íí
Nox4 is a major superoxide-producing enzyme localized in mitochondria in the cardiovascular system.
ATP-binding motif within the NADPH oxidase isoform, NOX4, and show that ATP directly binds and negatively regulates NOX4 activity.
íí
íí
https://www.nature.com/articles/hr2010201
íí
Mol Cell Endocrinol. Author manuscript; available in PMC 2010 Apr 29.
NADPH Oxidases and Angiotensin II Receptor Signaling
Abel Martin Garrido and Kathy K. Griendling
Author information Copyright and License information Disclaimer
Division of Cardiology, Emory University, Atlanta, GA 30322
Address correspondence to: Kathy K. Griendling, Ph.D., Emory University School of Medicine, Division of Cardiology, 319 WMB, 1639 Pierce Dr., Atlanta, GA 30322, USA, Phone: 404-727-3364, Fax: 404-727-3585, ude.yrome@dneirgk
Abstract
Over the last decade many studies have demonstrated the importance of reactive oxygen species (ROS) production by NADPH oxidases in angiotensin II (Ang II) signaling, as well as a role for ROS in the development of different diseases in which Ang II is a central component. In this review, we summarize the mechanism of activation of NADPH oxidases by Ang II and describe the molecular targets of ROS in Ang II signaling in the vasculature, kidney and brain. We also discuss the effects of genetic manipulation of NADPH oxidase function on the physiology and pathophysiology of the renin angiotensin system.
Keywords: Angiotensin II, NADPH oxidases, reactive oxygen species, vascular, kidney, brain
Over a decade ago, it was observed that angiotensin II (Ang II) is able to activate a NADPH oxidase (Griendling et al., 1994) that mediates the hypertensive response induced by Ang II infusion (Rajagopalan et al., 1996). Since then, numerous studies have demonstrated that NADPH oxidases regulate several Ang II functions in different tissues, many of which contribute to hypertension. In this review, we will describe the consequences of Ang II-mediated NADPH oxidase activation, with a focus on the cardiovascular, renal and central nervous systems.
NADPH Oxidases
Structure and activity
All types of cells have the ability to produce reactive oxygen species (ROS), including superoxide anion (O2.-), hydrogen peroxide (H2O2) and peroxynitrite. Among ROS, H2O2 is the most stable and has been most often implicated as a second messenger. It is mainly derived from dismutation of O2.- by superoxide dismutase (SOD), which in turn can be produced by multiple enzyme systems, of which NADPH oxidases are major sources in non-phagocytic cells.
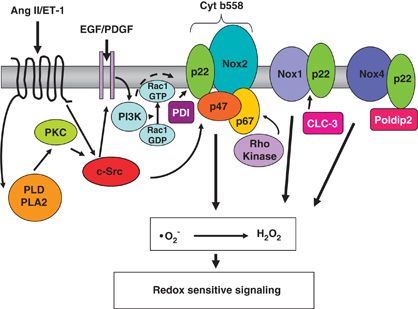
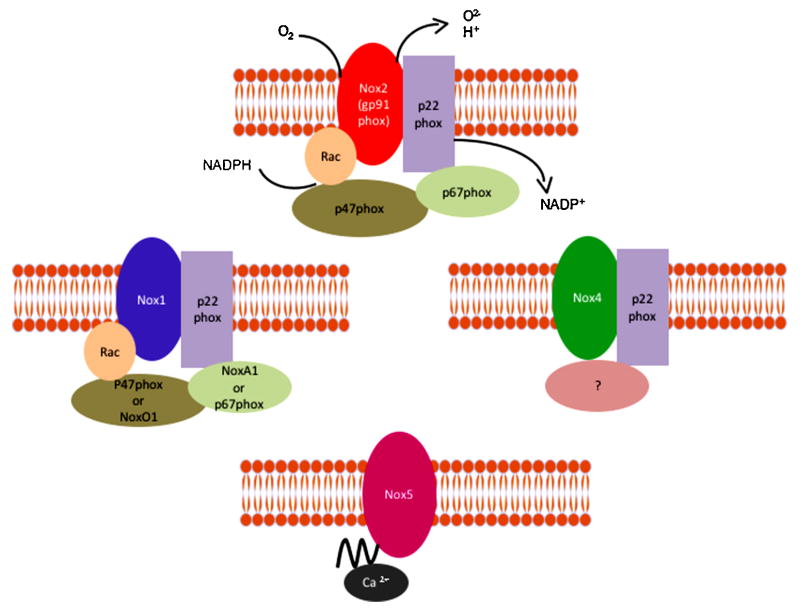
The classical NADPH oxidase is a multimeric enzyme that originally was described in neutrophils, where it plays an antimicrobial role. The phagocytic NADPH oxidase contains a heterodimeric membrane-bound cytochrome b558 complex, which consists of gp91phox (renamed Nox2) and p22phox, and 3 cytosolic subunits (p47phox, p67phox and the small G-protein Rac). Upon agonist stimulation, these cytosolic subunits translocate to the cytochrome complex, leading to an increase in enzymatic activity. In humans, there are five different NADPH oxidase homologues called Nox1 to Nox5, and two related oxidases, Duox1 and Duox2, as well as two additional homologues of the cytosolic units, NoxO1 and NoxA1 (Banfi et al., 2003; Geiszt et al., 2003; Takeya et al., 2003; Ueyama et al., 2007). Although all Nox enzymes are able to increase intracellular ROS, there are important differences regarding their activation, subunit composition, localization and expression (Figure 1). For more details please refer to the following comprehensive reviews (Bedard and Krause, 2007; Hordijk, 2006; Sumimoto, 2008). Ang II has been functionally linked to Nox1 (Dikalova et al., 2005; Lassegue et al., 2001; Matsuno et al., 2005), Nox2 (Bendall et al., 2002; Lavigne et al., 2001; Pagano et al., 1997) and variably to Nox4 (Wingler et al., 2001) in the vasculature, Nox2 and Nox4 in the kidney (Block et al., 2008; Gorin et al., 2003; Haque and Majid, 2004) and Nox2 in the brain (Wang et al., 2006).
Figure 1
Structure of the different vascular NADPH oxidases
Different homologues of NADPH oxidases have been found in the vasculature. NoxA1 (NADPH oxidase activator 1) and NoxO1 (NADPH oxidase organizer 1).
