íí
Published: 06 January 2003
Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: a role in organ dysfunction?
Benoît Vallet
Critical Care volume 7, Article number: 130 (2003) Cite this article
Abstract
During the past decade a unifying hypothesis has been developed to explain the vascular changes that occur in septic shock on the basis of the effect of inflammatory mediators on the vascular endothelium. The vascular endothelium plays a central role in the control of microvascular flow, and it has been proposed that widespread vascular endothelial activation, dysfunction and eventually injury occurs in septic shock, ultimately resulting in multiorgan failure. This has been characterized in various models of experimental septic shock. Now, direct and indirect evidence for endothelial cell alteration in humans during septic shock is emerging. The present review details recently published literature on this rapidly evolving topic.
Introduction
The vascular endothelium regulates the flow of nutrient substances, diverse biologically active molecules and the blood cells themselves. This role of endothelium is achieved through the presence of membrane-bound receptors for numerous molecules, including proteins, lipid transporting particles, metabolites and hormones, as well as through specific junction proteins and receptors that govern cellĘCcell and cellĘCmatrix interactions [1,2]. Endothelial dysfunction and/or injury with subendothelium exposure facilitates leucocyte and platelet aggregation, and aggravation of coagulopathy. Therefore, endothelium dysfunction and/or injury should favour impaired perfusion, tissue hypoxia and subsequent organ dysfunction.
The present review describes, within the context of sepsis, why altered endothelial properties may be suspected to be involved in organ failure (Table 1).
ííTable 1 Physiology and pathophysiology of endothelial cells
Properties of ECs In sepsis
Surface area: 1-7 m2 ECs become injured, prothrombotic and antifibrinolytic
Weight: 1 kg/70 kg body weight They promote platelet adhesion
Number: 1-6 í┴ 1013 cells They promote leucocyte adhesion and inhibit vasodilation
They line vessels in every organ: 'gate keeping role'
They favour vasodilatation
They promote antithrombosis and profibrinolysis
They inhibit platelet adhesion and leucocyte adhesion
íí
Full size table
Endothelial injury
Endothelial injury describes a state in which microscopically visible endothelial cell (EC) shape change or injury can be identified, as well as defects in endothelial lining or elevated soluble markers of endothelial injury [3]. Anatomical damage to the endothelium during septic shock has been assessed in several studies [4,5,6]. A single injection of bacterial lipopolysaccharide (LPS) has long been demonstrated to be a nonmechanical technique for removing endothelium [5]. In endotoxic rabbits, observations tend to demonstrate that EC surface modification occurs easily and rapidly [5,6], with ECs being detached from the internal elastic lamina with an indication of subendothelial oedema. As early as 15 min after LPS injection [7] cellular injuries are apparent, with nuclear vacuolization, cytoplasmic swelling and protrusion, cytoplasmic fragmentation, and various degrees of detachment of the endothelium from its underlying layer. This can also be observed 10 hours after the onset of sepsis in a caecal ligation and puncture rat model [8]. Proinflammatory cytokines increase permeability of the Ecs, and this is manifested approximately 6 hours after inflammation is triggered and becomes maximal over 12-24 hours as the combination of cytokines exert potentiating effects [8,9]. Endothelial physical disruption allows inflammatory fluid and cells to shift from the blood into the interstitial space.
Plasma levels of thrombomodulin (TM), intercellular adhesion molecule (ICAM)-1 and E-selectin may be measured in order to assess EC injury [10,11]. von Willebrand factor (vWF) and its propeptide can also be measured as circulating blood proteins to assess endothelial injury. It has been demonstrated that the half-life of mature vWF and that of its propeptide differ fourfold to fivefold [12]. The molar ratio of the propeptide to mature vWF can serve as a tool with which to assess the extent of EC injury and to distinguish between acute and chronic disease [13]. In patients with diabetes mellitus propeptide levels are only slightly elevated, whereas vWF levels are elevated twofold to threefold. In acute sepsis, both vWF and propeptide are elevated several fold. High levels of TM, ICAM-1 and vWF have been reported in several inflammatory diseases, sepsis and acute lung injury in patients with nonpulmonary sepsis, in which endothelial damage is thought to be important [11,14,15].
