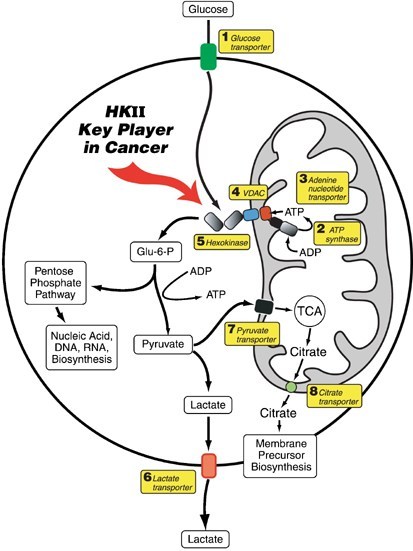
TARGET GLUCOSE TRANSPORTER AND GLYCOLYTIC ENZYMES
Key glycolytic enzymes: HK2,PDK,LDH
Inhibitors:
HK2:metformin, Dehydroascorbic acid,2-DG,curcumin, 2-BP,Oroxylin A(黄芩素),AMPK
PDK: Quercetin,DCA(dichlorocetate),
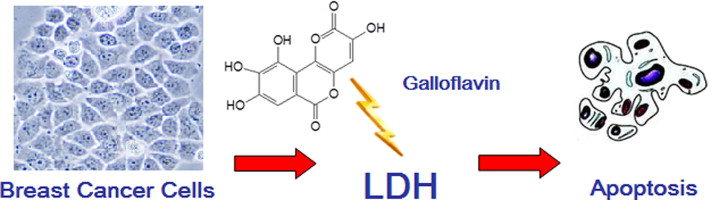
LDH: Lactic acid, Galloflavin (derived from gallic acid)
MCT: quercetin
Activators of PDK:ATP, NADH and acetyl‐CoA
Inhibitors of PDK: AMPK,exercise, sudden energy demand
Inhibitors of G6PD: 6-aminonicotiamide
Inhibitors of GAPDH:vitamin C/dehydroascorbic acid
Inhibitor of aldehyde dehydrogenase (of cancer stem cell): quercetin
Inhibitors of LDHA: oxamate and galloflavin, two inhibitors of LDH activity, and Cyclin G2
activators of pyruvate dehydrogenase: exercise, AMPKActivators of LDHA:Activation of the myc oncogene in cancer cells upregulates lactate dehydrogenase A (LDH-A) expression
Note:
Hexokinase 2 highly expressed in tumor cells (and muscles, fat and heart)
Hexokinase 2 attached on mitochondria inhibits apoptotic stimuli
Hexokinase 1 is constitutely expressed in all tissues
Curcumin: downregulate the expressions and activities of hexokinase (HK) and phosphofructokinase-2 (PFK2) within HSCs. The glucose transporter Glut4 and lactate transporter MCT4 are also concomitantly downregulated (Lian et al., 2015) and HIF-1a
Inflammatory cytokines TNF-α and IL-17, two pro-inflammatory cytokines, modifies LDH activity, causing a shift toward the A isoform which results in increased lactate production. Vitamin C blocks the signaling of TNF-α and IL-17.
Glycolysis and Cori's cycle Dr. Ashok Kumar Jeppu
https://www.slideshare.net/drashokkumarj/glycolysis-and-coris-cycle-dr-ashok-kumar-jeppu
http://chemistry.berea.edu/~biochemistry/2013/ma_kn_at/
Regulation
- activators of pyruvate dehydrogenase
- ↓ in energy status of the cell
- ↑ NAD+/NADH ratio
- ↑ ADP
- ↑ exercise
- ↑ Ca2+
- ↑ insulin
- inhibited by acetyl-CoA
Pyruvate Dehydrogenase Complex - Biochemistry - Medbullets Step 1
https://step1.medbullets.com/step1-biochemistry/102048/pyruvate-dehydrogenase-complex
Glycolysis - Chemistry LibreTexts
https://chem.libretexts.org/Bookshelves/Biological_Chemistry/Supplemental_Modules_(Biological_Chemistry)/Metabolism/Catabolism/Glycolysis
Cancer Metabolism | Oncohema Key
https://oncohemakey.com/cancer-metabolism-2/
http://www.cell.com/cancer-cell/abstract/S1535-6108(13)00288-2
https://www.nature.com/articles/1209603
Effects of luteolin and quercetin, inhibitors of tyrosine kinase, on cell growth and metastasis-associated properties in A431 cells overexpressing epidermal growth factor receptor
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1571723/
An overview of the molecular players in the metabolism of glucose, fatty acids, and glutamine. Pathways were divided in five different sections based on recognition as different entity in biochemical literature. Molecules labeled green were found to be upregulated in RCC specimens, while red molecules were downregulated
Novel drugs that target the metabolic reprogramming in renal cell cancer | Cancer & Metabolism | Full Text
https://cancerandmetabolism.biomedcentral.com/articles/10.1186/s40170-016-0154-8
Cell Death Induction by Targeting Tumor Metabolism | IntechOpen
https://www.intechopen.com/books/cell-death-autophagy-apoptosis-and-necrosis/cell-death-induction-by-targeting-tumor-metabolism
https://pubs.niaaa.nih.gov/publications/arh27-2/134-142.htm
脱氢抗坏血酸不可逆地抑制己糖激酶(HK2)活性
Dehydroascorbic acid irreversibly inhibits hexokinase activity
分子与细胞生物化学,2000Università degli Studi di Urbino "Carlo Bo"
维生素C(脱氢抗坏血酸,DHA)的氧化形式以伪一级顺序完全不可逆地灭活了重组人I型己糖激酶。失活反应没有饱和发生,表明DHA不会与己糖激酶形成可逆复合物。此反应的进一步特征表明,灭活不需要氧气,而二硫苏糖醇(dithiothreitol)虽然能够防止DHA介导的酶活性丧失,但却无法恢复DHA抑制酶的活性。灭活与肽链的断裂或交联无关。然而,酶活性的下降既依赖于残基用碱性pKa去质子化,又依赖于DHA与蛋白质的共价结合。另外,在底物葡萄糖或MgATP存在下,己糖激酶的失活分别减少或增加。最后,对DHA修饰的己糖激酶的氨基酸分析表明,半胱氨酸残基减少了。综上所述,以上结果与试剂与半胱氨酸的硫醇基团的共价结合是DHA介导的己糖激酶活性丧失的关键事件的可能性一致。Dehydroascorbic acid irreversibly inhibits hexokinase activity
Molecular and Cellular Biochemistry,2000
The oxidized form of vitamin C (dehydroascorbic acid, DHA) completely and irreversibly inactivates recombinant human hexokinase type I, in a pseudo-first order fashion. The inactivation reaction occurs without saturation, indicating that DHA does not form a reversible complex with hexokinase. Further characterization of this response revealed that the inactivation does not require oxygen and that dithiothreitol, while able to prevent the DHA-mediated loss of enzyme activity, failed to restore the activity of the DHA-inhibited enzyme. Inactivation was not associated with cleavage of the peptide chain or cross-linking. The decay in enzymatic activity was however both dependent on deprotonation of a residue with an alkaline pKa and associated with covalent binding of DHA to the protein. In addition, inactivation of hexokinase decreased or increased, respectively, in the presence of the substrates glucose or MgATP. Finally, amino acid analysis of the DHA-modified hexokinase revealed a decrease of cysteine residues. Taken together, the above results are consistent with the possibility that covalent binding of the reagent with a thiol group of cysteine is a critical event for the DHA-mediated loss of hexokinase activity....
... Intracellular levels of DHA are therefore kept always very low, but nevertheless several reports proposed its involvement in various biological reactions. For example, it has been suggested that DHA is an inhibitor of the activities of enzymes [16][17][18] and transcription factors [19], although the information available is insufficient to define the biological relevance of these effects, in particular since the con-centrations employed in these studies were far greater than those reasonably achieved inside the cells. ...
... The presence of thiols in SVCT2 is well documented [36][37] and the activity of both the plasma membrane [38] and mitochondrial (Fig. 3B) SVCT2 is indeed susceptible to inhibition by thiol-reactive agents. In addition, DHA is known to react with -SH groups [28] and to inhibit the activity of enzymes containing critical cysteines [17][18]. ...Intracellular dehydroascorbic acid inhibits SVCT2-dependent transport of ascorbic acid in mitochondria
... It was suggested that DHA could be responsible for growth inhibition, since it inhibited the activity of pyridine nucleotide dependent-dehydrogenases measured in cell-free extracts (). Recently, it has been shown that DHA at low concentrations inhibits the activity of several enzymes in vitro, including malate dehydrogenase , fructose 1,6-bisphosphatase (Morell et al. 1997) and hexokinase (Fiorani et al. 2000 ). Moreover, root growth inhibition has been observed in response to DHA administration in vivo (Cordoba-Pedregosa et al. 1996), whereas an increase in AA content stimulates growth (Cordoba-Pedregosa et al. 1996, Arrigoni et al. 1997). ...
... Furthermore, it has long been known that DHA, beside oxidising thiols, can form addition compounds as a consequence of the interaction of its carbonyl groups with amino acid residues (Drake et al. 1942). Recent results showed that DHA irreversibly inhibits human type I hexokinase, and the amino acid analysis of the DHA-modified enzyme reveals a decrease in the number of cysteine residues (Fiorani et al. 2000). It is conceivable that protein-thiol oxidation does not quantitatively contribute to the observed rate of DHA reduction to AA. ...
Dehydroascorbic acid irreversibly inhibits hexokinase activity | Request PDF
https://www.researchgate.net/publication/12378967_Dehydroascorbic_acid_irreversibly_inhibits_hexokinase_activity
Metabolic control by dehydroascorbic acid: Questions and controversies in cancer cells
Article
Apr 2019J CELL PHYSIOL
Luciano Ferrada
Katterine Andrea SalazarFrancisco Nualart
ViewShow abstract
... For example, the use of Apatone™ provokes an 80% decrease in the respiration of tumor cells due to a 30% inhibition in the activity of glyceraldehyde-3-phosphate dehydrogenase and 100% depletion of cellular NAD+ (36). The high level of dehydroascorbate has also been proposed to be involved in the inhibition of other enzymes in this metabolic pathway, including hexokinase and glucose-6-phosphate dehydrogenase (37). In addition to the high levels of ROS produced by BrQ/VC, we demonstrated that this combination, but not VK3/VC, can chemically deplete the coenzyme NADH, which could have direct implications for the glycolytic pathway. ...
Dehydroascorbic acid irreversibly inhibits hexokinase activity | Request PDF
https://www.researchgate.net/publication/12378967_Dehydroascorbic_acid_irreversibly_inhibits_hexokinase_activityMetabolic control by dehydroascorbic acid: Questions and controversies in cancer cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/30963581
己糖激酶2 (HK2)是肿瘤发生和维持所必需的,其系统性删除可治疗癌症小鼠模型
癌症细胞,2013年
伊利诺伊大学芝加哥分校
强调
•
HK2缺失对致癌转化产生抗性
•
HK2缺失选择性靶向癌细胞
•
HK2的系统性删除有良好的耐受性和治疗癌症
•
HK2是利用葡萄糖衍生物和谷氨酰胺用于合成代谢所必需。
摘要
加速的葡萄糖代谢是癌细胞的共同特征。己糖激酶(Hexokinase 2,HK2 )催化葡萄糖代谢的第一步。己糖激酶2(HK2)在癌细胞中高水平表达,但仅在有限数量的正常成人组织中表达。使用Hk2条件性基因敲除小鼠,我们显示HK2在KRas驱动的肺癌和ErbB2驱动的乳腺癌的小鼠模型中,即使HK1持续表达,对于肿瘤的发生和维持也是必需的。同样,在体外和体内,HK2消融抑制人肺癌和乳腺癌细胞的肿瘤表型。系统性Hk2缺失对患有肺肿瘤的小鼠具有治疗作用,而没有不利的生理后果。肺癌细胞中Hk2的缺失抑制了葡萄糖来源的核糖核苷酸,并削弱了合成代谢中谷氨酰胺来源的碳利用。
己糖激酶(HKs)催化葡萄糖代谢的第一步,即ATP依赖的葡萄糖(Glc)磷酸化以产生6-磷酸葡萄糖(G6P)。由单独的基因编码的四种主要己糖激酶同工型在哺乳动物组织中表达:HK1,HK2,HK3和HK4(也称为葡萄糖激酶)(Robey和Hay,2006年)。通过己糖激酶催化Glc磷酸化为G6P,促进并维持浓度梯度,从而促进葡萄糖进入细胞并启动所有主要的葡萄糖利用途径。因此,己糖激酶影响细胞内葡萄糖通量的大小和方向。尽管四个HK具有许多共同的生化特性,但它们固有的酶活性和组织分布使它们彼此区别。 HK1,HK2和HK3是高亲和力的亚型,但HK3被生理浓度的葡萄糖抑制(Wilson,2003年)。高亲和力的己糖激酶被过量的G6P抑制。葡糖激酶是一种低亲和力的己糖激酶,不受G6P抑制,主要在肝脏和胰腺中表达。两种高亲和力的己糖激酶HK1和HK2与线粒体有关,也与细胞存活有关(Gottlob et al。 ,2001,Majewski等,2004)。 HK1在大多数哺乳动物成年组织中组成性表达。然而,HK2尽管在胚胎组织中大量表达,但仅在有限的成年组织中,例如脂肪,骨骼和心肌中以高水平表达(Wilson,2003)。但是,癌细胞表达高水平的HK2(Mathupala等人,2001; Shinohara等人,1994),这使它们与正常细胞区别开来,并且至少部分负责加速葡萄糖通量。通过使用正电子发射断层扫描(PET)可视化肿瘤,可以证明糖酵解癌症中HK2的高表达和高活性。注射标记的葡萄糖类似物[18F]氟-2-脱氧葡萄糖(FDG)后使用PET,然后由糖酵解性癌细胞摄取并由己糖激酶磷酸化以形成FDG-磷酸,PET可以检测到。己糖激酶的磷酸化是将FDG保留在癌细胞中所必需的。
Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer
CANCER CELL, 2013
University of Illinois at Chicago
Highlights
•
HK2 deletion exerts resistance to oncogenic transformation
•
HK2 deletion selectively targets cancer cells
•
Systemic deletion of HK2 is well tolerated and therapeutic for cancer l
•
HK2 is required for glucose-derived and glutamine in utilization in anaplerosis
Summary
Accelerated glucose metabolism is a common feature of cancer cells. Hexokinases catalyze the first committed step of glucose metabolism. Hexokinase 2 (HK2) is expressed at high level in cancer cells, but only in a limited number of normal adult tissues. Using Hk2 conditional knockout mice, we showed that HK2 is required for tumor initiation and maintenance in mouse models of KRas-driven lung cancer, and ErbB2-driven breast cancer, despite continued HK1 expression. Similarly, HK2 ablation inhibits the neoplastic phenotype of human lung and breast cancer cells in vitro and in vivo. Systemic Hk2 deletion is therapeutic in mice bearing lung tumors without adverse physiological consequences. Hk2 deletion in lung cancer cells suppressed glucose-derived ribonucleotides and impaired glutamine-derived carbon utilization in anaplerosis.
Hexokinases (HKs) catalyze the first committed step in glucose metabolism, i.e., the ATP-dependent phosphorylation of glucose (Glc) to yield glucose-6-phosphate (G6P). Four major hexokinase isoforms, encoded by separate genes, are expressed in mammalian tissues: HK1, HK2, HK3, and HK4 (also known as glucokinase) (Robey and Hay, 2006). By
catalyzing the phosphorylation of Glc to G6P, hexokinases promote and sustain a concentration gradient that facilitates glucose entry into cells and the initiation of all major pathways of glucose utilization. Therefore, hexokinases influence both the magnitude and the direction of glucose flux within cells. Although the four HKs share many common biochemical properties, their intrinsic enzymatic activity and their tissue distribution distinguish them from each other. HK1, HK2, and HK3 are high-affinity isoforms, but HK3 is inhibited by physiological concentrations of glucose (Wilson, 2003). The high-affinity hexokinases are inhibited by excess of G6P. Glucokinase is a low-affinity hexokinase, which is not inhibited by G6P and is mainly expressed in liver and pancreas.The two high-affinity hexokinases, HK1 and HK2, are associated with mitochondria and were also implicated in cell survival (Gottlob et al., 2001, Majewski et al., 2004). HK1 is constitutively expressed in most mammalian adult tissues. HK2, however, although abundantly expressed in embryonic tissues, is expressed at high levels only in a limited number of adult tissues, such as adipose, skeletal, and cardiac muscles (Wilson, 2003). However, cancer cells express high levels of HK2 (Mathupala et al., 2001, Shinohara et al., 1994), which distinguishes them from the normal cells and is, at least in part, responsible for the accelerated glucose flux.
The high level of HK2 expression and activity in glycolytic cancers are manifested by the use of positron emission tomography (PET) to visualize tumors. PET is used following injection of the labeled glucose analog, [18F]fluoro-2-deoxyglucose (FDG), which is then taken up by glycolytic cancer cells and phosphorylated by hexokinase to form FDG-phosphate, which can be detected by PET. The phosphorylation by hexokinase is required for the retention of FDG in the cancer cells.The studies described here are aimed at elucidating the role of HK2 in tumor initiation and maintenance of KRas-driven non-small cell lung cancer (NSCLC) and ErbB2-driven breast cancer and to provide a proof of concept that HK2 can be systemically deleted for cancer therapy with no adverse physiological consequences.
Results
HK2 Is Required for Oncogenic Transformation;
HK2 Is Required for Lung and Breast Cancer Development in Mouse Models;
Taken together, these results provided compelling evidence that HK2 is required for both oncogenic KRas-driven lung tumorigenesis and ErbB2-driven mammary gland tumorigenesis in vivo.
