��
1. IL-6 is expressed and release in response to enhanced ROS induced stresses, like infectious inflammation and non-infectious injuries
2. expression of the NF-��B target gene IL-6 was significantly up-regulated in GFAP super-repressor mice,
3. inducible astrocytic NF-��B does not function in cell survival but exacerbates chemokine-driven defects in spinal-cord recovery
4. NF-��B signaling in astrocytes might regulate chemokine-induced infiltration of immune cells into the lesioned brain.
5. Vitamin C suppresses TNF alpha-induced NF-kappa B activation by inhibiting I Kappa B alpha phosphorylation
https://www.researchgate.net/publication/11072278_Vitamin_C_suppresses_TNF_alpha...
Vitamin C is an essential water-soluble vitamin with antioxidant and anti-inflammatory properties, in addition to being a cofactor in the synthesis of catecholamines in the human body.��
Signaling mechanisms of ROS-mediated nuclear factor kappa-B (NF-��B) activation. Inflammatory stimuli (proinflammatory cytokines, oxidative stress, etc.), and ROS produced by mitochondria, NADPH oxidase, and the endoplasmic reticulum triggers those kinase pathways that results in NF-��B activation. NF-��B can then translocate to the nucleus and induces target gene transcription, such as TNF-��, IL-1��, IL-6, and IL-8. Orz can reduce inflammation by scavenging ROS and consequently inhibiting the NF-��B pathways.
��
NF-��B�����Bϸ���ĺ����Ӧ�������ǿ�ӣ���һ�ֵ����ʸ�����ɿ���DNA��ת¼��ϸ�����ӵIJ�����ϸ���� NF-��B�������������ж���ϸ�������У�������ϸ���Դ̼��ķ�Ӧ������Ӧ����ϸ�����ӣ����ɻ����ؽ��������������䣬������LDL�Լ�ϸ������ԭ��NF-��B�ڵ��ڶԸ�Ⱦ�����߷�Ӧ����ؼ����á� NF-��B�����ʵ��방֢�����Ժ����������Լ�������Ѫ֢���ݿˣ�������Ⱦ�����߷��������йء� NF-��BҲ��ͻ�������Ժͼ�������йء�
NF-��B���û��� �ڸ�ͼ�У���Rel��p50������ɵ�NF-��B�������Ϊ��������ʧ��״̬ʱ��NF-��Bλ�������Ƶ���I��B�����ϵİ����ܽ��С�ͨ������Ĥ����Ľ鵼������ϸ�����źſ��Լ���øI��B��ø��IKK���� IKK�����������ữI��B�����ף��Ӷ����·��ػ���I��B����NF-��B�ķ����Լ�����ø�����ս���I��B����Ȼ���NF-��B��λ��ϸ�����У������Ϊ��ӦԪ����RE�����ض�DNA���н�ϡ�Ȼ��DNA /NF-��B������ļ�����������ʣ����繲�������Ӻ�RNA�ۺ�ø�����ǽ�����DNAת¼��mRNA����������mRNA������ɵ����ʣ�����ϸ�����ܸı䡣��
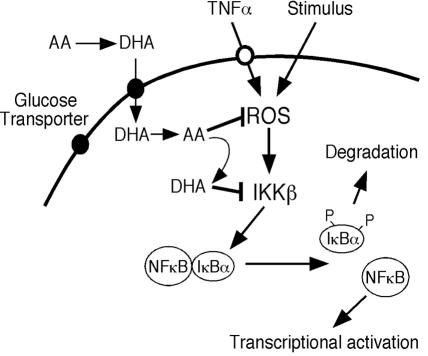
ά����C�����ź���Ӧ��ʾ��ͼ��ά����C��ΪDHAͨ��������ת�˵�����ϸ������Ѹ�ٻ�ԭΪAA�� ROSͨ������IKK���յ�NF-��B�źŴ���Ӧ�𣬶�AA���ROS������IKK�µļ������Щ�����У�AA������DHA����DHA����IKK�¡�
��������ø����ֳ�D���������PPAR-alpha����Ҳ��ΪNR1C1���������Ǽ���1��C�飬��Ա1������һ������������PPARA�������ĺ����嵰�ס�[5]���������ø����ֳ�D������ĺ�������ø����ֳ�D�������һ��PPAR-���ǹ�������ø����ֳ�D�������Ǽ����һ���֡�����1990��˹�ٷҡ����֣�Stephen Green����¡��PPAR����ĵ�һ����Ա�����ѱ�ȷ��Ϊ�����������ø����ֳ�Ķ���������ΰ��°���ĺ����塣[6]
��
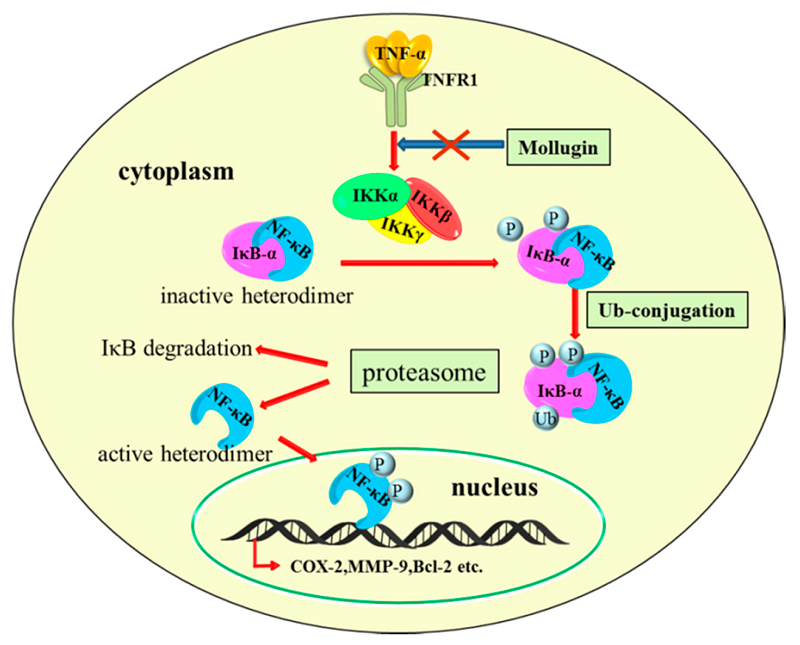
IJMS | Free Full-Text | Mollugin Has an Anti-Cancer Therapeutic Effect by Inhibiting TNF-��-Induced NF-��B Activation | HTML
https://www.mdpi.com/1422-0067/18/8/1619/htm��
Proposed working model for the role of the NF-kB signaling pathway in the regulation of ethanol-enhanced the progression of HCC. (1) ROS induced by alcohol mediated activation of the NF-kB pathway. This effect can be abolished by C3G or PDTC. (2) Nuclear translocation of NF-kB via phosphorylation by the IkB kinase, which then binds target DNA that regulates VEGF and MCP-1 gene expression. Ultimately, the changes stimulated by alcohol enhance angiogenesis, tumor growth and metastasis.
Activation of the NF-��B pathway as a mechanism of alcohol enhanced progression and metastasis of human hepatocellular carcinoma | Molecular Cancer | Full Text
https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-014-0274-0
Cold Spring Harb Perspect Biol. 2009 Sep; 1(3): a001271.
NF-��B in the Nervous System
Barbara Kaltschmidt1 and Christian Kaltschmidt2
Author information Copyright and License information Disclaimer
1Molecular Neurobiology, University of Bielefeld, Universitätsstr. 25, D-33501 Bielefeld
2Cell Biology, University of Bielefeld, Universitätsstr. 25, D-33501 Bielefeld
Abstract
The transcription factor NF-��B has diverse functions in the nervous system, depending on the cellular context. NF-��B is constitutively activated in glutamatergic neurons. Knockout of p65 or inhibition of neuronal NF-��B by super-repressor I��B resulted in the loss of neuroprotection and defects in learning and memory. Similarly, p50−/− mice have a lower learning ability and are sensitive to neurotoxins. Activated NF-��B can be transported retrogradely from activated synapses to the nucleus to translate short-term processes to long-term changes such as axon growth, which is important for long-term memory. In glia, NF-��B is inducible and regulates inflammatory processes that exacerbate diseases such as autoimmune encephalomyelitis, ischemia, and Alzheimer's disease. In summary, inhibition of NF-��B in glia might ameliorate disease, whereas activation in neurons might enhance memory. This review focuses on results produced by the analysis of genetic models.
