��
https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14181
��
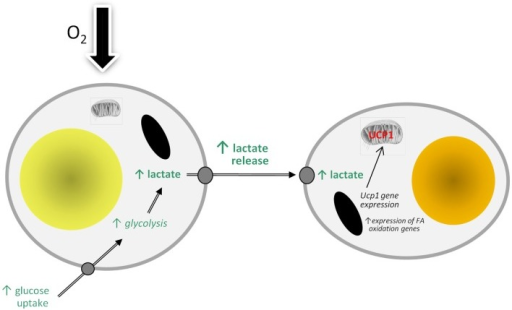
Schematic view of the potential effects of hypoxia on brown adipocyte function. FA, fatty acid; GLUT1, facilitative glucose transporter-1; HIF-1, hypoxia-inducible factor-1; IL-6, interleukin-6; MCT1, monocarboxylate transporter-1; TFs, transcription factors; Ucp1, uncoupling protein-1 (gene); VEGF, vascular endothelial growth factor.
https://mcb.asm.org/content/29/16/4467
��
https://diabetes.diabetesjournals.org/content/63/10/3169
��
Oxygen deprivation and the cellular response to hypoxia in adipocytes - perspectives on white and brown adipose tissues in obesity.
Relative hypoxia has been shown to develop in white adipose tissue depots of different types of obese mouse (genetic, dietary), and this leads to substantial changes in white adipocyte function. These changes include increased production of inflammation-related adipokines (such as IL-6, leptin, Angptl4, and VEGF), an increase in glucose utilization and lactate production, and the induction of fibrosis and insulin resistance. Whether hypoxia also occurs in brown adipose tissue depots in obesity has been little considered. However, a recent study has reported low pO2 in brown fat of obese mice, this involving mitochondrial loss and dysfunction. We suggest that obesity-linked hypoxia may lead to similar alterations in brown adipocytes as in white fat cells - particularly changes in adipokine production, increased glucose uptake and lactate release, and insulin resistance. This would be expected to compromise thermogenic activity and the role of brown fat in glucose homeostasis and triglyceride clearance, underpinning the development of the metabolic syndrome. Hypoxia-induced augmentation of lactate production may also stimulate the "browning" of white fat depots through recruitment of UCP1 and the development of brite adipocytes.
F2: Model of how hypoxia may lead to the recruitment of brite adipocytes and the ��browning�� of white adipose tissue depots through stimulating the production and release of lactate. FA, fatty acid; UCP1, uncoupling protein-1.
https://openi.nlm.nih.gov/detailedresult.php?img=PMC4333869_fendo-06-00019-g002&req=4
��
https://www.cherrybiotech.com/scientific-note/tumor-hypoxia-and-metastasis
��
Targeting Hypoxia-induced Inflammation
Mucosal Inflammation Program, Department of Anesthesiology, University of Colorado Denver, Aurora, Colorado.
Anesthesiology 2 2011, Vol.114, 239-242.
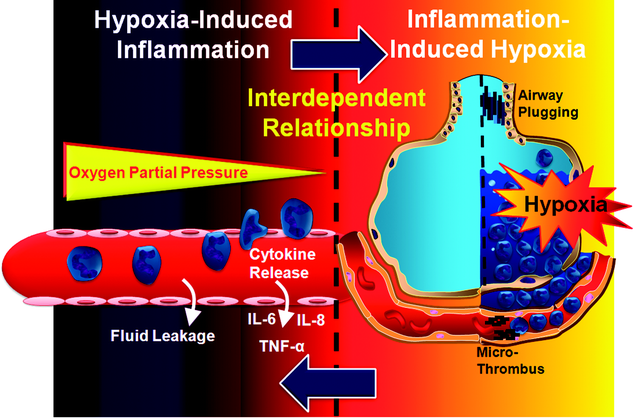
IN the current issue of Anesthesiology, an article by Kim et al. 1 from the research laboratory of H. Thomas Lee, M.D., Ph.D., investigates strategies to dampen ischemia-driven inflammation of the kidneys and subsequent multiorgan failure using experimental models of acute kidney injury (AKI). In fact, one of the leading causes of morbidity and mortality in surgical patients is perioperative organ failure, such as failure that occurs in the context of AKI, liver failure, intestinal or myocardial ischemia, stroke, or acute lung injury. A common feature of these perioperative diseases is the presence of hypoxia-induced inflammation.2,3 The relationship between hypoxia and inflammation under these conditions is interdependent. For example, exposure to ambient hypoxia, as seen during high-altitude mountaineering, is associated with edema of the lungs or brain and systemic inflammatory responses in humans.4,5 Similarly, short-term exposure of mice to ambient hypoxia (e.g. , 8% oxygen over 4�C8 h) leads to increased inflammatory cytokine concentrations and pulmonary edema.6 Moreover, prolonged donor organ exposure to ischemia during organ transplantation is known to enhance graft inflammation and early graft failure.7,8 For example, the endotoxin receptor toll-like receptor 4 (TLR4) is expressed in the donor kidney during kidney transplantation, and TLR4 expression concentrations increase with prolonged ischemia time. Donor kidneys with a loss-of-function mutation of the TLR4 receptor show attenuated kidney inflammation and an increased rate of immediate graft function.7 Together, these studies indicate that hypoxia represents an inflammatory stimulus (fig. 1) and suggest that targeting hypoxia-elicited inflammation could represent an important therapeutic approach in perioperative medicine.