HK2 Ablation Reverses Tumorigenesis In Vitro and In Vivo of NSCLC and Breast Cancer Cells
Systemic Hk2 Deletion in Adult Mice Does Not Cause an Overt Phenotype but Reduces Tumor Burden of Lung Cancer
As was shown above, HK2 is expressed at high levels in cancer cells but is hardly detectable in the corresponding normal tissues from which they were derived. HK2 is expressed as the predominant isoform in only a limited number of adult tissues, such as fat, muscles, and heart (Figure S4A), and mice with 50% deletion of Hk2 do not show an overt phenotype except of mildly impaired glucose homeostasis in response to exercise and high fat diet (Fueger et al., 2003).
HK2 Is Required for Ribonucleotide Synthesis, the Serine Biosynthesis Pathway, and the Flow of Carbons from Glycolysis into the TCA Cycle and Fatty Acid Synthesis in KRasG12D- NSCLC Cells
Glutamine-Derived Carbon Incorporation into TCA Cycle Intermediates Is Reduced in KRasG12D-NSCLC Cells following Hk2 Deletion
Oncogenic Ras-transformed cells are highly dependent on the availability of glutamine, which is heavily incorporated into TCA cycle intermediates in these cells (Gaglio et al., 2011). We therefore examined Hk2 deletion for the ability to alter glutamine requirements in oncogenic Ras-expressing cells. Interestingly, we found that exogenous glutamine restriction markedly reduced proliferation of both KRasG12D-NSCLC cells and MEFs transformed by oncogenic Ras. In contrast, proliferation of these cells following Hk2 deletion was not further affected by glutamine restriction (Figure S5F).In summary, HK2 plays a critical role in the diversion of glucose into pathways, which are required for the anabolic activities in cancer cells. Interestingly, it was shown and proposed previously that, in cancer cells, downstream glycolytic activities are attenuated to divert glucose into the branched pathways. Our results suggest that an increase in the most upstream step in glucose metabolism catalyzed by HK2 is also critical for diverting glucose into these pathways in cancer cells. Therefore, this could explain why cancer cells, in contrast to their normal counterparts, express high levels of HK2. The expression of HK1 is possibly sufficient for normal cell metabolism. The accelerated anabolic metabolism in cancer cells demands a robust hexokinase activity; therefore, the induction of HK2 expression is required.
Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer: Cancer Cell
https://www.cell.com/cancer-cell/fulltext/S1535-6108(13)00288-2
Inhibition of Liver Hexokinase by Dehydroascorbic Acid and Alloxan
Bhattacharya, S. K.
Abstract
THE structure and properties of dehydroascorbic acid are to a certain extent similar to those of alloxan, and both the substances are diabetogenic in rats1,2. It was previously reported by Bhattacharya, Robson and Stewart3 that dehydroascorbic acid causes a precipitous but temporary fall in the reduced glutathione content of rat liver. Their results suggested that the fall in reduced glutathione was not due to oxidation but due to a complex formation between the sulphydryl group of glutathione and dehydroascorbic acid. Hexokinase is an enzyme the activity of which probably depends on the presence of free sulphydryl group(s)4,5. It seemed desirable, therefore, to study the effect of dehydroascorbic acid on the hexokinase activity of rat liver homogenate. Although Griffiths6 reported that cysteine is capable of releasing the inhibition of muscle hexokinase by alloxan, in view of the results obtained with dehydroascorbic acid it was decided to study the effect of alloxan and cysteine on liver hexokinase.
Publication:
Nature
Pub Date: November 1959 DOI: 10.1038/1841638b0 Bibcode: 1959Natur.184.1638B
Inhibition of Liver Hexokinase by Dehydroascorbic Acid and Alloxan - NASA/ADS
https://ui.adsabs.harvard.edu/abs/1959Natur.184.1638B/abstract
Hexokinase Inactivation Induced by Ascorbic Acid/Fe(II) in Rabbit Erythrocytes Is Independent of Glutathione-Reductive Processes and Appears to Be Mediated by Dehydroascorbic Acid☆
Instituto di Chimica Biologica “Giorgio Fornaini,” Università degli Studi di Urbino, Via Saffi, 2, 61029, Urbino (PS), Italy
Abstract
Recent studies performed in our laboratory demonstrated that rabbit red blood cell hexokinase was remarkably inhibited by the cocktail ascorbic acid/Fe(II) (Stocchiet al.,1994,Arch. Biochem. Biophys.311, 160–167) and that the formation of dehydroascorbic acid was a key event in this process (Fioraniet al.,1996,Arch. Biochem. Biophys.334, 357–361). The present study was undertaken to determine the final hexokinase-inactivating species using cell-free extract as a model. Our results demonstrate superimposable kinetics of hexokinase decay promoted by either ascorbic acid/Fe(II) or dehydroascorbic acid in erythrocyte lysates in which the reduced glutathione (GSH) levels were variously manipulated. In particular, neither removal nor addition of this tripeptide was able to significantly alter the rate or extent of hexokinase inhibition. Thus, GSH-reductive processes are dispensable events in the process of hexokinase inhibition promoted by ascorbic acid/Fe(II) in red blood cells. As a consequence, dehydroascorbic acid appears to be the species which directly inhibits hexokinase. This inference is further supported by the observation that addition of dehydroascorbic acid to the purified enzyme leads to a remarkable inhibition in its activity.抗坏血酸/ Fe(II)引起的兔红细胞己糖激酶的失活与谷胱甘肽还原过程无关,并且似乎由脱氢抗坏血酸介导☆
乌尔比诺大学化学大学生物学研究所“ Giorgio Fornaini”,意大利乌尔比诺(PS),邮编:Via Saffi,2,61029
抽象
在我们实验室中进行的最新研究表明,鸡尾酒抗坏血酸/ Fe(II)能够显着抑制兔红细胞己糖激酶(Stocchiet等,1994; Arch。Biochem。Biophys.311,160-167)。脱氢抗坏血酸是该过程中的关键事件(Fioraniet等人,1996,Arch。Biochem。Biophys.334,357-361)。进行本研究以确定使用无细胞提取物作为模型的最终己糖激酶灭活物种。我们的研究结果表明,抗坏血酸/ Fe(II)或脱氢抗坏血酸在红血球溶解产物中促进了己糖激酶衰变的叠加动力学,在该溶血产物中谷胱甘肽(GSH)的含量被不同地控制。特别地,去除或添加该三肽均不能显着改变己糖激酶抑制的速率或程度。因此,在红细胞中由抗坏血酸/ Fe(II)促进的己糖激酶抑制过程中,GSH还原过程是必不可少的事件。结果,脱氢抗坏血酸似乎是直接抑制己糖激酶的物质。通过将脱氢抗坏血酸添加至纯化的酶导致其活性的显着抑制的观察进一步支持了这一推论。
Hexokinase Inactivation Induced by Ascorbic Acid/Fe(II) in Rabbit Erythrocytes Is Independent of Glutathione-Reductive Processes and Appears to Be Mediated by Dehydroascorbic Acid - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S000398619799963XHexokinase Inactivation Induced by Ascorbic Acid/Fe(II) in Rabbit Erythrocytes Is Independent of Glutathione-Reductive Processes and Appears to Be Mediated by Dehydroascorbic Acid, Archives of Biochemistry and Biophysics | 10.1006/abbi.1997.9963 | DeepDyve
https://www.deepdyve.com/lp/elsevier/hexokinase-inactivation-induced-by-ascorbic-acid-fe-ii-in-rabbit-gbDRbVjSG1
Inhibitors of glucose transport and glycolysis as novel anticancer therapeutics
Abstract
Metabolic reprogramming and altered energetics have become an emerging hallmark of cancer and an active area of basic, translational, and clinical cancer research in the recent decade. Development of effective anticancer therapeutics may depend on improved understanding of the altered cancer metabolism compared to that of normal cells. Changes in glucose transport and glycolysis, which are drastically upregulated in most cancers and termed the Warburg effect, are one of major focuses of this new research area. By taking advantage of the new knowledge and understanding of cancer’s mechanisms, numerous therapeutic agents have been developed to target proteins and enzymes involved in glucose transport and metabolism, with promising results in cancer cells, animal tumor models and even clinical trials. It has also been hypothesized that targeting a pathway or a process, such as glucose transport or glucose metabolism, rather than a specific protein or enzyme in a signaling pathway may be more effective. This is based on the observation that cancer somehow can always bypass the inhibition of a target drug by switching to a redundant or compensatory pathway. In addition, cancer cells have higher dependence on glucose. This review will provide background information on glucose transport and metabolism in cancer, and summarize new therapeutic developments in basic and translational research in these areas, with a focus on glucose transporter inhibitors and glycolysis inhibitors. The daunting challenges facing both basic and clinical researchers of the field are also presented and discussed.Core tip: Reprogramming of metabolism has been recognized at the beginning of 21st century as an emerging hallmark of cancer. The Warburg effect is one of the major focuses in the reprogramming. We cannot fully understand or more effectively treat cancer without a better understanding of cancer metabolism. Targeting cancer metabolism, particularly glucose transport and glycolysis, has been shown to be effective in inhibiting cancer growth. This review summarizes recent progresses in developments of therapeutics inhibiting glucose transporters and glycolytic enzymes, provides key information associated with each inhibitor, discusses their promises and problems as well as future challenges and directions of the basic and translational research of the field.
by Ohio University
World J Translation Medicine, published in 2014
Key Words: Cancer metabolism, Warburg effect, Glycolytic enzymes, Glucose transporters, Translational research
INTRODUCTION
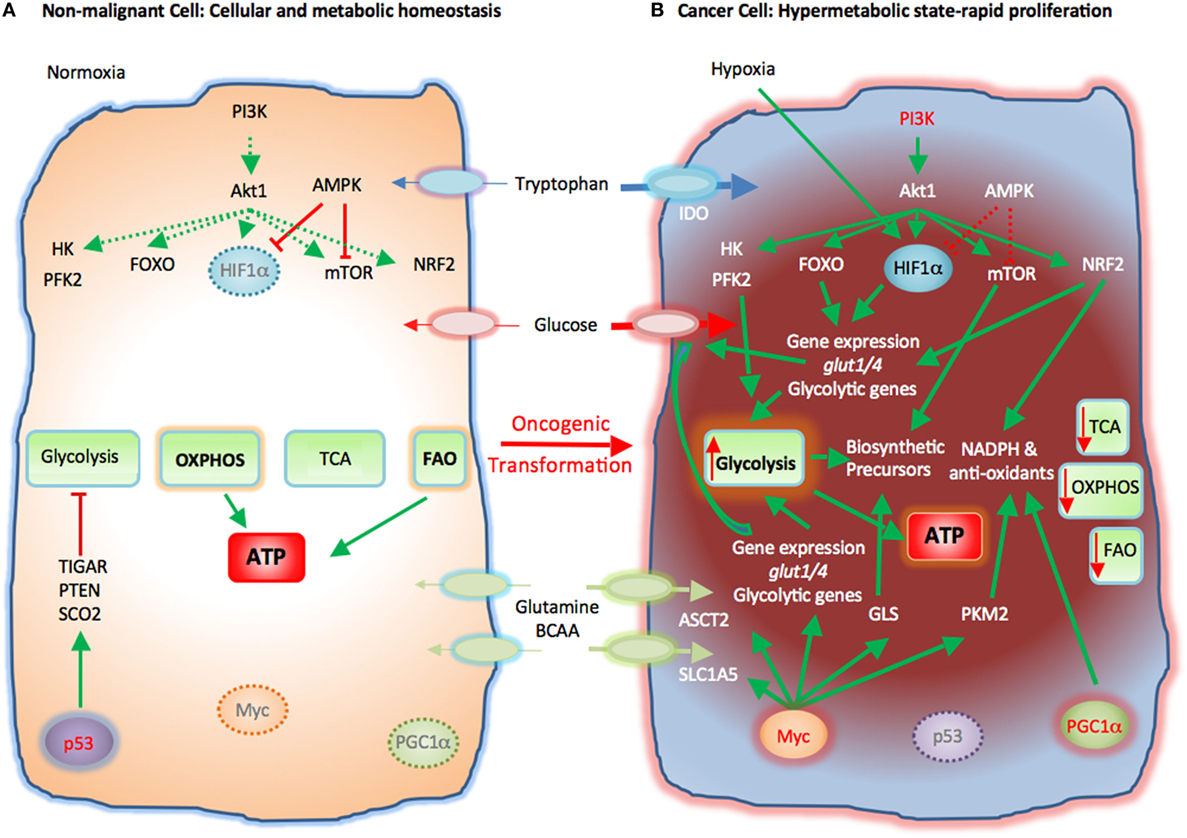
Cancer has long been considered a group of diseases caused by genetic mutations and genetic mutations only. However, in recent decades, extensive biochemical and biological studies have convincingly demonstrated that cancers exhibit significantly reprogrammed metabolism, which plays important roles in tumorigenesis[1-6]. In some cases, altered metabolism may be not only the consequence of genetic mutations, but also a contributing factor or cause of tumorigenesis[7-9]. Cancer metabolic reprogramming and altered energetics have been recognized now as a hallmark of cancer[10].
.
The importance of metabolism in cancer was actually recognized long time ago. In the 1920s, the German biochemist Otto Warburg, studied glucose metabolism in cancer tissues. He found that, unlike in normal tissue, incubated cancer samples always switched from mitochondrial oxidative phosphorylation (OXPHOS) to cytosolic glycolysis even when oxygen was abundant[11]. This phenomenon of so-called aerobic glycolysis has been known as the Warburg effect[12-15]. Warburg went so far as to claim that the altered glucose metabolism was the cause of cancer. This hypothesis is called the Warburg theory of cancer. He speculated that due to some mitochondrial dysfunctions, mitochondria could not synthesize ATP and thus cells must switch to cytosolic glycolysis, leading to cancer formation[14,16]. Biological studies in recent decades have found that Warburg’s view on the cause of the switch was largely incorrect: many cancers switch to glycolysis even without any mitochondrial defects. New biological and biochemical studies in the past decades revealed that the switch from OXPHOS to glycolysis is not just for ATP synthesis but also for biomass synthesis[15,17], production of NADPH[15,18], a reducing agent needed to remove reactive oxygen species (ROS) generated by cancer cells’ accelerated metabolism, as well as synthesis of amino acids[15,19]. The Warburg effect appears to be a strategic move made by cancer cells to deal with multiple requirements for growth, survival, and proliferation in a microenvironment with numerous constraints.Altered cancer metabolism has also been recognized as a potential target for cancer therapeutics. Glucose transport and glucose metabolism are significantly upregulated in cancer as revealed by the PET scan and other detection methods[20-24]. The reliance of cancer cells on glucose indicates that they are addicted to the Warburg effect or glucose[25-27]. As a result, cancer cells are more sensitive than normal cells to changes in glucose concentration and will die before normal cells[25-28]. The recognition of this vulnerability in cancer cells has led to targeting glucose transport and metabolism as a new anticancer strategy. Furthermore, although targeted anticancer drugs inhibit one or more proteins or enzymes, cancers demonstrate the ability to escape inhibition using redundant signaling pathway(s). It has been proposed that targeting a signaling pathway or a metabolic process, rather than a protein in a pathway, may be more effective in preventing drug resistance and prolonging treatment effectiveness[29,30]. Potential targets for this proposed new approach include glucose transport and glycolysis, the predominant glucose metabolic changes found in cancer cells.
It should be emphasized that targeting cancer metabolism is not an entirely novel strategy. Some of the earliest chemotherapy drugs, such as methotrexate, also target metabolism and show significant efficacy[31-33]. As we have accumulated more knowledge about cancer metabolism, we should be able to develop more successful anti-cancer-metabolism drugs. In the following sections, recently developed glucose transport and glycolysis inhibitors will be described.GLUCOSE TRANSPORT AND GLUCOSE METABOLISM IN CANCER CELLS–THE WARBURG EFFECT
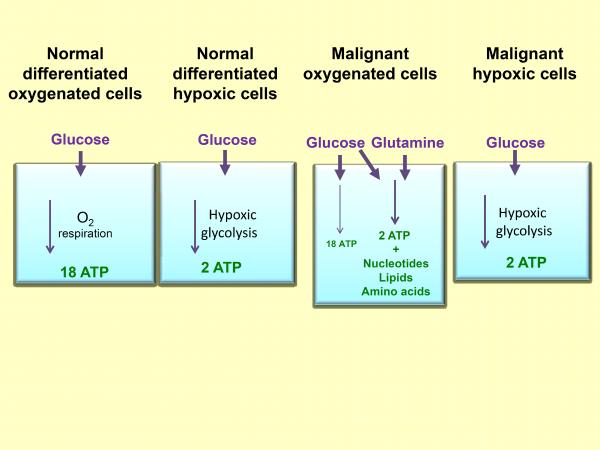
In normal cells under aerobic conditions, OXPHOS is used to make ATP, the universal energy currency in all living organisms[34]. OXPHOS is used because it is the most efficient way for making ATP. For each molecule of glucose, approximately 34 molecules of ATP can be produced by OXPHOS[34]. However, OXPHOS can proceed only when oxygen is present and abundant, a condition called normoxia. When oxygen is lacking, a condition called hypoxia, cells are forced to shift to anaerobic glycolysis to maintain ATP synthesis and energy metabolism[35]. Due to rapid growth and proliferation, a large proportion of the cancer cells in a tumor are in a hypoxic condition and thus use glycolysis to make ATP and other essential biomass molecules such as ribonucleotides. The phenomenon of OXPHOS-to-glycolysis shift in cancer cells is called the Warburg effect[12-16]. Although the Warburg effect was observed more than 80 years ago, its interpretation is still controversial and evolving. Warburg thought that the effect was caused by mitochondrial dysfunctions and the effect is a forced alternative strategy for ATP synthesis. However, research in recent decades largely disagrees with this interpretation. Recently, it has been found that the switch in cancer cells is primarily for the synthesis of biomass (e.g., of RNA precursor and others)[17], the reducing agent NADPH[18], which is needed for clearing ROS, and the amino acid serine[19]. ATP synthesis seems not to be a rate-limiting factor. This conclusion is very different from Warburg’s and is based on the observation that although cancer cells upregulate all glycolytic enzymes, they switch pyruvate kinase (PK), the last enzyme in the glycolytic pathway, from a form with higher higher activity (PKM1) to that with lower activity, PKM2[36-39]. This change suggests that cancer cells do not want all the glucose obtained from the upregulated glucose transport to be converted to pyruvate, but rather diverts some glucose metabolic intermediates to other connected metabolic pathways, such as pentose phosphate pathway (PPP) for synthesis of biomass and reducing agents[17-19,40]. This also suggests that ATP synthesis is not the top priority of the upregulation of glucose transport and metabolism. On the other hand, since glycolysis is about 18 times less efficient compared to OXPHOS, cancer cells must drastically upregulate glycolysis to compensate for the low ATP production.ANTICANCER THERAPEUTICS TARGETING GLYCOLYSIS AND ITS CONNECTED PATHWAYS
Currently, the Warburg effect is a very active cancer research area[13]. Targeting glucose metabolism and transport, has been proposed as an effective anticancer strategy[1,3]. Glycolysis, the key process of increased glucose metabolism in cancer cells, has been targeted both in vitro and in vivo[3,41,42]. Glycolysis genes are overexpressed in various cancers[35]. In addition to higher potentials for invasiveness and metastasis[43], the glycolytic switch in cancer also increases cancer’s sensitivity to external interference because of their higher dependence on aerobic glycolysis[25-28].
