Vitamin c is the vital cofactor in prophyl hydroxylase that degrade HIF-1
Uric acid is the main antioxidant in plasma, forms urate radical, called urate hydroperoxide~oxidizes peroxireduxins, consume nitric oxide,
vitamin c regenerate uric acid via reducing urate radical.
Urate hydroperoxide oxidizes endothelial cell surface protein disulfide isomerase-A1 and impairs adherence
Hypoxia-inducible factor (HIF)α homeostasis is controlled by its rate of translation and degradation. Under physiologic conditions, HIFα is constantly degraded in a process dependent on oxygen, prolyl hydroxylation (by PHD) and ubiquitylation (mediated by pVHL). In cancer, elevated HIFα levels can be seen owing to hypoxia (lack in oxygen) or pseudo-hypoxia (PHD inhibition or pVHL mutation) that inhibit HIFα degradation or owing to accelerated translation (potentially mediated by Akt or loss of Tsc complex or LKB1). Proteins coloured in green represent HIF downregulators whereas those in red represent HIF inducers. Interestingly, many oncogenes and tumour suppressor genes are involved in HIF regulation either positively or negatively, respectively, emphasizing the importance of HIF in tumour development and/or sustention. Solid arrows indicate direct interaction whereas dotted arrows indicate an indirect pathway.
Succinate dehydrogenase and fumarate hydratase : linking mitochondrial dysfunction and cancer | Oncogene
https://www.nature.com/articles/1209594
VITAMIN C & CANCER – HEALTH & DISEASE MASTERKEY (PART 3)
Dorish Loh
Cancer is fundamentally difficult to cure because of the diversity in tumor microenvironments. Hypoxia, or the lack of sufficient oxygen in the microenvironment, allows cancer cells to become heterogenous through epigenetic adaptation. Heterogeneity in cancer cells is probably one of the main reasons for cancer’s exceptional resilience against treatment [1].
Heterogeneity is also the reason why the same type of treatment across different patients with the same type of tumor would produce different responses [2]. Heterogeneity allows the main tumor cells to develop into a wide variety of cells with distinct molecular signatures. Together with the diverse tumor microenvironment, sensitivity to treatment becomes highly varied [1]. Heterogeneity in tumor is now viewed as an independent risk factor for poor survival in many tumor types.
Tumor heterogeneity is fueled by genomic alterations which results in genomic instability. Most common causes of genomic instability are mutations in the DNA mismatch repair system or in the proofreading polymerase enzymes. Mismatch repair (MMR) systems are critical in maintaining genetic stability because their function is to repair DNA replication errors [3].
Hypoxia is now accepted to be responsible for the induction of genetic instability in tumor microenvironments [4].
HIF – Master Regulator of Cancer Biology
Tumor microenvironments often are hypoxic. Tumor cells exhibit increased oxygen demand as a result of cell expansion in a background of decreased oxygen supply due to defective tumor vascularization [5]. In response to inadequate oxygen supply, complex mechanisms, like hypoxia-inducible factors (HIFs) and their downstream gene expression networks are activated to allow cells to adapt and survive in a hostile environment [6].
The HIF signaling pathway is regarded as the major regulator of cancer characteristics such as stemness, dormancy, invasion, metastasis, angiogenesis, immunity, metabolic reprogramming and resistance to anti-cancer therapies [7, 8]. By inducing stemness, HIF signaling enhances tumor heterogeneity, rendering the tumor resistant to treatment while developing increased capacity for metastasis [9].
Genetic instability, a hallmark of tumorigenesis, is induced by increased rate of DNA mutation brought on by hypoxia. Hypoxia can alter transcription and translation of DNA damage response and repair genes. The adaptation of tumor cells to the hypoxic microenvironment therefore drives increased genetic instability and malignant progression [10]. The ability of cancer cells to escape cell death allows them to proliferate in the presence of damaged DNA, and thus acquiring even more DNA mutations [11].
Nrf2 & Stabilization of HIF-1a – the Iron Connection
Hypoxia inducible factor 1α (HIF1α) is a transcription factor that can be activated under hypoxia. HIF-1a levels are normally kept low in the presence of oxygen. When there is adequate oxygen, HIF-1a is bound by the von Hippel-Lindau (VHL) protein, which then prepares HIF-1a for degradation [12]. If HIF-1a is not degraded properly, it will be able to bind with its heterodimeric component, HIF-1β, and becomes stabilized.
Once HIF-1a is stabilized, it can activate the expression of a large number of genes associated with survival, apoptosis, and metabolic reprogramming. HIF-1a can induce the upregulation of enzymes involved in glycolysis, and directly suppress mitochondrial metabolism by decreasing the efficiency of the electron transport chain by modulating expression of cytochrome c oxidase isoforms [13].
HIF-1a is crucial for adaptation to hypoxia. However, HIF-1a can be stabilized even under conditions of normoxia where there is sufficient oxygen. Loss of control of HIF-1a is often associated with a poor disease outcome [14].
The HIF signaling pathway is a stress response pathway. Environmental stress is not limited to low oxygen supply Mild hypothermia has been observed to remodel gene expression by activating transcription factors such as HIF1 [15]. Mild hypothermia also activates another evolutionary conserved stress response molecule, Nrf2.
The nuclear factor (erythroid-derived 2)-like 2 (NRF2) transcription factor was first discovered for its role in erythropoiesis. Since its discovery, Nrf2 research has been focused on its functions in detoxification and cancer prevention [16]. More recent works identified many genes involved in iron storage, iron export in addition to heme synthesis and hemoglobin catabolism are also under the control of NRF2 [17].
Since Nrf2 regulates iron homeostasis, it is not surprising that Nrf2 is deeply implicated in the activation of HIF-1a.
Activation of Nrf2 and its subsequent deregulation of iron metabolism are heavily implicated in cancer development. Nrf2 can upregulate iron storage protein ferritin, leading to enhanced tumor proliferation and therapy resistance [18]. When ferritin is upregulated by Nrf2, available iron can be lowered by about 25%. Lowering of available iron can decrease the activity of the HIF-1a degradation enzyme prolyl hydroxylase by an impressive 75%, resulting in the activation of HIF-1a [18].
