Ceruloplasmin - Definition, Test and Function | Biology ...
https://biologydictionary.net/ceruloplasmin
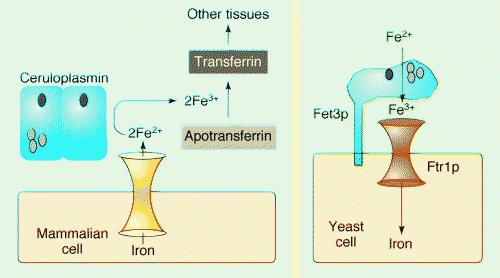
Jul 04, 2017 ・ Ceruloplasmin is a multi-copper serum ferroxidase that needs copper to oxidize ferrous iron (Fe 2+) into ferric iron (Fe 3+) to enable cellular iron transport. Ferrous iron is taken at the cell surface, it is oxidized by ceruloplasminand it is then transported by transferrin , which is an extracellular iron transporter that can carry iron only
in its ferric form (Fe 3+ ).
https://www.frontiersin.org/articles/10.3389/fnut.2018.00103/full
铁是一种新的胆固醇
Iron is the new cholesterol
“谚语:你吃的东西里,只有四分之一用来维持生命,另外四分之三用来养活商人和医生。”
铁过载(iron overload)
铁过载是从癌症到糖尿病等一系列疾病的中心。
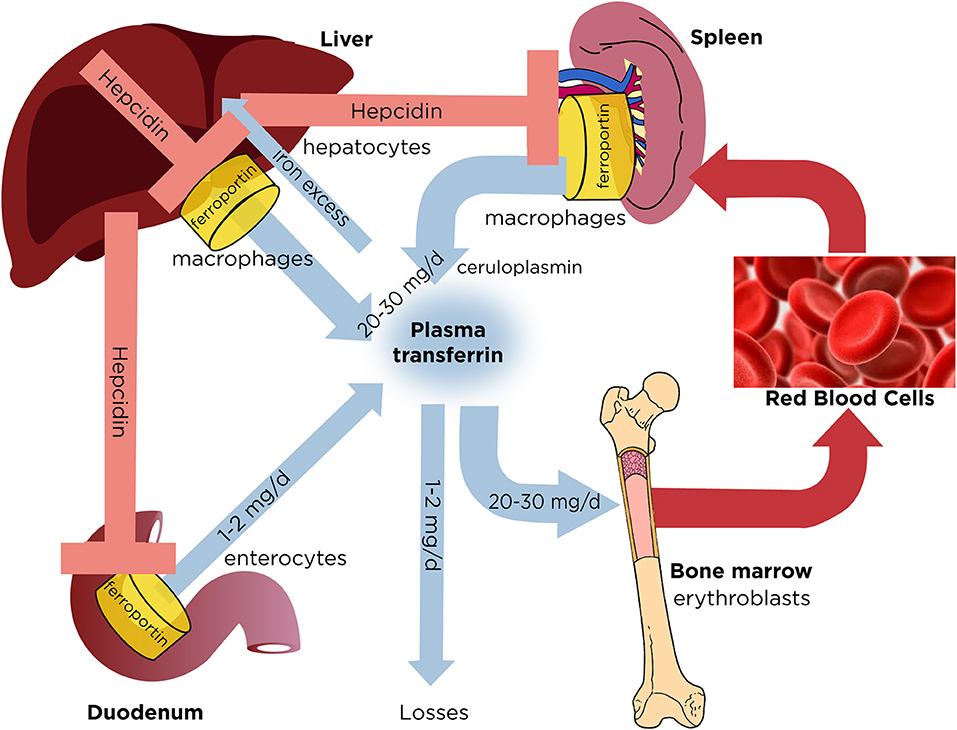
红细胞造血所需要的铁主要来源于三种途径:饮食摄入铁、体内贮存铁、循环血液中衰老的红细胞释放的铁。机体每天只从食物中吸收1-2mg 铁,因此健康成人主要是利用体内衰老的红细胞分解释放的铁。铁在体内的运输主要依靠转铁蛋白,转铁蛋白与细胞膜上的转铁蛋白受体(TfRs)结合,通过内吞作用将铁转运入细胞内。肝脏是储存铁的重要部位。铁的再循环是在网状内皮系统完成的。无论是单细胞还是多细胞动物,生理状态下都缺少排泄铁的机制,因此,体内铁稳态的维持只能通过调节铁的吸收来完成。目前认为,铁调素(hepcidin)是体内铁稳态的重要调节物质。铁调素是由肝脏产生的25个氨基酸构成的多肽。铁调素的代谢失衡是铁代谢异常的多种疾病的病因[2]。除长期大量输血之外,遗传突变是铁过载疾病的首要病因。当各种原因导致铁过载时,过多的细胞内铁会诱导活性氧(ROS)的产生。循环中的铁需与转铁蛋白结合,但是当血浆中的转铁蛋白饱和后,会出现非转铁蛋白结合铁(NTBI,non-transferrin-bound-iron),其中与血浆结合蛋白结合最弱的是LPI(labile plasma iron),LPI具有氧化还原活性,可以进入心脏、肝脏等器官,参与ROS的生成。ROS包括过氧化物、羟基自由基等,可以导致脂质过氧化、DNA氧化损伤、DNA双链断裂、基因不稳定等,造成细胞损伤,过量的铁沉积在不同的器官,临床上可出现肝纤维化、心力衰竭、内分泌功能紊乱、神经系统疾病等表现[4]。
Cheerios是美国最畅销的早餐麦片。根据标签上的说明,这种多谷物食品每份含有18毫克的铁。就像几乎所有由小麦粉制成的精制食品一样,它是用铁强化的。事实上,在防御过程中并没有太多的疏忽。一项研究测量了29种早餐谷物的实际铁含量,发现其中21种含有标签价值的120%,8种含有150%或更多。一个包含了标签价值的200%。
如果你的一碗麦片中含有的铁含量比广告上多20%,也就是22毫克。一个安全的假设是,人们倾向于同时食用至少两种分量的食物。得到44毫克。建议男性每日铁摄入量为8毫克,绝经前女性为18毫克。美国国立卫生研究院(National Institutes of health)认为,成年人每天可耐受的最大摄入量(美国国立卫生研究院认为是安全的)为45毫克。一名普通市民只要吃一碗被认为是相当健康的全麦早餐,就有可能非常接近每日铁摄入量的上限,而这被认为是安全的。
这只是早餐。
与此同时,我们的铁摄入量已经增长到安全的边缘,我们开始明白,高铁水平与从癌症到心脏病等各种疾病有关。波士顿儿童医院(Boston Children’s Hospital)研究铁与糖尿病之间关系的科学家克里斯蒂娜・埃勒维克(Christina Ellervik)这样说:“我们现在对铁的了解就像40年前对胆固醇的了解一样。”
能量代谢的故事――细胞层面生命的基本引擎――是一个电子流动的故事,就像水从山上流向大海一样。我们的细胞可以利用这种流动,通过调节这些电子的运动方式,并在它们运动的过程中从中获取能量。整个装置真的很像水电站。
这些电子流向的海洋就是氧气,而对于地球上的大多数生命来说,铁就是河流。(章鱼是奇怪的异类――它们用铜代替铁,这使得它们的血液呈蓝绿色而不是红色)。氧对电子的渴求使它成为一个理想的目的地。促进运输的蛋白质含有微小的铁核,当电子被运送到氧气时,铁核负责电子的处理。
这就是为什么铁和氧对生命都是必不可少的。然而,这种细胞田园也有其黑暗的一面。
氧和铁是产生能量所必需的,但也可能共同破坏我们细胞的微妙秩序。
细胞内正常的能量代谢产生低水平的有毒副产品。其中一个副产品是氧的衍生物,叫做超氧化物。幸运的是,细胞中含有几种酶,几乎可以立即清除大部分泄漏的超氧化物。他们通过将其转化为另一种叫做过氧化氢的介质来达到这一目的,过氧化氢可以在你的药柜里用来治疗伤口和擦伤。然后过氧化氢被解毒成水和氧气。
如果超氧化物或过氧化氢在解毒的过程中碰巧遇到一些铁,事情就会出错。接下来发生的是一系列化学反应(Haber-Weiss chemistry和Fenton chemistry描述了这些化学反应),它们产生一种叫做羟基自由基的强效活性氧衍生物。这种激进分子,也被称为自由激进分子,在任何地方都对生物分子造成破坏。巴里・哈利维尔的化学家和约翰Gutteridge-who写了一本关于铁biochemistry-put,“羟基自由基的反应如此之大,如果它们形成于生命系统,他们将立即与任何生物分子反应在他们的附近,产生二次变量反应的自由基。”2
这就是这个星球上的生命所达成的浮士德式的交易。氧和铁是产生能量所必需的,但也可能共同破坏我们细胞的微妙秩序。正如神经学家j・r・康纳(J.R. Connor)所说,“生命的存在本来就是存在于铁元素充足和缺乏之间的交界地带。
在20世纪末,铁在人体中的代谢仍然是一个谜。科学家们只知道身体能够排出铁血的两种方式,以及皮肤和胃肠细胞的常规脱落。但这些过程每天只相当于几毫克。这意味着身体必须有某种方式来严格控制饮食中的铁的吸收。2000年宣布了一项重大突破――发现了一种蛋白质,它具有铁的主调节功能。这个系统,就像许多生物系统一样,是完美优雅的。当铁含量足够时,这种被称为铁调素(hepcidin)的蛋白质被肝脏分泌到血液中。然后,它向胃肠道细胞发出信号,减少它们对铁的吸收,并向身体周围的其他细胞发出信号,将它们的铁隔离到铁蛋白中,铁蛋白是一种储存铁的蛋白质。当铁水平较低时,血液中的hepcidin水平下降,肠道细胞开始重新吸收铁。自那以后,铁调素Hepcidin被认为是人体铁稳态的主要调控因子。但是,如果hepcidin如此巧妙地调节饮食对铁的吸收,以满足身体的需要,那么任何人都有可能吸收过多的铁吗?
1996年,一组科学家宣布他们发现了导致遗传性血色素沉着症的基因,这是一种导致身体吸收过多铁的疾病。他们称之为HFE。随后的研究表明,HFE基因的产物在调节hepcidin方面起着重要作用。在hepcidin所协调的整个调节装置中,该基因具有可遗传突变的人实际上存在严重障碍。
这就为我们提供了一种可能性,那就是我们中的一些人实际上摄入的铁超过了身体所能承受的量。但是这些突变有多常见呢?甚至对阅读这些单词的少数人也有影响吗?
