ĪĪ
Mechanisms
1. it was not AA itself but the acidic conditions created by the acid.AA is a relatively strong acid (pKa1=4.1),easily lowering the pH. Indeed, the pH measured in the studied solutions had the acidic values between 3.52 and 3.66 (indicated in Figure 1A), that are the values lower than ~4.5 below which urease is known to irreversibly lose activity39.Further, to check if the AA-generated inactivation of urease was purely pH-dependent, we performed the inactivation of urease by low pHs in 20mM acetate buffer. The inactivation time courses are shown in Figure 1B. They are similar to those of the AA-induced inactivation, but intriguingly, the same amount of inactivation as in AA was brought about here by higher pHs from the range 4.18©C4.34.
2.
Indian J Pharmacol. 2011 Nov-Dec; 43(6): 624©C627.
Vitamin-C as anti-Helicobacter pylori agent: More prophylactic than curative- Critical review
Jagannath Pal, Madhusudana Girija Sanal,1 and Gopal Jee Gopal2
Department of Medical Oncology, Dana Farber Cancer Institute, USA
1Department of Research, Institute of Liver and Biliary Sciences, D1, Vasant Kunj, New Delhi - 110070, India
2Special Center for Molecular Medicine, Jawaharlal Nehru University, New Delhi - 110067, India
ĪĪ
Vitamin C: A Preventative, Therapeutic Agent Against Helicobacter Pylori - PubMed
https://pubmed.ncbi.nlm.nih.gov/30280058/ĪĪ
Abstract
Potential of nonantibiotic therapies for treatment of Helicobacter pylori-related acid peptic disease remains underexplored. Several clinical studies have shown that higher prevalence of H. pylori infection is associated with low Vitamin C (Vit C) level in serum and gastric juice. However, there is no consensus regarding the usefulness of Vit C supplementation in the management of H. pylori infection. Surveying the existing literature we conclude that high concentration of Vit C in gastric juice might inactivate H. pylori urease, the key enzyme for the pathogen's survival and colonization into acidic stomach. Once infection established, urease is not very important for its survival. The role of Vit-C as anti-H. pylori agent in peptic ulcer diseases appears to be preventive rather than curative. Rather than supplementing high dose of Vit C along with conventional triple therapy, it is preferable to complete the conventional therapy and thereafter start Vit C supplementation for extended period which would prevent reinfection in susceptible individuals, provided the patients are not achlorhydric. Further studies are required to prove the role of Vit C in susceptible population.
KEY WORDS: Helicobacter pylori, prophylactic, supplementation, urease, vitamin C
Go to:
Introduction
In spite of initial success of eradication treatment for Helicobacter pylori in peptic ulcer diseases using combination of a proton pump inhibitor and antibiotics, emergence of antibiotics resistance in recent years, leads to frequent treatment failure in at least 10-20% of the patients.[1] Nonantibiotic therapies, including phytomedicines, probiotics and antioxidants have been increasingly investigated as potential adjuvants for the treatment of H. pylori.[2] Several clinical studies have shown that high H. pylori infection rate is related to low ascorbic acid (AA)/Vitamin C (Vit C) level in serum as well as in gastric juice.[3©C5] On the other hand high dose of Vit C have been shown to inhibit H. pylori growth, colonization or even eradication of H. pylori infection in few studies while others gave nonconclusive results.[2,6©C11] But still it is not clear how Vit C level can affect the course of H. pylori infection in stomach and how the infection affects the level of Vit C in serum and gastric juice. Moreover doses of Vit C used in those experiments were extremely higher compared to the physiological concentration of Vit C in the stomach. These high concentrations cannot be achieved in physiological condition due to its pro-oxidant activity at high concentration (500 mg/day or more) particularly in presence of high body iron stores leading to several adverse effects including DNA damage and an increase risk of cancer.[12,13] So the actual role of Vit C at physiological or therapeutic level, in the course of H. pylori infection in the stomach is still inconclusive. One study shows that AA does not inhibit growth of H. pylori at a concentration (400 Mm) within the physiological range.[14] This experiment was carried out in buffered condition at pH 7.4, but in vivo H. pylori have to counter unbuffered acidic pH (pH 3) to colonize successfully into the stomach. At low pH the effect of AA on survival of H. pylori might be different. So these ex vivo studies also do not rule out the role of AA as anti-H. pylori agent.