Iron is actually one of the several cofactors critical for the degradation of HIF1-a. Even under normoxia, conditions like inflammation can increase ferritin, lowering free iron availability. Inadequate cytosolic free iron under normoxia has been demonstrated to activate HIF-1a. Once activated, HIF-1a will actively modulate the expression of genes that control iron homeostasis such as iron transporters (DMT1), ferroportin 1 (FPN1), duodenal cytochrome b (Dcytb), and transferrin receptor (TfR). Even central systemic mediators for iron homeostasis like hepcidin and iron regulatory proteins are regulated by HIFs once they become activated and stabilized [19].
The appropriate degradation of HIF-1a is perhaps vital in cancer pathology.
Prolyl Hydroxylase, HIF-1a & Cancer – A Tale of Oxygen, Iron and Ascorbic Acid
When there is enough oxygen (normoxia), an enzyme called prolyl hydroxylase (PHD) will add a hydroxyl group (hydroxylation) to the α-subunits of HIF. This hydroxylation allows HIF-1a to be bound to the von Hippel-Lindau protein (VHL), which then initiates the degradation process of HIF-1a.
In the HIF signaling pathway, prolyl hydroxylases (PHDs) are the true oxygen sensors. When there is enough oxygen, PHD uses oxygen as substrate for the hydroxylation of HIF where PHD inserts oxygen atoms into the prolyl residue. The hydroxylation process also requires PHD to obtain electrons from another co-substrate called α-ketoglutarate [20].
Prolyl hydroxylases (PHDs) belong to a class of non-heme iron α-ketoglutarate (αKG) dioxygenase. That means their enzymatic activities depend upon the energy derived from the conversion of iron between its oxidized and reduced states [21, 22]. PHDs also require ascorbic acid as a co-factor during the enzymatic process of hydroxylation.
In January of 2019, Kulper et al. demonstrated that intracellular ascorbate levels modulate the hypoxic HIF pathway in a dose dependent manner in human clear cell renal cell carcinoma (ccRCC). Increased intracellular ascorbate dramatically elevated activity levels of prolyl hydroxylase, resulting in lower stabilization of HIF-1a [23].
In the following chart, notice the complete inactivation of HIF-1a in 10% oxygen at higher ascorbate concentrations. The effect of ascorbate is reduced upon diminishing levels of oxygen.
HIF-1a Stabilization in Various Ascorbate and Oxygen Concentrations in human ccRCC
[Source: Wohlrab C, Kuiper C, Vissers MCM, Phillips E, Robinson BA, Dachs GU Ascorbate modulates the hypoxic pathway by increasing intracellular activity of the HIF hydroxylases in renal cell carcinoma cells Dove Press journal Hypoxia Volume 2019:7 Pages 17—31 DOI https://doi.org/10.2147/HP.S201643]
A truly astounding and groundbreaking landmark study released in April 2019 by Osipyants et al. revealed how Nature uses ascorbic acid as the master key that unlocks many doors in vital biochemical processes.
The Molecular Structure of Ascorbic Acid – MasterKey to Health & Disease
Most living organisms including plants, insects and animals produce ascorbic acid. Ascorbic acid exists naturally in the form of L-ascorbic acid [24]. In physiological pH, L-ascorbic acid exists predominantly in the ionic form of L-ascorbate.
In their 2019 landmark study, Osipyants et al. not only showed that prolyl hydroxylase depended upon ascorbic acid as substrate, but demonstrated definitively for the first time that ascorbic acid MUST BE IN THE SPECIFIC MOLECULAR STRUCTURE of L-ASCORBATE in order to suppress HIF-1a [25]!!
The discovery by Osipyants et al. brings into question the long-held understanding that the role of ascorbic acid in enzymatic reactions of non-heme iron α-ketoglutarate (αKG) dioxygenases such as prolyl hydroxylase is solely confined to its reducing capacity as an antioxidant in the conversion of ferric ions to ferrous ions.
When Osipyants et al. substituted L-ascorbate with the potent cell-permeable reducing agent, N-acetyl cysteine (NAC), they observed NO effect in the suppression of HIF-1a. These brilliant scientists then proceeded to experiment with D-ascorbate, an enantiomer of L-ascorbate. To their utmost surprise, the D-isomer, even though it possessed the same reductive potency with respect to ferric iron, was also completely ineffective when compared to L-ascorbate in the suppression of HIF-1a [25].
D-ascorbic acid is a mirror image of L-ascorbic acid. It does not exist in nature but can be synthesized artificially [26]. The binding sites in the enzyme active centers of PHDs allow for the natural docking of L-ascorbate molecules, but not its D-isomer. Look at how the insertion of D-Ascorbate into HIF prolyl hydroxylase crystal structures displayed molecular interference in diagram B, versus the perfect docking of L-ascorbate in diagram A.
L-Ascorbate Docked in Prolyl Hydroxylase
D-Ascorbate Inserted in Prolyl Hydroxylase
[Source: Andrey I. Osipyants et al. Biochimie. 2018 April ; 147: 46–54. doi:10.1016/j.biochi.2017.12.011. L-ascorbic acid: A true substrate for HIF prolyl hydroxylase?]
Ascorbic acid is known for its ability to convert iron between ferric and ferrous forms in other important biochemical processes such as the catalytic cycle during dopamine synthesis by tyrosine hydroxylase. Might L-ascorbate assume a similar identity as master key that facilitate these biological processes?
Optimal physiological intracellular ascorbate concentrations have been found to significantly increase expression of tyrosine hydroxylase proteins [27]. Deficiency of tyrosine hydroxylase can result in impaired synthesis of dopamine, epinephrine and norepinephrine. Deficiency in any of these important catecholamines will results in a wide range of diseases. For example, epinephrine and norepinephrine are signaling molecules that can activate the conversion of ATP molecules into secondary messengers known as cyclic AMP (cAMP). cAMP regulates multiple cellular functions including cell growth and specialization; protein expression; and even gene transcriptions [28].
