ĪĪ
PLoS Biol. 2010 Aug; 8(8): e1000465.
IL-6 and IL-10 Anti-Inflammatory Activity Links Exercise to Hypothalamic Insulin and Leptin Sensitivity through IKK”┬ and ER Stress Inhibition
Eduardo R. Ropelle, 1 Marcelo B. Flores, 1 Dennys E. Cintra, 1 Guilherme Z. Rocha, 1 Jos©” R. Pauli, 1 Joseane Morari, 1 Claudio T. de Souza, 1 Juliana C. Moraes, 1 Patr©¬cia O. Prada, 1 Dioze Guadagnini, 1 Rodrigo M. Marin, 1 Alexandre G. Oliveira, 1 Taize M. Augusto, 2 Hernandes F. Carvalho, 2 L©¬cio A. Velloso, 1 Mario J. A. Saad, 1 and Jos©” B. C. Carvalheira 1 , *
Antonio J. Vidal-Puig, Academic Editor
Author information Article notes Copyright and License information Disclaimer
1Department of Internal Medicine, Faculty of Medical Sciences, State University of Campinas (UNICAMP), Campinas, São Paulo, Brazil
2Department of Anatomy, Cell Biology, Physiology and Biophysics, State University of Campinas (UNICAMP), Campinas, São Paulo, Brazil
University of Cambridge, United Kingdom
ĪĪ
Abstract
Overnutrition caused by overeating is associated with insulin and leptin resistance through IKK”┬ activation and endoplasmic reticulum (ER) stress in the hypothalamus. Here we show that physical exercise suppresses hyperphagia and associated hypothalamic IKK”┬/NF-”╩B activation by a mechanism dependent upon the pro-inflammatory cytokine interleukin (IL)-6. The disruption of hypothalamic-specific IL-6 action blocked the beneficial effects of exercise on the re-balance of food intake and insulin and leptin resistance. This molecular mechanism, mediated by physical activity, involves the anti-inflammatory protein IL-10, a core inhibitor of IKK”┬/NF-”╩B signaling and ER stress. We report that exercise and recombinant IL-6 requires IL-10 expression to suppress hyperphagia-related obesity. Moreover, in contrast to control mice, exercise failed to reverse the pharmacological activation of IKK”┬ and ER stress in C3H/HeJ mice deficient in hypothalamic IL-6 and IL-10 signaling. Hence, inflammatory signaling in the hypothalamus links beneficial physiological effects of exercise to the central action of insulin and leptin.
Author Summary
The hypothalamus is a brain region that gathers information on the body's nutritional status and governs the release of multiple metabolic signaling molecules such as insulin and leptin to maintain homeostasis. Overeating and obesity are associated with insulin and leptin resistance in the hypothalamus, and recent studies provide an intriguing link between inflammation and dysfunction of hypothalamic insulin and leptin signaling through activation of IKK”┬, a key player in immune response, and endoplasmic reticulum (ER) stress. This means that strategies to reduce the aberrant activation of inflammatory signaling in the hypothalamus are of great interest to improve the central insulin and leptin action and prevent or treat related metabolic diseases. Using a combination of pharmacological, genetic, and physiological approaches, our study indicates that physical activity reorganizes the set point of nutritional balance through anti-inflammatory signaling mediated by interleukin (IL)-6 and IL-10 in the hypothalamus of rodents. Hence, IL-6 and IL-10 are important physiological contributors to the central insulin and leptin action mediated by exercise, linking it to hypothalamic ER stress and inflammation.
Introduction
Overnutrition and sedentary lifestyle are among the most important factors that lead to an unprecedented increase in the prevalence of obesity. In mammals, food intake and energy expenditure are tightly regulated by specific neurons localized in the hypothalamus. The hypothalamus can gather information on the body's nutritional status by integrating multiple signals, including potent hormonal signals such as insulin and leptin [1],[2]. The impairment of hypothalamic insulin and leptin signaling pathways is sufficient to promote hyperphagia, obesity, and type 2 diabetes (T2D) in different genetic rodent models with neuronal ablation of insulin and leptin signaling [1],[3],[4]. We and others have proposed that overnutrition induces the central insulin and leptin resistance through the aberrant hypothalamic activation of proinflammatory molecules, including TLR4 and IKK [5]©C[7].