In vertebrates, the nervous system is composed of the central nervous system (CNS) and the peripheral nervous system (PNS). The CNS comprises the brain and spinal cord, whereas peripheral nerves are part of the PNS. The CNS coordinates different tasks: integration of all stimuli that are presented from outside or inside the organism, coordination of all motor processes, regulation of hormone systems, and organ control. The most fascinating function of the brain is the coordination of learning and memory. This review will focus on the function of NF-��B in the nervous system, especially in learning and memory in rodent genetic models. The most abundant cell types in the vertebrate nervous system are neurons (about 100 billion) and glia (10�C50 times more). Typical glial cells are astrocytes, microglia, and the nerve fiber ensheathing cells, such as Schwann cells, which insulate nerves in the PNS and oligodendrocytes in the CNS. Neurons are highly polarized cells. Dendrites and/or soma receive electrochemical signals that are transmitted by axons to other neurons. Synapses are major sites of information input or output. These are formed by the presynaptic boutons derived from axons and the postsynaptic sites mainly localized on dendrites.
In a short introduction to the neurobiology of learning, we present a reductionist view (Dudai 1989). Although it is not easy to formally define learning (see Dudai 1989), here learning is defined simply as the capability of an animal to acquire novel skills. Learning can lead to a lasting modification of the internal representations of the outer world. Memory is thus the retention of the outer world experience-dependent internal representations. Memory retrieval is the use of memory in behavioral tasks. Comparison of learning capabilities in different species has suggested that learning has developed to provide organisms with an improved coping mechanism against adverse environments. Thus the ability to learn is encoded within the genome.
Over the years, a biochemical/cellular concept of memory has emerged that can be summarized as a dialogue between genes and synapses (Kandel 2001). Central to this concept is synaptic transmission, which is the release of, for example, excitatory neurotransmitters (axon potential-inducing) from a presynaptic release site. Transformation of an axon potential to neurotransmitter release takes place at presynaptic sites and is a unidirectional process. Two major transmitter systems in the CNS are predominant: glutamate, released from excitatory glutamatergic neurons, and ��-aminobutyric acid (GABA), released from inhibitory GABAergic neurons. Glutamate can direct the opening of Na+ ion channels at the postsynaptic site, inducing an excitatory neuronal response. In contrast, GABA directs the opening of Cl− channels, thus inducing an inhibitory response. Activity-dependent release of glutamate from presynaptic sites leads to the activation of AMPA receptors and to the depolarization of the postsynaptic neuron. Depolarization can occur by action potentials in the millisecond range locally at the synapse. Depolarization of postsynaptic neurons leads to the removal of NMDA receptor inhibition by Mg++ and then to Ca2+ influx through the receptor. This activates voltage-gated calcium channels (VGCCs), another source of synaptic Ca2+ ions. This short-lived membrane depolarization process can be transformed to changes in gene expression and to long-term morphological changes such as synaptic plasticity, leading to additional or more efficient synapses (Lamprecht and LeDoux 2004). It is thought that by the modification of synapses the internal representations are modified and thus memory is enhanced, retained, or lost.
The role of NF-��B in the nervous system has gained interest because of its involvement in synaptic processes, neurotransmission, and neuroprotection. Furthermore, inducible NF-��B plays a crucial role in brain inflammation and neural stem cell proliferation. The role of NF-��B in the cellular context of the nervous system is depicted in Figure 1.
Figure 1.
The role of NF-��B within the cellular context of the nervous system. Constitutively activated NF-��B is detected mostly in glutamatergic neurons (green nuclei), whereas NF-��B in glia has a lower basal activity and is heavily inducible (red nuclei). For details, see text.
All DNA-binding subunits of NF-��B have been detected within the CNS, and in the adult rodent brain the major DNA-binding complexes are p50/p65 (Kaltschmidt et al. 1993; Bakalkin et al. 1993; Schmidt-Ullrich et al. 1996; Meffert et al. 2003). In contrast, in the developing nervous system complexes consisting of cRel/p65, p50/p65, and p50 homodimers were reported (Bakalkin et al. 1993).
Go to:
NEURONS
Initially, constitutive (high basal) NF-��B activity was found in glutamatergic neurons of the CNS, such as the hippocampus (granule cells and pyramidal neurons of CA1 and CA3) and cerebral cortex (layers 2, 4, and 5), by antibody staining (Kaltschmidt et al. 1993; Kaltschmidt et al. 1994; Kaltschmidt et al. 1995), gelshift assays, and Western blotting. Transgenic reporter mouse models verified these data and also showed constitutive NF-��B activity in several rodent brain regions such as the cerebral cortex, hippocampus, amygdala, olfactory lobes, cerebellum, and hypothalamus (Schmidt-Ullrich et al. 1996; Bhakar et al. 2002). Here, constitutive NF-��B activity shall be defined as activity that makes cells blue in transgenic reporter mice (mainly in neurons, but also endothelial cells). Constitutive NF-��B activity can be suppressed by pharmacological inhibitors such as glutamate antagonists and L-type Ca2+ channel blockers (Lilienbaum and Israel 2003; Meffert et al. 2003). This suggests that constitutive NF-��B results from physiological basal synaptic transmission. Furthermore, constitutive NF-��B activity in endothelial cells, the roof plate (a dorsal signaling center of the developing nervous system), and floor plate (part of the neural tube) is dependent on TRAF6, as shown by crossing lacZ reporter mice with TRAF6−/− mice (Dickson et al. 2004). Inducible NF-��B was detected in biochemically purified synapses (synaptosomes) (Kaltschmidt et al. 1993; Meberg et al. 1996; Meffert et al. 2003). Interestingly, analysis of TNFRI−/−, p65−/− double knockout mice showed no presence of p65, p50, and I��B �� and �� in synapses. These data suggest that p65 is the driving subunit for synaptic localization and transport processes. It cannot be replaced by alternative subunits such as c-Rel or RelB.
A p65/GFP fusion protein can be retrogradely transported from active synaptic sites back to the nucleus (Wellmann et al. 2001; Meffert et al. 2003) after glutamatergic stimulation. This retrograde transport is dependent on the p65 nuclear localization signal (NLS) and involves a dynein�Cdynactin motor protein complex gliding on microtubules (Mikenberg et al. 2007; Shrum et al. 2009). p65 NLS is also important for the contact of p65 and the motor protein complexes. In summary, these observations indicate that NF-��B has a central role in translating short-term synaptic events into changes in gene expression.
Go to:
LEARNING AND MEMORY
Genetic evidence* for the involvement of NF-��B in learning and memory was provided for the first time in TNFRI−/−, p65−/− mice by Meffert et al. 2003. In a radial arm maze, TNFRI−/−, p65−/− mice made significantly more trial errors than control mice. A second, conditional mouse model uses a neuronal promoter-specific ablation of NF-��B in the basal forebrain (CamKII-tTA/ tetOtnI��B-��). In this model, all NF-��B subunits were repressed in vivo as measured by triple transgenic lacZ reporter mice in hippocampal neurons (Fridmacher et al. 2003). These mice showed impairments in spatial learning. The mice had to learn the position of a platform useful for resting that was submersed within a water basin (Morris water maze). Mice expressing the super-repressor (tnI��B-��) showed reduced long-term potentiation (LTP) and long term depression (LTD) (Kaltschmidt et al. 2006). Furthermore, learning during training sessions in a Morris water maze strongly induced NF-��B binding activity in hippocampal extracts of wild-type mice (O��Mahony et al. 2006). To obtain clues about NF-��B-regulated changes in gene expression, transcriptome profiling was performed. The catalytic subunit �� of the protein kinase A gene (PRKACA) was identified as a novel NF-��B target gene (Kaltschmidt et al. 2006). Recent research has shown that protein kinase A (PKA)�CCREB signaling is an essential pathway for learning and memory (Kandel 2001). Connected with this, repression of NF-��B in transgenic mice resulted in reduced PKA activity, leading to a strong reduction in forskolin-induced CREB phosphorylation (forskolin activates adenylcylase and thus raises intracellular cAMP levels). A functional NF-��B binding element was identified within intron 2 of mouse and human PKA genes (Kaltschmidt et al. 2006). These findings identified a novel transcriptional signaling cascade in neurons in which NF-��B regulates the PKA/CREB pathway function in learning and memory. Synapse density is an important parameter for learning processes. After NF-��B ablation in the hippocampus, the density of synapses was strongly reduced within a hippocampal subfield, the stratum lucidum (Kaltschmidt et al., unpubl.). In addition, in mice with neuronal NF-��B ablation the formation of axonal mossy fiber projections was impaired (Kaltschmidt et al. unpublished). These fibers normally connect granule cells with CA3 pyramidal cells. To our knowledge, this is the first in vivo model showing NF-��B dependent structural plasticity.
