradigms.
Although our study does not invalidate the other emergence routes, it does argue for a third paradigm in which circulating bat CoV pools maintain 'poised' spike proteins that are capable of infecting humans without mutation or adaptation (Fig. 4c). This hypothesis is illustrated by the ability of a chimeric virus containing the SHC014 spike in a SARS-CoV backbone to cause robust infection in both human airway cultures and in mice without RBD adaptation. Coupled with the observation of previously identified pathogenic CoV backbones3,20, our results suggest that the starting materials required for SARS-like emergent strains are currently circulating in animal reservoirs. Notably, although full-length SHC014-CoV probably requires additional backbone adaption to mediate human disease, the documented high-frequency recombination events in CoV families underscores the possibility of future emergence and the need for further preparation.
To date, genomics screens of animal populations have primarily been used to identify novel viruses in outbreak settings21. The approach here extends these data sets to examine questions of viral emergence and therapeutic efficacy. We consider viruses with the SHC014 spike a potential threat owing to their ability to replicate in primary human airway cultures, the best available model for human disease. In addition, the observed pathogenesis in mice indicates a capacity for SHC014-containing viruses to cause disease in mammalian models, without RBD adaptation. Notably, differential tropism in the lung as compared to that with SARS-MA15 and attenuation of full-length SHC014-CoV in HAE cultures relative to SARS-CoV Urbani suggest that factors beyond ACE2 binding—including spike processivity, receptor bio-availability or antagonism of the host immune responses—may contribute to emergence. However, further testing in nonhuman primates is required to translate these finding into pathogenic potential in humans. Importantly, the failure of available therapeutics defines a critical need for further study and for the development of treatments. With this knowledge, surveillance programs, diagnostic reagents and effective treatments can be produced that are protective against the emergence of group 2b–specific CoVs, such as SHC014, and these can be applied to other CoV branches that maintain similarly heterogeneous pools.
In addition to offering preparation against future emerging viruses, this approach must be considered in the context of the US government–mandated pause on gain-of-function (GOF) studies22. On the basis of previous models of emergence (Fig. 4a,b), the creation of chimeric viruses such as SHC014-MA15 was not expected to increase pathogenicity. Although SHC014-MA15 is attenuated relative to its parental mouse-adapted SARS-CoV, similar studies examining the pathogenicity of CoVs with the wild-type Urbani spike within the MA15 backbone showed no weight loss in mice and reduced viral replication23. Thus, relative to the Urbani spike–MA15 CoV, SHC014-MA15 shows a gain in pathogenesis (Fig. 1). On the basis of these findings, scientific review panels may deem similar studies building chimeric viruses based on circulating strains too risky to pursue, as increased pathogenicity in mammalian models cannot be excluded. Coupled with restrictions on mouse-adapted strains and the development of monoclonal antibodies using escape mutants, research into CoV emergence and therapeutic efficacy may be severely limited moving forward. Together, these data and restrictions represent a crossroads of GOF research concerns; the potential to prepare for and mitigate future outbreaks must be weighed against the risk of creating more dangerous pathogens. In developing policies moving forward, it is important to consider the value of the data generated by these studies and whether these types of chimeric virus studies warrant further investigation versus the inherent risks involved.除了为将来的新兴病毒提供准备外,还必须在美国政府强制暂停功能获得研究(GOF)的背景下考虑采用这种方法22。在以前出现的模型的基础上(图4a,b),嵌合病毒如SHC014-MA15的产生预计不会增加致病性。尽管SHC014-MA15相对于其亲本小鼠适应性SARS-CoV减毒,但类似的研究检查了MA15主链内野生型Urbani尖峰的CoV的致病性,显示小鼠无体重减轻和病毒复制减少23。因此,相对于Urbani峰值–MA15 CoV,SHC014-MA15的发病机理有所提高(图1)。基于这些发现,科学评论小组可能认为类似的研究基于无法冒险进行的循环株构建嵌合病毒,因为不能排除哺乳动物模型中致病性的增加。再加上对小鼠适应株的限制以及使用逃逸突变体开发单克隆抗体的研究,对CoV出现和治疗功效的研究可能会严重受限。这些数据和限制加在一起代表了GOF研究关注的十字路口。必须权衡准备和缓解未来爆发的潜力与创造更多危险病原体的风险。在制定向前发展的政策时,重要的是要考虑这些研究产生的数据的价值,以及这些类型的嵌合病毒研究是否值得进一步调查以及所涉及的固有风险。
Overall, our approach has used metagenomics data to identify a potential threat posed by the circulating bat SARS-like CoV SHC014. Because of the ability of chimeric SHC014 viruses to replicate in human airway cultures, cause pathogenesis in vivo and escape current therapeutics, there is a need for both surveillance and improved therapeutics against circulating SARS-like viruses. Our approach also unlocks the use of metagenomics data to predict viral emergence and to apply this knowledge in preparing to treat future emerging virus infections.Methods
Viruses, cells, in vitro infection and plaque assays.
Wild-type SARS-CoV (Urbani), mouse-adapted SARS-CoV (MA15) and chimeric SARS-like CoVs were cultured on Vero E6 cells (obtained from United States Army Medical Research Institute of Infectious Diseases), grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco, CA) and 5% fetal clone serum (FCS) (Hyclone, South Logan, UT) along with antibiotic/antimycotic (Gibco, Carlsbad, CA). DBT cells (Baric laboratory, source unknown) expressing ACE2 orthologs have been previously described for both human and civet; bat Ace2 sequence was based on that from Rhinolophus leschenaulti, and DBT cells expressing bat Ace2 were established as described previously8. Pseudotyping experiments were similar to those using an HIV-based pseudovirus, prepared as previously described10, and examined on HeLa cells (Wuhan Institute of Virology) that expressed ACE2 orthologs. HeLa cells were grown in minimal essential medium (MEM) (Gibco, CA) supplemented with 10% FCS (Gibco, CA) as previously described24. Growth curves in Vero E6, DBT, Calu-3 2B4 and primary human airway epithelial cells were performed as previously described8,25. None of the working cell line stocks were authenticated or tested for mycoplasma recently, although the original seed stocks used to create the working stocks are free from contamination. Human lungs for HAE cultures were procured under University of North Carolina at Chapel Hill Institutional Review Board–approved protocols. HAE cultures represent highly differentiated human airway epithelium containing ciliated and non-ciliated epithelial cells as well as goblet cells. The cultures are also grown on an air-liquid interface for several weeks before use, as previously described26. Briefly, cells were washed with PBS and inoculated with virus or mock-diluted in PBS for 40 min at 37 °C. After inoculation, cells were washed three times and fresh medium was added to signify time '0'. Three or more biological replicates were harvested at each described time point. No blinding was used in any sample collections nor were samples randomized. All virus cultivation was performed in a biosafety level (BSL) 3 laboratory with redundant fans in the biosafety cabinets, as described previously by our group2. All personnel wore powered air purifying respirators (Breathe Easy, 3M) with Tyvek suits, aprons and booties and were double-gloved.
