Stress Glucocorticoids Lymphatic System Studies
during inflammation, glucocorticoids are increased in the circulation and that these protect the host from over-shooting and ensuing self-inflicted injury.
Leukotrienes activate the immune system after infection, injury, or contact with allergens.
ˇˇ
Endogenous Glucocorticoids Inhibit Myocardial Inflammation Induced by Lipopolysaccharide: Involvement of Regulation of Histone Deacetylation
Journal of Cardiovascular Pharmacology: July 2012
Emerging evidence indicates that myocardial inflammation plays a key role in the pathogenesis of cardiac diseases. But the exact mechanisms for this chronic inflammatory disorder have not been elucidated. Glucocorticoids (GCs) are the most effective anti-inflammatory treatments available for many inflammatory diseases. However, it is unknown whether endogenous GCs are able to exert anti-inflammatory effect on myocardial inflammation. In this study, the potential role of endogenous GCs in the regulation of myocardial inflammation was investigated. We showed that the reduction of endogenous GC level by adrenalectomy promoted the production of basal and lipopolysaccharide (LPS)-induced proinflammatory cytokines, which could be partly reversed by supplementing with exogenous physiological level of hydrocortisone. Inhibition of GC receptor (GR) signaling pathway with GR antagonist mifepristone (RU486) or histone deacetylase inhibitor trichostatin A (TSA) also increased the levels of basal and LPS-induced proinflammatory cytokines. Moreover, blockade of GC¨CGR signaling pathway by adrenalectomy, RU486 or TSA enhanced LPS-induced myocardial nuclear factor-¦ĘB activation and histone acetylation but inhibited myocardial histone deacetylase expression and activity. Cardiac function studies demonstrated that blockade of the GC¨CGR signaling pathway aggravated inflammation-induced cardiac dysfunction. These findings indicate that endogenous GCs are able to inhibit myocardial inflammation induced by LPS. Endogenous GCs represent an important endogenous anti-inflammatory mechanism for myocardium in rats and such mechanism injury may be an important factor for pathogenesis of cardiac diseases.Endogenous Glucocorticoids Inhibit Myocardial Inflammation I... : Journal of Cardiovascular Pharmacology
https://journals.lww.com/cardiovascularpharm/Abstract/2012/07000/Endogenous_Glucocorticoids_Inhibit_Myocardial.5.aspxˇˇ
Therapeutic Exploitation of Endogenous Anti-Inflammatory Mechanisms:Old and New Leads
Understanding the way our body switches off host defence responses has yielded some of the most innovative recent discoveries in inflammation research. In reality the concept is not new, and was already implicit in early publications of the 1970s which showed that during inflammation, glucocorticoids are increased in the circulation and that these protect the host from over-shooting and ensuing self-inflicted injury. Stemming from the first example of drugs developed on an endogenous anti-inflammatory pathway, that of the glucocorticoid, we have here touched upon other counterregulatory breakpoints, such as those centred on melanocortins; the annexin 1 system; the polyunsaturated fatty acid derivatives lipoxins and resolvins; galectin-1 and selected others, including novel chemical entities engineered to release anti-inflammatory gases and factors originally discovered in the developmental field. We propose that understanding the molecular mechanisms switched on by a given anti-inflammatory mediator and the events it modulates in target cells will be of great help in developing innovative ways to control inflammatory pathologies. This seems quite articulated and with a degree of complexity in the group of developmental axonal guidance factors. We propose that drugs discovered along this philosophy will have a better compliance and would be theoretically devoid of side effects since they will be acting by mimicking the way our own body assures the inflammatory response is restricted both spatially and temporally.
