ˇˇ
ˇˇ
ŇČĎ٦Âϸ°űŁş´Ó˛úÉúµ˝ÔŮÉú
1. Autopsy studies have shown deficits in beta cell mass in approximately 70~100% and 0~65% in patients with T1D and T2D, respectively (1, 2).
The islet of Langerhans comprise to ~2% of the pancreatic mass, and the diameter of one islet is about 100 ¦Ěm, or 0.1 mm (1). The human pancreas has about 1-2 million islets (2), and the beta cells has a diameter of about 0.01 mm.
Pancreatic ¦Â cell mass is primarily regulated by replication and apoptosis (109). Transcriptional factors pancreatic duodenal homeobox 1 (PDX1) plays a pivotal role in proliferation, survival, and function of ¦Â cells and activation of insulin gene expression (3, 145). Meanwhile, ¦Â cell apoptosis in diabetic subjects is a more deciding factor than replication compared with control subjects (13). This event can be triggered by high glucose (30) and cytokines that induce ROS and RNS formations (91).
ˇˇ2. a series of inhibitors of DYRK1A-NFAT, GSK3, and NF-¦ĘB signaling pathways were shown to increase human pancreatic beta cell replication. Osteoprotegerin(OPG) and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-¦ĘB ligand pathway.
3. Differentiation of Pancreatic Progenitors Into beta Cells.
4. conversions of other pancreatic cells, including exocrine and endocrine pancreatic cells into beta cells.
5. Potential Drugs for Stimulation of beta Cells Conversion: ¦Ă-Aminobutyric acid (GABA), Artemisinin, a novel diet therapy with 4-day fasting-mimicking diet (FMD) cycles can reverse beta cell failure and can reverse diabetes in mice.
6. exercises induce osteoprotegerin
7. GABA receptor/GABA GABA increases beta-cell proliferation in vivo and in vitro, protects INS-1 cells from streptozotocin (STZ). The functional recovery of STZ-induced hyperglycemic mice may thus be caused by two effects: protection from ¦Â-cell apoptosis (anti-inflammation) and stimulation of beta-cell proliferation. GABA acts as a growth factor that regulates the survival and replication of islet beta-cells.
8. ROS induced apoptosis in beta cells: Oxidative stress of beta-cells was induced with STZ. The increase in superoxide preceded apoptosis. Sources of ROS: alloxan seletely enters beta cells and produces ROS superoxide and H2O2, with little catalase beta cells are vulnerable to ROS,especially H2O2 and hydroxyl radical when transient metals are present. Pancreatic islets contain low activities of catalase, selenium-dependent glutathione peroxidase 1 (GPX1), and Cu,Zn-superoxide dismutase 1 (SOD1).
The pancreatic islet beta cells are very sensitive to oxidative stress, probably due to the extremely low level of anti-oxidant enzymes, particularly catalase. In contrast to beta cells, pancreatic alpha cells are significantly more resistant to diabetogenic toxins. however, the intensive catalase expression in alpha cells of diabetic and non-diabetic mice.
9.OPG inhibits insulin secretion under acute inflammatory conditions
10. pancreatic progenitors are already specified at the endoderm stage -- the first step of differentiation.
11. Germany scientists found ways to use pluripotent stem cell differentiation protocol to generate beta cells
12. Beta cells also secret hormones amlyn and C-peptide
13. beta cells are destroyed by inflammation, saturated fats and trans fat, amyloid deposits, and exhaustion-making excess insulin, and immune cells attacks in T1D. Yale scientists found people without Type 2 Diabetes or AlzheimerˇŻs disease did not allow for the formation of amyloid deposits.
14. HDL the good cholesterol, protects beta cells and aid in insulin secretion
15. beta cells divide every 12 to 20 days. Our beta cells replicate the most from birth through five years, Proliferation is greatest up to age 20. Then it starts to taper off, with the least beta cell replication occurring in the elderly population.
16. They are about 20% responsible for producing and secreting glucagon. Glucagon is also produced in the liver.
17. Zinc is essential for storage of insulin and for processing of insulin in the body. In diabetic patients the content of zinc is greatly decreased in the pancreas. the zinc content of pancreatic beta cells is among the highest in the body. Insulin secretion from pancreatic beta-cells is dependent on zinc ions as essential components of insulin crystals, zinc transporters are thus involved in the insulin secretory process. the reduced levels of circulating zinc found in individuals with T2D. zinc is essential for superoxide dismutase.
18. since 1940s, Scientists have known that beta cells take in about 1,000 times more zinc than surrounding cells, and that trait has been used to help scientists visually identify beta cells in pancreatic tissues. Human pancreatic beta cells have exceptionally high zinc content. In beta cells the highest zinc concentration is in insulin secretory granules(ISGs), from which it is co-secreted with the hormone. At steady state in human ¦Â cells, ZnT8 shows a high degree of co-localization with insulin, consistent with its principal role of facilitating uptake of Zn2+ into ISGs. Zinc transporter 8 (ZnT8) transports zinc ions for crystallization and storage of insulin in pancreatic beta-cells and ZnT8 dysfunction is involved in pathogenesis of diabetes.
19. Important contributors to increases in intercellular ROS in beta cells are nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes. Several key observations have linked NADPH oxidase activity with regulation of insulin secretion. The product of NADPH oxidase activity, generation of H2O2, is required for insulin secretion. Elevated glucose leads to an increase in H2O2 generation, thus linking NADPH oxidase activity to regulation of insulin secretion .Inhibition of NADPH oxidase by the general inhibitor, diphenyleneiodonium (DPI), led to a decrease in H2O2 production and also impaired insulin secretion (Imoto et al. 2008). With global inhibition of NADPH oxidase activity, beta cells are protected from the effects of cytokine or FFA treatment.
20.Advanced glycation end products (AGE) contribute to oxidative stress and the development of diabetes . AGEs form when carbohydrates, such as glucose, react nonenzymatically (e.g., glycation and oxidation) with amino groups. Binding of AGE to its receptor, RAGE (receptor for advanced glycation end products), generates ROS leading to oxidative stress in beta cells. Evidence suggests that AGE may be increasing ROS generation through NADPH oxidase. The increase in superoxide preceded apoptosis. Pancreatic beta cell lines including INS-1, MIN6, and BTC-6 cells and isolated primary rat islets treated with AGE showed an increase in ROS generation that was followed by apoptosis
21.insulin-stimulated oxidants production
22.Nitric oxide (NO) contributes to carbohydrate metabolism and decreased NO bioavailability is involved in the development of type 2 diabetes mellitus (T2DM). via inhibition of angiotensin 2 and
23. Pancreatic beta cells express vitamin D receptorŁ¬Vitamin D supplementation restores insulin secretion from islets of deficient animal modelsŁ¬ vitamin D treatment or silencing of FoxO1 gene could reverse the ROS-induced cell apoptosis
24.The alpha cells produce glucagon, and make up 15¨C20% of total islet cells. The beta cells produce insulin and amylin, and make up 65¨C80% of the total islet cells. The delta cells produce somatostatin, and make up 3¨C10% of the total islet cells. The gamma cells produce pancreatic polypeptide, and make up 3¨C5% of the total islet cells.
25.blood supply to endocrine and exocrine compartments is integrated. Secretion of digestive enzymes and insulin is simultaneously activated when nutrients need to be absorbedˇ˘
26. Replication and proliferation of beta cells: Glucose increases the rate of beta cell proliferation in vitro and during short periods of glucose infusion in rodents with glucose identified as the key systemic factor controlling beta cell replication. Beta cell mass is dynamic and can respond to environmental cues such as glucose and insulin. Beta cell number increases markedly in the first year of rodent life , up to 1.5-fold during pregnancy and up to 10-fold in insulin resistant states. When not constrained by persistent autoimmune attack or the toxicity of persistent hyperglycemia (glucotoxicity) , beta cells inherently have the capacity to regenerate While the mechanism regulating beta cell expansion remains unclear, all beta cells are capable of replication (Brennand et al., 2007). Pluripotent stem cells also serve as sources of new beta cells. In human adult beta cells, replication is estimated at 10-fold less than in adult mice with the most replication in <5 year old children. A recent study identified a simple mechanism for homeostasis of beta cell proliferation and mass where beta cells adjust their proliferation rate according to the rate of glycolysis; this provides a system for sensitive measurement of organismal demand for beta cells, while normoglycemia is maintained. Beta cell proliferation progressively reduces with age. Glucose stimulates human beta cell replication in a transplant setting, and Beta cell death was not influenced by hyperglycaemia, donor age or donor BMI Ł¨due to the lasting time of the experiment and different metabolism and antioxidants system?)
In human type 2 diabetes, both increased apoptosis and reduced replication may contribute to beta cell loss and reduced beta cell mass
27. Dietary restriction using sharp reductions in caloric intake can result in improved beta-cell function (7). insulin sensitivity as a consequence of weight loss was the primary driver of this functional reversal.
28. TZDs are partial PPAR¦Ă agonists and primarily act as insulin sensitizers, with little effect on insulin secretion
29.Pancreatic ¦Â-cells are remarkably adaptable in response to metabolic status. For example, beta-cell insulin secretory capacity essentially is shut down during prolonged fasting as a protective measure against hypoglycemia, yet the fasted beta-cells remain poised to rapidly restore effective insulin production hours after refeeding (12).
30.Hopkins U: People with diabetes have long been known to have more zinc in their urine and less in their bloodstream.
31. ZnT8 protects against EMT-tubulointerstitial fibrosis though the restrain of TGF-beta1/Smads signaling activation in DKD
32.Insulin resistance: Resistance to signaling by the hormone insulin. Resistance to signaling by the hormone insulin is a major problem, not only in patients with type 2 diabetes but also in a broad range of other physiological states, including pregnancy, obesity, and septic shock..and systemic acute inflammation. both Tumor necrosis factor- (TNF-alpha) or dexamethasone produce insulin resistance
33.Inflammatory states produce insulin resistance: Over a hundred years ago, high doses of salicylates were shown to lower glucose levels in diabetic patients. Inflammatory mediators, specifically the cytokines TNF-alpha, IL-1, IL-6, and C-reactive protein, also induce peripheral insulin resistance.Concomitantly, stress and the inflammatory response result in decreased translocation of GLUT-4 to the cell membrane. It is likely that proinflammatory mediators, particularly TNF-alpha and IL-1, are responsible for the reciprocal effects on the surface expression of these glucose transporters (Figure (Figure2)2) [5]. Elevated TNF-alpha directly interferes with insulin signal transduction through the phosphorylation of various molecules along the insulin-signaling pathway. During infection, the upregulation of GLUT-1 and downregulation of GLUT-4 may play a role in redistributing glucose away from peripheral tissues towards immune cells and the nervous system. both dexamethasone and TNF-¦Á caused increased intracellular accumulation of ROS. Furthermore, treatment of cultured cells with antioxidant molecules or expression of enzymes that remove ROS suppressed insulin resistance induced by dexamethasone and TNF (vitamin C blocks TNFS signaling)
Phosphorylation by IKK¦Â targets I¦ĘB¦Á for proteasomal degradation, which liberates NF-¦ĘB for translocation into the nucleus, where it promotes the expression of numerous target genes whose products induce insulin resistance
34. Adipose tissues: main sources of inflammatory cytokines in obese people: TLRs, ACEs, saturated fats and adiposity activate both JNK and IKK¦Â
35. stress hyperglycemia: acute illness: hemorrhage, hypoxia and sepsis. Adrenal cortisol output increases up to ten-fold with severe stress (approximately 300 mg hydrocortisone per day) [12]. In patients with shock, plasma concentrations of epinephrine increase 50-fold and norepinephrine levels increase 10-fold [13]. The adrenal medulla is the major source of these released catecholamines [13]. Adrenalectomy eliminates the epinephrine response and blunts the norepinephrine response to hemorrhagic shock [13]. The increased release of stress hormones results in multiple effects (metabolic, cardiovascular and immune) aimed at restoring homeostasis during stress. The HPA axis, sympathoadrenal system and proinflammatory cytokines (TNF-¦Á, IL-1 and IL-6) act collectively and synergistically to induce stress hyperglycemia.
ICU mortality rates were however significantly different; 26.9 % of those without stress hyperglycemia died before discharge from ICU, this compared with just 14.8 % of those with stress hyperglycemia.
36.Glucose is largely utilized by tissues that are non-insulin dependent, and these include the central and peripheral nervous system, bone marrow, white and red blood cells and the reticuloendothelial system [20]. It has been estimated that, at rest, non-insulin mediated glucose uptake accounts for 75 to 85% of the total rate of whole glucose disposal. Glucose is the primary source of metabolic energy for the brain.
37.glucose transporter isoforms and distribution: Although 14 GLUT isoforms have been identified in the human genome, glucose uptake per se is facilitated by GLUT-1, GLUT-3 and GLUT-4 in various tissues. Insulin increases GLUT-4-mediated glucose transport by increasing translocation of GLUT-4 from intracellular stores to the cell membrane [20].
38. . NAD+ , the key player in glycolysis- NAMPT-mediated NAD+ biosynthesis is severely compromised in metabolic organs by high-fat diet (HFD). Strikingly, nicotinamide mononucleotide (NMN), a product of the NAMPT reaction and a key NAD+ intermediate, ameliorates glucose intolerance by restoring NAD+ levels in HFD-induced T2D mice.
39. Non-insulin dependent glucose uptake tissues via GLUT1: the pancreas, the anterior pituitary gland, the kidneys, the gonads, and osteoblasts and osteoclasts in bone. beta-cells, RBC, blood braine barrier, kidney, liver, instestinal cells, placenta. eye, BV, nerves, CNS, account for 80~ 85% of glucose uptake at rest.
40. Insulin-dependent tissues: skeletal muscles, cardiac muscles, smooth muscles, adipose tissues
41. Omega-3 fatty acids improve insulin sensitivity: EPA increases insulin sensitivity, DHA reduces inflammation, ALA reduces body weight
42. Effect of high fat diet on beta cells: mice fed the HF diet developed obesity and glucose intolerance,HF group developed hyperglycemia and hyperinsulinemia caused by insulin resistance
43. renalase expressed in beta cells, vulnerable to stress. renalase may protect beta cells against autoimmune attack, renalase probably increased the expression of Bcl-2.; renase is reduced in chronic kidney disease patients,with hypertension....Overexpression of bcl-2 protects epithelial cells from death. Bcl-2 did not alter the activation of T-helper cell 1 but inhibited the growth of T-helper cell 17
44. C-peptide is a byproduct to the body´s own production of insulin and is produced 1:1 to endogenous insulin (1). It can be measured in blood and urine and is often used as a diagnose tool, as well as following the progress of diabetes in an individual.
45. hypoglycemic actions of salicylates and identified the molecular target to be the I¦ĘB kinase-¦Â (IKK¦Â)/NF-¦ĘB axis. (same for dehydroascorbic acid )
46.VDR protects beta cells againest inflammation and apoptosis.
47. roles of liver on glucose homeostasis: glucose uptake, glycogen systhesis, glucagon production and a liver-beta cell axis, which is likely to be dependent on bile acids controlling beta cell secretion capacity
48. effect of chronic intermittent hypoxia (CIH): CIH leads to pancreatic beta cell dysfunction manifested by augmented basal insulin secretion, insulin resistance, defective proinsulin processing, impaired GSIS(glucose-stimulated-insulin-screction) and increased mitochondrial ROS
49. effect of visceral fat in liver and beta cells : "T2D is simply the result of too much fat overloading the liver and pancreas in people who happen to be susceptible to the fat-induced damage," The three-month initial weight loss stage is a liquid dietˇŞfour shakes a day, 200 calories each.ˇ± Taylor New castle u
50. TYPE 1 diabetes: type 1 diabetes is a T-cell mediated process marked by autoimmune destruction of ¦Â-cells,
ˇˇ
ˇˇ
https://www.sciencealert.com/new-breakthroughs-diabetes-research-treatments-2018
ˇˇ
The Pancreas | Boundless Anatomy and Physiology
The alpha cells produce glucagon, and make up 15¨C20% of total islet cells. The beta cells produce insulin and amylin, and make up 65¨C80% of the total islet cells. The delta cells produce somatostatin, and make up 3¨C10% of the total islet cells. The gamma cells produce pancreatic polypeptide, and make up 3¨C5% of the total islet cells.
ˇˇ
The pancreatic islets or islets of Langerhans are the regions of the pancreas that contain its endocrine (hormone-producing) cells, discovered in 1869 by German pathological anatomist Paul Langerhans. The pancreatic islets constitute 1¨C2% of the pancreas volume and receive 10¨C15% of its blood flow. The pancreatic islets are arranged in density routes throughout the human pancreas, and are important in the metabolism of glucose.
ˇˇ
islet microcirculation is open and not isolated from that of the surrounding exocrine tissue.
https://diabetes.diabetesjournals.org/content/69/7/1336
Structure of Islets and Vascular Relationship to the ...
The islets of Langerhans, which compromise nearly 2% of the total pancreatic volume, are highly vascularized structures within the pancreas, receiving approximately 10-fold the blood supply of the exocrine tissue by volume, with the intra-islet microcapillary network constituting 7-8% of the total islet volume (15, 27) (Figure 1).
ˇˇ
ˇˇ
https://www.thediabetescouncil.com/beta-cells-diabetes/
ˇˇ
Insulin has three disulfide bonds, two of which join the A-Chain and B-Chain of insulin together, and sufficiently high concentrations of GSH, such as those used in the previous assays, can reduce disulfide bonds.
http://www.pharmacology2000.com/Diabetes/diabetes6.htm
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3063182/
ˇˇ
Disulfide Bond - an overview | ScienceDirect Topics
www.sciencedirect.com/.../disulfide-bond
Disulfide-bond formation is a reversible process with numerous biological functions, including stabilization of protein fold, enzyme catalysis, and protection against oxidative damage. 85 The ability to form and break a disulfide-bond depends on the disulfide bond stability, the environmental redox state, and the nature of the oxidant and reductant.ˇˇ
Insulin has three disulfide bonds, two of which join the A-Chain and B-Chain of insulin together, and sufficiently high concentrations of GSH, such as those used in the previous assays, can reduce disulfide bonds.
http://sphweb.bumc.bu.edu/otlt/mph-modules/ph/ph709_basiccellbiology/PH709_BasicCellBIology7.html
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3063182/
Potential cellular mechanisms for activating inflammatory signaling.
Obesity and high-fat diet activate IKK¦Â/NF-¦ĘB and JNK pathways in adipocytes, hepatocytes, and associated macrophages. Stimuli that have been shown to activate these pathways during metabolic dysregulation include ligands for TNF-¦Á, IL-1, Toll, or AGE receptors (TNFR, IL-1R, TLR, or RAGE, respectively), intracellular stresses including ROS and ER stress, ceramide, and various PKC isoforms. Obesity-induced IKK¦Â activation leads to NF-¦ĘB translocation and the increased expression of numerous markers and potential mediators of inflammation that can cause insulin resistance. Obesity-induced JNK activation promotes the phosphorylation of IRS-1 at serine sites that negatively regulate normal signaling through the insulin receptor/IRS-1 axis. Examples include serine-302 (pS302) and serine-307 (pS307). By contrast, evidence has not been reported for obesity-induced effects on transcription factors such as AP-1 that are regulated by JNK. IKK¦Â and/or NF-¦ĘB are inhibited or repressed by the actions of salicylates, TZDs, and statins.Several drugs in current clinical practice have been shown to have antiinflammatory properties or side effects distinct from their major mechanisms of action, including members of the thiazolidinedione (TZD) class of PPAR¦Ă agonists and members of the statin class of HMG CoA reductase inhibitors. Both appear to have important antiinflammatory properties and potential benefits beyond their primary actions on glucose homeostasis and cholesterol lowering, respectively.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1483173/
depending on the stage of obesity, ROS can be generated by three distinct mechanisms: i.e., NOX4, NOX2, and mitochondria.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4995181/
ˇˇ
Local, portal, and systemic effects of inflammation in insulin resistance and atherogenesis.
Increasing adiposity activates inflammatory responses in fat and liver, with associated increases in the production of cytokines and chemokines. Immune cells including monocytes and macrophages are recruited and/or activated, and together these cause local insulin resistance. Portal delivery of abdominal fat¨Cderived cytokines and lipids contributes to hepatic inflammation and insulin resistance. Proinflammatory and proatherogenic mediators are produced in the adipose tissue and liver and associated immune cells. This creates a systemic inflammatory diathesis that promotes insulin resistance in skeletal muscle and other tissues and atherogenesis in the vasculature.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1483173/
ˇˇ
http://lllnutrition.com/mod_lll/TOPIC18/old/m182e.htm
ˇˇ
301 Moved Permanently
studyblue.com
ˇˇ
While both JNK and IKK¦Â/NF-¦ĘB play important roles in inflammation-induced insulin resistance, accumulated evidence suggests that they do so through dissimilar mechanisms. JNK is a stress kinase that normally phosphorylates the c-Jun component of the AP-1 transcription factor, but to date there are no known links between this well-established transcriptional pathway and JNK-induced insulin resistance. Instead, JNK has been shown to promote insulin resistance through the phosphorylation of serine residues in IRS-1 (41, 42, 51, 57, 58) (Figure (Figure1).1). Insulin receptor signaling that normally occurs through a tyrosine kinase cascade is inhibited by counterregulatory serine/threonine phosphorylations (59).