Sequence clustering and structural modeling.
The full-length genomic sequences and the amino acid sequences of the S1 domains of the spike of representative CoVs were downloaded from Genbank or Pathosystems Resource Integration Center (PATRIC), aligned with ClustalX and phylogenetically compared by using maximum likelihood estimation using 100 bootstraps or by using the PhyML (https://code.google.com/p/phyml/) package, respectively. The tree was generated using maximum likelihood with the PhyML package. The scale bar represents nucleotide substitutions. Only nodes with bootstrap support above 70% are labeled. The tree shows that CoVs are divided into three distinct phylogenetic groups defined as α-CoVs, β-CoVs and γ-CoVs. Classical subgroup clusters are marked as 2a, 2b, 2c and 2d for β-CoVs, and 1a and 1b for the α-CoVs. Structural models were generated using Modeller (Max Planck Institute Bioinformatics Toolkit) to generate homology models for SHC014 and Rs3367 of the SARS RBD in complex with ACE2 based on crystal structure 2AJF (Protein Data Bank). Homology models were visualized and manipulated in MacPyMol (version 1.3).
Construction of SARS-like chimeric viruses.
Both wild-type and chimeric viruses were derived from either SARS-CoV Urbani or the corresponding mouse-adapted (SARS-CoV MA15) infectious clone (ic) as previously described27. Plasmids containing spike sequences for SHC014 were extracted by restriction digest and ligated into the E and F plasmid of the MA15 infectious clone. The clone was designed and purchased from Bio Basic as six contiguous cDNAs using published sequences flanked by unique class II restriction endonuclease sites (BglI). Thereafter, plasmids containing wild-type, chimeric SARS-CoV and SHC014-CoV genome fragments were amplified, excised, ligated and purified. In vitro transcription reactions were then preformed to synthesize full-length genomic RNA, which was transfected into Vero E6 cells as previously described2. The medium from transfected cells was harvested and served as seed stocks for subsequent experiments. Chimeric and full-length viruses were confirmed by sequence analysis before use in these studies. Synthetic construction of chimeric mutant and full-length SHC014-CoV was approved by the University of North Carolina Institutional Biosafety Committee and the Dual Use Research of Concern committee.
Ethics statement.
This study was carried out in accordance with the recommendations for the care and use of animals by the Office of Laboratory Animal Welfare (OLAW), NIH. The Institutional Animal Care and Use Committee (IACUC) of The University of North Carolina at Chapel Hill (UNC, Permit Number A-3410-01) approved the animal study protocol (IACUC #13-033) used in these studies.
Mice and in vivo infection.
Female, 10-week-old and 12-month-old BALB/cAnNHsD mice were ordered from Harlan Laboratories. Mouse infections were done as previously described20. Briefly, animals were brought into a BSL3 laboratory and allowed to acclimate for 1 week before infection. For infection and live-attenuated virus vaccination, mice were anesthetized with a mixture of ketamine and xylazine and infected intranasally, when challenged, with 50 μl of phosphate-buffered saline (PBS) or diluted virus with three or four mice per time point, per infection group per dose as described in the figure legends. For individual mice, notations for infection including failure to inhale the entire dose, bubbling of inoculum from the nose, or infection through the mouth may have led to exclusion of mouse data at the discretion of the researcher; post-infection, no other pre-established exclusion or inclusion criteria are defined. No blinding was used in any animal experiments, and animals were not randomized. For vaccination, young and aged mice were vaccinated by footpad injection with a 20-μl volume of either 0.2 μg of double-inactivated SARS-CoV vaccine with alum or mock PBS; mice were then boosted with the same regimen 22 d later and challenged 21 d thereafter. For all groups, as per protocol, animals were monitored daily for clinical signs of disease (hunching, ruffled fur and reduced activity) for the duration of the experiment. Weight loss was monitored daily for the first 7 d, after which weight monitoring continued until the animals recovered to their initial starting weight or displayed weight gain continuously for 3 d. All mice that lost greater than 20% of their starting body weight were ground-fed and further monitored multiple times per day as long as they were under the 20% cutoff. Mice that lost greater than 30% of their starting body weight were immediately sacrificed as per protocol. Any mouse deemed to be moribund or unlikely to recover was also humanely sacrificed at the discretion of the researcher. Euthanasia was performed using an isoflurane overdose and death was confirmed by cervical dislocation. All mouse studies were performed at the University of North Carolina (Animal Welfare Assurance #A3410-01) using protocols approved by the UNC Institutional Animal Care and Use Committee (IACUC).
Histological analysis.
The left lung was removed and submerged in 10% buffered formalin (Fisher) without inflation for 1 week. Tissues were embedded in paraffin and 5-μm sections were prepared by the UNC Lineberger Comprehensive Cancer Center histopathology core facility. To determine the extent of antigen staining, sections were stained for viral antigen using a commercially available polyclonal SARS-CoV anti-nucleocapsid antibody (Imgenex) and scored in a blinded manner by for staining of the airway and parenchyma as previously described20. Images were captured using an Olympus BX41 microscope with an Olympus DP71 camera.
Virus neutralization assays.
Plaque reduction neutralization titer assays were performed with previously characterized antibodies against SARS-CoV, as previously described11,12,13. Briefly, neutralizing antibodies or serum was serially diluted twofold and incubated with 100 p.f.u. of the different infectious clone SARS-CoV strains for 1 h at 37 °C. The virus and antibodies were then added to a 6-well plate with 5 × 105 Vero E6 cells/well with multiple replicates (n ≥ 2). After a 1-h incubation at 37 °C, cells were overlaid with 3 ml of 0.8% agarose in medium. Plates were incubated for 2 d at 37 °C, stained with neutral red for 3 h and plaques were counted. The percentage of plaque reduction was calculated as (1 − (no. of plaques with antibody/no. of plaques without antibody)) × 100.
Statistical analysis.
All experiments were conducted contrasting two experimental groups (either two viruses, or vaccinated and unvaccinated cohorts). Therefore, significant differences in viral titer and histology scoring were determined by a two-tailed Student's t-test at individual time points. Data was normally distributed in each group being compared and had similar variance.
Biosafety and biosecurity.
Reported studies were initiated after the University of North Carolina Institutional Biosafety Committee approved the experimental protocol (Project Title: Generating infectious clones of bat SARS-like CoVs; Lab Safety Plan ID: 20145741; Schedule G ID: 12279). These studies were initiated before the US Government Deliberative Process Research Funding Pause on Selected Gain-of-Function Research Involving Influenza, MERS and SARS Viruses (http://www.phe.gov/s3/dualuse/Documents/gain-of-function.pdf). This paper has been reviewed by the funding agency, the NIH. Continuation of these studies was requested, and this has been approved by the NIH.
