ĪĪ
Viral-induced FasL kills viral-infected cells and immune cells
1. Fas (also called Apo1 or CD95) is a death domain-containing member of the TNFR
2. The Fas receptor induces an apoptotic signal by binding to FasL expressed on the surface of other cells
3. death receptor ligands include tumor necrosis factor (TNF) and Fas ligand (FaSL), that trigger apoptosis by binding to cell surface receptors. But TNF also activates survivial pathways.
4. antigen-presenting cells such as monocyte-derived human macrophages (MDM) but not monocyte-derived dendritic cells express basal levels of FasL.
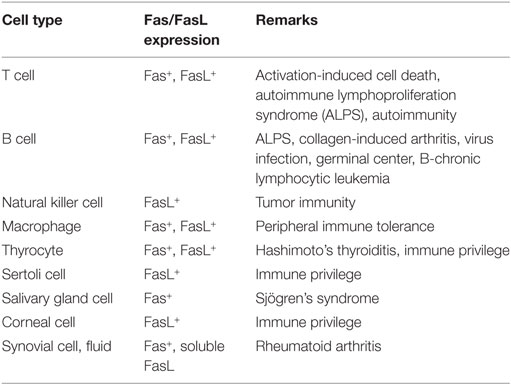
5. FasL as a cytotoxic molecule expressed by activated T cells6. FasL-expressing T cells kill the Fas-expressing activated B cells
7.FasL in activated T cells and NK (natural killer ) cells works as an effector of CTL and NK cells to remove the cells infected by virus, or cancerous cells.
8.FasL expression on activated T cells is induced by stimulation via TCR, costimulatory molecules, and cytokine receptors (65). FasL expression is regulated by several transcription factors, including NF-”╩B, nuclear factor of activated T cells (NF-AT), early growth response gene family transcription factors, c-Myc, AP-1, secretory protein-1, and interferon regulatory factors (66©C72).
9.FasL expression on the cell surface is specific to the immune system.
10. activation of mouse B cells leads to the expression of FasL and killing of Fas-expressing target cells by B cells
11. HSV-infected human monocytic cells were able to kill Fas-positive human CD4+ T cells, CD8+ T cells, and natural killer (NK) cells in in vitro co-culture assays.
12. Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death
13. immune previlage sites: cornea, testis which constitutely express FasL
ĪĪ
https://www.frontiersin.org/articles/10.3389/fimmu.2017.01220/full
ĪĪ
ĪĪ
Induction of Fas Ligand Expression by HIV Involves the Interaction of Nef with the T Cell Receptor ”Ų Chain | Journal of Experimental Medicine | Rockefeller University Press
https://rupress.org/jem/article/189/9/1489/25871/Induction-of-Fas-Ligand-Expression-by-HIV-InvolvesĪĪ
ĪĪ
ĪĪ
ĪĪ
ĪĪ
Figure 3Three Types of Killing by the Fas and FasL System
http://www.cell.com/fulltext/S0092-8674(00)81874-7
ĪĪ
http://www.lookfordiagnosis.com/mesh_info.php?term=Fas+Ligand+Protein&lang=1
ĪĪ
Fas (also called Apo1 or CD95) is a death domain-containing member of the TNFR (Tumor Necrosis Factor Receptor) superfamily. It has a central role in the physiological regulation of programmed cell death and has been implicated in the pathogenesis of various malignancies and diseases of the immune system. Although the FasL (Fas Ligand)-Fas system has been appreciated mainly with respect to its death-inducing function, it also transduces proliferative and activating signals through pathways that are still poorly defined. The Fas receptor induces an apoptotic signal by binding to FasL expressed on the surface of other cells. Fas is a Type-I transmembrane protein, where as FasL a Type-II transmembrane protein of TNF family and can be shed in a soluble form by action of metalloproteinase (Ref. 1).Fas Signaling Pathway | Thermo Fisher Scientific - CN
https://www.thermofisher.com/cn/zh/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/fas-signaling-pathway.htmlĪĪ
Prevention of immune cell apoptosis as potential therapeutic strategy for severe infections.
Parrino J, Hotchkiss RS, Bray M - Emerging infectious diseases (2007)
Some labile cell types whose numbers are normally controlled through programmed cell death are subject to markedly increased destruction during some severe infections. Lymphocytes, in particular, undergo massive and apparently unregulated apoptosis in human patients and laboratory animals with sepsis, potentially playing a major role in the severe immunosuppression that characterizes the terminal phase of fatal illness. Extensive lymphocyte apoptosis has also occurred in humans and animals infected with several exotic agents, including Bacillus anthracis, the cause of anthrax; Yersinia pestis, the cause of plague; and Ebola virus. Prevention of lymphocyte apoptosis, through either genetic modification of the host or treatment with specific inhibitors, markedly improves survival in murine sepsis models. These findings suggest that interventions aimed at reducing the extent of immune cell apoptosis could improve outcomes for a variety of severe human infections, including those caused by emerging pathogens and bioterrorism agents.
F1: Apoptotic pathways of cell death. The extrinsic pathway is mediated by a variety of death receptor ligands, including tumor necrosis factor (TNF) and Fas ligand (FaSL), that trigger apoptosis by binding to cell surface receptors. In the intrinsic pathway, several adverse factors act upon mitochondria to cause loss of the mitochondrial membrane potential, resulting in leakage into the cytosol of cytochrome C (Cyto C), which together with apoptotic protease activating factor 1 forms the apoptosome that activates caspase-9. Communication between the pathways exists through cleavage of Bcl-2 interacting domain (Bid) by active caspase-8 to form truncated Bid (tBid). Inhibitors of apoptosis (IAPs) can prevent caspase activation under certain conditions. Trail, tumor necrosis factor-”┴©Crelated apoptosis-inducing ligand; Bim/Puma, Bcl-2 interacting mediator of cell death/p53-upregulated modulator of apoptosis; FADD, Fas-associated death domain; FLIP, Fas-associated death domain-like interleukin-1”┬ converting enzyme-like inhibitory protein.Apoptotic pathways of cell death. The extrinsic pathway | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC2725847_06-0963-F1&req=4ĪĪ
Viral Immunol. 2011 Feb; 24(1): 11©C26.
Herpes Simplex Virus Type 1-induced FasL Expression in Human Monocytic Cells and Its Implications for Cell Death, Viral Replication, and Immune Evasion
Alexandre Iannello,1,,2,,3,,6,* Olfa Debbeche,1,,2,,3,,6,* Raoudha El Arabi,1,,2,,3,,6 Suzanne Samarani,1,,2,,3,,6 David Hamel,2,,5,,6 Flore Rozenberg,7 Nikolaus Heveker,2,,4,,6 and Ali Ahmadcorresponding author1,,2,,3,,6
Author information Article notes Copyright and License information Disclaimer
1Laboratory of Innate Immunity, Montreal, Quebec, Canada.
2CHU-Sainte-Justine Research Center, Montreal, Quebec, Canada.
3Department of Microbiology and Immunology, Montreal, Quebec, Canada.
4Department of Biochemistry, Montreal, Quebec, Canada.
5Department of Pharmacology, Montreal, Quebec, Canada.
6University of Montreal, Montreal, Quebec, Canada.
7Facult©” de M©”decine Ren©” Descartes, Universit©” de Paris V, Paris, France.
Abstract
Herpes simplex virus type 1 (HSV-1) is a ubiquitously occurring pathogen that infects humans early in childhood. The virus persists as a latent infection in dorsal root ganglia, especially of the trigeminal nerve, and frequently becomes reactivated in humans under conditions of stress. Monocytic cells constitute an important component of the innate and adaptive immune responses. We show here for the first time that HSV-1 stimulates human FasL promoter and induces de novo expression of FasL on the surface of human monocytic cells, including monocytes and macrophages. This virus-induced FasL expression causes death of monocytic cells growing in suspension, but not in monolayers (e.g., macrophages). The addition of a broad-spectrum caspase inhibitor, as well as anti-FasL antibodies, reduced cell death but increased viral replication in the virus-infected cell cultures. We also show here for the first time that the virus-induced de novo expression of FasL on the cell surface acts as an immune evasion mechanism by causing the death of interacting human CD4+ T cells, CD8+ T cells, and natural killer (NK) cells. Our study provides novel insights on FasL expression and cell death in HSV-infected human monocytic cells and their impact on interacting immune cells.Introduction
Herpes simplex virus type 1 (HSV-1; hereafter referred to as HSV) is a ubiquitously occurring human herpes virus that infects humans early in life (reviewed in 1©C3). It is a member of the ”┴-Herpesviridae subfamily. Primary infections with the virus usually occur in early childhood and are mild or symptomless. However, infected humans can never eliminate the virus and become lifelong carriers. The virus travels from the oral and facial skin nerve endings to dorsal root ganglia, especially of the trigeminal nerve, where it becomes latent. The latent infections frequently become reactivated under conditions of stress, immunosuppression, physical trauma, or exposure to UV radiation (4). These reactivations are often manifested as painful blisters or Ī░cold soresĪ▒ at the mucocutaneous junctions of the lips. The condition is called herpes labialis. The virus may also infect the cornea and cause keratitis. These conditions cause considerable discomfort and represent a serious health problem. Primary and reactivated latent infections may rarely cause encephalitis, especially in neonates and immunocompetent persons with unknown defects of the immune system (3). HSV infection is the most common cause of sporadic infectious encephalitis in apparently healthy individuals. Effective anti-HSV drugs have been developed; however, the emergence of drug-resistant viruses has also been documented, particularly in immunocompromised individuals (reviewed in 5). Unfortunately, effective vaccines against the virus are not yet available.
Monocytes and macrophages represent important cellular elements of the immune system. In response to a viral infection, they release a variety of proinflammatory cytokines and chemokines, and recruit inflammatory cells to the site of infection. Activated macrophages phagocytose pathogens and immune complexes, and present viral antigens to other immune cells. Unlike epithelial cells, in which HSV prevents apoptosis and causes cell death with predominant features of necrosis, HSV infects monocytic cells with different degrees of permissiveness, and appears to induce their cell death via apoptosis (6©C8). However, little is known about the mechanism of this virus-induced apoptosis, or its consequences for antiviral immunity as well as for viral replication. We addressed these questions and show here that HSV infection causes apoptosis in human monocytic cells by inducing expression of FasL on their surface. Our data provide experimental evidence showing for the first time that the virus induces FasL at the transcriptional level by stimulating FasL promoter. Interference with this apoptotic pathway prevents cell death, but enhances viral replication. Furthermore, HSV-infected human monocytic cells were able to kill Fas-positive human CD4+ T cells, CD8+ T cells, and natural killer (NK) cells in in vitro co-culture assays. These observations provide valuable insights about the relevance of apoptosis to viral replication and immune evasion in this viral infection.Herpes simplex virus infection induces death of THP-1 cells, but not of U937 cells
In order to learn about the interactions of HSV-1 with human monocytic cells, we infected THP-1 and U937 cells in vitro with the virus. As shown in Fig. 1A, HSV-1 infection resulted in the death of THP-1 cells, compared to the mock-infected cells (80% versus 18%). Interestingly, HSV infection of U937 cells did not affect their viability significantly. In order to confirm that this effect on cells was due to HSV infection and not due to the presence of any other soluble factor in the viral preparation, we infected THP-1 cells with UV-irradiated HSV-1 or a filtered viral preparation. As shown in Fig. 1B, the infection of THP-1 cells with the UV-irradiated virus abrogated the virus-induced cell death, resulting in a percentage of the dead cells equivalent to that seen in the mock-infected cells. Furthermore, we passed the viral preparation through a 0.1-”╠m filter, which should have retained the viral particles present in the preparation. As shown in Fig. 1B, the 0.1-”╠m filter-passed preparation failed to cause death in THP-1 cells compared to a 0.45-”╠m filter-passed preparation, in which the virus was not retained. These results show that HSV infection affects the two human monocytic cell lines differently; it infects and kills THP-1 but not U937 cells.
FIG. 1.
Infection with HSV induces THP-1 cell death in a dose- and time-dependent manner. (A) THP-1 and U937 cells were infected with HSV (MOI = 5) or mock-infected for 2 h at 37ĪŃC. The cells were then washed and put in culture for 24 h. After 24 h, total cells were counted and dead cells were discriminated using trypan blue. (B) Cells were infected as described above with HSV, with the HSV preparation previously passed through 0.45-”╠M and 0.1-”╠M filters, or with UV-treated viruses. (C) THP-1 and U937 cells were infected with different concentrations of HSV (MOI) for 24 h. For each condition, total cells were counted and dead cells were discriminated using trypan blue coloration. (D) THP-1 and U937 cells were mock- or HSV-infected with a low MOI (0.1), and the cells were incubated at 37ĪŃC for different time periods. For each time point total cells were counted, and the percentage of dead cells was calculated using trypan blue. All the experiments were repeated at least three times (***p < 0.001).
We next investigated if the cell death observed in THP-1 cells was dose- and time-dependent. We infected THP-1 cells with HSV-1 in vitro at different MOI (ranging from 0.001 to 100) for different lengths of time. Our data show that the percentages of dead cells correlate with the quantity of virus used for the infection (Fig. 1C). The numbers of dead cells were not significantly different in U937 cells between infected and mock-infected cells at all MOI. In order to know if the virus-induced cell death was time-dependent, we infected THP-1 cells with a low MOI (0.1) and counted dead cells at different time points (6 to 72 h). The cell death started 12 h post-infection and continued increasing until 72 h post-infection. It is noteworthy that by 24 h post-infection, during which time the virus completes its first round of infection, most of the cell death has already occurred. On the contrary, cell death in U937 cells remained stable during the entire culture period (Fig. 1D).
The HSV-induced cell death in THP-1 cells occurs via apoptosis
We performed experiments in order to determine whether the cell death in the THP-1 cells was due to apoptosis or necrosis. For this purpose, we stained the cells with DAPI and examined them under a fluorescence microscope. As shown in Fig. 2A, the nuclei of HSV-infected THP-1 cells showed chromatin condensation characteristic of apoptosis. We also stained cells with PI and FITC-conjugated annexin V to discriminate between cell death caused by apoptosis and that caused by necrosis. As shown in Fig. 2B, THP-1 cells infected with the virus, but not U937 cells, became positive for the two markers. It is noteworthy that the cells undergoing apoptosis and necrosis differ from each other temporally with respect to the sequence of expression of these two markers. The cells undergoing necrosis become positive for annexin V and PI simultaneously. However, the cells undergoing apoptosis show a time lag between staining for these two markers. First, they show staining for annexin V, and become PI-positive only several hours later (18). In order to determine whether HSV-infected THP-1 cells undergo sequential (a characteristic of apoptosis) or simultaneous (a characteristic of necrosis) staining with FITC-annexin V and PI, we harvested these cells at different time points (3, 9, and 18 h) after the infection, stained them with the two markers and examined them by flow cytometry. In this experiment, we treated THP-1 cells with anti-Fas-agonistic antibody and used them as positive controls for apoptosis. We also treated these cells separately with TNF-”┴ and used them as positive controls for necrosis, as previously described (18). As shown in Fig. 2C, the virus-infected THP-1 cells followed a pattern of sequential staining with FITC-annexin V and PI, as was seen in the case of cells undergoing apoptosis due to treatment with anti-Fas antibodies. They shifted progressively from the lower left quadrant (PI-negative/FITC-annexin V-negative cells) to the lower right quadrant (PI-negative/FITC-annexin V-positive cells; t = 9 h), and with time to the upper right quadrant (PI-positive/FITC-annexin V-positive cells; t = 18 h). These data show that HSV-1 induced apoptosis of THP-1 cells.
FIG. 2.
HSV-1 infection induces THP-1 cell apoptosis. (A) THP-1 and U937 cells were infected (MOI = 5) and put in culture. After 24 h, the cells were permeabilized and DNA was stained with DAPI. The fluorescence was observed by confocal microscopy (arrows indicate chromatin condensation). (B) THP-1 and U937 cells were mock- and HSV-infected (MOI = 5) for 2 h at 37ĪŃC and put in culture at 37ĪŃC. After 18 h, the cells were harvested and stained with annexin-V FITC and propidium iodide. (C) THP-1 cells were treated with 100 ng/mL TNF-”┴ (to induce necrosis), and 1 ”╠g/mL of a monoclonal anti-Fas agonist antibody (to induce apoptosis), or infected with HSV-1 (MOI = 5) for 3, 9, and 18 h. At each time point, the cells were harvested and stained with annexin V-FITC and propidium iodide. (D) THP-1 cells were infected with HSV (MOI = 0.1) or mock-infected for 2 h at 37ĪŃC. The cells were then washed and put in culture for different time periods, with or without 10 ”╠g/mL of a broad-spectrum caspase inhibitor (Z-VAD). For each time point, the total cells were counted and the dead cells were discriminated using trypan blue. (E) For each time point, cell culture supernatants from HSV infection with or without 10 ”╠g/mL of Z-VAD and anti-FasL neutralizing antibodies were also harvested and titered for their contents in newly-produced HSV particles using a standard TCID50 titration method as described in the materials and methods section. The figure shows data from one representative experiment out of three (Col Ab means control antibody).
Inhibition of HSV-induced apoptosis and its effects on viral replication
Caspases are the main effector molecules that execute apoptosis in human cells. Therefore, we investigated whether a broad-spectrum cell-permeable caspase inhibitor (Z-VAD-fmk) could block HSV-induced cell death in THP-1 cells. As shown in Fig. 2D, addition of the inhibitor reduced virus-induced cell death significantly (p < 0.01) at all tested time points.
Since HSV-induced apoptosis could be inhibited by caspase inhibitors, we sought to determine the effect of this inhibition on viral replication. As shown in Fig. 2E, addition of a broad-spectrum caspase inhibitor enhanced HSV production in THP-1 cells at all time points examined. Furthermore, similarly to the effect of the caspase inhibitor, addition of anti-FasL-antagonistic antibodies also enhanced HSV production in THP-1 cells at all tested time points (Fig. 2E). These results show that preventing apoptosis in HSV-infected THP-1 cells enhances viral replication.
HSV replicates in THP-1 cells with lower efficiency than in Vero cells
In order to determine if HSV infection was productive and resulted in the release of infectious virions, we infected THP-1, U937, and Vero cells with a recombinant HSV-1, KGFP-gB, as described in the materials and methods section, and analyzed them for the expression of the virus-encoded gB-GFP by confocal microscopy. Vero cells were used as a positive control for the viral replication due to their high permissivity for productive HSV-1 infection. When the infected cells were examined 3 h post-infection, no fluorescence signal was observed in any of the cell types, suggesting that any residual virus from the inoculum could not have given false-positive results (data not shown). When examined 18 h post-infection, green fluorescence was observed in Vero and THP-1 cells, but not in U937 cells (Fig. 3A). The numbers of GFP-positive cells were significantly lower (p < 0.01) in the THP-1 cultures than in the Vero cultures (5.0 Ī└ 4.6% versus 86.0 Ī└ 10.6% positive cells, respectively). Furthermore, the intensity of the signal emitted by the gB-GFP was much lower in the virus-infected THP-1 cells than in the infected Vero cells (Fig. 3A). No fluorescence was observed in U937 cells. These data suggest that HSV-1 replicates less efficiently in THP-1 cells than in Vero cells.
FIG. 3.
HSV replication occurring in THP-1 cells is needed to induce their apoptosis. (A) THP-1, U937, and Vero cells were infected with a fluorescent HSV-1 (KGFP-gB) (MOI = 5) for 2 h at 37ĪŃC. The cells were then extensively washed and put in culture for 18 h. After this time, the cells were harvested and permeabilized, and DNA was stained with DAPI. The fluorescence was observed by confocal microscopy. The representative figures from three sets of experiments are shown. (B) THP-1, U937, and Vero cells were infected with HSV-1 (MOI = 5). After 24 h, supernatants (SN) of these cell cultures were harvested and titrated using the standard plaque-forming unit method as described in the materials and methods section. The fourth wash after infection was kept at ©C20ĪŃC and was used as control for the inoculum background. (C) THP-1, U937, and Vero cells were infected with HSV-1 (MOI = 5). After 24 h, supernatants of these cell cultures were harvested, filtered through a 0.45-”╠M filter, and treated or not with UV light before infection with fresh THP-1 cells for 24 hours. After the infection, all cells were harvested and dead cells were counted using trypan blue. The fourth wash was kept at ©C20ĪŃC and was also used as control for the inoculum background. The results from three different experiments are shown. (D) THP-1 cells were infected as described above with a fluorescent HSV-1 (KGFP-gB) (MOI = 5). The cells were then extensively washed and put in culture for 18 h with or without 100 ”╠M of acyclovir. After this time, the cells were permeabilized and DNA was stained with DAPI. The fluorescence was observed by confocal microscopy. In the same experiment, cells were also harvested, stained with annexin V-FITC and propidium iodide, and examined by flow cytometry (E). Similar results were obtained in three independent experiments. Color images available online at www.liebertonline.com/vim.
We also verified these results by measuring viral titers in the cell culture supernatants obtained from HSV-infected Vero, THP-1, and U937 cells. For this purpose, the cells were infected with the virus for 24 h, and the culture supernatants were titrated by plaque-forming assay as described in the materials and methods section. The cells were washed four times after the infection, and the fourth wash was also titrated to determine if residual HSV particles remained in the cell cultures. As shown in Fig. 3B, THP-1 cells produced three times fewer viral particles than Vero cells (0.9 Ī└ 0.1 Ī┴ 106 PFU/mL versus 2.6 Ī└ 0.1 Ī┴ 106 PFU/mL, respectively). These data show that THP-1 cells produce infectious virions, but were less permissive to viral replication than the Vero cells. We also collected cell-free culture supernatants from the three sets of infected cells and determined their ability to cause cell death in THP-1 cells. As shown in Fig. 3C, the culture supernatant-caused cell death correlated with their viral titers.
Viral replication is needed for HSV-induced apoptosis of THP-1 cells
In order to know if the viral replication was needed to induce apoptosis in THP-1 cells, we infected cells with and without adding acyclovir for 24 h. Acyclovir, a guanine analogue, is one of the most commonly used drugs against HSV infections (19). It inhibits replication of the virus by inhibiting the activity of viral DNA polymerase. As shown in Fig. 3D, no fluorescence was seen in THP-1 and Vero cells infected with the KGFP-gB virus when they were treated with 300 ”╠g/mL of acyclovir, compared to untreated cells. These data confirm the efficacy of treatment with acyclovir in abrogating viral replication in THP-1 and Vero cells. We treated HSV-infected THP-1 cells with this drug, stained them with FICT-annexin V and PI, and examined by flow cytometry. The drug inhibited virus-induced cell death (1% versus 41% dead cells; Fig. 3E). These data clearly show that viral replication is needed to induce apoptosis in THP-1 cells.
The virus-induced apoptosis in THP-1 cells is mediated by Fas/FasL interactions
In order to understand the molecular mechanisms involved in the HSV-induced cell death in THP-1 cells, we attempted to block apoptosis by using anti-FasL antibodies that inhibit Fas/FasL interactions. The addition of anti-FasL antibody significantly reduced apoptosis in HSV-1-infected cells compared to isotype-matched control antibodies (Fig. 4A). Only 29% of the infected cells incubated with anti-FasL antibody were positively stained with FITC-annexin-V, compared to 68% of cells treated with the isotype control antibody. The experiment was repeated and dead cells were counted using the trypan blue exclusion assay. As shown in Fig. 4B, the viability of THP-1 cells infected with HSV-1 and treated with 1 ”╠g/mL anti-FasL antibody was increased by almost 30% compared to the isotype-treated cells (43 Ī└ 0.17% versus 73 Ī└ 1%, respectively; p < 0.01). We conclude from these experiments that the Fas/FasL pathway represents one of the mechanisms involved in viral-induced THP-1 apoptosis.
FIG. 4.
The Fas/FasL apoptotic pathway is involved in HSV-1-induced THP-1 apoptosis. (A) THP-1 cells were mock- or HSV-1-infected (MOI = 5) as described above for 18 h at 37ĪŃC, with and without 1 ”╠g/mL of a neutralizing monoclonal anti-FasL (Anti-FasL) antibody. Mouse IgG (1 ”╠g/mL; Control Ab) was also used as a control. After 18 h, the cells were stained with annexin V-FITC and propidium iodide and analyzed by flow cytometry. The experiment was repeated three times and the figure shows results from a representative experiment. (B) This graph shows percentages of trypan blue©Cretaining dead THP-1 cells after 18 h of infection with the virus from three different experiments. (**p < 0.01; ***p < 0.001).