Keywords: Glucocorticoids, anti-inflammation, lipocortin 1, neutrophil, melanocortins, galectin-1, lipoxins, resolvinsˇˇ
Therapeutic Exploitation of Endogenous Anti-Inflammatory Mechanisms:Old and New Leads | Bentham Science
https://www.eurekaselect.com/58004ˇˇ
Comparative effects of the & omega-3 polyunsaturated fatty acid derivatives resolvins E1 and D1 and protectin DX in models of inflammation and pain
The effects of Rvs E1 and D1 and protectin DX (PDX) were assessed in rat paws inflamed by the standard proinflammatory stimulus carrageenan or by histamine, 5-hydroxytryptamine, substance P or prostaglandin E2. The experimental outcomes were the mechanical nociceptive threshold and increase in paw volume as a measure of pain and edema formation, respectively. The analgesic and anti-inflammatory activities of the indicated specialized pro-resolving lipid mediators Ł¨SPMsŁ© were also compared with nonsteroidal (indomethacin and celecoxib) and steroidal (dexamethasone) anti-inflammatory drugs.Results: Only RvE1 and RvD1 presented analgesic and anti-inflammatory activities in the carrageenan model, and RvE1 was twice as potent as RvD1. Both substances tended to be better analgesics than anti-inflammatory agents, with a modeling profile similar to steroidal anti-inflammatory drugs. However, proinflammatory effects (edema formation) were also detected when the mediators histamine, 5-hydroxytryptamine or substance P replaced carrageenan as the proinflammatory stimuli. The analgesic and anti-inflammatory effects of resolvins were specifically prevented by an antagonist of the leukotriene B4 receptor 1 (BLT1).Conclusion: Rvs, as analgesic agents, may be better therapeutic agents than nonsteroidal anti-inflammatory drugs, the current choice in the relief of pain of an inflammatory origin. However, the possibility of developing adverse effects cannot be overlooked.¦Ř3¶ŕ˛»±ĄşÍÖ¬·ľËáŃÜÉúÎďÔÚŃ×Ö˘şÍĚŰÍ´ÄŁĐÍÖĐĎűÍËËŘE1şÍD1şÍ±Ł»¤ËŘDXµÄ±Č˝Ď×÷ÓĂ
ÔÚÓɱę׼´ŮŃ×ĐԽDzć˛Ë˝ş»ň×é°·Ł¬5-ôÇÉ«°·Ł¬ÎďÖĘP»ňÇ°ÁĐĎŮËŘE2ÓŐ·˘Ń×Ö˘µÄ´óĘóצÖĐĆŔąŔÁËĎűÍËËŘRvs E1şÍD1ŇÔĽ°±Ł»¤ËŘDXŁ¨PDXŁ©µÄ×÷ÓáŁĘµŃé˝áąű·Ö±đĘÇ»úеÉËş¦ăĐÖµşÍצĚĺ»ýÔöĽÓŁ¬·Ö±đ×÷ÎŞĚŰÍ´şÍË®Ö×ĐγɵÄÁż¶ČˇŁ»ą±Č˝ĎÁËÖ¸¶¨SPMµÄŐňÍ´şÍżąŃ×»îĐÔÓë·ÇçŢĚ壨ßĹßáĂŔĐÁşÍČűŔ´Îô˛ĽŁ©şÍçŢĚ壨µŘČűĂ×ËÉŁ©żąŃ×Ň©µÄ˝áąűˇŁÔڽDzć˛Ë˝şÄŁĐÍÖĐŁ¬Ö»ÓĐRvE1şÍRvD1ľßÓĐŐňÍ´şÍżąŃ×»îĐÔˇŁ RvE1µÄЧÁ¦ĘÇRvD1µÄÁ˝±¶ˇŁÁ˝ÖÖÎďÖʶĽÇăĎňÓڱȿąŃ×Ň©¸üşĂµÄŐňÍ´Ň©Ł¬ĆäÔěĐÍŔŕËĆÓÚŔŕąĚ´ĽżąŃ×Ň©ˇŁµ«ĘÇŁ¬µ±˝éÖĘ×é°·Ł¬5-ôÇÉ«°·»ňPÎďÖĘ´úĚć˝Ç˛ć˛Ë˝ş×÷ÎŞ´ŮŃ×´ĚĽ¤ĽÁʱŁ¬Ň˛Ľě˛âµ˝´ŮŃ××÷ÓĂŁ¨Ë®Ö×Đγɣ©ˇŁ°×ČýĎ©B4ĘÜĚĺ1Ł¨BLT1Ł©µÄŢ׿ąĽÁżÉĚŘŇěĐÔµŘŇÖÖĆĎűÍËËصÄŐňÍ´şÍżąŃ××÷Ó᣽áÂŰŁşĎűÍËËŘRvs×÷ÎŞŐňÍ´Ň©żÉÄܱȷÇçŢĚĺżąŃ×Ň©¸üşĂŁ¬ŐâĘÇĿǰµÄѡÔńˇŁĽőÇáŃ×Ö˘ĐÔĚŰÍ´ˇŁµ«ĘÇŁ¬˛úÉú˛»ÁĽÓ°ĎěµÄżÉÄÜĐÔ˛»ČÝşöĘÓˇŁComparative effects of the ω3 polyunsaturated fatty acid derivatives resolvins E1 and D1 and protectin DX in models of inflammation and pain - CORE
https://core.ac.uk/display/88066578ˇˇ
Leukotriene B4 receptor 1 exacerbates inflammation following myocardial infarction