IKK”┬ is a key player in controlling both innate and adaptive immunity. Activation of IKK”┬ by phosphorylation at S177 and S181 induces phosphorylation, ubiquitination, and subsequent proteosomal degradation of its substrate I”╩B”┴. The degradation of I”╩B”┴ allows NF-”╩B proteins to translocate to the nucleus and bind their cognate DNA binding sites to regulate the transcription of a large number of genes, including stress-response proteins and cytokines [8]. Growing evidence provides an intriguing link between metabolic inflammation and dysfunction of insulin and leptin signaling via activation of IKK”┬ and endoplasmatic reticulum (ER) stress [9]©C[14]. Examination of ER stress markers in different tissues of dietary (high-fat diet-induced) and genetic (ob/ob) mouse models of obesity demonstrated increased levels of PERK phosphorylation and JNK and IKK”┬ activity [7],[12]. In addition, a recent study showed the activation of hypothalamic IKK”┬/NF-”╩B, at least in part, through elevated endoplasmic reticulum stress in the hypothalamus and that these phenomena are associated with central insulin and leptin resistance, hyperphagia, and body weight gain in mice [7]. Thus, strategies to reduce the aberrant activation of inflammatory signaling and/or ER stress in hypothalamic neurons are of great interest to improve the central insulin and leptin action and prevent or treat obesity and related diseases.
Physical activity is considered a cornerstone of the treatment for obesity. Exercise has long been reported to reduce body weight and visceral adiposity, increasing the energy expenditure and improving glycaemic control in overweight or T2D patients [15],[16]. Since the discovery of interleukin (IL)-6 releases from contracting skeletal muscle, accumulating evidence indicates that exercise induces metabolic changes in other organs, such as the liver, the adipose tissue, and hypothalamus, in an IL-6 dependent manner. IL-6 is most often classified as a pro-inflammatory cytokine, although consistent data also demonstrate that IL-6 has an anti-inflammatory effect and may negatively regulate the inflammation of acute phase response by increasing IL-10, IL-1 receptor antagonist (IL-1ra), and soluble TNF-receptors (sTNF-R) [17]. Moreover, IL-6 appears to play a central role in the regulation of appetite, energy expenditure, and body composition [18],[19]. However, the effects of physical activity in the metabolic regulatory pathways in the central nervous system (CNS) remain unexplored. Thus, we hypothesized that exercise could exert its effects in the CNS by modulating the specific hypothalamic neurons responsible for the control of food consumption. In the present study, we investigated the effect of the anti-inflammatory response, mediated by IL-6, on hypothalamic IKK”┬ activation and ER stress, central insulin and leptin sensitivity, and food intake in diet-induced rats after physical activity.ĪĪ
Hypothalamic IL-10: A Core Anti-Inflammatory Cytokine Induced by IL-6
Although our findings clearly show that IL-6 diminished hypothalamic IKK”┬ and ER stress activation and restored the central insulin and leptin action in an animal model of obesity, the question remains as to how IL-6 promotes these events in the hypothalamus. Following exercise, the high circulating levels of IL-6 are followed by an increase in two anti-inflammatory molecules, IL-1ra and IL-10 [25]. Therefore, IL-6 induces an anti-inflammatory environment by inducing the production of IL-1ra and IL-10. In our study, we found that exercise increased the hypothalamic levels of IL-10 but did not change IL-1ra expression in this tissue. Thus, we showed that the anti-inflammatory response mediated by IL-6 involves the increase of IL-10 expression in the hypothalamus.
IL-10 is an important immunoregulatory cytokine with multiple biological effects. In the cytoplasm, it has been demonstrated that IL-10 blocks NF-”╩B activity at two levels: suppressing IKK activity and NF-”╩B DNA binding activity [26]. Moreover, IL-10 reduced ER stress in intestinal eptithelial cells, whereas IL-10−/− mice demonstrated that the expression of the ER stress response protein grp-78/BiP was increased in intestinal eptithelial cells under conditions of chronic inflammation [27].
