��
��
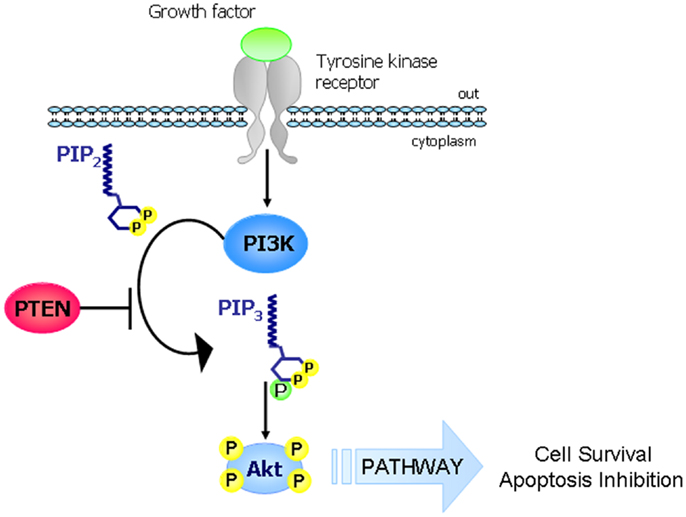
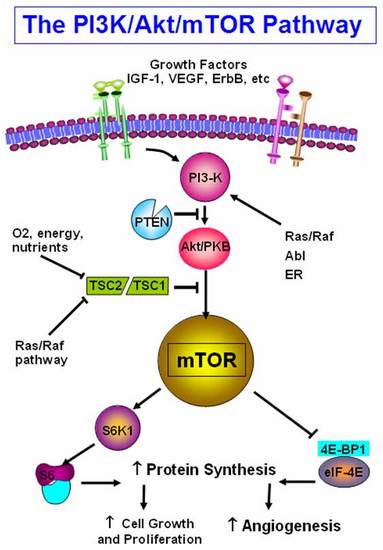
The PI3K/AKT/mTOR pathway is an intracellular signaling pathway important in regulating the cell cycle. Therefore, it is directly related to cellular quiescence, proliferation, cancer, and longevity. PI3K activation phosphorylates and activates AKT, localizing it in the plasma membrane.
1.Natural PI3K/AKT/mTOR inhibitors:
2.fish oil EPA/DHA diet significantly increased the level of PTEN protein in the breast tumors.
In addition, the fish oil diet attenuated the PI 3 kinase and Akt kinase activity in the tumors leading to significant inhibition of NF��B activation. Fish oil diet also prevented the expression of anti-apoptotic proteins Bcl-2 and Bcl-XL in the breast tumors with concomitant increase in caspase 3 activity.
��
��
��
��
Phosphatase and Tensin homolog (PTEN) is a tumor suppressor gene. The main function of PTEN is to block the PI3K pathway by dephosphorylating phosphatidylinositol (PI) 3,4,5-triphosphate to PI-4,5-bisphosphate thus counteracting PI3K function. PTEN inactivation is a frequent event in many cancer types and can occur through various genetic alterations including point mutations, large chromosomal deletions, and epigenetic mechanisms.
Frontiers | Functions and Regulation of the PTEN Gene in Colorectal Cancer | Oncology
https://www.frontiersin.org/articles/10.3389/fonc.2013.00326/full��
PI3K/Akt and HIF‑1 signaling pathway in hypoxia‑ischemia (Review)
https://www.spandidos-publications.com/mmr/18/4/3547��
Multiple roles and therapeutic implications of Akt signaling in cancer.
The prominence of the PI3K-Akt signaling pathway in several tumors indicates a relationship with tumor grade and proliferation. Critical cellular processes are driven through this pathway. More detailed knowledge of the pathogenesis of tumors would enable us to design targeted drugs to block both membrane tyrosine kinase receptors and the intracellular kinases involved in the transmission of the signal.The newly approved molecular inhibitors sunitinib (an inhibitor of vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and other tyrosine kinase receptors), sorafenib (a serine-threonine kinase inhibitor that acts against B-Raf) and temsirolimus (an mTOR inhibitor) shown clinical activity in advanced kidney cancer. Chronic myeloid leukemia has changed its natural history thanks to imatinib and dasatinib, both of which inhibit the intracellular bcr/abl protein derived from the alteration in the Philadelphia chromosome.
Intracellular pathways are still important in cancer development and their blockade directly affects outcome. Cross-talk has been observed but is not well understood. Vertical and horizontal pathway blockade are promising anticancer strategies.
Indeed, preclinical and early clinical data suggest that combining superficial and intracellular blocking agents can synergize and leverage single-agent activity. The implication of the Akt signaling pathway in cancer is well established and has led to the development of new anticancer agents that block its activation.
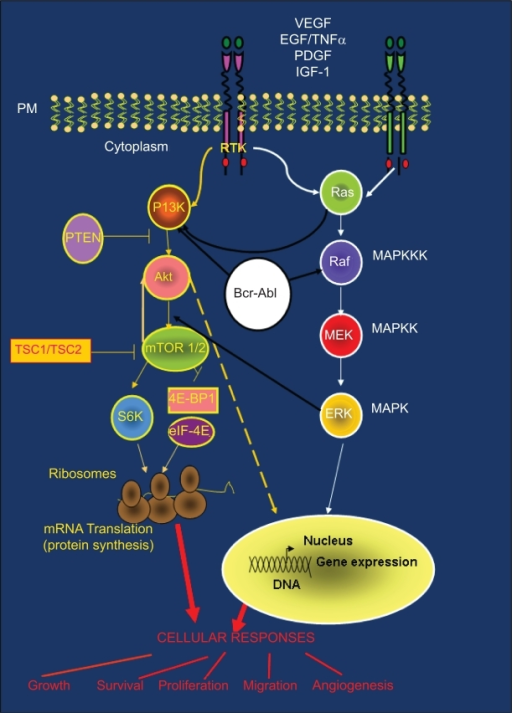
PI3K-Akt-mTOR pathway and cross-talk with other signaling cascades: (Ras/Raf/MAPK and BCR-ABL). PI3K-Akt and Ras/Raf/MAPK pathways are common routes that control key cellular responses. The large amount of cross-talk between these pathways is often responsible for treatment resistance.
��
https://openi.nlm.nih.gov/detailedresult.php?img=PMC2886325_ott-2-135f3&req=4
��
Figure 58. PI3K/Akt pathway integrates signals from growth factor receptors, stress and available nutrients. mTOR, an intracellular serine-threonine kinase is centrally located downstream of PI3K.mTOR is a master regulator involved in translation of proteins critical for cell growth, proliferation and angiogenesis in response to upstream signals.
figure58 - Endotext
https://www.endotext.org/chapter/carcinoid-tumors/figure58/��
https://link.springer.com/article/10.1007%2Fs10549-018-4697-y
��
Targeting the PI3K/Akt/mTOR pathway: effective ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2442829
The PI3K/Akt/mTOR pathway is a prototypic survival pathway that is constitutively activated in many types of cancer. Mechanisms for pathway activation include loss of tumor suppressor PTEN function, amplification or mutation of PI3K, amplification or mutation of Akt, activation of growth factor receptors, and exposure to carcinogens.
PI3K-PKB/Akt Pathway
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3428770
Abstract. The PI3K-PKB/Akt pathway is highly conserved, and its activation is tightly controlled via a multistep process (as shown in Fig. 1) Activated receptors directly stimulate class 1A PI3Ks bound via their regulatory subunit or adapter molecules such as the insulin receptor substrate (IRS) proteins.