令人惊讶的是,答案是肯定的。遗传性血色素沉着病的患病率实际上相当高――在美国,每200例中就有1例存在HFE基因的缺陷副本,并有铁超载的临床迹象。也许每40个人中就有一个人有两个缺陷的HFE基因而没有明显的血色素沉着。这意味着超过800万的美国人在调节铁的吸收和代谢方面可能会出现明显的短路。
如果你只有一个有缺陷的HFE基因,和一个完全正常的基因呢?这叫做杂合性。我们期望在这种情况下找到比纯合子更多的人,或者那些有两个坏拷贝基因的人。事实上,我们是这样做的。目前的估计表明,超过30%的美国人口可能是杂合子,带有一个功能失调的HFE基因。4 . That’s pretty close to一亿people.那接近一亿人。
这有关系吗?或者一个好的基因就足够了吗?目前还没有太多的研究,但到目前为止的证据表明,一些杂合子确实损害了铁的代谢。研究表明,HFE杂合子中铁蛋白和转铁蛋白的含量似乎有适度的升高,转铁蛋白是一种伴随铁通过血液的蛋白质,这表明铁含量升高。5、6和2001年发表的一项研究得出结论,HFE杂合子发生铁超载的风险可能高达四倍
许多研究文章都支持铁与癌症之间的联系。
也许更令人担忧的是,这些杂合子也被证明会增加患心脏病和中风等几种慢性病的风险。一项研究发现,吸烟的杂合子患心血管疾病的风险是对照组的3.5倍,而另一项研究发现,单是杂合子就会显著增加心脏病和中风的风险。8第三项研究发现杂合性使导致心力衰竭的心肌病的风险增加了近六倍过量的铁与心血管疾病之间的联系可能超出了杂合子的范围。最近的一项荟萃分析确定了55项关于这种联系的研究,这些研究足够严格,符合纳入标准。55项研究中,27项支持铁与心血管疾病之间存在正相关关系(铁越多,疾病越多),20项没有发现明显的关系,8项发现负相关关系(铁越多,疾病越少)
几个亮点:斯堪的纳维亚的一项研究比较了患有心脏病的男性和没有患心脏病的男性,发现高铁蛋白水平会使心脏病发作的风险增加两到三倍。另一项研究发现,高铁蛋白水平使心脏病发作的可能性是正常水平的5倍。一项针对2000名芬兰男性的大型研究发现,铁蛋白水平升高会使心脏病发作的风险增加两倍,铁蛋白水平每增加1%,心脏病发作的风险就会增加4%。在这项研究中发现的唯一一个比铁蛋白更强的风险因素是吸烟。
然而,铁蛋白并不是铁状态的完美标志,因为它也会受到任何引起炎症的因素的影响。为了解决这个问题,一组加拿大研究人员直接将血液中的铁含量与心脏病发作的风险进行了比较,发现较高的铁含量会使男性患心脏病的风险增加两倍,女性患心脏病的风险增加五倍。
如果心血管疾病是铁的疾病网中的一个点,那么糖尿病可能是另一个。铁与糖尿病之间的关系最早出现在上世纪80年代末,当时研究人员发现,定期输血(其中含有相当多的铁)的患者患糖尿病的风险显著增加。在血色素沉着症中,没有办法知道葡萄糖代谢的相关紊乱是由于铁本身的积累,还是潜在的遗传缺陷。这种频繁输血和糖尿病之间的新联系间接证明了铁本身可能是导致糖尿病的原因。
下一步是挖掘现有的数据,寻找铁含量与糖尿病之间的关联。第一个这样做的研究于1997年来自芬兰:在随机选择的1000名斯堪的纳维亚男性中,铁蛋白作为一种功能失调的葡萄糖代谢的强预测因子出现,仅次于身体质量指数作为一种风险因素。1999年,研究人员发现,高铁蛋白水平会使男性患糖尿病的几率增加5倍,而女性患糖尿病的几率则会增加近4倍――这与肥胖和糖尿病之间的关联程度相当。12五年后,另一项研究发现,铁蛋白水平升高会使代谢综合征的风险增加一倍。代谢综合征通常会导致糖尿病、高血压、肝病和心血管疾病。克里斯蒂娜・埃勒维克(Christina Ellervik)在该领域的首次贡献是在2011年,她的一项研究调查了转铁蛋白饱和度增加与糖尿病风险之间的关系。埃勒维克发现,在近35,000名丹麦人的样本中,转铁蛋白饱和度超过50%的人患糖尿病的风险增加了两到三倍。她还发现,当转铁蛋白饱和度超过50%时,死亡率会增加。
2015年,她领导的另一项研究发现,在6000人的样本中,铁蛋白水平最高的20%的人患糖尿病的几率是铁蛋白水平最低的20%的人的4倍。铁蛋白水平越高,血糖水平、胰岛素水平和胰岛素敏感性越高。
“令人难以置信的是,有这么多有前途的文献,却没有人做临床试验。”
但这里有个问题。所有这些研究都显示了关联。它们表明两件事往往同时发生。但它们没有告诉我们任何因果关系。要了解因果关系,你需要干预。对于铁,你需要降低铁的浓度,然后观察会发生什么。幸运的是,有一种非常简单、非常安全的方法可以降低铁的含量,这种方法每年都要做上百万次――静脉切开术,也就是献血。
最早使用静脉切开术检查铁和糖尿病之间关系的研究之一发表于1998年16月,作者发现,在健康和糖尿病患者中,静脉切开术都能改善胰岛素敏感性和葡萄糖代谢。2005年的一项研究发现,经常献血的人比不献血的人表现出更低的铁储量和更强的胰岛素敏感性。2012年,研究人员对糖尿病前期志愿者进行了静脉抽血,直到他们的铁蛋白水平显著下降,随后发现他们的胰岛素敏感性明显改善。同年,另一组科学家研究了献血对包括葡萄糖代谢在内的多种代谢综合征的影响。他们发现,一次献血可以在六周后改善血压、空腹血糖、糖化血红蛋白(平均血糖水平的标志)和血液胆固醇
许多警告适用于这一证据――相关性和因果关系之间的界限仍不清楚,一些研究使用相对较小的样本量,而献血可能导致除降低铁之外的其他变化。但综合来看,这些数据为铁在糖尿病曲折的病理生理过程中扮演重要角色的观点提供了支持。
随着越来越多的公开数据开始表明铁、心血管疾病和糖尿病之间的关系,研究人员开始撒网。
接下来是癌症。早在20世纪50年代末,科学家就已经知道向实验动物注入大量的铁会导致恶性肿瘤,但直到20世纪80年代,科学家才开始寻找铁与人类癌症之间的联系。1985年,欧内斯特・格拉夫和约翰・伊顿提出,各国结肠癌发病率的差异可以用当地饮食中纤维含量的差异来解释,而纤维含量的差异反过来又会影响铁的吸收
次年,理查德•史蒂文斯(Richard Stevens)发现,在2万名中国男性中,铁蛋白水平升高与死于癌症的风险增加了两倍。21两年后,史蒂文斯发现,美国癌症患者的转铁蛋白饱和度和血清铁含量高于未患癌症的患者。在1990年,一项针对瑞典献血者的大型研究发现,与非献血者相比,他们患癌症的可能性要低20%。23四年后,一组芬兰研究人员发现,4万斯堪的纳维亚人体内转铁蛋白饱和度的升高使结直肠癌的风险增加了3倍,肺癌的风险增加了1.5倍
自从Graf和伊顿公学的第一篇论文发表以来,已经发表了大量的研究论文,其中大多数支持铁与癌症之间的联系,尤其是结肠癌。2001年,一项对33篇研究铁与结直肠癌之间关系的出版物的综述发现,超过75%的出版物支持铁与结直肠癌之间的关系。2004年的一项研究发现,随着血清铁和转铁蛋白饱和度的升高,死于癌症的风险增加。癌症水平最高的人死于癌症的可能性是癌症水平最低的人的两倍。在2008年,另一项研究证实瑞典的献血者患癌症的风险降低了30%
还有其他一些证据支持铁与癌症之间的联系。HFE基因突变的人患结肠癌和血癌的风险更高。28 .相反,被诊断患有乳腺癌、血癌和结肠癌的人与健康对照组相比,HFE杂合子的可能性要高出一倍以上
也有一些介入试验研究铁和癌症之间的关系。首先发表在2007年由一群日本科学家曾发现铁减少通过放血基本上归一化标记丙型肝炎患者肝损伤的肝细胞癌(HCC)是一个可怕的后果丙型肝炎和肝硬化,他们推测,放血也可能降低患癌症的风险。结果非常显著――在5年的时间里,接受献血的组中只有5.7%的患者发生了HCC,而对照组的这一比例为17.5%。10年后的研究结果更加令人震惊,8.6%的被抽血的患者发生HCC,而对照组中这一比例高达39%。第二年,达特茅斯学院(Dartmouth)名誉教授利奥•扎卡尔斯基(Leo Zacharski)发表了第二项研究,旨在研究献血对癌症风险的影响。在一项多中心、随机化的研究中,原本是为了观察献血对血管疾病的影响,在4.5年后,减铁组患者患癌症的几率比对照组低35%左右。在所有的癌症患者中,献血组的患者在随访结束时死于癌症的可能性要低60%
大脑是一个饥饿的器官。虽然只占身体质量的2%到3%,但它消耗了身体总需氧量的20%。新陈代谢如此之快,大脑在消耗氧气的同时不可避免地会产生更多的自由基。令人惊讶的是,研究表明,大脑的抗氧化能力似乎比身体其他组织要低,这可能会使大脑更容易受到氧化应激的影响。正常细胞能量代谢和活性氧损伤之间的平衡,在大脑中可能比在身体的其他地方更为微妙。这反过来又表明了对铁的敏感性。
早在20世纪20年代,人们就知道阿尔茨海默氏症和帕金森氏症等神经退行性疾病与大脑铁沉积增加有关。1924年,一位杰出的巴黎神经学家Jean Lhermitte是第一个发现晚期帕金森氏症患者大脑某些区域会因异常的铁含量而充血的人之一。33三十年后的1953年,一位名叫路易斯・古德曼(Louis Goodman)的医生证明,阿尔茨海默氏症患者的大脑中存在明显异常的铁沉积水平,这些铁沉积的区域与定义这种疾病的著名斑块和缠结所在的区域相同。古德曼的工作在几十年里基本上被遗忘了,直到1992年的一篇论文重新发表,证实了他的发现,并引发了新的兴趣。两年后,一项令人兴奋的新技术――核磁共振(MRI)被应用于研究铁与疾病之间的关系,证实了早期尸检结果,即阿尔茨海默症患者的大脑组织铁存在明显的异常
Zacharski确信,铁超载是一个巨大的共同支点,在许多正在席卷西方国家的慢性代谢疾病的基础上。到上世纪90年代中期,有令人信服的证据表明,阿尔茨海默病和帕金森病与大脑中铁代谢的某些失调有关,但没有人知道这种关系是疾病过程的原因还是后果。与此同时,核磁共振成像的发现也陆续出现了一些蛛丝马迹。1993年的一篇论文报道称,铁促进淀粉样蛋白b的聚集,而淀粉样蛋白b是阿尔茨海默病斑块的主要成分。1997年,研究人员发现,与阿尔茨海默病斑块相关的异常铁具有高度反应性,能够自由地产生有毒的氧自由基。37到2010年,已经证明氧化损伤是与阿尔茨海默氏症相关的最早可检测到的变化之一,而且反应性铁存在于该病的早期阶段。38、39和2015年,一项为期7年的纵向研究表明,脑脊液铁蛋白水平是认知能力下降和阿尔茨海默氏症(Alzheimer’s痴呆)发展的一个强有力的预测因子
也许最令人惊讶的是1999年的发现,淀粉样蛋白b的前驱体受细胞铁水平的直接控制――周围的铁越多,产生的淀粉样蛋白就越多。41 .这就提出了一种诱人的可能性,即淀粉样斑块实际上可能代表一种适应性反应,而不是病因,这一想法得到了间接的支持,因为几乎所有直接针对淀粉样蛋白治疗该病的努力都以失败告终。
总之,这些发现表明,大脑中异常的铁代谢可能是阿尔茨海默氏症和其他神经退行性疾病的致病因素。如果这是真的,那么我们可能会认为,从基因上来说,有铁代谢异常倾向的人患痴呆的风险比其他人要高。事实也是如此。
在本世纪初,人们发现家族性阿尔茨海默氏症患者比健康对照组更有可能拥有HFE基因之一。42另一项研究发现,与对照组相比,这些基因型与疾病的早期发病有关,而且在HFE和ApoE4基因(阿尔茨海默氏症的主要遗传风险因素)的人群中,这种基因型的影响甚至更大。43 2004年的一项研究表明,HFE基因与一种已知的转铁蛋白基因变体同时出现,使患老年痴呆症的风险增加了5倍。44两年后,一组葡萄牙科学家发现HFE变异也与帕金森病的风险增加有关。
那么介入性试验呢?对于神经退行性疾病,只有一个。1991年,一组加拿大科学家发表了一项为期两年的随机试验结果,该试验对48名阿尔茨海默病患者进行铁螯合剂脱氧铁恶胺(desferrioxamine)治疗。螯合剂是一种药物,可以结合铁等金属阳离子,隔离它们,并促进它们从体内排出。随机分配患者接受去铁立沙明、安慰剂或不治疗。结果令人印象深刻――两年后,铁的减少使认知能力下降的速度降低了一半。
这项研究发表在世界上最负盛名的医学杂志之一《柳叶刀》上,但似乎在这20多年间被遗忘了。自那以后,还没有发表过任何一项测试铁在阿尔茨海默氏症中的作用的介入研究。
如果这么多的研究似乎表明铁水平与慢性病之间存在一致的联系,那么为什么没有做更多的工作来阐明这种风险呢?