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1483173/
ˇˇ
ˇˇ
ˇˇ
ˇˇ
Recombinant Insulin Human, Structure of human pro-insulin, 82-polypeptide hormone
ˇˇ
ˇˇ
ˇˇ
https://biology-forums.com/index.php?action=gallery;sa=view;id=10856
ˇˇ
Redox Regulation of Insulin Degradation by Insulin ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3063182
(A) Insulin degradation. (B) Amyloid ¦Â degradation. Background TCA solubility in the absence of enzyme was subtracted. Treatment with GSH after heat inactivation of the enzyme increases TCA solubility of insulin products by breaking disulfides. Amyloid-¦Â, not having any disulfide bonds, is unaffected either before or after enzyme inactivation.ˇˇ
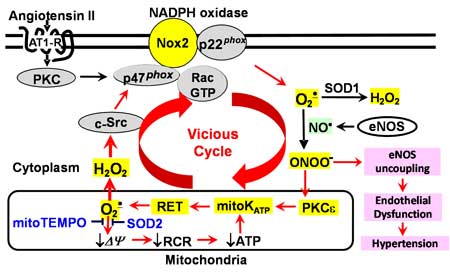
Scheme 1. Cross-talk between NADPH oxidase and mitochondria in endothelial dysfunction and hypertension.
stimulation of endothelial cells with angiotensin II increases production of mitochondrial O2ˇ¤.
http://www.mc.vanderbilt.edu/labs/dikalovlab/Research.html
ˇˇ
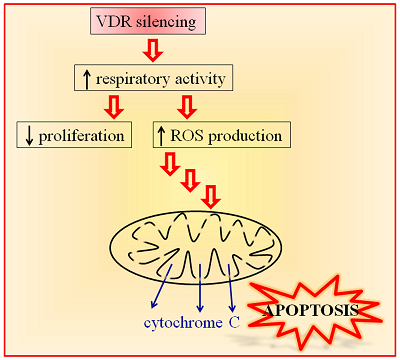
Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health
Vitamin D receptor (VDR) mediates many genomic and non-genomic effects of vitamin D. Recently, the mitochondrial effects of vitamin D have been characterized in many cell types. In this article, we investigated the importance of VDR not only in mitochondrial activity and integrity but also in cell health. The silencing of the receptor in different healthy, non-transformed, and cancer cells initially decreased cell growth and modulated the cell cycle. We demonstrated that, in silenced cells, the increased respiratory activity was associated with elevated reactive oxygen species (ROS) production. In the long run, the absence of the receptor caused impairment of mitochondrial integrity and, finally, cell death. Our data reveal that VDR plays a central role in protecting cells from excessive respiration and production of ROS that leads to cell damage. Because we confirmed our observations in different models of both normal and cancer cells, we conclude that VDR is essential for the health of human tissues.
https://www.mdpi.com/1422-0067/19/6/1672
ˇˇ
ˇˇ
ˇˇ
Beta-cell development and regeneration
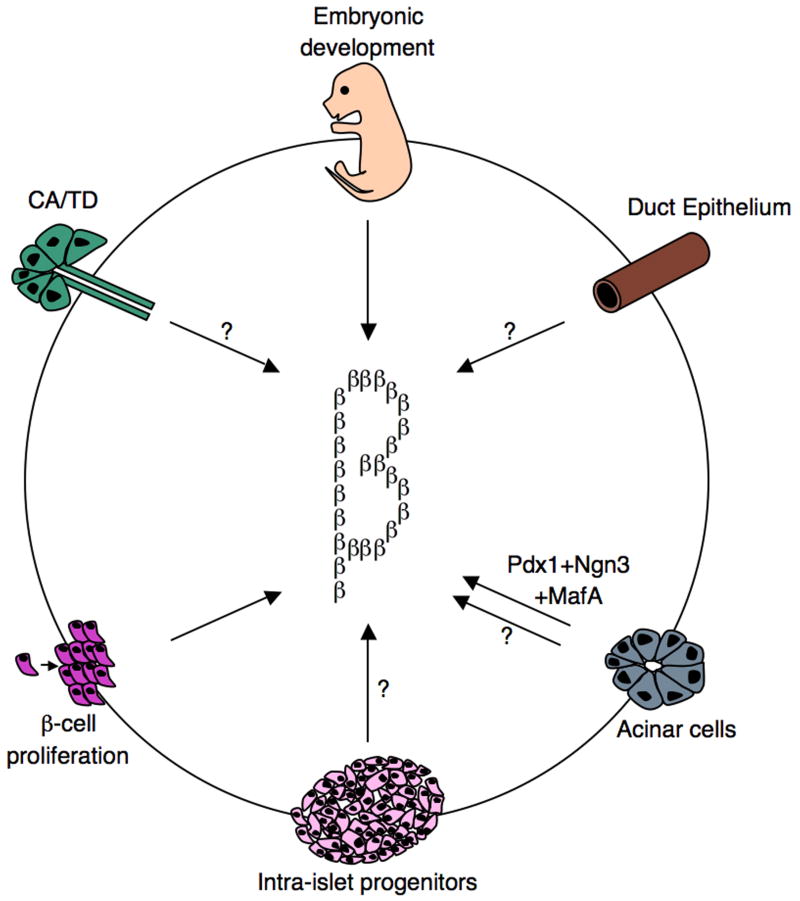
Schematics representing various roadmaps leading to the generation of functional insulin-producing cells. During embryonic development, beta-cells are generated from Ngn3+ progenitors. Moreover, beta-cells have the capacity to undergo self-replication to expand the beta-cell mass. This occurs during development, pregnancy, and following injury. Findings from several independent studies, using various pancreas injury models, are consistent with the idea that stem/progenitor cells do exist in the adult pancreas. Several sources of facultative stem/progenitor cells were suggested and are depicted: duct epithelium, acinar cells, centroacinar (CA)/terminal duct (TD), and intra-islet progenitors. Glucagon+, Pdx1+, and Ngn3+ were often found in the duct lining, suggesting that this may be a site where stem/progenitor cells at least transiently reside.
Semin Cell Dev BiolŁ¬ 2011 Oct 1.
Pancreatic beta-cells: from generation to regeneration
Patrick Collombat,1,2 Xiaobo Xu,2,3,4 Harry Heimberg,2,3,4,# and Ahmed Mansouri2,5,6,#
1Inserm U636, Diabetes Genetics team, Universit¨¦ de Nice Sophia-Antipolis, FR-06108 Nice, France
2Beta-cell Biology Consortium, 2213 Garland Avenue, 9465 MRB IV, Nashville, TN 37323-0494, USA
3JDRF Center for Beta-cell Therapy in Diabetes, Laarbeeklaan 103, B-1090 Brussels, Belgium
4Diabetes Research Center, Vrije Universiteit Brussel, Laarbeeklaan 103, B-1090 Brussels, Belgium
5Max-Planck Institute for Biophysical Chemistry, Department of Molecular Cell Biology, Am Fassberg, D-37077 Göttingen, Germany
6Department of Clinical Neurophysiology, University of Göttingen, Robert-Koch Strasse 40, D-37075 Göttingen, Germany
Summary
The pancreas is composed of two main compartments consisting of endocrine and exocrine tissues. The majority of the organ is exocrine and responsible for the synthesis of digestive enzymes and for their transport via an intricate ductal system into the duodenum. The endocrine tissue represents less than 2% of the organ and is organized into functional units called islets of Langerhans, comprising alpha-, beta-, delta-, epsilon- and PP¨Ccells, producing the hormones glucagon, insulin, somatostatin, ghrelin and pancreatic polypeptide (PP), respectively. Insulin-producing beta-cells play a central role in the control of the glucose homeostasis. Accordingly, absolute or relative deficiency in beta-cells may ultimately lead to type 1 and/or type 2 diabetes, respectively. One major goal of diabetes research is therefore to understand the molecular mechanisms controlling the development of beta-cells during pancreas morphogenesis, but also those underlying the regeneration of adult injured pancreas, and assess their significance for future cell-based therapy. In this review, we will therefore present new insights into beta-cell development with focus on beta-cell regeneration.
Keywords: pancreas, stem cells, regeneration, mouse, diabetes
Go to:
Introduction
Although insulin supplementation allows a reasonable control of blood sugar levels, diabetic patients still suffer from long-term side effects of blood glucose variations, too often resulting in severe alterations of various organ functions. Transplantation of islets isolated from the pancreases of organ donors alleviates insulin-dependence in type 1 diabetes patients, providing strong support to stem cell-based therapy. It is obvious that, due to the islet shortage to suffice the need for transplantation, alternative cell sources have to be explored. At first glance, human embryonic stem (h-ES) cells and induced pluripotent stem cells (iPS) potentially may represent an unlimited source for generating beta-cells in vitro. However, although several straightforward protocols were established, including procedures using chemical compounds, both the efficiency of in vitro programming and the function of h-ES-derived beta-cells remain unsatisfactory. Besides, safety concerns due to inherent risks of teratoma formation originating from residual stem cells remain a major hurdle [1-3].
It is now accepted that in various experimental animal models of injury to the (endocrine) pancreas, such as partial pancreatectomy (PPX), partial duct ligation (PDL), and chemically-or genetically induced beta-cell destruction, [4-10], islet cells actively regenerate. It is therefore of fundamental interest to consider the molecular mechanisms that control such regenerative programs to putatively open new avenues for an improved treatment of diabetes. The expansion of the beta-cell mass during early postnatal life, pregnancy, as well as in the pancreas of animal models for which beta-cells were genetically ablated, has been mainly attributed to beta-cell proliferation [7, 11, 12]. Despite these findings, it was demonstrated that beta-cells may also arise from alternative cell subtypes, such as duct-lining or acinar cells [13], as observed during embryonic development. Indeed, several studies in various animal and transgenic models support a process of duct-lining cell-derived beta-cell neogenesis [4, 14-22]. However, as we will discuss thereafter, while replication of preexisting beta-cells has been established as a major component of the expansion of the beta-cell mass in normal and injured pancreas, the existence of duct-derived progenitor cells is still controversially discussed [23-25]. Finally, intra-islet precursor cells promoting beta-cell neogenesis have been also suggested [26-28].
Go to:
Beta-cell development
The first obvious morphological signs of pancreas development appear at approximately 8.5 days post-coitum, as two protusions at the dorsal and ventral portion of the foregut/midgut junction [29-32]. The specification of this endodermal region towards a pancreatic fate is achieved through the concerted interplay of different signaling pathways emanating from the adjacent mesoderm [32-34]. During subsequent pancreas histogenesis, both the dorsal and the ventral buds develop under the control of distinct signaling machineries, their growth and branching being regulated by the surrounding mesenchyme [32, 35-37]. In the mouse, the ventral pancreatic bud rotates and eventually fuses with its dorsal counterpart between E17 and E18 of embryonic development [32].
During pancreas morphogenesis, the first hormone-expressing cells that are detected contain glucagon and appear at E9.5, some of which initiating insulin expression a day later [29, 38]. However, these few early scattered cells that produce both insulin and glucagon will not contribute to the mature endocrine pancreas [39]. Endocrine and exocrine cell proliferation peaks at the so-called secondary transition starting at about E13.5 [29]. Multipotent progenitor cells located at the distal tip of the growing epithelium are characterized by the expression of Ptf1a, c-myc, and carboxypeptidase a1 (cpa1) [40]. Lineage tracing experiments demonstrated that cpa1-positive cells delaminate from the epithelium to generate all pancreatic cell types, including Ngn3-marked cells that will adopt an endocrine cell fate. Accordingly, in the absence of Ngn3, endocrine cells fail to develop [41]. It is worth noticing that Ngn3-labeled endocrine progenitors are subjected to a window of competence for the generation of the different endocrine hormone-producing cell subtypes [42]. At about E14.5, cpa1-labeled progenitors are restricted towards the acinar lineage [40]. Concurrently, a significant number of maturing insulin- or glucagon-labeled endocrine and amylase-labeled acinar cells accumulate. At E15.5, the first somatostatin-expressing delta-cells appear. Lastly, shortly before birth, PP-labeled cells emerge and aggregation of endocrine cells to form mature islets of Langerhans is initiated [29].
The use of gain- and loss-of-function mutant mice as a tool to study gene function has allowed to gain further insights into the crucial role exerted by transcription factors in the processes underlying endocrine cell specification (excellently reviewed in [30-32, 36, 43]). However, gaining further insights into the molecular mechanisms controlling normal beta-cell development, as well as their replenishment in animal models of beta-cell injury, is of fundamental interest for the generation of insulin-producing cells from progenitor or embryonic stem cells.
Go to:
Expansion of the beta-cell mass through self-replication
Glucose-mediated insulin secretion is required for the proper control of glucose homeostasis. Interestingly, during pregnancy [11] or in obese individuals [44], a beta-cell mass expansion was outlined to compensate for the increased needs. Although several molecular mechanisms promoting adaptation of the functional beta-cell mass are under discussion, self-duplication of preexisting beta-cells unquestionably represent an important process to increase islet size. Accordingly, Cre recombinase-mediated lineage tracing analyses provided compelling evidences for beta-cell replication as the main source of beta-cell neogenesis under normal physiological conditions, but also following 70% PPX [45]. Another in vivo pulse-chase study, supplemented by a clonal analysis of dividing beta-cells, supported a model where insulin-expressing cells equally participate to the replication-mediated expansion of the beta-cell mass [12]. In addition, using a novel DNA analog-based lineage tracing approach, it was found that only beta-cells contribute to the beta-cell regeneration occurring under normal physiological conditions, following 50% PPX or treatment with the GLP-1 agonist Exendin-4, and during pregnancy, underscoring the importance of replication to sustain the beta-cell turn-over [46]. These findings were confirmed by studies in transgenic mice with beta-cell depletion induced by tetracycline-controlled diphtheria toxin expression or by c-myc activated overexpression leading to beta-cell apoptosis [7, 47].
In rodents, beta-cell replication appears to follow an age-dependent process for which the beta-cell mass expansion observed next to pancreas injury seems limited to young animals [48, 49]. Accordingly, the proliferation rate of insulin-producing cells was found extremely reduced in one-year old mice [49], with a Ki67 proliferation index significantly decreased in islets of 94-week old animals as compared to their 20-week old counterparts [50]. Furthermore, a clear decline in islet proliferation capability upon 90% PPX was also associated with age in rats [51].
Beta-cell replication obviously requires the activity of cell cycle regulators (for review see [52-54]). Accordingly, cyclin D2-deficient mice exhibit smaller islets, a dramatically reduced beta-cell mass, and compromised beta-cell proliferation capabilities [55, 56]. While the lack of Cdk4 activity results in diabetes, the expression of a constitutively active form of Cdk4 is accompanied by beta-cell hyperplasia [57-59]. Moreover, virus-mediated overexpression of Cdk4 in human islets also induces beta-cell proliferation [52, 58, 60]. Similarly, the adenovirus-mediated ectopic expression of E2F1, together with protein kinase B (Akt), in primary beta-cells, results in an increase in the absolute cell number provoked by an induction of proliferation and a concomitant inhibition of cell death [61]. On the other hand, the in vivo conditional overexpression of E2F1 in beta-cells stimulates their proliferation, but not sufficiently enough to increase the beta-cell mass. However, increased insulin contents and glucose-mediated insulin release were noticed and associated to protection against STZ (streptozotocin)-induced diabetes [61].
The diminished proliferation ability in aging mice was recently correlated with an increased expression of the cell cycle inhibitor p16INK4a in older animals [62-64]. Accordingly, the loss of p16INK4a activity in mutant mice results in improved regeneration capabilities in islets of older animals following chemical ablation of beta-cells using streptozotocin (STZ), while the overexpression of p16INK4a is accompanied by diminished islet proliferation [62]. Moreover, the propensity of beta-cells to multiply in younger animals strongly correlates with the expression levels of Bmi1 and Ezh2, two polycomb proteins that control the Ink4a/Arf locus through modulation of histone modifications [65, 66]. Hence, Bmi-1-deficient young mice display increased expression of p16INK4a, and thereby lose their capacity to expand their beta-cell mass in response to exendin-4 [63]. Similarly, in the absence of Ezh2, insulin-producing beta-cells exhibit reduced proliferation rates and mutant mice suffer from mild diabetes [66]. Interestingly, MLL, a member of trithorax TrxG protein family, was found associated to the activation of the Ink4a/Arf locus [65]. Menin, a factor encoded by the men1 locus, recruits MLL to the p27kip1 and p18Ink4c promoters [67] and thereby modulates pancreatic islet growth by inducing histone methylation and activating the transcription of cell cycle inhibitors [68]. These findings underscore the role of epigenetic control of the beta-cell mass expansion during life span. Finally, it is worth noticing that other signaling pathways and factors, such as STAT5, growth hormones, prolactin and FoxM1 also play a role in beta-cell proliferation during pregnancy [11, 30, 65].
Go to:
Do facultative stem cells exist in the pancreas?
Recent studies indicate that adult pancreatic cells are more ˇ°plasticˇ± than hitherto assumed, and provide evidence for islet neogenesis in several animal models. One attractive hypothesis favors injury-induced activation of facultative stem/progenitor cells to expand the beta-cell mass [4, 14, 69, 70]. Differentiation of facultative stem/progenitor cells might involve recapitulation of the program that controls the embryogenesis of the endocrine pancreas, including re-expression of the proendocrine factor Ngn3. As a common denominator to these reports, the ductal compartment seemingly represents the site where stem/progenitor cells at least transiently reside [4, 16-22]. Robust injury using partial pancreatic duct ligation created the appropriate microenvironment to unambiguously demonstrate the existence of multipotent endocrine progenitors in the adult mouse pancreas [19]. In this model, at least part of the embryonic endocrine program is reinitiated leading to reactivation of Ngn3 in a subset of Pdx1+ cells lining the duct. Ngn3-positive cells isolated from adult duct-ligated pancreata gave rise to the four main endocrine cell subtypes when implanted in pancreata of embryonic mice that were null mutant for Ngn3 and thus incapable of producing endogenous endocrine cells. Remarkably, the Ngn3-/- embryonic pancreata explants engrafted with adult Ngn3+ cells were glucose responsive and the newly formed beta-cells intensely proliferated [19]. Lineage-tracing experiments using the human carbon anhydrase II (CAII) promoter to drive the expression of cre recombinase and follow the progeny of pancreatic duct cells following birth or partial duct ligation showed that CAII cells can give rise to both endocrine and exocrine cells [20].
In yet another model, conditional expression of Pax4 allowed the transcription factor to be ectopically present in alpha-cells and initiate their conversion into functional beta-cells [21]. The ensuing glucagon shortage induced compensatory neogenesis of glucagon-producing cells. Along the same line of evidence, deficiency/alterations in glucagon signaling in glucagon receptor knockout or prohormone convertase-deficient mice were previously found to also trigger alpha-cell hyperplasia [71, 72]. Upon Pax4 misexpression, alpha-cells consequently adopted a beta-cell phenotype leading to oversized islets mainly comprised of insulin-producing cells [21]. Importantly, in transgenic mice misexpressing Pax4 in alpha-cells, a progressive normalization of the glycemia was observed in mice that underwent chemically-induced diabetes. The regenerated alpha-cells noted in Pax4 transgenic mice were found to originate from the reactivation of Ngn3, but not Pdx1, in the ductal lining. Moreover, knockdown experiments using Ngn3-specific interfering RNA demonstrated the requirement of Ngn3 re-expression for endocrine cell neogenesis in Pax4 transgenic mice [21].
Recently, mice expressing a constitutively active form of Cdk4 (Cdk4R24C) displaying beta-cell hyperplasia (see also above), were found to exhibit increased proliferation rates of beta-cells, but also of ductal cells, following 60% pancreatectomy [22]. Duct cells in injured pancreas of both Cdk4wt and Cdk4R24C mice contained Pdx1+ cells and were able to express insulin. In this study, however, no Ngn3+ cells were observed prior to or following pancreatectomy [22]. In contrast, wild type mice and FoxM1-deficient animals exhibit Ngn3 re-expression in the duct epithelium after 60% pancreatectomy [73]. This discrepancy may be due to the technical difficulties encountered using immunohistochemical detection of Ngn3 expression in the adult tissue. In the adult injured pancreas, the duct epithelium often contains insulin-, glucagon- or Glut-2-expressing cells, suggesting islet neogenesis [15, 74-76]. Finally, adult transgenic mice expressing the human diphtheria toxin receptor under the control of the insulin promoter to induce global beta-cell ablation by diphtheria toxin treatment were also found to undergo beta-cell regeneration through spontaneous conversion of alpha-cells [8].
Together these findings provide evidence that, besides beta-cell replication, additional mechanisms of islet regeneration operate in the adult pancreas, some involving facultative stem cells. The mechanism underlying such beta-cell neogenesis appears to depend on the extent and/or the method of beta-cell injury. The ductal origin of endocrine cell formation in adult injured pancreas still remains controversial. In contrast to the clear contribution of carbonic anhydrase II-positive cells to endocrine and exocrine cell neogenesis following birth and PDL [20], such ductal origin was not noticed for Hnf1b-marked cells following PDL- or alloxan/EGF/gastrin-induced injuries [23, 25]. Furthermore, no contribution of acinar and of duct cells to endocrine cell genesis in the early postnatal period was observed by conditional lineage tracing of Muc1+ cells [24]. It remains to be determined whether, in the injured pancreas, Muc1-labeled cells are involved in beta-cell regeneration.
It needs to be mentioned that cre recombinase-mediated lineage tracing strongly depends on the efficiency of recombination and that never all duct cells are labeled, increasing the chance to overlook rare stem cells residing in the duct epithelium or acinar cell compartment (see also [77]). Ngn3+ cells were clearly detected in the duct of mice subjected to PDL, or with ectopic Pax4 expression in alpha-cells [19, 21].