Cited by: 472
Publish Year: 2012
Author: Brian A. Hemmings, David F. Restuccia
PI3K/Akt/mTOR signaling pathway and targeted therapy for ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5078108
May 31, 2016 �� EGFR and PI3K/Akt/mTOR signal transduction pathway. Subsequently, PIP3 will recruit the downstream Akt to inner membranes and phosphorylates Akt on its serine/threonine kinasesites (Thr308 and Ser473) [ 27, 28 ]. Activated Akt is involved in the downstream mTORC1 mediated response to biogenesis of protein and ribosome.
Cited by: 140
Publish Year: 2016
��PI3K/Akt and HIF‑1 signaling pathway in hypoxia‑ischemia (Review)
Authors: Zhen Zhang Li Yao Jinhua Yang Zhenkang Wang Gang Du
Affiliations: Department of Cardiac Surgery, Guangdong Cardiovascular Institute, Guangdong General Hospital, Guangdong Academy of Medical Science, Guangzhou, Guangdong 510100, P.R. China, Department of Bioinformatics, Guangzhou GenCoding Lab, Guangzhou, Guangdong 510670, P.R. China, Department of Cardiovascular Surgery, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, P.R. China
Published online on: August 9, 2018
Abstract
Hypoxia‑ischemia (H‑I) is frequently observed in perinatal asphyxia and other diseases. It can lead to serious cardiac injury, cerebral damage, neurological disability and mortality. Previous studies have demonstrated that the phosphatidylinositol‑3 kinase (PI3K)/protein kinase B (Akt) signaling pathway, which regulates a wide range of cellular functions, is involved in the resistance response to H‑I through the activation of proteins associated with survival and inactivation of apoptosis‑associated proteins. It can also regulate the expression of hypoxia‑induced factor‑1�� (HIF‑1��). HIF‑1�� can further regulate the expression of downstream proteins involved in glucose metabolism and angiogenesis, such as vascular endothelial growth factor and erythropoietin, to facilitate ischemic adaptation. Notably, HIF‑1�� may also induce detrimental effects. The effects of HIF‑1 on ischemic outcomes may be dependent on the H‑I duration, animal age and species. Thus, further investigation of the PI3K/Akt signaling pathway may provide further insights of the potential targets for treating diseases accompanied by H‑I.
Introduction
Hypoxia-ischemia (H-I) commonly occurs during myocardial infarction, stroke and perinatal asphyxia. It can lead to severe injuries such as cerebral palsy (1), H-I brain damage, chronic neurological and neurodevelopmental disability in children, and even death (2). In addition, H-I may trigger massive cellular malfunction and cell death. On the other hand, the decline of cellular oxygen level during H-I also induces many compensatory responses, such as neovascularization (3), metabolic regulation and production of various neurotrophic mediators, which protect neurons from ischemic death. These processes also form part of an endogenous adaptive response that aims to defend and help tissues recover from ischemic injury (4). The rapid restoration of blood flow in the occluded coronary arteries following H-I is the most important aspect of the protective mechanism. Nevertheless, the early opening of an occluded coronary artery may lead to ischemia/reperfusion (I/R) injury (5,6).
It has been reported that phosphatidylinositol-3 kinase (PI3K)/Akt signaling pathway is involved in H-I. In this review, we have discussed the potential mechanism of PI3K/Akt signaling pathway in cellular responses for resisting H-I.
PI3K/Akt signaling pathway
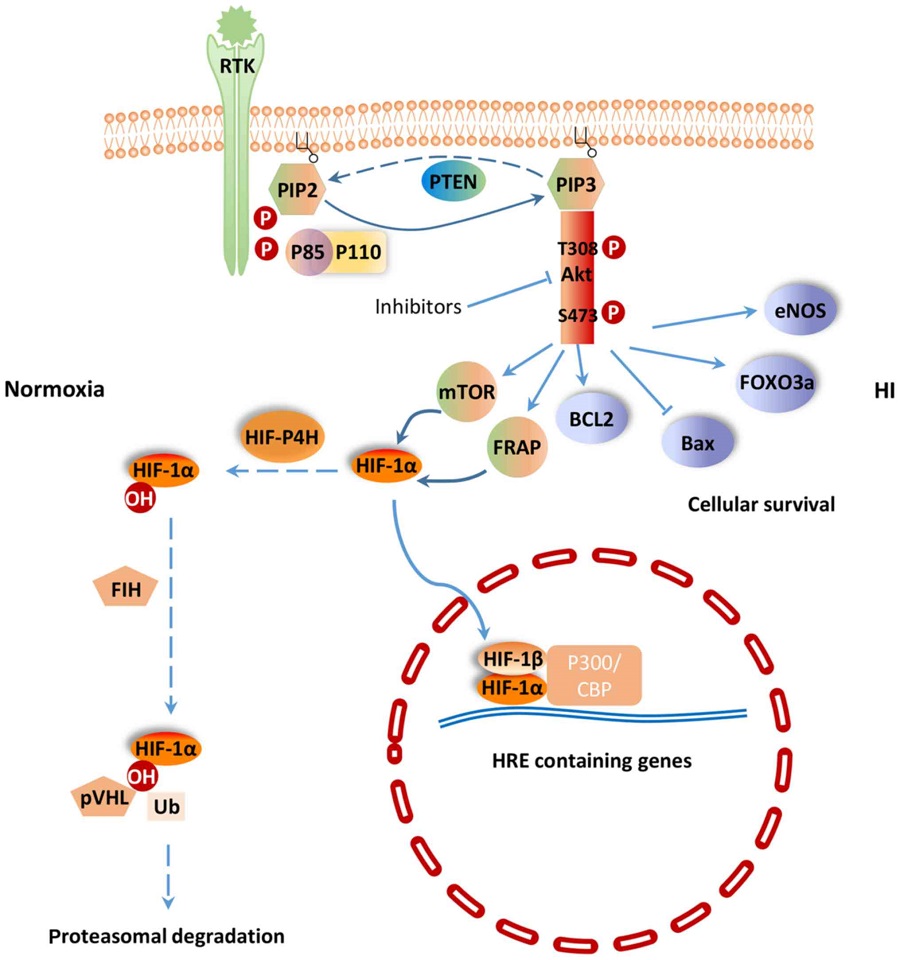
PI3K/Akt signaling pathway regulates a wide range of cellular activities including cell survival, proliferation, metabolism, neuroscience, motility and cancer progression (7). PI3K belongs to a lipid kinase family which is characterized by their ability to phosphorylate inositol ring 3��-OH group in inositol phospholipids in the plasma membrane (8). PI3Ks are divided into two classes: Class-I and II. The function of class-I PI3K is to phosphorylate PIP-2 to generate the second messenger PIP-3 within sec (9). PIP-3 can mediate different cellular functions of PI3K through specific interactions with pleckstrin homology (PH) domain containing proteins such as Akt (10). Akt is considered as the central mediator of the PI3K/Akt signaling pathway, which ultimately leads to the phosphorylation of some vital downstream targets (11). Furthermore, some negative regulators, such as phosphatase and tensin homologue (PTEN) inhibit PI3K/Akt signaling pathway. PTEN is a lipid phosphatase that negatively regulates the PI3K/Akt pathway by hydrolyzing PIP-3 to PIP-2, resulting in a lack of downstream p-Akt (12) (Fig. 1). PI3K and the downstream effector Akt belong to a conserved family of signal transduction enzymes, which are involved in regulating cellular activation, inflammatory responses and apoptosis (13).
Figure 1.
PI3K/Akt signaling pathway in HI. Dotted arrows represent the response under normoxia, and solid arrows represent the response under HI. PI3K, phosphatidylinositol 3 kinase; Akt, protein kinase B; HI, hypoxia-ischemia; RTK, receptor tyrosine kinase; HIF, hypoxia-induced factor; PTEN, phosphatase and tensin homologue; FoxO3a, Forkhead box O3; mTOR, mammalian target of rapamycin; FRAP, FKBP-rapamycin associated protein; pVHL, von Hippel-Lindau tumor suppressor protein; P300/CBP, cyclic-adenosine monophosphate-response element-binding protein binding protein; BCL2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; eNOS, endothelial cell nitric oxide synthase.