Leukotriene B4 receptor 1 (BLT1), a high©\affinity G©\protein©\coupled receptor for leukotriene B4 (LTB4), is expressed on various inflammatory cells and plays critical roles in several inflammatory diseases. In myocardial infarction (MI), various inflammatory cells are known to be recruited to the infarcted area, but the function of BLT1 in MI is poorly understood. Here, we investigated the role of BLT1 in MI and the therapeutic effect of a BLT1 antagonist, ONO©\4057, on MI. Mice with infarcted hearts showed increased BLT1 expression and LTB4 levels. BLT1©\knockout mice with infarcted hearts exhibited attenuated leukocyte infiltration, proinflammatory cytokine production, and cell death, which led to reduced mortality and improved cardiac function after MI. Bone©\marrow transplantation studies showed that BLT1 expressed on bone marrow©\derived cells was responsible for the exacerbation of inflammation in infarcted hearts. Furthermore, ONO©\4057 administration attenuated the inflammatory responses in hearts surgically treated for MI, which resulted in reduced mortality and improved cardiac function after MI. Our study demonstrated that BLT1 contributes to excessive inflammation after MI and could represent a new therapeutic target for MI.
ˇˇLeukotriene B4 receptor 1 exacerbates inflammation following myocardial infarction - Horii - 2020 - The FASEB Journal - Wiley Online Library
https://faseb.onlinelibrary.wiley.com/doi/full/10.1096/fj.202000041Rˇˇ
Am J Pathol 2020
Resolvin E1 Reduces Leukotriene B4-Induced Intracellular Calcium Increase and Mucin Secretion in Rat Conjunctival Goblet Cells
Harvard Medical School
Leukotriene B4 (LTB4) is a major proinflammatory mediator important in host defense, whereas resolvins (Rvs) are produced during the resolution phase of inflammation. The authors determined the actions of both RvE1 and RvD1 on LTB4-induced responses of goblet cells cultured from rat conjunctiva. The responses measured were an increase in the intracellular [Ca] ([Ca]) and high-molecular-weight glycoprotein secretion. Treatment with RvE1 or RvD1 for 30 minutes significantly blocked the LTB4-induced [Ca] increase. The actions of RvE1 on LTB4-induced [Ca] increase were reversed by siRNA for the RvE1 receptor, and the actions of RvD1 were reversed by an RvD1 receptor inhibitor. The RvE1 and RvD1 block of LTB4-stimulated increase in [Ca] was also reversed by an inhibitory peptide to ¦Â-adrenergic receptor kinase. LTB4 and block of the LTB4-stimulated increase in [Ca] by RvE1 and RvD1 were partially mediated by the depletion of intracellular Ca stores. RvE1, but not RvD1, counterregulated the LTB4-induced high-molecular-weight glycoprotein secretion. Thus, both RvE1 and RvD1 receptors directly inhibit LTB4 by phosphorylating the LTB4 receptor using ¦Â adrenergic receptor kinase. RvE1 receptor counterregulates the LTB4-induced increase in [Ca] and secretion, whereas RvD1 receptor only counterregulates LTB4-induced [Ca] increase.Resolvin E1ĽőÉŮ´óĘó˝áĤ±×´Ď¸°űÖĐ°×ČýĎ©B4ÓŐµĽµÄϸ°űÄÚ¸ĆÔöĽÓşÍŐłµ°°×·ÖĂÚ
ąţ·đҽѧԺ
°×ČýĎ©B4Ł¨LTB4Ł©ĘÇŇ»ÖÖÖ÷ŇŞµÄ´ŮŃ×˝éÖĘŁ¬ÔÚËŢÖ÷·ŔÓůÖĐşÜÖŘŇŞŁ¬¶řRESOLVINŁ¨RvsŁ©ĘÇÔÚŃ×Ö˘ĎűÍ˽׶βúÉúµÄˇŁ×÷ŐßČ·¶¨ÁËRvE1şÍRvD1¶ÔLTB4ÓŐµĽµÄ´óĘó˝áĤĹŕŃřµÄ±×´Ď¸°ű·´Ó¦µÄ×÷ÓᣲâµĂµÄ·´Ó¦ĘÇϸ°űÄÚ[Ca]Ł¨[Ca]Ł©şÍ¸ß·Ö×ÓÁżĚǵ°°×·ÖĂÚµÄÔöĽÓˇŁÓĂRvE1»ňRvD1´¦Ŕí30·ÖÖÓżÉĎÔ×Ĺ×čÖąLTB4ÓŐµĽµÄ[Ca]ÔöĽÓˇŁ¶ÔÓÚRvE1ĘÜĚ壬siRNAÄćתÁËRvE1¶ÔLTB4ÓŐµĽµÄ[Ca]ÔöĽÓµÄ×÷ÓĂŁ¬¶řRvD1ĘÜĚĺŇÖÖĆĽÁÄćתÁËRvD1µÄ×÷ÓᣠµÄRvE1şÍRvD1×čÖąLTB4´ĚĽ¤µÄ[Ca]ÔöĽÓҲ±»¦Â-ÉöÉĎĎŮËŘĘÜĚ弤øµÄŇÖÖĆëÄÄćתˇŁ RvE1şÍRvD1ŇýĆđµÄ[Ca]µÄLTB4şÍLTB4´ĚĽ¤µÄ×čÖͲż·ÖĘÇÓÉϸ°űÄÚCa´˘´ćµÄĎűşÄ˝éµĽµÄˇŁ RvE1Ł¬¶ř˛»ĘÇRvD1Ł¬·´µ÷˝ÚLTB4ÓŐµĽµÄ¸ß·Ö×ÓÁżĚǵ°°×·ÖĂÚˇŁŇň´ËŁ¬RvE1şÍRvD1ĘÜĚĺľůżÉͨąýĘąÓæÂÉöÉĎĎŮËŘÄÜĘÜĚ弤øʹLTB4ĘÜĚĺÁ×ËữŔ´Ö±˝ÓŇÖÖĆLTB4ˇŁ RvE1ĘÜĚĺµ÷˝ÚLTB4ÓŐµĽµÄ[Ca]µÄÔöĽÓŁ¬¶řRvD1ĘÜĚĺ˝öµ÷˝ÚLTB4ÓŐµĽµÄ[Ca]µÄÔöĽÓˇŁ