Besides the ductal lining, intra-islet precursor cells as well as acinar cells were suggested to contribute to beta-cell neogenesis [18, 26, 28, 70, 78, 79]. In mice treated with STZ and kept normoglycemic using exogenous insulin (STZ/IN), as well as in aging animals, two seemingly distinct beta-cell precursors were detected in islets [70]. These cells were characterized by the expression of Glut-2 and Pdx1/somatostatin, respectively. In RIPcreER and Z/AP reporter mice [45] that label insulin+ cells by placental alkaline phosphatase (PLAP) and allow identification of putative precursor cells, the islets of aging and STZ/IN-treated mice contained beta-cell precursors expressing either Pdx1 or Mafb [28]. These findings contrast with the beta-cell replication described earlier [45], and were attributed to differences in immunhistochemical techniques [28]. However, the source of these putative precursor cells remains unclear and, as the authors pointed out, they may also have a ductal origin. Interestingly, Glut-2-positive cells were also detected in the PANIC-ATTAC mouse model characterized by beta-cell injury provoked by the activation of caspase 8-mediated apoptosis, as well as in the pancreatic duct of PDL-treated rats [15, 75].
It is worth noticing that the Mafb+ cells, probably representing alpha-cells, detected in the islets of STZ/IN-treated or aging mice [28], are reminiscent of glucagon+ cells that spontaneously convert to beta-cells, following diphtheria toxin-mediated beta-cell depletion [8]. In both studies mice were treated with insulin to counter hyperglycemia and allow survival [8, 28]. It is conceivable that the Mafb- or glucagon-marked cells contributing to beta-cell replenishment in these mice may derive from the duct epithelium, not excluding that intra-islet precursor cells may exist as well. Duct-derived endocrine cell neogenesis requires the reactivation of Ngn3 in the duct epithelium following pancreatic duct ligation or forced expression of Pax4 in alpha-cells [19, 21]. Hence, duct-derived progenitors that differentiate into hormone producing cells may migrate in order to expand that beta-cell pool or, alternatively, form new islets adjacent to the duct epithelium.
Go to:
Reprogramming acinar cells into beta-cells
As the most abundant cell type in pancreas, acinar cells are considered as a rich supply for generating beta-cells [80]. Culturing the acinar cancer cell line AR42J in the presence of betacellulin, activin or glucagon-like peptide, was found to induce insulin or glucagon production [81-83]. Dexamethasone treatment of duct-ligated rat pancreas provoked acinoductal transdifferentiation and revealed an intermediate cell type coexpressing both acinar and duct markers [84]. The currently most successful approach consists in supplementing rat acinar cell culture with the cytokines epidermal growth factor (EGF) and leukemia inhibitory factor (LIF) to generate functional beta-cells that normalized hyperglycemia in immune-incompetent diabetic mice [85]. This transdifferentiation process was found to pass through an intermediary cell type that expresses both duct- and beta-cell markers [85], such results being confirmed by non-genetic lineage tracing [86]. Acinar- to beta-cell reprogramming uses Notch signaling as gatekeeper [86] and requires both Ngn3 expression and signaling through the JAK/STAT pathway [87]. The notion of acinar- to beta-cell differentiation was further supported by genetic lineage tracing in suspension cultures of adult pancreatic exocrine cells isolated from transgenic mice expressing the ROSA26-eCFP, infected with recombinant adenoviruses expressing Cre under the control of the promoter of either amylase-2 or elastase-1 in cell cultures supplemented with EGF and nicotinamide [88]. Furthermore, in vitro culture of pancreatic explants isolated from transgenic mice expressing TGF-alpha under the control of the metallothionein promoter, revealed that acinar-to-duct transdifferentiation occurs through a dedifferentiated nestin-positive intermediate, in an EGFR-dependent manner [89]. It has been speculated that EGF activation in pancreatic exocrine cell cultures could be triggered by cell dissociation and that exogenous EGF might enhance cell survival [88].
In addition, transgenic mice expressing IFN-gamma under the control of the insulin promoter countered STZ-mediated beta-cell depletion through the budding of newly formed islets from ducts, with acinar cells as putative precursors [90]. In contrast, during regeneration of the mouse exocrine pancreas after caerulein-induced pancreatitis, acinar cell dedifferentiation was observed without further redifferentiation into duct cells [91]. Replenishment of the exocrine tissue in caerulein-mediated pancreatitis depended on the Notch signaling pathway and was mediated by repression of beta-catenin signaling pathway [92]. In transgenic mice expressing the tamoxifen-inducible Cre recombinase (CreERT2) under the control of the acinar-specific promoter elastase I, and crossed with Rosa26LacZ mice, lineage tracing was performed in three models with pancreas injury, including PPX, PDL and caerulein-induced pancreatitis, but no evidence for the contribution of acinar cells to islet neogenesis was found [93]. Following PPX, self-replication of preexisting acinar cells was the predominant mechanism involved in regeneration of the newly formed acinar tissue [94]. This indicates that the capacity of adult acinar cells to transdifferentiate into endocrine cells in vivo is still a matter of debate und that more challenging studies are needed. On the other hand, the forced co-expression of Pdx1, Ngn3 and Mafa in acinar cells promoted the formation of insulin-producing beta-cells in vivo by direct conversion rather than dedifferentiation [95]. The reprogramming of acinar cells by the forced expression of transcription factors suggests that the transdifferentiation of acinar to endocrine cells is under the control of a repressive mechanism, like Notch signaling, in the normal pancreas. Accordingly, inhibition of Notch1 signaling results in a more efficient acinar- to beta-cell conversion [86].
Finally, the centroacinar and terminal duct cells of the pancreas are not well defined and their molecular characteristics poorly established [96, 97]. Only few studies have suggested that these cells have the capacity to actively proliferate in different injury models, including PPX, or treatment with caerulein or STZ [18, 98, 99]. However, it is not clear whether the centroacinar and terminal duct (CA/TD) cells consist of distinct cell types or are functionally equivalent. Recently, these cells were characterized by immunhistochemistry and were found to display high levels of ALDH1 enzymatic activity [100] enabling their isolation by FACS. Isolated CA/TD cells are able to differentiate into endocrine and exocrine cell types in vitro. Following caerulein administration to induce pancreatitis, ALDH1+ CA/TD cells were found expanded, as compared to controls, suggesting that CA/TD cells possess progenitor cell characteristics [100]. Along the same line, lineage tracing of Bmi1+ cells identified a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis [101]. Further studies are needed to examine whether CA/TD cells are able to contribute to the endocrine cell compartment, as previously suggested [18, 99].
Go to:
Conclusions
Beta-cell proliferation clearly appears as the fundamental mechanism involved for beta-cell turn-over. The induction of beta-cells in vitro to generate sufficient numbers of cells for transplantation would be an interesting alternative if at least the expanded beta-cell mass would remain glucose responsive. A similar approach in vivo should carefully take the risk of tumor formation, such as insulinomas, into account.
Islet transplantation demonstrated that stem cell-based therapy could represent a realistic option for the treatment of diabetes. Hence, the current islet shortage may be compensated in the future by the generation of insulin-producing beta-cells from pancreatic non-beta-cells, beta-cell progenitors or embryonic stem cells. Besides embryonic stem cells, it is now well accepted that the injured adult pancreas has the capacity to regenerate new beta-cells: several independent studies in various animal models of beta-cell injury provided strong evidences for the existence of facultative stem cells that are able to give rise to functional beta-cells. The source of such cells still is subject of controversial discussion. A molecular analysis of the current models is required to identify the factors implicated in the activation of such cells. Gaining further insights into the molecular mechanisms underlying regeneration processes may disclose the different cell sources that are implicated in endocrine cell neogenesis. It should be outlined that, depending on the type of pancreatic injury, different progenitor cells might be activated. In this context, it is of high interest to define how the application of insulin to streptozotocin-treated mice may activate the regeneration of significantly more beta-cells, as compared to streptozotocin-treated mice that remain hyperglycemic [28, 70]. One possible explanation is that high glucose levels in hyperglycemic animals may perturb or inhibit islet neogenesis and beta-cell regeneration.
Nevertheless, the hunting for tools that activate stem cells in vivo, or induce the transdifferentiation of non-beta hormone-producing cells, such as alpha-cells, into functional beta-cells, is now open. Future efforts should focus on the identification of markers for facultative stem cells in the injured pancreas and examine whether such molecules also do exist in the human pancreas.
An external file that holds a picture, illustration, etc.
Object name is nihms231790f1.jpg
Open in a separate window
Beta-cell development and regeneration
Schematics representing various roadmaps leading to the generation of functional insulin-producing cells. During embryonic development, beta-cells are generated from Ngn3+ progenitors. Moreover, beta-cells have the capacity to undergo self-replication to expand the beta-cell mass. This occurs during development, pregnancy, and following injury. Findings from several independent studies, using various pancreas injury models, are consistent with the idea that stem/progenitor cells do exist in the adult pancreas. Several sources of facultative stem/progenitor cells were suggested and are depicted: duct epithelium, acinar cells, centroacinar (CA)/terminal duct (TD), and intra-islet progenitors. Glucagon+, Pdx1+, and Ngn3+ were often found in the duct lining, suggesting that this may be a site where stem/progenitor cells at least transiently reside.ˇˇ
Front. Endocrinol., 20 February 2019 |
Endogenous Pancreatic ¦Â Cell Regeneration: A Potential Strategy for the Recovery of ¦Â Cell Deficiency in Diabetes
Fan Zhong1,2† and Yan Jiang2*†
1Department of Gastroenterology, Songjiang Hospital Affiliated First People's Hospital, Shanghai Jiao Tong University, Shanghai, China
2Institutes of Biomedical Sciences of Shanghai Medical College, Fudan University, Shanghai, China
Endogenous pancreatic ¦Â cell regeneration is a potential strategy for ¦Â cell expansion or neogenesis to treat diabetes. Regeneration can occur through stimulation of existing ¦Â cell replication or conversion of other pancreatic cells into ¦Â cells. Recently, various strategies and approaches for stimulation of endogenous ¦Â cell regeneration have been evaluated, but they were not suitable for clinical application. In this paper, we comprehensively review these strategies, and further discuss various factors involved in regulation of ¦Â cell regeneration under physiological or pathological conditions, such as mediators, transcription factors, signaling pathways, and potential pharmaceutical drugs. Furthermore, we discuss possible reasons for the failure of regenerative medicines in clinical trials, and possible strategies for improving ¦Â cell regeneration. As ¦Â cell heterogeneity and plasticity determines their function and environmental adaptability, we focus on ¦Â cell subtype markers and discuss the importance of research evaluating the characteristics of new ¦Â cells. In addition, based on the autoimmunologic features of type 1 diabetes, NOD/Lt-SCID-IL2rgnull (NSG) mice grafted with human immune cells and ¦Â cells are recommended for use in evaluation of antidiabetic regenerative medicines. This review will further understand current advances in endogenous ¦Â cell regeneration, and provide potential new strategies for the treatment of diabetes focused on cell therapy.Replication of Existing Pancreatic ¦Â Cells
Pancreatic ¦Â cells replicate readily in the fetal and neonatal stages. However, this ability to replicate rapidly declines after these stages. Furthermore, this ability to replicate is different in rodents and humans. Proliferation of ¦Â cells is precisely controlled by cell cycle regulators and circulating soluble factors. Studies have shown that many mitogenic agents could stimulate ¦Â cell replication in young rodents, but not in humans. However, using high-throughput chemical screening, a series of inhibitors of DYRK1A-NFAT, GSK3, and NF-¦ĘB signaling pathways were shown to increase human pancreatic ¦Â cell replication, suggesting that these inhibitors have unique potential for treatment of diabetes.Frontiers | Endogenous Pancreatic ¦Â Cell Regeneration: A Potential Strategy for the Recovery of ¦Â Cell Deficiency in Diabetes | Endocrinology
https://www.frontiersin.org/articles/10.3389/fendo.2019.00101/fullˇˇ
GABAergic System in ¦Â-Cells: From Autoimmunity Target to Regeneration Tool
Paolo Fiorina
-Author Affiliations
Nephrology Division, Boston ChildrenˇŻs Hospital, Harvard Medical School, Boston, Massachusetts
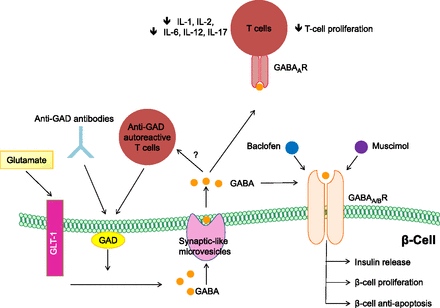
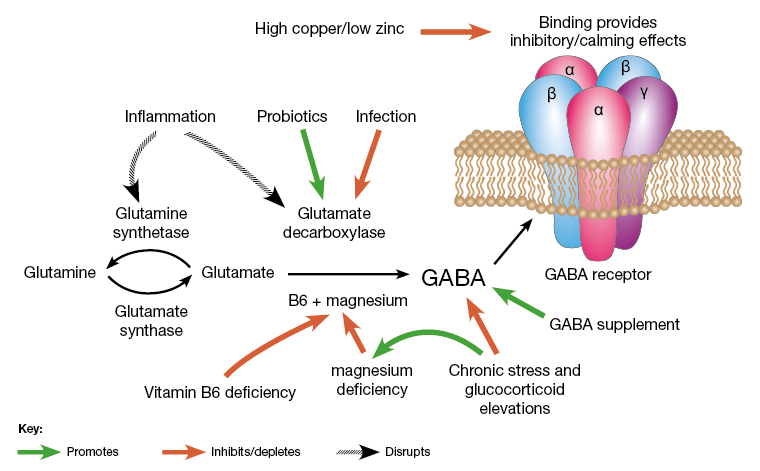
Department of Medicine, San Raffaele Hospital, Milan, Italy.The ¦Ă-aminobutyric acid (GABA) is a product of decarboxylation of the amino acid glutamate mediated by the synthesizing enzyme glutamic acid decarboxylase (GAD) (1¨C3). Although GABA is a major inhibitory neurotransmitter of the brain, it is produced at high levels in pancreatic islets (4). ¦Â-Cells store GABA in synaptic-like microvesicles, and upon its secretion, GABA exerts many paracrine functions in pancreatic islets (4). While the total function of GABA in ¦Â-cells is incompletely understood (4), its synthesizing enzyme GAD is possibly one of the most significant pancreatic islet ¦Â-cell autoantigens (5). GAD is a primary target of autoantibodies, and anti-GAD antibodies are associated with the development of type 1 diabetes (T1D) (5).
FIG. 1.
Regenerative and immunological abilities of the inhibitory neurotransmitter GABA. Extracellular glutamate, the precursor of GABA, enters ¦Â-cells through the glutamate transporter-1 (GLT-1), where it is converted to GABA by the enzyme GAD and is then stored in synaptic-like microvesicles. GABA signals through the GABABR expressed by ¦Â-cells, thus increasing insulin release, protecting ¦Â-cells from STZ-induced apoptosis, and stimulating ¦Â-cell proliferation. Baclofen and muscimol, both agonists of the GABABR, have the same effects. T cells are sensitive to GABA due to the presence of GABAARs on their cell surface. GABA exerts an immunomodulatory effect on T cells by decreasing the production of inflammatory cytokines (IL-1, IL-2, IL-6, IL-12, and IL-17) and inhibiting or reducing T-cell proliferation. Of note, GAD is targeted by autoreactive T cells and specific anti-GAD antibodies.https://diabetes.diabetesjournals.org/content/62/11/3674
ˇˇ
GABA: THE FORGOTTEN AMINO ACID
The results of the study were astonishing. After 12 weeks the group who used the whey and GABA had more than double the resting plasma growth hormone levels at week 4, 8, and 12 of the study when compared to their individual baseline values taken at the beginning as a baseline. By comparison, the ˇ°whey onlyˇ± group showed only an increase in resting plasma growth hormone levels at week 8 of the study. In addition, the group who used whey and GABA showed an increase in lean mass of 1,340g whereas the control group only gained 146g.
http://www.ironmagazine.com/2016/gaba-the-forgotten-amino-acid/
ˇˇ
J Surg Res. Author manuscript; available in PMC 2019 Apr 1.
GABA, ¦Ă-Aminobutyric Acid, Protects Against Severe Liver Injury
Dr Toshiyuki Hata, MD, Dr Fatima Rehman, PhD, Dr Tomohide Hori, MD, and Dr Justin H. Nguyen, MD
Dr Toshiyuki Hata, Department of Hepatobiliary-pancreatic and Transplant Surgery, Kyoto University Graduate School of Medicine, Kyoto, Japan;Background:
Acute liver failure (ALF) from severe acute liver injury is a critical condition associated with high mortality. The purpose of this study was to investigate the impact of preemptive administration of ¦Ă-aminobutyric acid (GABA) on hepatic injury and survival outcomes in mice with experimentally induced ALF.
Materials and Methods:
To induce ALF, C57BL/6NHsd mice were administered GABA, saline, or nothing for 7 days, followed by intraperitoneal administration of 500 ¦Ěg of tumor necrosis factor ¦Á and 20 mg of D-galactosamine. The study mice were humanely euthanized 4 to 5 hours after ALF was induced or observed for survival. Proteins present in the blood samples and liver tissue from the euthanized mice were analyzed using Western blot and immunohistochemical and histopathologic analyses. For inhibition studies, we administered the STAT3-specific inhibitor, NSC74859, 90 minutes before ALF induction.
Results:
We found that GABA-treated mice had substantial attenuation of TUNEL-positive hepatocytes and hepatocellular necrosis, decreased caspase-3, H2AX, and p38 MAPK protein levels; and increased expressions of Jak2, STAT3, Bcl-2, and Mn-SOD, with improved mitochondrial integrity. The reduced apoptotic proteins led to a significantly prolonged survival after ALF induction in GABA-treated mice. The STAT3-specific inhibitor NSC74859 eliminated the survival advantage in GABA-treated mice with ALF, indicating the involvement of the STAT3 pathway in GABA-induced reduction in apoptosis.
Conclusions:
Our results showed that preemptive treatment with GABA protected against severe acute liver injury in mice via GABA-mediated STAT3 signaling. Preemptive administration of GABA may be a useful approach to optimize marginal donor livers before transplantation.
ˇˇ
GABA (ƴ-aminobutyric acid) protects against liver injury via Stat-3 signal. In this model of severe liver injury that is induced with tumor necrosis factor (TNF) and D-galactosamine (Gal), pretreatment with GABA markedly prolongs TNF/Gal-induced mice with a median of 12.15 h as compared with saline-pretreated TNF/Gal-induced animals, 6.23 h. When the specific Stat-3 inhibitor NSC74859 is administered 90 minutes in the GABA-pretreated TNF/Gal¨Cinduced mice, the GABA-mediated survival advantage is abolished.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6420924/
ˇˇ
D-Galactosamine Intoxication in Experimental Animals: Is ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4451715May 22, 2015 ˇ¤ Background Galactosamine (GAL) is a 6-carbon amino sugar derivative of galactose. Under physiological conditions, it is a component of specific glycoprotein hormones, such as follicle-stimulating hormone or luteinizing hormone. GAL is a potent hepatotoxic substance, which can cause hepatocyte death both by necrosis and apoptosis.
ˇˇ
Diabetes. 2013 Nov;
¦Ă-Aminobutyric Acid Regulates Both the Survival and Replication of Human ¦Â-Cells
Jide Tian,1 Hoa Dang,1 Zheying Chen,1 Alice Guan,1 Yingli Jin,1 Mark A. Atkinson,2 and Daniel L. Kaufman1
1Department of Molecular and Medical Pharmacology, University of California, Los Angeles, Los Angeles, California
2Departments of Pathology and Pediatrics, University of Florida, Gainesville, Florida
¦Ă-Aminobutyric acid (GABA) has been shown to inhibit apoptosis of rodent ¦Â-cells in vitro. In this study, we show that activation of GABAA receptors (GABAA-Rs) or GABAB-Rs significantly inhibits oxidative stress¨Crelated ¦Â-cell apoptosis and preserves pancreatic ¦Â-cells in streptozotocin-rendered hyperglycemic mice. Moreover, treatment with GABA, or a GABAA-R¨C or GABAB-R¨Cspecific agonist, inhibited human ¦Â-cell apoptosis following islet transplantation into NOD/scid mice. Accordingly, activation of GABAA-Rs and/or GABAB-Rs may be a useful adjunct therapy for human islet transplantation. GABA-R agonists also promoted ¦Â-cell replication in hyperglycemic mice. While a number of agents can promote rodent ¦Â-cell replication, most fail to provide similar activities with human ¦Â-cells. In this study, we show that GABA administration promotes ¦Â-cell replication and functional recovery in human islets following implantation into NOD/scid mice. Human ¦Â-cell replication was induced by both GABAA-R and GABAB-R activation. Hence, GABA regulates both the survival and replication of human ¦Â-cells. These actions, together with the anti-inflammatory properties of GABA, suggest that modulation of peripheral GABA-Rs may represent a promising new therapeutic strategy for improving ¦Â-cell survival following human islet transplantation and increasing ¦Â-cells in patients with diabetes.In summary, we observed that activation of GABAA-R or GABAB-R inhibited oxidative stress¨Crelated ¦Â-cell apoptosis and preserved pancreatic ¦Â-cells in hyperglycemic mice. Similarly, treatment with either a GABAA-R¨C or GABAB-R¨Cspecific agonist inhibited human islet cell apoptosis in mice following islet transplantation. Furthermore, treatment with either a GABAA-R¨C or GABAB-R¨Cspecific agonist promoted mouse and human ¦Â-cell replication in mice. Hence, GABA acts as a growth factor that regulates the survival and replication of islet ¦Â-cells.