SARS-CoV is a select agent. All work for these studies was performed with approved standard operating procedures (SOPs) and safety conditions for SARS-CoV, MERs-CoV and other related CoVs. Our institutional CoV BSL3 facilities have been designed to conform to the safety requirements that are recommended in the Biosafety in Microbiological and Biomedical Laboratories (BMBL), the US Department of Health and Human Services, the Public Health Service, the Centers for Disease Control (CDC) and the NIH. Laboratory safety plans were submitted to, and the facility has been approved for use by, the UNC Department of Environmental Health and Safety (EHS) and the CDC. Electronic card access is required for entry into the facility. All workers have been trained by EHS to safely use powered air purifying respirators (PAPRs), and appropriate work habits in a BSL3 facility and active medical surveillance plans are in place. Our CoV BSL3 facilities contain redundant fans, emergency power to fans and biological safety cabinets and freezers, and our facilities can accommodate SealSafe mouse racks. Materials classified as BSL3 agents consist of SARS-CoV, bat CoV precursor strains, MERS-CoV and mutants derived from these pathogens. Within the BSL3 facilities, experimentation with infectious virus is performed in a certified Class II Biosafety Cabinet (BSC). All members of the staff wear scrubs, Tyvek suits and aprons, PAPRs and shoe covers, and their hands are double-gloved. BSL3 users are subject to a medical surveillance plan monitored by the University Employee Occupational Health Clinic (UEOHC), which includes a yearly physical, annual influenza vaccination and mandatory reporting of any symptoms associated with CoV infection during periods when working in the BSL3. All BSL3 users are trained in exposure management and reporting protocols, are prepared to self-quarantine and have been trained for safe delivery to a local infectious disease management department in an emergency situation. All potential exposure events are reported and investigated by EHS and UEOHC, with reports filed to both the CDC and the NIH.Change history
20 November 2015In the version of this article initially published online, the authors omitted to acknowledge a funding source, USAID-EPT-PREDICT funding from EcoHealth Alliance, to Z.-L.S. The error has been corrected for the print, PDF and HTML versions of this articleKey Laboratory of Special Pathogens and Biosafety, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, China
A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence | Nature Medicine
https://www.nature.com/articles/nm.3985
宿主可能是蝙蝠 武汉病毒所石正丽团队研究成果在NATURE在线刊出
原标题:宿主可能是蝙蝠 武汉病毒所石正丽团队研究成果在NATURE在线刊出
2月3日,NATURE杂志在线刊出一篇题为A pneumonia outbreak associated with a new coronavirus of probable bat origin的论文,这是中国科学院武汉病毒研究所石正丽团队针对新型冠状病毒发表的研究论文,据了解,该论文此前曾在在bioRxiv预印版平台上,以Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin刊发,后经修回后,如今正式在NATURE杂志发表。
该论文显示,武汉新型冠状病毒nCoV-2019的序列与一种蝙蝠中的冠状病毒序列一致性高达96%,也就是说,引发武汉新型冠状病毒的宿主可能仍然是蝙蝠。但目前不明确其间是否还有中间宿主。
石正丽团队在文章中指出,自18年前SARS爆发以来,大量与严重急性呼吸综合征相关的冠状病毒(SARSr-CoV)在它们的天然宿主蝙蝠中被发现。先前的研究表明,其中一些蝙蝠sarsr - cov有可能感染人类。本文报道了一种新型冠状病毒(2019-nCoV)的鉴定和特征,该病毒在中国武汉引起了急性呼吸综合征的流行。
该疫情从2019年12月12日开始,截至2020年1月26日,已造成2050例实验室确诊感染和56例死亡病例。在疫情早期,石正丽团队从5名患者中获得了全长基因组序列,它们之间几乎完全相同,共有79.5%的序列识别到SARS-CoV。此外,还发现2019-nCoV在全基因组水平上与蝙蝠冠状病毒96%相同。对7个保守的非结构蛋白的两两序列分析表明,该病毒属于SARSr-CoV。然后从一名危重病人的支气管肺泡灌洗液中分离出2019-nCoV病毒,该病毒可被几名病人的血清中和。重要的是,我们已经证实这种新的CoV使用与SARS-CoV相同的细胞进入受体ACE2。该研究成果为后续病毒致病机理、病毒溯源等研究提供了重要依据。
武汉病毒所官网信息也显示,2019年12月30日,在疫情爆发初期,武汉病毒所启动新型冠状病毒的样本收集和标准化入库工作。2020年1月2日,该所确定了新型冠状病毒(以下称2019新型冠状病毒)全基因组序列,并于1月5日成功分离到了病毒毒株。1月9日该毒株资源已按标准完成国家病毒资源库入库,并进行了标准化保藏(保藏编号:IVCAS 6.7512),这为当前2019新型冠状病毒的科学研究、疫苗开发、生物医药筛选等提供重要资源支撑。1月11日,武汉病毒所作为国家卫健委的指定机构之一,向世界卫生组织提交了2019新型冠状病毒基因组序列信息,在全球流感共享数据库(GISAID,Global Initiative on Sharing All Influenza Data)发布,实现全球共享。此后,在全球范围内,针对新型冠状病毒的研究论文已经超过50篇。