At this juncture a new query emerges; whether antioxidant effect of Vit C has any effect on the survival of H. pylori at low pH? Till date very limited studies have been carried out in this direction. A systematic analysis of the available information would lead to some interesting logical derivations which might explain the existing confusion associated with the role of Vit C in H. pylori infection. This would help us to design more clinical studies in a logical way and to formulate appropriate antipylori regimen.
Go to:
Mechanism of H. pylori Survival At Low Gastric pH
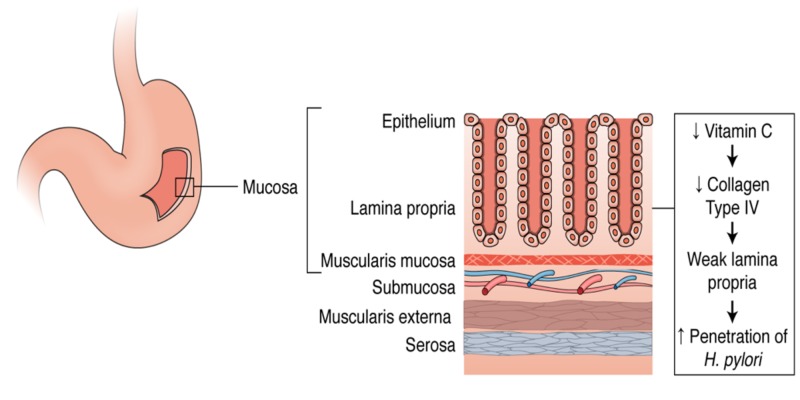
H. pylori synthesizes large amount of urease, which is found in its cytosol. The cytosolic urease is released into the gastric juice upon spontaneous autolysis of a subpopulation of H. pylori and subsequently it is adsorbed onto the surface of intact bacteria. The urease catalyses the hydrolysis of urea present in the gastric juice, to yield carbonic acid and ammonia. Thus H. pylori makes a cloud of ammonia on its surface to neutralize the gastric acid which enables it to colonize the gastric epithelium.[15,16] Once successfully colonized, H. pylori resides below the gastric mucus layer which has a higher pH than gastric lumen.[17,18] So in chronic infection, the role played by urease, in survival of the bacteria seems less important. However, besides protecting from acid, urease also aids in colonization by providing ammonia for bacterial protein synthesis.
Go to:
Structural Features of H. pylori Urease
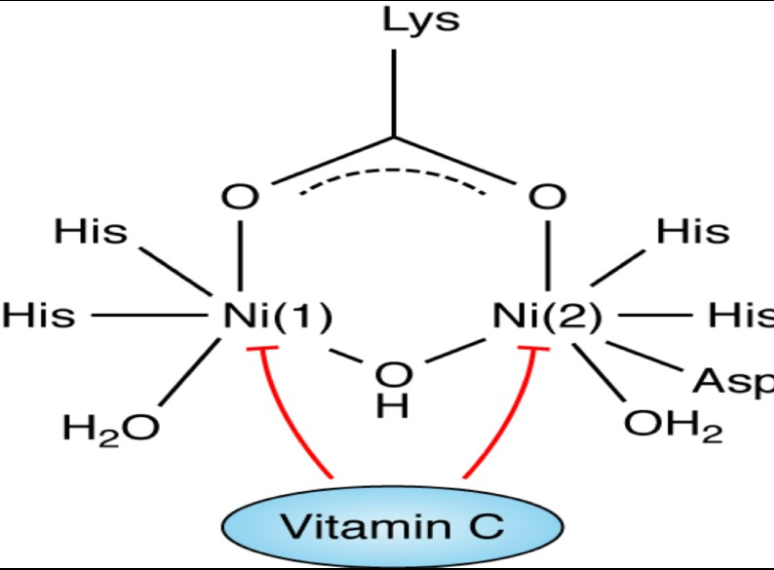
In the active site of urease there are two Ni (Ni++) centers held by co-ordination bonding. Ni(1) is coordinated by two imidazole ligands from two histidine residues and a water molecule. Ni(2) is coordinated by two histidine residues as well as with an aspartate and a water molecule.[19] It has been found that purified urease is inactivated at pH<5 (in buffered solution). So it had long been unanswered that how extracellular urease on H. pylori cell surface remains active in low gastric pH. Moreover cytosolic pH (pH 6) of H. pylori is also suboptimal for maximal urease activity. Ha et al; 2001 have convincingly shown that supramolecular assembly of urease create a pore within the complex which serves as a pathway for diffusion of urea toward the 12 clustered active sites which protect each other from acid inactivation by producing localized cloud of ammonia from breakdown of urea.[20]
Go to:
Vit C as Biological Antioxidant
Vit C is an acidic molecule with strong reducing activity and is an essential component of most living tissues. It has two major redox forms: AA and DHA, the reduced and oxidized form, respectively, and they are all interconvertible. Within the cell DHA is rapidly converted to AA by the specific enzyme systems like DHA reductase, glutaredoxins and protein disulfide isomerase in presence of glutathione or other thiols as electron donors[21,22] Unlike AA, DHA is relatively unstable and undergo rapid spontaneous irreversible hydrolysis particularly at a pH > 4.[23]
Go to:
Vit C as a Reducing Agent for Transition Metals
Few transition metals like Fe(III), Cu(II), Hg(II), Cr(VI) are able to accept electron from AA, resulting in their reduction and simultaneous oxidation of AA to DHA. In presence of strong biological oxidants like oxygen, hydrogen peroxide the oxidation of AA is accelerated, where the metal ions act predominantly as catalyst. In aqueous solutions at neutral pH, metal ions, having relatively low redox potential like Zn(II), Ni(II), Co(II), Pb(II) unable to oxidize AA by themselves nor serve as a catalyst for oxidation by molecular oxygen. However, in biological solutions (e.g.: tissue fluid, blood, serum) redox chemistry of these metal ions is different due to presence of different natural metal complexing agents like amino acids, peptides, proteins, nucleotides, etc. These natural ligands are also able to form ternary complexes with the metal ions and Vit C. Among all biological ligands, histidine or histidine containing peptides are shown to be more efficient in binding transition metals like Ni(II) and form stable ternary complexes with AA. The assembly favors oxidation of AA by the metal ion and the process is relatively faster in presence of oxygen.[24©C26]