Fig. 1. Interdependent relationship between hypoxia and inflammation. Exposure to ambient hypoxia triggers an acute inflammatory response in different organs, including the kidney, the intestine, the heart, or the lungs. Hypoxia-induced inflammation can manifest itself as increased vascular leakage, accumulation of inflammatory cells in hypoxic organs, or release of inflammatory mediators (e.g. , tumor necrosis factor [TNF]-��, interleukin [IL]-6, or IL-8). At the same time (and as shown herein for acute lung injury), inflammation is associated with tissue hypoxia caused by dramatic increases in the demand for metabolites and oxygen by resident cells (e.g. , pulmonary and vascular endothelia) and recruited inflammatory cells (e.g. , neutrophils). In addition, oxygen supply is attenuated due to pulmonary edema, airway plugging, or microthrombi.Targeting Hypoxia-induced Inflammation | Anesthesiology | ASA Publications
https://anesthesiology.pubs.asahq.org/Article.aspx?articleid=1933201��
Diabetologia. Author manuscript; available in PMC 2012 Jun 1.
Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells
Abstract
Aims
Hypoxia has been implicated as a cause of adipose tissue inflammation in obesity, although the inflammatory response of human adipose tissue to hypoxia is not well understood. The goal of this study was to define in vitro inflammatory responses of human adipose tissue to hypoxia and identify molecular mechanisms of hypoxia-induced inflammation.
Methods
The inflammatory milieu and responses of visceral (VAT) and subcutaneous (SAT) adipose tissue explants and purified stromovascular cells (SVFs) from obese and lean humans were studied in an in vitro hypoxic culture system using quantitative real-time PCR, ELISA, western blotting, immunofluorescence microscopy, flow cytometry and immunohistochemistry.
Results
Human adipose tissue in obesity demonstrates an increased leucocyte infiltrate that is greater in VAT than SAT and involves macrophages, T cells and natural killer (NK) cells. Hypoxic culture regulates inflammatory cytokine secretion and transcription of metabolic stress response genes in human adipose tissue SVF. Adipocyte diameter is increased and adipose tissue capillary density is decreased in obese participants. Inhibition of c-Jun terminal kinase (JNK) or p38 significantly attenuates hypoxia-induced SVF inflammatory responses. Hypoxia induces phosphorylation of p38 in adipose tissue.
Conclusions
Human adipose tissue in obesity is characterised by a depot-specific inflammatory cell infiltrate that involves not only macrophages, but also T cells and NK cells. Hypoxia induces inflammatory cytokine secretion by human adipose tissue SVF, the primary source of which is adipose tissue macrophages. These data implicate p38 in the regulation of hypoxia-induced inflammation and suggest that alterations in adipocyte diameter and adipose tissue capillary density may be potential underlying causes of adipose tissue hypoxia.
Keywords: Adipose tissue, Hypoxia, Inflammation, JNK, Macrophage, Obesity, p38, T cell
RNA fold change in transcript level in response to hypoxic culture
Obese VAT SVF transcriptional responses to hypoxic culture. RNA was prepared from VAT SVF from ten obese participants cultured in hypoxic or normoxic conditions for 24 h and subject to QRTPCR analysis. The ordinate shows fold difference in transcript level in hypoxic culture relative to normoxic culture referent. *p<0.05 derived from paired t tests of ��Ct values comparing hypoxic vs normoxic cultures for matched SVF specimens. Error bars show SEM. Similar data from matched SAT SVF from the same ten obese participants, and from VAT SVF from eight lean participants demonstrate that hypoxia did not induce upregulation of any transcripts studied except for GLUT1 in lean VAT SVF (data not shown, see the Results for details)��
Human adipose tissue SVF cytokine and transcriptional responses to hypoxia
Hypoxia regulated TNF-�� and IL-10 secretion in obese VAT SVF in a reciprocal manner, inducing TNF-�� secretion and inhibiting IL-10 secretion, consistent with a proinflammatory effect. Hypoxia also induced upregulation of GLUT1, VEGF, PERK, and C-JUN transcripts in obese VAT SVF. These hypoxia-induced responses were absent in lean VAT SVF and in obese SAT SVF, consistent with the decreased inflammatory infiltrate in these tissues.