达特茅斯大学的扎卡尔斯基对我说:“令人难以置信的是,有这么多有前途的文献,却没有人做临床试验。”“如果人们接受挑战,对铁假说进行精心设计、富有洞察力的研究,我们就会对这个问题有更坚定的理解。”想象一下,如果它被证实了!”
他对为什么还没有进行更多试验的观点很吸引人,与该领域其他专家的观点在很大程度上是一致的。信不信由你,“性感”这个词出现在许多谈话中――分子生物学和靶向药物很热门(而且有利可图),而铁肯定不是。“也许它不够性感,太过时了,太老套了,”我采访过的一位研究人员说。扎卡尔斯基在我们的谈话中也提到了这一点,他指出,许多现代试验都是由制药行业资助的,制药行业热衷于开发下一种价值10亿美元的药物。像NIH这样的政府机构可以介入填补营利性研究行业留下的空白,但是公共资助的科学家和其他人一样容易受到性别偏见的影响。正如一位资深大学科学家告诉我的,“NIH追求时尚。”
Zacharski确信,铁超载是一个巨大的共同支点,在许多正在席卷西方国家的慢性代谢疾病的基础上。他认为,即使是微量的铁含量升高也会导致自由基的形成,进而导致慢性炎症。我们知道,从心脏病到糖尿病,从癌症到老年痴呆症,慢性炎症与所有疾病都有密切联系。
“如果这不值得进行随机试验,”他告诉我,“那么我不知道什么值得。”在那些随机试验到来之前,我们在血库见。
克莱顿・道尔顿是波士顿麻省总医院的急诊医生。他曾与美国国家公共广播电台(NPR)、《永安》(Aeon)和《洛杉矶评论》(Los Angeles Review)发表过故事和文章。
References
1. Whittaker, P., Tufaro, P.R., & Rader. J.I. Iron and folate in fortified cereals. The Journal of the American College of Nutrition 20, 247-254 (2001).
2. Halliwell, B. & Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochemical Journal 219, 1-14 (1984).
3. Connor, J.R. & Ghio, A.J. The impact of host iron homeostasis on disease. Preface. Biochimica et Biophysica Acta 1790, 581-582 (2009).
4. Hanson, E.H., Imperatore, G., & Burke, W. HFE gene and hereditary hemochromatosis: a HuGE review. Human Genome Epidemiology. American Journal of Epidemiology 154, 193-206 (2001).
5. Beutler, E., Felitti, V.J., Koziol, J.A., Ho, N.J., & Gelbart, T. Penetrance of 845G―> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. The Lancet 359, 211-218 (2002).
6. Rossi, E., et al. Effect of hemochromatosis genotype and lifestyle factors on iron and red cell indices in a community population. Clinical Chemistry 47, 202-208 (2001).
7. Roest, M., et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation 100, 1268-1273 (1999).
8. Tuomainen, T.P., et al. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: A prospective cohort study in men in eastern Finland. Circulation 100, 1274-1279 (1999).
9. Pereira, A.C., et al. Hemochromatosis gene variants in patients with cardiomyopathy. American Journal of Cardiology 88, 388-391 (2001).
10. Muñoz-bravo, C., Gutiérrez-bedmar, M., Gómez-aracena, J., García-rodríguez, A., & Navajas, J.F. Iron: protector or risk factor for cardiovascular disease? Still controversial. Nutrients 5, 2384-2404 (2013).
11. Tuomainen, T.P., et al. Body iron stores are associated with serum insulin and blood glucose concentrations. Population study in 1,013 eastern Finnish men. Diabetes Care 20, 426-428 (1997).
12. Ford, E.S. & Cogswell, M.E. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 22, 1978-1983 (1999).
13. Jehn, M., Clark, J.M., & Guallar, E. Serum ferritin and risk of the metabolic syndrome in U.S. adults. Diabetes Care 27, 2422-2428 (2004).
14. Ellervik, C., et al. Elevated transferrin saturation and risk of diabetes: three population-based studies. Diabetes Care 34, 2256-2258 (2011).
15. Bonfils, L., et al. Fasting serum levels of ferritin are associated with impaired pancreatic beta cell function and decreased insulin sensitivity: a population-based study. Diabetologia 58, 523-533 (2015).
16. Facchini, F.S. Effect of phlebotomy on plasma glucose and insulin concentrations. Diabetes Care 21, 2190 (1998).
17. Fernández-real, J.M., López-bermejo, A., & Ricart, W. Iron stores, blood donation, and insulin sensitivity and secretion. Clinical Chemistry 51, 1201-1205 (2005).
18. Gabrielsen, J.S., et al. Adipocyte iron regulates adiponectin and insulin sensitivity. Journal of Clinical Investigation 122, 3529-3540 (2012).
19. Houschyar, K.S., et al. Effects of phlebotomy-induced reduction of body iron stores on metabolic syndrome: results from a randomized clinical trial. BMC Medicine 10:54 (2012).
20. Graf, E. & Eaton, J.W. Dietary suppression of colonic cancer. Fiber or phytate?. Cancer 56, 717-718 (1985).
21. Stevens, R.G., Beasley, R.P., & Blumberg, B.S. Iron-binding proteins and risk of cancer in Taiwan. Journal of the National Cancer Institute 76, 605-610 (1986).
22. Stevens, R.G., Jones, D.Y., Micozzi, M.S., & Taylor, P.R. Body iron stores and the risk of cancer. New England Journal of Medicine 319, 1047-1052 (1988).
23. Merk, K., et al. The incidence of cancer among blood donors. International Journal of Epidemiology 19, 505-509 (1990).
24. Knekt, P., et al. Body iron stores and risk of cancer. International Journal of Cancer 56, 379-382 (1994).
25. Nelson, R.L. Iron and colorectal cancer risk: human studies. Nutrition Review 59, 140-148 (2001).
26. Wu, T., Sempos, C.T., Freudenheim, J.L., Muti, P., & Smit, E. Serum iron, copper and zinc concentrations and risk of cancer mortality in US adults. Annals of Epidemiology 14, 195-201 (2004).
27. Edgren, G., et al. Donation frequency, iron loss, and risk of cancer among blood donors. Journal of the National Cancer Institute 100, 572-579 (2008). 28. Nelson, R.L., Davis, F.G., Persky, V., & Becker, E. Risk of neoplastic and other diseases among people with heterozygosity for hereditary hemochromatosis. Cancer 76, 875-879 (1995).
29. Weinberg, E.D. & Miklossy, J. Iron withholding: a defense against disease. Journal of Alzheimer’s Disease 13, 451-463 (2008).
30. Kato, J., et al. Long-term phlebotomy with low-iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. Journal of Gastroenterology 42, 830-836 (2007).
31. Zacharski, L.R., et al. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. Journal of the National Cancer Institute 100, 996-1002 (2008).
32. Lee, H.G., et al. Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses. Journal of Pharmacology and Experimental Therapeutics 321, 823-829 (2007).
33. Lhermitte, J., Kraus, W.M., & Mcalpine, D. Original Papers: On the occurrence of abnormal deposits of iron in the brain in Parkinsonism with special reference to its localisation. Journal of Neurology and Psychopathology 5, 195-208 (1924).
34. Goodman, L. Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. The Journal of Nervous and Mental Disease 118, 97-130 (1953).
35. Bartzokis, G., et al. In vivo evaluation of brain iron in Alzheimer’s disease and normal subjects using MRI. Biological Psychiatry 35, 480-487 (1994). 36. Mantyh, P.W., et al. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. Journal of Neurochemistry 61, 1171-1174 (1993).
37. Smith, M.A., Harris, P.L., Sayre, L.M., & Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proceedings of the National Academy of Sciences 94, 9866-9868 (1997).
38. Nunomura, A., et al. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology 60, 759-767 (2001).
39. Smith, M.A., et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. Journal of Alzheimer’s Disease 19, 363-372 (2010).
40. Ayton, S., Faux, N.G., & Bush, A.I. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nature Communications 6:6760 (2015).
41. Rogers, J.T., et al. Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5’-untranslated region sequences. Journal of Biological Chemistry 274, 6421-6431 (1999).
42. Moalem, S., et al. Are hereditary hemochromatosis mutations involved in Alzheimer disease? American Journal of Medical Genetics 93, 58-66 (2000).
43. Combarros, O., et al. Interaction of the H63D mutation in the hemochromatosis gene with the apolipoprotein E epsilon 4 allele modulates age at onset of Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders 15, 151-154 (2003).