Furthermore, dendritic arborization is regulated by NF-��B in peripheral and cortical neurons (Gutierrez et al. 2005). Thus both the receiving structure (dendrite) and the sending structure (axon) seem to be regulated by NF-��B. Current literature suggests that dendritic and axonal outgrowth can be regulated by NF-��B in different ways in different neurons. In cortical neurons, dendritic arborization is repressed by I��B (Gutierrez et al. 2005), whereas in hippocampal granule cells the axonal outgrowth depends on NF-��B activity (Kaltschmidt et al. unpublished). An important region of neuronal information processing is the axon initial segment (AIS). This region is extremely rich in membrane-embedded voltage-gated sodium channels and is the axon potential generating region. Interestingly, the phosphorylated forms of IKK-1, IKK-2, and I��B-�� are concentrated within the AIS (Schultz et al. 2006). The node of Ranvier is also enriched in action potential generating sodium channels and also in phosphorylated forms of IKK 1, 2, and I��B-�� (Politi et al. 2008). The localization of phosphorylated NF-��B regulators might indicate the major sites of intracellular NF-��B activation. Pharmacological blockade of IKK function in cultured hippocampal neurons interfered with the localization of phosphorylated I��B-�� and IKK within the AIS (Sanchez-Ponce et al. 2008). In summary, these data show that NF-��B is an important regulator of neuronal morphology and shapes brain structures that are important for learning and memory.
A critical function of NF-��B in inhibitory GABAergic interneurons was observed in a recent transgenic mouse model (O��Mahony et al. 2006). Cell-type specific expression of super-repressor I��B-�� (Prion promoter-driven tTA/tetO super-repressor I��B-��) in both inhibitory GABAergic interneurons (robust expression) and hippocampal excitatory glutamatergic neurons (low level expression) resulted in a phenotype opposite to that resulting from the inhibition of NF-��B in glutamatergic neurons only (Kaltschmidt et al. 2006). Expression of glutamate decarboxylase (GAD65), a rate-limiting enzyme required for the synthesis of the inhibitory neurotransmitter, GABA, was down-regulated in these mice. Super-repressor I��B mice completed a radial maze in less time and made fewer errors than control mice, indicating enhanced spatial learning and memory. Furthermore, LTP was enhanced. These findings might be explained by a model in which excitatory neurons are the motor of learning and GABAergic neurons provide the brake. A blockade of the brake (by expression of the super-repressor) would result in overactivation of the motor (excitatory neurons) and would explain the enhanced learning. Consistent with this is the observation that Baclofen, a pharmacological agonist of GABA, negatively affected learning (McNamara and Skelton 1996). A major advantage of super-repressor expression is the inhibition of all NF-��B subunits. On the other hand, overexpression might have unexpected gain of function effects. The p50�C/�C mice had defects in novel task acquisition (Kassed et al. 2002), decreased anxiety (Kassed and Herkenham 2004), and reduced short-term memory (Denis-Donini et al. 2008). Homodimers of p50, which lack a transactivation domain, might act as a repressor of gene transcription on a set of specific promoters. Thus, some of the effects observed in p50−/− animals might be in addition to the loss of p50 function, a result of the derepression of NF-��B target genes.
The function of NF-��B was analyzed in fear-conditioning paradigms, a form of learning in which an unconditioned stimulus like a tone is coupled to noxious stimulation such as electric shock. Fear-conditioning seems to depend on the amygdala, a region in which constitutive NF-��B activity has already been reported. Moreover, a c-Rel knockout was analyzed in a cued fear-conditioning paradigm and analyzed 24 h later. No amygdala-dependent changes in long-term memory were reported in c-Rel knockout animals (Levenson et al. 2004). The presentation of landmarks in the fear-conditioning paradigm gave quite different results, with deficits in freezing being observed. In addition, c-Rel−/− mice showed lower activity in an open field test. Bioinformatic analysis suggested an overrepresentation of NF-��B binding sites in the promoters of genes potentially involved in memory consolidation (Levenson et al. 2004), but this was not tested experimentally with, for example, ChIP. Likewise, a recent bioinformatic study has suggested an enrichment of NF-��B and E2F binding sites in genes potentially important for the development of neurons from neural precursors, and thus additional experiments are necessary (Greco et al. 2008).
Recently, a pharmacological study using DDTC (Diethyldithiocarbamate) and SN50 (a cyclic peptide, spanning the NLS of p50) showed impaired memory reconsolidation (Lubin and Sweatt 2007).
Overall, the results of behavioral analyses of mice with reduced NF-��B activity in brain (see Table 1) can be summarized as follows: Deletion of DNA-binding subunits in all cell types, including neurons and glia, resulted in lower performance in different behavioral tests. This phenotype was also seen following repression of NF-��B in glutamatergic neurons (CamKII tTA / tetO super-repressor), suggesting that NF-��B function in glutamatergic neurons is responsible for learning and memory. On the other hand, changing the balance of glutamatergic and GABAergic neurons by the higher expression of super-repressor in GABAergic neurons enhances the learning of spatial clues.
Table 1.
Genetic mouse models interfering with NF-��B activity in the nervous system
Genotype Cell type afflicted Cognitive defect Additional phenotype References
p50−/− All Defect in novel task acquisition; decreased anxiety; reduced short-term memory Reduced neuroprotection; hearing loss; reduced neurogenesis; reduced ischemic damage; impaired acute and inflammatory nociception Yu et al. 1999; Yu et al. 2000; Kassed et al. 2002; Kassed and Herkenham 2004; Duckworth et al. 2006; Denis-Donini et al. 2008; Schneider et al. 1999, Niederberger et al. 2007
p65−/− Isolated sensory neurons na* Reduced neuroprotection Middleton et al. 2000
p65−/− Isolated Schwann cells na* Reduced myelination of peripheral nerves; Nickols et al. 2003
p65−/− /TnfrI−/− All Delayed spatial learning in radial maze No synaptic NF-��B Meffert et al. 2003
CamKII tTA/tetO super-repressor I��B-�� Glutamatergic forebrain neurons Impairments in spatial memory; reduced LTP and LTD Reduced neuroprotection; decreased PKA expression and P-CREB Fridmacher et al. 2003; Kaltschmidt et al. 2006
Prion-tTA/tetO super-repressor I��B-�� Glutamatergic and inhibitory neurons Enhanced spatial learning; enhanced LTD Reduced GAD65 expression O��Mahony et al. 2006
GFAP- super-repressor I��B-�� Glia: astrocytes Deficits in learning only in females: delayed spatial learning, impaired cued fear memory LTP reduced in females; LTP enhanced in males; reduction of mGluR5 in females; better recovery after spinal cord injury; reduced pain sensitivity Bracchi-Ricard et al. 2008; Brambilla et al. 2005
c-Rel−/− All Impaired late phase LTD; impaired long-term memory; impaired cued fear memory Reduced neuroprotection Pizzi et al. 2002; Levenson et al. 2004; Ahn et al. 2008
LysM-Cre/IKK-2FL/FL Glia: microglia; macrophages na* 30% reduction of neuronal death: 10-fold reduced infarct size after MCAO Cho et al. 2008
Nestin-Cre/IKK-2FL/FL Neural (glia and neuron) na* 25% reduction of infarct size after MCAO; amelioration of EAE Herrmann et al. 2005; van Loo et al. 2006
Nestin-Cre/Ikk-1FL/FL Neural (glia and neuron) na* No effect on EAE van Loo et al. 2006
Nestin-Cre/NemoFL/FL Neural (glia and neuron) na* Amelioration of EAE van Loo et al. 2006
NSE-SR-I��B-�� Neuronal na* Improved LPS-induced hypothermia and survival Juttler et al. 2007
Open in a separate window
na*: not analyzed.
