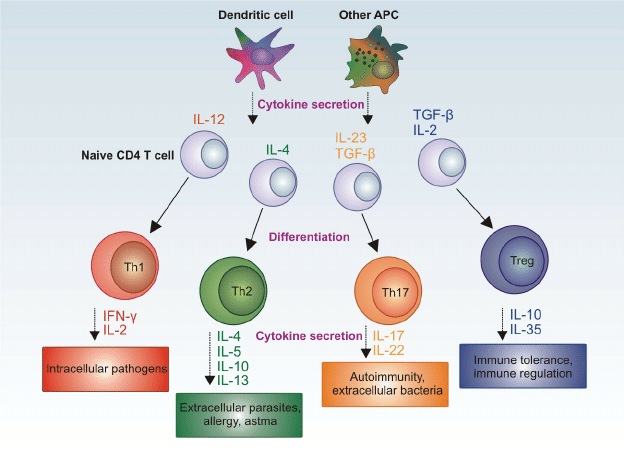
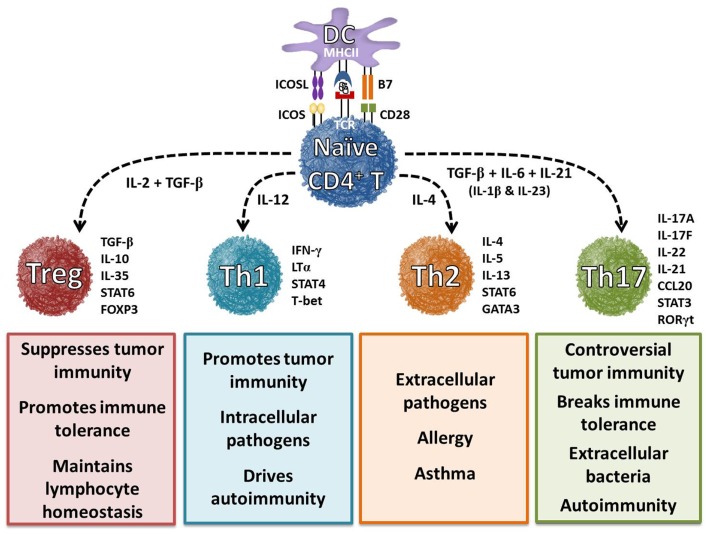
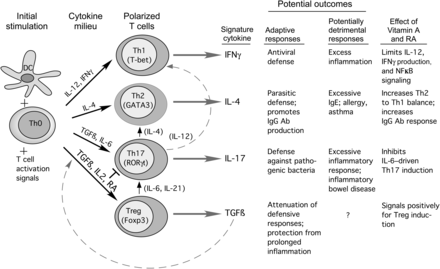
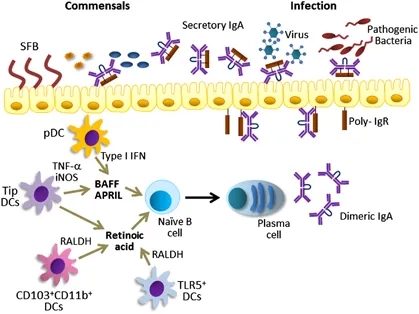
ˇˇ
Anti©\inflammatory effect of retinoic acid
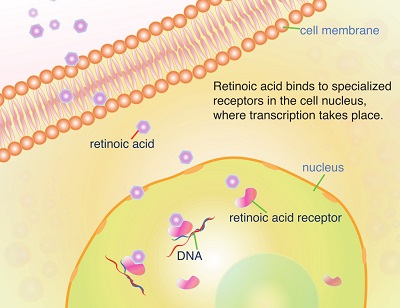
1. Retinoic acid (RA), a highly potent small molecule exhibiting a great variety of anti-inflammatory and neuroprotective properties in the adult central nervous system (CNS). RA homeostasis in the adult CNS is tightly controlled through local RA synthesis and cytochrome P450 (CYP450)-mediated inactivation of RA.
2. RA treatment significantly elevates the expression of Arginase 1 (Arg1)in macrophages, a gene product critical to wound healing process.
3. during an anti-inflammatory response when macrophages were alternatively (M2) polarized, retinoic acid (RA) dramatically activated arginase 1 gene (Arg1), a gene crucial for wound healing.
4.RA counteracted the inflammatory regulation of cyclooxygenase (COX)©\2 mRNA and protein in astrocytes and thereby reduced the synthesis of PGE2 by approximately 60%.
5. Retinoic acid dampens LPS-induced NF-kappaB activity
6.Glyburide enhances M2 polarization and anti-inflammation
7. Receptor Interacting Protein 140 (RIP140) is a protein found in metabolic tissues, such as liver, muscle and adipose tissue. RIP140 is also known to be expressed in the monocyte-macrophage lineage and can regulate inflammatory responses.
8. RIP140 is pro-inflammatory because it is a cofactor of NF-kB, facilitating M1 polarization21, and an inhibitor of STAT6, suppressing M2 polarization22.
9. RIP140 as a risk biomarker of, and a therapeutic target for, atherosclerosis.
10. Glyburide is anti-inflammatory because it activates CamKII, which modifies RIP140 protein for proteosome-mediated degradation.
11.The anti-inflammatory effects of minocycline on TNF-¦Á expression were completely abolished by a pharmacological blockage of retinoic acid receptors (RARs), RA-dependent, anti-inflammatory effect for minocycline in human microglial-like cells via inhibition of local RA turnover.
12.RIP140 promotes macrophage-derived foam cell formation.
RIP140 suppresses LXR-regulated expression of ABCA1 and ABCG1
Lowering RIP140 level in macrophage ameliorates atherosclerosis.13.ATRA(all-trans-retinoic acid) is an interferon-inducing agent with antiviral activity against EV71 in vitro
14. Northwestern Uni:Treatment with all-trans retinoic acid (1 mg/kg) prevented virus-induced hyperreactivity and M2 receptor dysfunction. However, retinoic acid also significantly reduced viral titers in the lungs and attenuated virus-induced lung inflammation.
15. The ADH©\ALDH Pathway Is a Potent Antiviral Host Factor. retinoic acid protects the liver against alcohol damage
16. retinoic acid enhances proliferation of B lymphatic cells while limiting T cells
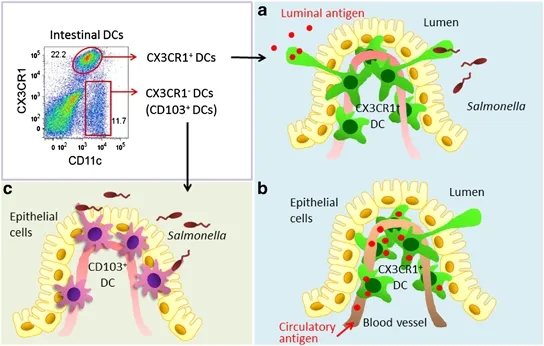
17. Vitamin A is necessary for maintaining intestinal integrity (41), regulating mucin gene expression (42), and normal production of intestinal IgA (43).
18Ł¬retinoic acid induction of neuronal differentiation.
ˇˇ
ˇˇ
ˇˇ
Mediators Inflamm. 2018; 2018: 3067126.
Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases
Luana de Mendonça Oliveira, Franciane Mouradian Emidio Teixeira, and Maria Notomi Satocorresponding author
Author information Article notes Copyright and License information Disclaimer
Laboratory of Dermatology and Immunodeficiencies, LIM-56, Department of Dermatology, School of Medicine, University of São Paulo, Institute of Tropical Medicine of São Paulo, São Paulo, SP, Brazil
Abstract
Vitamin A metabolite retinoic acid (RA) plays important roles in cell growth, differentiation, organogenesis, and reproduction and a key role in mucosal immune responses. RA promotes dendritic cells to express CD103 and to produce RA, enhances the differentiation of Foxp3+ inducible regulatory T cells, and induces gut-homing specificity in T cells. Although vitamin A is crucial for maintaining homeostasis at the intestinal barrier and equilibrating immunity and tolerance, including gut dysbiosis, retinoids perform a wide variety of functions in many settings, such as the central nervous system, skin aging, allergic airway diseases, cancer prevention and therapy, and metabolic diseases. The mechanism of RA is interesting to explore as both a mucosal adjuvant and a combination therapy with other effective agents. Here, we review the effect of RA on innate and adaptive immunity with a special emphasis on inflammatory status.Impact of Retinoic Acid on Immune Cells and Inflammatory Diseases
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6109577/
Anti©\inflammatory effect of retinoic acid on prostaglandin synthesis in cultured cortical astrocytes
Eric Kampmann Sonja Johann Sabien Van Neerven Cordian Beyer Jörg Mey
Institut f¨ąr Biologie II, RWTH Aachen, Germany
Abstract
Prostanoids are important mediators of inflammation and pain signaling. Although it is now well accepted that astrocytes participate in inflammatory reactions in the CNS, the molecular regulation of this activity is still largely unknown. Specifically, the regulation of prostanoid synthesis by this type of glia remains to be resolved.Recent evidence suggests that the transcriptional regulator retinoic acid (RA) is involved in regulation of the immune response. We have investigated the expression pattern of the enzymes that catalyze prostanoid and leukotriene synthesis in cultured cortical astrocytes, their stimulation by lipopolysaccharides (LPS) and their regulation by RA. The data indicate that astrocytes are an important source of prostaglandins (PGs) and that RA reduces their inflammatory biosynthesis. LPS treatment induced the expression of enzymes for the production of arachidonic acid and PGs but caused down©\regulation of a PG degrading enzyme and of leukotriene synthesizing enzymes that compete with PG synthesis. Consequently, the secretion of the PGE2 was highly increased after LPS exposure.
RA counteracted the inflammatory regulation of cyclooxygenase (COX)©\2 mRNA and protein in astrocytes and thereby reduced the synthesis of PGE2 by approximately 60%. In the absence of LPS, RA enhanced the expression of COX©\1 mRNA.
In conclusion, RA might be effective in suppressing inflammatory processes in the brain by inhibiting PG synthesis.
Anti©\inflammatory effect of retinoic acid on prostaglandin synthesis in cultured cortical astrocytes - Kampmann - 2008 - Journal of Neurochemistry - Wiley Online Library
https://onlinelibrary.wiley.com/doi/full/10.1111/j.1471-4159.2008.05395.xˇˇ
Clinical Immunology
Volume 119, Issue 3, June 2006, Pages 272-279
Anti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis
Author links open overlay panelYujiNozakiToshiakiYamagataMasafumiSugiyamaShinyaIkomaKojiKinoshitaMasanoriFunauchi
Show more
https://doi.org/10.1016/j.clim.2005.11.012Get rights and contentDepartment of Nephrology and Rheumatology, Kinki University School of Medicine, 377-2 Ohno-Higashi, Osaka-Sayama, Osaka, 589-8511, Japan
Abstract
Objective:
To determine whether all-trans-retinoic acid (ATRA) improves the destruction of joints and the effect of cytokines on DBA/1J mice with collagen-induced arthritis (CIA).
Methods:
Starting from the time of type II collagen injection, DBA/1J mice were injected intraperitoneally with PBS or 0.5 mg of ATRA 3 times per week for 35 days. The effects of treatment were monitored by determining arthritis and histological scores and measuring cellular proliferation, production of cytokines (IL-2, IL-10, IL-12, IL-6, IFN-¦Ă, and TNF-¦Á) and IgG, and the expression of mRNAs for inducible nitric oxide synthase (iNOS), monocyte chemoattractant protein-1 (MCP-1), and CXCR3.
Results:
The arthritis score and incidence of arthritis were lower in the mice treated with ATRA than in those treated with PBS. Histopathologic evidence of joint damage was 34% lower, and the infiltrations of macrophages were reduced in the mice treated with ATRA compared with those treated with PBS. Type II collagen- and ConA-stimulated proliferation of spleen cells, the production of cytokines (IL-6, IL-12, and TNF-¦Á), the serum levels of total IgG and IgG1 anti-collagen antibodies, and the expression of mRNAs for MCP-1 were significantly reduced in the mice treated with ATRA than in those treated with PBS.
Conclusion:
ATRA improved the clinical course and reduced the production of inflammatory cytokines, immunoglobulin, and chemokines in murine CIA. These data suggest that ATRA might be also effective for the treatment of inflammatory arthritis like human rheumatoid arthritis.
Keywords
All-trans-retinoic acidCytokinesTh1/Th2Collagen-induced arthritis miceAnti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S1521661605003906ˇˇ
Immunity. Author manuscript; available in PMC 2012 Aug 14.
The Role of Retinoic Acid in Tolerance and Immunity
J.A. Hall, J.R. Grainger, S.P. Spencer, and Y. Belkaid
Author information Copyright and License information Disclaimer
Mucosal Immunology Section, Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892
CORRESPONDENCE: Jason Hall: vog.hin.diain@sajllah; Yasmine Belkaid: vog.hin.liam@diakleby
ˇˇIntroduction
In the early 20th century E.V. McCollum and Thomas Osborne independently embarked on studies to identify dietary constituents that were essential for mammalian health and survival. Using different dietary supplements they arrived at the seminal conclusion that a single factor present in lipids was essential for growth and survival, which they coined ˇ°fat soluble factor Aˇ± (Wolf, 1996). Subsequently designated vitamin A, studies over the years have demonstrated the pleiotropic influence of this nutrient, ranging from eyesight and organogenesis to metabolism and immunological fitness (Acin-Perez et al.; Duester, 2008; Underwood, 2004; Ziouzenkova et al., 2007). Exposing its critical contribution to immunological health, vitamin A supplementation was shown to dramatically curb young-childhood mortality in endemic regions of malnutrition (Rahmathullah et al., 1990; Sommer, 2008; Sommer et al., 1986). The vitamin A metabolite, retinoic acid (RA), first received attention as an interventional therapy upon discovery that it could substitute for more toxic chemotherapeutic regimens to dramatically improve the prognosis of acute promyelocytic leukemia, a malignancy caused by genetic translocations with the retinoic acid receptor (RAR), RAR¦Á(de The and Chen, 2010). While numerous investigations of APL have highlighted the ability of RA to promote myeloid cell differentiation (Kastner et al., 2001), over the last 20 years it has become clear that this metabolite influences multiple immune cell lineages and an array immunological functions (Cantorna et al., 1995; Chun et al., 1992). In this review, we discuss recent advances that have established RA as central to both immunological tolerance and the elicitation of adaptive immune responses. Further, we provide a comprehensive overview of the cell types and factors that control the production of RA and discuss how host perturbations may affect the ability of this metabolite to control tolerance and immunity, or instigate pathology.The Role of Retinoic Acid in Tolerance and Immunity
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3418663/ˇˇ
Biochemical and Biophysical Research Communications
Volume 329, Issue 1, 1 April 2005, Pages 125-131
Biochemical and Biophysical Research Communications
Anti-inflammatory roles of retinoic acid in rat brain astrocytes: Suppression of interferon-¦Ă-induced JAK/STAT phosphorylationˇî
Author links open overlay panelWoo-HyuckChoiaKyung-AeJiaSae-BomJeonabMyung-SoonYangabHoKimcKyoung-jinMindMinhoShongcIloJoudEun-HyeJoeabd
Abstract
The anti-inflammatory effect of retinoic acid (RA) has been investigated for several decades. However, the underlying mechanisms responsible for this effect are largely unknown. In this study, we demonstrate that 9-cis-RA (cRA) and all-trans-RA (tRA) inhibit interferon-¦Ă (IFN-¦Ă)-induced inflammatory responses in astrocytes. In primary cultured rat brain astrocytes and C6 astroglioma cells, both cRA and tRA decreased IFN-¦Ă-induced expression of interferon regulatory factor-1. Both RA isoforms also reduced IFN-¦Ă-induced activation of signal transducers and activators of transcription (STAT)1, STAT3, Janus kinase (JAK)1, and JAK2. This inhibitory effect was significant when cells were pre-treated with RA prior to IFN-¦Ă. Furthermore, the effect of pre-treated RA was abolished in the presence of cycloheximide, indicating a requirement for de novo protein synthesis. Suppressors of cytokine signaling (SOCS), which are negative regulators of the JAK/STAT pathway, may be candidate mediators of the anti-inflammatory function of RA. Both cRA and tRA induced SOCS3 mRNA expression. These results suggest that RA induces an anti-inflammatory effect by suppressing the activation of the JAK/STAT pathway in IFN-¦Ă-treated astrocytes. SOCS3 may be at least one of the mechanisms that mediate the anti-inflammatory roles of RA.Anti-inflammatory roles of retinoic acid in rat brain astrocytes: Suppression of interferon-¦Ă-induced JAK/STAT phosphorylation - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0006291X05001749ˇˇ
Nutrients 2018, 10(8), 1016; https://doi.org/10.3390/nu10081016
Review
Retinoic Acid, Leaky Gut, and Autoimmune Diseases
by Leila Abdelhamid and Xin M. Luo *OrcID
Department of Biomedical Sciences and Pathobiology, College of Veterinary Medicine, Virginia Tech, Blacksburg, VA 24061, USA
*
Author to whom correspondence should be addressed.
Received: 12 June 2018 / Accepted: 27 July 2018 / Published: 3 August 2018
Abstract: A leaky gut has been observed in a number of autoimmune diseases including type 1 diabetes, multiple sclerosis, inflammatory bowel disease, and systemic lupus erythematosus. Previous studies from our laboratory have shown that lupus mice also bear a leaky gut and that the intestinal barrier function can be enhanced by gut colonization of probiotics such as Lactobacillus spp. Retinoic acid (RA) can increase the relative abundance of Lactobacillus spp. in the gut. Interestingly, RA has also been shown to strengthen the barrier function of epithelial cells in vitro and in the absence of probiotic bacteria. These reports bring up an interesting question of whether RA exerts protective effects on the intestinal barrier directly or through regulating the microbiota colonization.In this review, we will discuss the roles of RA in immunomodulation, recent literature on the involvement of a leaky gut in different autoimmune diseases, and how RA shapes the outcomes of these diseases.