1月23日,武汉病毒所牵头,由该所研究员石正丽任组长,与来自华中农业大学、华中科技大学、武汉大学、湖北省中医院、武汉金银潭医院等单位的13位专家共同组成科研攻关专家组,在快速检测技术产品研发、疾病发生、发展和传播规律及临床诊治、抗病毒应急药物和抗体类药物等8个方面开展联合攻关。
1月26日,由湖北省疾控中心等2家疾病预防控制机构、同济医院等9家医疗机构、以及武汉病毒所等2家专业机构,启动了2019新型冠状病毒肺炎病原学检测。目前,武汉病毒所已开展了部分2019新型冠状病毒临床样本检测,同时,由研究所开发的病毒检测试剂和方法,已应用于本次病原检测工作中,为后续诊断试剂盒的开发和推广使用奠定基础。
此外,武汉病毒所实现了2019新型冠状病毒相关抗原蛋白的原核和真核表达。通过与珠海丽珠试剂股份有限公司合作,在短时间内完成了2019新型冠状病毒IgG、IgM血清学诊断试剂盒(酶联免疫法),可作为除咽拭子病原核酸检测以外的重要辅助诊断手段。
当前,武汉病毒所还在开展新型冠状病毒感染的抗病毒药物筛选、动物模型建立、疫苗研发等工作。目前已筛选出几种有潜在临床应用价值的药物, 筛选结果已向国家和湖北省新型冠状病毒感染的肺炎疫情防控指挥部科技攻关组报告,供综合研判后指导医疗救治。在动物模型方面,已基本完成小鼠和非人灵长类动物模型的建立,将为后续研究提供关键支撑。
宿主可能是蝙蝠 武汉病毒所石正丽团队研究成果在NATURE在线刊出
2020年02月03日 21:34 21世纪经济报道
原标题:宿主可能是蝙蝠 武汉病毒所石正丽团队研究成果在NATURE在线刊出
2月3日,NATURE杂志在线刊出一篇题为A pneumonia outbreak associated with a new coronavirus of probable bat origin的论文,这是中国科学院武汉病毒研究所石正丽团队针对新型冠状病毒发表的研究论文,据了解,该论文此前曾在在bioRxiv预印版平台上,以Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin刊发,后经修回后,如今正式在NATURE杂志发表。
该论文显示,武汉新型冠状病毒nCoV-2019的序列与一种蝙蝠中的冠状病毒序列一致性高达96%,也就是说,引发武汉新型冠状病毒的宿主可能仍然是蝙蝠。但目前不明确其间是否还有中间宿主。
石正丽团队在文章中指出,自18年前SARS爆发以来,大量与严重急性呼吸综合征相关的冠状病毒(SARSr-CoV)在它们的天然宿主蝙蝠中被发现。先前的研究表明,其中一些蝙蝠sarsr - cov有可能感染人类。本文报道了一种新型冠状病毒(2019-nCoV)的鉴定和特征,该病毒在中国武汉引起了急性呼吸综合征的流行。
该疫情从2019年12月12日开始,截至2020年1月26日,已造成2050例实验室确诊感染和56例死亡病例。在疫情早期,石正丽团队从5名患者中获得了全长基因组序列,它们之间几乎完全相同,共有79.5%的序列识别到SARS-CoV。此外,还发现2019-nCoV在全基因组水平上与蝙蝠冠状病毒96%相同。对7个保守的非结构蛋白的两两序列分析表明,该病毒属于SARSr-CoV。然后从一名危重病人的支气管肺泡灌洗液中分离出2019-nCoV病毒,该病毒可被几名病人的血清中和。重要的是,我们已经证实这种新的CoV使用与SARS-CoV相同的细胞进入受体ACE2。该研究成果为后续病毒致病机理、病毒溯源等研究提供了重要依据。
武汉病毒所官网信息也显示,2019年12月30日,在疫情爆发初期,武汉病毒所启动新型冠状病毒的样本收集和标准化入库工作。2020年1月2日,该所确定了新型冠状病毒(以下称2019新型冠状病毒)全基因组序列,并于1月5日成功分离到了病毒毒株。1月9日该毒株资源已按标准完成国家病毒资源库入库,并进行了标准化保藏(保藏编号:IVCAS 6.7512),这为当前2019新型冠状病毒的科学研究、疫苗开发、生物医药筛选等提供重要资源支撑。1月11日,武汉病毒所作为国家卫健委的指定机构之一,向世界卫生组织提交了2019新型冠状病毒基因组序列信息,在全球流感共享数据库(GISAID,Global Initiative on Sharing All Influenza Data)发布,实现全球共享。此后,在全球范围内,针对新型冠状病毒的研究论文已经超过50篇。
1月23日,武汉病毒所牵头,由该所研究员石正丽任组长,与来自华中农业大学、华中科技大学、武汉大学、湖北省中医院、武汉金银潭医院等单位的13位专家共同组成科研攻关专家组,在快速检测技术产品研发、疾病发生、发展和传播规律及临床诊治、抗病毒应急药物和抗体类药物等8个方面开展联合攻关。
1月26日,由湖北省疾控中心等2家疾病预防控制机构、同济医院等9家医疗机构、以及武汉病毒所等2家专业机构,启动了2019新型冠状病毒肺炎病原学检测。目前,武汉病毒所已开展了部分2019新型冠状病毒临床样本检测,同时,由研究所开发的病毒检测试剂和方法,已应用于本次病原检测工作中,为后续诊断试剂盒的开发和推广使用奠定基础。
此外,武汉病毒所实现了2019新型冠状病毒相关抗原蛋白的原核和真核表达。通过与珠海丽珠试剂股份有限公司合作,在短时间内完成了2019新型冠状病毒IgG、IgM血清学诊断试剂盒(酶联免疫法),可作为除咽拭子病原核酸检测以外的重要辅助诊断手段。
当前,武汉病毒所还在开展新型冠状病毒感染的抗病毒药物筛选、动物模型建立、疫苗研发等工作。目前已筛选出几种有潜在临床应用价值的药物, 筛选结果已向国家和湖北省新型冠状病毒感染的肺炎疫情防控指挥部科技攻关组报告,供综合研判后指导医疗救治。在动物模型方面,已基本完成小鼠和非人灵长类动物模型的建立,将为后续研究提供关键支撑。
新型肺炎疫情地图-全国各地实时查询
关键词 : 疫情
武汉华南海鲜市场野味店全关 野味菜单含梅花鹿、活猴
2020年01月23日 22:31 作者:张晓梅 热度:116℃ 来源:特色美食
“武汉市华南海鲜市场陆续出现不明原因肺炎病人”引发广泛关注。红星新闻记者发现,有微博名为@萬謙寵愛_Yuki的网友在微博上发文称华南海鲜市场有野鸡、蛇、土拨鼠等动物宰杀出售。该网友称自己曾去过华南海鲜市场,“表面卖海鲜,深处除了待宰的猫狗,活蛇活鳖,各种野鸡,活蹦乱跳的土拨鼠,都在明晃晃地卖,梅花鹿、活猴之类的招牌也在明晃晃地挂……”
↑西区六街尽头遗弃的动物尸体和内脏
12月31日下午,红星新闻记者来到在位于武汉市江汉区的华南海鲜市场走访发现,有遗弃的兔子头及动物内脏散落在市场西区六街角落。附近一摊位店主大爷称,六街有几家卖野味的,有野鸡、蛇等很多品种,“你来晚了,(都)关门了。”在附近街区,红星新闻记者还发现了一些摊位附近有闲置的铁笼。对于记者询问是否有野味卖的问题,摊主显得非常警惕。红星新闻记者在西区入口处向几名卖干货的摊主和小卖部店主询问哪里能买到野味,都说“往里走”。
↑“卖野味”的店位于市场西区六街,目前都已关门
下午4点半左右,红星记者在市场发现,特色美食,数名武汉市疾控中心工作人员身着便衣,手持一份名单,分头对商户招牌进行拍照。针对红星记者“是否是在对患者摊位进行排查”的提问,工作人员拒绝回答。红星新闻记者从工作人员手中的名单发现上面有至少15人的名字及电话。红星新闻记者随后向西区入口处一摊主打听名单中一名叫李某环的人员,该摊主说其为市场里“卖野兽(野味)的”商家。
↑疾控中心工作人员在排查
红星新闻记者随后在华南海鲜市场管理办公室向相关工作人员求证上述说法,一工作人员称“无可奉告”,并称相关政府部门已组织他们开会,一切以相关部门发出消息为准。
调查发现,此次肺炎病例大部分为华南海鲜城经营户。目前,相关病毒分型检测、隔离治疗、舆情管控、终末消毒等工作正在进行。目前,所有病例均已隔离治疗,密切接触者的追踪调查和医学观察正在进行中,对华南海鲜城的卫生学调查和环境卫生处置正在进行中。
武汉大学医学部一位研究病毒的专家向红星新闻介绍,一般来说,很难想象肺炎病毒会从海鲜市场出来。海鲜市场最多的传染病是病从口入的甲肝、副溶血弧菌感染等。该专家称,即使是有,也是人的原因,跟那里的海产品关系较小。从目前得到的关于检测的结果看,有很小很小的可能,不能排除,但真的非常倾向于不是SARS冠状病毒。同源性非常低,不到4%的基因覆盖,无法进行判断。需要再检测。
该专家称,一般来说,纯粹海鲜市场肺炎病原体非常少见。一般能够造成肺炎的,比如说2003年的SARS病原体,“都是在野生动物当中会多一些。”
该专家提醒:流感高发期,应避免去人群多的地方,特别是小孩和老人。家里要注意通风,要加强锻炼。最近甲流、乙流蔓延,中小学停课的不少。流感极大地降低免疫力,也容易合并感染,造成肺炎。
武汉病毒研究所正开展新型冠状病毒疫苗研发等工作
2020-01-29中国新闻网
中新社北京1月29日电 中国科学院武汉分院29日发布文章称,武汉病毒所正开展新型冠状病毒感染的抗病毒药物筛选、疫苗研发等工作。目前已筛选出了几种有潜在临床应用价值的药物,筛选结果已向国家和湖北省新型冠状病毒感染的肺炎疫情防控指挥部科技攻关组报告。