Go to:
HYPOTHALAMUS
Previous reports analyzing transgenic NF-��B lacZ reporter mice have described constitutive NF-��B activity within the hypothalamus (Schmidt-Ullrich et al. 1996; Bhakar et al. 2002). The hypothalamus contains the dominant neuroendocrine center for the control of food intake and energy expenditure. Recently, it was reported that a high fat chow increased the already activated NF-��B in the hypothalamus neurons two- to fourfold (De Souza et al. 2005). Zhang and coworkers reported high expression of IKK-2 and I��B-�� in neurons of the mediobasal hypothalamus, a brain region involved in nutrition sensing. High fat chow, acute administration of glucose, or intraventricular injection of oleic acid hyper-activated NF-��B two- to fourfold (Zhang et al. 2008). Tissue-specific knockout of IKK-2 with Nestin CRE mice or virally transmitted CRE (lentivirus or adenovirus) injected into the hypothalamus reduced food consumption and weight gain in transgenic mice fed with high fat chow. In contrast, the neural expression of a constitutive form of IKK-2 enhanced weight gain after a high-fat diet and impaired hypothalamic sensitivity to insulin and leptin. Ablation of IKK-2 in specific AGRP hypothalamic neurons protected against induction of central insulin and leptin resistance after a high-fat diet. Zhang and coworkers identified SOCS3 (a suppressor of cytokine signaling) as a novel NF-��B target gene and a crucial regulator of diet-induced obesity. Hypothalamic neurons are now also considered to form a key center involved in sleep regulation (Mignot et al. 2002). Sleep-inducing substances include proinflammatory cytokines such as TNF and IL-1. TNFR1 knockout animals cannot sleep in response to TNF-�� application (Fang et al. 1997).
A unique population of neurons immunoreactive for the p65 subunit of NF-��B was previously localized within the caudal dorsolateral hypothalamus of rats.
In relation to this, analysis of NF-��B-driven lacZ reporter mice has shown that sleep deprivation increases the number of cells expressing NF-��B-dependent ��-galactosidase in the magnocellular lateral hypothalamus and zona incerta dorsal, as well as the adjacent subthalamus in transgenic mice (Brandt et al. 2004). Intracerebral injection of cell permeable NF-��B-inhibiting peptide spanning the NLS of p50 (SN50) inhibited IL-1-induced sleep in rats and rabbits (Kubota et al. 2000). LPS-induced hypothermia activated ��B lacZ expression in brain, and neuronal expression of the IkB[��] super-repressor suppressed hypothermia and increased survival (Juttler et al. 2007).
Go to:
NEUROPROTECTION
Initially observed in cerebellar granule neurons, a subtoxic dose of a neurotoxin (A�� or Fe++) led to long-lasting NF-��B activation. This was protective against a higher dose of neurotoxins (Kaltschmidt et al. 1999; Kaltschmidt et al. 2002). This process was described by the 16th-century physician Theophrastus Bombastus von Hohenheim (Paracelsus): ��All Ding sein Gift allein die Dosis machts�� (all things are poisons only the dose is important). Preconditioning or hormesis (Mattson 2008) was dependent on activated NF-��B and might be a subcellular vaccination strategy, as suggested by D. Baltimore (Baltimore 1988). Further in vitro studies provided evidence that activation of NF-��B can protect neurons against amyloid �� peptide toxicity (Barger et al. 1995) and excitotoxic or oxidative stress (Goodman and Mattson 1996; Mattson et al. 1997). Adenoviruses expressing super-repressor or dominant-negative NIK reduced survival of cortical neurons, whereas overexpression of p65 protected cortical neurons against apoptotic cell death induced by etopside (Bhakar et al. 2002). In vivo experiments in models of brain preconditioning with kainate or ischemia or linolenic acid showed NF-��B dependent protection against neuronal death (Blondeau et al. 2001). Later, transgenic inhibition of NF-��B by neuronal overexpression of the IkB[��] super-repressor decreased neuroprotection after kainic acid or Fe++ application (Fridmacher et al. 2003). In hippocampal acute slices derived from cRel−/− mice treatment with NMDA and IL-1�� increased neurodegeneration (Pizzi et al. 2002). Surprisingly, under wild-type conditions no c-Rel containing DNA binding complexes were detected in adult brain (Kaltschmidt et al. 1993; Schmidt-Ullrich et al. 1996; Bakalkin et al. 1993; Meffert et al. 2003). Only activation of metabotropic glutamate receptors with, for example, 1 mM (R,S)-2-chloro-5-hydroxyphenylglycine (CHPG) uncovered the protective action of c-Rel, whereas RNA against the c-Rel or c-Rel−/− genotype had no cell death enhancing effect in neurons without pharmacological activation of the metabotropic glutamate receptor (Pizzi et al. 2005). Injection of neurotoxins in p50−/− mice such as trimethyltin (Kassed et al. 2004), excitotoxins (Yu et al. 1999), or mitochondrial toxin 3-nitropropionic acid (Yu et al. 2000) increased neuronal damage. Furthermore, p50−/− animals suffered from hearing loss because of degeneration of spiral ganglion neurons (Lang et al. 2006). Ischemia induced in p50−/− mice showed a clear neuroprotective role of NF-��B in the hippocampus and striatum, in which degenerating neurons were detected 4 d after 1 h of ischemia (MCAO paradigm) (Duckworth et al. 2006). Degenerating neurons did not show NF-��B-dependent reporter gene expression, and in addition p50−/− mice experienced a 2.4-fold increase in postoperative death in comparison to controls (Duckworth et al. 2006). At a first glance, these data appear to conflict with reports of reduced infarct volume in p50−/− mice and a 25% reduction in models with neuronal- or brain-specific IKK-2 ablation (Schneider et al. 1999). However, a recent analysis showed a 10-fold reduced infarct size in mice with microglial-specific IKK-2 ablation (Cho et al. 2008). Thus NF-��B-dependent microglia activation might be a crucial contributor to ischemia. Unfortunately, neuroprotective strategies developed in animal models do not work in human stroke (Rother 2008). There could be many reasons for this, such as translation arrest and/or increased vulnerability in human white matter. A summary of the neuroprotective effects observed in genetic models is presented in Table 1.
Go to:
GLIA
No constitutive NF-��B activity was detected in glial cells of untreated animals (Schmidt-Ullrich et al. 1996; Bhakar et al. 2002), suggesting that inducible NF-��B activity might be linked to pathological events. Initially, it was reported that NF-��B activated in astrocytes via amyloid �� peptide (A��) led to the production of nitric oxide (Akama et al. 1998). This astroglial neurodegenerative role of A�� contrasts with the neuroprotective activation of NF-��B in neurons by nanomolar concentrations of A�� (see previous discussion) (Kaltschmidt et al. 1997; Kaltschmidt et al. 1999). GFAP promoter-driven super-repressor I��B expression was present in astrocytes of brain, spinal cord, and peripheral nerves (Brambilla et al. 2005). Spinal cord contusion injury increased GFAP expression in activated astrocytes at the lesion site. Activated astrocytes release inflammatory mediators such as chemokines and cytokines. Unexpectedly, expression of the NF-��B target gene IL-6 was significantly up-regulated in GFAP super-repressor mice, whereas TNF expression remained unchanged (Brambilla et al. 2005). However, the expression of the chemokines CXCL10 and CCL2 was significantly down-regulated in comparison to wild-type mice after spinal-cord injury in GFAP SR mice. Outcome after spinal-cord injury, as measured by locomoter activity, was significantly ameliorated, suggesting that inducible astrocytic NF-��B does not function in cell survival but exacerbates chemokine-driven defects in spinal-cord recovery. Likewise, NF-��B inhibition resulted in reduced glial scarring, which perhaps allows better axon regeneration (Brambilla et al. 2005). Axotomy of the entorhinal perforant path projection resulted in astrocytic STAT2 up-regulation and phosphorylation and concomitant expression of CCL2. Using the same mouse model (GFAP-SR), it was shown that the lesion-induced expression of CCL2 and STAT2 was significantly blunted (Khorooshi et al. 2008). These results suggest that NF-��B signaling in astrocytes might regulate chemokine-induced infiltration of immune cells into the lesioned brain. Similarly, Nemo and IKK-2 deletion within the CNS resulted in a significant reduction of infiltration of inflammatory cells (van Loo et al. 2006). Experimental autoimmune encephalomyelitis (EAE), a model of human multiple sclerosis, was ameliorated after Nemo or IKK-2 deletion in astrocytes. A 10-fold reduction of nuclear p65 in astrocytes after IL-1 or TNF treatment resulted in a more than 10% reduction in chemokine production (CXCL10). These data underscore the pivotal role of astrocytes in EAE and the recruitment of infiltrating inflammatory cells. The role of microglia in neuroprotection or neurodegeneration is heavily debated. Recently, analysis of LysM-Cre/IKKFL/FL mice showed 36% deletion of the IKK �� gene in cultured neonatal microglia (Cho et al. 2008), but only 4% deletion in adult microglia. Kainic acid injection resulted in the deletion of IKK-2 in 73% of microglial cells. Kainic acid-induced cell death was reduced, presumably by decreased activation of NF-��B-regulated microglial proinflammatory genes, such as TNF-�� and IL-1��. A remarkable result was the 10-fold reduction of the infarct area after MCAO (Cho et al. 2008). In summary, IKK-2-mediated microglia activation potentiated neuronal excitotoxicity.