In a recent report, Mutunga et al. [16] developed a method for detecting circulating ECs that provides direct evidence of EC shedding in human sepsis. Blood samples were subsequently taken from 11 healthy volunteers, nine ventilated intensive care unit (ICU) control patients without sepsis, eight patients with sepsis but without shock, and 15 patients with septic shock. EC were identified by indirect immunofluorescence, using antibodies to vWF and the vascular endothelial growth factor receptor EGFR. vWF-positive EC counts per millilitre were significantly greater in patients with sepsis (16.1 í└ 2.7 [mean í└ SEM]) and septic shock (30.1 í└ 3.3) than in healthy (1.9 í└ 0.5) or ICU control individuals (2.6 í└ 0.6). EGFR-positive EC counts per ongoing EC lesions were also significantly higher in patients with sepsis (4.2 í└ 1.1) and septic shock (10.4 í└ 1.2) than in healthy (0.7 í└ 0.3) or ICU control individuals (0.5 í└ 0.2). Cell counts measured using anti-vWF antibody were consistently higher than those measured using anti-EGFR antibody, but correlation between the two counts was high (r2 = 0.93). The number of circulating EGFR-positive ECs per millilitre was significantly higher in patients who died of septic shock than in survivors (12.0 í└ 1.6 versus 7.1 í└ 1.2; P = 0.026). An increase in circulating ECs can therefore be identified during sepsis and septic shock. That study was among the first to support the hypothesis that endothelial damage occurs in human sepsis.
An important point is that EC injury is sustained over time. In an endotoxic rabbit model, we demonstrated that endothelium denudation is present at the level of the abdominal aorta as early as after several hours following injury and persisted for at least 5 days afterward [6,17]. After 21 days we observed that the endothelial surface had recovered. The de-endothelialized surface accounted for approximately 25% of the total surface. Similarly, in 12 human volunteers receiving 4 ng/kg Escherichia coli LPS by intravenous injection, Taylor and coworkers [18] showed that the immediate symptomatic inflammatory stage (0-8 hours after LPS injection) was followed after 12 hours by an asymptomatic noninflammatory stage (volunteers were back at work). The latter stage was characterized by decreased tumour necrosis factor, interleukin-10, thrombinĘCantithrombin and plasminĘCantiplasmin complexes, and levels of TM peaked at 24 hours, suggesting ongoing EC lesions. Increased TM was associated with a level of tissue factor (TF) that was still increasing at 48 hours, suggesting risk for activated coagulation. Indeed, TF is the principal activator of the extrinsic coagulation pathway, and as such is responsible for an intravascular procoagulant state. Taylor and coworkers concluded that sustained injury to the vascular endothelium secondary to reperfusion of the microvasculature occurred in those asymptomatic individuals. In our endotoxic model, we also demonstrated that at 5 days the rabbits had maximal monocyte TF expression, which coincided with maximal endothelial injury [6,17]. This, together with altered coagulation modulation properties, may ultimately result in intravascular microthrombosis.
Endothelial injury associated abnormal coagulation and fibrinolysis
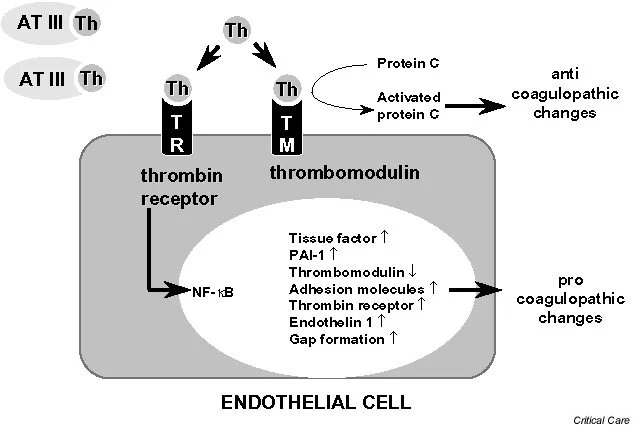
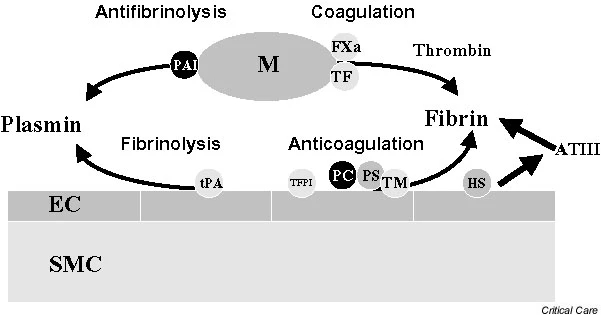
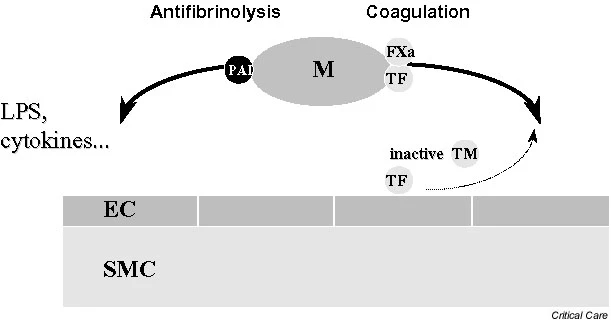