文章对该所近期工作进行了三方面总结。
一是提供资源支撑,牵头联合攻关。研究所于2020年1月2日确定了新型冠状病毒(以下称2019新型冠状病毒)全基因组序列,于1月5日成功分离到了病毒毒株。1月9日该毒株资源已按标准完成国家病毒资源库入库,并进行了标准化保藏,可依法依规提供给有关机构,将为当前2019新型冠状病毒的科学研究、疫苗开发、生物医药筛选等提供重要资源支撑。1月11日,武汉病毒所作为国家卫健委的指定机构之一,向世界卫生组织提交了2019新型冠状病毒基因组序列信息,在全球流感共享数据库发布,实现全球共享。
2020年1月23日,湖北省新型肺炎应急科研攻关专家组召开第一次工作会议。会议正式宣布成立由武汉病毒所牵头,石正丽研究员任组长,与来自华中农业大学、华中科技大学、武汉大学、湖北省中医院、武汉金银潭医院等单位的13位专家共同组成科研攻关专家组,着重在快速检测技术产品研发、疾病发生、发展和传播规律及临床诊治、抗病毒应急药物和抗体类药物等8个方面开展联合攻关,协同全省优势科研力量,全力打好科技防控攻关战。
二是主动参与,助力检测诊断。目前,武汉病毒所已开展部分2019新型冠状病毒临床样本检测,助力武汉市缓解防疫压力,并进一步提升新冠肺炎的检测及诊治能力。同时,由研究所开发的病毒检测试剂和方法,已应用于本次病原检测工作中,为后续诊断试剂盒的开发和推广使用奠定了坚实基础。
三是突破科学难题,取得重要进展。1月23日,武汉病毒所石正丽团队在bioRxiv预印版平台上发表文章《一种新型冠状病毒的发现及其可能的蝙蝠起源》,提出新型肺炎病毒或来源于蝙蝠。文章首次证实了该新型冠状病毒使用与SARS冠状病毒相同的细胞进入受体(ACE2),并发现新型冠状病毒与一种蝙蝠的冠状病毒的序列一致性高达96%,为后续病毒致病机理、病毒溯源等研究提供了重要依据。
同时武汉病毒所正开展新型冠状病毒感染的抗病毒药物筛选、动物模型建立、疫苗研发等工作。目前已筛选出了几种有潜在临床应用价值的药物,筛选结果已向国家和湖北省新型冠状病毒感染的肺炎疫情防控指挥部科技攻关组报告,供综合研判后指导医疗救治。在动物模型方面,已基本完成小鼠和非人灵长类动物模型的建立,将为后续研究提供关键支撑。(完)
What is Coronavirus
Coronaviruses are RNA enveloped viruses than can infect animals and humans. So far, there are six known human coronaviruses. Four of these coronaviruses are less pathogenic, generally causing only minor respiratory symptoms like the common cold. Two other coronaviruses—Severe Acute Respiratory Syndrome Coronavirus (SARS CoV) and Middle East Respiratory Syndrome Coronavirus (MERS CoV) —can cause serious respiratory diseases.
A new strain of Coronavirus (2019-nCoV) has emerged, is causing illnesses globally and is different from other coronaviruses.
To date, the virus has infected hundreds of people, with more than a dozen reported deaths. As surveillance proceeds, there may be more cases identified. Some, but not all, of the cases are linked to a live animal market in Wuhan City, in the Hubei Province of China. Public Health authorities are actively investigating this outbreak.
Because of this, public health recommendations focus on standard infection control practices to reduce exposure to and transmission of a range of illnesses
WHAT ARE THE SYMPTOMS?
Symptoms of 2019-nCoV are like other upper-respiratory infections, including fever, cough and difficulty breathing. They range from mild to severe and appear 2-14 days after exposure.
HOW IS IT TRANSMITTED?
Since coronaviruses can be extremely contagious and spread easily from person to person, medical diagnosis is required.
Transmission routes for 2019-nCoV, include:
- The air by coughing and sneezing
- Close, personal contact, such as touching or shaking hands
- Touching surfaces or objects contaminated with virus particles, then touching your mouth, nose or eyes before washing your hands.
HOW IS IT CONTROLLED?
There are currently no vaccines available to protect you against human coronavirus infection.
Infection risk can be reduced by doing the following:
- Wash your hands often and correctly. Washing your hands often with soap and water is one of the best ways to avoid transmission of emerging pathogens.
- Avoid touching your eyes, nose or mouth with unwashed hands
- Avoid close contact with people who are sick
- Do not touch animals or animal feces and do not eat wild animals
- Avoid farmers markets, live poultry markets or farms
- If you have fever or other symptoms after traveling home, you should wear a mask and immediately call a doctor. Be sure to let the doctor know where you were traveling.
Enveloped viruses are the least resistant to inactivation by disinfection. The structure of these viruses includes a lipid envelope, which is easily compromised by most disinfectants. Once the lipid envelope is damaged, the integrity of the virus is compromised, thereby neutralizing its infectivity.
As with any communicable disease, it is recommended to focus on proper hand hygiene and disinfection of high-touchpoint areas including railings, doorknobs and handles and restroom surfaces.