Glucose deprivation, a method traditionally used to reduce glucose concentration in cultured cells for metabolic studies, has been used frequently in cancer research[44-47]. Glucose deprivation limits glucose supply, forcing cancer cells to slow down proliferation or undergo apoptosis[48-50]. Blocking glucose transport or glycolysis is similar to glucose deprivation, suggesting the possibility of restricting glucose supply with glucose transport or glycolysis inhibitors as an anticancer strategy.
Various inhibitors of glycolytic enzymes have shown significant anticancer efficacy. Most of the reported glycolysis inhibitors are summarized (Table 1 and Figure 1). The enzymes targeted include hexokinase (HK), phosphofructokinase (PFK), pyruvate kinase (PK), lactate dehydrogenase (LDH), and pyruvate dehydrogenase kinase (PDK). Related studies revealed that these inhibitors induced apoptosis in cancer cells[51,52]. Moreover, inhibition of glycolysis has been shown to overcome drug resistance in multiple cancer cells associated with mitochondrial respiratory defect and hypoxia[53]. Although numerous attempts to block glycolysis by using various inhibitors in cancer cells and in animal models have been made, developing clinically effective and safe glucose metabolism-targeting therapeutics is still a challenging task.
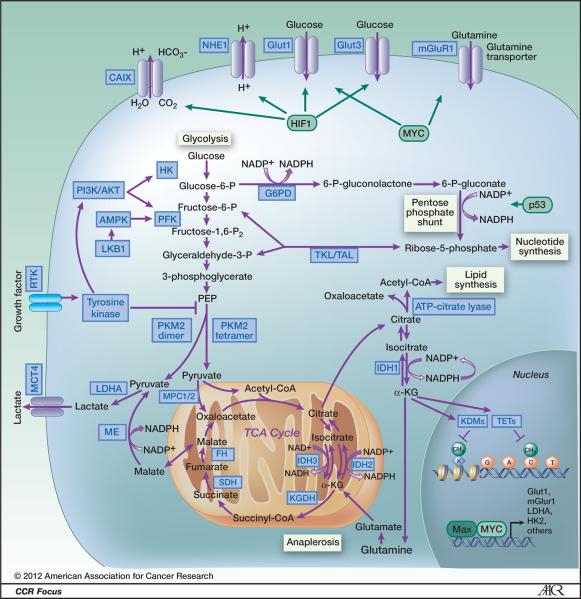
Figure 1 Glycolysis and inhibitors/activators of glycolysis as potential anti-cancer therapeutics. Glucose transporters and enzymes are shown in red and glycolytic intermediates are shown in blue. Inhibitors/activators are in black squares. PPP: Pentose phosphate pathway; OXPHOS: Oxidative phosphorylation; shown in green. GLUTs: Glucose transporters; HK: Hexokinase; GPI: Glucose-6-phosphate isomerase; PFK: Phosphofructokinase; ALD: Aldolase; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; PGK: Phosphoglycerate kinase; PGAM1: Phosphoglycerate mutase 1; ENO: Elonase; PKM2: Pyruvate kinase M2; LDH: Lactate dehydrogenase; PDK: Pyruvate dehydrogenase kinase; PDH: Pyruvate dehydrogenase; G6PD: Glucose-6-phosphate dehydrogenase; TKTL1: Transketolase-like enzyme 1; PHGDH: Phosphoglycerate dehydrogenase; G6P: Glucose 6-phosphate; F6P: Fructose 6-phosphate; F-1,6-bisP: Fructose 1,6-bisphosphate; 3-PG: 3-phosphoglycerate; 2-PG: 2-phosphoglycerate; 6-P-gluconolactone: 6-phosphogluconolactone; Xylulose-5-P: D-xylulose-5-phosphate; 2-DG: 2-deoxyglucose; 3-BP: 3-bromopyruvate; 3PO: 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one; DCA: Dichloroacetate; 6-AN: 6-aminonicotinamide.
Hexokinase (HK) as the first enzyme in glycolysis phosphorylates glucose to glucose-6-phophate (G6P) irreversibly, which is a rate-limiting step. In cancer cells, type II HK (HK2) is bound to mitochondria, facilitating a high glycolytic flux rate and preventing cancer cell from apoptosis[54]. HK2 is required for cancer initiation and maintenance and the systemic deletion of HK2 is therapeutic in mice bearing tumors[55]. Thus, targeting HK2 may be an effective anti-cancer strategy.
2-deoxy-D-glucose (2-DG) is one of the most widely studied HK inhibitors. 2-DG is a glucose analog with a hydrogen group instead of a hydroxyl group in position 2 of glucose. Due to its structural similarity, 2-DG competes with glucose and inhibits HK with a Ki of 0.25 mmol/L[56]. The product 2-deoxy-D-glucose-6-phosphate made from 2-DG cannot be processed in the following glycolytic steps and therefore blocks glycolysis, leading to ATP depletion, cell cycle arrest and cell death[57,58]. Synergistic studies combining 2-DG and other anti-cancer drugs, such as adriamycin and paclitaxel, indicated that 2-DG is effective in vivo in combination with other drugs[59]. 2-DG sensitizes glioblastoma cells to other anti-cancer treatments and radiation[60-63]. Though effective, 2-DG is relatively toxic with side effects when administered to patients[61,64]. This is at least in part because 2-DG has to be used at high concentrations, around and higher than 5 mmol/L, in order to compete with blood glucose[65].
3-bromopyruvate (3-BP) is another HK inhibitor which has been shown to inhibit the progression of tumors in vivo[66-68]. 3-BP also increases the total ROS in tumor cells[69,70]. A recent study demonstrated that 3-BP inactivates ABC transporters, restoring drug sensitivity in cancer cells[71]. 3-BP has also been studied in combination with various anti-cancer drugs for synergistic effects, and it has been found to be effective in vitro[72] and in vivo[73], although with some hepatotoxicity[74]. However, 3-BP inhibits other enzymes, such as GAPDH, as well[75]. Up to now, no clinical trials have been reported for 3-BP. This may be attributed to its low target specificity and relatively high toxicity.
Lonidamine specifically inhibits mitochondria-bound HK2, which is present mostly in cancer cells but not in normal cells[76]. It effectively inhibits the cell growth, decreasing lactate and ATP generation, in cancer cells[77,78]. Meanwhile, the combination of lonidamine with other anti-cancer agents reverts drug resistance and is effective in the treatment of various cancer cells in both pre-clinical and phase II/III studies[78-80]. However, the combination of lonidamine and epirubicine resulted in no improvement in patients’ survival[81]. Though lonidamine has been widely studied, its hepatotoxicity resulted in the termination of several clinical trials[82,83]. These studies of the HK2 inhibitors suggest that, although HK2 is a potential target, being the first and the rate-limiting step of glycolysis, inhibition of HK2 may result in severe side effects. However, the combination of HK2 inhibitors and other anti-cancer drugs may still be an alternative approach for HK2-overexpressing tumors.Pyruvate dehydrogenase kinase (PDK) favors glycolysis over mitochondrial oxidative phosphorylation (OXPHOS) by blocking the activity of pyruvate dehydrogenase (PDH) by phosphorylating it[142]. Under normal oxygen pressures, pyruvate goes to mitochondria and is converted to acetyl-CoA in a step catalyzed by PDH. Acetyl-CoA is an important metabolite involved in the citric acid cycle and OXPHOS. In studies in cancer cells, PDK1 expression was induced by HIF-1 in hypoxic conditions and shown to lead to increased glycolysis and suppressed OXPHOS[143,144]. The expression of PDK1 is associated with poor prognosis in head-and-neck squamous cancer[145]. Also, the upregulation of PDK in cancer was associated with a more aggressive phenotype[146]. For these reasons, PDK has been considered an attractive and promising anti-cancer target.
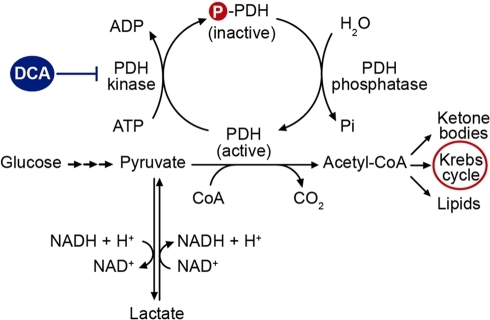
Dichloroacetate (DCA), an analog of pyruvate, has been identified as a PDK inhibitor and widely studied for its ability to inhibit lactate production and cancer growth[147-151]. DCA decreases lactate production by shifting the pyruvate metabolism from glycolytic fermentation towards mitochondrial OXPHOS, and restores mitochondrial function, thus potentially restoring apoptosis-induction, allowing cancer cells to undergo programmed cell death and shrink the tumor[53]. DCA’s research and clinical trials were based on the belief that cancer cells’ mitochondrial function is abnormal and therefore cancer cell growth will be reduced by upregulating and normalizing their OXPHOS. DCA was shown to be effective in suppressing the growth of cancer cells both in vitro and in vivo[152-155]. Several human clinical trials of DCA started after the successful cell and animal studies and still on-going. A phase II clinical trial for malignant glioblastoma has been completed and shows that DCA can be used safely in patients with glioblastoma, suggesting that DCA is a promising anti-cancer agent and inhibiting glycolysis is a potent and effective anti-cancer strategy[156] (Table 1). In addition, several clinical trials combining DCA and other anti-cancer drugs or therapies are in progress. On the other hand, human studies indicate that DCA’s anticancer effects, if any, may be cancer type-related. More basic biomedical studies need to be conducted on the compound before DCA’s anticancer activity can be better evaluated.
Inhibitors of glucose transport and glycolysis as novel anticancer therapeutics
https://www.wjgnet.com/2220-6132/full/v3/i2/37.htm
Dietary polyphenols decrease glucose uptake by human intestinal Caco‐2 cells - Johnston - 2005 - FEBS Letters - Wiley Online Library
https://febs.onlinelibrary.wiley.com/doi/full/10.1016/j.febslet.2004.12.099
New natural inhibitors of hexokinase 2 (HK2): Steroids from Ganoderma sinense
Abstract
Hexokinase 2 (HK2), a rate-limiting enzyme in the first step of glycolysis pathway, expresses at high level in cancer cells compared with normal cells. HK2 provides a new target for cancer therapy due to its pivotal role in tumor tumourigenic and metastatic process. The structure-based virtual ligand screening in a small in-house database of natural products predicted that a new steroid, (22E,24R)-6β-methoxyergosta-7,9(11),22-triene-3β,5α-diol (2) from Ganoderma sinense(黑灵芝) has high binding affinity to HK2 with significant calculated binding free energy. Based on this prediction, compound 2, together with the other 12 steroid analogues (1, 3–13) from this plant were selected for further in vitro microscale thermophoresis (MST), enzyme inhibition, and cell-based assays based on the HK2 target. And compound 2 was finally identified as an HK2 inhibitor. As the first natural HK2 inhibitor, compound 2 can be considered as a potential drug candidate targeting at HK2 for cancer therapy.
A total of 13 steroids (1–13) including two new ones (1–2) were isolated from the fruiting bodies of Ganoderma sinense. Among them, (22E,24R)-6β-methoxyergosta-7,9(11),22-triene-3β,5α-diol (2) was found to have the highest binding affinity to HK2. In vitro MST, enzyme inhibition, and cell-based assays were undertaken to identify compound 2 as a HK2 inhibitor using this method. In vitro MST, enzyme inhibition and cell-based assays further validated compound 2 as a potential HK2 inhibitor.
New natural inhibitors of hexokinase 2 (HK2): Steroids from Ganoderma sinense - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0367326X1731612X
Oroxylin A induces dissociation of hexokinase II from the ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3641353
Oroxylin A is a major active component of the Chinese traditional medicinal plant Scutellaria baicalensis Georgi(黄芩), which has been reported as a potential anticancer drug.We demonstrated that, Oroxylin A inhibited the glycolysis and the binding of hexokinase II (HK II) with mitochondria in human breast carcinoma cell lines, which was dependent on sirtuin-3 (SIRT3).
Cited by: 91
Publish Year: 2013
Metformin Repurposed Anti-Cancer Drug - Jeffrey Dach MD
https://jeffreydachmd.com/2017/07/metformin-repurposed-anti-cancer-drug/
https://jeffreydachmd.com/2017/07/metformin-repurposed-anti-cancer-drug/
Dehydroascorbic acid irreversibly inhibits hexokinase ...
https://link.springer.com/article/10.1023/A:1007168032289
Fiorani M, De Sanctis R, Scarlatti F, Stocchi V: Substrates of hexokinase, glucose-6-phosphate dehydrogenase, and glyceraldehyde-3-phosphate dehydrogenase prevent the inhibitory response induced by ascorbic acid/iron and dehydroascorbic acid in rabbit erythrocytes.
Cited by: 37
Publish Year: 2000
Author: Mara Fiorani, Roberta De Sanctis, Francesca Scarlatti, Luciana Vallorani, Roberta De Bellis, Giordan...
Dehydroascorbic acid irreversibly inhibits hexokinase ...
https://www.researchgate.net/publication/12378967_Dehydroascorbic_acid_irreversibly...
Dehydroascorbic acid irreversibly inhibits hexokinase activity Article in Molecular and Cellular Biochemistry 209(1-2):145-53 · July 2000 with 21 Reads How we measure 'reads'
Inhibition of Liver Hexokinase by Dehydroascorbic Acid and ...
http://adsabs.harvard.edu/abs/1959Natur.184.1638B
Abstract THE structure and properties of dehydroascorbic acid are to a certain extent similar to those of alloxan, and both the substances are diabetogenic in rats 1,2.It was previously reported by Bhattacharya, Robson and Stewart 3 that dehydroascorbic acid causes a precipitous but temporary fall in the reduced glutathione content of rat liver. Their results suggested that the fall in reduced ...
Published in:
Nature · 1959
Authors:
S K Bhattacharya
Affiliation:
University of Leeds
About:
Enzyme · Glutathione · Dehydroascorbic acid
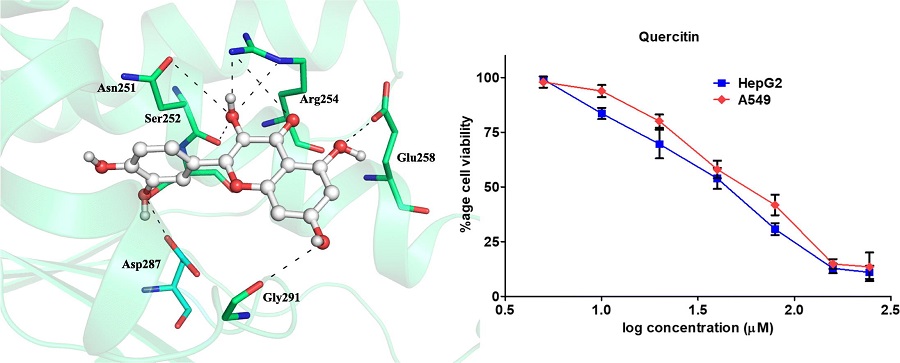
Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy
Centre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India
Received 3 May 2019, Revised 19 June 2019, Accepted 21 June 2019, Available online 22 June 2019.
Highlights
•
Inhibitory potential of quercetin to the PDK3 was investigated.
•
Quercetin binds to the active site of PDK3 and forms significant non-covalent interactions.
•
Quercetin shows significant binding affinity and enzyme inhibition potential for PDK3.
•
Quercetin reduce the proliferation of cancer cells and is non-toxic to normal cells.
Abstract
Pyruvate dehydrogenase kinase 3 (PDK3) is a mitochondrial protein, has recently been considered as a potential pharmacological target for varying types of cancer. Here, we report the binding mechanism of quercetin to the PDK3 by using molecular docking, simulation, fluorescence spectroscopy and isothermal titration calorimetric assays. Molecular docking along with simulation provided an in-depth analysis of protein-ligand interactions. We have observed that quercetin interacts to the important residues of active site cavity of PDK3 and shows a well-ordered conformational fitting. The stability of quercetin-PDK3 complex is maintained by several non-covalent interactions throughout the simulation. To complement in silico findings with the experiments, we have successfully expressed and purified human PDK3. Both fluorescence and isothermal titration calorimetric experiments showed excellent binding affinity of quercetin to the PDK3. Kinase inhibition assay further revealed a significant inhibitory potential of quercetin to the PDK3 with the IC50 values in μM range. Quercetin is non-toxic to HEK293, and significantly inhibits the proliferation of cancer (HepG2 and A549) cell lines. All these observations clearly indicate that quercetin may be further evaluated as promising therapeutic molecule for PDK3 with required modifications and in vivo validation.