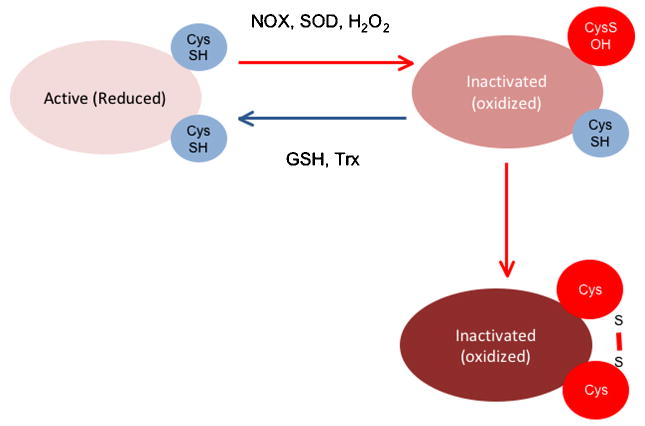
ROS derived from NADPH oxidases serve a signaling function by inducing specific biochemical changes in their molecular targets. H2O2 can oxidize the thiol group of protein (P) cysteines to sulfenic acid (P-SOH), disulfide (PSSP), sulfinic acid (P-SO2H), or sulfonic acid (P-SO3H), the latter of which is an irreversible state of thiol oxidation (Monteiro et al., 2008) (Figure 2). Thiol oxidation by H2O2 (Juarez et al., 2008) has been shown to inhibit protein tyrosine phosphatase (PTP) activity (Kwon et al., 2004; Mahadev et al., 2004; Tabet et al., 2008). Sensitivity to H2O2 is dictated largely by the pKa of the cysteines in the active site of these enzymes: they have an unusually low pKa, which renders them more susceptible to oxidation (Salmeen and Barford, 2005; Tonks, 2005). In contrast, superoxide reacts with iron-sulfur (Fe-S) centers of heme-containing molecules, resulting in altered activity. Two examples are inactivation of aconitase, leading to inhibition of mitochondrial function (Gardner et al., 1994), and activation of guanylate cyclase (Burke-Wolin et al., 1991; Burke and Wolin, 1987).
Figure 2
Inactivation of protein tyrosine phosphatases (PTP) by H2O2-induced thiol oxidation of cysteine residues
H2O2 can inactivate PTPs either irreversibly (right side) or reversibly (left side). Reactivation of the PTPs requires reduction via thioredoxin (Trx) or glutathione (GSH).
NADPH oxidase activation by Ang II
The mechanism of NADPH oxidase activation by Ang II is complex and still incompletely understood. It has been shown that Ang II-mediated ROS production in vascular smooth muscle cells (VSMCs) from large arteries is mediated by Nox1 activation, as shown by loss of the ROS response after treatment with an adenovirus expressing antisense Nox1 (Lassegue et al., 2001), while Nox2 appears to be the Ang II-responsive Nox in resistance vessels, as demonstrated in human VSMCs transfected with gp91phox antisense oligonucleotides (Touyz et al., 2002), in the heart, as shown using small interference RNA (siRNA) against Nox2 (Hingtgen et al., 2006) and in the kidney, as described in gp91phox knockout mice (Haque and Majid, 2004). Typically, there are two different phases of ROS production: one is rapid and transient and the other is delayed and sustained. The first peak is due to an acute activation of NADPH oxidases by Ang II, while the second is dependent on the initial burst and is a consequence of upregulation of different NADPH oxidase subunits by Ang II. For example, in VSMC, the sustained phase of ROS production is preceded by upregulation of Nox1 mRNA (Lassegue et al., 2001; Wingler et al., 2001), p22phox mRNA (Fukui et al., 1997)(Fukui et al., 1997)(Fukui et al., 1997)and protein (Touyz et al., 2002) and p47phox protein (Ni et al., 2007; Touyz et al., 2002). There is also one report that Ang II can upregulate Nox4 mRNA in the vasculature (Wingler et al., 2001), although this is not observed universally (Mollnau et al., 2002).
In VSMCs, rapid ROS release requires p47phox via its phosphorylation by protein kinase C (PKC) and subsequent translocation to the membrane (Lavigne et al., 2001; Seshiah et al., 2002; Touyz et al., 2002), where it organizes the activating subunits NoxA1 and Rac (Ambasta et al., 2006; Hassanain et al., 2007). While each of these subunits has been implicated in the response to Ang II, actual formation of this complex in VSMCs remains to be shown biochemically. Ang II can stimulate PKC-dependent NADPH oxidase activity through three different phospholipases (PL): PLC (Karathanassis et al., 2002), PLD (Touyz and Schiffrin, 1999), and PLA2 (Zafari et al., 1999), with PLD likely predominating. Another important mediator of p47phox phosphorylation is Src (Touyz et al., 2003), although it is not clear whether it regulates p47phox directly or by its actions on the cytoskeleton through cortactin, a Src substrate that binds p47phox (Agerer et al., 2005; Sangrar et al., 2007; Touyz et al., 2005) and facilitates the proper assembly and function of NADPH oxidases by Ang II (Touyz et al., 2005).
Ang II activation of Nox1 and Nox2 also requires Rac1 (Cheng et al., 2006; Miyano et al., 2006; Ueyama et al., 2006). Stimulation of Rac1 by Ang II seems to be mediated by activation of the guanine nucleotide exchange factor (GEF), SOS-1 (Zuo et al., 2005), although a relationship between SOS-1 and Nox activation remains to be demonstrated. Activation of the Rac-GEF is dependent upon Src-mediated transactivation of the epidermal growth factor receptor (EGFR), which then serves as a binding site for phosphatidylinositol 3-kinase (PI-3K), an immediate activator of the Rac-GEF (Seshiah et al., 2002). This initial activation step appears to occur in caveolae (Ushio-Fukai et al., 2001), the site of Nox1 localization (Hilenski et al., 2004). Of importance, the H2O2 that is produced as a result of initial NADPH oxidase activation then activates Src, creating an amplification of oxidase activity (Seshiah et al., 2002). Prolonged signal generation requires upregulation of the Nox subunit, p47phox and p22phox (Fukui et al., 1997; Lassegue et al., 2001; Touyz et al., 2002).