Keywords: retinoic acid; leaky gut; autoimmune diseasesNutrients | Free Full-Text | Retinoic Acid, Leaky Gut, and Autoimmune Diseases | HTML
https://www.mdpi.com/2072-6643/10/8/1016/htmˇˇ
Retinoic Acid Signaling in the Functioning Brain | Science ...
https://stke.sciencemag.org/content/2006/324/pe10
Feb 28, 2006 ˇ¤ Retinoic acid, an active form of vitamin A, regulates gene expression throughout the body, and many components of the signaling system through which it acts are present in the brain. Very little is known, however, about how retinoic acid functions in neurobiological systems.
Cited by: 60
Publish Year: 2006
Author: Ursula C. Dräger
The rhythm of retinoids in the brain
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4283048
We review the role of vitamin A and retinoic acid (RA) as mediators of rhythm in the brain. In the suprachiasmatic nucleus and hippocampus they control expression of circadian clock genes while in the cortex retinoic acid is required for delta oscillations of sleep.
Cited by: 31
Publish Year: 2014
Author: Jemma Ransom, Peter J. Morgan, Peter J. McCaffery, Patrick N. Stoney
Diverse Functions of Retinoic Acid in Brain Vascular ...
https://www.jneurosci.org/content/36/29/7786
Jul 20, 2016 ˇ¤ Using mouse genetic and in vitro approaches, we identified retinoic acid (RA) as an important regulator of brain vascular development via non-cell-autonomous and cell-autonomous regulation of endothelial WNT signaling.
Cited by: 15
Publish Year: 2016ˇˇ
BMC Neurosci. 2018 Sep 21;19(1):58. doi: 10.1186/s12868-018-0460-x.
Anti-inflammatory effects of minocycline are mediated by retinoid signaling.
Clemens V1, Regen F2, Le Bret N2, Heuser I2, Hellmann-Regen J2.
Author information
Abstract
BACKGROUND:
Minocycline is a lipophilic tetracycline of increasing appeal in neuroscience as it inhibits microglial activation, a mechanism involved in numerous neuropsychiatric disorders. Own data point towards retinoid-mediated effects of minocycline in murine brain and skin, and towards a vicious cycle of neuroinflammation which is driven by microglial activation-induced breakdown of local retinoids such as retinoic acid (RA). We therefore sought to study minocycline's anti-inflammatory effects on human microglial-like monocyte-derived cells in the context of retinoid signaling.
RESULTS:
As hypothesized, minocycline exposure resulted in a substantial increase of RA levels in the human monocytic cell line THP-1. While pro-inflammatory stimulation with lipopolysaccharides resulted in increased tryptophane-degrading indoleamine-2,3-dioxygenase IDO-expression and TNF-¦Á levels in primary human monocyte-derived microglial-like cells, this effect was attenuated by minocycline only in the presence of retinoids. The anti-inflammatory effects of minocycline on TNF-¦Á expression were completely abolished by a pharmacological blockage of retinoic acid receptors (RARs) using BMS-493 and unaffected by selectively blocking retinoid-X-receptors using UVI-3003.
CONCLUSIONS:
Our data indicate for the first time a RA-dependent, anti-inflammatory effect for minocycline in human microglial-like cells via inhibition of local RA turnover. The RA-dependent mode of action for minocycline appears to be predominantly mediated through RAR-signaling.
KEYWORDS:
Cytokines; Microglia; Minocycline; Neuroinflammation; Retinoic acid
PMID: 30241502 PMCID: PMC6151010 DOI: 10.1186/s12868-018-0460-xAnti-inflammatory effects of minocycline are mediated by retinoid signaling. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/30241502ˇˇ
J Nutr Biochem. 2009 Sep;20(9):726-34. doi: 10.1016/j.jnutbio.2008.07.002. Epub 2008 Oct 16.
Retinoic acid dampens LPS-induced NF-kappaB activity: results from human monoblasts and in vivo imaging of NF-kappaB reporter mice.
Austenaa LM1, Carlsen H, Hollung K, Blomhoff HK, Blomhoff R.
Author information
Department of Nutrition Research, Institute of Basic Medical Sciences, University of Oslo, Norway.
Abstract
Bacterial lipopolysaccharide (LPS) is a major inducer of systemic inflammatory reactions and oxidative stress in response to microbial infections and may cause sepsis.In the present study, we demonstrate that retinoic acid inhibits LPS-induced activation in transgenic reporter mice and human monoblasts through inhibition of nuclear factor kappaB (NF-kappaB).
By using noninvasive molecular imaging of NF-kappaB luciferase reporter mice, we showed that administration of retinoic acid repressed LPS-induced whole-body luminescence, demonstrating in vivo the dynamics of retinoic acid's ability to repress physiologic response to LPS. Retinoic acid also inhibited LPS-induced NF-kappaB activity in the human myeloblastic cell line U937. Retinoic-acid-receptor-selective agonists mimicked - while specific antagonists inhibited - the effects of retinoic acid, suggesting the involvement of nuclear retinoic acid receptors.
Retinoic acid also repressed LPS-induced transcription of NF-kappaB target genes such as IL-6, MCP-1 and COX-2.
The effect of retinoic acid was dependent on new protein synthesis, was obstructed by a deacetylase inhibitor and was partly eliminated by a signal transducer and activator of transcription-1 (STAT1)/methyltransferase inhibitor, indicating that retinoic acid induces a new protein, possibly STAT1, that is involved in inhibiting NF-kappaB.
This provides more evidence for retinoic acid's anti-inflammatory potential, which may have clinical implications in terms of fighting microbial infections.
PMID: 18926686 DOI: 10.1016/j.jnutbio.2008.07.002
[Indexed for MEDLINE]Anti-inflammatory effects of minocycline are mediated by retinoid signaling. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/30241502ˇˇ
World J Biol Psychiatry. 2016 Dec;17(8):634-640. Epub 2015 Jun 5.
Inhibition of brain retinoic acid catabolism: a mechanism for minocycline's pleiotropic actions?
Regen F1, Le Bret N1, Hildebrand M1, Herzog I1, Heuser I1, Hellmann-Regen J1.
Author information
Abstract
OBJECTIVES:
Minocycline is a tetracycline antibiotic increasingly recognized in psychiatry for its pleiotropic anti-inflammatory and neuroprotective potential. While underlying mechanisms are still incompletely understood, several lines of evidence suggest a relevant functional overlap with retinoic acid (RA), a highly potent small molecule exhibiting a great variety of anti-inflammatory and neuroprotective properties in the adult central nervous system (CNS). RA homeostasis in the adult CNS is tightly controlled through local RA synthesis and cytochrome P450 (CYP450)-mediated inactivation of RA. Here, we hypothesized that minocycline may directly affect RA homeostasis in the CNS via altering local RA degradation.
METHODS:
We used in vitro RA metabolism assays with metabolically competent synaptosomal preparations from murine brain and human SH-SY5Y neuronal cells as well as viable human SH-SY5Y neuroblastoma cell cultures.
RESULTS:
We revealed that minocycline potently blocks RA degradation as measured by reversed-phase high-performance liquid chromatography and in a viable RA reporter cell line, even at low micromolar levels of minocycline.
CONCLUSIONS:
Our findings provide evidence for enhanced RA signalling to be involved in minocycline's pleiotropic mode of action in the CNS. This novel mode of action of minocycline may help in developing more specific and effective strategies in the treatment of neuroinflammatory or neurodegenerative disorders.
KEYWORDS:
cytochrome p450 metabolism; major depression; minocycline; neuroprotection; retinoic acid
PMID: 26047390 DOI: 10.3109/15622975.2015.1036116
[Indexed for MEDLINE]Inhibition of brain retinoic acid catabolism: a mechanism for minocycline's pleiotropic actions? - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/26047390ˇˇ
Published: 04 March 2012
NF-¦ĘB-mediated degradation of the coactivator RIP140 regulates inflammatory responses and contributes to endotoxin tolerance
Ping-Chih Ho, Yao-Chen Tsui, Xudong Feng, David R Greaves & Li-Na Wei
Nature Immunology volume 13, pages379¨C386(2012)Cite this article
Abstract
Tolerance to endotoxins that is triggered by prior exposure to Toll-like receptor (TLR) ligands provides a mechanism with which to dampen inflammatory cytokines. The receptor-interacting protein RIP140 interacts with the transcription factor NF-¦ĘB to regulate the expression of genes encoding proinflammatory cytokines.Here we found lipopolysaccharide stimulation of kinase Syk¨Cmediated tyrosine phosphorylation of RIP140 and interaction of the NF-¦ĘB subunit RelA with RIP140. These events resulted in more recruitment of the E3 ligase SCF to tyrosine-phosphorylated RIP140, which degraded RIP140 to inactivate genes encoding inflammatory cytokines.
Macrophages expressing nondegradable RIP140 were resistant to the establishment of endotoxin tolerance for specific 'tolerizable' genes. Our results identify RelA as an adaptor with which SCF fine tunes NF-¦ĘB target genes by targeting the coactivator RIP140 and show an unexpected role for RIP140 degradation in resolving inflammation and endotoxin tolerance.
Published: 16 January 2018
Glyburide and retinoic acid synergize to promote wound healing by anti-inflammation and RIP140 degradation
Yi-Wei Lin, Pu-Ste Liu, Kasey Ah Pook & Li-Na Wei
Scientific Reports volume 8, Article number: 834 (2018) Cite this article
Abstract
Chronic inflammation underlies the development of metabolic diseases and individuals with metabolic disease often also suffer from delayed wound healing due to prolonged inflammation. Resolving inflammation provides a therapeutic strategy in treating metabolic diseases. We previously showed that during an anti-inflammatory response when macrophages were alternatively (M2) polarized, retinoic acid (RA) dramatically activated arginase 1 gene (Arg1), a gene crucial for wound healing. Here we report that a widely used sulfonylurea drug for type 2 diabetes mellitus (T2DM), glyburide, enhances the anti-inflammatory response and synergizes with RA to promote wound healing. Our data also delineate the mechanism underlying glyburideˇŻs anti-inflammatory effect, which is to stimulate the degradation of a pro-inflammatory regulator, Receptor Interacting Protein 140 (RIP140), by activating Ca2+/calmodulin-dependent protein kinase II (CamKII) that triggers specific ubiquitination of RIP140 for degradation. By stimulating RIP140 degradation, glyburide enhances M2 polarization and anti-inflammation. Using a high-fat diet induced obesity mouse model to monitor wound healing effects, we provide a proof-of-concept for a therapeutic strategy that combining glyburide and RA can significantly improve wound healing. Mechanistically, this study uncovers a new mechanism of action of glyburide and a new pathway modulating RIP140 protein degradation that is mediated by CamKII signaling.
Introduction
Metabolic diseases such as Type II diabetes mellitus (T2DM) cast a huge burden on global health. These patients exhibit multiple symptoms such as hyperglycemia, insulin resistance, hypertension and dyslipidemia1, as well as debilitating, delayed wound healing2,3. One common underlying condition of metabolic diseases is systemic inflammation4; as such, resolving inflammation is an important goal in managing these symptoms5. This is especially critical to aid patients of metabolic diseases in wound recovery.
Retinoic acid (RA) exerts pleiotropic effects and is widely used to treat various health conditions particularly those involving the immune system6,7. In a clinical setting, retinoids have been used to treat cancers such as acute promyelocytic leukemia, although systemic toxicity has caused wide concerns8,9. Additionally, there is a long history of topical application of RA in dermatology, such as to enhance wound healing, treat acne, and reverse skin aging caused by UV damage. These topical applications have proven relatively safe10,11,12,13. Recently, we have found that in macrophage polarization, a process essential to innate immunity, RA treatment significantly elevates the expression of Arginase 1 (Arg1), a gene product critical to wound healing process. This happens particularly in the phase of alternative (M2) macrophage polarization, and therefore, it can potentially boost the wound healing process14.
The management of metabolic diseases especially T2DM and gestational diabetes mellitus includes drugs that stimulate insulin secretion, such as glyburide (also known as glibenclamide)15,16, a drug belonged to the sulfonylurea class. Glyburide can also inhibit sulfonylurea receptor 1-transient receptor potential melastatin 4 (Sur1-Trpm4) channels to protect patients from ischemic and hemorrhagic strokes17. It can also inhibit Cryopyrin/Nalp3 inflammasome pathway18. Additionally, our unpublished preliminary data suggest an anti-inflammatory potential for glyburide. Given that RA can boost Arg1 expression, and that glyburide is potentially anti-inflammatory, we propose that combining RA and glyburide may be synergistically beneficial to the management of wounds, especially for patients suffering from, or in the process of developing, metabolic diseases. The study is to test this therapeutic strategy for wound healing in a high fat diet (HFD)-induced obesity mouse model that mimics the condition of chronic inflammation.
Our data show that glyburide and RA indeed synergize to facilitate wound healing in these animals. We also uncover a new mechanism of action of glyburide, which is by stimulating protein degradation of a key inflammatory coregulator named nuclear receptor coregulator RIP140 (Nrip1). RIP140 is a wide spectrum transcription co-regulator19,20. For innate immune cells especially macrophages, RIP140 is pro-inflammatory because it is a cofactor of NF-kB, facilitating M1 polarization21, and an inhibitor of STAT6, suppressing M2 polarization22. Therefore, silencing RIP140 expression or reducing its protein levels in macrophages generally leads to M2 polarization (anti-inflammation). Since RIP140 gene (Nrip1) expression is maintained largely constant in macrophages23,24, its protein level is primarily regulated by post-transcriptional control. We have previously determined that RIP140 can be degraded by Syk-mediated tyrosine phosphorylation on Tyr364, Tyr418 and Tyr436 in the pathological context of LPS-induced inflammation, which prevents septic shock21.
While the ability to control RIP140 protein level is highly desirable, especially for treating diseases related to, or caused by, inflammation, this strategy has remained a challenge due to the lack of therapeutic agents that could trigger effective and specific protein degradation of RIP140. The current study aims to identify potential therapeutic agents that are safe and can modulate RIP140 protein levels in order to promote resolution of a chronic inflammation-related condition. This effort has identified glyburide as a potential therapeutic agent. Given RAˇŻs effect in elevating Arg114, which is beneficial to wound healing, combining glyburide and RA would be predicted to synergistically facilitate the healing process of chronic wounds associated with inflammation. By examining signaling pathways, we also determine the mechanism of glyburideˇŻs action in regulating RIP140ˇäs protein level, which is mediated by CamKII signaling. This also represents a new regulatory pathway for protein quality control of RIP140.
Results
Glyburide improves wound healing in HFD-induced obese mice
We first determined the effects of glyburide in healing the wounds created in HFD-induced obesity mice. The results show that topical treatment of these mice with glyburide significantly improved their wound healing as compared to the control (Fig. 1A). We then examined if glyburide could alter RIP140 protein levels in the macrophage populations of wounded tissues. Indeed, glyburide treatment down-regulated RIP140 protein levels in macrophages collected from these wounds (Figs 1B and S1A, S1B). Further, glyburide treatment elevated their M2 (anti-inflammatory) markers and reduced their M1 (inflammatory) markers (Fig. 1C), confirming that glyburide is anti-inflammatory and can reduce RIP140 protein level in macrophages.
Figure 1
figure1
Glyburide improves wound healing in HFD-induced obese mice. (A) Daily record of wound closure in mice fed with ND, HFD, and HFD and treated with a control solvent DMSO (ND and HFD-Ctrl) or glyburide (HFD-glyburide). Data were presented as mean ˇŔ SD. A two-way ANOVA test was used *p < 0.05, **p < 0.01; ***p < 0.001. (ND vs. HFD-Ctrl); †p < 0.05, ††p < 0.01, †††p < 0.001 (HFD-Ctrl vs. HFD-glyburide) (N = 6 in each group), Ctrl = Control. (B) Immunofluorescence images of RIP140 in primary mouse macrophages isolated from wound. (C) qPCR analyses of M1 and M2 markers detected in wound tissues from HFD-Ctrl or HFD-glyburide animals. Student test was used and data were presented as means ˇŔ SD. ***p < 0.001 (N = 6 in each group).
Full size image
Combining glyburide and RA enhances wound healing
Our recent studies demonstrated that RA treatment in macrophage cultures or mice enhanced Arg1 gene expression, which also improved wound healing14. Given anti-inflammatory effects of glyburide and Arg1-elevating activity of RA, we suspected that combining RA and glyburide might be able to further improve wound healing in a condition of chronic inflammation such as a HFD-induced obese condition. To test this possibility, we employed the same cutaneous wound model in three cohorts of obese mice all treated with HFD for 8-weeks. These include a control group (treated with a control cream), a group treated with daily topical glyburide application, and a group treated with topical glyburide for 2 days followed by the combination of glyburide and 0.1% tretinoin cream (RA) daily. Closure of the wounds was then monitored. As shown in Fig. 2A, the group treated with the combination of glyburide and RA significantly improved their wound healing as compared to the control and the group treated with glyburide alone (Fig. 2A). Consistently, gene expression analysis showed that combining RA and glyburide boosted anti-inflammation, indicated by an increase in the M2 marker and a decrease in the M1 marker in the wound tissues (Fig. 2B).
figure2
Co-treatment with glyburide and RA improves wound healing as compared to treatment of glyburide alone. (A) Left: Daily record of wound closure in HFD-mice treated with ctrl, glyburide, or glyburide/RA co-treatment. Data were presented as mean ˇŔ SD. A two-way ANOVA test was used *p < 0.05 (Ctrl vs. glyburide); †p < 0.05, ††p < 0.01, †††p < 0.001 (Ctrl vs. glyburide + RA); #p < 0.05, ##p < 0.01 (glyburide vs. glyburide + RA) (N = 6 in each group), Ctrl = Control. Right: Representative cutaneous wound on day 1, 3 and 7 after wound creation. (B) qPCR analyses of M1 and M2 markers in wound tissues was presented as mean ˇŔ SD. A two-way ANOVA test was used *p < 0.05, ***p < 0.001 (Ctrl vs. glyburide); †p < 0.05, †††p < 0.001 (Ctrl vs. glyburide + RA); #p < 0.05 (glyburide vs. glyburide + RA) (N = 6 in each group).