Go to:
Vit-C as an Anti-H. pylori Agent
Normally AA is actively secreted from plasma to the gastric juice.[27,28] High concentration of AA in gastric juice favor reduction of Ni++ centers, coordinated to the histidine residues of the urease, secreted from H. pylori, leading to inactivation of the enzyme followed by acid denaturation [Figure 1]. This reduction of the transition metal ion by AA, also accelerated at low pH of gastric juice as in case of reduction of ferric iron by AA.[10,11] Recently Krajewska et al. showed that in a buffered system neither AA nor DHA themselves are inhibitors of urease. The inhibitory effect of AA and DHA was seen in the presence of Fe(3+) ions and, unlike reported in the literature, they found that it was mediated by H2O2. Interestingly the resulting inhibition by DHA-Fe(3+) consisted of enzyme thiol oxidation and its effectiveness grew with increasing pH.[29] This could be another interesting mechanism by which AA can act against H. pylori especially in the initial stages of infection.
An external file that holds a picture, illustration, etc.
Object name is IJPharm-43-624-g001.jpg
Open in a separate window
Figure 1
Inactivation of H. pylori urease by Vit.C in H. pylori-infected gastric mucosa. Abbreviation: Ni ©C nickel (reduced form), AA- ascorbic acid/Vit. C, DHA- dehydroascorbic acid, HP- H. pylori
In low gastric pH, once urease is inactive it becomes difficult for H. pylori to survive and colonize in stomach. But once it successfully colonizes into stomach wall, H. pylori stays within the gastric mucosa, where the pH is suitable for the survival of the bacteria, due to the bicarbonate buffer from gastric epithelium secreted into the luminal surface.[17,18] Moreover after chronic infection, a proportion of patients develop relative achlorhydria leading to higher pH of gastric juice.[30] So at this stage of infection, role of urease for its survival is relatively less. This might explain the fact that even a high dose AA supplementation is not able to eradicate H. pylori infection in a significant percentage of cases[11] though AA have been shown to inhibit urease in vitro.[29,31] In chronically infected stomach, the bacteria continue to produce urease, which capture the free Ni++ from the gastric juice. As discussed above, this bound Ni++ is more preferred substrate for reduction by AA than free Ni++. As a consequence of reduction of the Ni++ centers of urease, AA itself oxidized to DHA and rapidly exhausted by spontaneous hydrolysis particularly when passed through alkaline medium of intestine. This might be one of the explanations of low serum and gastric juice AA level in chronically H. pylori-infected patients. Though local ROS, generated by chronic gastritis may also be responsible partially for conversion of AA to DHA. At the site of inflammation extra cellular DHA is taken up by leucocytes and rapidly converted to AA and thus recycled.[32,33] So the local gastric inflammation may not have significant effect on Vit C deficiency in H. pylori induced gastritis. There is also evidence that, in Ni++ toxicity, there is depletion of serum AA and the toxic effect is reversed by addition of AA.[26] This seems to be due to reduction of Ni++ by AA in the biological solution in vivo. On the other hand, patients with already low serum AA likely to be more prone to get infection by H. pylori, because low AA in gastric juice might favor colonization by the bacteria as explained above. Moreover low antioxidant level in chronically infected gastric mucosa, causes elevation of ROS contributing perpetuation of inflammation and infection cycle.[34,35] Thus it is possible that AA can have an anti-H. pylori effect during initiation, spreading and perpetuation of the infection although major effect could be during initiation of colonization.