PI3K/Akt signaling pathway is involved in H-I
It has been shown that H-I-induced injuries could be treated by certain agents that act on the PI3K/Akt signaling pathway. In cerebral ischemia rats, p-Akt 473 and p-Akt 308 protein expression was significantly increased after treatment with silibinin, a compound of flavonolignan with anti-apoptotic, anti-inflammatory and anti-oxidative functions (14). Phosphorylated Akt promotes the phosphorylation of downstream molecules, including Bcl-2 apoptosis related family members, Forkhead box O3 (FoxO3a) transcription factor, mammalian target of rapamycin (mTOR) and glycogen synthase kinase-3, in order to protect cells from apoptosis. Bcl-2, an inhibitor of neuronal apoptosis, is significantly upregulated, while Bax, which can promote neuronal apoptosis, is significantly downregulated in cerebral ischemia rats treated with silibinin (15). Li et al (16) found that the PI3K/Akt/FoxO3a pathway is involved in neuronal apoptosis in the developing rat brain. Activated Akt phosphorylates FoxO3a, and leads to the cytoplasmic localization of FoxO3a and inhibition of apoptosis (17) (Fig. 1). In addition, sodium tanshinoneIIA sulfonate and bromelain protect the rat heart from I/R injury via the activation of PI3K/Akt/FoxO3a pathway (18). In the cytoplasm, mTOR, a phosphoinositide kinase-related kinase family member, serves as a Ser/Thr protein kinase (19). Previous study found that the regulatory mechanism of mTOR activity is related to the PI3K/Akt signaling pathway (14). Zhong et al (20) were among the first to show that activation of the epidermal growth factor receptor (EGFR)/PI3K/AKT/mTOR pathway could positively regulate hypoxia-induced factor-1�� (HIF-1��) at the protein level. Fibroblast growth factor-2 is a signaling molecular in the PI3K/Akt signaling pathway. Activation of PI3K/Akt pathway by fibroblast growth factor-2 prevents reactive oxygen species (ROS)-induced apoptosis and protects heart from I/R injury by decreasing infarct size and improving left ventricular function (21).
Hypoxia-inducible factor-1 (HIF-1)
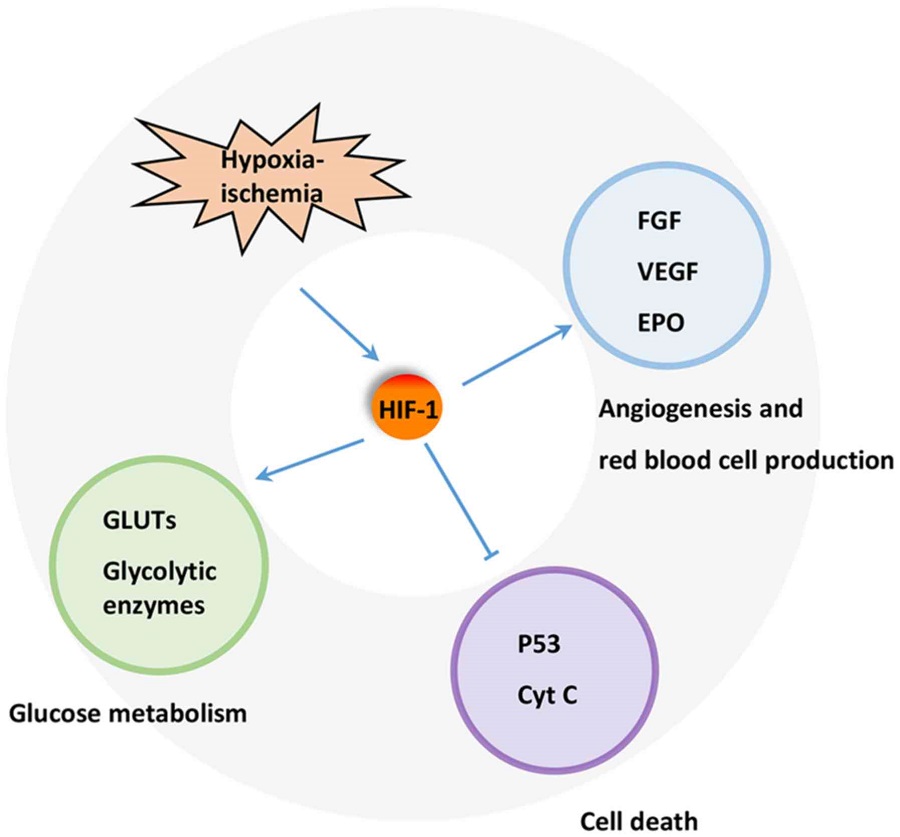
A key regulator of the response to HI is HIF-1. HIF-1 is a heterodimeric transcription factor composed of an oxygen sensitive subunit HIF-1�� and an aryl hydrocarbon nuclear translocator HIF-1��. Under normoxic condition, HIF-1�� is hydroxylated at prolines 402 and 564 by HIF prolyl-4-hydroxylase, leading to its ubiquitination and proteasomal degradation through the ubiquitin-proteasome (26S) pathway, which can continuously provoke proteasomal degradation. Its destruction is caused by the ubiquitin E3 ligase complex, in which the von Hippel-Lindau tumor suppressor protein (pVHL) is able to bind to the oxygen-dependent destruction domain on the subunit, resulting in a short half-life of the protein under normoxic conditions. In contrast, when HIF prolyl-4-hydroxylase is less active, HIF-1�� is more stable. This stabilization allows HIF-1�� to translocate to the nucleus and dimerize with its partner HIF-1�� (Fig. 1). The HIF-1 dimer subsequently binds to the hypoxia response element site on DNA, initiating the expression of more than 100 genes that participate in hypoxic adaptation (22,23). HIF-1�� is involved in pathologic conditions such as hypoxia or ischemia. HIF-1�� has also been shown to regulate the expression of vascular endothelial growth factor (VEGF), erythropoietin (EPO) and glycolytic enzymes (24) (Fig. 2).
Figure 2.
Mechanism underlying the protective effect of HIF-1 in HI. Blunt arrow represents inhibitory regulation, and pointed arrows represent stimulatory regulation. HIF1, hypoxia-induced factor 1; H-I, hypoxia-ischemia; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; EPO, erythropoietin; GLUTs, glucose transporters; Cyt C, cytochrome C.
HIF-1�� is regulated by PI3K/Akt signaling pathway
Previous studies have shown that HIF-1�� is subjected to regulation by the PI3K/Akt/mTOR (20,25) and PI3K/Akt/FRAP (26) signaling pathways. The p-Akt and HIF-1�� protein levels were shown to increase in response to hypoxia in human mesenchymal stem cells. Moreover, p-Akt expression peaked earlier than that of HIF-1��. Interestingly, the PI3K inhibitor LY294002 (27) and Dual PI3K/mTOR inhibitor NVP-BEZ235 (28) could suppress the activation of p-Akt and the expression of HIF-1�� and VEGF resulted from H-I. The Akt inhibitor, wortmannin, could also inhibit the expression of HIF-1�� at the protein, but not the mRNA level (7). mTOR is a hypoxia/nutrient sensor and a target of Akt during cell cycle regulation, glycogen metabolism and protein synthesis upon phosphorylation of its two main targets, eukaryotic initiation factor 4E-binding protein-1 and ribosomal protein S6 kinase (29). Moreover, mTOR is an upstream mediator of HIF-1�� activation (30). Based on these previous findings, the PI3K/Akt signaling pathway could potentially regulates HIF-1�� via mTOR, which could alter HIF-1�� post-transcriptional protein level, but not at the transcriptional mRNA level.