GABA can inhibit autoreactive Th1 cell responses directly ex vivo (15¨C17), increase regulatory T cells (2,18), inhibit antigen-presenting cell function (2,19), and inhibit inflammation in mouse models of T1D (2,16,20), rheumatoid arthritis (17), multiple sclerosis (19), and type 2 diabetes (18). Twenty-six weeks of GABA treatment did not significantly alter the numbers or percentages of splenic CD4+, CD8+, T, and B lymphocytes (16), nor did long-term GABA treatment desensitize T cells to GABA-mediated inhibition (16,17). Several studies have suggested that local inflammation helps promote ¦Â-cell replication (21 and references therein). Accordingly, although inflammation should be very limited in the C57Bl/6 and NOD/scid mouse models that we have studied, GABAˇŻs anti-inflammatory activity may have partially counteracted its pro¨C¦Â-cell replication activity.
GABAˇŻs anti-inflammatory properties, together with its ability to promote ¦Â-cell replication and functional recovery, suggest that modulation of peripheral GABA-Rs may be a promising strategy for preserving and increasing islet ¦Â-cells. GABA has little capacity to pass through the blood¨Cbrain barrier and is safe for human consumption (22¨C25). Therefore, our findings may provide a basis for the design of new therapies for patients with type I and II diabetes.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3806626/
ˇˇ
Osteoprotegerin - an overview | ScienceDirect Topics
https://www.sciencedirect.com/.../osteoprotegerin ˇ¤ ·Ňë´ËŇłOsteoprotegerin is the decoy receptor which binds and thereby opposes RANK ligand, another cytokine which activates osteoclasts and causes bone resorption (Riches et al., 2009). One report contains suggestive evidence of pathogenic autoantibodies against osteoprotegerin that cause osteoporosis ( Riches et al., 2009 ).
ˇˇ
Mechanical stretching induces osteoprotegerin in ...
https://asbmr.onlinelibrary.wiley.com/doi/full/10.1002/jbmr.9Apr 30, 2010 ˇ¤ Mechanical stretching induces a sustained increase in OPG expression and a transient decrease in the expression of OC and ALP. C2C12 cells were subjected to BM for 3 days prior to mechanical stretching. (A) RT©\PCR measurements of OPG and GAPDH expression in C2C12 cells taken before stretching (0) and after 24 or 48 hours of stretching (S24, S48) or from an unstretched control ˇ
ˇˇ
ˇˇ
Int J Clin Exp Pathol. 2019;
Osteoprotegerin promotes islet ¦Â cell proliferation in intrauterine growth retardation rats through the PI3K/AKT/FoxO1 pathway
Shi Tang, Ying Xin, Min Yang, Dan Zhang, Chunzhuo Xu
Department of Pediatrics, Shengjing Hospital, China Medical University, Shenyang, Liaoning, China,
Abstract
Osteoprotegerin (OPG) is a critical factor involved in bone metabolism. The level of OPG is increased in the serum of diabetic patients; however, there is no consensus in prior studies on the role of OPG in regulating the function of islet ¦Â cells. A rat model of intrauterine growth retardation (IUGR) was established in the present study to investigate whether OPG could enhance the proliferation of ¦Â cells; and an in vitro culture model of rat islet ¦Â cell line INS-1 was used, to confirm the effect of OPG supplementation and reveal the possible mechanism. The results showed that endogenous OPG expression was reduced and normal proliferation of ¦Â cells was impaired in the IUGR islets. Exogenous supplement of OPG restored ¦Â cell proliferation to an extent in the IUGR rats, possibly associated with regulation of the PI3K/AKT/FoxO1 signalling, as evidenced by the changes of protein expression in the pathway. Furthermore, treating rat islet INS-1 cells with a PI3K inhibitor, LY294002, blunted the effects of OPG supplement in promoting cell cycle and suppressing cell apoptosis. Taken together, the present work demonstrated that OPG supplementation could improve the proliferation of islet ¦Â cells in IUGR, and the PI3K/AKT/FoxO1 pathway is involved in the underlying mechanism.
Keywords: Osteoprotegerin, intrauterine growth retardation, islet ¦Â cell, INS-1 cell line, PI3K/AKT/FoxO1 signalling pathway
Int J Clin Exp Pathol. 2019;
ąÇ±Ł»¤ËŘͨąýPI3K/AKT/FoxO1ͨ·´Ů˝řą¬ÄÚÉúł¤łŮ»ş´óĘóŇȵşĎ¸°űÔöÖł
ĚĆĘŻĐŔŁ¬ŃîĂôŁ¬Őŵ¤Ł¬Đ촺׿
ÖĐąúŇ˝żĆ´óѧ¸˝Ęôʢľ©Ň˝Ôş¶ůżĆŁ¬ÁÉÄţÉňŃô
ŐŞŇŞ
ąÇ±Ł»¤ËŘ(Osteoprotegerin, OPG)ĘDzÎÓëąÇ´úĐ»µÄÖŘŇŞŇňËءŁĚÇÄň˛ˇ»ĽŐßŃŞÇĺÖĐOPGˮƽÉý¸ß;Č»¶řŁ¬¶ÔÓÚOPGÔÚµ÷˝ÚŇȵşĐˇĎ¸°űą¦ÄÜÖеÄ×÷ÓĂŁ¬ŇÔÍůµÄŃĐľż˛˘ÎŢą˛Ę¶ˇŁ˝¨Á˘ą¬ÄÚÉúł¤łŮ»ş´óĘóÄŁĐÍŁ¬Ě˝ĚÖOPGĘÇ·ńÄÜ´Ů˝řϸ°űÔöÖł;˛˘˛ÉÓĂ´óĘóŇȵşĎ¸°űĐÇĐνşÖĘϸ°űINS-1ĚĺÍâĹŕŃřÄŁĐÍŁ¬Ö¤ĘµOPGµÄ˛ąłä×÷Óò˘˝ŇĘľĆäżÉÄܵĻúÖơŁ˝áąűĎÔĘľŁ¬IUGRŇȵşÄÚÔ´ĐÔOPG±í´ďĽőÉŮŁ¬ŐýłŁĎ¸°űÔöÖłĘÜËđˇŁÍâÔ´ĐÔ˛ąłäOPGÔÚŇ»¶¨łĚ¶ČÉϻָ´ÁËIUGR´óĘóµÄϸ°űÔöÖłŁ¬ŐâżÉÄÜÓëµ÷żŘPI3K/AKT/FoxO1ĐĹşĹͨ·Óйأ¬żÉŇÔͨąýµ°°×±í´ďµÄ¸Ä±äŔ´Ö¤Ă÷ˇŁ´ËÍ⣬ÓĂPI3KŇÖÖĆĽÁLY294002´¦Ŕí´óĘóŇȵşINS-1ϸ°űŁ¬żÉŇÔĽőČőOPGµÄ´Ů˝řϸ°űÖÜĆÚşÍŇÖÖĆϸ°űµňÍöµÄ×÷ÓáŁ×ŰÉĎËůĘöŁ¬±ľŃĐľżÖ¤ĘµOPGµÄ˛ąłäżÉŇÔ´Ů˝řIUGRÖĐŇȵşĎ¸°űµÄÔöÖłŁ¬Ćä»úÖĆÓëPI3K/AKT/FoxO1ͨ·ÓйءŁ
ąŘĽü´Ę:ąÇ±Ł»¤Ëء˘ą¬ÄÚÉúł¤łŮ»şˇ˘ŇȵşĎ¸°űˇ˘INS-1ϸ°űϵˇ˘PI3K/AKT/FoxO1ĐĹşĹͨ·https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6949609/
ˇˇ
Diabetes Obes Metab, 2016 Sep;18
Novel factors modulating human ¦Â-cell proliferation
J Shirakawa 1 2, R N Kulkarni 3 4 5
1Islet Cell and Regenerative Biology, Joslin Diabetes Center, Boston, Massachusetts.
2Department of Medicine, Brigham and Women's Hospital, Boston, Massachusetts.
3Islet Cell and Regenerative Biology, Joslin Diabetes Center, Boston, Massachusetts. rohit.kulkarni@joslin.harvard.edu.
4Department of Medicine, Brigham and Women's Hospital, Boston, Massachusetts. rohit.kulkarni@joslin.harvard.edu.
5Harvard Stem Cell Institute, Harvard Medical School, Boston,
¦Â-Cell dysfunction in type 1 and type 2 diabetes is accompanied by a progressive loss of ¦Â-cells, and an understanding of the cellular mechanism(s) that regulate ¦Â-cell mass will enable approaches to enhance hormone secretion. It is becoming increasingly recognized that enhancement of human ¦Â-cell proliferation is one potential approach to restore ¦Â-cell mass to prevent and/or cure type 1 and type 2 diabetes. While several reports describe the factor(s) that enhance ¦Â-cell replication in animal models or cell lines, promoting effective human ¦Â-cell proliferation continues to be a challenge in the field. In this review, we discuss recent studies reporting successful human ¦Â-cell proliferation including WS6, an IkB kinase and EBP1 inhibitor; harmine and 5-IT, both DYRK1A inhibitors; GNF7156 and GNF4877, GSK-3¦Â and DYRK1A inhibitors; osteoprotegrin and Denosmab, receptor activator of NF-kB (RANK) inhibitors; and SerpinB1, a protease inhibitor. These studies provide important examples of proteins and pathways that may prove useful for designing therapeutic strategies to counter the different forms of human diabetes.
Keywords: GNF4877; SerpinB1; WS6; harmine; human ¦Â-cell proliferation; osteoprotegrin.
© 2016 John Wiley & Sons Ltd.https://pubmed.ncbi.nlm.nih.gov/27615134/
ˇˇ
PLoS One. 2011;
Receptor Activator of NF-kB (RANK) Expression in Primary Tumors Associates with Bone Metastasis Occurrence in Breast Cancer Patients
Daniele Santini,# 1 , * Gaia Schiavon,# 1 , 2 Bruno Vincenzi, 1 Laura Gaeta, 3 Francesco Pantano, 1 Antonio Russo, 4 Cinzia Ortega, 5 Camillo Porta, 6 Sara Galluzzo, 1 Grazia Armento, 1 Nicla La Verde, 7 Cinzia Caroti, 8 Isabelle Treilleux, 9 Alessandro Ruggiero, 10 Giuseppe Perrone, 3 Raffaele Addeo, 11 Philippe Clezardin, 9 Andrea Onetti Muda, 3 and Giuseppe Tonini 1
Abstract
Background
Receptor activator of NFkB (RANK), its ligand (RANKL) and the decoy receptor of RANKL (osteoprotegerin, OPG) play a pivotal role in bone remodeling by regulating osteoclasts formation and activity. RANKL stimulates migration of RANK-expressing tumor cells in vitro, conversely inhibited by OPG.
Materials and Methods
We examined mRNA expression levels of RANKL/RANK/OPG in a publicly available microarray dataset of 295 primary breast cancer patients. We next analyzed RANK expression by immunohistochemistry in an independent series of 93 primary breast cancer specimens and investigated a possible association with clinicopathological parameters, bone recurrence and survival.
Results
Microarray analysis showed that lower RANK and high OPG mRNA levels correlate with longer overall survival (P = 0.0078 and 0.0335, respectively) and disease-free survival (P = 0.059 and 0.0402, respectively). Immunohistochemical analysis of RANK showed a positive correlation with the development of bone metastases (P = 0.023) and a shorter skeletal disease-free survival (SDFS, P = 0.037). Specifically, univariate analysis of survival showed that ˇ°RANK-negativeˇ± and ˇ°RANK-positiveˇ± patients had a SDFS of 105.7 months (95% CI: 73.9¨C124.4) and 58.9 months (95% CI: 34.7¨C68.5), respectively. RANK protein expression was also associated with accelerated bone metastasis formation in a multivariate analysis (P = 0.029).
Conclusions
This is the first demonstration of the role of RANK expression in primary tumors as a predictive marker of bone metastasis occurrence and SDFS in a large population of breast cancer patients.
Frontiers | Osteoprotegerin: Relationship to Breast Cancer ...
https://www.frontiersin.org/articles/10.3389/fonc.2020.00462
Introduction. Osteoprotegerin (OPG; encoded by the TNFRSF11B gene) is a secreted member of the TNF receptor protein superfamily that was first characterized and named for its protective role in bone remodeling (3, 4).OPG acts as a decoy receptor for Receptor Activator of NF-kappaB Ligand (RANKL; TNFSF11), blocking interaction with RANK (TNFRSF11A) and thereby stimulation of osteoclast ˇ
Publish Year: 2020
Decoy receptors: a strategy to regulate inflammatory ...
https://www.sciencedirect.com/science/article/pii/S147149060101941X
Jun 01, 2001 ˇ¤ Decoy receptors recognize certain inflammatory cytokines with high affinity and specificity, but are structurally incapable of signaling or presenting the agonist to signaling receptor complexes. They act as a molecular trap for the agonist and for signaling receptor components.
Publish Year: 2001ˇˇ
Clin Exp Immunol. 2004 Aug;
Osteoprotegerin (OPG) acts as an endogenous decoy receptor in tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis of fibroblast-like synovial cells
T Miyashita,* A Kawakami,* T Nakashima,† S Yamasaki,* M Tamai,* F Tanaka,* M Kamachi,* H Ida,* K Migita,* T Origuchi,‡ K Nakao,ˇě and K Eguchi*
Author information Article notes Copyright and License information Disclaimer
*The First Department of Internal Medicine, Nagasaki University School of Medicine, Nagasaki, Japan
†Department of Hospital Pharmacy, Nagasaki University School of Medicine, Nagasaki, Japan
‡Department of Physical Therapy, Nagasaki University School of Health Sciences, Nagasaki, Japan
ˇěHealth Research Center, Nagasaki University, Nagasaki, Japan
We examined the role of osteoprotegerin (OPG) on tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in rheumatoid fibroblast-like synovial cells (FLS). OPG protein concentrations in synovial fluid from patients with rheumatoid arthritis (RA) correlated with those of interleukin (IL)-1¦Â or IL-6. A similar correlation was present between IL-1¦Â and IL-6 concentrations. Rheumatoid FLS in vitro expressed both death domain-containing receptors [death receptor 4 (DR4) and DR5] and decoy receptors [decoy receptor 1 (DcR1) and DcR2]. DR4 expression on FLS was weak compared with the expression of DR5, DcR1 and DcR2. Recombinant TRAIL (rTRAIL) rapidly induced apoptosis of FLS. DR5 as well as DR4 were functional with regard to TRAIL-mediated apoptosis induction in FLS; however, DR5 appeared be more efficient than DR4. In addition to soluble DR5 (sDR5) and sDR4, OPG administration significantly inhibited TRAIL-induced apoptogenic activity. OPG was identified in the culture supernatants of FLS, and its concentration increased significantly by the addition of IL-1¦Â in a time-dependent manner. Neither IL-6 nor tumour necrosis factor (TNF)-¦Á increased the production of OPG from FLS. TRAIL-induced apoptogenic activity towards FLS was reduced when rTRAIL was added without exchanging the culture media, and this was particularly noticeable in the IL-1¦Â-stimulated FLS culture; however, the sensitivity of FLS to TRAIL-induced apoptosis itself was not changed by IL-1¦Â. Interestingly, neutralization of endogenous OPG by adding anti-OPG monoclonal antibody (MoAb) to FLS culture restored TRAIL-mediated apoptosis. Our data demonstrate that OPG is an endogenous decoy receptor for TRAIL-induced apoptosis of FLS. In addition, IL-1¦Â seems to promote the growth of rheumatoid synovial tissues through stimulation of OPG production, which interferes with TRAIL death signals in a competitive manner.
Keywords: fibroblast-like synovial cells, IL-1¦Â, OPG, rheumatoid arthritis, TRAILhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC1809120/
ˇˇ
Med Sci Monit. 2018;
Effect of Resistance Exercise on Serum Osteoprotegerin Levels and Insulin Resistance in Middle-Aged Women with Metabolic Syndrome
Sun Hur,1,A,F,G Sung-Hyoun Cho,2,C,D Bo-Kyoung Song,3,D,F and Byung-Jun Chocorresponding author4,B,C,E
1Department of Sports Science, College of Art and Culture, Kangwon National University, Kangwon, South Korea
2Department of Physical Therapy, Nambu University, Gwangju, South Korea
3Department of Occupational Therapy, Kangwon National University, Kangwon, South Korea
4Department of Emergency Medical Technology, Kangwon National University, Kangwon, South Korea
Background
Osteoprotegerin (OPG) is a soluble glycoprotein that belongs to the tumor necrosis factor (TNF) receptor superfamily. OPG is mainly secreted by bone. The relationship between acute resistance training, serum OPG levels and metabolic syndrome, including insulin resistance, remains unclear. The purpose of this study was to determine the effect of resistance exercise on serum OPG levels and insulin resistance in middle-aged women with metabolic syndrome.
Material/Methods
Twenty-four middle-aged women were divided into those with metabolic syndrome (n=12) and a normal control group without metabolic syndrome or insulin resistance (n=12). Metabolic syndrome was diagnosed according to the National Cholesterol Education Program Adult Treatment Panel III (NCEP-ATP III) criteria. The quantitative insulin-sensitivity check index (QUICKI) and the homeostatic model assessment (HOMA) index for assessing beta-cell function and insulin resistance were used. The intensity of the resistance exercise was 60¨C70% of the repetition maximum, for 40 minutes with 10¨C12 repetitions, performed three times per week. Venous blood samples were tested using standard laboratory procedures.
Results
Before exercise, the metabolic syndrome group showed a significant increase in waist circumference (P=0.030) and serum triglyceride (TG) (P=0.014), and lower high-density lipoprotein-cholesterol (HDL-C) (P=0.010) compared with the control group. After the eight-week resistance exercise program, waist circumference, and the QUICKI decreased and OPG levels were significantly increased in the metabolic syndrome group compared with the normal control group.
Conclusions
A resistance exercise program was effective in reducing factors associated with metabolic syndrome including insulin resistance and increases serum levels of OPG in middle-aged women.
MeSH Keywords: Exercise, Insulin Resistance, Metabolic Syndrome X, Osteoprotegerinhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC6320661/
Osteoprotegerin Regulates Pancreatic ¦Â-Cell Homeostasis upon Microbial Invasion
Yukiko Kuroda,Kenta Maruyama,
Osteoprotegerin (OPG), a decoy receptor for receptor activator of NF-¦ĘB ligand (RANKL), antagonizes RANKLˇŻs osteoclastogenic function in bone. We previously demonstrated that systemic administration of lipopolysaccharide (LPS) to mice elevates OPG levels and reduces RANKL levels in peripheral blood. Here, we show that mice infected with Salmonella, Staphylococcus, Mycobacteria or influenza virus also show elevated serum OPG levels. We then asked whether OPG upregulation following microbial invasion had an effect outside of bone. To do so, we treated mice with LPS and observed OPG production in pancreas, especially in ¦Â-cells of pancreatic islets. Insulin release following LPS administration was enhanced in mice lacking OPG, suggesting that OPG inhibits insulin secretion under acute inflammatory conditions. Consistently, treatment of MIN6 pancreatic ¦Â-cells with OPG decreased their insulin secretion following glucose stimulation in the presence of LPS. Finally, our findings suggest that LPS-induced OPG upregulation is mediated in part by activator protein (AP)-1 family transcription factors, particularly Fos proteins. Overall, we report that acute microbial infection elevates serum OPG, which maintains ¦Â-cell homeostasis by restricting glucose-stimulated insulin secretion, possibly preventing microbe-induced exhaustion of ¦Â-cell secretory capacity.https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0146544
ˇˇ
Beta cells from stem cells: Potential for cell replacement therapy
Date: April 27, 2020
Source:
Helmholtz Zentrum M¨ąnchen - German Research Center for Environmental Health
Summary:
Researchers have developed an improved pluripotent stem cell differentiation protocol to generate beta cells via CD177 in vitro with superior glucose response and insulin secretion -- a major step towards beta cell replacement therapy.
https://www.sciencedaily.com/releases/2020/04/200427125132.htmˇˇ
Beta Cells and Diabetes
By Elisabeth Almekinder RN, BA, CDE
What hormones do the beta cells secrete?
Just when you thought that beta cells didnˇŻt have enough to do, it turns out they are also busy making other hormones, too. The beta cells secrete the hormones Amylin and C-peptide.
Amylin
Amylin is the short stop. It stops blood sugar, and makes it wait a while before it is allowed to enter the blood stream.
Amylin is made in the beta cells, along with C-peptide. It is another peptide hormone. People with diabetes have deficient amylin (Type 2), and absent amylin (Type 1). Amylin stops the sugar (glucagon) from being secreted, slows the gastric system, and curbs appetite.
C-peptide
C-peptide gets into your blood stream at about the same rate as insulin does. Its purpose is to come along behind blood sugar in your veins and arteries, and sweep up the mess that high blood sugars leave behind. C-peptide will also patch up any damage caused by high blood sugar from high carbohydrate meals that has been consumed.