44. Robson, K.J., et al. Synergy between the C2 allele of transferrin and the C282Y allele of the haemochromatosis gene (HFE) as risk factors for developing Alzheimer’s disease. Journal of Medical Genetics 41, 261-265 (2004).
45. Pulliam, J.F., et al. Association of HFE mutations with neurodegeneration and oxidative stress in Alzheimer’s disease and correlation with APOE. American Journal of Medical Genetics; Part B 119B, 48-53 (2003).
46. Crapper-McLachlan, D.R., et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. The Lancet 337, 1304-1308 (1991).
The role of iron metabolism as a mediator of macrophage inflammation and lipid handling in atherosclerosis
Iron is an essential mineral needed for normal physiologic processes. While its function in oxygen transport and other important physiologic processes is well known, less is understood about its role in inflammatory diseases such as atherosclerosis. Existing paradigms suggest iron as a driver of atherosclerosis through its actions as a pro-oxidant capable of causing lipid oxidation and tissue damage. Recently we and others have identified hemoglobin (Hb) derived iron as an important factor in determining macrophage differentiation and function in areas of intraplaque hemorrhage within human atherosclerosis. Hb associated macrophages, M(Hb), are distinct from traditional macrophage foam cells because they do not contain large amounts of lipid or inflammatory cytokines, are characterized by high levels of expression of mannose receptor (CD206) and CD163 in addition to producing anti-inflammatory cytokines such as IL-10. Despite the well-known role of iron as an catalyst capable of producing lipid peroxidation through generation of reactive oxygen species (ROS) such as hydroxyl radical, we and others have shown that macrophages in areas of intraplaque hemorrhage demonstrate reduced intracellular iron and ROS which triggers production of anti-inflammatory cytokines as well as genes involved in cholesterol efflux. These data suggest that manipulation of macrophage iron itself may be a promising pharmacologic target for atherosclerosis prevention through its effects on macrophage inflammation and lipid metabolism. In this review we will summarize the current understanding of iron as it relates to plaque inflammation and discuss how further exploration of this subject may lead to new therapies for atherosclerosis.
Frontiers | The role of iron metabolism as a mediator of macrophage inflammation and lipid handling in atherosclerosis | Pharmacology
https://www.frontiersin.org/articles/10.3389/fphar.2014.00195/full
铁蛋白和转铁蛋白受体更新 发布时间:2019-05-28 作者:DDM 来源:临床实验室 浏览:4470 收藏: 0 1. 铁、生命和转铁蛋白 地球上最早的生物诞生于古老的海洋。那时水是热的和酸的,并含有高浓度的铁和硫离子。环境中存在极少的氧气;因此,早期的生物开始利用铁和硫合成铁硫簇,用于各种生物活动,包括呼吸。大约24亿年前,蓝藻(一种光合生物)的大规模繁殖发生,极大地改变了全球环境。大量氧气产生,大部分铁被氧化和沉淀在海底。因为有氧呼吸比无氧呼吸更有效地产生能量,所以好氧生物开始扩大;一个好氧生物碰巧进入厌氧生物,形成共生关系并产生第一个线粒体。然后,厌氧生物形成了一种能力,能够利用线粒体产生的ATP,并产生核膜以保护其遗传信息免受线粒体产生的活性氧物质(ROS)影响。真核生物就这样诞生了。 有些真核生物最终演变为多细胞生物,然后成为多细胞动物和脊椎动物。脊椎动物从小肠中吸收铁,并通过血管运送至体内的所有细胞。由于铁介导高毒性ROS的产生,所以脊椎动物产生了一种铁载体蛋白,即转铁蛋白(transferrin,Tf),可以将铁安全携带至循环中。Tf及其细胞受体已得到广泛研究,近期很多发现增加了我们对铁转运和氧化应激调节的认识。 本文更新并概述了我们对于哺乳动物Tf及其两种受体(TfR1和TfR2)的认识,主要关注其功能、表达和临床用途。 2. 转铁蛋白 所有脊椎动物都有功能性Tf或卵铁传递蛋白,其在爬行类和鸟类中具有铁结合及抗菌特性。人Tf是一种76kDa的糖蛋白,主要在肝脏中产生,在血清中的半衰期约为8天。Tf由N-和C-末端的两个叶片(lobe)组成;这些叶片具有很高的同源性,并通过短连接区连接。每个叶片可以不同的亲和力结合一种金属离子,如铁、镓、铝、锌或镉;因此,一个Tf分子可携带两个三价铁离子(Fe3+)。Fe3+与Tf之间的相互作用取决于pH,它在pH 7.4时与Tf高效结合,并在酸性pH下与Tf分离,比如在核内体中。人血清Tf的浓度约为200-300mg/dL,在肝功能障碍或炎症条件下浓度下降。血清中的大多数铁与Tf共存,血清Tf可以是非铁结合(脱铁-Tf)、单铁或双铁(全铁-Tf)形式。Tf饱和度(TSAT)用血清铁浓度除以总铁结合力来计算,反映血清Tf的浓度。TSAT低于20%表示铁缺乏,而TSAT高于50%表示铁超载。当TSAT超过80%-85%时,具有高毒性的非Tf结合铁(NTBI)出现于血清中,并导致器官损伤。 先天缺乏Tf的小鼠,如果不用外源性Tf治疗或输注红细胞,则会死于严重贫血。虽然具有严重的缺铁性贫血,但肝脏、肾脏、心脏、肾上腺和胰腺等非造血组织出现严重铁超载,表明这些组织中存在与Tf无关的铁摄取机制。 3. TfR1 3.1 TfR1介导的细胞铁摄取 Tf结合铁主要通过TfR1介导的机制进入需要铁调节的细胞。基本上所有脊椎动物都有TfR1。TfR1是一种97-kDa的2型膜蛋白,作为同源二聚体表达于细胞膜中。在细胞表面,全铁-Tf(holo-Tf)与TfR1结合,该复合物在网格蛋白介导的内吞作用下开始内在化。TfR1与Tf之间的相互作用取决于pH;在pH 7.4时,TfR1与铁饱和的全铁-Tf结合,但不与无铁的脱铁-Tf(apo-Tf)结合。相反,在核内体的较低pH下,TfR1与脱铁-Tf结合但不与全体-Tf结合。在核内体的酸性环境下,Fe3+与Tf分离。Fe3+在金属还原酶比如前列腺六次跨膜上皮抗原3(Steap3)的作用下转变为Fe2+,并经由二价金属离子转运蛋白1(DMT1)转运至胞质溶胶中。然后,核内体中的Tf/TfR1复合物转运至再循环内体的细胞表面,脱铁-Tf从细胞表面释放到血流中(图1)。因而,细胞通过Tf/TfR1系统有效地吸收血清铁,这个过程完成后TfR1和Tf被循环利用,进入细胞摄取铁的另一个周期(图1)。 技术导航-转铁蛋白-图1.jpg 图1. 转铁蛋白受体1(TfR1)介导的转铁蛋白(Tf)和铁蛋白吸收。 (左)Tf吸收。在细胞表面,铁饱和的全铁-Tf与TfR1结合,该复合物通过网格蛋白介导的内吞作用内在化。在核内体的酸性环境下,Fe3+与Tf分离,并在金属还原酶比如前列腺六次跨膜上皮抗原3(STEAP3)的作用下还原为Fe2+,然后经由二价金属离子转运蛋白1(DMT1)转运至胞质溶胶中。核内体中的全铁-Tf/TfR1复合物转运至再循环内体的细胞表面,脱铁-Tf释放到血流中。STEAP3在有核红细胞中高表达并与TfR1相互作用,但不在淋巴细胞中高表达。