Go to:
PAIN
Recently, a role of glial NF-��B in pain has emerged. Pain can arise from the activation of specific high-threshold PNS neurons (nociceptors) and could serve as a sensing mechanism to prevent further damage. However, clinical pain can arise from damage to the nervous system (neuropathic pain) or chronic inflammation (inflammatory pain). Analysis of p50−/− mice revealed an impairment of acute and inflammatory nociception (Niederberger et al. 2007). Recent data suggest an important role of astroglial NF-��B in pain perception: Inhibition of the expression of pain mediators (TNF-��, IL-6, and iNOS) by lentiviral delivery of I��B-�� super-repressor injected into the dorsal horn reduced pain (Meunier et al. 2007). Similarly, GFAP super-repressor mice have decreased formalin pain sensation (Fu et al. 2007).
Go to:
NEURAL STEM CELLS
During adulthood, neural stem cells continue to proliferate in the subventricular zone (SVZ) and within the hilus of the hippocampus. In adult neural stem cells isolated from the SVZ, p65 regulates proliferation via NF-��B target genes c-myc and cylin D1 (Widera et al. 2006). Furthermore, proliferation was strongly inhibited in neural stem cells prepared from the ganglionic eminence of p50−/− p65−/− embryos (Young et al. 2006). Expression of p65 and p50 persists into adulthood, particularly in subventricular zone astrocyte-like cells and in migrating neuronal precursors, respectively.
In particular, p65 and p50 are expressed in radial glial cells, in migrating neuronal precursors, and in a population belonging to the astrocytic lineage (Shingo et al. 2001). RelB, on the other hand, is only expressed in migrating neuronal precursors, whereas c-Rel is present in a few cells located at the edges of the rostral migratory stream (Denis-Donini et al. 2005).
In p50−/− animals, no defects in proliferation of hippocampal stem cells was detected (Denis-Donini et al. 2008). However, survival of neural progenitors after 21 days post BrdU injection was significantly reduced in p50−/− animals.
Taken together, current literature suggests that NF-��B fulfils very different functions in different cell types. A lot of controversial results were obtained in the last decade by neglecting the cell type-specific effect of NF-��B and the interplay between different neuronal cell types with glia.
In this line, the generation and analysis of sophisticated cell type-specific knockout models might be necessary to unravel all of the mysteries of NF-��B in the nervous system.
ABBREVIATIONS
PNS:peripheral nervous systemCNS:central nervous systemMCAO:middle cerebral artery occlusionEAE:experimental autoimmune encephalomyelitisVGCC:voltage-gated calcium channelsGABA:��-aminobutyric acidAIS:axon initial segmentChIP:chromatin immunoprecipitationLTD:long term depressionLTP:long term potentiationNF-��B in the Nervous System
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2773634/��
Oxidized Omega-3 Fatty Acids Inhibit NF-��B Activation Via a PPAR��-Dependent Pathway
Archana Mishra , Ashok Chaudhary , and Sanjeev Sethi Originally published1 Jul 2004https://doi.org/10.1161/01.ATV.0000137191.02577.86
Abstract
Abstract
Objective�� The aim of this study was to determine the effects of oxidized versus native omega-3 fatty acids on the endothelial expression of chemokines MCP-1 and IL-8, and, if effective in inhibiting chemokine expression, to determine the mechanism for the inhibition of chemokine expression.
Methods and Results�� Using enzyme-linked immunosorbent assays, we show that oxidized EPA and DHA but not unoxidized EPA or DHA inhibit cytokine-induced endothelial expression of monocyte chemoattractant protein (MCP)-1 and, to a lesser extent, IL-8. In electrophoretic mobility shift assays, oxidized EPA but not unoxidized EPA potently inhibited cytokine-induced activation of endothelial nuclear factor-��B (NF-��B). Using Western blot analyses, we show that the inhibition of NF-��B activation was not caused by prevention of phosphorylation of I��B�� because oxidized EPA did not inhibit cytokine-induced phosphorylation and ubiquination of I��B��. Furthermore, oxidized EPA inhibited NF-��B activation in endothelial cells derived from wild-type mice but had no inhibitory effects on NF-��B activation in endothelial cells derived from peroxisome proliferator-activated receptor �� (PPAR��)-deficient mice, indicating that oxidized EPA requires PPAR�� for its inhibitory effects on NF-��B.
Conclusions�� These studies show that the antiinflammatory effects of fish oil may result from the inhibitory effects of oxidized omega-3 fatty acids on NF-��B activation via a PPAR��-dependent pathway.��
Discussion
Omega-3 fatty acids in fish oil has been reported to improve the prognosis of several chronic inflammatory diseases characterized by leukocyte accumulation, including atherosclerosis, inflammatory bowel disease, rheumatoid arthritis, psoriasis, etc.1�C4
Omega-3 fatty acids, such as EPA and DHA, are highly polyunsaturated and readily undergo oxidation at ambient and subambient temperatures, even in the absence of exogenous oxidizing reagents.5,6 In view of the ease with which omega-3 PUFA spontaneously oxidize and in vivo data suggesting extensive accumulation of oxidation products after fish oil consumption, we investigated the possibility that oxidized omega-3 fatty acids may be an important component of the observed antiinflammatory effects of fish oil. In our previous studies, we showed that oxidized EPA and not unoxidized EPA pretreatment of HUVEC inhibits leukocyte adhesion to cytokine-stimulated HUVEC, and this effect is mediated through a PPAR��-dependent pathway.11,12 In our present studies, we extend these observations and show that oxidized EPA is also effective in inhibiting cytokine-induced endothelial chemokine expression, particularly MCP-1, and propose a mechanism for these antiinflammatory effects.
The fact that oxidation of the omega-3 fatty acids is required for the aforementioned antiinflammatory effects is also pointed out by other studies. Nohe et al and De Caterina et al have shown that prolonged incubation of endothelial cells with native omega-3 fatty acids (26 hours of total exposure, 6 hours before and 20 hours after TNF�� stimulation) results in inhibition of cytokine and adhesion molecule expression, whereas shorter incubation periods (6 hours) has no effect.30�C32 This suggests that oxidation of omega-3 fatty acids takes place during the long incubation period, and that the oxidation product(s) and not the native omega-3 fatty acids are most likely responsible for the inhibition of leukocyte�Cendothelial interactions.
To ascertain that the inhibition of chemokine expression was not caused by the cytotoxic effects of oxidized EPA, we performed MTT assays, which showed that oxidized EPA in the doses used for our experiments had no effect on the viability of endothelial cells (data not shown). Also, oxidized EPA did not have any effect on the constitutively expressed surface proteins such as von Willebrand factor, endoglin, and human leukocyte antigen class I molecules.11,12
The expression of MCP-1 and IL-8 is regulated through activation of NF-��B. NF-��B activation appears to be necessary for the induction of chemokine genes, and deletion of NF-��B binding sites results in an inability to induce these genes.26 In these studies, we show that oxidized EPA and not unoxidized EPA inhibits cytokine-induced activation of NF-��B and promotes cytosolic retention of the p50 and p65 subunits. Hence, the oxidized EPA-mediated inhibition of MCP-1 and IL-8 in cytokine-stimulated endothelial cells could be explained by an inhibitory effect of oxidized EPA on NF-��B activity.