HSV-1 infection induces de novo expression of FasL expression on THP-1 cells
Based on these observations, we investigated whether HSV-1 infection induced FasL expression in THP-1 cells. For this purpose, we infected THP-1 and U937 cells for 18 h, and analyzed FasL expression by flow cytometry. As shown in Fig. 5A, the infection induced FasL expression on the surface of THP-1 cells, but not on U937 cells. Interestingly, no FasL expression was observed after treating HSV-infected THP-1 cells with acyclovir (Fig. 5B), suggesting that viral replication is required to induce the expression of FasL. These results also confirm our above-mentioned observations that the viral replication was needed to induce THP-1 cell apoptosis. We then investigated whether the viral infection was inducing expression of the FasL gene. For this purpose, we determined the effect of the viral infection on the transcription of a reporter gene placed under the control of the FasL promoter as described in the materials and methods section. As shown in Fig. 5C, the FasL promoter activity is significantly increased in HSV-infected cells compared to mock-treated cells (46,057 Ī└ 686 versus 6870 Ī└ 70 relative luciferase activity in arbitrary units, respectively; p < 0.01). Similarly to our flow cytometry data, no FasL promoter activity was observed after treating HSV-infected THP-1 cells with acyclovir. We also compared the levels of FasL expression in HSV- and mock-infected THP-1 cells by Western blots 18 h after infection. As shown in Fig. 5D, HSV-1 infection caused increased expression of FasL in the virus-infected cells. The results also suggest that both THP-1 and U937 constitutively express FasL intracellularly. Since FasL is known to be shed into culture medium by proteolytic cleavage of the surface-expressed FasL (20), we measured soluble (sFasL) using a commercial ELISA kit. In repeated experiments, no significant increase in the concentrations of the sFasL could be detected in the supernatants harvested from THP-1 and U937 cells that were mock- or HSV-infected (data not shown). Taken together, our data suggest that viral infection leads to FasL expression on the surface of THP-1 cells, which is implicated in the apoptosis of these cells. In separate experiments, we compared the expression of Bcl-2 and Bcl-XL between HSV-infected and mock-infected THP-1 cells 18 h post-infection by Western blots, and found very little difference between them (data not shown).
FIG. 5.
HSV-1 infection of THP-1 cells induces FasL expression. (A) THP-1 and U937 cells were infected with HSV-1 (MOI = 5). After 18 h, the cells were harvested and Fc-receptors were blocked with 1 ”╠g of mouse IgG. The cells were then stained using an anti-FasL PE-conjugated monoclonal antibody and analyzed by flow cytometry. (B) THP-1 and U937 cells were infected as described above and treated with or without 100 ”╠M of acyclovir. After 18 h, the cells were harvested and Fc-receptors were blocked with 1 ”╠g of mouse IgG before staining with a PE-conjugated anti-FasL monoclonal antibody. The cells were then analyzed by flow cytometry. Filled and empty histograms in A and B indicate staining with control and anti-FasL antibodies, respectively. (C) THP-1 cells were transfected with a FasL promoter-reporter construct containing the human FasL promoter region (©C511 before ATG), fused with the firefly luciferase gene. SV-40-luc (Positive control) and Basic-luc (Negative control) constructs were also transfected. Twelve hours after transfection, the cells were infected with HSV or the mock viral preparation with or without 100 ”╠M of acyclovir. After 18 h, the cells were washed and lysed in lysis buffer, and the luminescence was measured as described in the materials and methods section. (D) Cell lysates from THP-1 and U937 cells that were mock- (lanes 1 and 3) or HSV-infected (lanes 2 and 4) for 18 h were analyzed by Western blotting using anti-FasL monoclonal antibodies. Individual bands were quantified by densitometry, and the ratios between the band densities of the FasL proteins and GAPDH are also indicated in the figure panel below the Western blots. All experiments were repeated at least three times.
HSV infects and induces apoptosis in purified human monocytes via the Fas/FasL pathway
We further wanted to know whether HSV-1 infects and causes apoptosis in purified human monocytes. For this purpose, we isolated monocytes from PBMCs as described in the materials and methods section, and infected them with KGFP-gB and examined them under a fluorescence microscope. The virus underwent replicative cycles as evidenced by the expression of the protein GFP-gB (Supplementary Fig. 1; see online supplementary material at http://www.liebertonline.com). These results conclusively show permissivity of human monocytes to HSV. As shown in Fig. 6A, the viral infection caused apoptosis in these cells, and the neutralizing anti-FasL antibody significantly reduced this apoptosis in HSV-1-infected cells compared to control antibody-treated cells. Indeed, only 23% (7% + 16%) of the infected cells incubated with anti-FasL antibody were positively stained with annexin-V, compared to 92% (22% + 70%) of the infected cells alone, and 90% (23% + 67%) of the cells treated with the isotype control antibody. The experiments were repeated and dead cells were counted using the trypan blue exclusion assay. As shown in Fig. 6B, the viability of monocytes infected with HSV and treated with 1 ”╠g/mL of anti-FasL antibody was significantly increased (21 Ī└ 0.55% versus 86 Ī└ 3% for untreated and treated cells, respectively; p < 0.001). Furthermore, we also determined the expression of FasL on the surface of HSV-1-infected and mock-infected human monocytes. As shown in Fig. 6C, HSV-infected cells expressed FasL on their surface, and the caspase inhibitor significantly (p < 0.01) reduced cell death at 24, 48, and 72 h after infection (Fig. 6D). Taken together, these data show that HSV also induces apoptosis in human monocytes, and the Fas/FasL pathway represents the main mechanism of this apoptosis.
FIG. 6.
HSV infection of freshly isolated human monocytes induces their apoptosis via the Fas/FasL pathway. (A) Isolated human monocytes were mock- or HSV-infected (MOI = 5) with and without the addition of isotype-control and anti-FasL antibodies (1 ”╠g/mL each). After 18 h, the cells were stained with FITC-conjugated annexin V and propidium iodide, and analyzed by flow cytometry. (B) Cell death was also measured by counting dead cells using the trypan blue exclusion assay. The results from three different experiments are shown. (C) Monocytes were infected with HSV and treated with or without 100 ”╠M acyclovir. After 18 h, the cells were harvested and Fc-receptors were blocked with 1 ”╠g of mouse IgG. The cells were then stained using an anti-FasL PE-conjugated monoclonal antibody and analyzed by flow cytometry. The figure shows results from a typical experiment that was repeated three times. (D) Monocytes were infected with HSV at a low MOI (0.1), or mock-infected for 2 h at 37ĪŃC. The cells were then washed and put in culture for different time periods with or without 10 ”╠g/mL of a broad-spectrum caspase inhibitor (Z-VAD). The figure shows results from three different experiments. (***p < 0.001). Filled and empty histograms in C indicate staining with a control and anti-FasL antibodies, respectively.
In vitro infection with HSV induces FasL expression in infected cells, but not in bystander cells
We next wanted to know whether HSV-1 induces FasL expression in the virus-infected cells and/or in uninfected bystander cells. For this purpose, we infected THP-1 cells as well as freshly isolated monocytes from two healthy donors with KGFP-gB, and measured expression of FasL by flow cytometry on GFP-positive (infected) cells, and on GFP-negative (uninfected bystander) cells. In these experiments, productively-infected (GFP-positive) THP-1 cells and human monocytes, but not GFP-negative bystander cells, were found to express FasL (Fig. 7). The culture supernatants from these experiments did not differ in their soluble FasL content, when they were tested by a commercial ELISA kit (data not shown).
FIG. 7.
HSV induces FasL expression on the cell surface in infected, but not in uninfected bystander, cells. THP-1 cells and isolated human monocytes from two different healthy donors were infected with a fluorescent HSV-1 (KGFP-gB) at MOI of 1 for 2 h at 37ĪŃC. The cells were then extensively washed and cultured at 37ĪŃC in the incubator. After 18 h, the cells were incubated on ice with mouse IgG to block Fc receptors, and stained with a PE-conjugated anti-FasL monoclonal antibody, or with an isotype-matched control antibody. The stained cells were analyzed by flow cytometry. The left and right rectangles in the top panel show gated uninfected (GFP-negative) and infected (GFP-positive) cells. For each staining, 10,000 gated cells were analyzed for the expression of FasL. Dark solid-line histograms indicate staining with the PE-conjugated isotype-matched control antibody, and clear dashed-line histograms indicate staining with PE-conjugated anti-FasL antibody. Note the expression of FasL on the virus-infected, but not on uninfected bystander, cells.
HSV infection induces apoptosis in monocyte-derived macrophages
In vivo monocytes differentiate into macrophages, which play an important role in regulating inflammatory and immune responses in body tissues in response to pathogens. In order to investigate whether HSV infects and induces apoptosis in these cells, we generated macrophages from purified human monocytes as described in the materials and methods section. We infected the macrophages with HSV-1 or UV-irradiated HSV-1 for 24 h, and examined them with light microscopy to see if the virus was able to induce cytopathic effects in them. Interestingly, cytopathic effects (rounding, detachment, and cell death) were observed in the cells 24 h after infection with the virus, but not with the mock-infected or UV-inactivated viral preparations (Fig. 8A). In fact, many of the infected cells became detached and were floating in the culture medium, leaving visible empty surfaces in the cell monolayers, whereas the monolayers were intact in mock-inactivated HSV-1-infected cells. These data show that HSV is able to induce cell death in human macrophages. In contrast to monocytes, the addition of the antagonistic anti-FasL antibodies did not reduce cell death in HSV-infected monocyte-derived macrophages (data not shown), which suggests that the virus-induced cell death occurs due to viral replication, and was not due to Fas-FasL interactions. We further wanted to know whether monocyte-derived macrophages were permissive to HSV-1. For this purpose, we infected them with KGFP-gB and examined them under a fluorescence microscope. The virus underwent replicative cycles, as evidenced by the expression of GFP-gB and the shedding of virions into the culture medium, as was seen in the case of isolated human monocytes (Supplementary Fig. 2; see online supplementary material at http://www.liebertonline.com). We stained the macrophages 12 and 24 h post-infection, and determined the expression of FasL on their surface by flow cytometry. The infection induced expression of FasL on the surface of human macrophages (Fig. 8B). Western blots for FasL expression showed that mock-infected macrophages also expressed this molecule, and that HSV infection increased this expression (Fig. 8C). Finally, we determined whether HSV-infected and FasL-expressing human macrophages could induce death of Fas-positive human lymphocytes in co-cultures. For this purpose, we co-cultured purified human CD4+ T cells, CD8+ T cells, and NK cells in separate experiments with HSV or mock-infected macrophages. In separate wells, we also added anti-FasL or control antibodies to these co-cultures. After separating CD4+ T cells, CD8+ T cells and NK cells from these co-cultures, we determined their staining for anexin V and PI. As shown in Fig. 8D, HSV-infected macrophages induced apoptosis of Fas-positive human CD4+ T cells, CD8+ T cells, and NK cells. The death induced by the infected macrophages was inhibited by anti-FasL, but not by control, antibodies.
FIG. 8.
HSV-1 infection of human macrophages induces FasL expression and apoptosis of co-cultured Fas-positive cells. (A) Monocytes were isolated from PBMCs by their adhesion onto plastic dishes, and differentiated into macrophages by incubating them in RPMI 10% FCS, 5% human AB serum, and 2 ng/mL of GM-CSF. After 5 d, monocyte-derived macrophages were mock- or HSV-infected (MOI = 5) for 2 h at 37ĪŃC. Infection with UV-treated HSV-1 was performed as a control. After 2 h of infection, the cells were washed and put in culture for 24 h. The cytopathic effect was observed by light microscopy after 24 h of culture. (B) Macrophages were infected with HSV for 12 and 24 h as described above. The cells were then harvested and Fc-receptors were blocked with 1 ”╠g of mouse IgG. The cells were then stained using a PE-conjugated anti-FasL monoclonal antibody and analyzed by flow cytometry. Filled and empty histograms indicate staining with the control and anti-FasL antibodies, respectively. (C) Cell lysates from macrophages mock-infected (lane 1) or HSV-infected (lane 2) for 24 h were analyzed by Western blotting using a monoclonal anti-FasL antibody. Individual bands were quantified by densitometry, and the ratios between the band densities of the FasL and GAPDH proteins are also indicated. (D) Macrophages were mock-infected or HSV-infected (MOI = 5) for 15 h as previously described, washed with PBS to remove unbound dead cells, and fixed with PBS and 2% paraformaldehyde for 30 min. Purified autologous human NK cells, CD4+ T cells, or CD8+ T cells, with or without 1 ”╠g/mL anti-FasL or control antibodies (mouse IgG, Col Abody) were then added to the monolayers. The cells were allowed to settle and remain in contact with the monolayers for 4 h. Thereafter, floating cells were harvested by gentle washing of the monolayers with PBS, stained with annexin V-FITC and propidium iodide, and analyzed by flow cytometry. Percentages of apoptotic cells as determined by annexin V and propidium iodide positivity are shown. Each part of the figure shows results of a typical experiment, each of which was repeated at least three times.
Discussion
We have shown here that HSV-1 productively infects human monocytes and macrophages, as well as a monocytic human cell line THP-1. These conclusions were reached using both a wild-type HSV-1, as well as a recombinant virus in which the coding sequences for green fluorescent protein (GFP) were fused in frame with those of the viral gB (12). It is noteworthy that gB is transcribed as a late (”├1) gene. GFP expression could clearly be seen under a confocal microscope in human monocytes, macrophages, and THP-1 cells. Furthermore, these cell types also produced infectious virions, as shown by the ability of their culture supernatants to infect Vero cells, which are known to be very permissive for replication of HSV-1. Previous studies showed that monocytes were resistant to HSV-1 infection, and only become susceptible to it upon differentiation towards macrophages (21©C23). Another study showed that human peritoneal macrophages become partially permissive to viral infection upon treatment with thioglycollate, and are fully permissive after prior infection with Corynebacterium parvum (24). We show here that freshly isolated human monocytes and monocyte-derived macrophages, as well as the monocytic cell line THP-1, are susceptible to infection with the virus, albeit with different degrees of permissiveness. The rate of virus replication is much lower in these cells than in Vero cells. Furthermore, we show here that another human monocytic cell line, U937, is not permissive to HSV-1 replication. These results are in agreement with those of Feng et al. (25), who showed that the virus did not replicate in U937 cells, but was able to induce activation of NF-”╩B in them. The cell line, however, also becomes permissive to viral replication upon differentiation with phorbol myristate acetate, but not with dimethyl sulfoxide or all-trans retinoic acid (23). The cell line expresses the herpesvirus entry mediator (HVEM), a specific receptor for gD of the virion. The viral glycoprotein gD binds HVEM and activates NF-”╩B (26). Collectively, these data suggest that freshly isolated human CD14+ monocytes, macrophages, and certain monocytic cell lines can be productively infected with HSV-1.
We show here for the first time that monocytic cells undergo apoptosis upon infection with HSV-1, and that this occurs due to the virus-induced expression of FasL on the surface of these cells. The addition of anti-FasL antibodies reduced virus-induced cell death in our study. Similar effects of the virus were reported earlier on human monocyte-derived macrophages (27,28). The infected macrophages were reported to induce death of interacting CD8+ T cells. The induction of FasL by these cells served as an immune evasion strategy. We extend these observations and show here for the first time that FasL-expressing HSV-infected macrophages also kill human CD4+ T cells and NK cells. In this regard another human virus, the human immunodeficiency virus type 1 (HIV-1), has also been reported to induce FasL expression on infected macrophages (28a).
Given the widespread expression of Fas on the surface of human cells, the HSV-induced expression of FasL on human monocytic cells may induce death of the virus-infected cells via apoptosis, and the infected cells may also kill Fas-positive immune cells (e.g., T cells, neutrophils, and NK cells), and thus evade the antiviral immune response. The first consequence may benefit the host by causing early death of the infected cells, and hence may result in reduced viral production. Indeed our results show that treating HSV-1-infected monocytic cells with a broad-spectrum caspase inhibitor reduces cell death, and increases viral titers in the culture medium. In this respect we have shown that the virus-induced apoptosis is a host beneficial response, as its inhibition with a broad-spectrum caspase inhibitor or with antagonistic anti-FasL antibodies decreases cell death, but increases viral replication. This conclusion is supported by an in-vivo study with HSV-2, a virus that is very closely related to HSV-1, showing that the virus causes more deaths and replicates to higher titers in mice lacking Fas or the FasL gene than in wild-type mice (29). The FasL-positive CD4+ T cells were shown to play a protective role in this study.
We also show here for the first time that HSV-1 enhances FasL expression in monocytic cells at the transcriptional level, as the infection stimulates FasL promoter and increases expression of the human FasL promoter-driven reporter gene. Interestingly, replication of the virus seems to be absolutely essential for the virus-mediated induction of FasL expression and cell death in human monocytic cells. Treating the infected cells with acyclovir, which inhibits viral DNA polymerase and viral DNA replication (19), inhibited FasL expression and prevented cell death. Furthermore, we show here that the virus induces de novo FasL expression on the surface of virus-infected, but not on uninfected bystander, cells.
It is noteworthy that in addition to monocytic cells, HSV-1 has been shown to induce FasL expression in other cell types. For example, the virus was shown to induce FasL expression and fratricide death in activated human CD8+ T lymphocytes (30). The virus induced FasL expression on various cell types in the eye when mice were infected via its anterior chamber. The infection resulted in enhanced apoptosis of various cell types in the eye and brain of the infected mice (31). A relatively recent study has shown that HSV-1 induces expression of FasL on neonatal, but not on adult, neutrophils (32). The study demonstrated hastened death of the virus-infected neonatal neutrophils that could be inhibited with antagonistic anti-Fas or anti-FasL antibodies. Interestingly, HSV-1 does not induce FasL expression in all cell types. For example, the infection induced cell death in immature dendritic cells not by inducing expression of FasL, but rather by causing enhanced proteasomal degradation of the long form of the cellular FILCE (or pro-caspase 8) inhibitory protein (c-FLIP-L) (33,34).
Several early and immediate early proteins of HSV-1 have been shown to induce apoptosis in different human cell types, although their exact mode of action remains unknown. The pro-apoptotic action of these early viral proteins is countered by subsequently-expressed gene products (e.g., ”┴-2, US3, and gJ). The net result is that HSV-1-infected cells become resistant to several exogenous death-inducing stimuli (e.g., osmotic shock, thermal shock, Fas, TNF-”┴ and C2 ceramide; 35©C37). Furthermore, a micro RNA encoded by exon 1 of the LAT gene of HSV-1 protects neuroblastoma cells from apoptosis by downregulating TGF-”┬ and SMAD3 expression (38). Interestingly, the resistance of HSV-1-infected cells to the death-inducing stimuli depends upon the cell type. Furthermore, the pro-apoptotic and anti-apoptotic effects of different viral proteins also varied with the cell type. It appears that these viral proteins exert their effects by interacting with different cellular factors. The differential expression of these factors in different human cells may be responsible for the differential effects of the viral proteins with respect to their effect on cell death (reviewed in 39).
We have shown here that HSV-1 replicates in human monocyte-derived macrophages (MDM), and induces de novo FasL expression on their surface. The virus-infected cells become rounded and detach from monolayers and die. The cell death in these cells could not be prevented by the addition of anti-FasL antibodies (unpublished data), despite the fact that these cells express FasL. The virus-infected cells retain their sensitivity to death by anti-Fas agonistic antibodies (unpublished data). The lack of Fas-FasL-mediated cell death in these cells may be due to the fact that they grow in monolayers and do not interact with each other via Fas-FasL. The inability of anti-FasL antibodies to reduce their death suggests that their ultimate death may be due to virus-induced changes occurring intrinsically, as happens in epithelial cells (35). We have demonstrated in this study that the virus-infected FasL-expressing macrophages are able to induce apoptosis in Fas-positive cells that interact with them. Thus the virus-induced expression of FasL may be more important for the infected cells in evading natural and virus-specific adaptive immunity of the host.
It is noteworthy that because of its strong capacity to kill Fas-positive cells, FasL is stored in secretory lysosomes in hematopoietic cells, and is translocated to the cell surface upon activation of the cell, or upon its interaction with a target cell (reviewed in 39). In line with this paradigm, our Western blot and flow cytometry results showed that THP-1 cells constitutively express FasL intracellularly, but not on the cell surface. The HSV-mediated de novo expression of FasL on the cell surface shows that, in addition to increasing FasL expression at the transcriptional level, the virus is also able to translocate intracellular FasL to the cell surface. Further studies are required to understand the mechanism behind the virus-induced translocation of FasL to the cell surface.
Overall, our study provides novel insights into HSV-induced apoptosis in human monocytic cells and its impact on viral replication and antiviral immunity.Herpes Simplex Virus Type 1-induced FasL Expression in Human Monocytic Cells and Its Implications for Cell Death, Viral Replication, and Immune Evasion
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3117309/ĪĪ
Apoptosis by Death Factor
Shigekazu Nagata
Department of Genetics, Osaka University Medical School, 2-2 Yamada-oka, Suita, Osaka,565, Japan
Introduction
There is an old Japanese saying that Ī░Once we are in the land of the living, we will eventually die.Ī▒ This is true, not only for human beings, but also for the cells that constitute our bodies. By repeated cell division (mitosis) and differentiation, a fertilized egg produces billions of cells to create our bodies. During this process, many surplus or harmful cells are generated, and they must be removed or killed (Jacobson et al., 1997 [this issue of Cell ]. For example, thymocytes that have failed to rearrange their T cell©Creceptor gene, or whose T cell receptor may recognize their own tissues, must be eliminated. The magnitude of the cell death is staggering: more than 95% of thymocytes die in the thymus during maturation. Even in adults, senescent cells are removed and replaced by newly generated cells to maintain homeostasis. The cell death that occurs during embryogenesis, metamorphosis, endocrine-dependent tissue atrophy, and normal tissue turnover is Ī░programmed cell death,Ī▒ mediated by a process termed Ī░apoptosis.Ī▒
Here, I focus on apoptosis controlled by cytokines. Two death factors, Fas ligand (FasL) or tumor necrosis factor (TNF), bind to their receptors and induce apoptosis, killing the cells within hours. In a classic definition of apoptosis, cells die by Ī░suicide;Ī▒ that is, cells programmed to die would do so autonomously. However, the identification of death factor©Creceptor pairs that regulate apoptosis indicates that apoptosis can also be controlled by an external killer in some instances.
Death Factor and Receptor
Fas Ligand and the TNF Family
Cytokines are a family of proteins that regulate cellular proliferation and differentiation by binding to their specific receptors on target cells. Cytokines are grouped into at least three subfamilies based on structure, cysteine-knot growth factors, tumor necrosis factor, and helical cytokines. FasL belongs to the TNF family (66, 52), which includes TNF, lymphotoxin, CD30 ligand, 4-1BB ligand, CD40 ligand, CD27 ligand, and TRAIL (TNF-related apoptosis-inducing ligand). FasL is synthesized as a type II©Cmembrane protein; that is, its N terminus is in the cytoplasm and its C-terminal region extends into the extracellular space. The extracellular region of about 150 amino acids is well conserved (20©C25%) among members of the TNF family, while the length and sequence of the cytoplasmic segments differ significantly.
Proteolysis of membrane-associated TNF produces soluble TNF. The proteolysis is mediated by a membrane metalloproteinase (Gearing et al. 1994). Similarly, membrane-bound FasL undergoes metalloproteinase-mediated proteolytic cleavage to generate soluble cytokine (Tanaka et al. 1996). A specific metalloproteinase inhibitor blocks the processing of TNF as well as FasL, suggesting that a similar enzyme cleaves TNF and FasL. Since the CD40 ligand is also cleaved off from the membrane to become soluble, it is likely that all TNF family members are processed to a soluble form. The soluble form of human FasL is functional, but mouse FasL loses its activity when it is cleaved from the membrane. Furthermore, membrane-bound TNF is more active than soluble TNF in activating the type II TNF receptor (Grell et al. 1995). These results may indicate that FasL and TNF work locally via cell©Ccell interactions under physiological conditions and that the purpose of shedding TNF or FasL is to attenuate the process.
The functional, soluble forms of TNFs as well as human FasL exist as trimers (Tanaka et al. 1997). It has not yet been demonstrated whether membrane-bound TNF or FasL are trimers. However, lymphotoxin ”┬, a member of the TNF family, consists of a heterotrimer of one ”┴ (lymphotoxin-”┴, or TNF”┬) and two ”┬ chains (lymphotoxin-”┬) on the membrane (Androlewicz et al. 1992), suggesting that membrane-bound TNF and FasL have the potential to form trimeric structures. X-ray diffraction analyses of TNF”┴ and TNF”┬ have indicated that each monomer forms an elongated, antiparallel ”┬-pleated sheet sandwich with a jelly roll topology (Jones et al. 1989). Amino acids conserved among members of the TNF family are mainly within the ”┬ strands. Computer-assisted modeling of FasL based on the amino acid sequence suggests that FasL has a similar tertiary structure to TNF”┴ and TNF”┬.