The outer membrane of ECs normally expresses various membrane-associated components with anticoagulant properties, among which are cell surface heparin-like molecules. These molecules accelerate inactivation of coagulation proteases by antithrombin and represent a TF pathway inhibitor (TFPI) reserve [19]. The EC surface thrombin-binding protein TM is responsible for inhibition of thrombin activity. TM, when bound to thrombin, forms a potent protein C (PC) activator complex (Fig. 1). Whereas unperturbed ECs confer anticoagulant properties (Fig. 2), exposure to inflammatory and/or septic stimuli rapidly lead to procoagulant behaviour (Figs 1 and 3). Moreover, the profibrinolytic property of ECs is blunted, because of decreased release of tissue plasminogen activator. This occurs within the context of increased release of plasminogen activator inhibitor-1. During sepsis the procoagulant activity of TF increases, with transcriptional upregulation of its expression on monocytes and ECs among other cell types, whereas levels of endothelium anticoagulant membrane components decrease, with internalization of TM [20] and release of inactive TM into the bloodstream (Fig. 3). Loss of TM and associated PC activation represents a key event, namely decreased endothelial coagulation modulation ability. Cleavage of TM by neutrophil elastase and other proteases certainly participate in the reduced expression of TM.
íí
figure1
Thrombomodulin and protein C activation at the microcirculatory level. The endothelial cell surface thrombin (Th)-binding protein thrombomodulin (TM) is responsible for inhibition of thrombin activity. TM, when bound to Th, forms a potent protein C activator complex. Loss of TM and/or internalization results in Th-thrombin receptor (TR) interaction. Loss of TM and associated protein C activation represents the key event of decreased endothelial coagulation modulation ability and increased inflammation pathways. Adapted from Iba and coworkers [88]. ATIII, antithrombin III; NF-Ž╩, nuclear factor-Ž╩B; PAI, plasminogen activator inhibitor; Th, thrombin; TM, thrombomodulin.
figure2
Coagulation and fibrinolysis pathways. Unperturbed endothelial cells (ECs) provide anticoagulant (tissue factor pathway inhibitor [TFPI], protein C [PC], protein S [PS], thrombomodulin [TM], heparan sulphate [HS]) and fibrinolytic (tissue plasminogen activator [tPA]) properties. ATIII, antithrombin III; FXa, coagulation factor Xa; M, activated monocyte; PAI, plasminogen activator inhibitor; SMC, smooth muscle cell; TF, tissue factor.
figure3
Sepsis and coagulation-fibrinolysis pathways. Exposure to inflammatory and/or septic stimuli rapidly leads to procoagulant behaviour. The profibrinolytic property of endothelial cells (ECs) is blunted, due to decreased release of tissue plasminogen activator. This occurs in a context of increased plasminogen activator inhibitor (PAI)-1 release with antifibrinolysis. LPS, lipopolysaccharide; M, activated monocyte; SMC, smooth muscle cell; TF, tissue factor; TM, thrombomodulin.
Full size image
In severe meningococcal sepsis, Faust and coworkers [21] recently demonstrated that PC activation is impaired - a finding that is consistent with downregulation of the endothelial TMĘCendothelial PC receptor pathway. In 21 children (median age 41 months) with purpura fulminans (meningococcal sepsis), purpuric lesion skin biopsies exhibited decreased expression of endothelial TM and of the endothelial PC receptor as compared with control specimens, both in vessels with and in those without thrombosis. Plasma TM levels in the children with meningococcal sepsis (median 6.4 ng/l) were higher than those in the controls (median 3.6 ng/l; P = 0.002). Plasma levels of PC antigen, protein S antigen and antithrombin antigen were lower than those in the controls. In two patients treated with unactivated PC concentrate, activated PC (APC) was undetectable at the time of admission, and plasma levels remained low.