Full size image
Glyburide stimulates RIP140 protein degradation in macrophages
As shown in Fig. 1B, glyburide reduced RIP140 protein levels in macrophages collected from wound tissues. To determine the mechanism of glyburideˇŻs action specifically in macrophages, we employed both primary mouse peritoneal macrophage (PM) and a mouse macrophage cell line Raw 264.7 for the studies. In PM cultures, glyburide reduced RIP140 protein levels in a dose- and time-dependent manner (Fig. 3A left panel); whereas RIP140 mRNA levels remained relatively constant (Fig. 3A right panel). This led us to suspect that glyburide could reduce RIP140 protein levels by triggering its degradation. We previously identified that, in the patho-physiological context of LPS-induced inflammation, RIP140 was degraded by Syk-stimulated Tyr phosphorylation on Tyr364, Tyr418, and Tyr436 that stimulated its ubiquitination and degradation21. We then determined whether glyburide-stimulated RIP140 protein down regulation was mediated by Syk-stimulated degradation. As shown in Fig. 3B, the effect of glyburide was not related to Syk-stimulated degradation because a Syk inhibitor failed to block the effect of glyburide. The mechanism of action of glyburide in pancreatic beta cells has been attributed to, primarily, its activity in inhibiting ATP-sensitive potassium channel (KATP)25. However, a potassium channel opener, pinacidil, also failed to effectively prevent glyburide-induced RIP140 degradation (Fig. 3C), ruling out the effects through altering KATP. Interestingly, a proteasome inhibitor MG132 could effectively prevent glyburideˇŻs effect in down-regulating RIP140 protein level (Fig. 3D), suggesting that glyburide triggered RIP140 degradation via a proteasome-mediated degradation pathway that is different from Syk-stimulated Try phosphorylation on RIP140. As predicted, glyburide-treated macrophages were more prone to IL-4 stimulated M2 activation (for anti-inflammatory response) (Fig. 3E), because RIP140 level was reduced.
figure3
Glyburide stimulates RIP140 protein degradation in macrophage. (A) Western blot (left) and qPCR (right) analyses of RIP140 in mouse PMs treated with glyburide at three doses, and at three time intervals using 15 uM of glyburide. (B) Western blot of RIP140. The SYK inhibitor failed to block the effect of glyburide in Raw 264.7 cells. (C) Western blot of RIP140, showing the rescue of glyburide stimulated RIP140 degradation with pinacidil treatment in Raw 264.7 cells. (D) Western blot of RIP140, showing the rescue of glyburide stimulated RIP140 degradation with MG132 treatment in Raw 264.7 cells. (E) qPCR analyses of Arg-1 in Raw 264.7 cells treated with glyburide. Student test was used. All experiments were performed three times and presented as mean ˇŔ SD; ***P < 0.001.
Full size image
Glyburide stimulates RIP140 degradation by activating CamKII that phosphorylates RIP140
Glyburide is also known to elevate intracellular calcium concentration, we thus examined whether altering intracellular calcium concentration and/or calcium signaling in macrophage could affect its endogenous RIP140 protein level. The data (Fig. 4A) show that BayK8644, a calcium channel activator, induced RIP140 degradation, and a pan Ca2+/calmodulin-dependent protein kinase II (CamKII) inhibitor, KN-93, effectively blocked glyburide-induced RIP140 degradation. Further, glyburide indeed activated CamKII in this experimental system (Fig. 4B). In an in vitro CamKII assay, we found that CamKII could directly phosphorylate RIP140 (Fig. 4C). Based on the consensus sequence RXXS/TX of CamKII targets26, we predicted four possible CamKII target sites on RIP140 (Fig. 4C). We then generated a quadruple RIP140 mutant (S460A, S519A, S672A, S1141A, Fig. 4D) to eliminate CamKII substrate sites. This quadruple RIP140 mutant indeed was resistant to glyburide-induced degradation, confirming our prediction (Fig. 4E).
figure4
Glyburide stimulates RIP140 degradation through activating CamKII signaling pathway. (A) Western blot of RIP140, showing the effects of CamKII inhibitor and calcium channel activator in Raw 264.7 cells. (B) Western blot of phospho-CamKII demonstrating activation of CamKII in Raw 264.7 cells by glyburide treatment. (C) In vitro CamKII kinase assay showing CamKII-phosphorylation at serine residues of RIP140. (D) Predicted CamKII target sites on mouse RIP140. (E) Western blot of RIP140 showing glyburide-stimulated degradation of the wild type but not the quadruple mutant RIP140 in Raw 264.7 cells.
Full size image
Taken together, these data show that glyburide is anti-inflammatory because it activates CamKII, which modifies RIP140 protein for proteosome-mediated degradation. Further, combining glyburide and RA would boost would healing because this strategy elevates Arg1 level in an enhanced anti-inflammatory condition. A proof-of-concept for this strategy is provided in our results showing more effective management of the wounds created in the HFD-induced obese condition.
Discussion
This study provides a proof-of-concept for a new therapeutic regime in managing inflammation-associated chronic wounds, which is by combining two widely applied therapeutic agents, glyburide and RA. Glyburide promotes anti-inflammation, which sensitizes the system allowing more effective action of RA to elevate Arg1 level that is critical to wound healing. Both are widely applied therapeutics - glyburide is a relatively safe medication even for a long term use and tretinoin creams (RA) is safe when applied on the skin; therefore there should be little concern over toxicity when they are applied to manage topical wounds. The feasibility of developing this regime into an economical therapeutic to improve wound healing is quite high.
Calcium ion (Ca2+) plays a critical role in numerous physiological processes particularly in neuronal signal transmission, muscle contraction, and fertilization, etc.27,28,29. For the immune system, calcium signal contributes to the activation and differentiation of various immune cells including macrophages, monocytes, T cells, B cells, NK cells and dendritric cells30,31, etc. Study of Drosophila suggests that calcium acts as the earliest inflammatory signal to attract immune cell migration into the wound32. It is interesting that our current study shows that Ca2+ can also stimulate RIP140 protein degradation in macrophages, thereby contributing to anti-inflammation. RIP140 is an important regulator of inflammation; controlling its protein level is vital to the maintenance of immune homeostasis. The current study is the first to demonstrate protein quality control of RIP140 via calcium signaling.
Glyburide, as an anti-diabetic drug, is best known to stimulate insulin secretion in pancreatic beta cells. It has also been shown to be anti-inflammatory18,33,34, but the underlying mechanism was not clear. In the present study, we are able to delineate the mechanism of glyburideˇŻs action in anti-inflammation, which is to facilitate RIP140 protein degradation through activating CamKII. This subsequently triggers posttranslational modification of RIP140 on Ser460, Ser519, Ser672, and Ser1141 and elicits its proteasome-mediated protein degradation. Interestingly, this degradation pathway is different from the degradation pathway triggered by LPS-induced inflammation where RIP140 is tyrosine phosphorylated on Tyr364, Tyr418 and Tyr436, which also leads to proteasome-mediated degradation. Conceivably, the patho-physiological context is crucial for RIP140 protein quality control, and it involves distinct signaling pathways in various physiological or pathological conditions to differentially modify RIP140. But all these lead to proteasome-mediated RIP140 protein degradation. To this end, it remains to be determined as to the physiological context where RIP140 may be degraded through endogenously activated CamKII.
RA, one of the active metabolites of retinoids, is important for a wide spectrum of biological processes and functions35. For innate immunity, RA can decrease pro-inflammatory cytokines production and increase anti-inflammatory cytokines secretion7. This is supported by epidemiological studies which demonstrate that RA deficiency alters immune responses to vaccines, infectious agents and auto-antigens, etc.36. In a clinical setting, all-trans-RA (tretinoin), 13-cis-RA (isotretinoin) and 9-cis-RA have all been shown to promote wound healing37 and applied in treating skin conditions. We have recently shown that RA enhances Arg1 gene activation in IL4-stimulated M2, anti-inflammatory macrophages to facilitate wound healing14. The current study exploits this recent observation to develop a novel therapeutic regime for severe wounds such as those associated with chronic/systemic inflammation (using a HFD-induced obese mouse model). This regime should be safe for managing topical wounds; but its safety in systemic application for managing internal wounds remains to be further investigated.
RIP140 is a wide spectrum transcription co-regulator important for various biological processes19,20. Increasing evidences have revealed RIP140ˇäs critical roles in regulating innate immunity. While numerous studies have all suggested that targeting RIP140 in macrophage can be a therapeutic strategy for inflammation-related diseases, as demonstrated using experimental systems like macrophage-specific knockdown21,38, bone marrow transplantation39, local injection of therapeutic macrophages40, and fecal microbiome transplantation41, a major challenge has to do with the lack of safe reagents/compounds that can be applied exogenously as a drug to stimulate RIP140 degradation. This current study is the first to identify such a pharmacological candidate that can facilitate RIP140 degradation to improve anti-inflammation. This information will be very helpful in future studies to screen for compounds that can modulate protein quality of RIP140 and the innate immune status. However, to provide more genetic evidence for this mechanism in the future, it is desirable to use genetically manipulated mouse models, such as mice carrying CamKII-resistant RIP140 mutation.
Methods
Reagents
Anti-RIP140 (Ab-42126) antibody was obtained from Abcam. Anti-b-actin, anti-CamKII, anti-phospho-CamKII, anti-mouse-IgG-HRP and anti-rabbit-IgG-HRP antibodies were purchased from Santa Cruz. Anti-phospho-Ser/Thr (9631 S) antibody was obtained from Cell Signaling. Anti-flag antibody, BayK 8644 (B112), glyburide (G2539) was from Sigma-Aldrich.
Animals
All studies were performed using male C57Bl/6 mice purchased from The Jackson Laboratory. All experiments were approved by and in accordance with the guidelines and regulations of the University of Minnesota Institutional Animal Care and Use Committee. Animals were maintained in the animal facility of University of Minnesota on a 12 h light/dark photocycle. Mice were fed a normal diet (ND) (2018; Harlan Teklad, Madison, WI) or a high-fat diet (HFD) with 60% calories from fat (F3282; Bio-Serv, West Chester, PA).
Cutaneous wound healing assay
Cutaneous wound healing assay was carried out as described22. 5-mm round-shape cutaneous wounds were made on shaved mice back using biopsy punch under anesthesia, to create 2 wounds per animal (n = 6). The reagents (glyburide, control cream and 0.1% tretinoin cream) were applied topically on wounds and wound size was recorded daily and analyzed by Image J.
RNA Isolation and Gene Expression Analyses
Total RNA was isolated using TRIzol (Invitrogen) followed by manufacturerˇŻs instruction. Reverse transcription and quantitative real-time PCR (qPCR) was performed as described previously using High-Capacity cDNA Reverse Transcription Kit containing RNase Inhibitor (Applied Biosystems) and Maxima SYBR Green qPCR Master Mixes (Thermo Scientific). Each gene-expression experiment was performed in triplicate and normalized to ¦Â-actin. Primers for Tnf¦Á (QT00104006), IL1¦Â (QT01048355), iNOS (QT00100275), IL10 (QT00106169), Arg1 (QT00134288) and TGF¦Â (QT00145250) were purchased from Qiagen.
Flow Cytometry
Wound tissues were digested with 0.1% of collagenase and 0.01% of DNase I to disperse cells. Cell-surface antigens were blocked using Block (20 m g/mL; BD Biosciences). After blocking, cells were stained with antibodies or isotype control antibodies. Fluorophore-conjugated primary antibodies were purchased from BioLegend: F4/80-Alexa Fluor 647 (cat# 123122), and BD Bioscience: PE F(abˇŻ)2 Donkey anti-Rabbit IgG (cat# 558416). Cells were analyzed on a BD Acuri C6 using FlowJo 10.0.6.
In vitro CamKII assay
Flag-RIP140 and flag-CamKII fusion protein were made by TnT® Quick Coupled Transcription/Translation System (Promega) followed by manufactureˇŻs instruction. Flag-RIP140 protein was incubated alone or with flag-CaMKII protein in 30 ¦Ěl of in vitro kinase incubation buffer (25 mM HEPES/KOH, pH 7.4, 10 mM MgCl2, 1 mM CaCl2, 0.6 ¦ĚM calmodulin, 6 ¦ĚM ATP) at 30 ˇăC for 10 minutes. The reaction was immunoprecipitated with anti-flag antibody and determined by western blotting using anti-phospho-Ser/Thr antibody.
Cell culture
Primary peritoneal macrophages were isolated and maintained as described previously39. Raw 264.7 cells were maintained in DMEM medium as describe. Transfection was conducted using GenePorter 3000 (Genlatis) according to the manufacturerˇŻs instructions.
Plasmid Constructs
Point mutations involving residues Ser-460, Ser-520, Ser-672, and Ser1141 in mouse wild-type RIP140 (CMV/T7/FLAG) vector as template were made according to Q5 Site-Directed Mutagenesis Kit (NEB) according to the manufacturerˇŻs instructions.
Statistical Analysis
Experiments were carried out at least twice and presented as means ˇŔ SD. Unpaired two-tailed StudentˇŻs t test or two-way ANOVA was used for comparison between two groups. p values ˇÜ 0.05 were considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001).Glyburide and retinoic acid synergize to promote wound healing by anti-inflammation and RIP140 degradation | Scientific Reports
https://www.nature.com/articles/s41598-017-18785-xˇˇ
J Mol Cell Cardiol. Author manuscript; available in PMC 2016 Feb 1.
RIP140 contributes to foam cell formation and atherosclerosis by regulating cholesterol homeostasis in macrophages
Yi-Wei Lin,1 Pu-Ste Liu,1 Neeta Adhikari,2 Jennifer L. Hall,2 and Li-Na Wei1,2,*
Author information Copyright and License information Disclaimer
1Department of Pharmacology, University of Minnesota Medical School, Minneapolis, Minnesota, USA
2Lillehei Heart Institute, University of Minnesota, Minneapolis, MN 55455
Abstract
Atherosclerosis, a syndrome with abnormal arterial walls, is one of the major causes that lead to the development of various cardiovascular diseases. The key initiator of atherosclerosis is cholesterol accumulation. The uncontrolled cholesterol deposition, mainly involving low-density lipoprotein (LDL), causes atheroma plaque formation, which initiates chronic inflammation due to the recruitment of inflammatory cells such as macrophages. Macrophages scavenge excess peripheral cholesterol and transport intracellular cholesterol to high-density lipoprotein (HDL) for excretion or storage. Cholesterol-laden macrophage-derived foam cell formation is the main cause of atherogenesis. It is critical to understand the regulatory mechanism of cholesterol homeostasis in the macrophage in order to prevent foam cells formation and further develop novel therapeutic strategies against atherosclerosis. Here we identified a protein, RIP140 (receptor interacting protein 140), which enhances macrophage-derived foam cell formation by reducing expression of reverse cholesterol transport genes, A TP-binding membrane cassette transporter A-1 (ABCA1) and ATP-binding membrane cassette transporter G-1 (ABCG1). In animal models, we found that reducing RIP140 levels by crossing macrophage-specific RIP140 knockdown (MϕRIP140KD) mice with ApoE null mice effectively ameliorates high-cholesterol diet-induced atherosclerosis. Our data suggest that reducing RIP140 levels in macrophages significantly inhibits atherosclerosis, along with markers of inflammation and the number of macrophages in a western diet fed ApoE null mouse. This study provides a proof-of-concept for RIP140 as a risk biomarker of, and a therapeutic target for, atherosclerosis.
Keywords: RIP140, atherosclerosis, foam cell, reverse cholesterol transport
ˇˇ1. INTRODUCTION
Hallmarks of atherosclerosis are abnormal cholesterol metabolism and inflammation [1]. Macrophages are critically involved in cholesterol metabolism and inflammation in the progression of atherosclerosis [2]. Macrophages scavenge excess peripheral cholesterol by uptake of LDL, and transport intracellular cholesterol to high-density lipoprotein (HDL), which can be stored or excreted by the liver through reverse cholesterol transport (RCT) [3]. However, when cholesterol levels are pathologically elevated, cholesterol-laden macrophages become inflammatory and turn to active foam cells [2, 4]. Macrophage-derived foam cell formation marks the initiation of atherosclerosis. Cholesterol retention in the macrophage promotes foam cell formation. In macrophages, the ATP-binding membrane cassette transporter A-1 (ABCA1) and ATP-binding membrane cassette transporter G-1 (ABCG1) are the major transporters mediating RCT: ABCA1 regulates RCT to apolipoprotein A-I, and ABCG1 regulates RCT to mature HDL [5].
Receptor Interacting Protein 140 (RIP140) is a protein found in metabolic tissues, such as liver, muscle and adipose tissue [6, 7]. As a versatile co-regulator of various transcription factors, RIP140 regulates metabolism, such as fat accumulation in adipocytes, by affecting the expression of metabolic genes [8¨C11]. RIP140 also exerts various regulatory functions through its extensive post-translational modifications, including various forms of phosphorylation, lysine-acetylation, lysine methylation, arginine methylation, vitamin B6 conjugation, and ubiquitination, etc [12¨C16]. RIP140 is also known to be expressed in the monocyte-macrophage lineage and can regulate inflammatory responses [17¨C19]. Our recent study indicated that accumulation of intracellular cholesterol in the macrophage elevated RIP140 and that RIP140 expression was sufficient to enhance inflammatory cytokine production and the inflammatory potential of the macrophage [20].
In this study, we provide novel data showing that RIP140 promotes foam cell formation by reducing cholesterol efflux. This process is mediated through the repression of ABCA1 and ABCG1. Further, cholesterol loading stimulates RIP140ˇŻs post-translational modification that enhances its repressive activity. In vitro, augmenting RIP140 levels in the macrophage affects the efficiency of foam cell formation. Our preliminary study indicated that, fed a normal chow, mice with reduced RIP140 expression in macrophages, such as by macrophage specific RIP140 knockdown (mϕRIP140KD), exhibited no particular phenotypic changes. However, in ApoE null mice, lowering RIP140 levels specifically in the macrophage reduced the severity of western diet-induced atherosclerosisˇˇ
Taken together, these data support that reducing RIP140 levels in the macrophage can significantly inhibit the progression of atherosclerosis.