The Fas and TNF Receptor Family
Fas (also known as APO-1 or CD95), the receptor for FasL, is a type I©Cmembrane protein (33, 54) and a member of the TNF receptor (TNFR) family, which includes two TNFRs (TNFR1 and TNFR2), the receptor for lymphotoxin-”┬, the NGF receptor (p75), CD40, CD27, and CD30 (Nagata and Golstein 1995). This family is still growing, and three new members have recently been identified. They are human DR-3 (death receptor-3)/Wsl-1 (13, 39), human HVEM (herpes virus early mediator) (Montogomery et al. 1996), and chicken CAR1 (cytopathic avian leukosis-sarcoma virus receptor) (Brojatsch et al. 1996). The extracellular region of the TNF receptor family members carries 2©C6 repeats of a cysteine-rich subdomain that has about 25% similarity among various members. In contrast, the cytoplasmic regions have little similarity among the members, except for Fas, TNFR1, DR-3/Wsl-1, and CAR1, as discussed below.
TNF induces apoptosis and activates the transcription factor NF-”╩B. It can also stimulate the proliferation of thymocytes. Although both TNFR1 and TNFR2 can transduce the signal for apoptosis and NF-”╩B activation, TNFR1 is responsible for these signals in most cases (Vandenabeele et al. 1995). On the other hand, TNFR2 but not TNFR1 is responsible for the TNF-induced proliferation signal in thymocytes. Binding of FasL to Fas or cross-linking Fas with agonistic antibodies (IgM class anti-Fas antibody, or IgG3 class anti-APO1 antibody) induces apoptosis in Fas-bearing cells (74, 82, 33). Most other receptors in the TNF receptor family transduce activation or stimulatory signals, although some of them, such as CD40 and CD30, may also have the ability to inhibit growth, probably causing apoptosis. The presence of a homologous domain (about 80 amino acids) in the cytoplasmic regions of Fas and TNFR1 suggested that this region is responsible for transducing the death signal. In fact, subsequent mutational analyses in Fas and TNFR1 indicated that this is the case, and this domain has been designated a death domain (34, 71). DR-3/Wsl-1 also carries a death domain and has the potential to transduce an apoptotic signal, as well as to activate NF-”╩B (13, 39). CAR1, which also contains a death domain, has been shown to cause apoptosis in chicken cells when it is cross-linked by the envelope protein of ALSV (Brojatsch et al. 1996).
The death domain has a tendency to self-aggregate, and the tertiary structure of the Fas death domain, revealed by heteronuclear multidimensional NMR spectroscopy, shows that the death domain is a novel protein fold consisting of six antiparallel, amphipathic ”┴ helices (Huang et al. 1996). Many charged amino acids are present on the surface, which is probably responsible for mediating the interactions between death domains described below.
Signal for Apoptosis
Cascade Leading to ICE
Binding of ligand to a tyrosine kinase receptor, such as PDGF or EGF receptor, induces dimerization of the receptor and activates the intrinsic kinase activity in the cytoplasmic domain. The receptors for hematopoietic growth factors such as colony-stimulating factor and for interferons do not contain kinase domains in their cytoplasmic regions. Instead, the ligand-induced dimerization recruits a kinase(s) to the receptor and activates it, which then results in transduction of the proliferation and/or differentiation signals. In the case of Fas or TNFR1, however, dimerization with a divalent anti-Fas or TNFR1 monoclonal antibody is not sufficient to activate these receptors. Fas and TNFR1 must be oligomerized to be activated; that is, IgM class anti-Fas monoclonal antibody or IgG3 class anti-APO1 antibody that possess a tendency to aggregate function as potent agonists (74, 82). X-ray diffraction analysis of the TNF”┬©CTNF receptor complex has indicated that a TNF”┬ trimer makes a complex with three molecules of the extracellular region of the TNF receptor (Banner et al. 1993), suggesting that TNF induces trimerization of the receptor. The similarity between the structures of FasL and TNF and between Fas and the TNF receptors suggests that FasL also induces trimerization of Fas and that the trimerized cytoplasmic region then transduces the signal.
Fas- and TNFR1-mediated apoptosis occur in the presence of inhibitors of either RNA or protein synthesis (82, 33). Even enucleated cells undergo apoptosis upon Fas activation (Schulze-Osthoff et al. 1994), suggesting that all of the components necessary for apoptotic signal transduction are present and that Fas activation simply triggers this machinery. To dissect the signal-transducing machinery for Fas- and TNFR1-mediated apoptosis, two approaches have been used. In one approach, several groups have identified a molecule(s) that binds to the cytoplasmic region of Fas or TNFR1, while in the other, information gained from studying apoptosis in the nematode, C. elegans, was applied to the Fas and TNF system.
Utilization of the yeast two-hybrid system with the Fas cytoplasmic region as bait led to the identification of a molecule called FADD (Fas-associating protein with death domain) or MORT1, which contains a death domain at its C terminus (8, 12). FADD/MORT1 is recruited to Fas upon its activation (Kischkel et al. 1995) and binds to Fas via interactions between the death domains. The N-terminal region (termed the death effector domain [DED] or MORT1 domain) is responsible for downstream signal transduction. A similar death domain©Ccontaining protein (TRADD, TNFR1-associated death domain protein) binds to TNFR1 (Hsu et al. 1995). But unlike FADD/MORT1, TRADD does not carry a death effector domain, and its death domain is responsible for mediating apoptosis. This apparent discrepancy between FADD/MORT1 and TRADD is resolved by the finding that TRADD binds to FADD/MORT1 via interactions between their death domains (Hsu et al. 1996b). These results suggest that Fas and TNFR1 use FADD as a common signal transducer and share the signaling machinery downstream of FADD/MORT1 (Figure 1). In addition to this pathway, TNFR1 has another pathway leading to apoptosis. RIP (receptor interacting protein), originally identified as a Fas-binding protein, preferentially binds to TRADD (Hsu et al. 1996a). RIP is a serine/threonine kinase containing a death domain and binds to TRADD via interactions between their death domains. RIP induces apoptosis when overexpressed. The death domain of RIP, but not its kinase domain, is responsible for transduction of the death signal, indicating that RIP does not possess a death effector domain, but rather another downstream effector molecule may be recruited through the death domain of RIP (see below) (Figure 1).ĪĪ
ĪĪ
Figure 1 Models for Apoptosis Signaling by Death Factors
Show full caption
View Large Image Figure ViewerDownload Hi-res image Download (PPT)
Figure thumbnail fx1b
Figure 1Models for Apoptosis Signaling by Death Factors
Show full caption
View Large Image Figure ViewerDownload Hi-res image Download (PPT)
To find the signaling molecule downstream of FADD/MORT1, Wallach and his associates again used the yeast two-hybrid system, using the N-terminal DED/MORT1 domain of FADD/MORT1 as bait (Boldin et al. 1996). At the same time, a collaborative group, led by Dixit and Peter, continued the biochemical characterization of molecules recruited to the activated Fas receptor (Muzio et al. 1996). Both groups identified the same molecule, which was originally termed FLICE (FADD-like ICE) or MACH (MORT1-associated CED-3 homologue) and is now designated caspase-8 (Alnemri et al. 1996) (Table 1). Caspase-8 carries two DED/MORT1 domains at the N-terminal region, through which it binds FADD/MORT1. The C-terminal region of caspase-8 is related to ICE family members, more specifically, to members of the caspase-3 (CPP32) subfamily, and recombinant caspase-8 preferentially cleaves caspase-3 substrates over caspase-1 (ICE) substrates (Boldin et al. 1996).
Table 1Human ICE Protease Superfamily
Proteases Alternative Names Recognition Sequence Substrates
Math Eq ICE YVAD pro-IL1”┬, pro-caspase 3 and 4
caspase-4 ICErel-II, TX, ICH-2
caspase-5 ICErel-III, TY
Math Eq ICH-1 PARP
caspase-9 ICE-LAP6 PARP
Math Eq CPP32, Yama, apopain DEVD PARP, DNA-PK, SRE/BP, rho-GDI
caspase-6 Mch2 VEID lamin A
caspase-7 Mch3, ICE-LAP3, CMH-1 PARP, pro-caspase 6
caspase-8 FLICE, MACH, Mch5
caspase-9 ICE-LAP6, Mch6 PARP
caspase-10 Mch4
The caspase family members can be divided into three subfamilies: caspase-1 (ICE), caspase-2 (ICH-1), and caspase-3 (CPP32), according to Alnemri et al. 1996.
Open table in a new tab
Figure 1 presents the current model for Fas- and TNFR1-mediated apoptosis. Binding of a trimeric FasL to Fas induces trimerization of Fas, and FADD/MORT1 binds to the trimerized Fas cytoplasmic region through the interaction of the respective death domains. Caspase-8 is then recruited to FADD/MORT1 through binding of the DED domains, which in turn may induce self-activation of the protease domain. One apoptotic pathway from TNFR1 uses caspase-8 pathway through the interaction of TRADD with FADD/MORT1. TRADD additionally recruits RIP, which may trigger a second apoptotic pathway. The recently identified DR-3/Wsl-1 receptor is more similar to TNFR1 than to Fas. That is, DR-3 binds TRADD, which then recruits FADD and RIP (13, 39). The apoptotic signaling pathway downstream of RIP is currently unknown. However, another death domain©Ccontaining adaptor, termed RAIDD (RIP-associated Ich-1/CED-3 homologous protein with a death domain) has recently been identified (Duan and Dixit 1997). RAIDD binds RIP through its death domain and recruits caspase-2 (Ich-1) to RIP. Although an involvment of RAIDD in the TNFR1 or DR3/Wsl-1-mediated apoptotic pathway has not yet been demonstrated, it is possible that RAIDD plays a role in transducing an apoptotic signal from one of the death receptors.
The signal from Fas seems to be restricted to apoptosis, whereas other members of the TNF receptor family including TNFR1 activate NF-”╩B. NF-”╩B activation by TNF receptor family members is mediated by TRAF (TNF receptor©Cassociated factor) family (Rothe et al. 1994). So far, five members have been identified in this family, and all contain a TRAF domain of about 230 amino acids. Among members of this family, TRAF2 binds directly to TNFR2 and CD30 and indirectly to TNFR1 through TRADD and RIP. A dominant-negative TRAF2 blocks TNF-induced NF-”╩B activation, but not apoptosis (Liu et al. 1996). Instead, blocking NF-”╩B activation with the dominant-negative TRAF2 potentiates the cytotoxic activity of TNF in various cell types, suggesting that NF-”╩B activation leads to the expression of a protein(s) that inhibits TNF-induced cytotoxicity. NF-”╩B consists of two subunits (p50 and p65) and exists in a complex with I”╩B in resting cells. The signal from TRAF2 results in phosphorylation of I”╩B and subsequent degradation by the proteosome. NF-”╩B, thus released from I”╩B, enters the nucleus and activates various genes carrying the NF-”╩B response element. Cells lacking one component of NF-”╩B (65 kDa) or expressing I”╩B mutants that cannot be phosphorylated are more sensitive to TNF-induced cytotoxicity, confirming that one of the target genes for NF-”╩B is a gene encoding a survival factor (5, 46, 75, 78). These results are in good agreement with the fact that Fas, which cannot activate NF-”╩B, mediates a stronger apoptotic signal than TNFR1, which can activate NF-”╩B. The cytotoxicity of TNF can be potentiated by cycloheximide or actinomycin D, which is probably due to the inhibition of the NF-”╩B-induced gene expression.
ICE Protease Cascade
Genetic analysis of programmed cell death in C. elegans has revealed a number of gene products that regulate the cell death process (Ellis et al. 1991). Among them, the CED-3 product is required for cell death, and molecular cloning of the ced-3 gene revealed it to be a homologue of mammalian ICE (interleukin-1”┬ converting enzyme) (Yuan et al. 1993), which converts the IL-1”┬ precursor to the mature form. ICE is a cysteine protease consisting of two large (p17) and two small (p10) subunits, which are generated by proteolytic cleavage of the ICE precursor (a zymogen). Cross-hybridization with ICE cDNA and a search of the human genome database revealed at least 10 ICE homologues (see Table 1), which are divided into three subgroups (ICE-like, CPP32-like, and Ich1-like proteases), based on their sequence homology (Alnemri et al. 1996). All of these cause apoptosis when overexpressed in cells. They appear to be cysteine proteases, containing conserved sequences for substrate binding and catalysis; they cleave their substrates after aspartic acid. Therefore, they are now designated as caspases (cysteine aspases) (Table 1) (Alnemri et al. 1996). So far, recognition sequences for three ICE family members have been identified. That is, caspase-1 (ICE) recognizes the sequence Tyr©CVal©CAla©CAsp (YVAD) in the proform of IL-1”┬, caspase-3 (CPP32/Yama/apopain) recognizes Asp©CGlu©CVal-Asp (DEVD) and cleaves poly(ADP-ribose) polymerase, and caspase-6 (Mch2) recognizes Val©CGlu©CIle©CAsp (VEID) and cleaves lamin (53, 67). However, it is uncertain whether each ICE family member has a specific substrate for mediating apoptosis, or if some members of the subfamily are redundant, cleaving the same substrates. In this regard, it is noteworthy that caspase-1-null mice do not show any phenotype in programmed cell death (Li et al. 1995), while the mice lacking caspase-3 show hyperplasia and disorganized cell development in the brain (Kuida et al. 1996). These results suggest that caspase-1 is redundant in all cell types, while caspase-3 plays a major role in apoptosis in some cells of the brain.
Using what was known about the specific recognition sequences of the ICE proteases, specific competitive inhibitors and fluorescent substrates for caspase-1 and -3 have been designed (Thornberry et al. 1992). In addition, several proteins encoded by viral genes are known to inhibit members of the ICE family. These include crmA, a cytokine response©Cmodifier gene encoded by cowpox virus, and p35, coded for by Baculovirus. These viral proteins seem to inhibit protease activity by forming a stable complex. p35 has a broader specificity for ICE family members than crmA. That is, crmA preferentially inhibits caspase-1 over caspase-3, while p35 inhibits both caspase-1 and -3 equally well.
Inhibitors of caspase-1 or -3 block Fas- and TNF-induced apoptosis, which suggests that both cas- pase-1- and caspase-3-like proteases are involved in Fas- and TNFR1- mediated apoptosis (19, 47, 72, 20). Monitoring the protease activity with specific fluorescent substrates for caspase-1 and -3 demonstrates that a caspase-1-like protease is transiently activated, whereas the activation of a caspase-3-like protease gradually increases during Fas-induced apoptosis (Enari et al. 1996). A similar sequential activation of caspase-1- and caspase-3-like proteases was also found in vivo. When agonistic anti-Fas antibody was administered to mice, the livers were damaged (Ogasawara et al. 1993). As the damage proceeded, caspase-1-like activity was detected in the liver, followed by the gradual activation of a caspase-3-like protease (Rodriguez et al. 1996a). The activation of the caspase-3-like protease is dependent on the activation of a caspase-1-like protease (Enari et al. 1996), indicating that these proteases are sequentially activated. This sequential activation can also be seen in a cell-free system. That is, cell lysate from Fas-activated, but not from nonactivated cells, induced apoptotic morphological changes in intact nuclei (Enari et al. 1995a). However, when the cell lysates from growing, nonapoptotic cells were supplemented with recombinant caspase-1 or -3, the lysates induced apoptosis. This caspase-1-induced apoptosis was inhibited, not only by an inhibitor of caspase-1, but also by the inhibitor of caspase-3 (Enari et al. 1996), confirming the sequential activation of caspase-1- and caspase-3-like proteases. It is likely that other members of the ICE family are also activated in the cascade, cleaving their Ī░death substratesĪ▒ such as lamin, actin, poly(ADP)ribose polymerase, rho-GDI, SREBP, and DNA-dependent protein kinase, to cause the apoptotic morphological changes observed on cells and nuclei, as well as chromosomal DNA degradation.
As discussed above, Fas engagement recruits caspase-8 to the Fas receptor complex. How can this result be integrated into the model of sequential ICE protease activation? Here, I suggest two models. In the first model, the oligomerization of caspase-8 through the interaction with FADD/MORT1 leads to its autocatalytic activation, which then triggers the protease cascade by cleaving the caspase-1-like protease zymogen. In the second model, oligomerization does not activate caspase-8, but a caspase-1-like protease activates the oligomerized caspase-8, which then sequentially activates other members of the ICE family. In addition to ICE family proteases, other proteases such as cathepsin D aspartic protease and the serine protease AP24 (apoptosis protease 24) may be involved in Fas- and TNFR1-induced apoptosis. To understand how these proteases may be involved in the apoptotic process, it will be necessary to biochemically characterize the purified or recombinant proteins and determine their specific substrates.
The Bcl-2 Family
ced-9, a homologue of the mammalian protooncogene Bcl-2, prevents programmed cell death in C. elegans (Hengartner and Horvitz 1994). Similarly, overexpression of Bcl-2 blocks apoptosis of mammalian cells that is triggered by a number of different stimuli such as factor deprivation, irradiation, c-myc, or anti-cancer drugs. A number of CED-9/Bcl-2 family members have been identified in mammals: Bcl-2, Bcl-xL, Bcl-w, and Mcl-1 inhibit apoptosis, whereas others, such as Bax, Bik, Bak, Bad, and Bcl-xs, activate apoptosis. The various Bcl-2 family members can dimerize with one another, with one monomer antagonizing or enhancing the function of the other. In this way, the ratio of inhibitors to activators in a cell may determine the propensity of the cell to undergo apoptosis (Yang and Korsmeyer 1996). For example, if either bcl-x (Motoyama et al. 1995) or bcl-2 (Veis et al. 1993) is disrupted in mice, the animals die as embryos or postnatally, respectively, as the result of excessive programmed cell death in particular organs. Conversely, if bax is disrupted, some normal programmed cell death fails to occur (Knudson et al. 1995). Another attractive mechanism to regulate dimerization of Bcl-2 family members is phosphorylation (Gajewski and Thompson 1996). For example, Bad, a proapoptotic member of the Bcl-2 family, is phosphorylated by a putative kinase that can be activated by growth factor engagement. The phosphorylated Bad loses the ability to bind Bcl-xL. Instead, it binds to 14-3-3, a protein that can interact with several signaling enzymes. The Bcl-xL dissociated from Bad now can execute its antiapoptotic function (Zha et al. 1996).
How does Bcl-2 or Bcl-xL inhibit apoptosis? Genetic studies of ced-9, ced-4, and ced-3 mutants in C. elegans indicate that ced-9 controls programmed cell death upstream of ced-4 and ced-3 (Shaham and Horvitz 1996). However, little is known about the biochemical mechanism whereby CED-9/Bcl-2 and their family members inhibit apoptosis. Bcl-2 and Bcl-x are localized to outer mitochondrial membranes and endoplasmic reticulum as well as nuclear membranes. The tertiary structure of Bcl-xL has been determined by X-ray and NMR analyses (Muchmore et al. 1996). It consists of two central, hydrophobic ”┴ helices, which are similar to the pore-forming bacteria toxins such as diphtheria toxin and the colicins, suggesting that Bcl-xL also generates pores in the membrane. When mitochondria are damaged by an agent that causes permeability transition, nuclear apoptosis is induced (Zamzami et al. 1996). This permeability transition of mitochondrial membrane, and thus nuclear apoptosis, is blocked by Bcl-2, suggesting that the membrane pores in the mitochondria, generated by the Bcl-2 family members, play an important role in apoptosis, at least in this system.
Bcl-2 and Bcl-xL can also inhibit Fas-mediated apoptosis in vitro as well as in vivo (35, 7, 58). Fas activation damages mitochondrial function, but the damage is inhibited by ICE protease inhibitors (Krippner et al. 1996). These results suggest that the mitochondrial damage is downstream of the ICE protease cascade in Fas-induced apoptosis and is probably a secondary effect. Thus, it is not clear how Bcl-2/Bcl-xL located in mitochondria can modulate the Fas-induced apoptotic signaling pathway that seems to take place in the cytoplasm. One possible mechanism is that the damage of mitochondria by ICE protease may amplify the signal by releasing apoptosis-inducing molecules (41, 84).
Other Regulators in the Signaling Pathway
Ceramide, generated by sphingomyelinases, increases during Fas- or TNFR1-mediated apoptosis, and ceramide itself can induce cell death (Spiegel et al. 1996). Since ceramide activates the ras/MAP kinase pathway, it was postulated that activated ras is responsible for apoptotic cell death. However, the recent observation that generation of ceramide and activation of JNK during Fas activation is blocked by ICE protease inhibitors suggests that the production of ceramide occurs downstream of the ICE protease cascade (23, 44). An increase in ceramide during Fas activation is likely to be one of the changes that accompanies apoptosis and is unlikely to be a mediator of apoptosis. Many other proteins have been suggested as regulators of Fas-mediated apoptosis. For example, c-abl tyrosine kinase, FAP tyrosine phosphatase, and small stress proteins (HSP24) inhibit the process, whereas the Fas-associated proteins of p59fyn kinase and FAF seem to augment apoptotic signal induced by Fas. How these proteins regulate the process is currently unknown.
Physiological and Pathological Roles of Fas
Down-Regulation of the Immune Reaction
Apoptosis occurs in various processes in mammalian life (Jacobson et al. 1997). What kinds of apoptosis are regulated by the Fas system? Fas is ubiquitously expressed in various tissues with abundant expression in the thymus, liver, heart, and kidney. On the other hand, FasL is predominantly expressed in activated T lymphocytes and Natural Killer (NK) cells, although it is also expressed constitutively in the tissues of the Ī░immune-privilege sitesĪ▒ such as the testis and eye, as described below. The mouse mutations, lpr (lymphoproliferation) and gld (generalized lymphoproliferative disease), are spontaneous recessive mutations (Cohen and Eisenberg 1991). Mice carrying homozygous mutations in lpr or gld develop lymphadenopathy and splenomegaly by accumulating CD4−CD8− cells of T cell origin, and some strains of mice develop autoimmune diseases. Genetic and molecular analyses of lpr and gld mutations showed that they are loss-of-function mutations in the Fas and FasL genes, respectively (79, 68). The Fas-null mice, established by gene targeting (Adachi et al. 1995), also show lymphadenopathy and splenomegaly (Figure 2), which is much more pronounced than in mice carrying the leaky lpr mutation. Furthermore, when Fas was expressed in the lymphocytes of lpr mice as a transgene, the lymphoproliferative phenotype was rescued (Wu et al. 1994), confirming that Fas plays a role in the programmed cell death of T lymphocytes.ĪĪ
Figure 2 When Apoptosis Fails
T lymphocytes, which are responsible for removing virally infected and cancerous cells, die at various stages of their development. Most immature T cells are useless (incorrect rearrangement of the T cell receptor) or potentially detrimental (self-reactive) to the organism. More than 95% of thymocytes that immigrate into the thymus are eliminated by positive and negative selection during their development. In the periphery, mature T cells that recognize self antigens are also deleted (peripheral clonal deletion). When mature T cells encounter target cells, they are activated to proliferate. However, after the activated T cells accomplish their task, they must be removed to avoid accumulation. Mature T cells from lpr or gld mice do not die after activation, and activated cells accumulate in the lymph nodes and spleens of these mice. When T cell hybridomas are activated in the presence of a Fas-neutralizing molecule, they do not die. These results indicate that Fas is involved in activation-induced suicide of T cells, i.e., in down-regulation of the immune reaction (Figure 3a) (Nagata and Golstein 1995). Peripheral clonal deletion may also be mediated by the Fas system, because the cells to be deleted in this process are activated by interactions with cells expressing self antigens. However, thymic clonal deletion is apparently normal in mice lacking the functional Fas system (lpr-, gld-, or Fas-null mice) (Singer and Abbas 1994), even though thymocytes abundantly express Fas and are sensitive to Fas- induced apoptosis. These results suggest that Fas is not involved in the deletion process in the thymus, although one cannot rule out the possibility that this process is mediated by redundant mechanisms.ĪĪ
ĪĪ
ĪĪ
Figure 3Three Types of Killing by the Fas and FasL System
ĪĪ
In addition to T cells, the Fas-deficient mice accumulate B cells and have elevated levels of immunoglobulins of various classes that include anti-ssDNA and anti-dsDNA antibodies (Cohen and Eisenberg 1991), suggesting an involvement of the Fas system in the deletion of activated or autoreactive B lymphocytes. In fact, immunization of mice with antigens rapidly induces Fas expression in germinal centers. Furthermore, the activation of naive B cells through CD40 sensitizes them to Fas-mediated apoptosis, while their costimulation through CD40 and Ig receptor makes them resistant (Rothstein et al. 1995). Although these results suggest that FasL-expressing T cells kill the Fas-expressing activated B cells, the precise mechanism and physiological role of Fas in the deletion of B cells remains to be studied.
Children carrying a defect in the Fas gene have also been identified (21, 56). Most of these patients carry a heterozygous mutation in the Fas gene. The affected Fas protein seems to work in a dominant-negative fashion, and T cells from the patients do not die upon activation. The patients show phenotypes (ALPS, autoimmune lymphoproliferative syndrome) that are remarkably similar to those of lpr mice, including lymphadenopathy, splenomegaly, and hypergammaglobulinemia. Some patients show autoimmune diseases such as hemolytic anemia, thrombocytopenia, and neutropenia by producing autoantibodies against red blood cells and platelets. On the other hand, the fathers or mothers of the patients, who also carry the heterozygous mutation of the Fas gene, do not show an abnormal phenotype, suggesting that the patients carry mutations in other complementing genes. Alternatively, Fas could be required only for the perinatal period, and the parents may also have had a similar phenotype in childhood that was rescued later, since the heterozygous mutation is leaky.