In the CNS, the anti-inflammatory role of IL-10 has been extensively studied in experimental autoimmune encephalomyelitis, an animal model of human multiple sclerosis. The increase in IL-10 expression in the CNS during recovery from brain inflammation and the inability of IL-10 null mice to recover from acute CNS inflammation suggests that the presence of IL-10 within this target organ is required for disease remission [42],[43]. However, the role of hypothalamic IL-10 in the control of low-grade inflammation generated during obesity was unknown. Here, we discovered that intrahypothalamic infusion of recombinant IL-10 blocked IKK/NF-”╩B signaling and ER stress and restored Akt and STAT3 phosphorylation, promoting a re-balance in the energy intake in obese animals. On the other hand, the selective decrease in IL-10 expression in discrete hypothalamic nuclei of obese animals mediated by ASO treatment blunted the effects of both exercise and the intrahypothalamic infusion of recombinant IL-6 in the restoration of central insulin and leptin actions. In addition, we demonstrated that in mice that sustained significantly lower hypothalamic levels of IL-6 and IL-10 after exercise (C3H/HeJ), there was no reduction in pharmacological ER stress activation, in contrast to WT mice. These data are intriguing as IL-10 represents an important cytokine that may reduce both inflammation and ER stress in the hypothalamus. Thus, the modulation of hypothalamic IL-10 expression could be considered the direct target of exercise/IL-6 and constitutes a promising alternative to reduce hypothalamic inflammation and ER stress related to obesity.
The decrease in food intake induced by IL-10 in obese rats is not in accordance with the effects observed in IL-10 KO. It has been reported that mice with combined deficiency of leptin and IL-10 gain less body weight than mice lacking leptin only [44]. However, these discrepancies may be a consequence of methodological differences related to physiological versus genetic approaches and acute versus chronic situation investigated, and most important it may be consequence of IL-10 effects in the regulation of energy expenditure, likewise observed in mice lacking TNF-”┴ receptor [45]; thus, the role of IL-10 in the control of food intake and energy expenditure deserves further exploration.
The long-term reversal effects on body composition, mediated by exercise alone, are controversial. It should be acknowledged that it is often difficult to find long-term reversal effects on body fat in both experimental animals and humans by exercise alone without restrained diet [46]. In the chronic experiments, we observed that the obese animals lost weight during the same period in which a reduction in food intake was observed. After this period, no significant difference was observed in the body weight of exercised animals, although the obese animals presented a significant improvement in metabolic parameters after the chronic exercise protocol.
Since IKK”┬/NF-”╩B inhibition in the CNS represents a potential target therapy to combat obesity and most anti-inflammatory therapies have limited direct effects on IKK”┬/NF-”╩B and a limited capacity for concentration in the CNS, our study provides substantial evidence that physical activity could help to reorganize the set point of nutritional balance and therefore aid in counteracting the energy imbalance induced by overnutrition through the anti-inflammatory response in hypothalamic neurons. Hence, IL-6 and IL-10 are important physiological contributors to the central insulin and leptin action mediated by physical activity, linking it to hypothalamic ER stress and inflammation.IL-6 and IL-10 Anti-Inflammatory Activity Links Exercise to Hypothalamic Insulin and Leptin Sensitivity through IKK”┬ and ER Stress Inhibition
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2927536/ĪĪ
Blood. 1997 Jun 15;89(12):4461-9.
Interleukin-10 inhibits interferon-gamma-induced intercellular adhesion molecule-1 gene transcription in human monocytes.
Song S1, Ling-Hu H, Roebuck KA, Rabbi MF, Donnelly RP, Finnegan A.
Department of Medicine, Rush Presbyterian-St Luke's Medical Center, Chicago, IL 60612, USA.