(右)铁蛋白吸收的假设图示。铁蛋白是另一种TfR1配体。人胞质铁蛋白由24个H-和L-亚基以各种比率组成,但只有H-亚基可与TfR1相互作用。只有TfR1表达超过一定阈值水平的细胞,比如有核红细胞,才能通过TfR1吸收铁蛋白;因此,吸收铁蛋白复合物可能需要不止一个TfR1复合物。 3.2 TfR1表达的调节 由于TfR1是作为细胞吸收铁的受体,其表达受细胞铁状态的调控。在细胞铁缺乏状态下,TfR1表达升高,而在铁过量情况下,TfR1表达下降。快速增殖细胞和需能细胞,比如癌症细胞、破骨细胞和活化淋巴细胞,由于铁需求增加而导致TfR1高水平表达。有核红细胞需要大量铁进行血红蛋白合成,其TfR1表达水平也非常高(见表1)。 1.png TfR1的表达在转录和转录后水平都受到调节。缺氧反应元件(HRE)存在于该基因的启动子中。在缺氧或铁缺乏情况下,缺氧诱导因子(HIF-1α和HIF-2α)的表达升高,这些蛋白与TFRC启动子中的HRE结合,从而促进TFRC转录。在转录后水平,另一种复杂的机制被称为铁调节蛋白(IRP)-铁反应元件(IRE)系统,扮演着重要角色。转录后TFRC mRNA的3’-非翻译区(UTR)包含5个IRE。IRE是发夹环状结构,细胞内铁感应分子IRP1和IRP2可与其结合。在细胞内铁缺乏情况下,IRP与IRE结合以稳定TFRC mRNA并增强TfR1蛋白的表达。在铁过量情况下,IRP失去与IRE的相互作用;IRP1通过构象变化成为顺乌头酸酶,而IRP2在泛素化后降解,从而使TFRC mRNA不稳定和降解。值得注意的是,HRE存在于IRP1基因的启动子中;因此,缺氧和铁缺乏也使IRP1的表达在转录水平上得到增强,这进一步增强了TfR1的表达。因为一部分TfR1的细胞外结构域在脱落后释放到血液中,所以可溶性TfR1被广泛用作铁缺乏的生物标志物。 3.3 TfR1缺乏的动物模型 与Tf缺乏小鼠的非造血组织表现出铁超载相比,TfR1缺乏小鼠的造血和非造血组织均表现出铁缺乏。敲除小鼠的TfR1编码基因(tfrc)使其由于红细胞生成缺陷和神经系统异常而死于子宫中。肌肉中特异性缺乏TfR1的小鼠不仅显示肌肉铁缺乏,也显示脂肪组织和肝脏铁缺乏。肠上皮中tfrc基因被特异性灭活的小鼠表现出多效性影响,导致肠上皮细胞增殖和体内平衡的丧失,并诱导参与上皮-间质转化的基因表达;这些影响与铁摄取无关。心脏缺乏TfR1的小鼠较早死亡,并表现出心脏肥大、心功能差、线粒体呼吸失败和线粒体自噬无效。这种表型可以通过积极的铁疗法或通过给予NAD前体烟酰胺核糖苷来拯救。这些结果表明TfR1在细胞铁摄取和组织保护中起重要作用。 3.4 TFRC突变引起的先天性免疫缺陷 TfR1是哺乳动物发育必不可少的,Tfrc敲除小鼠显示胎死。然而,近期,发现了两个含有TFRC错义突变(Y20H)的家族。这一突变位于内在化基序并损害体外TfR1的内吞作用。具有这种突变纯合子的个体早年表现出腹泻、反复感染、低丙种球蛋白血症、周期性血小板减少症和轻度小红细胞性贫血。他们的外周淋巴细胞数量正常;但是,外周T细胞的功能严重受损,记忆B细胞数量减少,其产生抗体的能力受损,免疫球蛋白类别转换功能也受损。患者淋巴细胞和成纤维细胞的细胞膜中TfR1表达显著升高,反映了细胞铁缺乏,体外给予枸橼酸铁补充后患者淋巴细胞的受损功能恢复。存在这种突变的情况下,缺乏TfR1内在化导致有核红细胞铁缺乏。然而,目前仍不清楚如果有核红细胞无法通过TfR1吸收铁,那么具有这种突变的患者为何没有表现出严重贫血。 金属还原酶STEAP3,可与TfR1形成复合物,包含其细胞内结构域内吞作用必需的内在化基序(见图1)。因此,即使TfR1缺乏功能性内在化基序,TfR1/STEAP3复合物也可以内在化并根据需要将铁运送至细胞。由于STEAP3在有核红细胞而不是淋巴细胞中高表达,所以可以推测出具有这种突变纯合子的个体会表现出严重的免疫缺陷,但没有严重贫血。 3.5 另一种TfR1配体:铁蛋白 Tf是循环中的主要铁载体,而铁蛋白(ferritin)是细胞中的主要储铁蛋白。人胞质铁蛋白由24个H-和L-亚基以各种比率组成,形成一种笼状复合物。在胞质溶胶中,被称之为多聚胞嘧啶结合蛋白(poly-r(C)-binding protein)的伴侣蛋白,PCBP1和PCBP2,参与了从核内体到铁蛋白的铁运输。在进入铁蛋白的笼子之前,Fe2+在铁蛋白H-亚基的酶活性作用下被氧化成Fe3+。这个笼子可以安全储存多达4500个Fe3+离子。铁蛋白的表达受IRP调控,IRP与位于H-和L-亚基转录本的5’-UTR的IRE相互作用。在铁缺乏的情况下,IRP与IRE结合阻断翻译过程,预先存在的铁蛋白分子转移至溶酶体并通过一种特定的自我吞噬机制(被称之为铁自噬)降解。相反,在铁过量的情况下,IRP与IRE分离并且铁蛋白的翻译增加。 由于巨噬细胞吞噬衰老的红细胞并从中提取铁用于再循环,所以它们在系统性铁储存中起到中心作用。在铁过量的情况下,巨噬细胞合成大量铁蛋白且积极地分泌铁蛋白到循环中。因为血清铁蛋白的主要来源是巨噬细胞,而且血清铁蛋白水平反映了体内的铁储存状况,所以血清铁蛋白在临床上用作系统性铁状态的适宜标志物。 近期,Li等人证明了除了转铁蛋白,TfR1也可通过内吞作用介导细胞内铁蛋白摄取(见图1)。只有铁蛋白-H可与TfR1相互作用,铁蛋白-L不可以。铁蛋白-H均聚物(H-铁蛋白)与TfR1之间的相互作用,需要细胞表面的TfR1表达超过一定阈值水平。因而,摄取铁蛋白复合物(分子量大于470kDa)可能需要不止一个TfR1复合物。在血细胞中,有核红细胞,其TfR1表达水平非常高,可特异性吸收H-铁蛋白,而外周淋巴细胞和粒细胞则不能。H-铁蛋白通过内吞作用被吸收并转运至溶酶体进行降解。铁蛋白被这些细胞吸收后的生理学作用仍待阐明。然而,体外研究证明H-铁蛋白可抑制正常造血作用,并且通过调节树突细胞的功能来抑制免疫应答。因而,TfR1可与两种结构完全不同的铁载体蛋白即Tf和H-铁蛋白相互作用,并通过内吞作用将它们转运至细胞中。 4. TfR2 4.1 TfR2:另一种Tf受体 多年来,TfR1被认为是哺乳动物的唯一Tf受体。1999年,我们报道了另一种Tf受体,称之为TfR2,其一级结构与TfR1类似。人TfR2基因通过选择性启动子转录为至少两种异构体:全长型(α)和短型(β)。TfR2-α是一种2型膜蛋白,分子量为90-105kDa,选择性表达于肝细胞和红系前体细胞。相反,TfR2-β主要是胞质溶胶型,在各种细胞中低水平表达(表1)。TfR2-α的细胞外结构域与TfR1具有45%一致性和66%相似性。与TfR1类似,TfR2-α是一种糖蛋白并可在细胞膜中形成同源二聚体。体外实验表明TfR2-α像TfR1一样使细胞有效地吸收Tf结合铁。此外,TfR2-α以pH依赖性方式与Tf结合;TfR2-α在中性pH下以约为TfR1的1/25-1/30亲和力与全铁-Tf结合,并在酸性pH下与脱铁-Tf结合。人TfR2-α与人和牛Tf均可结合,而人TfR1具有物种特异性,无法与牛Tf结合。研究发现,表达外源性人TfR2-α的TfR缺乏中国仓鼠卵巢细胞,对铁螯合剂去铁敏具有抗性,并且与用空载体转染的细胞相比,在裸鼠中形成更大的肿瘤。 TfR1的表达随着铁超载而减少并且随着铁螯合作用而增加,但TfR2 mRNA的表达并未随着铁超载或铁螯合作用发生明显变化。TfR2启动子受转录因子刺激,比如C/EBP-α,其主要表达于髓细胞、肝细胞和脂肪细胞;GATA1,主要表达于早期红系细胞;和肝细胞核因子 4α。另外,TfR2-α蛋白被CD81下调,它与TfR2-α在脂筏中形成复合物并促使TfR2-α降解。相反,TfR2-α被全铁-Tf显著上调,它稳定了TfR2-α(见表1)。TfR2-α的这些复合物表达模式突出了一种可能性,即其主要功能不只有细胞铁摄取。 4.2 TfR2是肝脏铁感应机制的一部分 2000年,Camaschella等人报道了两个起源于西西里岛、含有TFR2基因突变(Y250X)的遗传性血色素沉着症家族。这种突变导致TfR2-α的一大部分细胞外结构域缺失。遗传性血色素沉着症是一种遗传性铁超载异常,有各种症状表现,包括皮肤色素沉着、肝功能障碍、糖尿病、甲状腺功能减退、性腺机能减退和心脏衰竭。彼时认为HFE是致病基因。人血色素沉着症蛋白(HFE),是表达于细胞膜的MHC I类分子;HFE与TfR1发生物理相互作用,降低Tf对TfR1的亲和力,并在内吞过程中与TfR1一起移动。后来证明TfR2与HFE在细胞膜中形成复合物。大多数遗传性血色素沉着症白人患者含有纯合性HFE C282Y突变;然而,Camaschella等人报道的含有TFR2基因突变的血色素沉着症家族并没有这种HFE突变。随后,很多含有各种TFR2突变的遗传性血色素沉着症家族被报道出来。但是,彼时尚不清楚HFE和TFR2突变通过何种机制导致系统性铁超载。 系统性铁稳态的中心调节蛋白是铁调素,其主要随着铁负载或炎症性刺激暴露从肝细胞中释放出来。在巨噬细胞、肝细胞和肠上皮细胞中,铁调素与哺乳动物细胞的唯一已知铁输出蛋白-膜铁转运蛋白(ferroportin)结合。膜铁转运蛋白与铁调素结合后,通过内吞作用内在化并在溶酶体中降解。在铁过量的情况下,肝窦上皮细胞分泌骨形态生成蛋白(BMP)-6。BMP6与肝细胞中的BMP受体(I和II型)和铁调素调节蛋白(BMPR/HJV)复合物相互作用,并通过SMAD蛋白的磷酸化作用开启对铁调素表达的细胞内信号传导。肝脏分泌的铁调素下调了肠上皮细胞中膜铁转运蛋白的表达,并使小肠铁吸收减少。相比之下,在铁缺乏条件下,铁调素表达减少,肠上皮细胞中的膜铁转运蛋白表达增加,铁吸收增加。因而,BMPR/HJV复合物是肝脏铁感应机制的核心,而铁调素在维持系统性铁稳态方面起到关键作用。 有些研究证明小鼠的Hfe或Tfr2敲除以及人的HFE或TFR2突变导致铁调素表达下调,铁调素异常低表达促使小肠铁吸收,导致系统性铁超载。HFE与TfR1在细胞膜发生物理相互作用,HFE与BMP I型受体ALK3相互作用以调节铁调素表达。因此,TfR2/HFE复合物连同BMPR/HJV复合物,似乎充当肝细胞中的铁感应分子并控制铁调素表达(见图2)。重要的是,这些分子中任一分子缺陷均可导致系统性铁超载。遗传性血色素沉着症目前被分为几种类型:1型由HFE突变导致;2A型由编码铁调素调节蛋白的HFE2突变导致;2B型由编码铁调素的HAMP突变导致;3型由TFR2突变导致;4型由编码膜铁转运蛋白的SLC40A1突变导致。Matriptase 2,一种由TMPRSS6编码的膜结合丝氨酸蛋白酶,通过分裂组成铁感应复合物的蛋白(包括TfR2)的细胞外结构域来调节铁调素表达。