Effector of Cytotoxic T Lymphocytes and Natural Killer Cells
Cytotoxic T lymphocytes (CTL) recognize and kill cells infected by viruses or bacteria, while NK cells kill cancerous cells. The professional CTL are CD8+ T cells, but Th1-type CD4+ T cells also show cytotoxicity. How these CTL and NK cells kill target cells was under debate for a long time, because a well-known perforin/granzyme-based mechanism could not account for all of the examples of CTL killing. However, the identification of FasL as a cytotoxic molecule expressed by activated T cells resolved this problem. Studies with mice deficient in either perforin/granzyme or FasL indicated that the perforin/granzyme and FasL systems are major pathways for CTL-mediated cytotoxicity (Nagata and Golstein 1995). Activation of CTLs through T cell©Creceptor interaction with viral antigens induces the expression of the FasL gene. The FasL expressed on the surface of the effector cells binds to Fas on the target cell and causes apoptosis by activating caspases, as described above (Figure 3b). A similar activation of CTLs through T cell receptor would release perforins and granzymes that were stored in granules. It is believed that perforin makes pores in the plasma membrane of the target cells, through which granzymes are introduced. One of the granzymes (granzyme B) is a serine protease aspase, which activates some of the caspase family members by proteolysis (Darmon et al. 1995). Thus, although perforin/granzyme and FasL can independently trigger the cell death program, the processes leading to apoptosis are similar in both cases. The CD8+ T cells and NK cells use both the perforin/granzyme and FasL/Fas pathways, whereas the Th1-type CD4 T cells preferentially use the FasL system. Whether particular CD8+ T cells and NK cells have any preference for using the perforin/granzyme or FasL/Fas system on specific target cells remains to be studied. Furthermore, CTLs in mice deficient in both the perforin and FasL systems show some residual cytotoxicity in long-term assays (Braun et al. 1996), suggesting that yet another death factor(s), perhaps TNF or TRAIL, functions as a CTL effector under these conditions.
Immune Privilege
Cellular immune response reactions and their associated inflammatory responses can cause nonspecific damage to nearby tissues. Although most organs can tolerate such inflammation, some, such as the eye and testis, cannot. These organs, therefore, have a mechanism to protect themselves against dangerous and unwanted immune reactions. These organs are called Ī░immune privilege sitesĪ▒ and are able to support allogenic and xenogeneic tissue transplants. Initially, it was thought that immune privilege is maintained by preventing the activated cells from entering the organs. However, another attractive mechanism has recently been proposed (6, 26). That is, although the activated inflammatory cells can enter these organs, they are immediately killed by FasL expressed in the organs (Figure 3c). The constitutive expression of functional FasL has been found in the corneal epithelium and endothelium, iris, and ciliary cells of the eye, as well as in the Sertoli cells of the testis. When the eyes of wild-type mice were infected with herpes simplex virus (HSV-1), very few inflammatory cells were found associated with the retina, while massive inflammation was observed in the retina of gld mice, which have a defect in FasL. Furthermore, while testes expressing functional FasL survived when transplanted under the kidney capsule of allogenic animals, testis grafts from gld mice were rejected. These findings indicate that FasL accounts for at least part of the immune-privileged nature of the eye and testis and suggest a use for FasL as an immunosuppressive agent to target activated effector cells in transplantation. In fact, when islets of Langerhans were cotransplanted with syngeneic myoblasts expressing functional FasL, they were protected from immune rejection and were able to maintain normoglycemia for a substantial period in a mouse model system for diabetes (Lau et al. 1996). Similarly, several groups have recently found that some tumor cells become resistant to Fas-induced apoptosis and constitutively express FasL (27, 65). FasL expressed on tumor cells then counterattacks CTL and NK cells by binding Fas on their surfaces to cause apoptosis. This mechanism may also account for the ability of tumor cells to evade immune destruction.
A Double-Edged Sword
As long as death factors are appropriately expressed, they will be useful in maintaining homeostasis. However, if the system under- or over-functions, it will have deleterious effects. Loss of function causes hyperplasia, such as lymphoproliferation. The lymphocytes accumulated in patients carrying the heterozygous mutation in the Fas gene are not tumorigenic. However, the families of these patients sometimes have histories of Hodgkin's lymphoma (Fisher et al. 1995). The abnormal survival of the lymphocytes may allow the cells to accumulate mutations that lead to malignancy. Genes for death factors and their receptors, such as FasL and Fas, may therefore be regarded as tumor suppressor genes. On the other hand, when the system overfunctions, it causes tissue destruction and kills the animals. When an agonistic anti-Fas antibody or recombinant FasL was injected into mice to activate the Fas system in vivo, the mice were quickly killed by liver failure with symptoms similar to fulminant human hepatitis (55, 70). Fulminant hepatitis is known to be caused by abnormally activated T cells, and the transformation of hepatocytes with hepatitis B virus or hepatitis C virus causes the up-regulation of Fas expression. These results suggest that under normal circumstances, CTLs recognizing viral antigens expressed on the cell surface of infected hepatocytes are activated through the T cell receptor and kill the hepatocytes via the Fas/FasL system. If this killing process works properly, it benefits the organism. However, when the system is exaggerated, it may lead to fulminant hepatitis. It is possible that other CTL-induced autoimmune diseases such as graft-versus-host disease, AIDS, and insulitis are also mediated by the Fas system.
Various cancer patients produce TNF”┴ in a soluble form, and it works like a cachectin to induce systemic tissue damage. Similarly, the soluble form of FasL was found in the sera of patients with NK lymphoma or large granular lymphocytic leukemia (LGL) of the NK or T cell type (Tanaka et al. 1996). The leukemic cells themselves were found to express functional FasL on their surfaces. These patients often show systemic tissue damage such as hepatitis and neutropenia. Since hepatocytes and neutrophils are particularly sensitive to Fas-mediated apoptosis, it is possible that the systemic tissue damage observed in these patients is due to FasL in their serum or to FasL expressed on the circulating leukemic cells.
Conclusions and Perspectives
Many growth and differentiation factors regulate proliferation and differentiation of mammalian cells during development. So far, three death factors (TNF, FasL, and TRAIL) and four death factor receptors (Fas, TNFR1, DR3/Wsl-1, and CAR1) have been identified. Loss-of-function mutations in the Fas system, lpr and gld mice, illustrated the importance of this death factor system in maintaining mammalian homeostasis, specifically in the life and death of lymphocytes. It is possible that many more death factor and receptor systems that regulate apoptosis in a tissue-specific manner will be found in the future. Growth and differentiation signals are mediated by the phosphorylation and dephosphorylation of proteins, as well as by small second-messenger molecules such as cAMP and phosphatidyl inositol. These signals are reversible in most cases. On the other hand, the apoptosis signal triggered by death factors is irreversible; that is, a protease cascade is activated by the death signal, and the proteases cleave various cellular components, which leads to morphological changes of the cells and nuclei that are typical for apoptosis. Since other apoptosis-inducing agents also activate caspase family proteases, their signal transduction system may be similar or identical. However, the apoptotic system in mammals seems to be more complicated and sophisticated than that in C. elegans. Instead of the single ICE/CED-3 and the single Bcl-2/CED-9 in C. elegans, the mammalian genome carries at least ten members of the caspase family and nine members of the Bcl-2 family. Whether they are just redundant or have different roles remains to be examined. Biochemical analysis of each family member and establishment of mice lacking each member will clarify these points. Both TNF and FasL induce apoptosis. However, since TNF can induce other signals, such as activation of NF-”╩B, it was thought likely that the signaling pathway through the TNF receptor would be more complicated. In fact, identification of the molecules involved in TNF and Fas signaling indicates that the Fas-mediated signal is simpler than that of the TNF receptor. The TNF receptor shares a signal cascade with Fas in one apoptotic pathway, but it also activates additional signaling pathways including one that activates a survival signal. It will be interesting to examine what kinds of survival genes are activated by NF-”╩B and how these molecules inhibit apoptosis. Identification of these survival genes may provide clues as to why some tumor cells are resistant to various apoptosis-inducing agents including FasL, TNF, and anti-cancer drugs.
As described above, the Fas death factor system is a double-edged sword. If this system is properly regulated, it is useful for down-regulating the immune reaction and for removing virally infected as well as cancerous cells; but, if this system is exaggerated, it can cause tissue destruction. How can modulation of this system be applied to human diseases? The first obvious application is the killing of tumor cells, since some cancer cells, particularly some lymphoid tumors, express functional Fas. However, since the systemic treatment of patients with FasL will cause deleterious side effects, methods of local administration and/or proper targeting of FasL to the tumor should be devised. FasL can also be used as an immune-suppressive agent. As discussed above, the rejection of transplants is mediated by activated T cells. If a transplanted tissue is engineered to express FasL or is cotransplanted with FasL-expressing cells, the transplant may be tolerated. The other application of this system is to block FasL-induced tissue destruction. If Fas is shown to play a role in human diseases such as fulminant hepatitis, AIDS, and other diseases involving CTL-induced tissue destruction, then neutralizing antibodies against Fas or FasL, or other inhibitors of Fas-mediated apoptosis, would have potential as therapeutic agents.
Acknowledgements
I thank Drs. D. V. Goeddel and P. Golstein for helpful comments on the manuscript. I am grateful to all members of my laboratory in Osaka University Medical School and Osaka Bioscience Institute, and to Dr. O. Hayaishi for encouragement and discussion. I thank Drs. L. Migletta and G. Gray of Clarify Editing for careful editing of the manuscript. The work in my laboratory was supported in part by Grants-in-Aid from the Ministry of Education, Science, and Culture of Japan and by a Research Grant from the Princess Takamatsu Cancer Research Fund, and performed in part through Special Coordination Funds of the Science and Technology Agency of the Japanese Government. Because of limitations on the number of references, I cannot cite many important papers; I apologize to their authors.Apoptosis by Death Factor: Cell
https://www.cell.com/fulltext/S0092-8674(00)81874-7ĪĪ
J Clin Invest. 1998 Jun 1; 101(11): 2394©C2405.
The expression of Fas Ligand by macrophages and its upregulation by human immunodeficiency virus infection.
D H Dockrell, A D Badley, J S Villacian, C J Heppelmann, A Algeciras, S Ziesmer, H Yagita, D H Lynch, P C Roche, P J Leibson, and C V Paya
Division of Infectious Diseases, Mayo Clinic, Rochester, Minnesota 55905, USA.
Abstract
Fas/Fas Ligand (FasL) interactions play a significant role in peripheral T lymphocyte homeostasis and in certain pathological states characterized by T cell depletion.In this study, we demonstrate that antigen-presenting cells such as monocyte-derived human macrophages (MDM) but not monocyte-derived dendritic cells express basal levels of FasL.
HIV infection of MDM increases FasL protein expression independent of posttranslational mechanisms, thus highlighting the virus-induced transcriptional upregulation of FasL. The in vitro relevance of these observations is confirmed in human lymphoid tissue. FasL protein expression is constitutive and restricted to tissue macrophages and not dendritic cells. Moreover, a significant increase in macrophage-associated FasL is observed in lymphoid tissue from HIV (+) individuals (P < 0.001), which is further supported by increased levels of FasL mRNA using in situ hybridization. The degree of FasL protein expression in vivo correlates with the degree of tissue apoptosis (r = 0.761, P < 0. 001), which is significantly increased in tissue from HIV-infected patients (P < 0.001).
These results identify human tissue macrophages as a relevant source for FasL expression in vitro and in vivo and highlight the potential role of FasL expression in the immunopathogenesis of HIV infection.
The expression of Fas Ligand by macrophages and its upregulation by human immunodeficiency virus infection.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC508829/ĪĪ
Genet Mol Res. 2016 Jun 20;15(2). doi: 10.4238/gmr.15027607.
Fas-FasL expression and myocardial cell apoptosis in patients with viral myocarditis.
Huang TF1, Wu XH2, Wang X3, Lu IJ4.
Author information
Abstract
The aim of the current study was to investigate Fas and FasL expression and myocardial cell apoptosis in viral myocarditis patients.Human heart specimens were selected from patients who were autopsied between February 2012 and February 2015; of these, 25 patients were diagnosed with viral myocarditis. Another 15 cases with no diagnosis of myocarditis were selected for the control group. All tissue specimens were divided into two parts, one for reverse transcription-polymerase chain reaction analysis and the other for immunohistochemical and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) analyses.
In situ detection of apoptosis was performed by the TUNEL method, which revealed that myocardial cells from the viral myocarditis group exhibited significant apoptosis, whereas no apoptotic cells were observed in the control group.
The number of cells staining positive for Fas and FasL protein in the viral myocarditis group was significantly higher than that in the control group (P < 0.05). There was also a correlation between Fas and FasL protein expression levels and scores (r = 0.92, P < 0.05). The mRNA expression of Fas and FasL was significantly higher in the viral myocarditis group than in the control group (P < 0.05). In conclusion, the Fas-FasL system may be involved in the pathogenesis of viral myocarditis.
Furthermore, cytotoxic T lymphocytes may mediate cardiac muscle cells apoptosis via Fas-FasL signaling, and thus participate in the pathogenesis of viral myocarditis.
Journal of the American College of Cardiology
Volume 39, Issue 8, April 2002
EXPERIMENTAL STUDY
Role of Fas/FasL pathway in the activation of infiltrating cells in murine acute myocarditis caused by Coxsackievirus B3
Yoshinori Seko, Nobuhiko Kayagaki, Ken-ichiro Seino, Hideo Yagita, K.o Okumura and Ryozo Nagai
Author + information
Abstract
Objectives This study was designed to investigate the roles of Fas/FasL pathway in myocardial damage in murine acute myocarditis caused by Coxsackie virus B3 (CVB3).
Background Cardiac myocyte apoptosis rarely occurs in murine acute myocarditis caused by CVB3. Fas/FasL belong to the tumor necrosis factor receptor/ligand superfamily of costimulatory molecules and are known to play a critical role in the induction of apoptosis, as well as in the cytotoxicty mediated by T-cells and natural killer cells.
Methods We first analyzed the expression of Fas on cardiac myoctyes in vivo and in vitro. Second, we examined the development of myocardial damage, in C3H/He mice treated with an anti-FasL monoclonal antibody (mAb), and in C3H/He-lpr/lpr mice and C3H/He-gld/gld mice infected with CVB3. Third, to investigate the effects of anti-FasL mAb treatment on the activation of the infiltrating cells, we examined the expression of interferon (IFN)-gamma and interleukin (IL)-2 as activation markers in the heart of mice by semiquantitative polymerase chain reaction.
Results Fas was markedly induced on cardiac myocytes with acute myocarditis. Myocardial inflammation was decreased in mice treated with anti-Fas L mAb, C3H/He-lpr/lpr mice and C3H/He-gld/gld mice. Anti-FasL mAb-treatment also decreased the expression of IFN-gamma, IL-2, inducible nitric oxide synthase and CVB3 genomes in myocardial tissue.
Conclusions Our findings strongly suggested that the Fas/FasL pathway played a critical role in the development of massive myocardial necrosis through activation of infiltrating cells, and raise the possibility of immunotherapy by blocking the Fas/FasL pathway to prevent myocardial damage and improve the prognosis of patients with viral myocarditis.
We previously reported that costimulatory molecules belonging to the immunoglobulin superfamily and the tumor necrosis factor receptor/ligand superfamilies play an important role in the development of myocardial injury involved in murine viral myocarditis (1©C7). Fas/FasL are the most well-characterized costimulatory molecules, playing an essential role in the induction of programmed cell death (apoptosis). They are also known to play an important role in the cytotoxicity mediated by T-cells and natural killer (NK) cells (8©C10). Although several reports have studied apoptosis in a murine model of myocarditis and in patients with myocarditis and dilated cardiomyopathy (11©C15), the percentage of cardiac myocytes affected by apoptosis was too low to explain the mechanism involved in massive myocardial damage induced by myocarditis. Colston et al. (11)reported that Coxsackie virus B3 (CVB3)-induced myocarditis did not significantly alter the expression of Fas on cardiac myocytes of C3H/He mice, supporting the minimal contribution of apoptosis to myocardial damage. However, the authors analyzed Fas expression by an ordinary immunohistochemical method using paraffin-embedded samples. The purpose of the present study was to investigate in more detail the role of the Fas/FasL pathway in the myocardial damage induced by myocarditis, with special focus on the activation of the infiltrating immune cells rather than apoptosis of cardiac myocytes.
Materials and methods
Animals
Five-week-old male C3H/He mice, C3H/He-lpr/lpr mice, C3H/He-gld/gld mice and pregnant female C3H/He mice were purchased from Shizuoka Laboratory Animal Center (Shizuoka, Japan).
Virus
The preparation of CVB3 (Nancy strain) was as described previously (3). Five-week-old mice were inoculated intraperitoneally with 1 Ī┴ 106plaque-forming unit of CVB3 in 0.2 ml phosphate-buffered saline.
Antibodies
Rabbit anti-Fas polyclonal antibody (sc-1886), which reacts with mouse Fas antigen, was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, California). The preparation of mouse anti-mouse FasL monoclonal antibody (mAb) (K10, IgG2b) (16)and anticardiac myosin mAb (CMA19) (17)was previously described. Polyclonal rabbit anti-asialo GM1 antibody was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). Rat anti-L3T4 mAb (RM4-5) and rat anti-Lyt 2 mAb (5H10-1) were purchased from PharMingen (San Diego, California).
In vivo treatment of mice with an anti-FasL mAb
Five-week-old C3H/He mice received the anti-FasL mAb (K10) (25 mg/kg intraperitoneally), on the day of virus inoculation (day 0) and on day 3. Control C3H/He mice received mouse immunoglobulin G (IgG) (Organon Teknika Corporation, Durham, North Carolina) in the same way. C3H/He-lpr/lpr mice and C3H/He-gld/gld mice were simply inoculated with CVB3 without treatment with anti-FasL mAb (eight mice were used for each group).
Mice were euthanized on day 7, and half of each heart was fixed in 10% buffered formalin and used for histologic study. The other half of each heart was freshly frozen in liquid nitrogen and used for immunohistochemistry and polymerase chain reaction (PCR).
Histology
The procedures for histologic analysis were as described previously (3).
Preparation of cultured cardiac myocytes
Cultured ventricular cardiac myocytes were prepared from fetal C3H/He mice and were treated with recombinant murine interferon (IFN)-gamma (105U/l) (Shionogi and Co., Ltd., Tokyo, Japan) for 48 h, then subjected to immunocytochemical study as described previously (3).
Immunohistochemistry
In this study, to amplify the specific signals of antigen-antibody reaction, we used tyramide signal amplification (TSA) technology for fluorescence (TSA-Direct [Green], NEN Life Science Products [Boston, Massachusetts], according to the manufacturerĪ»s instructions) for the staining of Fas. The procedures were as described previously (3).
Double-immunofluorescence
To analyze the phenotypes of the infiltrating cells expressing FasL, we did double-immunofluorescent stainings for FasL and asialo GM1 (as a marker for NK cells), L3T4 (as a marker for T-helper [Th]-cells) or Lyt 2 (as a marker for cytotoxic T lymphocytes [CTLs]). After fixation, the sections were incubated with rabbit anti-asialo GM1, rat anti-L3T4 or rat anti-Lyt 2 for 1 h at 37ĪŃC. After washing in PBS, the sections were incubated with tetramethylrhodamine isothiocyanate-conjugated donkey anti-rabbit IgG (Chemicon International Inc., Temecula, California) or goat anti-rat IgG antibody (EY Laboratories, Inc., San Mateo, California) for 1 h at 37ĪŃC, respectively. The subsequent procedures for the staining of FasL were the same as those for Fas.
Immunocytochemistry
For immunocytochemical analysis, to distinguish cardiac myocytes from nonmuscle cells (mainly fibroblasts), we performed double-staining for cardiac myosin heavy chain and Fas as described previously (3).
Preparation of ribonucleic acid (RNA) and complementary deoxyribonucleic acid (DNA) synthesis
Mice were euthanized on day 7 after virus inoculation. The procedures for preparation of total cytoplasmic RNA from the heart tissues and cDNA synthesis were as described previously (18).
Amplification of complementary DNA by PCR
To examine the expression of cytokine messenger RNA in the heart tissues semiquantitatively, we amplified the single-stranded complementary DNA in a DNA thermal cycler (Perkin-Elmer Cetus, Norwalk, Connecticut) with 1 U of AmpliTaq DNA polymerase (Perkin-Elmer Cetus) using 5"- and 3"-primers specific for CVB3, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IFN-gamma, interleukin (IL)-2 and inducible nitric oxide synthase (iNOS), respectively. The primer sequences, annealing temperature and the number of cycles for IFN-gamma, IL-2, iNOS and GAPDH were as described previously (19). For the detection of CVB3 genomes, we used a 5"-sense primer (5"-TCCTCCGGCCCCTGAATGCG-3") and a 3"-antisense primer (3"-ACCGACGAATACCACTGTTA-5") (20). The annealing temperature and the number of cycles for CVB3 were 60ĪŃC and 18 (cycles). The PCR was performed denaturation at 94ĪŃC for 1 min, primer annealing for 2 min and primer extension at 72ĪŃC for 3 min. Expression of these cytokine messenger RNAs was examined using ethidium bromide-stained agarose gel electrophoresis.
Statistical analysis
Mann-Whitney Utest (using p corrected by DunnĪ»s modulus for multiple comparison) was used to evaluate differences between the groups.
Results
Expression of Fas in ventricular tissue
In ventricular tissue of normal mice, Fas was weakly to moderately expressed by many interstitial cells, which were thought to be dendritic cells and fibroblasts, almost uniformly distributed over the myocardium (Fig. 1A). Only slight expression of Fas was seen on some of cardiac myocytes and almost no expression was seen on vascular endothelial cells (Fig. 1A[arrow]). There was also only weak or almost no expression of Fas on cardiac myocytes of mice on days 1 to 4 (data not shown). On day 5 after virus inoculation, just after massive cell infiltrations appeared, expression of Fas was weakly to moderately induced on the sarcolemma of cardiac myocytes around the cell infiltrations. On day 7, when inflammation reached a maximum level, expression of Fas was clearly induced on some of the cardiac myocytes around the cell infiltrations (Fig. 1B). The infiltrating cells also expressed Fas. The expression of Fas on cardiac myocytes reached a maximum level around two weeks after virus inoculation and continued for more than four weeks with gradual decrease. Figure 1C(lower magnification) shows that Fas was extensively expressed on cardiac myocytes over the myocardium at two weeks after virus inoculation. Higher magnification confirmed the expression of Fas on the sarcolemma of cardiac myocytes (Fig. 1D). Many vascular endothelial cells also strongly expressed Fas at two weeks after virus inoculation (Fig. 1E[arrows]). Strong expression of Fas on vascular endothelial cells continued for more than six weeks (Fig. 1F[arrows]), when the expression of Fas on cardiac myocytes returned to a low level.
Figure 1
Immunohistochemical study for Fas in ventricular tissue. Normal ventricular myocardium (A)and ventricular myocardium of mice on day 7 (B), at two weeks (C to E)and at six weeks (F)after Coxsackie virus B3 infection were stained with anti-Fas antibody. Bar = 20 ”╠m.
Induction of Fas on cultured cardiac myocytes by IFN-gamma
Next, to confirm the induction of Fas on cardiac myocytes, we examined the expression of Fas on cultured cardiac myocytes treated with IFN-gamma in vitro. Figure 2shows double-stained cardiac myocytes cultured in a medium with or without IFN-gamma for 48 h. Figures 2A and 2Bshow the staining pattern specific for Fas. Figures 2C and 2D, which correspond to Figures 2A and 2B, respectively, show the staining pattern specific for cardiac myosin heavy chain, and indicate that most of the cells were cardiac myocytes. There was very slight or no expression of Fas on the cardiac myocytes of the control group (Fig. 2A). After treatment with IFN-gamma, most of the cardiac myocytes moderately and clearly expressed Fas on their surfaces (Fig. 2B). Fas expression was also induced on some of the nonmuscle cells, which mainly consisted of fibroblasts, by treatment with IFN-gamma.
Figure 2
Immunocytochemical study of cultured cardiac myocytes for Fas. (A and B)Myocytes in control group (A)and interferon-gamma-treated group (B), stained with anti-Fas antibody and labeled with fluorescein isothiocyanate. (C and D)Myocytes stained with a mAb for cardiac myosin heavy chain (CMA19) and labeled with tetramethylrhodamine isothiocyanate, corresponding to A and B, respectively. Bar = 10 ”╠m.