ANG II Activation of NADPH Oxidases in the Cardiovascular System
Hypertension
Ang II is a major contributor to hypertension via its central, vascular and renal effects. It is clear that NADPH oxidase-derived ROS production plays a critical role in the hypertension induced by Ang II (Dikalova et al., 2005; Fukui et al., 1997; Landmesser et al., 2002; Rajagopalan et al., 1996; Weber et al., 2005). For example, our group demonstrated that p22phox is increased in hypertensive rats (Fukui et al., 1997), that smooth-muscle specific p22phox overexpressing mice have elevated blood pressure (Weber et al., 2005), and that smooth-muscle specific overexpression of Nox1 in mice induces a higher pressure response to Ang II (Dikalova et al., 2005). It should be noted that overexpression of one subunit is often accompanied by an increase in the others, and in most reported cases results in an increase in NADPH oxidase activity (Dikalova et al., 2005; Fukui et al., 1997; Laude et al., 2005; Weber et al., 2005). For example, overexpression of p22phox increases the expression of Nox1 protein and NADPH oxidase activity (Laude et al., 2005). In contrast, in p47phox- or Nox1ĘCdeficient mice, the Ang II effects on vascular tone and blood pressure are inhibited (Gavazzi et al., 2006; Landmesser et al., 2002; Matsuno et al., 2005). The infusion of Ang II does, however, induce hypertension in Nox2 deficient mice (Touyz et al., 2005; Wang et al., 2001). Other studies showed that different ROS scavengers, including catalase (Wilson, 1990; Yang et al., 2003) and SOD (Gongora et al., 2006; Wilson, 1990), blunt the response to Ang II. In seeming contrast to these results, a recent study demonstrated that in a model of chronic Ang II elevation, deletion of Nox1 has no effect on blood pressure, but does improve hypertension-related end-organ damage (Yogi et al., 2008). Similarly, Liu et al. (Liu et al., 2003) infusion of the Nox2 inhibitor gp91-dstat into rats co-infused with AngII for 1 week attenuated the increase in medial hypertrophy, but not blood pressure. It is thus possible that NADPH oxidases are necessary for the development, but not the maintenance, of hypertension by Ang II, and contribute to its pathophysiological effects.
With respect to the role of H2O2 and O2.- in the regulation of vascular tone per se, the observed response is tissue and concentration dependent (Barlow and White, 1998; Wei et al., 1996; Yang et al., 1999; Yang et al., 1998). For example, O2.- and H2O2 have vasodilator effects in cerebral arteries, while they have a vasoconstrictor effect in non-cerebral arteries (for more details, please refer to (Miller et al., 2006)). Superoxide exerts its effects mainly, but not exclusively, by inactivating nitric oxide, while H2O2 acts via upregulation of endothelial nitric oxide synthase (eNOS) (Drummond et al., 2000) and via direct effects on ion channels (Liu and Gutterman, 2002) or the contractile machinery (Jin and Rhoades, 1997). In coronary vessels, H2O2 is a potent vasodilator acting via modulation of K+ channels (Miura et al., 2003; Sato et al., 2003). The ROS produced by Ang II activation of NADPH oxidases thus modulate vascular tone by regulating both endothelial function and VSMC contraction.
Endothelial cells
Endothelial dysfunction is largely thought to be a consequence of hypertension, although recent studies suggest that endothelial dysfunction also may predispose to the development of hypertension (Schlaich et al., 2004; Taddei et al., 1996; Yang et al., 2007). Impaired endothelium-dependent relaxation results from a decrease of nitric oxide bioavailability, largely due to increased O2.-. The link between Ang II, ROS and endothelial dysfunction had been demonstrated in hypertensive animals. Acute infusion of Ang II results in impaired relaxation to acetylcholine, the calcium ionophore A23187, and nitroglycerin (Rajagopalan et al., 1996). Moreover, Ang II, through PKC activation, induces an upregulation of Nox1 (Lassegue et al., 2001) and uncouples eNOS (Mollnau et al., 2002), thus increasing O2.- production in a positive feedback fashion. Interestingly, these effects are accompanied by a decrease in soluble guanylate cyclase and a diminished activation of protein kinase G (Mollnau et al., 2002). In other models of hypertension such as the two kidney-one clip (2K-1C) model, it has been demonstrated that Ang II-induced NADPH oxidase activation is responsible for impaired vasodilator responses to endogenous and exogenous nitrovasodilators (Heitzer et al., 1999). Recently, Doughan et al. (Doughan et al., 2008) demonstrated that Ang II activation of NADPH oxidases leads to mitochondrial dysfunction in endothelial cells, amplifying the increase in ROS. This represents a new pathophysiological pathway of Ang II activation.
Vascular smooth muscle cells
Contraction
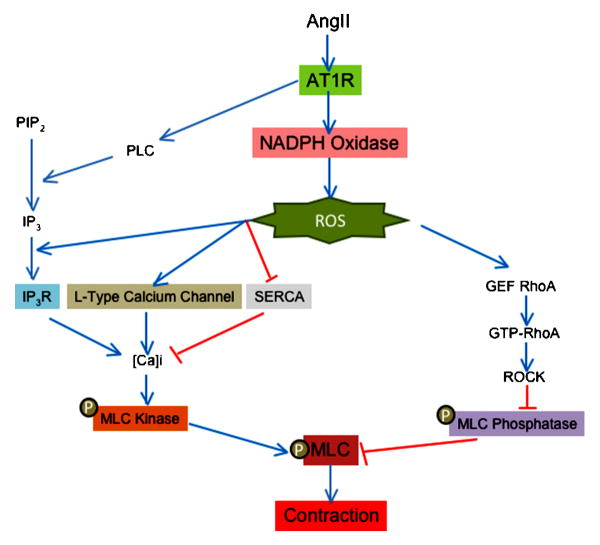
Ang II is one of the most powerful vasoconstrictor agents for vascular smooth muscle. As with other vasoconstrictors, phosphorylation of the myosin light chain (MLC) is a crucial step in force development to Ang II. The rate of MLC phosphorylation depends on a balance between Ca2+/calmodulin-dependent MLC kinase and myosin phosphatase activity (Somlyo and Somlyo, 2000). Thus, an increase in MLC kinase stimulation or a decrease in MLC phosphatase activity leads to VSMC contraction. ROS have been implicated in both of these pathways (Figure 3).
Figure 3
Role of ROS in contraction induced by angiotensin II in vascular smooth muscle cells
ROS production is implicated in calcium dependent and independent induced by angiotensin II. Red lines show inhibitory effects and blue lines depict activation. ROS (reactive oxygen species), AT1R (angiotensin II receptor type 1), PLC (phospholipase C), PIP2 (phosphoinositol biphosphate), IP3 (inositol 1,4,5 triphosphate), IP3R (inositol 1,4,5 triphosphate receptor), SERCA (sarco/endoplasmic reticulum Ca2+-ATPase), GEF (guanine nucleotide exchange factor), GTP (guanosin triphosphate), ROCK (Rho-associate kinase) and MLC (myosin light chain).