Resolvin E1 Reduces Leukotriene B4-Induced Intracellular Calcium Increase and Mucin Secretion in Rat Conjunctival Goblet Cells | Harvard Medical School Department of Ophthalmology
https://eye.hms.harvard.edu/publications/resolvin-e1-reduces-leukotriene-b-induced-intracellular-calcium-increase-and-mucinˇˇ
Glucocorticoids and Immunity
Denis Franchimont, in Encyclopedia of Endocrine Diseases, 2004
Cell Trafficking and Adhesion Molecules
Intravenous injection of glucocorticoids or acute cortisol release following stress greatly influence white cell trafficking during local or systemic inflammation. The redistribution of T cells, the increased neutrophilia, and the profound depletion of eosinophils and basophils result from the complex regulation of leukocyte and endothelial adhesion molecules and chemokines by glucocorticoids. These factors modulate bone marrow release, peripheral tissue infiltration, and clearance of leukocytes. The increase in neutrophilia and decrease in eosinophils after glucocorticoid administration are familiar to most clinicians. In fact, glucocorticoids promote the survival and proliferation of neutrophils and induce the apoptosis of eosinophils and basophils. Glucocorticoids enhance granulocyte colony-stimulating factor, which in turn participates in the proliferation and expansion of neutrophils. Leukotriene B4 (LTB4) seems to be a critical survival factor for neutrophils since inhibitors of 5-lipooxygenase or LTB4 antagonists prevent glucocorticoid-induced neutrophil survival. Interestingly, glucocorticoids up-regulate the high-affinity leukocyte LTB4 receptor (BLT1) expression and enhance LTB4-mediated neutrophil survival and chemotaxis. The glucocorticoid-induced neutrophilia is mostly explained by an increase in the release of polymorphonuclear cells from the bone marrow and inhibition of neutrophil transmigration to inflammatory sites. Inhibition of neutrophil transmigration by glucocorticoids occurs through inhibition of leukocyte adhesion molecules, such as L-selectin. L-Selectin seems to be essential for glucocorticoid-mediated actions on neutrophil migration. However, the immediate action of glucocorticoids on neutrophils is not related to a direct regulation of L-selectin on circulating peripheral neutrophils. In fact, glucocorticoids promote shedding of L-selectin from the neutrophils, preventing their attachment to the endothelial cells. Glucocorticoids enhance lipocortin 1 (annexin 1) expression, which in turn would activate a metalloprotease ˇ°sheddaseˇ± that frees L-selectin from neutrophils. This mechanism has been described and accounts for the early changes in neutrophilia during acute stress or following glucocorticoid administration. Also, glucocorticoids decrease in a timely manner the expression of L-selectin on bone marrow progenitors and on differentiating neutrophils that, then, reach the bloodstream with low L-selectin levels. Chemokines released from the inflamed tissue are important for activating neutrophils and initiating the program of cell transmigration. The inhibition of the release of chemokines, such as interleukin-8 (IL-8) and other cysteine-X-cysteine (CXC) chemokines, also participates in the inhibition of neutrophil transmigration by glucocorticoids.