Effects of blockade of Fas/FasL pathway on the development of myocardial inflammation
The incidence of myocarditis was 100% in all groups. Extensive cell infiltration and necrosis were seen in the mice from Group A (mouse IgG-treated control group), whereas both cell infiltration and necrosis were less severe in the mice from Group B (anti-FasL mAb-treated group), and were markedly less severe in the mice from Group C and D (C3H/He-lpr/lpr and C3H/He-gld/gld group, respectively). The (mean Ī└ SE) percent area of myocardium undergoing inflammation was decreased (but not statistically significant) in Group B (6.76 Ī└ 1.36), and was significantly decreased in Group C (2.98 Ī└ 0.24; p < 0.005) and Group D (3.41 Ī└ 0.28; p < 0.005) as compared with Group A (11.31 Ī└ 1.54). This suggests that anti-FasL mAb-treatment could not completely inhibit Fas/FasL pathway in vivo. Thus, blockade of Fas/FasL pathway significantly decreased the myocardial inflammation induced by CVB3.
Expression of FasL on the infiltrating cells
We analyzed the phenotypes of FasL-expressing infiltrating cells by double-immunostaining for FasL and for asialo GM1, L3T4 or Lyt 2, which were markers for NK cells, Th-cells and CTLs, respectively, representing most infiltrating cells at this stage of myocarditis. We found that most of the infiltrating NK cells, Th-cells and CTLs moderately to strongly expressed FasL, strongly suggesting that most of the infiltrating cells could be affected by blockade of Fas/FasL pathway (data not shown).
Effects of anti-FasL mAb-treatment on the expression of proinflammatory cytokine transcripts and CVB3 genomes in the ventricular tissues
Next, to investigate the effects of anti-FasL mAb treatment on the activation of infiltrating cells, we analyzed the expression of IFN-gamma and IL-2, which are mainly expressed by the infiltrating cells and can be good markers for activation (18), as well as iNOS, which is induced by inflammation and may have a cytotoxic effect (21,22)and CVB3 genomes. As shown in Figure 3, the expression of CVB3 genomes was remarkably decreased in the anti-FasL mAb-treated group (weakly expressed in only one sample out of six) as compared with the mouse IgG-treated control group (strongly expressed in four samples out of six). The expression of IFN-gamma, IL-2 and iNOS was also significantly decreased in the anti-FasL mAb-treated group as compared with the mouse IgG-treated control group. Expression of GAPDH transcripts as internal standard showed that almost equivalent amounts of RNA were prepared from each mouse.
Figure 3
Effects of anti-FasL monoclonal antibody (mAb) treatment on the expression of proinflammatory cytokine transcripts and Coxsackie virus B3 (CVB3) genomes in the ventricular tissues. Total ribonucleic acid was prepared from ventricular tissues of mice from mouse immunoglobulin G (IgG)-treated control group and anti-FasL mAb-treated group, and analyzed for CVB3, interferon (IFN)-gamma, interleukin (IL)-2, inducible nitric oxide synthase (iNOS) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts by a semiquantitative polymerase chain reaction method.
Discussion
Expression of Fas in myocarditis
In this study, we demonstrated that Fas was markedly induced on cardiac myocytes in murine acute viral myocarditis. The induction of Fas on cardiac myocytes was confirmed by treatment with IFN-gamma in vitro. We also found that the expression of Fas on cardiac myocytes reached a maximum level around two weeks after virus inoculation, significantly later than that of other costimulatory molecules and major histocompatibility complex antigens (1©C3,5,23), which reached a maximum level on day 7, along with inflammation itself. However, Fas was also significantly induced on cardiac myocytes around cell infiltrations on day 7. The reason peak induction of Fas on cardiac myocytes was delayed is unknown.
Mechanism of the role of Fas/FasL pathway in myocarditis
We showed that myocardial inflammation was significantly decreased in Fas-deficient C3H/He-lpr/lpr mice and FasL-deficient C3H/He-gld/gld mice infected with CVB3. This indicates that the Fas/FasL pathway played a critical role in the development of myocardial damage induced by viral myocaridits. However, because cardiac myocytes affected by apoptosis were rare in myocarditis, including this murine model, we thought that the Fas/FasL pathway might play a critical role in the activation and cytotoxicity of the infiltrating T cells and NK cells rather than in inducing apoptosis of cardiac myocytes. This is satrongly supported by the present data that anti-FasL mAb-treatment significantly decreased the expression of IFN-gamma and IL-2, which are mainly expressed by the infiltrating T cells and NK cells and can be good markers for activation. This is also supported by the data that enhanced expression of Fas and FasL in the hearts of transgenic mice did not cause cardiac myocyte apoptosis (24). We think that the Fas/FasL expression levels induced by myocarditis might be lower than the threshold levels for cardiac myocytes to undergo apoptosis. The induction of Fas on cardiac myocytes around cell infiltrations is thought to enhance the activation of FasL-expressing infiltrating NK cells and T cells. Therefore, blockade of Fas/FasL pathway may inhibit cardiac myocyte necrosis, directly mediated by cytolytic factors such as perforin (25), by these infiltrating cells. Especially for CTLs, it was shown that a costimulatory signal through FasL was essential for antigen-specific maximal proliferation to occur in vivo as well as in vitro (26,27). Furthermore, anti-FasL mAb treatment also significantly decreased the expression of iNOS and CVB3 genomes, indicating that blockade of the Fas/FasL pathway could decrease inflammation and perforin-mediated cardiac myocyte necrosis without interfering with the virus clearance. In the present study, virus clearance seemed to be accelerated rather than unaffected by blockade of the Fas/FasL pathway. This is strongly supported by the fact that perforin knockout mice infected with CVB3 developed less severe myocarditis than did wild type, and could control the infection and eradicate the virus (28).
Conclusions
Taken together, our findings strongly suggest that the Fas/FasL pathway plays a critical role in the development of massive myocardial necrosis rather than inducing cardiac myocyte apoptosis, and raise the possibility of immunotherapy by blocking the Fas/FasL pathway to prevent massive myocardial necrosis and improve the prognosis of patients with viral myocarditis.Role of Fas/FasL pathway in the activation of infiltrating cells in murine acute myocarditis caused by Coxsackievirus B3 | JACC: Journal of the American College of Cardiology
http://www.onlinejacc.org/content/39/8/1399ĪĪ
Experimental Eye Research
Volume 179, February 2019, Pages 47-54Soluble Fas ligand blocks destructive corneal inflammation in mouse models of corneal epithelial debridement and LPS induced keratitis
Author links open overlay panelMeredithGregory-KsanderaVictor L.PerezbAnnMarshak-Rothsteinc1Bruce R.Ksandera1
a
The Schepens Eye Research Institute, Department of Ophthalmology, Harvard Medical School, 20 Staniford Street, Boston, MA, USA
b
Department of Ophthalmology, Duke University School of Medicine, Durham, NC, USA
c
Department of Medicine, University of Massachusetts Medical School, Worcester, MA, USA
https://doi.org/10.1016/j.exer.2018.10.013Get rights and content
Highlights
•
Membrane-bound Fas ligand and soluble Fas ligand play opposing roles in corneal inflammation.
•
Membrane-bound Fas ligand is pro-inflammatory and exacerbates corneal inflammation.
•
Soluble Fas ligand is non-inflammatory and antagonizes the activity of membrane-bound Fas ligand.
•
Soluble Fas ligand specifically inhibits neutrophil-mediated corneal inflammation and associated tissue damage.
Abstract
Neutrophil-mediated inflammation plays a critical role in corneal damage following injury or infection. Previous studies demonstrated that membrane-bound FasL (mFasL) induces neutrophil chemokine production. However, the extracellular domain of mFasL is normally cleaved by matrix metalloproteinases to release a soluble form of FasL (sFasL) and sFasL antagonizes mFasL-mediated chemokine production. Therefore, we hypothesized that sFasL could be used to prevent neutrophil-mediated corneal inflammation associated with injury and bacterial keratitis. To test this hypothesis, GFP-only, sFasL-GFP, or mFasL-GFP were expressed in the corneal stroma of C57BL/6 mice, using intra-stromal injections of plasmid DNA or adenoviral vectors (AV) and the role of mFasL and sFasL in corneal inflammation was examined in models of corneal injury and LPS-induced keratitis. Our work addresses an important area of disagreement in the field of FasL, with regard to the mechanism by which sFasL regulates ocular inflammation. Herein, we demonstrate that an intrastromal injection of GFP-only, sFasL-GFP, or mFasL-GFP plasmid DNA resulted in GFP expression throughout the corneal stroma for up to two weeks with little to no evidence of inflammation in the GFP-only and sFasL-GFP groups and mild corneal inflammation in the mFasL-GFP group. Similarly, following epithelial debridement, corneas expressing GFP-only or sFasL-GFP showed no significant signs of corneal inflammation, with clear corneas at 15 days post debridement. By contrast, epithelial debridement of corneas expressing mFasL-GFP triggered persistent corneal inflammation and the development of central corneal opacities that was blocked by sFasL. Similar to the mFasL-GFP plasmid DNA, intrastromal injection of mFasL-GFP AV triggered mild corneal inflammation, but it was transient and resolved by day 10 with corneas remaining clear out to 30 days post injection. Nevertheless, intrastromal expression of mFasL-GFP AV exacerbated LPS-induced keratitis, corneal opacity, and neovascularization, while sFasL-GFP AV expression prevented LPS-induced keratitis, resulting in a clear cornea. Histological analysis of corneas with LPS-induced keratitis revealed a robust infiltration of macrophages and neutrophils and sFasL expression specifically blocked the neutrophil influx. Overall, our data demonstrate that stromal expression of mFasL is inflammatory, while sFasL is non-inflammatory, and opposes the effects of mFasL in mouse models of epithelial debridement and LPS-induced keratitis. These data demonstrate that a delicate balance between sFasL and mFasL regulates ocular inflammation. This study further identifies sFasL as a potent inhibitor of neutrophil-mediated corneal damage, and supports the potential use of sFasL in the treatment of neutrophil-mediated keratitis. These results strongly support the hypothesis that, in the immune privileged environment of the eye, the isoform of FasL regulates immune privilege and determines the extent of inflammation: mFasL promotes inflammation and sFasL blocks inflammation.Soluble Fas ligand blocks destructive corneal inflammation in mouse models of corneal epithelial debridement and LPS induced keratitis - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0014483518304640ĪĪ
Effects of Shenqi Fuzheng injection on Fas/FasL protein ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4840685
Effects of SFI on the expression levels of Fas and FasL proteins in the myocardial cells of a mouse model of VMC . A significant increase in the expression levels of Fas and FasL proteins was detected in the model group, as compared with the blank group at all time points (P<0.01).
Neutrophil Expression of FasL and Perforin Directs ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3424302
Sep 01, 2012 Īż However, examination of hapten-primed CD8 T cells has failed to demonstrate expression of FasL and the primed CD8 T cells do not exhibit cytolytic functions when co-cultured with hapten-labeled targets raising the possibility that the requirement for either FasL or perforin is mediated through expression by other cells participating in the elicitation of CHS responses (17©C19).
Cited by: 28
Publish Year: 2012
Author: Danielle D. Kish, Anton V. Gorbachev, Neetha Parameswaran, Neetu Gupta, Neetu Gupta, Robert L. Fairc...
Human Fas-ligand expression on porcine endothelial cells ...
https://www.ncbi.nlm.nih.gov/pubmed/14962292
FasL expressed on porcine EC induced apoptosis in human NK and T cells, but did not protect porcine EC against killing mediated by human NK cells. 2A2-FasL released soluble FasL, which induced strong chemotaxis in human PMN.
Cited by: 12
Publish Year: 2004
Author: Ulrike B. Matter®\Reissmann, Kai®\Christian Sonntag, Urs O. Gilli, Christian Leguern, Mårten K. J. Sch...
Fas/FasL and perforin©Cgranzyme pathways mediated T cell ...
https://www.sciencedirect.com/science/article/pii/S2211283912000147
In summary, in this study, we demonstrated the gene expression of cytolytic molecules Fas, FasL, caspase-3 and PFN and the infiltration of CD8 + T cells in bursal and splenic tissues of ĪŁ
Cited by: 14
Publish Year: 2012
Author: Abdul Rauf, Mahesh Khatri, MariaĪĪ
Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo
Peiyee Lee, Masataka Sata, David J. Lefer, Stephen M. Factor, Kenneth Walsh, and Richard N. Kitsis
Abstract
Fas is a widely expressed cell surface receptor that can initiate apoptosis when activated by its ligand (FasL). Whereas Fas abundance on cardiac myocytes increases in response to multiple pathological stimuli, direct evidence supporting its role in the pathogenesis of heart disease is lacking. Moreover, controversy exists even as to whether Fas activation induces apoptosis in cardiac myocytes. In this study, we show that adenoviral overexpression of FasL, but not ”┬-galactosidase, results in marked apoptosis both in cultures of primary neonatal cardiac myocytes and in the myocardium of intact adult rats. Myocyte killing by FasL is a specific event, because it does not occur in lpr (lymphoproliferative) mice that lack functional Fas. To assess the contribution of the Fas pathway to myocardial infarction (MI) in vivo, lpr mice were subjected to 30 min of ischemia followed by 24 h of reperfusion. Compared with wild-type mice, lpr mice exhibited infarcts that were 62.3% smaller with 63.8% less myocyte apoptosis. These data provide direct evidence that activation of Fas can induce apoptosis in cardiac myocytes and that Fas is a critical mediator of MI due to ischemia-reperfusion in vivo.
cardiac myocytesundergo apoptosis in a wide variety of pathophysiological situations, including ischemia (6, 14, 25, 44), ischemia-reperfusion (8, 14, 16), and hemodynamic overload and heart failure (33, 41, 51). These observations suggest that apoptosis may play a role in the pathogenesis of heart disease. In the case of ischemia-reperfusion, this possibility is supported by studies in which inhibition of apoptosis by a variety of pharmacological and genetic approaches results in smaller infarctions (7-9, 21, 36, 58). Whereas these experiments are important in that they demonstrate a causal role for apoptosis in myocardial infarction (MI), they provide relatively limited information about the pathways that mediate cardiac myocyte apoptosis in vivo.
Apoptosis is mediated by two central pathways. In the mitochondrial pathway (18), diverse stimuli, including nutrient and growth/survival factor deprivation, hypoxia, and oxidative stress, stimulate the translocation of cytochrome c from the mitochondrial intermembrane space and inner membrane to the cytoplasm. Cytochrome c, along with dATP, which is already present in the cytoplasm, then binds to and stimulates the oligomerization of Apaf-1 and the subsequent recruitment and activation of procaspase-9. This leads to the activation of downstream procaspases-3, -6, and -7, proteolysis of specific cellular substrates and cell death. In contrast, the death receptor pathway (2) involves the binding of soluble or cell membrane-bound ligands to cell surface receptors such as Fas and tumor necrosis factor receptor 1 (TNFR1). In the case of Fas, trimeric Fas ligand (FasL), an integral membrane protein, binds to a Fas trimer. This is presumed to induce a conformational change in Fas that enables its cytoplasmic tail to recruit Fas-associated death domain protein (FADD) through interactions involving death domains in both molecules. FADD, in turn, recruits procaspase-8 through homotypic interactions involving death effector motifs. The approximation of procaspase-8 stimulates its autoactivation, following which caspase-8 activates downstream caspases and induces apoptosis. There is cross talk between the death receptor and mitochondrial pathways as caspase-8 cleaves BID, the carboxyl fragment of which translocates to the mitochondria and stimulates cytochrome c release (32, 34).
Recent work has shown that the mitochondrial pathway is activated in cardiac myocytes in response to metabolic stress, hypoxia, and reoxygenation (5, 11, 26, 35). In addition, the mitochondrial pathway is important in cardiac myocyte apoptosis because its disruption by transgenic overexpression of Bcl-2 (7,9) or dominant negative procaspase-9 (42) markedly reduces infarct size during ischemia-reperfusion in vivo. In contrast, the role of the death receptor pathway in cardiac myocyte apoptosis is less well understood. Although Fas signaling has been implicated in cardiac hypertrophy (3, 38) and calcium signaling (13), its role in cardiac myocyte apoptosis has been less clear. Neonatal and adult cardiac myocytes are killed relatively inefficiently by FasL (55), unless costimulated by doxorubicin (56) or hypoxia (57). Moreover, mice treated systemically with an activating Fas antibody die from massive hepatocyte apoptosis but exhibit no cardiac pathology (40). These observations raise the possibility that Fas activation may not induce apoptosis efficiently in cardiac myocytes. Other observations, however, suggest that the Fas death pathway is functional in these cells. For example, interference of Fas signaling with neutralizing FasL antibodies and in lpr mice that lack Fas (1) decreases cardiac myocyte apoptosis in animal models of doxorubicin toxicity (37) and in isolated perfused hearts subjected to ischemia-reperfusion (23). In addition, the abundance of Fas in the heart increases in response to a variety of insults (23, 25, 37, 50,55, 59). These data suggest that the Fas-mediated apoptosis may be important in myocardial disease.
Accordingly, the objectives of this study were to test directly whether Fas activation kills cardiac myocytes and, if so, whether this pathway contributes to both myocyte apoptosis and MI following ischemia-reperfusion in vivo. Our data indicate that the Fas death pathway is indeed functional in cardiac myocytes and critical for myocardial damage due to ischemia-reperfusion in vivo.MATERIALS AND METHODS
Reagents and animals.
Chemicals were purchased from Sigma Chemical (St. Louis, MO) unless otherwise noted. Six- to eight-week-old C57Bl/6J, MRL/MpJ, and MRL/MpJ-Faslpr male mice were purchased from Jackson Laboratories (Bar Harbor, ME). One-day-old neonatal Sprague-Dawley rat pups were obtained from Taconic Farms (Germantown, NY). Six- to eight-week-old female Wistar rats were purchased from Charles River Laboratories (Wilmington, MA). All experimental protocols were approved by the review board of the Animal Institute of the Albert Einstein College of Medicine.
Primary cultures of neonatal rat cardiac myocytes and adenoviral infections.
Primary cardiac myocyte cultures were prepared from 1-day-old rat pups as previously described (5), except that cells were plated at a density of 380 cells/mm2. Recombinant adenoviruses encoding murine FasL (AdFasL) and ”┬-galactosidase (Ad”┬-gal) under the control of the cytomegalovirus promoter have been described previously (45). Approximately 24 h after being plated, cardiac myocytes were infected with the indicated adenovirus at multiplicity of infection of 200 plaque forming units (pfu)/cell. This concentration was chosen because infection with Ad”┬-gal at this multiplicity of infection resulted in transduction of 98©C100% of cells with no toxicity. Cells were harvested for ladder assays 48 h after infection.
DNA ladder assay.
This assay was performed on floating and adherent cells as previously described (5).
Direct cardiac injection of adenoviruses.
These were performed as described previously (27). The injectate for rats consisted of 50 ”╠l of phospate-buffered saline containing a total of 7 Ī┴ 107 pfu of the virus. This was delivered as a single injection into the anterolateral and apical aspect of the left ventricle through a 27-gauge needle attached to a Hamilton syringe. Experimental animals received a combination of 6 Ī┴ 107 pfu of AdFasL and 1 Ī┴ 107 pfu of Ad”┬-gal, the latter to identify the area of injection. Controls received 7 Ī┴ 107 pfu of Ad”┬-gal. Mouse hearts were injected in the same way except that 30-gauge needles were used and the injectate consisted of 15 ”╠l. Animals were euthanized 48 h after injection.
Immunohistochemistry for FasL.
Cryosections (5 ”╠m thick) were fixed in paraformaldehyde. Biotin-conjugated primary antibody (1:100, clone MFL3, Pharmingen, San Diego, CA; or 1:100, clone Kay10; Pharmingen), which recognizes C57BL/6J mouse FasL, was used, followed by horseradish peroxidase-conjugated streptavidin (BioGenex Laboratories; San Ramon, CA) and AEC chromagen (BioGenex Laboratories). Sections were counter-stained with Meyer's hematoxylin.
TUNEL assay.
Deoxynucleotidlytransferase-mediated dUTP nick end labeling (TUNEL) was performed on 5-”╠m frozen sections. The area of the injection was first identified by staining serial sections for ”┬-galactosidase, which was expressed by the coinjected Ad”┬-gal. TUNEL was performed with the TACS 2-terminal deoxynucleotidyl transferase Blue or the Fluorescent Apoptosis Detection Kit (Trevigen; Gaithersburg, MD) according to the manufacturer's recommendations utilizing 1-h labeling reaction in the presence of Mn2+. Sections stained with the TACS2 Blue reagent were counterstained with eosin, whereas fluorescent sections were counterstained with 1 ”╠g/ml 4Īõ,6-diamidino-2-phenylindole (DAPI). Only cells with clear striations were scored as cardiac myocytes.
Quantitation of TUNEL-positive cells.
An apical and a midventricular section taken from the same level of each of the hearts were quantitated by collecting images with an Olympus IX70 CCD digital camera (Roper Scientific; Tuscon, AZ). The left ventricular free wall was demarcated using IPLab (Scanlytics; Vienna, VA) and its area (”╠m2) calculated by Excel (Microsoft; Redmond, WA). The total number of nuclei per squared micrometer, as determined by staining with DAPI, was assessed in four 10-”╠m2 sections and averaged. From this density and the area of the left ventricle, the total number of nuclei in the entire left ventricular free wall was calculated. The total number of TUNEL-positive nuclei in cells with clear striations was counted directly over the entire left ventricular free wall (fluorescein). The percentage of TUNEL-positive myocyte nuclei per total nuclei was calculated by dividing the latter by the former and multiplying by 100. The results for the apical and midventricular sections were averaged.
Myocardial ischemia-reperfusion model and infarct size assessment.
MRL/MpJ and MRL/MpJ-Faslpr mice were subjected to 30 min of ischemia and 24 h of reperfusion in vivo (19) following which infarcts were quantitated (24) as previously described.
Statistical analysis.
Results are expressed as means Ī└ SE. Groups were compared using one-way ANOVA with Tukey's posttest. P < 0.05 was considered statistically significant.
RESULTS
FasL induces apoptosis in cultured neonatal cardiac myocytes.
We had previously determined that all components of the Fas pathway (FasL, Fas, FADD, and procaspase-8) are expressed in the adult heart (not shown). To test directly whether activation of this pathway can induce apoptosis in heart muscle cells, primary cultures of neonatal rat cardiac myocytes were infected with recombinant AdFasL or Ad”┬-gal. Microscopic examination of DAPI-stained cells in plates infected with AdFasL showed the development of classic apoptotic changes consisting of cytoplasmic shrinkage, nuclear condensation, and detachment of the cells from the plate. In contrast, cells on plates infected with Ad”┬-gal appeared relatively normal (not shown). DNA ladder assays were performed on lysates from adherent and floating cells harvested 48 h later. DNA was intact in lysates from uninfected plates (Fig. 1, lane 1) and plates infected with Ad”┬-gal (Fig. 1, lanes 4 and 5). In contrast, DNA from plates infected with AdFasL demonstrated a clear nucleosomal pattern of DNA degradation indicative of apoptosis (Fig. 1,lanes 2 and 3). Thus FasL induces apoptosis in neonatal cardiac myocytes.
Fig. 1.
Fas ligand (FasL) induces apoptosis in neonatal cardiac myocytes (M). Primary cultures of neonatal cardiac myocytes were uninfected (lane 1) or infected with adenoviruses encoding murine FasL (AdFasL) (lanes 2 and 3) or adenoviruses encoding ”┬-galactosidase (Ad”┬-gal) (lanes 4and 5), and DNA from lysates of adherent and floating cells collected 48 h later was analyzed on agarose gels. Nucleosomal DNA laddering is detected only in cultures infected with AdFasL. This experiment was performed three times, using independent preparations of cells, with similar results.
Download figureDownload PowerPoint
FasL induces apoptosis in adult cardiac myocytes in vivo.
To extend the above results to adult cardiac myocytes in the intact animal, AdFasL or Ad”┬-gal was injected into the myocardium of beating rat hearts, and FasL protein expression and TUNEL was assessed 48 h later. With the use of two different antibodies specific for murine FasL (encoded by the adenovirus), AdFasL-injected rat hearts exhibited immunostaining for mouse FasL in cardiac myocytes near the injection site (Fig. 2A, b andd). Mouse FasL staining was not detected in Ad”┬-gal-injected rat hearts (Fig. 2A,a and c). These control hearts were also negative for TUNEL staining (Fig. 2B, a). In contrast, hearts injected with AdFasL exhibited strong TUNEL staining, most of which was localized to the area surrounding the injection track and which included myocytes and nonmyocytes (Fig. 2B,b). Of the 15 hearts injected with AdFasL, 11 contained TUNEL-positive myocytes (Fig. 2C). In contrast, none of the 15 hearts injected with Ad”┬-gal contained TUNEL-positive cells.
Fig. 2.