It has been shown that the pVHL mutant fails to degrade HIF-1��, which implies that pVHL plays an important role in controlling the stability of HIF-1�� (31). In another word, the stabilization of HIF-1�� could be attributed to failure in pVHL-mediated ubiquitination and proteasomal degradation. The proteasomal degradation process is often controlled by phosphorylation (32). Therefore, it is speculated that HIF-1�� activity is under the control of protein kinase phosphorylation, potentially through the universal phosphorylation signal transduction pathway of the PI3K/Akt (33).
PI3K/HIF pathway and H-I
Protective effect
Under H-I condition, PI3K/HIF pathway plays important roles in cardio-protection and neuro-protection. The expression of HIF-1�� has been shown to increase significantly in various ischemic organs and tissues, including myocardium, nervous system and retina (34). The protection of HIF-1 has also been widely reported in various H-I models. For example, HIF-1 has been demonstrated to participate in neuroprotection during permanent focal ischemia in vivo (22). Various iron chelators, such as deferoxamine mesylate and mimosine, protect neurons from apoptosis through activating HIF-1 in vitro or in vivo (35,36). These results reveal that induction of HIF-1 by ischemia itself or via pharmacological channels can protect against H-I. Furthermore, HIF-1 can regulate the expression of various genes, including EPO, VEGF, inducible nitric oxide synthase, hemeoxygenase, and cardiotropin as well as those involved in glucose metabolism, mitochondrial function, cell apoptosis, and resistance to oxidative stress that protect or restore cell functions and facilitate cellular adaptation to H-I (37,38).
A number of mechanisms have been proposed for the protective effect of HIF-1. HIF-1 has been found to protect cells from hypoxic injury by promoting nutrient and O2 transport via inducing the expression of downstream proteins such as VEGF and EPO, which promote angiogenesis and erythropoiesis. This induction is partly PI3K/Akt inhibition-dependent, suggesting a close relationship between PI3K/Akt, HIF-1�� and the VEGF cascade in HI (7). EPO promotes the production and release of red blood cell into blood, thereby, enhancing the oxygen transport. Meanwhile, the increase in hemoglobin level also affects oxygen transport capacity, and ultimately reduces tissue damage. On the other hand, HIF-1 may prevent apoptotic cell death through inhibiting the release of cytochrome C, PARP cleavage and caspase activation. In addition, HIF-1 may maintain cell survival by suppressing p53 activation. Increased glucose transport and glycolytic flow consequential of HIF-1 activation by H-I has also been implicated in tissue viability and cell survival (39).
PI3K/HIF pathway regulates glucose metabolism
One important function of glucose metabolism is to sustain a reducing environment in cells by generating reducing equivalents through oxidative phosphorylation, glycolysis, and the pentose phosphate pathway (40). The switch from aerobic to anaerobic glucose metabolism by upregulating glucose transporters (GLUTs) and glycolysis-related enzymes, such as phosphofructokinase 1, fructose-bisphosphate aldolase, phosphoglycerate kinase 1, pyruvate dehydrogenase kinase 1, and lactate dehydrogenase, is one of the key mechanisms to maintain cellular energy production and cell survival during ischemia (39). The expression of these proteins is mainly controlled by HIFs. HIF-1 activation leads to increased oxygen and nutrient delivery via enhancing angiogenesis and erythropoiesis (41,42) and improving oxygen utilization in metabolism (43). Activated HIF-1 is either directly or indirectly associated with the upregulation of GLUTs (44) and glycolytic enzymes in glycolysis and lactate production (45,46) (Fig. 2). This effect ultimately leads to the upregulation of aerobic glycolysis in tumor cells, while dampening the oxidative phosphorylation pathway (47). GLUT1 is upregulated by H-RAS, at least in part, via PI3K/HIF (48). Some stimuli, such as insulin, insulin-like growth factor 1, epidermal growth factor and angiotensin II, are able to increase HIF-1�� level in cells (49). Other key enzymes involved in metabolism are also upregulated to further ensure cellular survival (50). Other studies have further demonstrated that HIF-1 activation could be attributed to cellular mutations under non-hypoxic conditions. This phenomenon is resulted from inactivation of various tumor suppression genes, along with activation of numerous oncogenes, which then lead to mutations in several growth factor pathways, such as the loss of pVHL. HIF-1�� inactivation is caused by a physical interaction with pVHL, which elicits the 26S proteasome response. Studies have also reported that the �� domain of pVHL interacts directly with the HIF-1�� subunits. Therefore, any mutation that affects the �� domain of pVHL may prevent its interaction between HIF-1�� and thereby lead to the constitutive activation of HIF-1 (51).
The glycolysis process is an important metabolic pathway in mammals. Similar to GLUT, Hexokinase (HK) acts as a rate-limiting enzyme and is the first glycolytic enzyme which facilitates the irreversible phosphorylation of glucose to glucose-6-phosphate in cells, thereby committing the glucose molecule to the glycolytic cycle. HIF-1 activation can upregulate the expression level of HK1 and HK2 (52). In addition, HIF-1 has been shown to effectively upregulate the expression of many other glycolytic enzymes, leading to enhanced glycolysis. The glycolytic flux triggered by HIF-1�� is also related to the kinetic patterns of the expressed isoforms of the key glycolytic enzymes, which can further promote glycolytic energetic capability. Moreover, HIF-1 induces the transcription of pyruvate dehydrogenase kinase 1, which effectively inhibits pyruvate dehydrogenase activity, thereby downregulating acetyl-CoA production and suppressing the TCA cycle (53). The level of Akt directly correlates with the rate of glucose uptake into the cell through the GLUT1 transporter (54). In addition, Akt can further influence glycolysis via HK2. Akt also activates FOXO3a to inhibit apoptosis and increases mitochondrial biogenesis to support a cellular survival (55). PKM2, an isoform of pyruvate kinase, harbors a hormone response element within its first intron, indicating that its transcriptional activity is regulated by HIF-1. PKM2 is also found to interact with HIF-1�� in the nucleus and is believed to act as a transcriptional co-activator. It has been shown that activated tyrosine kinase inhibits pyruvate kinase, which further prevents pyruvate from entering into mitochondria and participating in the TCA cycle (56). Other studies on the glycolytic cycle have shown that increased pyruvate and lactate result in an increased expression of the monocarboxylate transporter (MCT) and lactate dehydrogenase (57). Although the mechanism is still unknown, reducing the expression of lactate dehydrogenase may lead to a decrease in production of lactate. On the other hand, MCT provides rapid transportation of monocarboxylate compounds, such as pyruvate and lactate, across plasma membrane, providing essential support for energy metabolism. Furthermore, activation of these transporters is closely related to HIF-1��. Previous studies have revealed that the inhibition of MCT1 can suppress lactate-induced HIF-1 activation. Whereas, the expression of MCT4 is mainly regulated by HIF-1��. Taken together, metabolism via the aerobic glycolytic pathway appears to be favored over the oxidative phosphorylation pathway in the presence of activated HIF. TCA cycle intermediates oxygen molecules and ��-ketoglutarate is responsible for facilitating the degradation of HIF-1�� (58).