Graphical abstract
Showing interaction of quercetin with the PDK3 and inhibition of cancer cell proliferation.
Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0141813019332702
Site of action of dichloroacetate.DCA inhibits the mitochondrial enzyme PDH kinase, thereby maintaining the PDH complex in its unphosphorylated catalytically active state and facilitating the aerobic oxidation of glucose.
Site of action of dichloroacetate.DCA inhibits the mito | Open-i
https://openi.nlm.nih.gov/detailedresult?img=PMC3318006_pone.0034776.g001&req=4
Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer
Dichloroacetate (DCA), an orphan drug that promotes a shift from glycolysis to oxidative phosphorylation, has been repurposed for cancer therapy. The present study investigated whether DCA may overcome cisplatin resistance in head and neck cancer (HNC). Two cisplatin-resistant HNC cell lines (AMC-HN4R and -HN9R), their parental lines, and other human HNC lines were used.
The effect of DCA, alone and in combination with cisplatin, was assessed by measuring cell cycle, viability, death, reactive oxygen species (ROS) production, mitochondrial membrane potential (ΔΨm), and protein expression in preclinical mouse tumor xenograft models. Increased glycolysis correlated with decreased sensitivity to cisplatin and was reduced by DCA. Cisplatin-resistant cells overexpressed pyruvate dehydrogenase kinase 2 (PDK2). DCA induced HNC cell death by decreasing ΔΨm and promoting mitochondrial ROS production. This effect was decreased by the antioxidant N-acetyl-L-cysteine or by inhibition of caspase-mediated apoptosis. Activation of mitochondrial glucose oxidation by DCA eventually activated downstream mitochondrial apoptotic signaling, leading to the death of chemoresistant cancer cells. Therefore, DCA significantly sensitized resistant HNC cells to cisplatin in vitro and in vivo. High glycolysis and PDK2 overexpression are closely linked to cisplatin resistance in HNC cells; the latter can be overcome by DCA.
Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer | Request PDF
https://www.researchgate.net/publication/284434401_Activation_of_mitochondrial_oxidation_by_PDK2_inhibition_reverses_cisplatin_resistance_in_head_and_neck_cancer
Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells .. This in turn increases the flux of pyruvate into the mitochondria, thereby stimulating oxidative phosphorylation over glycolysis. DCA has shown in preclinical work to reverse glycolysis-related suppression of mitochondrial apoptosis in cancer cells (20,216) and boosted the effectiveness of hypoxia-specific che- motherapies in vitro (65,164) and in animal models (19). Resveratrol was reported recently to enhance PDH activity, likely via activation of AMPK, which in turn led to a shift from glycolysis to oxidative phosphorylation in HTC116 colon cancer cells (171). ...
Inhibition of Liver Hexokinase by Dehydroascorbic Acid and ...
adsabs.harvard.edu/abs/1959Natur.184.1638B
Abstract THE structure and properties of dehydroascorbic acid are to a certain extent similar to those of alloxan, and both the substances are diabetogenic in rats 1,2.It was previously reported by Bhattacharya, Robson and Stewart 3 that dehydroascorbic acid causes a precipitous but temporary fall in the reduced glutathione content of rat liver. Their results suggested that the fall in reduced ...
Published in:
Nature · 1959
Authors:
S K Bhattacharya
Affiliation:
University of Leeds
About:
Enzyme · Glutathione · Dehydroascorbic acid
Anti-cancer effects of vitamin C revisited
Vitamin C was first suggested to have cancer-fighting properties in the 1930s and has been the subject of controversy ever since. Despite repeated reports of selective cancer cell toxicity induced by high-dose vitamin C treatment in vitro and in mouse models, the mechanism of action has remained elusive.
Yun et al.1 have recently shed light on what was until now the elusive mechanism by which vitamin C (aka ascorbate) induces toxicity in selected oncogene-driven cancers. They reported that in cells with mutations of KRAS or BRAF, death is not caused by vitamin C itself, but rather by its oxidized form dehydroascorbate (DHA). Whereas vitamin C enters cells through sodium cotransporters, DHA competes with glucose for uptake through glucose transporters (particularly GLUT1 and GLUT4) and then is reduced back to vitamin C in cells2 (Figure 1). It was previously observed that while melanoma cell lines take up DHA at much higher rates than vitamin C, normal melanocytes do not, demonstrating that transformation-driven upregulation of GLUTs leads to increased uptake of DHA3. More recently, using Magnetic Resonance Spectroscopy Imaging, it was demonstrated that hyperpolarized 13C-labeled DHA is rapidly taken up by cancer cells and converted to vitamin C, illustrating the tumors' reducing state4. Yun et al.1 now show that the reduction of DHA back to vitamin C is at the crux of the vitamin C-induced cell death observed in these cancer cells.
Mechanistic overview of proposed vitamin C toxicity in CRCs driven by KRAS and BRAF mutations. KRAS and BRAF mutations induce metabolic reprogramming by upregulating GLUT1, glucose uptake, and glycolytic flux. Upon vitamin C treatment and its extracellular oxidation, DHA (the oxidized form of vitamin C) is taken up through GLUT1 and is reduced back to vitamin C in the cells, depleting GSH and NADPH. Consequently, an increase in ROS leads to GAPDH oxidation, and with it, to a decrease in glycolytic flux. In parallel, ROS-mediated oxidative DNA damage induces PARP activation and subsequently, NAD+ levels fall and cause additional inhibition of GAPDH and glycolysis, resulting in energy crisis and cell death.
Full size image
Mutations in KRAS or BRAF are found in approximately half of the cases of colorectal cancer (CRC) and their expression correlates with an increase in GLUT1 expression, glucose uptake and reliance on glycolysis. Yun et al.1 observed that vitamin C is oxidized to DHA in tissue culture media and that KRAS or BRAF mutated CRC cell lines take up more DHA compared to their wild-type counterparts. More importantly, they found that DHA induces death in the mutant lines, but not in wild-type counterparts overexpressing GLUT1, suggesting that additional oncogenic reprogramming is necessary for DHA-induced toxicity. The authors then profiled metabolic changes after treatment with vitamin C. In cells with KRAS or BRAF mutations, they found an accumulation of the glycolytic intermediates upstream of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) whereas those downstream of GAPDH were depleted. This indicated a decrease in GAPDH activity and a concomitant diversion of glucose into the oxidative phase of the pentose phosphate pathway (PPP), a metabolic shift that, upon oxidative stress, helps restore NADPH levels and cellular reducing potential (Figure 1). Indeed, Yun and co-workers found that the intracellular reduction of DHA back to vitamin C depleted the cellular stores of reduced glutathione (GSH, the major antioxidant in cells), leading to an increase in reactive oxygen species (ROS). Furthermore, they found that, upon exposure to DHA, GAPDH itself was oxidized (on Cys152), and consequently inhibited. Inhibition of GAPDH caused energy stress in the highly glycolytic mutant cell lines, leading to cell death. In mice harboring tumor xenografts with either KRAS or BRAF mutations, treatment with high doses of vitamin C reduced tumor size. Treatment also reduced the number and size of polyps in an Apc-driven transgenic mouse model of intestinal cancer, but again, only in tumors expressing mutant Kras. Moreover, in addition to showing the direct inhibition of GAPDH by oxidation, the authors demonstrated that its activity is further hindered by DHA-induced NAD+ depletion, since GAPDH activity relies on the availability of NAD+ as a co-substrate. The major NAD+ consumer, the DNA repairing enzyme poly ADP-ribose polymerase (PARP) was then investigated. It was found that the increase in ROS after high-dose vitamin C treatment also induced DNA damage and PARP activation in KRAS or BRAF transformed cells. Providing the cells with a PARP inhibitor or an NAD+ precursor partially rescued their viability. Thus, ROS cause the inhibition of GAPDH activity in cells on two fronts: first, via inducing its direct oxidation and second, by causing NAD+ depletion (Figure 1). Yun et al.1 thus demonstrated an intricate mechanism by which oncogenic reprogramming, which causes glycolysis addiction, induces a metabolic vulnerability which can be exploited with high doses of DHA that elevate intracellular ROS as it is converted back to vitamin C.
Despite numerous clinical studies, the anti-cancer property of vitamin C has remained controversial. Potential translation of the mechanism presented by Yun et al.1 to therapeutic application raises concerns regarding toxicities of high-dose vitamin C treatment. Though the authors do not report side effects in their mice (treated daily with 4-8 g/kg body weight IP), the upper dose equates to over half a kilogram for the average person. High-dose oral supplementation of vitamin C is associated with increased kidney stone incidence, and clinical studies demonstrated significant renal, cardiac, and metabolic toxicity upon vitamin C administration. Still, overall reports of toxicity are variable, poorly graded, and therefore inconclusive5,6. Affinity studies of DHA for GLUT1 may help establish a lower effective dose, though unwanted side effects in tissues that highly express GLUT1 need to be considered. The brain obtains vitamin C by uptake of DHA through GLUT1 at the blood-brain barrier and its subsequent reduction7. Erythrocytes express high levels of GLUT1 and are crucial for ascorbate recycling, keeping DHA levels low8. Importantly, erythrocytes rely solely on glycolysis for energy production. Thus, high DHA levels may induce brain toxicity and haemolysis via mechanisms similar to those described by Yun et al.1.
Though vitamin C oxidizes rapidly in tissue culture media, it acts mainly as an antioxidant in vivo. It remains unclear how and where circulating vitamin C is oxidized in vivo, an issue not addressed by Yun et al.1. Oxidation of vitamin C to DHA by tumor stroma has been suggested9, complicating the ability to predict tumor responsiveness to the treatment. As such, without being able to control the extent of vitamin C oxidation to DHA, effectiveness and toxicity of vitamin C treatment cannot be predicted.
Finally, the authors report that, following DHA uptake, NAD+ depletion by PARP activation contributes to the inhibition of glycolysis (and potentially to the stimulation of oxidative PPP flux). The observation that PARP inhibition partly rescues vitamin C-treated cells may suggest that the toxic effect of DHA uptake is not caused by ROS alone, since restoring NAD+ levels and glycolysis with the PARP inhibitor may actually decrease PPP flux and NADPH production, and aggravate the redox stress. It remains to be demonstrated that PARP inhibition indeed restores NAD+ levels in vitamin C-treated cells, and how this affects the balance between energy and redox metabolism. This also raises the question whether PARP activity in BRCA1/2-deficient tumors produces a similar metabolic phenotype via NAD+ depletion and whether the use of a PARP inhibitor (e.g., olaparib) to treat these tumors10 might restore NAD+ levels and counterbalance GAPDH inhibition by other oxidative agents.
In summary, Yun et al.1 show that, in glycolysis-addicted KRAS and BRAF driven cancer cells, high-dose vitamin C treatment induces cell death via the uptake and reduction of its oxidized form DHA back to vitamin C. DHA reduction, through scavenging GSH, induces oxidative stress, leading to GAPDH inactivation, inhibition of glycolysis and the subsequent energy crisis and cell death. This study further elucidates the mechanism by which ROS can induce cell death, and neatly shows how vitamin C, an antioxidant, can work as a double edged sword. However, further work is necessary to determine whether there is a therapeutic potential for vitamin C in cancer patients.
Anti-cancer effects of vitamin C revisited | Cell Research
https://www.nature.com/articles/cr20167Zhejiang Da Xue Xue Bao Yi Xue Ban. 2019 May 25;48(3):296-302.
[High dose vitamin C inhibits proliferation of breast cancer cells through reducing glycolysis and protein synthesis].
[Article in Chinese]
Wang Q1, Xu Q1, Wei A1, Chen S1, Zhang C1, Zeng L1.
Author information
1
School of Medicine, Zhejiang University City College, Hangzhou 310015, China.
Abstract
OBJECTIVE:
To investigate the effects of high dose vitamin C (VC) on proliferation of breast cancer cells and to explore its mechanisms.
METHODS:
Human breast cancer cells Bcap37 and MDA-MB-453 were treated with VC at low dose (0.01 mmol/L), medium dose (0.10 mmol/L) and high dose (2.00 mmol/L). Cell proliferation was determined with CCK-8 assay, protein expression was evaluated by Western blot, and the secretion of lactic acid in tumor cells was detected by colorimetric method. Bcap37 cells were inoculated in nude mice, and tumor baring nude mice were intraperitoneally injected with high VC(4 g/kg, VC group, n=5)or normal saline (control group, n=5) for 24 d. Tumor weight and body weight were calculated.
RESULTS:
In vitro experiments demonstrated that high dose VC significantly inhibited cell proliferation in Bcap37 and MDA-MB-453 cells (all P<0.01); the expressions of Glut1 and mTOR signaling pathway-related proteins were decreased (all P<0.05); and the secretion of lactic acid was also markedly reduced (all P<0.05). In vivo experiment showed that the tumor weight was decreased in mice treated with high-dose VC as compared with control group (P<0.05), but no difference in body weights between two groups was observed.
CONCLUSIONS:
High dose VC may inhibit proliferation of breast cancer cells both in vitro and in vivo through reducing glycolysis and protein synthesis.[High dose vitamin C inhibits proliferation of breast cancer cells through reducing glycolysis and protein synthesis]. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31496162
Glucose is necessary for stabilization of hypoxia-inducible factor-1α under hypoxia: Contribution of the pentose phosphate pathway to this stabilization
OkaaYasushiHashibaaSatoshiAkibaaSusumuImaokabTakashiSatoa
a
Department of Pathological Biochemistry, Kyoto Pharmaceutical University, Misasagi, Yamashina-ku, Kyoto 607-8414, Japan
b
Nanobiotechnology Research Center and Department of Bioscience, School of Science and Technology, Kwansei Gakuin University, Gakuen, Sanda 669-1337, Japan
Received 16 February 2010, Revised 10 April 2010, Accepted 22 May 2010, Available online 1 June 2010.
Abstract
In this study, we observed that low glucose or fructose reduces the increase in hypoxia-inducible factor-1α (HIF-1α) protein under hypoxic conditions. 6-Aminonicotinamide (6-AN), an inhibitor of the pentose phosphate pathway (PPP), also inhibited the increase of HIF-1α protein under hypoxic conditions, while the reduced protein levels of HIF-1α by low glucose were apparently recovered by the addition of MG-132 or NADPH. Moreover, siRNA for glucose-6-phosphate dehydrogenase, which produces NADPH, reduced the increase in HIF-1α protein. On the other hand, cobalt-induced expression of HIF-1α protein was not affected by low glucose or 6-AN under normoxic conditions. In conclusion, glucose metabolism through the PPP, but not in glycolysis, plays an important role in the stabilization of HIF-1α protein under hypoxic conditions.
Glucose- and glutamine-fueled stabilization of C-Myc is required for T-cell proliferation and malignant transformation | Cell Death Discovery
https://www.nature.com/articles/cddiscovery201647
The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, 3052, Victoria, Australia
The Department of Medical Biology, University of Melbourne, Parkville, 3010, Victoria, Australia
Published: 27 June 2016
Glucose- and glutamine-fueled stabilization of C-Myc is required for T-cell proliferation and malignant transformation
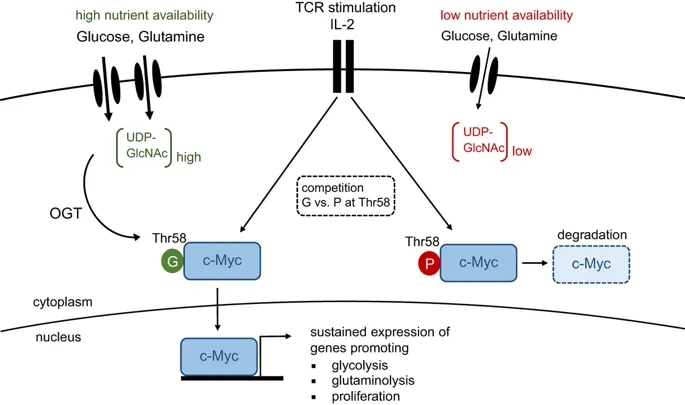
Dynamic regulation of cellular metabolism in response to stimulation or changes in the environment has a crucial role in many biological processes. In particular, T cells undergo dramatic changes in their metabolism when progressing from quiescence to proliferation and acquisition of effector function. This is associated with augmented flux of glucose and glutamine into cells, and is of fundamental importance to meet the increased energy demands associated with growth, proliferation, migration and effector molecule production. Importantly, a similar metabolic switch occurs in cancer cells during malignant transformation. In a study published recently in Nature Immunology, Cantrell et al. uncover another pathway that is tightly dependent on nutrient availability and critical for T cells.1 The authors show that increased and sustained import of glucose and glutamine into activated T cells is indispensable for O-GlcNAcylation, the enzymatically-mediated transfer of N-acetylglucosamine (GlcNAc) to proteins. Multiple intracellular proteins were found to be O-GlcNAcylated in response to T-cell receptor (TCR) signaling, including c-Myc, an important positive regulator of cellular metabolism and proliferation.1 O-GlcNAcylation resulted in stabilization of c-Myc, and thus revealed an unappreciated positive-feedback loop between maintenance of cellular metabolism and proliferation that is required for T-cell development, clonal expansion and malignant transformation (Figure 1).