4. Discussion
Cholesterol imbalance, especially in the macrophage, is a major culprit triggering atherosclerosis. Macrophages can engulf excess cholesterol via specific receptors and also exert reverse cholesterol transport to expel cholesterol[28, 29]. It has been suggested that reducing cholesterol retention in the macrophage can ameliorate atherosclerosis. The current study provides a proof of concept in animal models of atherosclerosis and identifies a new target, RIP140, for augmenting this disease. The study also determines the mechanism that RIP140 represses the expression of ABCA1 and ABCG1 to reduce RCT and that cholesterol can alter RIP140ˇŻs post-translational modification that further escalates its RCT-repressing effects. This current study also shows that reduction in macrophageˇŻs RIP140 level lowers the expression levels of pro-inflammatory cytokines. This is in line with our previous report that RIP140 activates pro-inflammatory cytokine production by serving as a coactivator of NF-¦ĘB [20]. Thus, lowering the level of RIP140 in the macrophage can reduce macrophage-derived foam cell formation and contribute to reduced inflammation, ultimately this helps to ameliorate atherosclerosis and dampen monocyte/macrophage accumulation in the plaque. Given that RIP140 affects steroid receptor inhibition, it would be interesting to see whether macrophage specific reduction in RIP140 level would similarly reduce foam cell formation and inflammation in female animals.
Studies have indicated that LDL triggers p42/44 MAPK (ERK1/2) activation[30, 31]. We previously reported that activated ERK1/2 stimulates RIP140 phosphorylation[12]. This current study shows that oxLDL activates ERK1/2, which can stimulate RIP140ˇŻs phosphorylation and subsequently induce its lysine-acetylation to enhance its transcriptional co-repressive activity, which is in line with previous observations, and validates that fat contents in the cell can affect RIP140ˇŻs biological activity. In the macrophage, RIP140 functions to repress the expression of ABCA1 and ABCG1 by co-repressing LXR-target genes such as ABCA1 and ABCG1. Thus, lowering RIP140 levels or altering RIP140 modification, such as blocking its phosphorylation or lysine-acetylation, can reduce its repressive effects on ABCA1 and ABCG1, and can be beneficial. In vivo animal study provides strong evidence that reducing RIP140 levels in the macrophage could be a therapeutic strategy for atherosclerosis.
We previously reported that MiR-33 targets RIP140ˇŻs 3ˇäUTR to repress its expression[18]. Other studies showed that MiR-33 also reduces the expression of ABCA1 and ABCG1[32, 33]. It has also been shown that Mir-33 deficiency raised HDL to increase cholesterol efflux[34]. Thus, while MiR-33 can effectively reduce RIP140 levels, it may not be a therapeutically viable strategy in the case of atherosclerosis because of its broad effects. Currently, there is no specific compound that can directly repress RIP140 levels or augment its post-translational modifications. Further worthy studies are needed such as to screen for compounds that can specifically target RIP140 to reduce its expression level or to augment its post-translational modifications. Furthermore, RIP140 can also serve as a disease maker, such as in assessing the risk or progression of atherosclerosis.
Highlights
RIP140 promotes macrophage-derived foam cell formation.
RIP140 suppresses LXR-regulated expression of ABCA1 and ABCG1
Lowering RIP140 level in macrophage ameliorates atherosclerosis.RIP140 contributes to foam cell formation and atherosclerosis by regulating cholesterol homeostasis in macrophages
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4302032/ˇˇ
PSYCHIATRY
Antibiotics Found Effective in Schizophrenia
Tetracyclines help treat psychosis as well as tick-borne disorders.A controlled clinical trial was just published in the psychiatric literature, showing that minocycline is effective in treating negative symptoms in early phase schizophrenia. A prior pilot study, published in 2010 in the Journal of Clinical Psychiatry, also showed that minocycline was effective in schizophrenia, helping executive functioning such as working memory. The authors postulate that the mechanism of action of minocycline would include affecting glutamate pathways in the central nervous system, blocking nitric oxide-induced neurotoxicity, or inhibiting microglial activation in the brain, causing inflammation. All of these are reasonable potential mechanisms of action. Neither author discusses the obvious fact however that minocycline is a tetracycline antibiotic and that it may be treating an occult infection. Have infections ever been reported to cause schizophrenia?
Lyme disease causes a wide range of psychiatric manifestations. Published research has shown a higher prevalence of antibodies to Borrelia burgdorferi in psychiatric patients than in healthy subjects. There is also a known geographic correlation of schizophrenia with ticks and tick-borne encephalitis, with peer reviewed literature showing an association of Lyme disease with schizophrenia. Other tick-borne infections, such as Bartonella (cat scratch disease) have also been reported to cause neurological and neurocognitive dysfunction, as well as causing agitation, panic disorder and treatment resistant depression. Minocycline, as well as other tetracycline antibiotics like doxycycline, are well known treatments for neurological manifestations of Lyme disease and associated co-infections like Bartonella. It is therefore plausible that a certain number of cases of severe psychiatric presentations are due to underlying infections, especially since Lyme disease is the number one spreading vector borne infection in the world.
I have seen several patients who came to my medical clinic with a diagnosis of schizophrenia, on anti-psychotic medications like Risperdal. Upon further testing, their Western Blots returned positive for exposure to Borrelia burgdorferi, the agent of Lyme disease. They were given doxycycline (a similar tetracycline antibiotic), and their psychotic symptoms and cognition improved significantly. Working with their psychiatrist, we were able to reduce, and in some cases eliminate, all of their antipsychotic medication. They remained clinically stable as long as they remained on antibiotics. Their psychiatric symptoms returned once they were no longer being treated for Lyme and associated tick-borne disorders, as these organisms have been shown to be able to establish a persistent infection in the body.
When should we suspect Lyme disease as a potential etiological co-factor in psychiatric symptoms? Lyme disease is a multisystemic illness. If a patient presents with a symptom complex that comes and goes with good and bad days, with associated fevers, sweats and chills, fatigue, migratory joint and muscle pain, migratory neuralgias with tingling, numbness and burning sensations, a stiff neck and headache, memory and concentration problems, a sleep disorder and associated psychiatric symptoms (that may or may not be of recent onset), then we should suspect Lyme disease and associated co-infections. Have these patients fill out a Lyme screening questionnaire that we developed in my medical office. Among 100 patients who filled out this form, a score above 46 was associated with a high probability of a tick-borne disorder. In that case, blood testing should be performed through a reliable laboratory to look for Lyme and co-infections, including Babesia and Bartonella, which can also significantly increase underlying symptomatology.
Lyme disease is a major cause of psychiatric symptoms. Psychiatric case reports, as reported by psychiatrist Dr Brian Fallon, have linked Lyme disease to paranoia, thought disorders, delusions with psychosis, schizophrenia, with or without visual, auditory or olfactory hallucinations, depression, panic attacks and anxiety, obsessive compulsive disorder, anorexia, mood lability with violent outbursts, mania, personality changes, catatonia and dementia. Other psychiatric disorders in adults due to Lyme disease include atypical bipolar disorder, depersonalization/derealization, conversion disorders, somatization disorders, atypical psychoses, schizoaffective disorder and intermittent explosive disorders. In children and adolescents, Lyme disease can also mimic Specific or Pervasive Developmental Delays, Attention-Deficit Disorder (Inattentive subtype), oppositional defiant disorder and mood disorders, obsessive compulsive disorder (OCD), anorexia, TouretteˇŻs syndrome, and pseudo-psychotic disorders. The take home message: Lyme is the ˇ°great imitatorˇ±. DonˇŻt exclude Lyme disease and associated infections as a possible underlying cause of psychiatric symptoms, and donˇŻt assume that a positive response to an antibiotic like minocycline is not treating an underlying infection.
article continues after advertisement
Dr Richard Horowitz
http://www.amazon.com/Why-Cant-Get-Better-Solving/dp/1250019400
www.cangetbetter.com
www.facebook.com/DrRichardHorowitz
Scientific references:
Liu, F., Guo, X., Wu, R., et al. (2014). Minocycline supplementation for treatment of negative symptoms in early-phase schizophrenia: A double blind, randomized, controlled trial. Schizophr Res. Published on line: http://www.schres-journal.com/article/S0920-9964%2814%2900014-0/fulltext
Levkovita, Y., Mendlovich, S., Riwkes, S., et al. (2010). A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J. Clin Psychiatry 71(2):138-49.
Fallon, B.A., & Nields, J.A. (1994). Lyme disease: A neuropsychiatric illness. Am J Psychiatry 151(11):1571-83.
Fallon, B.A., Kochevar, J.M., Gaito, A., & Nields, J.A. (1998). The underdiagnosis of neuropsychiatric Lyme disease in children and adults. Psychiatric Clinics of North America 21: 693-703.
Hajek, T., Paskova, B., Janovska, D., et al. (2002). Higher prevalence of antibodies to Borrelia burgdorferi in psychiatric patients than in healthy subjects.
Am J Psychiatry 159:297-301.
Hess, A., Buchmann, J., Zettl, U.K., Henschel, S., Schlaefke, D., Grau, G., & Benecke, R. (1999). Borrelia burgdorferi central nervous system infection presenting as an organic schizophrenialike disorder. Biol Psychiatry 45(6):795.
Brown, J.S. Jr. (1994). Geographic correlation of schizophrenia to ticks and tick-borne encephalitis. Schizophr Bull; 20(4):755-75
article continues after advertisement
Breitschwerdt, E.B., Maggi, R.G., Nicholson, W.L., Cherry, N.A., & Woods, C.W. Bartonella sp. bacteremia in patients with neurological and neurocognitive dysfunction. Journal of Clinical Microbiology. 46(9):2856¨C2861.
Schaller, J.L., Burkland, G.A., & Langhoff, P.J. (2007). Do bartonella infections cause agitation, panic disorder, and treatment-resistant depression? MedGenMed. 9(3):54.
Fallon, B.A., Levin, E.S., Schweitzer, P.J., & Hardesty, D. (2010). Inflammation and central nervous system Lyme disease. Neurobiology of Disease 37: 534-541
Sherr, V.T. (2000). Panic attacks may reveal previously unsuspected chronic disseminated Lyme disease. J. Psychiatric Practice, 6:352-356.
Benke, T., Gasse, T., Hittmair-Delazer, M., & Schmutzhard, E. (1995). Lyme encephalopathy: Long-term neuropsychological deficits years after acute neuroborreliosis. Acta Neurol Scand. 91(5):353-7;
Nicolson G., & Haier, J. (2009). Role of chronic bacterial and viral infections in neurodegenerative, neurobehavioral, psychiatric, autoimmune and fatiguing illnesses: Part I. BJMP 2(4) 20-28. http://www.bjmp.org/content/role-chronic-bacterial-and-viral-infections-neurodegenerative-neurobehavioral-psychiatric-auŔłÄ·˛ˇ Ëř¶¨
±ľ´ĘĚőÓÉąúĽŇÎŔ˝ˇÎŻČ¨ÍţҽѧżĆĆŐĎîÄż´«˛ĄÍřÂçƽ̨/°ŮżĆĂűŇ˝Íř Ěáą©ÄÚČÝ ˇŁ
ŔłÄ·˛ˇĘÇŇ»ÖÖŇÔňçÎŞĂ˝˝éµÄÂÝĐýĚĺ¸ĐČľĐÔĽ˛˛ˇŁ¬ĘÇÓɲ®ĘĎĘčÂÝĐýĚĺËůÖµÄ×ÔČ»ŇßÔ´ĐÔĽ˛˛ˇˇŁÎŇąúÓÚ1985ÄęĘ×´ÎÔÚşÚÁú˝ĘˇÁÖÇř·˘ĎÖ±ľ˛ˇ˛ˇŔýŁ¬ŇÔÉńľĎµÍłËđş¦ÎŞ¸Ă˛ˇ×îÖ÷ŇŞµÄÁŮ´˛±íĎÖˇŁĆäÉńľĎµÍłËđş¦ŇÔÄÔĤŃס˘ÄÔŃס˘ÂÉńľŃס˘Ô˶ŻşÍ¸ĐľőÉńľŃ××îÎŞłŁĽűˇŁĆäÖĐŇ»ĆÚŔłÄ·˛ˇ˝öÓĂżąÉúËŘĽ´żÉ×ŕЧŁ¬ÖÁ¶ţĆÚˇ˘ČýĆÚÓĂżąÉúËŘÎŢĽĂÓÚĘÂŁ¬ĚرđĘÇÉńľĎµÍłË𺦸ü·¦ĚŘЧÁĆ·¨ˇŁÔçĆÚŇÔƤ·ôÂýĐÔÓÎ×ßĐÔşě°ßÎŞĚص㣬ŇÔşółöĎÖÉńľˇ˘ĐÄÔŕ»ňąŘ˝Ú˛ˇ±äŁ¬Í¨łŁÔÚĎÄĽľşÍÔçÇď·˘˛ˇŁ¬żÉ·˘ÉúÓÚČÎşÎÄęÁ䣬ÄĐĐÔÂÔ¶ŕÓÚĹ®ĐÔˇŁ·˘˛ˇŇÔÇŕ׳ÄęľÓ¶ŕŁ¬ÓëְҵĎŕąŘĂÜÇСŁŇÔŇ°Í⹤×÷Őߡ˘ÁÖҵą¤Č˸ĐČľÂʽϸߡŁ
Ó˘ÎÄĂűłĆ Lyme disease
ľÍŐďżĆĘŇ ¸ĐČľżĆ
łŁĽű˛ˇŇň ÓÉňç´«˛ĄµÄ˛®ĘĎĘčÂÝĐýĚĺÎŞ˛ˇÔĚĺ
łŁĽű֢״ ·¦Á¦ˇ˘Î·ş®·˘Čȡ˘Í·Í´ˇ˘¶ńĐġ˘Ĺ»Í¡˘ąŘ˝ÚÍ´
Ó˘ÎÄĂűłĆ Lyme disease ľÍŐďżĆĘŇ ¸ĐČľżĆ łŁĽű˛ˇŇň ÓÉňç´«˛ĄµÄ˛®ĘĎĘčÂÝĐýĚĺÎŞ˛ˇÔĚĺ łŁĽű֢״ ·¦Á¦ˇ˘Î·ş®·˘Čȡ˘Í·Í´ˇ˘¶ńĐġ˘Ĺ»Í¡˘ąŘ˝ÚÍ´
˛ˇŇň
ÓÉňç´«˛ĄµÄ˛®ĘĎŁ¨BurgdorferiŁ©ĘčÂÝĐýĚĺÎŞ˛ˇÔĚ塣
ÁŮ´˛±íĎÖ
1.֢״
Ł¨1Ł©Ç±·üĆÚ 3ˇ«32Ě죬ƽľů7Ěě×óÓҡŁÁŮ´˛Ö˘×´żÉ·ÖČýĆÚˇŁ
Ł¨2Ł©µÚŇ»ĆÚ Ö÷ŇŞ±íĎÖΪƤ·ôµÄÂýĐÔÓÎ×ßĐÔşě°ßŁ¬ĽűÓÚ´ó¶ŕĘý˛ˇŔýˇŁ˛ˇłőłŁ°éÓĐ·¦Á¦ˇ˘Î·ş®·˘Čȡ˘Í·Í´ˇ˘¶ńĐġ˘Ĺ»Í¡˘ąŘ˝ÚşÍĽˇČâĚŰÍ´µČ֢״Ł¬ŇŕżÉłöĎÖÄÔĤ´ĚĽ¤Ő÷ˇŁľÖ˛żşÍČ«ÉíÁÜ°Í˝áżÉÖ×´óˇŁĹĽÓĐƢ´óˇ˘¸ÎŃס˘ŃĘŃס˘˝áĤŃס˘şçĤŃ×»ňŘşÍčÖ×Ő͡Ł
Ł¨3Ł©µÚ¶ţĆÚ ·˘˛ˇşóĘýÖÜ»ňĘýÔÂŁ¬15%şÍ8%µÄ»ĽŐß·Ö±đłöĎÖĂ÷ĎÔµÄÉńľĎµÍłÖ˘×´şÍĐÄÔŕĘÜŔ۵ÄŐ÷ĎóˇŁ
Ł¨4Ł©µÚČýĆÚ ¸ĐČľşóĘýÖÜÖÁ2ÄęÄÚŁ¬ÔĽ80%×óÓҵĻĽŐßłöĎ̶ֳȲ»µČµÄąŘ˝Ú֢״ČçąŘ˝ÚĚŰÍ´ˇ˘ąŘ˝ÚŃ×»ňÂýĐÔÇÖĎ®ĐÔ»¬Ä¤ŃסŁŇÔĎĄˇ˘Ö⡢÷ŵȴóąŘ˝Ú¶ŕ·˘Ł¬ĐˇąŘ˝ÚÖÜΧ×éÖŻŇŕżÉĘÜŔۡŁÖ÷Ҫ֢״ΪąŘ˝ÚĚŰÍ´Ľ°Ö×ŐÍŁ¬ĎĄąŘ˝ÚżÉÓĐÉŮÁż»ýŇşˇŁłŁ·´¸´·˘×÷ˇŁ
2.ĚĺŐ÷
Ł¨1Ł©Ć¤·ô˛ˇ±ä łŁÎŞĘ×·˘Ö˘×´Ł¬ĚŘŐ÷ĐÔ±íĎÖÎŞÂýĐÔÓÎ×ßĐÔşě°ßŁ¬łőĆđÎŞşěÉ«°ßŐî»ňÇđŐÖđ˝ĄŔ©´ółÉ»·×´Ë𺦡ŁŇ»°ăłöĎÖÔÚň綣ҧşó3ˇ«32Ě죬şĂ·˘ÓÚÇű¸Éˇ˘´óÍȡ˘¸ąąÉąµˇ˘Ň¸Ďµȴ¦ˇŁ
Ł¨2Ł©ÉńľĎµÍł˛ˇ±ä ÔĽĽűÓÚ15%µÄ»ĽŐߣ¬ÓëƤŐîͬʱ»ňĎűÍËşó1ˇ«6ÖÜłöĎÖˇŁ±íĎÖÎŞÄÔĤŃס˘ÄÔÉńľŃס˘Î赸֢ˇ˘ĐˇÄÔą˛ĽĂʧµ÷Ł¬łöĎÖÄÔĤ´ĚĽ¤Ő÷ˇ˘»čĂÔˇ˘Ăć̱»ňČý˛ćÉńľÍ´µČˇŁ