Few clinical studies have been carried out where different conventional anti-H. pylori regimens supplemented with Vit C were used. The results of these studies[36©C41] range from no benefit on Vit C supplementation with conventional anti-H. pylori regimens to studies which reported that Vit C decreased effectiveness of conventional anti-H. pylori particularly where a triple therapy containing metronidazol was used.[36,37] In all those anti-H. pylori regimens proton pump inhibitor was one of the critical components which increased the gastric pH. Ex vivo studies using physiological concentration of AA and pH (7.4) in H. pylori culture media also support these views. Moreover it have been reported that PPI reduce bioavailability and stability of Vit C in gastric juice.[42,43] Secondly in the anerobic micro-organisms, metmronidazol enter into the bacterial cell through passive diffusion and is reduced to generate active free radicals. Thus a flow of metronidazol is maintained into the cells along a concentration gradient.[9,44,45] In presence of high dose of AA extracelluar reduction of metronidazole might take place, hampering effective accumulation of the drug into the bacterial cells. More over active free radical forms of metronidazol might be stabilized by high concentration of Vit C which leads to reduced H. pylori eradication rates when this combined regimen is used. Vit C supplementation at a dose of 500 mg b.d. following triple therapy seems to be quite effective as shown from a preliminary study by Sezikli et al.[38] More clinical trials are required in this direction to determine the optimum dosage and regimen keeping in mind the increasing pro-oxidative role of Vit C at higher dose (500 mg/day) as observed by Podmore et al.[13] It is important to particularly consider long-term Vit C supplementation therapy. However, a few recent studies report an increased eradication of H. pylori when triple therapy was supplemented with Vit C.[38©C41] Yet there is no convincing evidence if supplementation of Vit C along with triple therapy could be beneficial in eradicating resistant H. pylori infection.[41] Interestingly, in one study Vit C was supplemented two more weeks following completion of triple therapy showing better eradication rate than achievable by triple therapy alone.[38]
Conclusions
The role of AA as anti-H. pylori agent in peptic ulcer diseases is most likely to be preventive rather than curative. Rather than supplementing high doses of AA along with conventional antipylori regimen it is preferable to complete the standard course of antipylori regimen, which might then be followed by Vit C supplementation therapy for extended period which would prevent from reinfection in susceptible subjects and also might eradicate the residual H. pylori infection, provided the patients are not achlorhydric or under prolonged acid suppressive therapy. It is also essential to maintain a constant protective level of Vit C in gastric juice or in serum to inhibit colonization or reinfection by H. pylori in gastric epithelium. Considering the short biological half life of this water soluble vitamin and the chances of toxicity from single daily bolus dose, split multiple doses/sustained release formulations would be more effective to maintain constant protective level of Vit C in plasma and gastric juice. More clinical trials are required in this direction to determine the actual dosage and regimen. It would be interesting to study the incidence of H. pylori infection among the large cohorts of population having varied concentrations of plasma Vit C. Future studies should be directed toward comparison of the reinfection rates in cohorts undergone the standard course of anti-H. ylori pylori regimen versus those who are maintained on Vit C supplementation after the completion of the standard therapy. It is clear from this review that well-controlled, well-designed studies are required in this direction as H. pylori infection and consequent gastric cancer is a major public health problem.Vitamin-C as anti-Helicobacter pylori agent: More prophylactic than curative- Critical review
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3229773/#ĪĪ
Journal of Molecular Catalysis B: Enzymatic