One of the most important early signals in the initiation of contraction by Ang II is thus a PLC-mediated increase in intracellular calcium concentration ([Ca]i). Torrecillas et al. (Torrecillas et al., 2001) demonstrated that H2O2 production by Ang II in primary aortic VSMCs (Rodriguez-Puyol et al., 2002) mediates this increase in intracellular calcium concentration ([Ca]i), and therefore MLC phosphorylation, without modifying inositol 1,4,5-trisphosphate (IP3) production by PLC. This suggests that ROS production by Ang II may modify the affinity of the IP3 receptor for its ligand. In support of this idea, Redondo et al. (Redondo et al., 2004) observed that the H2O2-induced increase in [Ca]i in platelets is abolished by preincubation with Xetospongin C (an IP3 receptor inhibitor). However, other studies have shown that activation of Src (Touyz et al., 2001) and ERK (Touyz et al., 1999) by Ang II is upstream of IP3 production and the increase in [Ca]i, suggesting that they may be the actual targets of H2O2 because they have previously been shown to be redox sensitive (Touyz et al., 2003; Ushio-Fukai et al., 2001; Viedt et al., 2000). Because the activation of ERK by ROS is not universally observed (Pinzar et al., 2005; Sano et al., 2001; Touyz et al., 2003; Viedt et al., 2000) and Src activation is only partially ROS dependent (Seshiah et al., 2002), it is likely that ROS generation by Ang II may also be necessary for the correct release of calcium by IP3. In fact, [Ca]i is enhanced in arteries from spontaneously hypertensive rats, and this increase is both ROS- and Ang II-dependent (Tabet et al., 2004). Another mechanism responsible for the ROS-sensitivity of the Ang II-induced increase in [Ca]i is calcium channel activation. Thus, it has been shown that increased expression of the Ž┴1C subunit of the L-type calcium channel by Ang II is ROS-, PKC- and CREB-dependent (Tsai et al., 2007), and the function of this channel is regulated by NADPH oxidases (Wang et al., 2006). Finally, it has been demonstrated that H2O2 inhibits sarcoendoplasmic reticulum Ca-ATPase (SERCA), thereby blocking the reuptake of calcium from the cytosol to the internal stores and increasing [Ca]i. Inhibition of SERCA occurs through oxidation of specific sulphydryl groups (Redondo et al., 2004), although the source of the responsible ROS is unclear. Thus, it is likely that ROS regulate the increase in [Ca]i induced by Ang II at multiple levels.
As noted above, the other factor that regulates the levels of phosphorylated and therefore activated MLC is the activity of MLC phosphatase, which is inhibited by RhoA/RhoA kinase (ROCK) (Seko et al., 2003)-mediated phosphorylation on Thr696 of MYPT1, a myosin binding subunit of MLC phosphatase. Importantly, Uehata et al. (Uehata et al., 1997) observed that ROCK is implicated in the pathophysiology of hypertension induced by Ang II. Recently, it has been shown that the Ang II-stimulated ROCK-mediated phosphorylation of MLC (Seko et al., 2003) is dependent on NADPH oxidases (Higashi et al., 2003). The mechanism of activation of RhoA/ROCK by ROS is not clear, but may involve PDZ- (Hilgers et al., 2007) and leukemia-associated (Ying et al., 2006) GEFs. More studies are necessary to define the actual molecular target of ROS in this pathway.
Hypertrophy
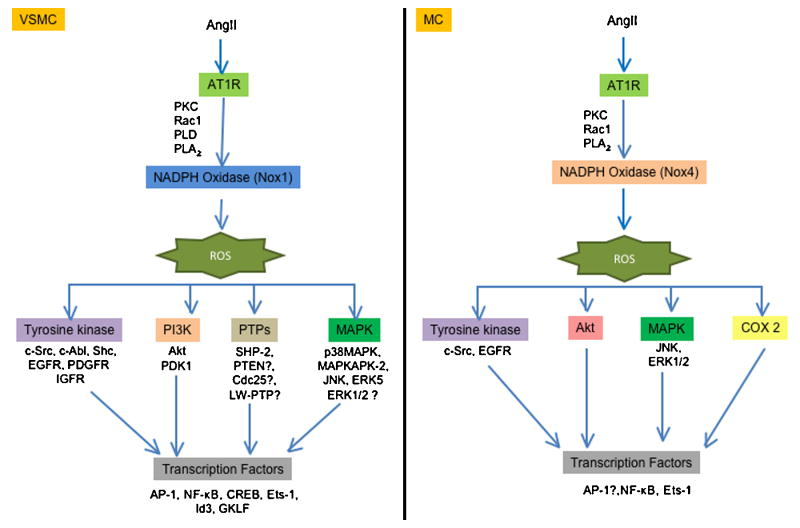
Many of the pathophysiological effects of Ang II are due to its pro-growth properties. In vivo proof that these events are ROS-dependent comes from studies in genetically modified animals. Thus, overexpression of Nox1 (Dikalova et al., 2005) or p22phox (Weber et al., 2005) potentiates the effect of Ang II on vascular hypertrophy, while knockout of Nox1 (Matsuno et al., 2005) or Nox2 (Bendall et al., 2002) inhibits it. Similarly, Ang II-induced cardiac hypertrophy requires the Nox activator Rac1 (Satoh et al., 2006). In both VSMCs and cardiomyocytes, multiple molecular targets that are activated by Ang II in a ROS-dependent manner contribute to hypertrophy, including tyrosine kinases, mitogen activated protein kinases (MAPK), the survival kinase Akt and redox-sensitive transcriptional factors (Taniyama et al., 2004; Touyz et al., 2003; Touyz et al., 2004; Ushio-Fukai et al., 1999) (Figure 4). In addition, ROS-mediated inactivation of protein tyrosine phosphatases (PTP) amplifies growth-related signaling. Classic PTPs include transmembrane receptor-like proteins (RPTPs) and non-transmembrane cytoplasmic PTPs. All members of the PTP family are characterized by the presence of a highly conserved sequence HCXXGXXRS/T, which contains a Cys residue that is necessary for catalysis. As mentioned earlier, the thiol oxidation in this Cys by addition of H2O2 leads to the inactivation of the PTP activity (Juarez et al., 2008). A recent study has shown that SOD1 is essential for PTP inactivation by growth factors, indicating that H2O2, but not O2.-, is implicated in PTP inactivation (Juarez et al., 2008). Tabet et al. (Tabet et al., 2008) demonstrated that Ang II oxidizes PTP/SHP-2 via Nox1 activation, and that PTP/SHP-2 inactivation regulates Ang II-induced activation of Akt, but not of ERK1/2 or p38 MAPK. Other studies have demonstrated that H2O2 inactivates phosphatase and tensin homolog (PTEN) (Lee et al., 2002; Leslie et al., 2003), thus increasing the concentration of PIP3 and activating Akt (Leslie et al., 2003), low molecular weight PTP (LMW-PTP) (Caselli et al., 1998), and Cdc25 (Savitsky and Finkel, 2002). However, whether Ang II similarly inactivates these phosphatases remains to be determined.