If the molecular structure of L-ascorbate must be maintained in order for these biochemical processes to proceed, then an open question would be how the supplementation with synthetic forms of buffered ascorbic acid impact health and disease. An examination of the molecular structures of several popular ascorbate supplements show that they do not resemble L-ascorbic acid at all.
Ascorbic Acid
Sodium Ascorbate
Magnesium Ascorbate
Calcium Ascorbate
The importance of the molecular structure of ascorbic acid in biological processes also brings into attention the emerging role of ascorbic acid as an effective epigenetic regulator. DNA methylation in cancer cells are often dysregulated. Recent advancements in the field of cancer epigenetics reveal extensive epigenetic reprogramming involving DNA methylation, histone modifications, nucleosome positioning, and non-coding mRNA expression [29]. Electromagnetic radiation has been observed to cause epigenetic changes involving chromatin accessibility [30]. The fact that ascorbic acid is used by Nature as primary quantum interface now makes even more sense than ever [31].
Have you had your AA today?
References:
[1] Tumour heterogeneity and resistance to cancer therapies | Nature Reviews Clinical Oncology https://www.nature.com/articles/nrclinonc.2017.166
[2] Tumor heterogeneity: a central foe in the war on cancer | Journal of Clinical Outcomes Management https://www.mdedge.com/jcomjournal/article/168663/immunotherapy/tumor-heterogeneity-central-foe-war-cancer
[4] HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/15780936
[5] Hypoxia-inducible factors and the response to hypoxic stress. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/20965423
[6] Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/28741521
[7] Regulation of angiogenesis by hypoxia: role of the HIF system. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/12778166?dopt=Abstract
[8] Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/25811878?dopt=Abstract
[9] Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/23301832?dopt=Abstract
[10] Tumor hypoxia as a driving force in genetic instability https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4016142/
[11] Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/20103649/
[12] Hypoxia-Inducible Factors in Physiology and Medicine https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3437543/
[13]Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies https://www.cell.com/trends/pharmacological-sciences/fulltext/S0165-6147%2817%2930110-4
[14] Targeting hypoxia in cancer therapy. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/21606941/
[15] Hypothermia augments stress response in mammalian cells – ScienceDirect https://www.sciencedirect.com/science/article/pii/S0891584918307482
[16] NRF2, a Key Regulator of Antioxidants with Two Faces towards Cancer https://www.hindawi.com/journals/omcl/2016/2746457/
[17] Emerging Regulatory Role of Nrf2 in Iron, Heme, and Hemoglobin Metabolism in Physiology and Disease https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6191506/
[18] Ferritin-Mediated Iron Sequestration Stabilizes Hypoxia-Inducible Factor-1α upon LPS Activation in the Presence of Ample Oxygen. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/26628374
[19] Regulation of iron metabolism by hypoxia-inducible factors http://www.actaps.com.cn/qikan/manage/wenzhang/2017-5-14.pdf
[20] HIF-independent role of prolyl hydroxylases in the cellular response to amino acids https://www.nature.com/articles/onc2012465.pdf?origin=ppub
[21] Insight into the mechanism of an iron dioxygenase by resolution of steps following the FeIV═O species https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2840172/
[22] Ferrous iron and α-ketoglutarate-dependent dioxygenases in the biosynthesis of microbial natural products. – PubMed – NCBI https://www.ncbi.nlm.nih.gov/pubmed/26845569
[23] Ascorbate modulates the hypoxic pathway by increasing intracellular activity of the HIF hydroxylases in renal cell carcinoma cells https://www.dovepress.com/ascorbate-modulates-the-hypoxic-pathway-by-increasing-intracellular-ac-peer-reviewed-fulltext-article-HP
[24] Ascorbic acid | HC6H7O6 – PubChem https://pubchem.ncbi.nlm.nih.gov/compound/L-ascorbic%20acid
[25] L-ascorbic acid: A true substrate for HIF prolyl hydroxylase? https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6460286/
[26] D-Ascorbic acid | C6H8O6 – PubChem https://pubchem.ncbi.nlm.nih.gov/compound/D-Ascorbic-acid
[27] Mechanisms of Ascorbic Acid Stimulation of Norepinephrine Synthesis in Neuronal Cells https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3449284/
[28] The Cyclic AMP Pathway https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3504441/
[29] Epigenetics in cancer https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2802667/
[30] Specific low frequency electromagnetic fields induce epigenetic and functional changes in U937 cells https://arxiv.org/ftp/arxiv/papers/1810/1810.06255.pdf
[31] Electromagnetic Radiation and Quantum Decoherence: Is Vitamin C the Ultimate Quantum Interface? | LinkedIn https://www.linkedin.com/pulse/electromagnetic-radiation-quantum-decoherence-vitamin-doris-loh/
(Visited 2.406 times, 1 visits today)
REDOX, DISEASE & EVOLUTION – PART 3 – HEARTS ON CLOCKS
Doris Loh
Bioarchaeological studies reveal unsettling evidence that cardiovascular diseases in the form of atherosclerosis was not uncommon thousands of years ago. Most people believe the decline of heart health is associated with the modern way of life. So why would a man from the Alps who lived 5,300 years ago, and a princess from Thebes who lived 3,500 years ago both develop atherosclerosis eating two drastically different diets in the ABSENCE of artificial LED lights, cell phones and electricity?
The right way to rephrase this question should be: what has not changed in human biology since the first identification of hominins some 2.5 million years ago?
Over the past 2.5 million years, the human genome has undergone quite a makeover. There appeared to have been an extreme burst of action in the human genome about 15,000 to 2000 years ago. A close examination of 15,336 genes in 6,515 individuals of European American and African American ancestry indicated that most human mutations happened within the past 5,000-10,000 years. The spectrum of protein-coding variation in modern humans has been found to differ considerably compared to those as recent as 200 to 400 generations ago. Yet diseases like atherosclerosis and cancer remained persistent and constant for close to 2 million years.