Ł¨3Ł©ĐÄÔಡ±ä ĽűÓÚ8%×óÓҵĻĽŐߣ¬łŁÓÚƤËđłöĎÖ3ÖÜşó·˘Éú·żĘŇ´«µĽ×čÖ͡˘ĐÄĽˇŃס˘ĐÄ°üŃ×»ňČ«ĐÄŃ׵ȡŁ
Ł¨4Ł©ąŘ˝Ú˛ˇ±ä ÔĽĽűÓÚ60%µÄ»ĽŐߣ¬¶ŕŔŰĽ°´óąŘ˝ÚŁ¬ÓČĆäĘÇĎĄąŘ˝ÚŁ¬·´¸´·˘×÷Ö×Ő͡˘ĚŰÍ´Ł¬10%µÄ»ĽŐßżÉת±äÎŞÂýĐԹؽÚŃסŁ
Ł¨5Ł©ĆäËű±íĎÖ ·˘Čȡ˘·¦Á¦ˇ˘ĽˇÍ´ˇ˘¶ńĐġ˘Ĺ»Í¡˘˝áĤŃס˘şçĤŃס˘Áܰͽἰ¸ÎƢ´óµČˇŁ
Ľě˛é
ÍâÖÜŃŞĎó»ů±ľŐýłŁŁ¬ŃŞłÁÇá¶ČÔöżěŁ¬ŃŞÇĺÖĐŔäłÁµíĂâŇßÇňµ°°×żÉŃôĐÔŁ¬×Ş°±Ă¸żÉÉý¸ßˇŁ
´ÓŃŞˇ˘ÄÔĽąŇşĽ°˛ˇ±äƤ·ôµČ±ę±ľÖпɼěłöÂÝĐýĚ塣˛ÉÓĂĂâŇßÓ«ąâˇ˘ĂâŇßתӡµČ·˝·¨żÉÔÚ»ĽŐßŃŞÖвâłöĚŘŇěĐÔżąĚ塣˛ˇÔĚĺ·ÖŔ뼰ĚŘŇěĐÔżąĚĺĽě˛âľßÓĐČ·ŐďŇâŇ塣ľŰşĎøÁ´·´Ó¦Ł¨PCRŁ©ĽĽĘőŁşĽě˛â»ĽŐßŃŞˇ˘Äňˇ˘ÄÔĽąŇşĽ°Ć¤·ô±ę±ľµČŔłÄ·˛ˇÂÝĐýĚĺDNAŁ¨Bb-DNAŁ©Ł¬˛˘Í¬Ę±żÉ˛âłöËů¸ĐČľľúÖęµÄ»ůŇňĐ͡Ł
Őď¶Ď
1.ŃŞłŁąć
°×ϸ°ű×ÜĘýŐýłŁŁ¬ÖĐĐÔÁŁĎ¸°űżÉÉÔÔö¶ŕˇŁŃŞłÁÔöżěŁ¬Ŕŕ·çĘŞŇň×ÓŇőĐÔˇŁŃ»·ĂâŇ߸´şĎÎďŃôĐÔˇŁ
2.XĎ߼ě˛é
żÉĽűĘÜŔ۹ؽÚÖÜΧČí×éÖŻÖ×ŐÍÓ°Ł¬ÉŮĘý»ĽŐßÓĐČíąÇşÍąÇÇÖĎ®±íĎÖˇŁ
3.˛ˇÔĚĺ·ÖŔ뼰ĚŘŇěĐÔżąĚĺĽě˛âÓĐÖúŐď¶ĎˇŁ
ÖÎÁĆ
1.żąÉúËŘ
¶ÔŔłÄ·˛ˇµÄ¸÷ÖÖ˛ˇ±äľůÓĐЧˇŁ
Ł¨1Ł©ËÄ»·ËŘĂżČŐ4´ÎŁ¬ÁĆłĚ10ˇ«20Ě졣ΪÔçĆÚ˛ˇŔýµÄĘ×ѡҩÎÔиľˇ˘˛¸ČéĆÚ¸ľĹ®şÍ¶ůÍŻ˝űÓáŁ
Ł¨2Ł©°˘ÄŞÎ÷ÁÖĂżČŐ3´ÎŁ¬ÁĆłĚ14ˇ«21Ě졣
Ł¨3Ł©ÇŕĂąËŘľ˛ÂöµÎעÿČŐ1ˇ«2´ÎŁ¬ÁĆłĚ14ˇ«21Ě졣
Ł¨4Ł©ĆäËű¶ŕÎ÷»·Ëء˘µÚ3´úÍ·ćßĂąËصȿÉѡÓáŁ
2.·ÇçŢĚĺżąŃ×Ň©
ÓĂÓÚŔłÄ·˛ˇąŘ˝ÚŃ×µÄÖÎÁĆŁ¬ČçĎűŃ×Í´ˇ˘·Ň±ŘµĂµČˇŁ
3.ĚÇƤÖĘĽ¤ËŘ
ĘĘÓĂÓÚŔłÄ·˛ˇÄÔĤŃ×»ňĐÄÔŕŃ×»ĽŐߡŁĆĂÄáËÉŁ¬Ö˘×´¸ÄÉĆşóÖ𽥼őÁżÖÁÍŁŇ©ˇŁ
4.ŃĎÖŘ·żĘŇ´«µĽ×čÖÍ»ĽŐß
Ó¦»ýĽ«¶ÔÖ˘´¦ŔíˇŁŃĎÖصĹؽÚŃ׿ÉĐĐ»¬Ä¤ÇĐłýˇŁˇˇ
»®´Ę
Ľě˛âµ˝ŁşÓ˘Óď » ÖĐÎÄ
·Ňë ČËą¤·Ň롡
Biochemical and Biophysical Research Communications
Volume 206, Issue 1, 5 January 1995, Pages 223-229
Biochemical and Biophysical Research Communications
Regular Article
Effects of Retinoic Acid (Vitamin A) on Tumor Necrosis Factor Cytolytic Action
Author links open overlay panelHughesT.K.FulepE.
Univ Texas, Med Branch, Dept Microbiol & Immunol, J 19, Galveston, TX 77550, USA
Available online 25 May 2002.
Tumor necrosis factor (TNF) is a monokine produced primarily by macrophages. TNF has a number of activities including direct lysis of certain transformed cells and induction of antiviral activity. One of the protoypical transformed cell lines used for studying TNF cytolysis is murine L-929 cells. Because of the lysis, TNF has not been shown to have antiviral activity in these cells. Since retinoic acid (RA) induces a normal phenotype in the L-929 cells, we sought to determine if their conversion to a normal phenotype would 1) render them insensitive to the cytolytic effect and 2) allow for the development of an antiviral state. We present evidence that both the cis- and trans- forms of RA and to a lesser extent, the RA precursor beta-carotene, can inhibit recombinant human TNF cytolytic activity in mouse L-929 cells. However, blockage of the cytolytic activity does not allow development of an antiviral state.
Sci Rep. 2016; 6: 25835.Effects of Retinoic Acid (Vitamin A) on Tumor Necrosis Factor Cytolytic Action - ScienceDirect
https://www.sciencedirect.com/science/article/abs/pii/S0006291X85710315ˇˇ
9-cis Retinoic acid enhances the antiviral effect of interferon on hepatitis C virus replication through increased expression of type I interferon receptor
February 2003Journal of Laboratory and Clinical Medicine 141(1):58-66
DOI: 10.1067/mlc.2003.8
SourcePubMed
Sachiko Hamamoto
Ryo Fukuda
Norihisa IshimuraShow all 9 authors
Yoshikazu Kinoshita
The concentration of type I interferon receptor (IFN-Rc) in the liver is a crucial factor in determining the efficacy of interferon (IFN) therapy in patients with chronic hepatitis C. Retinoic acids (RAs) can enhance the expression of type I IFN-Rc expression. The aim of this study was to investigate whether RAs increase the anti-hepatitis C virus (HCV) effect of IFN through an increase in IFN-Rc. The hepatocellular carcinoma cell line HuH-7 was treated with 10(-7) mol/L all-trans RA (ATRA) and 9-cis RA (9-CRA). Expression of type I IFN-Rc was investigated at both the mRNA and protein levels with the use of real-time quantitative polymerase chain reaction and flow cytometry, respectively. We investigated the anti-HCV effect, using in vitro HCV transfection, by monitoring the level of HCV RNA in the culture medium. ATRA and 9-CRA enhanced the expression of type I IFN-Rc at both the mRNA and protein levels. After IFN-alpha treatment, the activity of 2,5'-oligoadenylate synthetase was enhanced by RAs, and this enhancement was abolished when blocking antibodies had previously been bound to the surface receptors. IFN treatment decreased the concentration of HCV RNA, and this effect was enhanced by treatment with RAs. Our findings suggest that RAs enhance the anti-HCV replication effect of IFN-alpha through up-regulation of type I IFN-Rc in HuH-7 cells.ˇˇ
... Retinoic acid binding to retinoic acid receptor (RAR) and retinol X receptor (RXR) leads to some transcription activities[10][11][12]. Our group previously reported that some retinoic acids raise IFN activity via increasing IFN receptors on hepatoma cells in vitro[13]. However, no study to date has examined clinically the effect of retinoic acids on IFN treatment. ...
... Retinoids are a group of vitamin A-related compounds that influence proliferation of and induce differentiation in epithelial tissue and cells of the immune system[11,12,14]. Hamamoto et al.[13]reported that retinoic acids can enhance the anti-HCV replication effect of IFN-¦Á through up-regulation of type I IFN receptors in a hepatoma cell-line. They showed that retinoic acids enhanced IFN-induced 2,5'AS augmentation. ...
... This dose is less than the conventional clinical dose. However, the putative concentration of retinoic acid in bodily fluids is 2 ˇÁ 10 −6 M if a dose of retinol equivalent to 3 mg/day retinoic acid is administered[13]. This concentration is more than the concentration of retinoic acid which increased the anti-HCV replication effect of IFN-¦Á through up-regulation of type I IFN receptors in a hepatoma cell-line. ...ˇˇ
... Vitamin A has been shown to both promote and inhibit HCV replication in vitro. Retinoic acid binding to CRABP1 can activate lipid metabolism gene expression in hepatocytes, thus providing a platform for HCV replication complexes and subsequent viral propagation [124,125]. A separate study has demonstrated that administration of all-trans-retinoic acid (ATRA), an active metabolite of vitamin A, results in the down-regulation of the HCV replicon, at least in part via the induction of GPx [94]. ...
... HCV replication is up-regulated in cells expressing CRABP1 via lipid droplet formation [125]. ATRA reduces viral replication [94]. ..ˇˇ
9-cis Retinoic acid enhances the antiviral effect of interferon on hepatitis C virus replication through increased expression of type I interferon receptor
https://www.researchgate.net/publication/10959437_9-cis_Retinoic_acid_enhances_the_antiviral_effect_of_interferon_on_hepatitis_C_virus_replication_through_increased_expression_of_type_I_interferon_receptorˇˇ
The Role of Micronutrients in the Infection and Subsequent Response to Hepatitis C Virus
Article
Full-text available
Jun 2019
Sunil Gupta
Scott Read
Nicholas A Shackel
Golo Ahlenstiel... Retinoid acids have been implicated in clinical applications, both as a potential antitumor agent and for the treatment of skin diseases (15). In addition, there have been a number of studies suggest that the retinoic acid or its analogues exhibit antiviral activities against a variety of pathogens, including human immunodeficiency virus type 1 (HIV-1) (16,17), measles virus (18), human herpesvirus 8 (HHV-8) (19), hepatitis C virus (HCV) (20)(21)(22), and adenovirus (23). Particularly, acitretin has been used to treat patients with HIV who have psoriasis (17,24), and the drug is well tolerated. ...
... As indicated previously, retinoic acids have been reported to be able to induce the pattern recognition sensor RIG-I expression and activate a type I interferon-mediated innate immune response. Several reports have explained the antiviral effect of retinoic acid against HIV and HCV from the perspective of innate immune activation (17,21,34). Here, our results showed that compared to the cells treated with the positive controls, including pppRNA and pegylated IFN-, tazarotene did not significantly induce genes of IFN responses, such as RIG-I, MX2, and IFIT1, suggesting that tazarotene inhibits HBV in our system in an IFN-independent manner. ...Micronutrient deficiencies develop for a variety of reasons, whether geographic, socioeconomic, nutritional, or as a result of disease pathologies such as chronic viral infection. As micronutrients are essential for a strong immune response, deficiencies can significantly dampen both the innate and the adaptive arms of antiviral immunity. The innate immune response in particular is crucial to protect against hepatitis C virus (HCV), a hepatotropic virus that maintains chronic infection in up to 80% of individuals if left untreated. While many micronutrients are required for HCV replication, an overlapping group of micronutrients are also necessary to enact a potent immune response. As the liver is responsible for the storage and metabolism of many micronutrients, HCV persistence can influence the micronutrientsˇŻ steady state to benefit viral persistence both directly and by weakening the antiviral response. This review will focus on common micronutrients such as zinc, iron, copper, selenium, vitamin A, vitamin B12, vitamin D and vitamin E. We will explore their role in the pathogenesis of HCV infection and in the response to antiviral therapy. While chronic hepatitis C virus infection drives deficiencies in micronutrients such as zinc, selenium, vitamin A and B12, it also stimulates copper and iron excess; these micronutrients influence antioxidant, inflammatory and immune responses to HCV.
(PDF) The Role of Micronutrients in the Infection and Subsequent Response to Hepatitis C Virus
https://www.researchgate.net/publication/333839545_The_Role_of_Micronutrients_in_the_Infection_and_Subsequent_Response_to_Hepatitis_C_Virusˇˇ
Original Papers
Published: March 1989
Differential modulating effects of retinoic acid on interferon antiviral activity
Chi-Kuan Ho, Bor-Rung Ou, Ching-Yun Wang, Hour-Young Chen & Tsuguo Kuwata
Archives of Virology volume 109, pages25¨C34(1989)Cite this article
Summary
The effect of retinoic acid (RA) on the antiviral activity of interferons (IFNs) ¦Á and ¦Â in the U 937 and WISH cells was examined to ascertain whether or not RA could reduce the effectiveness of IFN-induced resistance to viral infection. Our results indicate that in the U 937 cells, RA (0.1¨C1.0 µM) had neither enhancing nor suppressive effect on the antiviral activity of IFN-¦Á or ¦Â against the Semliki Forest virus (SFV). However, in the WISH cells, RA had different effects on IFNs ¦Á and ¦Â. Thus, while RA (0.1¨C50 µM) invariably suppressed the activity of IFN-¦Á, it enhanced the action of IFN-¦Â at low dose (0.1¨C1.0 µM) but became suppressive at higher concentrations (ˇÝ 10 µM). Furthermore, higher antiviral activity was consistently obtained when RA (0.1¨C10 µM) was added prior to either IFN-¦Á or IFN-¦Â comparing to cultures with IFN alone. In addition, direct correlation between antiviral activity and the amplitude of 2¨C5 oligoadenylate (A) synthetase activity was not observed. These results suggest that modulation of IFN antiviral activity by RA varies with different systems and is dependent on the sequence of treatment.Differential modulating effects of retinoic acid on interferon antiviral activity | SpringerLink
https://link.springer.com/article/10.1007/BF01310515ˇˇ
Access Volume 111, Issue 9 14 May 2014 , pp. 1586-1593
Effect of all-trans-retinoic acid on enterovirus 71 infection in vitro
Siyuan Chen (a1), Yi Yang (a1), Jin Xu (a1), Liyun Su (a1) ...
DOI: https://doi.org/10.1017/S0007114513004133Published online by Cambridge University Press: 04 February 2014
Abstract
Our previous studies have shown that vitamin A (VA) status is associated with antiviral immunity and pathogenic conditions in enterovirus 71 (EV71)-infected children. In the present study, we established an in vitro model to investigate the effects and potential mechanism of the antiviral activity of VA. Human monocytic U937 cells were cultured in vitro and infected with EV71. All-trans-retinoic acid (ATRA), the active metabolite of VA, and Ro 41-5253, a retinoic acid receptor-¦Á (RAR-¦Á) antagonist, were used as the experimental treatment agents. The percentage of EV71-infected cells and apoptosis induced by EV71 were determined using flow cytometry. The level of interferon-¦Á (IFN-¦Á) in the supernatants of the cultures was detected using ELISA. The expression of retinoid-induced gene I (RIG-I) and its downstream genes was examined with real-time quantitative PCR. The results indicated that ATRA reduced the percentage of EV71-infected cells and protected cells against EV71-induced apoptosis. Correspondingly, ATRA increased the production of IFN-¦Á one of the most important antiviral cytokines, at both mRNA and protein levels in EV71-infected cells. In addition, the expression of RIG-I mRNA and its downstream genes was up-regulated by ATRA in EV71-infected cells. Ro 41-5253 abrogated the inhibitory effects of ATRA on EV71.The present findings suggest that ATRA is an interferon-inducing agent with antiviral activity against EV71 in vitro and that its actions are mediated at least in part by RAR-¦Á activity and the RIG-I signalling pathway.