Volume 68, Issues 3©C4, March 2011, Pages 262-269
Hydrogen peroxide-induced inactivation of urease: Mechanism, kinetics and inhibitory potency
Evidence has been collected that H2O2 resulting from redox reactions taking place in some inhibitor systems, is responsible for the inactivation of urease. Accordingly, in this study the inactivation by H2O2 exogenously added to urease (jack bean) was investigated. The reaction accountable for the inactivation was the oxidation of the enzyme thiol groups. The reaction was studied at pHs over 6.2©C8.2. At each pH, the first-order kinetics was obeyed, the resulting dependences of kobs on H2O2 concentration being hyperbolic. Analyzed with Kitz-Wilson method, the kinact appeared to be invariant with pH, while the inhibitor binding constant KI decreased with increasing pH. Consequently, the process grew faster with an increase in pH, the second-order rate constants increasing from 0.0032 M−1 s−1 at pH 6.2 to 0.0615 M−1 s−1 at pH 8.2, consistent with higher susceptibility of thiolate anions to oxidation by H2O2 compared to reduced thiols. The reactions always resulted in irreversible inactivation of urease, indicative of the oxidation of thiol groups to either sulfinic or sulfonic acid, independent of the extent of inactivation. Analysis of the numbers of urease thiol groups modified with H2O2 revealed that for the complete inactivation, out of 36 thiols/molecule available under non-denaturating conditions, the rapid oxidation of 30 highly reactive ©CSH groups was responsible for a 50% loss in enzyme activity, and the slower oxidation of the remaining six ©CSH groups, for the other 50%. The enzyme was protected by active-site binding inhibitors, boric acid and fluoride, from the inactivation, suggesting that the six thiols are the active-site flap Cys-592s. For comparison with other urease inhibitors, the IC50 was determined for 20 min incubation with H2O2, its value changing from 242 mM at pH 6.2 to 11 mM at pH 8.2.
Graphical abstract
The rate of oxidative inactivation of urease by H2O2 grows with increasing pH. The inactivation is irreversible.
Download : Download full-size image
Research highlights
▶ The inactivation of jack bean urease by H2O2 was investigated. ▶ It involved the oxidation of enzyme thiol groups and was found irreversible with pH-dependent kinetics. ▶ To entirely abolish the enzyme activity, the oxidation of the active-site flap cysteine residue was required.Hydrogen peroxide-induced inactivation of urease: Mechanism, kinetics and inhibitory potency - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S1381117710002985ĪĪ
Published: 26 February 1944
Urease Activity and Ascorbic Acid
K. V. GIRI & P. SESHAGIRI RAO
Department of Biochemistry, Indian Institute of Science, Bangalore
Abstract
IN a recent note, Elson1 has shown that ascorbic acid at low concentrations inhibits urease activity, and that this inhibition is prevented by addition of cysteine. Quastel2 suggests that the inhibition of the activity by ascorbic acid is due to the oxidized form of ascorbic acid, namely, the diketone (dehydro-ascorbic acid) and not to the ascorbic acid itself. The disappearance of the inhibition on the addition of cysteine has been attributed to the reduction of the dehydroascorbic acid to the inert ascorbic acid. It seemed to us more probable that the action of ascorbic acid is not connected with either ascorbic acid or dehydroascorbic acid, but with the oxidation of the vitamin by traces of heavy metals like copper present in the reaction mixture. For several years we have been investigating the influence of ascorbic acid on enzymes, and we have obtained results showing that the vitamin as such or the dehydroascorbic acid has very little influence on the enzymes (phosphatases and amylase); but when the vitamin is oxidized by traces of copper present in the reaction mixture the enzyme is inhibited to a considerable extent. It was also found that substances which inhibit the oxidation of ascorbic acid annul the inhibition by the vitamin. In the light of these results we have now investigated the influence of ascorbic acid, dehydroascorbic acid and substances which inhibit the oxidation of the vitamin, on urease. The experimental method adopted was as follows.Urease Activity and Ascorbic Acid | Nature