TMPRSS6的种系突变通过上调铁调素表达而导致铁剂难治性缺铁性贫血。 4.3 TfR2在红细胞生成中的作用 TfR2-α在肝细胞和红系前体细胞中均高水平表达。TfR2-α的表达高峰出现在红系细胞分化的早期,早于TfR1的表达高峰。在红系细胞中,TfR2-α与红细胞生成素(EPO)受体(EPOR)形成复合物(图2)。EPO通过支持正常红系前体细胞的存活,在红细胞生成中担当关键细胞因子。自然情况下,EPOR在红系前体细胞中高水平表达。Tfr2敲除小鼠的红系祖细胞对EPO的灵敏度下降,使循环EPO水平升高。人红系细胞中的TfR2敲减抑制EPOR运输至细胞表面。骨髓特异性Tfr2敲除小鼠随着铁剥夺(iron deprivation)表现出严重的小红细胞性贫血,但红细胞数量没有减少。虽然TfR2在红细胞生成中的具体作用尚未完全阐明,但这些发现表明TfR2可能通过与EPOR相互作用参与了红细胞数量的调节,特别是在铁限制性条件下(见图2)。 技术导航-转铁蛋白-图2.jpg 图2. 转铁蛋白受体2(TfR2)的细胞类型依赖性功能。 (左)TfR2-α是肝细胞中铁感应机制的组成部分。TfR2-α与一种血色素沉着症蛋白HFE在脂筏中发生物理相互作用。在铁超载情况下,肝窦上皮细胞分泌BMP-6,其与骨形态生成蛋白(BMP)I及II型受体和铁调素调节蛋白复合物(BMPR/HJV)相互作用,通过SMAD蛋白的磷酸化作用,开启对系统性铁稳态的中心调节蛋白-铁调素表达的细胞内信号传导。磷酸化SMAD蛋白复合物转移至核,并与铁调素(HAMP)基因启动子中的BMP-反应元件结合。TfR2-α与铁饱和的全铁-Tf结合后变稳定,通过BMP/SMAD途径增强铁调素表达。CD81与TfR2-α相互作用,促使TfR2-α降解并增强铁调素表达。(右)TfR2是红系前体细胞中红细胞生成素(EPO)受体(EPOR)的搭档。TfR2-α与EPOR形成复合物,促使复合物运输至细胞表面。该复合物参与了红细胞生成并调节红细胞数量,尤其在铁限制性条件下。 4.4 TfR2促使铁运输至线粒体 被TfR1或TfR2介导的内吞作用吸收后,一大部分Tf结合铁前往铁需求量最高的细胞器-线粒体。线粒体是细胞呼吸的中心以及氧化磷酸化的主要部位。参与此过程的细胞色素C和酶,比如NADH-辅酶Q氧化还原酶(复合物I)和琥珀酸盐-Q氧化还原酶(复合物II),均使用铁作为主要辅因子。此外,线粒体利用铁来合成血红素和铁硫簇。在有核红细胞中,血红蛋白合成需要大量铁,丰富的铁应从核内体有效地转运至线粒体。然而,由DMT1从核内体输出的Fe2+离子在转运至线粒体的过程中可产生有毒性的ROS;因此,必须有一个安全而有效的途径将铁转运至线粒体。TfR2-α在有核红细胞的这个转运过程中起到一定作用。与全铁-Tf在细胞表面结合后,全铁-Tf/TfR2-α复合物通过内吞作用进入细胞,含有该复合物的核内体将被转移至溶酶体。线粒体靶向基序存在于TfR2-α的细胞内结构域;通过此基序,含有TfR2-α的溶酶体移向线粒体并与之发生物理相互作用来促进铁转移(见图3)。 技术导航-转铁蛋白-图3.jpg 图3. 转铁蛋白受体2(TfR2)介导铁直接从溶酶体转运至线粒体的假设图示。功能性线粒体靶向基序存在于 TfR2-α的细胞内结构域。在细胞表面与全铁-Tf结合后,全铁-Tf/ TfR2复合物通过内吞作用进入细胞,含有该复合物的核内体被转移至溶酶体。在TfR2-α的线粒体靶向基序作用下,溶酶体移向线粒体并与之发生物理相互作用来促进铁转移。该机制在有核红细胞和神经元中得到证实。粘脂蛋白是溶酶体的铁输出蛋白,线粒体转铁蛋白是线粒体内膜中的铁转运蛋白。 据Mastroberardino等人报道,TfR2-α在神经元中也有相似功能。在大脑中,铁累积于黑质中,且TfR2在这一区域的多巴胺能神经元中高水平表达。研究发现转染了TfR2-α表达质粒的人细胞会吸收细胞外Tf结合铁,并将这种铁转运至线粒体。在多巴胺能神经元中Tf 和TfR2在某种程度上均与线粒体共存。另外,在由复合物I-抑制剂鱼藤酮诱导的帕金森病动物模型及帕金森病患者的大脑组织中,观察到多巴胺能神经元中的铁和氧化Tf显著增加。因而,Tf/TfR2系统在铁转运至多巴胺能神经元的线粒体的过程中似乎起到重要作用,并且由于该系统导致过多铁累积于线粒体中可能促使了帕金森病的形成。 4.5 TfR2-β的功能 与几乎仅表达于肝细胞和红系前体细胞的TfR2-α相比,TfR2-β在各种组织和细胞中均低水平表达,包括脾脏、心脏、大脑、肝脏、前列腺、髓细胞、T淋巴细胞和各种癌症细胞,比如成神经细胞瘤和结肠癌细胞。TfR2-β缺乏信号肽和跨膜结构域,因此被认为是一种胞质蛋白。TfR2-β蛋白的功能是未知的;但是,研究表明这种蛋白可能在巨噬细胞的铁稳态中起到重要作用。建立了一个特异性缺乏 Tfr2β的小鼠模型;这些小鼠的TSAT、肝脏铁浓度和铁调素水平均正常,但在早年出现短暂性贫血并在成年后其脾脏中有严重过量的铁累积。这些动物脾脏中的细胞铁输出蛋白-膜铁转运蛋白水平显著下降。巨噬细胞特异性 Tfr2缺失也导致外周巨噬细胞中的膜铁转运蛋白表达下降,但不影响系统性铁稳态。此外,Tfr2β缺乏使小鼠脾脏中的不成熟红细胞生成增加。总的来说,这些研究表明TfR2-β可通过抑制编码膜铁转运蛋白的SLC40A1基因的转录,针对性地控制脾脏的铁外排(iron efflux),并在控制巨噬细胞的铁稳态上起到关键作用。另外,TfR2-β在局部缺血/再灌注野生型小鼠的心脏中表达升高。Tfr2β缺失小鼠的心脏中铁蛋白-H、血红素加氧酶1和HIF-2-α表达升高使其免受这类损伤。TfR2-β靶向疗法可保护组织,可能是这类损伤的有效治疗方法。 5. Tf/TfR1系统的治疗用途 5.1 人脱铁-Tf的治疗用途 无转铁蛋白血症是一种非常罕见的遗传性疾病,表现为早发性及严重小红细胞性贫血和铁超载;这些表型表明了Tf在红细胞生成和铁稳态中的重要性。在无转铁蛋白血症(Atransferrinemia)或低转铁蛋白血症(hypotransferrinemia)患者以及此类疾病小鼠模型中观察到的铁超载,至少在一定程度上是由于肝脏中铁调素产生下调导致的,在肝脏中TfR2充当血浆全铁-Tf的感应分子。人Tf和含有Tf的新鲜冷冻血浆被成功用于改善这类疾病患者的贫血状况。在各种病理条件下比如无转铁蛋白血症、血色素沉着症、脊髓发育不良和组织缺血,以及某些治疗干预之后比如化疗和造血干细胞移植,循环中的NTBI(可导致器官损伤)均会升高。由于脱铁-Tf是可清除游离铁和潜在阻止宿主形成毒性ROS的内源性铁螯合剂,在这些情况下给予脱铁-Tf可具有组织保护作用。的确,肾缺血小鼠模型发现,腹膜内注射脱铁-Tf可减少循环中氧化还原活性铁的数量、阻止肾超氧化物形成和抑制肾功能下降。由氧过多诱导的早产高氧肺损伤兔子模型表明,气管内或静脉内注射脱铁-Tf可增加表面活性剂活性并降低肺泡-毛细血管渗透性。此外,局部给予Tf可以改善由缺血引起的脱髓鞘动物模型的症状,以及保护视网膜退化大鼠模型的视网膜功能,表明在这些疾病的发生过程中产生了铁诱导的ROS。脱铁-Tf还可以改善β-地中海贫血动物模型的贫血状况。地中海贫血是由球蛋白基因突变导致的一组遗传性贫血疾病,表现为骨髓中红细胞生成无效以及由于铁调素的不当低表达引起的铁超载。给予脱铁-Tf可增强铁调素表达,改善组织铁超载情况,提高有核红细胞去核效力,减小脾脏大小,降低平均红细胞容积,和改善贫血状况。虽然并不完全了解脱铁-Tf如何改善这些β-地中海贫血模型的贫血状况,但已证明脱铁-Tf可下调有核红细胞中的 TfR1表达,潜在减少可用于血红素合成的铁,限制α-球蛋白合成,纠正α和β球蛋白合成之间的平衡,减少α-球蛋白链沉积,以及提高红细胞生成。因而,给予脱铁-Tf可用于治疗β-地中海贫血。 5.2 TfR1靶向治疗 由于铁需求增加TfR1在癌症细胞中高水平表达,因此TfR1是癌症的潜在治疗靶标。体外实验和小鼠异种移植物模型表明,抗人TfR1单克隆抗体,通过干扰TfR1与Tf之间的相互作用,抑制成人T细胞白血病/淋巴瘤细胞的细胞生长。另外,抗TfR1抗体(ch128.1)对很多骨髓瘤异种移植物小鼠模型均有抗肿瘤效果。TfR1也可以作为传染性疾病的靶标,因为各种病原体均利用TfR1进入细胞。例如,某些新世界沙粒病毒(人类感染后产生威胁生命的出血热),利用TfR1进入宿主细胞。丙肝病毒也利用TfR1进入人肝细胞,TfR1敲除可抑制人肝细胞感染丙肝病毒。间日疟原虫是最广泛分布的人疟原虫,这种疟原虫进入网织红细胞需要TfR1。因而,各种病原体均利用TfR1,这些病原体与TfR1之间的相互作用成为治疗这些严重疾病的潜在靶标。 5.3 利用TfR1配体的给药系统 鉴于TfR1可介导细胞外分子的内吞作用,所以正通过靶定该受体来开发有效的给药系统。抗TfR1抗体、Tf结合物以及合成H-铁蛋白可经由TfR1介导的机制而被内吞。例如,已开发出了以TfR1为靶标的可变区单链片段,其具有灵活的药物偶联连接子;也正进行临床试验来评价这些结构对于癌症患者的安全性和功效,结果证明应用前景广阔。各种Tf结合物被用于药物和基因递送系统;其中,阿霉素与Tf结合对各种癌症细胞具有选择毒性。该系统可能对大脑肿瘤有效,因为TfR1表达于血脑屏障且通常在大脑肿瘤中过表达。由于Tf和H-铁蛋白可被 TfR1内吞,合成H-铁蛋白纳米笼也被用于递送各种药物,包括顺铂、阿霉素和奥拉帕尼,以治疗乳腺癌、结肠癌、胰腺癌和肺癌等癌症。由于细胞H-铁蛋白摄取需要TfR1表达超过一定阈值水平,所以H-铁蛋白纳米笼仅将药物递送至TfR1表达水平非常高的选定细胞,比如癌症细胞和有核红细胞。然而,需要谨慎使用这类 TfR1靶向给药系统,因为有核红细胞可能是这些治疗的主要靶标。 6. 结论 Tf/TfR系统在铁转运及其他依赖于组织和细胞类型的多效性功能中起到复杂而又关键的作用。然而,需要进一步研究来明确新鉴别的TfR1-配体-铁蛋白的生理作用;且TfR2-α和TfR2-β在红细胞生成和巨噬细胞中的功能仍需阐明。临床证明,脱铁-Tf能够防止氧化应激产生的组织损伤,因此可用于治疗β-地中海贫血患者。 本文链接:http://www.ddm360.com/article/detail/869 版权所有,转载时请以链接的形式注明来源! 相关主题: 炎症性肠病
转铁蛋白和转铁蛋白受体更新 - 临床实验室
http://ivdchina.com.cn/article/detail/869
Anemia of Inflammation with An Emphasis on Chronic Kidney ...