FasL induces apoptosis in adult cardiac myocytes in vivo. A: mouse FasL protein expression in sections from adult rat hearts injected in vivo 48 h earlier with adenoviruses encoding ”┬-galactosidase (d”┬-gal, a andc) or recombinant adenoviruses encoding murine FasL (AdFasL,b and d). FasL was detected with two antibodies specific for mouse FasL (encoded by AdFasL), MFL3 (a andb), and Kay 10 (c and d). Both antibodies detected mouse FasL expression (brown-red signal) near the injection track of AdFasL-injected rat hearts but not of Ad”┬-gal-injected rat hearts. B: terminal deoxynucleotidlytransferase-mediated dUTP nick end labeling (TUNEL) assays on sections from adult rat hearts injected in vivo 48 h earlier with Ad”┬-gal (a) or AdFasL (b). Sections counterstained with eosin. Multiple TUNEL-positive myocyte and nonmyocyte nuclei (blue) are seen in AdFasL-, but not Ad”┬-gal-injected hearts. C: number of TUNEL− and TUNEL+ hearts injected with Ad”┬-gal (n = 15) or AdFasL (n = 15). A heart was considered TUNEL+ if it contained any number of TUNEL+ myocytes. Eleven of 15 AdFasL-injected hearts were TUNEL+. In contrast, all of the Ad”┬-gal-injected hearts were TUNEL−.FasL-induced cardiac myocyte apoptosis requires Fas.
To determine whether FasL-induced cardiac myocyte apoptosis is a specific event, we tested whether it is receptor dependent. To accomplish this, we assessed whether intramyocardial injection of AdFasL would induce apoptosis in the hearts of lprmice, a naturally occurring mutant deficient in Fas due to an insertion of an early transposable element that results in premature mRNA termination (1). Because lpr mice develop lymphoproliferative disease in later life (46, 54), they were used in the current studies before developing any detectable abnormalities. Wild-type mice of the same genetic background (MRL/MpJ) were studied in parallel. Injection with AdFasL induced cardiac myocyte apoptosis in 3 of 3 wild-type mice (Table1). In contrast, TUNEL-positive cells were absent in 2 of 2 AdFasL-injected lpr mice, although the injections were successful as evidenced by positive ”┬-galactosidase staining resulting from the coinjected Ad”┬-gal. This experiment demonstrates that induction of myocyte apoptosis by FasL is a specific event that requires Fas. Taken together, these rat and mouse studies demonstrate that activation of Fas can result in apoptosis in adult cardiac myocytes in vivo.
Table 1. Specificity of FasL-induced cardiac myocyte apoptosis: requirement for Fas
Enlarge table
Fas pathway is critical for cardiac myocyte apoptosis and infarct development due to ischemia-reperfusion.
To test the importance of the Fas pathway for myocyte death and infarction during ischemia-reperfusion injury in vivo, wild-type (MRL/MpJ) and lpr mice were subjected to sham operation or left anterior descending coronary artery occlusion for 30 min, followed by 24 h of reperfusion. At the conclusion of reperfusion, the sizes of the region at risk and infarct were quantitated using Evans blue and 2,3,5-triphenyltetrazolium chloride staining, respectively, as previously described (24). Figure 3A depicts representative stained sections from wild-type (a) andlpr (b) hearts following ischemia-reperfusion in vivo. Despite similar regions at risk (denoted by the absence of blue), the lpr heart has a markedly smaller infarct (denoted by white) than the wild-type heart. These parameters were quantitated for 9 wild-type and 8 lpranimals in Fig. 3B. The region at risk was similar between genotypes (wild-type, 51.8 Ī└ 4.4%; lpr, 52.3 Ī└ 3.1%; P is not significant). Within these similar regions at risk, however, the mean infarct size was 62.3% smaller in thelpr mice compared with wild-type mice (wild-type, 62.9 Ī└ 5.1%; lpr, 23.7 Ī└ 5.4%; P < 0.01). As shown in Fig. 3C, this reduction in infarct size was accompanied by a 63.8% reduction in myocyte apoptosis in a parallel cohort of mice (wild-type, 3.62 Ī└ 1.25%;lpr, 1.31 Ī└ 0.5%; P < 0.01). These data indicate that the Fas pathway plays a critical role in myocyte death and the development of MI during ischemia-reperfusion in vivo.
Fig. 3.
Critical role of the Fas pathway in cardiac myocyte apoptosis and myocardial infarction due to ischemia-reperfusion. A: Evans blue (blue) and tetrazolium (red) staining of representative heart sections from wild-type (a) and lpr (b) mice subjected to 30 min of left anterior descending coronary artery occlusion, followed by 24 h of reperfusion. Within similar risk regions (absence of blue), the lpr heart shows a markedly smaller infarct (white) compared with the wild-type heart.B: quantitation of infarct sizes in wild-type (n = 9) and lpr (n = 8) mice. Regions at risk were similar between the genotypes (wild-type, 51.8 Ī└ 4.4%; lpr, 52.3 Ī└ 3.1%, Pnot significant). Within these similar at-risk regions, the infarct was 62.3% smaller in lpr mice compared with wild-type mice (wild-type, 62.9 Ī└ 5.1%, lpr, 23.7% Ī└ 5.4%,P < 0.01). C: percentage of TUNEL+ myocyte nuclei per total cardiac nuclei in wild-type (n = 3) and lpr (n = 4) mice subjected to 30 min of ischemia and 24 h reperfusion and wild-type (n = 3) and lpr (n = 3) mice subjected to sham operation, followed by 24 h. The percentages of TUNEL+ myocytes in the sham-operated groups were similar between genotypes (wild-type, 0.0074 Ī└ 0.0017%; lpr, 0.0083 Ī└ 0.0021%, P, not significant). In contrast, in the ischemia-reperfusion groups, the percentage of TUNEL+ myocytes in the lpr mice was 63.8% lower than the wild-type mice (wild-type, 3.62 Ī└ 1.25%; lpr, 1.31 Ī└ 0.5%, P < 0.01).
Download figureDownload PowerPoint
DISCUSSION
The significance of the death receptor pathway in cardiac myocyte apoptosis has been in question. This is appropriate, because Ī░death ligandsĪ▒ do not uniformly activate death pathways in all cell types. For example, tumor necrosis factor (TNF)-”┴ is inefficient at killing many cell types because of its simultaneous activation of survival pathways (4, 53). In fact, although TNF-”┴ can kill cardiac myocytes under some conditions (29, 48), its net effect in the heart during ischemia-reperfusion is prosurvival as illustrated by increased infarcts in TNFR1 and TNFR2 double knockout mice (30). Our results show that activation of the Fas pathway can induce apoptosis in neonatal and adult cardiac myocytes. Moreover, the marked reduction in infarct size and abundance of apoptotic cardiac myocytes in mice lacking Fas demonstrates the importance of this pathway in ischemia-reperfusion injury in vivo.
Fas activation can kill cardiac myocytes.
Although Fas signaling has been suggested to modulate cardiac hypertrophy (3, 38) and calcium signaling (13), the ability of this pathway to kill cardiac myocytes efficiently has not been shown. This study demonstrates directly that activation of Fas can induce apoptosis in both cultured neonatal cardiac myocytes and adult cardiac myocytes in vivo. Moreover, this killing is specific as evidenced by the inability of AdFasL to induce apoptosis in the hearts of lpr mice. Although these data establish that the Fas death pathway is functional in cardiac myocytes, it is not known whether Fas activation brings about apoptosis in this cell type primarily via the direct activation of downstream caspases by caspase-8, through activation of the mitochondrial death pathway, or both.
Our results are in agreement with previous indirect data showing that the survival of fetal mouse cardiac myocytes decreases when they are cocultured with FasL-expressing lymphocytes (47). It is interesting to consider why only low levels of cardiac myocyte apoptosis have been observed in response to soluble FasL in some previous studies (17, 55-57). We speculate that this may be the result of inadequate levels and/or presentation of the ligand. Perhaps the high-level expression of FasL from closely neighboring cells achieved with a recombinant adenovirus facilitated killing in our studies.
Critical role of the Fas death pathway in ischemia-reperfusion in vivo.
Strong proof for the importance of the Fas death pathway in vivo is provided by the 62.3% reduction in infarct size during ischemia-reperfusion in lpr mice. This reduction in overall infarct size was accompanied by a 63.8% decrease in apoptotic cardiac myocytes. A previous study using isolated, perfused preparations showed a 56% reduction in apoptotic myocytes in lpr hearts compared with wild-type hearts (23). Our study extends these ex vivo experiments to the in vivo setting. In addition, we demonstrate for the first time that the decrease in cardiac myocyte apoptosis translates into an actual reduction in infarct size. The similarity between the magnitude of the reductions in myocyte apoptosis in the ex vivo study (performed in the absence of blood) and our in vivo study suggests that Fas deficiency in heart cells, rather than blood cells, is responsible for the reduced death. Although not yet formally proven, the deficiency of Fas on cardiac myocytes themselves is the most likely mechanism.
The quantitatively similar decreases in infarct size and cardiac myocyte apoptosis in our study demonstrate that total and apoptotic cell death were reduced in parallel but do not necessarily indicate that the reduction in cell death resulted solely from less apoptosis. To determine this, one would need to know1) the proportions of total cell death attributable to necrosis versus apoptosis in this model and 2) the abundance of necrotic cells in lpr versus wild-type mice following ischemia-reperfusion. Because necrosis was not assessed directly in this study, it remains to be determined whether disruption of Fas signaling in the myocardium decreases necrosis as well as apoptosis. There is a precedent for this possibility in T lymphocytes, however, where Fas has been implicated in the regulation of both forms of cell death (20).
Whereas ablation of Fas signaling strikingly reduces infarct size during ischemia-reperfusion in vivo, it is noteworthy that it does not result in a complete rescue. The most likely explanations for this are direct activation of the mitochondrial pathway and possibly activation of the death receptor pathway through death receptors other than Fas.
Modulating factors.
Whereas the experiments in this study provide strong genetic evidence for activation of the Fas death pathway during ischemia-reperfusion, they do not identify the actual triggering events. The most likely initiating event would be an increase in the abundance of FasL. Although we were unable to document this in our in vivo model, upregulation of FasL could be demonstrated in the concentrated coronary effluents from isolated, perfused hearts subjected to ischemia-reperfusion (23). A second potential trigger could be increases in Fas. There is precedent for Fas being limiting in the activation of the death receptor pathway during the pathogenesis of autoimmune thyroiditis (15) and diabetes (10). In fact, there are significant increases in Fas protein during ischemia-reperfusion (49, 59). A third potential regulatory mechanism involves two proteins that modulate Fas signaling by inhibiting caspase-8: Fas-associated death domain protein-like-interleukin-1 ”┬-converting enzyme inhibitory protein (FLIP) (22) functions as a dominant negative inhibitor of procaspase-8, whereas apoptosis repressor with a caspase recruitment domain (ARC) (28) binds to caspase-8 and inhibits its activity. Of note, during ischemia-reperfusion, levels of FLIP (43) and ARC (12, 39) decrease dramatically, which could potentially contribute to activation of the Fas pathway.
Fas: to grow or die?
Recently, it has become clear that Fas signaling plays a role in cellular growth as well as death. In T cell receptor-stimulated lymphocyte proliferation, this growth response requires FADD and appears to involve activation of extracellular signal-regulated kinase and nuclear factor-”╩B pathways by FLIP (31, 52). The Fas pathway has also been implicated in cardiac hypertrophy. Transgenic mice with cardiac-specific overexpression of FasL have been found to exhibit mild cardiac hypertrophy (38). Conversely, mice that lack Fas (lpr) fail to undergo hypertrophy in response to pressure overload, although surprisingly FasL appears to be dispensable (3). FasL-induced hypertrophy in cultured cardiac myocytes requires phosphorylation and inactivation of glycogen synthase kinase 3”┬, which occurs in a phosphatidylinositol-3 (PI-3)-kinase-dependent manner. The molecular link between activation of Fas and PI-3-kinase is presently unclear as are the mechanisms that determine whether Fas activation will result in hypertrophy or death. On the basis of cell culture studies (3, 56), it has been previously suggested that the decision to undergo hypertrophy or death is regulated in part by the concentration of FasL in the extracellular space (3). In keeping with this notion, it is likely that significantly higher FasL levels would be achieved with injections of AdFasL into the myocardium in the present study where apoptosis was observed than through transgenesis where it was absent (38). Given the complexities of pathways unveiled thus far, however, there are likely to be multiple mechanisms downstream of Fas that regulate the differential effects of activating this receptor in a cardiac myocyte.
In conclusion, the experiments in this study provide direct evidence that Fas activation by FasL can induce apoptosis in neonatal and adult cardiac myocytes and that the Fas death pathway is critical for myocyte killing and the full development of MI during ischemia-reperfusion in vivo.Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo | American Journal of Physiology-Heart and Circulatory Physiology
https://journals.physiology.org/doi/full/10.1152/ajpheart.00777.2002ĪĪ
Loss of Functional Fas Ligand Enhances Intestinal ...
https://cancerres.aacrjournals.org/content/67/10/4800
May 15, 2007 Īż Fas ligand (FasL/CD95L), a member of the tumor necrosis factor family, interacts with a specific receptor Fas, ultimately leading to cell death. Tumor expression of FasL has been proposed to aid in immune evasion through a Ī░Fas counterattackĪ▒ mechanism but has also been described as a proinflammatory factor.
Cited by: 18
Publish Year: 2007
Author: Barbara Fingleton, Kathy J. Carter, Lynn M. Matrisian
[PDF]Differential Expression of Fas (CD95) and Fas Ligand on ...
https://pdfs.semanticscholar.org/25da/e3ec2bd3cc290834c03c466f1ff1e4990ce6.pdf
FasL system in normal human phagocytes. Although Fas expression was detected on neutro- phils, monocytes, and eosinophils, constitutive expression of FasL was restricted to neutrophils. The three types of phagocytes demonstrated differential sensitivity to Fas-induced apoptosis.
Neutrophil Suppresses Tumor Cell Proliferation via Fas ...
www.ijbs.com/v14p2103.htm
The high expression of FasL on melanoma cells leads to neutrophil inactivation and assists tumor development and local transfer of PMA-treated neutrophils delayed tumor formation by melanoma cells . In this work, neutrophils function different depend on the expression level of tumor CD95L and the activity of neutrophils in the tumor-bearing mice.
Cited by: 3
Publish Year: 2018
Author: Bingwei Sun, Weiting Qin, Mingming Song, Lu Liu,ĪĪ
ĪĪ
http://www.lookfordiagnosis.com/mesh_info.php?term=Fas+Ligand+Protein&lang=1
ĪĪ
Published: December 1998
Newly discovered role for Fas ligand in the cell-cycle arrest of CD4+ T cells
Julie Desbarats, Richard C. Duke & M. Karen Newell
Nature Medicine volume 4, pages1377©C1382(1998)Cite this article
Abstract
Fas Ligand (FasL) can induce apoptosis of Fas-bearing cells. It is expressed on the cell surface of many tumor cells, immune-privileged tissues and activated lymphocytes. We report here that FasL can itself transduce signals, leading to cell-cycle arrest and cell death in CD4+ T cells. In vitro, FasL engagement inhibited CD4+ T-cell proliferation, cell-cycle progression, and IL-2 secretion. In vivo, FasL engagement prevented superantigen-mediated CD4+, but not CD8+, T-cell expansion. These findings demonstrate that FasL engagement regulates cell-cycle progression, and show that FasL engagement in vivo has a potent anti-inflammatory effect specific for CD4+ T cells.Newly discovered role for Fas ligand in the cell-cycle arrest of CD4 + T cells | Nature Medicine
https://www.nature.com/articles/nm1298_1377ĪĪ
Front. Immunol., 05 April 2017 | https://doi.org/10.3389/fimmu.2017.00403
Dual Role of Fas/FasL-Mediated Signal in Peripheral Immune Tolerance
imageAkiko Yamada, imageRieko Arakaki, imageMasako Saito, imageYasusei Kudo and imageNaozumi Ishimaru*
Department of Oral Molecular Pathology, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan
Fas-mediated apoptosis contributes to physiological and pathological cellular processes, such as differentiation and survival. In particular, the roles of Fas in immune cells are complex and critical for the maintenance of immune tolerance. The precise pathways and unique functions associated with Fas/FasL-mediated signaling in the immune system are known. The dual character of Fas/FasL-mediated immune regulation that induces beneficial or harmful effects is associated with the onset or development of immune disorders. Studies on mutations in genes encoding Fas and FasL gene of humans and mice contributed to our understanding of the pathogenesis of autoimmune diseases. Here, we review the opposing functions of Fas/FasL-mediated signaling, bilateral effects of Fas/FasL on in immune cells, and complex pathogenesis of autoimmunity mediated by Fas/FasL.
Introduction
Fas receptor (CD95, tumor necrosis factor receptor superfamily member 6) is a death receptor (DR) localized on the surface of various cells, which triggers a signal transduction pathway leading to apoptosis (1, 2). The interaction of Fas with its ligand FasL (FasL/CD95L) regulates numerous physiological and pathological processes that are mediated through programmed cell death (3). The beneficial and harmful effects of Fas-mediated apoptosis on the immune system were identified after the discovery (4©C6). Moreover, signaling downstream of Fas is intricately regulated by numerous pathway components (7©C9).
MRL-lpr/lpr mice bear mutations in the gene encoding Fas and serve as a widely used model for autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), SjögrenĪ»s syndrome (SS), and autoimmune lymphoproliferation syndrome (ALPS) (10, 11). Fas-mediated apoptosis of peripheral T cells, which represents a key mechanism that maintains immunological tolerance, is impaired in MRL-lpr/lpr mice. This mechanism, known as activation-induced cell death (AICD), deletes overactivated or autoreactive T cells in the periphery (12). The deletion of peripheral T cells by AICD is impaired in MRL-lpr/lpr mice, leading to increased autoreactive T cells that trigger the induction of autoimmune lesions in multiple organs (10, 13). Moreover, mutations in the gene encoding Fas occur in patients with ALPS (14, 15).
By contrast, FasL expression on the cell surface is specific to the immune system. For example, FasL expression by T cells is associated with their activation (4). FasL is unstable because it is shed from the cell surface through the action of certain enzymes (16, 17). When soluble FasL (sFasL) binds to Fas, cell proliferation, but not apoptosis, is induced (18). Mice with the gld/gld genotype bear mutations in the gene encoding FasL, and they are widely used as a model of autoimmune disease (19, 20). Moreover, mutations of the gene encoding FasL occur in patients with ALPS (14, 15, 21), and FasL is expressed by thyroid, endothelial, and corneal cells (22©C24). Expression of FasL by cells residing in Ī░immunoprivileged siteĪ▒ protects them from attack by activated or autoreactive lymphocytes (5).
This review describes the multiple functions of Fas/FasL in the immune system with focus on duality of Fas/FasL signaling in immune regulation and autoimmunity.
Fas-Mediated Apoptosis
Fas protein has 319 amino acids and the predicted molecular weight is 48 kDa. The mature protein is divided into three domains: an extracellular domain, a transmembrane domain, and a cytoplasmic domain. The extracellular domain consists of 157 amino acids with cysteine-rich domain. The transmembrane and cytoplasmic domains have 17 and 145 amino acids, respectively. Exons 1 through 5 encode the extracellular region. Exon 6 encodes the transmembrane region. Exons 7©C9 encode the intracellular region (1, 2).
Extrinsic and intrinsic pathways are the major DR-mediated pathways of apoptosis. Engagement of the DRs with cognate ligands including FasL induces recruitment and activation of the apoptosis-initiating proteases, such as caspase-8 and caspase-10, and then induces apoptosis through various molecules. By contrast, the binding of some cognate ligands to DR triggers transcriptional events leading to nuclear factor-kappa ”┬ (NF-”╩B)- or activator protein-1 (AP-1)-dependent pro-inflammatory cytokine expression (Figure 1). DRs, which are members of a subset of the TNF receptor superfamily known as death receptors, possess a cytoplasmic death domain (DD). Fas-mediated apoptosis proceeds through the extrinsic pathway via the binding to their respective receptors of ligands, such as FasL, tumor necrosis factor-alpha (TNF-”┴), lymphotoxin-alpha (LT-”┴), TNF-like protein-1A (TL1A), and Apo2L/TNF-related apoptosis-inducing ligand (TRAIL) (Figure 1). FasL is the ligand for the Fas receptor. TNF-”┴ and LT-”┴ are ligands for the TNF superfamily member 1A (TNFR1), TL1A is a ligand for TNF receptor superfamily member 25 (DR3), and TRAIL is a ligand for the TNF receptor superfamily member 10a (DR4/TRAIL-R1) or tumor necrosis factor receptor superfamily member 10b (DR5/TRAIL-R2). These receptors are members of a subset of the TNF receptor super family known as DRs. Engagement of DRs of their cognate ligands promotes recruitment and activation of the apoptosis-initiating proteases caspase-8 and caspase-10 within membrane receptor complexes (25). Apoptosis induced by the engagement of the DRs by their cognate ligands is well understood (26©C29) (Figure 1).
ĪĪ
FIGURE 1
www.frontiersin.org
Figure 1. Apoptotic signaling via Fas/FasL. Engagement of the death receptors (DRs) with their cognate ligands, such as FasL/CD95L, tumor necrosis factor-alpha (TNF-”┴), lymphotoxin-alpha (LT-”┴), TNF-like protein-1A (TL1A), and Apo2L/TNF-related apoptosis-inducing ligand (TRAIL), and their receptors promotes recruitment and activation of the apoptosis-initiating proteases, caspase-8, and caspase-10 and induces transcriptional events leading to nuclear factor-kappa ”┬ (NF-”╩B)-dependent pro-inflammatory cytokine expression.
Binding of DRs to their cognate ligands recruits one of two pivotal DD-containing adaptor proteins: Fas-associated protein with DD (FADD) or TNF receptor-associated DD (TRADD) (Figure 1). FADD controls cell death by recruiting caspase-8 and caspase-10, TRADD controls non-apoptotic functions by recruiting the DD-containing kinase receptor-interacting protein-1 (30), and the E3 ubiquitin ligases TNF receptor-associating factor 2 (TRAF2) and cellular inhibitor of apoptosis proteins (31). TRADD signaling activates phosphorylation cascades comprising the IKK kinase complex, which phosphorylates the inhibitor of NF-”╩B and the mitogen-activated protein kinases, c-jun N-terminal kinase (JNK), and p38 (Figure 1). The recruiting events initiate a transcriptional program to express genes that induce the synthesis of mediators of inflammation (7©C9). DRs can be divided into two categories based on the primary adaptor protein they bind to Fas/CD95/APO-1, DR4/TRAIL-R1 and DR5/TRAIL-R2 bind FADD and mediate mainly proapoptotic functions (Figure 1). By contrast, TNFR1 and DR3 bind TRADD and mediate mainly pro-inflammatory and immune-stimulatory activities (7©C9) (Figure 1).
In the Fas-mediated apoptotic pathway, binding of FasL drives Fas clustering and binding of Fas to FADD. FADD recruits caspase-8 and caspase-10 to form the death-inducing signaling complex (DISC) (25). DISC is activated by specific post-translational modifications of the DR, such as palmitoylation and O-linked glycosylation (32, 33). The DISC mediates autocatalytic processing and activation of caspase-8 and caspase-10, which propagate the death signal either through proteolysis of effector caspases such as caspases-3, caspase-6, and caspase-7. In type I cells such as thymocytes, processing by effector caspases is sufficient to induce apoptosis (Figure 1). By contrast, apoptosis requires caspase-8-mediated cleavage of BH3-interacting domain death agonist (Bid), which is a BH3-only protein that can promote the permeabilization of mitochondrial outer membranes and release of cytochrome c in type II cells such as B cells (Figure 1). Upon release from mitochondria, cytochrome c acts as a cofactor for the assembly of a cytosolic caspase-activating complex called the apoptosome, which propagates the caspase activation cascade (33). Thus, Fas signal has dual pathways of apoptosis and non-apoptosis in various cells.
Fas-Mediated T Cell Immune Regulation
The negative selection of autoreactive T cells in thymus is regulated by strong T cell signals that induce apoptosis through Bcl-2-interacting mediator of cell death (Bim), but not Fas (34). By contrast, apoptosis of activated peripheral T cells is controlled by the intrinsic and extrinsic pathways (34). Antigen stimulation via the T-cell receptor (TCR) contributes to antigen-specific T cell responses related to cell survival. TCR and Fas signaling are linked, and the AICD of peripheral T cells is controlled by interaction between TCR and Fas signaling (34).
Following TCR ligation, signaling through CD3 complex leads to recruitment of signaling molecules such as lymphocyte-specific protein tyrosine kinase (LCK), phosphorylation of ”Ų-chain-associated protein kinase 70 (ZAP70), linker for activation of T cells (LAT), phospholipase C”├1, VAV, SH2-domain-containing leukocyte protein 76, and protein kinase C”╚, in coordination with costimulatory molecules such as CD28 and CD4 (35, 36) (Figure 2).