Fig 4
Role of ROS in the hypertrophy induced by angiotensin II in vascular smooth muscle cells (left pannel) or messangial cells (right pannel)
Vascular smooth muscle and messangial cells exhibit differences in the targets activated in the hypertrophy induced by angiotensin II. ROS (reactive oxygen species), AT1R (angiotensin II receptor type 1), PKC (protein kinase C), PLD (phospholipase D), PLA2 (phospholipase A2), EGFR (epidermal growth factor receptor), PDGFR (platelet derived growth factor receptor), IGFR (insulin like growth factor receptor) PI3K (phosphatidylinositol 3-kinase), PDK1 (phosphoinositide-dependent protein kinase 1), SHP-2 (Src homology 2 domain-containing protein tyrosine phosphatase), PTEN (phosphatase and tensin homolog), LMW-PTP (low molecular weight phosphatase), p38MAPK (p38 mitogen-activated protein kinase), MAPKAPK-2 (MAPK-activated protein kinase-2), JNK (C-Jun N-terminal kinases), ERK (extracellular signal-regulated kinases), AP-1 (activator protein 1), NF-Ž╩Ž┬ (nuclear factor-kappa Ž┬), CREB (cAMP response element-binding), Ets-1 (E26 transformation-specific sequence), GFLK (Gut-enriched KrĘ╣ppel-like factor) and COX-2 (ciclooxigenase 2).
One of the early effects of Ang II stimulation is c-Src activation, which is inhibited by catalase overexpression or treatment with pharmacological antioxidants (Ushio-Fukai et al., 2001). Phosphorylated Src mediates the recruitment of the AT1R to caveolae, which is required for proper organization of ROS-dependent Ang II signaling. Ang II also transactivates the IGF-1R (Touyz et al., 2003) and PDGFR via ROS and Shc (Heeneman et al., 2000). Transactivation of these receptors leads to the recruitment and stimulation of different proteins, including phosphatidylinositol 3-kinase, which activates Akt (Ushio-Fukai et al., 1999). Whether the ROS sensitivity of this pathway is conferred by Src, or if there are redox modifications of these proteins or their associated phosphatases, remains to be determined. Because Akt is phosphorylated by MAPKAPK-2, a substrate of the ROS-sensitive p38MAPK (Taniyama et al., 2004), it is possible that it is p38MAPK activation that is the actual target of ROS. One intriguing hypothesis is that ROS mediate the dissociation of the p38MAPK kinase Ask-1 from redox-active thioredoxin (Hsieh and Papaconstantinou, 2006; Matsuzawa et al., 2005), although this remains to be investigated.
Activation of other MAPKs by Ang II also requires ROS derived from NADPH oxidases. Thus, c-Jun NH2 terminal kinase (JNK) activation in response to Ang II is blunted by different antioxidants (Kyaw et al., 2001) or by transfection with antisense against the p22phox subunit (Viedt et al., 2000). Moreover, Shc is implicated in the phosphorylation of JNK (Yoshizumi et al., 2001). In contrast to p38MAPK and JNK, whose redox sensitivity to Ang II is nearly universal in the vasculature, the sensitivity of ERK1/2 to ROS is controversial. Some studies found that activation of ERK1/2 is ROS independent (Kyaw et al., 2001; Touyz et al., 2003; Viedt et al., 2000), while others found it to be ROS sensitive (Frank et al., 2000; Pinzar et al., 2005; Sano et al., 2001). This suggests that ERK1/2 is not an actual target of ROS; rather, when redox-sensitivity is observed, it is likely conferred by an upstream kinase or phosphatase and may occur in a specific subcellular compartment.A number of years ago, we suggested that some aspects of Ang II signaling may occur subsequent to internalization of the AT1R in endosomes (Griendling et al., 1986). The apparent role of caveolae in organizing redox signals (Zuo et al., 2005) suggests that ROS-dependent signaling is intimately linked to vesicular trafficking. A recent series of elegant papers by the Englehardt and Miller labs shows that Nox-dependent ROS generation in response to IL-1Ž┬ or TNF-Ž┴ occurs in the early endosome and is released into the cytosol by a pathway requiring activation of chloride channels (Miller et al., 2007; Mumbengegwi et al., 2008), although the origin of the vesicles is unknown. This provides a molecular explanation for the finding that ROS generation in nonphagocytic cells is intracellular, even though the Nox enzymes are oriented to the outside of the plasma membrane. Although this has not been studied for Ang II, it is likely that a similar mechanism explains the robust intracellular generation of H2O2 observed in VSMCs, and the apparent compartmentalization of redox signaling in these cells. Both caveolin-mediated and clathrin-dependent internalization have been shown for AT1R internalization (Gaborik et al., 2001; Ishizaka et al., 1998), but have not been studied with respect to Ang II stimulated ROS generation. However, Choi et al. (Choi et al., 2008) demonstrated that Ž┬-arrestins are not necessary for Nox1 activation by Ang II, which may indicate that activation occurs prior to internalization. Because this latter study was conducted entirely in a reconstituted system, these results must be interpreted with caution.