TCR activation and IL-2 signaling results in c-Myc expression, which coordinates the metabolic switch from oxidative phosphorylation to aerobic glycolysis. This process is accompanied by an increased flux of glucose and glutamine into the cell, resulting in elevated levels of UDP-GlcNAc, the end product of the hexamine biosynthetic pathway (HBP). O-GlcNAc transferase (OGT) covalently links GlcNAc sugars to proteins. Under nutrient-rich conditions, O-GlcNAcylation of Thr-58 in c-Myc outcompetes phosphorylation at the same residue, thereby preventing its degradation. Stable c-Myc expression is required for maintenance of aerobic glycolysis and proliferation upon T-cell activation. G, O-GlcNAcylation; P, phosphorylation.
In summary, the study by Cantrell et al. sheds light on a previously unappreciated role of glycolytic metabolism in T cells. Besides being critical for the anabolic metabolism of proliferating T cells and their effector functions, glycolysis is also required for stabilizing c-Myc, which itself is a critical regulator of aerobic glycolysis. Thus, O-GlcNAcylation generates a positive-feedback loop that efficiently sustains cellular metabolism and proliferation (Figure 1). These findings add an additional layer of complexity to the signaling network that controls metabolic adaptation upon immune activation and guides the differentiation of effector T cells.
Glucose- and glutamine-fueled stabilization of C-Myc is required for T-cell proliferation and malignant transformation | Cell Death Discovery
https://www.nature.com/articles/cddiscovery201647
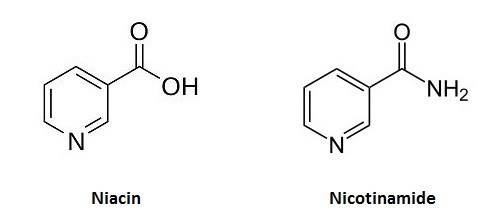
Niacin analogue, 6-Aminonicotinamide, a novel inhibitor of hepatitis B virus replication and HBsAg production
6-AN is a well-known inhibitor of G6PD, to exclude the possibility that its anti-HBV function is due to inhibiting G6PD.
a
The Key Laboratory of Molecular Biology of Infectious Diseases designated by the Chinese Ministry of Education, Institute for Viral Hepatitis, Department of Infectious Diseases, The Second Affiliated Hospital, Chongqing Medical University, Room 617, College of Life Sciences Building, 1 YiXueYuan Road, YuZhong District, Chongqing 400016, China
b
State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Room 704a-02, Block H, Macau, China
Received 27 April 2019, Revised 28 September 2019, Accepted 13 October 2019, Available online 31 October 2019.
Abstract
Background: Hepatitis B surface antigen (HBsAg) is one of the important clinical indexes for hepatitis B virus (HBV) infection diagnosis and sustained seroconversion of HBsAg is an indicator for functional cure. However, the level of HBsAg could not be reduced by interferons and nucleoside analogs effectively. Therefore, identification of a new drug targeting HBsAg is urgently needed.
Methods: In this study, 6-AN was screened out from 1500 compounds due to its low cytotoxicity and high antiviral activity. The effect of 6-AN on HBV was examined in HepAD38, HepG2-NTCP and PHHs cells. In addition, the antivirus effect of 6-AN was also identified in mouse model.
Findings: 6-AN treatment resulted in a significant decrease of HBsAg and other viral markers both in vitro and in vivo. Furthermore, we found that 6-AN inhibited the activities of HBV SpI, SpII and core promoter by decreasing transcription factor PPARα, subsequently reduced HBV RNAs transcription and HBsAg production.
Interpretation: We have identified a novel small molecule to inhibit HBV core DNA, HBV RNAs, HBsAg production, as well as cccDNA to a minor degree both in vitro and in vivo. This study may shed light on the development of a novel class of anti-HBV agent.Taken together, the findings in this study has revealed that 6-AN exhibits a potent anti-HBV effect both in vitro and in vivo, via affecting HBsAg production as well as HBV transcription and replication, thus may provide a valuable alternative or complementary therapy for the current and future antiviral treatments.
Niacin analogue, 6-Aminonicotinamide, a novel inhibitor of hepatitis B virus replication and HBsAg production - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S2352396419306863
Frontiers | Targeting T Cell Metabolism for Improvement of Cancer Immunotherapy | Oncology
https://www.frontiersin.org/articles/10.3389/fonc.2018.00237/full
Biomed Pharmacother. 2017 Feb;86:502-513. doi: 10.1016/j.biopha.2016.12.056. Epub 2016 Dec 23.
Ascorbic acid, but not dehydroascorbic acid increases intracellular vitamin C content to decrease Hypoxia Inducible Factor -1 alpha activity and reduce malignant potential in human melanoma.
Fischer AP1, Miles SL2.
Author information
1
Department of Biomedical Sciences, Joan C. Edwards School of Medicine, Marshall University, One John Marshall Drive, Huntington, WV 25755, United States. Electronic address: fischer35@marshall.edu.
2
Department of Biomedical Sciences, Joan C. Edwards School of Medicine, Marshall University, One John Marshall Drive, Huntington, WV 25755, United States. Electronic address: kittlaus1@marshall.edu.
Abstract
INTRODUCTION:
Accumulation of hypoxia inducible factor-1 alpha (HIF-1α) in malignant tissue is known to contribute to oncogenic progression and is inversely associated with patient survival. Ascorbic acid (AA) depletion in malignant tissue may contribute to aberrant normoxic activity of HIF-1α. While AA supplementation has been shown to attenuate HIF-1α function in malignant melanoma, the use of dehydroascorbic acid (DHA) as a therapeutic means to increase intracellular AA and modulate HIF-1α function is yet to be evaluated. Here we compared the ability of AA and DHA to increase intracellular vitamin C content and decrease the malignant potential of human melanoma by reducing the activity of HIF-1α.
METHODS:
HIF-1α protein accumulation was evaluated by western blot and transcriptional activity was evaluated by reporter gene assay using a HIF-1 HRE-luciferase plasmid. Protein expressions and subcellular localizations of vitamin C transporters were evaluated by western blot and confocal imaging. Intracellular vitamin C content following AA, ascorbate 2-phosphate (A2P), or DHA supplementation was determined using a vitamin C assay. Malignant potential was accessed using a 3D spheroid Matrigel invasion assay. Data was analyzed by One or Two-way ANOVA with Tukey's multiple comparisons test as appropriate with p<0.05 considered significant.
RESULTS:
Melanoma cells expressed both sodium dependent vitamin C (SVCT) and glucose (GLUT) transporters for AA and DHA transport respectively, however advanced melanomas responded favorably to AA, but not DHA. Physiological glucose conditions significantly impaired intracellular vitamin C accumulation following DHA treatment. Consequently, A2P and AA, but not DHA treated cells demonstrated lower HIF-1α protein expression and activity, and reduced malignant potential. The ability of AA to regulate HIF-1α was dependent on SVCT2 function and SVCT2 was not significantly inhibited at pH representative of the tumor microenvironment.
CONCLUSIONS:
The use of ascorbic acid as an adjuvant cancer therapy remains under investigated. While AA and A2P were capable of modulating HIF-1α protein accumulation/activity, DHA supplementation resulted in minimal intracellular vitamin C activity with decreased ability to inhibit HIF-1α activity and malignant potential in advanced melanoma. Restoring AA dependent regulation of HIF-1α in malignant cells may prove beneficial in reducing chemotherapy resistance and improving treatment outcomes.
Copyright © 2016 Elsevier Masson SAS. All rights reserved.
KEYWORDS:
Ascorbic acid; Dehydroascorbic acid; Hypoxia inducible factor-1 alpha; Melanoma; SVCT2
Ascorbic acid, but not dehydroascorbic acid increases intracellular vitamin C content to decrease Hypoxia Inducible Factor -1 alpha activity and re... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28012930
Targeting Cancer MetabolismClin Cancer Res. 2012 Oct 15; 18(20): 5537–5545.
National Cancer Institute Bethesda
Abstract The understanding that oncogenes can have profound effects on cellular metabolism and the discovery of mutations and alterations in several metabolism-related enzymes (IDH1, IDH2, SDH, FH, PKM2) has renewed interest in cancer metabolism and renewed hope of taking therapeutic advantage of cancer metabolism. Otto Warburg observed that aerobic glycolysis was a characteristic of cancer cells. More than 50-years later, we understand that aerobic glycolysis and uptake of glutamine and glycine allow cancer cells to produce energy (ATP) and the nucleotides, amino acids and lipids required for proliferation. Expression of the MYC oncogene drives the increase in cellular biomass facilitating proliferation. PKM2 expression in cancer cells stimulates aerobic glycolysis. Amongst intermediary metabolism enzyme, mutations in succinate dehydrogenase (SDH) occur in gastointestinal stromal tumors and result in a pseudohypoxic metabolic milieu. Fumarate hydratase (FH) mutations lead to a characteristic renal cell carcinoma. Isocitrate dehydrogenase (IDH1/2) mutations have been found in leukemias, gliomas, prostate cancer, colon cancer, thyroid cancer and sarcomas. These recently recognized oncogenic metabolic lesions may be selective targets for new anticancer therapeutics. Keywords: Cancer metabolism, IDH1/2, PKM2, MYC, succinate dehydrogenase, fumarate hydratase
Figure 1
Schematic illustrating the sources of ATP energy used by normal and malignant cells under varied conditions of oxygenation.
Figure 2
Schematic illustrating metabolic pathways prominent in malignant cells. Blue boxes indicate enzymes and transporters that may be useful therapeutic targets in cancer. Green boxes represent transcription factors that alter metabolic pathways. Glycolysis is the ten-step metabolic pathway that converts glucose into pyruvate. The free energy released in this process is used to form the high-energy compounds ATP and NADH. The pentose phosphate pathway is a process that generates NADPH and 5-carbon sugars as alternative to glycolysis. Nucleotide synthesis provides molecules that make up the individual structural units of the nucleic acids RNA and DNA. In addition, nucleotides participate in cell signaling (cGMP and cAMP), are cofactors of enzymatic reactions, and nucleoside triphosphates are sources of chemical energy. Fatty acid synthesis is the synthesis of fatty acids from acetyl-CoA and malonyl-CoA precursors via fatty acid synthases. Anaplerotic reactions are those that replenish and maintain intermediates of a metabolic pathway. The abbreviations are: CAIX, Carbonic anhydrase IX; NHE1, Na+/H+ exchanger; Glut1, glucose transporter 1; Glut3, Glucose transporter 3; mGluR1, metabotropic glutamate receptor 1; PI3K/AKT, phophotidylinositol-3-kinase/Protein kinase B; G6PD, glucose-6-phosphate dehydrogenase; AMPK, AMP-activated protein kinase; PFK, phosphofructokinase; LKB1, serine/threonine kinase 11 (STK11), liver kinase B1; TKL/TAL, transketolase-transaldolase; RTK, receptor tyrosine kinase; PKM2, pyruvate kinase M2; MCT4, monocarboxylate transporter 4; IDH1 isocitrate dehydrogenase 1; IDH2, isocitrate dehydrogenase 2; IDH3, isocitrate dehydrogenase 3; LDH-A, lactate dehydrogenase A; MPC1/2, mitochondrial pyruvate carrier 1/2; ME, malic enzyme, malate dehydrogenase; FH, fumarate hydratase; SDH, succinate dehydrogenase; KGDH, 2-keto-glutarate dehydrogenase; TETs, DNA hydroxylases; KDMs, lysine histone demethylases; HK, hexokinase; HIF1, hypoxia-inducible factor 1; α-KG, α-ketoglutarate; TCA cycle, tricarboxylic acid cycle.
Targeting Cancer Metabolism
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3475613/
Cell Chem Biol. 2017 Sep 21;24(9):1161-1180. doi:
Targeting Metabolism for Cancer Therapy.
1
The Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
2
The Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Dana-Farber Cancer Institute, Boston, MA 02115, USA. Electronic address: mvh@mit.edu.
Abstract
Metabolic reprogramming contributes to tumor development and introduces metabolic liabilities that can be exploited to treat cancer. Chemotherapies targeting metabolism have been effective cancer treatments for decades, and the success of these therapies demonstrates that a therapeutic window exists to target malignant metabolism. New insights into the differential metabolic dependencies of tumors have provided novel therapeutic strategies to exploit altered metabolism, some of which are being evaluated in preclinical models or clinical trials. Here, we review our current understanding of cancer metabolism and discuss how this might guide treatments targeting the metabolic requirements of tumor cells.
https://www.ncbi.nlm.nih.gov/pubmed/28938091
Sci Rep. 2019 Nov 8;9(1):16313. doi: 10.1038/s41598-019-52814-1.
Gastric cancer depends on aldehyde dehydrogenase 3A1 for fatty acid oxidation.
Lee JS1, Kim SH2, Lee S1, Kang JH1, Lee SH1, Cheong JH3, Kim SY4.
Author information
1
Division of Cancer Biology, Research Institute, National Cancer Center, Goyang, 10408, Republic of Korea.
2
Department of Surgery, Yonsei University Health System, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722, Republic of Korea.
3
Department of Surgery, Yonsei University Health System, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722, Republic of Korea. JHCHEONG@yuhs.ac.
4
Division of Cancer Biology, Research Institute, National Cancer Center, Goyang, 10408, Republic of Korea. kimsooyoul@gmail.com.
Abstract
The major source of ATP in cancer cells remains unclear. Here, we examined energy metabolism in gastric cancer cells and found increased fatty acid oxidation and increased expression of ALDH3A1. Metabolic analysis showed that lipid peroxidation by reactive oxygen species led to spontaneous production of 4-hydroxynonenal, which was converted to fatty acids with NADH production by ALDH3A1, resulting in further fatty acid oxidation. Inhibition of ALDH3A1 by knock down using siRNA of ALDH3A1 resulted in significantly reduced ATP production by cancer cells, leading to apoptosis. Oxidative phosphorylation by mitochondria in gastric cancer cells was driven by NADH supplied via fatty acid oxidation. Therefore, blockade of ALDH3A1 together with mitochondrial complex I using gossypol and phenformin led to significant therapeutic effects in a preclinical gastric cancer model.
Gastric cancer depends on aldehyde dehydrogenase 3A1 for fatty acid oxidation. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/31705020
Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers13
ABSTRACT Quercetin is a dietary antioxidant that prevents oxidation of low-density lipoproteins in vitro. Intake of quercetin was inversely associated with coronary heart disease mortality in elderly Dutch men. However, the extent of absorption of quercetin in humans is unclear. The aim of this study was to quantify absorption of various forms of quercetin. Nine healthy ileostomy subjects were studied, to avoid losses caused by colonic bacteria. They followed a quercetin-free diet for 12 d; on days 4, 8, and 12 they received a supplement of fried onions at breakfast (rich in quercetin glucosides) equivalent to 89 mg aglycone, pure quercetin rutinoside (the major quercetin compound in tea) equivalent to 100 mg aglycone, or I 00 mg pure quercetin aglycone, in random order. Subsequently, participants collected ileostomy effluent and urine for 13 h. In vitro incubations of quercetin or its glycosides with gastrointestinal fluids showed minimal degradation. Absorption of quercetin, defined as oral intake minus ileostomy excretion and corrected for 14 % degradation within the ileostomy bag, was 52 ± 15 % for quercetin glucosides from onions, 17 ± 15 % for quercetin rutinoside, and 24 ± 9 % for quercetin aglycone. Mean excretion of quercetin or its conjugates in urine was 0.5 % of the amount absorbed; quercetin excretion in urine was negatively correlated with excretion in ileostomy effluent (r =-0.78, n = 27). We conclude that humans absorb appreciable amounts of quercetin and that absorption is enhanced by conjugation with glucose. Am
Keyphrases
healthy ileostomy volunteers13 dietary quercetin glycoside quercetin glucoside ileostomy effluent mg aglycone colonic bacteria appreciable amount dietary antioxidant mean excretion vitro incubation nine healthy ileostomy subject random order oral intake minus ileostomy excretion minimal degradation quercetin excretion fried onion coronary heart disease mortality quercetin rutinoside abstract quercetin quercetin aglycone ileostomy bag low-density lipoprotein mg pure quercetin aglycone pure quercetin rutinoside elderly dutch men various form quercetin-free diet gastrointestinal fluid major quercetin compoundCiteSeerX — Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers13
http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.319.4267
Conjugated quercetin glucuronides as bioactive metabolites and precursors of aglyconein vivo
共轭槲皮素葡糖醛酸苷作为生物活性代谢产物和糖苷配基体内的前体
槲皮素是一种普遍存在于蔬菜中的典型抗氧化类黄酮。它可能通过发挥清除活性氧(ROS)的活性和/或与特定蛋白(例如信号转导途径中的氧化酶和转录因子)结合而充当生物活性化合物。从其疾病预防潜力的观点出发,对其吸收和代谢(及其分子靶标)进行了广泛的研究。已知在摄入含有槲皮素的饮食后,具有或不具有O-甲基化的葡糖醛酸苷和/或硫酸盐缀合物仅在人血中循环。我们建议槲皮素的葡萄糖醛酸苷共轭物不仅作为解毒代谢产物起作用,而且对各种ROS产生系统和疏水糖苷配基的前体具有亲水性生物活性剂。槲皮素糖苷配基被认为是在氧化应激(例如炎症)下通过β-葡萄糖醛酸苷酶活性的作用而出现在靶部位的。心血管系统和中枢神经系统似乎是人类血液中循环的共轭槲皮素葡萄糖醛酸苷的主要靶标。
Quercetin is a typical anti-oxidative flavonoid ubiquitously distributed in vegetables. It is likely to act as a bioactive compound by exerting reactive oxygen species (ROS)-scavenging activity and/or binding to specific proteins such as oxidative enzymes and transcriptional factors in signal transduction pathways. Its absorption and metabolism (as well as its molecular targets) have been extensively explored from the viewpoint of its potential for disease prevention. It is known that glucuronide and/or sulfate conjugates with or without O-methylation exclusively circulate in the human bloodstream after intake of a quercetin-containing diet. We propose that glucuronide conjugates of quercetin function not only as detoxified metabolites but hydrophilic bioactive agents to various ROS-generating systems and precursors of hydrophobic aglycone. Quercetin aglycone is assumed to emerge in the target site by the action of β-glucuronidase activity under oxidative stress such as inflammation. The cardiovascular system and central nervous system seem to be the major targets of conjugated quercetin glucuronides circulating in the human bloodstream.