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6835368
Oct 11, 2019 ・ High levels of hepcidin recorded in CKD patients act to impair absorption and mobilization of iron. Furthermore, the chronic inflammation which manifests in CKD patients results in the production of pro-inflammatory cytokines including interleukin-6 (IL-6), which has been shown to stimulate the synthesis of hepcidin.
Hepcidin: inflammation's iron curtain | Rheumatology ...
https://academic.oup.com/rheumatology/article/43/11/1323/2389966
Jul 27, 2004 ・ Hepcidin mRNA moves with the body's iron levels, increasing as they increase and decreasing as they decrease . More pertinently, hepcidin rises with infection or inflammation and falls with hypoxia or anaemia . The anaemia of chronic disease has long confounded physicians.
Cited by: 20
Publish Year: 2004
Author: H. McGrath, P. G. Rigby
J Biol Chem. 2001 Mar 16;276(11):7806-10. Epub 2000 Dec 11.
Hepcidin, a urinary antimicrobial peptide synthesized in the liver.
Park CH1, Valore EV, Waring AJ, Ganz T.
Abstract
Cysteine-rich antimicrobial peptides are abundant in animal and plant tissues involved in host defense. In insects, most are synthesized in the fat body, an organ analogous to the liver of vertebrates. From human urine, we characterized a cysteine-rich peptide with three forms differing by amino-terminal truncation, and we named it hepcidin (Hepc) because of its origin in the liver and its antimicrobial properties. Two predominant forms, Hepc20 and Hepc25, contained 20 and 25 amino acid residues with all 8 cysteines connected by intramolecular disulfide bonds. Reverse translation and search of the data bases found homologous liver cDNAs in species from fish to human and a corresponding human genomic sequence on human chromosome 19. The full cDNA by 5' rapid amplification of cDNA ends was 0.4 kilobase pair, in agreement with hepcidin mRNA size on Northern blots. The liver was the predominant site of mRNA expression. The encoded prepropeptide contains 84 amino acids, but only the 20-25-amino acid processed forms were found in urine. Hepcidins exhibited antifungal activity against Candida albicans, Aspergillus fumigatus, and Aspergillus niger and antibacterial activity against Escherichia coli, Staphylococcus aureus, Staphylococcus epidermidis, and group B Streptococcus. Hepcidin may be a vertebrate counterpart of cysteine-rich antimicrobial peptides produced in the fat body of insects.
Hepcidin, a urinary antimicrobial peptide synthesized in the liver. - PubMed - NCBI
https://www.ncbi.nlm.nih.gov/pubmed/11113131
Low hepcidin in liver fibrosis and cirrhosis; a tale of progressive disorder and a case for a new biochemical marker
Driton Vela
Molecular Medicine volume 24, Article number: 5 (2018) Cite this article
Abstract
Liver fibrosis is a precursor of liver cirrhosis, which is associated with increased mortality. Though liver biopsy remains the gold standard for the diagnosis of fibrosis, noninvasive biochemical methods are cost-effective, practical and are not linked with major risks of complications. In this respect, serum hepcidin, has emerged as a new marker of fibrosis and cirrhosis. In this review the discussion uncovers molecular links between hepcidin disturbance and liver fibrosis/cirrhosis. The discussion also expands on clinical studies that suggest that hepcidin can potentially be used as a biochemical parameter of fibrosis/cirrhosis and target of therapeutic strategies to treat liver diseases. The debatable issues such as the complicated nature of hepcidin disturbance in non-alcoholic liver disease, serum levels of hepcidin in acute hepatitis C virus infection, cause of hepcidin disturbance in autoimmune hepatitis and hepatic insulin resistance are discussed, with potential solutions unveiled in order to be studied by future research.
Hepcidin expression and mode of action. BMP6, bone morphogenetic protein 6; BMPR, BMP receptor; ERFE, erythroferrone; Fe, iron; FPN, ferroportin; HAMP, hepcidin antimicrobial peptide; HFE, hemochromatosis protein; IL-6, interleukin 6; IL-6R, IL-6 receptor; JAK2, janus kinase 2; SMAD proteins, s-mothers against decapentaplegic proteins; STAT3, signal transducer and activator of transcription 3; TFR2, transferrin receptor 2. Hepcidin expression is primary regulated by iron-status, inflammation and erythropoietic drive. Iron-status induces hepcidin expression by through BMPR ligands such as BMP6. BMPR activates SMAD pathway which increases hepcidin expression through HAMP transcription. Inflammation induces hepcidin expression through cytokines like IL-6 which activates JAK2/STAT3 pathway in heptocytes. This pathway increases hepcidin expression. Erythropoiesis suppresses hepcidin expression probably through erythroferrone produced by erythrocyte precursors. Once released by hepatocytes in plasma, hepcidin reaches target cells like enterocytes and macrophages, where hepcidin induces FPN degradation. This action reduces iron efflux from cells, because FPN is the major exporter of iron out of cells
Background
Hepcidin is an ubiquitous antimicrobial peptide found in different species, including humans (Segat et al. 2008). Though initially discovered for its antimicrobial properties, in 2001 scientists found that, in fact, hepcidin is the major regulator of iron metabolism (Ganz 2011; Nicolas et al. 2001). Since then we have learned that hepcidin is mostly produced by hepatocytes in response to iron-load in cells. Whenever this load increases, hepcidin expression goes up in hepatocytes, which results in increased serum hepcidin levels (Nicolas et al. 2001). Hepcidin main mode of action is realized through its binding with ferroportin (FPN) in target cells, like enterocytes, macrophages, hepatocytes (Fig. 1). FPN is the major protein channel that regulates iron export from cells. Its complex with hepcidin induces FPN degradation inside cells (Nemeth et al. 2004). This means that high levels of hepcidin reduce the levels of iron in serum. In this way hepcidin protects us from iron-overload. This role of hepcidin is important since there is no known excretory pathway for body iron. Keeping iron in check through hepcidin is vital for our cells, because high levels of iron saturate the capacity of the proteins to keep iron in bound form (Loréal et al. 2000). Excess iron can cause oxidative damage, but also can be used by microbes to maintain their survival (Puntarulo 2005; Skaar 2010). This is the reason why infection with bacteria can cause increased mortality in patients with high iron-overload (Skaar 2010). On the other hand, low levels of iron cause anemia, but paradoxically whenever the immune system of patients is damaged in chronic diseases (autoimmune and inflammatory) our organism sets “priorities” by choosing anemia as the “norm” in these situations, which seems to protect us from potentially dangerous infections (Zarychanski and Houston 2008; Roy 2010). In this everlasting fight of our cells to use iron efficiently and as discreetly as possible, maintaining hepcidin balance is crucial to prevent disease and organ damage.
Hepcidin (in)actions in normal liver and liver fibrosis. Fe, iron; FPN, ferroportin. In normal liver, hepcidin produced by heptocytes controls iron levels in plasma by preventing excessive iron absorption from enterocytes. In this way hepcidin protects liver form iron-load. But hepcidin can protect liver by inactivating hepatic stellate cells as well. In liver fibrosis low hepcidin causes high iron-load and oxidative stress. Oxidative stress and lack of hepcidin-induced supression of hepatic stellate cells induces their activation, which results in deposition of scar tissue and liver fibrosis
Low hepcidin in alcoholic liver disease (ALD)
Alcohol is an already established inducer of hepatocyte damage, which can progress to overt liver fibrosis. Suspected mechanisms of alcohol-induced liver fibrosis include increased levels of lipopolysaccharide (LPS), activation of HSCs and inhibition of antifibrotic actions (Gao and Bataller 2011).
Alcohol is also linked with disturbances in levels of hepcidin. It is interesting to notice that, in alcoholic patients, low levels of hepcidin are observed even with preserved liver function (Costa-Matos et al. 2012). This would suggest that alcohol is a primary cause of low levels of hepcidin, and not a consequence of alcohol-induced liver damage. The rationale behind this observation is the direct effect of alcohol on hepcidin expression. Alcohol can inhibit hepcidin expression through its suppressive effects on CCAAT-enhancer-binding protein (C/EBP) in hepatocytes, at the same time counteracting iron-induced activity of this transcription factor, thus rendering iron-induced hepcidin expression ineffective (Harrison-Findik et al. 2007) (Fig. 3). In addition, the upregulation of divalent metal transporter 1 (DMT1) and FPN in enterocytes increases serum iron levels and cellular iron-load, which is linked with liver fibrosis (Harrison-Findik et al. 2007). This effect of alcohol can be reversed with treatment by antioxidants, which is not surprising since alcohol induces oxidative stress (Harrison-Findik et al. 2006). This is the reason why progression rate of fibrosis is twice as high in steatotic drinkers compared to steatotic nondrinkers (Serfaty et al. 2002). Another mechanism of hepcidin suppression by alcohol includes suppression through toll-like receptor 4 (TLR4) pathway. TLR4 is a transmembrane protein involved in innate immune responses. In mice with defective TLR4 receptor alcohol cannot suppress hepcidin expression (Zmijewski et al. 2014). It is interesting to notice that TLR4 deficiency protects from liver fibrosis, making it an interesting candidate to be studied in the context of alcohol-induced hepcidin down-regulation (Weber et al. 2016; Seki et al. 2007). The mediator cell of TLR4 signaling remains to be found, and it seems that Kupffer cells are not involved in alcohol-induced hepcidin expression (Harrison-Findik et al. 2008). Hepatocytes are unlikely candidates as well, since their expression of TLRs is low, while their reaction to TLR ligands is weak. A plausible candidate seems to be HSCs, since they express different TLRs and react in response to their actions (Yang and Seki 2012).
Fig. 3
figure3
Mechanisms behind low levels of hepcidin in liver disease. BMPR, bone morphogenetic protein receptor; C/EBP alpha, CCAAT enhancer binding protein alpha; HAMP, hepcidin antimicrobial peptide; HCV, hepatitis C virus; HFE, hemochromatosis protein; HH, hemochromatosis; HJV, hemojuvelin; IL-6, interleukin 6; InR, insulin receptor; STAT3, signal transducer and activator of transcription 3; TFR2, transferrin receptor 2; TLR4, toll-like receptor 4. Different pathogenic factors are responsible for low values of hepcidin. In HH mechanisms behind low levels of hepcidin are already known and they include defective signalling though HJV, HFE, TFR2 or through direct inhibition of HAMP gene. Alcohol inhibits hepcidin expression through its actions on C/EBP, but also indirectly through TLR4 pathway. It is believed that TLR4 mediates this action of alcohol through nonparenchymal liver cells, but the paracrine signal responsible for this effect is unknown. HCV also suppresses hepcidin expression through oxidative stress, which inhibits C/EBP and STAT3 actions on HAMP. Cholestasis, on the other hand, suppresses hepcidin expression by inhibiting IL-6/STAT3 pathway. In AILD mechanisms behind low levels of hepcidin are unknown. In hepatic insulin resistance defective insulin signaling is linked with defective hepcidin expression, partially through STAT3 pathway
Full size image
Alcohol might disrupt canonical hepcidin pathways such as BMPR/SMAD pathway, but also can suppress hepcidin via hypoxic signals, though the importance of these alcohol-induced actions on hepcidin expression remain to be confirmed (Gerjevic et al. 2012; Heritage et al. 2009).