Abstract
Interleukin-10 (IL-10) is a potent monocyte regulatory cytokine that inhibits gene expression of proinflammatory mediators. In this study, we investigated the mechanism by which IL-10 downregulates expression of intercellular adhesion molecule-1 (ICAM-1) on the cell surface of normal human monocytes activated with interferon-gamma (IFN-gamma). IL-10 inhibition of IFN-gamma-induced ICAM-1 expression was apparent as early as 3 hours and was blocked by an anti-IL-10 antibody but not by an isotype-matched control antibody. Northern blot analysis showed that IL-10 reduced the accumulation of ICAM-1 mRNA in IFN-gamma-stimulated monocytes. IL-10 inhibition of ICAM-1 steady-state mRNA was detected at 3 hours and remained at 24 hours. Nuclear run-on transcription assays showed that IL-10 inhibited the rate of IFN-gamma-induced transcription of the ICAM-1 gene, and mRNA stability studies showed that IL-10 did not alter the half-life of IFN-gamma-induced ICAM-1 message. Thus, IL-10 inhibits IFN-gamma-induced ICAM-1 expression in monocytes primarily at the level of gene transcription. Activation of IFN-gamma-responsive genes requires tyrosine phosphorylation of the transcriptional factor STAT-1alpha (signal transducer and activator of transcription-1alpha). However, IL-10 did not affect IFN-gamma-induced tyrosine phosphorylation of STAT-1alpha or alter STAT-1alpha binding to the IFN-gamma response element (IRE) in the ICAM-1 promoter. Instead, IL-10 prevented IFN-gamma-induced binding activity at the NF-kappaB site of the tumor necrosis factor alpha (TNF-alpha)-responsive NF-kappaB/C-EBP composite element in the ICAM-1 promoter. These data indicate that IL-10 inhibits IFN-gamma-induced transcription of the ICAM-1 gene by a regulatory mechanism that may involve NF-kappaB.
PMID: 9192770
[Indexed for MEDLINE]Interleukin-10 inhibits interferon-gamma-induced intercellular adhesion molecule-1 gene transcription in human monocytes. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/9192770ĪĪ
Pro-Resolving MoleculesĪ¬New Approaches to Treat Sepsis?
by Christa Buechler 1,*OrcID, Rebekka Pohl 1 and Charalampos Aslanidis 2
1 Department of Internal Medicine I, Regensburg University Hospital, 93042 Regensburg, Germany
2 Institute of Clinical Chemistry and Laboratory Medicine, Regensburg University Hospital, 93042 Regensburg, Germany
Int. J. Mol. Sci. 2017, 18(3), 476; https://doi.org/10.3390/ijms18030476
Abstract
Inflammation is a complex response of the body to exogenous and endogenous insults. Chronic and systemic diseases are attributed to uncontrolled inflammation. Molecules involved in the initiation of inflammation are very well studied while pathways regulating its resolution are insufficiently investigated. Approaches to down-modulate mediators relevant for the onset and duration of inflammation are successful in some chronic diseases, while all of them have failed in sepsis patients.Inflammation and immune suppression characterize sepsis, indicating that anti-inflammatory strategies alone are inappropriate for its therapy.
Heme oxygenase 1 is a sensitive marker for oxidative stress and is upregulated in inflammation. Carbon monoxide, which is produced by this enzyme, initiates multiple anti-inflammatory and pro-resolving activities with higher production of omega-3 fatty acid-derived lipid metabolites being one of its protective actions. Pro-resolving lipids named maresins, resolvins and protectins originate from the omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid while lipoxins are derived from arachidonic acid.
These endogenously produced lipids do not simply limit inflammation but actively contribute to its resolution, and thus provide an opportunity to combat chronic inflammatory diseases and eventually sepsis.