If one cannot attribute the development of atherosclerosis and cancer to genetic mutations, then is there a common thread that spanned this entire time? Is this factor intrinsic to the pathological development of diseases like atherosclerosis and cancer? What did hominids, hominins and humans all have in common since 61 million years ago?
Humans and our great ape ancestors all have pseudogenes in L-gulonolactone oxidase (GULO) and Urate Oxidase (UOX). We cannot produce ascorbic acid, and we produce relatively high amounts of uric acid compared to other animals with functional UOX.
The evolutionary pressure to eliminate the function of urate oxidase has been clearly elucidated in “Uric Acid & Vitamin C: Devolution of Evolution in a 5G World” However, the relatively exorbitant price paid for this necessary genetic adaptation that facilitated the survival of our species has not been truly defined.
Hyperuricemia: Cancer Past and Present
Hyperuricemia is a condition where the level of serum uric acid (SUA) is greater than 6.8 mg/dL. Hyperuricemia has been definitively correlated to carcinogenesis in the largest study ever conducted that involved close to half a million cancer free participants aged 20 years or older. 72,349 persons developed cancer during the following 19.47 years. Males had a higher incidence rate of cancer (56.96%). Serum uric acid levels were higher in the group who developed cancer, compared to the cancer-free group.
My article “Cancer on Clocks” presented scientific documentations of cancer recorded from samples as far back as 1.98 million years ago. The archeological discovery of metastatic carcinoma in a young man from Ancient Nubia around 1200 BC provided scientists with further irrefutable evidence that cancer is not a product of modern living. The remains of the young man from over 3000 years ago showed multiple osteolytic lesions on the vertebrae, ribs, sternum, clavicles, scapula, pelvis, and humeral and femoral heads. The inferred metastatic malignant soft-tissue carcinoma that had spread across large regions of the body represented the first complete example in the world of a human who suffered metastatic tissue cancer.
In my paper on “Uric Acid & Vitamin C: Evolution of Devolution in a 5G World”, I connected the evolutionary need for Uric acid to activate mTOR as a stress response. Unfortunately, mTOR activation can also be carcinogenic, as sustained increases in mTORC1 activity can sensitize cells to stress, and even induce irreversible damages when mTORC1 or p53 becomes dysregulated.
The strong correlation between hyperuricemia and cancer may indicate that uric acid’s role in carcinogenesis may not be limited to the activation of mTOR only.
Hyperuricemia: Heart Disease, Past and Present
Uric acid is now accepted to be an accurate biochemical marker of endothelial function and atherosclerosis. It has also been used to assess risk factors for the development of cardiovascular diseases, as well as predicting the occurrence of cardiovascular events. Even though there has been considerable controversy regarding the association of coronary heart disease (CHD), a systematic review and dose-dependent meta-analysis of a total of 29 prospective cohort studies representing data from 958,410 participants showed that hyperuricemia was associated with increased risk of CHD morbidity. This 2016 study revealed that for each increase of 1 mg/dl in uric acid level, the pooled multivariate risk ratio of CHD mortality was 1.13, with a higher risk ratio being observed in females.
Hyperuricemia has been correlated to higher medium and long-term mortality rates as well as major cardiovascular event rates in patients following acute coronary syndrome. It has been determined that patients exhibiting gout, who show no sign of hyperuricemia, and lack clinical evidence of cardiovascular disease actually have a high prevalence of SUBCLINICAL atherosclerosis. The outstanding question remains- how do we know humans in antiquity had hyperuricemia?
Hyperuricemia in Antiquity
Science now believes humans evolved genetic mutations that favored insulin resistance as a result of selection pressure during the course of evolution in the past 2 million years. The fact that these polymorphisms are still retained in many humans indicate the tremendous survival edge conferred by these genetic adaptations in the past. However, a person who is genetically insulin resistant needs to follow the path of hominins who lived during the Ice Ages, and adopt a low carbohydrate diet. Cold temperatures together with periodic starvation during the Ice Ages created the necessity to maintain steady blood glucose via insulin resistance. Eating a diet rich in carbohydrates will result in hyperglycemia, and increase the risk for developing metabolic syndrome, including diabetes and cardiovascular dysfunction.
Princess Ahmose-Meryet-Amon who developed atherosclerosis lived in Thebes some 3,500 years ago. Thebes was an ancient Egyptian city located along the Nile about 500 miles south of the Mediterranean. Being close to the Nile meant that controlled irrigation would have yielded surplus crops. Ancient Egypt is well known for its irrigation systems and agricultural production techniques. Might the princess be genetically insulin resistant, and her high carbohydrate intake caused her to become diabetic?
The development of cardiovascular disease (CVD) is intimately related to diabetes
Science has now established that hyperglycemia, coronary artery calcification, and diagnostic HbA1c are predictive indicators for cardiovascular disease (CVD). For every 1% increase in HbA1c, the risk associated with atherosclerotic coronary vascular disease has been observed to increase by 11% to 16%. If hominins were genetically insulin resistant, did humans in antiquity also suffer from diabetes like modern humans?
Diabetes in Antiquity
Diabetes was first documented around 5th century BC by the famous Indian surgeon Sushruta. In his famous work Samhita, Sushruta referred to diabetes by the term ‘madhumeha’ or honey-like urine, and indicated that the urine had the ability to attract ants due to its sweetness and the texture of the liquid was sticky to the touch. Sushruta noted that diabetes mostly affected the rich castes who were able to afford high consumption of carbohydrates such as rice, cereals and sweets.
Diabetes was frequently mentioned in ancient Egyptian papyri, as well as ancient Indian and Chinese medical literature. Ancient Greek physician Aretaeus of Cappadocia who lived in the 2nd century AD provided the first accurate description of diabetes. He was the first to use the term “diabetes”. The term “mellitus” was added in the 17th century by Thomas Willis, in order to emphasize the extreme sweet taste of the urine.