Activation of these ROS-sensitive signal transduction pathways ultimately leads to stimulation of specific transcription factors. One of the most well-studied is AP-1, a heterodimer of Fos and Jun. Viedt et al. (Viedt et al., 2004) and Wu et al. (Wu et al., 2005) demonstrated that activation of a p22phox- and gp91phox-containing oxidase is essential for Ang II-stimulated AP-1 binding to DNA in VSMCs and cardiomyocytes. Conversely, activation of AP-1 is necessary for increased transcription of p22phox by Ang II, suggesting a feed-forward mechanism (Manea et al., 2008). AP-1 is implicated in diverse biological functions, such as cell proliferation, protein synthesis, apoptosis and secretion of pro-fribotic factors via its induction of specific genes. With respect to Ang II, the increase in TGF-Ž┬, endothelin, MMP-1 and fibronectin mRNA expression is mediated by ROS- and AP-1 dependent mechanisms (Browatzki et al., 2005; Cheng et al., 2003; Moriguchi et al., 1999; Wenzel et al., 2001). Another important ROS-dependent transcription factor activated by Ang II is NFŽ╩B. The prototype of the NFŽ╩B family is the p50/p65 heterodimer expressed constitutively in most mammalian cells. It is well established that NFŽ╩B is complexed with and sequestered in the cytoplasm by inhibitory IŽ╩B (inhibitor of NFŽ╩B) proteins, and that many activating stimuli induce phosphorylation of IŽ╩B by the IŽ╩B-kinase complex (IKKŽ┴, IKKŽ┬, and IKKŽ├), initiate IŽ╩B ubiquitination and degradation by means of the 26S proteasome, and allow translocation of NFŽ╩B to the nucleus (Chen et al., 1996; DiDonato et al., 1996; DiDonato et al., 1995; Ghosh and Karin, 2002). Although many investigators have shown that NFŽ╩B is redox sensitive, there are just a few studies that demonstrated the activation of this factor by ROS in VSMCs (Browatzki et al., 2005; Ortego et al., 1999; Shang et al., 2008). Among the different targets regulated by Ang II in a NFŽ╩B dependent manner is monocyte chemoattractant proteinĘC1 (MCP-1) (Capers et al., 1997; Chen et al., 1998). Other Ang II- and ROS-sensitive transcription factors are cyclic AMP response element-binding protein (CREB) (Funakoshi et al., 2002; Ichiki et al., 2003; Tsai et al., 2007), hypoxia inducible factor 1-Ž┴ (Gorlach et al., 2001), Id3 and GKLF (Nickenig et al., 2002). These latter two factors have opposite effects on cell cycle progression, suggesting that ROS must be compartmentalized. Recently, it has been shown that And II-induced activation of Ets-1, which is important in vascular remodeling (Zhan et al., 2005), is redox sensitive and its activation induces an increase in p47phox expression (Ni et al., 2007). Ets-1 is also an important regulator of the cyclin-dependent kinase inhibitor p21CIP(Zhang et al., 2003), plasminogen activator inhibitorĘC1 (PAI-1) and MCP-1 (Zhan et al., 2005).
íí
ANG II Activation of NADPH Oxidases in the Kidney
The kidney also expresses NADPH oxidases, the most abundant of which is Nox4 (originally called Renal NADPH oxidase or Renox)(Geiszt et al., 2000). The effect of Ang II on NADPH oxidase activity in this organ is well described, although the molecular mechanisms are less clear and have been investigated mainly at the level of gene expression. Thus, infusion of Ang II upregulates the expression of p22phox and Nox1 and downregulates the expression of extracellular superoxide dismutase (EC-SOD) (Chabrashvili et al., 2003). The decrease of EC-SOD seems to participate in the upregulation of p22phox and Nox1. Moreover, the resulting increase in ROS production by Ang II in the kidney is important in the regulation of renal function (Chabrashvili et al., 2003; Jaimes et al., 2005; Lopez et al., 2003; Modlinger et al., 2006; Nouri et al., 2007). Thus, LĘ«pez et al. (Lopez et al., 2003) observed that renal infusion of Ang II in rats reduced renal blood flow (RBF), glomerular filtration (GF), sodium excretion, and NO levels, all of which were blunted by the AT1R antagonist valsartan, the superoxide scavenger tempol and the NADPH oxidase inhibitor apocynin. Furthermore, treatment of rats with siRNA against p22phox prevents the increase of renal cortical NADPH oxidase activity and increases the excretion of the lipid peroxidation marker 8-isoprostane PGF2Ž┴ induced by perfusion with Ang II (Modlinger et al., 2006). The role of superoxide in regulating renal function during Ang II infusion has been confirmed in a number of studies (Kopkan et al., 2006; Mori and Cowley, 2003; Mori et al., 2007; Wang et al., 2003; Welch et al., 2005).
Similar to its effects in VSMCs, Ang II can stimulate protein synthesis in mesangial cells (MC) and tubular cells (Hannken et al., 1998; Jaimes et al., 1998), leading to hypertrophy (Figure 4) and synthesis of extracellular components. Both processes contribute to the pathogenesis of fibrosis of the glomerular microvascular bed (Ardaillou et al., 1999). However, whereas in VSMC Ang II activation of the NADPH oxidase has been studied extensively in renal cells, it is less well understood. Similar to VSMC, the activation of the NADPH oxidase is dependent on the production of arachidonic acid (Block et al., 2006; Gorin et al., 2001), PKC (Jaimes et al., 1998) and Rac1 (Gorin et al., 2003); however, the principal Nox that is coupled to Ang II is Nox4 (Gorin et al., 2003). Because Nox4 has been demonstrated to be Rac1 independent (Martyn et al., 2006), the significance of Rac1 in this system requires further study. As in VSMC, multiple targets are downstream of Nox activation by Ang II, including Akt (Gorin et al., 2003), EGFR (Ding et al., 2007), JNK (Ding et al., 2007), ERK1/2 (Yu et al., 2005) (Gorin et al., 2004), ETS-1 (Pearse et al., 2008), cyclooxigenase-2 (COX2) (Jaimes et al., 2005; Jaimes et al., 2008; Pearse et al., 2008) and PDK1 (Block et al., 2008). Some of the extracellular components regulated by ROS in Ang II-stimulated MC include fibronectin (Block et al., 2006; Pearse et al., 2008), collagen I (Zhao et al., 2008) and TGF-Ž┬ (Lodha et al., 2002; Zhao et al., 2008), a profibrotic cytokine.
ANG II Activation of NADPH Oxidases in the Central Nervious System
Ang II in the brain participates in regulation of blood pressure, sympathetic activity, vasopressin release, thirst, and sodium appetite (Phillips, 1987). In the last several years, numerous studies have demonstrated that ROS generation by Ang II modulates some of these effects in the CNS, acting in different regions of the brain. Zimmerman et al. observed that the acute intracerebroventricular injection (Zimmerman et al., 2002) or chronic systemic perfusion (Zimmerman et al., 2004) of Ang II induced O2.- production in the subfornical organ (SFO), leading to an increase in blood pressure and dipsogenesis. Notably, transfection with adenovirus that expresses Mn-SOD or Cu/Zn SOD blocked these effects of Ang II. Moreover, studies from Zucker's group (Gao et al., 2004) demonstrated that the Ang II-NADPH oxidase axis is regulated by sympathetic excitation in the rostral ventrolateral medulla (RVLM), which is implicated in chronic heart failure (Gao et al., 2005). Ang II increases gp91phox and p47phox expression in the RVLM as well (Gao et al., 2004).