The difference in the extent of inhibition of MCP-1 and IL-8 expression by oxidized EPA is most likely caused by the differential effects of oxidized EPA on endothelial NF-��B and AP-1 activation. Oxidized EPA almost completely inhibits endothelial NF-��B activation, whereas it had minimal effects in preventing AP-1 activation. Because AP-1 activation alone can result in IL-8 expression,28 oxidized EPA had only a mild inhibitory effect on IL-8 expression.
What is the mechanism for the oxidized EPA-mediated inhibition of cytokine-induced NF-��B activation? We first hypothesized that oxidized EPA inhibits phosphorylation of I��B�� by inhibiting the IKK (kinase) complex. However, it is unlikely that oxidized EPA inhibits IKK (kinase) activity because phosphorylation and ubiquination of IkB�� is noted in oxidized EPA-treated endothelial cells. In fact, when normalized to actin controls, oxidized EPA pretreatment before TNF�� stimulation for 60 minutes resulted in 30% more p-IkB�� when compared with TNF��-treated cells. Also, it unlikely that oxidized EPA induces IkB�� expression because after 15 minutes of cytokine stimulation, no IkB�� is noted in the cytoplasm of cells pretreated with oxidized EPA. After 60 minutes, I��B�� starts to appear in the cytoplasm of oxidized EPA pretreated cells and its concentration is, in fact, somewhat less than the TNF��-treated cells. Thus, it is likely that oxidized EPA does not prevent NF-��B activation by increasing the expression of I��B��.DISCUSSION
In our previous studies we noted that oxidized EPA is a potent activator of PPAR�� and that PPAR�� is needed for the inhibitory effects of oxidized EPA on leukocyte�Cendothelial interactions,12 which lead us to hypothesize that oxidized EPA might inhibit cytokine-induced NF-��B activation through a PPAR��-dependent pathway. Here, we show that although oxidized EPA inhibits cytokine-induced NF-��B activation in wild-type cells, it has no inhibitory effects in PPAR��-deficient endothelial cells, suggesting that oxidized EPA mediates its inhibitory effects on NF-��B through PPAR��. The PPAR��-mediated inhibitory effects of oxidized EPA on NF-��B activation are possibly through direct interactions of PPAR�� with the p50/p65 subunits. Delerive et al (1999) have shown, using glutathione S-transferase pull-down experiments, that after fibrate (PPAR�� agonist) treatment, PPAR�� physically interacts with p65 in vitro.33
Taken together with our previous studies,11,12 we show that auto-oxidation of omega-3 fatty acids results in the generation of oxidized compounds with potent antiinflammatory properties that inhibit proinflammatory responses such as leukocyte adhesion receptor and chemokine expression. The central theme for the antiinflammatory effects of oxidized omega-3 fatty acids is likely through inhibition of NF-��B via a PPAR��-dependent pathway. The oxidation of omega-3 fatty acids is likely to occur in areas of active inflammation caused by increased expression of oxidative enzymes (eg, NADPH oxidase, myeloperoxidase, cyclooxygenase, lipoxygenase) and the generation of reactive oxygen species in these areas, which are capable of oxidizing PUFAs. The identification of these products could result in potent, low-toxicity, proinflammatory response inhibitors with potent PPAR�� agonist and anti-NF-��B properties for the treatment of inflammatory diseases.
��Oxidized Omega-3 Fatty Acids Inhibit NF-��B Activation Via a PPAR��-Dependent Pathway | Arteriosclerosis, Thrombosis, and Vascular Biology
https://www.ahajournals.org/doi/10.1161/01.atv.0000137191.02577.86��
NF-kB Beyond Inflammation
Posted on March 8, 2017
At different times, I take about 8 different anti-inflammatory supplements, including aspirin, ibuprofen, omega 3 oils, curcumin, berberine, resveratrol, ashwaghanda, and boswellia, in addition to eating foods such as ginger, rosemary, tea and several mushroom species with anti-inflammatory effects. There is good evidence for benefits from each of these individually, but I have no idea how they interact with one another. Just last week, I learned that they all act (in part) through inhibition of NF-kB.
It��s certain that the separate benefits of each of these don��t just add up in combination. It could be that all of them together are no better than just one of them individually. It might even be that they interfere destructively with one another, competing for a common receptor, so that piling on more supplements is counter-productive. There is no research on interactions among longevity supplements.
Prelude
In this (2007) study, a multidisciplinary team performed a systematic search for blood factors that change most consistently with age over a sample of mammalian models, and organized these into modules that tend to vary in a coordinated way. They then searched for transcription factors that can turn each module on or off. Their most prominent finding was that NF-��B turns on the suite of factors characteristic of old age. Out on a teleological limb, they were bold enough to call it ��Enforcement of aging by continual NF-��B activity�� in the title of the article. (In this perspective, senescence is an active process, coordinated by the genome. I agree there is good evidence for this.)
After a long path leading to NF-��B as their prime subject, the authors go on to test whether inhibiting NF-��B can have anti-aging effects. The first obstacle that they encounter: NF-��B has important developmental functions (in young animals) and also is essential for regulating apoptosis (in older animals as well). Mice with genes for NF-��B knocked out don��t survive gestation. So they arranged to selectively ��blockade�� the binding of NF-��B to DNA in old mice, in skin cells only. The result was a dramatic rejuvenation of the skin.
Background
Research with rodents and humans suggest that there are factors in the blood that keep us young and, more important, factors that make us old. Prime suspects in the latter category are the signals that dial up inflammation. It��s my hunch that the most effective anti-aging strategy over the next 10 years will be to re-adjust signal molecules in the blood, adding what we lose with age but, more important, neutralizing or inhibiting pro-aging factors.
For various reasons, NF-��B is a good place to start.
��NF-kB has been termed the central mediator of the immune response. Gene knockout and other studies establish roles for NF-kB in the ontogeny of the immune system but also demonstrate that NF-kB participates at multiple steps during oncogenesis [ref] and the regulation of programmed cell death [ref].�� [John Hiscott]
It is a complex of different molecules that acts as a master transcription factor. It is always resident in the periphery of the cell, waiting so that it can be activated quickly when needed. Latent, NF-��B is bound to an inhibitor molecule called I��B. When a stimulus comes along that phosphorylates the I��B, the NF-��B is freed to enter the cell nucleus and switch on a variety of different genes, which varies from one cell type to another. The best-known activity of NF-��B is in white blood cells (T and B cells) where it activates an inflammatory response involving TNFa and IL-6. Overactivity of NF-��B with age is a mediator of the systemic inflammation that contributes so much to cancer, heart disease and dementia.
NF-��B itself is not circulated in the blood, but signals in the blood can cause it to be turned on. The Conboys early recognized NF-��B as one of the pathways that promote aging in parabiosis and transfusion experiments, where blood from older mice is introduced into younger mice. It is a very good bet that inhibiting NF-��B would slow inflammaging, perhaps relieving arthritic and other auto-immune symptoms immediately, while reducing long-term risk of mortality and disease.
Inflammaging and Auto-immunity
Exponential amplification is a basic principle of the body��s immune response. When the signal is received announcing an invader or an infection, there are just a few cells involved. These send signals that trigger an immune response in other cells, triggering a chain reaction.
The beauty of such a system is that it ramps up quickly, and can mobilize a response throughout the body in short order. This is also the danger of the system. It requires an accurate and reliable switch to turn it off; otherwise, it can become like the Sorceror��s Apprentice, each magic broomstick producing two more to carry water until the workshop is flooded.
Note: Dr. Katcher, in a note below, makes an important point about this positive feedback loop.
Senescent cells spit out inflammatory cytokines
This activates NF-��B, which blocks apoptosis that could get rid of the senescent cells.
Inflammation from NF-��B turns more cells senescent, beginning the cycle over again.