https://www.nature.com/articles/153253b0ĪĪ
Published: 21 August 1943
Urease Activity and Ascorbic Acid
J. H. QUASTEL
Nature volume 152, page215(1943)Cite this article
Abstract
WITH reference to Mr. Elson's observation1 that ascorbic acid at low concentrations inhibits urease activity, and that this inhibition disappears in the presence of cysteine, it is worth noting that certain polyhydric phenols, for example, catechol and quinol, also exert at low concentrations (one part in two millions) highly inhibitory effects on urease activity, this inhibition disappearing in presence of thiol compounds such as cysteine2. It has been shown that the inhibition in this case is due, not to the phenol, but to the corresponding oxidized product, that is, the quinone present in solution with the phenol. The alleviating action of cysteine is due to the reduction of the quinone to the inert phenol. It may be suggested that, in an analogous manner, the toxicity of ascorbic acid in dilute solution is due to the oxidized form of ascorbic acid, namely, the diketone, present in solution, and not to the ascorbic acid itself. The effect of cysteine will be to reduce the dehydro-ascorbic acid to the inert ascorbic acid, a phenomenon which is known to take place, as shown by Crook3.Urease Activity and Ascorbic Acid | Nature
https://www.nature.com/articles/152215b0ĪĪ
Urease activity and l-ascorbic acid
Barbara Krajewska, and Małgorzata Brindell
Jagiellonian University, Faculty of Chemistry, 30-060 Krak©«w, Ingardena 3, Poland
Abstract
In this work, we studied the behaviours of urease in the presence of l-ascorbic acid (AA) and dehydroascorbic acid
(DHA) in different conditions. The inactivations of urease were carried out in an unbuffered and buffered system. We
show that in the unbuffered system AA inactivated urease in a biphasic manner by denaturation brought about by
AA-lowered pH. Further, we show that in the buffered system neither AA nor DHA themselves are inhibitors of urease.
The inhibitory action of AA and DHA was revealed in the presence of Fe3+ ions and most importantly, unlike reported
in the literature, it was found to be primarily mediated by H2O2. The resulting inhibition by DHA-Fe3+ consisted of
enzyme thiol oxidation and its effectiveness grew with increasing pH. The results may shed light on the roles of AA in therapies applied in ureolytic bacteria infections, notably those with Helicobacter pylori.Chemical properties of AA
it was not AA itself but the acidic conditions created by the acid.AA is a relatively strong acid (pKa1=4.1),easily lowering the pH. Indeed, the pH measured in the studied solutions had the acidic values between 3.52 and 3.66 (indicated in Figure 1A), that are the values lower than ~4.5 below which urease is known to irreversibly lose activity39.Further, to check if the AA-generated inactivation of urease was purely pH-dependent, we performed the inactivation of urease by low pHs in 20mM acetate buffer. The inactivation time courses are shown in Figure 1B. They are similar to those of the AA-induced inactivation, but intriguingly, the same amount of inactivation as in AA was brought about here by higher pHs from the range 4.18©C4.34.
AA, the formula of which is C6H8O6 (Scheme 1), is a sugar
acid with redox properties. As an acid it shows strength
comparable with the lower monocarboxylic acids, its pKa
for dissociation of the first hydrogen ion being 4.1 and
for the second, 11.826. As a redox agent by contrast, AA
exhibits a dual nature27,28. First, it is best known for being an antioxidant with powerful reducing properties. In vivo the properties are exploited at the cellular level for scavenging reactive oxygen species (ROS), including superoxide O2•©C, hydroxyl •OH and hydrogen peroxide H2O2,thereby providing protection to biomolecules against oxidative damage. During the process (Scheme 1), AA is initially oxidized to ascorbyl radical and further to its first chemically stable oxidized form, dehydroascorbic acid
(DHA). This can be reduced back to AA or be irreversibly hydrolyzed to 2,3-diketogulonic acid, which then is converted primarily to oxalic and threonic acids among other numerous five or less carbon species29.