The mechanism of NADPH activation by Ang II in the CNS is incompletely understood. Even so, it has been demonstrated that NADPH oxidase activation requires Rac1 (Zimmerman et al., 2004) and PKC (Wang et al., 2006). More information is available concerning the downstream consequences of NADPH oxidase-derived ROS in the CNS (Figure 5). Thus, Wang et al. (Wang et al., 2006) observed that inhibition of NADPH oxidases by apocynin or an inhibitory Nox2 peptide docking sequence decreased Ang II-stimulated increases in [Ca]i, and, more recently, it was observed that O2.- production by Ang II is implicated in the elevation of [Ca]i (Zimmerman et al., 2005) in neuroblastoma Neuro 2A cells. In addition, ROS production by Ang II has been shown to be critical for the inhibition of the delayed rectifier potassium current (I(Kv)), which contributes to neuronal activity (Sun et al., 2005). Other targets of Ang II stimulated NADPH oxidases in the CNS are p38 and ERK1/2. Chan et al. (Chan et al., 2005) demonstrated that Ang II injection in the RVLM increased the phosphorylation of ERK1/2 and p38MAPK, but not in JNK. These effects were attenuated by application into the RVLM of a flavin oxidase inhibitor, DPI, antisense oligonucleotides that target p22phox or p47phox mRNA, or Tempol. Moreover, they observed that DPI, Tempol and the p38MAPK inhibitor decreased the frequency of glutamate-sensitive spontaneous excitatory postsynaptic currents and the effects on the blood pressure induced by Ang II (Chan et al., 2005).
Fig 5
Role of ROS in modulation of angiotensin II signaling in the brain
ROS are implicated in the dipsogenic action and blood pressure modulated by angiotensin II in the brain. ROS (reactive oxygen species), AT1R (angiotensin II receptor type 1), PKC (protein kinase C), p38MAPK (p38 mitogen-activated protein kinase) and ERK (extracellular signal-regulated kinases).
Conclusions and Future Directions
Multiple studies have demonstrated a role for NADPH oxidases in Ang II function. Although many downstream targets have been identified, the temporal sequence of events that lead to the proper activation and function of NADPH oxidase is not completely understood, and the compartmentalization of signaling remains to be fully investigated. In addition, more work is necessary to identify the precise molecular modifications of the putative signaling targets of ROS after Ang II stimulation, with particular attention to phosphatases. Understanding which NADPH oxidases are activated by Ang II in specific tissues, in normal physiology and in disease development is also important, and may help us to define better interventions aimed at blocking the deleterious effects of Ang II activation while supporting those necessary for survival.
Acknowledgments
This work was supported by NIH grants HL38206, HL075209, HL058000, and HL05863.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.NADPH Oxidases and Angiotensin II Receptor Signaling
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2835147/íí
Hypertension Research, Review Series
Published: 28 October 2010
Reactive oxygen species and vascular biology: implications in human hypertension
Rhian M Touyz & Ana M Briones
Kidney Research Centre, Ottawa Health Research Institute, University of Ottawa, Ottawa, Ontario, Canada
Abstract
Increased vascular production of reactive oxygen species (ROS; termed oxidative stress) has been implicated in various chronic diseases, including hypertension. Oxidative stress is both a cause and a consequence of hypertension. Although oxidative injury may not be the sole etiology, it amplifies blood pressure elevation in the presence of other pro-hypertensive factors. Oxidative stress is a multisystem phenomenon in hypertension and involves the heart, kidneys, nervous system, vessels and possibly the immune system. Compelling experimental and clinical evidence indicates the importance of the vasculature in the pathophysiology of hypertension and as such much emphasis has been placed on the (patho)biology of ROS in the vascular system. A major source for cardiovascular, renal and neural ROS is a family of non-phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox), including the prototypic Nox2 homolog-based NADPH oxidase, as well as other Noxes, such as Nox1 and Nox4. Nox-derived ROS is important in regulating endothelial function and vascular tone. Oxidative stress is implicated in endothelial dysfunction, inflammation, hypertrophy, apoptosis, migration, fibrosis, angiogenesis and rarefaction, important processes involved in vascular remodeling in hypertension. Despite a plethora of data implicating oxidative stress as a causative factor in experimental hypertension, findings in human hypertension are less conclusive. This review highlights the importance of ROS in vascular biology and focuses on the potential role of oxidative stress in human hypertension.
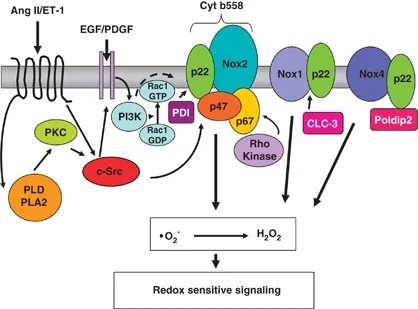
Regulation of Noxes in vascular cells. Activation of NADPH oxidase involves multiple subunits and many signaling pathways involving c-Src p21Ras, protein kinase C (PKC), phospholipase D (PLD), phospholipase A2 (PLA2) and Rho kinase. All Noxes, except Nox5, appear to have an obligatory need for p22phox. Nox2 requires p47phox and p67phox for its activity. Oxidase activation involves Rac translocation, phosphorylation of p47phox and its translocation, possibly with p67phox, and p47phox association with cytochrome b558. Induction of Nox mRNA expression is observed in response to many stimuli including vasoactive agents (angiotensin II (Ang II), endothelin 1 (ET-1)), growth factors (epidermal growth factor (EGF), platelet-derived growth factor (PDGF)), amongst others. Recently identified Nox regulators include ClC-3, Poldip2 and protein disulfide isomerase (PDI). PDI associates with p22phox to regulate Nox2. Poldip2 associates with p22phox to activate Nox4, leading to regulation of focal adhesion turnover and vascular smooth muscle cell migration. Nox1 is regulated by the chloride/proton exchanger ClC-3. Cyt, cytochrome; GDP, guanosine diphosphate; GTP, guanosine triphosphate; PI3K, phosphoinositide 3-kinase
íí
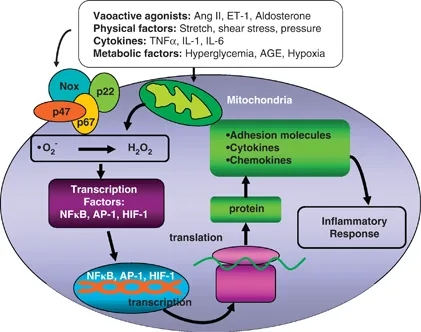
Induction of cellular inflammation by reactive oxygen species (ROS). Generation of ROS in vascular cells by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase or mitochondria, leads to redox signaling that activates transcription factors important in inflammation. AGE, advanced glycation end product; Ang II, angiotensin II; AP-1, activator protein-1; ET-1, endothelin-1; HIF-1; hypoxia-inducible factor-1; IL, interleukin; NF-Ž╩B, nuclear factor-Ž╩B; TNFŽ┴, transforming growth factor Ž┴.