PI3K/HIF pathway regulates angiogenesis
Angiogenesis is a key step in oxygen and nutrient transport. Therapeutic angiogenesis is an attractive approach for curing or alleviating ischemic cardiovascular disease (59). Angiogenesis plays an important role in the repair of tissues subjected to ischemic insult. Neovascularization is expected to reduce ventricular dysfunction and remodeling after myocardial infarction (MI) (60). The PI3K/Akt signaling pathway is crucial to inducing vascularization of heart and inhibiting cardiomyocyte apoptosis after MI (61,62). PI3K has several different isoforms (p110��, p110��, and p110��), but only p110�� is selectively required for angiogenesis (63). Interestingly, the protein kinase, Akt, has also been implicated as a mediator of cardio-protection (64). The activation of Ras and EGFR, a transmembrane receptor tyrosine kinase (RTK) that belongs to the HER family of receptors, upregulates HIF-1�� via the PI3K/Akt signaling pathway (65). EGFR/PI3K/AKT/mTOR pathway increases VEGF and endothelial cell NO synthase (eNOS) expression by upregulating HIF-1��. VEGF, an endothelial-specific mitogen and survival factor, is one of the most potent angiogenic factors, and plays key roles in both angiogenesis and vasculogenesis. Hypoxia can increase eNOS phosphorylation by activating the PI3K/AKT pathway (66). HIF-1�� can also directly influence the expression of eNOS, which can be activated by phosphorylation of the serine 1177 residue, thereby, triggering migration and angiogenesis (67) (Fig. 2). Accumulating evidence has shown that HIF-1�� acts as a potential therapeutic proangiogenic molecule in experimental models (68,69). Furthermore, EGFR amplification and PTEN mutation exert an additive effect on increasing VEGF promoter activity in human glioblastoma cells. A recent study that explored the role of PTEN in hepatocellular carcinoma also found similar inhibition of angiogenesis (70). Elevated levels of VEGF can increase vascular permeability, leading to vessel leakage, sluggish blood flow, and elevated interstitial pressure. One of the potent stimuli for increased VEGF production is hypoxia (71). Binding of both STAT3 and HIF-1�� to the VEGF promoter has been demonstrated to be essential for maximum transcription of VEGF mRNA under hypoxia (72). Therefore, therapies that affect HIF-1�� expression could potentially induce neoangiogenesis in ischemic heart.
PI3K/HIF and I/R injury
The reintroduction of oxygen after H-I is inevitable, nevertheless, reperfusion is associated with exacerbation of I/R tissue injury caused by inflammatory responses and ROS production. Therefore, the alleviation of I/R injury is a popular strategy for treating diseases associated with H-I. Factors such as high mobility group box 1 (HMGB1) may exert its protective effect by upregulating the protein expression of HIF-1�� in the ischemic myocardium via enhancing Akt phosphorylation through the PI3K/Akt signaling pathway. Treatment with LY294002 inhibits HMGB1-induced expression of HIF-1�� and eliminates the cardioprotective effects exerted by intravenous HMGB1 in an I/R rat model. ROS can directly damage the cell membrane and cause cell death during I/R. Furthermore, ROS-mediated apoptosis and necrosis can be a determinant of infarct size. HMGB1 reduces the myocardial content of MDA and increases the activity of SOD induced by I/R, whereas LY294002 eliminates these effects (34). Guo et al (73) demonstrated that inhibiting HIF-1�� expression by HIF-1��-specific small interfering RNA transfection increases ROS generation and promotes cell death. Cardiomyocyte-specific HIF-1�� gene deletion leads to reduced contractility and vascularization, along with altering the expression of multiple genes in normoxic heart. I/R significantly increases the myocardial expression of HIF-1��, while HMGB1 also markedly upregulates the expression of HIF-1��. Furthermore, consistent with the increased expression of HIF-1��, the myocardial injury induced by I/R was inhibited by HMGB1. It was also found that intravenous HMGB1 increases SOD activity in the I/R myocardium, which suggests that these changes may be occurring downstream of its effects on HIF-1�� overexpression. Thus, intravenous HMGB1 may exert its cardioprotective effects through increasing the expression of HIF-1�� (34). In addition, increasing HIF-1�� expression by drugs such as desferrioxamine, can induce a more reducing environment and decrease cell death. These results suggest that maintenance of cellular redox status via HIF-1 can protect cells from H-I mediated injuries (74).
Detrimental effects
Although HIF-1 exerts protective effects, it may also contribute to cellular and tissue damage. It has been reported that HIF-1 may mediate apoptosis in embryonic stem cells under hypoxic conditions (75). Similarly, it has been observed that HIF-1 signaling elicits delayed death via p53 in ischemic primary cortical neurons in vivo (76) and in vitro (77). Chen et al (78) has shown that inhibition of HIF-1 decreases the expression of VEGF and BCL2 interacting protein 3 (BNIP3) and thereby offering protection against delayed cell death. BNIP3 reduces increased levels of ROS via HIF-1-inducible mitochondrial autophagy (79), meanwhile causing mitochondrial dysfunction, opening of the mitochondrial permeability transition pores, membrane depolarization and cell death. Two h of ischemia has been shown to result in damage of brain cortex and blood-brain barrier in the non-infarcted ventromedial striatum and preoptic area. BNIP3 is induced in the brain under H-I condition as a master regulator in hypoxia. Suppression of HIF-1�� and VEGF has been shown to reduce acute hyperglycemia-induced HT in the ischemic brain (80). Moreover, the various protective effects through PI3K/AKT and HIF-1 pathways may become reverse in cancer hypoxic microenvironment. Multiple members of the lysyl oxidase family induced in an HIF-1-dependent manner are involved in Metastatic niche formation (81,82). It was shown that HIF-1 is involved in almost every key step of the breast cancer metastatic process including epithelial-mesenchymal transition, invasion, intravasation, extravasation, and metastatic niche formation (83).
Time pattern
Using the same neuron-specific HIF-1�� knock-out mice, Baranova et al (84) and Helton et al (85) have reported distinct HIF-1 effects on neuronal injuries following ischemia. Baranova et al (84) found that the neuron-specific knockdown of HIF-1�� increases tissue damage and reduces the survival rate of middle cerebral artery occlusion mice, suggesting that HIF-1 is neuroprotective in their ischemic model. On the other hand, Helton et al (85) observed that the knocking out of HIF-1�� reduces ischemic injury, indicating that HIF-1 may lead to tissue damage in brain ischemia. Interestingly, the ischemic model that Baranova et al (84) used was subjected to 30 min ischemia with unilateral common carotid artery occlusion (mild ischemia), while Helton's model was exposed to 75 min ischemia with bilateral occlusion (severe ischemia). Studies have demonstrated an enhanced survival and migration capability of dendritic cells (86) and transplanted stem cells in the ischemic myocardium (87,88) via short-term hypoxic preconditioning. Similarly, Jian et al (89) observed that exposure of endothelial progenitor cells to hypoxia for 24 h showed an increase in tube formation and cell motility, while prolonged hypoxia of endothelial progenitor cells for 48 and 72 h were reversed in these effects. Meanwhile, mRNA expressions of Akt and PI3K demonstrated similarly tend in a time-dependent manner. Interestingly, hypoxic preconditioning at 1% O2 in various cell lines accumulated HIF1/2�� protein after 4 h followed by a markedly reduce after 24 h to 7 days (90), and the significantly enhancement of HIF2�� protein was contrasted by a dramatic reduce of HIF2�� under hypoxia within 24 h (91). The varied observations support a notion that hypoxia may induce cell death in severe and prolonged ischemia, while promote cell survival following mild ischemic insults via HIF-1�� and PI3K/Akt pathways. Therefore, effects of HIF-1 on ischemic outcomes may be dependent on the duration of H-I, animal age and species (7).
Future directions
Despite the relatively high incidence of ischemic cerebrovascular and cardiovascular disease, limited therapies are currently available for its prevention and treatment (92,93). Although the survival rate for pre-term infants has been increased, neurological conditions such as cerebral palsy still occur in most survivors (94). PI3K/HIF pathway is important for both the mechanistic understanding and therapeutic intervention of diseases associate with H-I such as stroke, cardiovascular disease, cerebral ischemia and perinatal asphyxia. Interestingly, HIF-1 and PI3K/Akt appears to be involved in the cellular responses to H-I, but with a double-edged sword effect, which could possibly be dependent on the degree and duration of H-I. Therefore, therapies for hypoxic injury should be selected with this caveat in mind, and further study is necessary to find the optimal hypoxic pattern of different cell types. Understanding the mechanism of HIF-1 and PI3K/Akt accumulation would undoubtedly provide important insight into its role in H-I and provide potential approaches to regulate its expression.PI3K/Akt and HIF‑1 signaling pathway in hypoxia‑ischemia (Review)
https://www.spandidos-publications.com/mmr/18/4/3547��
Inhibition of the PI3K Pathway: Hope We Can Believe in?