Second, AA also has prooxidant properties, which are revealed when the
acid is oxidized in reactions catalyzed by transition metal ions, notably cupric and ferric27,28,30. It is because the metal ion-catalyzed oxidation promotes the generation of ROS, paradoxically the same AA is known to destroy. The model currently adopted for the process assumes that on
its oxidation AA reduces the metal ions Men+ Ī· Me(n©C1)+, which is accompanied by the generation of H2O2. Further, the reduced metal ions react with H2O2 driving the formation of hydroxyl radicals •OH via the Fenton reaction:
Me(n©C1)+ + H2
O2 Ī· Men+ + OH−
+ •OH29,31
. The resulting ROS
bring about inactivation of biomolecules by oxidation.ĪĪ
Urease activity and AA
Intriguingly, there is no consensus of opinion whether at all, and if yes, how AA interacts with ureases. In the early works the acid was reported to be a strong urease inhibitor (IC50=5.7 ”╠M) with the action prevented by cysteine32 . Later it was suggested that the inhibition was due to a reaction of DHA with the enzyme thiol groups33 . DHA, however, was later proved not to be an inhibitor, and instead it was proposed that the inhibition was provoked by the presence of traces of Cu2+ ions, which when reduced by AA, formed an inhibitory product Cu2 O34 . In refs. 32©C33, the buffers applied were not specified. A subsequent study performed in 0.7M phosphate buffer pH 6.835 , showed that neither ascorbic (up to 1.1mM), nor dehydroascorbic or 2,3-diketogulonic acid themselves were urease inhibitors, but that ascorbic and 2,3-diketogulonic acid inhibited the enzyme in the presence of Cu2+ (15.6 ”╠M). The inhibition by AA was ascribed to the reduction of Cu2+ Ī· Cu+ , and the involvement of Cu+ in the inhibition could not be ruled out. Both cysteine and glutathione protected the enzyme. In contrast to these findings, in a more recent study of Helicobacter pylori urease by electron microscopy and electrophoresis24 , AA was found to completely inhibit the enzyme at a concentration of 2.8mM within 30min, and the authors suggested a DHA-mediated mechanism. In another recent work, where a 13 C-NMR screening for urease inhibitors was presented36 , the inhibition of urease by AA in 70mM phosphate buffer pH 6.5 was determined to be noncompetitive with Ki =16.7mM (IC50=16.7mM). The reduction of Ni2+ ions at urease active site by AA or a covalent modification of the protein by DHA were hypothesized as mechanisms of this inhibition. In view of these conflicting data, the aim of this work was to present an overview of behaviours of urease in the presence AA in different conditions to resolve the question of AA inactivation of the enzyme. Given the properties of AA, the inactivation was studied in an unbuffered and buffered system. In the unbuffered system the inactivation was compared with that by acetate buffer of low pH. In the buffered system on the other hand, the inactivation was studied at pH 7.2 in the absence and presence of Fe3+ ions, and was further extended into the inactivation study by DHA, also in the absence and presence of Fe3+ ions, the latter at different pHs. Being a much weaker inhibitor of urease37 , Fe3+ ions were chosen as a catalyst in place of commonly applied Cu2+ ions to avoid their interference with the activity of urease. The urease inactivation results were correlated with UV-vis spectral kinetic data recorded for AA and DHA conversion.
ĪĪ
Inactivation of urease by AA in a buffered system in the absence and presence of Fe3+ ions
...
The correlations prove that neither AA nor DHA are the direct inactivators of urease and that the slow inactivation of the enzyme is provoked by a common product generated in the course of AA oxidation downstream of DHA, possibly ROS, H2 O2 included. To check this possibility we performed the same inactivations in the presence of Fe3+ ions. A concentration of 10 ”╠M Fe3+ was chosen as submaximal noninhibitory for urease37 . As can be seen from Figure 3, the addition of Fe3+ considerably speeded up the reactions, the halflives of the inactivations being now 18h and 2h for AAFe3+ and DHA-Fe3+ systems, respectively. In a separate experiment we also observed that the activity of urease was protected by catalase practically entirely (98 %) from the inactivation by AA-Fe3+ and in 82% by DHA-Fe3+, this being strongly supportive of the involvement of ROS in the process, notably H2 O2 27,28,30 . To clarify the mechanism of these inactivations we further examined the quickest system, urease-DHA-Fe3+.
ĪĪ
Inactivation of urease by DHA in the presence of Fe3+ ions
The inactivation of urease in 200mM phosphate buffer by 10mM DHA in the presence of 10 ”╠M Fe3+ was studied at five pHs from the range 6.2©C8.2. The results are shown in Figure 5A, where RA of urease is plotted
...