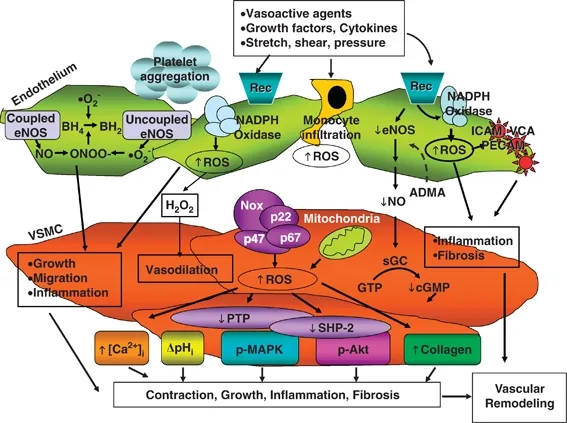
Redox-sensitive mechanisms underlying vascular changes in hypertension. Activation of reactive oxygen species (ROS)-generating enzymes, such as NAD(P)H oxidase, uncoupling of NOS and mitochondrial enzymes in endothelial and vascular smooth muscle cells results in decreased nitric oxide (NO) production and increased generation of •O2− and H2O2, which in turn influence redox-sensitive signaling molecules including MAPKs, PTPs, ion channels, transcription factors as well as induction of pro-inflammatory adhesion molecules such as platelet/endothelial cell adhesion molecule (PECAM), intercellular adhesion molecules (ICAM) and vascular cell adhesion molecules (VCAM). These processes lead to vascular growth, fibrosis, contraction/dilation, inflammation and platelet aggregation, which underlie vascular damage and structural remodeling in hypertension and other cardiovascular diseases. ADMA, asymmetric N(G), N(G)-dimethyl-L-arginine, endogenous NOS inhibitor; BH4, tetrahydrobiopterin; cGDP, cyclic guanosine diphosphate; eNOS, endothelial nitric oxide synthase; GTP, guanosine triphosphate; MAPK, mitogen-activated protein kinases; p-, phosphorylated protein; NADPH, nicotinamide adenine dinucleotide phosphate; PTP, protein tyrosine phosphatase; Rec, receptor; sGC, soluble guanylyl cyclase; SHP-2, SH2 domain-containing protein tyrosine phosphatase-2; VSMC, vascular smooth muscle cells.
Reactive oxygen species and vascular biology: implications in human hypertension | Hypertension Research
https://www.nature.com/articles/hr2010201íí
Exp Mol Med. 2019 Dec;
Mitochondrial dysfunction and oxidative stress in heart disease
Jessica N. Peoples,1 Anita Saraf,2 Nasab Ghazal,1 Tyler T. Pham,3 and Jennifer Q. Kwongcorresponding author1
1Department of Pediatrics, Division of Cardiovascular Biology, Emory University School of Medicine, Atlanta, GA 30322 USA
2Department of Medicine, Division of Cardiology, Emory University School of Medicine, Atlanta, GA 30322 USA
3Emory College of Arts and Sciences, Emory University, Atlanta, GA 30322 USA
Beyond their role as a cellular powerhouse, mitochondria are emerging as integral players in molecular signaling and cell fate determination through reactive oxygen species (ROS). While ROS production has historically been portrayed as an unregulated process driving oxidative stress and disease pathology, contemporary studies reveal that ROS also facilitate normal physiology. Mitochondria are especially abundant in cardiac tissue; hence, mitochondrial dysregulation and ROS production are thought to contribute significantly to cardiac pathology. Moreover, there is growing appreciation that medical therapies designed to mediate mitochondrial ROS production can be important strategies to ameliorate cardiac disease. In this review, we highlight evidence from animal models that illustrates the strong connections between mitochondrial ROS and cardiac disease, discuss advancements in the development of mitochondria-targeted antioxidant therapies, and identify challenges faced in bringing such therapies into the clinic.
Subject terms: Cardiovascular diseases, Cardiomyopathieshttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC6923355/
íí
Mitochondrial dysfunction and oxidative stress in heart ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6923355
Dec 19, 2019 íĄ NOXs are a family of proteins involved in intracellular ROS production and catalyze the transfer of NADPH to molecular oxygen to generate superoxide and hydrogen peroxide. Of particular interest is NOX4, which partially localizes to the mitochondrial inner membrane in cardiomyocytes and renal cells 20ĘC22.íí
Published: 19 October 2017
NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance
Karthigayan Shanmugasundaram, Bijaya K. Nayak, William E. Friedrichs, Dharam Kaushik, Ronald Rodriguez & Karen BlockDepartment of Medicine, UT Health, San Antonio, TX, 78229, USA
Abstract
The molecular mechanisms that couple glycolysis to cancer drug resistance remain unclear. Here we identify an ATP-binding motif within the NADPH oxidase isoform, NOX4, and show that ATP directly binds and negatively regulates NOX4 activity. We find that NOX4 localizes to the inner mitochondria membrane and that subcellular redistribution of ATP levels from the mitochondria act as an allosteric switch to activate NOX4. We provide evidence that NOX4-derived reactive oxygen species (ROS) inhibits P300/CBP-associated factor (PCAF)-dependent acetylation and lysosomal degradation of the pyruvate kinase-M2 isoform (PKM2). Finally, we show that NOX4 silencing, through PKM2, sensitizes cultured and ex vivo freshly isolated human-renal carcinoma cells to drug-induced cell death in xenograft models and ex vivo cultures. These findings highlight yet unidentified insights into the molecular events driving cancer evasive resistance and suggest modulation of ATP levels together with cytotoxic drugs could overcome drug-resistance in glycolytic cancers.https://www.nature.com/articles/s41467-017-01106-1
íí