Think about that for a minute. If the beta cells are damaged or worse yet, completely non-functioning, then they canˇŻt make C-peptide that repairs the damage, either.https://www.thediabetescouncil.com/beta-cells-diabetes/
ˇˇ
Role of Zinc Supplementation on Diabetes https://www.imedpub.com/articles/role-of-zinc-supplementation-on-diabetes.pdf
Zinc is essential for storage of insulin and for processing of insulin in the body. In diabetic patients the content of zinc is greatly decreased in the pancreashttps://medium.com/@leonardjpmail/think-zinc-9c114fde64bf
ˇˇ
NATURE, 13 June 2019
The zinc transporter Zip14 (SLC39a14) affects Beta-cell Function: Proteomics, Gene expression, and Insulin secretion studies in INS-1E cells
Trine Maxel, Kamille Smidt, Charlotte C. Petersen, Bent Honor¨¦, Anne K. Christensen, Per B. Jeppesen, Birgitte Brock, Jørgen Rungby, Johan Palmfeldt & Agnete LarsenDepartment of Biomedicine, Faculty of Health, Aarhus University, Aarhus, Denmark
Abstract
Insulin secretion from pancreatic beta-cells is dependent on zinc ions as essential components of insulin crystals, zinc transporters are thus involved in the insulin secretory process. Zip14 (SLC39a14) is a zinc importing protein(ZIP) that has an important role in glucose homeostasis. Zip14 knockout mice display hyperinsulinemia and impaired insulin secretion in high glucose conditions. Endocrine roles for Zip14 have been established in adipocytes and hepatocytes, but not yet confirmed in beta-cells. In this study, we investigated the role of Zip14 in the INS-1E beta-cell line. Zip14 mRNA was upregulated during high glucose stimulation and Zip14 silencing led to increased intracellular insulin content. Large-scale proteomics showed that Zip14 silencing down-regulated ribosomal mitochondrial proteins, many metal-binding proteins, and others involved in oxidative phosphorylation and insulin secretion. Furthermore, proliferation marker Mki67 was down-regulated in Zip14 siRNA-treated cells. In conclusion, Zip14 gene expression is glucose sensitive and silencing of Zip14 directly affects insulin processing in INS-1E beta-cells. A link between Zip14 and ribosomal mitochondrial proteins suggests altered mitochondrial RNA translation, which could disturb mitochondrial function and thereby insulin secretion. This highlights a role for Zip14 in beta-cell functioning and suggests Zip14 as a future pharmacological target in the treatment of beta-cell dysfunction.ˇˇ
ZIP14 is highly expressed in human liver and pancreas33, and it is present in both the alpha- and beta-cells of the human pancreas
ˇˇ
https://www.nature.com/articles/s41598-019-44954-1
ˇˇ
Diabetologia. 2012 Mar 1.
Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD¨Csevere combined immunodeficiency (SCID) mice
H. E. Levitt, Division of Endocrinology and Metabolism, University of Pittsburgh School of Medicine, 200 Lothrop St, BST E1140, Pittsburgh, PA 15261, USA;
Abstract
Aims/hypothesis
We determined whether hyperglycaemia stimulates human beta cell replication in vivo in an islet transplant model
Methods
Human islets were transplanted into streptozotocin-induced diabetic NOD¨Csevere combined immunodeficiency mice. Blood glucose was measured serially during a 2 week graft revascularisation period. Engrafted mice were then catheterised in the femoral artery and vein, and infused intravenously with BrdU for 4 days to label replicating beta cells. Mice with restored normoglycaemia were co-infused with either 0.9% (wt/vol.) saline or 50% (wt/vol.) glucose to generate glycaemic differences among grafts from the same donors. During infusions, blood glucose was measured daily. After infusion, human beta cell replication and apoptosis were measured in graft sections using immunofluorescence for insulin, and BrdU or TUNEL.
Results
Human islet grafts corrected diabetes in the majority of cases. Among grafts from the same donor, human beta cell proliferation doubled in those exposed to higher glucose relative to lower glucose. Across the entire cohort of grafts, higher blood glucose was strongly correlated with increased beta cell replication. Beta cell replication rates were unrelated to circulating human insulin levels or donor age, but tended to correlate with donor BMI. Beta cell TUNEL reactivity was not measurably increased in grafts exposed to elevated blood glucose.
Conclusions/interpretation
Glucose is a mitogenic stimulus for transplanted human beta cells in vivo. Investigating the underlying pathways may point to mechanisms capable of expanding human beta cell mass in vivo.
Keywords: Glucose, Human islets, Hyperglycaemia, Insulin, Mitosis, NOD-SCID mice, Proliferation, Replication, Transplantation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3034833/ˇˇ
Insulin and Zinc ˇŞ Two Peas in a Pancreas - dummies
Zinc in pancreatic beta cells binds to several insulin molecules, six to be exact, forming whatˇŻs called an insulin hexamer for storage. In fact, long acting insulin formulations for injection ˇŞ NPH or Lantus, to name two ˇŞ contain zinc so that the insulin is bound in hexamers that convert to the active insulin monomer (a single insulin molecule) slowly.
Regenerating insulin-producing cells with zinc could ...
ˇˇ
Role of NADPH Oxidase in Beta Cell Dysfunction
Dysfunction of pancreatic beta cells and loss of beta cell mass is a major factor in the development of diabetes. Currently there is no cure for diabetes, and available therapies do not focus on halting or reversing the loss of beta cell function. New strategies to preserve beta cells in diabetes are needed. Conferring protection to the beta cells against the effects of sustained intracellular reactive oxygen species (ROS) presents a novel approach to preserve beta cell mass. Important contributors to increases in intercellular ROS in beta cells are nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes. Discussed in this review are the roles of NADPH oxidases in the beta cell, their contribution to beta cell dysfunction, and new emerging selective inhibitors of NADPH oxidase.
Keywords
Beta cell Diabetes Inhibitors Islet NADPH oxidase NOX Proinflammatory cytokines ROSNADPH Oxidases and Beta Cells
Select NOX family members are expressed in pancreatic beta cells. NOX-1, NOX-2, NOX-4, NOXO1 (homolog of p47 phox ), NOXA1 (homolog of p67 phox ), and p40 phox have been described in a variety of pancreatic and islet studies, including isolated rat beta cells (Oliveira et al. 2003; Nakayama et al. 2005; Lupi et al. 2006; Shao et al. 2006; Uchizono et al. 2006; Rebelato et al. 2012). Expression of NOX-5, DUOX-1 and DUOX-2, as determined by RT-PCR, has additionally been described in the pancreas, though the functional relevance of their expression has yet to be determined (Cheng et al. 2001; Edens et al. 2001). Historically, expression of NADPH oxidase family members in beta cells has been associated with regulation of glucose-stimulated insulin secretion (Morgan et al. 2007; Pi et al. 2007; Morgan et al. 2009). More recently, activity of NOX enzymes has been linked to beta cell dysfunction. Beta cell damage likely arises from generation of intracellular ROS stimulating redox signaling pathways and, more chronically, oxidative stress. In the following sections, the roles of the specific NOXs expressed in the beta cell are discussed.
NOX and Glucose-Stimulated Insulin Secretion
Several key observations have linked NADPH oxidase activity with regulation of insulin secretion. The product of NADPH oxidase activity, generation of H2O2, is required for insulin secretion (Pi et al. 2007). Elevated glucose leads to an increase in H2O2 generation, thus linking NADPH oxidase activity to regulation of insulin secretion (Morgan et al. 2007, 2009). Inhibition of NADPH oxidase by the general inhibitor, diphenyleneiodonium (DPI), led to a decrease in H2O2 production and also impaired insulin secretion (Imoto et al. 2008). Antisense-mediated decrease in the expression of p47 phox , an important subunit for the active NOX enzyme, reduced glucose-stimulated insulin secretion (Morgan et al. 2009). Activation and translocation of p47 phox is required for an active NADPH complex. Translocation of proteins to membranes or subunits is supported since inhibitors of protein prenylation or protein farnesyltransferase significantly decrease NOX-2-induced ROS generation and decrease glucose-stimulated insulin secretion (Syed et al. 2011b; Matti et al. 2012). These results suggest NOX activity is necessary for the transient increase in ROS that is needed for glucose-stimulated insulin secretion.
Which NOX family isotypes are involved in insulin secretion and what exactly their role is remains an unanswered question. A major limitation to resolving this question has been the lack of isoform-specific inhibitors of the NOX enzymes.
NOX and Beta Cell Dysfunction
Associated with a diabetic state is an increase in the serum levels of proinflammatory cytokines, free-fatty acids (FFA), and glucose. These serum mediators have been shown to elevate the expression and activity of NADPH oxidases (Morgan et al. 2007). Additionally, deposition of fibrillar human islet amyloid polypeptide (IAPP) in the beta cell line RIN5mF cells increases NADPH oxidase activity and intracellular lipid peroxidation (Janciauskiene and Ahren 2000). Accumulation of amyloid in islets is a pathogenic state associated with type 2 diabetes (Marzban and Verchere 2004). In a small sample group, we showed that NOX-1 expression is elevated in islets from human type 2 diabetic donors (Weaver et al. 2012). Animal models of type 2 diabetes have also reported an increase in NOXs. For example, the role of NOX-2 in beta cell dysfunction has been explored in the Zucker diabetic fatty rat (Syed et al. 2011a). The Zucker diabetic fatty (ZDF) rat is a model of type 2 diabetes where rats become obese and develop hyperinsulinemia, hyperglycemia, and beta cell dysfunction. Examination of islets isolated from ZDF rats showed an increase in intracellular ROS levels that corresponded with elevated expression of the NOX enzyme subunits p47 phox , gp91 phox , and Rac1 (Syed et al. 2011a). Exposure of islets from Wistar rats to the free-fatty acid palmitate resulted in elevation of p47 phox protein and an increase in the mRNA levels of p22 phox , gp91 phox , p47 phox , proinsulin, and the G protein-coupled protein receptor 40, a signaling receptor that activates the phospholipase C signaling pathway (Graciano et al. 2011). With global inhibition of NADPH oxidase activity, beta cells are protected from the effects of cytokine or FFA treatment (Michalska et al. 2010).
Advanced glycation end products (AGE) contribute to oxidative stress and the development of diabetes (Kaneto et al. 1996; Hofmann et al. 2002; Peppa et al. 2003; Cai et al. 2008; Zhao et al. 2009; Coughlan et al. 2011). AGEs form when carbohydrates, such as glucose, react nonenzymatically (e.g., glycation and oxidation) with amino groups. Binding of AGE to its receptor, RAGE (receptor for advanced glycation end products), generates ROS leading to oxidative stress in beta cells. Evidence suggests that AGE may be increasing ROS generation through NADPH oxidase. In isolated rat islets, NADPH-dependent superoxide generation in homogenates increased after treatment with high glucose plus glycolaldehyde (Costal et al. 2013); addition of the NADPH oxidase inhibitor DPI decreased superoxide production. VAS2870, a NADPH oxidase inhibitor, also decreased intracellular superoxide production in islets treated with glucose plus glycolaldehyde (Costal et al. 2013). The increase in superoxide preceded apoptosis. Pancreatic beta cell lines including INS-1, MIN6, and BTC-6 cells and isolated primary rat islets treated with AGE showed an increase in ROS generation that was followed by apoptosis (Lim et al. 2008). INS-1 beta cells exposed to varying concentrations of AGE showed a time-dependent increase in intracellular ROS and apoptosis that was dependent upon NADPH oxidase (Lin et al. 2012). Since superoxide production in vascular smooth muscle cells exposed to AGE has been linked to an increase in the transcription of NOX-1, a similar pathway linking AGE and NADPH oxidase-1 in beta cells may occur (San Martin et al. 2007).Role of NADPH Oxidase in Beta Cell Dysfunction | SpringerLink
https://link.springer.com/referenceworkentry/10.1007/978-94-007-6884-0_46-3ˇˇ
Front. Endocrinol., 03 April 2013
NOX, NOX who is there? The contribution of NADPH oxidase one to beta cell dysfunction
David A. Taylor-Fishwick1,2*
1Department of Internal Medicine, Strelitz Diabetes Center, Eastern Virginia Medical School, Norfolk, VA, USA
2Department of Microbiology and Molecular Cell Biology, Eastern Virginia Medical School, Norfolk, VA, USA
Predictions of diabetes prevalence over the next decades warrant the aggressive discovery of new approaches to stop or reverse loss of functional beta cell mass. Beta cells are recognized to have a relatively high sensitivity to reactive oxygen species (ROS) and become dysfunctional under oxidative stress conditions. New discoveries have identified NADPH oxidases in beta cells as contributors to elevated cellular ROS. Reviewed are recent reports that evidence a role for NADPH oxidase-1 (NOX-1) in beta cell dysfunction. NOX-1 is stimulated by inflammatory cytokines that are elevated in diabetes. First, regulation of cytokine-stimulated NOX-1 expression has been linked to inflammatory lipid mediators derived from 12-lipoxygenase activity. For the first time in beta cells these data integrate distinct pathways associated with beta cell dysfunction. Second, regulation of NOX-1 in beta cells involves feed-forward control linked to elevated ROS and Src-kinase activation. This potentially results in unbridled ROS generation and identifies candidate targets for pharmacologic intervention. Third, consideration is provided of new, first-in-class, selective inhibitors of NOX-1. These compounds could have an important role in assessing a disruption of NOX-1/ROS signaling as a new approach to preserve and protect beta cell mass in diabetes.¶ÔδŔ´Ľ¸Ę®ÄęĚÇÄň˛ˇ»Ľ˛ˇÂʵÄÔ¤˛âÖµµĂ»ýĽ«Ě˝Ë÷ÍŁÖą»ňÄćתą¦ÄÜĐÔ¦Âϸ°űÁżËđʧµÄĐ·˝·¨ˇŁ Betaϸ°ű±»ČĎÎŞ¶Ô»îĐÔŃőŁ¨ROSŁ©ľßÓĐĎŕ¶Ô˝Ď¸ßµÄĂô¸ĐĐÔŁ¬˛˘ÔÚŃő»ŻÓ¦Ľ¤ĚőĽţĎÂą¦ÄÜʧµ÷ˇŁĐ·˘ĎÖŇŃČ·¶¨¦Âϸ°űÖеÄNADPHŃő»ŻĂ¸ĘǵĽÖÂϸ°űROSÉý¸ßµÄÔŇňˇŁÉó˛éÁË×î˝üµÄ±¨µŔŁ¬ŐâĐ©Ö¤ľÝ±íĂ÷NADPHŃő»ŻĂ¸-1Ł¨NOX-1Ł©ÔÚ¦Âϸ°űą¦ÄÜŇ쳣ÖĐĆđ×÷ÓáŁÔÚĚÇÄň˛ˇÖĐÉý¸ßµÄŃ×ĐÔϸ°űŇň×Ó´ĚĽ¤NOX-1ˇŁĘ×ĎČŁ¬Ď¸°űŇň×Ó´ĚĽ¤µÄNOX-1±í´ďµÄµ÷˝ÚÓëÔ´×Ô12-Ö¬ŃőşĎø»îĐÔµÄŃ×Ö˘Ö¬ÖĘ˝éĚĺÓйءŁŐâĐ©ĘýľÝĘ×´ÎÔÚ¦Âϸ°űÖĐŐűşĎÁËÓë¦Âϸ°űą¦ÄÜŐĎ°ĎŕąŘµÄ¶ŔĚŘÍľľ¶ˇŁĆä´ÎŁ¬¦Âϸ°űÖĐNOX-1µÄµ÷˝ÚÉ漰ÓëÉý¸ßµÄROSşÍSrcĽ¤Ă¸Ľ¤»îĎŕąŘµÄÇ°ŔˇżŘÖơŁŐâżÉÄܻᵼÖÂÎŢĎŢÖƵÄROSÉúłÉŁ¬˛˘Č·¶¨Ň©Îď¸ÉÔ¤µÄşňѡ°Đ±ęˇŁµÚČýŁ¬żĽÂÇÁËеģ¬Ň»Á÷µÄѡÔńĐÔNOX-1ŇÖÖĆĽÁˇŁŐâĐ©»ŻşĎÎďżÉÄÜÔÚĆŔąŔNOX-1 / ROSĐźŵÄĆĆ»µÖĐĆđÖŘŇŞ×÷ÓĂŁ¬ŐâĘDZŁ´ćşÍ±Ł»¤ĚÇÄň˛ˇ¦Âϸ°űÖĘÁżµÄĐ·˝·¨ˇŁ
Figure 2. Feed-forward regulation of NOX-1 expression in beta cells.
...In terms of disease pathology, self-sustaining NOX-1 would lead to unbridled ROS generation, oxidative stress, and induce beta cell dysfunction/destruction. Consequently, this pathway has potential for high impact therapeutic intervention.
ˇˇ
Frontiers | NOX, NOX Who is There? The Contribution of NADPH Oxidase One to Beta Cell Dysfunction | Endocrinology
https://www.frontiersin.org/articles/10.3389/fendo.2013.00040/fullˇˇ
J Cell Physiol
. 2011 Apr;226(4):1110-7. doi: 10.1002/jcp.22432.
NAD(P)H oxidase participates in the palmitate-induced superoxide production and insulin secretion by rat pancreatic islets
Maria Fernanda R Graciano 1, Laila R B Santos, Rui Curi, Angelo R Carpinelli
...
Palmitic acid oxidation was demonstrated to contribute for the fatty acid induction of superoxide production in the presence of 5.6 mM glucose. In fact, palmitate caused p47(PHOX) translocation to plasma membrane,https://pubmed.ncbi.nlm.nih.gov/20857410/
ˇˇ
Antioxid Redox Signal. 2019 Dec 20; 31(18): 1371¨C1410.
The Emerging Roles of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2 in Skeletal Muscle Redox Signaling and Metabolism
1Section of Molecular Physiology, Department of Nutrition, Exercise and Sports (NEXS), Faculty of Science, University of Copenhagen, Copenhagen, Denmark.
Significance: Skeletal muscle is a crucial tissue to whole-body locomotion and metabolic health. Reactive oxygen species (ROS) have emerged as intracellular messengers participating in both physiological and pathological adaptations in skeletal muscle. A complex interplay between ROS-producing enzymes and antioxidant networks exists in different subcellular compartments of mature skeletal muscle. Recent evidence suggests that nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are a major source of contraction- and insulin-stimulated oxidants production, but they may paradoxically also contribute to muscle insulin resistance and atrophy.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6859696/ˇˇ
Trends Endocrinol Metab. 2020 Feb;
Role of Nitric Oxide in Insulin Secretion and Glucose Metabolism
Zahra Bahadoran 1, Parvin Mirmiran 2, Asghar Ghasemi 3
Nitric oxide (NO) contributes to carbohydrate metabolism and decreased NO bioavailability is involved in the development of type 2 diabetes mellitus (T2DM). NO donors may improve insulin signaling and glucose homeostasis in T2DM and insulin resistance (IR), suggesting the potential clinical importance of NO-based interventions. In this review, site-specific roles of the NO synthase (NOS)-NO pathway in carbohydrate metabolism are discussed. In addition, the metabolic effects of physiological low levels of NO produced by constitutive NOS (cNOS) versus pathological high levels of NO produced by inducible NOS (iNOS) in pancreatic ¦Â-cells, adipocytes, hepatocytes, and skeletal muscle cells are summarized. A better understanding of the NOS-NO system in the regulation of glucose homeostasis can hopefully facilitate the development of new treatments for T2DM.
Keywords: carbohydrate metabolism; nitric oxide; nitric oxide synthase; type 2 diabetes mellitus.https://pubmed.ncbi.nlm.nih.gov/31690508/
ˇˇ
Biomed Pharmacother. 2019 Sep;
The vitamin D receptor (VDR) protects pancreatic beta cells against Forkhead box class O1 (FOXO1)-induced mitochondrial dysfunction and cell apoptosis
Chen Chen 1, Yuming Luo 2, Yajuan Su 3, Lichen Teng 3
1Department of Clinical Laboratory, Women's Hospital of Nanjing Medical University, Nanjing Maternity and Child Health Care Hospital, Nanjing, China. Electronic address: gg20180101wh@163.com.
2Department of Gynecology, Qixia District Maternal and Child Health Care Hospital, Nanjing, China.
Abstract
Vitamin D deficiency is identified as a risk factor for gestational diabetes mellitus (GDM). Forkhead box class O1 (FoxO1) is closely related to GDM; however, the role of vitamin D deficiency and the underlying pathogenesis of GDM has not been elucidated. Serum vitamin D level was detected using chemiluminescence immunoassay. FOXO1 expression was examined using Real-time polymerase chain reaction (PCR), western blot and immunocytochemistry analysis. Apoptosis of cells was assessed by flow cytometry. Mitochondrial function was assessed via reactive oxygen species (ROS) generation and changes in the mitochondrial membrane potential (¦¤¦·m). Our study demonstrated that vitamin D levels were significantly lower in 40 GDM patients. The silencing of the vitamin D receptor (VDR) decreased cell survival and increased both FoxO1 mRNA and protein expression. Overexpression of FoxO1 could cause the mitochondrial dysfunction (including production of ROS and decrease of mitochondrial membrane potential (¦¤¦·m)) and cell apoptosis. However, Overexpression of VDR and vitamin D treatment could induce the cell survival and alleviate the FoxO1-induced cell apoptosis, furthermore, vitamin D treatment or silencing of FoxO1 gene could reverse the ROS-induced cell apoptosis. Therefore, our results support that vitamin D may protect FoxO1-induced pancreatic beta cell apoptosis, which suggests that vitamin D may have beneficial effects in preventing and treating GDM.
Keywords: Forkhead box class O1 (FOXO1); Gestational diabetes mellitus (GDM); Vitamin D; Vitamin D receptor (VDR).