Michiel S. van der Heijden and Ren�� Bernards
DOI: 10.1158/1078-0432.CCR-09-3004 Published June 2010
Abstract
The phosphoinositide 3-kinase (PI3K) pathway is one of the most commonly activated pathways in human cancer and has roles in cell proliferation, apoptosis, protein synthesis, and metabolism.The PI3K pathway can be activated by amplification or activating mutation of upstream receptor tyrosine kinases, and by mutations or deletions downstream in the pathway.
Trastuzumab, a monoclonal antibody targeting the human epidermal growth factor receptor 2 (HER2), has been one of the most successful and most widely used targeted therapies. However, many HER2-positive cancers are not sensitive to HER2-based therapies or become resistant during treatment; downstream activation of the pathway is one of the causes of resistance.
Because of the common activation of the PI3K pathway in cancer, compounds targeting proteins downstream in the pathway have been developed in recent years. The mammalian target of rapamycin (mTOR) inhibitors everolimus and temsirolimus have been shown to be beneficial in certain cancer types; many other inhibitors of the PI3K pathway are in various stages of clinical development. Ongoing research should clarify which molecular cancer subtypes are most susceptible to specific compounds and explore combinatorial approaches, ultimately leading to individualized patient treatment. Clin Cancer Res; 16(12); 3094�C9. ©2010 AACR.
��
Inhibition of the PI3K Pathway: Hope We Can Believe in? | Clinical Cancer Research
https://clincancerres.aacrjournals.org/content/16/12/3094
��Natural Mtor Inhibitors �C Potentially The Holy Grail ...
https://www.acneeinstein.com/natural-mtor-inhibitors-potentially-the-holy-grail
EGCG, found in green tea.
Vitamin D and synthetic derivatives.
Curcumin, found in turmeric.
Reservatrol, found in red grapes and red wine.
Image: openi.nlm.nih.gov
Natural Inhibitors of mTOR
Glutamine? [ 27].
Calorie restriction [ 28].
Ketogenic Diets [ 29].
Intermittent Calorie Restriction [ 28].
Exercise [ 30, 31] �C Inhibited in liver and fat cells.
Cortisol / Glucocorticoids [ 5].
Metformin [ 32 ], (by enhancing PRAS40��s association...
More ...
All About mTOR + Natural mTOR Inhibitors & Activators ...
selfhacked.com/blog/mtor-natural-mtor-inhibitors/��
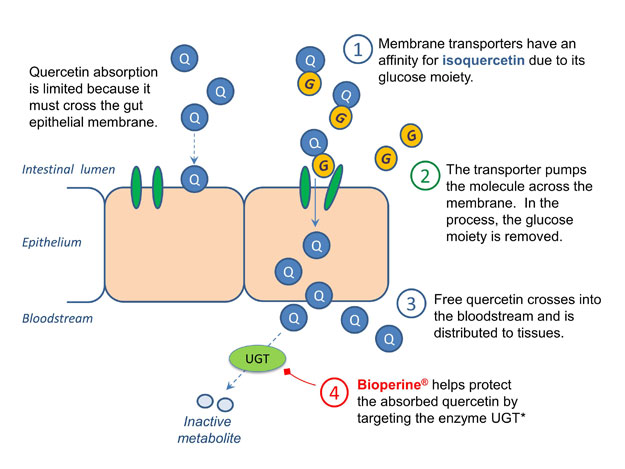
CiteSeerX �� Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers13
http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.319.4267��
Alkylation of the Tumor Suppressor PTEN Activates Akt and ��-Catenin Signaling: A Mechanism Linking Inflammation and Oxidative Stress with Cancer
PTEN, a phosphoinositide-3-phosphatase, serves dual roles as a tumor suppressor and regulator of cellular anabolic/catabolic metabolism. Adaptation of a redox-sensitive cysteinyl thiol in PTEN for signal transduction by hydrogen peroxide may have superimposed a vulnerability to other mediators of oxidative stress and inflammation, especially reactive carbonyl species, which are commonly occurring by-products of arachidonic acid peroxidation. Using MCF7 and HEK-293 cells, we report that several reactive aldehydes and ketones, e.g. electrophilic ��,��-enals (acrolein, 4-hydroxy-2-nonenal) and ��,��-enones (prostaglandin A2, ��12-prostaglandin J2 and 15-deoxy-��-12,14-prostaglandin J2) covalently modify and inactivate cellular PTEN, with ensuing activation of PKB/Akt kinase; phosphorylation of Akt substrates; increased cell proliferation; and increased nuclear ��-catenin signaling. Alkylation of PTEN by ��,��-enals/enones and interference with its restraint of cellular PKB/Akt signaling may accentuate hyperplastic and neoplastic disorders associated with chronic inflammation, oxidative stress, or aging.Introduction
Inflammation and cancer are intricately linked [1], [2]. ��Smoldering�� inflammation [3], also called para-inflammation [4], occurs in many types of pre-malignant and malignant tumors, e.g. colorectal adenoma and adenocarcinoma where the content of inflammatory leukocytes and the inflammatory enzyme cyclooxygenase-2 (COX-2) influence progression, prognosis and survival [5], [6]. Non-steroidal anti-inflammatory drugs (NSAIDs) that inhibit COX-2 can prevent certain, but not all, cancers [7]; and some NSAIDs, such as sulindac sulfone, act independently of COX and prostaglandin E2 (PGE2) inhibition [8]. Other NSAIDs, e.g. celecoxib, can paradoxically enhance tumor progression in APCMin/+ mice, which model intestinal tumorigenesis [9]. While COX-2 and its metabolite PGE2 are undoubtedly important, para-inflammation may enhance tumorigenesis by mechanisms that are incompletely understood. Innate immune mechanisms are prime candidates for investigation.
Usually, innate immune inflammation consists of a ��wounding�� phase to annihilate pathogens, and a ��healing�� phase to repair and regenerate damaged host tissue [10]. The transition between phases depends on gradual exhaustion of inflammatory mediators and conversion of certain pro-inflammatory mediators, e.g. PGD2, into anti-inflammatory metabolites, ��12-PGJ2 [11], [12], [13]. Elements of the inflamed site itself, e.g. reactive oxygen species (ROS), albumin, fibroblasts and neutrophils, orchestrate this conversion [14], [15], [16], [17]. For example, reactive oxygen species (ROS) cause non-enzymatic peroxidation of essential fatty acids, like arachidonic acid (AA) [18]. AA hydroperoxides transform readily into reactive products containing an ��,�¨Cunsaturated carbonyl [19], [20] that include acrolein (2-propenal) [21], [22], 4-hydroxy-2-nonenal (4-HNE) [23], and cyclopentenone prostaglandins (cyPGs), PGA2 and ��12-PGJ2 [24]. Covalent modification of NF��B and IKK��/�� proteins by these ��, ß�Cunsaturated carbonyl metabolites (i.e. protein alkylation) seems to be a ��switch�� to terminate inflammation [25]. Following this precedent, we hypothesized that alkylation may also act as a ��switch�� to initiate repair and regeneration of tissue damaged by inflammation.
Alkylation of the Tumor Suppressor PTEN Activates Akt and ��-Catenin Signaling: A Mechanism Linking Inflammation and Oxidative Stress with Cancer
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0013545��
Ann N Y Acad Sci. 2017 Aug;1401(1):136-149. doi: 10.1111/nyas.13422.