ˇˇ
ˇˇ
Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome
Heetae Lee1,* and GwangPyo Koa,1,2
1Center for Human and Environmental Microbiome, Institute of Health and Environment, School of Public Health, Seoul National University, Gwanak-ro, Gwanak-gu, Seoul 151-742, Korea
2N-BIO, Seoul National University, Gwanak-ro, Gwanak-gu, Seoul 151-742, Koreaˇˇ
Abstract
The effect and underlying mechanism of vitamin A on norovirus infection are largely unknown. This study aimed to investigate how vitamin A administration affects the gut microbiome after norovirus infection. Here, we demonstrate that treatment with either retinol or retinoic acid (RA) inhibits murine norovirus (MNV) replication using both in vitro and in vivo models. Compositional changes in the gut microbiome associated with RA administration and/or norovirus infection were also investigated. Oral administration of RA and/or MNV significantly altered intestinal microbiome profiles. Particularly, bacterial species belonging to the Lactobacillaceae families were remarkably increased by MNV inoculation and RA administration, suggesting that the antiviral effects of RA occur via the modulation of specific microbiota. The antiviral causal effect of Lactobacillus was identified and demonstrated using in vitro models in RAW264.7 cells. The antiviral immune response to MNV was mediated by IFN-¦Â upregulation. This study represents the first comprehensive profiling of gut microbiota in response to RA treatment against norovirus infection. Moreover, we conclude that the abundance of Lactobacillus through gut microbiota modulation by RA is at least partially responsible for norovirus inhibition. Norovirus is the most frequent etiological viral agent of acute gastroenteritis among all age groups worldwide.
Norovirus causes approximately 90% of all epidemic nonbacterial outbreaks of gastroenteritis around the world, and is responsible for approximately 50% of all foodborne outbreaks of gastroenteritis in the United States and many other countries1,2. Viral infection causes various clinical symptoms, including diarrhea, vomiting, nausea, abdominal pain, and fever lasting one to three days. Unfortunately, there is no current treatment or vaccine effective against norovirus infection.
A previous epidemiological study suggested that vitamin A supplementation decreases norovirus infection rates and clinical symptoms3. Moreover, the responses of various intestinal cytokines were modified by vitamin A supplementation during norovirus infection4. Retinoic acid (RA), the metabolite of dietary vitamin A, contributes to both innate and adaptive immune responses5. Additional evidence also indicates that RA deficiency impairs immunity, whereas RA excess can induce inflammatory disorders5. Retinoic acid-inducible gene 1 (RIG-1) and melanoma differentiation-associated gene 5 (MDA5) signaling play crucial roles in antiviral responses to viral RNA by producing type I interferons (IFNs)6. Moreover, a recent study reported that sufficient vitamin A supplementation reduced both mortality and morbidity associated with infectious gastrointestinal and respiratory diseases7.
The gut microbiota plays a pivotal role in pathogen infection and mucosal immune responses through cross-talk with mucosal immune systems8,9. For example, an altered gut microbiome in mice lacking Toll-like receptors (TLRs) and myeloid differentiation primary response gene 88 (Myd88) was strongly associated with metabolic syndrome, type 1 diabetes (T1D) and host defense against microbial infection10,11,12,13. Above all, dietary foods and drugs play a crucial role in modulating gut microbiome diversity, with changes that are directly linked to health conditions. For example, recent studies suggested that Akkermansia muciniphila, which increased significantly in the gut environment as a result of metformin treatment, may improve metabolic diseases such as type 2 diabetes14,15,16. In this study, we evaluated: 1) compositional changes in the gut microbiota and host immune responses following RA treatment, and 2) the anti-norovirus effects of specific gut microbiota whose abundance was increased by RA treatment (Lactobacillus spp.).
ˇˇAntiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4867650/ˇˇ
Viral Hepatitis Free Access
Retinoid regulation of antiviral innate immunity in hepatocytes
Noell E. Cho Bo©\Ram Bang Purnima Gurung Meng Li Dahn L. Clemens T. Michael Underhill Laura P. James Jenifer R. Chase Takeshi Saito
First published: 05 December 2015 https://doi.org/10.1002/hep.28380 Citations: 9
Potential conflicts of interest: Dr. James is part owner of Acetaminophen Toxicity Diagnostics, LLC. ATD is a recipient of grant R42DK079387 from NIDDK to develop a clinical laboratory test for acetaminophen protein adducts.
This work was supported by funds from AASLD/ALF Liver Scholar Award, Baxter Foundation Award, SCRC for ALPD & Cirrhosis Pilot Project Grant (5P50AA011999) and USC RCLD pilot grant (5P30DK048522), NIAAA (R21AA022751), NIDDK (RO1DK101773), ACS IRG (to T.S.), and NIGMS (P20GM103408; to J.R.C.).
Abstract
Persistent infection of hepatitis C virus (HCV) is one of the leading causes of end©\stage liver disease (ESLD), such as decompensated cirrhosis and liver cancer. Of particular note, nearly half of HCV©\infected people in the United States are reported to be heavy drinkers. This particular group of patients is known to rapidly progress to the ESLD. Although accelerated disease progression among alcohol abusers infected with HCV is clinically well recognized, the molecular pathophysiology behind this manifestation has not been well elucidated.Hepatocytes metabolize ethanol (EtOH) primarily through two steps of oxidative catabolism in which alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) play central roles. The ADH©\ALDH pathway also governs the metabolism of retinol (vitamin A) to its transcriptionally active metabolite, retinoic acid (RA).
In this study, we defined that the ADH©\ALDH pathway serves as a potent antiviral host factor in hepatocytes, which regulates the expression of interferon (IFN)©\stimulated genes (ISGs) by biogenesis of RA. ISGs constitute over 300 antiviral effectors, which cooperatively govern intracellular antiviral innate immunity.
Our study revealed that intracellular RA levels greatly influence ISG expression under basal conditions. Moreover, RA augments ISG induction in response to viral infection or exposure to IFN in a gene©\specific manner.
Lastly, our results demonstrated that EtOH attenuates the antiviral function of the ADH©\ALDH pathway, which suggests the possibility that EtOH©\retinol metabolic competition is one of the molecular mechanisms for the synergism between HCV and alcohol abuse in liver disease progression.
Conclusions: RA plays a critical role in the regulation of intracellular antiviral innate immunity in hepatocytes. (Hepatology 2016;63:1783©\1795)
The ADH©\ALDH Pathway Is a Potent Antiviral Host Factor
Restoration of EtOH metabolic capacity in Huh7 cells enabled us to investigate the association between EtOH metabolic pathways and the HCV life cycle. Surprisingly, transient ADH1B overexpression in Huh7 cells harboring HCV subgenomic replicon (HCV©\SGR) dramatically suppressed HCV replication (Fig. 2A,B). In contrast, overexpression of CYP2E1 was associated with an enhancement of HCV replication. Moreover, the antiviral effect of ADH1B was reduced in the presence of EtOH (Fig. 2B). Next, we tested whether ADH1B affects the efficiency in establishing viral life cycle by HCV©\SGR replicon transduction assay. Transfection of HCV subgenome in Huh7 cells that stably express ADH1B demonstrated a significantly lesser number of HCV©\replicating foci formation; however, the difference between control and ADH1B cells diminished upon EtOH treatment (Fig. 2C). These results indicate that the ADH©\ALDH pathway serves as an antiviral host factor in the absence of EtOH. Furthermore, our results suggest that either (1) the EtOH metabolic by©\products generated by ADH1B may offer a suitable environment for efficient HCV replication or (2) EtOH prohibits the biogenesis of antiviral molecules produced by the ADH©\ALDH pathway. To distinguish between these possibilities, we addressed whether EtOH metabolites increase HCV replication efficiency. The results showed that neither acetaldehyde nor acetate treatment changed viral replication efficiency (Supporting Fig. 2A,B).
To further understand how the ADH©\ALDH pathway suppresses HCV, we assessed the effect of ADH1B expression on each stage of the viral life cycle. First, an HCV pseudo particle containing a green fluorescent protein (GFP) reporter was employed to test the effect of ADH1B expression on viral entry. Under these conditions, ADH1B expression in Huh7 cells was found to have a negligible effect on viral entry (Fig. 2D). Next, we examined the effect of ADH1B expression on genome replication using reverse©\transcription polymerase chain reaction (RT©\PCR). The abundance of each region of the viral genome decreased with distance from the replication initiation site (5ˇäUTR [untranslated region]) of the viral genome, especially in ADH1B©\expressing cells (Fig. 2E). This result may suggest that the ADH©\ALDH pathway restricts HCV by promoting premature termination of genome replication. Taken together, these results indicate that the ADH©\ALDH pathway restricts HCV at the genome replication/translation stage.
Abbreviations
ADH
alcohol dehydrogenase
APAP
acetaminophen
ATRA
all trans retinoic acid
ALD
alcoholic liver disease
bp
base pairs
CYP2E1
cytochrome P450©\2E1
DMEM
Dulbecco's modified Eagle's medium
DR5
direct repeat 5
EtOH
ethanol
GC
gas chromatography
GFP
green fluorescent protein
HCV
hepatitis C virus
HCV©\SGR
HCV subgenomic replicon
HPLC©\EC
high©\performance liquid chromatography with electrochemical detection
HSCs
hepatic stellate cells
IFN
interferon
ISGs
interferon©\stimulated genes
IV
intravenous
Jak
Janus kinase
MOI
multiplicity of infection
NAD+
nicotinamide adenine dinucleotide
NAPQI
N©\acetyl©\p©\benzoquinone imine
PAMPs
pathogen©\associated molecular patterns
PHHs
primary human hepatocytes
RA
retinoic acid
RAL
retinaldehyde
RAR
retinoic acid receptor
RARE
retinoic acid response element
ROL
retinol
RT©\PCR
reverse©\transcription polymerase chain reaction
RE
retinyl ester
RIG©\I
retinoic acid©\inducible gene I
RXR
retinoid X receptor
SeV
Sendai virus
STAT
signal transducer and activator of transcription
UTR
untranslated regionˇˇ
Retinoid regulation of antiviral innate immunity in hepatocytes - Cho - 2016 - Hepatology - Wiley Online Library
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.28380ˇˇ
American Journal of Physiology - Lung Cellular and Molecular PhysiologyRetinoic acid prevents virus-induced airway hyperreactivity and M 2 receptor dysfunction via anti-inflammatory and antiviral effects
Liliana Moreno-Vinasco, Norah G. Verbout, Allison D. Fryer, David B. JacobyNorthwestern University
Abstract
Inhibitory M2 muscarinic receptors on airway parasympathetic nerves normally limit acetylcholine release. Viral infections decrease M 2 receptor function, increasing vagally mediated bronchoconstriction.Since retinoic acid deficiency causes M2 receptor dysfunction, we tested whether retinoic acid would prevent virus-induced airway hyperreactivity and prevent M2 receptor dysfunction. Guinea pigs infected with parainfluenza virus were hyperreactive to electrical stimulation of the vagus nerves, but not to intravenous acetylcholine, indicating that hyperreactivity was due to increased release of acetylcholine from parasympathetic nerves. The muscarinic agonist pilocarpine, which inhibits vagally mediated bronchoconstriction in control animals, no longer inhibited vagally induced bronchoconstriction, demonstrating M2 receptor dysfunction.
Treatment with all-trans retinoic acid (1 mg/kg) prevented virus-induced hyperreactivity and M2 receptor dysfunction. However, retinoic acid also significantly reduced viral titers in the lungs and attenuated virus-induced lung inflammation. In vitro, retinoic acid decreased M2 receptor mRNA expression in both human neuroblastoma cells and primary cultures of airway parasympathetic neurons.
Thus, the protective effects of retinoic acid on airway function during viral infection appear to be due to anti-inflammatory and antiviral mechanisms, rather than to direct effects on M2 receptor gene expression.
Retinoic acid prevents virus-induced airway hyperreactivity and M 2 receptor dysfunction via anti-inflammatory and antiviral effects ˇŞ Northwestern Scholars
https://www.scholars.northwestern.edu/en/publications/retinoic-acid-prevents-virus-induced-airway-hyperreactivity-and-mˇˇ
Beyond sensing: Retinoic acid inducible gene©\I (RIG©\I) continues to expand its antiviral effector functions
Amy E.L. Stone Ph.D. Michael J. Gale Jr. Ph.D.
First published: 20 March 2017 https://doi.org/10.1002/hep.29161 Citations: 1
Center for Innate Immunity and Immune Disease,
Department of Immunology
University of Washington School of Medicine
Seattle, WA
Abbreviations
HEV
hepatitis E virus
IFN
interferon
IRF
IFN regulatory factor
ISG
IFN©\stimulated gene
MAVS
mitochondrial antiviral signaling
NF©\¦ĘB
nuclear factor kappa B
PAMP
pathogen©\associated molecular pattern
RIG©\I
retinoic acid inducible gene©\I
STAT
signal transducer and activator of transcription
Retinoic acid gene©\I (RIG©\I) is a cytosolic RNA helicase and pathogen recognition receptor that recognizes pathogen©\associated molecular patterns (PAMPs) within viral RNA to trigger downstream interferon (IFN) production and innate immune activation during RNA virus infection. Analysis of RIG©\I during hepatitis E virus (HEV) infection now reveals a role in IFN©\independent innate immunity to suppress HEV replication.
It has long been accepted that there exists complex signaling and crosstalk among innate immune response pathways to direct Types I and III IFN defenses against microbial pathogens.1 During RNA virus infection IFN is induced as a result of pathogen recognition receptor (PRR) recognition of viral RNA PAMPs. For many RNA viruses PAMP recognition is mediated by RIG©\I1 wherein RIG©\I signaling induces the downstream activation of transcription factors, including IFN regulatory factor 3 (IRF3) and IRF7 as well as nuclear factor kappa B (NF©\¦ĘB),2 and drives the expression of virus©\responsive genes, including IFN (reviewed in Loo and Gale1). Secreted IFN then binds to its cognate receptor on the infected cell and on bystander cells to signal through the Janus kinase (JAK)¨Csignal transducer and activator of transcription (STAT) pathway and induce hundreds of IFN©\stimulated genes (ISGs) (Fig. 1). Among their many actions, virus©\responsive genes and ISGs direct antiviral actions to suppress viral replication and spread, and serve to modulate the adaptive immune response to drive antiviral immunity. While RIG©\I can also serve as an antiviral effector gene to bind to viral RNA and block RNA/protein associations,3 it is widely recognized for trigging this innate immune antiviral signaling cascade as a result of PAMP recognition and binding.4 Xu and colleagues5 now reveal a role for RIG©\I in mediating innate immune effector actions against HEV that show roles for RIG©\I in IFN©\independent innate immunity. Remarkably, RIG©\I is observed to drive the expression of a subset of ISGs independently of IRF3, IRF7, and IFN but requiring NF©\¦ĘB actions, thus providing evidence for a novel RIG©\I©\directed innate immune effector response pathway in hepatocytes (the target cell of HEV infection) that uncouples IRF and NF©\¦ĘB signaling programs to operate outside of the canonical IRF/IFN/ISG innate immune effector pathway (Fig. 1).ˇˇ
Figure 1
RIG©\I acts in multiple ways to inhibit virus infection. RIG©\I recognizes PAMP RNA, leading to multiple divergent pathways. (1) MAVS located on the mitochondria©\associated membrane induces IRF3/7 dimerization, leading to the induction of IFN genes. (2) MAVS located on peroxisomes induces NF©\¦ĘB activation, leading to the expression of proinflammatory genes. (3) RIG©\I sterically hinders the interaction of viral proteins with viral RNAs. (4) RIG©\I induces STAT1 phosphorylation independent of IFN induction. (5) RIG©\I induces ISG production independent of IRF3/7 and STAT1 signaling, as describes by Xu et al.5 This action may be mediated by MAVS and NF©\¦ĘB or potentially other transcription factors.
On the surface, this article shows that RIG©\I, an RNA helicase and pathogen recognition receptor, controls HEV infection in human cells.5 While this finding has implications for clinical treatment of patients with HEV by targeting RIG©\I with activator molecules, the authors elucidate that this outcome is mediated independently of IFN.5 This main observation serves as a launching point for a mechanistic study in the downstream signaling of RIG©\I and its role in IFN©\independent ISG induction. Other groups have shown that RIG©\I overexpression, followed by its activation by viral RNA transfection or actual virus infection can induce IRF3 activation and IFN and ISG expression in hepatoma cells and hepatocytes during infection by hepatitis C virus, another liver©\tropic virus (reviewed in Loo and Gale1). Now Xu et al. report that overexpression of RIG©\I and activation of its signaling actions through transfection of cells with PAMP RNA ligand fails to induce IFN production.5 This observation is validated through bioassays that include assessment of a sensitive IFN©\induced promoter©\luciferase reporter gene (ISRE©\Luc)ˇŞin this case when RIG©\I was expressed and activated the ISRE©\Luc promoter was highly induced and ISGs were expressed, but no IFN was produced when RIG©\I was overexpressed and activated.5 Examination of tyrosine phosphorylation of STAT1, a marker of STAT activation, showed that in this context STAT1 was phosphorylated/activated, thus linking RIG©\I to IFN©\independent STAT activation and ISG expression.5
RIG©\I signals downstream by forming a complex with the mitochondrial antiviral signaling (MAVS) adaptor protein.3 Of note is that MAVS located on peroxisomes directs signaling independently of IFN and occurring through NF©\¦ĘB, AP©\1, IRF1, and IRF3.6 Because Xu et al. also show that cells lacking IRF3 induce ISGs through the RIG©\I/IFN©\independent pathway, NF©\¦ĘB could likely serve as the central transcription factor for gene induction in the IFN©\independent RIG©\I signaling pathway. The presence of NF©\¦ĘB binding sites in the promoter of many ISGs, including those whose expression is responsive to this novel RIG©\I pathway as shown by Xu et al., support this notion. Unfortunately, Xu et al. do not address the role of NF©\¦ĘB or other transcription factors following RIG©\I activation or overexpression in this IFN©\independent pathway. Importantly, tumor necrosis factor©\alpha (TNFalpha), a major inflammatory cytokine, has been shown toz suppress HEV infection/replication through NF¦ĘB©\dependent ISRE activation,7 such that it could also play a role here to suppress HEV infection. Further, the genes up©\regulated by RIG©\I overexpression independent of IRF3/7 are distinct from those found to be induced by the IFN©\independent peroxisomal MAVS signaling.6 Thus, it is notable that the authors also did not examine the possibility of direct RIG©\I/HEV RNA interaction as an innate immune effector mechanism suppressing HEV infection. We cannot simply conclude that the antiviral effects of RIG©\I against HEV occur exclusively through the ISGs that are induced by IFN©\independent, possibly NF©\¦ĘB©\driven, RIG©\I signaling alone, as RIG©\I itself might bind HEV RNA to disrupt viral replication. However, we can conclude that the innate immune action of RIG©\I goes beyond IRF signaling and IFN induction into a more direct role linking RIG©\I and STAT1.