REFERENCES AND FURTHER INFORMATION
1. WHO - https://www.who.int/health-topics/coronavirus
2. CDC - https://www.cdc.gov/coronavirus/2019-ncov/index.html
Thank you to Ecolab for this information. Source: https://en-id.ecolab.com/expertise-and-innovation/resources/microbial-risks/coronavirus
Coronavirus Information
https://www.connect2cleanrooms.com/news/coronavirus-2019-ncov-information
2019 Novel Coronavirus (2019-nCoV) and Plasma Protein Therapies
Recent international reports have highlighted the emergence of a new coronavirus in Wuhan, Hubei Province, China.1 The 2019 Novel Coronavirus (2019-nCoV) was first identified by Chinese authorities in December 2019 and since then has been associated with pneumonia in over 2000 persons in China and more than 50 deaths.1, 2
In addition, there has been a growing number of cases identified outside of Hubei Province and internationally.1, 2, 3 On January 21, 2020 the U.S. Centers for Disease Control and Prevention (CDC) confirmed the first case of a person being infected with 2019-nCoV in the U.S.4 More cases have since then been confirmed internationally.1, 2, 3
PPTA considers that the 2019-nCoV is not a concern for the safety of plasma protein therapies manufactured by PPTA member companies based on the following information:
To date, the majority of cases detected has been in the Hubei province in China. Individuals diagnosed with the virus in other countries acquired the disease through travel to Wuhan City, Hubei Province. Based on the current epidemiological evidence, it is unlikely that the virus is present in U.S. and European populations. Moreover, donor screening procedures are in place to prevent donations from individuals showing disease symptoms typical of a coronavirus infection (raised temperature/ fever, cough, difficulty breathing) from donating plasma.
The 2019-nCoV is a large sized virus (approximately 120 nm in diameter).5, 6 The relatively large size and lipid envelope makes it highly susceptible to steps with virus inactivation and removal capacity used during the manufacturing processes, such as solvent-detergent (S/D),7 low pH incubation, caprylate, pasteurization8 or dry-heat treatments,9 nanofiltration or fractionation processes and others.10 The effectiveness of these processes has been demonstrated on other lipid-enveloped model viruses which are quite similar to 2019-nCoV, e.g. human coronavirus 229E and OC43, SARS-CoV, and porcine coronavirus TGEV.8, 11, 12
Based on these data, PPTA is convinced that existing manufacturing methods provide significant safety margins against the 2019-nCoV.
Public health bodies in the US (CDC) and in Europe (ECDC), as well as the WHO and Chinese authorities, are continuously monitoring the situation and have put in place proactive measures to monitor 2019-nCoV infection in Europe and in the U.S, as well as internationally, including issuing travel guidance for Wuhan City, Hubei Province, China1, 13, 14, 15 testing16, 17 and reporting guidance for 2019-nCoV, and adding entry health screening at major U.S.1 and international airports18 for passengers coming from Wuhan City.
Based on strict screening procedures for plasma donors and the established processes of virus inactivation and removal during manufacturing of plasma-derived products, PPTA concludes that the 2019-nCoV is not a concern for the safety margins of plasma protein therapies manufactured by PPTA member companies.
Background:
The 2019 Novel Coronavirus (2019-nCoV) belongs to the family of Coronaviridae, which are known to infect animals and humans, causing respiratory and gastrointestinal illness. Seven different coronaviruses are known to infect humans, causing mild to moderate illness. In rare cases, animal coronaviruses can evolve and infect humans. This has been observed in the past with Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS), both known to cause severe illness.5, 6 There is no published data documenting transmission of respiratory coronaviruses by blood transfusion.5, 6
It appears that the 2019-nCoV can be spread through human-human contact via respiratory droplets. More research is needed, however, to fully understand the mode of transmission, clinical course of disease and epidemiology, and whether particular population groups are at a higher risk of illness.5, 62019 Novel Coronavirus (2019-nCoV) and Plasma Protein Therapies - Plasma Protein Therapeutics Association (PPTA)
https://www.pptaglobal.org/media-and-information/ppta-statements/1055-2019-novel-coronavirus-2019-ncov-and-plasma-protein-therapies
Clinical evidence does not support corticosteroid ...
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30317-2/fulltext
Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury Next Article 2019-nCoV transmission through the ocular surface must not be ignored The 2019 novel coronavirus (2019-nCoV) outbreak is a major challenge for clinicians.
[Comment] Clinical evidence does not support ...
https://www.lifescience.net/entries/355043/comment-clinical-evidence-does-not-support...
[Comment] Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Journal content | Feb 07, 2020 Recommendations: n/a. Published in. The Lancet, The Lancet (Reed Elsevier) Content. The 2019 novel coronavirus (2019-nCoV) outbreak is a …
Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury
Clark D Russell
Jonathan E Millar
J Kenneth Baillie
Published:February 07, 2020DOI:https://doi.org/10.1016/S0140-6736(20)30317-2
The 2019 novel coronavirus (2019-nCoV) outbreak is a major challenge for clinicians. The clinical course of patients remains to be fully characterised, little data are available that describe the disease pathogenesis, and no pharmacological therapies of proven efficacy yet exist.
Corticosteroids were widely used during the outbreaks of severe acute respiratory syndrome (SARS)-CoV1 and Middle East respiratory syndrome (MERS)-CoV,2 and are being used in patients with 2019-nCoV in addition to other therapeutics.3 However, current interim guidance from WHO on clinical management of severe acute respiratory infection when novel coronavirus (2019-nCoV) infection is suspected (released Jan 28, 2020) advises against the use of corticosteroids unless indicated for another reason.4 Understanding the evidence for harm or benefit from corticosteroids in 2019-nCoV is of immediate clinical importance. Here we discuss the clinical outcomes of corticosteroid use in coronavirus and similar outbreaks (table).
TableSummary of clinical evidence to date
Outcomes of corticosteroid therapy* Comment
MERS-CoV Delayed clearance of viral RNA from respiratory tract2 Adjusted hazard ratio 0·4 (95% CI 0·2–0·7)
SARS-CoV Delayed clearance of viral RNA from blood5 Significant difference but effect size not quantified
SARS-CoV Complication: psychosis6 Associated with higher cumulative dose, 10 975 mg vs 6780 mg hydrocortisone equivalent
SARS-CoV Complication: diabetes7 33 (35%) of 95 patients treated with corticosteroid developed corticosteroid-induced diabetes
SARS-CoV Complication: avascular necrosis in survivors8 Among 40 patients who survived after corticosteroid treatment, 12 (30%) had avascular necrosis and 30 (75%) had osteoporosis
Influenza Increased mortality9 Risk ratio for mortality 1·75 (95% CI 1·3–2·4) in a meta-analysis of 6548 patients from ten studies
RSV No clinical benefit in children10, 11 No effect in largest randomised controlled trial of 600 children, of whom 305 (51%) had been treated with corticosteroids
CoV=coronavirus. MERS=Middle East respiratory syndrome. RSV=respiratory syncytial virus. SARS=severe acute respiratory syndrome.
* Hydrocortisone, methylprednisolone, dexamethasone, and prednisolone.
Open table in a new tab
Acute lung injury and acute respiratory distress syndrome are partly caused by host immune responses. Corticosteroids suppress lung inflammation but also inhibit immune responses and pathogen clearance. In SARS-CoV infection, as with influenza, systemic inflammation is associated with adverse outcomes.12 In SARS, inflammation persists after viral clearance.13, 14 Pulmonary histology in both SARS and MERS infections reveals inflammation and diffuse alveolar damage,15 with one report suggesting haemophagocytosis.16 Theoretically, corticosteroid treatment could have a role to suppress lung inflammation.