It seems that alcohol consumption in the setting of iron-overload can serve as a strong inducer of liver fibrosis. In HH patients, alcohol consumption of more than 60 g/day increases the risk of cirrhosis by 9 fold (Fletcher and Powell 2003). This increase in risk of progressive liver damage in alcoholic HH patients is in-line with the so-called “multiple hit” scenario, where two or more pathophysiological factors induce hepatocyte damage in a complementary manner, which eventually leads to liver fibrosis (Takaki et al. 2013). There are other similar examples of “multiple hit” factors adding to the liver damage; in mice fed with long term high cholesterol diet and alcohol, liver shows signs of early fibrosis, compared to individual effects of high cholesterol and alcohol (Krishnasamy et al. 2016). The risk of liver cirrhosis is dramatically increased in patients with hepatitis C virus (HCV) infection, when HCV infection is accompanied with heavy alcohol consumption (Harris et al. 2001).
Recently, the effect of alcohol in hepcidin has been suggested to be more complex than previously thought (Harrison-Findik and Lu 2015). Also, alcohol might cause a reduction in hepcidin values through ubiquitous proteins involved in liver regeneration, but the importance of this finding remains to be evaluated (Kumar et al. 2016).
As we can see the evidence for alcohol-induced low levels of hepcidin is considerable and some of the mechanisms behind these disturbances in hepcidin levels have already been elucidated, but the full picture remains to be solved.Low hepcidin in HBV- and HCV-induced liver disease
Infection with hepatitis B virus (HBV) and HCV is a known causative factor in liver fibrosis (Ohkoshi et al. 2016; Bataller et al. 2004).
The mechanisms behind HCV-induced liver fibrosis include induction of reactive oxygen species (ROS) which impairs C/EBP and STAT3 binding to hepcidin promoter (Miura et al. 2008). Low levels of hepcidin in HCV infection deteriorate liver function by serving as a strong factor in inducing liver fibrosis (Angelucci et al. 2002; Chapoutot et al. 2000; Horl and Schmidt 2014). It has been suggested that by lowering hepcidin, HCV protects itself from antiviral innate immune responses, since hepcidin can inhibit HCV replication (Liu et al. 2012). In chronic HCV infection, patients mRNA of liver hepcidin is correlated with iron status and not with virus load or fibrosis stage (Aoki et al. 2005). This would suggest that hepcidin regulation is preserved in response to iron status. Low hepcidin in HCV is more clearly linked with end-stage liver disease than with early fibrosis (Nagashima et al. 2006). Data suggest that though hepcidin expression in chronic HCV infection is induced by iron status, this upregulation is not sufficient, which is consistent with the finding of the impaired binding of hepcidin upregulators to HAMP seen in HCV infection due to increased oxidative stress (Miura et al. 2008; Fujita et al. 2007). What is more, HCV is known to induce more pronounced oxidative stress than other viruses that cause hepatitis (Valgimigli et al. 2000; Farinati et al. 1995). Consolidating this argument is the success seen during treatment of HCV infection with antiviral therapy which restores hepcidin levels by increasing the expression of STAT3 (Ryan et al. 2012), while keeping in mind that loss of STAT3 is linked with increased susceptibility to oxidative stress (Barry et al. 2009). Iron reduction that ensues after antiviral therapy is associated with reduced viral load. It has to be noted that phlebotomy reduces iron-load and markers of oxidative stress which is why long-term treatment with phlebotomy improves liver histology, but also reduces the risk of progression to hepatocellular carcinoma (Kato et al. 2001; Yano et al. 2002; Kato et al. 2007). But, according to one study, phlebotomy does not correct the inappropriate hepcidin response to ferritin load (Sugimoto et al. 2009). This might be explained by differences in short-term vs long-term treatment success, but also by timing of the interventions, which means that phlebotomy in advanced liver disease induced by HCV might not recuperate hepcidin expression, though this hypothesis need to be evaluated by further studies.
Hepcidin expression during HCV infection is complicated by the effect of genetic factors. These factors have been shown to predispose patients with HCV to a different treatment potential of anti-HCV drugs (Wróblewska et al. 2017).
According to a paper from Foka et al., HCV in acute setting upregulates hepcidin expression to increase iron availability for viral replication, while in chronic setting hepcidin levels go down, but do get increased in response to viral load (Foka et al. 2016). But, this and other in-vitro studies are in contradiction with other studies where hepcidin is clearly low in HCV replicon and infected cell lines (Miura et al. 2008; Liu et al. 2012; Bartolomei et al. 2011). These contradictions might have occurred because of differences in models of study. Clinical data suggest that hepcidin expression is unchanged or goes down in acute HCV infection during peak viremia, but this observation should be validated by larger studies (Armitage et al. 2014). Similarly, Foka et al. results from their clinical study confirm that hepcidin levels go down in chronic HCV infection, while results from patients with acute HCV infection showed increased levels of hepcidin, which was also observed in chronic HCV patients with high viremic load. Acutely infected HCV patients were all males (compared to other groups) which could affect the reliability of hepcidin results. Hepcidin levels show gender differences and they should be taken into account to avoid false results (Galesloot et al. 2011). At the same time, the group of patients with acute HCV infection showed nearly double the levels of ferritin compared to other groups, which could mean that increased levels of hepcidin in this group was a result of reactive response to increased iron-load. Unfortunately, this study did not provide detailed correlation reports between the examined variables. Still, this would not explain the differences seen between chronic HCV patients with low and high viral load. On closer look average hepcidin values between these groups did not show great differences compared to more prominent increased levels in acute HCV infection. Also, we have to be careful when examining differences in hepcidin levels in such a small group of patients because small differences in levels of hepcidin might be e result of chance and could reflect variations in normal range values.
In chronic HBV infection levels of hepcidin also change; they rise in early stages of the disease, only to be reduced in the cirrhotic stage of the disease, reflecting the inability of hepatocytes to control hepcidin levels caused by destruction of liver architecture during fibrosis (Wang et al. 2016; Lin et al. 2013). Differences in pathophysiological mechanisms induced by HBV and HCV explain why hepcidin levels have a specific mode of fluctuation in these infections; they include differences in the level of oxidative stress, co-infection with hepatitis D, level of viral load, presence of inflammation (Aoki et al. 2005; Fujita et al. 2008; Sebastiani et al. 2012; Wang et al. 2013). On the other hand, HBV mode of action in the liver is to evade innate immune recognition, while HCV counteracts the already activated immune response. Therefore, low hepcidin expression could be a defensive strategy of HCV through which HCV counteracts hepcidin role in suppressing HCV replication (Liu et al. 2012; Wieland and Chisari 2005).
Low hepcidin in autoimmune liver disease (AILD)
Levels of hepcidin are low in newly diagnosed patients with liver autoimmune disease (Lyberopoulou et al. 2015; Tan et al. 2012). The reason behind this observation remains a mystery, but it is pertinent to speculate that the immune system disturbances that cause the liver disease are behind low levels of hepcidin.
In mouse models of autoimmune diabetes, the autoimmune process can be dampened by beta-cells of the pancreas by inducing cathelin-related antimicrobial peptide (CRAMP) expression, which is an antimicrobial peptide “cousin” to hepcidin (Sun et al. 2015; Kościuczuk et al. 2012). This suppression of autoimmunity by beta-cells of the pancreas is mediated through gut microbiota signaling, which is an often overlooked factor in hepcidin expression (Shanmugam et al. 2014). Similarly, it might be that the autoimmune process in the liver could disrupt hepcidin expression due to dysregulation of immune response. This hypothesis would mean that hepcidin has a role in immune responses, similar to other antimicrobial peptides, like the aforementioned CRAMP. Other circumstantial evidence might help us understand disturbances of hepcidin levels in AILD; IL-22 has been shown to induce hepcidin expression during its control of early immune response (Armitage et al. 2011), while disruption of this cytokine in T-cell mediated hepatitis causes progressive damage in the liver (Pan et al. 2014). In conclusion, data from studies on the role of autoimmune process in AILD in lowering hepcidin expression are sparse, and remain to be studied by future research.Hepcidin role in fibrosis goes beyond control of iron-load?
Recent studies suggest that hepcidin has additional important protective features in liver fibrosis. Hepcidin can serve as a paracrine signal from hepatocytes to suppress hepatic stellate cell (HSC) activation. HSCs activation and subsequent release of profibrotic cytokines is one of the main features of liver fibrosis. By restoring hepcidin levels we can curb the process of HSC activation and subsequent liver fibrosis (Han et al. 2016). Similarly, BMP6 as one of the main inducers of hepcidin expression has been shown to have a protective role in liver fibrosis by inhibiting hepatic stellate cells activation (Arndt et al. 2015). More studies are needed to explain this new role of hepcidin in liver protection during fibrosis, but the idea is intriguing, and it might expand the importance of hepcidin in liver fibrosis.
Low hepcidin in liver fibrosis and cirrhosis; a tale of progressive disorder and a case for a new biochemical marker | Molecular Medicine | Full Text
https://molmed.biomedcentral.com/articles/10.1186/s10020-018-0008-7
Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice
Bone morphogenetic protein (BMP)6 signaling in hepatocytes is a central transcriptional regulator of the iron hormone hepcidin to control systemic iron balance. How iron levels are sensed to regulate hepcidin production is not known, but local induction of liver BMP6 expression by iron is proposed to have a critical role. To identify the cellular source of BMP6 responsible for hepcidin and iron homeostasis regulation, we generated mice with tissue specific ablation of Bmp6 in different liver cell populations and evaluated their iron phenotype. Efficiency and specificity of Cre-mediated recombination was assessed using Cre reporter mice, PCR of genomic DNA and quantitation of Bmp6 mRNA expression from isolated liver cell populations. Localization of the BMP co-receptor hemojuvelin was visualized by immunofluorescence microscopy. Analysis of the Bmp6 conditional knockout mice revealed that liver endothelial cells expressed Bmp6, whereas resident liver macrophages (Kupffer cells) and hepatocytes did not. Loss of Bmp6 in endothelial cells recapitulated the hemochromatosis phenotype of global Bmp6 knockout mice, whereas hepatocyte and macrophage Bmp6 conditional knockout mice exhibited no iron phenotype. Hemojuvelin was localized on the hepatocyte sinusoidal membrane immediately adjacent to Bmp6-producing sinusoidal endothelial cells. Together, these data demonstrate that endothelial cells are the predominant source of BMP6 in the liver, and support a model where endothelial cell BMP6 has paracrine actions on hepatocyte hemojuvelin to regulate hepcidin transcription and maintain systemic iron homeostasis.
https://www.researchgate.net/publication/310589162_Endothelial_cells_produce_bone...
Liver sinusoidal endothelial cells produce bone morphogenetic protein 6 (BMP6), which acts on the hepatocyte coreceptor hemojuvelin (HJV) to regulate hepcidin production in a paracrine fashion.