It is understandable that high carbohydrate diets caused metabolic syndrome in humans thousands of years ago, resulting in the possible development of atherosclerosis. But how do we explain the evidence of major calcifications in the carotid arteries, distal aorta and right iliac artery of the Tyrolean Iceman named Ötzi who grew up and lived in different valleys in the southern region of the Alps 5,300 years ago? If he were insulin resistant, surely his diet that was high in animal products as supported by evidence of degenerative arthritis, and gallbladder stones, would have prevented the development of hyperglycemia and diabetic complications.
A diet high in animal products would also be high in purines. Uric acid is the end product of the metabolism of purine compounds in humans. A diet high in purines would have naturally resulted in high serum uric acid for our Tyrolean Iceman. Princess Ahmose-Meryet-Amon, if she were insulin-resistant, would also have a high level of serum uric acid. How does uric acid cause atherosclerosis and cancer? For that answer, we will have to understand the roles of the ancient circadian clocks, peroxiredoxins.
Peroxiredoxin are REDOX Circadian Regulators
The previous chapter, “Cancer on Clocks”, introduced peroxiredoxins as ancient transcript-independent circadian time-keepers from perhaps 3 billion years ago that functioned in concert with transcriptional circadian clocks. Peroxiredoxins also serve pivotal roles as antioxidant enzymes and REDOX signaling regulators. As important regulators of cellular homeostasis, peroxiredoxins can have a wide range of influence on different stages of growth and development in cells and tissues, as well as disease and cancer progression. How do peroxiredoxins regulate REDOX?
Peroxiredoxin Binds Hydrogen Peroxide
The reactive oxygen species hydrogen peroxide has been known for its important role as secondary messenger in REDOX signaling. Yet the mechanism involved has never been fully elucidated. Recent understanding of peroxiredoxin (PRX) show that they participate in the DIRECT regulation of intracellular signal transduction pathways that involve H2O2.
Mammalian cells have various means to eliminate H2O2, including enzymes like catalase and glutathione peroxidase (GPx). Yet peroxiredoxins, known for their antioxidant capacity in the catalytic reduction of hydrogen peroxide and other reactive oxygen species like peroxynitrite, possess a high-affinity binding site for H2O2 that is lacking in catalase and GPx. The target for H2O2 in Prx is actually the cysteine component, which is highly susceptible to oxidation by H2O2.
Mammals have six different isoforms of peroxiredoxins, PRDX1-6, and are distributed at sites of ROS production, such as the cytosol, mitochondria, as well as peroxisomes. Structurally, PRX enzymes belong to either the 1-cys or 2-cys groups. These two groups have the same mechanism of activity, but the way the Prx enzymes are recycled back from the oxidized to the reduced states are different.
Prx6 is the only mammalian peroxiredoxin that belong to the 1-cys group. Prx 2-5 in mammals are unique in that they undergo an additional step of oxidation called hyperoxidation that can reversibly, or irreversibly inactivate the enzyme.
It is the inactivation of peroxiredoxins in the hyperoxidized state that allows for circadian REDOX regulation.
Hyperoxidation: REDOX Circadian Regulation
If peroxiredoxins are not recycled back into their reduced state after the cysteine component is oxidized by H2O2, they can be reversibly, or irreversibly INACTIVATED when they are further oxidized by H2O2 into a hyperoxidized state. This oxidative inactivation of PRXs is now recognized as the key mechanism that allows local H2O2 levels to accumulate and initiate redox-signaling events.
The continuous cycle of hyperoxidation and reduction of peroxiredoxins is the basis of REDOX circadian regulation that oversees many cellular pathways. Most 2-cys Prx that are hyperoxidzed can be regenerated by an ATP-consuming process involving another molecule called sulfiredoxin (Srx).
Prx3 is a 2-cys peroxiredoxin. Its hyperoxidation and subsequent regeneration generates a REDOX circadian rhythm that regulates cellular functions like steroid biosynthesis. In the adrenal steroidogenesis of corticosterone, the extent of the hyperoxidation of Prx3 is proportional to the number of H2O2 molecules removed by Prx3, which in turn affects the number of corticosterone molecules synthesized. Signals generated from H2O2 buildup in the hyperoxidation of Prx3 will reflect to the organism that sufficient corticosterone has been produced.
Different isoforms of peroxiredoxins are found to exert different effects on signaling in humans. Hyperoxidized Prx2 levels are found to be higher in patients with sleep apnea compared to those who only snored. Human keratinocytes redox balance has been found to be regulated by the circadian oscillations of Prx2. Prx2 is now recognized as a potent suppressor of melanoma.
Uric Acid and the Hyperoxidation of Peroxiredoxins
The formation of hydrogen peroxide begins with the generation of superoxide, the one-electron reduction of molecular oxygen. Superoxide is easily generated in the human body, such as during the autooxidation of hemoglobin; during the production of ATP in mitochondria electron transport chain; as well as by exposure to ultraviolet radiation.
In the absence of adequate ascorbic acid, uric acid is one of the major antioxidants in plasma. The plasma level of urate in humans is about 300 microM. This level is considerably higher than that of ascorbate. Yet uric acid will preferentially react with nitric oxide instead of neutralizing superoxide, leaving superoxide to produce hydrogen peroxide under inadequate reducing environments.