Copyright © 2019 The Authors. Published by Elsevier Masson SAS.. All rights reserved.https://pubmed.ncbi.nlm.nih.gov/31261027/
ˇˇ
Vitamin D Receptor Overexpression in ¦Â-Cells Ameliorates Diabetes in Mice
1Center of Animal Biotechnology and Gene Therapy, Universitat Aut¨°noma de Barcelona, Bellaterra, Spain
2Department of Biochemistry and Molecular Biology, Universitat Aut¨°noma de Barcelona, Bellaterra, Spain
3CIBER de Diabetes y Enfermedades Metab¨®licas Asociadas (CIBERDEM), Instituto de Salud Carlos III, Madrid, Spain
4Bellvitge Biomedical Research Institute, Universitat de Barcelona, Barcelona, Spain
Abstract
Vitamin D deficiency has been associated with increased incidence of diabetes, both in humans and in animal models. In addition, an association between vitamin D receptor (VDR) gene polymorphisms and diabetes has also been described. However, the involvement of VDR in the development of diabetes, specifically in pancreatic ¦Â-cells, has not been elucidated yet. Here, we aimed to study the role of VDR in ¦Â-cells in the pathophysiology of diabetes. Our results indicate that Vdr expression was modulated by glucose in healthy islets and decreased in islets from both type 1 diabetes and type 2 diabetes mouse models. In addition, transgenic mice overexpressing VDR in ¦Â-cells were protected against streptozotocin-induced diabetes and presented a preserved ¦Â-cell mass and a reduction in islet inflammation. Altogether, these results suggest that sustained VDR levels in ¦Â-cells may preserve ¦Â-cell mass and ¦Â-cell function and protect against diabetes.
https://diabetes.diabetesjournals.org/content/69/5/927ˇˇ
Apoptosis. 2006 Feb
1,25-Dihydroxyvitamin D3 protects human pancreatic islets against cytokine-induced apoptosis via down-regulation of the Fas receptor
1Facult¨¦ de M¨¦decine, Cellular Therapy of Diabetes, Institut National de la Sant¨¦ et de la Recherche M¨¦dicale, ERIT-M 0106, 59045, Lille, France.
Beta cell loss occurs at the onset of type 1 diabetes and after islet graft. It results from the dysfunction and destruction of beta cells mainly achieved by apoptosis. One of the mediators believed to be involved in beta cell apoptosis is Fas, a transmembrane cell surface receptor transducing an apoptotic death signal and contributing to the pathogenesis of several autoimmune diseases. Fas expression is particularly induced in beta cells by inflammatory cytokines secreted by islet-infiltrating mononuclear cells and makes cells susceptible to apoptosis by interaction with Fas-ligand expressing cells.ˇˇ
We have previously demonstrated that 1,25(OH)2D3, the active metabolite of vitamin D, known to exhibit immunomodulatory properties and prevent the development of type 1 diabetes in NOD mice, is efficient against apoptosis induced by cytokines in human pancreatic islets in vitro. The effects were mainly mediated by the inactivation of NF-kappa-B. In this study we demonstrated that 1,25(OH)2D3 was also able to counteract cytokine-induced Fas expression in human islets both at the mRNA and protein levels. These results were reinforced by our microarray analysis highlighting the beneficial effects of 1,25(OH)2D3 on death signals induced by Fas activation. Our results provides additional evidence that 1,25(OH)2D3 may be an interesting tool to help prevent the onset of type 1 diabetes and improve islet graft survival.
https://pubmed.ncbi.nlm.nih.gov/16502254/ˇˇ
Pathophysiology
Pancreatic ¦Â-Cell Rest Replenishes Insulin Secretory Capacity and Attenuates Diabetes in an Extreme Model of Obese Type 2 DiabetesBrandon B. Boland1,2⇑, Charles Brown Jr.1, Michelle L. Boland1,2, Jennifer Cann1, Michal Sulikowski1, Gitte Hansen2, Rikke V. Grønlund2, Wanda King1, Cristina Rondinone1, James Trevaskis1, Christopher J. Rhodes1 and Joseph S. Grimsby1
-Author Affiliations
1Division of Cardiovascular and Metabolic Disease, MedImmune LLC, Gaithersburg, MD
2Gubra ApS, Hørsholm, Denmark
The onset of common obesity-linked type 2 diabetes (T2D) is marked by exhaustive failure of pancreatic ¦Â-cell functional mass to compensate for insulin resistance and increased metabolic demand, leading to uncontrolled hyperglycemia.Here, the ¦Â-cell¨Cdeficient obese hyperglycemic/hyperinsulinemic KS db/db mouse model was used to assess consequential effects on ¦Â-cell functional recovery by lowering glucose homeostasis and/or improving insulin sensitivity after treatment with thiazolidinedione therapy or glucagon-like peptide 1 receptor agonism alone or in combination with sodium/glucose cotransporter 2 inhibition (SGLT-2i). SGLT-2i combination therapies improved glucose homeostasis, independent of changes in body weight, resulting in a synergistic increase in pancreatic insulin content marked by significant recovery of the ¦Â-cell mature insulin secretory population but with limited changes in ¦Â-cell mass and no indication of ¦Â-cell dedifferentiation.
Restoration of ¦Â-cell insulin secretory capacity also restored biphasic insulin secretion. These data emphasize that by therapeutically alleviating the demand for insulin in vivo, irrespective of weight loss, endogenous ¦Â-cells recover significant function that can contribute to attenuating diabetes. Thus, this study provides evidence that alleviation of metabolic demand on the ¦Â-cell, rather than targeting the ¦Â-cell itself, could be effective in delaying the progression of T2D.
ÔÚ·ĘĹÖµÄ2ĐÍĚÇÄň˛ˇĽ«¶ËÄŁĐÍÖĐŁ¬ŇČĎ٦Âϸ°űľ˛Ď˘żÉ»Ö¸´ŇȵşËصķÖĂÚÄÜÁ¦˛˘ĽőÇáĚÇÄň˛ˇˇŁ
1ÂíŔďŔĽÖݸÇÉŞËą±¤ĘĐMedImmune LLCŁ¬ĐÄŃŞąÜÓë´úĐ»Ľ˛˛ˇżĆ
2 Gubra ApSŁ¬µ¤Âó»ô¶ű˹ķ
łŁĽűµÄÓë·ĘĹÖĎŕąŘµÄ2ĐÍĚÇÄň˛ˇŁ¨T2DŁ©µÄ·˘×÷ŇÔŇČĎ٦Âϸ°űą¦ÄÜĐÔą¦ÄÜËĄ˝ßÍęČ«ÎŢ·¨ĂÖ˛ąŇȵşËصֿąşÍÔöĽÓµÄ´úĐ»ĐčÇóÎŞĚŘŐ÷Ł¬´Ó¶řµĽÖÂŃŞĚÇʧżŘˇŁ
ÔÚŐâŔʹÓĂŕçßňÍé¶ţÍŞÖÎÁĆ»ňŇȸßŃŞĚÇËŘŃůëÄÖÎÁĆşóŁ¬Í¨ąý˝µµÍĆĎĚŃĚÇÎČ̬şÍ/»ň¸ÄÉĆŇȵşËŘĂô¸ĐĐÔŁ¬ĘąÓæÂϸ°űȱĎÝĐÍ·ĘĹֵĸßŃŞĚÇ/¸ßŇȵşËŘKS db / dbСĘóÄŁĐÍĆŔąŔ¶Ô¦Âϸ°űą¦Äָܻ´µÄĎŕÓ¦Ó°Ď졣 1ĘÜĚ弤¶ŻĽÁµĄ¶Ŕ»ňÓëÄĆ/ĆĎĚŃĚÇą˛×ŞÔ˵°°×2ŇÖÖĆ˝áşĎĘąÓĂŁ¨SGLT-2iŁ©ˇŁ SGLT-2iÁŞşĎÁĆ·¨¸ÄÉĆÁËĆĎĚŃĚǵÄĚĺÄÚÎČ̬Ł¬ÓëĚĺÖر仯Î޹أ¬µĽÖÂŇČĎŮŇȵşËŘş¬ÁżĐͬÔöĽÓŁ¬ĆäĚŘŐ÷ĘǦÂϸ°űłÉĘěŇȵşËŘ·ÖĂÚČşµÄĎÔׯָ´Ł¬µ«¦Âϸ°űÖĘÁż±ä»ŻÓĐĎŢÇŇÎŢČκΦÂϸ°űČĄ·Ö»ŻµÄĎÔĘľˇŁ
¦Âϸ°űŇȵşËŘ·ÖĂÚÄÜÁ¦µÄ»Ö¸´Ň˛»Ö¸´ÁËË«ĎŕŇȵşËصķÖĂÚˇŁŐâĐ©ĘýľÝÇżµ÷Ł¬ÎŢÂŰĚĺÖŘĽőÇᣬͨąýÖÎÁĆĐԵؼőÇáĚĺÄÚ¶ÔŇȵşËصÄĐčÇóŁ¬ÄÚÔ´ĐÔ¦Âϸ°ű»Ö¸´ÁËżÉÓĐÖúÓÚĽőÇáĚÇÄň˛ˇµÄÖŘŇŞą¦ÄܡŁŇň´ËŁ¬ŐâĎîŃĐľżĚáą©ÁËÖ¤ľÝŁ¬Ľ´ĽőÇá¦Âϸ°űµÄ´úĐ»ĐčÇó¶ř˛»ĘÇŐë¶Ô¦Âϸ°ű±ľÉíŁ¬żÉŇÔÓĐЧµŘŃÓłŮT2DµÄ·˘ŐąˇŁ
https://diabetes.diabetesjournals.org/content/68/1/131ˇˇ
Trends Endocrinol Metab. 2014 Aug;
ZINC TRANSPORTER 8 (ZNT8) AND BETA CELL FUNCTION
1Barbara Davis Center for Diabetes, University of Colorado Denver Anschutz Medical Campus, Aurora, CO, 80045
2Integrated Department of Immunology, University of Colorado Denver Anschutz Medical Campus, Aurora, CO, 80045
3Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, TN 37232
Abstract
Human pancreatic ¦Â cells have exceptionally high zinc content. In ¦Â cells the highest zinc concentration is in insulin secretory granules, from which it is co-secreted with the hormone. Uptake of zinc into secretory granules is mainly mediated by zinc transporter 8 (ZnT8), the product of the SLC30A8 gene. The minor alleles of several single nucleotide polymorphisms (SNPs) in SLC30A8 are associated with decreased risk of type 2 diabetes (T2D), but the precise mechanisms underlying the protective effects remain uncertain. In this article we review current knowledge of the role of ZnT8 in maintaining zinc homeostasis in ¦Â cells, its role in glucose metabolism based on knockout mouse studies, and current theories regarding the link between ZnT8 function and T2D.
ˇˇ...
Compared to other cell types pancreatic ¦Â cells have exceptionally high zinc content [11]. Within ¦Â cells the highest levels of zinc are located in insulin secretory granules (ISGs), which may contain up to 70% of the total ¦Â cell zinc, and where the total concentration is ~10¨C20 mM [12]. ZnT8, the product of the SLC30A8 gene (UniGene Hs.532270) is responsible for the very high level of zinc accumulation in ISGs [13, 14]. The major intra-granular ligand for zinc is insulin, which is stored in a crystalline lattice of insoluble hexamers in which 6 insulin molecules are complexed with 2 Zn2+ ions and 1 Ca2+ ion (reviewed by [15, 16]). The high capacity binding provided by nascent (pro)insulin hexamers likely acts as a ˇ°sinkˇ± to drive uptake, evidenced by the fact that guinea pigs, that lack the insulin B10 His that coordinates Zn2+, accumulate only low levels of zinc in their islets [17], although whether guinea pigs actually express a ZnT8 isoform is currently unclear. Additional ISG Zn2+ ligands include inorganic ions such as phosphate, and other proteins that are co-secreted together with insulin [12]. Of particular note is islet amyloid polypeptide (IAPP) [18, 19], which is the major constituent of the amyloid plaques that are present in the pancreas of the majority of individuals with T2D [20] and have been implicated in ¦Â-cell apoptosis [21] and islet inflammation [22]. Intriguingly, ISG Zn2+ may play a key role in preventing amyloidogenesis, acting both to increase the lag time of fiber formation and decrease the rate of addition of monomers to existing fibrils [23, 24]. Moreover, IAPP can also interact with monomeric and crystalline insulin [25], which may also influence amyloidogenesis [18]. At rest, ¦Â cell cytoplasmic free Zn2+ is estimated to be approximately 400¨C450 pM [26]. Zinc in ISGs only slowly exchanges with Zn2+ in the cytoplasm [27], thus the increases in cytoplasmic Zn2+ concentrations that occur during GSIS [26, 28] are unlikely to involve internal release of the ion from ISGs. However, since upon exocytosis the elevated pH (~5.5 in granules, ~7.4 in blood) destabilizes insulin crystals releasing the monomeric hormone and free Zn2+ and Ca2+, re-uptake of co-secreted zinc could contribute to the increases observed. Co-secreted Zn2+ might potentially act in a paracrine manner to regulate glucagon secretion from ¦Á-cells [29, 30], although this remains a subject of controversy [31], but may also act in an autocrine fashion to potentiate GSIS [32].
Keywords: Islet, SLC30A8, Slc30a8ZINC TRANSPORTER 8 (ZNT8) AND BETA CELL FUNCTION
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4112161/#:~:text=Human%20pancreatic%20%CE%B2%20cells%20have%20exceptionally%20high%20zinc,8%20%28ZnT8%29%2C%20the%20product%20of%20the%20SLC30A8%20gene.ˇˇ
NATURE. Cell Death & Disease
Published: 17 July 2020
Effects of ZnT8 on epithelial-to-mesenchymal transition and tubulointerstitial fibrosis in diabetic kidney disease
Xiuli Zhang, Tingwen Guan, Boxuan Yang, Harvest F. Gu & Zhihong Chi
Abstract
Zinc transporter 8 (ZnT8) transports zinc ions for crystallization and storage of insulin in pancreatic beta-cells and ZnT8 dysfunction is involved in pathogenesis of diabetes.https://www.nature.com/articles/s41419-020-2731-6
ˇˇ
PLoS One. 2011;
Redox Regulation of Insulin Degradation by Insulin-Degrading Enzyme
Abstract
Insulin-degrading enzyme (IDE) is a thiol sensitive peptidase that degrades insulin and amyloid ¦Â, and has been linked to type 2 diabetes mellitus and Alzheimer's disease.We examined the thiol sensitivity of IDE using S-nitrosoglutathione, reduced glutathione, and oxidized glutathione to distinguish the effects of nitric oxide from that of the redox state. The in vitro activity of IDE was studied using either partially purified cytosolic enzyme from male Sprague-Dawley rats, or purified rat recombinant enzyme.
We confirm that nitric oxide inhibits the degrading activity of IDE, and that it affects proteasome activity through this interaction with IDE, but does not affect the proteasome directly. Oxidized glutathione inhibits IDE through glutathionylation, which was reversible by dithiothreitol but not by ascorbic acid. Reduced glutathione had no effect on IDE, but reacted with partially degraded insulin to disrupt its disulfide bonds and accelerate its breakdown to trichloroacetic acid soluble fragments. Our results demonstrate the sensitivity of insulin degradation by IDE to the redox environment and suggest another mechanism by which the cell's oxidation state may contribute to the development of, and the link between, type 2 diabetes and Alzheimer's disease.
...
IDE is a ubiquitously expressed zinc metalopeptidase that is inhibited by thiol reactive agents [9]. The cysteines most likely responsible for this thiol sensitivity have been identified [10]. We and others have shown that nitric oxide (NO) is capable of reacting with IDE and inhibiting activity [11], [12]. Both T2DM and AD have been associated with a chronic inflammatory state, which may result in locally increased iNOS expression and NO release [13], [14], [15], [16], [17]. In further study of this phenomenon, we have used the NO donor S-nitrosoglutathione (GSNO), which may be a physiological sink for NO in cells [18], [19]. Our results further characterize our previous report, but also demonstrate a role for glutathione in the control of insulin degradation by interaction with both IDE and partially degraded insulin itself. This is significant because of the change in cellular redox state found in both diabetes and AD.
...
IDE has been linked to T2DM and AD, thus control of its function is of interest regarding the etiology of these diseases. A number of compounds have been reported to alter IDE activity, including fatty acids, ATP, hydrogen peroxide, and NO [11], [12], [34], [35], [36]. Given the thiol sensitivity of IDE, hydrogen peroxide and NO likely react with one or more of the cysteines previously identified to have an effect on activity, namely C178, C789, C812, C819, or C966 [10], [37]. Unlike alkylating reagents such as N-ethylmaleimide, the reaction of these compounds with cysteine residues is reversible. This provides the cell with a potential mechanism to regulate the activity of IDE, depending on the redox state of the cell. But the redox control of IDE may be complex as cysteine C178 has been suggested to provide protection from inactivation by preventing nitrosylation of C110 [37]. However, this effect was seen in a mutated form of IDE with limited cysteines, and whether it is physiologically relevant is uncertain as C110 is not nitrosylated in wild type IDE containing all 13 cysteines. The results presented here add to that complexity and are significant for several reasons.Third, while GSSG inhibits IDE , reduced glutathione (GSH) appears to increase insulin degradation.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3063182/ˇˇ
Skeletal Muscle Glucose Metabolism and Insulin Resistance
L. Norton, R. DeFronzo, in Pathobiology of Human Disease, 2014
Insulin Resistance and IR Tyrosine Kinase Activity
IR tyrosine kinase activity has been examined in the skeletal muscle, adipocytes, and hepatocytes from normal-weight and obese diabetic subjects. Most, but not all, investigators have found a reduction in tyrosine kinase activity that cannot be explained by alterations in IR number or IR binding affinity (see in the succeeding text). However, restoration of normoglycemia by weight loss has been shown to correct the defect in IR tyrosine kinase activity, suggesting that the defect in tyrosine kinase is acquired secondary to some combination of hyperglycemia, distributed intracellular glucose metabolism, hyperinsulinemia, and insulin resistance ¨C all of which improved after weight loss. Exposure of cultured fibroblasts to high glucose concentration also inhibits IR tyrosine kinase activity. Since IR tyrosine kinase activity assays are performed in vitro, the results of these assays could provide misleading information with regard to IR function in vivo. To circumvent this problem, investigators have employed the euglycemic hyperinsulinemic clamp with muscle biopsies and antiphosphotyrosine immunoblot analysis to provide a ˇ®snapshotˇŻ of the insulin-stimulated tyrosine phosphorylation state of the receptor in vivo. In insulin-resistant obese nondiabetic and T2DM subjects, a substantial decrease in IR tyrosine phosphorylation has been demonstrated. However, when insulin-stimulated IR tyrosine phosphorylation was examined in normal glucose-tolerant, insulin-resistant individuals (offspring of two diabetic parents) at high risk of developing T2DM, a normal increase in tyrosine phosphorylation of the IR was observed. These findings are consistent with the concept that impaired IR tyrosine kinase activity in T2DM patients is acquired secondary to hyperglycemia or some other metabolic disturbance.https://www.sciencedirect.com/topics/neuroscience/insulin-receptor
ˇˇ
Insulin Resistance Due to Phosphorylation of IRS-1 at Serine 302
Insulin Resistance Due to Phosphorylation of IRS-1 at Serine 302 Inhibitory serine phosphorylat Inhibitory serine phosphorylation is a potential molecular mechanism for insulin resistance. We have developed a new variant of the yeast 2-hybrid method, referred to as disruptive yeast tri-hybrid (Y3H), to identify inhibitory kinases and sites of phosphorylation in insulin receptors (IR) and IRS-1. Using IR and IRS-1 as bait and prey, respectively, and JNK1 as the disruptor, we now show that phosphorylation of IRS-1 S307, a previously identified site, is necessary but not sufficient for JNK1-mediated disruption of IR/IRS-1 binding. We further identify a new phosphorylation site, S302, and show that this too is necessary for JNK1-mediated disruption. Seven additional kinases potentially linked to insulin resistance similarly block IR/IRS-1 binding in the disruptive Y3H, but through distinct, S302- and S307-independent mechanisms. Phosphospecific antibodies that recognize sequences surrounding pS302 or pS307 were used to determine whether the sites were phosphorylated under relevant conditions. Interestingly, efficient S307 phosphorylation appears to require prior phosphorylation at S302. Phosphorylation was promoted at both sites in Fao hepatoma cells by reagents known to promote S/T phosphorylation, including the phorbol ester PMA, anisomycin, calyculin A, and insulin. The antibodies further showed that pS302 and pS307 are increased in animal models of obesity and insulin resistance, including genetically obese [italic]ob/ob[/italic] mice, diet-induced obesity, and upon induction of hyperinsulinemia. These findings suggest that S302 and S307 phosphorylation are both required for JNK1-mediated inhibition of IR/IRS-1 interactions, that S302 is phosphorylated in cultured cells and [italic]in vivo[/italic] under conditions of insulin resistance, and that phosphorylations at S302 and S307 occur sequentially and under parallel conditions. JONGSOON LEE, ERIC D. WERNER, LONE HANSEN, MINSHENG YUAN, STEVEN E. SHOELSON 1289-P Boston, MA Insulin Action - Insulin Resistance
Author:
JONGSOON LEE
Congress:
64th Scientific Sessions (2004)
Category:
Insulin Action - Insulin Resistancehttps://professional.diabetes.org/abstract/insulin-resistance-due-phosphorylation-irs-1-serine-302
ˇˇ
Redox-Regulated Insulin Resistance
Science's STKE 18 Apr 2006:
Resistance to signaling by the hormone insulin is a major problem, not only in patients with type 2 diabetes but also in a broad range of other physiological states, including pregnancy, obesity, and septic shock. Houstis et al. wondered how these varied conditions might all produce a common effect on insulin signaling and whether there might be a common pathway by which they impact cellular sensitivity to insulin.Thus, they compared gene expression profiles in cultured mouse adipocytes exposed to tumor necrosis factor-¦Á (TNF-¦Á) or dexamethasone--two stimuli that produce insulin resistance but act through very different signaling mechanisms (the former through a cytokine receptor at the cell surface and the latter through a nuclear hormone receptor). Their analyses indicated that 18% of the genes that were similarly regulated in response to both treatments encoded products that influenced the abundance of reactive oxygen species (ROS) (radical forms of oxygen that are produced during cellular respiration or other enzymatic reactions and are known both to damage other molecules and to act as signaling molecules).