How does RIG©\I directly link with the JAK¨CSTAT pathway? Jiang et al. demonstrated that overexpression of RIG©\I in U937 cells leads to increased STAT1 phosphorylation,8 and Hou et al. showed that knocking down RIG©\I reduced the levels of IFN©\¦Á©\induced STAT1 phosphorylation due to increased association of SHP1 and STAT1.9 Here, the STAT1 tyrosine phosphorylation is occurring independently of IFN. While other cytokines were not investigated, it has been shown that STAT1 can be phosphorylated through noncanonical pathways, such as a toll©\like receptor (TLR)¨Cinduced, TRAF6©\dependent mechanism.10 RIG©\I has also been associated with TRAF611; but a RIG©\I/TRAF6/STAT1 pathway has yet to be established, and the authors did not examine such an axis, leaving the RIG©\I/STAT1 connection and resulting STAT1 activation undefined.
Xu et al. clearly demonstrated the power of activating RIG©\I with ligand RNA to drive antiviral defenses to suppress HEV infection as transfection of cells with PAMP RNA served to trigger RIG©\I activation and signaling through the IFN©\independent RIG©\I pathway. Overall, this work underscores the important role of RIG©\I in innate antiviral immunity and defines RIG©\I as a therapeutic target to control virus infection. Shifting away from IFN©\¦Á as the frontline antiviral is critical to reducing harmful side effects and additional pathogenesis in patients most in need of antiviral treatments, such as transplant recipients. Targeting RIG©\I to engage this novel IFN©\independent pathway therefore could have utility for development of antiviral therapeutics that dispense with IFN altogether. Many groups and biotech companies are developing RIG©\I agonists for clinical applications. Additional studies, like this one by Xu et al., are required to fully define the nature and players within the IFN©\independent pathway for understanding its full application to antiviral immunity. As the authors state, ˇ°RIG©\I and its downstream pathways form a large complex web.ˇ± We need to understand this web and the crosstalk among the various signaling pathways to effectively treat viral diseases. We need new frontline treatments to combat emerging infectious diseases, which are largely RNA viruses. Elucidating the mechanism of action of ISGs will help define these webs and inform us about generating new antiviral treatments.
ˇˇBeyond sensing: Retinoic acid inducible gene©\I (RIG©\I) continues to expand its antiviral effector functions - Stone - 2017 - Hepatology - Wiley Online Library
https://aasldpubs.onlinelibrary.wiley.com/doi/full/10.1002/hep.29161ˇˇ
Clinical and experimental immunology 2013-7-20
Retinoic acid-producing, ex-vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive B lymphocytes.
[V Di Caro, B Phillips, C Engman, J Harnaha, M Trucco, N Giannoukakis]
Abstract
While much is known about tolerogenic dendritic cell effects on forkhead box protein 3 (FoxP3)⁺ regulatory T cells, virtually nothing is known about their effects on another arm of immunoregulation that is mediated by a subpopulation of immunosuppressive B cells. These cells suppress rheumatoid arthritis, lupus and inflammatory bowel disease in mice, and functional defects have been reported in human lupus. We show that co-stimulation-impaired tolerogenic dendritic cells that prevent and reverse type 1 diabetes mellitus induce the proliferation of human immunosuppressive B cells in vitro. We also show that the suppressive properties of these B cells concentrate inside the CD19⁺ CD24⁺ B cell population and more specifically inside the CD19⁺ CD24⁺ CD38⁺ regulatory B cell population.We discovered that B cell conversion into suppressive cells in vitro is partially dependent on dendritic cell production of retinoic acid and also that CD19⁺ CD24⁺ CD38⁺ B regulatory cells express retinoic acid receptors. Taken together, our data suggest a model whereby part of the immunosuppressive properties of human tolerogenic dendritic cells could be mediated by retinoic acid which, in addition to its known role in favouring T cell differentiation to FoxP3⁺ regulatory T cells, acts to convert B cells into immunosuppressive cells.
Retinoic acid-producing, ex-vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive B lymphocytes. | Sigma-Aldrich
https://www.sigmaaldrich.com/catalog/papers/23865694ˇˇ
Hepatobiliary Surg Nutr. 2013 Oct; 2(5): 244¨C247.
Carotenoids and alcoholic liver disease
Camilla P. Stice and Xiang-Dong Wangcorresponding author
Abstract
Chronic and excessive consumption of alcohol leads to the development of alcoholic liver disease. The depletion of vitamin A is a well-known consequence of alcohol consumption, and may be associated with the observed alcohol-induced hepatic injury. The provitamin A carotenoid ¦Â-carotene has been demonstrated to increase alcohol-induced hepatic injury when given in high doses, while low dose supplementation provides protection against hepatic injury. However, it is unknown if the hepatoprotective effects of low dose ¦Â-carotene are due to the protective actions of ¦Â-carotene itself or if the alterations are due to restored vitamin A levels. Future studies are needed to provide further insight into the specific mechanisms by which ¦Â-carotene exerts its protective effect. Further, supplementation studies utilizing high doses of ¦Â-carotene in the presence of alcohol must be done with caution.
Figure 1
Potential mechanisms of how excessive alcohol interferes with retinoid metabolism. Specifically: (I) both retinol and ethanol have similar metabolic pathways, and ethanol can act as a competitive inhibitor of vitamin A oxidation to retinoic acid involving alcohol dehydrogenases (ADHs) and aldehyde dehydrogenases (ALDHs); (II) Ethanol enhances catabolism of vitamin A and retinoic acid by inducing cytochrome P450 enzymes, particularly cytochrome P450 2E1 (CYP2E1) which contributes greatly to alcohol-related liver injury; and (III) ethano alters retinoid homeostasis via increased vitamin A mobilization from liver to extrahepatic tissues. Supplementation with the provitamin A carotenoid, ¦Â-carotene, may restore the vitamin A status to within a normal range, thereby providing protection against alcohol-related liver injury.
Keywords: Carotenoids, ¦Â-carotene, vitamin A, alcoholic liver disease, hepatic injuryCarotenoids and alcoholic liver disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3924697/ˇˇ
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease
Volume 1772, Issue 1, January 2007, Pages 66-71
Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease
Effects of retinoic acid on the development of liver fibrosis produced by carbon tetrachloride in mice
Department of Medicine, The Johns Hopkins University School of Medicine, The Johns Hopkins University, Baltimore, MD 21205-2195, USA
The role of retinoic acid (RA) in liver fibrogenesis was previously studied in cultured hepatic stellate cells (HSCs). RA suppresses the expression of ¦Á2(I) collagen by means of the activities of specific nuclear receptors RAR¦Á, RXR¦Â and their coregulators. In this study, the effects of RA in fibrogenesis were examined in carbon tetrachloride (CCl4) induced liver fibrosis in mice. Mice were treated with CCl4 or RA and CCl4, along side control groups, for 12 weeks. RA reduced the amount of histologically detectable fibrosis produced by CCl4. This was accompanied by a attenuation of the CCl4 induced increase in ¦Á2(I) collagen mRNA and a lower (2-fold versus 3-fold) increase in liver hydroxyproline. Furthermore, RA reduced the levels of 3-nitrotyrosine (3-NT) protein adducts and thiobarbituric acid (TBA) reactive substance (TBARS) in the liver, which are formed as results of oxidative stress induced by CCl4 treatment. These in vivo findings support our previous in vitro studies in cultured HSC of the inhibitory effect of RA on type I collagen expression. The data also provide evidence that RA reduces CCl4 induced oxidative stress in liver, suggesting that the anti-fibrotic role of RA is not limited to the inhibition of type I collagen expression.Effects of retinoic acid on the development of liver fibrosis produced by carbon tetrachloride in mice - ScienceDirect
https://www.sciencedirect.com/science/article/pii/S0925443906001670ˇˇ
ˇˇ
https://www.researchgate.net/publication/263704518_Th17_Cells_in_Cancer_The_Ultimate_Identity_Crisis
ˇˇ
Vitamin A and retinoic acid in T cell¨Crelated immunity
A Catharine Ross
The American Journal of Clinical Nutrition, Volume 96, Issue 5, November 2012,
ABSTRACT
Interest in vitamin A as a regulator of immune function goes back to the early 1900s. Recently, several lines of evidence have converged to show that retinoic acid (RA), a major oxidative metabolite of vitamin A, plays a key role in the differentiation of T cell subsets, the migration of T cells into tissues, and the proper development of T cell¨Cdependent antibody responses.This review discusses evidence from experimental studies that RA promotes the differentiation of regulatory T cells, which help to suppress inflammatory reactions, and plays a significant role in normal mucosal immunity by modulating T cell activation and regulating cell trafficking. RA also promotes antibody responses to T cell¨Cdependent antigens.
Conversely, in a state of vitamin A deficiency, inflammatory T cell reactions may be inadequately opposed and therefore become dominant.
Although data from human studies are still needed, the framework now developed from studies in mice and rat models suggests that adequate vitamin A status, whether derived from ingestion of preformed retinol or ¦Â-carotene, is important for maintaining a proper balance of well-regulated T cell functions and for preventing excessive or prolonged inflammatory reactions.
INTRODUCTION
The idea that vitamin A is important for immunity goes back to the early 20th century when Edward Mellanby and his colleague Harry Green reported on vitamin A and ¦Â-carotene as ˇ°anti-infectiveˇ± agents (1, 2). Mellanby later recollected that while they were conducting studies on bone health in dogs fed diets lacking in fat-soluble vitamins A and D, they noted the development of bronchopulmonary infections, which they believed were unrelated to the status of the bones, and thus independent of a deficiency of the antirachitic factor, vitamin D (3). Green and Mellanby then undertook studies in rats and reported in 1929¨C1930 that vitamin A conferred protection against infection (1), as did ¦Â-carotene (2).
By the turn of the 21st century, several randomized clinical trials of vitamin A intervention (reviewed in references 4¨C8) had shown that correcting a deficiency of vitamin A in at-risk populations can improve overall outcomes in terms of reduced morbidity and mortality. In young children, reductions in deaths from measles and diarrheal disease are believed to underlie much of this effect (4, 9¨C11). Today, dietary vitamin A, acting through its active metabolite, retinoic acid (RA)5, is recognized as an essential factor for normal immune system development and regulation. Whereas many immune cell types are involved in nearly every disease process, T lymphocytes are central to intestinal mucosal immunity. In the past few years, substantial progress has been made in understanding the roles of vitamin A in the regulation of T cell¨Cdependent responses. This review highlights new information regarding vitamin A and RA in T cell differentiation, regulation of immune responses in the intestine, and the antibody response.
RA IN THE REGULATION OF T CELL DIFFERENTIATION
T cells orchestrate a wide variety of immune responses. For a mature but naive T cell to become an effector T cell, the naive cell must receive multiple signals and integrate them effectively. Signals are derived by cell-cell contacts with antigen-presenting cells, including dendritic cells (DCs), macrophages, and B cells, which are themselves regulated in part by vitamin A and RA (12), and by signals delivered by cytokines present in the cell's immediate environment that bind to receptors on the T cell surface and initiate signal transduction events. The concept that T helper (Th) cells can undergo a process of stable differentiation along 2 distinct pathways, leading alternatively to Th1 and Th2 cells, was proposed by Mosmann et al (13) and Mosmann and Coffman (14) in the late 1980s as a framework for understanding discrete patterns of cytokine secretion observed in cloned, activated CD4+ T cells. Further studies showed that when uncommitted CD4+ T cells are activated through the T cell receptor in a microenvironment rich in the proinflammatory cytokine IL-12, which is produced by activated DCs and macrophages in response to infection or inflammation, as well as the cytokine interferon ¦Ă, produced by natural killer cells and T cells, they become polarized into ˇ°Th1 cells.ˇ± The Th1 cells themselves become producers of interferon ¦Ă as their ˇ°signature cytokine.ˇ± Alternatively, when uncommitted CD4+ T cells are activated in a microenvironment rich in IL-4, a cytokine produced by a variety of cell types, they become polarized into ˇ°Th2 cells.ˇ± These cells then produce IL-4 as their signature cytokine, along with IL-5 and other cytokines. The concept of stably differentiated effector Th1 and Th2 cell subsets stimulated great interest in understanding how pathogens and host environmental factors, including micronutrients, interact to regulate T cell activation and differentiation. Vitamin A deficiency was shown to result in an environment conducive to the differentiation of naive precursor CD4+ T cells into interferon ¦Ă¨Csecreting Th1 cells (15, 16). Conversely, vitamin A and RA generally promote differentiation toward Th2 cells and the production of IL-4 and IL-5 (17¨C21) or increase the ratio of Th2 cytokines relative to Th1 cytokines by reducing the Th1 response (22).
Subsequent molecular studies have shown that the differentiation of Th0 cells into either Th1 or Th2 cells is orchestrated by specific transcription factors. These factors act as master regulators that drive the expression of key genes that, in turn, determine the phenotype of the resulting differentiated T cells. For Th1 cells, the master regulator is T-bet, which, together with another transcription factor, runt-related transcription factor 3, drives the expression of the IFN¦Ă gene and is required for Th1 cell differentiation, while at the same time silencing the IL-4 gene (23). For Th2 cells, the master transcriptional regulator is GATA3, which drives expression of the IL-4 gene and is required for Th2 cell differentiation. Epigenetic changes further stabilize the polarized state of these T cell subsets (24). The cytokines produced by committed Th1 and Th2 cells act in an autocrine-paracrine manner to further regulate local immune responses. In experimental studies, animals or isolated naive T cells treated with appropriate stimuli in the presence of RA have generally expressed higher levels of Th2-associated genes and produced higher amounts of Th2 cytokines, such as IL-4 (19, 20). Alternatively, the relative amount of IL-4 compared with interferon ¦Ă or IL-12 may also be elevated as a result of downregulation of Th1 cytokines, as shown in RA-treated immunized mice (22), which also exhibited reduced expression of the Th1 factors T-bet and interferon regulatory factor 1 (22). In a mouse model of autoimmune insulitis (type 1 diabetes), RA reduced the T cell expression of T-bet as well as Signal Transducers and Activators of Transcription 4, a transducer of IL-12 signals. However, the induction of T-regulatory (Treg) cells, discussed below, was also required for suppression of the islet-infiltrating T cells responsible for this autoimmune disorder (25).
Several additional Th cell lineages have now been identified, and others have been proposed (26). A third major subset, the Th17 cell (27, 28), has emerged as playing a major role in the inflammatory response, especially at mucosal barriers where these cells are most abundant and where they function as effector cells against intracellular pathogens (27). Th17 cells also play an important role in the pathology of autoimmune and allergic disorders, and therefore it is important for multiple reasons to understand what controls their differentiation. Th17 cells are induced when T cells are activated in the presence of transforming growth factor ¦Â (TGF-¦Â) combined with IL-6, which provides a distinct proinflammatory signal. Th17 cells produce IL-17 as their signature cytokine, as well as IL-23, IL-22, IL-6, TNF-¦Á, and chemokines (27, 29), which help recruit neutrophils to sites where the Th17 cells were initially activated. The Th17 cell master regulators are 2 orphan nuclear receptors, retinoid orphan receptor (ROR) ¦Ăt and ROR¦Á, named for their homology to the nuclear retinoid receptor proteins (28, 30). RA has been shown to oppose Th17 cell commitment (29) whereas, as noted below, RA induces regulatory T cells that are critical for maintaining immune homeostasis and for preventing the induction of autoimmune T cells.
Whereas the rapid induction of effector T cell responses, discussed above, is important for host defense, Th1 and especially Th17 cells can cause excessive activation and tissue damage when their effects are prolonged or are not appropriately attenuated. Another subset of CD4+ Th cells, Treg cells, which also are prominent at mucosal surfaces (27), typically perform a suppressive function. Although Treg cells are functionally and phenotypically diverse, they produce cytokines that modulate inflammatory responses, such as IL-10. These cells are indispensable for self-tolerance and have been shown to suppress autoimmune disorders (31). Whereas ˇ°naturalˇ± Treg cells develop in the thymus, Treg cells can also develop in nonthymic tissues in response to infection or inflammation; this subset is referred to as induced Treg (iTreg) cells (31, 32) or adaptive Treg cells (33). As noted later, one crucial role for iTreg cells is in the maintenance of intestinal homeostasis, which is required for tolerance to commensal bacteria and food proteins. iTreg cells are marked by their expression of the Forkhead/winged helix family transcription factor forkhead box P3 (Foxp3), which functions as their master regulator and is essential for iTreg formation (28, 29, 31). Where does vitamin A fit in? Within the past few years, it has become recognized that RA is one of the critical factors that provides signals for the differentiation of iTreg cells (33¨C36). In the presence of TGF-¦Â and adequate amounts of RA, but low IL-6, iTreg differentiation is favored. Moreover, Foxp3 can directly bind to and inhibit the transcriptional activity of ROR¦Á and ROR¦Ăt through protein-protein interactions (27), aiding the induction of iTreg cells. RA also suppresses Th17 cell development by increasing TGF-¦Â signaling and reducing the level of expression of the IL-6 receptor (35). The relative increase in iTreg and the inhibition of Th17 development can shift the balance of these cells toward more effective immune homeostasis. Overall, the balance between ROR¦Ăt+ Th17 and Foxp3+ iTreg cells and their ability to cross-regulate one another (27, 28, 34) have emerged as crucial factors for normal mucosal immunity.