Taken together, the results of the above experiments imply that the inactivation of urease in the DHA-Fe3+ system consisted of generation of ROS, notably H2 O2 that oxidized the enzyme thiol groups. Apparently, half of the groups were oxidized to sulfenic acid and the remaining half, most likely, to further thiol oxidation states. The generation of H2 O2 was favored by pH values above 7.
Concluding remarks
Previous studies on the effects of AA on urease yielded conflicting results ranging from strongly inhibitory32 through moderately inhibitory24,36 to entirely noninhibitory35 . The discrepancies may be due to the choice of experimental conditions in which the inhibition was studied, including different unbuffered or bufferred systems, and the presence or absence of metal ions, such as Cu2+ that are known for their interference both with AA and urease. As a result, different mechanisms of AA interactions with urease were hypothesized, including DHA-modification of the protein24,33,36 , AA-reduction of Ni2+ ions in the urease active site25,36 , some reactions resulting from the reduction of Cu2+ by AA35 , and finally the reactions of Cu2+ and Cu+ with urease34,35 . In this work, we showed that in the unbuffered system AA inactivated urease by denaturation brought about by AA-lowered pH. Further we showed that in the buffered system neither AA nor DHA inhibited urease. Their inhibitory action, weak by AA and stronger by DHA, was revealed in the presence of Fe3+ ions and most importantly, unlike previously reported in the literature, it was found to be primarily mediated by H2 O2 . Thiol groups of urease appeared to be the target of this inhibition, as observed in some previous reports32,35 , but the reaction involved was determined to be the oxidation. Its effectiveness in the DHA-Fe3+-urease system was found to grow with increasing pH. The results collected in this work clarify the behaviours of urease in the presence of AA. If they may be of practical value under physiological conditions in therapeutic suppression of ureolytic bacteria apparently requires further considerations. For instance, in the conditions of the Helicobacter pylori-infected stomach, low pH-inactivation of urease by denaturation may be limited by the fact that the pH adjacent to the bacteria in the stomach is maintained close to neutrality. The prooxidant activity of AA- and DHA systems, by contrast, to be effective requires the presence of free metal ions (Cu2+, Fe3+). These, however, in the human body primarily occur bound to proteins. Iron for instance, is essential in the human body and its overall concentration is comparatively high, but it is almost entirely complexed with proteins, such as ferritin, transferrin or hemoglobin, hence practically unavailable for the catalysis of ROS generation. Ascorbate can, however, under certain conditions release iron from some of these complexes by reduction. Then under the condition of iron availability, the locally increased pH, such as the one around Helicobacter pylori in the stomach, would facilitate the inactivation of urease. The exact biological implications of these findings and considerations, however, for urease-producing bacteria and their possible eradication remain to be established.
14756366.2010.504675
https://www.tandfonline.com/doi/pdf/10.3109/14756366.2010.504675ĪĪ
Proteus mirabilis ureaseŲµęņ▒õą╬Ė╦Š·ļÕ├Ė: genetic organization, regulation, and expression of structural genes.
B D Jones and H L Mobley
Abstract
Proteus mirabilis, a cause of serious urinary tract infection, produces urease, an important virulence factor for this species. The enzyme hydrolyzes urea to CO2 and NH3, which initiates struvite or apatite stone formation. Genes encoding urease were localized on a P. mirabilis chromosomal DNA gene bank clone in Escherichia coli by deletion analysis, subcloning, Bal31 nuclease digestion, transposon Tn5 mutagenesis, and in vitro transcription-translation. A region of DNA between 4.0 and 5.4 kilobases (kb) in length was necessary for urease activity and was located within an 18.5-kb EcoRI fragment. The operon was induced by urea and encoded a multimeric, cytoplasmic enzyme comprising subunit polypeptides of 8,000, 10,000, and 73,000 daltons that were encoded by a single polycistronic mRNA and transcribed in that order. Seventeen urease-negative transposon insertions were isolated that synthesized either none of the structural subunit polypeptides, the 8,000-dalton polypeptide alone, or both the 8,000- and 10,000-dalton subunit polypeptides. The molecular weight of the native enzyme was estimated to be 212,000 by Superose-6 chromatography. Homologous sequences encoding the urease of Providencia stuartii synthesized subunit polypeptides of similar sizes and showed a similar genetic arrangement. However, restriction maps of the operons from the two species were distinct, indicating significant divergence.Proteus mirabilis urease: genetic organization, regulation, and expression of structural genes.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC211300/ĪĪ
ĪĪ
ĪĪ