Activation of coagulation concomitant with impaired fibrinolysis is associated with fibrin deposition, tissue ischaemia and tissue necrosis [22], and in critically ill patients with increased risk for death [23,24]. Conversely, inhibition of coagulation is associated with prevention of organ dysfunction [25,26]. Three therapeutic strategies that employ coagulation modulation - TFPI, antithrombin and APC - were recently proposed to reduce organ dysfunction and mortality in septic shock. It has clearly been shown in various animal models of septic shock that these treatments reduce organ dysfunction and mortality [27,28]. This was associated with a reduction in cytokine production [25,26,29]. With APC, it was further demonstrated that leucocyteĘCendothelial interactions were reduced [30]. Of note is the demonstration that aPC was also able to improve fibrinolysis by inhibiting plasminogen activator inhibitor-1 [31]. Clinical phase II trials suggested that mortality might be reduced by using these coagulation modulators in critically ill septic patients [32,33,34,35]. Three phase III trials of antithrombin, TFPI and APC were subsequently performed and recently completed in large populations of patients with severe sepsis, the net effect being an overall lack of efficacy with antithrombin [36] and TFPI (unpublished results), and a 19.43% reduction in relative risk for death with APC [37].
Although ECs probably have an important role in disseminated intravascular coagulation, there is also some evidence favouring a major role for monocytes in the cellular mechanisms of coagulation activation. We recently assessed the relative impact of endothelium injury and monocyte activation on coagulation disorders in our rabbit endotoxic shock model. L-arginine and the angiotensin-converting enzyme inhibitor perindopril were tested in that model for their demonstrated ability to treat endothelium injury [38,39]. We found that both L-arginine supplementation and perindopril could prevent septic-shock-associated deterioration in endothelium-dependent relaxation [40,41]. However, this preventive effect was not associated with any reduction in TF expression, suggesting that these two sepsis-associated abnormalities are not strictly linked. In a subsequent study [42] we used an antiglycoprotein IIb/IIIa, which attenuated endotoxin-induced monocyte TF expression through decreased platelet activation. This was associated with marked reduction in endothelial injury, increased endothelium-derived relaxation and improved survival rates in the treated animals. Those findings suggest that monocyte activation and TF expression may be of importance in sepsis-associated injuries, and that coagulation activation may itself contribute to the EC injury observed during sepsis.
Endothelial injury, in turn, exacerbates sepsis-induced coagulation abnormalities. Indeed, release of endothelium-derived factors such as nitric oxide (NO) and prostacyclin (PGI2) is impaired. Because NO and PGI2 not only control vascular tone but also have antiadhesive and tissue plasminogen activator-like properties, loss of NO and PGI2 release facilitates leucocyte and platelet aggregation, and aggravation of coagulopathy. Furthermore, when ECs generate adhesion molecules during endotoxaemia that bind leucocytes and monocytes, they favour enhancement in local procoagulant reactions. The relationship between activation of innate immunity and coagulation is phylogenetically ancient [43,44]. Localized activation of the coagulation system, as with the innate immune response, serves to protect against a discrete traumatic injury [43]. However, generalized intravascular coagulation, as a generalized inflammatory response, is detrimental to the host, favouring widespread fibrin deposition and altered tissue perfusion.
Endothelial activation
As a prelude to their migration into tissues, monocytes and leucocytes must adhere to endothelium. Both adhesion to and migration across endothelium are governed by the interaction of complementary adhesion molecules on the polymorphonuclear cells and endothelium [45]. The surface expression, adhesion avidity and surface modulation of these molecules are highly regulated by biological mediators such as cytokines. Local synthesis of platelet-activating factor and EC-derived cytokines such as interleukin-8, along with tumour necrosis factor and interleukin-1, are important in promoting neutrophilĘCEC interactions. 'Endothelial activation' refers to increased expression or release of endothelial adhesion molecules.