ĪĪ
FIGURE 2
www.frontiersin.org
Figure 2. Interaction between Fas receptor and T-cell receptor (TCR) pathway. Fas-mediated apoptosis is initiated by the recruitment of Fas-associated protein with DD (FADD). FADD then recruits caspase-8 and forms a homodimer with caspase-8. Caspase-8 homodimers lead to the activation of caspase-3 and induce apoptosis. FADD also recruits caspase-8-like inhibitory protein (cFLIP) and form a heterodimer with caspase-8. cFLIP interacts with the IKK complex leading to nuclear factor-kappa ”┬ (NF-”╩B) activation. By contrast, ligation of TCR leads to recruitment of LCK and activation of ”Ų-chain-associated protein kinase 70 (ZAP70), which in turn induces tyrosine phosphorylation of linker for activation of T cells (LAT) and SH2-domain-containing leukocyte protein 76 (SLP76). SLP76 also induces the formation of complex that contains CARD-MAGUK protein-1 (CARMA1), BCL-10, and MALT1. BCL-10 and MALT1 are associated with active caspase-8 and cFLIP to promote the induction of antiapoptotic NF-”╩B target genes.
Homeostasis of peripheral T cells is maintained by three mechanisms as follows: unresponsiveness (anergy) of T cells, suppression by regulatory T cells, and AICD (37). AICD is initiated by repeated stimulation of the TCR through Fas-mediated apoptosis to control the effector T-cell population (38). Further, AICD is induced through the interaction between Fas and FasL, and activated T cells expressing Fas and FasL are killed either though suicide or by mutual interaction (39, 40). During the immune responses, antigen-stimulated effector T cells are activated to produce various inflammatory cytokines and growth factors. Although the tuning of TCR signaling is controlled by costimulatory molecules such as CD28 and programmed cell death protein-1 (41), the system that declines the activated T cells maintains peripheral tolerance. Overactivated effector T cells are harmful to the immune system, and should be deleted from the periphery. Therefore, AICD induced by Fas-mediated apoptosis plays a potent role in the peripheral immune system (37).
Moreover, impairment of AICD contributes to the onset or development of autoimmunity. Numerous studies of lpr/lpr or gld/gld mice focused on the relationship between AICD of peripheral T cells and autoimmunity (10, 11, 19, 20). These studies found that impaired AICD in lpr/lpr or gld/gld mice leads to T cell dysfunctions and the onset of autoimmune lesions in multiple organs (10, 11, 19, 20). Evidence indicates that autoreactive T cells are abundant in the periphery of lpr/lpr or gld/gld mice as well as in patients with ALPS (10, 11, 19, 20). By contrast, Fas-independent T cell apoptosis is induced by the direct interaction between TRAIL-R2 on T cells and TRAIL on Fas-deficient dendritic cells in MRL-lpr/lpr mice (42). Thus, member of the TNFR family, including Fas and TRAIL-R2, likely play a key role in the maintenance of peripheral immune tolerance.
By contrast, Fas mediates apoptotic and non-apoptotic pathways (28). Acting downstream of cellular caspase-8-like inhibitory protein (cFLIP) and TRAF2 in the Fas signaling pathway, T-cell proliferation is induced through NF-”╩B activation (43) (Figure 2). The cFLIP N-terminal cleavage products p43-FLIP and p22-FLIP induce NF-”╩B activation by binding to the IKK complex (44, 45) (Figure 2). Further, overexpression of cFLIP inhibits Fas-induced apoptosis of activated T cells (46, 47). Moreover, Fas signaling regulates peripheral T cell homeostasis by modulating the equilibrium between proliferation and cell death, for example, in naive and memory T cell subsets (48). Therefore, homeostasis of peripheral T cells may be maintained by the dual outocomes of FasL/Fas signaling.
Fas-Mediated B Cell Immune Regulation
The first report of the expression of FasL on the surface of B cells of wild-type mice but not from gld/gld mice demonstrated that activation of mouse B cells leads to the expression of FasL and killing of Fas-expressing target cells by B cells (49). Further, surface-localized FasL is expressed by human and mouse B cells, and the abnormal function of FasL+ killer B cells leads to a novel target for immune modulation in many disease settings (50©C52). Moreover, reduction of the number of FasL+CD5+ B cells is associated with exacerbated severity of arthritis and inhibits T-cell death in a TCR transgenic mouse model with collagen-induced arthritis (53). Deficiency of the FasL/Fas signaling pathway in humans leads to ALPS, which is most often manifested as autoimmune hemolytic anemia, thrombocytopenia, or leukocytopenia, caused by cell-type specific autoantibody production (54, 55). Thus, FasL/Fas interactions play a critical role in regulating the production of pathogenic autoantibodies. The killer B cells that produce these autoantibodies may represent a novel modality for inducing T-cell death to treat autoimmunity.
Moreover, the expression of FasL on B cells of individuals infected by human immunodeficiency virus (HIV) (56) and the induction of Epstein©CBarr virus of FasL expression by B cells markedly increases the sensitivity of Th cells to Fas-mediated T cell apoptosis (57). Although these data suggest that FasL expression by B cells might play a pathogenic role by inducing T cell death during viral infections, this hypothesis was not explored.
Findings that FasL+ B-chronic lymphocytic leukemia (B-CLL) cells kill a susceptible CD4+ T-cell leukemia cell line provided the first direct evidence implicating B cells in the induction of T cell apoptosis (52). FasL expression frequently occurs in patients with the aggressive form of B-CLL as well as in other human B-cell leukemias and lymphomas, most notably, multiple myeloma (58, 59). These results suggest that killer B cells induced by viral infections and tumor cells play a crucial role in the ability of virus-infected or cancer cells to evade the hostĪ»s immune system.
By contrast, Fas is highly expressed in activated and germinal center (GC) B cells. B cell-specific Fas deficiency is associated with the onset of autoimmunity (60), suggesting that Fas prevents the development of self-reactive GC B cells that escape normal regulatory controls and produce high amounts of immunoglobulin and autoantibodies (60, 61). Further, MRL-lpr/lpr mice produce circulating autoantibodies, such as rheumatoid factor as well as those against a single- and double-strand DNA and nuclear antigens (62, 63), indicating the contribution of FasL/Fas signaling in B cells to cellular processes such as differentiation, proliferation, death, and immunoglobulin production. Thus, Fas signal in B cell plays potent roles in the function and the pathogenesis of immune disorders.Unique Roles of FasL
FasL is a homotrimeric membrane protein that belongs to the TNF superfamily including CD178, CD95L, and apoptosis antigen-1 (Apo-1) ligand. Human FasL shares 81 and 78% amino acid sequence identity with its mouse and rat homologs, respectively. FasL comprises a single transmembrane domain, an intracellular domain harboring a proline-rich domain, and an extracellular domain. The latter contains an oligomerization domain required for oligomerization (64).
FasL expression on activated T cells is induced by stimulation via TCR, costimulatory molecules, and cytokine receptors (65). FasL expression is regulated by several transcription factors, including NF-”╩B, nuclear factor of activated T cells (NF-AT), early growth response gene family transcription factors, c-Myc, AP-1, secretory protein-1, and interferon regulatory factors (66©C72).
Matrix metalloproteinase-7 (matrilysin) cleaves FasL to generate sFasL, which is released into the extracellular milieu (73). Abnormal levels of trimeric sFasL (73, 74) are detected in sera from patients with large granular lymphocytic leukemia and natural killer (NK) cell lymphoma (3), and other cancers as well as patients with SLE (75). The level of serum sFasL correlated with disease progression (3, 75) suggesting that sFasL induces the apoptosis of Fas+ T cells to evade immune surveillance. Thus, sFasL possesses proapoptotic and antiapoptotic properties depending of cell type and the microenvironment.
Apoptosis induced by FasL requires extensive oligomerization of the Fas receptor to activate the DISC (76), and membrane-bound FasL (mFasL) and sFasL can bind the Fas receptor. However, the naturally cleaved form of sFasL does not form oligomers with the Fas receptor, and therefore sFasL fails to induce apoptosis. Further, sFasL inhibits mFasL-mediated apoptosis through steric hindrance of its binding to the Fas receptor, indicating that mFas and sFasL have opposite functions that affect cell survival (77©C79). For example, sFasL shed by a disintegrin and metalloproteinases or MMPs induces proliferation of fibroblast-like synoviocytes in patients with RA through activation of v-akt murine thymoma viral oncogene homolog (Akt1), extracellular signal-regulated kinase, and JNK (Figure 3) (80). Thus, sFasL has dual functions for cell death and survival.
ĪĪ
FIGURE 3
www.frontiersin.org
Figure 3. Differential signaling pathway by membrane-bound FasL (mFasL) and soluble FasL (sFasL). mFasL leads to the recruitment of FADD and activates caspase pathways. mFas promotes apoptosis. mFasL is shed by a disintegrin and metalloproteinases (ADAM) or MMPs in the pathological or physiological condition. For instance, sFasL strongly activates extracellular signal-regulated kinase (ERK), PI3K/Akt, and c-jun N-terminal kinase (JNK) to induce proliferation and inhibit apoptotic activity of the fibroblast-like synoviocytes. Differential signal by the engagement of sFasL is determined by the cell type and the pathologic condition.
Loss-of-function mutations in the genes encoding murine and human FasL causes a phenotype similar to that of patients with lymphadenopathy and autoimmune disease because of decreased apoptosis in Fas+CD4−CD8− T lymphocytes and the production of autoantibodies (81, 82). In patients with ALPS, germline mutations of FasL gene are associated with defective apoptosis (83). Patients with rare heterozygous FasL gene mutations are classified as type Ib-ALPS (84).
The tissue distribution of FasL, which is predominantly expressed by activated T cells and NK cells, is limited (3). FasL is constitutively expressed by stromal cells of the retina and Sertoli cells of the testis, respectively, which are immunoprivileged tissues. In such tissues, FasL expression leads to the death of invading Fas+ cells. The susceptibility of the eyes of gld/gld mice to inflammation (85) and their rejection of corneal allografts prove that FasL plays an important role in sites of immune privilege (24). The ocular immune privilege is believed to be one mechanism by which the visual axis is protected from dangerous immune reactions. Therefore, immunoprivileged site is maintained by the sequestration of any antigens, the lack of lymphatic damage, and the blood-tissue barrier (5). In addition, both Fas and FasL are present on thyroid cells in patients with autoimmune thyroiditis (HashimotoĪ»s thyroiditis) (22). This suggests that Fas/FasL interactions among thyroid cells contribute to the pathogenesis of autoimmune thyroiditis with tissue destruction (22).
FasL is expressed in tumors such as colorectal carcinomas, melanomas, head and neck carcinomas, and myelomas (86). The level of FasL produced by non-immune cells can induce apoptosis in Fas+ T cells that recognize tissue-specific antigens to evade immune surveillance (86). Moreover, a variety of cell types can express FasL in response to different stimulatory conditions, including macrophages infected with HIV, hepatocytes treated with ethanol, leukemia cells exposed to chemotherapy drugs, as well as cancer cells (87, 88). Thus, the unique functions of FasL contribute to various physiological or pathological processes that are not associated with the immune system.
The Fas/FasL System in Autoimmunity
The contribution of Fas-mediated apoptosis to the onset and development of autoimmunity was established by studies of patients with autoimmune diseases and animal models (14, 15, 89, 90). ALPS is an inherited disorder of the systemic immune system that involves a spontaneous mutation in the Fas or FasL gene (14, 15, 21). Approximately two-thirds of patients with ALPS bear mutations in the gene encoding Fas (type Ia) (91, 92). By contrast, there are few reports of mutations in the genes encoding FasL gene (type Ib) and caspase-10 gene (ALPS type IIa) (92©C95), and these mutations are not detected in patients with ALPS type III (96).
The clinical features of patients with ALPS are splenomegaly, lymphadenopathy, and hepatomegaly, which are caused by the accumulation of polyclonal lymphocytes as well as autoimmune lesions in multiple organs. The risk of malignant lymphoma is increased in patients with ALPS (97). Peripheral T cells expressing TCR”┴/”┬, but not CD4 and CD8 [CD4−CD8− double negative (DN)], proliferate, and their population expands in the patients with ALPS due to an impaired AICD caused by a defect in the Fas/FasL system (98). Further, the abnormal programming of Fas-deficient T cells before the DN T-stage is caused by impaired signaling through the mTOR pathway (99). Moreover, a defect in B-cell selection occurs in patients with ALPS, which is caused by impaired class-switch recombination and somatic hypermutation of the genes encoding immunoglobulins (100). Polymorphisms in the genes encoding Fas and FasL are associated with the susceptibility and severity of autoimmune lesions in patients with RA (101, 102) as well as in patients with primary SS (103).
To understand the mechanism of Fas/FasL-mediated apoptosis in vivo, MRL-lpr/lpr mice were used as a model of susceptibility to autoimmune disease before spontaneous mutation of the gene encoding Fas was discovered to affect the onset of autoimmunity in the mice (10). Further, gld/gld mice bearing a spontaneous mutation of the gene encoding FasL are employed as a model for autoimmune disease as well (19, 20). Lymphoproliferative lesions in lpr/lpr and gld/gld mice demonstrate that Fas/FasL-mediated apoptosis plays a critical role in controlling the maintenance of peripheral lymphocytes. In particular, Fas/FasL-mediated apoptosis contributes to the AICD of T cells in the periphery (4). Moreover, peripheral T cell apoptosis is induced by FasL expression by macrophages, and apoptotic T cells are promptly phagocytosed by these macrophages, depending on their level of Fas expression (104). In these experiments, normal T cells of C57BL/6 (B6) mice were engulfed by macrophages of B6-lpr/lpr mice, which lead to enhanced Fas expression by donor T cells through IFN-”├/IFN-”├-receptor signaling (104). These findings revealed that the control of Fas expression by macrophages plays an essential role in maintaining T cell homeostasis in the peripheral immune system.
By contrast, the expression of Fas or FasL occurs in target organs of patients with autoimmune diseases and in animal models (105, 106). High levels of sFasL are present in the synovial fluid of patients with RA (107), which may be associated with Fas-mediated apoptosis of synovial cells, but not the AICD of T cells. Further, sFasL present in the synovial fluid of patients with RA inhibits angiogenesis in RA lesions (108). These observations are consistent with decreased levels of FasL mRNA expression in lacrimal gland tissues and peripheral blood lymphocytes as well as increased levels of Fas and FasL in salivary gland tissues of patients with SS (109, 110). These findings implicate Fas-mediated apoptosis in the destruction of target salivary gland tissue.
When we treated SS model mice with an anti-murine FasL-specific monoclonal antibody to protect against Fas-mediated apoptosis of the target salivary gland cells (111), we unexpectedly observed exacerbation of the autoimmune lesions in the salivary and lacrimal glands (111). We found that sFasL is processed by autoantigen-specific CD4+ T cells concomitant with metalloproteinase-9 expression (65, 111), indicating that increased sFasL expression inhibits the normal AICD of T cells and leads to the proliferation of autoreactive T cells in this SS model. Moreover, although mFasL, but not sFasL, is essential for cytotoxic activity that guards against lymphadenopathy and autoimmunity (29), it remains unclear whether the relationship between mFasL and sFasL contributes to the molecular mechanisms of AICD. Thus, the impairment of Fas/FasL system in the peripheral immune tolerance considerably contributes to the onset or development of a lot of autoimmune diseases.
Concluding Remarks
Some examples regarding the relationship between various cells and Fas/FasL expression are listed in Table 1. Numerous molecules precisely regulate Fas-mediated apoptosis through complicated signaling cascades. Apoptotic and antiapoptotic signaling pathways in T cells are controlled by the interaction between TCR and Fas signaling pathways and the expression of FasL. Further, the Fas/FasL system plays potent roles in B cell biology. The unique functions of FasL contribute to tumorgenesis, infection, immune disorders as well as to the outcomes of tissue transplantation. Finally, the multiple functions of Fas/FasL-mediated regulation maintain immune tolerance.
Frontiers | Dual Role of Fas/FasL-Mediated Signal in Peripheral Immune Tolerance | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2017.00403/fullĪĪ
Apoptosis induced by a death factor
Shigekazu Nagata
Medical Chemistry
Research output: Contribution to journal › Article
Abstract
Fas is a type I membrane protein belonging to the TNF receptor family. The Fas ligand (FasL) is a member of the TNF family, and induces apoptosis in Fas-bearing cells. The Fas-mediated apoptosis occurs in enucleated cells, and is executed by members of the ICE (interleukin-l”┬-converting cn/ymc)/Ccd-3 protease family (caspases), and can be inhibited by Bcl2/Ced-9. The Fas cytoplasmic region carries a domain (a death domain) which is necessary and sufficient to mediate apoptosis. In the current model, the Fas engagement causes trimeriyation of the Fas death domain, which recruits a caspase (caspase 8) to the receptor via an adaptor molecule called FADD/ MORT 1. This seems to cause self-activation of the protease, and leads to the sequential activation of members of the caspase family. The Fas system is involved in peripheral clonal deletion, and in down-regulation of the immune reaction. Mouse mutations of lymphoproliferation (Ipr) and generalized lymphoproliferative disease (gld) which cause lymphadenopathy and splenomegaly, and accelerate autoimmune disease, are loss-of-function mutations in the Fas and FasL genes, respectively. FasL in activated T cells and NK (natural killer ) cells works as an effector of CTL and NK cells to remove the cells infected by virus, or cancerous cells. If mice were injected by the rccombinant FasL, the mice died with hours by the liver damage, suggesting that overfunction of this system causes disease such as hepatitis. In fact, the Fas ligand-neutralizing molecules efficiently prevented deelopment of hepatitis in mouse model systems. Using an ELISA system, we detected a significant amount of the soluble FasL in the serum of human patients who carry NK lymphoma or large granular lymphocytes leukemia, showing systemic tissue damage such as hepatitis and neutropenia.Apoptosis induced by a death factor Ī¬ MD Anderson Cancer Center
https://mdanderson.elsevierpure.com/en/publications/apoptosis-induced-by-a-death-factorĪĪ
A subclass of dendritic cells kills CD4 T cells via Fas ...
https://rupress.org/.../183/4/1789/58376/A-subclass-of-dendritic-cells-kills-CD4-T-cells
Dendritic cells (DC), the most efficient antigen-presenting cells, are well equipped for activation of naive CD4+ T cells by their expression of high levels of major histocompatibility complex and costimulator molecules. We now demonstrate that some DC are equally well equipped for killing these same T cells.ĪĪ
HYPOTHESIS AND THEORY ARTICLE
Front. Immunol., 12 July 2012 | https://doi.org/10.3389/fimmu.2012.00196
The potential of Fas ligand (apoptosis-inducing molecule) as an unconventional therapeutic target in type 1 diabetes
Abdel Rahim A. R. Hamad1*, Kristin Arcara2, Sophia Uddin1 and, Thomas Donner3
1 Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, USA
2 Department of Pediatrics, Johns Hopkins University School of Medicine, Baltimore, MD, USA
3 Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA
The development of type 1 diabetes (T1D) is driven by autoreactive T cells that attack and destroy the insulin-producing ”┬-cells in pancreatic islets, forcing patients to take multiple daily insulin injections. Insulin therapy, however, is not a cure and diabetic patients often develop serious long-term microvascular and cardiovascular complications. Therefore, intensive efforts are being directed toward developing safe immunotherapy for the disease that does not impair host defense and preserves ”┬-cells, leading to better glycemic control than exogenous insulin therapy. Engineering therapies that differentially cripple or tolerate autoreactive diabetogenic T cells while sparing protective T cells necessary for maintaining a competent immune system has proven challenging. Instead, recent efforts have focused on modulating or resetting the immune system through global but transient deletion of T cells or B cells using anti-CD3 or anti-CD20 mAb, respectively. However, phase III clinical trials have shown promising but modest efficacy so far with these approaches. Therefore, there is a need to identify novel biological targets that do not fit the classic properties of being involved in adaptive immune cell activation. In this prospective, we provide preclinical evidence that targeting Fas ligand (FasL) may provide a unique opportunity to prevent or cure T1D and perhaps other organ-specific autoimmune diseases without causing immune suppression. Unlike conventional targets that are involved in T and B lymphocyte activation (such as CD3 and CD20, respectively), FasL is an apoptosis-inducing surface molecule that triggers cell death by binding to Fas (also known as CD95 Apo-1). Therefore, targeting FasL is not expected to cause immune suppression, the Achilles Heel of conventional approaches. We will discuss the hypothesis that targeting FasL has unique benefits that are not offered by current immunomodulatory approaches.
FIGURE 2. FasL interaction with its receptor (Fas) triggers cell death. Binding of FasL to Fas induces the apoptosis of Fas-bearing cells by activation of the caspase signaling cascade leading to cleavage of nuclear DNA and proteolysis of vital cellular proteins.
Frontiers | The potential of Fas ligand (apoptosis-inducing molecule) as an unconventional therapeutic target in type 1 diabetes | Immunology
https://www.frontiersin.org/articles/10.3389/fimmu.2012.00196/fullĪĪ
Proc Natl Acad Sci U S A. 1997 Jun 10; 94(12): 5986©C5990.
Fas-ligand: Privilege and peril
Douglas R. Green and Carl F. Ware
La Jolla Institute for Allergy and Immunology, San Diego, CA 92121
For over 100 years, immunologists have recognized that there are sites in the body in which immune responses do not occur. These immunologically Ī░privilegedĪ▒ sites have long held the promise of solving the problems of autoimmunity and graft rejection, because somehow they violate the accepted rules of immunology, allowing foreign agents and tissues to persist. In the last year, interest has focused on a molecule called Fas-ligand (FasL; also called CD95L or Apo-1L), which appears to be required for some tissues to display such a privileged status (discussed below). FasL functions to induce apoptotic cell death in most cells that express its receptor, Fas (1). Fas-bearing cells include cells of the immune system, and thus FasL functions in immunological privilege in this way: tissues that naturally express FasL kill infiltrating lymphocytes and inflammatory cells. If simply placing such a molecule into any tissue of choice would confer privilege by killing off any invading immune cells, such that the tissue would not be destroyed in a transplant rejection, then any recipient could accept such a graft. Investigators set out to bring home this Holy Grail of transplant biology.
Meanwhile, FasL was shown to play other roles in the body. As discussed in more detail below, activated cytotoxic T lymphocytes (and other cells) often express high levels of FasL and the ability of FasL to kill cells bearing Fas accounts for some destructive effects mediated by these cells. FasL not only protects tissues from immune assault, but also can damage those tissues that express Fas. Privilege and peril. Blocking the function of FasL is clearly one key to preventing tissue damage under a number of different circumstances.
Despite the promise afforded by this remarkable receptor/ligand pair, things have become less simple, and two papers previously published in the Proceedings (2, 3) add still more levels of complexity to what is already a complicated story. One of these papers (2) shows that naturally occurring alleles of FasL have dramatically different abilities to trigger apoptosis through Fas, suggesting that in different settings stronger or weaker FasL function might be favored. The other paper (3) challenges the idea that FasL can confer immunological privilege, in that expression of FasL in the pancreatic islets of FasL-trangenic mice appears to induce an inflammatory infiltrate, and these engineered islets are not protected from graft rejection in allogenic recipients.
Before FasL was characterized, it was suspected that it would be intriguing. Ligation of the Fas molecule with antibodies is a potent inducer of apoptosis in different cell types (4). Mice (5) and humans (6, 7) with a defect in Fas expression or function show a profound lymphoaccumulative disorder (the lpr phenotype), associated (in mice) with a dramatic acceleration of age-associated autoimmune phenomena. Genetic evidence suggested defects in the FasL gene result in a similar phenotype called gld (8). This was, in fact, the case (9), and suggested a role for FasL in immune homeostasis rather than surveillance, because these animals display a lymphoaccumulative disorder with accelerated autoimmune syndromes, but no obvious defects in antiviral or antitumor defenses. Thus, while FasL expression on T lymphocytes (1) and natural killer cells (10) is clearly a mechanism by which these cells can kill other cells, it seemed likely that the targets were susceptible lymphocytes, i.e., those lymphocytes that accumulate in animals with defects in either the ligand or receptor. The conclusion was that a major function of FasL/Fas interactions is to limit lymphoid expansion via lymphoid-lymphoid interactions.
This view was strongly supported by a number of studies on activation-induced apoptosis in T cells. Engaging the T cell receptor on previously activated or transformed T cells (11©C14) up-regulates expression of FasL and Fas, and the cells then undergo apoptosis as a consequence of FasL/Fas interactions. Activation-induced apoptosis also accounts for the phenomenon of peripheral deletion in vivo, in which T cells responding to a strong antigenic stimulus decrease in number over time, a process that may be important in immune homeostasis (Fig. (Fig.11 Left). Peripheral deletion is at least partially defective in animals deficient in Fas (15, 16), lending support to the view that expression of this ligand-receptor pair is important for depleting excess lymphocytes after an immune response.
ĪĪFigure 1
Some immunological effects of FasL. Chronically activated T lymphocytes express both Fas and FasL, and in conventional tissues (Left) this can result in apoptotic death of the T cells (peripheral deletion) and induction of apoptosis in other Fas-expressing cells. Immunologically privileged tissues (Right) constitutively express FasL, and infiltrating T cells and granulocytes rapidly undergo apoptosis. Thus, the tissue is protected from any damage that might result from an immune response. In some tissues, however (Center), FasL induces a granulocytic infiltration, which can damage the tissue. The conditions that favor one or the other of these contrasting effects of tissue FasL are unknown.