NF-��B is a master switch that sets in motion a chain of events that is specific to a cell type and its environment. Some auto-immune diseases (e.g., arthritis, type 1 diabetes, asthma, Crohn��s disease and irritable bowel) are associated with an excess of NF-��B [ref]. Its activation generally rises with age [in mice, in humans], but it is necessary at all ages, particularly for its contribution to the regulation of apoptosis (the selective elimination of cells that are potentially damaging). Animals lacking NF-��B are not viable; so it will probably be necessary to strongly but selectively inhibit NF-��B, beginning in middle age.
Inflammatory responses are complex and focused on the immediate threat at hand. NF-��B is a master switch that sets in motion a chain of events that is specific to a cell type and its environment It��s true that without NF-��B this response doesn��t happen, but the response to NF-��B varies from cel to cell. In this sense, inhibiting NF-��B is a kind of blunt instrument. It works to damp the body��s inflammatory response globally, but even better would be if we could selectively shut off the body��s attack on itself. It��s true that NF-��B activation rises with age [in mice, in humans]. But the real problem is not too much NF-��B expression, but the fact that NF-��B becomes defocused, so that the inflammatory response is not focused on a particular threat, but generalized throughout the body [ref].
Here��s an angle I learned about recently from Steve Cole: While inflammation is an important defense against bacterial infection, it is actually counter-productive against viruses. Inflammation can create an environment that invites viral infection, perhaps because apoptosis is suppressed. (NF-��B suppresses apoptosis.) Viruses aren��t so dumb, and some of them have learned the advantage of promoting NF-��B. Some of the reason that NF-��B is upregulated with age may be a residue of chronic viral infections. [Hiscott, again] (Just to confuse us, NF-��B can also promote apoptosis in other contexts.)
Two pathways
All the anti-inflammatory agents that I have been able to catalog work by one or both of these two pathways: NF-��B and COX2. By most accounts, NF-��B is upstream of COX2, but the two are interrelated. NF-��B regulates COX2, and also COX2 feeds back to regulate NF-��B.
NSAID drugs (aspirin, ibuprofen, naproxen, celecoxib, etc.) target cyclooxygenase-2=COX2. Common herbal anti-inflammatories, including curcumin, resveratrol, vitamin D and omega 3 oils (the last two not exactly herbs) are active both against COX2 and NF-��B. Inhibiting COX2 is a classic strategy for combatting arthritis. The more potent COX2 inhibitors have a tendency to decrease cancer risk, while increasing cardiovascular risk. This doesn��t necessarily mean, ��it��s a wash���Crather the stronger NSAID��s are right for people with some genetic risk profiles and should be avoided by others. Aspirin is the cheapest and oldest of the NSAIDs, for which there is copious data available on tens of millions of individuals. There is reasonably good evidence that aspirin leads to lower heart risk as well, probably because of anti-clotting rather than anti-inflammatory action [read more]. Daily aspirin also lowers risk of several cancers.
Inhibiting NF-��B
Intermittent fasting or caloric restriction tends to prevent NF��B binding to chromosomes.. There��s also a long list of natural products that inhibit NF��B.
source of this chart
There are a few pharmaceutical products that inhibit NF��B, though none has been developed explicitly for this purpose. These include emetine, fluorosalan, sunitinib malate, bithionol, narasin, tribromsalan, and lestaurtinib. Emetine (as the name suggests) is used to induce vomiting and also to treat amoebic diseases. It is the most potent inhibitor of NF��B among the listed drugs. Sunitinib and Lestaurtinib are cancer drugs. Bithionol is used in de-worming animals. Narasin is an uncommon antibiotic. Tribromsalan is used externally as an antiseptic. None of these is marketed to inhibit NF-��B, and none have (to my knowledge) been tested for anti-aging properties. The larger pool of prescription drugs that affect NF-��B are all steroids. For example, dexamethasoneis a glucocorticoid (steroid) drug that was one of the earliest inhibitors of NF-��B to be discovered.
Many items in the list of natural products have multiple benefits. Silymarin has been reported to promote telomerase. Rosemary and cloves protect against infection. Berberine helps maintain insulin sensitivity, and was found to be as good as metformin in one test. Tea polyphenols and resveratrol have been promoted as generally anti-aging. Too much has already been written about curcumin.
New to me in this list is celastrol, an ingredient in thunder god vine (Tripterygium wilfordii). This is a Chinese herb (leigong teng = ����), that has been prescribed for centuries in formulas to relieve arthritis, along with lupus, MS and other autoimmune disorders. It is reported to be a powerful appetite suppressant and weight loss aid. The trouble is that it is toxic, and the thunder god root must be prepared carefully in order to exclude triptolide, which is yet more toxic. Experienced practitioners of traditional Chinese medicine know how to mix with other herbs and control dosage to minimize side-effects. In the absence of this kind of expertise, I can only counsel experimenting gingerly with tiny quantities of thunder god vine in order to guage your personal response.
How important is inflammation?
It would be interesting (from a theoretical and a practical vantage) to know what is the maximum benefit available from manipulating the inflammatory pathway. Inflammaging is linked to all the diseases of old age. Suppose we dialed the systemic inflammation in a 80-year old back to where it was when he was 20, but we made no other change in the body. What would be the effect on mortality and morbidity? On vitality, resistance to infection, and stamina? In other words, how much of the aging process is directly attributable to inflammation?
We might try to get a handle on this question via an epidemiological calculation: What is the correlation between inflammation and all-cause mortality? If we extrapolate back to the inflammation level of a 20-year-old, how far does that go toward restoring the mortality rates of a 20-year-old?
We might think to look at genetically modified mice without NF-��B; however, they die in utero. (There are no pure aging genes; aging is caused by re-balancing hormones and proteins, all of which have life-supporting as well as life-denying functions.) There is a genetic variant of NF-��B that tends to be more common in centennarians than the rest of us [ref].
How much does inflammation rise with age?
Erythrocyte Sedimentation Rate and C-Reactive Protein
120 years old, the ESR test is still the most basic (and cheapest) measure of systemic inflammation. The quantity measured is the number of red blood cells that clump together and fall out of solution in one hour. Inflammation makes red blood cells sticky, and I��ve seen two explanations for the reason. One is that fibrinogen, the clotting protein, rises with inflammation; the other is that the negative charge (zeta potential) naturally associated with oxygen-carrying red blood cells decreases with inflammation, so there is less mutual electrostatic repulsion. The increase in blood��s tendency to clot that is associated with inflammation is part of the reason that inflammation is a risk for heart attacks and stroke.
C-Reactive Protein is a protein created in the liver as part of the response to inflammation. It is easily measured with an antibody, so it has become the second most common blood test for inflammation.
ESR rises with age, but not dramatically compared to interpersonal variation:
In fact, the difference between women and men is more than the difference between an 80-year-old and a 20-year-old of either sex. Increase in CRP with age is even more subtle. (This article claims it doesn��t rise at all, in a small sample of <400 patients.)
This article claims that CRP does rise with age (using a sample of 21,000):
A Glasgow study of 160,000 patients found strong correlation between CRP and near term mortality (within a year, HR=20) but not much for longer term. This is not what I was expecting.
In 26,000 patients, inflammatory markers were associated with a 1.5-fold increase in all-cause mortality (ACM) over 8 years.
A Norwegian study of 7,000 men and women found that high levels of CRP raised ACM only by a factor 1.25 (equivalent to just 2 years of aging). For comparison, the ACM risk for an 80-year-old male is 60 times higher than a 20-year-old male. The corresponding number for females is nearly 120.
The implication is that either inflammation is a minor (though significant) cause of mortality, or else the markers that we have for inflammation (including ESR, CRP and leucocytes) are not capturing the rise in systemic inflammation.
Hint: ��Centennarians, on the other hand, manage to stave off these deleterious sequelae.Despite signs of inflammation, such as high levels of interleukin-6 (IL-6), fibrinogen, and coagulation factors, they are remarkably free of most age-related diseases that have an inflammatory component.�� [ref]
NF-��B in the Brain
It is my favorite hypothesis that aging is mediated through hormonal signaling, under control of a clock in the neuroendocrine regions of the brain. So I am interested in changing NF-��B activity in the aging brain. This paper describes roles for NF-��B in brain development, regeneration after injury, and also evidence that NF-��B can be activated in response to nerve signals. In the other causal direction, neural signaling (and presumably behavior) can change in response to NF-��B. Directly on target (in my book) is this paper from Nature (2013). ��By systematically controlling NF-kB activity in the hypothalamus alone, the authors are able to increase the healthspan as well as the lifespan of mice.��
The Big Picture
In the Prelude above, we found evidence that NF-��B is a master regulator that turns on a suite of genes that ��enforces aging��. But in the section, How important is inflammation?, we found evidence that, while inflammation certainly increases with age, the increase is not large compared to the scatter among individuals. Centennarians commonly have high levels of inflammation, along with robust health. Large increases in inflammation are common just in the last year of life, but they are not well correlated with the gradual increase in mortality with age.