A current model of T cell development and its regulation by RA is shown in Figure 1. Whereas early models suggested that Th cell differentiation resulted in stable, terminally differentiated T cell subsets, there is now increasing evidence for T cell plasticity, ie, the potential for one type of differentiated T cell to develop some of the characteristics of another (26, 37). Plasticity is supported by studies of the epigenetic modification of chromatin, which have shown, for example, that the Foxp3 promoter in some iTreg cells is only partially demethylated, whereas in contrast the Foxp3 promoter in natural thymic Treg cells is fully demethylated, which is indicative of their full commitment to Foxp3 expression (24). Similarly, some iTreg cell histones possess ˇ°bivalentˇ± modifications, such lysine methylation patterns, which suggest that the gene is poised for further modification of its expression, rather than having reached a fixed, terminal state (24). Th cell plasticity is thought to be relatively high in the early stages of CD4+ T cell differentiation toward a given Th subset and then reduced as the T cells are further stimulated with polarizing cytokines and become more strongly committed to their particular subset type (26). It has been stated that plasticity of the T cell compartment allows the immune response to be tailored to the local milieu (28). However, the detection of Th cells that express both Foxp3 and ROR¦Ăt has added complexity and may suggest a kind of ˇ°hybridˇ± T cell or an intermediate stage in T cell differentiation in mucosal tissues. It could also be that iTreg cells can take on some of the characteristics of Th17 cells, albeit with lower IL-17 gene expression, depending on the amount of IL-6 or other factors, including IL-1¦Â, that are present in the local environment (27, 28, 37, 38). In this regard, the ability of the local environment to provide an adequate supply of RA to promote iTreg maintenance, or to be conducive to the conversion of Th17 cells into iTreg cells, could be very important. In current models, RA and IL-6 [and also IL-1¦Â in human T cells (27, 28)] function as binary switches that, on top of the necessary signals provided by TGF-¦Â for both of these differentiation pathways, direct naive T cells along the iTreg pathway or Th17 pathway, respectively (27¨C29). Whereas there is evidence that iTreg cells can convert to Th17-producing cells, evidence for the reverse is lacking (37). The ˇ°reprogrammingˇ± or ˇ°redifferentiationˇ± of iTreg cells into effector Th cells other than Th17 cells has also been reported (37, 39). Fate mapping studies (40) have shown that a substantial number of IL-4¨C and interferon ¦Ă¨Cproducing cells had once expressed Foxp3, which also suggests plasticity over time. It has been cautioned that most studies of T cell plasticity have been conducted at the population level, and thus the programs of differentiation of individual cells need further clarification (37).ˇˇ
Model of T cell differentiation, after contact with antigen-activated DCs, from uncommitted naive T cells into different T cell subsets that produce different cytokines and thus promote different functional activities. Black arrows indicate the cytokines that mainly drive the differentiation to specific subsets. RA together with TGF-¦Â promotes the differentiation of Treg cells, which generally are antiinflammatory, whereas RA also decreases Th1 activity and often promotes Th2 functions (see text). Upward arrows show proposed ˇ°plasticityˇ± or interconversion among some of the T cell subsets (see references 26, 37, and 39 for detailed reviews). The signature cytokines and potential outcomes, which also are determined by the environment and type of pathogen or antigen encountered, are listed. Effects of vitamin A or RA differ contextually but generally limit Th1 and Th17 processes while increasing Th2 and Treg-mediated processes. Ab, antibody; DC, dendritic cells; Fox, forkhead; IFN¦Ă, interferon ¦Ă; NF¦ĘB, nuclear factor kappa-light-chain-enhancer of activated B cells; RA, retinoic acid; ROR¦Ăt, retinoid orphan receptor ¦Ăt; TGF¦Â, transforming growth factor ¦Â; Th, T helper; Treg, T-regulatory.
VITAMIN A IS A NECESSARY FACTOR IN THE HOMING OF T CELLS AND B CELLS IN THE INTESTINE
One way in which a deficiency of vitamin A may increase the risk of morbidity and mortality is through an impaired response to diarrheal infections (4, 7). Vitamin A is necessary for maintaining intestinal integrity (41), regulating mucin gene expression (42), and normal production of intestinal IgA (43). Studies on the status of T cells in the intestine of vitamin A¨Cdeficient mice showed that the submucosal lamina propria region was nearly devoid of CD4+ and CD8+ T cells (44). A lack of lamina propria T cells would likely impair the immune response of the intestine to pathogens that have breeched the epithelium. Next, the expression of chemokine (C-C motif) receptor (CCR) 9 and ¦Á4¦Â7 integrin on the T cell surface was examined in vitamin A¨Cdeficient mice because previous work had shown that the cell-surface chemokine receptor CCR9, involved in T cell migration, and the integrin family adhesion molecule ¦Á4¦Â7, which binds to MAdCAM-1 (mucosal addressin cell adhesion molecule 1), are important for appropriate migration of gut-homing T cells. Both molecules were reduced significantly in vitamin A¨Cdeficient mice (44). In contrast, RA and an analog of RA that functions as a retinoid nuclear receptor ligand increased the expression of CCR9 and increased T cell chemotactic activity. It was also shown that the mesenteric lymph nodes (MLNs) express the gene for retinal dehydrogenase Raldh1a2 (RALDH2) and that 3H-labeled retinol was metabolized to RA by MLN cells ex vivo (44). These studies provided the first line of evidence that RA is essential for ˇ°imprintingˇ± gut-homing specificity on T cells activated by intestinal DCs and suggested that MLN DCs are a source of RA that drives T cell differentiation toward the gut-homing phenotype (45).
Further studies (46) have shown that the iTreg cells formed in the presence of RA are more effective in vivo in suppressing an acute small intestinal inflammation, but they were not more effective in a model of chronic colitis. In other studies, the gut-homing T cells formed in the presence of RA were, after transfer in vivo, more effective than untreated cells in suppressing an acute small intestinal inflammation, but they were not more effective in a model of chronic colitis (47). Thus, high amounts of certain cytokines may overcome the regulatory properties of T cells that are induced by RA. Others have reported finding that the conditions of vitamin A deficiency and vitamin A excess resulted in the induction of different subsets of Foxp3+ T cells, yet, surprisingly, both sets were effective in reversing an intestinal inflammation (48). The differentiation of intestinal mucosal Treg cell responses is initiated by a subset of CD103+ DCs and dependent on TGF-¦Â and RA (49). These results newly identify RA as a cofactor in Treg cell generation, providing a mechanism via which functionally specialized gut-associated lymphoid tissue DCs can extend the repertoire of Treg cells focused on the intestine.
Other studies that have compared MLN cells and cells from peripheral tissues (spleen, cutaneous lymph nodes, or other sites) have also indicated that MLN cells express higher levels of genes likely to be involved in RA metabolism (50). In a study of human CD8+ T cells stimulated with DCs from various sites, T cells cultured with MLN DCs in the presence of RA strongly expressed ¦Á4¦Â7 integrin and CCR9 proteins on their surface, whereas incubation with the retinoid receptor antagonist LE540 completely blocked this expression (51). Furthermore, treatment of activated murine CD8+ T cells in vitro with DCs from different tissue sources, with or without RA, resulted in much higher gut-homing of T cells activated in the presence of MLN DCs and RA (51). However, the increased expression of ¦Á4¦Â7 and CCR9 seems to require continued treatment with RA, because cells that were first stimulated in the presence of RA and later restimulated in its absence were less effective in a gut-homing migration assay (51). These cells therefore might not be fully imprinted but rather plastic in their phenotype.
Oral tolerance to foreign antigens requires a form of immune suppression. The relation between vitamin A and oral tolerance has been investigated in only a few studies, and the results at present are intriguing but complex. Chang et al (52) reported no differences between vitamin A¨Cadequate and vitamin A¨Cdeficient mice in response to oral challenge with ovalbumin, a nonself protein, followed by systemic immunization with the same protein. It was reported that depletion of CCR7+ langerin+ DCs, which were shown to express RALDH2, also resulted in reduced oral tolerance to ovalbumin. This suggests these DCs are important for the immune suppression that is necessary for tolerance to ingested antigens. MLN DCs from vitamin A¨Cdeficient mice, when cocultured with T cells, generated fewer Foxp3+ iTreg cells and more IL-17¨Cproducing T cells, compared with cocultures with MLN DCs from control mice (52). Another study used mice carrying an IL-15 transgene in their DCs, as a model of celiac disease (gluten enteropathy) in humans in which IL-15 expression is elevated, and evaluated whether RA exerted an anti- or proinflammatory effect on isolated cells and in intact mice (53). In the presence of IL-15, RA produced a more inflammatory response, shown by fewer Foxp3+ iTreg cells and a higher production of IL-12p70 and interferon ¦Ă, indicative of an inflammatory Th1 cell response. Both RA and an agonist selective for binding to the nuclear receptor RAR increased c-Jun terminal kinase signaling, which is also consistent with a proinflammatory response. In contrast, the elimination of the RAR¦Á receptor prevented the expression of interferon ¦Ă and IL-12p70 proteins in DCs cultured ex vivo. RA also synergized with IL-15 in intact mice to increase the proinflammatory response to gliadin feeding (53). The results of these 2 studies suggest that whether RA promotes or impairs tolerance to exogenous proteins in the intestine may depend exquisitely on the cytokine environment. It can also be inferred that when RA is used for therapeutic purposes, it should be used cautiously in subjects with various inflammatory bowel conditions and sensitivities to dietary antigens.
OTHER ACTIONS OF VITAMIN A AND RA ON T CELL¨CDEPENDENT IMMUNITY
RA is also an important factor in the response to immunization, which requires collaboration among antigen-presenting cells, T cells, and B cells. Retinoids apparently have opposing effects on T cell and B cell proliferation, limiting the former but stimulating the latter (21, 54¨C57). Several reports have shown that vitamin A deficiency results in a poor response to immunization, with generally low antibody responses to immunization with T cell¨Cdependent antigens (58, 59). The IgA response was lower and the IgG response elevated in a mouse model of viral infection, but nonetheless both responses were dissimilar to the normal response of vitamin A¨Cadequate mice (60). In various animal models, vitamin A deficiency causes abnormalities in nearly all of the lymphocyte populations examined (61¨C63). RA affects numerous B cell processes, both in isolated B cells in vitro and in intact animals, and it interacts with other costimulatory signals, such as cytokines and adjuvants, in a manner that is additive or synergistic (21). Several aspects of the B cell response that are influenced by all-trans-RA are as follows: 1) germline-immunoglobulin gene transcription (56); 2) expression of coreceptor molecules necessary for B and T cell activation (22); 3) rate of cell proliferation as noted above; 4) expression of enzymes such as activation-induced cytidine deaminase (56, 57), an enzyme essential for antibody diversification by class switching and somatic hypermutation (64); 5) formation of the germinal center in which class switching and affinity maturation of antibodies take place (65); and 6) expression of B cell surface markers indicative of plasma cell development (57). In studies conducted in adult and neonatal mice, the antibody titers produced after immunization with tetanus toxoid, a classical T cell¨Cdependent antigen, were higher when RA was administered orally at the time of priming (first immunization). Moreover, the recall response to a second immunization several weeks later was much higher in animals that had received RA with their initial antigen priming (22, 66). This may represent another form of ˇ°imprinting,ˇ± in which memory T and B cells that developed in the presence of RA during the initial response to immunization continue to express differentiated characteristics that permit them to produce a more robust secondary antibody response when they are later stimulated with antigen alone. The results of these studies are relevant to understanding the role of vitamin A in the humoral antibody response to immunization, which is the hallmark of successful vaccination.
THE FIELD CONTINUES TO MOVE AHEAD RAPIDLY
The keen interest in retinoids and their ability to interact with growth factors and cytokines to regulate immune cell differentiation continues, and several interesting reports have appeared since this conference. A few highlights include a study that showed that TGF-¦Â and RA induce the expression of a microRNA, miR-10a, in Treg cells, and that an outcome of this induction is reduction of the transcriptional repressor Bcl-6 and the corepressor N-Cor, which functions with a number of nuclear receptors (67). The expression of miR-10a was shown to maintain the pool of inducible Treg cells and to limit the production of Th17 cells. It also was reported recently that CYP26b1, a cytochrome P450 implicated in RA catabolism, is expressed in antigen-experienced CD44+ T cells. Whereas RA induced CYP26b1 and limited the RA-induced expression of the gut-homing molecule CCR9, the presence of TGF-¦Â limited the expression of CYP26b1 (68). This would be expected to maintain or prolong higher RA concentrations in the cells, although RA concentrations have yet to be measured experimentally. Overall, new pathways are beginning to come into focus through which the RA signal is regulated, which may in turn influence the balance of T cell types and their tissue-homing ability. Other articles not cited here have also contributed recently to the understanding of regulatory lymphocyte populations, and more are expected in the future.
QUESTIONS FOR FUTURE RESEARCH
It is now understood that vitamin A through RA can alter the levels of expression of several hundred genes (50), thus regulating numerous physiologic processes. The discovery that vitamin A has a major impact on intestinal immunity adds to this understanding, and it also suggests important questions for future research. Vitamin A deficiency is an extreme condition, but marginal vitamin A status is relevant in many parts of the world. Does the immune system adapt to marginal vitamin A status, and if so how? Is marginal vitamin A deficiency a risk factor for poor response to infection, or poor tolerance to commensal organisms, or allergic responses to food proteins? As noted elsewhere in this supplement, vitamin A supplementation programs for children and women have been promoted by the WHO and instituted in numerous participating countries (69). Does a large, bolus dose of vitamin A differ in its effect from that of a continuous, adequate dietary intake of vitamin A? Systemic immunity is also affected by vitamin A status, and similar questions about the sources of vitamin A and metabolic processes that generate RA in immune system microenvironments are also relevant. Recent studies have begun to shed light on RA synthesis and degradation pathways in immune system cells (44, 70). As of yet, few studies have addressed vitamin A and immunity in the very young or the elderly, and yet infants, young children, and the elderly are the populations most vulnerable to infections and may have low vitamin A status. ¦Â-Carotene metabolism may also play a role, and it is interesting to note that duodenal infusion of ¦Â-carotene results in higher concentrations of RA in the portal vein (71). This suggests that the mesenteric-splanchnic region may be a significant source of RA in the immediate postprandial period. RA is also a drug, and the impact of pharmacologic doses of RA on intestinal immune function needs to be further evaluated.
Other potential physiologic sources of vitamin A for RA production, both in the intestine and periphery, could include plasma retinol bound to retinol-binding protein (RBP) and chylomicron remnants. If RBP is involved, are there receptors for RBP, such as Stra6 (stimulated by retinoic acid 6) (72), on gut immune cells? Are there chylomicron remnant receptors on antigen-presenting cells that intercept newly absorbed chylomicron retinyl esters as a source of retinol for production of RA, as suggested by studies of bone marrow and blood leukocytes (73)? A challenge for the future is to better understand the connections between vitamin A nutrition, vitamin A metabolism, and immune function in contexts that are relevant to human nutrition. Future experimental studies that combine good animal models studied under conditions of dietary control with broad-based, systems-based informatics approaches are likely to better define the complex influence that vitamin A and RA have on immune system regulation, homeostasis, and response to infectious disease.
The author did not declare any conflicts of interest.ˇˇ
Vitamin A and retinoic acid in T cell¨Crelated immunity | The American Journal of Clinical Nutrition | Oxford Academic
https://academic.oup.com/ajcn/article/96/5/1166S/4577097ˇˇ
Mucosal dendritic cells shape mucosal immunity
Sun-Young Chang, Hyun-Jeong Ko & Mi-Na Kweon
Experimental & Molecular Medicine volume 46, pagee84(2014)Cite this article
Abstract
Dendritic cells (DCs) are key modulators that shape the immune system. In mucosal tissues, DCs act as surveillance systems to sense infection and also function as professional antigen-presenting cells that stimulate the differentiation of naive T and B cells. On the basis of their molecular expression, DCs can be divided into several subsets with unique functions. In this review, we focus on intestinal DC subsets and their function in bridging the innate signaling and adaptive immune systems to maintain the homeostasis of the intestinal immune environment. We also review the current strategies for manipulating mucosal DCs for the development of efficient mucosal vaccines to protect against infectious diseases.ˇˇ
RA is essential for maintaining the intestinal immune environment because it is a determinant for antibody isotype, TH cell and DC subsets.32, 33 The main goal of a mucosal vaccine is to elicit vaccine antigen-specific IgA production in the mucosal tissue of the infection route.
ˇˇ
ˇˇRA is essential for maintaining the intestinal immune environment because it is a determinant for antibody isotype, TH cell and DC subsets.32, 33 The main goal of a mucosal vaccine is to elicit vaccine antigen-specific IgA production in the mucosal tissue of the infection route.
ˇˇ
Secretory IgA production by intestinal DCs. Intestinal pDCs and tip DCs induce IgA production by expressing BAFF and APRIL. Intestinal CD103+CD11b+ DCs, tip DCs and TLR5+ DCs express RALDH2 that is converted into RA and can be used for IgA production.ˇˇ
Antigen uptake using intraepithelial dendrites of lamina propria DCs. (a) CX3CR1+ DCs can sample Salmonella organisms as well as luminal soluble bacterial antigens by extending long dendrites across the epithelium via a CX3CR1-dependent mechanism. (b) CX3CR1+ DCs facilitate the surveillance of circulatory antigens. (c) Intraepithelial CD103+ DCs can be recruited into the intestinal epithelium by luminal bacteria to sample bacterial antigens.Mucosal dendritic cells shape mucosal immunity | Experimental & Molecular Medicine
https://www.nature.com/articles/emm201416\ˇˇ
ˇˇ
ˇˇ
ˇˇ
ˇˇ
ˇˇ
ˇˇ
ˇˇ
ˇˇ