Substantial evidence indicates that FasL is a trimer with global structural features in common with related ligands in the tumor necrosis factor (TNF) superfamily. The interaction of FasL with its receptor is not fully elucidated, but we can make reasonable guesses about its nature based on the crystal structure of the ligand-receptor complex of the related ligand lymphotoxin-”┴ (LT”┴) and one of its receptors, TNFR60 (17). As shown in Fig. Fig.2,2, we expect that FasL functions as a trimer, clustering three Fas molecules. The face formed by two FasL subunits interacts with one chain of Fas (Fig. (Fig.22B). This model becomes especially interesting in light of the finding by Kayagaki et al. (2), who found that a polymorphism in FasL dramatically affects its ability to induce apoptosis in target cells. The two residues affected by this polymorphism are identified in the structure in Fig. Fig.22B. One of these, residue E/G218, is predicted to interact directly with residues on Fas (based on its counterpart in LT”┴). These probably include charged residues, D and K, based on proximity analysis, and the presence or absence of a negative charge on residue 218 therefore might affect its interactions. In contrast, the other residue, T/A184, is not predicted to interact with residues on Fas, although it is possible that the N-terminal structure of Fas (which is not as restricted by disulfide bonds as in the TNFR60 molecule upon which this model is based) might be available for interaction with residues (including 184) on the ligand. However, the likelihood that E/G218 directly interacts with the receptor suggests that the difference in the function of the two forms of FasL reflect differences in affinity of interaction. A possible reason such a polymorphism might exist is discussed below.
Figure 2
The E218G allelic substitution lies in the receptor binding region of Fas Ligand. (Upper) Space-filling depiction of LT”┴ TNFR60 ligand©Creceptor complex from the crystal structure derived by Banner et al. (17) (viewed with RasMol). The three receptor (R) chains (gold) surround the LT”┴ subunits (green) that form the trimeric ligand. The upper left panel (top) is viewed from the perspective of the receptor-expressing cell with the receptorĪ»s N terminus extending away from the reader; in the right panel (side) the N terminus of the elongated receptor protrudes away from the cell surface. (Lower) Location of the T184A and E218G polymorphisms of FasL in the structure of LT”┴. Residues Phe-110 (red) and Ser-70 (blue) of LT”┴ are equivalent to FasL 218 and 184 as identified by sequence alignment of TNF, LT”┴, and FasL (Pam250 matrix) and constrained by positions of conserved residues in the D-E and B-C ”┬-strands of LT”┴. [”┬-strand nomenclature is that defined by Eck (37)]. Left side shows the ligand-receptor complex (side view) and right side depicts the binding site (with R1 removed) rotated 90ĪŃ clockwise, exposing the contact residues. Amino acids that contact receptor with a surface area >20 Å2 (17) in the Ī░aĪ▒ and Ī░cĪ▒ LT”┴ subunits (Ī░cĪ▒ subunit, dark gray; Ī░aĪ▒ subunit, light gray).
Our comfortable notions of the primary role of FasL in immune regulation were upset (albeit happily) by the realization that functional FasL plays an essential role in the phenomenon of immune privilege mentioned above (Fig. (Fig.11 Right). After viral inoculation into a classically Ī░privilegedĪ▒ tissue, the anterior chamber of the eye, lymphocytes, and granulocytes that are recruited undergo apoptosis via exposure to resident FasL on the epithelial surfaces such that no tissue injury occurs (18, 19). This apoptosis does not occur in the eyes of animals with defective FasL (the gld defect), and the resulting uncontrolled inflammation destroys the tissue. FasL is thus necessary for the maintenance of the privileged status of the eye.
A striking protective effect of FasL expressed in the testes was observed by transplantation of allogenic testis to the kidney capsule of recipient mice (20). If the donor animal was defective in FasL or the recipient in Fas, the foreign tissue was rapidly rejected by a vigorous immune response, but when both ligand and receptor were functional the graft was maintained. This remarkable observation (and that concerning the anterior chamber of the eye) raised expectations that allogeneic tissues could be protected by ectopic expression of FasL, with obvious consequences for transplantation. Soon thereafter, it was shown that syngeneic myoblasts expressing ectopic FasL effectively protected allogeneic pancreatic islets coimplanted under the kidney capsule of animals made diabetic by streptozotocin treatment (21). These grafts, which were quickly rejected if myoblasts did not express FasL, maintained function for an extended period of time. Consistent with this was the observation that murine or allogeneic rat islets showed delayed rejection when coimplanted with FasL-expressing testes tissue in rats (22).
One of the most successful forms of transplantation in humans is that of corneas, with less than one-third rejection after 5 years, without tissue matching or immunosuppression. Recently, it was shown that human corneas express functional FasL (23), raising the possibility that this molecule acts to protect these grafts in humans. Examination of corneal transplants in mice supported this idea; while approximately 45% of allogeneic cornea transplants survived for an extended period (as described by others), no graft survival was seen if the cornea expressed defective FasL (gld) or the recipients had a defect in Fas expression (lpr). As with the testes, protection of allogeneic grafts was dependent upon the presence of functional FasL.
One other recent example of FasL-dependent immune privilege was described in a different context. A number of different murine and human tumors, including many nonlymphoid tumors, have been observed to constitutively express functional FasL (24©C26). For example, a FasL-expressing melanoma was capable of inducing potent antitumor immunity, providing that the host was defective in Fas expression (25). This suggested that the mechanism responsible for protecting tissues from autoimmune destruction during inflammatory responses, or during graft rejection, also could function to protect cells from that tissue from immune surveillance after transformation.
These observations strongly implicate nonlymphoid FasL in the control of immune responses, via induction of apoptosis in infiltrating lymphocytes and granulocytes. However, we also know that FasL can induce tissue damage. In graft-vs.-host disease, the ability of the graft effector cells to express functional FasL contributes to the destructive assault (27, 28). Anti-Fas antibody induces apoptosis in hepatocytes in vivo (29), and this has led to the idea that FasL-induced apoptosis of these cells contributes to some forms of hepatitis.
Recently, another interesting twist on FasL-induced apoptosis was reported in Hashimoto thyroiditis (30). Thyrocytes express functional FasL constitutively (as a mechanism of immune privilege?), but normally do not express Fas. However, in Hashimoto thyoiditis patients, the thyrocytes do express Fas, and these cells undergo apoptosis. In vitro, normal thyrocytes express Fas after exposure to interleukin-1 (IL-1), and the ensuing apoptosis is blocked by antibodies that disrupt Fas/FasL interactions. The possible protective nature of FasL on thyrocytes becomes the mechanism of their destruction. Why the system is Ī░wiredĪ▒ in this way is unclear, but it is likely that this effect contributes to the disease process.
All of these functions of FasL, whether involved in protection or promotion of tissue destruction, are consistent with the idea that FasL engages Fas to induce apoptosis, and all can be explained on this basis. However, a new perspective on these studies came with the observations of Seino et al. (31), who reported that FasL on tumor cells can induce a granulocyte-mediated rejection reaction (Fig. (Fig.11 Center). Tumors expressing FasL, implanted subcutaneously or intraperitoneally, induced a granulocyte infiltrate that was dependent upon functional Fas on the bone marrow-derived population, and the rejection was followed by a T cell-dependent anti-tumor immunity that persisted. These observations are inconsistent with an immunosuppressive effect of FasL on tumors discussed above, despite the fact that in some cases the same tumor lines were used in the contrasting studies.
Now, a new study further challenges the immunoprotective effect of FasL for graft rejection. Allison et al. (3) report that expression of functional FasL in the pancreatic islets of transgenic mice failed to protect these islets from allogenic transplant rejection when placed under the kidney capsule of recipient mice. As with the tumor cells discussed above, the presence of FasL induced a granulocytic infiltrate in the transgenic animals themselves, which damaged (but did not destroy) the islets. Because this observation was incompatible with the results of others (20, 21), the authors re-examined the fate of allogeneic testes grafts. In these studies they failed to observe differences in the rejection of grafts from normal versus FasL-defective gld mice. However, previous studies have shown that age of the testes graft is an important variable in this effect, such that grafts from young mice can resist rejection whereas testicular tissue from adult mice may be rejected (32). Thus, slight age differences might account for the acceptance or rejection of testes grafts in the different studies. It will be interesting to examine the influence of age on FasL expression (and its relation to immune privilege) in this tissue.
We are nevertheless still left with what appear to be irreconcilable differences in the results from different laboratories. These differences cannot be readily attributed to the polymorphism in FasL described by Kayagaki et al. (2), because, for example, Bellgrau et al. (20) used testes bearing either allele (BALB/c or C57BL/6) and observed protection from rejection in both cases. Cornea grafts with the less potent allelic form of FasL were accepted at a reasonable frequency (23), and this less active FasL expressed in myoblasts was used by Lau et al. (21) to protect islets from rejection. All of the studies on immunologic privilege in the eye were performed in animals bearing the less potent FasL. Nevertheless, a more careful comparison of the efficacy of the two allelic forms of FasL in these systems should be informative.
While the polymorphism in FasL does not appear to explain the discrepancy in the above results, these different effects of FasL might help to explain the polymorphism. Taken together, the studies suggest that FasL can prevent immune responses by inducing death of lymphocytes, cause damage by killing nonlymphoid Fas+ cells of the tissues, or induce potent granulocytic inflammatory responses, depending on the circumstances. Thus, any benefit of FasL expression is offset by the damage it can cause. If the ability to endow a tissue with immunologic privilege correlates with the induction of granulocytic inflammatory responses in the two forms of FasL reported by Kayagaki et al. (2) (that is, if the same ligand-receptor interaction is involved in both types of effects, as expected), then a Ī░weakerĪ▒ FasL avoids damaging inflammatory responses at the expense of less immune privilege. Clearly, one or the other alleles will be favored in different settings. From this point of view, it may be interesting that several autoimmune-prone mouse stains appear to carry the Ī░weakerĪ▒ form of FasL.
Returning to the issue of whether FasL protects grafts from rejection or not, one possible explanation for the differences in the effects of FasL observed in these different studies might involve the site of transplant. For example, while corneas grafted to the eyes of recipients often are accepted, heterotopic cornea grafts to the skin are rejected (33). However, this cannot simply be due to differences in the effects of Fas ligation on different cell types (e.g., skin), because the proinflammatory effect of FasL appears to depend upon the presence of Fas on bone marrow-derived cells, not stroma (31). (Of course, this does not rule out the possibility that bone marrow-derived cells in the skin are important for this effect.) Ligation of Fas can induce secretion of IL-8 (34), which might contribute to the ensuing inflammation. Interestingly, the Fas-mediated intracellular signaling events leading to IL-8 secretion versus apoptosis appear to be different, suggesting that other factors might favor one outcome of Fas-ligation over the other. Thus, at some sites (or in some animal colonies?) additional signals to lymphoid and myeloid cells might result in FasL-induced cytokine/chemokine release rather than apoptosis. In that setting, FasL will be proinflammatory. On the other hand, FasL in sites such as the eye can induce a remarkably rapid apoptosis in normal splenocytes (18, 19), which might suggest that other additional factors contribute to increased susceptibility to Fas-mediated apoptosis. One candidate for such a Ī░sensitizerĪ▒ is interferon ”├ (35). Thus, FasL may either promote or inhibit inflammation (depending on whether it induces chemokines or apoptosis), and the choice between these outcomes may be determined by the presence or absence of other factors.
While this argument can result in repetitive motion injury due to excessive hand waving, it is testable and makes some sense. The possible requirement for a sensitizing factor necessary to promote immune privilege by FasL might account for the strikingly different observations on FasL protection of islet grafts. If syngeneic myoblasts but not islet cells provide such a second signal, then myoblasts expressing FasL will protect from graft rejection while FasL-transgenic islet cells will not. FasL-bearing tumors that promote IL-8 production rather than apoptosis will express different surface or soluble mediators than those that induce cell death in targets. Identification of the responsible secondary factors will be important for the manipulation of FasL effects.
There is another explanation for the differences in the outcome of islet cell transplants that may involve the treatment of the recipients. In the studies by Lau et al. (21) animals had been treated with streptozotocin to induce diabetes, a treatment that is known to be immunosuppressive in some situations (36). The role of this treatment in these studies may have to be assessed more carefully.
We have gotten used to referring to the Fas molecule as a Ī░death receptor,Ī▒ and to thinking of the function of FasL entirely in terms of inducing apoptosis. Until recently, TNF and lymphotoxin were thought of in the same way (and still carry their sinister monikers). However, there is a side to FasL that involves promotion of inflammatory responses, and an understanding of where and when this function dominates its effects is critically important. We will not easily give up our dreams of using FasL, perhaps with necessary partner molecules, to control the rejection of grafts or limit autoimmune destruction. But itĪ»s not going to be as easy as we might have thought.Fas-ligand: Privilege and peril
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC33671/ĪĪ
Adenovirus-Mediated Expression of Fas Ligand Induces ...
https://www.liebertpub.com/doi/10.1089/hum.1997.8.8-955
Fas ligand (FasL) mediates apoptosis of Fas-bearing cells and is expressed on a limited number of tissues, predominantly activated T lymphocytes. We describe the construction and biological activity of a replication-deficient type-5 adenovirus encoding murine FasL under the control of the cytomegalovirus (CMV) promoter (adCMV-FasL).ĪĪ
Large Granular Lymphocyte - an overview | ScienceDirect Topics
https://www.sciencedirect.com/topics/medicine-and-dentistry/large-granular-lymphocyte
In addition, NK cells can kill target cells using Fas/Fas ligand (FasL)©Cmediated apoptotic pathways. NK cells bear FasL on their surface and can kill Fas-bearing target cells. NK cells do not express specific antigen receptors. Rather, NK cells use two categories of receptors to deliver either activation or inhibition signals.ĪĪ
Transcriptomic and epigenetic mechanisms underlying ...
https://www.nature.com/articles/s41590-019-0582-z
Jan 20, 2020 Īż The lung is inhabited by resident alveolar and interstitial macrophages as well as monocytic cells that survey lung tissues.
Cited by: 1
Publish Year: 2020
Author: Eniko Sajti, Verena M. Link, Verena M. Link, ZhengĪĪ
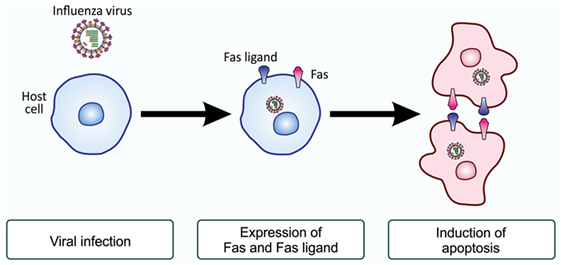
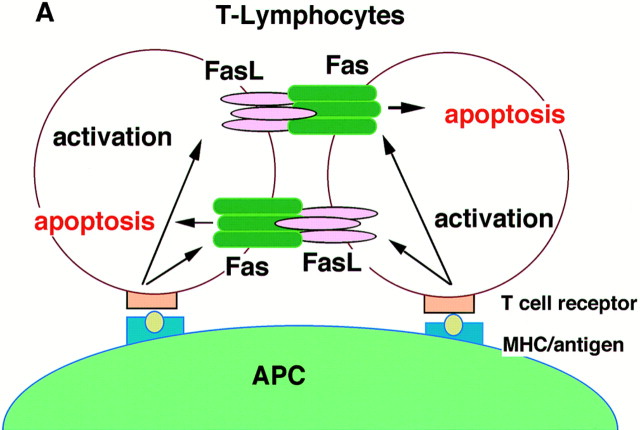
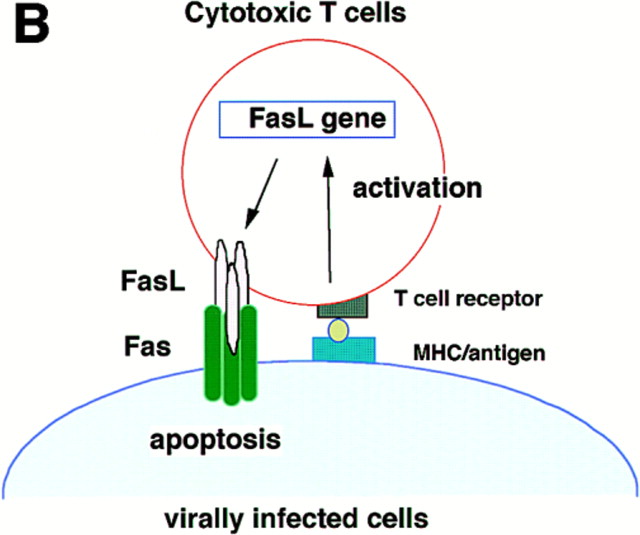
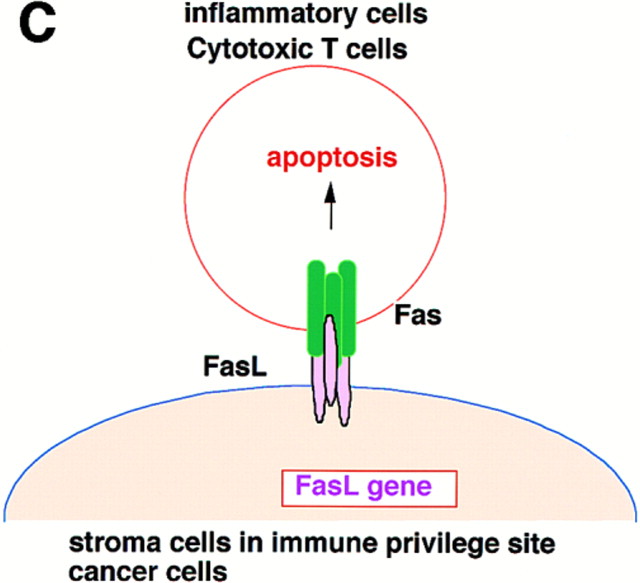
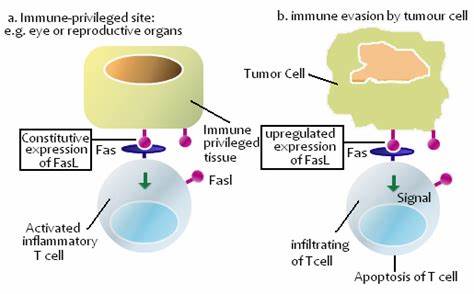
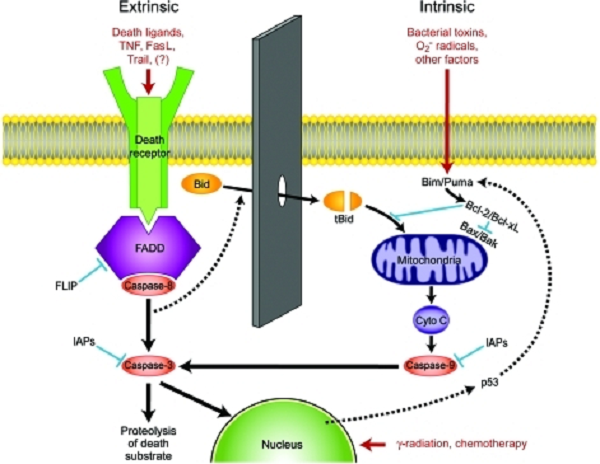
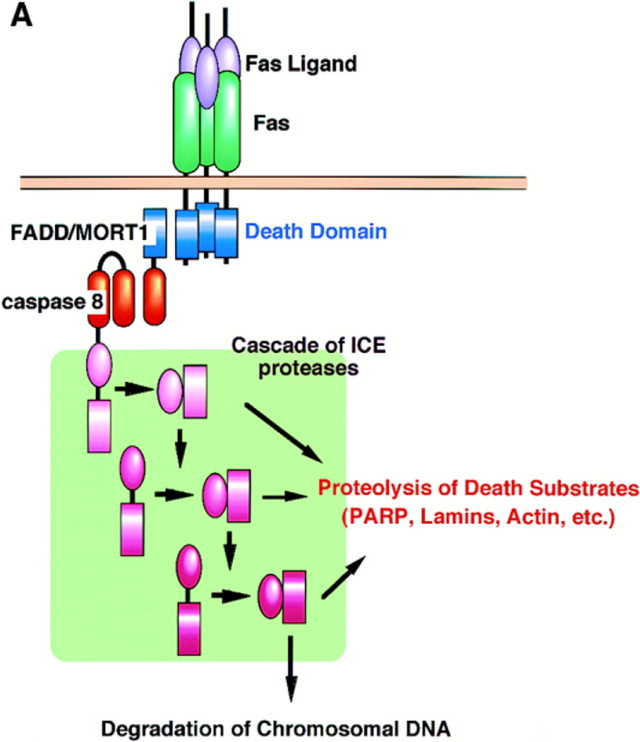

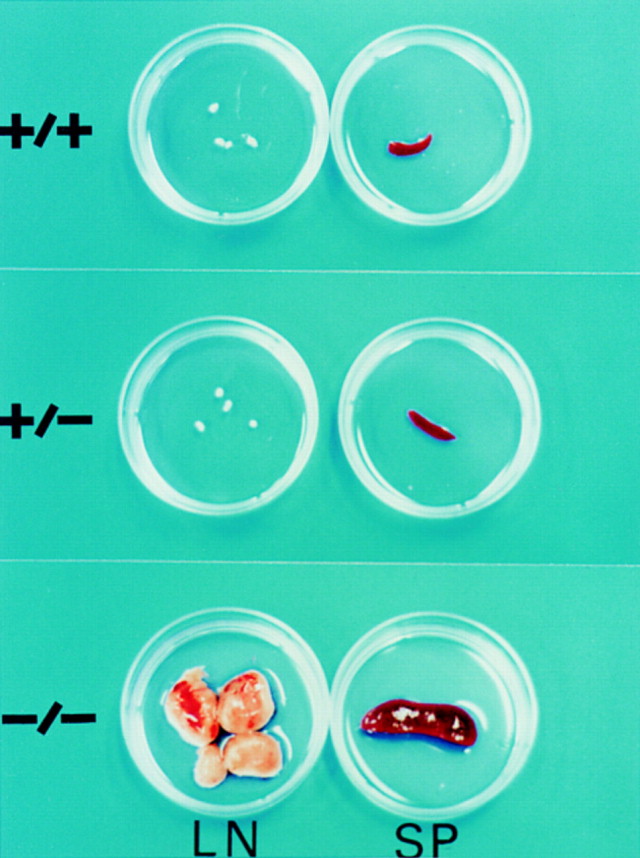
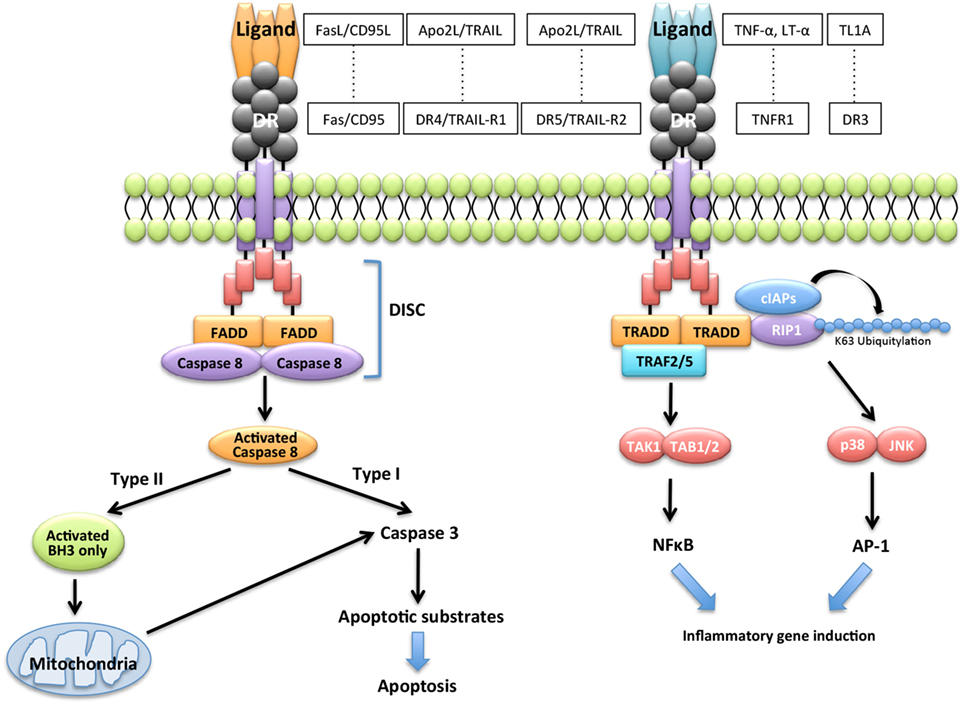
%20pathway..jpg)
%20and%20soluble%20FasL%20(sFasL)..jpg)