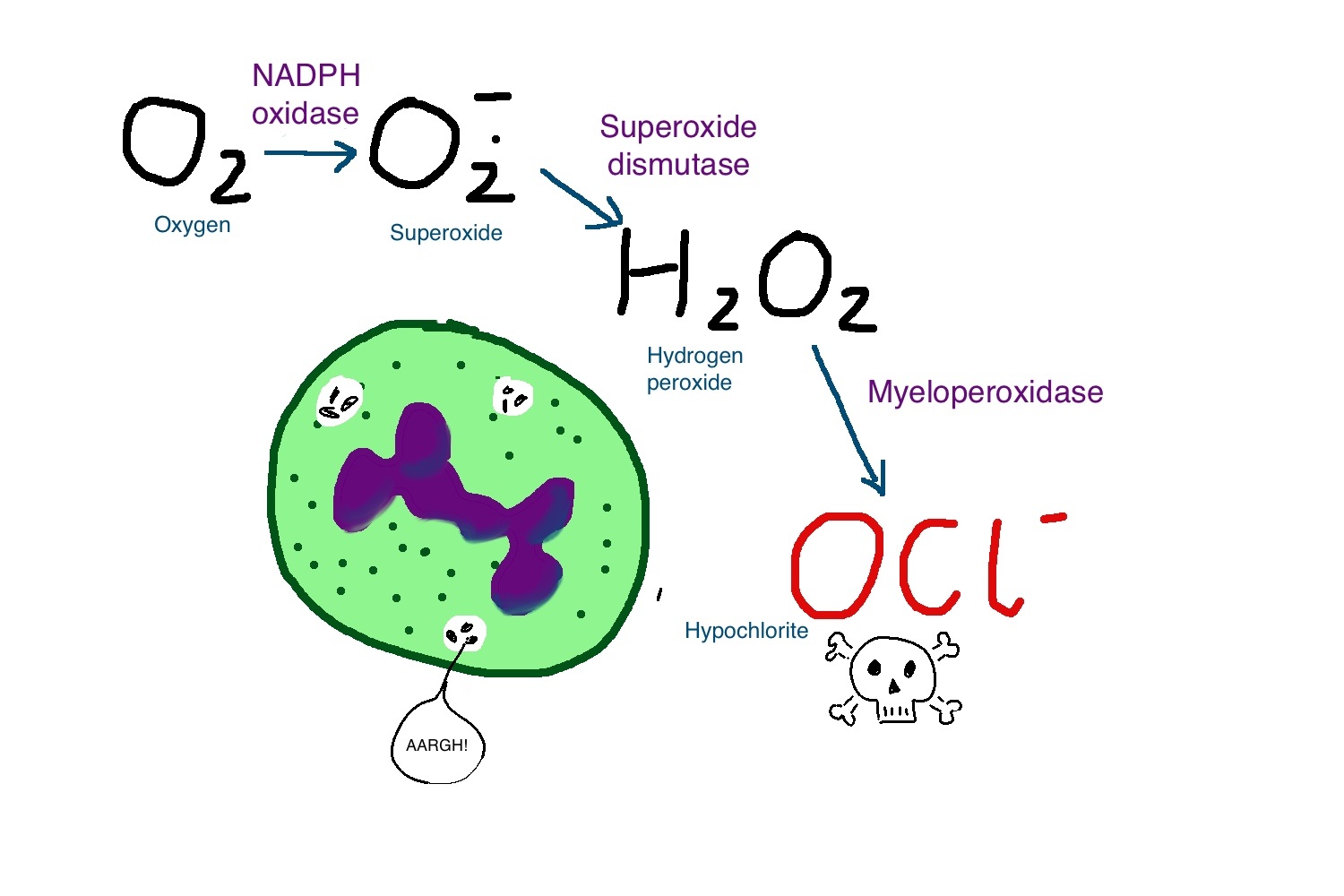
Xanthine oxidoreductase, responsible for the production of uric acid in the human body, generates superoxide and hydrogen peroxide instead of NADH under oxidative inflammatory environments.
Most interesting of all, is that when urate, the anionic form of uric acid, has been found to cause hyperoxidation of peroxiredoxins. Urate can accumulate in plasma at levels ranging from 50 to 420 microM in health individuals. When the urate free radical combines with superoxide, it becomes a strong oxidant called urate hydroperoxide.
A study in 2017 showed that urate hyrdoperoxide can cause hyperoxidation of Prx 1 and Prx2. Prx2, which is abundant in plasma, is as sensitive to urate hydroperoxide as hydrogen peroxide.
It is entirely possible that uric acid affects the proper functioning of peroxiredoxins, leading to pathogenesis of disease in ancient humans. The question we need to ask is why. Why did nature fashion the reactions of uric acid to hyperoxidize peroxiredoxins? How do peroxiredoxins affect pathogenesis of atherosclerosis and cancer? The last, but most important question: what role does Ascorbic acid, Vitamin C, play in the regulation of peroxiredoxins? —- To Be Continued —-
(Visited 910 times, 1 visits today)REDOX, Disease & Evolution - Part 3 - Hearts on Clocks - EvolutaMente.it
https://www.evolutamente.it/redox-disease-evolution-part-3-hearts-on-clocks/
Mitochondria Supercomplex - The Untold Story - EvolutaMente.it
https://www.evolutamente.it/mitochondria-supercomplex-the-untold-story/
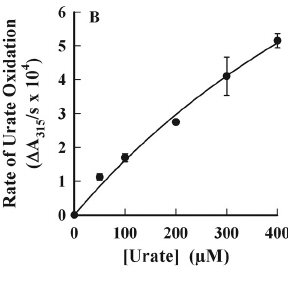
Urate as a physiological substrate for myeloperoxidase: Implications for hyperuricemia and inflammation
Apr 2011
Flavia Meotti
Guy N L Jameson
Rufus Turner[...]
Anthony J Kettle
Abstract
Urate and myeloperoxidase (MPO) are associated with adverse outcomes in cardiovascular disease. In this study, we assessed whether urate is a likely physiological substrate for MPO and if the products of their interaction have the potential to exacerbate inflammation. Urate was readily oxidized by MPO and hydrogen peroxide to 5-hydroxyisourate, which decayed to predominantly allantoin.The redox intermediates of MPO were reduced by urate with rate constants of 4.6 × 10(5) M(-1) s(-1) for compound I and 1.7 × 10(4) M(-1) s(-1) for compound II. Urate competed with chloride for oxidation by MPO and at hyperuricemic levels is expected to be a substantive substrate for the enzyme.
Oxidation of urate promoted super-stoichiometric consumption of glutathione, which indicates that it is converted to a free radical intermediate. In combination with superoxide and hydrogen peroxide, MPO oxidized urate to a reactive hydroperoxide. This would form by addition of superoxide to the urate radical. Urate also enhanced MPO-dependent consumption of nitric oxide.