Following this lead, the authors found that both dexamethasone and TNF-¦Á caused increased intracellular accumulation of ROS. Furthermore, treatment of cultured cells with antioxidant molecules or expression of enzymes that remove ROS suppressed insulin resistance induced by dexamethasone and TNF. In a mouse model of obesity, chronic treatment of mice with a chemical antioxidant resulted in improved glucose homeostasis and sensitivity to insulin.
The authors suggest that the relation of ROS to insulin sensitivity may have evolved because increased accumulation of ROS could be interpreted by the cell as an imbalance between substrate availability and oxidative capacity to which decreased insulin signaling (and thus decreased glucose uptake) would be an appropriate response. How various stimuli might converge on ROS generation and how ROS actually influence insulin signaling remain to be worked out, but in the meantime, the authors suggest that antioxidant therapy should be considered a therapeutic strategy to manage insulin resistance in the clinic.
N. Houstis, E. D. Rosen, E. S. Lander, Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944-948 (2006). [Online Journal]https://stke.sciencemag.org/content/2006/331/tw126.abstract
ˇˇ
J Clin Invest. 2006 Jul 3;
Inflammation and insulin resistance
Steven E. Shoelson, Jongsoon Lee, and Allison B. Goldfine
Joslin Diabetes Center and Department of Medicine, Harvard Medical School, Boston, Massachusetts, USA.
Abstract
Over a hundred years ago, high doses of salicylates were shown to lower glucose levels in diabetic patients. This should have been an important clue to link inflammation to the pathogenesis of type 2 diabetes (T2D), but the antihyperglycemic and antiinflammatory effects of salicylates were not connected to the pathogenesis of insulin resistance until recently. Together with the discovery of an important role for tissue macrophages, these new findings are helping to reshape thinking about how obesity increases the risk for developing T2D and the metabolic syndrome. The evolving concept of insulin resistance and T2D as having immunological components and an improving picture of how inflammation modulates metabolism provide new opportunities for using antiinflammatory strategies to correct the metabolic consequences of excess adiposity.Historical perspectives on the link between inflammation and insulin resistance
Clues to the involvement of inflammation in diabetes date back to more than a century ago, when high doses of sodium salicylate (5.0¨C7.5 g/d) were first demonstrated to diminish glycosuria in diabetic patients having ˇ°the milder form of the disease,ˇ± presumably type 2 diabetes (T2D) (1¨C3). In 1876 Ebstein concluded that sodium salicylate could make the symptoms of diabetes mellitus totally disappear (1, 3). Similarly, in 1901 Williamson found that ˇ°sodium salicylate had a definite influence in greatly diminishing the sugar excretionˇ± (2). The effect was rediscovered in 1957 when an insulin-treated diabetic, given high-dose aspirin to treat the arthritis associated with rheumatic fever, no longer required daily insulin injections (4). Fasting and postchallenge glucose concentrations were nearly normal, despite the discontinuation of insulin and treatment with aspirin alone. Upon resolution of the joint symptoms, the aspirin was discontinued, and a repeat glucose tolerance test was grossly abnormal. Intrigued by these findings, Reid and colleagues prospectively studied 7 additional patients, 4 the ˇ°overweight mild typeˇ± and three ˇ°lean more severe diabeticsˇ± (4). Over a 2-week course of high-dose aspirin (5.0¨C8.0 g/d), fasting blood glucose levels fell from an average of more than 190 mg/dl before treatment to 92 mg/dl; every patient responded. Additional trials showed equivalent efficacy (5, 6). Mechanistic studies focused on insulin secretion, undoubtedly because of the established importance of insulin secretion in the pathogenesis of diabetes, but the findings were inconclusive (7). Insulin resistance and its role in the pathogenesis of T2D were less well appreciated, and, as a result, insulin sensitization was not considered as a potential mechanism for glucose lowering at the time. It wasnˇŻt until much later that studies looking at a role for inflammation in the pathogenesis of insulin resistance reinvestigated the hypoglycemic actions of salicylates and identified the molecular target to be the I¦ĘB kinase-¦Â (IKK¦Â)/NF-¦ĘB axis (8¨C10).Inflammation and insulin resistance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1483173
NF-¦ĘB-dependent airway inflammation triggers systemic ...
https://medschool.vanderbilt.edu/mpb/publication/nf...
NF-¦ĘB-dependent airway inflammation triggers systemic insulin resistance.. Inflammatory lung diseases (e.g., pneumonia and acute respiratory distress syndrome) are associated with hyperglycemia, even in patients without a prior diagnosis of Type 2 diabetes.ˇˇ
Non-steroidal anti-inflammatory drugs activate NADPH ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3671242
May 30, 2013 ˇ¤ Non-steroidal anti-inflammatory drugs activate NADPH oxidase in adipocytes and raise the H 2 O 2 pool to prevent cAMP-stimulated protein kinase a activation and inhibit lipolysis
Cited by: 12
Publish Year: 2013
Author: H¨¦ctor V¨˘zquez-Meza, Martha Zentella de Piña, Ju
Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue
Chang Yeop Han
Division of Metabolism, Endocrinology & Nutrition, Department of Medicine and Diabetes and Obesity Center of Excellence, University of Washington, Seattle, WA, USA.
Abstract
Obesity resulting from the delivery of an excess amount of energy to adipose tissue from glucose or free fatty acids is associated with insulin resistance and adipose tissue inflammation. Reactive oxygen species (ROS) have been implicated as contributors to both the onset and the progression of insulin resistance.ROS can be generated by overloading the mitochondrial oxidative phosphorylation system, and also by nicotinamide adenine dinucleotide phosphate oxidases (NOX) produced by either adipocytes, which only produce NOX4, or by macrophages, which produce mainly NOX2. The source of the ROS might differ in the early, intermediate and late stages of obesity, switching from NOX4-dependence in the early phases to NOX2-dependence, in the intermediate phase, and transiting to mitochondria-dependence later in the time course of obesity. Thus, depending on the stage of obesity, ROS can be generated by three distinct mechanisms: i.e., NOX4, NOX2, and mitochondria.
In this review, we will discuss whether NOX4-, NOX2-, and/or mitochondria-derived ROS is/are causal in the onset of adipocyte insulin resistance as obesity progresses. Moreover, we will review the pathophysiological roles of NOX4, NOX2, and mitochondria-derived ROS on adipose tissue inflammation.
Keywords: Adipocytes, Insulin resistance, Mitochondria, NADPH oxidase, Obesity, Reactive oxygen specieshttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC4995181/
ˇˇ
Insulin-independent glucose transport regulates insulin ...
https://www.sciencedirect.com/science/article/pii/S0014579398011491
Oct 09, 1998 ˇ¤ Thus, GLUT4 is insulin-dependent and mediates the insulin-stimulated glucose disposal in muscle tissue. GLUT1 protein may be responsible, at least in part, for the constitutive, insulin-independent glucose uptake in all cells including muscle . In addition to its traditional role, muscle tissue has other functions, such as a role in immunology.
Cited by: 105
Publish Year: 1998
Author: Pertti Ebeling, Heikki A Koistinen, Veikko A Koivistoˇˇ
Nutritional Support in Intensive Care Patients
Key Messages
Glucose utilization is increased in non-insulin dependent organs and decreased in insulin-dependent organs and tissues;
Lipolysis is activated by the critical illness, particularly in patients with sepsis and acute inflammatory dieases;
Fat utilisation is stimulated in fasted and septic patients, reduced in patients with circulatory failure;
Protein catabolism is increased, and exceeds protein synthesis, promoting an erosion of the fat-free mass. Glucose and insulin decrease protein catabolism;
Formulas for critically ill patients should include 1.5 ¨C 2.0 g/kg protein per day, carbohydrate and lipids. Lipid supply should be reduced in patients with acute ischemic heart diseases and major burns.Insulin resistance
Insulin resistance is a hallmark of the critical illness (Fig. 1), leading to hyperglycemia and major changes in glucose, fat and protein metabolism (see module 17.2 for further details). This has important nutritional consequences, since it may be associated with a decreased efficacy of nutritional support.
Insulin resistance influences glucose plasma level, glucose uptake in skeletal muscle and adipose tissue, as well as the endogenous glucose production in the liver and kidney.
Figure 1
In healthy subjects, insulin is a major regulator of glucose endogenous production, to achieve a constant level of blood glucose: glucose production is suppressed by carbohydrate-rich meals and stimulated in the post-absorptive state (Fig. 2). This is not the case in surgical and critically ill patients, in whom the endogenous production of glucose stays high despite carbohydrate administration, as a consequence of insulin resistance (Fig. 3).
Figure 2
Figure 3
Studies performed in critically ill patients receiving isocaloric nutrition with various proportion of glucose and fat, show that the endogenous production of glucose stays constant for glucose supply ranging from 28 to 75% of total energy (Fig. 4). Such mechanism allows a large supply of glucose to the glucose-dependent tissues like the immune, inflammatory cells and the wounds.http://lllnutrition.com/mod_lll/TOPIC18/old/m182e.htm
ˇˇ
Which tissues are insulin dependent for glucose uptake? Is ...
https://www.quora.com/Which-tissues-are-insulin...
In brief, Skeletal muscle accounts for approximately 70% of insulin mediated glucose uptake. Adipose tissue accounts for about 10% of insulin dependent glucose uptake. While intracellular glucose uptake in the liver is not insulin dependent, insulin does modulate key metabolic processes in the liver through signalling cascades.ˇˇ
Developmental Physiology of Carbohydrate Metabolism and the Pancreas
Kathryn Beardsall, Amanda L. Ogilvy-Stuart, in Maternal-Fetal and Neonatal Endocrinology, 2020
34.4.2.1 Glucose Uptake
Glucose uptake is dependent on a family of eight developmentally regulated glucose transporters, each of which has a specific tissue distribution. GLUT 1 has a high affinity for glucose and is insulin independent, being responsible for basal glucose uptake.73¨C75 It is found in most tissues, but particularly in the brain, and maintains glucose transport across the blood-brain barrier. GLUT 1 appears to be downregulated by high glucose concentrations and upregulated by low glucose concentrations.76 After birth, GLUT 1 levels decrease, and the other isoforms increase: GLUT 2 in the liver, GLUT 3 in the brain, and GLUT 4 in the muscle.74, 76 InsulinˇŻs actions on glucose uptake are predominantly mediated by GLUT 4, which is found in muscle, fat, and heart, where glucose uptake is insulin-mediated. GLUT 4 is usually found in intracellular vesicles, but in adults, insulin stimulates the migration of GLUT 4 transporters to the plasma membrane.77 However, in the newborn, upregulation of GLUT 4 transporters in muscle only respond modestly to insulin.78https://www.sciencedirect.com/topics/medicine-and-dentistry/glucose-uptake
ˇˇ
On the foundation of this interesting finding, Dr L Boominathan PhD, Director-cum-chief Scientist of GBMD, reports that: Omega-3 PUFA-based therapy for Diabetes Mellitus: ¦Ř-PUFAs (DHA+EPA) augment the expression of FGF19 and FGF1, attenuate hepatic glucose production, decrease hepatic acetyl CoA content, bring down the levels of plasma ACTH, and corticosterone, augment insulin sensitivity, promote weight loss and alleviate TIDM, via up regulation of its target gene
ˇˇ
J Nutr Biochem, 2015 Jun;26(6):571-84. doi: 10.1016/j.jnutbio.2015.02.001. Epub 2015 Feb 26.
Mechanisms of enhanced insulin secretion and sensitivity with n-3 unsaturated fatty acids
Maharshi Bhaswant 1, Hemant Poudyal 2, Lindsay Brown 3
1Centre for Chronic Disease Prevention & Management, College of Health and Biomedicine, Victoria University, Melbourne VIC 3021, Australia; School of Health and Wellbeing, University of Southern Queensland, Toowoomba QLD 4350, Australia.
2Department of Diabetes, Endocrinology and Nutrition, Graduate School of Medicine and The Hakubi Center for Advanced Research, Kyoto University, Kyoto 606-8302, Japan.
3School of Health and Wellbeing, University of Southern Queensland,
The widespread acceptance that increased dietary n-3 polyunsaturated fatty acids (PUFAs), especially ¦Á-linolenic acid (ALA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), improve health is based on extensive studies in animals, isolated cells and humans. Visceral adiposity is part of the metabolic syndrome, together with insulin resistance, dyslipidemia, hypertension and inflammation. Alleviation of metabolic syndrome requires normalization of insulin release and responses.This review assesses our current knowledge of the mechanisms that allow n-3 PUFAs to improve insulin secretion and sensitivity. EPA has been more extensively studied than either ALA or DHA. The complex actions of EPA include increased G-protein-receptor-mediated release of glucagon-like peptide 1 (GLP-1) from enteroendocrine L-cells in the intestine, up-regulation of the apelin pathway and down-regulation of other control pathways to promote insulin secretion by the pancreatic ¦Â-cells, together with suppression of inflammatory responses to adipokines, inhibition of peroxisome proliferator-activated receptor ¦Á actions and prevention of decreased insulin-like growth factor-1 secretion to improve peripheral insulin responses. The receptors involved and the mechanisms of action probably differ for ALA and DHA, with antiobesity effects predominating for ALA and anti-inflammatory effects for DHA. Modifying both GLP-1 release and the actions of adipokines by n-3 PUFAs could lead to additive improvements in both insulin secretion and sensitivity.
Keywords: Adipokines; Inflammation; Insulin; Obesity; n-3 polyunsaturated fatty acids.https://pubmed.ncbi.nlm.nih.gov/25841249/
ˇˇ
J Nutr. 2010 Nov;
Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation
Nishan S Kalupahana 1, Kate Claycombe, Shelley J Newman, Taryn Stewart, Nalin Siriwardhana, Nirupa Matthan, Alice H Lichtenstein, Naima Moustaid-Moussa
1Department of Animal Science, Obesity Research Center, University of Tennessee, Knoxville, TN 37996, USA.
Abstract
We investigated the effects of eicosapentaenoic acid (EPA) on prevention (P) and reversal (R) of high saturated-fat (HF) diet-induced obesity and glucose-insulin homeostasis.Male C57BL/6J mice were fed low-fat (LF; 10% energy from fat), HF (45% energy from fat), or a HF-EPA-P (45% energy from fat; 36 g/kg EPA) diet for 11 wk. A 4th group was initially fed HF for 6 wk followed by the HF-EPA-R diet for 5 wk.
As expected, mice fed the HF diet developed obesity and glucose intolerance. In contrast, mice fed the HF-EPA-P diet maintained normal glucose tolerance despite weight gain compared with the LF group. Whereas the HF group developed hyperglycemia and hyperinsulinemia, both HF-EPA groups (P and R) exhibited normal glycemia and insulinemia. Further, plasma adiponectin concentration was lower in the HF group but was comparable in the LF and HF-EPA groups, suggesting a role of EPA in preventing and improving insulin resistance induced by HF feeding. Further analysis of adipose tissue adipokine levels and proteomic studies in cultured adipocytes indicated that dietary EPA supplementation of HF diets was associated with reduced adipose inflammation and lipogenesis and elevated markers of fatty acid oxidation. In C57BL/6J mice, EPA minimized saturated fat-induced insulin resistance and this is in part mediated by its effects on fatty acid oxidation and inflammation.