Conjugated quercetin glucuronides as bioactive metabolites and precursors of aglycone in vivo - Food & Function (RSC Publishing) DOI:10.1039/C0FO00106F
https://pubs.rsc.org/en/content/articlehtml/2011/fo/c0fo00106f
Quercetin and quercetin glucosides are the major flavonols present in onion (Allium cepa L.) and are predominantly present as quercetin, quercetin-3,4′-diglucoside and quercetin-4′-glucoside.
Effect of different exposed lights on qu
Utilization of quercetin and quercetin glycosides from ...
https://www.sciencedirect.com/science/article/pii/S030881461730821X
Nov 15, 2017 · Quercetin exists in a free as well as in a conjugate form with carbohydrate moiety, mainly as glucoside that represents approximately 80% of the total flavonol content of onion (Ko et al., 2015, Sharma et al., 2014). Quercetin and its glucoside content in OSW were quantified by using HPLC analytical method.
Cited by: 26
Publish Year: 2017
Utilization of quercetin and quercetin glycosides from onion (Allium cepa L.) solid waste as an antioxidant, urease and xanthine oxidase inhibitors
Highlights
•
Onion solid waste (OSW) used as cheap and valuable source for production of value-added products.
•
HPLC revealed significant amount of quercetin and quercetin glycosides in OSW.
•
OSW extracts inhibited the urease and xanthine oxidase activity in vitro.
•
OSW showed potent antioxidant activity with in vitro scavenging assays.
Abstract
This study aimed to determine the flavonol glycosides from onion solid waste (OSW) using HPLC analysis, with antioxidant and enzyme inhibitory activities. We found considerable amount of quercetin-4′-O-monoglucoside (QMG: 254.85), quercetin-3,4′-O-diglucoside (QDG: 162.34), quercetin (Q: 60.44), and isorhamnetin-3-glucoside (IMG: 23.92) (mg/100 g) dry weight (DW) of OSW. For OSW, the methanol and ethanol showed the strongest antioxidant activities, followed by ethyl acetate, chloroform, and n-hexane extracts. Among the flavonols, Q and QDG possessed higher antioxidant activities. OSW and flavonol glycosides displayed significant enzyme inhibitory activity, with IC50 values ranging from 12.5 ± 0.11 to 32.5 ± 0.28 for OSW, 8.2 ± 0.07 to 16.8 ± 0.02 for flavonol glycosides, and 4.2 ± 0.05 μg/mL for thiourea (positive control) towards urease; while 15.2 ± 0.8 to 35.8 ± 0.2 (μg/mL) for OSW, 10.5 ± 0.06 to 20.8 ± 0.05 (μg/mL) for flavonol glycosides, and 6.5 ± 0.05 μg/mL for allopurinol (positive control) towards xanthine oxidase, respectively. The OSW and flavonol glycosides may thus be considered as potential antioxidant and antigout agents.
Graphical abstract
Utilization of quercetin and quercetin glycosides from onion (Allium cepa L.) solid waste as an antioxidant, urease and xanthine oxidase inhibitors - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S030881461730821X
Metformin use and its effect on gastric cancer in patients with type 2 diabetes: A systematic review of observational studies
https://www.spandidos-publications.com/10.3892/ol.2017.7370
Original Article
Published: 20 February 2017
Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis
L Jin, J Chun, C Pan, G N Alesi, D Li, K R Magliocca, Y Kang, Z G Chen, D M Shin, F R Khuri, J Fan & S Kang
Oncogene volume 36, pages3797–3806(2017)Cite this article
Abstract
Metastases remain the major cause of death from cancer. Recent molecular advances have highlighted the importance of metabolic alterations in cancer cells, including the Warburg effect that describes an increased glycolysis in cancer cells. However, how this altered metabolism contributes to tumour metastasis remains elusive. Here, we report that phosphorylation-induced activation of lactate dehydrogenase A (LDHA), an enzyme that catalyses the interconversion of pyruvate and lactate, promotes cancer cell invasion, anoikis resistance and tumour metastasis. We demonstrate that LDHA is phosphorylated at tyrosine 10 by upstream kinases, HER2 and Src. Targeting HER2 or Src attenuated LDH activity as well as invasive potential in head and neck cancer and breast cancer cells. Inhibition of LDH activity by small hairpin ribonucleic acid or expression of phospho-deficient LDHA Y10F sensitized the cancer cells to anoikis induction and resulted in attenuated cell invasion and elevated reactive oxygen species, whereas such phenotypes were reversed by its product lactate or antioxidant N-acetylcysteine, suggesting that Y10 phosphorylation-mediated LDHA activity promotes cancer cell invasion and anoikis resistance through redox homeostasis. In addition, LDHA knockdown or LDHA Y10F rescue expression in human cancer cells resulted in decreased tumour metastasis in xenograft mice. Furthermore, LDHA phosphorylation at Y10 positively correlated with progression of metastatic breast cancer in clinical patient tumour samples. Our findings demonstrate that LDHA phosphorylation and activation provide pro-invasive, anti-anoikis and pro-metastatic advantages to cancer cells, suggesting that Y10 phosphorylation of LDHA may represent a promising therapeutic target and a prognostic marker for metastatic human cancers.Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis | Oncogene
https://www.nature.com/articles/onc20176
Tyrosine phosphorylation of lactate dehydrogenase A is ...
https://www.citeab.com/publication/116199-21969607-tyrosine-phosphorylation-of-lactate...
We found that the oncogenic receptor tyrosine kinase FGFR1 directly phosphorylates LDH-A. Phosphorylation at Y10 and Y83 enhances LDH-A activity by enhancing the formation of active, tetrameric LDH-A and the binding of LDH-A substrate NADH, respectively.Fibroblast growth factor receptor 1 (FGFR1) - DiscoverX
https://www.discoverx.com/target-data-sheets/kinase/fgfr1
Fibroblast growth factor receptor 1 (FGFR1) A full-length representative protein consists of an extracellular region, composed of three immunoglobulin-like domains, a single hydrophobic membrane-spanning segment and a cytoplasmic tyrosine kinase domain. The extracellular portion of the protein interacts with fibroblast growth factors,...
FGFR1 fusion kinase regulation of MYC expression drives ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6168426
Dual effect of FGFR1 fusion kinase activation of MYC expression (A) Introduction of chimeric kinase (BCR-FGFR1), truncated kinase (nFGFR1) or Asp432 mutant chimeric kinase (BCR-FGFR1m) (A) shows an increase in MYC expression in three model cell systems compared with …
Outcome and Human Epithelial Growth Factor Receptor (HER ...
https://breast-cancer-research.biomedcentral.com/articles/10.1186/bcr783
Mar 04, 2004 · The Human Epithelial Growth Factor Receptor (HER) family of receptor tyrosine kinases form part of a complex signal cascade modulating cell proliferation, survival, adhesion, migration and differentiation. The family is comprised of 4 homologous receptors HER1 (EGFR/ErbB1), HER2 (ErbB2/neu), HER3 (ErbB3) and HER4 (ErbB4) .
Publish Year: 2004
AuthHuman epidermal growth factor receptor (HER2) inhibitors are either tyrosine kinase inhibitors or monoclonal antibodies that slow down or stop cell growth. Human epidermal growth factor receptors are transmembrane receptors; they have an extracellular binding component, a transmembrane component and an intracellular tyrosine kinase component.
List of HER2 inhibitors - Drugs.com
www.drugs.com/drug-class/her2-inhibitors.html
EGFR/HER2 in Breast Cancer: A Biological Approach for ...
https://www.medscape.com/viewarticle/585224_7
Oct 29, 2019 · EGF receptor (EGFR) and HER2 are two members of the ErbB/HER-family of tyrosine-kinase receptors that are frequently altered in breast cancer …
HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1868927
Mar 02, 2007 · Tyrosine kinase inhibitors that block HER-2 kinase that are in clinical development However, many so-called HER specific inhibitors are promiscuous kinase inhibitors. A recent study [ 27 ] investigated the specificity of 20 TKIs that are approved by the US Food and Drug Administration or are currently in clinical trials.
Cited by: 65
Publish Year: 2007
Author: Neil L. Spector, Wenle Xia, Iman El‐Hariry, Yossi YaHER kinase inhibition in patients with HER2- and HER3 ...
https://www.nature.com/articles/nature25475
Jan 31, 2018 · HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Efficacy in HER2-mutant cancers varied as a function of both tumour type and mutant allele to a degree not predicted by preclinical models, with the greatest activity seen in breast, cervical and biliary cancers and with tumours that contain kinase domain missense mutations.
Cited by: 153
Publish Year: 2018
AuthoHER kinase inhibition in patients with HER2- and HER3 ...
https://www.nature.com/articles/nature25475
Jan 31, 2018 · HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Efficacy in HER2-mutant cancers varied as a function of both tumour type and mutant allele to a degree not predicted by preclinical models, with the greatest activity seen in breast, cervical and biliary cancers and with tumours that contain kinase domain missense mutations.
Tyrosine-protein kinase CSK also known as C-terminal Src kinase is an enzyme that, in humans, is encoded by the CSK gene. This enzyme phosphorylates tyrosine residues located in the C-terminal end of Src-family kinases (SFKs) including SRC, HCK, FYN, LCK, LYN and YES1. Jul 23 2019
Tyrosine-protein kinase CSK - Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Src_kinase
Overexpression of HER2 initiates intracellular signaling pathways involved in cell proliferation, differentiation, migration, and apoptosis. In breast cancer, HER2-positive status is associated with poor prognosis, and also identifies patients who could benefit from anthracycline-based regimens.
HER2 Overexpression and Trastuzumab Treatment in Advanced O…
www.medscape.com/viewarticle/580150
Cytoplasmic Overexpression of HER2: a Key Factor in ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3583442
Feb 21, 2013 · But the data on HER2 expression in colorectal cancer are ambiguous, with reported overexpression of HER2 varying between zero and 84%. In this review these studies are evaluated and compared. It shows that many factors influence the determination of HER2-expression, especially of the intracellular fraction of HER2.It shows that many factors influence the determination of HER2-expression, especially of the intracellular fraction of HER2. It is concluded that although membranous overexpression of HER2 is low in colorectal cancer with only 5% of all patients being positive, a significant proportion of the patients (30%) shows cytoplasmic HER2 overexpression.
Cited by: 67
Publish Year: 2013
Author: EHuman Epidermal Growth Factor Receptor 2 (HER2) in Cancers ...
https://www.hindawi.com/journals/mbi/2014/852748
HER2 in Other Cancers. In lung cancers, overexpression of HER2 has been reported in about 20% [49, 50]. Apart from overexpression, mutations of HER2 were also reported in lung adenocarcinomas. The mutations targeted never or light smokers, oriental ethnicity, and female gender [51, 52].
Cited by: 266
Publish Year: 2014
Aut
HER2-positive breast cancer
Summary
Anti-HER2 treatment for HER2-positive breast cancer has changed the natural biology of this disease. This Series article reviews the main achievements so far in the treatment of both metastatic and early HER2-positive breast cancer. The success of neoadjuvant therapy in HER2-positive early breast cancer is especially acknowledged, as pertuzumab has been approved on the basis of a higher proportion of patients achieving a pathological complete response with pertuzumab and trastuzumab than with trastuzumab alone in a neoadjuvant study. Event-free survival after the confirmatory adjuvant trial completed recruitment was numerically better with pertuzumab plus trastuzumab than with trastuzumab alone. With survival rates of almost 5 years in women with metastatic HER2-positive breast cancer and 75% of patients achieving a pathological complete response, new treatments in the past decade have clearly improved the prognosis of HER2-positive breast cancer. Despite these achievements, however, the persisting high toll of deaths resulting from HER2-positive breast cancer calls for continued, intensive clinical research of newer therapies and combinations.
HER2-positive breast cancer - The Lancet
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(16)32417-5/fulltext
Vitamin C Is a Kinase Inhibitor: Dehydroascorbic Acid ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC444845
DHA, the oxidized form of vitamin C, inhibits the activity of IKKβ and IKKα in vitro. To further investigate the mechanism of kinase inhibition by DHA, endogenous IKKβ immobilized to protein G was incubated with DHA or AA, and beads were washed to remove AA and DHA prior to the kinase reaction …
Cited by: 107
Vitamin C and Tyrosine Kinase Inhibitor in Lung Cancer ...
https://clinicaltrials.gov/ct2/show/NCT03799094
Jan 10, 2019 · Vitamin C and Tyrosine Kinase Inhibitor in Lung Cancer Patients With Epidermal Growth Factor Receptor Mutations The safety and scientific validity of this study is the responsibility of the study sponsor and investigators.
Vitamin C and Tyrosine Kinase Inhibitor in Lung Cancer Patients With Epidermal Growth Factor Receptor Mutations
Brief Summary:
This trial was to explore whether intravenous vitamin C can prolong resistance time of Tyrosine Kinase Inhibitor(TKI) on lung adenocarcinoma patients with Epidermal Growth Factor Receptor(EGFR) mutations, and can benefit NSCLC patients.
Detailed Description:
The effects of vitamin C in combination with tyrosine kinase inhibitor on tumor size, tumor markers, inflammatory factor levels, quality of life, duration of resistance, progression-free survival, and overall survival time were evaluated.
This trial is a low risk treatment, and has developed the appropriate safety measures and contingency plans to ensure patients' safety in the whole process.
Patients will be followed up after the end of the trial, and follow-up observations will be performed every month during the first year. Followed every 3 months in the second year for 3 years.
Study Type : Interventional (Clinical Trial)
Estimated Enrollment : 150 participants
Allocation: Randomized
Intervention Model: Parallel Assignment
Masking: Single (Outcomes Assessor)
Primary Purpose: Treatment
Official Title: Clinical Outcomes of Intravenous Vitamin C Synergy With Tyrosine Kinase Inhibitor in Lung Adenocarcinoma Patients With Epidermal Growth Factor Receptor Mutations
Actual Study Start Date : December 5, 2018
Estimated Primary Completion Date : December 31, 2020
Estimated Study Completion Date : December 31, 2022
Arm Intervention/treatment
Experimental: Experimental group
75 patients received a weekly intravenous Vitamin C injection (dose: 30 g / time, once a week, treatment termination when the disease progress is confirmed) in combination with daily taking tyrosine kinase inhibitor.
Drug: Vitamin C
Participants will receive intravenous vitamin C therapy at the indicated dose.
Other Name: Sodium Ascorbate
Drug: Tyrosine kinase inhibitor
Participants will receive Tyrosine kinase inhibitor therapy in cycles: continuous treatment at the indicated dose.
Experimental: Control group
75 patients received tyrosine kinase inhibitor daily. (dose: Osimertinib 80 mg/d, or Tarceva 150 mg/d, or Iressa 0.25 g/d.)
Arms and Interventions
Arm Intervention/treatment
Experimental: Experimental group
75 patients received a weekly intravenous Vitamin C injection (dose: 30 g / time, once a week, treatment termination when the disease progress is confirmed) in combination with daily taking tyrosine kinase inhibitor.
Drug: Vitamin C
Participants will receive intravenous vitamin C therapy at the indicated dose.
Other Name: Sodium Ascorbate
Drug: Tyrosine kinase inhibitor
Participants will receive Tyrosine kinase inhibitor therapy in cycles: continuous treatment at the indicated dose.
Experimental: Control group
75 patients received tyrosine kinase inhibitor daily. (dose: Osimertinib 80 mg/d, or Tarceva 150 mg/d, or Iressa 0.25 g/d.)
Drug: Tyrosine kinase inhibitor
Participants will receive Tyrosine kinase inhibitor therapy in cycles: continuous treatment at the indicated dose.
Outcome Measures
Go to sections
Primary Outcome Measures :
Progression free survival [ Time Frame: From the start date of treatment until the date of first documented progression or death, assessed up to 2 years ]
From the start of treatment until the patient has tumor progression or death
Secondary Outcome Measures :
Overall survival [ Time Frame: From the start date of treatment until the date of death from any cause, assessed up to 2 years. ]
The length of time from the start of treatment for a disease until death
Other Outcome Measures:
Changes in Health Related Quality of Life: European Organisation for Research and Treatment of Cancer(EORTC) quality of life questionnaire(QLQ)-C30 [ Time Frame: From the start date of treatment until the date of first documented progression or death, assessed up to 2 years ]
Using QLQ-C30: Function subscales, (include physical, role, emotional, cognitive, social, global) (averaged, 0-100, higher value represent a better outcome); Symptoms subscales (include fatigue, nausea, pain, dyspnea, insomnia, appetite loss, constipation, diarrhea) (averaged, 0-100, lower value represent a better outcome). Given at basal, 1, 2, 3, 6, 12 months.
Information from the National Library of Medicine
Choosing to participate in a study is an important personal decision. Talk with your doctor and family members or friends about deciding to join a study. To learn more about this study, you or your doctor may contact the study research staff using the contacts provided below. For general information, Learn About Clinical Studies.
Ages Eligible for Study: 18 Years to 75 Years (Adult, Older Adult)
Sexes Eligible for Study: All
Accepts Healthy Volunteers: No
Criteria
Inclusion Criteria:
Primary non-small cell lung cancer (adenocarcinoma) with EGFR mutations on exons 19 and 21.
18 years old to 75 years old.
During the trial, patients were prescribed TKI drugs(received initial treatment within 2 months, or change medication within 2 months) and did not receive chemotherapy or radiotherapy at the same time.