Our data confirm that CD14+ ATMs are the dominant source of both basal and hypoxia-induced inflammatory cytokine secretion within the SVF. Others studies of hypoxia-induced adipose tissue inflammatory responses have focused primarily on adipocytes differentiated in vitro from pre-adipocytes [11, 31�C33]. We focused study on the SVF because multiple reports suggest that it is the primary source of most inflammatory cytokines in adipose tissue [3�C10]. Nonetheless, adipocytes represent an alternative source of cytokines, producing similar levels of select cytokines compared with the SVF, most notably IL-6 [6�C8]. Our data in the SVF, therefore, not unexpectedly differ from studies of hypoxia-induced cytokine production in adipocytes. For example, Wang et al. demonstrated minimal hypoxia-induced TNF-�� secretion in adipocytes, but increases in IL-6 [33], a pattern that we did not observe in the SVF, consistent with data demonstrating that adipocytes secrete significant levels of IL-6, but low levels of TNF-�� [6�C8], in part possibly due to lack of adipocyte production of TNF-�� converting enzyme [34]. These observations aside, however, and with the exception of IL-6, previous data demonstrate that SVFs are the primary source of the cytokines studied in this manuscript [3�C10].��
Conclusion
Hypoxia induces inflammatory cytokine secretion and metabolic stress gene transcription in obese adipose tissue ATMs together with increased phosphorylation of p38 in whole adipose tissue. Human obesity is associated with increased adipocyte size and decreased adipose tissue capillary density, suggesting possible mechanisms of adipose tissue hypoxia. These data suggest that hypoxia may regulate adipose tissue inflammation in vivo, and implicate p38 in the regulation of hypoxia-induced adipose tissue inflammatory responses.
Discussion
Hypoxia is a potential root cause of adipose tissue inflammation. We used in vitro hypoxic culture to study human adipose tissue inflammatory responses to hypoxia and identify mediators of hypoxia-induced adipose tissue inflammation. Our data implicate p38 in the regulation of hypoxia-induced inflammation, and suggest that adipocyte size and adipose tissue capillary density may underlie adipose tissue hypoxia in obesity.
...
TNF-�� is a central proinflammatory and diabetogenic mediator in obesity [23�C25]. CCL2 and IL-6 have also been linked to adipose tissue inflammation [26�C28]. IL-10, in contrast, attenuates inflammation and is decreased in obesity and the metabolic syndrome [29]. We observed increased basal secretion of TNF-�� and IL-10 by the obese SVF compared with the lean SVF, consistent with the observed increased leucocyte infiltrate.
Finally, we demonstrate that CD14+ ATMs are the primary source of basal inflammatory cytokine production within the SVF, consistent with previous data [7, 30]. The increased basal levels of IL-10 secretion in the obese SVF are not consistent with the observed lower serum levels of IL-10 reported in obesity [29].Putative underlying mechanisms of hypoxias
Adipocyte enlargement may contribute to hypoxia by presenting a cellular oxygen diffusion barrier [1]. Adipocyte diameter was increased in obese compared with lean participants, and VAT adipocytes were smaller than SAT adipocytes, consistent with previous data [39�C41]. Alterations in adipose tissue blood flow have also been implicated as a potential cause of hypoxia in human obesity based on systemic radionuclide washout techniques [42, 43]. Consistent with these data, adipose tissue capillary density was decreased in obese participants, similar to data from other researchers in mice [44] and humans [45], although no depot-specific differences were observed. These data suggest that, while adipocyte enlargement and decreased adipose tissue capillary density may contribute to hypoxia in obesity, these phenomena do not explain depot-specific differences in inflammation.Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3159546/��
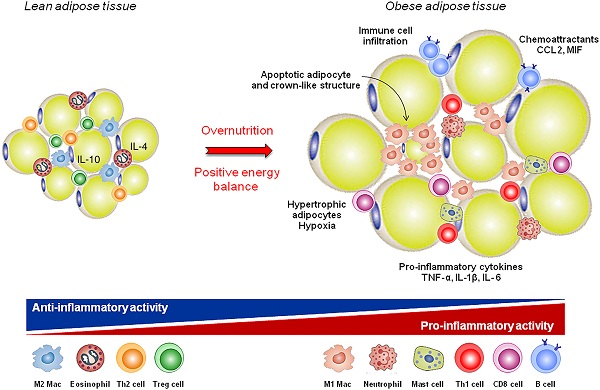
The "Big Bang" in Obese Fat: Events Initiating Obesity-Induced Adipose Tissue Inflammation
Abstract
Obesity is associated with the accumulation of pro-inflammatory cells in visceral adipose tissue (VAT), which is an important underlying cause of insulin resistance and progression to diabetes mellitus type 2 (DM2). Although the role of pro-inflammatory cytokines in disease development is established, the initiating events leading to immune cell activation remain elusive. Lean adipose tissue is predominantly populated with regulatory cells, such as eosinophils and type 2 innate lymphocytes. These cells maintain tissue homeostasis through the excretion of type 2 cytokines, such as IL-4, IL-5, and IL-13, which keep adipose tissue macrophages (ATMs) in an anti-inflammatory, M2-like state. Diet-induced obesity is associated with the loss of tissue homeostasis and development of type 1 inflammatory responses in VAT, characterized by IFN-��. A key event is a shift of ATMs toward an M1 phenotype. Recent studies show that obesity-induced adipocyte hypertrophy results in upregulated surface expression of stress markers. Adipose stress is detected by local sentinels, such as NK cells and CD8(+) T cells, which produce IFN-��, driving M1 ATM polarization. A rapid accumulation of pro-inflammatory cells in VAT follows, leading to inflammation. In this review, we provide an overview of events leading to adipose tissue inflammation, with a special focus on adipose homeostasis and the obesity-induced loss of homeostasis which marks the initiation of VAT inflammation.