The first step in migration consists of a 'rolling' of leucocytes on endothelium, which involves the selectin family. Selectins are molecules expressed on leucocytes (L-selectin), or even on platelets (P-selectin) and on ECs (E-selectin); these act as receptors that permit loose binding, which in turn facilitates rolling. Selectins allow leucocytes to roll in the direction of flow into the proximity of activating signals exhibited by ECs.
The second step involves receptors from the integrin family (Ž┬2-integrin) and immunoglobulin-like receptors. These receptors allow leucocyte arrest and adhesion strengthening. Three heterodimers of Ž┬2-integrin are present on the outer cell membrane of activated leucocytes and are collectively termed the CD11/CD18 complex. Stimulation of ECs induces expression of cell surface adhesion molecules, which are members of the immunoglobulin superfamily. Monoclonal antibodies to these molecules have been shown to block leucocyteĘCEC interactions and to improve sepsis-associated organ dysfunction [46,47]. Endothelial adhesion molecules include ICAMs, endothelial leucocyte adhesion molecules (E-selectins), platelet EC adhesion molecules, and vascular cell adhesion molecules.
In the third step, activated leucocytes migrate to the borders of ECs to interact with ICAMs, endothelial leucocyte adhesion molecules, platelet EC adhesion molecules or vascular cell adhesion molecules (for review [48]). A large number of experimental studies have documented the consequences of inhibition of adhesion molecules. Inhibition of neutrophil adherence to the ECs exerts significant protective effects in these conditions [46,49].
Interestingly, decreased reactive hyperaemia (suggesting modified endothelial-derived relaxation) was also demonstrated to coexist with increased leucocyte aggregation and ICAM-1 levels [50]. It is also important to emphasize that recent evidence suggested that adhesion can occur independent of adhesion molecules in organs such as lung or liver. This led to the hypothesis that stimulus-induced increases in actin-containing stress fibres (such as LPS) at the cell periphery lead to decreased deformability, preventing neutrophils from trafficking through the capillary bed and therefore increasing their sequestration at sites of inflammation [51].
Sessler and coworkers [52] measured blood level of the adhesion molecule ICAM-1 as a potential marker of EC activation in septic adults and healthy volunteers. Those investigators established a relationship between increased ICAM-1 levels and consequences of sepsis (i.e. multiple organ failure and death). Watanabe and coworkers [53] prevented endotoxin shock in rabbits by administering a specific monoclonal antibody against CD18 (integrin Ž┬2). In a mouse lethal septic shock model, Xu and coworkers [54] observed that animals deficient in ICAM-1 were markedly protected against death. Whereas 80% of wild-type animals died within 48 hours after receiving 40 mg/kg LPS, more than 90% of ICAM-1-deficient animals survived for longer than 4 days. Interestingly, EC dysfunction was found to involve a CD18-dependent neutrophil adherent mechanism. Consistently, Matsukawa and coworkers [55] recently provided evidence on the contributions of E-selectin and P-selectin to lethality in septic peritonitis. Mice that genetically lacked endothelial selectins were shown to be resistant. The experiments demonstrated that endothelial selectin mediated leucocyte rolling impacts on mouse survival by influencing the serum level of cytokines and by preventing renal dysfunction - a potential cause of death in that context.
Endothelial dysfunction
The term 'endothelial dysfunction' refers to decreased endothelial-dependent vascular relaxation or NO release, and decreased expression or activity of endothelial constitutive NO synthase (ecNOS). Endothelium-derived relaxation and/or production of endothelium-derived NO from the amino acid L-arginine by ecNOS may be used as an indicator of EC function. For example, the relaxing response of in vitro isolated vascular rings to picomolar concentrations of acetylcholine is dependent on the presence and integrity of ECs [56]. In vivo endothelial function can be determined by measurement of forearm blood flow responses to intra-arterial infusions of endothelium-dependent (i.e. acetylcholine) and endothelium-independent vasodilators (i.e. sodium nitroprusside). Drugs are infused at a constant rate (1 ml/min) with an infusion pump. Forearm blood flow is recorded for 10 s at 15-s intervals during the last 3 min of the drug and saline infusion period using venous occlusion plethysmography combined with a rapid cuff inflator [57,58,59].
Abnormal endothelial-dependent vascular relaxation has been recognized in multiple sepsis conditions. Several investigations, including our own [17,60,61], have demonstrated attenuated acetylcholine-induced relaxation in vascular rings isolated from large arteries. Apart from anatomical injuries, such abnormalities observed in these vessels may result from several mechanisms: alteration in EC surface receptors; modified signal transduction pathways (receptor-ecNOS coupling); altered function and/or density of the ecNOS; changes in pathways that lead to release of NO; and/or changes in mechanisms that participate in subsequent degradation of NO.