ĪĪ
Molecules Involved in Resolution of Inflammation
2.1. The Antiinflammatory Cytokine Interleukin (IL)-10
To prevent tissue damage, chronic inflammation and fibrosis, it is essential to timely shut down pro-inflammatory pathways. Mechanisms of immune response termination are activated early during initiation of inflammation [12,18]. Blockade of inflammation may also impair the induction of anti-inflammatory and pro-resolving pathways and delay its termination [12]. A well studied anti-inflammatory cytokine is interleukin (IL)-10 which binds to IL-10 receptor R1 thereby activating signal transducer and activator of transcription 3 (STAT3) [18,19]. Macrophages, dendritic cells and neutrophils respond to IL-10 and altered levels of its receptor during cell maturation and its regulation by inflammatory and anti-inflammatory mediators are mechanisms to control IL-10 sensitivity [18]. IL-10 contributes to endotoxin tolerance which arises upon secondary exposure to this toxin [20]. One of the underlying mechanisms is the accumulation of the p50 NF-”╩B subunit and formation of p50 homodimers. These homodimers block binding of the active p65©Cp50 heterodimer to the respective promoters [21,22]. Endotoxin tolerance is associated with a switch of inflammatory M1 macrophages to anti-inflammatory M2-like cells after toxin challenge [23].
Macrophages deficient in p21 do not develop endotoxin tolerance and binding of the p50©Cp50 dimer to the promoter regions is impaired. High expression of p21 in monocytes of sepsis patients is supposed to antagonize hyper-inflammation. These insensitive immune cells do not appropriately respond to secondary infections and promote sepsis progression [24]. Therefore, IL-10 contributes to immune suppression in sepsis and propagates secondary infections. In line with this, increased systemic IL-10 levels correlate with the sepsis score and death in patients [25].
Pro-resolving molecules discussed in the following paragraphs not only enhance anti-inflammatory effectors, but also boost the host immune response [26] and this may improve sepsis outcome.
2.2. Carbon Monoxide and Heme Oxygenase
Carbon monoxide is one of the molecules that enhance immune cell function. Carbon monoxide is produced by the constitutively expressed heme oxygenase (HO)-2, and by HO-1, which is upregulated upon cellular stress. HO-1 catalyzes the degradation of heme to biliverdin, carbon monoxide and iron (which binds to ferritin), all exerting anti-oxidative and anti-inflammatory activities [27]. Cluster of differentiation 163 (CD163) is the macrophage-specific receptor for hemoglobin©Chaptoglobin uptake and binding of its ligand enhances IL-10 synthesis which subsequently upregulates HO-1 levels [28,29].
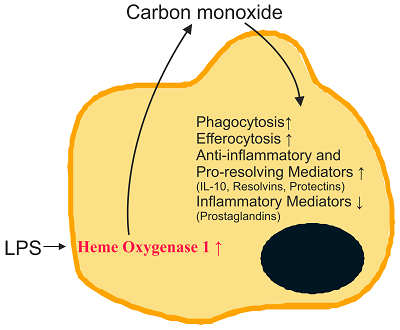
Endogenously formed carbon monoxide acts as a signaling molecule and induces antioxidant genes [27,30]. Protective effects of carbon monoxide include activation of pathways contributing to the elimination of microorganisms. Carbon monoxide promotes killing of bacteria and increases their clearance by macrophages [31]. Engulfment of apoptotic cells by efferocytosis is enhanced [14]. Furthermore, anti-inflammatory cytokines such as IL-10 are induced by carbon monoxide, while TLR2, -4, -5 and -9 activated pro-inflammatory pathways are repressed in macrophages [32] (Figure 1). The latter is attributed to blockade of nicotinamide adenosine dinucleotide phosphate (NADPH) oxidase-mediated production of reactive oxygen species which are involved in TLR translocation to lipid rafts [32].ĪĪ
Figure 1. Role of carbon monoxide in the resolution of inflammation. Heme oxygenase 1 in macrophages is induced in inflammation and carbon monoxide is produced. Carbon monoxide increases phagocytosis, efferocytosis and synthesis of anti-inflammatory cytokines and pro-resolving lipids. It suppresses the production of inflammatory lipids and cytokines. IL-10: interleukin-10; LPS: lipopolysaccharide. Ī³, induced; Ī², reduced.