The combination of these two findings suggests that NF-��B has other powerful roles in promoting senescence, in addition to its well-known role as effector of inflammation. Maybe it is a master regulator of development and aging, akin to mTOR and FOXO. It is a hypothesis worth testing that carefully tailored inhibition of NF-��B is a life extension strategy, so long as we can preserve apoptosis at an appropriate level.
This entry was posted in Uncategorized by Josh Mitteldorf. Bookmark the permalink.NF-kB Beyond Inflammation | Josh Mitteldorf
https://joshmitteldorf.scienceblog.com/2017/03/08/nf-kb-beyond-inflammation/��
Inflammatory Signaling and Cellular Senescence
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2870712
Using a mouse liver carcinoma model, a recent study demonstrated that in vivo, senescent tumor cells are cleared by an innate immune response triggered by the inflammatory cytokines secreted by the senescent cells . In this study, a chimeric liver cancer mouse model was created in which the tumor cells contained a repressible p53 shRNA.
Cited by: 120
Publish Year: 2009
Author: Jian-Lin Ren, Jin-Shui Pan, Ya-Pi Lu, Peiqing Sun, Jiahuai Han
Senescent cells as a source of inflammatory factors for ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2865636
Apr 13, 2010 �� Senescent cells oversecrete an array of chemokines that can create a gradient to promote cell migration and invasion. In pancreatic cancer, hepatocyte growth factor (HGF), and to a lesser degree bFGF, promote cancer cell invasion in culture and potentially can drive cancer dissemination in ��
Cited by: 464
Publish Year: 2010
Author: Albert R. Davalos, Jean-Philippe Coppe, Judith Campisi, Pierr��
Biochemistry, November, 2002
Vitamin C suppresses TNF alpha-induced NF-kappa B activation by inhibiting I Kappa B alpha phosphorylation
Extracellular stimuli signal for activation of the transcription factor NFkappaB, leading to gene expression regulating processes involved in immune responses, inflammation, and cell survival. Tumor necrosis factor-alpha (TNFalpha) activates NFkappaB via a well-defined kinase pathways involving NFkappaB-inducing kinase (NIK), which activates downstream multisubunit IkappaB kinases (IKK). IKK in turn phosphorylates IkappaB, the central regulator of NFkappaB function.
We found that intracellular vitamin C inhibits TNFalpha-induced activation of NFkappaB in human cell lines (HeLa, monocytic U937, myeloid leukemia HL-60, and breast MCF7) and primary endothelial cells (HUVEC) in a dose-dependent manner.
Vitamin C is an important antioxidant, and most cells accumulate ascorbic acid (AA) intracellularly by transporting the oxidized form of the vitamin, dehydroascorbic acid (DHA). Because ascorbic acid is a strong pro-oxidant in the presence of transition metals in vitro, we loaded cells with vitamin C by incubating them with DHA. Vitamin C-loaded cells showed significantly decreased TNFalpha-induced nuclear translocation of NFkappaB, NFkappaB-dependent reporter transcription, and IkappaBalpha phosphorylation.
Our data point to a mechanism of vitamin C suppression of NFkappaB activation by inhibiting TNFalpha-induced activation of NIK and IKKbeta kinases independent of p38 MAP kinase. These results suggest that intracellular vitamin C can influence inflammatory, neoplastic, and apoptotic processes via inhibition of NFkappaB activation.
Vitamin C suppresses TNF alpha-induced NF-kappa B activation by inhibiting I Kappa B alpha phosphorylation | Request PDF
https://www.researchgate.net/publication/11072278_Vitamin_C_suppresses_TNF_alpha-induced_NF-kappa_B_activation_by_inhibiting_I_Kappa_B_alpha_phosphorylation��
Published: 21 September 2006
Chronic Granulomatous Disease; fundamental stages in our understanding of CGD
Tracy Assari
Medical Immunology volume 5, Article number: 4 (2006) Cite this article
Abstract
It has been 50 years since chronic granulomatous disease was first reported as a disease which fatally affected the ability of children to survive infections. Various milestone discoveries from the insufficient ability of patients' leucocytes to destroy microbial particles to the underlying genetic predispositions through which the disease is inherited have had important consequences. Longterm antibiotic prophylaxis has helped to fight infections associated with chronic granulomatous disease while the steady progress in bone marrow transplantation and the prospect of gene therapy are hailed as long awaited permanent treatment options. This review unearths the important findings by scientists that have led to our current understanding of the disease.
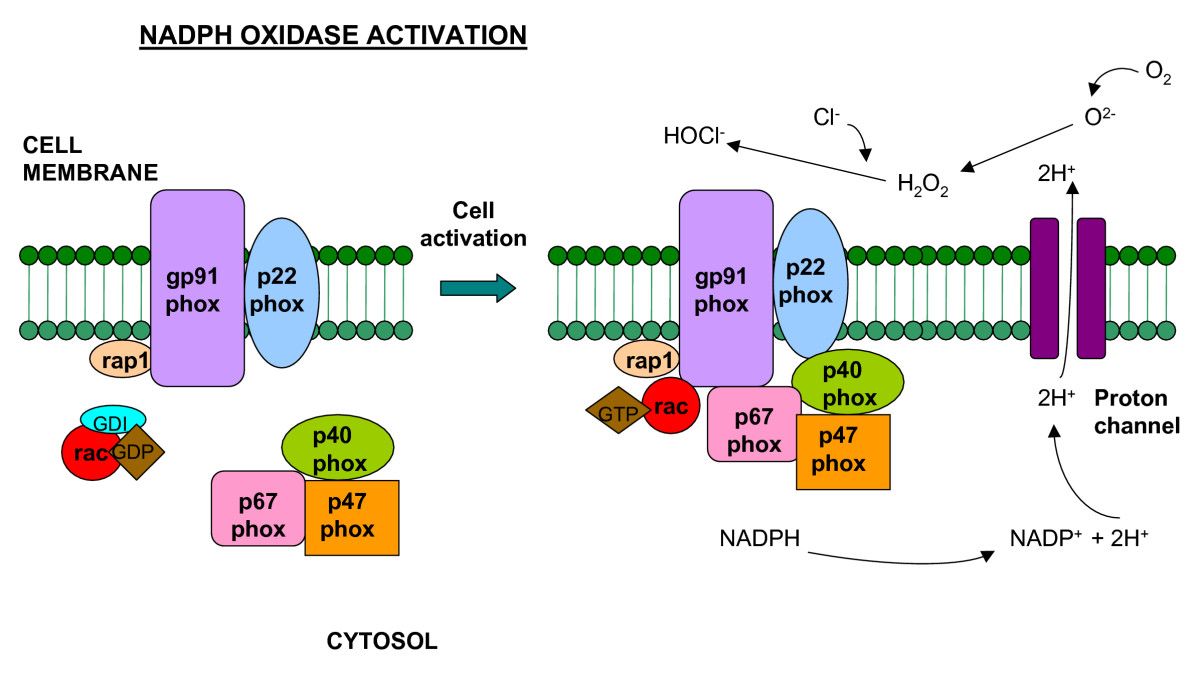
Schematic representation of the NADPH oxidase enzyme. The integral membrane of the phagocyte consists of two subunits: p22phox and gp91phox which respectively produce the smaller and larger chain of the cytochrome-b558. Two cytosolic subunits: p67phox and p47phox; a p40phox accessory protein and a Rac-GTP binding protein then translocate to the cell membrane upon cell activation to form the NADPH oxidase complex which generates a respiratory burst. Superoxide can react to form hydrogen peroxide and hypochlorus acid, which together participate in bacterial killing.
http://medimmunol.biomedcentral.com/articles/10.1186/1476-9433-5-4
��