In healthy volunteers, even brief exposure to endotoxin or certain cytokines impairs endothelium-dependent relaxation for many days [62,63]. This effect has been termed 'endothelial stunning'. After recovery from the acute insult, the endothelium may remain dysfunctional ('stunned') for a long period of time before full recovery. Hingorani and coworkers [59] also demonstrated that a mild inflammatory response, such as that generated by Salmonella typhi vaccine, is associated with temporary but profound dysfunction of arterial endothelium in both resistance and conduit vessels following application of both physical and pharmacological dilator stimuli. According to the concept of intrinsic metabolic regulation, vasodilatation in tissues with relatively high metabolic rates competes with sympathetic vasoconstrictor tone, thereby adjusting the balance between local tissue oxygen supply and demand. Although the nature of the oxygen-sensitive structures that act at the local tissue level is not completely understood, ECs in direct contact with blood have a number of properties that render them effective sensors. The endothelium and smooth muscle of arteries and arterioles appear to be coupled both structurally and functionally. Sensing involves local depolarization/hyperpolarization of the capillary EC, and communication is achieved by electronic spread via endothelium/smooth muscle cellĘCcell gap junctions [2,64]. During an hypoxic challenge, the ability of a tissue to extract oxygen - and to minimize shunting through areas with a high rate of perfusion relative to their oxygen uptake - may therefore be considered an integrative test of endothelial function and microcirculatory coordination [65].
We investigated the role of the endothelium in regulating the balance between oxygen demand and supply within an individual organ in an in vivo model of endothelial stripping in the dog hind limb [66]. The hind limb vascular endothelium was removed by injecting deoxycholate into the perfusing artery before ischaemic challenge. Deoxycholate - a detergent used to remove endothelium in in vitro studies - removes vascular endothelium within arteries, arterioles, capillaries and veins. It achieves this without causing apparent damage to either the vascular smooth muscle layer or the skeletal muscle parenchyma, as assessed by in vitro and in vivo studies of pharmacological vascular reactivity, tissue histology and electron microscopy. Hind limb oedema or capillary plugging by endothelial fragments was not observed. During progressive limitation of oxygen supply to the limb, a profound and significant impairment in limb oxygen extraction ability (41.7% versus 81% in controls) at critical oxygen delivery (at which oxygen uptake begins to decrease) was observed. We concluded that this severe limitation in the increase in oxygen extraction capabilities during ischaemia suggested that vascular endothelium plays an important role in matching oxygen supply to demand.
In order to test the role of the endothelial-derived relaxing factors NO and PGI2, we investigated, in a third group of dogs, the influence of a combination of NG-nitro-L-arginine methyl ester (an inhibitor of NO synthesis) and indomethacin (an inhibitor of PGI2 synthesis) [66]. In these dogs treated with indomethacin plus NG-nitro-L-arginine methyl ester, the severity of the oxygen extraction defect was lower than that observed in the deoxycholate-treated dogs, suggesting that other mediators and/or mechanisms may be involved in microcirculatory control during hypoxia. As suggested above, one of these mediators or mechanisms could be related to hyperpolarization. Membrane potential is an important determinant of vascular smooth muscle tone through its influence on calcium influx via voltage-gated calcium channels. Hyperpolarization (as well as depolarization) has been shown to be a means of cellĘCcell communication in upstream vasodilatation and microcirculatory coordination [67]. It is important to emphasize that intercell coupling exclusively involves ECs. Interestingly, it was recently shown that sepsis, a situation that is characterized by impaired tissue perfusion and abnormal oxygen extraction, is associated with abnormal inter-EC coupling and reduction in the arteriolar conducted response [68].
An intra-organ defect in blood flow related to abnormal vascular reactivity, cell adhesion and coagulopathy may account for impaired organ oxygen regulation and function. If specific classes of microvessels must or must not be perfused to achieve efficient oxygen extraction during limitation in oxygen delivery, then impaired vascular reactivity and vessel injury might produce a pathological limitation in supply. In sepsis, the inflammatory response profoundly alters circulatory homeostasis, and this has been referred to as a 'malignant intravascular inflammation' that alters vasomotor tone and the distribution of blood flow among and within organs [69]. These mechanisms might coexist with other types of sepsis-associated cell dysfunction. For example, data suggest that endotoxin directly impairs oxygen uptake in ECs and indicate the importance of endothelium respiration in maintaining vascular homeostasis under conditions of sepsis [70].