In human plasma, stimulated neutrophils produced allantoin in a reaction dependent on the NADPH oxidase, MPO and superoxide. We propose that urate is a physiological substrate for MPO that is oxidized to the urate radical. The reactions of this radical with superoxide and nitric oxide provide a plausible link between urate and MPO in cardiovascular disease.
尿酸盐和髓过氧化物酶(MPO)与心血管疾病的不良结局有关。
在这项研究中,我们评估了尿酸盐是否可能是MPO的生理底物,以及它们相互作用的产物是否具有加剧炎症的潜能。尿酸盐很容易被MPO和过氧化氢氧化成5-羟基异羟乙酸(5-hydroxyisourate),后者分解成尿囊素(allantoin)。尿酸盐还原MPO的氧化还原中间体,化合物I的速率常数为4.6×10(5)M(-1)s(-1),而化合物I的速率常数为1.7×10(4)M(-1)s(-1)化合物II。
尿酸盐与氯化物竞争被MPO氧化,并且在高尿酸水平下被认为是该酶的实质底物。尿酸盐的氧化促进了谷胱甘肽的超化学计量消耗,这表明它被转化为自由基中间体。与超氧化物和过氧化氢结合,MPO将尿酸盐氧化为反应性氢过氧化物。这将通过在尿酸盐自由基中添加超氧化物而形成。尿酸盐还增加了MPO依赖的一氧化氮消耗量。
在人血浆中,受刺激的中性粒细胞在依赖于NADPH氧化酶,MPO和超氧化物的反应中产生尿囊素。我们建议尿酸盐是被氧化成尿酸盐自由基的MPO的生理底物。该自由基与超氧化物和一氧化氮的反应在心血管疾病中提供了尿酸盐和MPO之间的合理联系。
Urate as a Physiological Substrate for Myeloperoxidase
https://www.jbc.org/content/286/15/12901
Biochimica et Biophysica Acta (BBA) - General Subjects
Volume 1864, Issue 3, March 2020, 129481
Biochimica et Biophysica Acta (BBA) - General Subjects
Urate hydroperoxide oxidizes endothelial cell surface protein disulfide isomerase-A1 and impairs adherence
a Department of Biochemistry, Institute of Chemistry, University of São Paulo, São Paulo, Brazil
b Heart Institute (Incor), University of São Paulo School of Medicine, São Paulo, Brazil
Highlights
• Urate hydroperoxide oxidizes PDI at a rate of constant of 6 × 103 M−1 s−1.
• Urate hydroperoxide oxidized cell surface PDI in HUVECs
• PDI oxidation, alkylation or inhibition decreased HUVECs adherence.
Abstract
Background
Extracellular surface protein disulfide isomerase-A1 (PDI) is involved in platelet aggregation, thrombus formation and vascular remodeling. PDI performs redox exchange with client proteins and, hence, its oxidation by extracellular molecules might alter protein function and cell response. In this study, we investigated PDI oxidation by urate hydroperoxide, a newly-described oxidant that is generated through uric acid oxidation by peroxidases, with a putative role in vascular inflammation.
Methods
Amino acids specificity and kinetics of PDI oxidation by urate hydroperoxide was evaluated by LC-MS/MS and by stopped-flow. Oxidation of cell surface PDI and other thiol-proteins from HUVECs was identified using impermeable alkylating reagents. Oxidation of intracellular GSH and GSSG was evaluated with specific LC-MS/MS techniques. Cell adherence, detachment and viability were assessed using crystal violet staining, cellular microscopy and LDH activity, respectively.
Results
Urate hydroperoxide specifically oxidized cysteine residues from catalytic sites of recombinant PDI with a rate constant of 6 × 103 M−1 s−1. Incubation of HUVECs with urate hydroperoxide led to oxidation of cell surface PDI and other unidentified cell surface thiol-proteins. Cell adherence to fibronectin coated plates was impaired by urate hydroperoxide, as well as by other oxidants, thiol alkylating agents and PDI inhibitors. Urate hydroperoxide did not affect cell viability but significantly decreased GSH/GSSG ratio.
Conclusions
Our results demonstrated that urate hydroperoxide affects thiol-oxidation of PDI and other cell surface proteins, impairing cellular adherence.
General significance
These findings could contribute to a better understanding of the mechanism by which uric acid affects endothelial cell function and vascular homeostasis.
Urate hydroperoxide oxidizes endothelial cell surface protein disulfide isomerase-A1 and impairs adherence - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0304416519302703
J Biol Chem
. 2017 May 26;292(21):8705-8715. doi: 10.1074/jbc.M116.767657. Epub 2017 Mar 27.
Urate Hydroperoxide Oxidizes Human Peroxiredoxin 1 and Peroxiredoxin 2
Larissa A C Carvalho 1, Daniela R Truzzi 1, Thamiris S Fallani 1, Simone V Alves 2, José Carlos Toledo Jr 3, Ohara Augusto 1, Luís E S Netto 2, Flavia C Meotti 4Universidade de São Paulo, São Paulo-SP CEP 05508-000, Brazil.
Abstract
Urate hydroperoxide is a product of the oxidation of uric acid by inflammatory heme peroxidases. The formation of urate hydroperoxide might be a key event in vascular inflammation, where there is large amount of uric acid and inflammatory peroxidases. Urate hydroperoxide oxidizes glutathione and sulfur-containing amino acids and is expected to react fast toward reactive thiols from peroxiredoxins (Prxs). The kinetics for the oxidation of the cytosolic 2-Cys Prx1 and Prx2 revealed that urate hydroperoxide oxidizes these enzymes at rates comparable with hydrogen peroxide. The second-order rate constants of these reactions were 4.9 × 105 and 2.3 × 106 m-1 s-1 for Prx1 and Prx2, respectively. Kinetic and simulation data suggest that the oxidation of Prx2 by urate hydroperoxide occurs by a three-step mechanism, where the peroxide reversibly associates with the enzyme; then it oxidizes the peroxidatic cysteine, and finally, the rate-limiting disulfide bond is formed. Of relevance, the disulfide bond formation was much slower in Prx2 (k3 = 0.31 s-1) than Prx1 (k3 = 14.9 s-1). In addition, Prx2 was more sensitive than Prx1 to hyperoxidation caused by both urate hydroperoxide and hydrogen peroxide. Urate hydroperoxide oxidized Prx2 from intact erythrocytes to the same extent as hydrogen peroxide. Therefore, Prx1 and Prx2 are likely targets of urate hydroperoxide in cells. Oxidation of Prxs by urate hydroperoxide might affect cell function and be partially responsible for the pro-oxidant and pro-inflammatory effects of uric acid.