Eosinophils, basophils, and T helper 2 (Th2) cells are recruited by chemokines, which are released by immune and nonimmune cells, such as epithelial cells or airway smooth muscle cells. These ¦Â-chemokines, or cysteine-cysteine (CC) chemokines, enhance adhesion molecule expression on endothelial cells to allow eosinophils and basophils to roll through the vessel wall. In fact, glucocorticoids prevent tissue invasion of eosinophils by inhibiting the release of CC chemokines, such as eotaxin, eotaxin 2, MCP4 (macrophage inflammatory protein 4), and RANTES (regulated on activated normal T-cell expressed and secreted), released by bronchial epithelial cells. Simultaneously, whereas glucocorticoids decrease the expression of some chemokine receptors, they enhance the expression of others on eosinophils, further complicating the overall picture of eosinophil trafficking.
The flow and movement of monocytes seem tightly regulated by glucocorticoids via similar mechanisms, namely, through the repression of monocytes and endothelial adhesion molecules and the regulation of chemokines and their receptors.ˇˇ
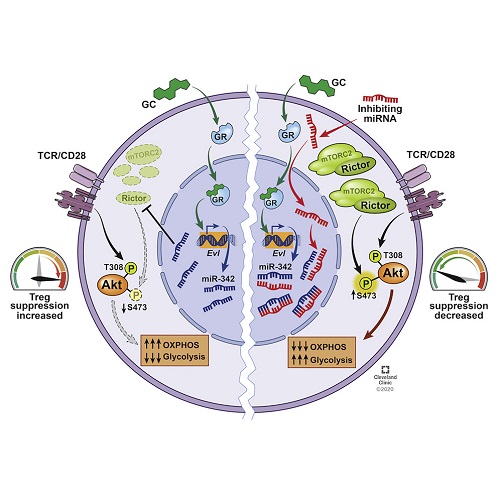
Anti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3+ Regulatory T Cells via a miR-342-Dependent Mechanism
1 Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH 44195
2 Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77230
3 Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, OH 44106
Published: July 23, 2020
Highlights
•
Without Foxp3+ Treg cells, glucocorticoid loses its therapeutic effects
•
Glucocorticoid induces miR-342-3p expression specifically in Treg cells
•
miR-342-3p targets Rictor and regulates the metabolic programming in Treg cells
•
miR-342-3p and Rictor expression controls Treg cell function in vivo
Summary
Glucocorticoids (GC) are the mainstay treatment option for inflammatory conditions. Despite the broad usage of GC, the mechanisms by which GC exerts its effects remain elusive. Here, utilizing murine autoimmune and allergic inflammation models, we report that Foxp3+ regulatory T (Treg) cells are irreplaceable GC target cells in vivo. Dexamethasone (Dex) administered in the absence of Treg cells completely lost its ability to control inflammation, and the lack of glucocorticoid receptor in Treg cells alone resulted in the loss of therapeutic ability of Dex. Mechanistically, Dex induced miR-342-3p specifically in Treg cells and miR-342-3p directly targeted the mTORC2 component, Rictor. Altering miRNA-342-3p or Rictor expression in Treg cells dysregulated metabolic programming in Treg cells, controlling their regulatory functions in vivo. Our results uncover a previously unknown contribution of Treg cells during glucocorticoid-mediated treatment of inflammation and the underlying mechanisms operated via the Dex-miR-342-Rictor axis.