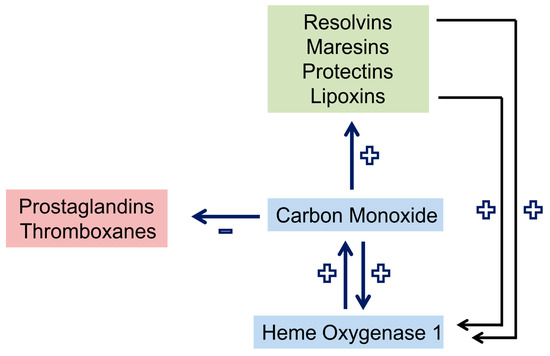
Carbon monoxide lowers the production of inflammatory prostaglandins and thromboxanes. Moreover, it enhances the expression of lipoxygenases which are crucial for the synthesis of pro-resolving lipids from arachidonic acid (lipoxins), eicosapentaenoic acid (EPA) (E-series resolvins) and docosahexaenoic acid (DHA) (D-series resolvins, maresins, and protectins) [14]. Resolvins and lipoxin in turn upregulate HO-1 in macrophages [14]. This cooperative cross-talk (Figure 1 and Figure 2) is highly effective in the resolution of inflammation.
Figure 2. Crosstalk and positive feedback loop of pro-resolving pathways in macrophages. Carbon monoxide upregulates heme oxygenase 1 and the production of pro-resolving lipids, which by themselves induce heme oxygenase 1. Synthesis of prostaglandins and thromboxanes is reduced by carbon monoxide. +, upregulation; −, downregulation.
2.3. Lipid Mediators Derived from Arachidonic Acid
Lipids are involved in the initiation and resolution of inflammation. Pro-inflammatory lipid species originate mostly from the omega-6 polyunsaturated fatty acid arachidonic acid which is released from the membrane by the activity of phospholipases. Cyclooxygenases (COX) 1 and 2 catalyze the synthesis of thromboxanes and prostaglandins, collectively termed prostanoids (Figure 3). Synthesis of prostanoids is strongly increased in inflamed tissues where COX2 is induced [33,34]. Prostaglandin E2 is produced in high quantities and exerts mostly pro-inflammatory effects. This lipid mediator is involved in all processes characteristic for an inflammatory response, namely swelling, redness and pain [33]. Prostaglandin E2 was shown to induce 15-lipoxygenase (15-LOX) which catalyzes the production of the pro-resolving lipoxins and thereby contributes to the resolution of an inflammatory response [35].
Figure 3. Lipids derived from arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid. Arachidonic acid is metabolized by cyclooxygenases (COX) 1/2 to prostaglandins and thromboxanes and by 5-lipoxygenase (5-LOX) to leukotrienes which are involved in the initiation of the inflammatory response (red boxes).Hydroxyeicosatetraenoic acids (HETEs) and lipoxins are also synthesized from arachidonic acid and here 5-, 12- and 15-LOX and cytochrome (Cyt) P450 are involved. Eicosapentaenoic acid is metabolized to E-series resolvins by Cyt P450 and 5-LOX. Resolvins of the D-series, protectins and maresins are derived from docosahexaenoic acid. Lipoxins, resolvins, protectins and maresins (faint green boxes) contribute to the resolution of inflammation.
Aspirin acetylates COX2 thereby preventing prostaglandin synthesis and enhancing the production of pro-resolving lipid mediators (Figure 4). Early shutdown of inflammation may prolong the inflammatory process because of the inappropriate induction of anti-inflammatory and pro-resolving pathways [36]. Low dose aspirin does, however, not reduce cytokine and prostaglandin levels while enhancing 15-epi-lipoxin synthesis [37]. Therefore, dosage and timing of therapy may affect outcome of treatment with aspirin.
Figure 4. Aspirin-triggered pro-resolving lipids. Aspirin-acetylated cyclooxygenase 2 synthesizes 15(R)-hydroxyeicosatetraenoic acid from arachidonic acid and 18(S)- and 18(R)-hydroxyeicosapentaenoic acid from eicosapentaneoic acid (EPA). These lipids are released from endothelial and epithelial cells and are taken up by polymorphonuclear leukocytes. Here, they are converted to 15-epi-lipoxin, resolvin E1 and resolvin E2 by 5-lipoxygenase. For resolvin E1 synthesis, leukotriene A4 (LTA4) hydrolase is also needed.