Keywords: hydrogen peroxide; inflammation; oxidation-reduction (redox); peroxiredoxin; urate hydroperoxide; uric acid.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.Urate Hydroperoxide Oxidizes Human Peroxiredoxin 1 and Peroxiredoxin 2 - PubMed
https://pubmed.ncbi.nlm.nih.gov/28348082/
Peroxiredoxin 1 - Wikipedia
https://en.wikipedia.org/wiki/Peroxiredoxin_1
OverviewClinical significanceFunctionInteractionsFurther readingExternal links
As enzymes that combat oxidative stress, peroxiredoxins play an important role in health and disease. Peroxiredoxin 1 and peroxiredoxin 2 have been shown to be released by some cells when stimulated by LPS or TNF-alpha. The released peroxiredoxin can then act to produce inflammatory cytokines. The levels of peroxiredoxin 1 are elevated in pancreatic cancer and it can potentially act as a marker for the diagnosis and prognosis of this disease. In some types of cancer, peroxiredoxin 1 has been determined …
Peroxiredoxins in: Biological Chemistry Volume 383 Issue …翻译此页
https://www.degruyter.com/view/journals/bchm/383/3-4/article-p347.xml?language=en
Peroxiredoxins are low efficiency peroxidases using thiols as reductants. They appear to be fairly promiscuous with respect to the hydroperoxide substrate; the specificities for the donor substrate vary considerably between the subfamilies, comprising GSH, thioredoxin, tryparedoxin and the analogous CXXC motifs in bacterial AhpF proteins.
The role of peroxiredoxins in cancer.
https://www.ncbi.nlm.nih.gov/pubmed/28357082
Peroxiredoxins (PRDXs) are a ubiquitously expressed family of small (22-27 kDa) non-seleno peroxidases that catalyze the peroxide reduction of H2O2, organic hydroperoxides and peroxynitrite. They are highly involved in the control of various physiological functions, including cell growth, differentiation, apoptosis, embryonic development, lipid metabolism, the immune response, as well as ...
Cited by: 63
Publish Year: 2017
Author: Ar
Peroxidase Activity of Hemoglobin·Haptoglobin Complexes
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2781594/
Proc Natl Acad Sci U S A
. 1989 Aug;86(16):6377-81. doi: 10.1073/pnas.86.16.6377.
Ascorbate Is an Outstanding Antioxidant in Human Blood Plasma
B Frei 1, L England, B N Ames
1Department of Biochemistry, University of California, Berkeley 94720.
PMID: 2762330 PMCID: PMC297842 DOI: 10.1073/pnas.86.16.6377
Free PMC article
Abstract
We have shown recently that the temporal order of antioxidant consumption in human blood plasma exposed to a constant flux of aqueous peroxyl radicals is ascorbate = protein thiols greater than bilirubin greater than urate greater than alpha-tocopherol and that detectable lipid peroxidation starts only after ascorbate has been consumed completely. In this paper, we show that it is indeed ascorbate that completely protects plasma lipids against detectable peroxidative damage induced by aqueous peroxyl radicals and that ascorbate is the only plasma antioxidant that can do so. Plasma devoid of ascorbate, but no other endogenous antioxidant, is extremely vulnerable to oxidant stress and susceptible to peroxidative damage to lipids. The plasma proteins' thiols, although they become oxidized immediately upon exposure to aqueous peroxyl radicals, are inefficient radical scavengers and appear to be consumed mainly by autoxidation. Our data demonstrate that ascorbate is the most effective aqueous-phase antioxidant in human blood plasma and suggest that in humans ascorbate is a physiological antioxidant of major importance for protection against diseases and degenerative processes caused by oxidant stress.美国国家科学研究院
。 1989年8月; 86(16):6377-81。 doi:10.1073 / pnas.86.16.6377。
抗坏血酸是人血浆中的最重要的抗氧化剂
B Frei 1,L英格兰,B N Ames
1加利福尼亚大学生物化学系,伯克利94720。
最近我们发现,暴露于恒定的过氧化氢自由基通量的人血浆中抗氧化剂消耗的时间顺序为抗坏血酸=蛋白质硫醇大于胆红素大于尿酸盐大于α-生育酚,并且可检测到的脂质过氧化作用仅在抗坏血酸被完全消耗掉了后才开始产生。在本文中,我们证明了完全保护血浆脂质免受水性过氧自由基引起的可检测到的过氧化损伤的确实是抗坏血酸盐,而抗坏血酸盐是唯一可以做到的血浆抗氧化剂。
血浆中抗坏血酸耗竭,而不是其他内源性抗氧化剂耗竭,极易受到氧化应激,并易受脂质的过氧化损伤。血浆蛋白的硫醇虽然在暴露于过氧化氢自由基后会立即被氧化,但却是效率低下的自由基清除剂,似乎主要被自氧化作用所消耗。我们的数据表明,抗坏血酸是人体血浆中最有效的水相抗氧化剂,并表明在人体中,抗坏血酸是对预防由氧化应激引起的疾病和退化过程具有重要意义的生理抗氧化剂。
抗坏血酸是人体血浆中的最重要的抗氧化剂。Ascorbate is an outstanding antioxidant in human blood plasma.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC297842/Ascorbate is an outstanding antioxidant in human blood plasma.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC297842/