Eastern Cooperative Oncology Group (ECOG) performance status are 0 to 2.
Expected survival over 3 months.
Household registration is Guangdong Province.
Exclusion Criteria:
Co-morbid conditions that affect survival: end stage congestive heart failure, unstable angina, myocardial infarction (within the past 6 weeks), and uncontrolled blood sugars of greater than 300 mg/dL, known chronic active hepatitis or cirrhosis.
Glucose-6-phosphate dehydrogenase deficiency (G6PD) (a relative contraindication).
Patients who are allergic to vitamin C.
Patients with HIV and other infectious diseases.
Patients who are taking anticoagulants and have coagulopathy;
Combine dysfunction of important organs such as heart, lung, liver and kidney;
Patients with impaired renal function (serum creatinine content > 1.2 mg/dL)
Compromised liver function with evidence of Serum total bilirubin content, Serum alanine aminotransferase(ALT) and aspartate transaminase(AST)> 2 times normal reference value.
Pregnant or lactating female.
Smoking and alcohol abuse patients;
Anti-infective treatment is required for systemic or localized serious infections;
Patients with hyperuricacidemia (normal: 91-456 μmol / 24h (8-40mg / 24h));
Wilson's disease.
Evidence of significant psychiatric disorder by history or examination that would prevent completion of the study or preclude informed consent.
Any condition that impairs the patients' ability to swallow, which impairs drug absorption or drug kinetic parameters, including any kind of gastrointestinal resection or surgery;
History of surgery of visceral organs within 6 weeks before the study.Vitamin C and Tyrosine Kinase Inhibitor in Lung Cancer Patients With Epidermal Growth Factor Receptor Mutations - Full Text View - ClinicalTrials.gov
https://clinicaltrials.gov/ct2/show/NCT03799094
Eur J Pharm Sci. 2017 Jan 1;96:37-44. doi: 10.1016/j.ejps.2016.09.014. Epub 2016 Sep 10.
Lactate dehydrogenase inhibitors can reverse inflammation induced changes in colon cancer cells.
Manerba M1, Di Ianni L1, Govoni M1, Roberti M2, Recanatini M2, Di Stefano G3.
Author information
1
Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Italy.
2
Department of Pharmacy and Biotechnology (FABIT), University of Bologna, Italy.
3
Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Italy. Electronic address: giuseppina.distefano@unibo.it.
Abstract
The inflammatory microenvironment is an essential component of neoplastic lesions and can significantly impact on tumor progression. Besides facilitating invasive growth, inflammatory cytokines were also found to reprogram cancer cell metabolism and to induce aerobic glycolysis. Previous studies did not consider the possible contribution played in these changes by lactate dehydrogenase (LDH). The A isoform of LDH (LDH-A) is the master regulator of aerobic glycolysis; it actively reduces pyruvate and causes enhanced lactate levels in tumor tissues. In cancer cells, lactate was recently found to directly increase migration ability; moreover, when released in the microenvironment, it can facilitate matrix remodeling. In this paper, we illustrate that treatment of human colon adenocarcinoma cells with TNF-α and IL-17, two pro-inflammatory cytokines, modifies LDH activity, causing a shift toward the A isoform which results in increased lactate production. At the same time, the two cytokines appeared to induce features of epithelial-mesenchymal transition in the treated cells, such as reduction of E-cadherin levels and increased secretion of metalloproteinases. Noteworthy, oxamate and galloflavin, two inhibitors of LDH activity which reduce lactate production in cells, were found to relieve the inflammation-induced effects. These results suggest LDH-A and/or lactate as common elements at the cross-road between cancer cell metabolism, tumor progression and inflammation. At present, LDH inhibitors suitable for clinical use are actively searched as possible anti-proliferative agents; our data lead to hypothesize for these compounds a wider potential in anticancer treatment.
Copyright © 2016 Elsevier B.V. All rights reserved.
KEYWORDS:
Cancer cell metabolism; Epithelial-mesenchymal transition; Inflammation; Lactate dehydrogenase
Lactate dehydrogenase inhibitors can reverse inflammation induced changes in colon cancer cells. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/27622920
Anticancer Drugs. 2013 Sep;24(8):862-70. doi: 10.1097/CAD.0b013e328363ae50.
Galloflavin suppresses lactate dehydrogenase activity and causes MYC downregulation in Burkitt lymphoma cells through NAD/NADH-dependent inhibition of sirtuin-1.
Vettraino M1, Manerba M, Govoni M, Di Stefano G.
Author information
1
Department of Experimental Diagnostic and Specialty Medicine, University of Bologna, Bologna, Italy.
Abstract
Activation of the myc oncogene in cancer cells upregulates lactate dehydrogenase A (LDH-A) expression, leading to a sustained glycolytic flux that is needed to produce ATP under hypoxic conditions. We studied the effects of galloflavin (GF), a recently identified LDH inhibitor, on myc overexpressing Burkitt lymphoma (BL) cells. Epstein-Barr virus-infected lymphoblasts were used as a non-neoplastic control. Our results showed that myc overactivation induced a two- to seven-fold increase in LDH-A expression in BL cells compared with non-neoplastic lymphoblasts; this result is consistent with previously reported data. Moreover, GF treatment suppressed LDH activity and inhibited BL cell replication but did not affect lymphoblast viability. Surprisingly, we found that increased levels of the MYC and LDH-A proteins did not lead to a metabolic shift in BL cells toward glycolytic ATP generation. BL cells were treated with GF at doses that achieved 50% inhibition of cell growth and lactate production, and ATP levels were scarcely affected after GF treatment. The same results were also obtained by suppressing LDH activity with oxamate, an LDH specific inhibitor. Our data suggest that LDH activity is important for maintaining a correct NAD/NADH balance in BL cells. LDH inhibition led to decreased NAD cellular levels, which resulted in sirtuin-1 inhibition. Confirming previous studies, sirtuin-1 inhibition caused a reduction in MYC protein levels, depriving BL cells of their most important survival signal. This study further describes the biological functions of the LDH enzyme and suggests that LDH inhibition could be useful for the treatment of cancer.
PMID: 23797802 DOI: 10.1097/CAD.0b013e328363ae50
[Indexed for MEDLINE]Galloflavin suppresses lactate dehydrogenase activity and causes MYC downregulation in Burkitt lymphoma cells through NAD/NADH-dependent inhibition... - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/23797802
Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways
Author links open overlay panelF.Farabegolia1M.Vettrainoa1M.ManerbaaL.FiumeaM.RobertibG.Di Stefanoa a Department of Experimental Pathology, University of Bologna, via San Giacomo 14, 40126 Bologna, Italy b Department of Pharmaceutical Sciences, University of Bologna, via Belmeloro 6, 40126 Bologna, Italy Received 16 January 2012, Revised 31 July 2012, Accepted 16 August 2012, Available online 29 August 2012. Show less https://doi.org/10.1016/j.ejps.2012.08.012Get rights and content Abstract Galloflavin (GF), a recently identified lactate dehydrogenase inhibitor, hinders the proliferation of cancer cells by blocking glycolysis and ATP production. The aim of the present experiments was to study the effect of this compound on breast cancer cell lines reproducing different pathological subtypes of this tumor: MCF-7 (the well differentiated form), MDA-MB-231 (the aggressive triple negative tumor) and MCF-Tam (a sub-line of MCF-7 with acquired tamoxifen resistance). We observed marked differences in the energetic metabolism of these cell lines. Compared to MCF-7 cells, both MDA-MB-231 and MCF-Tam cells exhibited higher LDH levels and glucose uptake and showed lower capacity of oxygen consumption. In spite of these differences, GF exerted similar growth inhibitory effects. This result was explained by the finding of a constitutively activated stress response in MDA-MB-231 and MCF-Tam cells, which reproduce the poor prognosis tumor forms. As a further proof, different signaling pathways were found to be involved in the antiproliferative action of GF. In MCF-7 cells we observed a down regulation of the ERα-mediated signaling needed for cell survival. On the contrary, in MCF-Tam and MDA-MB-231 cells growth inhibition appeared to be contributed by an oxidative stress condition. The prevalent mechanism of cell death was found to be apoptosis induction. Because of the clinical relevance of breast cancer forms having the triple negative and/or chemoresistant phenotype, our results showing comparable effects of GF even on aggressively growing cells encourage further studies to verify the potential of this compound in improving the chemotherapy of breast cancer.
Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0928098712003235
Int J Biol Sci 2019; 15(3):544-555. doi:10.7150/ijbs.30297
Research Paper
Cyclin G2 Inhibits the Warburg Effect and Tumour Progression by Suppressing LDHA Phosphorylation in Glioma
Sen Li, Jinlan Gao, Xinbin Zhuang, Chenyang Zhao, Xiaoyu Hou, Xuesha Xing, Chen Chen, Qi Liu, Shuang Liu, Yang Luo Corresponding address
The Research Center for Medical Genomics, Key Laboratory of Cell Biology, Ministry of Public Health, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, China
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.
Citation:
Li S, Gao J, Zhuang X, Zhao C, Hou X, Xing X, Chen C, Liu Q, Liu S, Luo Y. Cyclin G2 Inhibits the Warburg Effect and Tumour Progression by Suppressing LDHA Phosphorylation in Glioma. Int J Biol Sci 2019; 15(3):544-555. doi:10.7150/ijbs.30297. Available from http://www.ijbs.com/v15p0544.htm
Go File import instruction
Abstract
Cyclin G2 has been identified as a tumour suppressor in several cancers. However, its regulatory roles and underlying mechanisms in tumours are still unknown. In this study, we demonstrated that cyclin G2 was expressed at low levels in glioma, which was as a poor prognostic factor for this disease. We also found that, cyclin G2 could suppress cell proliferation, initiate cell apoptosis and reduce aerobic glycolysis, suggesting that cyclin G2 plays a tumour suppressive role in glioma. Mechanistically, cyclin G2 could negatively regulate tyrosine-10 phosphorylation of a critical glycolytic enzyme, lactate dehydrogenase A, through direct interaction. Taken together, these results indicate that cyclin G2 acts as a tumour suppressor in glioma by repressing glycolysis and tumour progression through its interaction with LDHA.
Keywords: glioma, Warburg effect, cyclin G2, lactate dehydrogenase ACyclin G2 Inhibits the Warburg Effect and Tumour Progression by Suppressing LDHA Phosphorylation in Glioma
https://www.ijbs.com/v15p0544.htm
Oncogene. 2017 Jul 6;36(27):3797-3806. doi: 10.1038/onc.2017.6. Epub 2017 Feb 20.
Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis.
Jin L1, Chun J1, Pan C1, Alesi GN1, Li D1, Magliocca KR2, Kang Y3, Chen ZG1, Shin DM1, Khuri FR1, Fan J4, Kang S1.
Author information
1
Winship Cancer Institute, Department of Hematology/Medical Oncology, Emory University School of Medicine, Atlanta, GA, USA.
2
Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, USA.
3
Department of Molecular Biology, Princeton University, Princeton, NJ, USA.
4
Winship Cancer Institute, Department of Radiation Oncology, Emory University School of Medicine, Atlanta, GA, USA.
Abstract
Metastases remain the major cause of death from cancer. Recent molecular advances have highlighted the importance of metabolic alterations in cancer cells, including the Warburg effect that describes an increased glycolysis in cancer cells. However, how this altered metabolism contributes to tumour metastasis remains elusive. Here, we report that phosphorylation-induced activation of lactate dehydrogenase A (LDHA), an enzyme that catalyses the interconversion of pyruvate and lactate, promotes cancer cell invasion, anoikis resistance and tumour metastasis. We demonstrate that LDHA is phosphorylated at tyrosine 10 by upstream kinases, HER2 and Src. Targeting HER2 or Src attenuated LDH activity as well as invasive potential in head and neck cancer and breast cancer cells. Inhibition of LDH activity by small hairpin ribonucleic acid or expression of phospho-deficient LDHA Y10F sensitized the cancer cells to anoikis induction and resulted in attenuated cell invasion and elevated reactive oxygen species, whereas such phenotypes were reversed by its product lactate or antioxidant N-acetylcysteine, suggesting that Y10 phosphorylation-mediated LDHA activity promotes cancer cell invasion and anoikis resistance through redox homeostasis. In addition, LDHA knockdown or LDHA Y10F rescue expression in human cancer cells resulted in decreased tumour metastasis in xenograft mice. Furthermore, LDHA phosphorylation at Y10 positively correlated with progression of metastatic breast cancer in clinical patient tumour samples. Our findings demonstrate that LDHA phosphorylation and activation provide pro-invasive, anti-anoikis and pro-metastatic advantages to cancer cells, suggesting that Y10 phosphorylation of LDHA may represent a promising therapeutic target and a prognostic marker for metastatic human cancers.
PMID: 28218905 PMCID: PMC5501759 DOI: 10.1038/onc.2017.6Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28218905
Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells
Oncotarget. 2011; 2:551-556. https://doi.org/10.18632/oncotarget.299
Metrics: PDF 5474 views | HTML 5471 views ?
Weibo Luo1,2 and Gregg L. Semenza1,2,3,4,5,6,7
1 Vascular Program, Institute for Cell Engineering, The Johns Hopkins University School of Medicine, Baltimore, USA
2 McKusick-Nathans Institute of Genetic Medicine, The Johns Hopkins University School of Medicine, Baltimore, USA
3 Department of Biological Chemistry, The Johns Hopkins University School of Medicine, Baltimore, USA
4 Department of Oncology, The Johns Hopkins University School of Medicine, Baltimore, USA
5 Department of Pediatrics, The Johns Hopkins University School of Medicine, Baltimore, USA
6 Department of Medicine, The Johns Hopkins University School of Medicine, Baltimore, USA
7 Department of Radiation Oncology, The Johns Hopkins University School of Medicine, Baltimore, USA
Received: June 24, 2011; Accepted: June 24, 2011; Published: June 25, 2011;
Figure 1: Regulation of glucose metabolism by HIF-1. HIF-1 controls transcription of genes encoding glucose transporters, which transport glucose from the extracellular (ext) to the intracellular (int) milieu, and glycolytic enzymes, which convert glucose to lactate as glycolytic end-product or acetyl coenzyme A (Acetyl-CoA) that is metabolized in the tricarboxylic acid cycle (TCA). Pyruvate kinase M2 converts phosphoenolpyruvate (PEP) into pyruvate, which is upstream of the decision point for glycolytic vs oxidative metabolism. Arrow indicates direction of glucose metabolism; blocked line indicates inhibition.
Figure 2: PKM2 contributes to metabolic reprogramming and cancer progression by serving as a PHD3-dependent coactivator for HIF-1. Prolyl hydroxylation of PKM2 by PHD3 promotes the interaction of PKM2 with HIF-1α, thereby stabilizing HIF-1 binding to the HRE of target genes, recruitment of coactivator p300, histone acetylation, and subsequent transcription of HIF-1 target genes, which encode proteins that are involved in metabolic reprogramming, angiogenesis, and many other critical aspects of cancer progression.
Figure 3: PKM2 and PHD3 are components of a positive feedback loop that amplifies HIF-1 transcriptional activity. HIF-1 activates transcription of genes encoding PKM2 and PHD3, which interact with HIF-1α to stimulate transactivation of HIF-1 target genes that promote cancer progression.
ABSTRACT
Cancer cells feature altered glucose metabolism that allows their rapid growth. They consume large amounts of glucose to produce lactate, even in the presence of ample oxygen, which is known as the Warburg effect. Pyruvate kinase M2 (PKM2) contributes to the Warburg effect by previously unknown mechanisms. Hypoxia-inducible factor 1 (HIF-1) mediates PKM2 gene transcription and glucose reprogramming in cancer cells. The recent discovery of novel physical and functional interactions between PKM2 and HIF-1 in cancer cells has provided insight into molecular mechanisms underlying the Warburg effect.
INTRODUCTION
Altered glucose metabolism is a key feature that distinguishes cancer cells from normal cells. Most cancer cells consume higher amounts of glucose and subsequently produce much more lactate than normal cells, even in the presence of ample O2. This phenomenon is known as the Warburg effect [1]. Since Otto Warburg made this important observation in 1924 [2], many researchers have attempted to elucidate the underlying molecular mechanisms in cancer cells. Hypoxia-inducible factor 1 (HIF-1), Myc, p53, Ras, Akt, Src, pyruvate kinase (PK) M2, and lactate dehydrogenase A (LDHA) have been implicated in the Warburg effect [3-8]. We recently discovered that the glycolytic enzyme PKM2 promotes the Warburg effect by serving as a transcriptional coactivator for HIF-1 in cancer cells [9]. This research perspective will discuss these recent findings regarding physical and functional interactions of HIF-1 and PKM2.
HIF-1 AND METABOLIC REPROGRAMMING IN CANCER CELLS
HIF-1 is a heterodimeric transcription factor, consisting of an O2-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit [10, 11]. HIF-1 is a master regulator of transcriptional responses to reduced O2 availability (hypoxia), which is found in the majority of advanced human cancers [12, 13]. In well-oxygenated human cells, HIF-1α is hydroxylated at proline-402 and/or proline-564 by the prolyl hydroxylase domain proteins, PHD1-3 [14]. PHD2 is primarily responsible for regulating basal HIF-1α levels in cancer cells [15]. Prolyl-hydroxylated HIF-1α is bound by the von Hippel-Lindau (VHL) tumor suppressor protein, which is the substrate recognition component of an E3 ubiquitin-ligase complex, leading to HIF-1α protein degradation by the 26S proteasome [16]. Under hypoxic conditions, HIF-1α prolyl hydroxylation is inhibited, thereby stabilizing HIF-1α protein [17]. HIF-1α protein levels are also increased in normoxic cancer cells with loss of function of certain tumor suppressors, most notably VHL in the clear cell type of renal cell carcinoma [16, 18, 19]. HIF-2α is a paralog of HIF-1α that is also O2-regulated, dimerizes with HIF-1β, and transactivates a group of target genes that partially overlaps the battery of genes regulated by HIF-1 [20, 21].