Keywords: Adiponectin; Adipose tissue; Diabetes mellitus type 2; IFN-��; Inflammation; Insulin resistance; Macrophages; NK cells; Obesity; TNF.The "Big Bang" in Obese Fat: Events Initiating Obesity-Induced Adipose Tissue Inflammation - PubMed
https://pubmed.ncbi.nlm.nih.gov/26220361/��
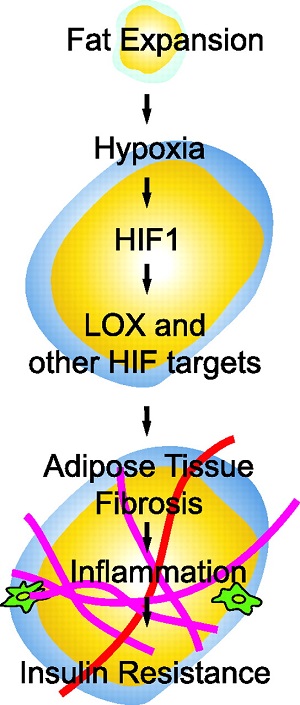
Adipose Tissue Hypoxia in Obesity and Its Impact on Preadipocytes and Macrophages: Hypoxia Hypothesis
Abstract
Obese subjects exhibit lower adipose tissue oxygen consumption in accordance with the lower adipose tissue blood flow. Thus, compared with lean subjects, obese subjects have 44% lower capillary density and 58% lower vascular endothelial growth factor (VEGF). The VEGF expression together with hypoxia-inducible transcription factor-1 (HIF-1) activity also requires phosphatidylinositol 3-kinase (PI3K)- and target of rapamycin (TOR)-mediated signaling. HIF-1alpha is an important signaling molecule for hypoxia to induce the inflammatory responses. Hypoxia affects a number of biological functions, such as angiogenesis, cell proliferation, apoptosis, inflammation and insulin resistance. Additionally, reactive oxygen radical (ROS) generation at mitochondria is responsible for propagation of the hypoxic signal. Actually mitochondrial ROS (mtROS) production, but not oxygen consumption is required for hypoxic HIF-1alpha protein stabilization. Adipocyte mitochondrial oxidative capacity is reduced in obese compared with non-obese adults. In this respect, mitochondrial dysfunction of adipocyte is associated with the overall adiposity. Furthermore, hypoxia also inhibits macrophage migration from the hypoxic adipose tissue. Alterations in oxygen availability of adipose tissue directly affect the macrophage polarization and are responsible from dysregulated adipocytokines production in obesity. Hypoxia also inhibits adipocyte differentiation from preadipocytes. In addition to stressed adipocytes, hypoxia contributes to immune cell immigration and activation which further aggravates adipose tissue fibrosis. Fibrosis is initiated in response to adipocyte hypertrophy in obesity.
Keywords: Adipocyte differentiation; Adipose tissue blood flow; Angiogenesis; CAAT/enhancer binding protein (C/EBP); CAAT/enhancer binding protein (C/EBP)-homologous protein (CHOP); Hypoxia-inducible transcription factor-1 (HIF-1) alpha; Inducible nitric oxide synthase (iNOS); Mitochondrial ROS (mtROS); Obesity; Phosphatidylinositol 3-kinase (PI3K); Reactive oxygen species (ROS); Vascular endothelial growth factor (VEGF).Adipose Tissue Hypoxia in Obesity and Its Impact on Preadipocytes and Macrophages: Hypoxia Hypothesis - PubMed
https://pubmed.ncbi.nlm.nih.gov/28585205/��