ĚÇƤÖĘĽ¤ËصĿąŃ××÷ÓĂĘÇÓÉFoxp3 +µ÷˝ÚĐÔTϸ°űͨąýmiR-342ŇŔŔµĐÔ»úÖƽ鵼µÄ
1żËŔű·ňŔĽŐďËů»ů˝đ»áŔŐÄÉŃĐľżËůŃ×Ö˘ÓëĂâŇßϵŁ¬¶íşĄ¶íÖÝżËŔű·ňŔĽ44195
2µÂżËČřËą´óѧMD°˛µÂÉ°©Ö˘ÖĐĐÄϵͳÉúÎďѧϵŁ¬µÂżËČřËąÖÝĐÝËą¶Ř77230
3żËąÎ÷´˘´óѧČËżÚÓ붨Áż˝ˇżµżĆѧϵŁ¬¶íşĄ¶íÖÝżËŔű·ňŔĽ44106
·˘˛ĽĘ±ĽäŁş2020Äę7ÔÂ23ČŐ
Çżµ÷
• Ă»ÓĐFoxp3 + Tregϸ°űŁ¬ĚÇƤÖĘĽ¤ËŘ˝«Ę§ČĄÖÎÁĆ×÷ÓĂ
• ĚÇƤÖĘĽ¤ËŘÔÚTregϸ°űÖĐĚŘŇěĐÔÓŐµĽmiR-342-3p±í´ď
• miR-342-3p°ĐĎňRictor˛˘µ÷˝ÚTregϸ°űÖеĴúĐ»łĚĐň
• miR-342-3pşÍRictor±í´ďÔÚĚĺÄÚżŘÖĆTregϸ°űµÄą¦ÄÜ
¸ĹŇŞ
ĚÇƤÖĘĽ¤ËŘŁ¨GCŁ©ĘÇŃ×ĐÔĽ˛˛ˇµÄÖ÷ŇŞÖÎÁĆѡÔńˇŁľˇąÜGCµÄĘąÓĂąă·şŁ¬µ«GC·˘»ÓĆä×÷ÓõĻúÖĆČÔČ»ÄŃŇÔ×˝ĂţˇŁÔÚŐâŔŔűÓĂĘóŔŕ×ÔÉíĂâŇߺ͹ýĂôĐÔŃ×֢ģĐÍŁ¬ÎŇĂDZ¨µŔFoxp3 +µ÷˝ÚĐÔTŁ¨TregŁ©Ď¸°űĘÇĚĺÄÚ˛»żÉĚć´úµÄGC°Đϸ°űˇŁÔÚĂ»ÓĐTregϸ°űµÄÇéżöϸřŇ©µÄµŘČűĂ×ËÉŁ¨DexŁ©ÍęȫɥʧÁËżŘÖĆŃ×Ö˘µÄÄÜÁ¦Ł¬˝öÔÚTregϸ°űÖĐȱ·¦ĚÇƤÖĘĽ¤ËŘĘÜĚ嵼ÖÂDexµÄÖÎÁĆÄÜÁ¦ÉĄĘ§ˇŁ´Ó»úŔíÉĎ˝˛Ł¬DexÔÚTregϸ°űÖĐĚŘŇěĐÔÓŐµĽmiR-342-3pŁ¬¶řmiR-342-3pÖ±˝Ó°ĐĎňmTORC2łÉ·ÖRictorˇŁ¸Ä±äTregϸ°űÖеÄmiRNA-342-3p»ňRictor±í´ď»áŇ쳣µ÷˝ÚTregϸ°űµÄ´úĐ»łĚĐňŁ¬´Ó¶řżŘÖĆËüĂÇÔÚĚĺÄڵĵ÷˝Úą¦ÄܡŁÎŇĂǵÄŃĐľż˝áąű˝ŇĘľÁËĚÇƤÖĘĽ¤Ëؽ鵼µÄŃ×Ö˘ÖÎÁĆąýłĚÖĐTregϸ°űµÄδ֪ą±Ďף¬ŇÔĽ°Í¨ąýDex-miR-342-RictorÖáĆđ×÷ÓõÄDZÔÚ»úÖơŁAnti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3+ Regulatory T Cells via a miR-342-Dependent Mechanism - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S107476132030282Xˇˇ
Metabolism, Nov 2001
A combined stress hormone infusion decreases in vivo protein synthesis in human T lymphocytes in healthy volunteers
From the Departments of Anaesthesiology and Intensive Care and Clinical Immunology, Huddinge University Hospital, Karolinska Institutet, Stockholm, Sweden; and the Department of Surgery, State University of New York, Stony Brook, NY.
In vivo protein synthesis decreases in mononuclear cells following a combined stress hormone infusion given to healthy volunteers as a human trauma model. Here, the purpose was to further investigate this finding and to measure in vivo protein synthesis in isolated T lymphocytes. Furthermore, the effects of stress hormones on the lymphocyte subpopulations and mononuclear cells, characterized by flow cytometry and phytohemagglutinin (PHA)-induced and unstimulated proliferative responses in vitro, were elucidated. Healthy volunteers (n = 16) were randomized into 2 groups to receive either a stress hormone or a saline