Activation of HIF-1 commonly occurs in many cancer types and is a driving force regulating many steps in cancer progression [18, 22]. HIF-1 activates the transcription of genes encoding proteins that mediate angiogenesis, invasion, metastasis, and the shift from oxidative to glycolytic metabolism [12, 18, 19, 22, 23]. By activating the transcription of genes encoding glucose transporters and glycolytic enzymes, HIF-1 enhances glucose uptake and glycolysis in cells [23-27]. HIF-1 also controls expression of LDHA and pyruvate dehydrogenase kinase 1 (PDK1) [25, 26, 28, 29]. LDHA catalyzes the conversion of pyruvate to lactate (Figure 1), thereby decreasing mitochondrial utilization of pyruvate as a substrate for pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl coenzyme A (AcCoA). PDK1 phosphorylates the catalytic subunit of PDH, leading to its inactivation, which shunts pyruvate away from the mitochondria. HIF-1 activation shifts the balance of metabolism from oxidative phosphorylation toward glycolysis and mediates the Warburg effect in VHL-null renal carcinoma cells [30].
Figure 1: Regulation of glucose metabolism by HIF-1. HIF-1 controls transcription of genes encoding glucose transporters, which transport glucose from the extracellular (ext) to the intracellular (int) milieu, and glycolytic enzymes, which convert glucose to lactate as glycolytic end-product or acetyl coenzyme A (Acetyl-CoA) that is metabolized in the tricarboxylic acid cycle (TCA). Pyruvate kinase M2 converts phosphoenolpyruvate (PEP) into pyruvate, which is upstream of the decision point for glycolytic vs oxidative metabolism. Arrow indicates direction of glucose metabolism; blocked line indicates inhibition.
REGULATION OF PKM2 EXPRESSION IN CANCER CELLS
PK catalyzes the conversion of phosphoenolpyruvate to pyruvate (Figure 1) and is composed of M1-/M2-type and L-/R-type isoforms, which are encoded by PKM2 and PKLR genes, respectively [31]. Tissue-specific promoters control expression of PKL, which is expressed in liver and kidney, and PKR, which is expressed in erythrocytes. PKM1 and PKM2 are the alternatively spliced products of the PKM2 primary RNA transcript with PKM1 and PKM2 mRNA containing sequences encoded by exon 9 or exon 10, respectively [32]. PKM1 is expressed in muscle and brain, whereas PKM2 is expressed in the embryo and in cancer cells. The transcription factors Sp1 and Sp3 bind to a GC-rich element in the promoter of the human PKM2 gene [33]. Sp1 constitutively activates transcription of PKM2, whereas Sp3 functions as a transcriptional repressor that dissociates from the PKM2 gene under hypoxic conditions.
We recently identified a hypoxia response element (HRE) within the first intron of the human PKM2 gene [9]. Heterodimer complexes of HIF-1β with HIF-1α, but not HIF-2α, bound to the PKM2 HRE and increased the activity of a luciferase reporter gene driven by the PKM2 HRE in hypoxic HeLa cells. Mutation of the HIF-1 binding site in the PKM2 HRE or knockdown of HIF-1α protein expression suppressed reporter gene activity. Hypoxia induced the expression of PKM1 and PKM2 mRNA in wild-type, but not in HIF-1α-knockout, mouse embryo fibroblasts [9].
PKM2 expression in cancer cells is also regulated by microRNAs (miRs). miR-326 matches two regions in the 3’-untranslated region (UTR) of PKM2 mRNA and transfection of miR-326 precursor decreased PKM2 3’-UTR-luciferase reporter activity and PKM2 protein levels in glioma cells [34]. miR-133a and miR-133b are also implicated in PKM2 expression. PKM2 overexpression is associated with downregulation of miR-133a and miR-133b, whereas transfection of miR-133a or miR-133b precursors inhibited PKM2 expression in tongue squamous cell carcinoma cells [35]. The significance of this mutual antagonism between miR-133a/b and PKM2 has not been determined.
Recent studies revealed the molecular mechanism underlying PKM2 mRNA splicing. Heterogeneous nuclear ribonucleoproteins (hnRNP) I, A1, and A2 bind to RNA sequences encoded by exon 9 and inhibit PKM1-specific mRNA splicing [36, 37]. The c-Myc oncoprotein regulates transcription of hnRNPI, hnRNPA1 and hnRNPA2, resulting in preferential PKM2 isoform expression in cancer cells overexpressing c-Myc [36]. Mammalian target of rapamycin (mTOR), a serine/threonine protein kinase that regulates cell growth, cell survival, and protein synthesis, also stimulates PKM2 expression through activation of HIF-1 and c-Myc [38]. Thus, activation of transcription factors and kinases, and downregulation of microRNAs results in high expression of PKM2 in cancer cells. However, analysis of human tumor and non-tumor tissues from kidney, liver, lung, and thyroid using an absolute quantification approach by mass spectrometry revealed that PKM2 protein expression is predominant in both human tumor tissues and tissue-matched normal controls, suggesting that no switch from PKM1 to PKM2 is required for tumor development [39]. These findings challenge the conclusion, which was based on the analysis of cancer cell lines, that PKM2 overexpression is a hallmark of cancer cells [3]. The proteomic study found that total PKM expression (PKM1 + PKM2) is increased 3-fold in tumor tissue compared to normal tissue [39]. HIF-1α is overexpressed in solid tumors due to intratumoral hypoxia, genetic alterations, or both [12, 18, 22, 23], and thus may be a predominant regulator that contributes to elevated levels of PKM2 in human tumor tissues.
PKM2 AND THE WARBURG EFFECT IN CANCER CELLS
Christofk et al. demonstrated that PKM2 expression was associated with increased glucose uptake and lactate production, but decreased O2 consumption in cancer cells [3]. Genetic manipulation of cancer cells that switched them from PKM2 to PKM1 expression reversed the Warburg effect, suggesting that high expression of PKM2 is required for aerobic glycolysis in cancer cells. Moreover, expression of PKM2, but not PKM1, induced tumor xenograft growth in nude mice [3]. The binding of tyrosine-phosphorylated peptides to PKM2 at lysine-433 was found to inhibit PKM2 enzymatic activity through release of the allosteric activator fructose-1,6-bisphosphate (FBP) and to promote cell growth and glycolytic metabolism in cancer cells [40]. Tyrosine kinases play critical roles in cell growth, cell metabolism, and angiogenesis in cancer [41, 42]. Hitosugi et al. reported that fibroblast growth factor receptor type 1 (FGFR1) phosphorylated PKM2 at tyrosine residues-83, -105, -148, -175, -370, and -390 in murine Ba/F3 hematopoietic cells [43]. Phosphorylation of PKM2 at tyrosine-105 induced FBP release from active tetrameric PKM2, promoted formation of less active dimeric PKM2, and subsequently decreased PKM2 enzymatic activity. In contrast, the phosphorylation of PKM2 at other tyrosine residues caused by FGFR1 failed to regulate PKM2 activity. However, it remained unclear how alterations in PKM2 activity could determine whether the product of the PKM2 reaction, pyruvate, was converted to lactate or to AcCoA (Figure 1).
We recently delineated a molecular mechanism by which PKM2 mediates the Warburg effect in cancer cells [9]. PKM2 was found to interact with HIF-1α in the nucleus and to function as a transcriptional coactivator in HeLa cervical carcinoma and Hep3B hepatoblastoma cells. PKM2 increased HIF-1 binding to HREs at target genes, recruitment of coactivator p300, histone acetylation, and subsequent transactivation of HIF-1 target genes including SLC2A1 (which encodes glucose transporter 1), LDHA, and PDK1 in HeLa and Hep3B cells. PKM2-stimulated expression of HIF-1 target genes promotes the shift from oxidative phosphorylation to glycolytic metabolism. PKM2 also binds to HIF-2α and promotes HIF-2-mediated transactivation in cancer cells [9]. In addition to its effects on genes encoding metabolic enzymes, PKM2 stimulates HIF-1- and HIF-2-mediated expression of the VEGF gene (which encodes vascular endothelial growth factor), thereby promoting angiogenesis. Thus, PKM2 may play a far broader role in promoting cancer progression than was previously appreciated (Figure 2).
Figure 2: PKM2 contributes to metabolic reprogramming and cancer progression by serving as a PHD3-dependent coactivator for HIF-1. Prolyl hydroxylation of PKM2 by PHD3 promotes the interaction of PKM2 with HIF-1α, thereby stabilizing HIF-1 binding to the HRE of target genes, recruitment of coactivator p300, histone acetylation, and subsequent transcription of HIF-1 target genes, which encode proteins that are involved in metabolic reprogramming, angiogenesis, and many other critical aspects of cancer progression.
The enzymatic activity of PKM2 is not required for HIF-1 transactivation [9]. Interestingly, PKM2 is prolyl hydroxylated in the PKM2-specific domain encoded by exon 10, and prolyl hydroxylation of PKM2 is required for HIF-1-mediated transactivation in cancer cells. PHD3 catalyzes hydroxylation of PKM2 and PHD3 knockdown reduced expression of the HIF-1 target genes SLC2A1, LDHA, and PDK1, and reversed the Warburg effect in VHL-null RCC4 renal carcinoma cells [9]. PKM2, but not PKM1, is prolyl hydroxylated and PKM2, but not PKM1, interacts with HIF-1α, thus providing a molecular basis for the selective effect of PKM2 on HIF-1-mediated transactivation and the Warburg effect in cancer cells [9].
Recently, the interaction of PHD3 with PKM2 was reported to increase the formation of dimeric PKM2, which has decreasd activity compared to the tetrameric form of the enzyme [44]. Chen et al. concluded that the hydroxylase activity of PHD3 was not required for its effect on PKM2 oligomerization/enzyme activity because mutant PHD3 (R205K) behaved similarly to wild-type PHD3. However, we found that mutation of arginine-205 alone was not sufficient to inactivate the hydroxylase activity of PHD3 (W.L. and G.L.S., unpublished) and thus it would be interesting to repeat these experiments using the PHD3 (H135A/D137A) mutant that lacks hydroxylase activity [9]. PHD3 is encoded by a HIF-1 target gene and increased PHD3 mRNA and protein expression is induced by HIF-1 under hypoxic conditions [45], which compensates for the reduced hydroxylase activity [9]. Thus, PHD3 and PKM2 exert a positive feedback loop in cancer cells that amplifies HIF-1 activity, which may play a major role in driving metabolic reprogramming, angiogenesis, and other critical aspects of cancer progression (Figure 3).
Figure 3: PKM2 and PHD3 are components of a positive feedback loop that amplifies HIF-1 transcriptional activity. HIF-1 activates transcription of genes encoding PKM2 and PHD3, which interact with HIF-1α to stimulate transactivation of HIF-1 target genes that promote cancer progression.
UNANSWERED QUESTIONS, FUTURE DIRECTIONS
Does PKM2 have other functions in the nucleus that promote cancer progression? PKM2 also binds to the transcription factor Oct-4 and enhances Oct-4-dependent gene transcription [46]. Oct-4 is a key mediator of pluripotency in embryonic stem cells [47] and induced pluripotent stem cells [48]. Oct-4 is also expressed in human breast cancer stem cells [49]. Hoshino et al. also found that nuclear translocation of PKM2 is induced by interleukin-3 and stimulates cell proliferation [50], although the nuclear target of PKM2 was not identified. It is likely that the stimulation of cell proliferation by PKM2 is independent of its regulation of HIF-1α transactivation.
Post-translational modification by prolyl hydroxylation and tyrosine phosphorylation regulate PKM2 activity as a transcriptional coactivator and glycolytic enzyme, respectively. PKM2 is also subjected to sumoylation [51] and lysine acetylation [52] and further studies are required to determine whether these post-translational modifications also regulate the role of PKM2 as a HIF-1 coactivator.
Several compounds have been shown to inhibit PKM2 enzymatic activity [53, 54]. However, those inhibitors may not suppress HIF-1 transactivation in cancer cells because the enzymatic activity of PKM2 is not required for its coactivator functions [9]. Combination therapy with HIF inhibitors [12, 18, 19] may prove to be more efficacious.Oncotarget | Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells
http://www.oncotarget.com/index.php?journal=oncotarget&page=article&op=view&path%5B%5D=299&path%5B%5D=473
LPS诱导的PKM2表达引起酪氨酸105的同时磷酸化,其程度与增加的表达水平相当或更高。这种磷酸化仅在1小时后就很明显,在48小时后诱导最强(图1A,中间,相对带强度增加了10.3倍)。丙酮酸激酶M2调节Hif-1α...
LPS-induced expression of PKM2 causes concurrent phosphorylation of Tyrosine 105 to an extent comparable or greater to the increased expression levels. This phosphorylation is evident after just 1 hr, with the strongest induction after 48 hr (Figure 1 A, middle, 10.3-fold increase in relative band intensity).
Pyruvate Kinase M2 Regulates Hif-1α ... - ScienceDirect
www.sciencedirect.com/science/article/pii/S1550413114005567

.jpg)


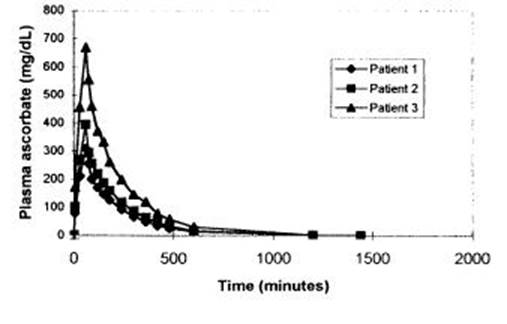
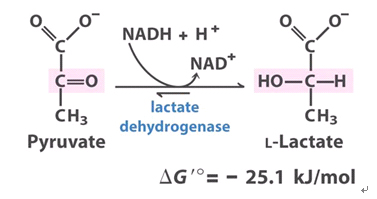


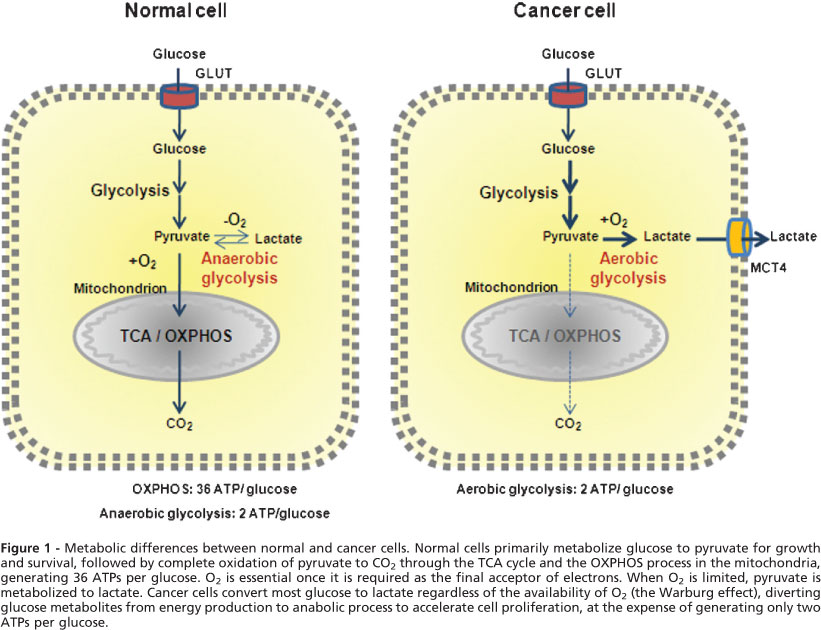
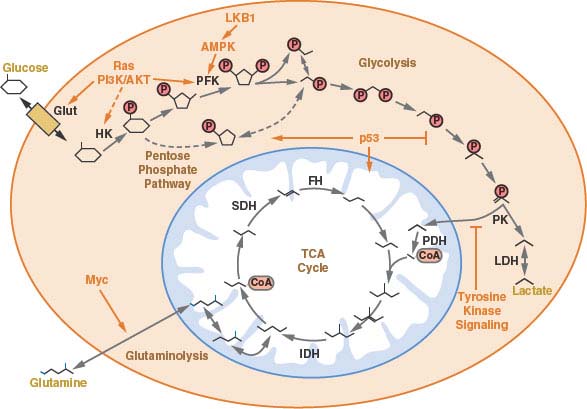

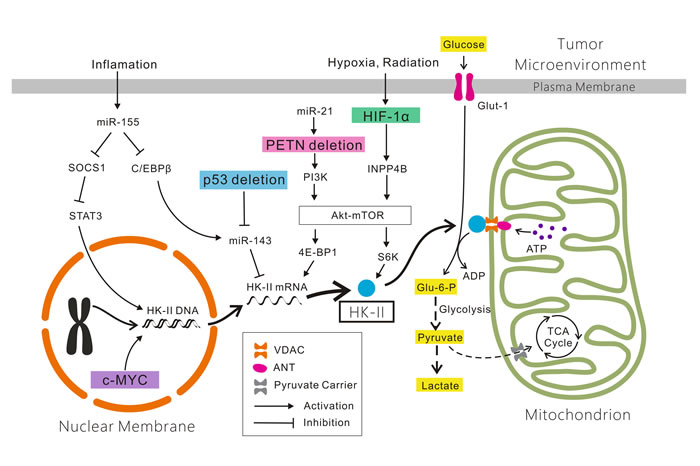
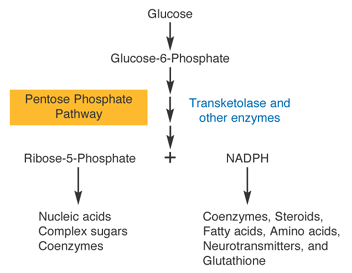


.jpg)
.jpg)