Acetylated COX2 maintains oxygenase activity and converts arachidonic acid to 15(R)-hydroxyeicosatetraenoic acid (HETE), which is a substrate for 5‑LOX to form 15‑epi-lipoxins also referred to as aspirin-triggered (AT) lipoxins [38] (Figure 4). Low affinity 15-HETE receptors have been described which mediate activation of 5-LOX in mast cells [39] indicating that HETE activates pathways involved in the formation of pro-resolving lipids.
The production of lipoxins can take place in a single cell. Activation of TLR4 in macrophages results in the accumulation of 15-HETE in the form of membrane phospholipid esters. The activation of purinergic receptor for extracellular adenosine triphosphate (ATP), P2X7, causes group IVA cytosolic phospholipase A2 catalyzed hydrolysis of the 15-HETE ester and its conversion to lipoxin by 5-LOX [40].
Intermediate metabolites may also be formed in one cell type and are subsequently transferred to another type of cell where the final end-product is synthesized. This transcellular process involves LOX enzymes in leukocytes, epithelial cells and platelets [41,42].
5-LOX is further involved in the synthesis of leukotrienes which exert inflammatory effects (Figure 3). Leukotriene B4, a dihydroxy derivative of arachidonic acid, initiates and augments neutrophil chemotaxis, release of granule products and superoxide anions mostly by binding to its high affinity receptor BLT1 [43].
Cytochrome P450 enzymes are capable of metabolizing arachidonic acid to epoxyeicosatrienoic acids and HETEs [27]. Epoxyeicosatrienoic acids induce HO-1 and most of the beneficial activities of these lipids are blocked by HO-1 inhibition [27]. Biologic functions of these lipids have been nicely summarized in a recent article [27] and are not further addressed in the present review. It is essential to note that epoxyeicosatrienoic acid exerts anti-inflammatory activities in murine sepsis while effects on the host immune system have not been described [44]. Upregulation of HO-1 nevertheless suggests a role in stimulating immune cell function.
One of the isomers produced by cytochrome P450 is 15(R)-HETE which is converted to 15-epi-lipoxin (Figure 3) [27,34].
In summary, lipids derived from the omega-6 polyunsaturated fatty acid arachidonic acid display pro- and anti-inflammatory properties (Figure 3). Of note, inflammatory lipids released early during inflammation induce production of pro-resolving lipids such as lipoxins which down-modulate inflammatory response and resolve inflammation [35].
2.4. Lipid Mediators Derived from Omega-3 Fatty Acids
Anti-inflammatory and pro-resolving molecules are synthesized from the omega-3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). E-series resolvins (RvE1 and RvE2) are derived from EPA by multistep biosynthesis involving cytochrome P450 and 5-LOX. D-series resolvins (RvD1©CRvD6) are generated from DHA by the enzymes 5-LOX and 15-LOX [13,45]. Additional pro-resolution lipids from DHA are protectins and maresins, and 15‑LOX and 12-LOX are involved in their production, respectively [11,45] (Figure 3). Aspirin-acetylated COX2 produces 18(S)- and 18(R)-hydroxyeicosapentaenoic acid (HEPE) from EPA which are converted to RvE2 enantiomers by 5-LOX and RvE1 enantiomers by 5-LOX and leukotriene A4 hydrolase [46,47] (Figure 4).
Acetylated COX2 further converts DHA to 17(R)-hydroxy-DHA which is oxidized to 17-epi-RvD1 by 5-LOX, also named aspirin-triggered RvD1 (AT-RvD1) [13]. Additional AT-RvD and AT-protectin D1 pro-resolving lipids have been described and biologic activities for most of them have been demonstrated [48].
Formation of these pro-resolving lipids usually is a transcellular process involving different cell types. For instance, synthesis of AT-RvDs starts with hydroxylation of DHA in vascular endothelial cells which is followed by transfer to nearby neutrophils where these lipids are being oxygenated to AT-RvDs [49].
Keywords: carbon monoxide; resolvin; cyclooxygenase; lipoxygenaseIJMS | Free Full-Text | Pro-Resolving MoleculesĪ¬New Approaches to Treat Sepsis? | HTML
https://www.mdpi.com/1422-0067/18/3/476/htmĪĪ