Abnormal oxygen extraction is a key feature of severe sepsis and septic shock. In an experimental study in dogs, an ablated reactive hyperaemia was associated with endotoxaemia-induced impaired oxygen extraction at the level of the gastrointestinal tract [71]. NeviĘĘre and coworkers [72] showed that reactive hyperaemia is attenuated in critically ill patients with septic shock, despite normal or elevated whole-body oxygen delivery. Proposed mechanisms to explain blunted hyperaemia in septic patients might include impaired vascular reactivity and/or microvascular obstruction that limits the number of recruitable capillaries. In critically ill patients with sepsis, it has been shown that decreased reactive hyperaemia coexists with increased leucocyte adhesion and increased release in surrogate markers of endothelium injury [50,73].
Thus, assessment of reactive hyperaemia might be used in the near future to evaluate the effects of treatments aimed at restoring endothelial function and tissue perfusion, such as coagulation modulators or leucocyte adhesion antagonists.
Conclusion
How do all of these altered properties contribute to altered perfusion and organ dysfunction? The combining effect of altered vascular relaxation, altered blood flow distribution, increased leucocyte adhesion and decreased coagulation modulation should significantly contribute to microcirculatory heterogeneity and lowered perfusion. Studies in the isolated perfused rabbit heart [74], autoperfused rat cremaster [75] and rat mesentery [76,77] suggested that these mechanisms are operative in the microvasculature. On an intravital microscopy extensor digitorum longus muscle model in rats with peritonitis [78], it was shown that sepsis is associated with a reduction in tissue perfused capillary density of up to 36%, increased perfusion heterogeneity and mean intercapillary distance, contributing to functional shunting. In another study [79], endotoxin administration resulted in a significant enhancement in leucocyteĘCEC interaction, as indicated by transiently increased number of leucocytes firmly attaching to the microvascular endothelium of arterioles and venules [79]. Microvascular injury and/or the appearance of greater heterogeneity of microvascular distribution of oxygen supply with respect to oxygen demand in endotoxin-treated animals is consistent with the observation that endotoxin impairs oxygen extraction [80,81,82]. The direct relationship between heterogeneity, decreased oxygen extraction and tissue acidosis was recently confirmed in a pig model of endotoxic shock [83].
Consistent with the hypothesis that alteration in endothelium plays a major in the pathophysiology of sepsis, it was observed that chronic ecNOS overexpression in the endothelium of mice resulted in resistance to LPS-induced hypotension, lung injury and death [84]. This observation was confirmed by another group of investigators, who used transgenic mice overexpressing adrenomedullin [85] - a vasodilating peptide that acts at least in part via an NO-dependent pathway. They demonstrated resistance of these animals to LPS-induced shock, and lesser declines in blood pressure and less severe organ damage than occurred in the control animals. It might therefore be of importance to favour ecNOS expression and function during sepsis. The recent negative results obtained with therapeutic strategies aimed at blocking inducible NOS with the nonselective NOS inhibitor NG-monomethyl-L-arginine in human septic shock [86] further confirm the overall importance of favoring vessel dilatation. In contrast, positive results obtained with corticosteroids [87] and APC [37] suggest that improving haemodynamics while decreasing vasopressor agents (corticosteroids) and limiting coagulation activation are logical strategies that may greatly favour tissue perfusion and improved oxygen delivery.
Abbreviations
APC:
APC = activated protein C
EC:
EC = endothelial cell
ecNOS:
ecNOS = endothelial constitutive nitric oxide synthase
ICAM:
ICAM = intercellular adhesion molecule
ICU:
ICU = intensive care unit
LPS:
LPS = lipopolysaccharide
NO:
NO = nitric oxide
PGI:
PGI2 = prostacyclin
TF:
TF = tissue factor
TFPI:
TFPI = tissue factor pathway inhibitor TM = thrombomodulin
vWF:
vWF = von Willebrand factor.
Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? | Critical Care | Full Text
https://ccforum.biomedcentral.com/articles/10.1186/cc1864