In a retrospective observational study reporting on 309 adults who were critically ill with MERS,2 almost half of patients (151 [49%]) were given corticosteroids (median hydrocortisone equivalent dose [ie, methylprednisolone 1:5, dexamethasone 1:25, prednisolone 1:4] of 300 mg/day). Patients who were given corticosteroids were more likely to require mechanical ventilation, vasopressors, and renal replacement therapy. After statistical adjustment for immortal time and indication biases, the authors concluded that administration of corticosteroids was not associated with a difference in 90-day mortality (adjusted odds ratio 0·8, 95% CI 0·5–1·1; p=0·12) but was associated with delayed clearance of viral RNA from respiratory tract secretions (adjusted hazard ratio 0·4, 95% CI 0·2–0·7; p=0·0005). However, these effect estimates have a high risk of error due to the probable presence of unmeasured confounders.
In a meta-analysis of corticosteroid use in patients with SARS, only four studies provided conclusive data, all indicating harm.1 The first was a case-control study of SARS patients with (n=15) and without (n=30) SARS-related psychosis; all were given corticosteroid treatment, but those who developed psychosis were given a higher cumulative dose than those who did not (10 975 mg hydrocortisone equivalent vs 6780 mg; p=0·017).6 The second was a randomised controlled trial of 16 patients with SARS who were not critically ill; the nine patients who were given hydrocortisone (mean 4·8 days [95% CI 4·1–5·5] since fever onset) had greater viraemia in the second and third weeks after infection than those who were given 0·9% saline control.5 The remaining two studies reported diabetes and avascular necrosis as complications associated with corticosteroid treatment.7, 8
A 2019 systematic review and meta-analysis9 identified ten observational studies in influenza, with a total of 6548 patients. The investigators found increased mortality in patients who were given corticosteroids (risk ratio [RR] 1·75, 95% CI 1·3–2·4; p=0·0002). Among other outcomes, length of stay in an intensive care unit was increased (mean difference 2·1, 95% CI 1·2–3·1; p<0·0001), as was the rate of secondary bacterial or fungal infection (RR 2·0, 95% CI 1·0–3·8; p=0·04).
Corticosteroids have been investigated for respiratory syncytial virus (RSV) in clinical trials in children, with no conclusive evidence of benefit and are therefore not recommended.10 An observational study of 50 adults with RSV infection, in which 33 (66%) were given corticosteroids, suggested impaired antibody responses at 28 days in those given corticosteroids.17
Life-threatening acute respiratory distress syndrome occurs in 2019-nCoV infection.18 However, generalising evidence from acute respiratory distress syndrome studies to viral lung injury is problematic because these trials typically include a majority of patients with acute respiratory distress syndrome of non-pulmonary or sterile cause. A review of treatments for acute respiratory distress syndrome of any cause, based on six studies with a total of 574 patients,19 concluded that insufficient evidence exists to recommend corticosteroid treatment.20
Septic shock has been reported in seven (5%) of 140 patients with 2019-nCoV included in published reports as of Jan 29, 2020.3, 18 Corticosteroids are widely used in septic shock despite uncertainty over their efficacy. Most patients in septic shock trials have bacterial infection, leading to vasoplegic shock and myocardial insufficiency.21, 22 In this group, there is potential that net benefit might be derived from steroid treatment in severe shock.21, 22 However, shock in severe hypoxaemic respiratory failure is often a consequence of increased intrathoracic pressure (during invasive ventilation) impeding cardiac filling, and not vasoplegia.23 In this context, steroid treatment is unlikely to provide a benefit.
No clinical data exist to indicate that net benefit is derived from corticosteroids in the treatment of respiratory infection due to RSV, influenza, SARS-CoV, or MERS-CoV. The available observational data suggest increased mortality and secondary infection rates in influenza, impaired clearance of SARS-CoV and MERS-CoV, and complications of corticosteroid therapy in survivors. If it is present, the effect of steroids on mortality in those with septic shock is small, and is unlikely to be generalisable to shock in the context of severe respiratory failure due to 2019-nCoV.
Overall, no unique reason exists to expect that patients with 2019-nCoV infection will benefit from corticosteroids, and they might be more likely to be harmed with such treatment. We conclude that corticosteroid treatment should not be used for the treatment of 2019-nCoV-induced lung injury or shock outside of a clinical trial.
JKB is a member of the WHO panel on clinical management for 2019-nCoV. CDR and JEM declare no competing interests.Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury - The Lancet
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30317-2/fulltext
镁客网
来自雪球发布于02-07 14:03
《柳叶刀》最新发文称:应避免使用激素作为新冠肺炎的治疗方法关于皮质类固醇激素治疗新冠肺炎
文章中给出了不建议使用皮质类固醇激素治疗的原因,即在SARS和MERS感染的肺组织学检查中都显示出炎症和严重急性呼吸综合症,这是由宿主免疫反应引起的。而皮质类固醇激素虽然可以抑制肺部炎症、但也抑制了免疫反应和人体免疫系统对于病毒的清除作用。
比如针对309名MERA危重患者的回顾性观察研究报告中显示,有151名患者在接受皮质类固醇激素治疗后,还需要其他的治疗方式,比如呼吸协助、药物增加血压以及透析形式。报告中还发现,使用类固醇激素治疗的人需要更长的时间才能从体内清除病毒。
对于国人比较熟知的是,在2003年SARS疫情发生时,在完全没有经验的情况下,使用大剂量糖皮质激素注射治疗非典病人,取得了意想不到的效果。不过后来许多患者在治愈后出现了股骨头坏死症状、肺部功能障碍等。
在新型冠状病毒爆发后,世界卫生组织在1月28日发布的最新版新型冠状病毒诊疗指南中提到,建议不要使用皮质类固醇,除非有原因表明使用皮质类固醇激素有益或者有害。