Regulation of the tumor suppressor PTEN by natural anticancer compounds.
Kim DH1, Suh J1, Surh YJ1,2,3, Na HK4.
Author information
1
Tumor Microenvironment Global Core Research Center, College of Pharmacy, Seoul National University, Seoul, South Korea.
2
Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science, Seoul National University, Seoul, South Korea.
3
Cancer Research Institute, Seoul National University, Seoul, South Korea.
4
Department of Food Science and Biotechnology, College of Knowledge-Based Services Engineering, Sungshin Women's University, Seoul, South Korea.
Abstract
The tumor suppressor phosphatase and tensin homologue (PTEN) has phosphatase activity, with phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a product of phosphatidylinositol 3-kinase (PI3K), as one of the principal substrates. PTEN is a negative regulator of the Akt pathway, which plays a fundamental role in controlling cell growth, survival, and proliferation. Loss of PTEN function has been observed in many different types of cancer. Functional inactivation of PTEN as a consequence of germ-line mutations or promoter hypermethylation predisposes individuals to malignancies. PTEN undergoes posttranslational modifications, such as oxidation, acetylation, phosphorylation, SUMOylation, and ubiquitination, which influence its catalytic activity, interactions with other proteins, and subcellular localization. Cellular redox status is crucial for posttranslational modification of PTEN and its functional consequences. Oxidative stress and inflammation are major causes of loss of PTEN function. Pharmacologic or nutritional restoration of PTEN function is considered a reliable strategy in the management of PTEN-defective cancer. In this review, we highlight natural compounds, such as curcumin, indol-3 carbinol, and omega-3 fatty acids, that have the potential to restore or potentiate PTEN expression/activity, thereby suppressing cancer cell proliferation, survival, and resistance to chemotherapeutic agents.
© 2017 New York Academy of Sciences.
Regulation of the tumor suppressor PTEN by natural anticancer compounds. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/28891094��
Dietary regulation of PI3K/AKT signaling via the modified PTEN expression
cited from Effective PI3K modulators for improved therapy against malignant tumors and for neuroprotection of brain damage after tumor therapy (Review)
https://www.spandidos-publications.com/10.3892/ijo.2016.3710
The PI3K/AKT and PTEN signaling plays various cellular key roles under normal and/or pathological conditions. Activation of both pathways can impact functional consequences in cancer therapy and the subsequent multiple CNS disorders. Of course, pharmacological interference of signaling molecules provides convenient method. For example, bisperoxovanadium compounds inhibit PTEN signaling, and have been used for elevation of neuroprotection in many CNS damage studies (43,59,60). In addition, small molecules have prevalent use by means of experimental tools as well as therapeutics (61). Now, it is challenging to describe appropriate strategies to achieve cost effective benefits from easy diets to control the PTEN signaling molecules. Dietary and/or therapeutic interventions could not only contribute to the prevention of diseases but reduce the rate of its development. Actually, several herbs had been established to have characteristics for antitumor activity (62). In addition, dietary regulation of PI3K/AKT signaling via the modulating PTEN expression might be effective both for cancer inhibition and for a neuroprotection. For example, dietary intake of isothiocyanate sulforaphane, a dietary isothiocyanate derived from broccoli, modulates PTEN gene expression (63). Dietary exposure to the soy isoflavone such as genistein also induces PTEN expression at physiologically relevant concentrations (64). Phytoestrogen exposure might result in an increase in PTEN expression and the subsequent decrease in cellular responses including AKT phosphorylation. Accordingly, consumption of moderate levels of soy, vegetables, some fruits, and red wine can be protective against cancers. A high-fat diet raises circulating fatty acids, which also increases PTEN expression in prostate epithelial cells (65). In addition, dietary intake of indole-3-carbinol upregulates PTEN in an animal model (66). Indole-3-carbinol is a promising cancer-preventive phytochemical found in some vegetables such as broccoli. PTEN expression at the level of mRNA and protein is elevated in experimental animals fed with whey protein (67) which has been shown to possess multiple health benefits (67). In addition, it has been reported that DHA and EPA raise the level of PTEN in breast cancer cells, providing a mechanism for the beneficial effects of fish oils on breast cancer cells (68,69). Fish oil rich in polyunsaturated fatty acids may induce the PTEN expression by activation of peroxisome proliferator-activated receptor (PPAR) (68,69), which also attenuate neuron cellular damage after a brain ischemia and appear to play an important role in the activation of anti-apoptotic signaling (70). Schematic structures of human PPAR and PTEN are shown in Fig. 3. PUFA ethanolamides DHA and EPA induce autophagy through PPAR activation in cancer cells (68). In contrast, high fat diet attenuates the neuroprotection because of decreased survival of AKT signaling (71). A lignan Honokiol isolated from the bark of Magnolia officinalis could attenuate PI3K/AKT signaling by upregulation of PTEN expression (72), which is a potential antitumor compound. Honokiol has been reported to improve the learning and memory impairments in experimental animals (73). A food ingredient curcumin, derived from the root of the plant Curcuma longa, repairs PTEN expression (74). Curcumin inhibits cell proliferation in human osteoclastoma cells (75). Furthermore, curcumin is a potential therapeutic mediator for neuro-cognition (76). In contrast, certain component of rosemary herb decreases the expression of PTEN in K562 human myeloid cells (77). Resveratrol, an ingredient in grapes, has been reported to exhibit anticancer activity, anti-inflammatory activity and cardiovascular protection property. Notably, resveratrol has been recently reported to have neuroprotective effect (78). Supplementation of these natural compounds may provide an innovative therapeutic approach to brain disorders after cancer therapy (31).
����
Figure 2 - Potential molecular targets based on the predominant PI3K/AKT/GSK3�� pathway, suggesting certain diets and medications may contribute to neuro-protection via modulating the function of AKT and GSK3��. Note that some critical events have been omitted for clarity. BDNF, brain-derived neurotrophic factor; GABA, gamma-aminobutyric acid; 5-HT, 5-hydroxytryptamine, serotonin; PUFA, polyunsaturated fatty acid; SSRI, selective serotonin reuptake inhibitors.
��
Figure 3
Schematic structures of human PPAR and PTEN protein. The predicted consensual domain structures for each protein are depicted. Note that the sizes of protein are modified for clarity. C2 domain, a structural domain involved in targeting proteins to cell membranes; PDZ, a common structural domain in signaling proteins (PSD95, Dlg, ZO-1).��
Effective PI3K modulators for improved therapy against malignant tumors and for neuroprotection of brain damage after tumor therapy (Review)
https://www.spandidos-publications.com/10.3892/ijo.2016.3710��
Breast Cancer Res Treat. Author manuscript; available in PMC 2010 Nov 1.
Published in final edited form as:
Breast Cancer Res Treat. 2009 Nov; 118(1): 213�C228.
Published online 2008 Oct 26. doi: 10.1007/s10549-008-0227-7
Triparna Ghosh-Choudhury, Department of Pathology, University of Texas Health Science, Center at San Antonio, San Antonio, TX, USA;
Fish oil targets PTEN to regulate NF��B for downregulation of anti-apoptotic genes in breast tumor growth
Abstract
The molecular mechanism for the beneficial effect of fish oil on breast tumor growth is largely undefined. Using the xenograft model in nude mice, we for the first time report that the fish oil diet significantly increased the level of PTEN protein in the breast tumors.In addition, the fish oil diet attenuated the PI 3 kinase and Akt kinase activity in the tumors leading to significant inhibition of NF��B activation. Fish oil diet also prevented the expression of anti-apoptotic proteins Bcl-2 and Bcl-XL in the breast tumors with concomitant increase in caspase 3 activity.