ˇˇ
JʳƷ2010Äę11ÔÂŁ»
¶ţʮ̼ÎĺĎ©Ëáͨąýµ÷˝ÚÖ¬·ľ×éÖŻŃ×Ö˘Ŕ´Ô¤·ŔşÍÄćת¸ßÖ¬ŇűĘłÓŐµĽµÄ·ĘĹÖСĘóµÄŇȵşËصֿą
Nishan S Kalupahana 1Ł¬Kate ClaycombeŁ¬Shelley J NewmanŁ¬Taryn StewartŁ¬Nalin SiriwardhanaŁ¬Nirupa MatthanŁ¬Alice H LichtensteinŁ¬Naima Moustaid-Moussa
1ĚďÄÉÎ÷´óѧ·ĘĹÖŃĐľżÖĐĐĶŻÎďżĆѧϵŁ¬ĹµżË˹ά¶űŁ¬ĚďÄÉÎ÷ÖÝ37996Ł¬ĂŔąúˇŁ
łéĎó
ÎŇĂǵ÷˛éÁ˶ţʮ̼ÎĺĎ©ËᣨEPAŁ©¶Ô¸ß±ĄşÍÖ¬·ľŁ¨HFŁ©ŇűĘłŇýĆđµÄ·ĘĹÖÖ˘şÍĆĎĚŃĚÇ-ŇȵşËŘÎČ̬µÄÔ¤·ŔŁ¨PŁ©şÍÄćתŁ¨RŁ©µÄÓ°Ď졣¸řĐŰĐÔC57BL / 6JСĘóµÍÖ¬ŇűĘłŁ¨LF; 10ŁĄµÄÖ¬·ľÄÜÁżŁ©Ł¬HFŁ¨45ŁĄµÄÖ¬·ľÄÜÁżŁ©»ňHF-EPA-PŁ¨45ŁĄµÄÖ¬·ľÄÜÁż; 36 g / kg EPAŁ©ŇűĘł11ÖܡŁµÚËÄ×é×îłőŇÔHFÎąŃř6ÖÜŁ¬Č»şóŇÔHF-EPA-RŇűʳιŃř5ÖܡŁ
ČçÔ¤ĆÚµÄÄÇŃůŁ¬ÎąĘłHFŇűĘłµÄСĘółöĎÖ·ĘĹÖşÍĆĎĚŃĚDz»ÄÍÖ˘ˇŁĎŕ·´Ł¬ÓëLF×éĎŕ±ČŁ¬ÎąŃřHF-EPA-PČŐÁ¸µÄСĘ󾡹ÜĚĺÖŘÔöĽÓŁ¬µ«ČÔάłÖŐýłŁµÄĆĎĚŃĚÇÄÍÁżˇŁ HF×éłöĎÖ¸ßŃŞĚǺ͸ßŇȵşËŘŃŞÖ˘Ł¬¶řHF-EPA×飨PşÍRŁ©ľůĎÔĘľŐýłŁµÄŃŞĚÇşÍŇȵşËŘŃŞÖ˘ˇŁ´ËÍ⣬HF×éµÄŃŞ˝¬Ö¬ÁŞËŘŨ¶Č˝ĎµÍŁ¬¶řLFşÍHF-EPA×éµÄŃŞ˝¬Ö¬ÁŞËŘŨ¶ČĎŕµ±Ł¬Őâ±íĂ÷EPAÔÚÔ¤·ŔşÍ¸ÄÉĆHFÎąŃřÓŐµĽµÄŇȵşËصֿąÖеÄ×÷Ó᣶ÔÖ¬·ľ×éÖŻÖ¬·ľŇň×ÓˮƽµÄ˝řŇ»˛˝·ÖÎöşÍĹŕŃřµÄÖ¬·ľĎ¸°űÖеĵ°°×ÖĘ×éѧŃĐľż±íĂ÷Ł¬ÉĹĘłEPA˛ąłäHFŇűĘłÓëĽőÉŮÖ¬·ľŃ×Ö˘şÍÖ¬·ľÉúłÉŇÔĽ°Ö¬·ľËáŃő»Ż±ęĽÇÎďÉý¸ßÓйءŁÔÚC57BL / 6JСĘóÖĐŁ¬EPAĘą±ĄşÍÖ¬·ľÓŐµĽµÄŇȵşËصֿą×îС»ŻŁ¬˛ż·ÖÔŇňĘÇËü¶ÔÖ¬·ľËáŃő»ŻşÍŃ×Ö˘µÄÓ°Ď졣
https://pubmed.ncbi.nlm.nih.gov/20861209/
ˇˇ
Biochem Soc Trans. 2017 Oct
Omega-3 fatty acids and inflammatory processes: from molecules to man
Philip C Calder 1 2
1Human Development and Health Academic Unit, Faculty of Medicine, University of Southampton
2NIHR Southampton Biomedical Research Centre, University Hospital Southampton NHS Foundation Trust and University of Southampton,
Abstract
Inappropriate, excessive or uncontrolled inflammation contributes to a range of human diseases. Inflammation involves a multitude of cell types, chemical mediators and interactions. The present article will describe nutritional and metabolic aspects of omega-6 (n-6) and omega-3 (n-3) fatty acids and explain the roles of bioactive members of those fatty acid families in inflammatory processes. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are n-3 fatty acids found in oily fish and fish oil supplements. These fatty acids are capable of partly inhibiting many aspects of inflammation including leucocyte chemotaxis, adhesion molecule expression and leucocyte-endothelial adhesive interactions, production of eicosanoids like prostaglandins and leukotrienes from the n-6 fatty acid arachidonic acid and production of pro-inflammatory cytokines. In addition, EPA gives rise to eicosanoids that often have lower biological potency than those produced from arachidonic acid, and EPA and DHA give rise to anti-inflammatory and inflammation resolving mediators called resolvins, protectins and maresins. Mechanisms underlying the anti-inflammatory actions of EPA and DHA include altered cell membrane phospholipid fatty acid composition, disruption of lipid rafts, inhibition of activation of the pro-inflammatory transcription factor nuclear factor ¦ĘB so reducing expression of inflammatory genes and activation of the anti-inflammatory transcription factor peroxisome proliferator-activated receptor ¦Ă. Animal experiments demonstrate benefit from EPA and DHA in a range of models of inflammatory conditions. Human trials demonstrate benefit of oral n-3 fatty acids in rheumatoid arthritis and in stabilizing advanced atherosclerotic plaques. Intravenous n-3 fatty acids may have benefits in critically ill patients through reduced inflammation. The anti-inflammatory and inflammation resolving actions of EPA, DHA and their derivatives are of clinical relevance.Omega-3Ö¬·ľËáşÍŃ×Ö˘ąýłĚŁş´Ó·Ö×Óµ˝ČË
˛»Ęʵ±Ł¬ąý¶Č»ňÎŢ·¨żŘÖƵÄŃ×Ö˘»áµĽÖÂһϵÁĐČËŔ༲˛ˇˇŁŃ×Ö˘É漰¶ŕÖÖϸ°űŔŕĐ͡˘»ŻŃ§˝éÖĘşÍĎ໥×÷ÓáŁ
±ľÎÄ˝«ĂčĘöomega-6Ł¨n-6Ł©şÍomega-3Ł¨n-3Ł©Ö¬·ľËáµÄÓŞŃřşÍ´úĐ»·˝Ă棬˛˘˝âĘÍŐâĐ©Ö¬·ľËáĽŇ×ĺµÄÉúÎď»îĐÔłÉÔ±ÔÚŃ×Ö˘ąýłĚÖеÄ×÷ÓáŁ
¶ţʮ̼ÎĺĎ©ËᣨEPAŁ©şÍ¶ţĘ®¶ţĚĽÁůĎ©ËᣨDHAŁ©ĘÇÔÚÓÍĐÔÓăşÍÓăÓͲąĆ·ÖĐ·˘ĎÖµÄn-3Ö¬·ľËᡣ
ŐâĐ©Ö¬·ľËáÄÜą»˛ż·ÖŇÖÖĆŃ×Ö˘µÄĐí¶ŕ·˝Ă棬°üŔ¨°×ϸ°űÇ÷»ŻĐÔŁ¬Őł¸˝·Ö×Ó±í´ďşÍ°×ϸ°ű-ÄÚƤճ¸˝Ď໥×÷ÓĂŁ¬´Ón-6Ö¬·ľËỨÉúËÄĎ©Ëá˛úÉúŔŕÇ°ÁĐĎŮËغͰ×ČýĎ©µÄŔ໨ÉúËáŇÔĽ°´ŮŃ×ĐÔϸ°űŇň×ӵIJúÉúˇŁ´ËÍ⣬EPA˛úÉúµÄŔ໨ÉúËáµÄÉúÎďЧÁ¦Í¨łŁµÍÓÚ»¨ÉúËÄĎ©Ëá˛úÉúµÄŔ໨ÉúËᣬ¶řEPAşÍDHAÔň˛úÉúłĆÎŞĎűŃ×ËŘ(resolvins)Ł¬±Ł»¤ËŘ(protectins)şÍÂíÁÖËŘ( maresins)µÄżąŃ׺ÍĎűŃ×˝éÖʡŁ
EPAşÍDHAµÄżąŃ××÷ÓõÄDZÔÚ»úÖĆ°üŔ¨¸Ä±äϸ°űĤÁ×Ö¬Ö¬·ľËá×éłÉŁ¬ĆĆ»µÖ¬·¤Ł¬ŇÖÖĆ´ŮŃ×ת¼Ňň×ÓşËŇň×Ó¦ĘBŁ¨NF-kB)µÄĽ¤»îŁ¬´Ó¶řĽőÉŮŃ×Ö˘»ůŇňµÄ±í´ďşÍĽ¤»îżąŃ×»îĐÔת¼Ňň×ÓąýŃő»ŻÎďøĚĺÔöÖłÎďĘÜĚĺ¦ĂŁ¨PPAR gamma)ˇŁ
¶ŻÎďʵŃéÖ¤Ă÷Ł¬ÔÚ¶ŕÖÖŃ×֢״żöÄŁĐÍÖĐŁ¬EPAşÍDHAµÄŇć´¦ˇŁČËĚĺĘÔŃéÖ¤Ă÷Ł¬żÚ·ţn-3Ö¬·ľËá¶ÔŔŕ·çĘŞąŘ˝ÚŃ׺ÍÎȶ¨ÍíĆÚ¶ŻÂöÖŕŃůÓ˛»Ż°ßżéľßÓĐŇć´¦ˇŁľ˛ÂöÄÚn-3Ö¬·ľËáżÉÄÜͨąýĽőÇáŃ×Ö˘¶ÔÖŘÖ˘»ĽŐßÓĐŇ档
EPAŁ¬DHAĽ°ĆäŃÜÉúÎďµÄżąŃ׺ÍĎűŃ××÷ÓĂľßÓĐÁŮ´˛ŇâŇ塣
Keywords: cytokine; disease; eicosanoid; fish oil; inflammation; polyunsaturated fatty acid.https://pubmed.ncbi.nlm.nih.gov/28900017/
ˇˇ
PREDIABETES PATIENTS IMPROVE FASTING GLUCOSE WITH ZINC
Six-month regimen of 30 mg zinc sulfate once daily found effective compared with those on placebo, according to study. In Diabetes Research and Clinical Practice, Australian researchers used a cohort of 55 adults, mean age of 44, to assess whether participants would improve fasting glucose with zinc supplementation. Adults with prediabetes randomly assigned daily zinc supplementation saw improved fasting plasma glucose over 6 months vs. those assigned a placebo, study findings show. They found those in the zinc supplement group also had statistically significant improvements in beta-cell function, insulin resistance and insulin sensitivity. Many genetic variants have been associated with glucose homeostasis and type 2 diabetes in genome-wide association studies. Zinc is an essential micronutrient that is important for ¦Â-cell function and glucose homeostasis. So they tested the hypothesis that zinc intake could influence the glucose-raising effect of specific variants. Chronic elevations in fasting or postprandial glucose levels are the cardinal features of type 2 diabetes (T2D), a common complex disease caused by the interplay of genetic and lifestyle factors. Genome-wide association studies (GWAS) have identified genetic loci reproducibly associated with glycemic traits or T2D. These studies improved our understanding of the mechanisms underlying impaired glucose homeostasis and T2D, potentially aiding the development of novel and individualized medical therapies.https://diabetestalk.net/blood-sugar/zinc-and-blood-sugar
ˇˇ
Renalase as a Novel Biomarker for Evaluating the Severity ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5102749H 2 O 2 is a mild oxidant but is converted into hydroxyl radicals, which are extremely reactive and more toxic than other ROS [49, 50]. Therefore, to investigate the responsiveness of renalase in hepatic cells under the condition of oxidative stress, H 2 O 2 was used to mimic the ROS-burst in oxidative stress.
ˇˇ
ˇˇ
Oxid Med Cell Longev. 2016;
Renalase as a Novel Biomarker for Evaluating the Severity of Hepatic Ischemia-Reperfusion Injury
Huili Li, 1 Jianrong Guo, 1 Hongli Liu, 2 Yanfeng Niu, 1 Lixia Wang, 3 Kun Huang, 4 , * and Jiliang Wang 1 , *
Abstract
Hepatic ischemia-reperfusion (I/R) injury is a serious complication in clinical practice. However, no efficient biomarkers are available for the evaluation of the severity of I/R injury. Recently, renalase has been reported to be implicated in the I/R injury of various organs. This protein is secreted into the blood in response to increased oxidative stress. To investigate the responsiveness of renalase to oxidative stress, we examined the changes of renalase in cell and mouse models. We observed a significant increase of renalase expression in HepG2 cells in a time- and dose-dependent manner when treated with H2O2. Renalase expression also increased significantly in liver tissues that underwent the hepatic I/R process. The increased renalase levels could be efficiently suppressed by antioxidants in vitro and in vivo. Furthermore, serum renalase levels were significantly increased in the mouse models and also efficiently suppressed by antioxidants treatment. The variation trends are consistent between renalase and liver enzymes in the mouse models. In conclusion, renalase is highly sensitive and responsive to oxidative stress in vitro and in vivo. Moreover, renalase can be detected in the blood. These properties make renalase a highly promising biomarker for the evaluation of the severity of hepatic I/R injury.Renalase as a Novel Biomarker for Evaluating the Severity of Hepatic Ischemia-Reperfusion Injury
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5102749/ˇˇ
8 Amazing Breakthroughs in Diabetes Research That Are Giving Us Hope
SIGNE DEAN & MIKE MCRAE2 APRIL 2018
According to recent research, we're not entirely sure how many diseases the label 'diabetes' covers. But no matter what causes our bodies to struggle with their blood sugar levels, it's a serious condition that requires daily care.Scientists have been working hard to find cures, new treatments, and better management techniques for the millions of people worldwide dealing with diabetes.
Here are some of the latest developments you need to know about.
1. Insulin producing implants made from stem cells
Clinical trials began last year for testing for ViaCyte's PEC-Direct device; a credit-card sized implant containing insulin-producing cells derived from stem cells.
Previous research had shown the implants could mature and function inside patients. Together with a cohort of volunteers who started testing in January, the new research should tell us soon whether the technology can help people with type-1 diabetes.
2. Brand new beta cells
Type 1 diabetes develops when a person's immune system wipes out insulin-producing beta cells in the pancreas.
But it turns out that another type of immature beta cell has been hiding in our pancreases all along, and scientists think it might be possible to use these 'virgin beta cells' to restore the functionality of the pancreas.
3. A common blood pressure medication
A drug on the World Health Organisation's list of essential drugs could have another purpose; blocking a molecule implemented in the autoimmune response that can give rise to type-1 diabetes.
Called methyldopa, the compound already has an important job treating high blood pressure in pregnant women and children. It's left to be seen if it could help reduce the incidence of diabetes in some way, but the fact it's already being used - rather than being stuck in the lab - makes for a promising find.
4. A unique transplant Doctors implanted insulin-producing cells into a fatty membrane in the stomach cavity,
One woman with severe type 1 diabetes has spent a year without insulin injections thanks to an experimental transplant.
Doctors implanted insulin-producing cells into a fatty membrane in the stomach cavity, and the success of the operation is paving the way towards more people receiving artificial pancreases.
5. An extreme diet
A clinical trial conducted on just 298 volunteers in the UK last year found an intensive weight management program could put type-2 diabetes into remission for those who lose a significant amount of weight.
The subjects were limited to roughly 850 calories a day for three to five months, consuming mostly soups and health shakes, before having more food introduced.
A similar study conducted on rats last year in the US also showed low calorie diets might help those who can stick to it reverse their condition.
6. Glucose-monitoring contact lenses
Until we can nail down a cure, there will always been a need to monitor those messy blood glucose levels.
Checking your tears for glucose using a smart contact lens, or monitoring your sweat with colour changing ink, could be a whole lot less invasive than drawing blood.
They're not new ideas, but constant improvements in miniaturising technology could mean these kinds of devices aren't too far off.
7. Loneliness could make us prone
While we can list a variety of genetic and lifestyle factors that affect a body's growing resistance to insulin, there's still a lot to learn.
A study published late last year involving nearly 3,000 subjects aged 40 to 75 found there seems to be a significant relationship between social isolation and type-2 diabetes.
It's not clear what the link might be, but having a few housemates or a local social group could make all the difference.
8. Mexican cavefish evolved to be diabetic
While developing a resistance to insulin is bad news in humans, the pale, eyeless animal known as a Mexican cavefish evolved a new version of the insulin receptor that makes it harder for the hormone to bind.
This isn't exactly a problem for the fish, which have also evolved other features to help it compensate. Studying its biology might help shine a light on how diabetes evolved in humans, and maybe even lead to new treatments.https://www.sciencealert.com/new-breakthroughs-diabetes-research-treatments-2018
ˇˇ
Catalase expression in pancreatic alpha cells of diabetic ...
https://pubmed.ncbi.nlm.nih.gov/17102991The pancreatic islet beta cells are very sensitive to oxidative stress, probably due to the extremely low level of anti-oxidant enzymes, particularly catalase. In contrast to beta cells, pancreatic alpha cells are significantly more resistant to diabetogenic toxins. However, whether alpha cells express a different level of catalase is not known.
ˇˇ
ˇˇ
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3026656/
ˇˇ

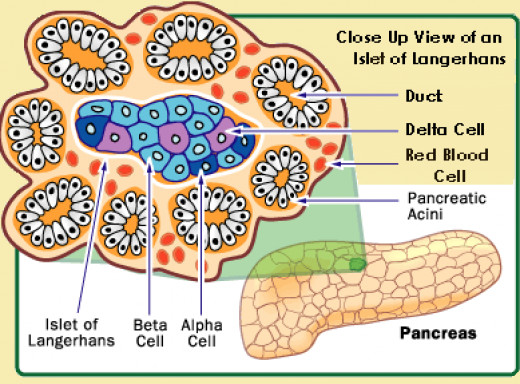
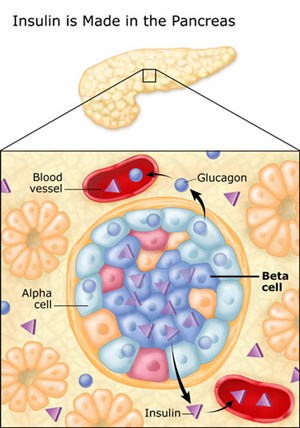
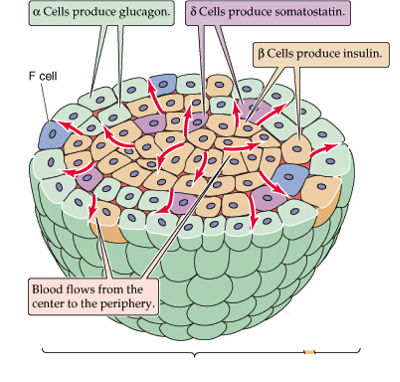
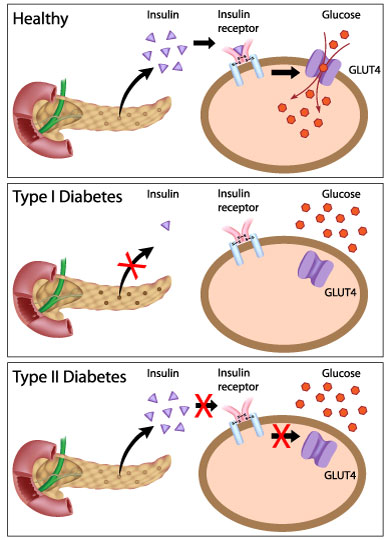
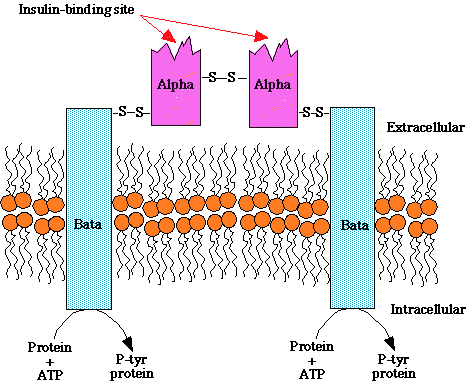
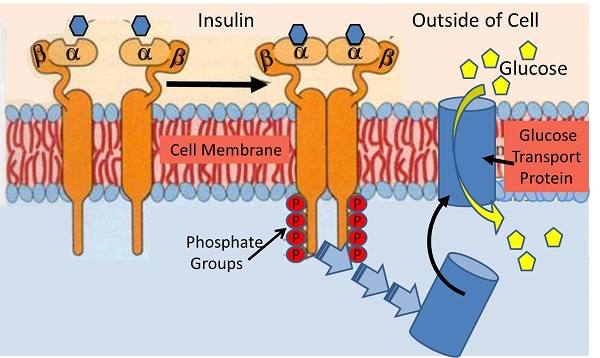
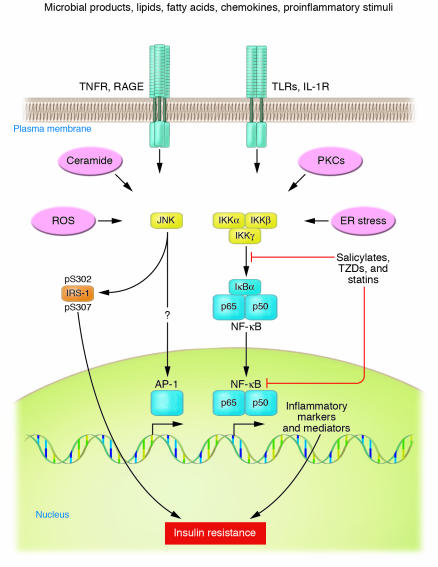
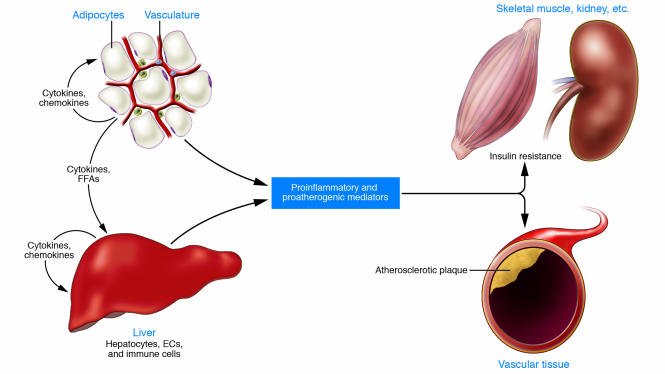
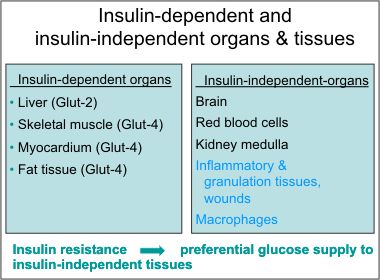
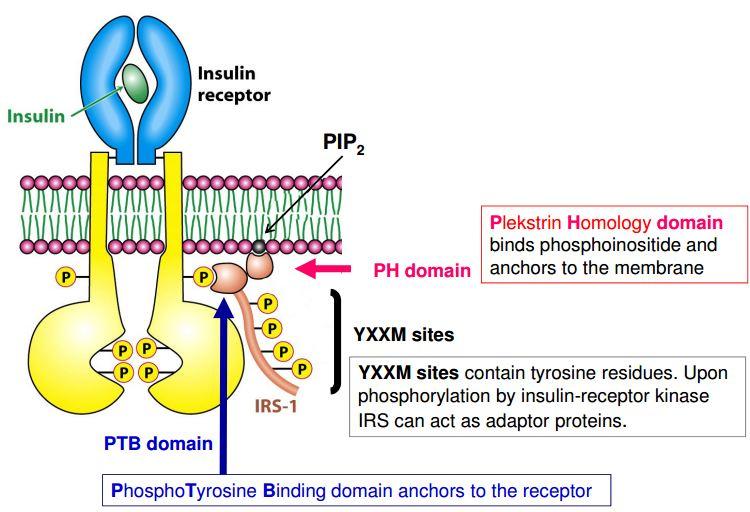
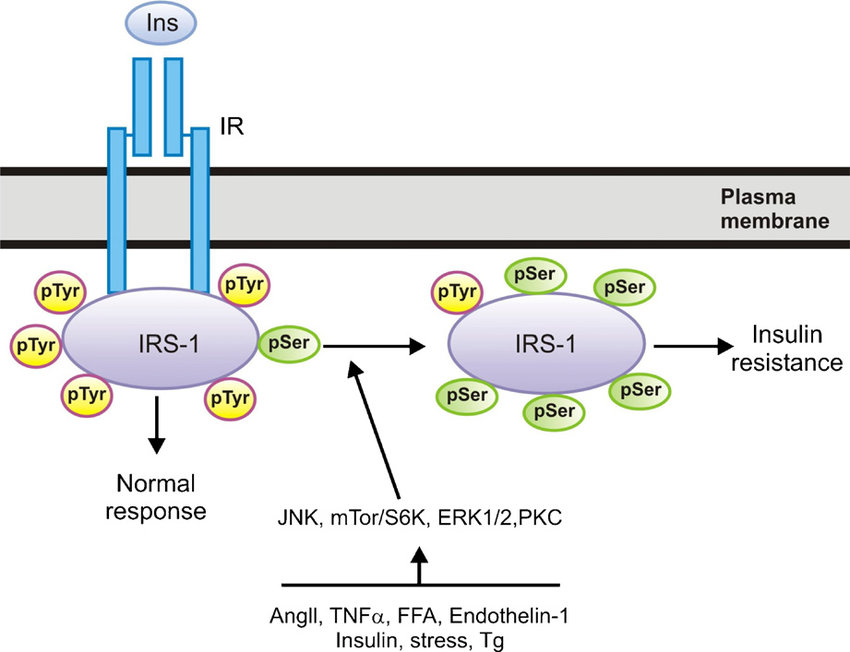
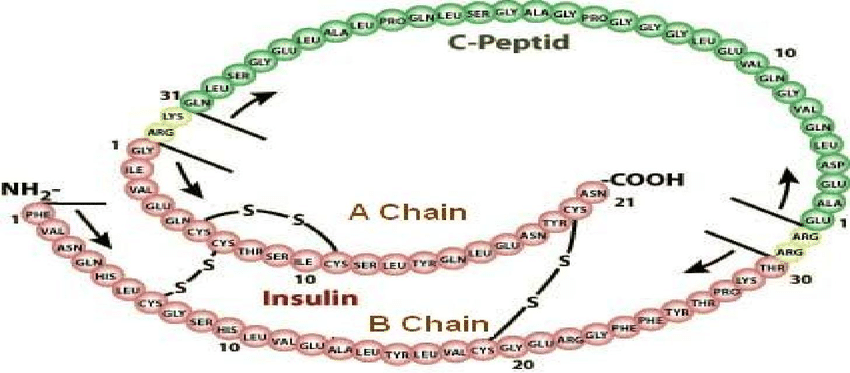

.png)