《柳叶刀》发布的这篇文章提到,新冠病毒感染会引发危及生命的急性呼吸窘迫症候群。然而,从急性呼吸窘迫症候群研究到病毒性肺损伤的一般性证据是有问题的,因为这些实验通常包括大多患有非肺病或者无菌性急性呼吸窘迫症候群的病人。而根据对共计574名患者的6项研究,对任何原因引发的感染性休克治疗进行回顾可以看到,没有足够的证据推荐使用皮质类固醇治疗。
其以在2020年1月29日发表的报告为例,在140例新冠肺炎患者中有7例发生了败血性休克。虽然有318种皮质类固醇激素已经广泛应用于败血性休克,但其治疗效果还尚不明确。然而严重低氧血症性休克通常是由于胸内压增高导致的,而不是血管麻痹,这种情况下,使用皮质类固醇激素治疗不太合适。
不过在2月5日,国家卫生委和国家中医药局联合印发了最新版《新型冠状病毒感染的肺炎诊疗方案(试行第五版)》中,结合了此前的四版诊疗方案,均包括针对重型、危重型病例的治疗,其中提出可以根据患者呼吸困难程度、胸部影像学进展情况,酌情短期内(3-5天)使用糖皮质激素,且建议剂量不超过相当于甲泼尼龙1~2mg/kg·d。
多种方式正在尝试治疗新冠肺炎
自疫情爆发以来,国内外多个机构和研究人员都在加紧进行关于新冠病毒的疫苗研发和抗病毒药物试验。
三天前,中国工程院院士、国家卫健委高级别专家组成员李兰娟及其团队表示,阿比朵尔、达芦那韦两种药物被证明可以有效抑制新型冠状病毒。李兰娟院士建议将这两种药物列入国家卫健委《新型冠状病毒感染的肺炎诊疗方案(试行第六版)》。不过需要强调的是,这两种药为处方药,患者一定要在医生的指导下服用。
国家卫健委新闻发言人、宣传司司长宋树立表示,他们在救治工作上进行了很多探索,如克力芝、瑞德西韦和中药治疗等。
不过克力芝已经被李兰娟院士认为有毒副作用,而瑞德西韦可以说是这几天的热点。2月6日瑞德西韦的临床试验的注册审批工作完成,武汉金银潭医院启动了这一临床试验,据该临床试验牵头人曹彬教授介绍总计拟入组761例患者,采用随机、双盲、安慰剂对照方法展开。如果III期临床试验结束,其有效性得到验证,该药预计能马上应用。
此外据媒体报道,泰国曾使用抗流感和抗艾滋的组合药物,洛匹那韦与利托那韦,使一名新型冠状病毒感染的肺炎患者病情在48小时内迅速好转,病毒检测结果也在48小时内由阳性转为阴性。
总的来说,在抗病毒药物试验方面,近日来取得了不少进展,关于新冠肺炎的诊疗方案的讨论也有很多,这一切或许都将会在新一版新冠肺炎诊疗方案中有所体现。
- 点击“阅读原文”,获取《柳叶刀》文档。(提取码:4xqw)
作者:镁客网
链接:https://xueqiu.com/4004656270/140711908
来源:雪球
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。《柳叶刀》最新发文称:应避免使用激素作为新冠肺炎的治疗方法 倾听创业故事,对接融资需求 欢迎做客【镁客·请讲】栏目 点击菜单栏-联系我们,让更多人看到你们 在治疗新冠肺炎中使用皮质... - 雪球
https://xueqiu.com/4004656270/140711908
Published: 06 August 2015
Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases
K. D. Rainsford, Ann L. Parke, Matthew Clifford-Rashotte & W. F. Kean
Inflammopharmacology volume 23, pages231–269(2015)Cite this article
Abstract
Objectives
This review examines the pharmacokinetics, modes of action and therapeutic properties of the anti-malarial drugs, hydroxychloroquine (HCQ) and chloroquine (CQ), in the treatment of systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and related conditions, as well as osteoarthritis (OA).
Key findings
Both HCQ and CQ have historically been employed successfully for the treatment of SLE and RA for over 70 years. HCQ has been used extensively for SLE where it has a good reputation for controlling the dermatological complications in SLE. It has also been reported to effectively control the symptoms of Sjøgren’s syndrome, as well as preventing thrombosis in phospholipid antibody (aPL) syndrome. In RA and SLE, HCQ is preferred because of the lower incidence of gastrointestinal adverse reactions compared with CQ and it might have a lower risk of ocular adverse reactions. There is increasing evidence that HCQ may reduce atherosclerosis and risks of cardiovascular disease in rheumatic patients. Both HCQ and CQ have been shown to improve glycaemia and reduce the risks of type II diabetes mellitus. Although both HCQ and CQ are effective in low-moderate RA, HCQ is now preferred as part of combination therapy for more severe disease. The advantages of combination therapy are that the doses of the individual drugs may be lowered so reducing adverse reactions. Both HCQ and CQ are diastereoisomers, have basic properties and are given as the sulphate and phosphate salts. While being relatively well absorbed orally and with good bioavailability, they have long and variable plasma terminal elimination half-lives (approximately 40–60 days). This reflects their high volume of distribution, V D (HCQ 44,000L; CQ 65,000L) which extends into aqueous compartments, long mean residence time (HCQ 1300 h; CQ 900 h) and with about half the drugs (metabolites) undergoing renal clearance. The strong binding to melanin reflects the ocular injury and dermatological properties of these drugs. The consensus is that the occurrence of ocular adverse reactions can be minimised by close attention to the dose (which should be set on a body weight basis) with regular (e.g. quarterly) retinal examination. Although HCQ and CQ can pass through the placenta, the use of these drugs during pregnancy does not appear to risk harm to the baby and might be beneficial to the mother with SLE and her child by controlling the SLE disease activity, which is known to be an important factor affecting pregnancy outcome. The modes of action of HCQ and CQ in these arthritides represent somewhat of an enigma. Undoubtedly, these drugs have multiple actions related, in part, their ability to accumulate in lysosomes and autophagosomes of phagocytic cells as well as affecting MHC Class II expression and antigen presentation; actions of the production of pro-inflammatory cytokines [e.g. interleukin-1 (IL-1) tumour necrosis factor-α (TNFα)]; control of toll-like receptor-9 activation; and leucocyte generation of reactive oxygen species (ROS); i.e. antioxidant activity. The actions of these drugs on T and B cells are less clear but may depend on these leucocyte-mediated actions. Anti-malarials also protect against cytokine-mediated cartilage resorption. This and other actions may underlie the potential benefits in treating OA. The exact relationships of these various actions, mostly determined in vitro, have not been specifically defined in vivo or ex vivo in relation to clinical efficacy.
Outcomes
HCQ and CQ have a good reputation for being effective and relatively safe treatments in SLE, mild-moderate RA and Sjøgren’s syndrome. There is need for (a) more information on their mode of action in relation to the control of these diseases, (b) scope for developing formulations that have improved pharmacokinetic and therapeutic properties and safety, and (c) further exploring their use in drug combinations not only with other disease-modifying agents but also with biologics.Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases | SpringerLink
https://link.springer.com/article/10.1007/s10787-015-0239-y







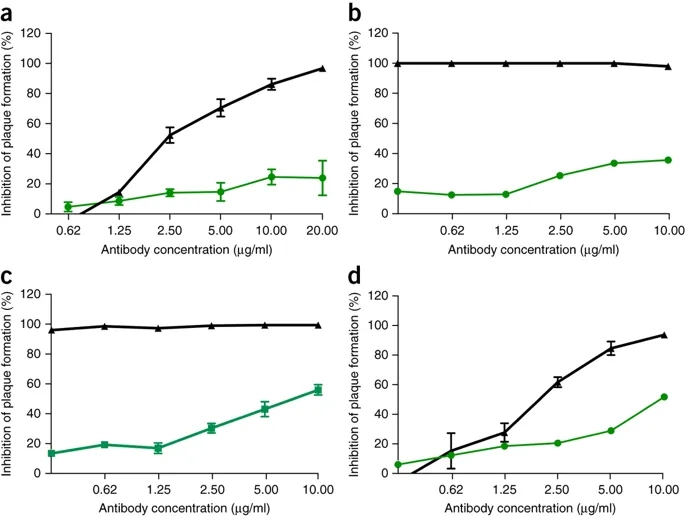
.jpg)
.jpg)