To extend these findings we tested the functional effects of DHA and EPA, the two active ��-3 fatty acids of fish oil, on cultured MDA MB-231 cells. In agreement with our in vivo data, DHA and EPA treatment increased PTEN mRNA and protein expression and inhibited the phosphorylation of p65 subunit of NF��B in MDA MB-231 cells. Furthermore, DHA and EPA reduced expression of Bcl-2 and Bcl-XL. NF��B DNA binding activity and NF��B-dependent transcription of Bcl-2 and Bcl-XL genes were also prevented by DHA and EPA treatment. Finally, we showed that PTEN expression significantly inhibited NF��B-dependent transcription of Bcl-2 and Bcl-XL genes.
Taken together, our data reveals a novel signaling pathway linking the fish oil diet to increased PTEN expression that attenuates the growth promoting signals and augments the apoptotic signals, resulting in breast tumor regression.
Keywords: PTEN, NF��B, DHA, EPA, Breast tumor growth, Apoptotic signal
Fish oil targets PTEN to regulate NF��B for downregulation of anti-apoptotic genes in breast tumor growth
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2847452/��
Fish oil targets PTEN to regulate NF��B for downregulation of anti-apoptotic genes in breast tumor growth
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2847452/��
miR-21 is targeted by omega-3 polyunsaturated fatty acid ...
https://academic.oup.com/carcin/article/33/10/1897/2463452
Jun 07, 2012 �� Doxorubicin-resistant breast cancer cells express elevated levels of miR-21 with concomitant reduction in PTEN protein, indicating a link between chemoresistance and miR-21 ( 51). Similarly, breast cancer cells resistant to trastuzumab therapy express enhanced levels of miR-21 ( 52). Consequently, these cells display decreased PTEN protein.
Cited by: 93
Publish Year: 2012
AIncreasing evidence shows the beneficial effects of fish oil on breast cancer growth and invasion in vitro and in animal models. Expression of CSF-1 (colony stimulating factor-1) by breast cancer cells acts as potent activator of malignancy and metastasis. In this report, we used two human breast cancer cell lines, MDA-MB-231 and MCF-7, to show that the bioactive fish oil component DHA (docosahexaenoic acid) inhibits expression of CSF-1 and its secretion from these cancer cells. We found that the tumor suppressor protein PTEN regulates CSF-1 expression through PI 3 kinase/Akt signaling via a transcriptional mechanism. The enhanced abundance of microRNA-21 (miR-21) in breast cancer cells contributes to the growth and metastasis. Interestingly, DHA significantly inhibited expression of miR-21. miR-21 Sponge, which derepresses the miR-21 targets, markedly decreased expression of CSF-1 and its secretion. Furthermore, miR-21-induced upregulation of CSF-1 mRNA and its transcription were prevented by expression of PTEN mRNA lacking 3��-untranslated region (UTR) and miR-21 recognition sequence. Strikingly, miR-21 reversed DHA-forced reduction of CSF-1 expression and secretion. Finally, we found that expression of miR-21 as well as CSF-1 was significantly attenuated in breast tumors of mice receiving a diet supplemented with fish oil. Our results reveal a novel mechanism for the therapeutic function of fish oil diet that blocks miR-21, thereby increasing PTEN levels to prevent expression of CSF-1 in breast cancer.
��
��
Regulation and function of miRNA-21 in health and disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3256347
Sep 01, 2011 �� The small regulatory RNA microRNA-21 (miR-21) plays a crucial role in a plethora of biological functions and diseases including development, cancer, cardiovascular diseases and inflammation. The gene coding for pri-miR-21 (primary transcript containing miR-21) is located within the intronic region of the TMEM49 gene.RNA Biol. 2011 Sep-Oct; 8(5): 706�C713.
Regulation and function of miRNA-21 in health and disease
Regalla Kumarswamy, Ingo Volkmann, and Thomas Thumcorresponding
Abstract
The small regulatory RNA microRNA-21 (miR-21) plays a crucial role in a plethora of biological functions and diseases including development, cancer, cardiovascular diseases and inflammation. The gene coding for pri-miR-21 (primary transcript containing miR-21) is located within the intronic region of the TMEM49 gene. Despite pri-miR-21 and TMEM49 are overlapping genes in the same direction of transcription, pri-miR-21 is independently transcribed by its own promoter regions and terminated with its own poly(A) tail. After transcription, primiR-21 is finally processed into mature miR-21. Expression of miR-21 has been found to be deregulated in almost all types of cancers and therefore was classified as an oncomiR. During recent years, additional roles of miR-21 in cardiovascular and pulmonary diseases, including cardiac and pulmonary fibrosis as well as myocardial infarction have been described. miR-21 additionally regulates various immunological and developmental processes. Due to the critical functions of its target proteins in various signaling pathways, miR-21 has become an attractive target for genetic and pharmacological modulation in various disease conditions.
Key words: EMT, fibrosis, gene promoter, microRNAs, miR-21
Inhibition of miR-21 Regulates Mutant KRAS Effector ...
https://cancerpreventionresearch.aacrjournals.org/...
Jun 18, 2020 �� Importantly, early systemic miR-21 inhibition completely intercepted premalignant progression. Finally, an evaluation of miR-21 expression in the PDA cohort of The Cancer Genome Atlas identified a correlation between tumor epithelial cell content and miR-21 expression in human tumors providing further rationale for conducting human studies.Inhibition of miR-21 Regulates Mutant KRAS Effector Pathways and Intercepts Pancreatic Ductal Adenocarcinoma Development
Abstract
Almost all pancreatic ductal adenocarcinomas (PDA) develop following KRAS activation, which triggers epithelial transformation and recruitment of desmoplastic stroma through additional transcriptional and epigenetic regulation, but only a few of these regulatory mechanisms have been described. We profiled dysregulated miRNAs starting with the earliest premalignant pancreatic intraepithelial neoplasias (PanIN) in genetically engineered mutated KRAS and P53 (KPC) mice programmed to recapitulate human PDA tumorigenesis. We identified miR-21 and miR-224 as cell-specific and compartment-specific regulators in PanINs and PDA. miR-21 is overexpressed in tumor epithelial cells of premalignant ducts, while miR-224 is overexpressed in cancer-associated fibroblasts in PDA stroma. Inhibition of miR-21 reverted protumorigenic functionalities to baseline levels. Overexpression of miR-224 induced activated phenotypes in normal fibroblasts. In vivo miR-21 inhibition improved survival in established PDA. Importantly, early systemic miR-21 inhibition completely intercepted premalignant progression. Finally, an evaluation of miR-21 expression in the PDA cohort of The Cancer Genome Atlas identified a correlation between tumor epithelial cell content and miR-21 expression in human tumors providing further rationale for conducting human studies. Thus, miR-21 may be useful for early PanIN detection, and for intercepting developing premalignant pancreatic lesions and other KRAS-driven premalignancies.��
Colony Stimulating Factor 1 - an overview | ScienceDirect ...
https://www.sciencedirect.com/topics/medicine-and...
Colony-stimulating factor-1 (CSF-1) is the major hematopoietic growth factor released by osteoblasts (37). PTH induces CSF-1 expression in osteoblasts by a transcriptional mechanism (37,38). CSF-1, in turn, enhances osteoclast formation in cocultures of mouse spleen cells and stromal cells (39).
Colony-Stimulating Factor-1 (CSF-1) Directly Inhibits ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2775986
Colony-stimulating factor-1 (CSF-1), released by osteoblasts, stimulates the proliferation of osteoclast progenitors via the c-fms receptor (CSF-1R) and, in combination with receptor activator of nuclear factor-��B ligand (RANKL), leads to the formation of mature osteoclasts.
Cited by: 21
Publish Year: 2009��
Colony Stimulating Factor 1 - an overview | ScienceDirect Topics
https://www.sciencedirect.com/topics/medicine-and-dentistry/colony-stimulating-factor-1��
��

.jpg)
.jpg)