infusion for 6 hours. In vivo protein synthesis was studied before and after the treatment by measuring the incorporation of stable isotopically-labeled phenylalanine into lymphocyte and mononuclear cell proteins. Protein synthesis decreased after stress hormone infusion in both cell populations: in T lymphocytes from 13.0% [plusmn] 0.7%/d (mean [plusmn] SD) to 8.6% [plusmn] 2.1%/d (P [lt ] .01) and in mononuclear cells from 13.3% [plusmn] 1.2%/d to 6.3 [plusmn] 2.0%/d (P [lt ] .001). No change in proliferative responsiveness in vitro was observed. The stress hormone infusion produced a decrease in the percentage of T helper CD3/CD4 from 41% to 18% (P [lt ] .001), T cytotoxic CD3/CD8 from 27% to 15% (P [lt ] .001), as well as total T CD3 cells from 69% to 35% (P [lt ] .001). There was an increase in the percentage of natural killer (NK) cells CD16/CD56 from 17% to 55% (P [lt ] .001). Determination of phenotypes expressed on activated T lymphocytes showed that CD3/HLA-DR was unchanged and CD3/CD25 decreased from 14% to 7% (P [lt ] .01) in the stress hormone group. The study showed that the decrease of in vivo protein synthesis was 34% in T lymphocytes as compared with 53% in mononuclear cells, when determined immediately after a 6-hour stress hormone infusion. This change was associated with a pronounced decrease in all lymphocyte subpopulations, except for the NK cells, which increased substantially.A combined stress hormone infusion decreases in vivo protein synthesis in human T lymphocytes in healthy volunteers - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0026049501688061ˇˇ
The Silent but Deadly Impact of Stress on the Lymphatic System. | elephant journal
https://www.elephantjournal.com/2017/05/the-silent-but-deadly-impact-of-stress-on-the-lymphatic-system/ˇˇ
Stress Weakens the Immune System
https://www.apa.org/research/action/immune
They also provided saliva samples for measuring levels of the stress hormone cortisol. Small networks and loneliness each independently weakened immunity to a core vaccine component. Immune response was most weakened by the combination of loneliness and small social networks, an obvious health stress facing shy new students who have yet to ...ˇˇ